User login
Black Dots on the Scalp of a Child
Black Dots on the Scalp of a Child
THE DIAGNOSIS: Terra Firma-Forme Dermatosis
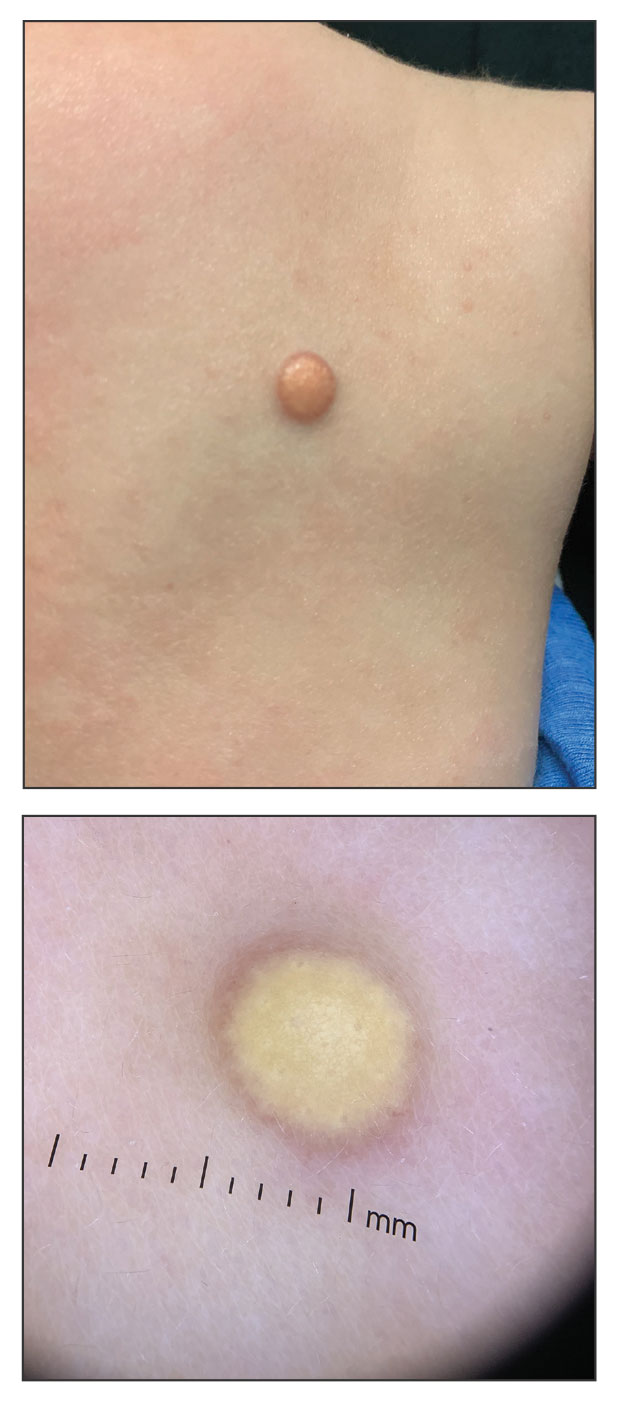
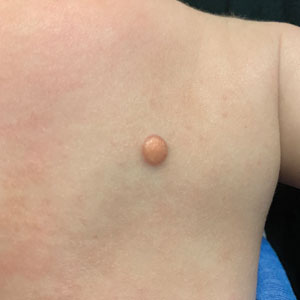
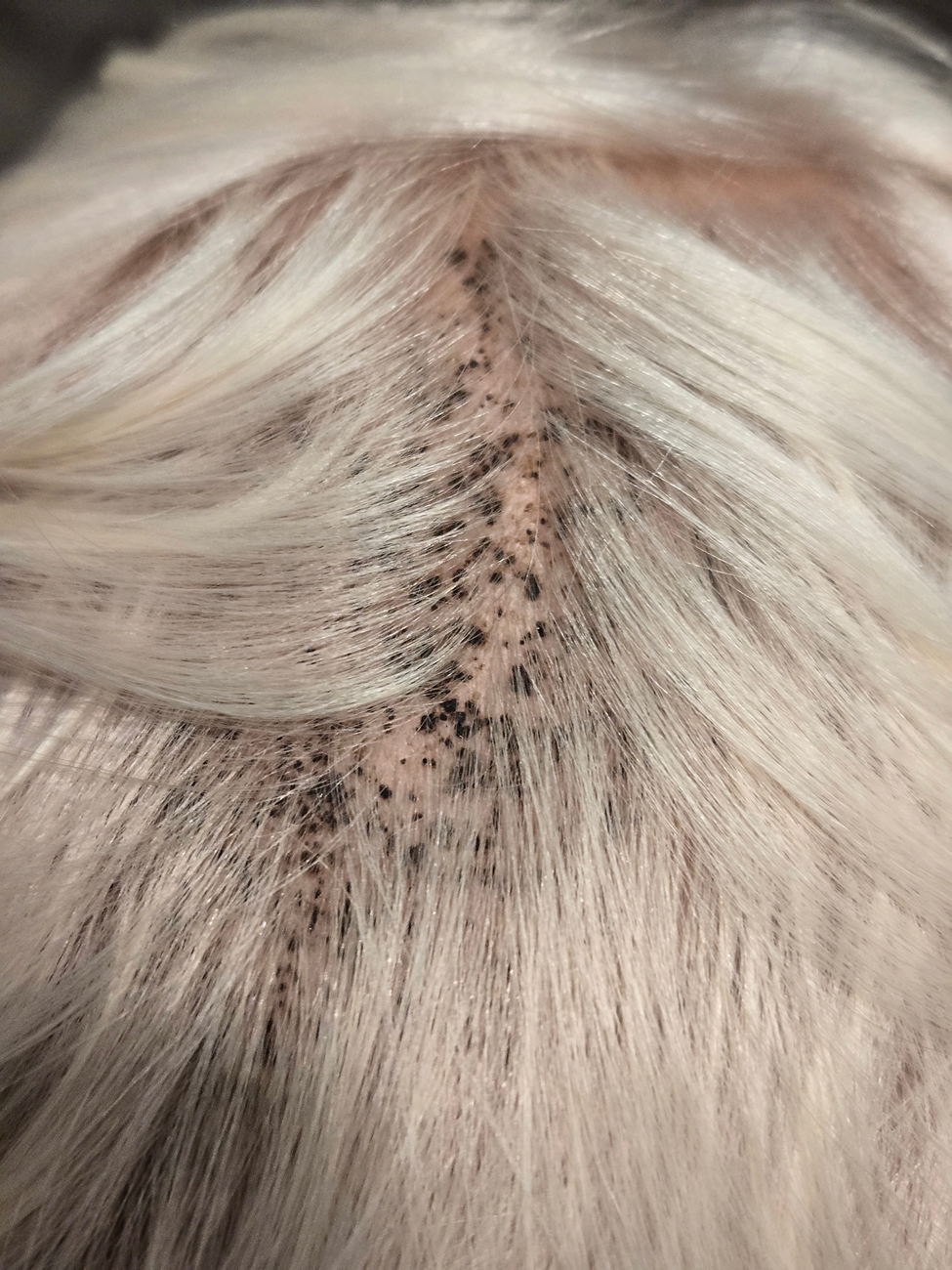
During clinical examination, a 70% alcohol swab was utilized to gently rub several of the lesions, which were successfully removed. This confirmed a diagnosis of terra firma-forme dermatosis (TFFD)(also known as Duncan’s dirty dermatosis). The patient’s mother was counseled about the diagnosis and was instructed on how to use alcohol pads to remove the remaining lesions. Three days later, after several treatment sessions at home, the mother reported complete resolution of the lesions with no residual pigmentary changes, ulceration, or scarring (Figures 1 and 2).
Terra firma-forme dermatosis was first described in 1987 in a 12-year-old girl with hyperpigmented plaques on the neck that cleared when rubbing alcohol was applied before biopsy.1,2 The term terra firma is Latin for “firm land” (or essentially “dirt”) in reference to what often is described as a characteristically “dirty” clinical appearance.2 Terra firmaforme dermatosis can manifest anywhere on the body but shows a predilection for the neck, arms and legs, axillae, inguinal region, and umbilicus.3 Lesions typically are described as asymptomatic, smooth, well-circumscribed, reticular papules or patches that are brown or black. Terra firma-forme dermatosis also may demonstrate secondary features such as hyperkeratotic, scaly, velvety, or verrucous plaques and nodules.3
The etiology of this condition is theorized to be a result of abnormal or delayed keratinization and prolonged keratinocyte adhesion.3,4 There are limited epidemiologic data, but TFFD has shown a predominance in children younger than 18 years (average age of onset, 10 years) with no known predilection for sex or race and no recognized pattern of inheritance.3-5
Histopathology typically demonstrates epidermal atrophy, hyperkeratosis, and often a component of trapping and compaction of melanin, sebum, microorganisms, and environmental debris.5
Management of TFFD is straightforward and generally consists of rubbing with 70% isopropyl alcohol to remove the lesions. For more adherent lesions or for extensive involvement, other keratolytics such as salicylic acid or alpha-hydroxy acids may be used.5 For TFFD manifesting in infants and young children, widespread involvement, or lesions involving the face or genitals, a urea-based keratolytic with or without a topical anti-inflammatory is suggested.5 Other treatment options include other alpha-hydroxy acids, topical retinoids, and nonpolar solvents such as acetone or CO2 laser for recalcitrant cases.4,5 Fortunately, most TFFD lesions respond well to conservative therapies, with recurrence reported only in 6.3% (5/79) of patients in one study.3
Dermatosis neglecta is clinically similar to TFFD and often is considered on the same spectrum of disease6; however, this entity is associated with decreased bathing or limited hygiene, which could be related to child or elder abuse/neglect or comorbid psychiatric disorders. These conditions can be distinguished by attempting to remove the lesions using soap and water; lesions of dermatosis neglecta will clear, whereas those of TFFD will not.
Metastatic melanoma in pediatric patients has a polymorphous appearance and may or may not be pigmented. Lesions often may be associated with lymphadenopathy of the draining lymph node basins, and nodules and lesions may be firm on palpation.7 Linear configurations of metastatic melanoma may represent a satellite or in-transit metastasis. Fortunately, melanoma is extraordinarily rare in children, with an estimated incidence of 2.1 per million for individuals younger than 20 years.8
Acanthosis nigricans is characterized by velvety plaques most commonly affecting the posterior neck, axillae, and flexor extremities. These lesions commonly are associated with obesity and insulin resistance but occasionally can be associated with underlying malignancy. In the latter association, acanthosis nigricans lesions tend to manifest more abruptly, often are pruritic, and can involve the mucous membranes. Fortunately, acanthosis nigricans related to malignancy in the pediatric population is rare.9
Epidermal nevi may exhibit clinical similarities to TFFD, particularly in lesions with brown/black pigment or with a reticulated or verrucous appearance; however, epidermal nevi often are congenital or manifest within the first few years of life. They commonly are distributed over the lines of Blaschko and have a linear appearance; they also enlarge and thicken as the patient ages.10
Black-dot tinea capitis, a classic manifestation of endothrix infection, manifests as alopecia with broken hairs and is most commonly caused by Tinea tonsurans.11 The black dots refer to the appearance of the infected hair shafts, which have been weakened and broken off at the follicular ostia. As such, lesions typically are monomorphic and may be interspersed with uninvolved hair shafts. There often is associated scale and a lack of inflammation.11,12
Additional differential diagnoses to consider include seborrheic keratoses and confluent and reticulated papillomatosis. Further workup (eg, potassium hydroxide preparation of skin scrapings or skin biopsy) may help elucidate the diagnosis.5 A simple and cost-effective initial diagnostic tool involves wiping suspicious lesions with a 70% isopropyl alcohol pad to confirm this diagnosis.
- Duncan WC. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567. doi:10.1001/archderm.1987.01660290031009
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015:29-33. doi:10.5826/dpc.0503a08
- Aslan NÇ, Güler S, Demirci K, et al. Features of terra firma-forme dermatosis. Ann Fam Med. 2018;16:52-54. doi:10.1370/afm.2175
- Sechi A, Patrizi A, Savoia F, et al. Terra firma-forme dermatosis. Clin Dermatol. 2021;39:202-205. doi:10.1016/j.clindermatol.2020.10.019
- Mohta A, Sarkar R, Narayan RV, et al. Terra firma-forme dermatosis—more than just dirty. Indian Dermatol Online J. 2024;15:99-104. doi:10.4103/idoj.idoj_424_23
- Erkek E, Çetin E, Sahin S, et al. Terra firma-forme dermatosis. Indian J Dermatol Venereol Leprol. 2012;78:358. doi:10.4103 /0378-6323.95455
- McMullan P, Grant-Kels JM. Childhood and adolescent melanoma: an update. Clin Dermatol. 2025;43:16-23. doi:10.1016 /j.clindermatol.2025.01.010
- NCCR*Explorer: An interactive website for NCCR cancer statistics. National Cancer Institute website. Accessed January 10, 2025. https://nccrexplorer.ccdi.cancer.gov/data-products.html
- Sinha S, Schwartz RA. Juvenile acanthosis nigricans. J Am Acad Dermatol. 2007;57:502-508. doi:10.1016/j.jaad.2006.08.016
- Waldman AR, Garzon MC, Morel KD. Epidermal nevi: what is new. Dermatol Clin. 2022;40:61-71. doi:10.1016/j.det.2021.09.006
- Wang X. Black dot tinea capitis. N Engl J Med. 2024; 391:E7. doi:10.1056/NEJMicm2401964
- Gupta AK, Summerbell RC. Tinea capitis. Med Mycol. 2000; 38:255-287. doi:10.1080/mmy.38.4.255.287
THE DIAGNOSIS: Terra Firma-Forme Dermatosis
During clinical examination, a 70% alcohol swab was utilized to gently rub several of the lesions, which were successfully removed. This confirmed a diagnosis of terra firma-forme dermatosis (TFFD)(also known as Duncan’s dirty dermatosis). The patient’s mother was counseled about the diagnosis and was instructed on how to use alcohol pads to remove the remaining lesions. Three days later, after several treatment sessions at home, the mother reported complete resolution of the lesions with no residual pigmentary changes, ulceration, or scarring (Figures 1 and 2).
Terra firma-forme dermatosis was first described in 1987 in a 12-year-old girl with hyperpigmented plaques on the neck that cleared when rubbing alcohol was applied before biopsy.1,2 The term terra firma is Latin for “firm land” (or essentially “dirt”) in reference to what often is described as a characteristically “dirty” clinical appearance.2 Terra firmaforme dermatosis can manifest anywhere on the body but shows a predilection for the neck, arms and legs, axillae, inguinal region, and umbilicus.3 Lesions typically are described as asymptomatic, smooth, well-circumscribed, reticular papules or patches that are brown or black. Terra firma-forme dermatosis also may demonstrate secondary features such as hyperkeratotic, scaly, velvety, or verrucous plaques and nodules.3
The etiology of this condition is theorized to be a result of abnormal or delayed keratinization and prolonged keratinocyte adhesion.3,4 There are limited epidemiologic data, but TFFD has shown a predominance in children younger than 18 years (average age of onset, 10 years) with no known predilection for sex or race and no recognized pattern of inheritance.3-5
Histopathology typically demonstrates epidermal atrophy, hyperkeratosis, and often a component of trapping and compaction of melanin, sebum, microorganisms, and environmental debris.5
Management of TFFD is straightforward and generally consists of rubbing with 70% isopropyl alcohol to remove the lesions. For more adherent lesions or for extensive involvement, other keratolytics such as salicylic acid or alpha-hydroxy acids may be used.5 For TFFD manifesting in infants and young children, widespread involvement, or lesions involving the face or genitals, a urea-based keratolytic with or without a topical anti-inflammatory is suggested.5 Other treatment options include other alpha-hydroxy acids, topical retinoids, and nonpolar solvents such as acetone or CO2 laser for recalcitrant cases.4,5 Fortunately, most TFFD lesions respond well to conservative therapies, with recurrence reported only in 6.3% (5/79) of patients in one study.3
Dermatosis neglecta is clinically similar to TFFD and often is considered on the same spectrum of disease6; however, this entity is associated with decreased bathing or limited hygiene, which could be related to child or elder abuse/neglect or comorbid psychiatric disorders. These conditions can be distinguished by attempting to remove the lesions using soap and water; lesions of dermatosis neglecta will clear, whereas those of TFFD will not.
Metastatic melanoma in pediatric patients has a polymorphous appearance and may or may not be pigmented. Lesions often may be associated with lymphadenopathy of the draining lymph node basins, and nodules and lesions may be firm on palpation.7 Linear configurations of metastatic melanoma may represent a satellite or in-transit metastasis. Fortunately, melanoma is extraordinarily rare in children, with an estimated incidence of 2.1 per million for individuals younger than 20 years.8
Acanthosis nigricans is characterized by velvety plaques most commonly affecting the posterior neck, axillae, and flexor extremities. These lesions commonly are associated with obesity and insulin resistance but occasionally can be associated with underlying malignancy. In the latter association, acanthosis nigricans lesions tend to manifest more abruptly, often are pruritic, and can involve the mucous membranes. Fortunately, acanthosis nigricans related to malignancy in the pediatric population is rare.9
Epidermal nevi may exhibit clinical similarities to TFFD, particularly in lesions with brown/black pigment or with a reticulated or verrucous appearance; however, epidermal nevi often are congenital or manifest within the first few years of life. They commonly are distributed over the lines of Blaschko and have a linear appearance; they also enlarge and thicken as the patient ages.10
Black-dot tinea capitis, a classic manifestation of endothrix infection, manifests as alopecia with broken hairs and is most commonly caused by Tinea tonsurans.11 The black dots refer to the appearance of the infected hair shafts, which have been weakened and broken off at the follicular ostia. As such, lesions typically are monomorphic and may be interspersed with uninvolved hair shafts. There often is associated scale and a lack of inflammation.11,12
Additional differential diagnoses to consider include seborrheic keratoses and confluent and reticulated papillomatosis. Further workup (eg, potassium hydroxide preparation of skin scrapings or skin biopsy) may help elucidate the diagnosis.5 A simple and cost-effective initial diagnostic tool involves wiping suspicious lesions with a 70% isopropyl alcohol pad to confirm this diagnosis.
THE DIAGNOSIS: Terra Firma-Forme Dermatosis
During clinical examination, a 70% alcohol swab was utilized to gently rub several of the lesions, which were successfully removed. This confirmed a diagnosis of terra firma-forme dermatosis (TFFD)(also known as Duncan’s dirty dermatosis). The patient’s mother was counseled about the diagnosis and was instructed on how to use alcohol pads to remove the remaining lesions. Three days later, after several treatment sessions at home, the mother reported complete resolution of the lesions with no residual pigmentary changes, ulceration, or scarring (Figures 1 and 2).
Terra firma-forme dermatosis was first described in 1987 in a 12-year-old girl with hyperpigmented plaques on the neck that cleared when rubbing alcohol was applied before biopsy.1,2 The term terra firma is Latin for “firm land” (or essentially “dirt”) in reference to what often is described as a characteristically “dirty” clinical appearance.2 Terra firmaforme dermatosis can manifest anywhere on the body but shows a predilection for the neck, arms and legs, axillae, inguinal region, and umbilicus.3 Lesions typically are described as asymptomatic, smooth, well-circumscribed, reticular papules or patches that are brown or black. Terra firma-forme dermatosis also may demonstrate secondary features such as hyperkeratotic, scaly, velvety, or verrucous plaques and nodules.3
The etiology of this condition is theorized to be a result of abnormal or delayed keratinization and prolonged keratinocyte adhesion.3,4 There are limited epidemiologic data, but TFFD has shown a predominance in children younger than 18 years (average age of onset, 10 years) with no known predilection for sex or race and no recognized pattern of inheritance.3-5
Histopathology typically demonstrates epidermal atrophy, hyperkeratosis, and often a component of trapping and compaction of melanin, sebum, microorganisms, and environmental debris.5
Management of TFFD is straightforward and generally consists of rubbing with 70% isopropyl alcohol to remove the lesions. For more adherent lesions or for extensive involvement, other keratolytics such as salicylic acid or alpha-hydroxy acids may be used.5 For TFFD manifesting in infants and young children, widespread involvement, or lesions involving the face or genitals, a urea-based keratolytic with or without a topical anti-inflammatory is suggested.5 Other treatment options include other alpha-hydroxy acids, topical retinoids, and nonpolar solvents such as acetone or CO2 laser for recalcitrant cases.4,5 Fortunately, most TFFD lesions respond well to conservative therapies, with recurrence reported only in 6.3% (5/79) of patients in one study.3
Dermatosis neglecta is clinically similar to TFFD and often is considered on the same spectrum of disease6; however, this entity is associated with decreased bathing or limited hygiene, which could be related to child or elder abuse/neglect or comorbid psychiatric disorders. These conditions can be distinguished by attempting to remove the lesions using soap and water; lesions of dermatosis neglecta will clear, whereas those of TFFD will not.
Metastatic melanoma in pediatric patients has a polymorphous appearance and may or may not be pigmented. Lesions often may be associated with lymphadenopathy of the draining lymph node basins, and nodules and lesions may be firm on palpation.7 Linear configurations of metastatic melanoma may represent a satellite or in-transit metastasis. Fortunately, melanoma is extraordinarily rare in children, with an estimated incidence of 2.1 per million for individuals younger than 20 years.8
Acanthosis nigricans is characterized by velvety plaques most commonly affecting the posterior neck, axillae, and flexor extremities. These lesions commonly are associated with obesity and insulin resistance but occasionally can be associated with underlying malignancy. In the latter association, acanthosis nigricans lesions tend to manifest more abruptly, often are pruritic, and can involve the mucous membranes. Fortunately, acanthosis nigricans related to malignancy in the pediatric population is rare.9
Epidermal nevi may exhibit clinical similarities to TFFD, particularly in lesions with brown/black pigment or with a reticulated or verrucous appearance; however, epidermal nevi often are congenital or manifest within the first few years of life. They commonly are distributed over the lines of Blaschko and have a linear appearance; they also enlarge and thicken as the patient ages.10
Black-dot tinea capitis, a classic manifestation of endothrix infection, manifests as alopecia with broken hairs and is most commonly caused by Tinea tonsurans.11 The black dots refer to the appearance of the infected hair shafts, which have been weakened and broken off at the follicular ostia. As such, lesions typically are monomorphic and may be interspersed with uninvolved hair shafts. There often is associated scale and a lack of inflammation.11,12
Additional differential diagnoses to consider include seborrheic keratoses and confluent and reticulated papillomatosis. Further workup (eg, potassium hydroxide preparation of skin scrapings or skin biopsy) may help elucidate the diagnosis.5 A simple and cost-effective initial diagnostic tool involves wiping suspicious lesions with a 70% isopropyl alcohol pad to confirm this diagnosis.
- Duncan WC. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567. doi:10.1001/archderm.1987.01660290031009
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015:29-33. doi:10.5826/dpc.0503a08
- Aslan NÇ, Güler S, Demirci K, et al. Features of terra firma-forme dermatosis. Ann Fam Med. 2018;16:52-54. doi:10.1370/afm.2175
- Sechi A, Patrizi A, Savoia F, et al. Terra firma-forme dermatosis. Clin Dermatol. 2021;39:202-205. doi:10.1016/j.clindermatol.2020.10.019
- Mohta A, Sarkar R, Narayan RV, et al. Terra firma-forme dermatosis—more than just dirty. Indian Dermatol Online J. 2024;15:99-104. doi:10.4103/idoj.idoj_424_23
- Erkek E, Çetin E, Sahin S, et al. Terra firma-forme dermatosis. Indian J Dermatol Venereol Leprol. 2012;78:358. doi:10.4103 /0378-6323.95455
- McMullan P, Grant-Kels JM. Childhood and adolescent melanoma: an update. Clin Dermatol. 2025;43:16-23. doi:10.1016 /j.clindermatol.2025.01.010
- NCCR*Explorer: An interactive website for NCCR cancer statistics. National Cancer Institute website. Accessed January 10, 2025. https://nccrexplorer.ccdi.cancer.gov/data-products.html
- Sinha S, Schwartz RA. Juvenile acanthosis nigricans. J Am Acad Dermatol. 2007;57:502-508. doi:10.1016/j.jaad.2006.08.016
- Waldman AR, Garzon MC, Morel KD. Epidermal nevi: what is new. Dermatol Clin. 2022;40:61-71. doi:10.1016/j.det.2021.09.006
- Wang X. Black dot tinea capitis. N Engl J Med. 2024; 391:E7. doi:10.1056/NEJMicm2401964
- Gupta AK, Summerbell RC. Tinea capitis. Med Mycol. 2000; 38:255-287. doi:10.1080/mmy.38.4.255.287
- Duncan WC. Terra firma-forme dermatosis. Arch Dermatol. 1987;123:567. doi:10.1001/archderm.1987.01660290031009
- Greywal T, Cohen PR. Terra firma-forme dermatosis: a report of ten individuals with Duncan’s dirty dermatosis and literature review. Dermatol Pract Concept. 2015:29-33. doi:10.5826/dpc.0503a08
- Aslan NÇ, Güler S, Demirci K, et al. Features of terra firma-forme dermatosis. Ann Fam Med. 2018;16:52-54. doi:10.1370/afm.2175
- Sechi A, Patrizi A, Savoia F, et al. Terra firma-forme dermatosis. Clin Dermatol. 2021;39:202-205. doi:10.1016/j.clindermatol.2020.10.019
- Mohta A, Sarkar R, Narayan RV, et al. Terra firma-forme dermatosis—more than just dirty. Indian Dermatol Online J. 2024;15:99-104. doi:10.4103/idoj.idoj_424_23
- Erkek E, Çetin E, Sahin S, et al. Terra firma-forme dermatosis. Indian J Dermatol Venereol Leprol. 2012;78:358. doi:10.4103 /0378-6323.95455
- McMullan P, Grant-Kels JM. Childhood and adolescent melanoma: an update. Clin Dermatol. 2025;43:16-23. doi:10.1016 /j.clindermatol.2025.01.010
- NCCR*Explorer: An interactive website for NCCR cancer statistics. National Cancer Institute website. Accessed January 10, 2025. https://nccrexplorer.ccdi.cancer.gov/data-products.html
- Sinha S, Schwartz RA. Juvenile acanthosis nigricans. J Am Acad Dermatol. 2007;57:502-508. doi:10.1016/j.jaad.2006.08.016
- Waldman AR, Garzon MC, Morel KD. Epidermal nevi: what is new. Dermatol Clin. 2022;40:61-71. doi:10.1016/j.det.2021.09.006
- Wang X. Black dot tinea capitis. N Engl J Med. 2024; 391:E7. doi:10.1056/NEJMicm2401964
- Gupta AK, Summerbell RC. Tinea capitis. Med Mycol. 2000; 38:255-287. doi:10.1080/mmy.38.4.255.287
Black Dots on the Scalp of a Child
Black Dots on the Scalp of a Child
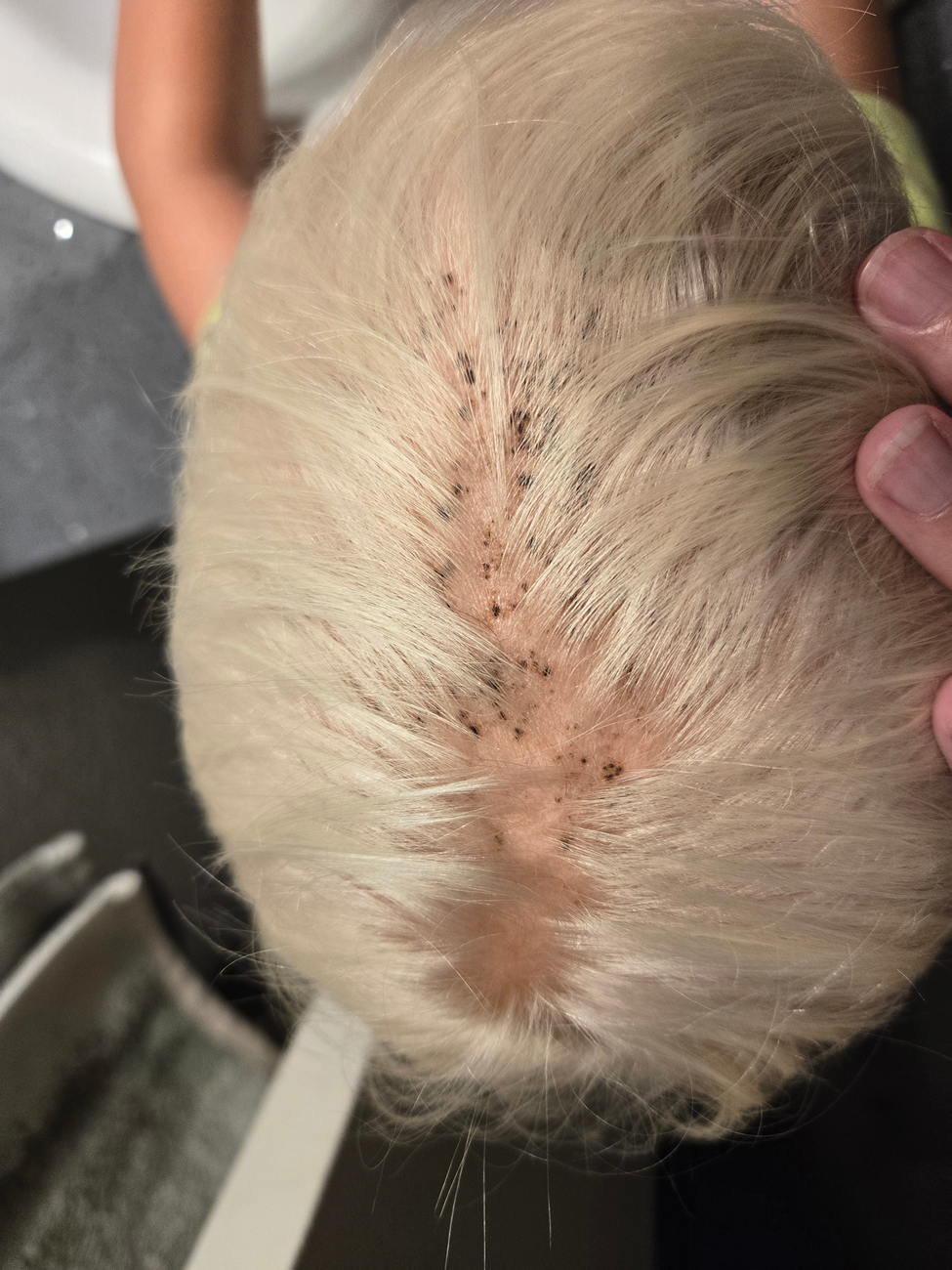
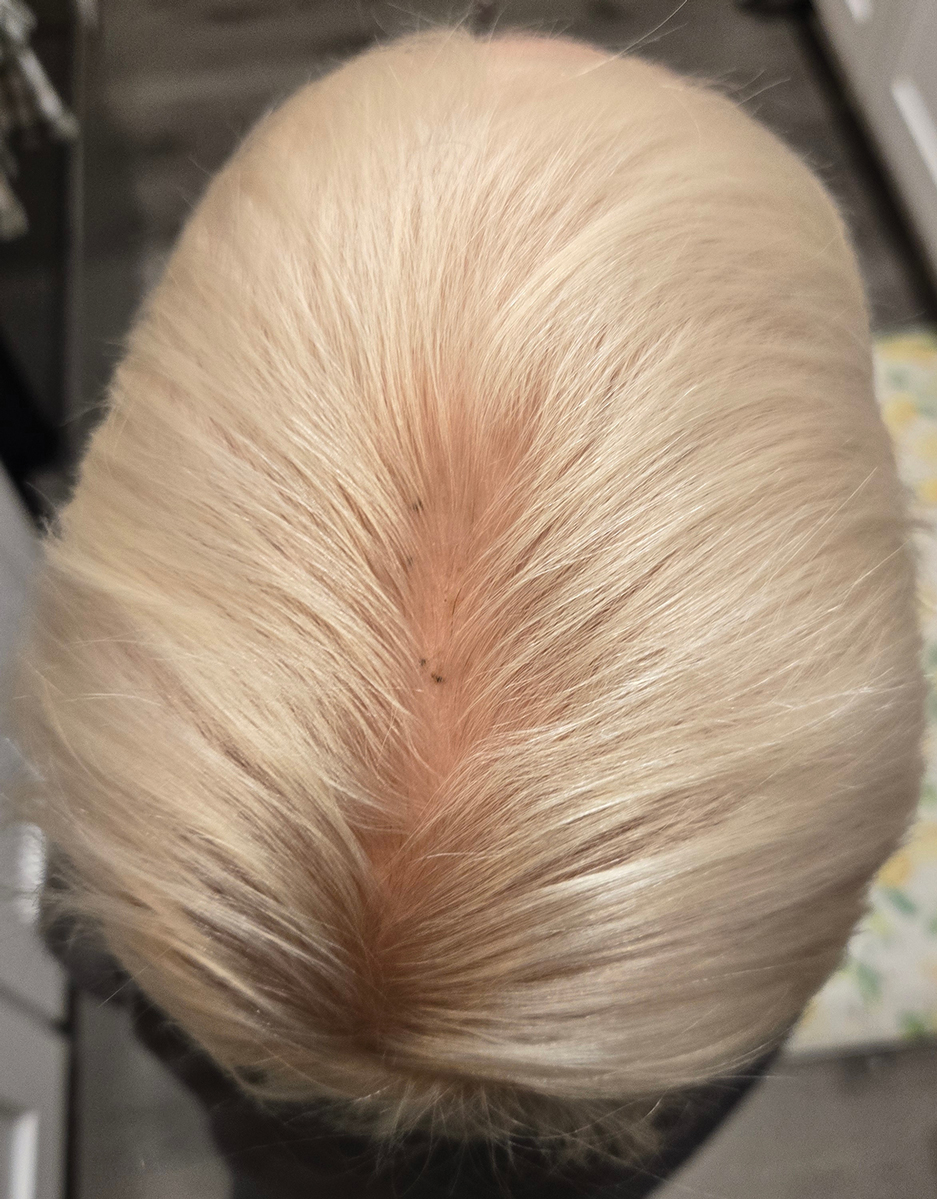
A 4-year-old boy was referred to the dermatology clinic by his pediatrician for evaluation of persistent black spots on the scalp of 1 month’s duration. The patient was otherwise healthy, and his mother stated that the lesions had appeared gradually, were not tender or pruritic, and did not wash off with shampoo and scrubbing. The patient had no history of any systemic illness, recent travel, genetic disorders, or genodermatoses. Physical examination revealed multiple well-circumscribed, 1- to 2-mm black papules and macules with confluence scattered over the vertex scalp. No erythema, scale, or induration was noted.

