User login
FDA approves first targeted drug for bile duct cancer
The U.S. Food and Drug Administration (FDA) has granted accelerated approval of a new targeted therapy for use in some patients with cholangiocarcinoma, a rare cancer of the bile ducts.
The product is pemigatinib (Pemazyre, Incyte), an oral kinase inhibitor.
It was approved specifically for use in patients with advanced cholangiocarcinoma who have received prior treatment and who have tumors that have a fusion or other rearrangement of the fibroblast growth factor receptor 2 (FGFR2) gene.
These FGFR2 genetic abnormalities are found in about 9% to 14% of patients with cholangiocarcinoma, notes the FDA.
At diagnosis, a majority of patients with cholangiocarcinoma have advanced disease, meaning that the disease is no longer treatable with surgery, the agency also notes. Until now, a combination of chemotherapy drugs has been the standard initial treatment.
Now the subgroup of patients with FGFR2 tumors have the option of a targeted therapy.
“Although cholangiocarcinoma is considered a rare disease, it has been on the rise over the past three decades,” commented Ghassan Abou-Alfa, MD, of Memorial Sloan Kettering Cancer Center, New York City, in a press release. “It is encouraging to have a new targeted treatment option for patients who historically have had limited options after first-line chemotherapy or surgery, in which relapse rates remain high.”
The accelerated approval was based on the overall response rate (ORR) and duration of response in an open-label clinical trial that involved 107 patients (the FIGHT-202 study).
As a condition of the accelerated approval, the manufacturer will complete and submit the results of a randomized trial demonstrating an improvement in progression-free survival or overall survival, the FDA noted.
Results from open-label clinical trial
The FIGHT-202 study enrolled 107 patients with locally advanced or metastatic cholangiocarcinoma who had received prior treatment and who had tumors with an FGFR2 fusion or rearrangement.
All patients received pemigatinib once a day for 14 days, followed by 7 days off, in 21-day cycles until the disease progressed or the patient experienced an unreasonable level of side effects. Patients underwent scanning every 8 weeks to assess ORR.
The ORR was 36% (38 of 107 patients), which included 2.8% of patients with a complete response and 33% with partial response.
Among the 38 patients who had a response, 24 patients (63%) had a response that lasted 6 months or longer, and 7 patients (18%) had a response that lasted 12 months or longer.
“With pemigatinib, we considered the observed efficacy results to be clinically meaningful and the overall risk to benefit assessment for patients with tumors harboring FGFR2 gene fusions and other rearrangements to be favorable, particularly when we considered that these patients have no other good options following first-line treatment with chemotherapy,” commented Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The most common adverse reactions, which occurred in 20% or more of patients who received pemigatinib, were electrolyte disorders (hyperphosphatemia and hypophosphatemia), alopecia, diarrhea, nail toxicity, fatigue, dysgeusia, nausea, constipation, stomatitis, dry eye, dry mouth, decreased appetite, vomiting, joint pain, abdominal pain, back pain, and dry skin. Ocular toxicity was seen rarely, the agency notes.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration (FDA) has granted accelerated approval of a new targeted therapy for use in some patients with cholangiocarcinoma, a rare cancer of the bile ducts.
The product is pemigatinib (Pemazyre, Incyte), an oral kinase inhibitor.
It was approved specifically for use in patients with advanced cholangiocarcinoma who have received prior treatment and who have tumors that have a fusion or other rearrangement of the fibroblast growth factor receptor 2 (FGFR2) gene.
These FGFR2 genetic abnormalities are found in about 9% to 14% of patients with cholangiocarcinoma, notes the FDA.
At diagnosis, a majority of patients with cholangiocarcinoma have advanced disease, meaning that the disease is no longer treatable with surgery, the agency also notes. Until now, a combination of chemotherapy drugs has been the standard initial treatment.
Now the subgroup of patients with FGFR2 tumors have the option of a targeted therapy.
“Although cholangiocarcinoma is considered a rare disease, it has been on the rise over the past three decades,” commented Ghassan Abou-Alfa, MD, of Memorial Sloan Kettering Cancer Center, New York City, in a press release. “It is encouraging to have a new targeted treatment option for patients who historically have had limited options after first-line chemotherapy or surgery, in which relapse rates remain high.”
The accelerated approval was based on the overall response rate (ORR) and duration of response in an open-label clinical trial that involved 107 patients (the FIGHT-202 study).
As a condition of the accelerated approval, the manufacturer will complete and submit the results of a randomized trial demonstrating an improvement in progression-free survival or overall survival, the FDA noted.
Results from open-label clinical trial
The FIGHT-202 study enrolled 107 patients with locally advanced or metastatic cholangiocarcinoma who had received prior treatment and who had tumors with an FGFR2 fusion or rearrangement.
