User login
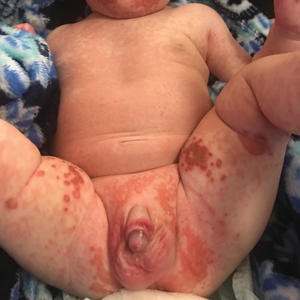
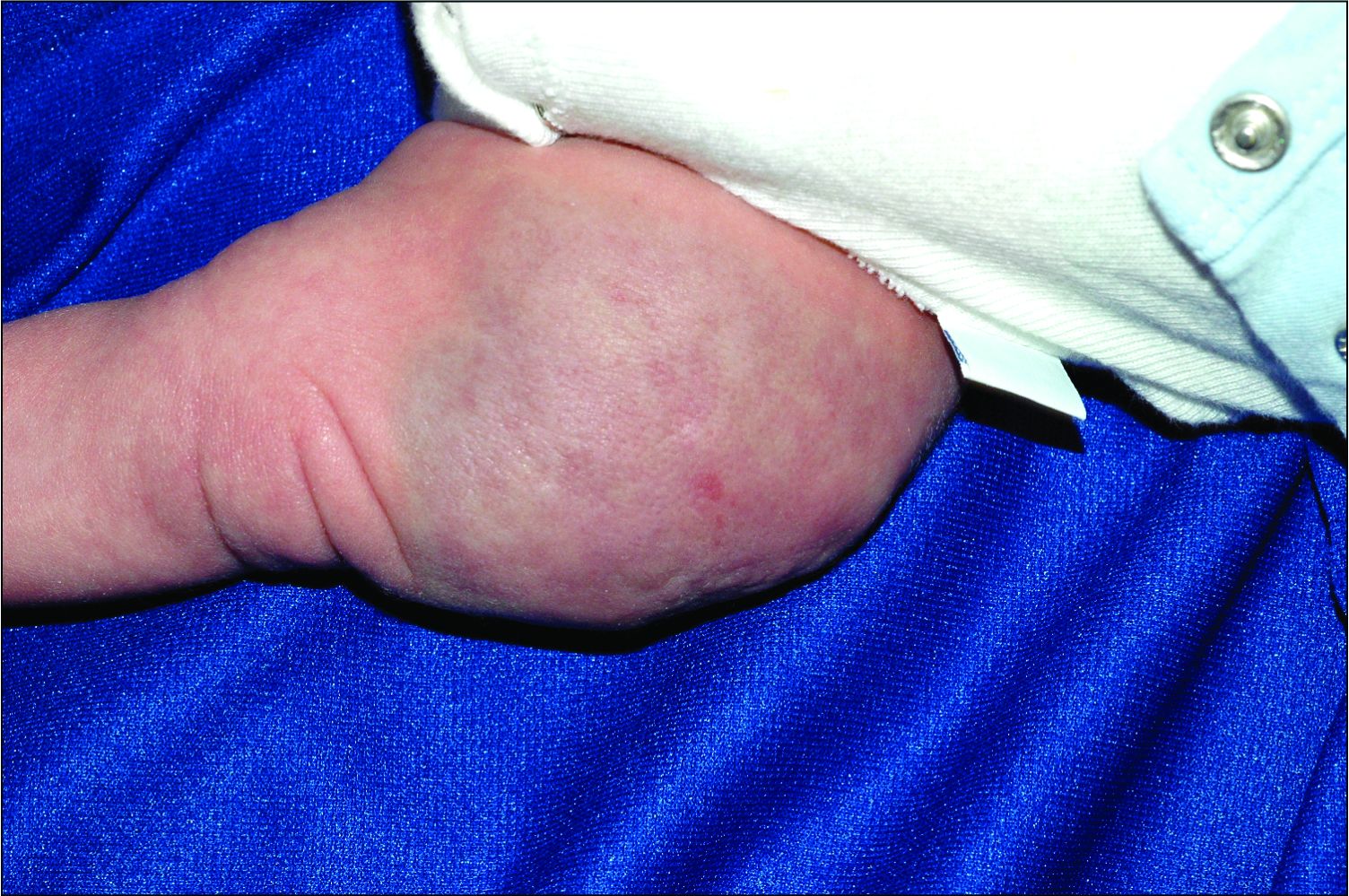
An infant girl presents with a growing pink-red leg nodule
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.

A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.

Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.
A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.
Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
The history of a brownish to pink patch with color change and rapid growth within the first year combined with the exam findings, are suggestive of a tufted angioma, though the findings presented may be nonspecific.
A tufted angioma is a rare vascular tumor of infancy or early childhood, that is present at birth in approximately half of cases. It may initially present as a faint pink to brown plaque, but develops as a firm, red to violaceous nodule or plaque, usually with “lumpiness” or nodularity.1-3 Lesions usually are infiltrative with indistinct borders. They are named for their histologic appearance, with lobules of capillaries which appear as “tufts” in the dermis and subdermis with “cannonball” appearance, and are considered to be on a spectrum with another vascular tumor called kaposiform hemangioendothelioma (KHE).4 These vascular tumors can trigger Kasabach-Merritt syndrome, a disease process in which vascular tumors trap platelets and clotting factors, resulting in a life-threatening thrombocytopenia and consumptive coagulopathy with a high risk of bleeding and high-output heart failure.5
What’s the differential diagnosis?
The differential diagnosis of tufted angioma includes other potentially large vascular lesions including infantile hemangioma, congenital hemangioma, port-wine birth marks (capillary malformations), hemangioendotheliomas, and rhabdomyosarcomas.
Infantile hemangiomas (IH) are common vascular tumors of infancy seen in 4%-5% of infants that are characterized by a growth and involution phase. Classically, lesions can be absent or minimally evident at birth, becoming noticeable within the first months of life with a rapid growth phase and typical progression to bright red papules, nodules, or plaques. Deeper hemangiomas may appear more skin colored on the surface with a bluish coloration underneath. They are usually more discreet, with relatively defined borders. Diagnosis is typically clinical and many IHs self-resolve, albeit with residual findings including skin atrophy, scarring, and telangiectasia. Observation or topical timolol are first-line treatment options for more superficial lesions while systemic propranolol is the treatment of choice for deeper IHs or those resulting in possible airway or vision compromise.
Congenital hemangiomas (CH) are another type of vascular growth characterized by a solitary erythematous to violaceous plaque or nodule present at birth with overlying telangiectasia. CHs can be subdivided into categories including rapidly involuting (RICH), partially involuting (PICH), and noninvoluting (NICH). Diagnosis is usually clinical and, depending on the subtype, treatment can involve watchful waiting (for RICHs) or more active intervention such as pulse dye laser or surgical resection (for PICHs or NICHs). The growing nature of this patient’s mass makes a diagnosis of CH unlikely.
Port-wine birth mark, also known as nevus flammeus, is a vascular malformation that appears at birth as a nonpalpable irregular erythematous to violaceous macular plaque. Port-wine stains may be isolated birthmarks, or associated with Sturge-Weber syndrome, complex vascular malformations, or soft-tissue overgrowth. Klippel-Trenauny syndrome (KTS) describes capillary-venous malformations with limb overgrowth, with or without lymphatic malformations, and many are associated with somatic mutations in the PIK3CA gene. While KTS could be considered in this patient, the nodular appearance with lumpy texture and rapid growth makes a vascular tumor more likely.
Rhabdomyosarcoma is a malignancy of skeletal muscle lineage and the most common soft tissue tumor in pediatrics. Cutaneous rhabdomyosarcomas present as erythematous nodules, markedly firm, often “fixed” to deep tissue. A rapidly growing atypical, firm tumor of infancy should raise the consideration of rhabdomyosarcoma and imaging and biopsy are appropriate for evaluation.
What should the evaluation and management of this patient be?
Initial workup should include a complete blood count with platelet count as well as coagulation studies including D-dimer, fibrinogen, prothrombin time, and activated partial thromboplastin time, to assess for any thrombocytopenia or coagulopathy.6 Ultrasound and/or MRI may also be performed to determine lesion extent. While typical MRI findings might be suggestive of a tufted angioma or hemangioendothelioma, biopsy for histologic examination is usually the approach to diagnosis, which will demonstrate stereotypic round lobules of capillaries in a “tufted” distribution.2,7 Biopsy may be performed by a surgeon or dermatologist but bleeding at time of biopsy needs to be considered before moving forward with the procedure.
Tufted angiomas of early life may regress spontaneously, though lesions with symptoms, with functional significance, or associated with KHE may require therapy. Surgical excision is one option, but it may be difficult to execute given that these lesions often have poorly defined margins.1 Other treatment choices include but are not limited to aspirin, systemic corticosteroids, vincristine, interferon-alpha, embolization, and sirolimus.8 No specific expert-directed consensus guidelines exist for these lesions, and suspicion of this lesion should prompt urgent referral to a pediatric dermatologist. Concern for Kasabach-Merritt syndrome should trigger immediate referral for rapid evaluation and management.
Complete blood count with platelet count and coagulation studies were normal in our patient. This infant underwent biopsy to confirm the diagnosis of tufted angioma and MRI to determine lesion extent. The lesion slowly involuted spontaneously without recurrence.
Mr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is MS4 at the University of Rochester, N.Y. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital, San Diego. Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Herron MD et al. Pediatr Dermatol. 2002;19(5):394-401.
2. Jones EW and Orkin M. J Am Acad Dermatol. 1989;20(2 Pt 1):214-25.
3. Wong SN and Tay YK. Pediatr Dermatol. 2002;19(5):388-93.
4. Croteau SE and Gupta D. Semin Cutan Med Surg. 2016;35(3):147-52.
5. Kelly M. Pediatr Clin North Am. 2010;57(5):1085-9.
6. Osio A et al. Arch Dermatol. 2010;146(7):758-63.
7. Padilla RS et al. Am J Dermatopathol. 1987;9(4):292-300.
8. Liu XH et al. Int J Cancer. 2016;139(7):1658-66.
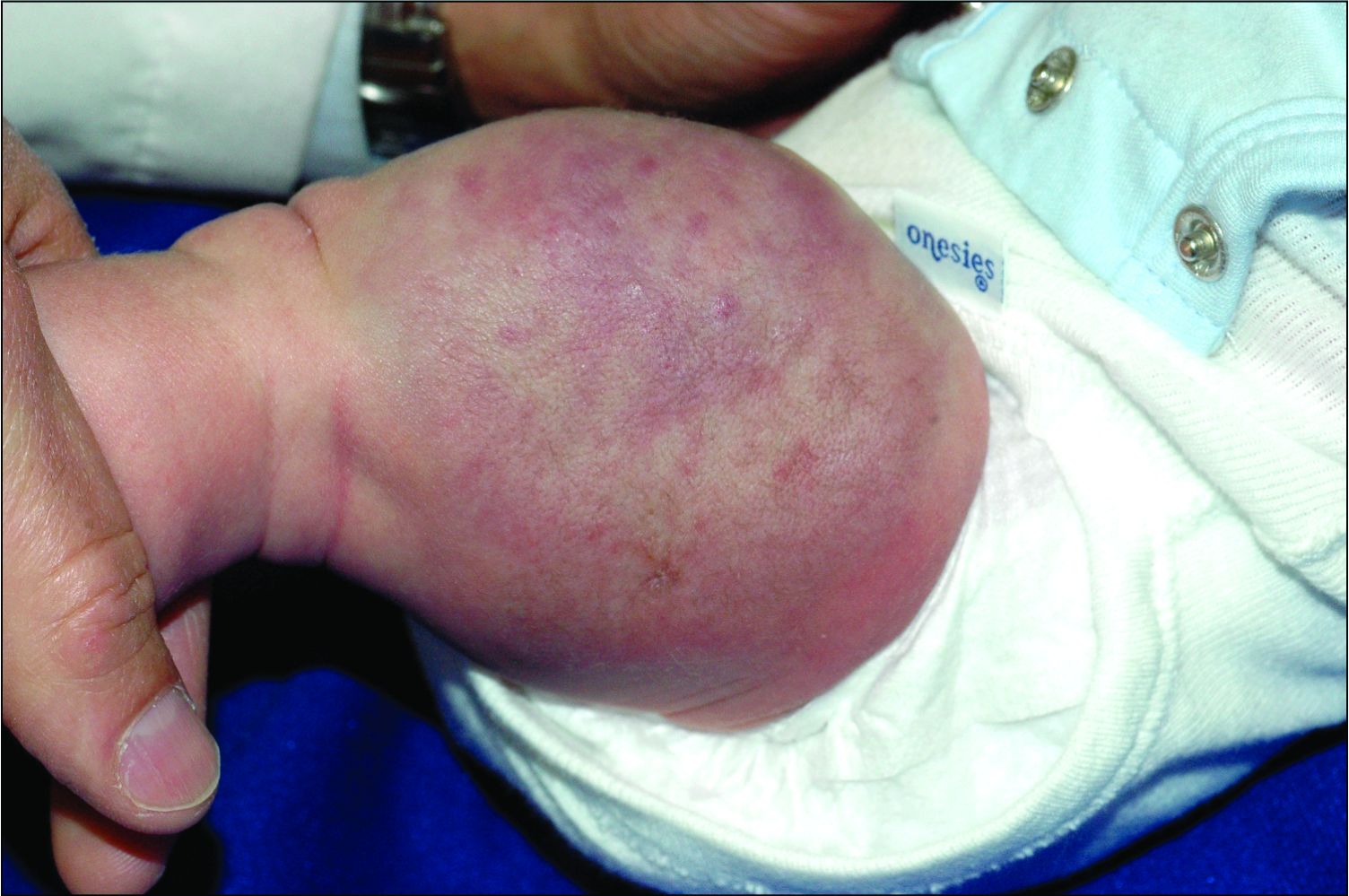
On physical exam, you see an infant with a mass of the left lower extremity. Close examination reveals an approximately 7 cm x 8 cm poorly defined mass with overlying central erythematous to violaceous color of the left anterior upper leg with a lumpy texture. The lesion is moderately firm and mildly tender on palpation.
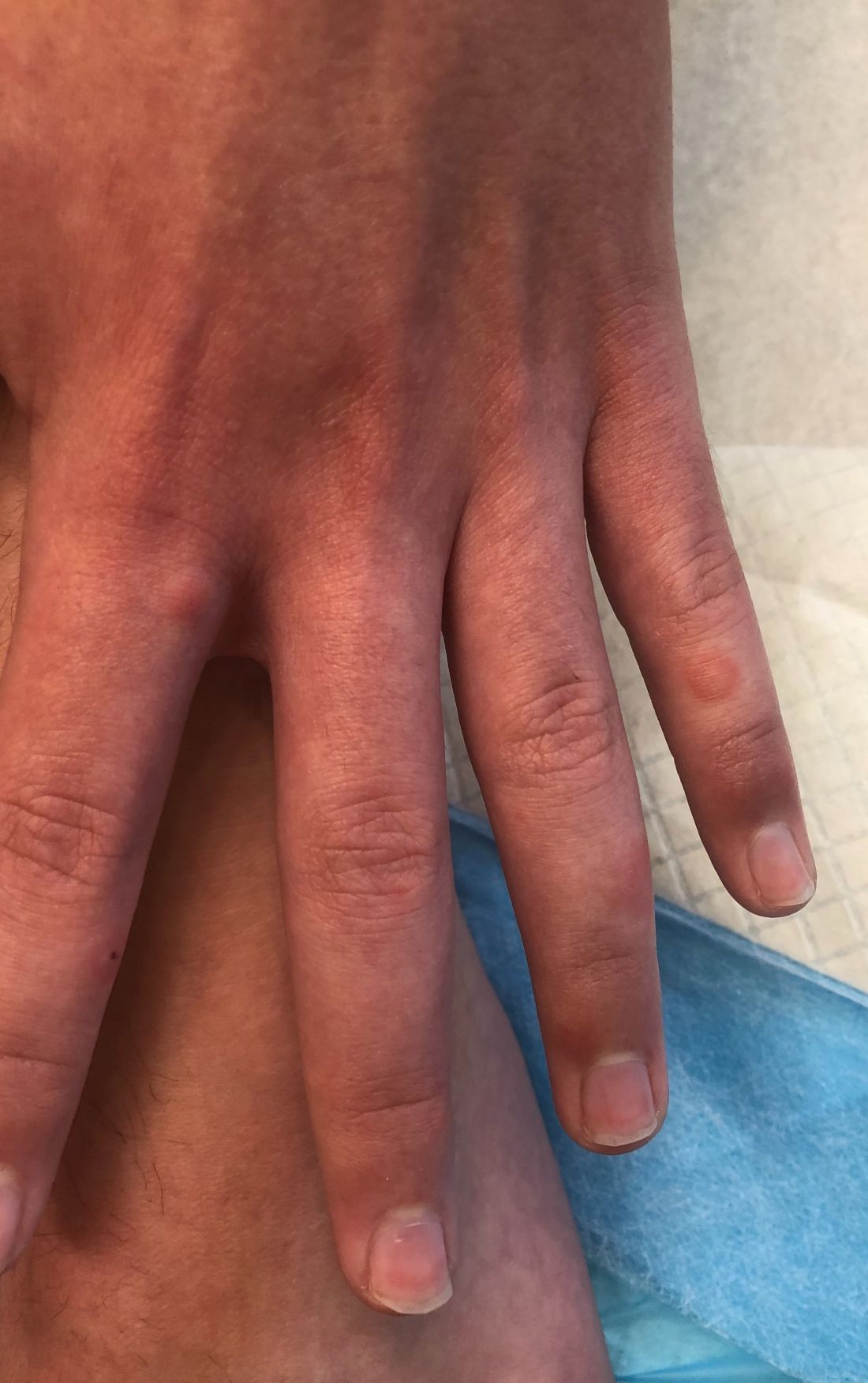
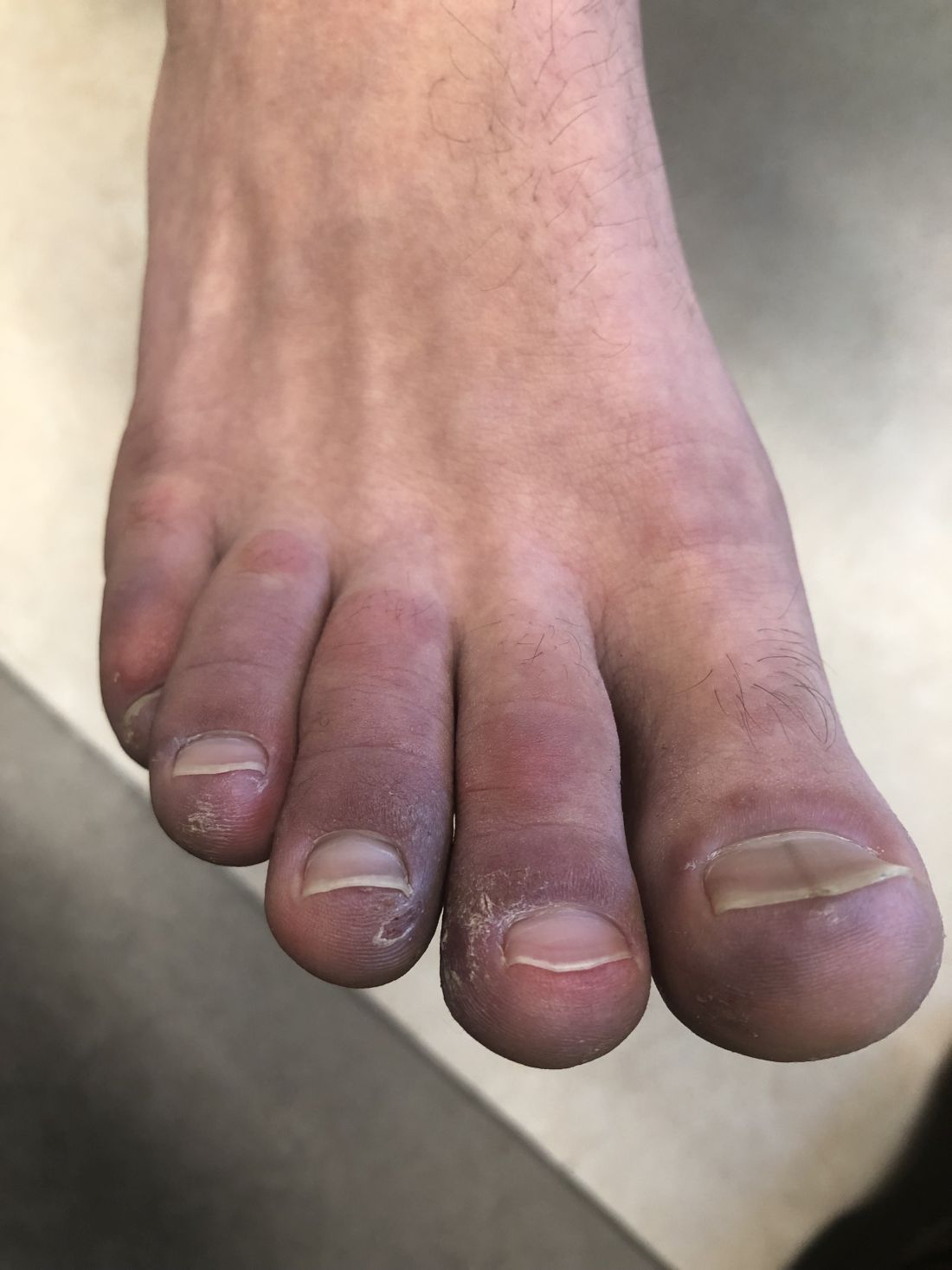
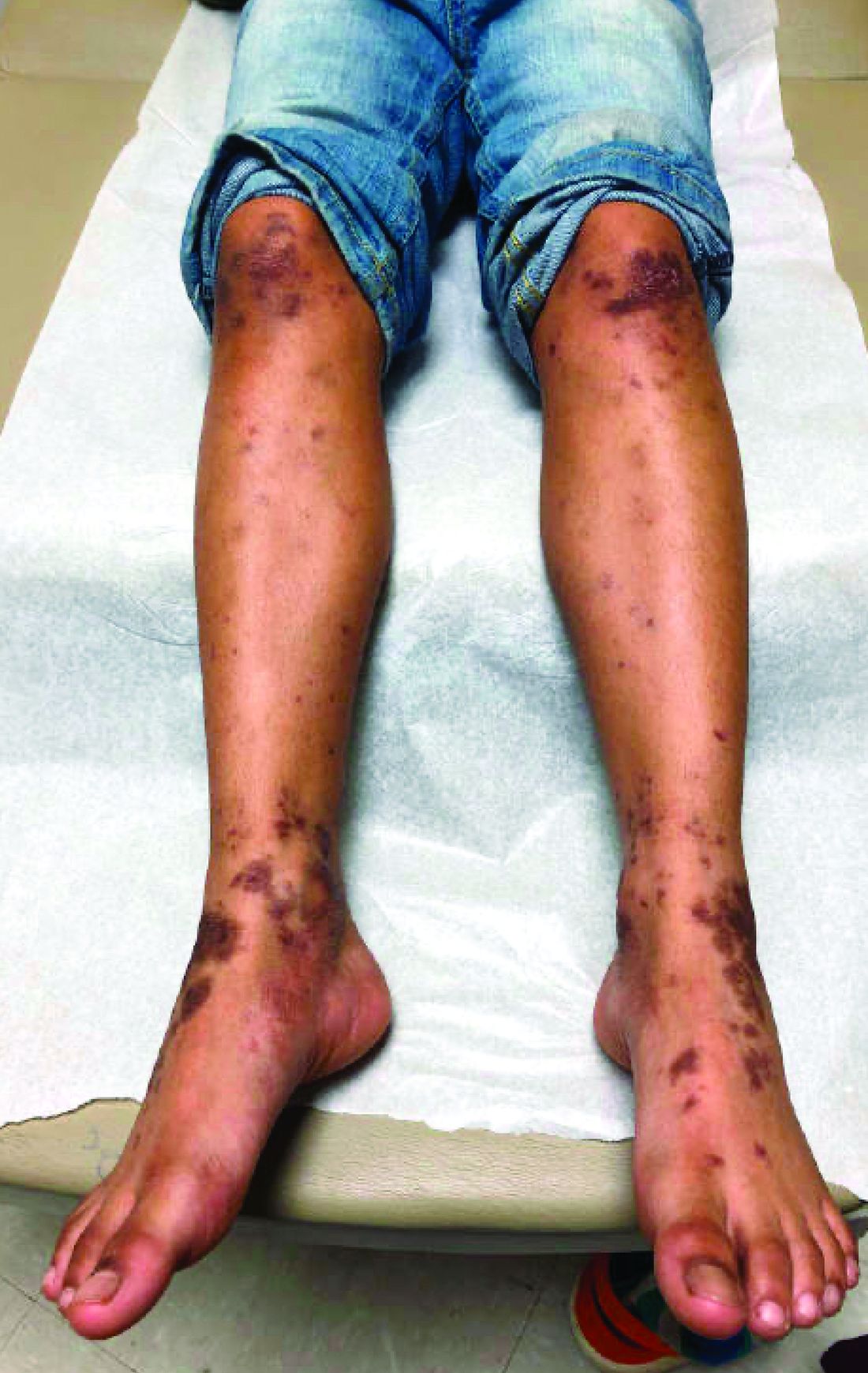
A 12-year-old male has persistent purple toes and new red lesions on his hands
A punch biopsy from one of the lesions on the feet showed subtle basal vacuolar interface inflammation on the epidermis and rare apoptotic keratinocytes. There was an underlying dermal lymphocytic inflammatory infiltrate around the vascular plexus. Dermal mucin appeared slightly increased. The histologic findings are consistent with pernio. He had a negative direct immunofluorescence study.