Progressive Erythematous Facial Rash
Progressive Erythematous Facial Rash
THE DIAGNOSIS: Follicular Mucinosis
Histologic examination of the hematoxylin and eosin–stained sections of the biopsy revealed an overall moderately dense, perivascular, and perifollicular lymphocytic infiltrate with follicular intraepidermal mucin (Figure). Immunohistochemical staining showed that the lymphocytic infiltrate was predominantly CD4+ over CD8+, with moderate loss of CD7 and absence of CD20 expression. Positive T-cell receptor (TCR) gene rearrangements were detected for both TCRγ and TCRΒ. The clinical features along with the histopathologic findings suggested a diagnosis of follicular mucinosis (FM) with concern in the differential for folliculotropic mycosis fungoides.
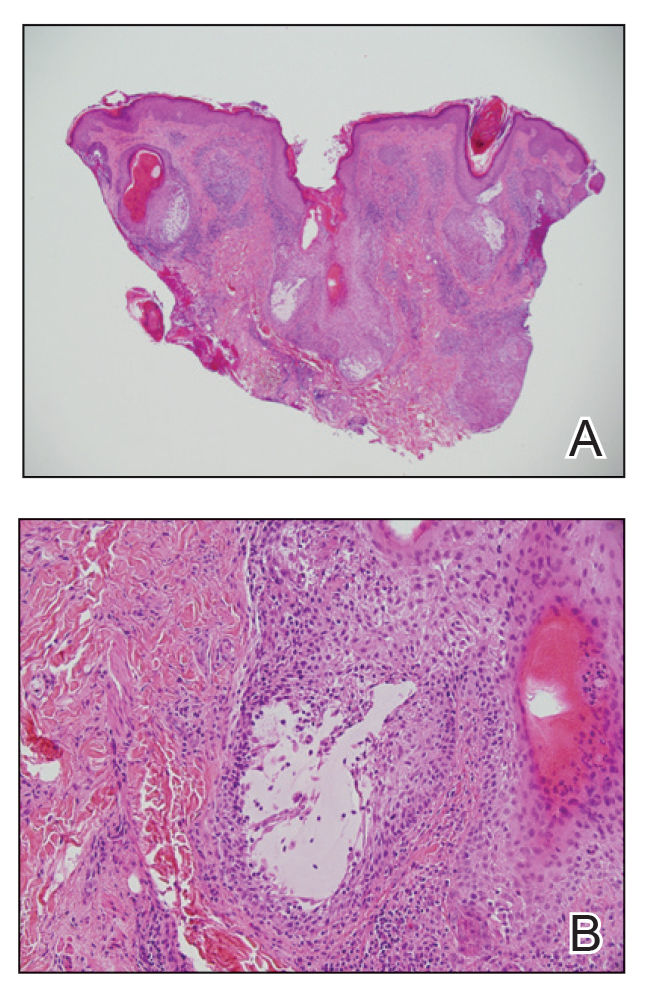
Follicular mucinosis, also known as alopecia mucinosa, is an uncommon inflammatory disorder characterized by follicular degeneration due to the accumulation of mucin within the pilosebaceous unit.1 This condition manifests clinically as indurated plaques and/or follicular papules most often on the face, neck, and scalp.2 It is further categorized as primary vs secondary FM. Primary idiopathic FM, which can further be subdivided into acute or chronic, tends to follow a more benign course, whereas secondary FM usually is associated with underlying inflammatory or neoplastic conditions, most commonly mycosis fungoides, a cutaneous T-cell lymphoma.1,2 In cases of secondary FM, treatment of the underlying cause often leads to resolution of symptoms. Regular follow-up is warranted in either classification.1,3
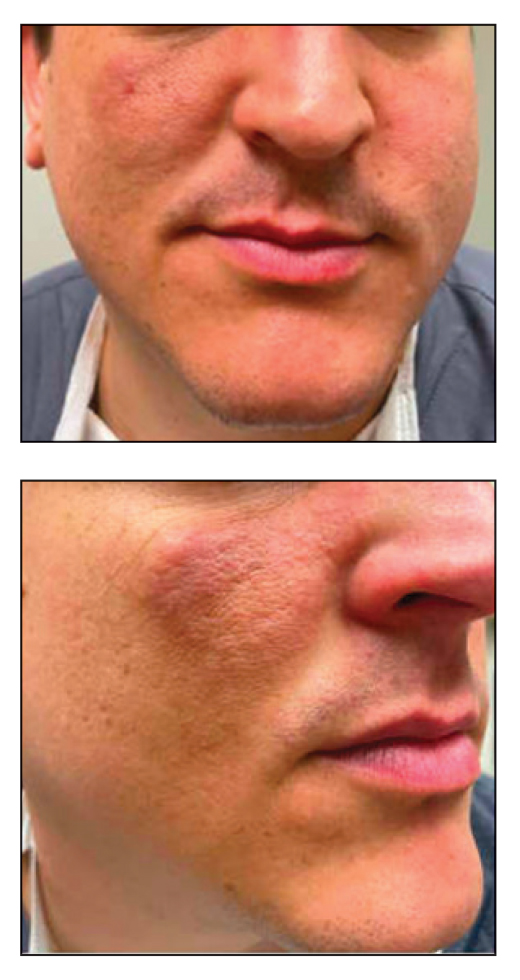
The initial differential diagnosis for this patient included contact dermatitis associated with mask use, with possible underlying seborrheic dermatitis or rosacea; however, the rash persisted and worsened after treatment with topical triamcinolone and ketoconazole. After the diagnosis of FM was made, the patient was started on topical betamethasone and tacrolimus with good response.
A referral to hematology/oncology revealed that the patient had primary FM and possible stage 1A folliculotropic mycosis fungoides with limited skin involvement (<10% body surface area). On physical examination, no palpable cervical or axillary lymphadenopathy were noted. Flow cytometry for lymphoma was negative with no lymphoid or blast population detected. Laboratory workup and positron emission tomography/computed tomography were unremarkable. The patient had rapid improvement with a more potent topical steroid but also was given tacrolimus ointment 0.1% for residual findings. His disease remained stable without progression at 1-year follow-up.
Contact dermatitis typically manifests as an eczematous eruption that appears on an anatomic location that was exposed to or came into contact with allergens or irritants.4 Contact dermatitis was less likely in our patient due to the lack of acute or subacute spongiosis and lymphocyte exocytosis. Rosacea is a chronic inflammatory dermatosis that presents as recurrent episodes of flushing or transient erythema, persistent erythema, phyphymatous changes, papules, pustules, and telangiectasia5; however, rosacea was less likely in our patient due to the histopathologic and immunohistochemical findings that were suggestive of FM on punch biopsy. Cutaneous lupus generally is associated with photosensitivity and manifests as erythema over the malar eminences and bridge of the nose with sparing of the nasolabial folds.6 Seborrheic dermatitis manifests as erythematous macules or patches with scale and associated pruritis on the scalp, eyebrows, eyelids, and nasolabial folds.7 This condition was less likely in our patient due to the persistence and worsening of the facial erythematous dermatitis despite the use of ketoconazole cream as well as no evidence of spongiosis, shoulder parakeratosis, vascular changes, or presence of microorganisms such as Malassezia species.
Due to the relatively rare nature of this condition as well as a wide variety of other more common etiologies for an erythematous dermatitis of the cheeks, the diagnosis of FM may be delayed or missed entirely. Physicians must have a high index of suspicion to diagnose properly and biopsy if necessary. This photoquiz serves as an important reminder to physicians to keep uncommon diseases on their differential, especially when the patient’s symptoms do not respond to treatment.
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165.
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746.
- Miyagaki T. Diagnosis of early mycosis fungoides. Diagnostics (Basel). 2021;1:1721.
- Elmas ÖF, Akdeniz N, Atasoy M, et al. Contact dermatitis: a great imitator. Clin Dermatol. 2020;38:176-192.
- van Zuuren EJ, Arents BWM, van der Linden MMD, et al. Rosacea: new concepts in classification and treatment. Am J Clin Dermatol. 2021;22:457-465.
- Rothfield N, Sontheimer RD, Bernstein M. Lupus erythematosus: systemic and cutaneous manifestations. Clin Dermatol. 2006;24:348-362.
- Borda LJ, Perper M, Keri JE. Treatment of seborrheic dermatitis: a comprehensive review. J Dermatolog Treat. 2019;30:158-169.
THE DIAGNOSIS: Follicular Mucinosis
Histologic examination of the hematoxylin and eosin–stained sections of the biopsy revealed an overall moderately dense, perivascular, and perifollicular lymphocytic infiltrate with follicular intraepidermal mucin (Figure). Immunohistochemical staining showed that the lymphocytic infiltrate was predominantly CD4+ over CD8+, with moderate loss of CD7 and absence of CD20 expression. Positive T-cell receptor (TCR) gene rearrangements were detected for both TCRγ and TCRΒ. The clinical features along with the histopathologic findings suggested a diagnosis of follicular mucinosis (FM) with concern in the differential for folliculotropic mycosis fungoides.
Follicular mucinosis, also known as alopecia mucinosa, is an uncommon inflammatory disorder characterized by follicular degeneration due to the accumulation of mucin within the pilosebaceous unit.1 This condition manifests clinically as indurated plaques and/or follicular papules most often on the face, neck, and scalp.2 It is further categorized as primary vs secondary FM. Primary idiopathic FM, which can further be subdivided into acute or chronic, tends to follow a more benign course, whereas secondary FM usually is associated with underlying inflammatory or neoplastic conditions, most commonly mycosis fungoides, a cutaneous T-cell lymphoma.1,2 In cases of secondary FM, treatment of the underlying cause often leads to resolution of symptoms. Regular follow-up is warranted in either classification.1,3
The initial differential diagnosis for this patient included contact dermatitis associated with mask use, with possible underlying seborrheic dermatitis or rosacea; however, the rash persisted and worsened after treatment with topical triamcinolone and ketoconazole. After the diagnosis of FM was made, the patient was started on topical betamethasone and tacrolimus with good response.
A referral to hematology/oncology revealed that the patient had primary FM and possible stage 1A folliculotropic mycosis fungoides with limited skin involvement (<10% body surface area). On physical examination, no palpable cervical or axillary lymphadenopathy were noted. Flow cytometry for lymphoma was negative with no lymphoid or blast population detected. Laboratory workup and positron emission tomography/computed tomography were unremarkable. The patient had rapid improvement with a more potent topical steroid but also was given tacrolimus ointment 0.1% for residual findings. His disease remained stable without progression at 1-year follow-up.
Contact dermatitis typically manifests as an eczematous eruption that appears on an anatomic location that was exposed to or came into contact with allergens or irritants.4 Contact dermatitis was less likely in our patient due to the lack of acute or subacute spongiosis and lymphocyte exocytosis. Rosacea is a chronic inflammatory dermatosis that presents as recurrent episodes of flushing or transient erythema, persistent erythema, phyphymatous changes, papules, pustules, and telangiectasia5; however, rosacea was less likely in our patient due to the histopathologic and immunohistochemical findings that were suggestive of FM on punch biopsy. Cutaneous lupus generally is associated with photosensitivity and manifests as erythema over the malar eminences and bridge of the nose with sparing of the nasolabial folds.6 Seborrheic dermatitis manifests as erythematous macules or patches with scale and associated pruritis on the scalp, eyebrows, eyelids, and nasolabial folds.7 This condition was less likely in our patient due to the persistence and worsening of the facial erythematous dermatitis despite the use of ketoconazole cream as well as no evidence of spongiosis, shoulder parakeratosis, vascular changes, or presence of microorganisms such as Malassezia species.
Due to the relatively rare nature of this condition as well as a wide variety of other more common etiologies for an erythematous dermatitis of the cheeks, the diagnosis of FM may be delayed or missed entirely. Physicians must have a high index of suspicion to diagnose properly and biopsy if necessary. This photoquiz serves as an important reminder to physicians to keep uncommon diseases on their differential, especially when the patient’s symptoms do not respond to treatment.
THE DIAGNOSIS: Follicular Mucinosis
Histologic examination of the hematoxylin and eosin–stained sections of the biopsy revealed an overall moderately dense, perivascular, and perifollicular lymphocytic infiltrate with follicular intraepidermal mucin (Figure). Immunohistochemical staining showed that the lymphocytic infiltrate was predominantly CD4+ over CD8+, with moderate loss of CD7 and absence of CD20 expression. Positive T-cell receptor (TCR) gene rearrangements were detected for both TCRγ and TCRΒ. The clinical features along with the histopathologic findings suggested a diagnosis of follicular mucinosis (FM) with concern in the differential for folliculotropic mycosis fungoides.
Follicular mucinosis, also known as alopecia mucinosa, is an uncommon inflammatory disorder characterized by follicular degeneration due to the accumulation of mucin within the pilosebaceous unit.1 This condition manifests clinically as indurated plaques and/or follicular papules most often on the face, neck, and scalp.2 It is further categorized as primary vs secondary FM. Primary idiopathic FM, which can further be subdivided into acute or chronic, tends to follow a more benign course, whereas secondary FM usually is associated with underlying inflammatory or neoplastic conditions, most commonly mycosis fungoides, a cutaneous T-cell lymphoma.1,2 In cases of secondary FM, treatment of the underlying cause often leads to resolution of symptoms. Regular follow-up is warranted in either classification.1,3
The initial differential diagnosis for this patient included contact dermatitis associated with mask use, with possible underlying seborrheic dermatitis or rosacea; however, the rash persisted and worsened after treatment with topical triamcinolone and ketoconazole. After the diagnosis of FM was made, the patient was started on topical betamethasone and tacrolimus with good response.
A referral to hematology/oncology revealed that the patient had primary FM and possible stage 1A folliculotropic mycosis fungoides with limited skin involvement (<10% body surface area). On physical examination, no palpable cervical or axillary lymphadenopathy were noted. Flow cytometry for lymphoma was negative with no lymphoid or blast population detected. Laboratory workup and positron emission tomography/computed tomography were unremarkable. The patient had rapid improvement with a more potent topical steroid but also was given tacrolimus ointment 0.1% for residual findings. His disease remained stable without progression at 1-year follow-up.
Contact dermatitis typically manifests as an eczematous eruption that appears on an anatomic location that was exposed to or came into contact with allergens or irritants.4 Contact dermatitis was less likely in our patient due to the lack of acute or subacute spongiosis and lymphocyte exocytosis. Rosacea is a chronic inflammatory dermatosis that presents as recurrent episodes of flushing or transient erythema, persistent erythema, phyphymatous changes, papules, pustules, and telangiectasia5; however, rosacea was less likely in our patient due to the histopathologic and immunohistochemical findings that were suggestive of FM on punch biopsy. Cutaneous lupus generally is associated with photosensitivity and manifests as erythema over the malar eminences and bridge of the nose with sparing of the nasolabial folds.6 Seborrheic dermatitis manifests as erythematous macules or patches with scale and associated pruritis on the scalp, eyebrows, eyelids, and nasolabial folds.7 This condition was less likely in our patient due to the persistence and worsening of the facial erythematous dermatitis despite the use of ketoconazole cream as well as no evidence of spongiosis, shoulder parakeratosis, vascular changes, or presence of microorganisms such as Malassezia species.
Due to the relatively rare nature of this condition as well as a wide variety of other more common etiologies for an erythematous dermatitis of the cheeks, the diagnosis of FM may be delayed or missed entirely. Physicians must have a high index of suspicion to diagnose properly and biopsy if necessary. This photoquiz serves as an important reminder to physicians to keep uncommon diseases on their differential, especially when the patient’s symptoms do not respond to treatment.
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165.
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746.
- Miyagaki T. Diagnosis of early mycosis fungoides. Diagnostics (Basel). 2021;1:1721.
- Elmas ÖF, Akdeniz N, Atasoy M, et al. Contact dermatitis: a great imitator. Clin Dermatol. 2020;38:176-192.
- van Zuuren EJ, Arents BWM, van der Linden MMD, et al. Rosacea: new concepts in classification and treatment. Am J Clin Dermatol. 2021;22:457-465.
- Rothfield N, Sontheimer RD, Bernstein M. Lupus erythematosus: systemic and cutaneous manifestations. Clin Dermatol. 2006;24:348-362.
- Borda LJ, Perper M, Keri JE. Treatment of seborrheic dermatitis: a comprehensive review. J Dermatolog Treat. 2019;30:158-169.
- Khalil J, Kurban M, Abbas O. Follicular mucinosis: a review. Int J Dermatol. 2021;60:159-165.
- Akinsanya AO, Tschen JA. Follicular mucinosis: a case report. Cureus. 2019;11:E4746.
- Miyagaki T. Diagnosis of early mycosis fungoides. Diagnostics (Basel). 2021;1:1721.
- Elmas ÖF, Akdeniz N, Atasoy M, et al. Contact dermatitis: a great imitator. Clin Dermatol. 2020;38:176-192.
- van Zuuren EJ, Arents BWM, van der Linden MMD, et al. Rosacea: new concepts in classification and treatment. Am J Clin Dermatol. 2021;22:457-465.
- Rothfield N, Sontheimer RD, Bernstein M. Lupus erythematosus: systemic and cutaneous manifestations. Clin Dermatol. 2006;24:348-362.
- Borda LJ, Perper M, Keri JE. Treatment of seborrheic dermatitis: a comprehensive review. J Dermatolog Treat. 2019;30:158-169.
Progressive Erythematous Facial Rash
Progressive Erythematous Facial Rash
A 32-year-old man presented to the dermatology clinic for evaluation of a progressive erythematous facial rash of 4 years’ duration. The patient reported some worsening with increased face mask wear during the COVID-19 pandemic. On occasion, fluid could be expressed when the area on the right cheek was compressed. Physical examination revealed a well-demarcated erythematous plaque on the right cheek. The patient also reported intermittent mild involvement of the nose and left cheek. He initially was treated with triamcinolone and ketoconazole cream for several months, but the rash persisted. Given the chronicity and worsening of the eruption, a punch biopsy from the right cheek with immunohistochemical staining was obtained.

Acute Pustular Eruption on the Hands
Acute Pustular Eruption on the Hands
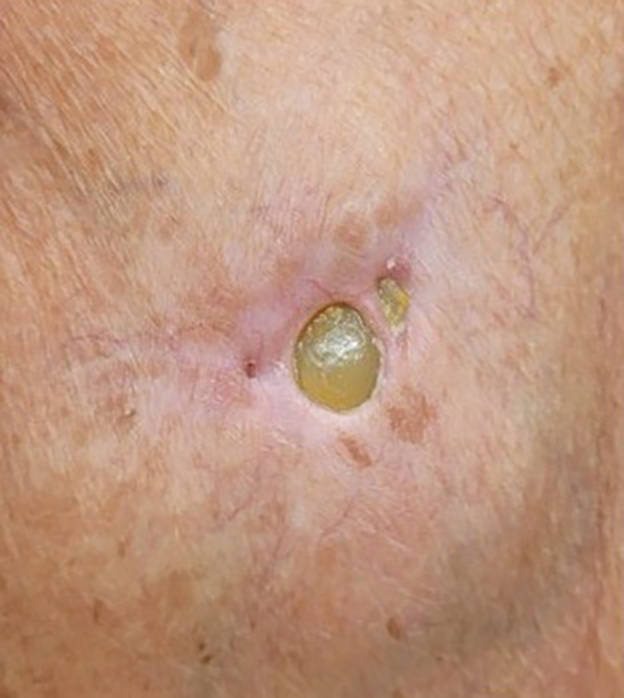
THE DIAGNOSIS: Neutrophilic Dermatosis of the Dorsal Hands
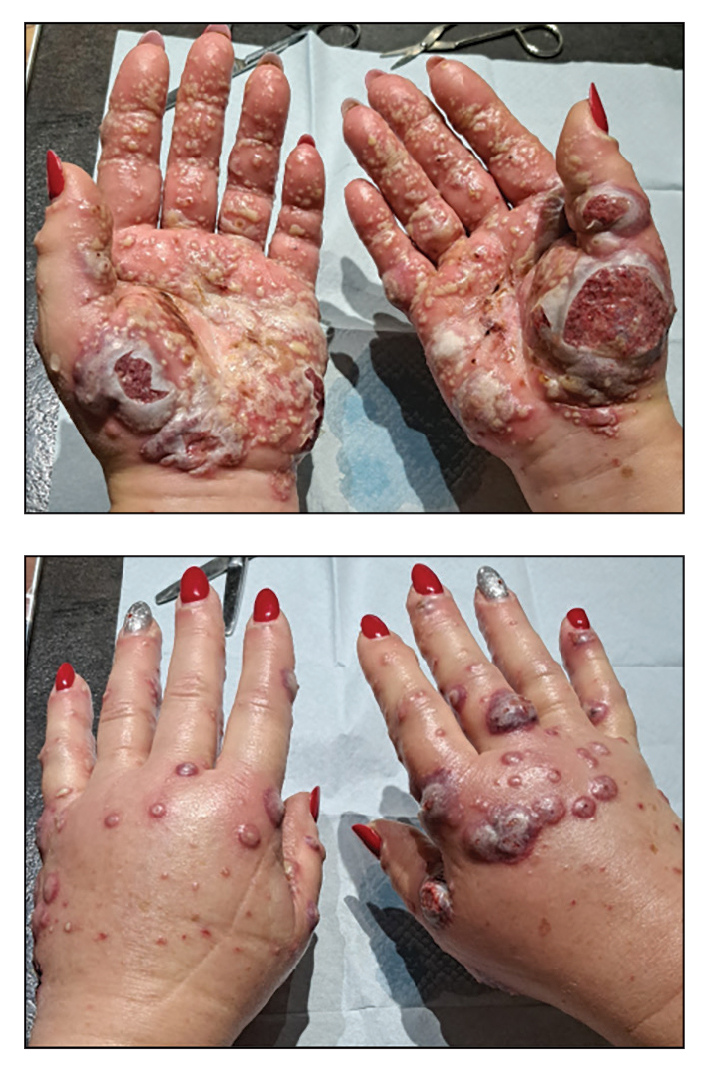
Histopathology showed a unilocular pustule with a dense neutrophilic infiltrate of the superficial dermis. Minimal vascular alterations also were observed. These findings were consistent with a diagnosis of neutrophilic dermatosis of the dorsal hands (NDDH). Our patient was treated successfully with systemic corticosteroids (1 mg/kg/d) with rapid improvement after 10 days of treatment.
Neutrophilic dermatosis of the dorsal hands is an evolving disease concept that was first described as pustular vasculitis by Strutton et al1 in 1995. Galaria et al2 subsequently identified NDDH as a clinical entity associating tender erythematous plaques, pustules, bullae, and/or ulcers on the dorsal hands with histologic features of Sweet syndrome (SS). After reviewing 9 cases of NDDH—all of which demonstrated clinical, laboratory, and histologic characteristics of SS—Walling et al3 concluded that NDDH was best understood as a distributional variant of SS.
Our patient presented with vascular alterations described as a reactive response to the neutrophilic infiltration. The presence of vasculitis in SS and NDDH biopsies is considered as an occasional epiphenomenon and should not rule out the diagnosis of NDDH.3 A literature review of 123 cases of NDDH revealed the presence of vasculitis in 36 (29.5%) patients.4 With regard to other clinical findings, it has been suggested that an increased white blood cell count and elevated C-reactive protein level, as was seen in our patient, may be observed in NDDH, albeit less frequently than in classical SS.4
While palmar involvement of NDDH is considered rare, the recent review of 123 cases of NDDH identified palmar lesions in 5 patients (4.1%).4 Earlier reviews had identified 12 historical cases.5 Palmar manifestations of NDDH have been shown to be associated with erythematous nonulcerated lesions (as opposed to the classical ulcerative or pustular plaques) and a lower association with hematologic malignancies.5
In our patient’s case, dyshidrosis was excluded due to the presence of painful ulcerative plaques rather than pruritic, deep-seated vesicles. Pustular psoriasis typically manifests with sterile pustules on the palms and soles; however, the rapid onset of ulcerative, necrotic plaques and substantial edema are more specific to NDDH. Poststreptococcal pustulosis generally follows a streptococcal infection and lacks the violaceous undermined borders seen in NDDH. Reactive arthritis manifests with hyperkeratotic plaques and is associated with the clinical triad of urethritis, conjunctivitis, and arthritis, which were absent in our patient.