All patients received pemigatinib once a day for 14 days, followed by 7 days off, in 21-day cycles until the disease progressed or the patient experienced an unreasonable level of side effects. Patients underwent scanning every 8 weeks to assess ORR.
The ORR was 36% (38 of 107 patients), which included 2.8% of patients with a complete response and 33% with partial response.
Among the 38 patients who had a response, 24 patients (63%) had a response that lasted 6 months or longer, and 7 patients (18%) had a response that lasted 12 months or longer.
“With pemigatinib, we considered the observed efficacy results to be clinically meaningful and the overall risk to benefit assessment for patients with tumors harboring FGFR2 gene fusions and other rearrangements to be favorable, particularly when we considered that these patients have no other good options following first-line treatment with chemotherapy,” commented Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The most common adverse reactions, which occurred in 20% or more of patients who received pemigatinib, were electrolyte disorders (hyperphosphatemia and hypophosphatemia), alopecia, diarrhea, nail toxicity, fatigue, dysgeusia, nausea, constipation, stomatitis, dry eye, dry mouth, decreased appetite, vomiting, joint pain, abdominal pain, back pain, and dry skin. Ocular toxicity was seen rarely, the agency notes.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration (FDA) has granted accelerated approval of a new targeted therapy for use in some patients with cholangiocarcinoma, a rare cancer of the bile ducts.
The product is pemigatinib (Pemazyre, Incyte), an oral kinase inhibitor.
It was approved specifically for use in patients with advanced cholangiocarcinoma who have received prior treatment and who have tumors that have a fusion or other rearrangement of the fibroblast growth factor receptor 2 (FGFR2) gene.
These FGFR2 genetic abnormalities are found in about 9% to 14% of patients with cholangiocarcinoma, notes the FDA.
At diagnosis, a majority of patients with cholangiocarcinoma have advanced disease, meaning that the disease is no longer treatable with surgery, the agency also notes. Until now, a combination of chemotherapy drugs has been the standard initial treatment.
Now the subgroup of patients with FGFR2 tumors have the option of a targeted therapy.
“Although cholangiocarcinoma is considered a rare disease, it has been on the rise over the past three decades,” commented Ghassan Abou-Alfa, MD, of Memorial Sloan Kettering Cancer Center, New York City, in a press release. “It is encouraging to have a new targeted treatment option for patients who historically have had limited options after first-line chemotherapy or surgery, in which relapse rates remain high.”
The accelerated approval was based on the overall response rate (ORR) and duration of response in an open-label clinical trial that involved 107 patients (the FIGHT-202 study).
As a condition of the accelerated approval, the manufacturer will complete and submit the results of a randomized trial demonstrating an improvement in progression-free survival or overall survival, the FDA noted.
Results from open-label clinical trial
The FIGHT-202 study enrolled 107 patients with locally advanced or metastatic cholangiocarcinoma who had received prior treatment and who had tumors with an FGFR2 fusion or rearrangement.
All patients received pemigatinib once a day for 14 days, followed by 7 days off, in 21-day cycles until the disease progressed or the patient experienced an unreasonable level of side effects. Patients underwent scanning every 8 weeks to assess ORR.
The ORR was 36% (38 of 107 patients), which included 2.8% of patients with a complete response and 33% with partial response.
Among the 38 patients who had a response, 24 patients (63%) had a response that lasted 6 months or longer, and 7 patients (18%) had a response that lasted 12 months or longer.
“With pemigatinib, we considered the observed efficacy results to be clinically meaningful and the overall risk to benefit assessment for patients with tumors harboring FGFR2 gene fusions and other rearrangements to be favorable, particularly when we considered that these patients have no other good options following first-line treatment with chemotherapy,” commented Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence and acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research.
The most common adverse reactions, which occurred in 20% or more of patients who received pemigatinib, were electrolyte disorders (hyperphosphatemia and hypophosphatemia), alopecia, diarrhea, nail toxicity, fatigue, dysgeusia, nausea, constipation, stomatitis, dry eye, dry mouth, decreased appetite, vomiting, joint pain, abdominal pain, back pain, and dry skin. Ocular toxicity was seen rarely, the agency notes.
This article first appeared on Medscape.com.
FDA approves first new breast cancer drug with international group
The U.S. Food and Drug Administration has approved the oral therapy tucatinib (Tukysa, Seattle Genetics) for the treatment of advanced HER2-positive breast cancer. This is the first new drug approved under Project Orbis, an international collaboration.
Tucatinib, which is a small-molecule tyrosine kinase inhibitor, is approved in combination with trastuzumab and capecitabine to treat patients who have received one or more prior treatments for advanced disease.
The FDA collaborated with the regulatory authorities of Australia, Canada, Singapore, and Switzerland on this review. However, only the FDA has approved tucatinib; the application is still under review at the other agencies.