Laboratory work-up showed an elevated antinuclear antibody (ANA) of 1:620; positive anticardiolipin IgM was at 15.2. A complete blood count showed no anemia or lymphopenia, he had normal complement C3 and C4 levels, normal urinalysis, negative cryoglobulins and cold agglutinins, and a normal protein electrophoresis.
Given the chronicity of his lesions, the lack of improvement with weather changes, the histopathologic findings of a vacuolar interface dermatitis and the positive ANA titer he was diagnosed with chilblain lupus.
Chilblain lupus erythematosus (CLE) is an uncommon form of chronic cutaneous lupus erythematosus that presents with tender pink to violaceous macules, papules, and/or nodules that sometimes can ulcerate and are present on the fingers, toes, and sometimes the nose and ears. The lesions are usually triggered by cold exposure.1 These patients also have clinical and laboratory findings consistent with lupus erythematosus.
Even though more studies are needed to clarify the clinical and histopathologic features of chilblain lupus, compared with idiopathic pernio, some authors suggest several characteristics: CLE lesions tend to persist in summer months, as occurred in our patient, and histopathologic evaluation usually shows vacuolar and interface inflammation on the basal cell layer and may also have a positive lupus band on direct immunofluorescence.2 About 20% of patient with CLE may later develop systemic lupus erythematosus.3
There is also a familial form of CLE which is usually inherited as an autosomal-dominant trait. Mutations in TREX1, SAMHD1, and STING have been described in these patients.4 Affected children present with skin lesions at a young age and those with TREX1 mutations are at a higher risk to develop systemic lupus erythematosus.
The differential diagnosis of chilblain lupus includes idiopathic pernio or pernio secondary to other conditions. Other conditions that are thought to be associated with pernio, besides lupus erythematosus, include infectious causes (hepatitis B, COVID-19 infection),5 autoimmune conditions, malignancy and hematologic disorders (paraproteinemia).6 In histopathology, pernio lesions present with dermal edema and superficial and deep lymphocytic infiltrate.
The pathogenesis of pernio is not fully understood but is thought be related to vasospasm with secondary poor perfusion and ischemia and type I interferon (INF1) immune response. A recent review of the published studies trying to explain the causality between COVID 19 and pernio-like lesions, from January 2020 to December 2020, speculate several possible mechanisms: an increase in the vasoconstrictive, prothrombotic, and proinflammatory effects of the angiotensin II pathway through activation of the ACE2 by the virus; COVID-19 triggers a robust INF1 immune response in predisposed patients; pernio as a sign of mild disease, may be explained by genetic and hormonal differences in the patients affected.7
Another condition that can be confused with CLE is Raynaud phenomenon, were patients present with white to purple to red patches on the fingers and toes after exposure to cold, but in comparison with pernio, the lesions improve within minutes to hours after rewarming. Secondary Raynaud phenomenon can be seen in patients with systemic lupus erythematosus and in patients with other connective tissue disorders. The skin lesions in our patient were persistent and were not triggered by cold exposure, making Raynaud phenomenon less likely. Children with vasculitis can present with painful red, violaceous, or necrotic lesions on the extremities, which can mimic pernio. Vasculitis lesions tend to be more purpuric and angulated, compared with pernio lesions, though in severe cases of pernio with ulceration it may be difficult to distinguish between the two entities and a skin biopsy may be needed.
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, is a rare skin condition in which children present with edematous tender nodules on the hands and with less frequency in other parts of the body with associated fever, malaise, conjunctivitis, or joint pain and it is usually associated with infection or malignancy. Our patient denied any systemic symptoms and had no conjunctivitis nor arthritis.
Most patients with idiopathic pernio do not require a biopsy or further laboratory evaluation unless the lesions are atypical, chronic, or there is a suspected associated condition. The workup for patients with prolonged or atypical pernio-like lesions include a skin biopsy with direct immunofluorescence, ANA, complete blood count, complement levels, antiphospholipid antibodies, cold agglutinins, and cryoglobulins.
Treatment of mild CLE is with moderate- to high-potency topical corticosteroids. In those patients not responding to topical measures and keeping the extremities warm, the use of hydroxychloroquine has been reported to be beneficial in some patients as well as the use of calcium-channel blockers.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Su WP et al. Cutis. 1994 Dec;54(6):395-9.
2. Boada A et al. Am J Dermatopathol. 2010 Feb;32(1):19-23.
3. Patel et al. SBMJ Case Rep. 2013;2013:bcr2013201165.
4. Genes Yi et al. BMC. 2020 Apr 15;18(1):32.
5. Battesti G et al. J Am Acad Dermatol. 2020;83(4):1219-22.
6. Cappel JA et al. Mayo Clin Proc. 2014 Feb;89(2):207-15.
7. Cappel MA et al. Mayo Clin Proc. 2021;96(4):989-1005.
A punch biopsy from one of the lesions on the feet showed subtle basal vacuolar interface inflammation on the epidermis and rare apoptotic keratinocytes. There was an underlying dermal lymphocytic inflammatory infiltrate around the vascular plexus. Dermal mucin appeared slightly increased. The histologic findings are consistent with pernio. He had a negative direct immunofluorescence study.
Laboratory work-up showed an elevated antinuclear antibody (ANA) of 1:620; positive anticardiolipin IgM was at 15.2. A complete blood count showed no anemia or lymphopenia, he had normal complement C3 and C4 levels, normal urinalysis, negative cryoglobulins and cold agglutinins, and a normal protein electrophoresis.
Given the chronicity of his lesions, the lack of improvement with weather changes, the histopathologic findings of a vacuolar interface dermatitis and the positive ANA titer he was diagnosed with chilblain lupus.
Chilblain lupus erythematosus (CLE) is an uncommon form of chronic cutaneous lupus erythematosus that presents with tender pink to violaceous macules, papules, and/or nodules that sometimes can ulcerate and are present on the fingers, toes, and sometimes the nose and ears. The lesions are usually triggered by cold exposure.1 These patients also have clinical and laboratory findings consistent with lupus erythematosus.
Even though more studies are needed to clarify the clinical and histopathologic features of chilblain lupus, compared with idiopathic pernio, some authors suggest several characteristics: CLE lesions tend to persist in summer months, as occurred in our patient, and histopathologic evaluation usually shows vacuolar and interface inflammation on the basal cell layer and may also have a positive lupus band on direct immunofluorescence.2 About 20% of patient with CLE may later develop systemic lupus erythematosus.3
There is also a familial form of CLE which is usually inherited as an autosomal-dominant trait. Mutations in TREX1, SAMHD1, and STING have been described in these patients.4 Affected children present with skin lesions at a young age and those with TREX1 mutations are at a higher risk to develop systemic lupus erythematosus.
The differential diagnosis of chilblain lupus includes idiopathic pernio or pernio secondary to other conditions. Other conditions that are thought to be associated with pernio, besides lupus erythematosus, include infectious causes (hepatitis B, COVID-19 infection),5 autoimmune conditions, malignancy and hematologic disorders (paraproteinemia).6 In histopathology, pernio lesions present with dermal edema and superficial and deep lymphocytic infiltrate.
The pathogenesis of pernio is not fully understood but is thought be related to vasospasm with secondary poor perfusion and ischemia and type I interferon (INF1) immune response. A recent review of the published studies trying to explain the causality between COVID 19 and pernio-like lesions, from January 2020 to December 2020, speculate several possible mechanisms: an increase in the vasoconstrictive, prothrombotic, and proinflammatory effects of the angiotensin II pathway through activation of the ACE2 by the virus; COVID-19 triggers a robust INF1 immune response in predisposed patients; pernio as a sign of mild disease, may be explained by genetic and hormonal differences in the patients affected.7
Another condition that can be confused with CLE is Raynaud phenomenon, were patients present with white to purple to red patches on the fingers and toes after exposure to cold, but in comparison with pernio, the lesions improve within minutes to hours after rewarming. Secondary Raynaud phenomenon can be seen in patients with systemic lupus erythematosus and in patients with other connective tissue disorders. The skin lesions in our patient were persistent and were not triggered by cold exposure, making Raynaud phenomenon less likely. Children with vasculitis can present with painful red, violaceous, or necrotic lesions on the extremities, which can mimic pernio. Vasculitis lesions tend to be more purpuric and angulated, compared with pernio lesions, though in severe cases of pernio with ulceration it may be difficult to distinguish between the two entities and a skin biopsy may be needed.
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, is a rare skin condition in which children present with edematous tender nodules on the hands and with less frequency in other parts of the body with associated fever, malaise, conjunctivitis, or joint pain and it is usually associated with infection or malignancy. Our patient denied any systemic symptoms and had no conjunctivitis nor arthritis.
Most patients with idiopathic pernio do not require a biopsy or further laboratory evaluation unless the lesions are atypical, chronic, or there is a suspected associated condition. The workup for patients with prolonged or atypical pernio-like lesions include a skin biopsy with direct immunofluorescence, ANA, complete blood count, complement levels, antiphospholipid antibodies, cold agglutinins, and cryoglobulins.
Treatment of mild CLE is with moderate- to high-potency topical corticosteroids. In those patients not responding to topical measures and keeping the extremities warm, the use of hydroxychloroquine has been reported to be beneficial in some patients as well as the use of calcium-channel blockers.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Su WP et al. Cutis. 1994 Dec;54(6):395-9.
2. Boada A et al. Am J Dermatopathol. 2010 Feb;32(1):19-23.
3. Patel et al. SBMJ Case Rep. 2013;2013:bcr2013201165.
4. Genes Yi et al. BMC. 2020 Apr 15;18(1):32.
5. Battesti G et al. J Am Acad Dermatol. 2020;83(4):1219-22.
6. Cappel JA et al. Mayo Clin Proc. 2014 Feb;89(2):207-15.
7. Cappel MA et al. Mayo Clin Proc. 2021;96(4):989-1005.
A punch biopsy from one of the lesions on the feet showed subtle basal vacuolar interface inflammation on the epidermis and rare apoptotic keratinocytes. There was an underlying dermal lymphocytic inflammatory infiltrate around the vascular plexus. Dermal mucin appeared slightly increased. The histologic findings are consistent with pernio. He had a negative direct immunofluorescence study.
Laboratory work-up showed an elevated antinuclear antibody (ANA) of 1:620; positive anticardiolipin IgM was at 15.2. A complete blood count showed no anemia or lymphopenia, he had normal complement C3 and C4 levels, normal urinalysis, negative cryoglobulins and cold agglutinins, and a normal protein electrophoresis.
Given the chronicity of his lesions, the lack of improvement with weather changes, the histopathologic findings of a vacuolar interface dermatitis and the positive ANA titer he was diagnosed with chilblain lupus.
Chilblain lupus erythematosus (CLE) is an uncommon form of chronic cutaneous lupus erythematosus that presents with tender pink to violaceous macules, papules, and/or nodules that sometimes can ulcerate and are present on the fingers, toes, and sometimes the nose and ears. The lesions are usually triggered by cold exposure.1 These patients also have clinical and laboratory findings consistent with lupus erythematosus.
Even though more studies are needed to clarify the clinical and histopathologic features of chilblain lupus, compared with idiopathic pernio, some authors suggest several characteristics: CLE lesions tend to persist in summer months, as occurred in our patient, and histopathologic evaluation usually shows vacuolar and interface inflammation on the basal cell layer and may also have a positive lupus band on direct immunofluorescence.2 About 20% of patient with CLE may later develop systemic lupus erythematosus.3
There is also a familial form of CLE which is usually inherited as an autosomal-dominant trait. Mutations in TREX1, SAMHD1, and STING have been described in these patients.4 Affected children present with skin lesions at a young age and those with TREX1 mutations are at a higher risk to develop systemic lupus erythematosus.
The differential diagnosis of chilblain lupus includes idiopathic pernio or pernio secondary to other conditions. Other conditions that are thought to be associated with pernio, besides lupus erythematosus, include infectious causes (hepatitis B, COVID-19 infection),5 autoimmune conditions, malignancy and hematologic disorders (paraproteinemia).6 In histopathology, pernio lesions present with dermal edema and superficial and deep lymphocytic infiltrate.
The pathogenesis of pernio is not fully understood but is thought be related to vasospasm with secondary poor perfusion and ischemia and type I interferon (INF1) immune response. A recent review of the published studies trying to explain the causality between COVID 19 and pernio-like lesions, from January 2020 to December 2020, speculate several possible mechanisms: an increase in the vasoconstrictive, prothrombotic, and proinflammatory effects of the angiotensin II pathway through activation of the ACE2 by the virus; COVID-19 triggers a robust INF1 immune response in predisposed patients; pernio as a sign of mild disease, may be explained by genetic and hormonal differences in the patients affected.7
Another condition that can be confused with CLE is Raynaud phenomenon, were patients present with white to purple to red patches on the fingers and toes after exposure to cold, but in comparison with pernio, the lesions improve within minutes to hours after rewarming. Secondary Raynaud phenomenon can be seen in patients with systemic lupus erythematosus and in patients with other connective tissue disorders. The skin lesions in our patient were persistent and were not triggered by cold exposure, making Raynaud phenomenon less likely. Children with vasculitis can present with painful red, violaceous, or necrotic lesions on the extremities, which can mimic pernio. Vasculitis lesions tend to be more purpuric and angulated, compared with pernio lesions, though in severe cases of pernio with ulceration it may be difficult to distinguish between the two entities and a skin biopsy may be needed.
Sweet syndrome, also known as acute febrile neutrophilic dermatosis, is a rare skin condition in which children present with edematous tender nodules on the hands and with less frequency in other parts of the body with associated fever, malaise, conjunctivitis, or joint pain and it is usually associated with infection or malignancy. Our patient denied any systemic symptoms and had no conjunctivitis nor arthritis.
Most patients with idiopathic pernio do not require a biopsy or further laboratory evaluation unless the lesions are atypical, chronic, or there is a suspected associated condition. The workup for patients with prolonged or atypical pernio-like lesions include a skin biopsy with direct immunofluorescence, ANA, complete blood count, complement levels, antiphospholipid antibodies, cold agglutinins, and cryoglobulins.
Treatment of mild CLE is with moderate- to high-potency topical corticosteroids. In those patients not responding to topical measures and keeping the extremities warm, the use of hydroxychloroquine has been reported to be beneficial in some patients as well as the use of calcium-channel blockers.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Su WP et al. Cutis. 1994 Dec;54(6):395-9.
2. Boada A et al. Am J Dermatopathol. 2010 Feb;32(1):19-23.
3. Patel et al. SBMJ Case Rep. 2013;2013:bcr2013201165.
4. Genes Yi et al. BMC. 2020 Apr 15;18(1):32.
5. Battesti G et al. J Am Acad Dermatol. 2020;83(4):1219-22.
6. Cappel JA et al. Mayo Clin Proc. 2014 Feb;89(2):207-15.
7. Cappel MA et al. Mayo Clin Proc. 2021;96(4):989-1005.
He denied any hair loss, mouth sores, sun sensitivity, headaches, gastrointestinal complaints, joint pain, or muscle weakness.
He is not taking any medications.
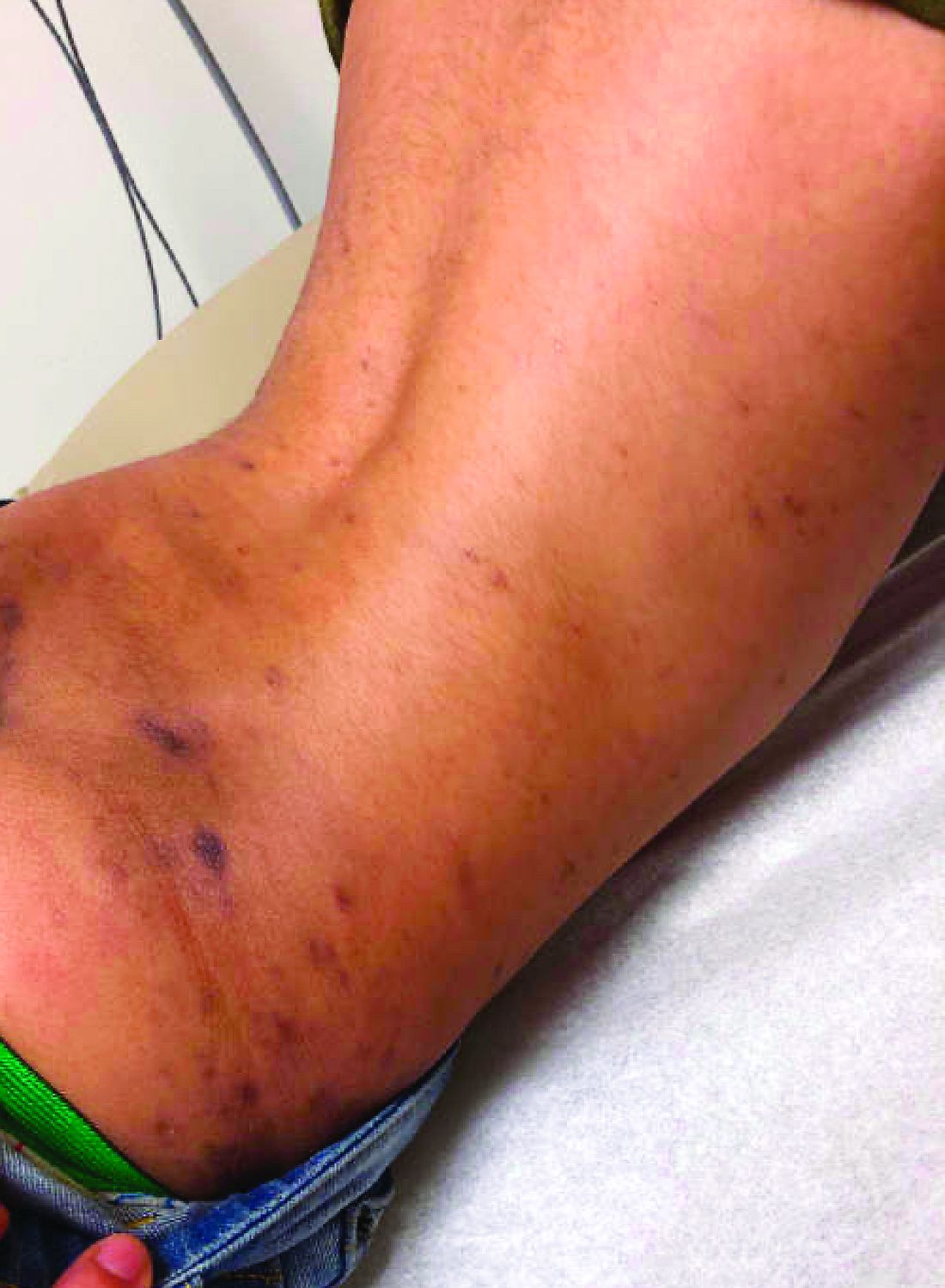
He has been at home doing virtual school and has not traveled. He likes to play the piano. There is no family history of similar lesions, connective tissue disorder, or autoimmunity.
On physical exam he has purple discoloration on the toes with some violaceous and pink papules. On the fingers he has pink to violaceous papules and macules.
There is no joint edema or pain.
A young girl presents with ‘itchy, rashy’ hands
Given the presence of erythema, lichenification, fissuring, and scale of the hands over the course of more than 3 months with the absence of nail findings is most consistent with a diagnosis of chronic hand eczema.
Chronic hand eczema (CHE) is an inflammatory dermatitis of the hands or wrists that persists for longer than 3 months or recurs twice or more in a 12-month timespan.1,2 Hand eczema can be a manifestation of atopic dermatitis, allergic contact dermatitis, or irritant contact dermatitis. Its multifactorial pathogenesis includes epidermal injury and disturbed epidermal barrier function from exogenous factors such as irritants or contact allergens, as well as endogenous factors including atopic dermatitis.3 In pediatrics, it often presents after an acute phase of hand dermatitis with chronic pruritus, erythema, and dry skin with scale.4 Examination findings vary widely with erythema, vesicles, scale, fissures, crusting, hyperkeratosis, and/or lichenification.3,5 Diagnosis is often achieved with careful history, asking about potential exposures that may induce lesions, and physical exam of the entire skin, including the feet. Based upon clinical history or persistent dermatitis, allergic contact dermatitis patch testing should be considered.2
What’s the treatment plan?