The histologic differential diagnosis of NDDH includes infection, pyoderma gangrenosum, bowel-associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatitis, and erythema elevatum diutinum3,4; however, these conditions typically manifest with distinct clinical features that allow for differentiation, despite histologic similarities. The wide histologic spectrum of neutrophilic dermatosis may contribute to variable clinical manifestations and an evolving disease concept, as the classification of NDDH has changed from a primary vasculitis to a variant of SS. However, this evolution does not affect the appropriate management, as they all have shown good response to corticosteroid treatment.4,6
- Strutton G, Weedon D, Robertson I. Pustular vasculitis of the hands. J Am Acad Dermatol. 1995;32(2 pt 1):192-198.
- Galaria NA, Junkins-Hopkins JM, Kligman D, et al. Neutrophilic dermatosis of the dorsal hands: pustular vasculitis revisited. J Am Acad Dermatol. 2000;43(5 pt 1):870-874.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63
- Micallef D, Bonnici M, Pisani D, et al. Neutrophilic dermatosis of the dorsal hands: a review of 123 cases. J Am Acad Dermatol. 2023;88:1338-1344.
- Arandes-Marcocci J, Altemir-Vidal A, Iglesias-Plaza A, et al. Neutrophilic dermatosis of the hands with palmar involvement: does it have clinical implication? Int J Dermatol. 2020;59:736-738.
- Del Pozo J, Sacristán F, Martínez W, et al. Neutrophilic dermatosis of the hands: presentation of eight cases and review of the literature. J Dermatol. 2007;34:243-247.
THE DIAGNOSIS: Neutrophilic Dermatosis of the Dorsal Hands
Histopathology showed a unilocular pustule with a dense neutrophilic infiltrate of the superficial dermis. Minimal vascular alterations also were observed. These findings were consistent with a diagnosis of neutrophilic dermatosis of the dorsal hands (NDDH). Our patient was treated successfully with systemic corticosteroids (1 mg/kg/d) with rapid improvement after 10 days of treatment.
Neutrophilic dermatosis of the dorsal hands is an evolving disease concept that was first described as pustular vasculitis by Strutton et al1 in 1995. Galaria et al2 subsequently identified NDDH as a clinical entity associating tender erythematous plaques, pustules, bullae, and/or ulcers on the dorsal hands with histologic features of Sweet syndrome (SS). After reviewing 9 cases of NDDH—all of which demonstrated clinical, laboratory, and histologic characteristics of SS—Walling et al3 concluded that NDDH was best understood as a distributional variant of SS.
Our patient presented with vascular alterations described as a reactive response to the neutrophilic infiltration. The presence of vasculitis in SS and NDDH biopsies is considered as an occasional epiphenomenon and should not rule out the diagnosis of NDDH.3 A literature review of 123 cases of NDDH revealed the presence of vasculitis in 36 (29.5%) patients.4 With regard to other clinical findings, it has been suggested that an increased white blood cell count and elevated C-reactive protein level, as was seen in our patient, may be observed in NDDH, albeit less frequently than in classical SS.4
While palmar involvement of NDDH is considered rare, the recent review of 123 cases of NDDH identified palmar lesions in 5 patients (4.1%).4 Earlier reviews had identified 12 historical cases.5 Palmar manifestations of NDDH have been shown to be associated with erythematous nonulcerated lesions (as opposed to the classical ulcerative or pustular plaques) and a lower association with hematologic malignancies.5
In our patient’s case, dyshidrosis was excluded due to the presence of painful ulcerative plaques rather than pruritic, deep-seated vesicles. Pustular psoriasis typically manifests with sterile pustules on the palms and soles; however, the rapid onset of ulcerative, necrotic plaques and substantial edema are more specific to NDDH. Poststreptococcal pustulosis generally follows a streptococcal infection and lacks the violaceous undermined borders seen in NDDH. Reactive arthritis manifests with hyperkeratotic plaques and is associated with the clinical triad of urethritis, conjunctivitis, and arthritis, which were absent in our patient.
The histologic differential diagnosis of NDDH includes infection, pyoderma gangrenosum, bowel-associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatitis, and erythema elevatum diutinum3,4; however, these conditions typically manifest with distinct clinical features that allow for differentiation, despite histologic similarities. The wide histologic spectrum of neutrophilic dermatosis may contribute to variable clinical manifestations and an evolving disease concept, as the classification of NDDH has changed from a primary vasculitis to a variant of SS. However, this evolution does not affect the appropriate management, as they all have shown good response to corticosteroid treatment.4,6
THE DIAGNOSIS: Neutrophilic Dermatosis of the Dorsal Hands
Histopathology showed a unilocular pustule with a dense neutrophilic infiltrate of the superficial dermis. Minimal vascular alterations also were observed. These findings were consistent with a diagnosis of neutrophilic dermatosis of the dorsal hands (NDDH). Our patient was treated successfully with systemic corticosteroids (1 mg/kg/d) with rapid improvement after 10 days of treatment.
Neutrophilic dermatosis of the dorsal hands is an evolving disease concept that was first described as pustular vasculitis by Strutton et al1 in 1995. Galaria et al2 subsequently identified NDDH as a clinical entity associating tender erythematous plaques, pustules, bullae, and/or ulcers on the dorsal hands with histologic features of Sweet syndrome (SS). After reviewing 9 cases of NDDH—all of which demonstrated clinical, laboratory, and histologic characteristics of SS—Walling et al3 concluded that NDDH was best understood as a distributional variant of SS.
Our patient presented with vascular alterations described as a reactive response to the neutrophilic infiltration. The presence of vasculitis in SS and NDDH biopsies is considered as an occasional epiphenomenon and should not rule out the diagnosis of NDDH.3 A literature review of 123 cases of NDDH revealed the presence of vasculitis in 36 (29.5%) patients.4 With regard to other clinical findings, it has been suggested that an increased white blood cell count and elevated C-reactive protein level, as was seen in our patient, may be observed in NDDH, albeit less frequently than in classical SS.4
While palmar involvement of NDDH is considered rare, the recent review of 123 cases of NDDH identified palmar lesions in 5 patients (4.1%).4 Earlier reviews had identified 12 historical cases.5 Palmar manifestations of NDDH have been shown to be associated with erythematous nonulcerated lesions (as opposed to the classical ulcerative or pustular plaques) and a lower association with hematologic malignancies.5
In our patient’s case, dyshidrosis was excluded due to the presence of painful ulcerative plaques rather than pruritic, deep-seated vesicles. Pustular psoriasis typically manifests with sterile pustules on the palms and soles; however, the rapid onset of ulcerative, necrotic plaques and substantial edema are more specific to NDDH. Poststreptococcal pustulosis generally follows a streptococcal infection and lacks the violaceous undermined borders seen in NDDH. Reactive arthritis manifests with hyperkeratotic plaques and is associated with the clinical triad of urethritis, conjunctivitis, and arthritis, which were absent in our patient.
The histologic differential diagnosis of NDDH includes infection, pyoderma gangrenosum, bowel-associated dermatosis-arthritis syndrome, rheumatoid neutrophilic dermatitis, and erythema elevatum diutinum3,4; however, these conditions typically manifest with distinct clinical features that allow for differentiation, despite histologic similarities. The wide histologic spectrum of neutrophilic dermatosis may contribute to variable clinical manifestations and an evolving disease concept, as the classification of NDDH has changed from a primary vasculitis to a variant of SS. However, this evolution does not affect the appropriate management, as they all have shown good response to corticosteroid treatment.4,6
- Strutton G, Weedon D, Robertson I. Pustular vasculitis of the hands. J Am Acad Dermatol. 1995;32(2 pt 1):192-198.
- Galaria NA, Junkins-Hopkins JM, Kligman D, et al. Neutrophilic dermatosis of the dorsal hands: pustular vasculitis revisited. J Am Acad Dermatol. 2000;43(5 pt 1):870-874.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63
- Micallef D, Bonnici M, Pisani D, et al. Neutrophilic dermatosis of the dorsal hands: a review of 123 cases. J Am Acad Dermatol. 2023;88:1338-1344.
- Arandes-Marcocci J, Altemir-Vidal A, Iglesias-Plaza A, et al. Neutrophilic dermatosis of the hands with palmar involvement: does it have clinical implication? Int J Dermatol. 2020;59:736-738.
- Del Pozo J, Sacristán F, Martínez W, et al. Neutrophilic dermatosis of the hands: presentation of eight cases and review of the literature. J Dermatol. 2007;34:243-247.
- Strutton G, Weedon D, Robertson I. Pustular vasculitis of the hands. J Am Acad Dermatol. 1995;32(2 pt 1):192-198.
- Galaria NA, Junkins-Hopkins JM, Kligman D, et al. Neutrophilic dermatosis of the dorsal hands: pustular vasculitis revisited. J Am Acad Dermatol. 2000;43(5 pt 1):870-874.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63
- Micallef D, Bonnici M, Pisani D, et al. Neutrophilic dermatosis of the dorsal hands: a review of 123 cases. J Am Acad Dermatol. 2023;88:1338-1344.
- Arandes-Marcocci J, Altemir-Vidal A, Iglesias-Plaza A, et al. Neutrophilic dermatosis of the hands with palmar involvement: does it have clinical implication? Int J Dermatol. 2020;59:736-738.
- Del Pozo J, Sacristán F, Martínez W, et al. Neutrophilic dermatosis of the hands: presentation of eight cases and review of the literature. J Dermatol. 2007;34:243-247.
Acute Pustular Eruption on the Hands
Acute Pustular Eruption on the Hands
A 56-year-old woman was referred to the dermatology department for a painful acral pustular eruption of 6 days’ duration. Her medical history was otherwise unremarkable. Physical examination revealed multiple pustules on the hands with large blisters on an erythematous base and painful surface ulceration (top). Papulonodular infiltrated lesions also were observed on the dorsal aspect of the hands (bottom). There were no additional systemic symptoms. Routine laboratory tests showed hyperleukocytosis at 17.9×103/mm3 (reference range, 4-10×103/mm3) with neutrophils at 12.3×103/mm3 (1.8-7.5×103/mm3) and elevated C-reactive protein at 67 mg/L (<5 mg/L). Screening for hematologic neoplasms, solid tumors, and inflammatory bowel disease was negative. An incisional biopsy was performed on a pustule on the palm of the left hand.

Spreading Ulcerations and Lymphadenopathy in a Traveler Returning from Costa Rica
Spreading Ulcerations and Lymphadenopathy in a Traveler Returning from Costa Rica
THE DIAGNOSIS: Cutaneous Leishmaniasis
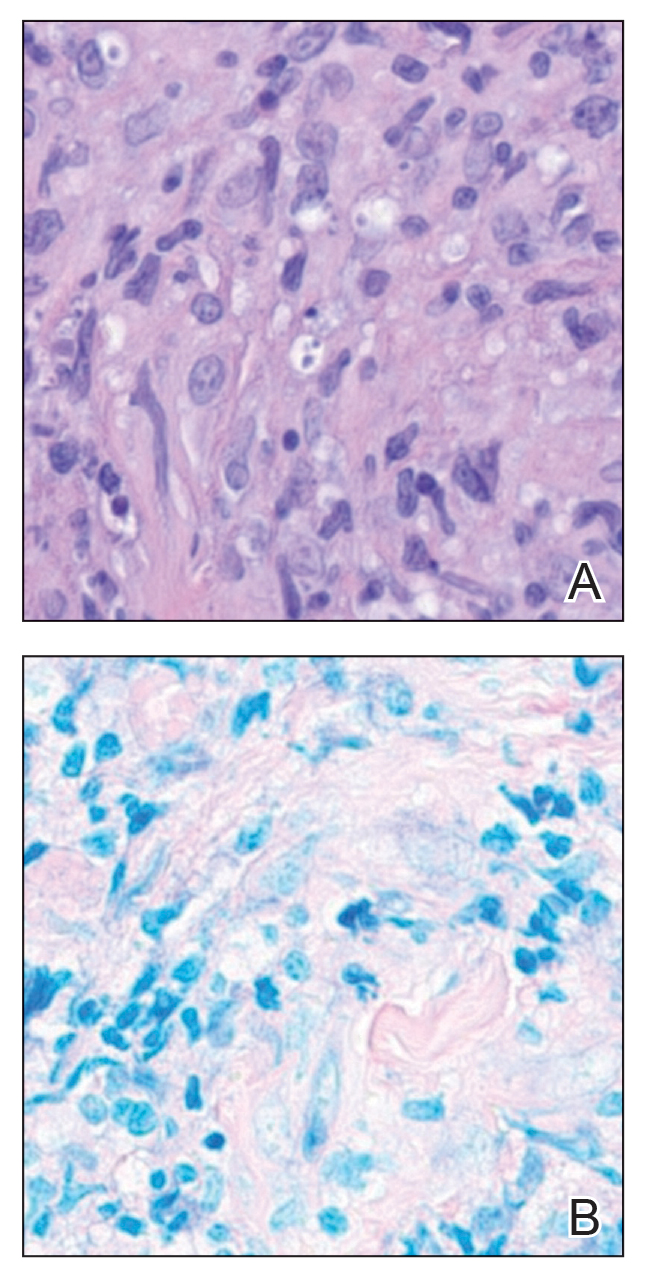
The biopsy results revealed amastigotes at the periphery of parasitized histiocytes, consistent with a diagnosis of cutaneous leishmaniasis. Polymerase chain reaction analysis revealed Leishmania guyanensis species complex, which includes both L guyanensis and Leishmania panamensis. In this case of disseminated cutaneous leishmaniasis (Figure 1), our patient received a prolonged course of systemic therapy with oral miltefosine 50 mg 3 times daily. At the most recent follow-up appointment, she showed ongoing resolution of ulcerations, subcutaneous plaques, and lymphadenopathy on the trunk and face, but development of subcutaneous nodules continued on the arms and legs. At the next follow-up, physical examination revealed that the lesions slowly started to fade.
Leishmania species are parasites transmitted by bites of female sand flies, which belong to the genera Phlebotomus (Old World, Eastern Hemisphere) and Lutzomyia (New World, Western Hemisphere) genera.1 Leishmania species have a complex life cycle, propagating within human macrophages, ultimately leading to cutaneous, mucocutaneous, and visceral disease manifestations.2 Cutaneous leishmaniasis manifests classically as scattered, painless, slow-healing ulcers.3 A biopsy taken from the edge of a cutaneous ulcer for hematoxylin and eosin processing is recommended for initial diagnosis, and subsequent polymerase chain reaction of the sample is required for speciation, which guides therapeutic options.4,5 Classic hematoxylin and eosin and Giemsa stain findings include amastigotes lining the edges of parasitized histiocytes (Figure 2).
Systemic treatment options include sodium stibogluconate, amphotericin B, pentamidine, paromomycin, miltefosine, and azole antifungals.2,5 Geography often plays a critical role in selecting treatment options due to resistance rates of individual Leishmania species; for example, paromomycin compounds are more effective for cutaneous disease caused by Leishmania major than Leishmania tropica. Miltefosine is not effective for treating Leishmania braziliensis which can be acquired outside Guatemala, and higher doses of amphotericin B are recommended for visceral disease from East Africa.2,5 In patients with cutaneous leishmaniasis caused by L guyanensis, miltefosine remains a first-line option due to its oral formulation and long half-life within organisms, though there is a risk for teratogenicity.2 Amphotericin B remains the most effective treatment for visceral leishmaniasis and can be used off label to treat mucocutaneous disease or when cutaneous disease is refractory to other treatment options.3
Given the potential of L guyanensis to progress to mucocutaneous disease, monitoring for mucosal involvement should be performed at regular intervals for 6 months to 1 year.2 Treatment may be considered efficacious if no new skin lesions occur after 4 to 6 weeks of therapy; existing skin lesions should be re-epithelializing and reduced by 50% in size, with most cutaneous disease adequately controlled after 3 months of therapy.2
- Olivier M, Minguez-Menendez A, Fernandez-Prada C. Leishmania viannia guyanensis. Trends Parasitol. 2019;35:1018-1019. doi:10.1016 /j.pt.2019.06.008
- Singh R, Kashif M, Srivastava P, et al. Recent advances in chemotherapeutics for leishmaniasis: importance of the cellular biochemistry of the parasite and its molecular interaction with the host. Pathogens. 2023;12:706. doi:10.3390/pathogens12050706
- Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin Infect Dis. 2016;63: 1539-1557. doi:10.1093/cid/ciw742
- Specimen Collection Guide for Laboratory Diagnosis of Leishmaniasis. Centers for Disease Control and Prevention. Accessed October 14, 2025. https://www.cdc.gov/dpdx/diagnosticprocedures /other/leish.html
- Aronson NE, Joya CA. Cutaneous leishmaniasis: updates in diagnosis and management. Infect Dis Clin North Am. 2019;33:101-117. doi:10.1016/j.idc.2018.10.004
THE DIAGNOSIS: Cutaneous Leishmaniasis
The biopsy results revealed amastigotes at the periphery of parasitized histiocytes, consistent with a diagnosis of cutaneous leishmaniasis. Polymerase chain reaction analysis revealed Leishmania guyanensis species complex, which includes both L guyanensis and Leishmania panamensis. In this case of disseminated cutaneous leishmaniasis (Figure 1), our patient received a prolonged course of systemic therapy with oral miltefosine 50 mg 3 times daily. At the most recent follow-up appointment, she showed ongoing resolution of ulcerations, subcutaneous plaques, and lymphadenopathy on the trunk and face, but development of subcutaneous nodules continued on the arms and legs. At the next follow-up, physical examination revealed that the lesions slowly started to fade.
Leishmania species are parasites transmitted by bites of female sand flies, which belong to the genera Phlebotomus (Old World, Eastern Hemisphere) and Lutzomyia (New World, Western Hemisphere) genera.1 Leishmania species have a complex life cycle, propagating within human macrophages, ultimately leading to cutaneous, mucocutaneous, and visceral disease manifestations.2 Cutaneous leishmaniasis manifests classically as scattered, painless, slow-healing ulcers.3 A biopsy taken from the edge of a cutaneous ulcer for hematoxylin and eosin processing is recommended for initial diagnosis, and subsequent polymerase chain reaction of the sample is required for speciation, which guides therapeutic options.4,5 Classic hematoxylin and eosin and Giemsa stain findings include amastigotes lining the edges of parasitized histiocytes (Figure 2).
Systemic treatment options include sodium stibogluconate, amphotericin B, pentamidine, paromomycin, miltefosine, and azole antifungals.2,5 Geography often plays a critical role in selecting treatment options due to resistance rates of individual Leishmania species; for example, paromomycin compounds are more effective for cutaneous disease caused by Leishmania major than Leishmania tropica. Miltefosine is not effective for treating Leishmania braziliensis which can be acquired outside Guatemala, and higher doses of amphotericin B are recommended for visceral disease from East Africa.2,5 In patients with cutaneous leishmaniasis caused by L guyanensis, miltefosine remains a first-line option due to its oral formulation and long half-life within organisms, though there is a risk for teratogenicity.2 Amphotericin B remains the most effective treatment for visceral leishmaniasis and can be used off label to treat mucocutaneous disease or when cutaneous disease is refractory to other treatment options.3
Given the potential of L guyanensis to progress to mucocutaneous disease, monitoring for mucosal involvement should be performed at regular intervals for 6 months to 1 year.2 Treatment may be considered efficacious if no new skin lesions occur after 4 to 6 weeks of therapy; existing skin lesions should be re-epithelializing and reduced by 50% in size, with most cutaneous disease adequately controlled after 3 months of therapy.2
THE DIAGNOSIS: Cutaneous Leishmaniasis
The biopsy results revealed amastigotes at the periphery of parasitized histiocytes, consistent with a diagnosis of cutaneous leishmaniasis. Polymerase chain reaction analysis revealed Leishmania guyanensis species complex, which includes both L guyanensis and Leishmania panamensis. In this case of disseminated cutaneous leishmaniasis (Figure 1), our patient received a prolonged course of systemic therapy with oral miltefosine 50 mg 3 times daily. At the most recent follow-up appointment, she showed ongoing resolution of ulcerations, subcutaneous plaques, and lymphadenopathy on the trunk and face, but development of subcutaneous nodules continued on the arms and legs. At the next follow-up, physical examination revealed that the lesions slowly started to fade.
Leishmania species are parasites transmitted by bites of female sand flies, which belong to the genera Phlebotomus (Old World, Eastern Hemisphere) and Lutzomyia (New World, Western Hemisphere) genera.1 Leishmania species have a complex life cycle, propagating within human macrophages, ultimately leading to cutaneous, mucocutaneous, and visceral disease manifestations.2 Cutaneous leishmaniasis manifests classically as scattered, painless, slow-healing ulcers.3 A biopsy taken from the edge of a cutaneous ulcer for hematoxylin and eosin processing is recommended for initial diagnosis, and subsequent polymerase chain reaction of the sample is required for speciation, which guides therapeutic options.4,5 Classic hematoxylin and eosin and Giemsa stain findings include amastigotes lining the edges of parasitized histiocytes (Figure 2).
Systemic treatment options include sodium stibogluconate, amphotericin B, pentamidine, paromomycin, miltefosine, and azole antifungals.2,5 Geography often plays a critical role in selecting treatment options due to resistance rates of individual Leishmania species; for example, paromomycin compounds are more effective for cutaneous disease caused by Leishmania major than Leishmania tropica. Miltefosine is not effective for treating Leishmania braziliensis which can be acquired outside Guatemala, and higher doses of amphotericin B are recommended for visceral disease from East Africa.2,5 In patients with cutaneous leishmaniasis caused by L guyanensis, miltefosine remains a first-line option due to its oral formulation and long half-life within organisms, though there is a risk for teratogenicity.2 Amphotericin B remains the most effective treatment for visceral leishmaniasis and can be used off label to treat mucocutaneous disease or when cutaneous disease is refractory to other treatment options.3
Given the potential of L guyanensis to progress to mucocutaneous disease, monitoring for mucosal involvement should be performed at regular intervals for 6 months to 1 year.2 Treatment may be considered efficacious if no new skin lesions occur after 4 to 6 weeks of therapy; existing skin lesions should be re-epithelializing and reduced by 50% in size, with most cutaneous disease adequately controlled after 3 months of therapy.2
- Olivier M, Minguez-Menendez A, Fernandez-Prada C. Leishmania viannia guyanensis. Trends Parasitol. 2019;35:1018-1019. doi:10.1016 /j.pt.2019.06.008
- Singh R, Kashif M, Srivastava P, et al. Recent advances in chemotherapeutics for leishmaniasis: importance of the cellular biochemistry of the parasite and its molecular interaction with the host. Pathogens. 2023;12:706. doi:10.3390/pathogens12050706
- Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin Infect Dis. 2016;63: 1539-1557. doi:10.1093/cid/ciw742
- Specimen Collection Guide for Laboratory Diagnosis of Leishmaniasis. Centers for Disease Control and Prevention. Accessed October 14, 2025. https://www.cdc.gov/dpdx/diagnosticprocedures /other/leish.html
- Aronson NE, Joya CA. Cutaneous leishmaniasis: updates in diagnosis and management. Infect Dis Clin North Am. 2019;33:101-117. doi:10.1016/j.idc.2018.10.004
- Olivier M, Minguez-Menendez A, Fernandez-Prada C. Leishmania viannia guyanensis. Trends Parasitol. 2019;35:1018-1019. doi:10.1016 /j.pt.2019.06.008
- Singh R, Kashif M, Srivastava P, et al. Recent advances in chemotherapeutics for leishmaniasis: importance of the cellular biochemistry of the parasite and its molecular interaction with the host. Pathogens. 2023;12:706. doi:10.3390/pathogens12050706
- Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin Infect Dis. 2016;63: 1539-1557. doi:10.1093/cid/ciw742
- Specimen Collection Guide for Laboratory Diagnosis of Leishmaniasis. Centers for Disease Control and Prevention. Accessed October 14, 2025. https://www.cdc.gov/dpdx/diagnosticprocedures /other/leish.html
- Aronson NE, Joya CA. Cutaneous leishmaniasis: updates in diagnosis and management. Infect Dis Clin North Am. 2019;33:101-117. doi:10.1016/j.idc.2018.10.004
Spreading Ulcerations and Lymphadenopathy in a Traveler Returning from Costa Rica
Spreading Ulcerations and Lymphadenopathy in a Traveler Returning from Costa Rica
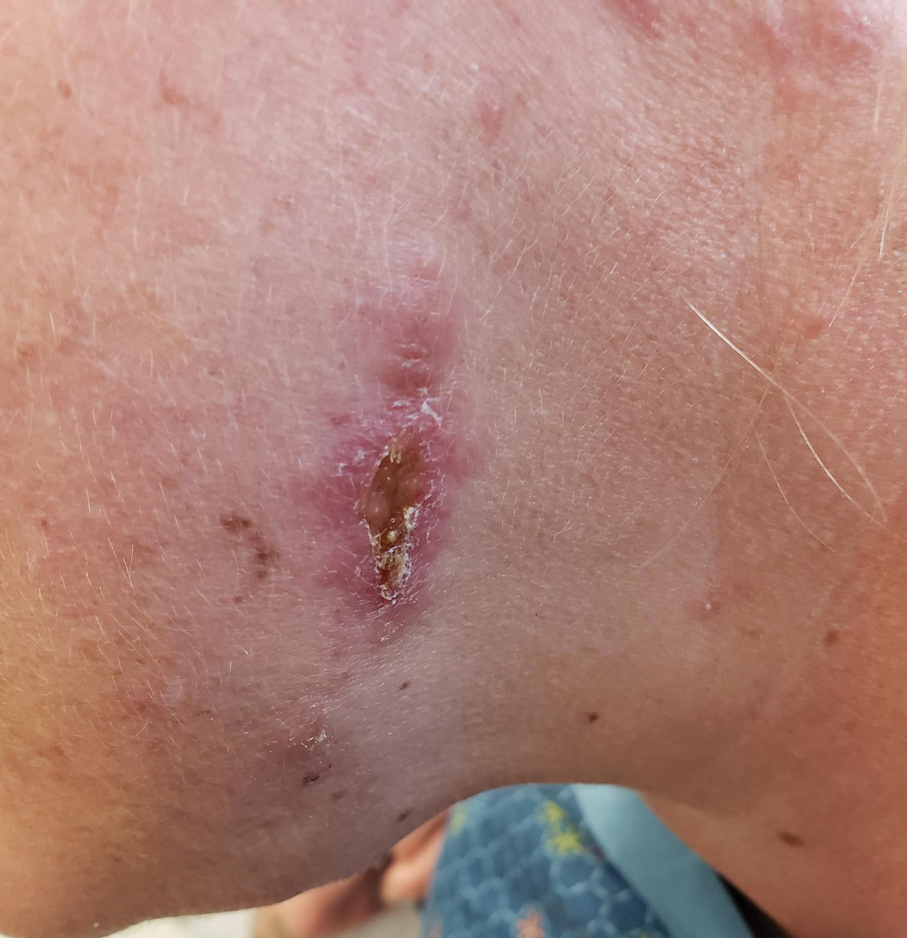
A 43-year-old woman presented to the dermatology clinic with widespread scaly plaques and ulcerations of 2 months’ duration. Her medical history was otherwise unremarkable. The patient reported that the eruption began after returning from a vacation to Costa Rica, during which she spent time on the beach and white-water rafting. She noted that she had been exposed to numerous insects during her trip, and that her roommate, who had accompanied her, had similar exposure history and lesions. The plaques were refractory to multiple oral antibiotics previously prescribed by primary care. Physical examination revealed submental lymphadenopathy and painless ulcerations with indurated borders without purulent drainage alongside scattered scaly papules and plaques on the face, neck, arms, and legs. A biopsy was taken from an ulceration edge on the left thigh.