While working with Project Orbis in 2019, the FDA granted an accelerated, conditional approval to a drug combination that included previously approved agents.
“The FDA’s Project Orbis provides a framework for concurrent submission and review of oncology drug applications among the FDA’s international collaborators,” said Richard Pazdur, MD, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in a statement.
Collaboration among regulators may allow patients with cancer to receive earlier access to products in other countries where there may be significant delays in regulatory submissions, according to the FDA.
The new drug is a “valuable addition” to the roster of treatments for advanced HER2-positive breast cancer, said study investigator Eric Winer, MD, Dana-Farber Cancer Institute, Boston, Massachusetts, in a company press statement.
“With highly significant and clinically important results for overall and progression-free survival, the addition of [tucatinib] to trastuzumab and capecitabine has the potential to become a standard of care for people with HER2-positive metastatic breast cancer after having received one or more previous anti-HER2 therapies in the metastatic setting,” he said.
The new approval is based on safety and efficacy results from the phase 2 HER2CLIMB trial that enrolled 612 patients with HER2-positive unresectable locally advanced or metastatic breast cancer who had previously received, either separately or in combination, trastuzumab, pertuzumab, and ado-trastuzumab emtansine.
Nearly half (48%) of patients in the study had brain metastases at the start of the trial. The primary outcome measure was progression-free survival (PFS). All patients received trastuzumab and capecitabine and were randomly assigned to either tucatinib or placebo.
Median PFS in the tucatinib patient group was 7.8 months, compared with 5.6 months in the placebo group. The PFS results in the subgroup of patients with brain metastases were nearly the same.
Median overall survival in the tucatinib patient group was 21.9 months versus 17.4 months in the placebo group.
The new drug is a rare success in the treatment of breast cancer brain metastases, said Jawad Fares, MD, of Northwestern University, Chicago, Illinois, who spoke to Medscape Medical News when the phase 3 trial data were first presented at the 2019 San Antonio Breast Cancer Symposium.
“Outcomes in the field have been pretty dismal,” summarized Fares, who was not involved in the study.
The results of the HER2CLIMB study, which was funded by Seattle Genetics, were published in the New England Journal of Medicine last year.
According to the FDA, common side effects with tucatinib were diarrhea, palmar-plantar erythrodysesthesia syndrome, nausea, fatigue, hepatotoxicity, vomiting, stomatitis, decreased appetite, abdominal pain, headache, anemia, and rash.
Tucatinib can cause serious side effects, including diarrhea associated with dehydration, acute kidney injury, and death. Health care professionals should start antidiarrheals as clinically indicated if diarrhea occurs and should interrupt treatment or reduce the dosage. Tucatinib can also cause severe hepatotoxicity; patients should be monitored with liver tests.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration has approved the oral therapy tucatinib (Tukysa, Seattle Genetics) for the treatment of advanced HER2-positive breast cancer. This is the first new drug approved under Project Orbis, an international collaboration.
Tucatinib, which is a small-molecule tyrosine kinase inhibitor, is approved in combination with trastuzumab and capecitabine to treat patients who have received one or more prior treatments for advanced disease.
The FDA collaborated with the regulatory authorities of Australia, Canada, Singapore, and Switzerland on this review. However, only the FDA has approved tucatinib; the application is still under review at the other agencies.
While working with Project Orbis in 2019, the FDA granted an accelerated, conditional approval to a drug combination that included previously approved agents.
“The FDA’s Project Orbis provides a framework for concurrent submission and review of oncology drug applications among the FDA’s international collaborators,” said Richard Pazdur, MD, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in a statement.
Collaboration among regulators may allow patients with cancer to receive earlier access to products in other countries where there may be significant delays in regulatory submissions, according to the FDA.
The new drug is a “valuable addition” to the roster of treatments for advanced HER2-positive breast cancer, said study investigator Eric Winer, MD, Dana-Farber Cancer Institute, Boston, Massachusetts, in a company press statement.
“With highly significant and clinically important results for overall and progression-free survival, the addition of [tucatinib] to trastuzumab and capecitabine has the potential to become a standard of care for people with HER2-positive metastatic breast cancer after having received one or more previous anti-HER2 therapies in the metastatic setting,” he said.
The new approval is based on safety and efficacy results from the phase 2 HER2CLIMB trial that enrolled 612 patients with HER2-positive unresectable locally advanced or metastatic breast cancer who had previously received, either separately or in combination, trastuzumab, pertuzumab, and ado-trastuzumab emtansine.
Nearly half (48%) of patients in the study had brain metastases at the start of the trial. The primary outcome measure was progression-free survival (PFS). All patients received trastuzumab and capecitabine and were randomly assigned to either tucatinib or placebo.