Given that CHE is an inflammatory disease process, the goal of treatment is to reduce inflammation and allow for skin barrier repair. Unfortunately, only one study has investigated therapeutics for pediatric CHE,6 with the remainder of the literature based on adult CHE. Current CHE guidelines recommend avoidance of allergens, irritants, or other triggers of the disease as well as liberal and regular use of emollients. Because of the relative thickness of hand skin, higher-potency topical corticosteroids are often used as first-line therapy, with lower-strength topical steroids, calcineurin inhibitors, or crisaborole used as maintenance therapy. Other treatment options include phototherapy, and rarely, systemic therapies are utilized for atopic dermatitis.
What’s the differential diagnosis?
The differential diagnosis of CHE includes other scaling or hyperkeratotic skin conditions including psoriasis and tinea manuum. Other skin conditions that localize to extremities including scabies and hand-foot-and-mouth disease are discussed below.
Psoriasis can present on the hands with erythematous, well-demarcated, silver scaling plaques. However, additional plaques may be found on the elbows, knees, scalp, umbilicus, and sacrum. Nails can demonstrate pitting, oil drops, splinter hemorrhages, or onycholysis. First-line treatment includes a combination of topical steroids, topical vitamin D analogues, and keratolytics.
Tinea mannum is a dermatophyte infection of the skin of the hands. Typically, only one hand is affected with concomitant bilateral tinea pedis. It results in a white scaly plaque with dorsal hand involvement demonstrating an annular appearance, elevated edge, and central clearing. KOH prep will demonstrate septate hyphae, and cultures will grow dermatophyte colonies. Treatment includes topical antifungals or systemic antifungals for recalcitrant disease.
Scabies presents with short linear hypopigmented lesions with a black dot on one end as well as erythematous pruritic papules. These appear on the interdigital web spaces, wrists, axilla, buttocks, and genital region. Skin scraping prep with mineral oil can show mites and eggs. All individuals in an affected household should be treated with either topical permethrin or oral ivermectin to avoid reinfection or parasitic spread. All contacted linens must be cleaned with hot water and dried on high heat.
Hand-foot-and-mouth disease, classically caused by coxsackievirus, is an acute viral illness that results in an eruption of erythematous macules, papules, and vesicles on the ventral hands, soles of the feet, and oral mucosa. Diagnosis is achieved clinically and treatment is symptomatic as the lesions are self-limited.
Our patient underwent patch testing but did not return positive to any allergens. She was started on potent topical corticosteroids, educated on trigger avoidance, and gradually achieved good disease control.
Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
Michael Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is a 4th year medical student at the University of Rochester (N.Y.). Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital.
References
1. Diepgen TL et al. Br J Dermatol. 2009;160(2):353-8.
2. Diepgen TL et al. J Dtsch Dermatol Ges. 2015;13(1):e1-22.
3. Agner T and Elsner P. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12.
4. Mortz CG et al. Br J Dermatol. 2001;144(3):523-32.
5. Silvestre Salvador JF et al. Actas Dermosifiliogr. 2020;111(1):26-40.
6. Luchsinger I et al. J Eur Acad Dermatol Venereol. 2020;34(5):1037-42.
7. English J et al. Clin Exp Dermatol. 2009;34(7):761-9.
8. Elsner P and Agner T. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:13-21.
Given the presence of erythema, lichenification, fissuring, and scale of the hands over the course of more than 3 months with the absence of nail findings is most consistent with a diagnosis of chronic hand eczema.
Chronic hand eczema (CHE) is an inflammatory dermatitis of the hands or wrists that persists for longer than 3 months or recurs twice or more in a 12-month timespan.1,2 Hand eczema can be a manifestation of atopic dermatitis, allergic contact dermatitis, or irritant contact dermatitis. Its multifactorial pathogenesis includes epidermal injury and disturbed epidermal barrier function from exogenous factors such as irritants or contact allergens, as well as endogenous factors including atopic dermatitis.3 In pediatrics, it often presents after an acute phase of hand dermatitis with chronic pruritus, erythema, and dry skin with scale.4 Examination findings vary widely with erythema, vesicles, scale, fissures, crusting, hyperkeratosis, and/or lichenification.3,5 Diagnosis is often achieved with careful history, asking about potential exposures that may induce lesions, and physical exam of the entire skin, including the feet. Based upon clinical history or persistent dermatitis, allergic contact dermatitis patch testing should be considered.2
What’s the treatment plan?
Given that CHE is an inflammatory disease process, the goal of treatment is to reduce inflammation and allow for skin barrier repair. Unfortunately, only one study has investigated therapeutics for pediatric CHE,6 with the remainder of the literature based on adult CHE. Current CHE guidelines recommend avoidance of allergens, irritants, or other triggers of the disease as well as liberal and regular use of emollients. Because of the relative thickness of hand skin, higher-potency topical corticosteroids are often used as first-line therapy, with lower-strength topical steroids, calcineurin inhibitors, or crisaborole used as maintenance therapy. Other treatment options include phototherapy, and rarely, systemic therapies are utilized for atopic dermatitis.
What’s the differential diagnosis?
The differential diagnosis of CHE includes other scaling or hyperkeratotic skin conditions including psoriasis and tinea manuum. Other skin conditions that localize to extremities including scabies and hand-foot-and-mouth disease are discussed below.
Psoriasis can present on the hands with erythematous, well-demarcated, silver scaling plaques. However, additional plaques may be found on the elbows, knees, scalp, umbilicus, and sacrum. Nails can demonstrate pitting, oil drops, splinter hemorrhages, or onycholysis. First-line treatment includes a combination of topical steroids, topical vitamin D analogues, and keratolytics.
Tinea mannum is a dermatophyte infection of the skin of the hands. Typically, only one hand is affected with concomitant bilateral tinea pedis. It results in a white scaly plaque with dorsal hand involvement demonstrating an annular appearance, elevated edge, and central clearing. KOH prep will demonstrate septate hyphae, and cultures will grow dermatophyte colonies. Treatment includes topical antifungals or systemic antifungals for recalcitrant disease.
Scabies presents with short linear hypopigmented lesions with a black dot on one end as well as erythematous pruritic papules. These appear on the interdigital web spaces, wrists, axilla, buttocks, and genital region. Skin scraping prep with mineral oil can show mites and eggs. All individuals in an affected household should be treated with either topical permethrin or oral ivermectin to avoid reinfection or parasitic spread. All contacted linens must be cleaned with hot water and dried on high heat.
Hand-foot-and-mouth disease, classically caused by coxsackievirus, is an acute viral illness that results in an eruption of erythematous macules, papules, and vesicles on the ventral hands, soles of the feet, and oral mucosa. Diagnosis is achieved clinically and treatment is symptomatic as the lesions are self-limited.
Our patient underwent patch testing but did not return positive to any allergens. She was started on potent topical corticosteroids, educated on trigger avoidance, and gradually achieved good disease control.
Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
Michael Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is a 4th year medical student at the University of Rochester (N.Y.). Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital.
References
1. Diepgen TL et al. Br J Dermatol. 2009;160(2):353-8.
2. Diepgen TL et al. J Dtsch Dermatol Ges. 2015;13(1):e1-22.
3. Agner T and Elsner P. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12.
4. Mortz CG et al. Br J Dermatol. 2001;144(3):523-32.
5. Silvestre Salvador JF et al. Actas Dermosifiliogr. 2020;111(1):26-40.
6. Luchsinger I et al. J Eur Acad Dermatol Venereol. 2020;34(5):1037-42.
7. English J et al. Clin Exp Dermatol. 2009;34(7):761-9.
8. Elsner P and Agner T. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:13-21.
Given the presence of erythema, lichenification, fissuring, and scale of the hands over the course of more than 3 months with the absence of nail findings is most consistent with a diagnosis of chronic hand eczema.
Chronic hand eczema (CHE) is an inflammatory dermatitis of the hands or wrists that persists for longer than 3 months or recurs twice or more in a 12-month timespan.1,2 Hand eczema can be a manifestation of atopic dermatitis, allergic contact dermatitis, or irritant contact dermatitis. Its multifactorial pathogenesis includes epidermal injury and disturbed epidermal barrier function from exogenous factors such as irritants or contact allergens, as well as endogenous factors including atopic dermatitis.3 In pediatrics, it often presents after an acute phase of hand dermatitis with chronic pruritus, erythema, and dry skin with scale.4 Examination findings vary widely with erythema, vesicles, scale, fissures, crusting, hyperkeratosis, and/or lichenification.3,5 Diagnosis is often achieved with careful history, asking about potential exposures that may induce lesions, and physical exam of the entire skin, including the feet. Based upon clinical history or persistent dermatitis, allergic contact dermatitis patch testing should be considered.2
What’s the treatment plan?
Given that CHE is an inflammatory disease process, the goal of treatment is to reduce inflammation and allow for skin barrier repair. Unfortunately, only one study has investigated therapeutics for pediatric CHE,6 with the remainder of the literature based on adult CHE. Current CHE guidelines recommend avoidance of allergens, irritants, or other triggers of the disease as well as liberal and regular use of emollients. Because of the relative thickness of hand skin, higher-potency topical corticosteroids are often used as first-line therapy, with lower-strength topical steroids, calcineurin inhibitors, or crisaborole used as maintenance therapy. Other treatment options include phototherapy, and rarely, systemic therapies are utilized for atopic dermatitis.
What’s the differential diagnosis?
The differential diagnosis of CHE includes other scaling or hyperkeratotic skin conditions including psoriasis and tinea manuum. Other skin conditions that localize to extremities including scabies and hand-foot-and-mouth disease are discussed below.
Psoriasis can present on the hands with erythematous, well-demarcated, silver scaling plaques. However, additional plaques may be found on the elbows, knees, scalp, umbilicus, and sacrum. Nails can demonstrate pitting, oil drops, splinter hemorrhages, or onycholysis. First-line treatment includes a combination of topical steroids, topical vitamin D analogues, and keratolytics.
Tinea mannum is a dermatophyte infection of the skin of the hands. Typically, only one hand is affected with concomitant bilateral tinea pedis. It results in a white scaly plaque with dorsal hand involvement demonstrating an annular appearance, elevated edge, and central clearing. KOH prep will demonstrate septate hyphae, and cultures will grow dermatophyte colonies. Treatment includes topical antifungals or systemic antifungals for recalcitrant disease.
Scabies presents with short linear hypopigmented lesions with a black dot on one end as well as erythematous pruritic papules. These appear on the interdigital web spaces, wrists, axilla, buttocks, and genital region. Skin scraping prep with mineral oil can show mites and eggs. All individuals in an affected household should be treated with either topical permethrin or oral ivermectin to avoid reinfection or parasitic spread. All contacted linens must be cleaned with hot water and dried on high heat.
Hand-foot-and-mouth disease, classically caused by coxsackievirus, is an acute viral illness that results in an eruption of erythematous macules, papules, and vesicles on the ventral hands, soles of the feet, and oral mucosa. Diagnosis is achieved clinically and treatment is symptomatic as the lesions are self-limited.
Our patient underwent patch testing but did not return positive to any allergens. She was started on potent topical corticosteroids, educated on trigger avoidance, and gradually achieved good disease control.
Neither Mr. Haft nor Dr. Eichenfield have any relevant financial disclosures.
Michael Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology at the University of California, San Diego, and Rady Children’s Hospital, San Diego. He is a 4th year medical student at the University of Rochester (N.Y.). Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital.
References
1. Diepgen TL et al. Br J Dermatol. 2009;160(2):353-8.
2. Diepgen TL et al. J Dtsch Dermatol Ges. 2015;13(1):e1-22.
3. Agner T and Elsner P. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:4-12.
4. Mortz CG et al. Br J Dermatol. 2001;144(3):523-32.
5. Silvestre Salvador JF et al. Actas Dermosifiliogr. 2020;111(1):26-40.
6. Luchsinger I et al. J Eur Acad Dermatol Venereol. 2020;34(5):1037-42.
7. English J et al. Clin Exp Dermatol. 2009;34(7):761-9.
8. Elsner P and Agner T. J Eur Acad Dermatol Venereol. 2020;34 Suppl 1:13-21.
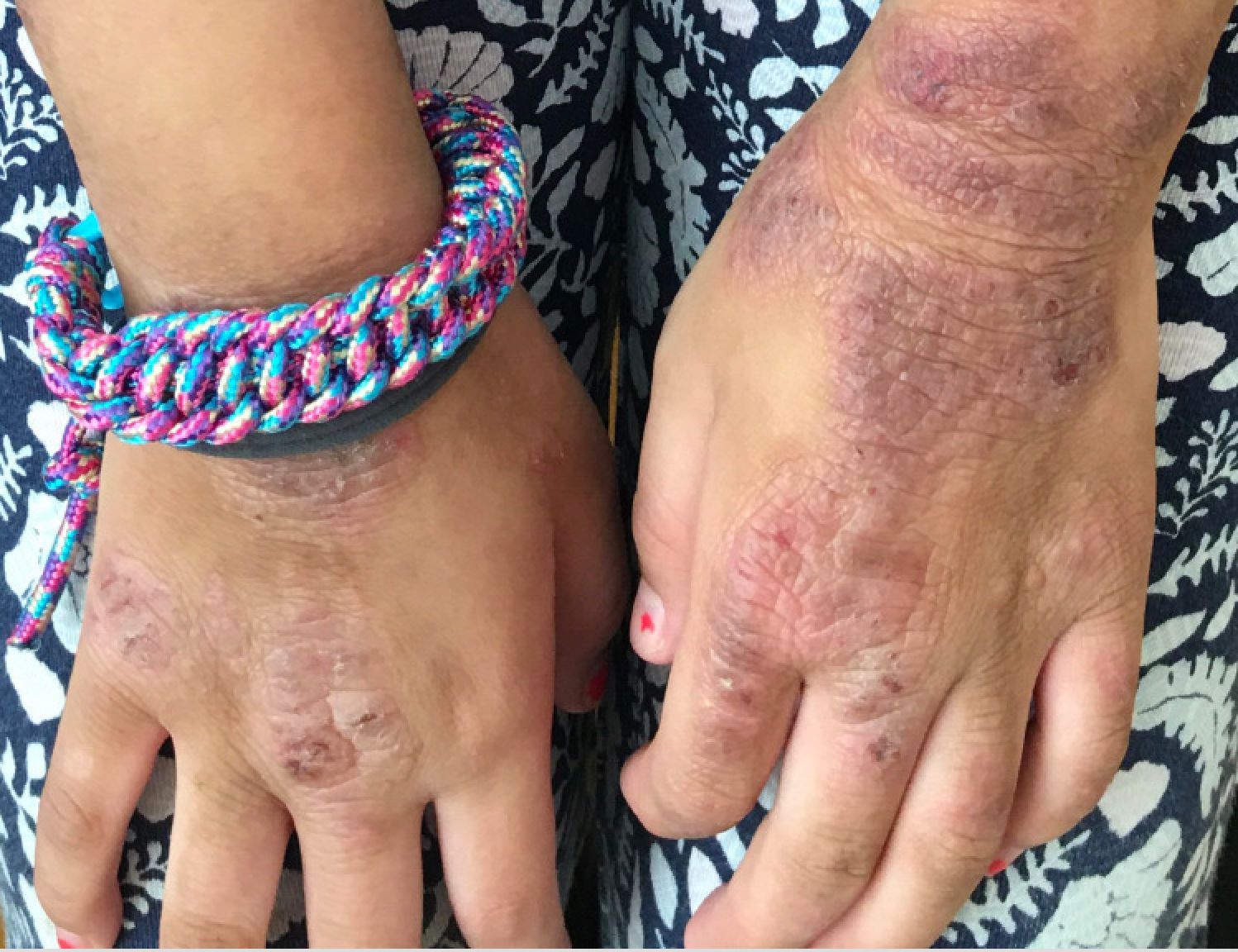
Examination findings of the bilateral hands and wrists demonstrate plaques of erythema, lichenification, and scale of the dorsal surfaces of the hands and digits. Closer inspection reveals fissuring and erythematous crust of the affected skin but normal nails. The rest of the skin exam is unremarkable.
Make the Diagnosis - March 2021
Because of the lack of improvement with topical corticosteroids, a skin biopsy was performed from a lesion on the lower back which showed an epidermis with compact hyperkeratosis and a thickened granular layer. Within the dermis, there was a lichenoid infiltrate of lymphocytes with a prominent interface change and rare dyskeratotic keratinocytes consistent with lichen planus.
Lichen planus is an inflammatory condition of the skin seen mainly in the adult population and is rare in children. This condition affects 0.5%-1% of the population, with maybe a higher prevalence in woman with no racial predilection in the adult or pediatric population. Most patients diagnosed are described to be over 40 years of age, but in children, the mean age for presentation is reported between the ages of 7 and 11.8 years.1 Interestingly, most of the published larger studies of lichen planus in children originate from India. In a U.K. study, about 80% of the cases reported were from children of Indian descent, as is our patient; so it is possible that lichen planus may be more prevalent in India.1 In a study based in the United States, cases were more prevalent in African American children.2
The exact cause of this condition is not known but studies have suggested that activated T cells, particularly CD8+, attack and cause apoptosis of the basal keratinocytes.3 There appears to be an up-regulation of Th1 cytokines such as interferon‐gamma, tumor necrosis factor–alpha, interleukin‐1 alpha, IL‐6, and IL‐8, as well as other apoptosis-related molecules.3
Lichen planus has been associated with other systemic conditions especially liver disease (chronic active hepatitis C and primary biliary cirrhosis). Children and adults may also have coexistence of other autoimmune diseases such as autoimmune polyendocrinopathy, myasthenia gravis, autoimmune thyroid disease, vitiligo, and thymoma. Some reports have also found a higher prevalence of atopic dermatitis in children with lichen planus.4
The lesions are typically described as the four “Ps” for pruritic, polygonal, purpuric flat-topped papules, and plaques. The papules of lichen planus have characteristically dry fine white streaks known as Wickham’s striae. The lesions can occur anywhere on the body, but they tend to occur more commonly on the flexures of the forearms, the wrists, ankles, shins, knees, and the torso. The face is rarely affected. In some patients oral, scalp (lichen planopilaris), nails, and rarely conjunctival, genital, and esophageal involvement can occur.2
In histopathology, the lesions are characterized by a wedge-shaped hypergranulosis, marked hyperkeratosis, and irregular sawtooth-like acanthosis of rete ridges on the epidermis. The dermal-epidermal junction typically shows an interstitial dermatitis. Civatte bodies may also be seen. On direct immunofluorescence, IgM-staining of the cytoid bodies in the dermal papilla or peribasilar areas are suggestive of lichen planus.1
The differential diagnosis of lichen planus includes severe lichenified atopic dermatitis, drug-induced lichen planus, graft-versus-host disease, psoriasis, pityriasis rosea, subacute cutaneous lupus, discoid lupus, secondary syphilis, and lichen simplex chronicus. Interestingly, our patient presented with lesions that were not pruritic and more generalized. Compared with eczema, were flexures are commonly affected, our patient’s lesions were localized to the ankles, wrists, extensor knees, and elbows, and no pruritus was reported. Lichenification of skin lesions occurs as a response to chronic scratching as it occurs in atopic dermatitis and lichen simplex chronicus, was considered in our patient, but the lack of pruritus and the more acute presentation made it unlikely.
Lichen planus is considered a self-limiting disease, so treatment is focused on the control of pruritus and to accelerate resolution. The first-line therapy for classic cutaneous lichen planus is the use of potent or superpotent topical corticosteroids for localized disease on the body and extremities and mild to mid-potency for intertriginous areas and the face. Clinical response should be assessed after 2-3 weeks of treatment. For patients with more generalized or recalcitrant disease like our patient, other treatment modalities like phototherapy (narrow-band UVB), a 4- to 6-week course of oral glucocorticoids, or acitretin may be considered. Our patient recently started narrow-band UVB. Other medications that have been reported beneficial for more severe cases include methotrexate, cyclosporine, griseofulvin, hydroxychloroquine, metronidazole, dapsone, and mycophenolate. Recent studies in the adult population have shown apremilast, a phosphodiesterase inhibitor, to be a promising medication for patients with cutaneous lichen planus, though this medication has not been approved yet for use in the pediatric population.5
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Payette MJ et al. Clin Dermatol. 2015 Nov-Dec;33(6):631-43.
2. Walton KE et al. Pediatr Dermatol. 2010;27:34-8.
3. Lehman JS et al. Int J Dermatol. 2009 Jul;48(7):682-94.
4. Laughter D et al. J Am Acad Dermatol. 2000;43:649-55.
5. Paul J et al. J Am Acad Dermatol. 2013 Feb;68(2):255-61.
Because of the lack of improvement with topical corticosteroids, a skin biopsy was performed from a lesion on the lower back which showed an epidermis with compact hyperkeratosis and a thickened granular layer. Within the dermis, there was a lichenoid infiltrate of lymphocytes with a prominent interface change and rare dyskeratotic keratinocytes consistent with lichen planus.
Lichen planus is an inflammatory condition of the skin seen mainly in the adult population and is rare in children. This condition affects 0.5%-1% of the population, with maybe a higher prevalence in woman with no racial predilection in the adult or pediatric population. Most patients diagnosed are described to be over 40 years of age, but in children, the mean age for presentation is reported between the ages of 7 and 11.8 years.1 Interestingly, most of the published larger studies of lichen planus in children originate from India. In a U.K. study, about 80% of the cases reported were from children of Indian descent, as is our patient; so it is possible that lichen planus may be more prevalent in India.1 In a study based in the United States, cases were more prevalent in African American children.2
The exact cause of this condition is not known but studies have suggested that activated T cells, particularly CD8+, attack and cause apoptosis of the basal keratinocytes.3 There appears to be an up-regulation of Th1 cytokines such as interferon‐gamma, tumor necrosis factor–alpha, interleukin‐1 alpha, IL‐6, and IL‐8, as well as other apoptosis-related molecules.3
Lichen planus has been associated with other systemic conditions especially liver disease (chronic active hepatitis C and primary biliary cirrhosis). Children and adults may also have coexistence of other autoimmune diseases such as autoimmune polyendocrinopathy, myasthenia gravis, autoimmune thyroid disease, vitiligo, and thymoma. Some reports have also found a higher prevalence of atopic dermatitis in children with lichen planus.4
The lesions are typically described as the four “Ps” for pruritic, polygonal, purpuric flat-topped papules, and plaques. The papules of lichen planus have characteristically dry fine white streaks known as Wickham’s striae. The lesions can occur anywhere on the body, but they tend to occur more commonly on the flexures of the forearms, the wrists, ankles, shins, knees, and the torso. The face is rarely affected. In some patients oral, scalp (lichen planopilaris), nails, and rarely conjunctival, genital, and esophageal involvement can occur.2
In histopathology, the lesions are characterized by a wedge-shaped hypergranulosis, marked hyperkeratosis, and irregular sawtooth-like acanthosis of rete ridges on the epidermis. The dermal-epidermal junction typically shows an interstitial dermatitis. Civatte bodies may also be seen. On direct immunofluorescence, IgM-staining of the cytoid bodies in the dermal papilla or peribasilar areas are suggestive of lichen planus.1
The differential diagnosis of lichen planus includes severe lichenified atopic dermatitis, drug-induced lichen planus, graft-versus-host disease, psoriasis, pityriasis rosea, subacute cutaneous lupus, discoid lupus, secondary syphilis, and lichen simplex chronicus. Interestingly, our patient presented with lesions that were not pruritic and more generalized. Compared with eczema, were flexures are commonly affected, our patient’s lesions were localized to the ankles, wrists, extensor knees, and elbows, and no pruritus was reported. Lichenification of skin lesions occurs as a response to chronic scratching as it occurs in atopic dermatitis and lichen simplex chronicus, was considered in our patient, but the lack of pruritus and the more acute presentation made it unlikely.