Crusted Lesion at the Implantation Site of a Pacemaker
Crusted Lesion at the Implantation Site of a Pacemaker
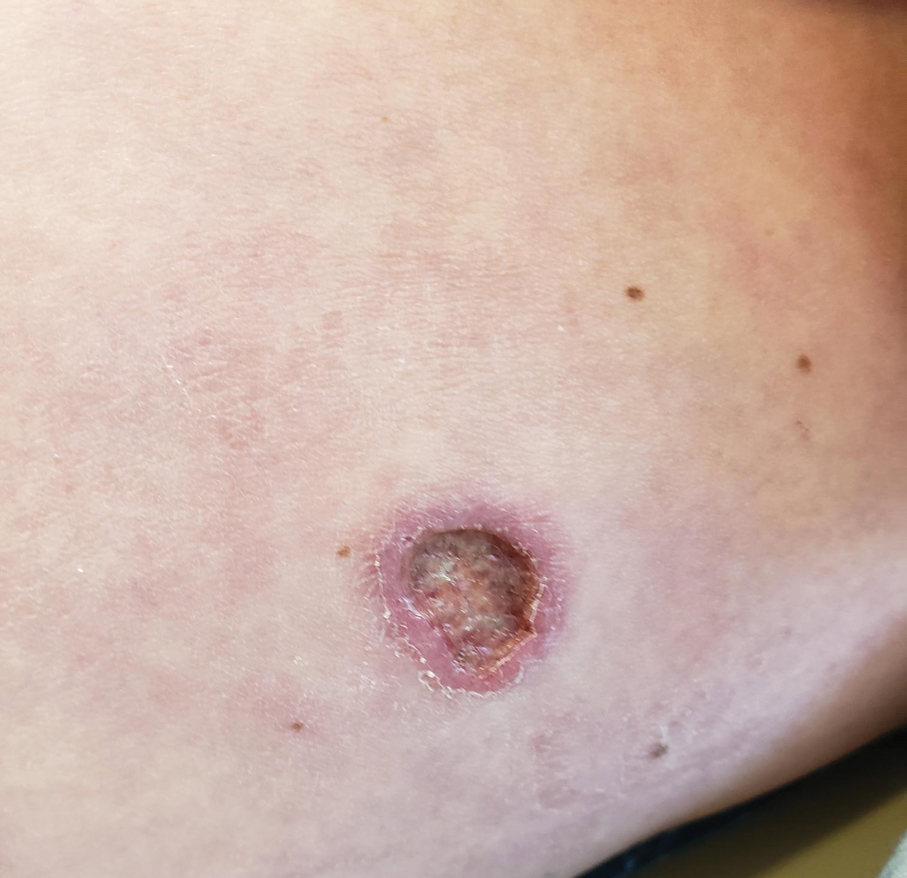
THE DIAGNOSIS: Pacemaker Extrusion
The lesion crust was easily scraped away to reveal extrusion of the permanent pacemaker (PPM) through the skin with a visible overlying gelatinous biofilm (Figure). The patient subsequently completed a 2-week course of clindamycin 300 mg 3 times daily followed by generator and lead removal, with reimplantation of the PPM into the right chest, as is the standard of care in the treatment of pacemaker extrusion.1
Ours is the first known reported case of pacemaker extrusion referred to dermatology with a primary concern for cutaneous malignancy. Pacemaker extrusion through the skin is not common, but it is the most common complication of PPM implantation, followed by infection.1 Pacemaker extrusion results from pressure necrosis and occurs when the PPM emerges through erythematous skin.1,2 Pacemaker extrusions generally are diagnosed by cardiology; however, it is important for dermatologists to recognize this phenomenon and differentiate it from other cutaneous pathologies, as the morphology of skin changes related to pacemaker extrusion through the skin can mimic cutaneous malignancy or other primary skin disease, especially if the outer layer of a biofilm that forms around the PPM hardens to form a crust. Our case emphasizes the importance of removing crusts when evaluating lesions.3
- Harcombe AA, Newell SA, Ludman PF, et al. Late complications following permanent pacemaker implantation or elective unit replacement. Heart. 1998;80:240-244. doi:10.1136/hrt.80.3.240
- Sanderson A, Hahn B. Pacemaker extrusion. Ann Emerg Med. 2013;62:648. doi:10.1016/j.annemergmed.2013.04.022
- Andrade AC, Hayashida MZ, Enokihara MMSES, et al. Dermoscopy of crusted lesion: diagnostic challenge and choice of technique for the analysis. An Bras Dermatol. 2021;96:387-388. doi:10.1016/j.abd.2020.06.016
THE DIAGNOSIS: Pacemaker Extrusion
The lesion crust was easily scraped away to reveal extrusion of the permanent pacemaker (PPM) through the skin with a visible overlying gelatinous biofilm (Figure). The patient subsequently completed a 2-week course of clindamycin 300 mg 3 times daily followed by generator and lead removal, with reimplantation of the PPM into the right chest, as is the standard of care in the treatment of pacemaker extrusion.1
Ours is the first known reported case of pacemaker extrusion referred to dermatology with a primary concern for cutaneous malignancy. Pacemaker extrusion through the skin is not common, but it is the most common complication of PPM implantation, followed by infection.1 Pacemaker extrusion results from pressure necrosis and occurs when the PPM emerges through erythematous skin.1,2 Pacemaker extrusions generally are diagnosed by cardiology; however, it is important for dermatologists to recognize this phenomenon and differentiate it from other cutaneous pathologies, as the morphology of skin changes related to pacemaker extrusion through the skin can mimic cutaneous malignancy or other primary skin disease, especially if the outer layer of a biofilm that forms around the PPM hardens to form a crust. Our case emphasizes the importance of removing crusts when evaluating lesions.3
THE DIAGNOSIS: Pacemaker Extrusion
The lesion crust was easily scraped away to reveal extrusion of the permanent pacemaker (PPM) through the skin with a visible overlying gelatinous biofilm (Figure). The patient subsequently completed a 2-week course of clindamycin 300 mg 3 times daily followed by generator and lead removal, with reimplantation of the PPM into the right chest, as is the standard of care in the treatment of pacemaker extrusion.1
Ours is the first known reported case of pacemaker extrusion referred to dermatology with a primary concern for cutaneous malignancy. Pacemaker extrusion through the skin is not common, but it is the most common complication of PPM implantation, followed by infection.1 Pacemaker extrusion results from pressure necrosis and occurs when the PPM emerges through erythematous skin.1,2 Pacemaker extrusions generally are diagnosed by cardiology; however, it is important for dermatologists to recognize this phenomenon and differentiate it from other cutaneous pathologies, as the morphology of skin changes related to pacemaker extrusion through the skin can mimic cutaneous malignancy or other primary skin disease, especially if the outer layer of a biofilm that forms around the PPM hardens to form a crust. Our case emphasizes the importance of removing crusts when evaluating lesions.3
- Harcombe AA, Newell SA, Ludman PF, et al. Late complications following permanent pacemaker implantation or elective unit replacement. Heart. 1998;80:240-244. doi:10.1136/hrt.80.3.240
- Sanderson A, Hahn B. Pacemaker extrusion. Ann Emerg Med. 2013;62:648. doi:10.1016/j.annemergmed.2013.04.022
- Andrade AC, Hayashida MZ, Enokihara MMSES, et al. Dermoscopy of crusted lesion: diagnostic challenge and choice of technique for the analysis. An Bras Dermatol. 2021;96:387-388. doi:10.1016/j.abd.2020.06.016
- Harcombe AA, Newell SA, Ludman PF, et al. Late complications following permanent pacemaker implantation or elective unit replacement. Heart. 1998;80:240-244. doi:10.1136/hrt.80.3.240
- Sanderson A, Hahn B. Pacemaker extrusion. Ann Emerg Med. 2013;62:648. doi:10.1016/j.annemergmed.2013.04.022
- Andrade AC, Hayashida MZ, Enokihara MMSES, et al. Dermoscopy of crusted lesion: diagnostic challenge and choice of technique for the analysis. An Bras Dermatol. 2021;96:387-388. doi:10.1016/j.abd.2020.06.016
Crusted Lesion at the Implantation Site of a Pacemaker
Crusted Lesion at the Implantation Site of a Pacemaker
A 78-year-old woman was referred to dermatology from the cardiology clinic with concerns of a nonhealing, scablike lesion on the left chest over the implantation site of a dual-chamber permanent pacemaker (PPM). Eight months prior, the patient underwent successful PPM implantation for symptomatic bradycardia and second-degree atrioventricular block. Her cardiologists subsequently noticed an oozing crusting scab at the site of implantation and eventually referred her to dermatology with concerns for squamous cell carcinoma. Physical examination at the current presentation revealed an exophytic serous crust overlying the PPM implantation site on the left chest.
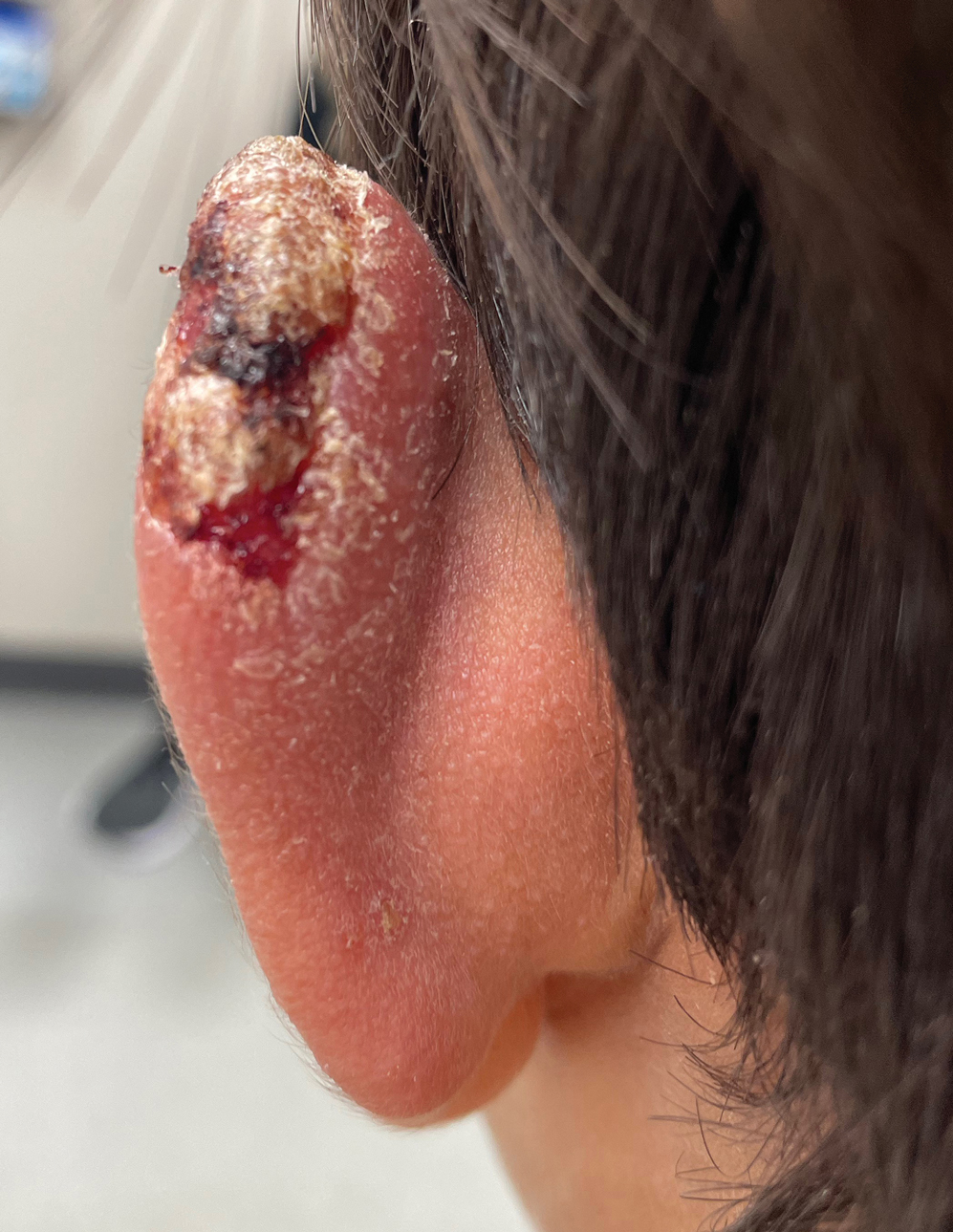
Nonhealing Lesion on the Ear in a Child
Nonhealing Lesion on the Ear in a Child
THE DIAGNOSIS: Cutaneous Leishmaniasis
The biopsy results demonstrated a nonspecific chronic granulomatous inflammatory infiltrate, including few multinucleated histiocytes, a surrounding mixed inflammatory infiltrate, mostly mature lymphocytes, few plasma cells, and fragmented neutrophils. A special stain panel was negative for acid-fast bacilli (AFB), Fite, and periodic acid–Schiff for fungi. Bacterial cultures from biopsy tissue grew normal skin flora, and both fungal and AFB cultures were negative. A second punch biopsy was recommended by infectious disease due to clinical suspicion of cutaneous leishmaniasis (CL). Histopathology showed nonnecrotizing granulomas with dense lymphoplasmacytic inflammation and negative Giemsa staining for Leishmania amastigotes; however, it was concluded by pathology that the reason for the negative Leishmania staining was the late stage of the disease, indicated by the presence of granulomas, which can make visualization of organisms difficult. Nonetheless, universal polymerase chain reaction (PCR) testing was positive for Leishmania tropica. Thus, although microscopic analysis was negative for visualization of Leishmania amastigotes, molecular analysis via PCR ultimately demonstrated a positive result and confirmed the diagnosis of CL (Figure 1). The variance in diagnostic accuracy exemplified in our case reinforces the need for multimodal diagnosis.
Multiple factors needed to be considered with regard to treatment in our patient, including but not limited to the location of the lesion on a slow-healing cartilaginous surface and the patient’s age. Considering the recalcitrant nature of the lesion and the L tropica strain exhibiting resistance to topical treatments, systemic therapies were the only option. Furthermore, parenteral routes of administration were confounded by the patient’s age, decreasing the likelihood of compliance with therapy. With these variables in mind and recommendations from the Infectious Diseases Society of America and the Centers for Disease Control and Prevention, the best treatment for our patient was deemed to be a 28-day course of oral miltefosine 50 mg twice daily. Compared to the initial presentation, a 1-month follow-up visit after completing the 28-day course of treatment demonstrated flattening of the lesion (Figure 2).
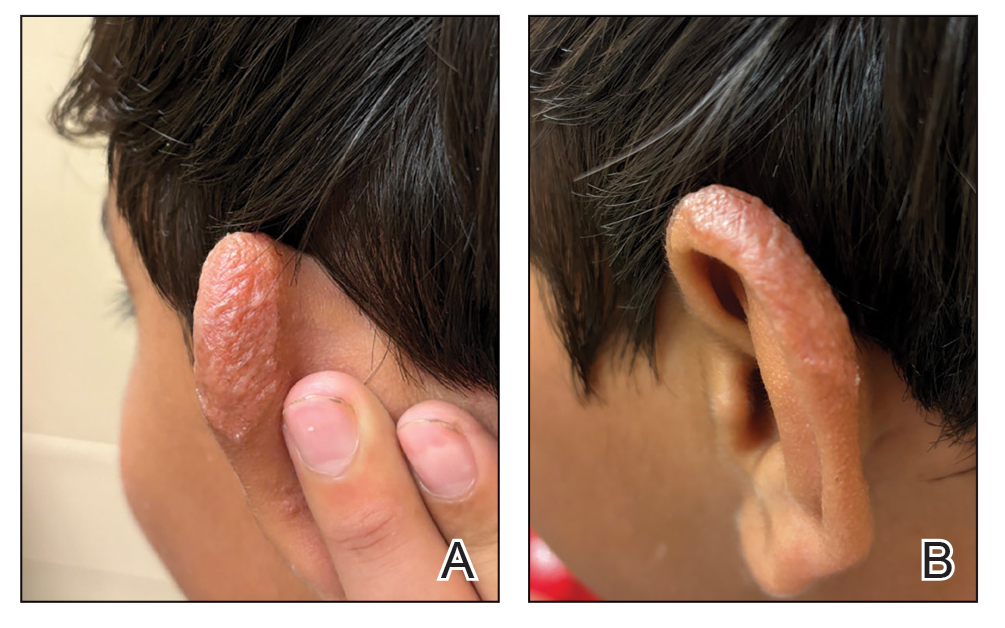
Leishmaniasis is a disease caused by a protozoan parasite of the Leishmania genus, spread via inoculation from the bite of sandfly vectors.1 Cutaneous leishmaniasis is the most common clinical manifestation of leishmaniasis. Other clinical manifestations include mucocutaneous leishmaniasis and visceral leishmaniasis.1,2 Cutaneous leishmaniasis typically manifests as open wounds on areas of skin that may have been exposed to sandfly bites.3 The lesion may not appear until weeks to months or even years after the initial inoculation.2 Initially, CL manifests as papules that may progress to nodular plaques, with eventual evolution to volcanic ulcerations with raised borders and central crateriform indentations covered by scabs or crusting.2,3 The infection may be localized or diffuse—in either case, development of satellite lesions, regional lymphadenopathy, and/or nodular lymphangitis is not uncommon. Generally, CL is not lethal, but the severity of the lesions may vary and can lead to permanent scarring and atrophy.2 Many cases of CL remain undiagnosed because of its appearance as a nonspecific ulcer that can mimic many other cutaneous lesions and because it generally heals spontaneously, leaving only scarring as an indicator of prior infection.4 Thus, CL requires a high diagnostic suspicion, as it can have a nonspecific presentation and is rare in nonendemic regions.
Diagnosis of CL is accomplished via microscopy, isoenzyme analysis, or serology or is made molecularly.3 Microscopic diagnosis includes visualization of Leishmania amastigotes, the stage of replication that occurs after the promastigote stage is phagocytosed by macrophages.3 Amastigote is the only stage that can be visualized in human tissue and is stained via Giemsa and/or hematoxylin and eosin.3 However, Leishmania amastigotes are morphologically indistinguishable from Trypanosoma cruzi amastigotes on microscopy, thus limiting diagnostic accuracy.3 Moreover, there is potential for missed diagnosis of persistent CL caused by L tropica due to fewer parasites being present, further complicating the diagnosis.5 In these cases, molecular diagnostics are helpful as they have higher sensitivity and quicker results. Additionally, DNA technologies can differentiate strains, which is beneficial for guiding treatment. Isoenzyme analysis also can help identify Leishmania species, although results can take weeks to return.3 Serologic testing is useful for suspected visceral leishmaniasis despite negative definitive diagnoses or conflicts with conducting definitive studies; however, there is not a strong antibody response in CL, thus serology is ineffective.3,5 Furthermore, serology can have cross-reactivity with T cruzi and cannot be used to assess for treatment response.3,5 The Infectious Diseases Society of America guidelines for diagnosis of leishmaniasis recommend using multiple methods to ensure a positive result, with molecular assays being the most sensitive.5
Differential diagnoses include any cause of cutaneous ulcerated lesions, including but not limited to mycobacterial or fungal infections. Leprosy often initially manifests with a hypopigmented macule with a raised border, although there often are associated neuropathic symptoms.6 Cutaneous tuberculosis is an extremely rare manifestation that occurs via direct inoculation of the mycobacterium, occurring primarily in children. Initially, it may manifest as a firm red papule that progresses to a painless shallow ulcer with a granular base.7 Cutaneous chromoblastomycosis is a fungal infection resulting from an initial cutaneous injury, similar to our patient, followed by a slow-developing warty lesion that may heal into ivory scars or spread as plaques on normal skin.8 The verrucous lesions seen in cutaneous chromoblastomycosis tend to manifest on the lower extremities and are unlikely to manifest on the head. Sarcoidosis is another granulomatous skin eruption that can be clinically nonspecific.9 Histologically, lesions may demonstrate noncaseating granulomatous inflammation, as seen with cutaneous leishmaniasis, with a broad presentation; for example, lupus pernio, a sarcoid variant, manifests as large blue-red dusky nodules/plaques on the face, ears, or digits.9 Other sarcoid lesions include red/brown, thickened, circular plaques; variably discolored papulonodular lesions; or mucosal involvement.9 Ultimately, it is important to differentiate these nonspecific and similarly appearing lesions through diagnostic techniques such as AFB culture and smear, fungal staining, tuberculosis testing, and PCR in more challenging cases.
Treatment of cutaneous leishmaniasis should be individualized to each case.5 A more than 50% reduction in lesion size within 4 to 6 weeks indicates successful treatment. Ulcerated lesions should be fully re-epithelialized and healed by 3 months posttreatment. Treatment failure is categorized by failure of reepithelization, incomplete healing by 3 months, or worsening of the lesion at any time, each necessitating additional treatment, such as a second course of miltefosine or a different medication regimen.5 Careful monitoring is required throughout treatment, assessing for treatment failure, adding to the challenges of leishmaniasis.
In conclusion, CL requires a high index of suspicion in nonendemic areas to ensure successful diagnosis and treatment. Our case highlights the importance of using multimodal diagnostic techniques for CL, as a single modality may not exhibit a positive result due to variations in diagnostic accuracy. Our case also exhibits the complex treatment of CL, and the considerations that should be made when choosing a treatment modality.
- Leishmaniasis. World Health Organization. Accessed September 14, 2024. https://www.who.int/news-room/fact-sheets/detail/leishmaniasis
- Clinical overview of leishmaniasis. Centers for Disease Control and Prevention. Accessed September 14, 2024. https://www.cdc.gov/leishmaniasis/hcp/clinical-overview/index.html
- CDC DPDx Leishmaniasis. Centers for Disease Control and Prevention. Accessed September 15, 2024. https://www.cdc.gov/dpdx/leishmaniasis/index.html
- Stark CG. Leishmaniasis differential diagnoses. Medscape. March 21, 2023. Accessed October 12, 2024. https://emedicine.medscape.com/article/220298-differential
- Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin Infect Dis. 2016;63:E202-E264. doi:10.1093/cid/ciw670
- Lewis FS. Dermatologic manifestations of leprosy. Medscape. June 19, 2023. Accessed October 12, 2024. https://emedicine.medscape.com/article/1104977-overview
- Ngan V. Cutaneous tuberculosis. DermNet. Accessed October 12, 2024. https://dermnetnz.org/topics/cutaneous-tuberculosis
- Schwartz RA. Chromoblastomycosis. Medscape. May 13, 2023. Accessed October 12, 2024. https://emedicine.medscape.com/article/1092695-overview#a4
- Elyoussfi S, Coulson I. Sarcoidosis. DermNet. May 31, 2024. Accessed October 25, 2024. https://dermnetnz.org/topics/sarcoidosis
THE DIAGNOSIS: Cutaneous Leishmaniasis
The biopsy results demonstrated a nonspecific chronic granulomatous inflammatory infiltrate, including few multinucleated histiocytes, a surrounding mixed inflammatory infiltrate, mostly mature lymphocytes, few plasma cells, and fragmented neutrophils. A special stain panel was negative for acid-fast bacilli (AFB), Fite, and periodic acid–Schiff for fungi. Bacterial cultures from biopsy tissue grew normal skin flora, and both fungal and AFB cultures were negative. A second punch biopsy was recommended by infectious disease due to clinical suspicion of cutaneous leishmaniasis (CL). Histopathology showed nonnecrotizing granulomas with dense lymphoplasmacytic inflammation and negative Giemsa staining for Leishmania amastigotes; however, it was concluded by pathology that the reason for the negative Leishmania staining was the late stage of the disease, indicated by the presence of granulomas, which can make visualization of organisms difficult. Nonetheless, universal polymerase chain reaction (PCR) testing was positive for Leishmania tropica. Thus, although microscopic analysis was negative for visualization of Leishmania amastigotes, molecular analysis via PCR ultimately demonstrated a positive result and confirmed the diagnosis of CL (Figure 1). The variance in diagnostic accuracy exemplified in our case reinforces the need for multimodal diagnosis.
Multiple factors needed to be considered with regard to treatment in our patient, including but not limited to the location of the lesion on a slow-healing cartilaginous surface and the patient’s age. Considering the recalcitrant nature of the lesion and the L tropica strain exhibiting resistance to topical treatments, systemic therapies were the only option. Furthermore, parenteral routes of administration were confounded by the patient’s age, decreasing the likelihood of compliance with therapy. With these variables in mind and recommendations from the Infectious Diseases Society of America and the Centers for Disease Control and Prevention, the best treatment for our patient was deemed to be a 28-day course of oral miltefosine 50 mg twice daily. Compared to the initial presentation, a 1-month follow-up visit after completing the 28-day course of treatment demonstrated flattening of the lesion (Figure 2).
Leishmaniasis is a disease caused by a protozoan parasite of the Leishmania genus, spread via inoculation from the bite of sandfly vectors.1 Cutaneous leishmaniasis is the most common clinical manifestation of leishmaniasis. Other clinical manifestations include mucocutaneous leishmaniasis and visceral leishmaniasis.1,2 Cutaneous leishmaniasis typically manifests as open wounds on areas of skin that may have been exposed to sandfly bites.3 The lesion may not appear until weeks to months or even years after the initial inoculation.2 Initially, CL manifests as papules that may progress to nodular plaques, with eventual evolution to volcanic ulcerations with raised borders and central crateriform indentations covered by scabs or crusting.2,3 The infection may be localized or diffuse—in either case, development of satellite lesions, regional lymphadenopathy, and/or nodular lymphangitis is not uncommon. Generally, CL is not lethal, but the severity of the lesions may vary and can lead to permanent scarring and atrophy.2 Many cases of CL remain undiagnosed because of its appearance as a nonspecific ulcer that can mimic many other cutaneous lesions and because it generally heals spontaneously, leaving only scarring as an indicator of prior infection.4 Thus, CL requires a high diagnostic suspicion, as it can have a nonspecific presentation and is rare in nonendemic regions.
Diagnosis of CL is accomplished via microscopy, isoenzyme analysis, or serology or is made molecularly.3 Microscopic diagnosis includes visualization of Leishmania amastigotes, the stage of replication that occurs after the promastigote stage is phagocytosed by macrophages.3 Amastigote is the only stage that can be visualized in human tissue and is stained via Giemsa and/or hematoxylin and eosin.3 However, Leishmania amastigotes are morphologically indistinguishable from Trypanosoma cruzi amastigotes on microscopy, thus limiting diagnostic accuracy.3 Moreover, there is potential for missed diagnosis of persistent CL caused by L tropica due to fewer parasites being present, further complicating the diagnosis.5 In these cases, molecular diagnostics are helpful as they have higher sensitivity and quicker results. Additionally, DNA technologies can differentiate strains, which is beneficial for guiding treatment. Isoenzyme analysis also can help identify Leishmania species, although results can take weeks to return.3 Serologic testing is useful for suspected visceral leishmaniasis despite negative definitive diagnoses or conflicts with conducting definitive studies; however, there is not a strong antibody response in CL, thus serology is ineffective.3,5 Furthermore, serology can have cross-reactivity with T cruzi and cannot be used to assess for treatment response.3,5 The Infectious Diseases Society of America guidelines for diagnosis of leishmaniasis recommend using multiple methods to ensure a positive result, with molecular assays being the most sensitive.5
Differential diagnoses include any cause of cutaneous ulcerated lesions, including but not limited to mycobacterial or fungal infections. Leprosy often initially manifests with a hypopigmented macule with a raised border, although there often are associated neuropathic symptoms.6 Cutaneous tuberculosis is an extremely rare manifestation that occurs via direct inoculation of the mycobacterium, occurring primarily in children. Initially, it may manifest as a firm red papule that progresses to a painless shallow ulcer with a granular base.7 Cutaneous chromoblastomycosis is a fungal infection resulting from an initial cutaneous injury, similar to our patient, followed by a slow-developing warty lesion that may heal into ivory scars or spread as plaques on normal skin.8 The verrucous lesions seen in cutaneous chromoblastomycosis tend to manifest on the lower extremities and are unlikely to manifest on the head. Sarcoidosis is another granulomatous skin eruption that can be clinically nonspecific.9 Histologically, lesions may demonstrate noncaseating granulomatous inflammation, as seen with cutaneous leishmaniasis, with a broad presentation; for example, lupus pernio, a sarcoid variant, manifests as large blue-red dusky nodules/plaques on the face, ears, or digits.9 Other sarcoid lesions include red/brown, thickened, circular plaques; variably discolored papulonodular lesions; or mucosal involvement.9 Ultimately, it is important to differentiate these nonspecific and similarly appearing lesions through diagnostic techniques such as AFB culture and smear, fungal staining, tuberculosis testing, and PCR in more challenging cases.
Treatment of cutaneous leishmaniasis should be individualized to each case.5 A more than 50% reduction in lesion size within 4 to 6 weeks indicates successful treatment. Ulcerated lesions should be fully re-epithelialized and healed by 3 months posttreatment. Treatment failure is categorized by failure of reepithelization, incomplete healing by 3 months, or worsening of the lesion at any time, each necessitating additional treatment, such as a second course of miltefosine or a different medication regimen.5 Careful monitoring is required throughout treatment, assessing for treatment failure, adding to the challenges of leishmaniasis.
In conclusion, CL requires a high index of suspicion in nonendemic areas to ensure successful diagnosis and treatment. Our case highlights the importance of using multimodal diagnostic techniques for CL, as a single modality may not exhibit a positive result due to variations in diagnostic accuracy. Our case also exhibits the complex treatment of CL, and the considerations that should be made when choosing a treatment modality.
THE DIAGNOSIS: Cutaneous Leishmaniasis
The biopsy results demonstrated a nonspecific chronic granulomatous inflammatory infiltrate, including few multinucleated histiocytes, a surrounding mixed inflammatory infiltrate, mostly mature lymphocytes, few plasma cells, and fragmented neutrophils. A special stain panel was negative for acid-fast bacilli (AFB), Fite, and periodic acid–Schiff for fungi. Bacterial cultures from biopsy tissue grew normal skin flora, and both fungal and AFB cultures were negative. A second punch biopsy was recommended by infectious disease due to clinical suspicion of cutaneous leishmaniasis (CL). Histopathology showed nonnecrotizing granulomas with dense lymphoplasmacytic inflammation and negative Giemsa staining for Leishmania amastigotes; however, it was concluded by pathology that the reason for the negative Leishmania staining was the late stage of the disease, indicated by the presence of granulomas, which can make visualization of organisms difficult. Nonetheless, universal polymerase chain reaction (PCR) testing was positive for Leishmania tropica. Thus, although microscopic analysis was negative for visualization of Leishmania amastigotes, molecular analysis via PCR ultimately demonstrated a positive result and confirmed the diagnosis of CL (Figure 1). The variance in diagnostic accuracy exemplified in our case reinforces the need for multimodal diagnosis.