Median PFS in the tucatinib patient group was 7.8 months, compared with 5.6 months in the placebo group. The PFS results in the subgroup of patients with brain metastases were nearly the same.
Median overall survival in the tucatinib patient group was 21.9 months versus 17.4 months in the placebo group.
The new drug is a rare success in the treatment of breast cancer brain metastases, said Jawad Fares, MD, of Northwestern University, Chicago, Illinois, who spoke to Medscape Medical News when the phase 3 trial data were first presented at the 2019 San Antonio Breast Cancer Symposium.
“Outcomes in the field have been pretty dismal,” summarized Fares, who was not involved in the study.
The results of the HER2CLIMB study, which was funded by Seattle Genetics, were published in the New England Journal of Medicine last year.
According to the FDA, common side effects with tucatinib were diarrhea, palmar-plantar erythrodysesthesia syndrome, nausea, fatigue, hepatotoxicity, vomiting, stomatitis, decreased appetite, abdominal pain, headache, anemia, and rash.
Tucatinib can cause serious side effects, including diarrhea associated with dehydration, acute kidney injury, and death. Health care professionals should start antidiarrheals as clinically indicated if diarrhea occurs and should interrupt treatment or reduce the dosage. Tucatinib can also cause severe hepatotoxicity; patients should be monitored with liver tests.
This article first appeared on Medscape.com.
The U.S. Food and Drug Administration has approved the oral therapy tucatinib (Tukysa, Seattle Genetics) for the treatment of advanced HER2-positive breast cancer. This is the first new drug approved under Project Orbis, an international collaboration.
Tucatinib, which is a small-molecule tyrosine kinase inhibitor, is approved in combination with trastuzumab and capecitabine to treat patients who have received one or more prior treatments for advanced disease.
The FDA collaborated with the regulatory authorities of Australia, Canada, Singapore, and Switzerland on this review. However, only the FDA has approved tucatinib; the application is still under review at the other agencies.
While working with Project Orbis in 2019, the FDA granted an accelerated, conditional approval to a drug combination that included previously approved agents.
“The FDA’s Project Orbis provides a framework for concurrent submission and review of oncology drug applications among the FDA’s international collaborators,” said Richard Pazdur, MD, acting director of the Office of Oncologic Diseases in the FDA’s Center for Drug Evaluation and Research, in a statement.
Collaboration among regulators may allow patients with cancer to receive earlier access to products in other countries where there may be significant delays in regulatory submissions, according to the FDA.
The new drug is a “valuable addition” to the roster of treatments for advanced HER2-positive breast cancer, said study investigator Eric Winer, MD, Dana-Farber Cancer Institute, Boston, Massachusetts, in a company press statement.
“With highly significant and clinically important results for overall and progression-free survival, the addition of [tucatinib] to trastuzumab and capecitabine has the potential to become a standard of care for people with HER2-positive metastatic breast cancer after having received one or more previous anti-HER2 therapies in the metastatic setting,” he said.
The new approval is based on safety and efficacy results from the phase 2 HER2CLIMB trial that enrolled 612 patients with HER2-positive unresectable locally advanced or metastatic breast cancer who had previously received, either separately or in combination, trastuzumab, pertuzumab, and ado-trastuzumab emtansine.
Nearly half (48%) of patients in the study had brain metastases at the start of the trial. The primary outcome measure was progression-free survival (PFS). All patients received trastuzumab and capecitabine and were randomly assigned to either tucatinib or placebo.
Median PFS in the tucatinib patient group was 7.8 months, compared with 5.6 months in the placebo group. The PFS results in the subgroup of patients with brain metastases were nearly the same.
Median overall survival in the tucatinib patient group was 21.9 months versus 17.4 months in the placebo group.
The new drug is a rare success in the treatment of breast cancer brain metastases, said Jawad Fares, MD, of Northwestern University, Chicago, Illinois, who spoke to Medscape Medical News when the phase 3 trial data were first presented at the 2019 San Antonio Breast Cancer Symposium.
“Outcomes in the field have been pretty dismal,” summarized Fares, who was not involved in the study.
The results of the HER2CLIMB study, which was funded by Seattle Genetics, were published in the New England Journal of Medicine last year.
According to the FDA, common side effects with tucatinib were diarrhea, palmar-plantar erythrodysesthesia syndrome, nausea, fatigue, hepatotoxicity, vomiting, stomatitis, decreased appetite, abdominal pain, headache, anemia, and rash.
Tucatinib can cause serious side effects, including diarrhea associated with dehydration, acute kidney injury, and death. Health care professionals should start antidiarrheals as clinically indicated if diarrhea occurs and should interrupt treatment or reduce the dosage. Tucatinib can also cause severe hepatotoxicity; patients should be monitored with liver tests.