Lichen planus is considered a self-limiting disease, so treatment is focused on the control of pruritus and to accelerate resolution. The first-line therapy for classic cutaneous lichen planus is the use of potent or superpotent topical corticosteroids for localized disease on the body and extremities and mild to mid-potency for intertriginous areas and the face. Clinical response should be assessed after 2-3 weeks of treatment. For patients with more generalized or recalcitrant disease like our patient, other treatment modalities like phototherapy (narrow-band UVB), a 4- to 6-week course of oral glucocorticoids, or acitretin may be considered. Our patient recently started narrow-band UVB. Other medications that have been reported beneficial for more severe cases include methotrexate, cyclosporine, griseofulvin, hydroxychloroquine, metronidazole, dapsone, and mycophenolate. Recent studies in the adult population have shown apremilast, a phosphodiesterase inhibitor, to be a promising medication for patients with cutaneous lichen planus, though this medication has not been approved yet for use in the pediatric population.5
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Payette MJ et al. Clin Dermatol. 2015 Nov-Dec;33(6):631-43.
2. Walton KE et al. Pediatr Dermatol. 2010;27:34-8.
3. Lehman JS et al. Int J Dermatol. 2009 Jul;48(7):682-94.
4. Laughter D et al. J Am Acad Dermatol. 2000;43:649-55.
5. Paul J et al. J Am Acad Dermatol. 2013 Feb;68(2):255-61.
Because of the lack of improvement with topical corticosteroids, a skin biopsy was performed from a lesion on the lower back which showed an epidermis with compact hyperkeratosis and a thickened granular layer. Within the dermis, there was a lichenoid infiltrate of lymphocytes with a prominent interface change and rare dyskeratotic keratinocytes consistent with lichen planus.
Lichen planus is an inflammatory condition of the skin seen mainly in the adult population and is rare in children. This condition affects 0.5%-1% of the population, with maybe a higher prevalence in woman with no racial predilection in the adult or pediatric population. Most patients diagnosed are described to be over 40 years of age, but in children, the mean age for presentation is reported between the ages of 7 and 11.8 years.1 Interestingly, most of the published larger studies of lichen planus in children originate from India. In a U.K. study, about 80% of the cases reported were from children of Indian descent, as is our patient; so it is possible that lichen planus may be more prevalent in India.1 In a study based in the United States, cases were more prevalent in African American children.2
The exact cause of this condition is not known but studies have suggested that activated T cells, particularly CD8+, attack and cause apoptosis of the basal keratinocytes.3 There appears to be an up-regulation of Th1 cytokines such as interferon‐gamma, tumor necrosis factor–alpha, interleukin‐1 alpha, IL‐6, and IL‐8, as well as other apoptosis-related molecules.3
Lichen planus has been associated with other systemic conditions especially liver disease (chronic active hepatitis C and primary biliary cirrhosis). Children and adults may also have coexistence of other autoimmune diseases such as autoimmune polyendocrinopathy, myasthenia gravis, autoimmune thyroid disease, vitiligo, and thymoma. Some reports have also found a higher prevalence of atopic dermatitis in children with lichen planus.4
The lesions are typically described as the four “Ps” for pruritic, polygonal, purpuric flat-topped papules, and plaques. The papules of lichen planus have characteristically dry fine white streaks known as Wickham’s striae. The lesions can occur anywhere on the body, but they tend to occur more commonly on the flexures of the forearms, the wrists, ankles, shins, knees, and the torso. The face is rarely affected. In some patients oral, scalp (lichen planopilaris), nails, and rarely conjunctival, genital, and esophageal involvement can occur.2
In histopathology, the lesions are characterized by a wedge-shaped hypergranulosis, marked hyperkeratosis, and irregular sawtooth-like acanthosis of rete ridges on the epidermis. The dermal-epidermal junction typically shows an interstitial dermatitis. Civatte bodies may also be seen. On direct immunofluorescence, IgM-staining of the cytoid bodies in the dermal papilla or peribasilar areas are suggestive of lichen planus.1
The differential diagnosis of lichen planus includes severe lichenified atopic dermatitis, drug-induced lichen planus, graft-versus-host disease, psoriasis, pityriasis rosea, subacute cutaneous lupus, discoid lupus, secondary syphilis, and lichen simplex chronicus. Interestingly, our patient presented with lesions that were not pruritic and more generalized. Compared with eczema, were flexures are commonly affected, our patient’s lesions were localized to the ankles, wrists, extensor knees, and elbows, and no pruritus was reported. Lichenification of skin lesions occurs as a response to chronic scratching as it occurs in atopic dermatitis and lichen simplex chronicus, was considered in our patient, but the lack of pruritus and the more acute presentation made it unlikely.
Lichen planus is considered a self-limiting disease, so treatment is focused on the control of pruritus and to accelerate resolution. The first-line therapy for classic cutaneous lichen planus is the use of potent or superpotent topical corticosteroids for localized disease on the body and extremities and mild to mid-potency for intertriginous areas and the face. Clinical response should be assessed after 2-3 weeks of treatment. For patients with more generalized or recalcitrant disease like our patient, other treatment modalities like phototherapy (narrow-band UVB), a 4- to 6-week course of oral glucocorticoids, or acitretin may be considered. Our patient recently started narrow-band UVB. Other medications that have been reported beneficial for more severe cases include methotrexate, cyclosporine, griseofulvin, hydroxychloroquine, metronidazole, dapsone, and mycophenolate. Recent studies in the adult population have shown apremilast, a phosphodiesterase inhibitor, to be a promising medication for patients with cutaneous lichen planus, though this medication has not been approved yet for use in the pediatric population.5
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego.
References
1. Payette MJ et al. Clin Dermatol. 2015 Nov-Dec;33(6):631-43.
2. Walton KE et al. Pediatr Dermatol. 2010;27:34-8.
3. Lehman JS et al. Int J Dermatol. 2009 Jul;48(7):682-94.
4. Laughter D et al. J Am Acad Dermatol. 2000;43:649-55.
5. Paul J et al. J Am Acad Dermatol. 2013 Feb;68(2):255-61.
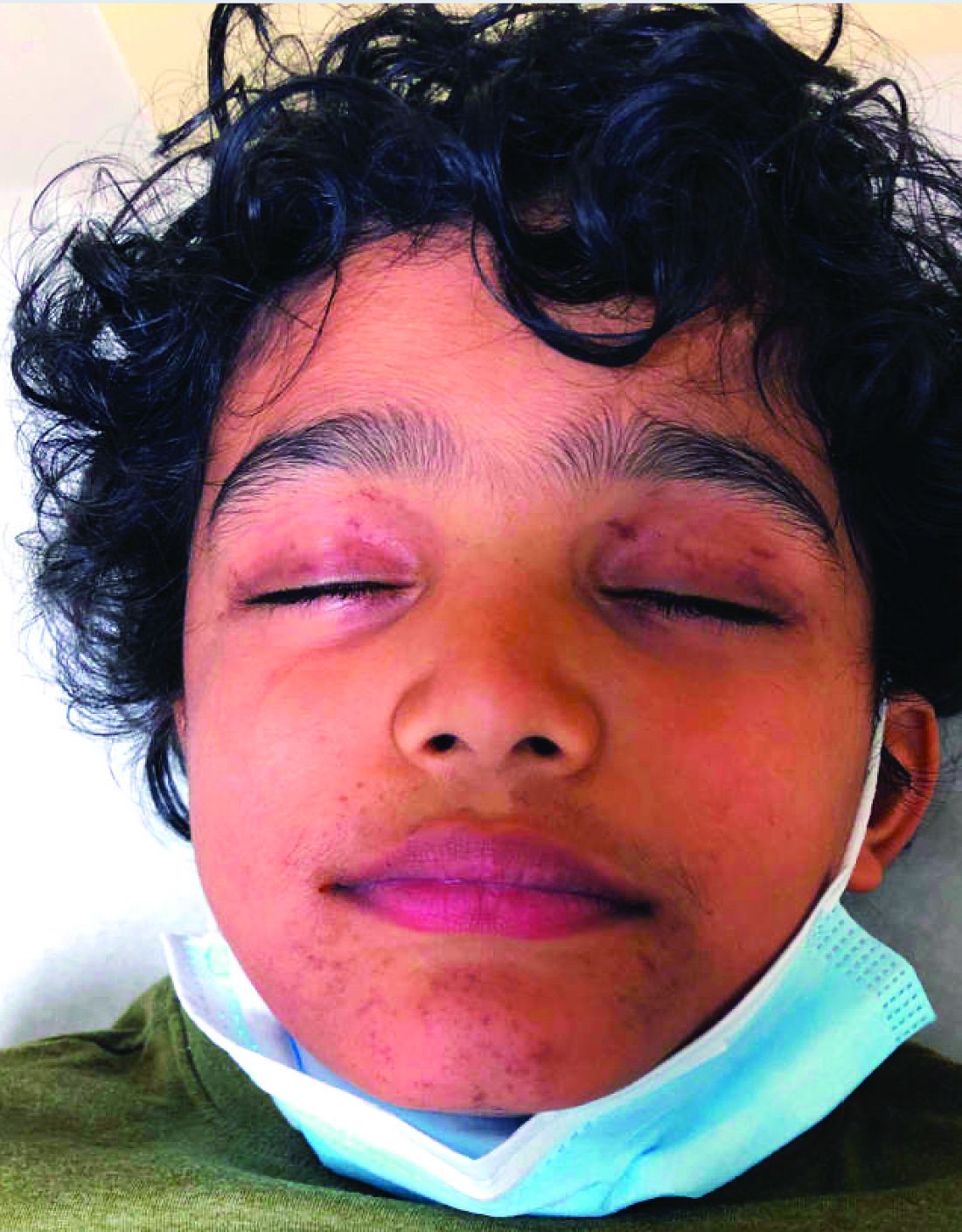
There was no prior personal or family history of atopic dermatitis or psoriasis. He has no other medical conditions and is not taking any medications.
He denied any joint pain, sun sensitivity, mouth sores, or other symptoms. After the initial consultation he was treated with fluocinonide 0.05% ointment for 2 weeks with slight improvement on the lesions.
On physical exam he presented with hyperpigmented and violaceous lichenified papules and plaques on the extremities and the torso. (photos 1 and 2). He also had hyperpigmented violaceous macules on the eyelids and around the mouth (photos 1 and 2).
What's the diagnosis?
Nipple eczema is a dermatitis of the nipple and areola with clinical features such as erythema, fissures, scaling, pruritus, and crusting.1,2 It is classically associated with atopic dermatitis (AD), though it may occur as an isolated condition less commonly. While it may affect female adolescents, nipple eczema has also been reported in boys and breastfeeding women.3,4 The overall risk of incidence of nipple dermatitis has also been shown to increase with age.5 Nipple eczema is considered a cutaneous finding of AD, and is listed as a minor diagnostic criteria for AD in the Hanifin-Rajka criteria.6 The patient had not related his history of AD, which was elicited after finding typical antecubital eczematous dermatitis, and he had not been actively treating it.
Diagnosis and differential

Nipple eczema may be a challenging diagnosis for various reasons. For example, a unilateral presentation and the changes in the eczematous lesions overlying the nipple and areola’s varying textures and colors can make it difficult for clinicians to identify.3 Many children and adolescents, including our patient, are initially diagnosed as having impetigo and treated with antibiotics. The diagnosis of nipple eczema is made clinically, and management straightforward (see below). However, additional testing may be appropriate including patch testing for allergic contact dermatitis or bacterial cultures if bacterial infection or superinfection is considered.7,8 The differential diagnosis for nipple eczema includes impetigo, gynecomastia, scabies, and allergic contact dermatitis.
Impetigo typically presents with honey-colored crusts or pustules caused by infection with Staphylococcus aureus or Streptococcus. Patients with AD have higher rates of colonization with S. aureus and impetiginized eczema in common. Impetigo of the nipple and areola is more common in breastfeeding women as skin cracking from lactation can lead to exposure to bacteria from the infant’s mouth.9 Treatments involve topical or oral antibiotics.
Gynecomastia is the development of male breast tissue with most cases hypothesized to be caused by an imbalance between androgens and estrogens.10 Some other causes include direct skin contact with topical estrogen sprays and recreational use of marijuana and heroin.11 It is usually a benign exam finding in adolescent boys. However, clinical findings such as overlying skin changes, rapidly enlarging masses, and constitutional symptoms are concerning in the setting of gynecomastia and warrant further evaluation.
Scabies, which is caused by the infestation of scabies mites, is a common infectious skin disease. The classic presentation includes a rash that is intensely itchy, especially at night. Crusted scabies of the nipples may be difficult to distinguish from nipple eczema. Areas of frequent involvement of scabies include palms, between fingers, armpits, groin, between toes, and feet. Treatments include treating all household members with permethrin cream and washing all clothes and bedding in contact with a scabies-infected patient in high heat, or oral ivermectin in certain circumstances.12
Allergic contact dermatitis is a common cause of breast and nipple dermatitis and should be considered within the differential diagnosis of nipple eczema with atopic dermatitis, or as an exacerbator.7,9 Patients in particular who present with bilateral involvement extending to the periareolar skin, or unusual bilateral focal patterns suggestive for contact allergy should be considered for allergic contact dermatitis evaluation with patch tests. A common causative agent for allergic contact dermatitis of the breast and nipple includes Cl+Me-isothiazolinone, commonly found in detergents and fabric softeners.7 Primary treatment includes avoidance of the offending agents.
Treatment
Topical corticosteroids are first-line treatment for treating nipple eczema. Low-potency topical steroids can be used for maintenance and mild eczema while more potent steroids are useful for more severe cases. In addition to topical medication therapy, frequent emollient use to protect the skin barrier and the elimination of any irritants are essential to a successful treatment course. Unilateral nipple eczema can also be secondary to inadequate treatment of AD, demonstrating the importance of addressing the underlying AD with therapy.3
Our patient was diagnosed with nipple eczema based on clinical presentation of an eczematous left nipple in the setting of active atopic dermatitis and minimal improvement on topical antibiotic. He was started on a 3-week course of fluocinonide 0.05% topical ointment (a potent topical corticosteroid) twice daily for 2 weeks with plans to transition to triamcinolone 0.1% topical ointment several times a week.
Ms. Park is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital. Neither Ms. Park nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Pediatr Dermatol. 2005;22(1):64-6.
2. Am J Dermatopathol. 2015;37(4):284-8.
3. Pediatr Dermatol. 2015;32(5):718-22.
4. J Cutan Med Surg. 2004;8(2):126-30.
5. Pediatr Dermatol. 2012;29(5):580-3.
6. Dermatologica. 1988;177(6):360-4.
7. Ann Dermatol. 2014;26(3):413-4.
8. BMJ Case Rep. 2020;13(8).
9. J Am Acad Dermatol. 2019;80(6):1483-94.
10. Pediatr Endocrinol Rev. 2017;14(4):371-7.
11. JAMA. 2010;304(9):953.
12. JAMA. 2018;320(6):612.
Nipple eczema is a dermatitis of the nipple and areola with clinical features such as erythema, fissures, scaling, pruritus, and crusting.1,2 It is classically associated with atopic dermatitis (AD), though it may occur as an isolated condition less commonly. While it may affect female adolescents, nipple eczema has also been reported in boys and breastfeeding women.3,4 The overall risk of incidence of nipple dermatitis has also been shown to increase with age.5 Nipple eczema is considered a cutaneous finding of AD, and is listed as a minor diagnostic criteria for AD in the Hanifin-Rajka criteria.6 The patient had not related his history of AD, which was elicited after finding typical antecubital eczematous dermatitis, and he had not been actively treating it.
Diagnosis and differential
Nipple eczema may be a challenging diagnosis for various reasons. For example, a unilateral presentation and the changes in the eczematous lesions overlying the nipple and areola’s varying textures and colors can make it difficult for clinicians to identify.3 Many children and adolescents, including our patient, are initially diagnosed as having impetigo and treated with antibiotics. The diagnosis of nipple eczema is made clinically, and management straightforward (see below). However, additional testing may be appropriate including patch testing for allergic contact dermatitis or bacterial cultures if bacterial infection or superinfection is considered.7,8 The differential diagnosis for nipple eczema includes impetigo, gynecomastia, scabies, and allergic contact dermatitis.
Impetigo typically presents with honey-colored crusts or pustules caused by infection with Staphylococcus aureus or Streptococcus. Patients with AD have higher rates of colonization with S. aureus and impetiginized eczema in common. Impetigo of the nipple and areola is more common in breastfeeding women as skin cracking from lactation can lead to exposure to bacteria from the infant’s mouth.9 Treatments involve topical or oral antibiotics.
Gynecomastia is the development of male breast tissue with most cases hypothesized to be caused by an imbalance between androgens and estrogens.10 Some other causes include direct skin contact with topical estrogen sprays and recreational use of marijuana and heroin.11 It is usually a benign exam finding in adolescent boys. However, clinical findings such as overlying skin changes, rapidly enlarging masses, and constitutional symptoms are concerning in the setting of gynecomastia and warrant further evaluation.
Scabies, which is caused by the infestation of scabies mites, is a common infectious skin disease. The classic presentation includes a rash that is intensely itchy, especially at night. Crusted scabies of the nipples may be difficult to distinguish from nipple eczema. Areas of frequent involvement of scabies include palms, between fingers, armpits, groin, between toes, and feet. Treatments include treating all household members with permethrin cream and washing all clothes and bedding in contact with a scabies-infected patient in high heat, or oral ivermectin in certain circumstances.12
Allergic contact dermatitis is a common cause of breast and nipple dermatitis and should be considered within the differential diagnosis of nipple eczema with atopic dermatitis, or as an exacerbator.7,9 Patients in particular who present with bilateral involvement extending to the periareolar skin, or unusual bilateral focal patterns suggestive for contact allergy should be considered for allergic contact dermatitis evaluation with patch tests. A common causative agent for allergic contact dermatitis of the breast and nipple includes Cl+Me-isothiazolinone, commonly found in detergents and fabric softeners.7 Primary treatment includes avoidance of the offending agents.
Treatment
Topical corticosteroids are first-line treatment for treating nipple eczema. Low-potency topical steroids can be used for maintenance and mild eczema while more potent steroids are useful for more severe cases. In addition to topical medication therapy, frequent emollient use to protect the skin barrier and the elimination of any irritants are essential to a successful treatment course. Unilateral nipple eczema can also be secondary to inadequate treatment of AD, demonstrating the importance of addressing the underlying AD with therapy.3
Our patient was diagnosed with nipple eczema based on clinical presentation of an eczematous left nipple in the setting of active atopic dermatitis and minimal improvement on topical antibiotic. He was started on a 3-week course of fluocinonide 0.05% topical ointment (a potent topical corticosteroid) twice daily for 2 weeks with plans to transition to triamcinolone 0.1% topical ointment several times a week.
Ms. Park is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital. Neither Ms. Park nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Pediatr Dermatol. 2005;22(1):64-6.
2. Am J Dermatopathol. 2015;37(4):284-8.
3. Pediatr Dermatol. 2015;32(5):718-22.
4. J Cutan Med Surg. 2004;8(2):126-30.
5. Pediatr Dermatol. 2012;29(5):580-3.
6. Dermatologica. 1988;177(6):360-4.
7. Ann Dermatol. 2014;26(3):413-4.
8. BMJ Case Rep. 2020;13(8).
9. J Am Acad Dermatol. 2019;80(6):1483-94.
10. Pediatr Endocrinol Rev. 2017;14(4):371-7.
11. JAMA. 2010;304(9):953.
12. JAMA. 2018;320(6):612.
Nipple eczema is a dermatitis of the nipple and areola with clinical features such as erythema, fissures, scaling, pruritus, and crusting.1,2 It is classically associated with atopic dermatitis (AD), though it may occur as an isolated condition less commonly. While it may affect female adolescents, nipple eczema has also been reported in boys and breastfeeding women.3,4 The overall risk of incidence of nipple dermatitis has also been shown to increase with age.5 Nipple eczema is considered a cutaneous finding of AD, and is listed as a minor diagnostic criteria for AD in the Hanifin-Rajka criteria.6 The patient had not related his history of AD, which was elicited after finding typical antecubital eczematous dermatitis, and he had not been actively treating it.
Diagnosis and differential
Nipple eczema may be a challenging diagnosis for various reasons. For example, a unilateral presentation and the changes in the eczematous lesions overlying the nipple and areola’s varying textures and colors can make it difficult for clinicians to identify.3 Many children and adolescents, including our patient, are initially diagnosed as having impetigo and treated with antibiotics. The diagnosis of nipple eczema is made clinically, and management straightforward (see below). However, additional testing may be appropriate including patch testing for allergic contact dermatitis or bacterial cultures if bacterial infection or superinfection is considered.7,8 The differential diagnosis for nipple eczema includes impetigo, gynecomastia, scabies, and allergic contact dermatitis.
Impetigo typically presents with honey-colored crusts or pustules caused by infection with Staphylococcus aureus or Streptococcus. Patients with AD have higher rates of colonization with S. aureus and impetiginized eczema in common. Impetigo of the nipple and areola is more common in breastfeeding women as skin cracking from lactation can lead to exposure to bacteria from the infant’s mouth.9 Treatments involve topical or oral antibiotics.
Gynecomastia is the development of male breast tissue with most cases hypothesized to be caused by an imbalance between androgens and estrogens.10 Some other causes include direct skin contact with topical estrogen sprays and recreational use of marijuana and heroin.11 It is usually a benign exam finding in adolescent boys. However, clinical findings such as overlying skin changes, rapidly enlarging masses, and constitutional symptoms are concerning in the setting of gynecomastia and warrant further evaluation.
Scabies, which is caused by the infestation of scabies mites, is a common infectious skin disease. The classic presentation includes a rash that is intensely itchy, especially at night. Crusted scabies of the nipples may be difficult to distinguish from nipple eczema. Areas of frequent involvement of scabies include palms, between fingers, armpits, groin, between toes, and feet. Treatments include treating all household members with permethrin cream and washing all clothes and bedding in contact with a scabies-infected patient in high heat, or oral ivermectin in certain circumstances.12
Allergic contact dermatitis is a common cause of breast and nipple dermatitis and should be considered within the differential diagnosis of nipple eczema with atopic dermatitis, or as an exacerbator.7,9 Patients in particular who present with bilateral involvement extending to the periareolar skin, or unusual bilateral focal patterns suggestive for contact allergy should be considered for allergic contact dermatitis evaluation with patch tests. A common causative agent for allergic contact dermatitis of the breast and nipple includes Cl+Me-isothiazolinone, commonly found in detergents and fabric softeners.7 Primary treatment includes avoidance of the offending agents.