Multiple factors needed to be considered with regard to treatment in our patient, including but not limited to the location of the lesion on a slow-healing cartilaginous surface and the patient’s age. Considering the recalcitrant nature of the lesion and the L tropica strain exhibiting resistance to topical treatments, systemic therapies were the only option. Furthermore, parenteral routes of administration were confounded by the patient’s age, decreasing the likelihood of compliance with therapy. With these variables in mind and recommendations from the Infectious Diseases Society of America and the Centers for Disease Control and Prevention, the best treatment for our patient was deemed to be a 28-day course of oral miltefosine 50 mg twice daily. Compared to the initial presentation, a 1-month follow-up visit after completing the 28-day course of treatment demonstrated flattening of the lesion (Figure 2).
Leishmaniasis is a disease caused by a protozoan parasite of the Leishmania genus, spread via inoculation from the bite of sandfly vectors.1 Cutaneous leishmaniasis is the most common clinical manifestation of leishmaniasis. Other clinical manifestations include mucocutaneous leishmaniasis and visceral leishmaniasis.1,2 Cutaneous leishmaniasis typically manifests as open wounds on areas of skin that may have been exposed to sandfly bites.3 The lesion may not appear until weeks to months or even years after the initial inoculation.2 Initially, CL manifests as papules that may progress to nodular plaques, with eventual evolution to volcanic ulcerations with raised borders and central crateriform indentations covered by scabs or crusting.2,3 The infection may be localized or diffuse—in either case, development of satellite lesions, regional lymphadenopathy, and/or nodular lymphangitis is not uncommon. Generally, CL is not lethal, but the severity of the lesions may vary and can lead to permanent scarring and atrophy.2 Many cases of CL remain undiagnosed because of its appearance as a nonspecific ulcer that can mimic many other cutaneous lesions and because it generally heals spontaneously, leaving only scarring as an indicator of prior infection.4 Thus, CL requires a high diagnostic suspicion, as it can have a nonspecific presentation and is rare in nonendemic regions.
Diagnosis of CL is accomplished via microscopy, isoenzyme analysis, or serology or is made molecularly.3 Microscopic diagnosis includes visualization of Leishmania amastigotes, the stage of replication that occurs after the promastigote stage is phagocytosed by macrophages.3 Amastigote is the only stage that can be visualized in human tissue and is stained via Giemsa and/or hematoxylin and eosin.3 However, Leishmania amastigotes are morphologically indistinguishable from Trypanosoma cruzi amastigotes on microscopy, thus limiting diagnostic accuracy.3 Moreover, there is potential for missed diagnosis of persistent CL caused by L tropica due to fewer parasites being present, further complicating the diagnosis.5 In these cases, molecular diagnostics are helpful as they have higher sensitivity and quicker results. Additionally, DNA technologies can differentiate strains, which is beneficial for guiding treatment. Isoenzyme analysis also can help identify Leishmania species, although results can take weeks to return.3 Serologic testing is useful for suspected visceral leishmaniasis despite negative definitive diagnoses or conflicts with conducting definitive studies; however, there is not a strong antibody response in CL, thus serology is ineffective.3,5 Furthermore, serology can have cross-reactivity with T cruzi and cannot be used to assess for treatment response.3,5 The Infectious Diseases Society of America guidelines for diagnosis of leishmaniasis recommend using multiple methods to ensure a positive result, with molecular assays being the most sensitive.5
Differential diagnoses include any cause of cutaneous ulcerated lesions, including but not limited to mycobacterial or fungal infections. Leprosy often initially manifests with a hypopigmented macule with a raised border, although there often are associated neuropathic symptoms.6 Cutaneous tuberculosis is an extremely rare manifestation that occurs via direct inoculation of the mycobacterium, occurring primarily in children. Initially, it may manifest as a firm red papule that progresses to a painless shallow ulcer with a granular base.7 Cutaneous chromoblastomycosis is a fungal infection resulting from an initial cutaneous injury, similar to our patient, followed by a slow-developing warty lesion that may heal into ivory scars or spread as plaques on normal skin.8 The verrucous lesions seen in cutaneous chromoblastomycosis tend to manifest on the lower extremities and are unlikely to manifest on the head. Sarcoidosis is another granulomatous skin eruption that can be clinically nonspecific.9 Histologically, lesions may demonstrate noncaseating granulomatous inflammation, as seen with cutaneous leishmaniasis, with a broad presentation; for example, lupus pernio, a sarcoid variant, manifests as large blue-red dusky nodules/plaques on the face, ears, or digits.9 Other sarcoid lesions include red/brown, thickened, circular plaques; variably discolored papulonodular lesions; or mucosal involvement.9 Ultimately, it is important to differentiate these nonspecific and similarly appearing lesions through diagnostic techniques such as AFB culture and smear, fungal staining, tuberculosis testing, and PCR in more challenging cases.
Treatment of cutaneous leishmaniasis should be individualized to each case.5 A more than 50% reduction in lesion size within 4 to 6 weeks indicates successful treatment. Ulcerated lesions should be fully re-epithelialized and healed by 3 months posttreatment. Treatment failure is categorized by failure of reepithelization, incomplete healing by 3 months, or worsening of the lesion at any time, each necessitating additional treatment, such as a second course of miltefosine or a different medication regimen.5 Careful monitoring is required throughout treatment, assessing for treatment failure, adding to the challenges of leishmaniasis.
In conclusion, CL requires a high index of suspicion in nonendemic areas to ensure successful diagnosis and treatment. Our case highlights the importance of using multimodal diagnostic techniques for CL, as a single modality may not exhibit a positive result due to variations in diagnostic accuracy. Our case also exhibits the complex treatment of CL, and the considerations that should be made when choosing a treatment modality.
- Leishmaniasis. World Health Organization. Accessed September 14, 2024. https://www.who.int/news-room/fact-sheets/detail/leishmaniasis
- Clinical overview of leishmaniasis. Centers for Disease Control and Prevention. Accessed September 14, 2024. https://www.cdc.gov/leishmaniasis/hcp/clinical-overview/index.html
- CDC DPDx Leishmaniasis. Centers for Disease Control and Prevention. Accessed September 15, 2024. https://www.cdc.gov/dpdx/leishmaniasis/index.html
- Stark CG. Leishmaniasis differential diagnoses. Medscape. March 21, 2023. Accessed October 12, 2024. https://emedicine.medscape.com/article/220298-differential
- Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin Infect Dis. 2016;63:E202-E264. doi:10.1093/cid/ciw670
- Lewis FS. Dermatologic manifestations of leprosy. Medscape. June 19, 2023. Accessed October 12, 2024. https://emedicine.medscape.com/article/1104977-overview
- Ngan V. Cutaneous tuberculosis. DermNet. Accessed October 12, 2024. https://dermnetnz.org/topics/cutaneous-tuberculosis
- Schwartz RA. Chromoblastomycosis. Medscape. May 13, 2023. Accessed October 12, 2024. https://emedicine.medscape.com/article/1092695-overview#a4
- Elyoussfi S, Coulson I. Sarcoidosis. DermNet. May 31, 2024. Accessed October 25, 2024. https://dermnetnz.org/topics/sarcoidosis
- Leishmaniasis. World Health Organization. Accessed September 14, 2024. https://www.who.int/news-room/fact-sheets/detail/leishmaniasis
- Clinical overview of leishmaniasis. Centers for Disease Control and Prevention. Accessed September 14, 2024. https://www.cdc.gov/leishmaniasis/hcp/clinical-overview/index.html
- CDC DPDx Leishmaniasis. Centers for Disease Control and Prevention. Accessed September 15, 2024. https://www.cdc.gov/dpdx/leishmaniasis/index.html
- Stark CG. Leishmaniasis differential diagnoses. Medscape. March 21, 2023. Accessed October 12, 2024. https://emedicine.medscape.com/article/220298-differential
- Aronson N, Herwaldt BL, Libman M, et al. Diagnosis and treatment of leishmaniasis: clinical practice guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin Infect Dis. 2016;63:E202-E264. doi:10.1093/cid/ciw670
- Lewis FS. Dermatologic manifestations of leprosy. Medscape. June 19, 2023. Accessed October 12, 2024. https://emedicine.medscape.com/article/1104977-overview
- Ngan V. Cutaneous tuberculosis. DermNet. Accessed October 12, 2024. https://dermnetnz.org/topics/cutaneous-tuberculosis
- Schwartz RA. Chromoblastomycosis. Medscape. May 13, 2023. Accessed October 12, 2024. https://emedicine.medscape.com/article/1092695-overview#a4
- Elyoussfi S, Coulson I. Sarcoidosis. DermNet. May 31, 2024. Accessed October 25, 2024. https://dermnetnz.org/topics/sarcoidosis
Nonhealing Lesion on the Ear in a Child
Nonhealing Lesion on the Ear in a Child
A 10-year-old boy who recently emigrated from Afghanistan presented to his pediatrician for evaluation of a painless nonhealing plaque on the posterior left pinna of more than 1 year's duration. The lesion reportedly started as a small scratch following an ear injury, initially improved with an unknown topical treatment administered in Afghanistan, and then recurred with no other associated lesions and no known insect bite. The lesion persisted for more than 1 year postemigration before the patient presented to his pediatrician, who noted signs of excoriation, which was confirmed by the patient's father. The patient was started on a 7-day course of cephalexin oral suspension and topical mupirocin 2%. After 2 months without improvement, a 2-week course of oral trimethoprim/sulfamethoxazole was initiated; however, the lesion continued to grow with no signs of healing, and he was referred to dermatology.
The patient presented to pediatric dermatology 3 months after the initial presentation to his pediatrician and 2 weeks after he completed the course of oral trimethoprim/sulfamethoxazole. Physical examination demonstrated a papulosquamous eruption with swelling and blistering on the helix of the left ear. Based on these findings, the patient was started on a 1-month trial of topical triamcinolone 1% followed by the addition of topical pimecrolimus 1%. Due to no improvement of the lesion and subsequent progression to ulceration, a punch biopsy was performed.
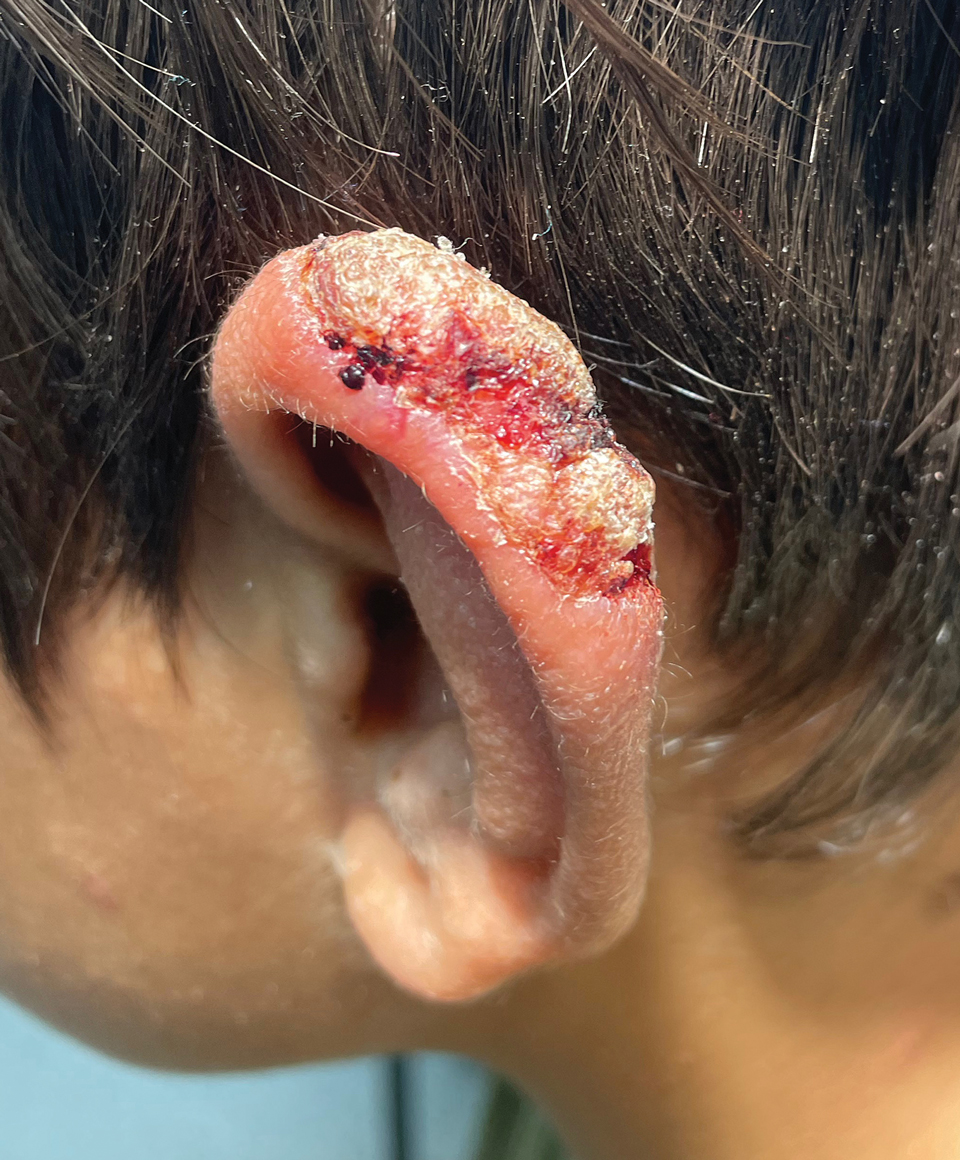

Progressive Dystrophy of the Fingernails and Toenails
Progressive Dystrophy of the Fingernails and Toenails
THE DIAGNOSIS: Nail Lichen Planus
The biopsy results showed features of hypergranulosis of the matricial epithelium, irregular acanthosis, apoptotic keratinocytes along the basal layer, and a lichenoid infiltrate consistent with nail lichen planus. The patient was started on topical clobetasol propionate 0.05% applied once daily under overnight occlusion. Additionally, intramatricial triamcinolone acetonide (2.5 mg/mL; 0.1 mL per injection) was administered into the affected nail matrix at 4-week intervals for a total of 2 sessions. At the 2-month follow-up visit, the patient reported improvement in longitudinal ridging; however, he subsequently was lost to follow-up.
Nail lichen planus is a chronic inflammatory disorder that occurs in 10% to 15% of patients with lichen planus worldwide and is more common in adults than children.1 It can manifest independently or concurrently with cutaneous and/or oral mucosal involvement. The fingernails are more commonly affected than the toenails.2 The clinical features of nail lichen planus can be classified based on involvement of the nail matrix (longitudinal ridging, red lunula, thinning of the nail plate, koilonychia, trachyonychia, pterygium, and anonychia) or nail bed (onycholysis, subungual hyperkeratosis, and splinter hemorrhages).1
In our patient, who presented with chronic progressive nail dystrophy affecting all 20 nails, onychomycosis, nail psoriasis, onychotillomania, and idiopathic trachyonychia were included in the differential.1
Onychomycosis manifests as white or yellow-brown discoloration of the nail, onycholysis, subungual hyperkeratosis, and thickening of the nail plate. Diagnosis is confirmed by the presence of septate hyphae (dermatophytes) or budding yeast cells (Candida species) on a potassium hydroxide mount. Other diagnostic modalities include dermoscopy, fungal culture, and histopathology of nail clippings, with demonstration of fungal elements identified on periodic acid-Schiff staining (eFigure 1).3
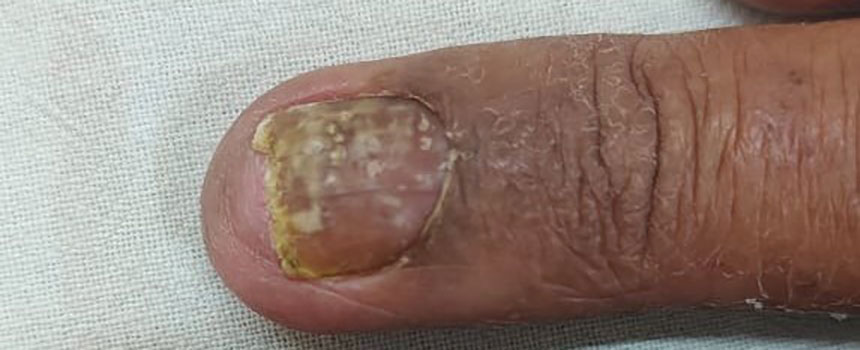
Nail psoriasis characteristically manifests as deep irregular pitting of the nails. Other features favoring psoriasis include involvement of the nail matrix manifesting as leukonychia, red lunula, and crumbling, as well as involvement of the nail bed manifesting as onycholysis, subungual hyperkeratosis, salmon patches/oil spots, and splinter hemorrhages (eFigure 2).4 Diagnosis primarily is clinical, supported by histopathology when uncertainty exists.
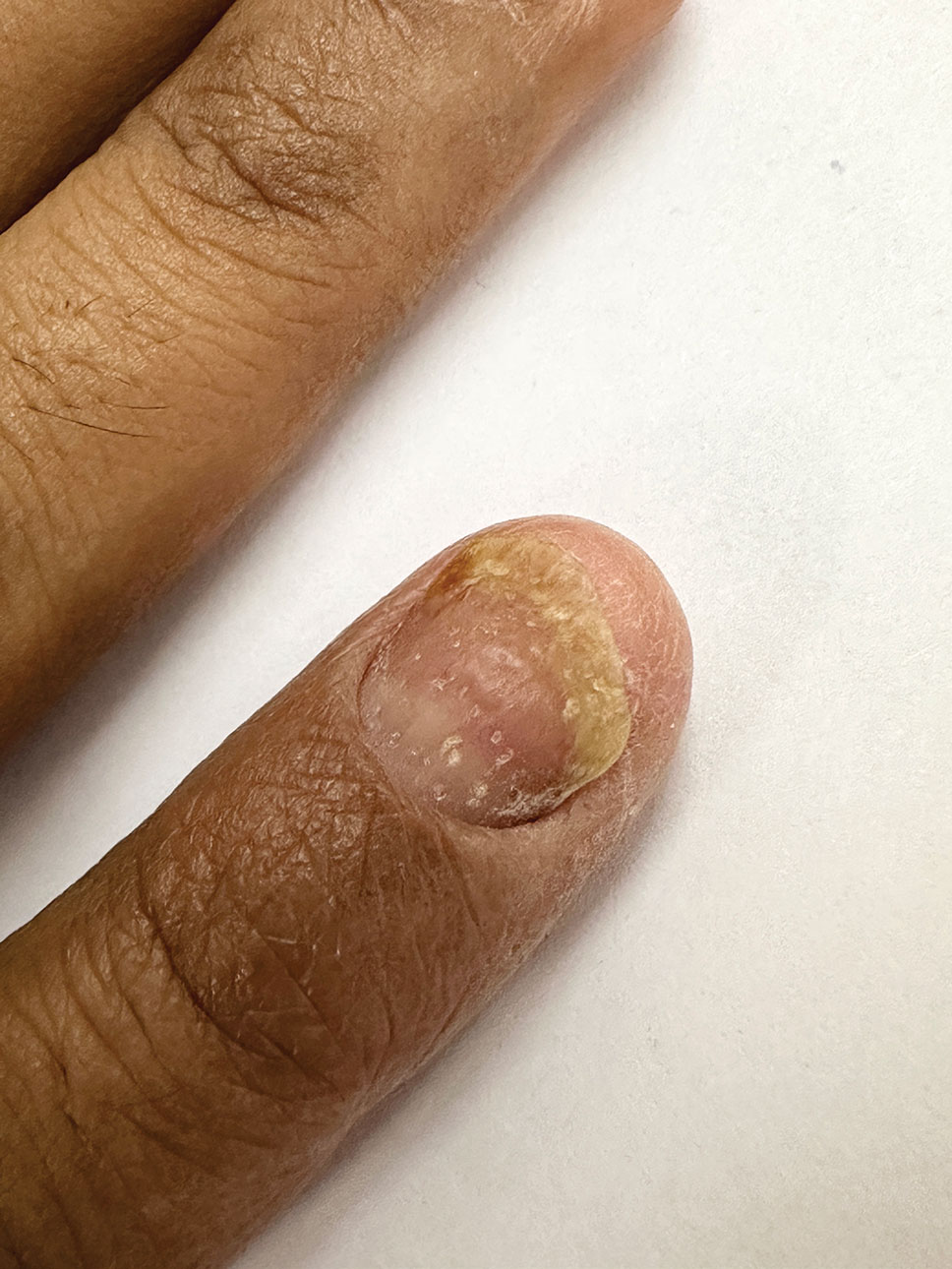
Onychotillomania is a behavioral disorder characterized by an irresistible urge or impulse in patients to either pick or pull at their fingernails and/or toenails. Clinicopathologic features of the involved nails are nonspecific and atypical, with possible involvement of periungual and digital skin. Diagnosis of onychotillomania is challenging.5 Dermoscopic features including anonychia with multiple obliquely arranged nail bed hemorrhages, gray pigmentation of the nail bed, and wavy lines, has been proposed to aid the diagnosis of onychotillomania.6
Idiopathic trachyonychia is isolated nail involvement characterized by rough, ridged, and thin nails affecting multiple or all of the fingernails and toenails without an underlying systemic or dermatologic condition (eFigure 3). The terms trachyonychia and 20-nail dystrophy have been used interchangeably in the literature; however, trachyonychia does not always involve all 20 nails. Other conditions causing widespread dystrophy of all 20 nails cannot be diagnosed as 20-nail dystrophy or trachyonychia without the distinct morphologic features of thin brittle nails with pronounced longitudinal ridging.7
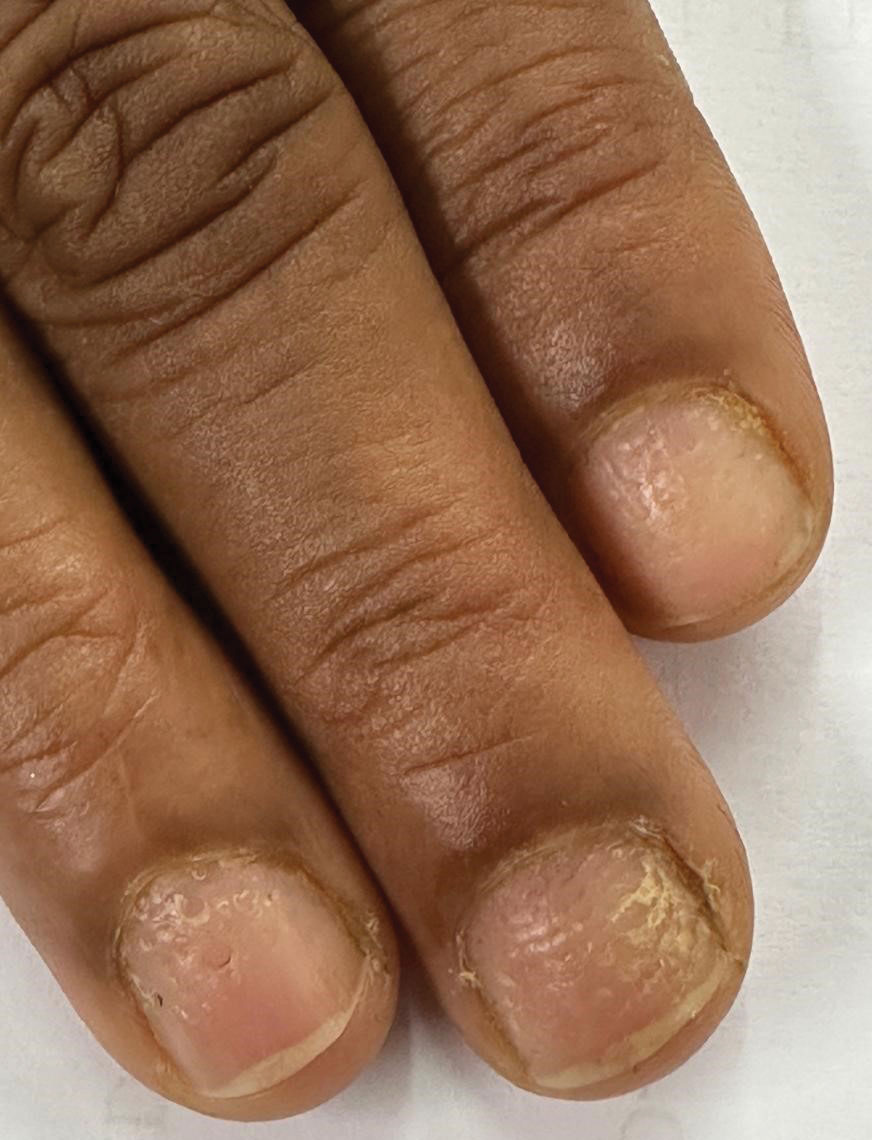
Prompt diagnosis and early intervention in nail lichen planus is crucial due to the potential for irreversible scarring. First-line treatment options include intramatricial and intramuscular triamcinolone acetonide for 3 to 6 months.4 Second-line therapies include oral retinoids such as acitretin and alitretinoin and immunosuppressive agents such as azathioprine, mycophenolate mofetil, and cyclosporine. Other reported treatment options include clobetasol propionate, tacrolimus, dapsone, griseofulvin, etanercept, hydroxychloroquine, methotrexate, and UV therapy.4
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Leung AKC, Lam JM, Leong KF, et al. Onychomycosis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:32-45. doi:10.2174/1872213X13666191026090713
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Sidiropoulou P, Sgouros D, Theodoropoulos K, et al. Onychotillomania: a chameleon-like disorder: case report and review of literature. Skin Appendage Disord. 2019;5:104-107. doi:10.1159/000489941
- Maddy AJ, Tosti A. Dermoscopic features of onychotillomania: a study of 36 cases. J Am Acad Dermatol. 2018;79:702-705. doi:10.1016 /j.jaad.2018.04.015
- Haber JS, Chairatchaneeboon M, Rubin AI. Trachyonychia: review and update on clinical aspects, histology, and therapy. Skin Appendage Disord. 2017;2:109-115. doi:10.1159/000449063
THE DIAGNOSIS: Nail Lichen Planus
The biopsy results showed features of hypergranulosis of the matricial epithelium, irregular acanthosis, apoptotic keratinocytes along the basal layer, and a lichenoid infiltrate consistent with nail lichen planus. The patient was started on topical clobetasol propionate 0.05% applied once daily under overnight occlusion. Additionally, intramatricial triamcinolone acetonide (2.5 mg/mL; 0.1 mL per injection) was administered into the affected nail matrix at 4-week intervals for a total of 2 sessions. At the 2-month follow-up visit, the patient reported improvement in longitudinal ridging; however, he subsequently was lost to follow-up.
Nail lichen planus is a chronic inflammatory disorder that occurs in 10% to 15% of patients with lichen planus worldwide and is more common in adults than children.1 It can manifest independently or concurrently with cutaneous and/or oral mucosal involvement. The fingernails are more commonly affected than the toenails.2 The clinical features of nail lichen planus can be classified based on involvement of the nail matrix (longitudinal ridging, red lunula, thinning of the nail plate, koilonychia, trachyonychia, pterygium, and anonychia) or nail bed (onycholysis, subungual hyperkeratosis, and splinter hemorrhages).1
In our patient, who presented with chronic progressive nail dystrophy affecting all 20 nails, onychomycosis, nail psoriasis, onychotillomania, and idiopathic trachyonychia were included in the differential.1
Onychomycosis manifests as white or yellow-brown discoloration of the nail, onycholysis, subungual hyperkeratosis, and thickening of the nail plate. Diagnosis is confirmed by the presence of septate hyphae (dermatophytes) or budding yeast cells (Candida species) on a potassium hydroxide mount. Other diagnostic modalities include dermoscopy, fungal culture, and histopathology of nail clippings, with demonstration of fungal elements identified on periodic acid-Schiff staining (eFigure 1).3
Nail psoriasis characteristically manifests as deep irregular pitting of the nails. Other features favoring psoriasis include involvement of the nail matrix manifesting as leukonychia, red lunula, and crumbling, as well as involvement of the nail bed manifesting as onycholysis, subungual hyperkeratosis, salmon patches/oil spots, and splinter hemorrhages (eFigure 2).4 Diagnosis primarily is clinical, supported by histopathology when uncertainty exists.
Onychotillomania is a behavioral disorder characterized by an irresistible urge or impulse in patients to either pick or pull at their fingernails and/or toenails. Clinicopathologic features of the involved nails are nonspecific and atypical, with possible involvement of periungual and digital skin. Diagnosis of onychotillomania is challenging.5 Dermoscopic features including anonychia with multiple obliquely arranged nail bed hemorrhages, gray pigmentation of the nail bed, and wavy lines, has been proposed to aid the diagnosis of onychotillomania.6
Idiopathic trachyonychia is isolated nail involvement characterized by rough, ridged, and thin nails affecting multiple or all of the fingernails and toenails without an underlying systemic or dermatologic condition (eFigure 3). The terms trachyonychia and 20-nail dystrophy have been used interchangeably in the literature; however, trachyonychia does not always involve all 20 nails. Other conditions causing widespread dystrophy of all 20 nails cannot be diagnosed as 20-nail dystrophy or trachyonychia without the distinct morphologic features of thin brittle nails with pronounced longitudinal ridging.7
Prompt diagnosis and early intervention in nail lichen planus is crucial due to the potential for irreversible scarring. First-line treatment options include intramatricial and intramuscular triamcinolone acetonide for 3 to 6 months.4 Second-line therapies include oral retinoids such as acitretin and alitretinoin and immunosuppressive agents such as azathioprine, mycophenolate mofetil, and cyclosporine. Other reported treatment options include clobetasol propionate, tacrolimus, dapsone, griseofulvin, etanercept, hydroxychloroquine, methotrexate, and UV therapy.4
THE DIAGNOSIS: Nail Lichen Planus
The biopsy results showed features of hypergranulosis of the matricial epithelium, irregular acanthosis, apoptotic keratinocytes along the basal layer, and a lichenoid infiltrate consistent with nail lichen planus. The patient was started on topical clobetasol propionate 0.05% applied once daily under overnight occlusion. Additionally, intramatricial triamcinolone acetonide (2.5 mg/mL; 0.1 mL per injection) was administered into the affected nail matrix at 4-week intervals for a total of 2 sessions. At the 2-month follow-up visit, the patient reported improvement in longitudinal ridging; however, he subsequently was lost to follow-up.