This article first appeared on Medscape.com.
Mitomycin approved for low-grade upper tract urothelial cancer
pyelocalyceal

“This is the first approval specifically for patients with low-grade [upper tract urothelial cancer] and provides an option for some patients who may otherwise require a nephroureterectomy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
“Due to substantial treatment challenges associated with the complex anatomy of the upper urinary tract, many patients need to be treated with radical surgery – usually complete removal of the affected kidney, ureter, and bladder cuff," Dr. Pazdur added. "Jelmyto gives patients, for the first time, an alternative treatment option for low-grade [upper tract urothelial cancer].”
The FDA’s approval of mitomycin is based on results from the phase 3 OLYMPUS trial (NCT02793128). This ongoing, single-arm trial enrolled 71 patients with treatment-naive or recurrent low-grade noninvasive upper tract urothelial cancer with at least one measurable papillary tumor located above the ureteropelvic junction. Patients with larger tumors were allowed to have prior tumor debulking.
The patients received mitomycin weekly for 6 weeks at the recommended dose of 4 mg/mL, instilled via ureteral catheter or nephrostomy tube, with the total instillation volume based on volumetric measurements using pyelography, not exceeding 15 mL (60 mg mitomycin).
Patients who achieved a complete response at 3 months could receive monthly instillations up to a maximum of 11 additional instillations.
At 3 months, 41 patients (58%) achieved a complete response (CR). At 12 months after CR determination, 19 patients were still in CR, and 7 patients had documented recurrences. The median duration of CR was not reached.
The most common adverse events (occurring in at least 20% of patients) were ureteric obstruction, flank pain, urinary tract infection, hematuria, renal dysfunction, fatigue, nausea, abdominal pain, dysuria, and vomiting. Ureteric obstruction occurred in 58% of patients, and 88% of those patients required ureteral stent placement.
In all, 23% of patients discontinued mitomycin due to adverse events, and 34% had dose interruptions due to adverse events.
The approval of mitomycin was granted to UroGen Pharma. The FDA granted the application priority review, fast track designation, and breakthrough therapy designation.
The full prescribing information for mitomycin is available for download from the FDA website.
pyelocalyceal
“This is the first approval specifically for patients with low-grade [upper tract urothelial cancer] and provides an option for some patients who may otherwise require a nephroureterectomy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
“Due to substantial treatment challenges associated with the complex anatomy of the upper urinary tract, many patients need to be treated with radical surgery – usually complete removal of the affected kidney, ureter, and bladder cuff," Dr. Pazdur added. "Jelmyto gives patients, for the first time, an alternative treatment option for low-grade [upper tract urothelial cancer].”
The FDA’s approval of mitomycin is based on results from the phase 3 OLYMPUS trial (NCT02793128). This ongoing, single-arm trial enrolled 71 patients with treatment-naive or recurrent low-grade noninvasive upper tract urothelial cancer with at least one measurable papillary tumor located above the ureteropelvic junction. Patients with larger tumors were allowed to have prior tumor debulking.
The patients received mitomycin weekly for 6 weeks at the recommended dose of 4 mg/mL, instilled via ureteral catheter or nephrostomy tube, with the total instillation volume based on volumetric measurements using pyelography, not exceeding 15 mL (60 mg mitomycin).
Patients who achieved a complete response at 3 months could receive monthly instillations up to a maximum of 11 additional instillations.
At 3 months, 41 patients (58%) achieved a complete response (CR). At 12 months after CR determination, 19 patients were still in CR, and 7 patients had documented recurrences. The median duration of CR was not reached.
The most common adverse events (occurring in at least 20% of patients) were ureteric obstruction, flank pain, urinary tract infection, hematuria, renal dysfunction, fatigue, nausea, abdominal pain, dysuria, and vomiting. Ureteric obstruction occurred in 58% of patients, and 88% of those patients required ureteral stent placement.
In all, 23% of patients discontinued mitomycin due to adverse events, and 34% had dose interruptions due to adverse events.
The approval of mitomycin was granted to UroGen Pharma. The FDA granted the application priority review, fast track designation, and breakthrough therapy designation.
The full prescribing information for mitomycin is available for download from the FDA website.
pyelocalyceal
“This is the first approval specifically for patients with low-grade [upper tract urothelial cancer] and provides an option for some patients who may otherwise require a nephroureterectomy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a statement.
“Due to substantial treatment challenges associated with the complex anatomy of the upper urinary tract, many patients need to be treated with radical surgery – usually complete removal of the affected kidney, ureter, and bladder cuff," Dr. Pazdur added. "Jelmyto gives patients, for the first time, an alternative treatment option for low-grade [upper tract urothelial cancer].”