Treatment
Topical corticosteroids are first-line treatment for treating nipple eczema. Low-potency topical steroids can be used for maintenance and mild eczema while more potent steroids are useful for more severe cases. In addition to topical medication therapy, frequent emollient use to protect the skin barrier and the elimination of any irritants are essential to a successful treatment course. Unilateral nipple eczema can also be secondary to inadequate treatment of AD, demonstrating the importance of addressing the underlying AD with therapy.3
Our patient was diagnosed with nipple eczema based on clinical presentation of an eczematous left nipple in the setting of active atopic dermatitis and minimal improvement on topical antibiotic. He was started on a 3-week course of fluocinonide 0.05% topical ointment (a potent topical corticosteroid) twice daily for 2 weeks with plans to transition to triamcinolone 0.1% topical ointment several times a week.
Ms. Park is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego, and Rady Children’s Hospital. Neither Ms. Park nor Dr. Eichenfield have any relevant financial disclosures.
References
1. Pediatr Dermatol. 2005;22(1):64-6.
2. Am J Dermatopathol. 2015;37(4):284-8.
3. Pediatr Dermatol. 2015;32(5):718-22.
4. J Cutan Med Surg. 2004;8(2):126-30.
5. Pediatr Dermatol. 2012;29(5):580-3.
6. Dermatologica. 1988;177(6):360-4.
7. Ann Dermatol. 2014;26(3):413-4.
8. BMJ Case Rep. 2020;13(8).
9. J Am Acad Dermatol. 2019;80(6):1483-94.
10. Pediatr Endocrinol Rev. 2017;14(4):371-7.
11. JAMA. 2010;304(9):953.
12. JAMA. 2018;320(6):612.
A 12-year-old boy presents to the dermatology clinic with a 1-month history of crusting and watery sticky drainage from the left nipple. Given concern for a possible skin infection, the patient was initially treated with mupirocin ointment for several weeks but without improvement. The affected area is sometimes itchy but not painful. He reports no prior history of similar problems.
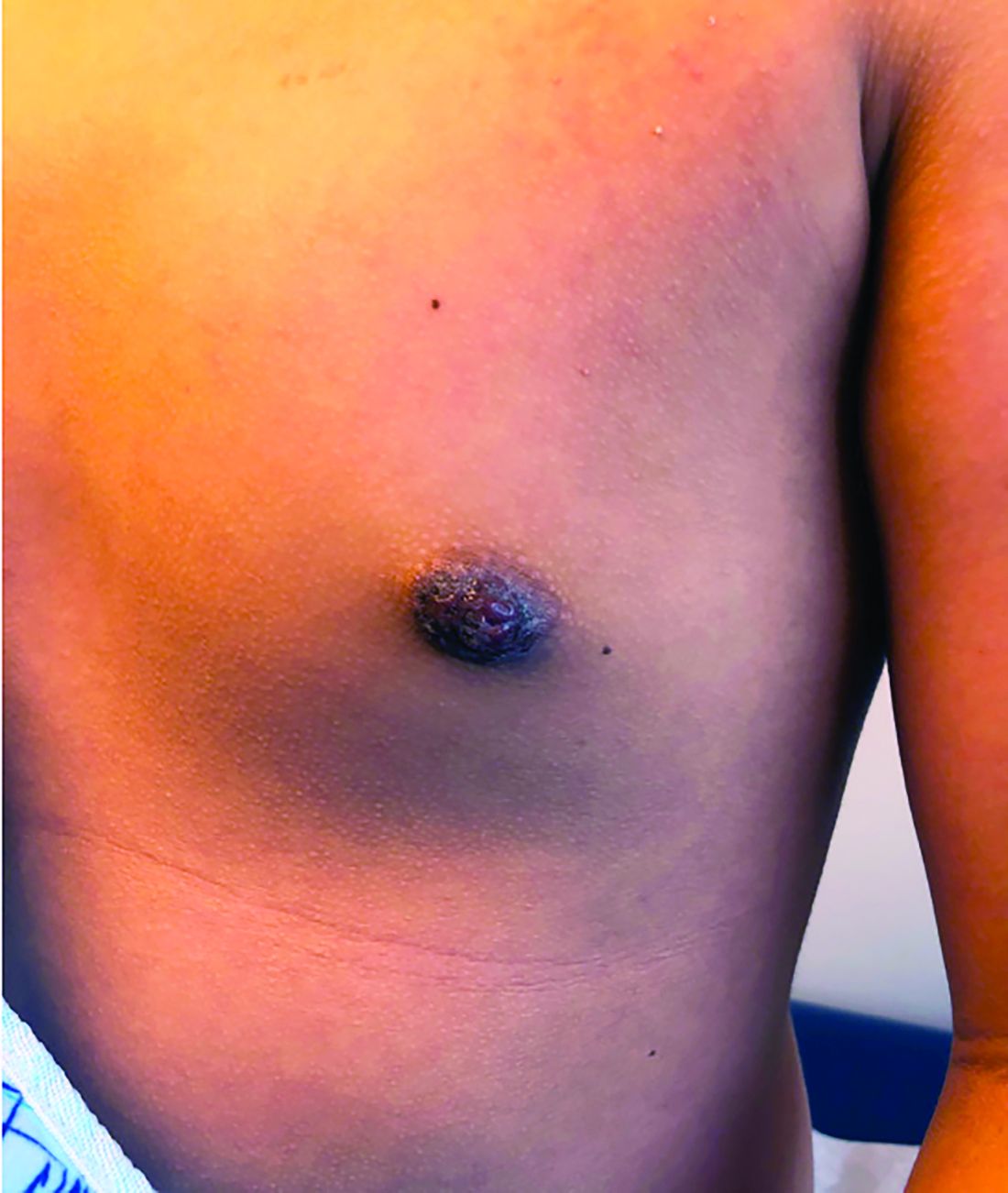
On physical exam, he is noted to have an eczematous left nipple with edema, xerosis, and scaling overlying the entire areola. There is no evidence of visible discharge, pustules, or honey-colored crusts in the area. The extensor surfaces of his arms bilaterally have skin-colored follicular papules, and his antecubital fossa display erythematous scaling plaques with mild lichenification and excoriations.
Atrophic Lesion on the Abdomen
The Diagnosis: Anetoderma of Prematurity
Anetoderma is a rare benign cutaneous disorder characterized by atrophic patches of skin due to dermal thinning. The term anetoderma is derived from the Greek words anetos (relaxed) and derma (skin).1 The physical appearance of the skin is associated with a reduction or loss of elastic tissue in the dermal layer, as seen on histolopathology.2
Two forms of anetoderma have been described. Primary anetoderma is an idiopathic form with no preceding inflammatory lesions. Secondary anetoderma is a reactive process linked to a known preceding inflammatory, infectious, autoimmune, or drug-induced condition.3 On histopathology, both primary and secondary anetoderma are characterized by a loss of elastic tissue or elastin fibers in the superficial to mid dermis.2
Anetoderma of prematurity was first described in 1996 by Prizant et al4 in 9 extremely premature (24-29 weeks' gestation) infants in neonatal intensive care units (NICUs). Although the exact mechanism behind anetoderma of prematurity is still unknown, Prizant et al4 and other investigators5 postulated that application of adhesive monitoring leads in the NICU played a role in the development of the lesions.
Iatrogenic anetoderma of prematurity is clinically characterized by circumscribed areas of either wrinkled macular depression or pouchlike herniations, ranging from flesh-colored to violaceous hues. Lesion size varies from a few millimeters to several centimeters in diameter, and they often are oval or round in shape.2 Although not common, it is possible for the atrophic patches to be preceded by an area of ecchymosis without necrosis or atrophy and, if present, they usually evolve within a few days to the characteristic appearance of anetoderma.3 They are found at discrete sites where monitoring leads or other medical devices are commonly placed, such as the forehead, abdomen, chest, and proximal limbs.
Lesions of anetoderma of prematurity are not present at birth, which distinguishes them from congenital anetoderma.6 It is unclear if the lesions are associated with the degree of prematurity, extremely low birth weight, or other associated factors of preterm birth. Although often clinically diagnosed, the diagnosis can be confirmed by a loss of elastic fibers on histopathology when stained with Verhoeff-van Gieson stain.1 Over time, the atrophic patches have the potential to evolve into herniated forms of anetoderma. Self-healing or improvement of the lesions often does not occur. Although the lesion is benign, it often requires surgical correction later in life for cosmesis.
Infants in the NICU are at risk for iatrogenic cutaneous injuries, which rarely may include anetoderma. Anetoderma of prematurity has been linked to the use of monitoring leads, adhesive tape, and other medical devices placed on the skin. Prizant et al4 postulated that the cause of anetoderma in these infants was irritants such as skin cleansers, urine, or sweat that may be trapped under the electrodes. Other hypotheses include local hypoxemia due to prolonged pressure from the electrodes on immature skin or excessive traction used when removing adhesive tape from the skin.7,8 Premature infants may be more susceptible to these lesions because of the reduced epidermal thickness of premature skin; immaturity of skin structure; or functional immaturity of elastin deposition regulators, such as elastase, lysyl oxidase, the complement system, and decay-accelerating factor.3 The diagnosis should be differentiated from congenital anetoderma, which also has been described in premature neonates but is characterized by lesions that are present at birth. Its origins are still unclear, despite having histopathologic features similar to iatrogenic anetoderma.9
Focal dermal hypoplasia (FDH) is the hallmark cutaneous finding in Goltz syndrome, a rare set of congenital abnormalities of the skin, oral structures, musculoskeletal system, and central nervous system. Similar to congenital anetoderma, FDH also is characterized by atrophic cutaneous lesions; however, the cutaneous lesions in FDH appear as linear, streaky atrophic lesions often with telangiectasias that follow Blaschko lines.10 The cutaneous lesions in FDH often are associated with other noncutaneous signs such as polydactyly or asymmetric limbs.10 Cutis laxa is caused by an abnormality in the elastic tissue resulting in a loose sagging appearance of the skin and frequently results in an aged facial appearance. There are both acquired and inherited forms that can be either solely cutaneous or present with extracutaneous features, such as cardiac abnormalities or emphysema.11
In contrast to the atrophic appearance of anetodermas, connective tissue nevi and nevus lipomatosus superficialis present as hamartomas that either can be present at birth or arise in infancy. Connective tissue nevi are hamartomas of dermal connective tissue that consist of excessive production of collagen, elastin, or glycosaminoglycans and appear as slightly elevated, flesh-colored to yellow nodules or plaques.12 Connective tissue nevi often are described in association with other diseases, most commonly tuberous sclerosis (shagreen patches) or familial cutaneous collagenoma. Nevus lipomatosus superficialis is an asymptomatic connective tissue hamartoma composed of mature adipocytes in the dermis. The lesions consist of clusters of flesh-colored to yellow, soft, rubbery papules or nodules with a smooth or verrucoid surface that do not cross the midline and may follow Blaschko lines.11
With advances in neonatal infant medical care, survival of extremely premature infants is increasing, and it is possible that this rare cutaneous disorder may become more prevalent. Care should be taken to avoid unnecessary pressure on surfaces where electrodes are placed and tightly applied adhesive tape. When electrodes are placed on the ventral side, the child should be placed supine; similarly, place electrodes on the dorsal side when the child is lying prone.5 A diagnosis of anetoderma of prematurity later in childhood may be difficult, so knowledge and awareness can help guide pediatricians and dermatologists to a correct diagnosis and prevent unnecessary evaluations and/or concerns.
- Misch KJ, Rhodes EL, Allen J, et al. Anetoderma of Jadassohn. J R Soc Med.1988;81:734-736.
- Venencie PY, Winkelmann RK. Histopathologic findings in anetoderma. Arch Dermatol. 1984;120:1040-1044.
- Maffeis L, Pugni L, Pietrasanta C, et al. Case report iatrogenic anetoderma of prematurity: a case report and review of the literature. 2014;2014:781493.
- Prizant TL, Lucky AW, Frieden IJ, et al. Spontaneous atrophic patches in extremely premature infants: anetoderma of prematurity. Arch Dermatol. 1996;132:671-674.
- Goujon E, Beer F, Gay S, et al. Anetoderma of prematurity: an iatrogenic consequence of neonatal intensive care anetoderma of prematurity from NICU. Arch Dermatol. 2010;146:565-567.
- Wain EM, Mellerio JE, Robson A, et al. Congenital anetoderma in a preterm infant. Pediatr Dermatol. 2008;25:626-629.
- Colditz PB, Dunster KR, Joy GJ, et al. Anetoderma of prematurity in association with electrocardiographic electrodes. J Am Acad Dermatol. 1999;41:479-481.
- Goujan E, Beer F, Gay S, et al. Study supervision. Arch Dermatol. 2010;146:565-567.
- Aberer E, Weissenbacher G. Congenital anetoderma induced by intrauterine infection? Arch Dermatol. 1997;133:526-527.
- Mallory SB, Krafchik BR, Moore DJ, et al. Goltz syndrome. Pediatr Dermatol. 1989;6:251-253.
- Bolognia J, Schaffer J, Cerroni L. Dermatology. Elsevier Saunders; 2017.
- Uitto J, Santa Cruz DJ, Eisen AZ. Connective tissue nevi of the skin. clinical, genetic, and histopathologic classification of hamartomas of the collagen, elastin, and proteoglycan type. J Am Acad Dermatol. 1980;3:441-461.
The Diagnosis: Anetoderma of Prematurity
Anetoderma is a rare benign cutaneous disorder characterized by atrophic patches of skin due to dermal thinning. The term anetoderma is derived from the Greek words anetos (relaxed) and derma (skin).1 The physical appearance of the skin is associated with a reduction or loss of elastic tissue in the dermal layer, as seen on histolopathology.2
Two forms of anetoderma have been described. Primary anetoderma is an idiopathic form with no preceding inflammatory lesions. Secondary anetoderma is a reactive process linked to a known preceding inflammatory, infectious, autoimmune, or drug-induced condition.3 On histopathology, both primary and secondary anetoderma are characterized by a loss of elastic tissue or elastin fibers in the superficial to mid dermis.2
Anetoderma of prematurity was first described in 1996 by Prizant et al4 in 9 extremely premature (24-29 weeks' gestation) infants in neonatal intensive care units (NICUs). Although the exact mechanism behind anetoderma of prematurity is still unknown, Prizant et al4 and other investigators5 postulated that application of adhesive monitoring leads in the NICU played a role in the development of the lesions.
Iatrogenic anetoderma of prematurity is clinically characterized by circumscribed areas of either wrinkled macular depression or pouchlike herniations, ranging from flesh-colored to violaceous hues. Lesion size varies from a few millimeters to several centimeters in diameter, and they often are oval or round in shape.2 Although not common, it is possible for the atrophic patches to be preceded by an area of ecchymosis without necrosis or atrophy and, if present, they usually evolve within a few days to the characteristic appearance of anetoderma.3 They are found at discrete sites where monitoring leads or other medical devices are commonly placed, such as the forehead, abdomen, chest, and proximal limbs.
Lesions of anetoderma of prematurity are not present at birth, which distinguishes them from congenital anetoderma.6 It is unclear if the lesions are associated with the degree of prematurity, extremely low birth weight, or other associated factors of preterm birth. Although often clinically diagnosed, the diagnosis can be confirmed by a loss of elastic fibers on histopathology when stained with Verhoeff-van Gieson stain.1 Over time, the atrophic patches have the potential to evolve into herniated forms of anetoderma. Self-healing or improvement of the lesions often does not occur. Although the lesion is benign, it often requires surgical correction later in life for cosmesis.
Infants in the NICU are at risk for iatrogenic cutaneous injuries, which rarely may include anetoderma. Anetoderma of prematurity has been linked to the use of monitoring leads, adhesive tape, and other medical devices placed on the skin. Prizant et al4 postulated that the cause of anetoderma in these infants was irritants such as skin cleansers, urine, or sweat that may be trapped under the electrodes. Other hypotheses include local hypoxemia due to prolonged pressure from the electrodes on immature skin or excessive traction used when removing adhesive tape from the skin.7,8 Premature infants may be more susceptible to these lesions because of the reduced epidermal thickness of premature skin; immaturity of skin structure; or functional immaturity of elastin deposition regulators, such as elastase, lysyl oxidase, the complement system, and decay-accelerating factor.3 The diagnosis should be differentiated from congenital anetoderma, which also has been described in premature neonates but is characterized by lesions that are present at birth. Its origins are still unclear, despite having histopathologic features similar to iatrogenic anetoderma.9
Focal dermal hypoplasia (FDH) is the hallmark cutaneous finding in Goltz syndrome, a rare set of congenital abnormalities of the skin, oral structures, musculoskeletal system, and central nervous system. Similar to congenital anetoderma, FDH also is characterized by atrophic cutaneous lesions; however, the cutaneous lesions in FDH appear as linear, streaky atrophic lesions often with telangiectasias that follow Blaschko lines.10 The cutaneous lesions in FDH often are associated with other noncutaneous signs such as polydactyly or asymmetric limbs.10 Cutis laxa is caused by an abnormality in the elastic tissue resulting in a loose sagging appearance of the skin and frequently results in an aged facial appearance. There are both acquired and inherited forms that can be either solely cutaneous or present with extracutaneous features, such as cardiac abnormalities or emphysema.11
In contrast to the atrophic appearance of anetodermas, connective tissue nevi and nevus lipomatosus superficialis present as hamartomas that either can be present at birth or arise in infancy. Connective tissue nevi are hamartomas of dermal connective tissue that consist of excessive production of collagen, elastin, or glycosaminoglycans and appear as slightly elevated, flesh-colored to yellow nodules or plaques.12 Connective tissue nevi often are described in association with other diseases, most commonly tuberous sclerosis (shagreen patches) or familial cutaneous collagenoma. Nevus lipomatosus superficialis is an asymptomatic connective tissue hamartoma composed of mature adipocytes in the dermis. The lesions consist of clusters of flesh-colored to yellow, soft, rubbery papules or nodules with a smooth or verrucoid surface that do not cross the midline and may follow Blaschko lines.11
With advances in neonatal infant medical care, survival of extremely premature infants is increasing, and it is possible that this rare cutaneous disorder may become more prevalent. Care should be taken to avoid unnecessary pressure on surfaces where electrodes are placed and tightly applied adhesive tape. When electrodes are placed on the ventral side, the child should be placed supine; similarly, place electrodes on the dorsal side when the child is lying prone.5 A diagnosis of anetoderma of prematurity later in childhood may be difficult, so knowledge and awareness can help guide pediatricians and dermatologists to a correct diagnosis and prevent unnecessary evaluations and/or concerns.
The Diagnosis: Anetoderma of Prematurity
Anetoderma is a rare benign cutaneous disorder characterized by atrophic patches of skin due to dermal thinning. The term anetoderma is derived from the Greek words anetos (relaxed) and derma (skin).1 The physical appearance of the skin is associated with a reduction or loss of elastic tissue in the dermal layer, as seen on histolopathology.2
Two forms of anetoderma have been described. Primary anetoderma is an idiopathic form with no preceding inflammatory lesions. Secondary anetoderma is a reactive process linked to a known preceding inflammatory, infectious, autoimmune, or drug-induced condition.3 On histopathology, both primary and secondary anetoderma are characterized by a loss of elastic tissue or elastin fibers in the superficial to mid dermis.2
Anetoderma of prematurity was first described in 1996 by Prizant et al4 in 9 extremely premature (24-29 weeks' gestation) infants in neonatal intensive care units (NICUs). Although the exact mechanism behind anetoderma of prematurity is still unknown, Prizant et al4 and other investigators5 postulated that application of adhesive monitoring leads in the NICU played a role in the development of the lesions.
Iatrogenic anetoderma of prematurity is clinically characterized by circumscribed areas of either wrinkled macular depression or pouchlike herniations, ranging from flesh-colored to violaceous hues. Lesion size varies from a few millimeters to several centimeters in diameter, and they often are oval or round in shape.2 Although not common, it is possible for the atrophic patches to be preceded by an area of ecchymosis without necrosis or atrophy and, if present, they usually evolve within a few days to the characteristic appearance of anetoderma.3 They are found at discrete sites where monitoring leads or other medical devices are commonly placed, such as the forehead, abdomen, chest, and proximal limbs.
Lesions of anetoderma of prematurity are not present at birth, which distinguishes them from congenital anetoderma.6 It is unclear if the lesions are associated with the degree of prematurity, extremely low birth weight, or other associated factors of preterm birth. Although often clinically diagnosed, the diagnosis can be confirmed by a loss of elastic fibers on histopathology when stained with Verhoeff-van Gieson stain.1 Over time, the atrophic patches have the potential to evolve into herniated forms of anetoderma. Self-healing or improvement of the lesions often does not occur. Although the lesion is benign, it often requires surgical correction later in life for cosmesis.
Infants in the NICU are at risk for iatrogenic cutaneous injuries, which rarely may include anetoderma. Anetoderma of prematurity has been linked to the use of monitoring leads, adhesive tape, and other medical devices placed on the skin. Prizant et al4 postulated that the cause of anetoderma in these infants was irritants such as skin cleansers, urine, or sweat that may be trapped under the electrodes. Other hypotheses include local hypoxemia due to prolonged pressure from the electrodes on immature skin or excessive traction used when removing adhesive tape from the skin.7,8 Premature infants may be more susceptible to these lesions because of the reduced epidermal thickness of premature skin; immaturity of skin structure; or functional immaturity of elastin deposition regulators, such as elastase, lysyl oxidase, the complement system, and decay-accelerating factor.3 The diagnosis should be differentiated from congenital anetoderma, which also has been described in premature neonates but is characterized by lesions that are present at birth. Its origins are still unclear, despite having histopathologic features similar to iatrogenic anetoderma.9
Focal dermal hypoplasia (FDH) is the hallmark cutaneous finding in Goltz syndrome, a rare set of congenital abnormalities of the skin, oral structures, musculoskeletal system, and central nervous system. Similar to congenital anetoderma, FDH also is characterized by atrophic cutaneous lesions; however, the cutaneous lesions in FDH appear as linear, streaky atrophic lesions often with telangiectasias that follow Blaschko lines.10 The cutaneous lesions in FDH often are associated with other noncutaneous signs such as polydactyly or asymmetric limbs.10 Cutis laxa is caused by an abnormality in the elastic tissue resulting in a loose sagging appearance of the skin and frequently results in an aged facial appearance. There are both acquired and inherited forms that can be either solely cutaneous or present with extracutaneous features, such as cardiac abnormalities or emphysema.11
In contrast to the atrophic appearance of anetodermas, connective tissue nevi and nevus lipomatosus superficialis present as hamartomas that either can be present at birth or arise in infancy. Connective tissue nevi are hamartomas of dermal connective tissue that consist of excessive production of collagen, elastin, or glycosaminoglycans and appear as slightly elevated, flesh-colored to yellow nodules or plaques.12 Connective tissue nevi often are described in association with other diseases, most commonly tuberous sclerosis (shagreen patches) or familial cutaneous collagenoma. Nevus lipomatosus superficialis is an asymptomatic connective tissue hamartoma composed of mature adipocytes in the dermis. The lesions consist of clusters of flesh-colored to yellow, soft, rubbery papules or nodules with a smooth or verrucoid surface that do not cross the midline and may follow Blaschko lines.11
With advances in neonatal infant medical care, survival of extremely premature infants is increasing, and it is possible that this rare cutaneous disorder may become more prevalent. Care should be taken to avoid unnecessary pressure on surfaces where electrodes are placed and tightly applied adhesive tape. When electrodes are placed on the ventral side, the child should be placed supine; similarly, place electrodes on the dorsal side when the child is lying prone.5 A diagnosis of anetoderma of prematurity later in childhood may be difficult, so knowledge and awareness can help guide pediatricians and dermatologists to a correct diagnosis and prevent unnecessary evaluations and/or concerns.