Nail lichen planus is a chronic inflammatory disorder that occurs in 10% to 15% of patients with lichen planus worldwide and is more common in adults than children.1 It can manifest independently or concurrently with cutaneous and/or oral mucosal involvement. The fingernails are more commonly affected than the toenails.2 The clinical features of nail lichen planus can be classified based on involvement of the nail matrix (longitudinal ridging, red lunula, thinning of the nail plate, koilonychia, trachyonychia, pterygium, and anonychia) or nail bed (onycholysis, subungual hyperkeratosis, and splinter hemorrhages).1
In our patient, who presented with chronic progressive nail dystrophy affecting all 20 nails, onychomycosis, nail psoriasis, onychotillomania, and idiopathic trachyonychia were included in the differential.1
Onychomycosis manifests as white or yellow-brown discoloration of the nail, onycholysis, subungual hyperkeratosis, and thickening of the nail plate. Diagnosis is confirmed by the presence of septate hyphae (dermatophytes) or budding yeast cells (Candida species) on a potassium hydroxide mount. Other diagnostic modalities include dermoscopy, fungal culture, and histopathology of nail clippings, with demonstration of fungal elements identified on periodic acid-Schiff staining (eFigure 1).3
Nail psoriasis characteristically manifests as deep irregular pitting of the nails. Other features favoring psoriasis include involvement of the nail matrix manifesting as leukonychia, red lunula, and crumbling, as well as involvement of the nail bed manifesting as onycholysis, subungual hyperkeratosis, salmon patches/oil spots, and splinter hemorrhages (eFigure 2).4 Diagnosis primarily is clinical, supported by histopathology when uncertainty exists.
Onychotillomania is a behavioral disorder characterized by an irresistible urge or impulse in patients to either pick or pull at their fingernails and/or toenails. Clinicopathologic features of the involved nails are nonspecific and atypical, with possible involvement of periungual and digital skin. Diagnosis of onychotillomania is challenging.5 Dermoscopic features including anonychia with multiple obliquely arranged nail bed hemorrhages, gray pigmentation of the nail bed, and wavy lines, has been proposed to aid the diagnosis of onychotillomania.6
Idiopathic trachyonychia is isolated nail involvement characterized by rough, ridged, and thin nails affecting multiple or all of the fingernails and toenails without an underlying systemic or dermatologic condition (eFigure 3). The terms trachyonychia and 20-nail dystrophy have been used interchangeably in the literature; however, trachyonychia does not always involve all 20 nails. Other conditions causing widespread dystrophy of all 20 nails cannot be diagnosed as 20-nail dystrophy or trachyonychia without the distinct morphologic features of thin brittle nails with pronounced longitudinal ridging.7
Prompt diagnosis and early intervention in nail lichen planus is crucial due to the potential for irreversible scarring. First-line treatment options include intramatricial and intramuscular triamcinolone acetonide for 3 to 6 months.4 Second-line therapies include oral retinoids such as acitretin and alitretinoin and immunosuppressive agents such as azathioprine, mycophenolate mofetil, and cyclosporine. Other reported treatment options include clobetasol propionate, tacrolimus, dapsone, griseofulvin, etanercept, hydroxychloroquine, methotrexate, and UV therapy.4
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Leung AKC, Lam JM, Leong KF, et al. Onychomycosis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:32-45. doi:10.2174/1872213X13666191026090713
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Sidiropoulou P, Sgouros D, Theodoropoulos K, et al. Onychotillomania: a chameleon-like disorder: case report and review of literature. Skin Appendage Disord. 2019;5:104-107. doi:10.1159/000489941
- Maddy AJ, Tosti A. Dermoscopic features of onychotillomania: a study of 36 cases. J Am Acad Dermatol. 2018;79:702-705. doi:10.1016 /j.jaad.2018.04.015
- Haber JS, Chairatchaneeboon M, Rubin AI. Trachyonychia: review and update on clinical aspects, histology, and therapy. Skin Appendage Disord. 2017;2:109-115. doi:10.1159/000449063
- Gupta MK, Lipner SR. Review of nail lichen planus: epidemiology, pathogenesis, diagnosis, and treatment. Dermatol Clin. 2021;39:221-230. doi:10.1016/j.det.2020.12.002
- Iorizzo M, Tosti A, Starace M, et al. Isolated nail lichen planus: an expert consensus on treatment of the classical form. J Am Acad Dermatol. 2020;83:1717-1723. doi:10.1016/j.jaad.2020.02.056
- Leung AKC, Lam JM, Leong KF, et al. Onychomycosis: an updated review. Recent Pat Inflamm Allergy Drug Discov. 2020;14:32-45. doi:10.2174/1872213X13666191026090713
- Hwang JK, Grover C, Iorizzo M, et al. Nail psoriasis and nail lichen planus: updates on diagnosis and management. J Am Acad Dermatol. 2024;90:585-596. doi:10.1016/j.jaad.2023.11.024
- Sidiropoulou P, Sgouros D, Theodoropoulos K, et al. Onychotillomania: a chameleon-like disorder: case report and review of literature. Skin Appendage Disord. 2019;5:104-107. doi:10.1159/000489941
- Maddy AJ, Tosti A. Dermoscopic features of onychotillomania: a study of 36 cases. J Am Acad Dermatol. 2018;79:702-705. doi:10.1016 /j.jaad.2018.04.015
- Haber JS, Chairatchaneeboon M, Rubin AI. Trachyonychia: review and update on clinical aspects, histology, and therapy. Skin Appendage Disord. 2017;2:109-115. doi:10.1159/000449063
Progressive Dystrophy of the Fingernails and Toenails
Progressive Dystrophy of the Fingernails and Toenails
A 35-year-old man presented to the dermatology department with gradually progressive dystrophy of the fingernails and toenails of 20 years’ duration. The patient reported no history of other dermatologic conditions. Physical examination revealed longitudinal ridging of all 20 nails and discoloration of the nail plates, as well as a few nails showing pterygium and anonychia; the skin and mucosal surfaces were otherwise normal, and nail plate thinning was not observed. A potassium hydroxide mount was negative. A biopsy of the nail matrix on the left thumbnail was performed.
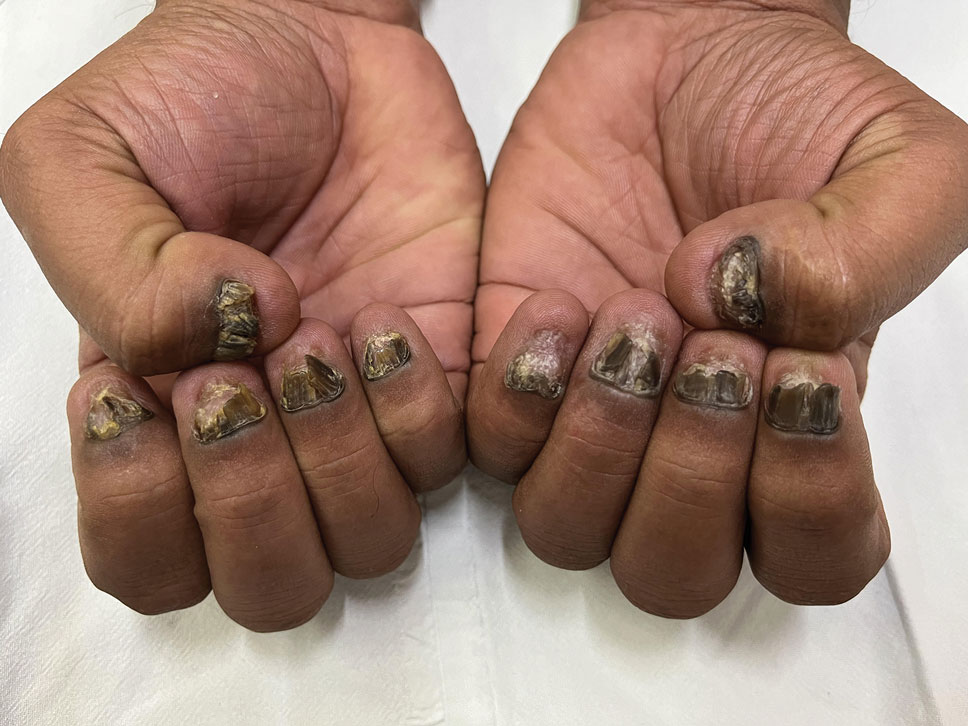
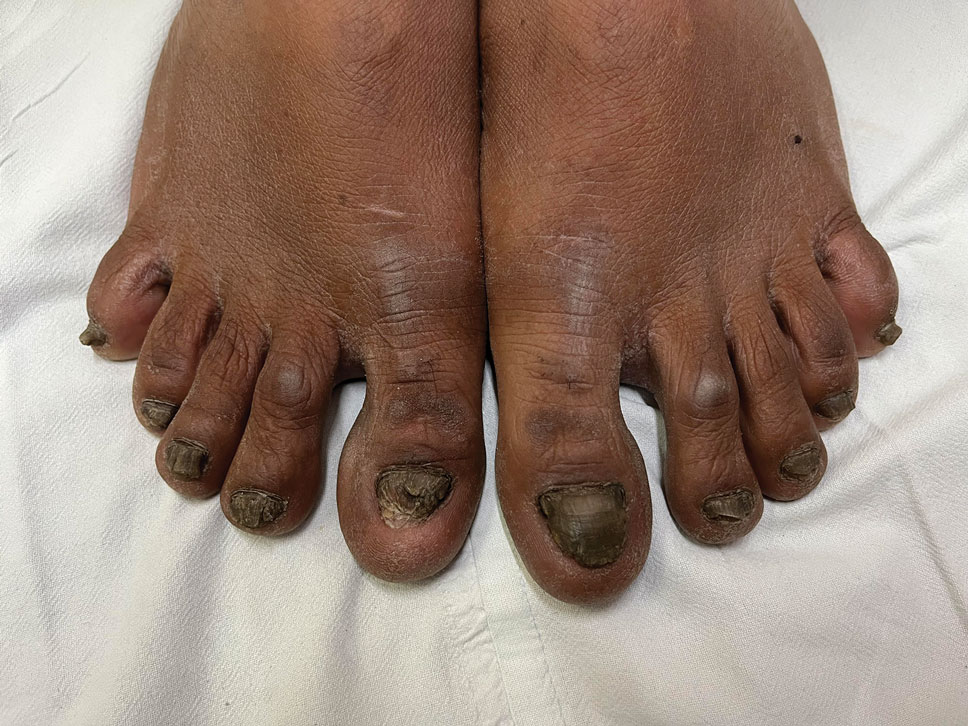

Cobblestonelike Papules on the Neck
The Diagnosis: Fibroelastolytic Papulosis
Histopathology demonstrated decreased density and fragmentation of elastic fibers in the superficial reticular and papillary dermis consistent with an elastolytic disease process (Figure). Of note, elastolysis typically is visualized with Verhoeff-van Gieson stain but cannot be visualized well with standard hematoxylin and eosin staining. Additional staining with Congo red was negative for amyloid, and colloidal iron did not show any increase in dermal mucin, ruling out amyloidosis and scleromyxedema, respectively. Based on the histopathologic findings and the clinical history, a diagnosis of fibroelastolytic papulosis (FP) was made. Given the benign nature of the condition, the patient was prescribed a topical steroid (clobetasol 0.05%) for symptomatic relief.
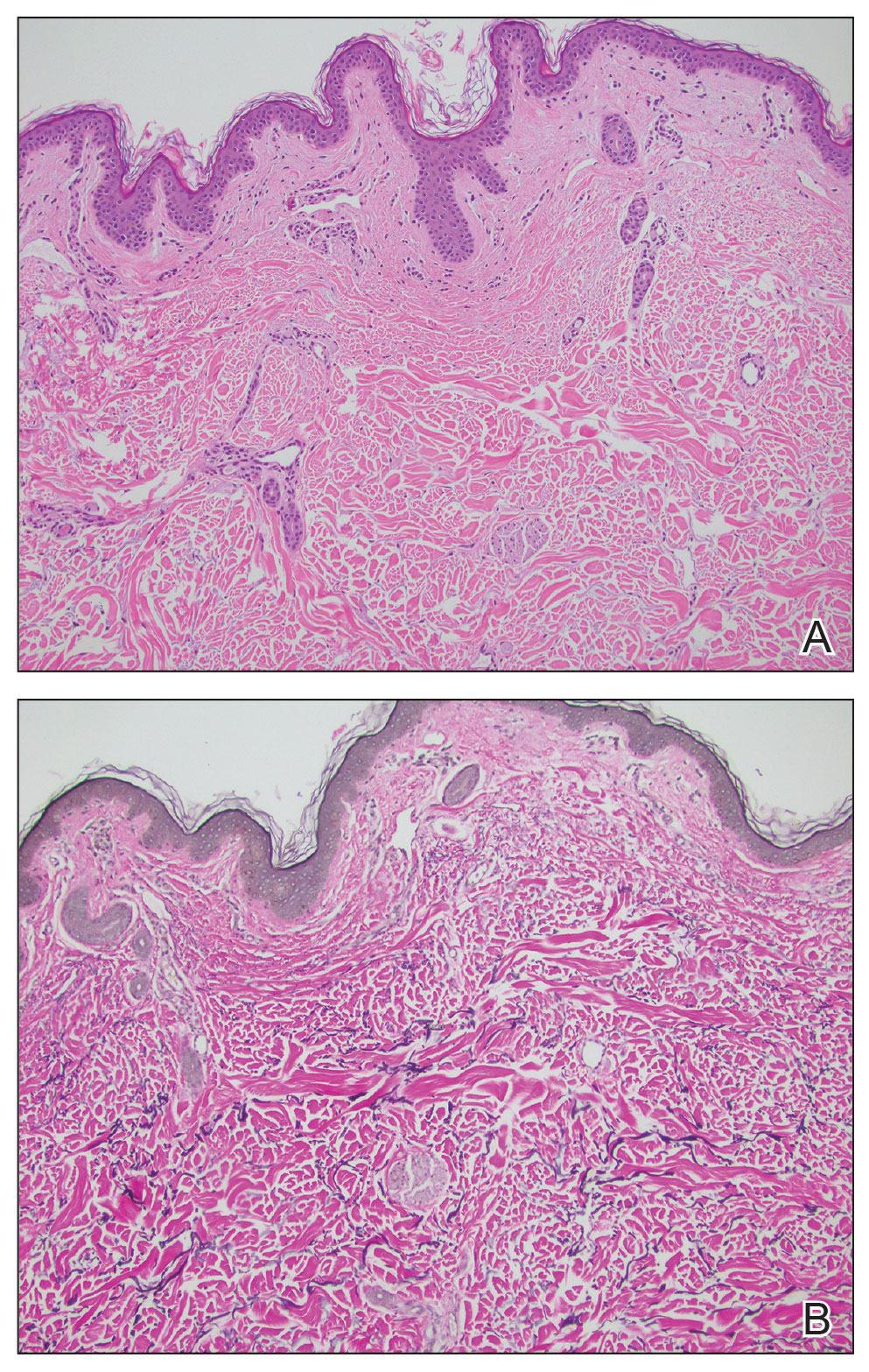
Cutaneous conditions can arise from abnormalities in the elastin composition of connective tissue due to abnormal elastin formation or degradation (elastolysis).1 Fibroelastolytic papulosis is a distinct elastolytic disorder diagnosed histologically by a notable loss of elastic fibers localized to the papillary dermis.2 Fibroelastolytic papulosis is an acquired condition linked to exposure to UV radiation, abnormal elastogenesis, and hormonal factors that commonly involves the neck, supraclavicular area, and upper back.1-3 Predominantly affecting elderly women, FP is characterized by soft white papules that often coalesce into a cobblestonelike plaque.2 Because the condition rarely is seen in men, there is speculation that it may involve genetic, hereditary, and hormonal factors that have yet to be identified.1
Fibroelastolytic papulosis can be classified as either pseudoxanthoma elasticum–like papillary dermal elastolysis or white fibrous papulosis.2,3 White fibrous papulosis manifests with haphazardly arranged collagen fibers in the reticular and deep dermis with papillary dermal elastolysis and most commonly develops on the neck.3 Although our patient’s lesion was on the neck, the absence of thickened collagen bands on histology supported classification as the pseudoxanthoma elasticum– like papillary dermal elastolysis subtype.
Fibroelastolytic papulosis can be distinguished from other elastic abnormalities by its characteristic clinical appearance, demographic distribution, and associated histopathologic findings. The differential diagnosis of FP includes pseudoxanthoma elasticum (PXE), anetoderma, scleromyxedema, and lichen amyloidosis.
Pseudoxanthoma elasticum is a hereditary or acquired multisystem disease characterized by fragmentation and calcification of elastic fibers in the mid dermis.1,4 Its clinical presentation resembles that of FP, appearing as small, asymptomatic, yellowish or flesh-colored papules in a reticular pattern that progressively coalesce into larger plaques with a cobblestonelike appearance.1 Like FP, PXE commonly affects the flexural creases in women but in contrast may manifest earlier (ie, second or third decades of life). Additionally, the pathogenesis of PXE is not related to UV radiation exposure. The hereditary form develops due to a gene variation, whereas the acquired form may be due to conditions associated with physiologic and/or mechanical stress.1
Anetoderma, also known as macular atrophy, is another condition that demonstrates elastic tissue loss in the dermis on histopathology.1 Anetoderma commonly is seen in younger patients and can be differentiated from FP by the antecedent presence of an inflammatory process. Anetoderma is classified as primary or secondary. Primary anetoderma is associated with prothrombotic abnormalities, while secondary anetoderma is associated with systemic disease including but not limited to sarcoidosis, systemic lupus erythematous, and Graves disease.1
Neither lichen myxedematosus (LM) nor lichen amyloidosis (LA) are true elastolytic conditions. Lichen myxedematosus is considered in the differential diagnosis of FP due to the associated loss of elastin observed with disease progression. An idiopathic cutaneous mucinosis, LM is a localized form of scleromyxedema, which is characterized by small, firm, waxy papules; mucin deposition in the skin; fibroblast proliferation; and fibrosis. On histologic analysis, typical findings of LM include irregularly arranged fibroblasts, diffuse mucin deposition within the upper and mid reticular dermis, increased collagen deposition, and a decrease in elastin fibers.5
Lichen amyloidosis is a subtype of primary localized cutaneous amyloidosis, a rare condition characterized by the extracellular deposition of amyloid proteins in the skin and a lack of systemic involvement. Although it is not an elastolytic condition, LA is clinically similar to FP, often manifesting as multiple localized, pruritic, hyperpigmented papules that can coalesce into larger plaques; it tends to develop on the shins, calves, ankles, and thighs.6,7 The condition commonly manifests in the fifth and sixth decades of life; however, in contrast to FP, LA is more prevalent in men and individuals from Central and South American as well as Middle Eastern and non-Chinese Asian populations.8 Lichen amyloidosis is a keratin-derived amyloidosis with cytokeratin-based amyloid precursors that only deposit in the dermis.6 Histopathology reveals colloid bodies due to the presence of apoptotic basal keratinocytes. The etiology of LA is unknown, but on rare occasions it has been associated with multiple endocrine neoplasia 2A rearranged during transfection mutations.6
In summary, FP is an uncommonly diagnosed elastolytic condition that often is asymptomatic or associated with mild pruritus. Biopsy is warranted to help differentiate it from mimicker conditions that may be associated with systemic disease. Currently, there is no established therapy that provides successful treatment. Research suggests unsatisfactory results with the use of topical tretinoin or topical antioxidants.3 More recently, nonablative fractional resurfacing lasers have been evaluated as a possible therapeutic strategy of promise for elastic disorders.9
- Andrés-Ramos I, Alegría-Landa V, Gimeno I, et al. Cutaneous elastic tissue anomalies. Am J Dermatopathol. 2019;41:85-117. doi:10.1097/DAD.0000000000001275
- Valbuena V, Assaad D, Yeung J. Pseudoxanthoma elasticum-like papillary dermal elastolysis: a single case report. J Cutan Med Surg. 2017;21:345-347. doi:10.1177/1203475417699407
- Dokic Y, Tschen J. White fibrous papulosis of the axillae and neck. Cureus. 2020;12:E7635. doi:10.7759/cureus.7635
- Recio-Monescillo M, Torre-Castro J, Manzanas C, et al. Papillary dermal elastolysis histopathology mimicking folliculotropic mycosis fungoides. J Cutan Pathol. 2023;50:430-433. doi:10.1111/cup.14402
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497. doi:10.1016/j.clindermatol.2006.07.011
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642. doi:10.1007/s40257-017-0278-9
- Ladizinski B, Lee KC. Lichen amyloidosis. CMAJ. 2014;186:532. doi:10.1503/cmaj.130698
- Chen JF, Chen YF. Answer: can you identify this condition? Can Fam Physician. 2012;58:1234-1235.
- Foering K, Torbeck RL, Frank MP, et al. Treatment of pseudoxanthoma elasticum-like papillary dermal elastolysis with nonablative fractional resurfacing laser resulting in clinical and histologic improvement in elastin and collagen. J Cosmet Laser Ther. 2018;20:382-384. doi:10.1080/14764172.2017.1358457
The Diagnosis: Fibroelastolytic Papulosis
Histopathology demonstrated decreased density and fragmentation of elastic fibers in the superficial reticular and papillary dermis consistent with an elastolytic disease process (Figure). Of note, elastolysis typically is visualized with Verhoeff-van Gieson stain but cannot be visualized well with standard hematoxylin and eosin staining. Additional staining with Congo red was negative for amyloid, and colloidal iron did not show any increase in dermal mucin, ruling out amyloidosis and scleromyxedema, respectively. Based on the histopathologic findings and the clinical history, a diagnosis of fibroelastolytic papulosis (FP) was made. Given the benign nature of the condition, the patient was prescribed a topical steroid (clobetasol 0.05%) for symptomatic relief.
Cutaneous conditions can arise from abnormalities in the elastin composition of connective tissue due to abnormal elastin formation or degradation (elastolysis).1 Fibroelastolytic papulosis is a distinct elastolytic disorder diagnosed histologically by a notable loss of elastic fibers localized to the papillary dermis.2 Fibroelastolytic papulosis is an acquired condition linked to exposure to UV radiation, abnormal elastogenesis, and hormonal factors that commonly involves the neck, supraclavicular area, and upper back.1-3 Predominantly affecting elderly women, FP is characterized by soft white papules that often coalesce into a cobblestonelike plaque.2 Because the condition rarely is seen in men, there is speculation that it may involve genetic, hereditary, and hormonal factors that have yet to be identified.1
Fibroelastolytic papulosis can be classified as either pseudoxanthoma elasticum–like papillary dermal elastolysis or white fibrous papulosis.2,3 White fibrous papulosis manifests with haphazardly arranged collagen fibers in the reticular and deep dermis with papillary dermal elastolysis and most commonly develops on the neck.3 Although our patient’s lesion was on the neck, the absence of thickened collagen bands on histology supported classification as the pseudoxanthoma elasticum– like papillary dermal elastolysis subtype.
Fibroelastolytic papulosis can be distinguished from other elastic abnormalities by its characteristic clinical appearance, demographic distribution, and associated histopathologic findings. The differential diagnosis of FP includes pseudoxanthoma elasticum (PXE), anetoderma, scleromyxedema, and lichen amyloidosis.
Pseudoxanthoma elasticum is a hereditary or acquired multisystem disease characterized by fragmentation and calcification of elastic fibers in the mid dermis.1,4 Its clinical presentation resembles that of FP, appearing as small, asymptomatic, yellowish or flesh-colored papules in a reticular pattern that progressively coalesce into larger plaques with a cobblestonelike appearance.1 Like FP, PXE commonly affects the flexural creases in women but in contrast may manifest earlier (ie, second or third decades of life). Additionally, the pathogenesis of PXE is not related to UV radiation exposure. The hereditary form develops due to a gene variation, whereas the acquired form may be due to conditions associated with physiologic and/or mechanical stress.1
Anetoderma, also known as macular atrophy, is another condition that demonstrates elastic tissue loss in the dermis on histopathology.1 Anetoderma commonly is seen in younger patients and can be differentiated from FP by the antecedent presence of an inflammatory process. Anetoderma is classified as primary or secondary. Primary anetoderma is associated with prothrombotic abnormalities, while secondary anetoderma is associated with systemic disease including but not limited to sarcoidosis, systemic lupus erythematous, and Graves disease.1
Neither lichen myxedematosus (LM) nor lichen amyloidosis (LA) are true elastolytic conditions. Lichen myxedematosus is considered in the differential diagnosis of FP due to the associated loss of elastin observed with disease progression. An idiopathic cutaneous mucinosis, LM is a localized form of scleromyxedema, which is characterized by small, firm, waxy papules; mucin deposition in the skin; fibroblast proliferation; and fibrosis. On histologic analysis, typical findings of LM include irregularly arranged fibroblasts, diffuse mucin deposition within the upper and mid reticular dermis, increased collagen deposition, and a decrease in elastin fibers.5
Lichen amyloidosis is a subtype of primary localized cutaneous amyloidosis, a rare condition characterized by the extracellular deposition of amyloid proteins in the skin and a lack of systemic involvement. Although it is not an elastolytic condition, LA is clinically similar to FP, often manifesting as multiple localized, pruritic, hyperpigmented papules that can coalesce into larger plaques; it tends to develop on the shins, calves, ankles, and thighs.6,7 The condition commonly manifests in the fifth and sixth decades of life; however, in contrast to FP, LA is more prevalent in men and individuals from Central and South American as well as Middle Eastern and non-Chinese Asian populations.8 Lichen amyloidosis is a keratin-derived amyloidosis with cytokeratin-based amyloid precursors that only deposit in the dermis.6 Histopathology reveals colloid bodies due to the presence of apoptotic basal keratinocytes. The etiology of LA is unknown, but on rare occasions it has been associated with multiple endocrine neoplasia 2A rearranged during transfection mutations.6
In summary, FP is an uncommonly diagnosed elastolytic condition that often is asymptomatic or associated with mild pruritus. Biopsy is warranted to help differentiate it from mimicker conditions that may be associated with systemic disease. Currently, there is no established therapy that provides successful treatment. Research suggests unsatisfactory results with the use of topical tretinoin or topical antioxidants.3 More recently, nonablative fractional resurfacing lasers have been evaluated as a possible therapeutic strategy of promise for elastic disorders.9
The Diagnosis: Fibroelastolytic Papulosis
Histopathology demonstrated decreased density and fragmentation of elastic fibers in the superficial reticular and papillary dermis consistent with an elastolytic disease process (Figure). Of note, elastolysis typically is visualized with Verhoeff-van Gieson stain but cannot be visualized well with standard hematoxylin and eosin staining. Additional staining with Congo red was negative for amyloid, and colloidal iron did not show any increase in dermal mucin, ruling out amyloidosis and scleromyxedema, respectively. Based on the histopathologic findings and the clinical history, a diagnosis of fibroelastolytic papulosis (FP) was made. Given the benign nature of the condition, the patient was prescribed a topical steroid (clobetasol 0.05%) for symptomatic relief.
Cutaneous conditions can arise from abnormalities in the elastin composition of connective tissue due to abnormal elastin formation or degradation (elastolysis).1 Fibroelastolytic papulosis is a distinct elastolytic disorder diagnosed histologically by a notable loss of elastic fibers localized to the papillary dermis.2 Fibroelastolytic papulosis is an acquired condition linked to exposure to UV radiation, abnormal elastogenesis, and hormonal factors that commonly involves the neck, supraclavicular area, and upper back.1-3 Predominantly affecting elderly women, FP is characterized by soft white papules that often coalesce into a cobblestonelike plaque.2 Because the condition rarely is seen in men, there is speculation that it may involve genetic, hereditary, and hormonal factors that have yet to be identified.1
Fibroelastolytic papulosis can be classified as either pseudoxanthoma elasticum–like papillary dermal elastolysis or white fibrous papulosis.2,3 White fibrous papulosis manifests with haphazardly arranged collagen fibers in the reticular and deep dermis with papillary dermal elastolysis and most commonly develops on the neck.3 Although our patient’s lesion was on the neck, the absence of thickened collagen bands on histology supported classification as the pseudoxanthoma elasticum– like papillary dermal elastolysis subtype.
Fibroelastolytic papulosis can be distinguished from other elastic abnormalities by its characteristic clinical appearance, demographic distribution, and associated histopathologic findings. The differential diagnosis of FP includes pseudoxanthoma elasticum (PXE), anetoderma, scleromyxedema, and lichen amyloidosis.