The FDA’s approval of mitomycin is based on results from the phase 3 OLYMPUS trial (NCT02793128). This ongoing, single-arm trial enrolled 71 patients with treatment-naive or recurrent low-grade noninvasive upper tract urothelial cancer with at least one measurable papillary tumor located above the ureteropelvic junction. Patients with larger tumors were allowed to have prior tumor debulking.
The patients received mitomycin weekly for 6 weeks at the recommended dose of 4 mg/mL, instilled via ureteral catheter or nephrostomy tube, with the total instillation volume based on volumetric measurements using pyelography, not exceeding 15 mL (60 mg mitomycin).
Patients who achieved a complete response at 3 months could receive monthly instillations up to a maximum of 11 additional instillations.
At 3 months, 41 patients (58%) achieved a complete response (CR). At 12 months after CR determination, 19 patients were still in CR, and 7 patients had documented recurrences. The median duration of CR was not reached.
The most common adverse events (occurring in at least 20% of patients) were ureteric obstruction, flank pain, urinary tract infection, hematuria, renal dysfunction, fatigue, nausea, abdominal pain, dysuria, and vomiting. Ureteric obstruction occurred in 58% of patients, and 88% of those patients required ureteral stent placement.
In all, 23% of patients discontinued mitomycin due to adverse events, and 34% had dose interruptions due to adverse events.
The approval of mitomycin was granted to UroGen Pharma. The FDA granted the application priority review, fast track designation, and breakthrough therapy designation.
The full prescribing information for mitomycin is available for download from the FDA website.
FROM FDA
FDA approves emergency use of saliva test to detect COVID-19
As the race to develop rapid testing for COVID-19 expands, the Food and Drug Administration has granted emergency approval for an approach that uses saliva as the primary test biomaterial.

According to a document provided to the FDA, the Rutgers Clinical Genomics Laboratory TaqPath SARS-CoV-2 Assay is intended for the qualitative detection of nucleic acid from SARS-CoV-2 in oropharyngeal (throat) swab, nasopharyngeal swab, anterior nasal swab, mid-turbinate nasal swab from individuals suspected of COVID-19 by their health care clinicians. To expand on this assay, Rutgers University–based RUCDR Infinite Biologics developed a saliva collection method in partnership with Spectrum Solutions and Accurate Diagnostic Labs.
The document states that Samples are transported for RNA extraction and are tested within 48 hours of collection. In saliva samples obtained from 60 patients evaluated by the researchers, all were in agreement with the presence of COVID-19.

“If shown to be as accurate as nasopharyngeal and oropharyngeal samples, saliva as a biomatrix offers the advantage of not generating aerosols or creating as many respiratory droplets during specimen procurement, therefore decreasing the risk of transmission to the health care worker doing the testing,” said Matthew P. Cheng, MDCM, of the division of infectious diseases at McGill University Health Centre, Montreal, who was not involved in development of the test but who has written about diagnostic testing for the virus.
“Also, it may be easy enough for patients to do saliva self-collection at home. However, it is important to note that SARS-CoV-2 tests on saliva have not yet undergone the more rigorous evaluation of full FDA authorization, and saliva is not a preferred specimen type of the FDA nor the [Centers for Disease Control and Prevention] for respiratory virus testing.”

In a prepared statement, Andrew I. Brooks, PhD, chief operating officer at RUCDR Infinite Biologics, said the saliva collection method enables clinicians to preserve personal protective equipment for use in patient care instead of testing. “We can significantly increase the number of people tested each and every day as self-collection of saliva is quicker and more scalable than swab collections,” he said. “All of this combined will have a tremendous impact on testing in New Jersey and across the United States.”
The tests are currently available to the RWJBarnabas Health network, based in West Orange, N.J., which has partnered with Rutgers University.
As the race to develop rapid testing for COVID-19 expands, the Food and Drug Administration has granted emergency approval for an approach that uses saliva as the primary test biomaterial.
According to a document provided to the FDA, the Rutgers Clinical Genomics Laboratory TaqPath SARS-CoV-2 Assay is intended for the qualitative detection of nucleic acid from SARS-CoV-2 in oropharyngeal (throat) swab, nasopharyngeal swab, anterior nasal swab, mid-turbinate nasal swab from individuals suspected of COVID-19 by their health care clinicians. To expand on this assay, Rutgers University–based RUCDR Infinite Biologics developed a saliva collection method in partnership with Spectrum Solutions and Accurate Diagnostic Labs.
The document states that Samples are transported for RNA extraction and are tested within 48 hours of collection. In saliva samples obtained from 60 patients evaluated by the researchers, all were in agreement with the presence of COVID-19.