- Misch KJ, Rhodes EL, Allen J, et al. Anetoderma of Jadassohn. J R Soc Med.1988;81:734-736.
- Venencie PY, Winkelmann RK. Histopathologic findings in anetoderma. Arch Dermatol. 1984;120:1040-1044.
- Maffeis L, Pugni L, Pietrasanta C, et al. Case report iatrogenic anetoderma of prematurity: a case report and review of the literature. 2014;2014:781493.
- Prizant TL, Lucky AW, Frieden IJ, et al. Spontaneous atrophic patches in extremely premature infants: anetoderma of prematurity. Arch Dermatol. 1996;132:671-674.
- Goujon E, Beer F, Gay S, et al. Anetoderma of prematurity: an iatrogenic consequence of neonatal intensive care anetoderma of prematurity from NICU. Arch Dermatol. 2010;146:565-567.
- Wain EM, Mellerio JE, Robson A, et al. Congenital anetoderma in a preterm infant. Pediatr Dermatol. 2008;25:626-629.
- Colditz PB, Dunster KR, Joy GJ, et al. Anetoderma of prematurity in association with electrocardiographic electrodes. J Am Acad Dermatol. 1999;41:479-481.
- Goujan E, Beer F, Gay S, et al. Study supervision. Arch Dermatol. 2010;146:565-567.
- Aberer E, Weissenbacher G. Congenital anetoderma induced by intrauterine infection? Arch Dermatol. 1997;133:526-527.
- Mallory SB, Krafchik BR, Moore DJ, et al. Goltz syndrome. Pediatr Dermatol. 1989;6:251-253.
- Bolognia J, Schaffer J, Cerroni L. Dermatology. Elsevier Saunders; 2017.
- Uitto J, Santa Cruz DJ, Eisen AZ. Connective tissue nevi of the skin. clinical, genetic, and histopathologic classification of hamartomas of the collagen, elastin, and proteoglycan type. J Am Acad Dermatol. 1980;3:441-461.
- Misch KJ, Rhodes EL, Allen J, et al. Anetoderma of Jadassohn. J R Soc Med.1988;81:734-736.
- Venencie PY, Winkelmann RK. Histopathologic findings in anetoderma. Arch Dermatol. 1984;120:1040-1044.
- Maffeis L, Pugni L, Pietrasanta C, et al. Case report iatrogenic anetoderma of prematurity: a case report and review of the literature. 2014;2014:781493.
- Prizant TL, Lucky AW, Frieden IJ, et al. Spontaneous atrophic patches in extremely premature infants: anetoderma of prematurity. Arch Dermatol. 1996;132:671-674.
- Goujon E, Beer F, Gay S, et al. Anetoderma of prematurity: an iatrogenic consequence of neonatal intensive care anetoderma of prematurity from NICU. Arch Dermatol. 2010;146:565-567.
- Wain EM, Mellerio JE, Robson A, et al. Congenital anetoderma in a preterm infant. Pediatr Dermatol. 2008;25:626-629.
- Colditz PB, Dunster KR, Joy GJ, et al. Anetoderma of prematurity in association with electrocardiographic electrodes. J Am Acad Dermatol. 1999;41:479-481.
- Goujan E, Beer F, Gay S, et al. Study supervision. Arch Dermatol. 2010;146:565-567.
- Aberer E, Weissenbacher G. Congenital anetoderma induced by intrauterine infection? Arch Dermatol. 1997;133:526-527.
- Mallory SB, Krafchik BR, Moore DJ, et al. Goltz syndrome. Pediatr Dermatol. 1989;6:251-253.
- Bolognia J, Schaffer J, Cerroni L. Dermatology. Elsevier Saunders; 2017.
- Uitto J, Santa Cruz DJ, Eisen AZ. Connective tissue nevi of the skin. clinical, genetic, and histopathologic classification of hamartomas of the collagen, elastin, and proteoglycan type. J Am Acad Dermatol. 1980;3:441-461.

An 18-month-old child presented with a 4-cm, atrophic, flesh-colored plaque on the left lateral aspect of the abdomen with overlying wrinkling of the skin. There was no outpouching of the skin or pain associated with the lesion. No other skin abnormalities were noted. The child was born premature at 30 weeks’ gestation (birth weight, 1400 g). The postnatal course was complicated by respiratory distress syndrome requiring prolonged ventilator support. The infant was in the neonatal intensive care unit for 5 months. The atrophic lesion first developed at 5 months of life and remained stable. Although the lesion was not present at birth, the parents noted that it was preceded by an ecchymotic lesion without necrosis that was first noticed at 2 months of life while the patient was in the neonatal intensive care unit.
A girl presents with blotchy, slightly itchy spots on her chest, back
On close evaluation of the picture on her chest, she has pale macules and patches surrounded by erythematous ill-defined patches consistent with nevus anemicus. The findings of the picture raise the suspicion for neurofibromatosis, and it was recommended for her to be evaluated in person.
She comes several days later to the clinic. The caretaker, who is her aunt, reports she does not know much of the girl’s medical history as she recently moved from South America to live with her. The girl is a very nice and pleasant 8-year-old. She reports noticing the spots on her chest for about a year and that they seem to get a little itchier and more noticeable when she is hot or when she is running. She also reports increasing headaches for several months. She is being home schooled, and according to her aunt she is at par with her cousins who are about the same age. There is no history of seizures. She has had back surgery in the past. There is no history of hypertension. There is no family history of any genetic disorder or similar lesions.
On physical exam, her vital signs are normal, but her head circumference is over the 90th percentile. She is pleasant and interactive. On skin examination, she has slightly noticeable pale macules and patches on the chest and back that become more apparent after rubbing her skin. She has multiple light brown macules and oval patches on the chest, back, and neck. She has no axillary or inguinal freckling. She has scars on the back from her prior surgery.
As she was having worsening headaches, an MRI of the brain was ordered, which showed a left optic glioma. She was then referred to ophthalmology, neurology, and genetics.
Neurofibromatosis type 1 (NF1) is a common genetic autosomal dominant disorder cause by mutations on the NF1 gene on chromosome 17, which encodes for the protein neurofibromin. This protein works in the Ras-mitogen–activated protein kinase pathway as a negative regulator. Based on the National Institute of Health criteria, children need two or more of the following to be diagnosed with NF1: more than six café au lait macules larger than 5 mm in prepubescent children and 2.5 cm after puberty; axillary or inguinal freckling; two or more Lisch nodules; optic gliomas; two or more neurofibromas or one plexiform neurofibroma; or a first degree relative with a diagnosis of NF1. With these criteria, about 70% of the children can be diagnosed before the age of 1 year.1
Nevus anemicus is an uncommon birthmark, sometimes overlooked, that is characterized by pale, hypopigmented, well-defined macules and patches that do not turn red after trauma or changes in temperature. Nevus anemicus is usually localized on the torso but can be seen on the face, neck, and extremities. These lesions are present in 1%-2% of the general population. They are thought to occur because of increased sensitivity of the affected blood vessels to catecholamines, which causes permanent vasoconstriction, which leads to hypopigmentation on the area.2 These lesions are usually present at birth and have been described in patients with tuberous sclerosis, neurofibromatosis, and phakomatosis pigmentovascularis.
Recent studies of patients with neurofibromatosis and other RASopathies have noticed that nevus anemicus is present in about 8.8%-51% of the patients studied with a diagnosis NF1, compared with only 2% of the controls.3,4 The studies failed to report any cases of nevus anemicus in patients with other RASopathies associated with café au lait macules. Bulteel and colleagues recently reported two cases of non-NF1 RASopathies also associated with nevus anemicus in a patient with Legius syndrome and a patient with Noonan syndrome with multiple lentigines.5 The nevus anemicus was reported to occur most commonly on the anterior chest and be multiple, as seen in our patient.
The authors of the published studies advocate for the introduction of nevus anemicus as part of the diagnostic criteria for NF1, especially because it can be an early finding seen in babies, which can aid in early diagnosis of NF1.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. Pediatrics. 2000 Mar. doi: 10.1542/peds.105.3.608.
2. Nevus Anemicus. StatPearls [Internet] (Treasure Island, Fla.: StatPearls Publishing; 2020 Jan).
3. J Am Acad Dermatol. 2013 Nov. doi: 10.1016/j.jaad.2013.06.039.
4. Pediatr Dermatol. 2015 May-Jun. doi: 10.1111/pde.12525.
5. JAAD Case Rep. 2018 Apr 5. doi: 10.1016/j.jdcr.2017.09.037.
On close evaluation of the picture on her chest, she has pale macules and patches surrounded by erythematous ill-defined patches consistent with nevus anemicus. The findings of the picture raise the suspicion for neurofibromatosis, and it was recommended for her to be evaluated in person.
She comes several days later to the clinic. The caretaker, who is her aunt, reports she does not know much of the girl’s medical history as she recently moved from South America to live with her. The girl is a very nice and pleasant 8-year-old. She reports noticing the spots on her chest for about a year and that they seem to get a little itchier and more noticeable when she is hot or when she is running. She also reports increasing headaches for several months. She is being home schooled, and according to her aunt she is at par with her cousins who are about the same age. There is no history of seizures. She has had back surgery in the past. There is no history of hypertension. There is no family history of any genetic disorder or similar lesions.
On physical exam, her vital signs are normal, but her head circumference is over the 90th percentile. She is pleasant and interactive. On skin examination, she has slightly noticeable pale macules and patches on the chest and back that become more apparent after rubbing her skin. She has multiple light brown macules and oval patches on the chest, back, and neck. She has no axillary or inguinal freckling. She has scars on the back from her prior surgery.
As she was having worsening headaches, an MRI of the brain was ordered, which showed a left optic glioma. She was then referred to ophthalmology, neurology, and genetics.
Neurofibromatosis type 1 (NF1) is a common genetic autosomal dominant disorder cause by mutations on the NF1 gene on chromosome 17, which encodes for the protein neurofibromin. This protein works in the Ras-mitogen–activated protein kinase pathway as a negative regulator. Based on the National Institute of Health criteria, children need two or more of the following to be diagnosed with NF1: more than six café au lait macules larger than 5 mm in prepubescent children and 2.5 cm after puberty; axillary or inguinal freckling; two or more Lisch nodules; optic gliomas; two or more neurofibromas or one plexiform neurofibroma; or a first degree relative with a diagnosis of NF1. With these criteria, about 70% of the children can be diagnosed before the age of 1 year.1
Nevus anemicus is an uncommon birthmark, sometimes overlooked, that is characterized by pale, hypopigmented, well-defined macules and patches that do not turn red after trauma or changes in temperature. Nevus anemicus is usually localized on the torso but can be seen on the face, neck, and extremities. These lesions are present in 1%-2% of the general population. They are thought to occur because of increased sensitivity of the affected blood vessels to catecholamines, which causes permanent vasoconstriction, which leads to hypopigmentation on the area.2 These lesions are usually present at birth and have been described in patients with tuberous sclerosis, neurofibromatosis, and phakomatosis pigmentovascularis.
Recent studies of patients with neurofibromatosis and other RASopathies have noticed that nevus anemicus is present in about 8.8%-51% of the patients studied with a diagnosis NF1, compared with only 2% of the controls.3,4 The studies failed to report any cases of nevus anemicus in patients with other RASopathies associated with café au lait macules. Bulteel and colleagues recently reported two cases of non-NF1 RASopathies also associated with nevus anemicus in a patient with Legius syndrome and a patient with Noonan syndrome with multiple lentigines.5 The nevus anemicus was reported to occur most commonly on the anterior chest and be multiple, as seen in our patient.
The authors of the published studies advocate for the introduction of nevus anemicus as part of the diagnostic criteria for NF1, especially because it can be an early finding seen in babies, which can aid in early diagnosis of NF1.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. Pediatrics. 2000 Mar. doi: 10.1542/peds.105.3.608.
2. Nevus Anemicus. StatPearls [Internet] (Treasure Island, Fla.: StatPearls Publishing; 2020 Jan).
3. J Am Acad Dermatol. 2013 Nov. doi: 10.1016/j.jaad.2013.06.039.
4. Pediatr Dermatol. 2015 May-Jun. doi: 10.1111/pde.12525.
5. JAAD Case Rep. 2018 Apr 5. doi: 10.1016/j.jdcr.2017.09.037.
On close evaluation of the picture on her chest, she has pale macules and patches surrounded by erythematous ill-defined patches consistent with nevus anemicus. The findings of the picture raise the suspicion for neurofibromatosis, and it was recommended for her to be evaluated in person.
She comes several days later to the clinic. The caretaker, who is her aunt, reports she does not know much of the girl’s medical history as she recently moved from South America to live with her. The girl is a very nice and pleasant 8-year-old. She reports noticing the spots on her chest for about a year and that they seem to get a little itchier and more noticeable when she is hot or when she is running. She also reports increasing headaches for several months. She is being home schooled, and according to her aunt she is at par with her cousins who are about the same age. There is no history of seizures. She has had back surgery in the past. There is no history of hypertension. There is no family history of any genetic disorder or similar lesions.
On physical exam, her vital signs are normal, but her head circumference is over the 90th percentile. She is pleasant and interactive. On skin examination, she has slightly noticeable pale macules and patches on the chest and back that become more apparent after rubbing her skin. She has multiple light brown macules and oval patches on the chest, back, and neck. She has no axillary or inguinal freckling. She has scars on the back from her prior surgery.
As she was having worsening headaches, an MRI of the brain was ordered, which showed a left optic glioma. She was then referred to ophthalmology, neurology, and genetics.
Neurofibromatosis type 1 (NF1) is a common genetic autosomal dominant disorder cause by mutations on the NF1 gene on chromosome 17, which encodes for the protein neurofibromin. This protein works in the Ras-mitogen–activated protein kinase pathway as a negative regulator. Based on the National Institute of Health criteria, children need two or more of the following to be diagnosed with NF1: more than six café au lait macules larger than 5 mm in prepubescent children and 2.5 cm after puberty; axillary or inguinal freckling; two or more Lisch nodules; optic gliomas; two or more neurofibromas or one plexiform neurofibroma; or a first degree relative with a diagnosis of NF1. With these criteria, about 70% of the children can be diagnosed before the age of 1 year.1
Nevus anemicus is an uncommon birthmark, sometimes overlooked, that is characterized by pale, hypopigmented, well-defined macules and patches that do not turn red after trauma or changes in temperature. Nevus anemicus is usually localized on the torso but can be seen on the face, neck, and extremities. These lesions are present in 1%-2% of the general population. They are thought to occur because of increased sensitivity of the affected blood vessels to catecholamines, which causes permanent vasoconstriction, which leads to hypopigmentation on the area.2 These lesions are usually present at birth and have been described in patients with tuberous sclerosis, neurofibromatosis, and phakomatosis pigmentovascularis.
Recent studies of patients with neurofibromatosis and other RASopathies have noticed that nevus anemicus is present in about 8.8%-51% of the patients studied with a diagnosis NF1, compared with only 2% of the controls.3,4 The studies failed to report any cases of nevus anemicus in patients with other RASopathies associated with café au lait macules. Bulteel and colleagues recently reported two cases of non-NF1 RASopathies also associated with nevus anemicus in a patient with Legius syndrome and a patient with Noonan syndrome with multiple lentigines.5 The nevus anemicus was reported to occur most commonly on the anterior chest and be multiple, as seen in our patient.
The authors of the published studies advocate for the introduction of nevus anemicus as part of the diagnostic criteria for NF1, especially because it can be an early finding seen in babies, which can aid in early diagnosis of NF1.
Dr. Matiz is a pediatric dermatologist at Southern California Permanente Medical Group, San Diego. She has no relevant financial disclosures. Email Dr. Matiz at pdnews@mdedge.com.
References
1. Pediatrics. 2000 Mar. doi: 10.1542/peds.105.3.608.
2. Nevus Anemicus. StatPearls [Internet] (Treasure Island, Fla.: StatPearls Publishing; 2020 Jan).
3. J Am Acad Dermatol. 2013 Nov. doi: 10.1016/j.jaad.2013.06.039.
4. Pediatr Dermatol. 2015 May-Jun. doi: 10.1111/pde.12525.
5. JAAD Case Rep. 2018 Apr 5. doi: 10.1016/j.jdcr.2017.09.037.
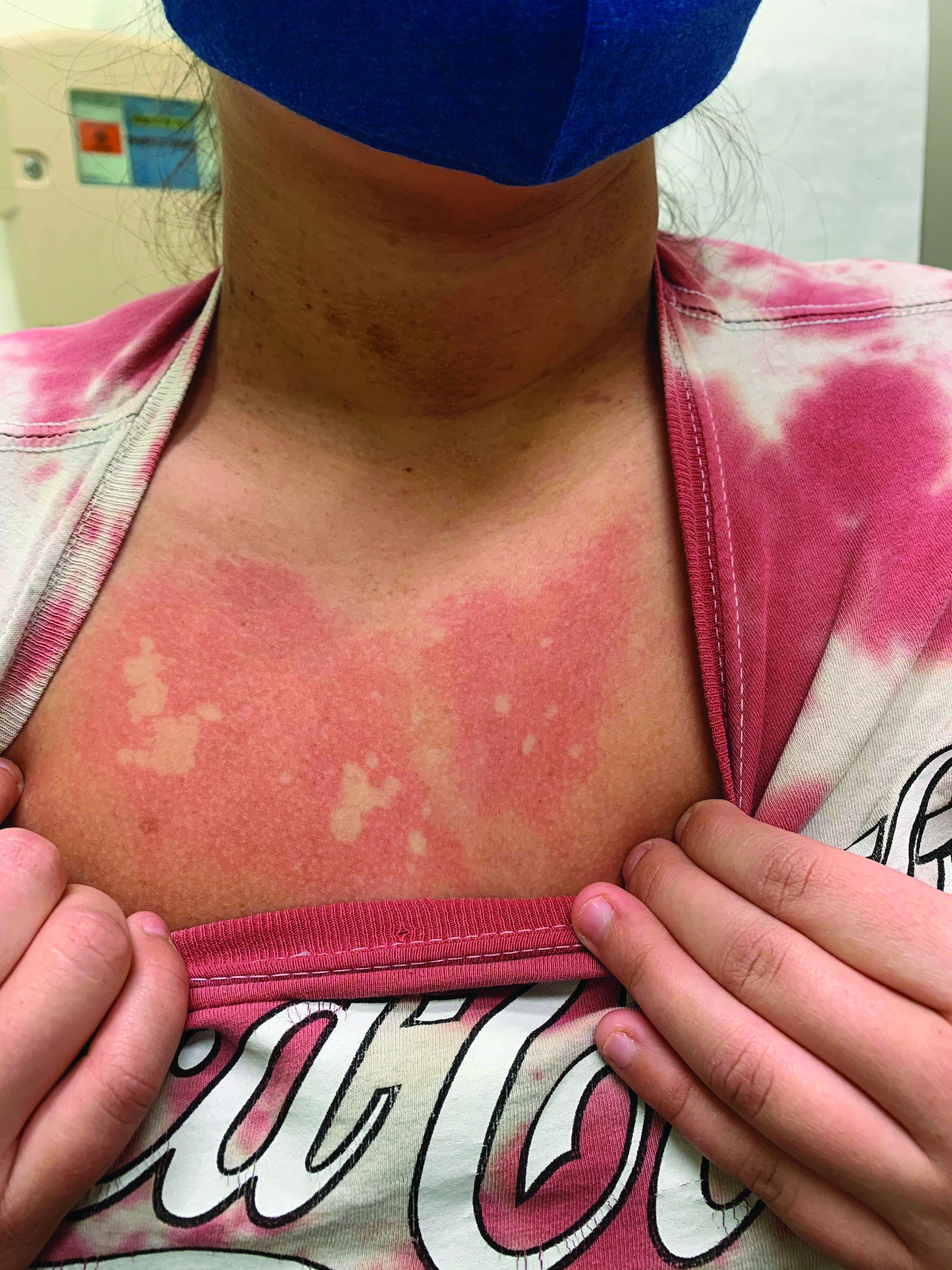
An 11-year-old female with a 3-year history of alopecia
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
Given the longstanding scarring alopecia, with negative fungal cultures and with perifollicular erythema and scaling, this diagnosis is most consistent with lichen planopilaris.
Lichen planopilaris (LPP) is considered one of the primary scarring alopecias, a group of diseases characterized by inflammation and subsequent irreversible hair loss.1 LPP specifically is believed to be caused by dysfunction of cell-mediated immunity, resulting in T lymphocytes attacking follicular hair stem cells.2 It typically presents with hair loss, pruritus, scaling, burning pain, and tenderness of the scalp when active,1,3 with exam showing perifollicular scale and erythema on the borders of the patches of alopecia.4,5 Over time, scarring of the scalp develops with loss of follicular ostia.1 Definitive diagnosis typically requires punch biopsy of the affected scalp, as such can determine the presence or absence of inflammation in affected areas of the scalp.1
What’s the treatment plan?
Given that LPP is an autoimmune inflammatory disease process, the goal of treatment is to calm down the inflammation of the scalp to prevent further progression of a patient’s hair loss. This is typically achieved with superpotent topical corticosteroids, such as clobetasol applied directly to the scalp, and/or intralesional corticosteroids, such as triamcinolone acetonide suspension injected directly to the affected scalp.3,6,7 Other treatment options include systemic agents, such as hydroxychloroquine, methotrexate, mycophenolate mofetil, pioglitazone, and doxycycline.3,6 Hair loss is not reversible as loss of follicular ostia and hair stem cells results in permanent scarring.1 Management often requires a referral to dermatology for aggressive treatment to prevent further hair loss.
What’s the differential diagnosis?
The differential diagnosis of lichen planopilaris includes other scarring alopecias, including central centrifugal cicatricial alopecia, discoid lupus erythematosus, folliculitis decalvans. While nonscarring, alopecia areata, trichotillomania, and telogen effluvium are discussed below as well.
Central centrifugal cicatricial alopecia is very rare in pediatrics, and is a type of asymptomatic scarring alopecia that begins at the vertex of the scalp, spreading centrifugally and resulting in shiny plaque development. Treatment involves reduction of hair grooming as well as topical and intralesional steroids.
Discoid lupus erythematosus presents as scaling erythematous plaques on the face and scalp that result in skin pigment changes and atrophy over time. Scalp involvement results in scarring alopecia. Treatment includes the use of high-potency topical corticosteroids, topical calcineurin inhibitors, and hydroxychloroquine.