Pseudoxanthoma elasticum is a hereditary or acquired multisystem disease characterized by fragmentation and calcification of elastic fibers in the mid dermis.1,4 Its clinical presentation resembles that of FP, appearing as small, asymptomatic, yellowish or flesh-colored papules in a reticular pattern that progressively coalesce into larger plaques with a cobblestonelike appearance.1 Like FP, PXE commonly affects the flexural creases in women but in contrast may manifest earlier (ie, second or third decades of life). Additionally, the pathogenesis of PXE is not related to UV radiation exposure. The hereditary form develops due to a gene variation, whereas the acquired form may be due to conditions associated with physiologic and/or mechanical stress.1
Anetoderma, also known as macular atrophy, is another condition that demonstrates elastic tissue loss in the dermis on histopathology.1 Anetoderma commonly is seen in younger patients and can be differentiated from FP by the antecedent presence of an inflammatory process. Anetoderma is classified as primary or secondary. Primary anetoderma is associated with prothrombotic abnormalities, while secondary anetoderma is associated with systemic disease including but not limited to sarcoidosis, systemic lupus erythematous, and Graves disease.1
Neither lichen myxedematosus (LM) nor lichen amyloidosis (LA) are true elastolytic conditions. Lichen myxedematosus is considered in the differential diagnosis of FP due to the associated loss of elastin observed with disease progression. An idiopathic cutaneous mucinosis, LM is a localized form of scleromyxedema, which is characterized by small, firm, waxy papules; mucin deposition in the skin; fibroblast proliferation; and fibrosis. On histologic analysis, typical findings of LM include irregularly arranged fibroblasts, diffuse mucin deposition within the upper and mid reticular dermis, increased collagen deposition, and a decrease in elastin fibers.5
Lichen amyloidosis is a subtype of primary localized cutaneous amyloidosis, a rare condition characterized by the extracellular deposition of amyloid proteins in the skin and a lack of systemic involvement. Although it is not an elastolytic condition, LA is clinically similar to FP, often manifesting as multiple localized, pruritic, hyperpigmented papules that can coalesce into larger plaques; it tends to develop on the shins, calves, ankles, and thighs.6,7 The condition commonly manifests in the fifth and sixth decades of life; however, in contrast to FP, LA is more prevalent in men and individuals from Central and South American as well as Middle Eastern and non-Chinese Asian populations.8 Lichen amyloidosis is a keratin-derived amyloidosis with cytokeratin-based amyloid precursors that only deposit in the dermis.6 Histopathology reveals colloid bodies due to the presence of apoptotic basal keratinocytes. The etiology of LA is unknown, but on rare occasions it has been associated with multiple endocrine neoplasia 2A rearranged during transfection mutations.6
In summary, FP is an uncommonly diagnosed elastolytic condition that often is asymptomatic or associated with mild pruritus. Biopsy is warranted to help differentiate it from mimicker conditions that may be associated with systemic disease. Currently, there is no established therapy that provides successful treatment. Research suggests unsatisfactory results with the use of topical tretinoin or topical antioxidants.3 More recently, nonablative fractional resurfacing lasers have been evaluated as a possible therapeutic strategy of promise for elastic disorders.9
- Andrés-Ramos I, Alegría-Landa V, Gimeno I, et al. Cutaneous elastic tissue anomalies. Am J Dermatopathol. 2019;41:85-117. doi:10.1097/DAD.0000000000001275
- Valbuena V, Assaad D, Yeung J. Pseudoxanthoma elasticum-like papillary dermal elastolysis: a single case report. J Cutan Med Surg. 2017;21:345-347. doi:10.1177/1203475417699407
- Dokic Y, Tschen J. White fibrous papulosis of the axillae and neck. Cureus. 2020;12:E7635. doi:10.7759/cureus.7635
- Recio-Monescillo M, Torre-Castro J, Manzanas C, et al. Papillary dermal elastolysis histopathology mimicking folliculotropic mycosis fungoides. J Cutan Pathol. 2023;50:430-433. doi:10.1111/cup.14402
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497. doi:10.1016/j.clindermatol.2006.07.011
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642. doi:10.1007/s40257-017-0278-9
- Ladizinski B, Lee KC. Lichen amyloidosis. CMAJ. 2014;186:532. doi:10.1503/cmaj.130698
- Chen JF, Chen YF. Answer: can you identify this condition? Can Fam Physician. 2012;58:1234-1235.
- Foering K, Torbeck RL, Frank MP, et al. Treatment of pseudoxanthoma elasticum-like papillary dermal elastolysis with nonablative fractional resurfacing laser resulting in clinical and histologic improvement in elastin and collagen. J Cosmet Laser Ther. 2018;20:382-384. doi:10.1080/14764172.2017.1358457
- Andrés-Ramos I, Alegría-Landa V, Gimeno I, et al. Cutaneous elastic tissue anomalies. Am J Dermatopathol. 2019;41:85-117. doi:10.1097/DAD.0000000000001275
- Valbuena V, Assaad D, Yeung J. Pseudoxanthoma elasticum-like papillary dermal elastolysis: a single case report. J Cutan Med Surg. 2017;21:345-347. doi:10.1177/1203475417699407
- Dokic Y, Tschen J. White fibrous papulosis of the axillae and neck. Cureus. 2020;12:E7635. doi:10.7759/cureus.7635
- Recio-Monescillo M, Torre-Castro J, Manzanas C, et al. Papillary dermal elastolysis histopathology mimicking folliculotropic mycosis fungoides. J Cutan Pathol. 2023;50:430-433. doi:10.1111/cup.14402
- Cokonis Georgakis CD, Falasca G, Georgakis A, et al. Scleromyxedema. Clin Dermatol. 2006;24:493-497. doi:10.1016/j.clindermatol.2006.07.011
- Weidner T, Illing T, Elsner P. Primary localized cutaneous amyloidosis: a systematic treatment review. Am J Clin Dermatol. 2017;18:629-642. doi:10.1007/s40257-017-0278-9
- Ladizinski B, Lee KC. Lichen amyloidosis. CMAJ. 2014;186:532. doi:10.1503/cmaj.130698
- Chen JF, Chen YF. Answer: can you identify this condition? Can Fam Physician. 2012;58:1234-1235.
- Foering K, Torbeck RL, Frank MP, et al. Treatment of pseudoxanthoma elasticum-like papillary dermal elastolysis with nonablative fractional resurfacing laser resulting in clinical and histologic improvement in elastin and collagen. J Cosmet Laser Ther. 2018;20:382-384. doi:10.1080/14764172.2017.1358457
A 76-year-old woman presented to the dermatology clinic for evaluation of a pruritic rash on the posterior lateral neck of several years’ duration. The rash had been slowly worsening and was intermittently symptomatic. Physical examination revealed monomorphous flesh-colored papules coalescing on the neck, yielding a cobblestonelike texture. The patient had been treated previously by dermatology with topical steroids, but symptoms persisted. A punch biopsy of the left lateral neck was performed.
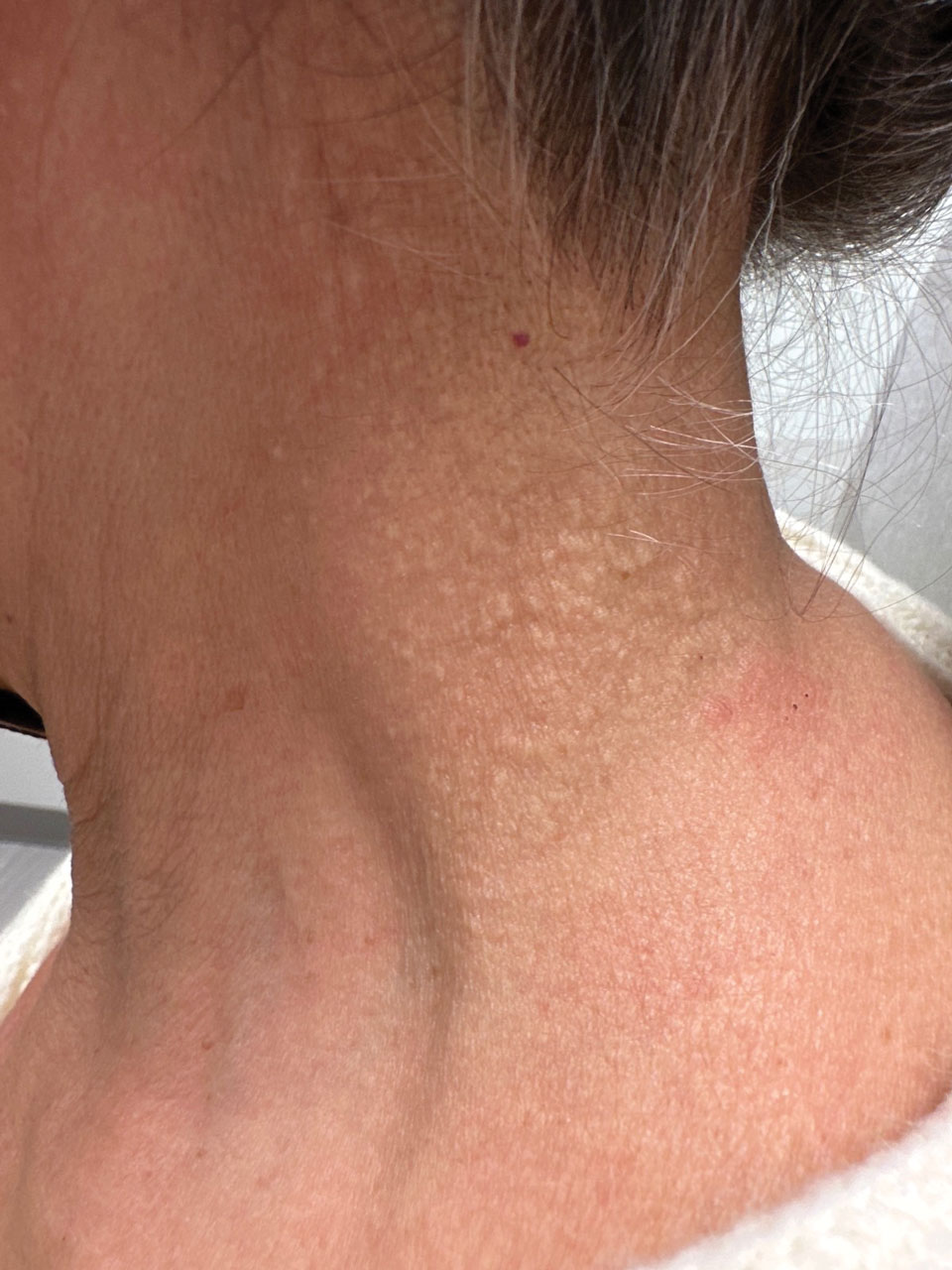
Alopecia and Pruritic Rash on the Forehead and Scalp
Alopecia and Pruritic Rash on the Forehead and Scalp
THE DIAGNOSIS: Folliculitis Decalvans
Biopsy results revealed a brisk perifollicular and intrafollicular mixed inflammatory infiltrate comprising lymphocytes, neutrophils, and plasma cells filling the upper dermis and encircling dilated hair follicles. Elastic stain (Verhoeff-van Gieson) demonstrated loss of elastic fibers in areas of scarring. Periodic acid–Schiff with diastase staining was negative for fungal elements, while Gram staining revealed colonies of bacterial cocci in the stratum corneum and within the hair follicles. Immunofluorescence was unremarkable, and culture revealed methicillin-sensitive Staphylococcus aureus, leading to a diagnosis of folliculitis decalvans (FD). The patient was treated with doxycycline 100 mg twice daily and received intralesional triamcinolone 2.5 mg/mL (total volume, 2 mL) every 6 weeks with considerable improvement in pustules, erythema, and scaling (Figure). While not yet in complete remission, our patient demonstrated short regrowing hairs in areas of incomplete scarring and focal remaining perifollicular erythema and scale along the midline frontal scalp 5 months after initial presentation.
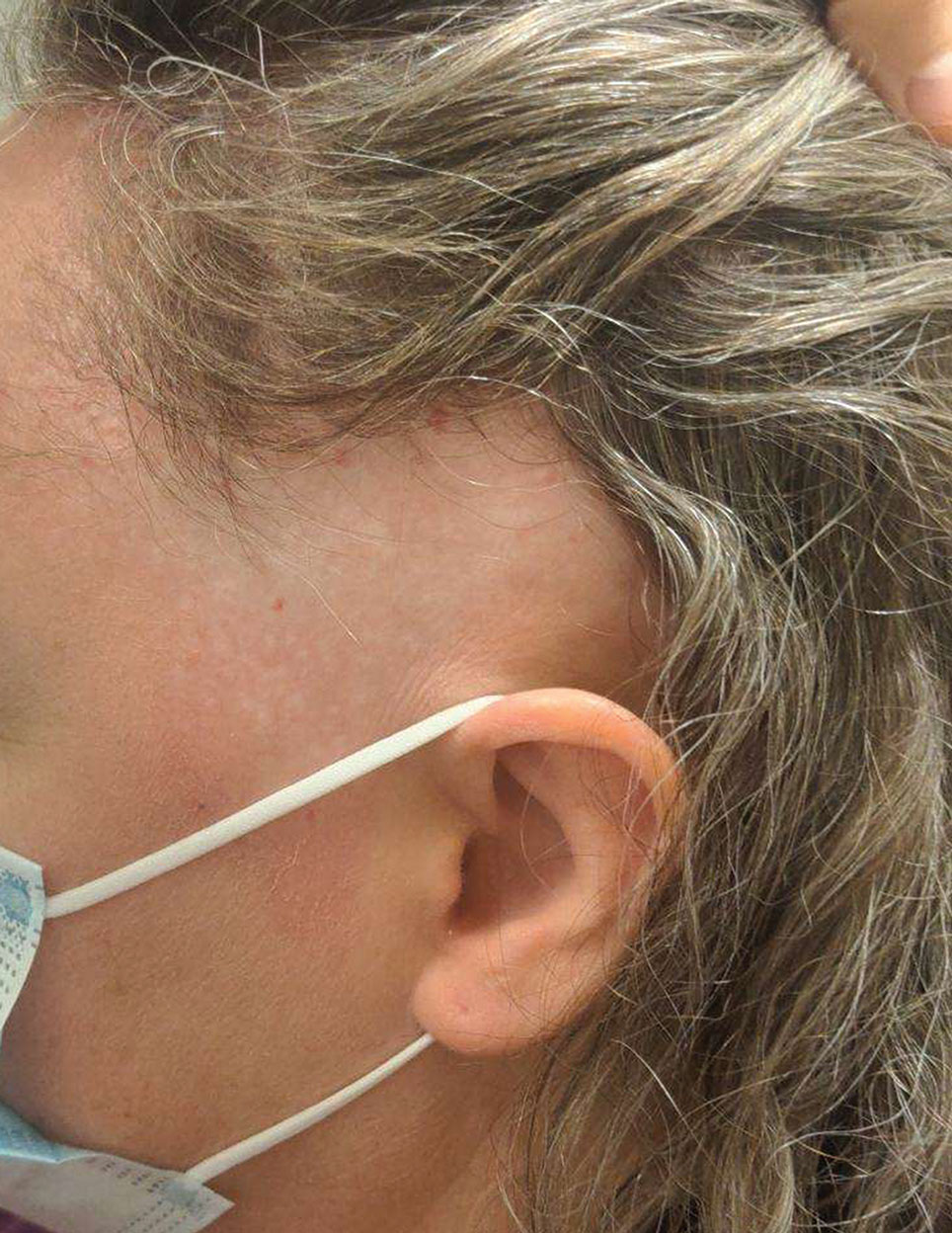
Folliculitis decalvans is an uncommon subtype of cicatricial alopecia that may mimic other forms of alopecia. Cicatricial alopecia often is difficult to diagnose due to its overlapping clinical characteristics, but early diagnosis is essential for appropriate management and prevention of further permanent hair loss. Traditionally classified as a primary neutrophilic cicatricial alopecia, lymphocyte-predominant variants of FD now are recognized.1
Patients with FD typically present with patchy scarring alopecia at the vertex scalp that gradually expands and may demonstrate secondary features of follicular tufting and pustules.1-3 While the epidemiology of FD is poorly characterized, Vañó-Galván et al4 reported that FD accounted for 2.8% of all alopecia cases and 10.5% of cicatricial alopecia cases in a multicenter study of 2835 patients. The pathophysiology of FD still is under investigation but is thought to result from a dysregulated immune response to a chronic bacterial infection (eg, S aureus), with resulting neutrophilpredominant inflammation in early stages.1-3 Vañó-Galván et al4 reported that, among 35 patients with FD cultured for bacteria, 74% (26/35) returned positive results, 96% (25/26) of which grew S aureus.5
A systematic review of 20 studies that included 263 patients found rifampin and clindamycin to be the most common treatments for FD; however, there is insufficient evidence to determine if this treatment is the most effective.6 In our patient, clindamycin was avoided due to its propensity to negatively alter the gut microbiome long term.7 Other therapies such as oral tetracyclines, high-potency topical steroids, and intralesional triamcinolone also can be used to achieve disease remission.5,6 Other treatments such as isotretinoin, red-light photodynamic therapy, tacrolimus, and external beam radiation have been reported in the literature but vary in efficacy.6 Our patient improved on a regimen of topical benzoyl peroxide wash, oral doxycycline, and intralesional triamcinolone.
Notably, FD may share clinical features with other causes of cicatricial alopecia. In our patient, FD mimicked other entities including discoid lupus erythematosus, frontal fibrosing alopecia, dissecting cellulitis, and erosive pustular dermatosis (Table).1-14 Discoid lupus erythematosus manifests as round hypopigmented and hyperpigmented plaques with associated atrophy, perifollicular erythema, and follicular plugging. Frontal fibrosing alopecia is a primary lymphocytic scarring alopecia that manifests in a bandlike linear distribution over the frontal scalp and may involve the temporal scalp, posterior hairline, and/or eyebrows. Isolated hairs (known as lonely hairs) often are seen. Dissecting cellulitis is characterized by boggy nodules associated with alopecia on the scalp without notable epidermal change, although pustules and sinus tracts may develop.9 Erosive pustular dermatosis is a diagnosis of exclusion but often is seen in older adults with chronic sun damage and clinically manifests with eroded plaques with adherent crusts.10
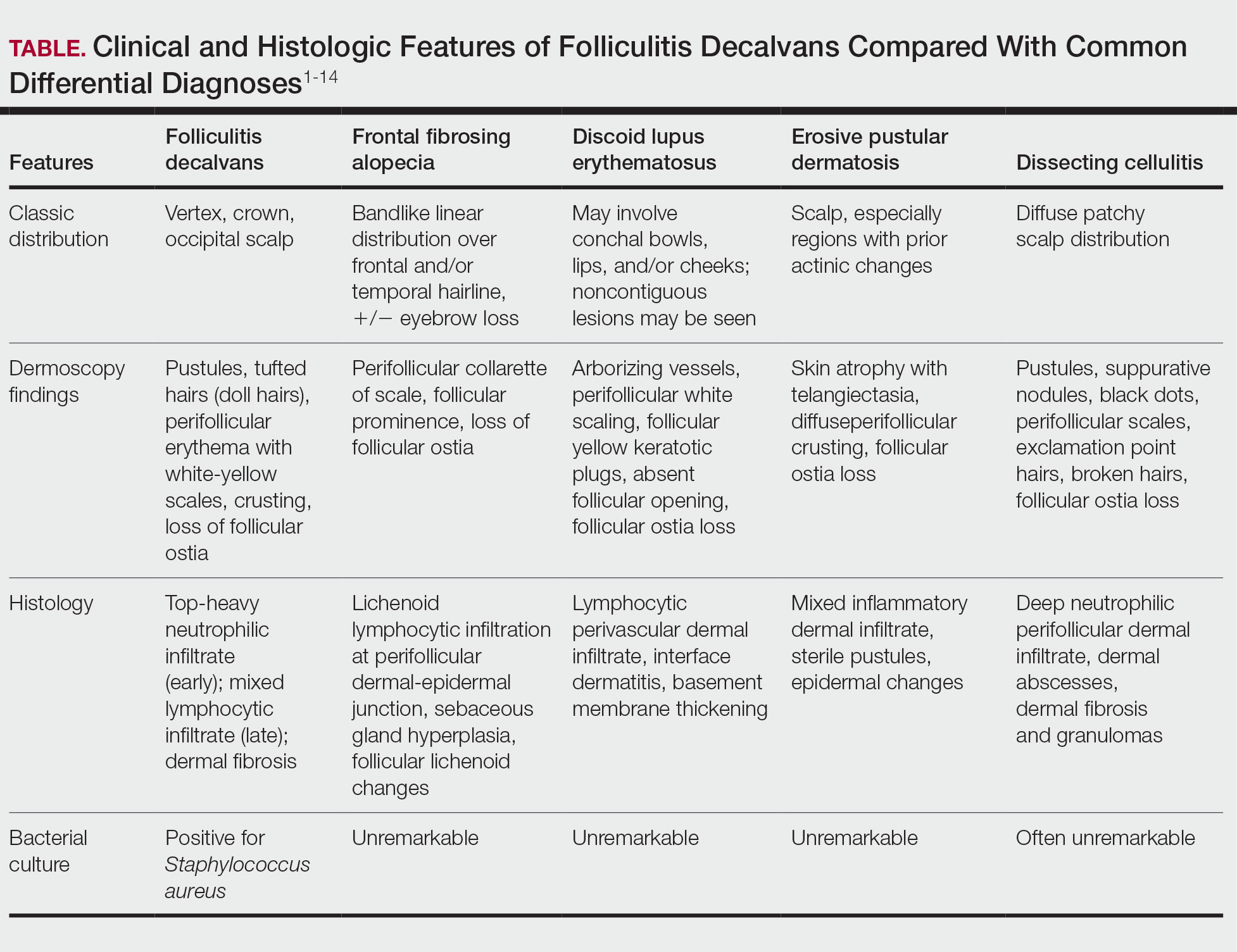
While our patient presented with several overlapping clinical features, including progressive hair loss along the frontal scalp in a bandlike pattern suspicious for frontal fibrosing alopecia as well as atrophic depigmented plaques with adherent peripheral scaling suspicious for discoid lupus erythematosus, the presence of pustules was an important clue. The biopsy demonstrating a mixed infiltrate inclusive of neutrophils confirmed the diagnosis of FD.
- Olsen EA, Bergfeld WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)-sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110. doi:10.1067/mjd.2003.68
- Filbrandt R, Rufaut N, Jones L. Primary cicatricial alopecia: diagnosis and treatment. CMAJ. 2013;185:1579-1585. doi:10.1503/cmaj.111570
- Otberg N, Kang H, Alzolibani AA, et al. Folliculitis decalvans. Dermatol Ther. 2008;21:238-244. doi:10.1111/j.1529-8019.2008.00204.x
- Vañó-Galván S, Saceda-Corralo D, Blume-Peytavi U, et al. Frequency of the types of alopecia at twenty-two specialist hair clinics: a multicenter study. Skin Appendage Disord. 2019;5:309-315. doi:10.1159/000496708
- Vañó-Galván S, Molina-Ruiz AM, Fernández-Crehuet P, et al. Folliculitis decalvans: a multicentre review of 82 patients. J Eur Acad Dermatol Venereol. 2015;29:1750-1757. doi:10.1111/jdv.12993
- Rambhia PH, Conic RRZ, Murad A, et al. Updates in therapeutics for folliculitis decalvans: a systematic review with evidence-based analysis. J Am Acad Dermatol. 2019;80:794-801. doi:10.1016/j.jaad.2018.07.050
- Zimmermann P, Curtis N. The effect of antibiotics on the composition of the intestinal microbiota - a systematic review. J Infect. 2019;79:471-489. doi:10.1016/j.jinf.2019.10.008
- Kanti V, Röwert-Huber J, Vogt A, et al. Cicatricial alopecia. J Dtsch Dermatol Ges. 2018;16:435-461. doi:10.1111/ddg.13498
- Melo DF, Slaibi EB, Siqueira TMFM, et al. Trichoscopy findings in dissecting cellulitis. An Bras Dermatol. 2019;94:608-611. doi:10.1016/j.abd.2019.09.006
- Anzai A, Pirmez R, Vincenzi C, et al. Trichoscopy findings of frontal fibrosing alopecia on the eyebrows: a study of 151 cases. J Am Acad Dermatol. 2021;85:1130-1134. doi:10.1016/j.jaad.2019.12.023
- Starace M, Loi C, Bruni F, et al. Erosive pustular dermatosis of the scalp: clinical, trichoscopic, and histopathologic features of 20 cases. J Am Acad Dermatol. 2017;76:1109-1114. doi:10.1016/j.jaad.2016.12.016
- Rongioletti F, Christana K. Cicatricial (scarring) alopecias: an overview of pathogenesis, classification, diagnosis, and treatment. Am J Clin Dermatol. 2012;13:247-260. doi:10.2165/11596960-000000000-00000
- Badaoui A, Reygagne P, Cavelier-Balloy B, et al. Dissecting cellulitis of the scalp: a retrospective study of 51 patients and review of literature. Br J Dermatol. 2016;174:421-423. doi:10.1111/bjd.13999
- Michelerio A, Vassallo C, Fiandrino G, et al. Erosive pustular dermatosis of the scalp: a clinicopathologic study of fifty cases. Dermatopathology (Basel). 2021;8:450-462. doi:10.3390/dermatopathology8040048
THE DIAGNOSIS: Folliculitis Decalvans
Biopsy results revealed a brisk perifollicular and intrafollicular mixed inflammatory infiltrate comprising lymphocytes, neutrophils, and plasma cells filling the upper dermis and encircling dilated hair follicles. Elastic stain (Verhoeff-van Gieson) demonstrated loss of elastic fibers in areas of scarring. Periodic acid–Schiff with diastase staining was negative for fungal elements, while Gram staining revealed colonies of bacterial cocci in the stratum corneum and within the hair follicles. Immunofluorescence was unremarkable, and culture revealed methicillin-sensitive Staphylococcus aureus, leading to a diagnosis of folliculitis decalvans (FD). The patient was treated with doxycycline 100 mg twice daily and received intralesional triamcinolone 2.5 mg/mL (total volume, 2 mL) every 6 weeks with considerable improvement in pustules, erythema, and scaling (Figure). While not yet in complete remission, our patient demonstrated short regrowing hairs in areas of incomplete scarring and focal remaining perifollicular erythema and scale along the midline frontal scalp 5 months after initial presentation.
Folliculitis decalvans is an uncommon subtype of cicatricial alopecia that may mimic other forms of alopecia. Cicatricial alopecia often is difficult to diagnose due to its overlapping clinical characteristics, but early diagnosis is essential for appropriate management and prevention of further permanent hair loss. Traditionally classified as a primary neutrophilic cicatricial alopecia, lymphocyte-predominant variants of FD now are recognized.1
Patients with FD typically present with patchy scarring alopecia at the vertex scalp that gradually expands and may demonstrate secondary features of follicular tufting and pustules.1-3 While the epidemiology of FD is poorly characterized, Vañó-Galván et al4 reported that FD accounted for 2.8% of all alopecia cases and 10.5% of cicatricial alopecia cases in a multicenter study of 2835 patients. The pathophysiology of FD still is under investigation but is thought to result from a dysregulated immune response to a chronic bacterial infection (eg, S aureus), with resulting neutrophilpredominant inflammation in early stages.1-3 Vañó-Galván et al4 reported that, among 35 patients with FD cultured for bacteria, 74% (26/35) returned positive results, 96% (25/26) of which grew S aureus.5
A systematic review of 20 studies that included 263 patients found rifampin and clindamycin to be the most common treatments for FD; however, there is insufficient evidence to determine if this treatment is the most effective.6 In our patient, clindamycin was avoided due to its propensity to negatively alter the gut microbiome long term.7 Other therapies such as oral tetracyclines, high-potency topical steroids, and intralesional triamcinolone also can be used to achieve disease remission.5,6 Other treatments such as isotretinoin, red-light photodynamic therapy, tacrolimus, and external beam radiation have been reported in the literature but vary in efficacy.6 Our patient improved on a regimen of topical benzoyl peroxide wash, oral doxycycline, and intralesional triamcinolone.
Notably, FD may share clinical features with other causes of cicatricial alopecia. In our patient, FD mimicked other entities including discoid lupus erythematosus, frontal fibrosing alopecia, dissecting cellulitis, and erosive pustular dermatosis (Table).1-14 Discoid lupus erythematosus manifests as round hypopigmented and hyperpigmented plaques with associated atrophy, perifollicular erythema, and follicular plugging. Frontal fibrosing alopecia is a primary lymphocytic scarring alopecia that manifests in a bandlike linear distribution over the frontal scalp and may involve the temporal scalp, posterior hairline, and/or eyebrows. Isolated hairs (known as lonely hairs) often are seen. Dissecting cellulitis is characterized by boggy nodules associated with alopecia on the scalp without notable epidermal change, although pustules and sinus tracts may develop.9 Erosive pustular dermatosis is a diagnosis of exclusion but often is seen in older adults with chronic sun damage and clinically manifests with eroded plaques with adherent crusts.10
While our patient presented with several overlapping clinical features, including progressive hair loss along the frontal scalp in a bandlike pattern suspicious for frontal fibrosing alopecia as well as atrophic depigmented plaques with adherent peripheral scaling suspicious for discoid lupus erythematosus, the presence of pustules was an important clue. The biopsy demonstrating a mixed infiltrate inclusive of neutrophils confirmed the diagnosis of FD.