“If shown to be as accurate as nasopharyngeal and oropharyngeal samples, saliva as a biomatrix offers the advantage of not generating aerosols or creating as many respiratory droplets during specimen procurement, therefore decreasing the risk of transmission to the health care worker doing the testing,” said Matthew P. Cheng, MDCM, of the division of infectious diseases at McGill University Health Centre, Montreal, who was not involved in development of the test but who has written about diagnostic testing for the virus.
“Also, it may be easy enough for patients to do saliva self-collection at home. However, it is important to note that SARS-CoV-2 tests on saliva have not yet undergone the more rigorous evaluation of full FDA authorization, and saliva is not a preferred specimen type of the FDA nor the [Centers for Disease Control and Prevention] for respiratory virus testing.”
In a prepared statement, Andrew I. Brooks, PhD, chief operating officer at RUCDR Infinite Biologics, said the saliva collection method enables clinicians to preserve personal protective equipment for use in patient care instead of testing. “We can significantly increase the number of people tested each and every day as self-collection of saliva is quicker and more scalable than swab collections,” he said. “All of this combined will have a tremendous impact on testing in New Jersey and across the United States.”
The tests are currently available to the RWJBarnabas Health network, based in West Orange, N.J., which has partnered with Rutgers University.
As the race to develop rapid testing for COVID-19 expands, the Food and Drug Administration has granted emergency approval for an approach that uses saliva as the primary test biomaterial.
According to a document provided to the FDA, the Rutgers Clinical Genomics Laboratory TaqPath SARS-CoV-2 Assay is intended for the qualitative detection of nucleic acid from SARS-CoV-2 in oropharyngeal (throat) swab, nasopharyngeal swab, anterior nasal swab, mid-turbinate nasal swab from individuals suspected of COVID-19 by their health care clinicians. To expand on this assay, Rutgers University–based RUCDR Infinite Biologics developed a saliva collection method in partnership with Spectrum Solutions and Accurate Diagnostic Labs.
The document states that Samples are transported for RNA extraction and are tested within 48 hours of collection. In saliva samples obtained from 60 patients evaluated by the researchers, all were in agreement with the presence of COVID-19.
“If shown to be as accurate as nasopharyngeal and oropharyngeal samples, saliva as a biomatrix offers the advantage of not generating aerosols or creating as many respiratory droplets during specimen procurement, therefore decreasing the risk of transmission to the health care worker doing the testing,” said Matthew P. Cheng, MDCM, of the division of infectious diseases at McGill University Health Centre, Montreal, who was not involved in development of the test but who has written about diagnostic testing for the virus.
“Also, it may be easy enough for patients to do saliva self-collection at home. However, it is important to note that SARS-CoV-2 tests on saliva have not yet undergone the more rigorous evaluation of full FDA authorization, and saliva is not a preferred specimen type of the FDA nor the [Centers for Disease Control and Prevention] for respiratory virus testing.”
In a prepared statement, Andrew I. Brooks, PhD, chief operating officer at RUCDR Infinite Biologics, said the saliva collection method enables clinicians to preserve personal protective equipment for use in patient care instead of testing. “We can significantly increase the number of people tested each and every day as self-collection of saliva is quicker and more scalable than swab collections,” he said. “All of this combined will have a tremendous impact on testing in New Jersey and across the United States.”
The tests are currently available to the RWJBarnabas Health network, based in West Orange, N.J., which has partnered with Rutgers University.
Suicide increased 35% during 1999-2018 in the U.S.
Age-adjusted suicide rate rose from 10.5 per 100,000 to 14.2 from 1999 to 2018, according to trends reported by the Centers for Disease Control and Prevention in a data brief.
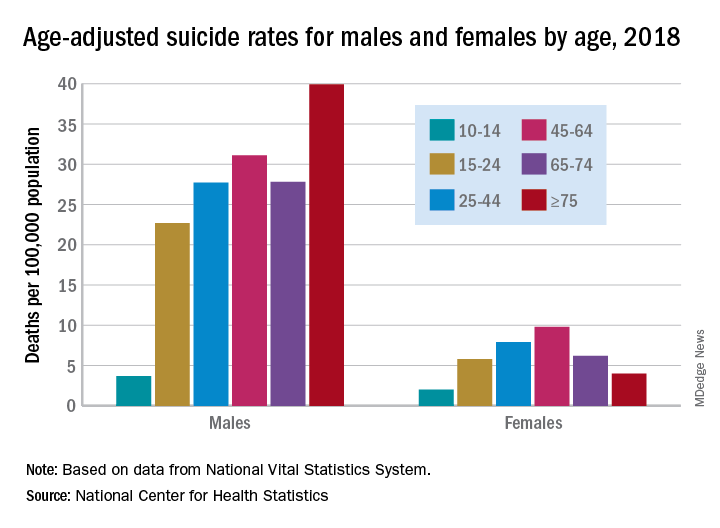
Holly Hedegaard, MD, and colleagues from the National Center for Health Statistics within the CDC analyzed final mortality data from the National Vital Statistics System. As the second most common cause of death among Americans aged 10-34 years and the fourth most common among those aged 35-54 years, suicide is a major contributer to premature mortality.
at 22.8 and 6.2 per 100,000, respectively. Young people aged 10-14 years among both genders had the lowest rates of completing suicide, but it was men aged 75 years and older and women aged 45-64 years who had the highest rates. All of these trends were consistent throughout the study period.