Folliculitis decalvans is another form of scarring alopecia believed to be caused by an inflammatory response to Staphylococcus aureus in the scalp, resulting in the formation of scarring of the scalp and perifollicular pustules. Treatment is topical antibiotics and intralesional steroids.
Alopecia areata is a form of nonscarring alopecia resulting in small round patches of partially reversible hair loss characterized by the pathognomonic finding of so-called exclamation point hairs that are broader distally and taper toward the scalp on physical exam. Considered an autoimmune disorder, it varies greatly in extent and course. While focal hair loss is the hallmark of this disease, usually hair follicles are present.
Trichotillosis, also known as trichotillomania (hair pulling), results in alopecia with irregular borders and broken hairs of different lengths secondary to the urge to remove or pull one’s own hair, resulting in nonscarring alopecia. It may be associated with stress or anxiety, obsessive-compulsive disorders, or other repetitive body-altering behaviors. Treatments include reassurance and education as it can be self-limited in some, behavior modification, or systemic therapy including tricyclic antidepressants or SSRIs.
Our patient underwent scalp punch biopsy to confirm the diagnosis and was started on potent topical corticosteroids with good disease control.
Dr. Haft is a pediatric dermatology research associate in the division of pediatric and adolescent dermatology, University of California, San Diego, and Rady Children’s Hospital, San Diego. Dr. Eichenfield is the vice chair of the department of dermatology and a professor of dermatology and pediatrics at the university, and he is chief of pediatric and adolescent dermatology at the hospital. Neither of the doctors had any relevant financial disclosures. Email them at pdnews@mdedge.com.
References
1. J Am Acad Dermatol. 2005 Jul. doi: 10.1016/j.jaad.2004.06.015.
2. J Pathol. 2013 Oct. doi: 10.1002/path.4233.
3. Pediatr Dermatol. 2015 Sep-Oct. doi: 10.1111/pde.12624.
4. J Am Acad Dermatol. 2004 Jan. doi: 10.1016/j.jaad.2003.04.001.
5. J Am Acad Dermatol. 1992 Dec. doi: 10.1016/0190-9622(92)70290-v.
6. Clin Cosmet Investig Dermatol. 2018 Feb 27. doi: 10.2147/CCID.S137870.
7. Semin Cutan Med Surg. 2009 Mar. doi: 10.1016/j.sder.2008.12.006.
An 11-year-old female is seen in clinic with a 3-year history of alopecia. The patient recently immigrated to the United States from Afghanistan. Prior to immigrating, she was evaluated for "scarring alopecia" and had been treated with oral and topical steroids as well as oral and topical antifungals. When active, she had itching and tenderness. She is not actively losing any hair at this time, but she has not regrown any of her hair. The patient has no family members with alopecia. She reports some burning pain and itching of her scalp, and denies any muscle pain or weakness or sun sensitivity.
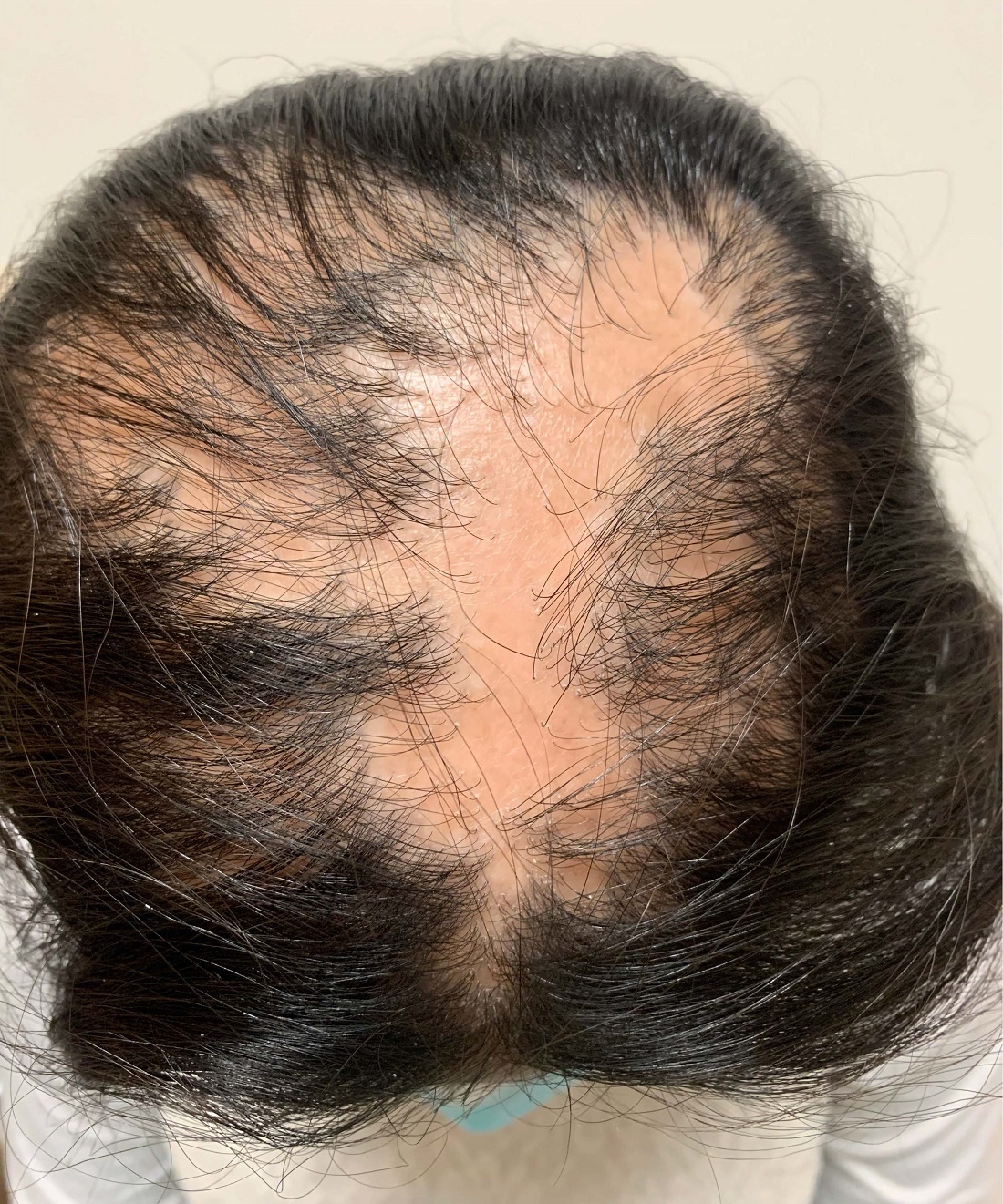
On physical exam, you see 50% loss of hair on the superior scalp with preservation of the anterior hair line. Patches of hair can be seen throughout, with segments of smooth-skinned alopecia, without pustules. There is a loss of the follicle pattern in scarred areas, and magnification or "dermoscopy" shows perifollicular erythema and scaling at the border of the affected scalp. Labs are all within normal limits. Bacterial and fungal cultures of the scalp do not grow organisms.
Sparse Hair on the Scalp
The Diagnosis: Monilethrix
Trichoscopy showed a beaded appearance of the hair shafts (Figure, A). Light microscopy demonstrated normal medullated nodes of hair coupled with internodal, thin, nonmedullated hair at regular intervals (Figure, B). Clinical and trichoscopic findings led to a diagnosis of monilethrix.
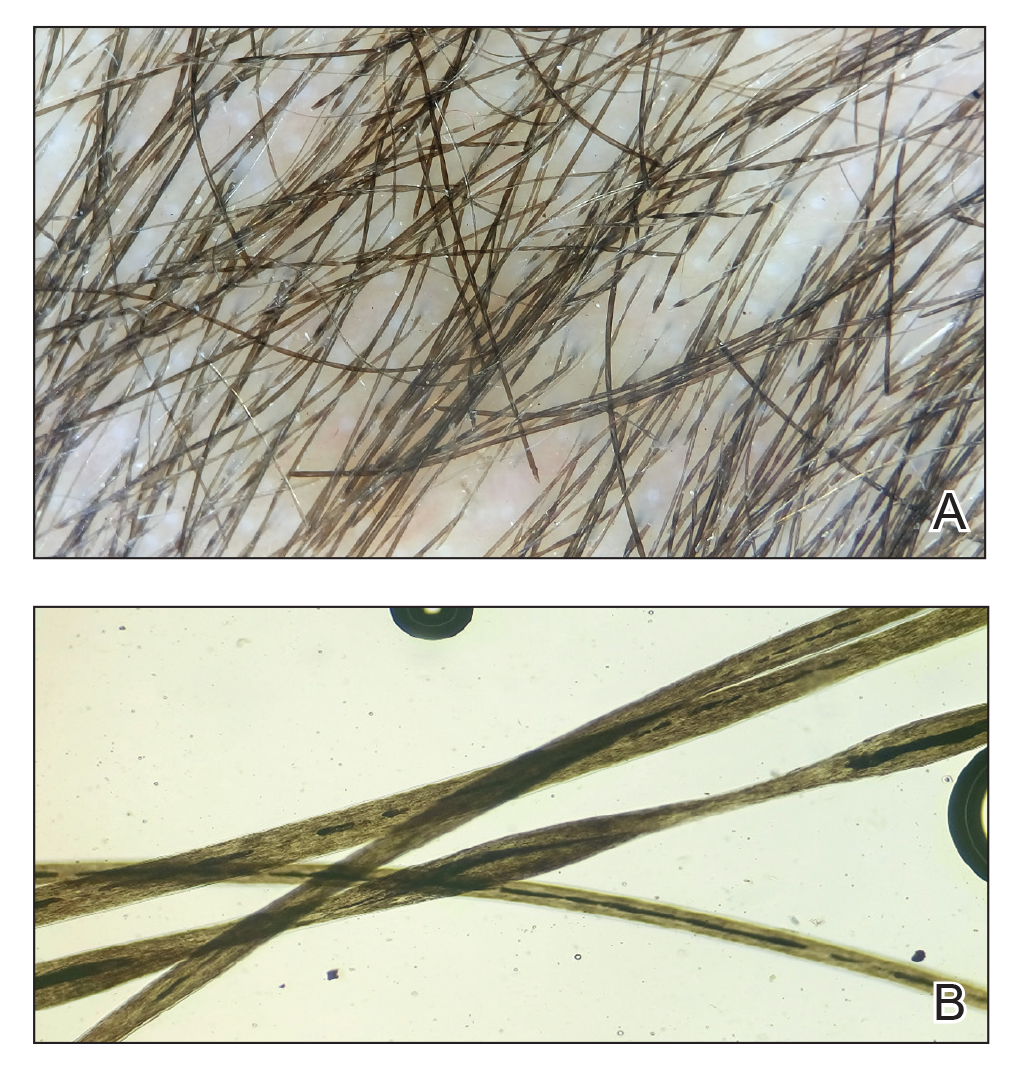
Monilethrix is a genetic hair disorder characterized by regular periodic thinning of the hair shafts, giving the strands a beaded appearance. The hair tends to break at these constricted parts, resulting in short hairs. Nodosities represent the normal hair shaft, whereas the constricted points are the site of the defect. The hair tends to be normal at birth and then becomes short, fragile, and brittle within months, leading to hypotrichosis, particularly on the occipital scalp.1 Monilethrix also may involve the eyebrows and eyelashes in addition to scalp hair. Follicular hyperkeratotic papules with perifollicular erythema frequently are noted on the occipital area. Monilethrix can be inherited in an autosomal-dominant fashion with mutations involving KRT81, KRT83, and KRT86, which code for the type II hair keratins Hb1, Hb3, and Hb6, respectively. The autosomal-recessive form is caused by mutations in the DSG4 gene, coding for the desmoglein 4 protein.2 Trichoscopy or light microscopy is essential to establish a diagnosis of monilethrix. Trichoscopy is an easy and rapid tool that is utilized to illustrate the beaded appearance of the hair shafts.3 Light microscopy shows the distinctive nodes that are medullated, with a normal hair diameter alternating with the internodes, or constrictions, that are nonmedullated and represent the sites of fracture.1 Monilethrix can improve by puberty. There is no definitive treatment; however, some patients show considerable improvement on minoxidil.4 Treatment with minoxidil was initiated in this patient; however, she was lost to follow-up.
Genetic hair disorders are rare and can be an isolated phenomenon or part of concurrent genetic syndromes. Therefore, thorough clinical examination of other ectodermal structures such as the nails and teeth is crucial as well as obtaining a detailed family history and review of systems to exclude other syndromes.2 Hypotrichosis simplex is characterized by hair loss exclusively on the scalp, sparing other ectodermal structures and with no systemic abnormalities. Ectodermal dysplasia is a heterogeneous group of disorders affecting not only the hair but also the teeth, nails, and sweat glands.2 Pili torti is another rare genetic hair disorder that is characterized by twisting of the hair fiber on its own axis. It presents clinically as sparse, depigmented, lusterless hair that is easily broken. Light microscopy demonstrates twists of hair at irregular intervals. Pili annulati is characterized by bright and dark bands when viewed with reflected light. Unlike monilethrix, there is no fragility, and the hair can grow long.5
- Mirmirani P, Huang KP, Price VH. A practical, algorithmic approach to diagnosing hair shaft disorders. Int J Dermatol. 2011;50:1-12.
- Ahmed A, Almohanna H, Griggs J, et al. Genetic hair disorders: a review. Dermatol Ther. 2019;9:421-448.
- Liu C-I, Hsu C-H. Rapid diagnosis of monilethrix using dermoscopy. Br J Dermatol. 2008;159:741-743.
- Rossi A, Iorio A, Fortuna MC, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol. 2011;24:239-242.
- Singh G, Miteva M. Prognosis and management of congenital hair shaft disorders with fragility—part I. Pediatr Dermatol. 2016;33:473-480.
The Diagnosis: Monilethrix
Trichoscopy showed a beaded appearance of the hair shafts (Figure, A). Light microscopy demonstrated normal medullated nodes of hair coupled with internodal, thin, nonmedullated hair at regular intervals (Figure, B). Clinical and trichoscopic findings led to a diagnosis of monilethrix.
Monilethrix is a genetic hair disorder characterized by regular periodic thinning of the hair shafts, giving the strands a beaded appearance. The hair tends to break at these constricted parts, resulting in short hairs. Nodosities represent the normal hair shaft, whereas the constricted points are the site of the defect. The hair tends to be normal at birth and then becomes short, fragile, and brittle within months, leading to hypotrichosis, particularly on the occipital scalp.1 Monilethrix also may involve the eyebrows and eyelashes in addition to scalp hair. Follicular hyperkeratotic papules with perifollicular erythema frequently are noted on the occipital area. Monilethrix can be inherited in an autosomal-dominant fashion with mutations involving KRT81, KRT83, and KRT86, which code for the type II hair keratins Hb1, Hb3, and Hb6, respectively. The autosomal-recessive form is caused by mutations in the DSG4 gene, coding for the desmoglein 4 protein.2 Trichoscopy or light microscopy is essential to establish a diagnosis of monilethrix. Trichoscopy is an easy and rapid tool that is utilized to illustrate the beaded appearance of the hair shafts.3 Light microscopy shows the distinctive nodes that are medullated, with a normal hair diameter alternating with the internodes, or constrictions, that are nonmedullated and represent the sites of fracture.1 Monilethrix can improve by puberty. There is no definitive treatment; however, some patients show considerable improvement on minoxidil.4 Treatment with minoxidil was initiated in this patient; however, she was lost to follow-up.
Genetic hair disorders are rare and can be an isolated phenomenon or part of concurrent genetic syndromes. Therefore, thorough clinical examination of other ectodermal structures such as the nails and teeth is crucial as well as obtaining a detailed family history and review of systems to exclude other syndromes.2 Hypotrichosis simplex is characterized by hair loss exclusively on the scalp, sparing other ectodermal structures and with no systemic abnormalities. Ectodermal dysplasia is a heterogeneous group of disorders affecting not only the hair but also the teeth, nails, and sweat glands.2 Pili torti is another rare genetic hair disorder that is characterized by twisting of the hair fiber on its own axis. It presents clinically as sparse, depigmented, lusterless hair that is easily broken. Light microscopy demonstrates twists of hair at irregular intervals. Pili annulati is characterized by bright and dark bands when viewed with reflected light. Unlike monilethrix, there is no fragility, and the hair can grow long.5
The Diagnosis: Monilethrix
Trichoscopy showed a beaded appearance of the hair shafts (Figure, A). Light microscopy demonstrated normal medullated nodes of hair coupled with internodal, thin, nonmedullated hair at regular intervals (Figure, B). Clinical and trichoscopic findings led to a diagnosis of monilethrix.
Monilethrix is a genetic hair disorder characterized by regular periodic thinning of the hair shafts, giving the strands a beaded appearance. The hair tends to break at these constricted parts, resulting in short hairs. Nodosities represent the normal hair shaft, whereas the constricted points are the site of the defect. The hair tends to be normal at birth and then becomes short, fragile, and brittle within months, leading to hypotrichosis, particularly on the occipital scalp.1 Monilethrix also may involve the eyebrows and eyelashes in addition to scalp hair. Follicular hyperkeratotic papules with perifollicular erythema frequently are noted on the occipital area. Monilethrix can be inherited in an autosomal-dominant fashion with mutations involving KRT81, KRT83, and KRT86, which code for the type II hair keratins Hb1, Hb3, and Hb6, respectively. The autosomal-recessive form is caused by mutations in the DSG4 gene, coding for the desmoglein 4 protein.2 Trichoscopy or light microscopy is essential to establish a diagnosis of monilethrix. Trichoscopy is an easy and rapid tool that is utilized to illustrate the beaded appearance of the hair shafts.3 Light microscopy shows the distinctive nodes that are medullated, with a normal hair diameter alternating with the internodes, or constrictions, that are nonmedullated and represent the sites of fracture.1 Monilethrix can improve by puberty. There is no definitive treatment; however, some patients show considerable improvement on minoxidil.4 Treatment with minoxidil was initiated in this patient; however, she was lost to follow-up.
Genetic hair disorders are rare and can be an isolated phenomenon or part of concurrent genetic syndromes. Therefore, thorough clinical examination of other ectodermal structures such as the nails and teeth is crucial as well as obtaining a detailed family history and review of systems to exclude other syndromes.2 Hypotrichosis simplex is characterized by hair loss exclusively on the scalp, sparing other ectodermal structures and with no systemic abnormalities. Ectodermal dysplasia is a heterogeneous group of disorders affecting not only the hair but also the teeth, nails, and sweat glands.2 Pili torti is another rare genetic hair disorder that is characterized by twisting of the hair fiber on its own axis. It presents clinically as sparse, depigmented, lusterless hair that is easily broken. Light microscopy demonstrates twists of hair at irregular intervals. Pili annulati is characterized by bright and dark bands when viewed with reflected light. Unlike monilethrix, there is no fragility, and the hair can grow long.5
- Mirmirani P, Huang KP, Price VH. A practical, algorithmic approach to diagnosing hair shaft disorders. Int J Dermatol. 2011;50:1-12.
- Ahmed A, Almohanna H, Griggs J, et al. Genetic hair disorders: a review. Dermatol Ther. 2019;9:421-448.
- Liu C-I, Hsu C-H. Rapid diagnosis of monilethrix using dermoscopy. Br J Dermatol. 2008;159:741-743.
- Rossi A, Iorio A, Fortuna MC, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol. 2011;24:239-242.
- Singh G, Miteva M. Prognosis and management of congenital hair shaft disorders with fragility—part I. Pediatr Dermatol. 2016;33:473-480.
- Mirmirani P, Huang KP, Price VH. A practical, algorithmic approach to diagnosing hair shaft disorders. Int J Dermatol. 2011;50:1-12.
- Ahmed A, Almohanna H, Griggs J, et al. Genetic hair disorders: a review. Dermatol Ther. 2019;9:421-448.
- Liu C-I, Hsu C-H. Rapid diagnosis of monilethrix using dermoscopy. Br J Dermatol. 2008;159:741-743.
- Rossi A, Iorio A, Fortuna MC, et al. Monilethrix treated with minoxidil. Int J Immunopathol Pharmacol. 2011;24:239-242.
- Singh G, Miteva M. Prognosis and management of congenital hair shaft disorders with fragility—part I. Pediatr Dermatol. 2016;33:473-480.
A 5-year-old girl presented to our clinic with sparse scalp hair. Her mother reported thinning of the hair and breakage that appeared shortly after birth. She also reported that the patient’s hair was dull, dry, and unable to be grown long. The patient was otherwise healthy. She was born to nonconsanguineous parents, and her family history was unremarkable. Physical examination revealed dry, brittle, and short hairs. The hair was sparser on the occipital area of the scalp, and multiple keratotic papules were noted in this area. No abnormalities were detected on the teeth or nails, and a review of systems was unremarkable. Trichoscopy and light microscopy were performed.
Irritable Baby With Weight Loss and a Periorificial and Truncal Rash
The Diagnosis: Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) was the presumptive diagnosis. Oral supplementation with zinc sulfate 3 mg/kg/d was started immediately after a zinc level was ordered. A low zinc level of 15 µg/dL (reference range, 56-134 µg/dL) eventually was obtained. The lesions began to fade in 2 days along with return of normal feeding and disposition, and the patient was discharged with continued zinc supplementation.
Acrodermatitis enteropathica is an autosomal-recessive condition resulting in severe zinc deficiency caused by a defect of dietary zinc absorption in the duodenum and jejunum.1 It occurs in 1 in 500,000 individuals with no gender or racial predilection. It can be acquired or inherited.2 Recognition of clinical symptoms is essential due to potential death if untreated. Zinc is an important trace element required for the proper functioning of all cells and plays a large role in the metabolism of protein, carbohydrates, and vitamin A. Zinc deficiency impairs immune function, leading to bacterial infections. It also is a cofactor of numerous metal enzymes such as alkaline phosphatase, RNA polymerase, and numerous digestive enzymes.3
Our laboratory analysis revealed low alkaline phosphatase and zinc levels, which led to the diagnosis of AE; unfortunately, these levels can be ambiguous.4 There are many causes of acquired zinc deficiency, including premature birth, low birth weight, zinc deficiency in maternal milk, exclusive parenteral nutrition, malabsorption syndromes such as Crohn disease and celiac disease, alcoholism, low calcium and phytate (cereal grain) diet, and kwashiorkor.5 The hereditary deficiency of zinc classically is known as AE and is caused by an autosomal-recessive mutation of the SLC39A4 gene on chromosome arm 8q24.3, which determines a congenital partial or total deficiency of the zinc transporter protein ZIP4.6
The clinical manifestations of acquired zinc deficiency and AE are similar and consist of 3 essential symptoms: periorificial dermatitis, alopecia, and diarrhea. Unfortunately, this clinical triad is complete in only 20% of patients with AE.3 For example, our patient was too young for an alopecia determination. The disease typically presents with eczematous papules and sometimes vesiculobullous or pustular lesions located around perioral and acral areas (Figure 1) as well as the anogenital region (Figures 2 and 3). The severity of the skin lesions is variable.7 Our patient also presented with eczematous truncal papules on the chest (Figure 4). Acrodermatitis enteropathica usually presents during childhood after weaning. Along with the aforementioned skin findings, other symptoms in infancy can include diarrhea, mood changes, and anorexia. In school-aged children and toddlers, zinc deficiency is characterized by growth retardation, alopecia, weight loss, and recurrent infections.