THE DIAGNOSIS: Folliculitis Decalvans
Biopsy results revealed a brisk perifollicular and intrafollicular mixed inflammatory infiltrate comprising lymphocytes, neutrophils, and plasma cells filling the upper dermis and encircling dilated hair follicles. Elastic stain (Verhoeff-van Gieson) demonstrated loss of elastic fibers in areas of scarring. Periodic acid–Schiff with diastase staining was negative for fungal elements, while Gram staining revealed colonies of bacterial cocci in the stratum corneum and within the hair follicles. Immunofluorescence was unremarkable, and culture revealed methicillin-sensitive Staphylococcus aureus, leading to a diagnosis of folliculitis decalvans (FD). The patient was treated with doxycycline 100 mg twice daily and received intralesional triamcinolone 2.5 mg/mL (total volume, 2 mL) every 6 weeks with considerable improvement in pustules, erythema, and scaling (Figure). While not yet in complete remission, our patient demonstrated short regrowing hairs in areas of incomplete scarring and focal remaining perifollicular erythema and scale along the midline frontal scalp 5 months after initial presentation.
Folliculitis decalvans is an uncommon subtype of cicatricial alopecia that may mimic other forms of alopecia. Cicatricial alopecia often is difficult to diagnose due to its overlapping clinical characteristics, but early diagnosis is essential for appropriate management and prevention of further permanent hair loss. Traditionally classified as a primary neutrophilic cicatricial alopecia, lymphocyte-predominant variants of FD now are recognized.1
Patients with FD typically present with patchy scarring alopecia at the vertex scalp that gradually expands and may demonstrate secondary features of follicular tufting and pustules.1-3 While the epidemiology of FD is poorly characterized, Vañó-Galván et al4 reported that FD accounted for 2.8% of all alopecia cases and 10.5% of cicatricial alopecia cases in a multicenter study of 2835 patients. The pathophysiology of FD still is under investigation but is thought to result from a dysregulated immune response to a chronic bacterial infection (eg, S aureus), with resulting neutrophilpredominant inflammation in early stages.1-3 Vañó-Galván et al4 reported that, among 35 patients with FD cultured for bacteria, 74% (26/35) returned positive results, 96% (25/26) of which grew S aureus.5
A systematic review of 20 studies that included 263 patients found rifampin and clindamycin to be the most common treatments for FD; however, there is insufficient evidence to determine if this treatment is the most effective.6 In our patient, clindamycin was avoided due to its propensity to negatively alter the gut microbiome long term.7 Other therapies such as oral tetracyclines, high-potency topical steroids, and intralesional triamcinolone also can be used to achieve disease remission.5,6 Other treatments such as isotretinoin, red-light photodynamic therapy, tacrolimus, and external beam radiation have been reported in the literature but vary in efficacy.6 Our patient improved on a regimen of topical benzoyl peroxide wash, oral doxycycline, and intralesional triamcinolone.
Notably, FD may share clinical features with other causes of cicatricial alopecia. In our patient, FD mimicked other entities including discoid lupus erythematosus, frontal fibrosing alopecia, dissecting cellulitis, and erosive pustular dermatosis (Table).1-14 Discoid lupus erythematosus manifests as round hypopigmented and hyperpigmented plaques with associated atrophy, perifollicular erythema, and follicular plugging. Frontal fibrosing alopecia is a primary lymphocytic scarring alopecia that manifests in a bandlike linear distribution over the frontal scalp and may involve the temporal scalp, posterior hairline, and/or eyebrows. Isolated hairs (known as lonely hairs) often are seen. Dissecting cellulitis is characterized by boggy nodules associated with alopecia on the scalp without notable epidermal change, although pustules and sinus tracts may develop.9 Erosive pustular dermatosis is a diagnosis of exclusion but often is seen in older adults with chronic sun damage and clinically manifests with eroded plaques with adherent crusts.10
While our patient presented with several overlapping clinical features, including progressive hair loss along the frontal scalp in a bandlike pattern suspicious for frontal fibrosing alopecia as well as atrophic depigmented plaques with adherent peripheral scaling suspicious for discoid lupus erythematosus, the presence of pustules was an important clue. The biopsy demonstrating a mixed infiltrate inclusive of neutrophils confirmed the diagnosis of FD.
- Olsen EA, Bergfeld WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)-sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110. doi:10.1067/mjd.2003.68
- Filbrandt R, Rufaut N, Jones L. Primary cicatricial alopecia: diagnosis and treatment. CMAJ. 2013;185:1579-1585. doi:10.1503/cmaj.111570
- Otberg N, Kang H, Alzolibani AA, et al. Folliculitis decalvans. Dermatol Ther. 2008;21:238-244. doi:10.1111/j.1529-8019.2008.00204.x
- Vañó-Galván S, Saceda-Corralo D, Blume-Peytavi U, et al. Frequency of the types of alopecia at twenty-two specialist hair clinics: a multicenter study. Skin Appendage Disord. 2019;5:309-315. doi:10.1159/000496708
- Vañó-Galván S, Molina-Ruiz AM, Fernández-Crehuet P, et al. Folliculitis decalvans: a multicentre review of 82 patients. J Eur Acad Dermatol Venereol. 2015;29:1750-1757. doi:10.1111/jdv.12993
- Rambhia PH, Conic RRZ, Murad A, et al. Updates in therapeutics for folliculitis decalvans: a systematic review with evidence-based analysis. J Am Acad Dermatol. 2019;80:794-801. doi:10.1016/j.jaad.2018.07.050
- Zimmermann P, Curtis N. The effect of antibiotics on the composition of the intestinal microbiota - a systematic review. J Infect. 2019;79:471-489. doi:10.1016/j.jinf.2019.10.008
- Kanti V, Röwert-Huber J, Vogt A, et al. Cicatricial alopecia. J Dtsch Dermatol Ges. 2018;16:435-461. doi:10.1111/ddg.13498
- Melo DF, Slaibi EB, Siqueira TMFM, et al. Trichoscopy findings in dissecting cellulitis. An Bras Dermatol. 2019;94:608-611. doi:10.1016/j.abd.2019.09.006
- Anzai A, Pirmez R, Vincenzi C, et al. Trichoscopy findings of frontal fibrosing alopecia on the eyebrows: a study of 151 cases. J Am Acad Dermatol. 2021;85:1130-1134. doi:10.1016/j.jaad.2019.12.023
- Starace M, Loi C, Bruni F, et al. Erosive pustular dermatosis of the scalp: clinical, trichoscopic, and histopathologic features of 20 cases. J Am Acad Dermatol. 2017;76:1109-1114. doi:10.1016/j.jaad.2016.12.016
- Rongioletti F, Christana K. Cicatricial (scarring) alopecias: an overview of pathogenesis, classification, diagnosis, and treatment. Am J Clin Dermatol. 2012;13:247-260. doi:10.2165/11596960-000000000-00000
- Badaoui A, Reygagne P, Cavelier-Balloy B, et al. Dissecting cellulitis of the scalp: a retrospective study of 51 patients and review of literature. Br J Dermatol. 2016;174:421-423. doi:10.1111/bjd.13999
- Michelerio A, Vassallo C, Fiandrino G, et al. Erosive pustular dermatosis of the scalp: a clinicopathologic study of fifty cases. Dermatopathology (Basel). 2021;8:450-462. doi:10.3390/dermatopathology8040048
- Olsen EA, Bergfeld WF, Cotsarelis G, et al. Summary of North American Hair Research Society (NAHRS)-sponsored Workshop on Cicatricial Alopecia, Duke University Medical Center, February 10 and 11, 2001. J Am Acad Dermatol. 2003;48:103-110. doi:10.1067/mjd.2003.68
- Filbrandt R, Rufaut N, Jones L. Primary cicatricial alopecia: diagnosis and treatment. CMAJ. 2013;185:1579-1585. doi:10.1503/cmaj.111570
- Otberg N, Kang H, Alzolibani AA, et al. Folliculitis decalvans. Dermatol Ther. 2008;21:238-244. doi:10.1111/j.1529-8019.2008.00204.x
- Vañó-Galván S, Saceda-Corralo D, Blume-Peytavi U, et al. Frequency of the types of alopecia at twenty-two specialist hair clinics: a multicenter study. Skin Appendage Disord. 2019;5:309-315. doi:10.1159/000496708
- Vañó-Galván S, Molina-Ruiz AM, Fernández-Crehuet P, et al. Folliculitis decalvans: a multicentre review of 82 patients. J Eur Acad Dermatol Venereol. 2015;29:1750-1757. doi:10.1111/jdv.12993
- Rambhia PH, Conic RRZ, Murad A, et al. Updates in therapeutics for folliculitis decalvans: a systematic review with evidence-based analysis. J Am Acad Dermatol. 2019;80:794-801. doi:10.1016/j.jaad.2018.07.050
- Zimmermann P, Curtis N. The effect of antibiotics on the composition of the intestinal microbiota - a systematic review. J Infect. 2019;79:471-489. doi:10.1016/j.jinf.2019.10.008
- Kanti V, Röwert-Huber J, Vogt A, et al. Cicatricial alopecia. J Dtsch Dermatol Ges. 2018;16:435-461. doi:10.1111/ddg.13498
- Melo DF, Slaibi EB, Siqueira TMFM, et al. Trichoscopy findings in dissecting cellulitis. An Bras Dermatol. 2019;94:608-611. doi:10.1016/j.abd.2019.09.006
- Anzai A, Pirmez R, Vincenzi C, et al. Trichoscopy findings of frontal fibrosing alopecia on the eyebrows: a study of 151 cases. J Am Acad Dermatol. 2021;85:1130-1134. doi:10.1016/j.jaad.2019.12.023
- Starace M, Loi C, Bruni F, et al. Erosive pustular dermatosis of the scalp: clinical, trichoscopic, and histopathologic features of 20 cases. J Am Acad Dermatol. 2017;76:1109-1114. doi:10.1016/j.jaad.2016.12.016
- Rongioletti F, Christana K. Cicatricial (scarring) alopecias: an overview of pathogenesis, classification, diagnosis, and treatment. Am J Clin Dermatol. 2012;13:247-260. doi:10.2165/11596960-000000000-00000
- Badaoui A, Reygagne P, Cavelier-Balloy B, et al. Dissecting cellulitis of the scalp: a retrospective study of 51 patients and review of literature. Br J Dermatol. 2016;174:421-423. doi:10.1111/bjd.13999
- Michelerio A, Vassallo C, Fiandrino G, et al. Erosive pustular dermatosis of the scalp: a clinicopathologic study of fifty cases. Dermatopathology (Basel). 2021;8:450-462. doi:10.3390/dermatopathology8040048
Alopecia and Pruritic Rash on the Forehead and Scalp
Alopecia and Pruritic Rash on the Forehead and Scalp
A 52-year-old woman presented to the dermatology department with an intermittently pruritic rash in a bandlike distribution on the left upper forehead and the frontal and temporal scalp of 4 years’ duration. The rash initially was diagnosed as psoriasis at an outside facility. Treatment over the year prior to presentation included tildrakizumab-asmn; topical crisaborole 2%; and excimer laser, which was complicated by blistering. The patient reported no history of topical or injected steroid use in the involved areas. Physical examination at the current presentation revealed arcuate erythematous plaques with follicular prominence, perifollicular scaling, pustules, and lone hairs. There also were porcelain-white atrophic plaques with loss of follicular ostia that were most prominent over the temporal scalp. A biopsy of the left lateral forehead was performed.
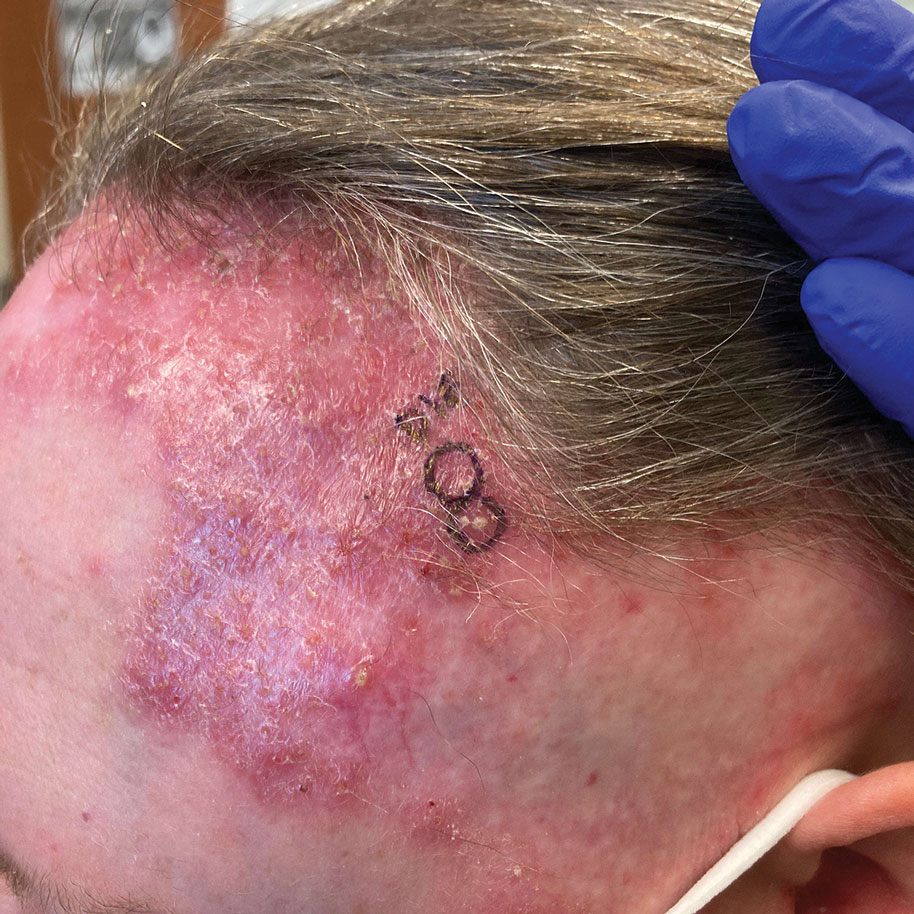

Solitary Yellow Papule on the Upper Back in an Infant
The Diagnosis: Juvenile Xanthogranuloma
Given the patient’s age, clinical features of the lesion, and characteristic setting-sun pattern on dermoscopy, a diagnosis of juvenile xanthogranuloma (JXG) was made. The patient showed no other signs of neurofibromatosis type 1 (NF1) or systemic disease and was managed conservatively with observation and routine follow-up. Minimal growth of the lesion was noted at 1-year follow-up, and he was meeting all age-appropriate developmental milestones and showed no other symptoms consistent with NF1.
Juvenile xanthogranuloma is the most common childhood non–Langerhans cell histiocytosis. While it typically manifests as an isolated condition, JXG also can be associated with NF1 as well as juvenile myelomonocytic leukemia.1-3 Neurofibromatosis type 1 is a multisystem disorder with variable clinical manifestations that commonly is associated with skin findings such as café au lait macules, intertriginous freckling, and neurofibromas, in addition to JXG.2,3 Diagnosis of JXG should prompt noninvasive evaluation for further signs and symptoms of NF1, including thorough patient and family history and physical examination to identify other characteristic cutaneous findings, and can include consideration of slit lamp eye examination and radiography for identification of osseous findings.
The pathogenesis of JXG is not fully known, though there is evidence that it may be associated with a mutation in the mitogen-activated protein kinase pathway.1 The majority of cases appear in the first year of life.4 Clinically, JXG can manifest with extracutaneous lesions, including on the eyes and lungs.5-7 Juvenile xanthogranuloma can be noninvasively diagnosed with dermoscopy. As seen in our patient, dermoscopic findings include a red-yellow or yellow-orange background with an erythematous border, typically described as a setting-sun pattern.4,8 Biopsy can confirm the diagnosis; however, given the usually benign course, this often is unnecessary. Most pediatric patients with cutaneous manifestations have a self-limited course with regression over several months to years. Generally, no treatment is required for cutaneous manifestations alone; however, lesions can be removed for aesthetic concerns. For those with systemic involvement, a range of other treatments have been used, including chemotherapy, radiotherapy, systemic corticosteroids, and cyclosporine.6,7
The differential diagnosis for JXG includes Brooke-Spiegler syndrome, Fabry disease, solitary cutaneous mastocytoma, and tuberous sclerosis complex. Brooke-Spiegler syndrome is an autosomal-dominant condition characterized by the growth of adnexal neoplasms, including trichoepitheliomas, cylindromas, and spiradenomas. These lesions usually manifest on the face but can include other areas such as the trunk.9 Fabry disease is an X-linked recessive lysosomal storage disorder with cutaneous manifestations such as angiokeratoma corporis diffusum and hypohidrosis. Patients also may present with systemic symptoms including hypertension and renal and cardiovascular disease.10 Mastocytosis encompasses several clinical disorders defined by mast cell hyperplasia and accumulation in various organ systems, and solitary cutaneous mastocytoma is the most common manifestation in children.11,12 Cutaneous mastocytoma can manifest as a single red-brown or yellow papule, usually located on the arms or legs.13 Solitary cutaneous mastocytomas in pediatric patients typically are diagnosed based on clinical appearance and the formation of a wheal upon firm palpation (Darier sign).11-13 Our patient did not demonstrate the Darier sign, and the lesion was asymptomatic. Tuberous sclerosis complex is an autosomal-dominant neurocutaneous disorder with neurologic and skin findings that occur early in the disease course and include facial angiofibromas, hypomelanotic macules, shagreen patches, and café-au-lait macules.14
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839-1842.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1998.
- Miraglia E, Laghi A, Moramarco A, et al. Juvenile xanthogranuloma in neurofibromatosis type 1. Prevalence and possible correlation with lymphoproliferative diseases: experience of a single center and review of the literature. Clin Ther. 2022;173:353-355.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 8, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
- Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129:227-237.
- Murphy JT, Soeken T, Megison S, et al. Juvenile xanthogranuloma: diverse presentations of noncutaneous disease. J Pediatr Hematol Oncol.2014;36:641-645.
- Xu J, Ma L. Dermoscopic patterns in juvenile xanthogranuloma based on the histological classification. Front Med (Lausanne). 2021;7:618946.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bokhari SRA, Zulfiqar H, Hariz A. Fabry disease. StatPearls [Internet]. StatPearls Publishing; 2025. Update July 4, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK435996/
- Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45.
- Klaiber N, Kumar S, Irani AM. Mastocytosis in children. Curr Allergy Asthma Rep. 2017;17:80.
- Sławin´ ska M, Kaszuba A, Lange M, et al. Dermoscopic features of different forms of cutaneous mastocytosis: a systematic review. J Clin Med. 2022;11:4649.
- Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150:1095-1101.
The Diagnosis: Juvenile Xanthogranuloma
Given the patient’s age, clinical features of the lesion, and characteristic setting-sun pattern on dermoscopy, a diagnosis of juvenile xanthogranuloma (JXG) was made. The patient showed no other signs of neurofibromatosis type 1 (NF1) or systemic disease and was managed conservatively with observation and routine follow-up. Minimal growth of the lesion was noted at 1-year follow-up, and he was meeting all age-appropriate developmental milestones and showed no other symptoms consistent with NF1.
Juvenile xanthogranuloma is the most common childhood non–Langerhans cell histiocytosis. While it typically manifests as an isolated condition, JXG also can be associated with NF1 as well as juvenile myelomonocytic leukemia.1-3 Neurofibromatosis type 1 is a multisystem disorder with variable clinical manifestations that commonly is associated with skin findings such as café au lait macules, intertriginous freckling, and neurofibromas, in addition to JXG.2,3 Diagnosis of JXG should prompt noninvasive evaluation for further signs and symptoms of NF1, including thorough patient and family history and physical examination to identify other characteristic cutaneous findings, and can include consideration of slit lamp eye examination and radiography for identification of osseous findings.
The pathogenesis of JXG is not fully known, though there is evidence that it may be associated with a mutation in the mitogen-activated protein kinase pathway.1 The majority of cases appear in the first year of life.4 Clinically, JXG can manifest with extracutaneous lesions, including on the eyes and lungs.5-7 Juvenile xanthogranuloma can be noninvasively diagnosed with dermoscopy. As seen in our patient, dermoscopic findings include a red-yellow or yellow-orange background with an erythematous border, typically described as a setting-sun pattern.4,8 Biopsy can confirm the diagnosis; however, given the usually benign course, this often is unnecessary. Most pediatric patients with cutaneous manifestations have a self-limited course with regression over several months to years. Generally, no treatment is required for cutaneous manifestations alone; however, lesions can be removed for aesthetic concerns. For those with systemic involvement, a range of other treatments have been used, including chemotherapy, radiotherapy, systemic corticosteroids, and cyclosporine.6,7
The differential diagnosis for JXG includes Brooke-Spiegler syndrome, Fabry disease, solitary cutaneous mastocytoma, and tuberous sclerosis complex. Brooke-Spiegler syndrome is an autosomal-dominant condition characterized by the growth of adnexal neoplasms, including trichoepitheliomas, cylindromas, and spiradenomas. These lesions usually manifest on the face but can include other areas such as the trunk.9 Fabry disease is an X-linked recessive lysosomal storage disorder with cutaneous manifestations such as angiokeratoma corporis diffusum and hypohidrosis. Patients also may present with systemic symptoms including hypertension and renal and cardiovascular disease.10 Mastocytosis encompasses several clinical disorders defined by mast cell hyperplasia and accumulation in various organ systems, and solitary cutaneous mastocytoma is the most common manifestation in children.11,12 Cutaneous mastocytoma can manifest as a single red-brown or yellow papule, usually located on the arms or legs.13 Solitary cutaneous mastocytomas in pediatric patients typically are diagnosed based on clinical appearance and the formation of a wheal upon firm palpation (Darier sign).11-13 Our patient did not demonstrate the Darier sign, and the lesion was asymptomatic. Tuberous sclerosis complex is an autosomal-dominant neurocutaneous disorder with neurologic and skin findings that occur early in the disease course and include facial angiofibromas, hypomelanotic macules, shagreen patches, and café-au-lait macules.14
The Diagnosis: Juvenile Xanthogranuloma
Given the patient’s age, clinical features of the lesion, and characteristic setting-sun pattern on dermoscopy, a diagnosis of juvenile xanthogranuloma (JXG) was made. The patient showed no other signs of neurofibromatosis type 1 (NF1) or systemic disease and was managed conservatively with observation and routine follow-up. Minimal growth of the lesion was noted at 1-year follow-up, and he was meeting all age-appropriate developmental milestones and showed no other symptoms consistent with NF1.
Juvenile xanthogranuloma is the most common childhood non–Langerhans cell histiocytosis. While it typically manifests as an isolated condition, JXG also can be associated with NF1 as well as juvenile myelomonocytic leukemia.1-3 Neurofibromatosis type 1 is a multisystem disorder with variable clinical manifestations that commonly is associated with skin findings such as café au lait macules, intertriginous freckling, and neurofibromas, in addition to JXG.2,3 Diagnosis of JXG should prompt noninvasive evaluation for further signs and symptoms of NF1, including thorough patient and family history and physical examination to identify other characteristic cutaneous findings, and can include consideration of slit lamp eye examination and radiography for identification of osseous findings.
The pathogenesis of JXG is not fully known, though there is evidence that it may be associated with a mutation in the mitogen-activated protein kinase pathway.1 The majority of cases appear in the first year of life.4 Clinically, JXG can manifest with extracutaneous lesions, including on the eyes and lungs.5-7 Juvenile xanthogranuloma can be noninvasively diagnosed with dermoscopy. As seen in our patient, dermoscopic findings include a red-yellow or yellow-orange background with an erythematous border, typically described as a setting-sun pattern.4,8 Biopsy can confirm the diagnosis; however, given the usually benign course, this often is unnecessary. Most pediatric patients with cutaneous manifestations have a self-limited course with regression over several months to years. Generally, no treatment is required for cutaneous manifestations alone; however, lesions can be removed for aesthetic concerns. For those with systemic involvement, a range of other treatments have been used, including chemotherapy, radiotherapy, systemic corticosteroids, and cyclosporine.6,7
The differential diagnosis for JXG includes Brooke-Spiegler syndrome, Fabry disease, solitary cutaneous mastocytoma, and tuberous sclerosis complex. Brooke-Spiegler syndrome is an autosomal-dominant condition characterized by the growth of adnexal neoplasms, including trichoepitheliomas, cylindromas, and spiradenomas. These lesions usually manifest on the face but can include other areas such as the trunk.9 Fabry disease is an X-linked recessive lysosomal storage disorder with cutaneous manifestations such as angiokeratoma corporis diffusum and hypohidrosis. Patients also may present with systemic symptoms including hypertension and renal and cardiovascular disease.10 Mastocytosis encompasses several clinical disorders defined by mast cell hyperplasia and accumulation in various organ systems, and solitary cutaneous mastocytoma is the most common manifestation in children.11,12 Cutaneous mastocytoma can manifest as a single red-brown or yellow papule, usually located on the arms or legs.13 Solitary cutaneous mastocytomas in pediatric patients typically are diagnosed based on clinical appearance and the formation of a wheal upon firm palpation (Darier sign).11-13 Our patient did not demonstrate the Darier sign, and the lesion was asymptomatic. Tuberous sclerosis complex is an autosomal-dominant neurocutaneous disorder with neurologic and skin findings that occur early in the disease course and include facial angiofibromas, hypomelanotic macules, shagreen patches, and café-au-lait macules.14
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839-1842.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1998.
- Miraglia E, Laghi A, Moramarco A, et al. Juvenile xanthogranuloma in neurofibromatosis type 1. Prevalence and possible correlation with lymphoproliferative diseases: experience of a single center and review of the literature. Clin Ther. 2022;173:353-355.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 8, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
- Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129:227-237.
- Murphy JT, Soeken T, Megison S, et al. Juvenile xanthogranuloma: diverse presentations of noncutaneous disease. J Pediatr Hematol Oncol.2014;36:641-645.
- Xu J, Ma L. Dermoscopic patterns in juvenile xanthogranuloma based on the histological classification. Front Med (Lausanne). 2021;7:618946.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bokhari SRA, Zulfiqar H, Hariz A. Fabry disease. StatPearls [Internet]. StatPearls Publishing; 2025. Update July 4, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK435996/
- Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45.
- Klaiber N, Kumar S, Irani AM. Mastocytosis in children. Curr Allergy Asthma Rep. 2017;17:80.
- Sławin´ ska M, Kaszuba A, Lange M, et al. Dermoscopic features of different forms of cutaneous mastocytosis: a systematic review. J Clin Med. 2022;11:4649.
- Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150:1095-1101.
- Durham BH, Lopez Rodrigo E, Picarsic J, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in histiocytic neoplasms. Nat Med. 2019;25:1839-1842.
- Friedman JM. Neurofibromatosis 1. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. University of Washington, Seattle; 1998.
- Miraglia E, Laghi A, Moramarco A, et al. Juvenile xanthogranuloma in neurofibromatosis type 1. Prevalence and possible correlation with lymphoproliferative diseases: experience of a single center and review of the literature. Clin Ther. 2022;173:353-355.
- Collie JS, Harper CD, Fillman EP. Juvenile xanthogranuloma. StatPearls [Internet]. StatPearls Publishing; 2025. Updated August 8, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK526103/
- Newman B, Hu W, Nigro K, et al. Aggressive histiocytic disorders that can involve the skin. J Am Acad Dermatol. 2007;56:302-316.
- Freyer DR, Kennedy R, Bostrom BC, et al. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129:227-237.
- Murphy JT, Soeken T, Megison S, et al. Juvenile xanthogranuloma: diverse presentations of noncutaneous disease. J Pediatr Hematol Oncol.2014;36:641-645.
- Xu J, Ma L. Dermoscopic patterns in juvenile xanthogranuloma based on the histological classification. Front Med (Lausanne). 2021;7:618946.
- Kazakov DV. Brooke-Spiegler syndrome and phenotypic variants: an update. Head Neck Pathol. 2016;10:125-130.
- Bokhari SRA, Zulfiqar H, Hariz A. Fabry disease. StatPearls [Internet]. StatPearls Publishing; 2025. Update July 4, 2023. Accessed November 4, 2025. https://www.ncbi.nlm.nih.gov/books/NBK435996/
- Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol. 2016;137:35-45.
- Klaiber N, Kumar S, Irani AM. Mastocytosis in children. Curr Allergy Asthma Rep. 2017;17:80.
- Sławin´ ska M, Kaszuba A, Lange M, et al. Dermoscopic features of different forms of cutaneous mastocytosis: a systematic review. J Clin Med. 2022;11:4649.
- Teng JM, Cowen EW, Wataya-Kaneda M, et al. Dermatologic and dental aspects of the 2012 International Tuberous Sclerosis Complex Consensus Statements. JAMA Dermatol. 2014;150:1095-1101.
A 6-month-old male infant with a history of cradle cap and an infantile hemangioma on the left shoulder presented to the dermatology clinic for evaluation of a slow-growing yellow papule on the upper back of 3 months’ duration. The lesion initially was noted 2 months prior to the current presentation by the patient’s pediatrician, who recommended follow-up with dermatology after an unsuccessful attempt at incision and drainage. Physical examination revealed a 7-mm, yellow, dome-shaped papule with a red collarette on the right upper back. No axillary freckling, ocular findings, or other skin findings were found. The patient was born at term with no complications, and his mother reported that he was otherwise healthy. There were no developmental concerns or known allergies, and his family history was negative for any similar lesions. Dermoscopic examination of the lesion revealed a well-circumscribed, circular, yellow-orange papule with an erythematous border and setting-sun appearance.