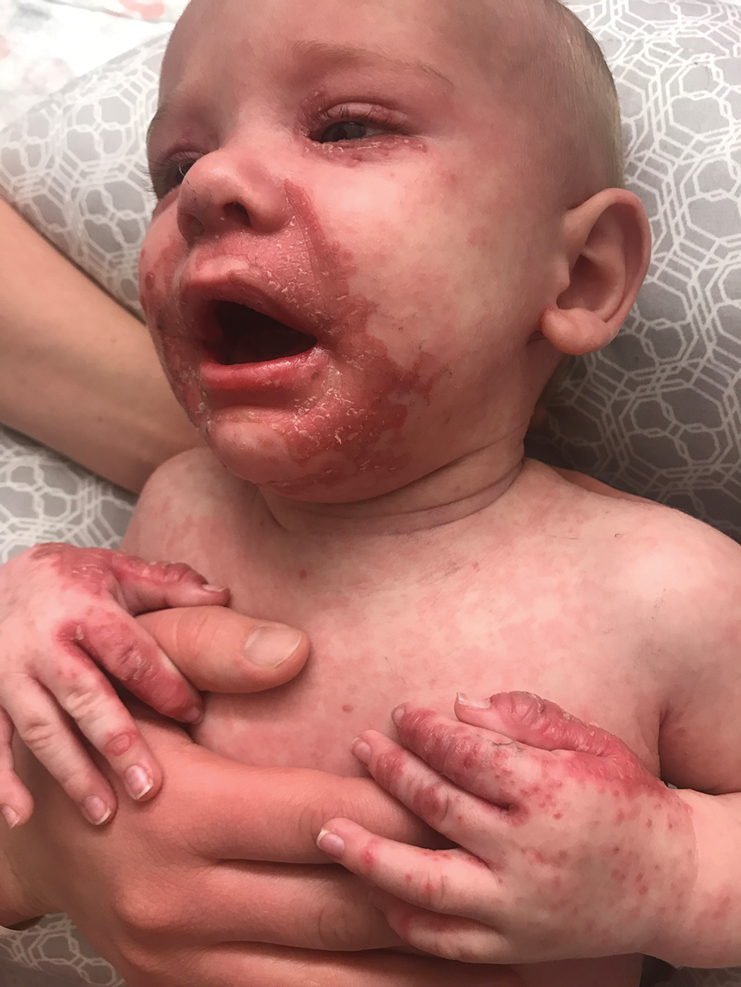

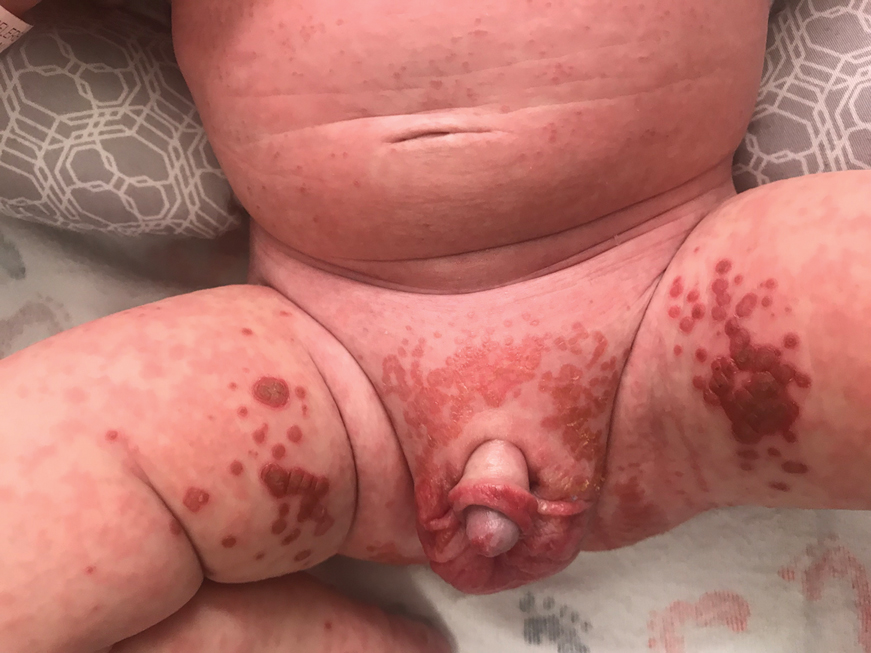
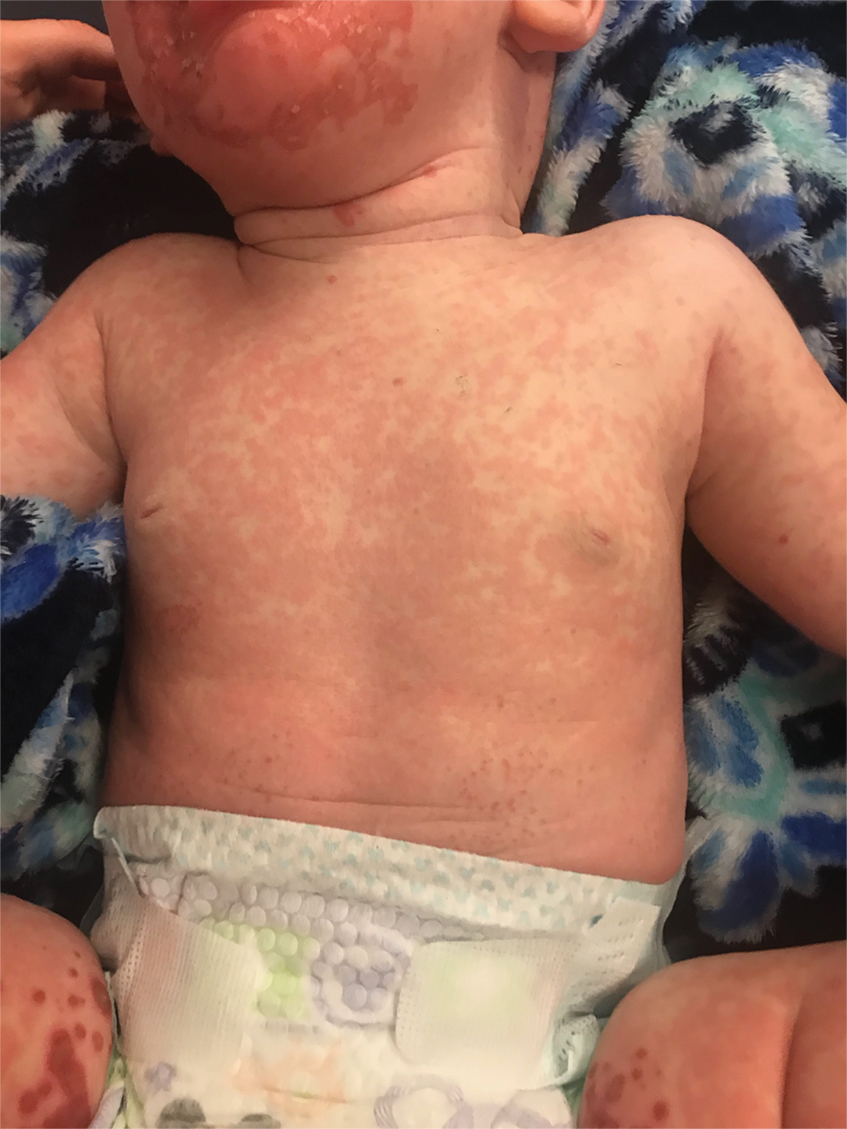
In the differential diagnosis, the clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and central nervous system (eg, seizures, ataxia, deafness).8 Cystic fibrosis presentation depends on the multiorgan involvement, but neonates often present with failure to thrive.9 Essential fatty acid deficiency presents clinically as dermatitis, alopecia, and thrombocytopenia, but a complete blood cell count with platelets was within reference range in our patient.10 Langerhans cell histiocytosis presents with perineal and postauricular lesions, but the skin biopsy did not confirm this diagnosis in our patient.11 Histopathologic examination of the buttock biopsy in our patient revealed nonspecific epidermal hyperplasia with acanthosis as well as clustered necrotic keratinocytes with vacuolization and parakeratosis.
Most clinicians who suspect AE treat with a therapeutic supplementation of zinc sulfate 3 mg/kg/d while awaiting laboratory results. Acrodermatitis enteropathica is a rare condition, and early recognition of skin findings is important because misdiagnosis can lead to infections, malnutrition, and possibly death.
- Sehgal VN, Jain S. Acrodermatitis enteropathica. Clin Dermatol. 2000;18:745-748.
- Van Wouwe JP. Clinical and laboratory assessment of zinc deficiency in Dutch children: a review. Biol Trace Elem Res. 1995;49:211-225.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr. 1989;149:2-8.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitisenteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kury S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239-240.
- Nistor N, Ciontu L, Frasinariu OE, et al. Acrodermatitis enteropathica: a case report. Medicine. 2016;95:E3553.
- Gratias T. Biotin deficiency. Medscape website. https://emedicine.medscape.com/article/984803-overview. Updated October 22, 2018. Accessed October 15, 2020.
- Sharma G. Cystic fibrosis. Medscape website. https://emedicine.medscape.com/article/1001602-overview. Updated September 28, 2018. Accessed October 15, 2020.
- Morley JE. Essential fatty acid deficiency. Merck Manual website. https://www.merckmanuals.com/professional/nutritional-disorders/undernutrition/essential-fatty-acid-deficiency. Updated January 2020. Accessed October 15, 2020.
- Shea CR. Langerhans cell histiocytosis. Medscape website. https://emedicine.medscape.com/article/1100579-overview. Updated June 12, 2020. Accessed October 15, 2020.
The Diagnosis: Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) was the presumptive diagnosis. Oral supplementation with zinc sulfate 3 mg/kg/d was started immediately after a zinc level was ordered. A low zinc level of 15 µg/dL (reference range, 56-134 µg/dL) eventually was obtained. The lesions began to fade in 2 days along with return of normal feeding and disposition, and the patient was discharged with continued zinc supplementation.
Acrodermatitis enteropathica is an autosomal-recessive condition resulting in severe zinc deficiency caused by a defect of dietary zinc absorption in the duodenum and jejunum.1 It occurs in 1 in 500,000 individuals with no gender or racial predilection. It can be acquired or inherited.2 Recognition of clinical symptoms is essential due to potential death if untreated. Zinc is an important trace element required for the proper functioning of all cells and plays a large role in the metabolism of protein, carbohydrates, and vitamin A. Zinc deficiency impairs immune function, leading to bacterial infections. It also is a cofactor of numerous metal enzymes such as alkaline phosphatase, RNA polymerase, and numerous digestive enzymes.3
Our laboratory analysis revealed low alkaline phosphatase and zinc levels, which led to the diagnosis of AE; unfortunately, these levels can be ambiguous.4 There are many causes of acquired zinc deficiency, including premature birth, low birth weight, zinc deficiency in maternal milk, exclusive parenteral nutrition, malabsorption syndromes such as Crohn disease and celiac disease, alcoholism, low calcium and phytate (cereal grain) diet, and kwashiorkor.5 The hereditary deficiency of zinc classically is known as AE and is caused by an autosomal-recessive mutation of the SLC39A4 gene on chromosome arm 8q24.3, which determines a congenital partial or total deficiency of the zinc transporter protein ZIP4.6
The clinical manifestations of acquired zinc deficiency and AE are similar and consist of 3 essential symptoms: periorificial dermatitis, alopecia, and diarrhea. Unfortunately, this clinical triad is complete in only 20% of patients with AE.3 For example, our patient was too young for an alopecia determination. The disease typically presents with eczematous papules and sometimes vesiculobullous or pustular lesions located around perioral and acral areas (Figure 1) as well as the anogenital region (Figures 2 and 3). The severity of the skin lesions is variable.7 Our patient also presented with eczematous truncal papules on the chest (Figure 4). Acrodermatitis enteropathica usually presents during childhood after weaning. Along with the aforementioned skin findings, other symptoms in infancy can include diarrhea, mood changes, and anorexia. In school-aged children and toddlers, zinc deficiency is characterized by growth retardation, alopecia, weight loss, and recurrent infections.
In the differential diagnosis, the clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and central nervous system (eg, seizures, ataxia, deafness).8 Cystic fibrosis presentation depends on the multiorgan involvement, but neonates often present with failure to thrive.9 Essential fatty acid deficiency presents clinically as dermatitis, alopecia, and thrombocytopenia, but a complete blood cell count with platelets was within reference range in our patient.10 Langerhans cell histiocytosis presents with perineal and postauricular lesions, but the skin biopsy did not confirm this diagnosis in our patient.11 Histopathologic examination of the buttock biopsy in our patient revealed nonspecific epidermal hyperplasia with acanthosis as well as clustered necrotic keratinocytes with vacuolization and parakeratosis.
Most clinicians who suspect AE treat with a therapeutic supplementation of zinc sulfate 3 mg/kg/d while awaiting laboratory results. Acrodermatitis enteropathica is a rare condition, and early recognition of skin findings is important because misdiagnosis can lead to infections, malnutrition, and possibly death.
The Diagnosis: Acrodermatitis Enteropathica
Acrodermatitis enteropathica (AE) was the presumptive diagnosis. Oral supplementation with zinc sulfate 3 mg/kg/d was started immediately after a zinc level was ordered. A low zinc level of 15 µg/dL (reference range, 56-134 µg/dL) eventually was obtained. The lesions began to fade in 2 days along with return of normal feeding and disposition, and the patient was discharged with continued zinc supplementation.
Acrodermatitis enteropathica is an autosomal-recessive condition resulting in severe zinc deficiency caused by a defect of dietary zinc absorption in the duodenum and jejunum.1 It occurs in 1 in 500,000 individuals with no gender or racial predilection. It can be acquired or inherited.2 Recognition of clinical symptoms is essential due to potential death if untreated. Zinc is an important trace element required for the proper functioning of all cells and plays a large role in the metabolism of protein, carbohydrates, and vitamin A. Zinc deficiency impairs immune function, leading to bacterial infections. It also is a cofactor of numerous metal enzymes such as alkaline phosphatase, RNA polymerase, and numerous digestive enzymes.3
Our laboratory analysis revealed low alkaline phosphatase and zinc levels, which led to the diagnosis of AE; unfortunately, these levels can be ambiguous.4 There are many causes of acquired zinc deficiency, including premature birth, low birth weight, zinc deficiency in maternal milk, exclusive parenteral nutrition, malabsorption syndromes such as Crohn disease and celiac disease, alcoholism, low calcium and phytate (cereal grain) diet, and kwashiorkor.5 The hereditary deficiency of zinc classically is known as AE and is caused by an autosomal-recessive mutation of the SLC39A4 gene on chromosome arm 8q24.3, which determines a congenital partial or total deficiency of the zinc transporter protein ZIP4.6
The clinical manifestations of acquired zinc deficiency and AE are similar and consist of 3 essential symptoms: periorificial dermatitis, alopecia, and diarrhea. Unfortunately, this clinical triad is complete in only 20% of patients with AE.3 For example, our patient was too young for an alopecia determination. The disease typically presents with eczematous papules and sometimes vesiculobullous or pustular lesions located around perioral and acral areas (Figure 1) as well as the anogenital region (Figures 2 and 3). The severity of the skin lesions is variable.7 Our patient also presented with eczematous truncal papules on the chest (Figure 4). Acrodermatitis enteropathica usually presents during childhood after weaning. Along with the aforementioned skin findings, other symptoms in infancy can include diarrhea, mood changes, and anorexia. In school-aged children and toddlers, zinc deficiency is characterized by growth retardation, alopecia, weight loss, and recurrent infections.
In the differential diagnosis, the clinical presentation of biotin deficiency involves abnormalities of the hair, skin, nails, and central nervous system (eg, seizures, ataxia, deafness).8 Cystic fibrosis presentation depends on the multiorgan involvement, but neonates often present with failure to thrive.9 Essential fatty acid deficiency presents clinically as dermatitis, alopecia, and thrombocytopenia, but a complete blood cell count with platelets was within reference range in our patient.10 Langerhans cell histiocytosis presents with perineal and postauricular lesions, but the skin biopsy did not confirm this diagnosis in our patient.11 Histopathologic examination of the buttock biopsy in our patient revealed nonspecific epidermal hyperplasia with acanthosis as well as clustered necrotic keratinocytes with vacuolization and parakeratosis.
Most clinicians who suspect AE treat with a therapeutic supplementation of zinc sulfate 3 mg/kg/d while awaiting laboratory results. Acrodermatitis enteropathica is a rare condition, and early recognition of skin findings is important because misdiagnosis can lead to infections, malnutrition, and possibly death.
- Sehgal VN, Jain S. Acrodermatitis enteropathica. Clin Dermatol. 2000;18:745-748.
- Van Wouwe JP. Clinical and laboratory assessment of zinc deficiency in Dutch children: a review. Biol Trace Elem Res. 1995;49:211-225.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr. 1989;149:2-8.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitisenteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kury S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239-240.
- Nistor N, Ciontu L, Frasinariu OE, et al. Acrodermatitis enteropathica: a case report. Medicine. 2016;95:E3553.
- Gratias T. Biotin deficiency. Medscape website. https://emedicine.medscape.com/article/984803-overview. Updated October 22, 2018. Accessed October 15, 2020.
- Sharma G. Cystic fibrosis. Medscape website. https://emedicine.medscape.com/article/1001602-overview. Updated September 28, 2018. Accessed October 15, 2020.
- Morley JE. Essential fatty acid deficiency. Merck Manual website. https://www.merckmanuals.com/professional/nutritional-disorders/undernutrition/essential-fatty-acid-deficiency. Updated January 2020. Accessed October 15, 2020.
- Shea CR. Langerhans cell histiocytosis. Medscape website. https://emedicine.medscape.com/article/1100579-overview. Updated June 12, 2020. Accessed October 15, 2020.
- Sehgal VN, Jain S. Acrodermatitis enteropathica. Clin Dermatol. 2000;18:745-748.
- Van Wouwe JP. Clinical and laboratory assessment of zinc deficiency in Dutch children: a review. Biol Trace Elem Res. 1995;49:211-225.
- Maverakis E, Fung MA, Lynch PJ, et al. Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol. 2007;56:116-124.
- Van Wouwe JP. Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr. 1989;149:2-8.
- Perafán-Riveros C, França LF, Alves AC, et al. Acrodermatitisenteropathica: case report and review of the literature. Pediatr Dermatol. 2002;19:426-431.
- Kury S, Dréno B, Bézieau S, et al. Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet. 2002;31:239-240.
- Nistor N, Ciontu L, Frasinariu OE, et al. Acrodermatitis enteropathica: a case report. Medicine. 2016;95:E3553.
- Gratias T. Biotin deficiency. Medscape website. https://emedicine.medscape.com/article/984803-overview. Updated October 22, 2018. Accessed October 15, 2020.
- Sharma G. Cystic fibrosis. Medscape website. https://emedicine.medscape.com/article/1001602-overview. Updated September 28, 2018. Accessed October 15, 2020.
- Morley JE. Essential fatty acid deficiency. Merck Manual website. https://www.merckmanuals.com/professional/nutritional-disorders/undernutrition/essential-fatty-acid-deficiency. Updated January 2020. Accessed October 15, 2020.
- Shea CR. Langerhans cell histiocytosis. Medscape website. https://emedicine.medscape.com/article/1100579-overview. Updated June 12, 2020. Accessed October 15, 2020.
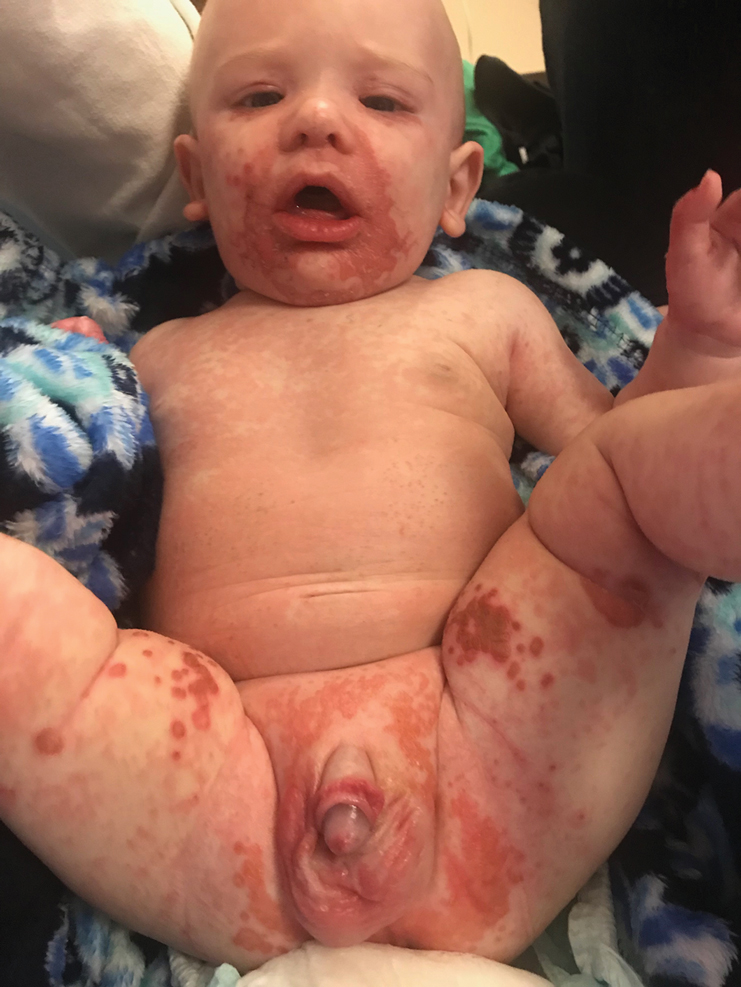
A 4-month-old infant boy presented to the pediatric hospital unit with a rash, fever, and failure to thrive. Prior to admission, the patient was treated for impetigo by a community dermatologist. After not responding to treatment, he was admitted and given intravenous acyclovir for 1 day by the pediatric hospitalist, and the dermatology service was consulted. The parents reported the patient had diarrhea for 1 month and a worsening rash over the last 2 weeks. The mother was breastfeeding. Physical examination revealed a fever (temperature, 38.9°C [102°F]) and an irritable infant whose growth curve had fallen from the 50th to 15th percentile since the 2-month well-baby examination. He had a fine, red, papular truncal rash with confluent plaques in a periorificial distribution that spared the inguinal skin folds, with some vesicles in a herpetiform presentation on the thighs as well as inflammation on the feet and hands. A complete blood cell count was within reference range, but the alkaline phosphatase level was low at 53 U/L (reference range, 72–307 U/L). A herpes simplex virus test was negative. A human immunodeficiency virus test and skin biopsy were performed.