Drawing from the 2013 National Center for Health Statistics Urban-Rural Classification Scheme for Counties, the researchers found that rural counties had significantly higher rates of suicide than did urban counties in 2018, and this was true for men and women. That said, suicide rates were still 3.5-3.9 times higher among men than among women regardless of urbanicity or rurality that year.
The full data brief can be found on the CDC website.
Age-adjusted suicide rate rose from 10.5 per 100,000 to 14.2 from 1999 to 2018, according to trends reported by the Centers for Disease Control and Prevention in a data brief.
Holly Hedegaard, MD, and colleagues from the National Center for Health Statistics within the CDC analyzed final mortality data from the National Vital Statistics System. As the second most common cause of death among Americans aged 10-34 years and the fourth most common among those aged 35-54 years, suicide is a major contributer to premature mortality.
at 22.8 and 6.2 per 100,000, respectively. Young people aged 10-14 years among both genders had the lowest rates of completing suicide, but it was men aged 75 years and older and women aged 45-64 years who had the highest rates. All of these trends were consistent throughout the study period.
Drawing from the 2013 National Center for Health Statistics Urban-Rural Classification Scheme for Counties, the researchers found that rural counties had significantly higher rates of suicide than did urban counties in 2018, and this was true for men and women. That said, suicide rates were still 3.5-3.9 times higher among men than among women regardless of urbanicity or rurality that year.
The full data brief can be found on the CDC website.
Age-adjusted suicide rate rose from 10.5 per 100,000 to 14.2 from 1999 to 2018, according to trends reported by the Centers for Disease Control and Prevention in a data brief.
Holly Hedegaard, MD, and colleagues from the National Center for Health Statistics within the CDC analyzed final mortality data from the National Vital Statistics System. As the second most common cause of death among Americans aged 10-34 years and the fourth most common among those aged 35-54 years, suicide is a major contributer to premature mortality.
at 22.8 and 6.2 per 100,000, respectively. Young people aged 10-14 years among both genders had the lowest rates of completing suicide, but it was men aged 75 years and older and women aged 45-64 years who had the highest rates. All of these trends were consistent throughout the study period.
Drawing from the 2013 National Center for Health Statistics Urban-Rural Classification Scheme for Counties, the researchers found that rural counties had significantly higher rates of suicide than did urban counties in 2018, and this was true for men and women. That said, suicide rates were still 3.5-3.9 times higher among men than among women regardless of urbanicity or rurality that year.
The full data brief can be found on the CDC website.
FDA approves Koselugo for pediatric neurofibromatosis treatment
The Food and Drug Administration has approved selumetinib (Koselugo) for the treatment of pediatric patients aged 2 years and older with type 1 neurofibromatosis (NF1) with symptomatic, inoperable plexiform neurofibromas.

FDA approval was based on results from the phase 2 SPRINT Stratum 1 trial, in which 50 patients with NF1 received selumetinib as twice-daily oral monotherapy. Of this group, 33 (66%) patients had a partial response of at least a 20% reduction in tumor volume. There were no complete responses, according to a press release.
The most common adverse events were vomiting, rash, abdominal pain, diarrhea, nausea, dry skin, fatigue, musculoskeletal pain, pyrexia, rash acneiform, stomatitis, headache, paronychia, and pruritus. Dose interruptions, dose reductions, and permanent drug discontinuation occurred in 80%, 24%, and 12% of patients, respectively.
Serious adverse reactions included cardiomyopathy, ocular toxicity, gastrointestinal toxicity, increased creatinine phosphokinase, and increased vitamin E levels and risk of bleeding, according to the press release.
“Previously, there were no medicines approved for this disease. This approval has the potential to change how symptomatic, inoperable NF1 plexiform neurofibromas are treated and provides new hope to these patients,” Roy Baynes, MD, PhD, senior vice president, head of global clinical development, and chief medical officer of Merck Research Laboratories, said in the press release.
The Food and Drug Administration has approved selumetinib (Koselugo) for the treatment of pediatric patients aged 2 years and older with type 1 neurofibromatosis (NF1) with symptomatic, inoperable plexiform neurofibromas.
FDA approval was based on results from the phase 2 SPRINT Stratum 1 trial, in which 50 patients with NF1 received selumetinib as twice-daily oral monotherapy. Of this group, 33 (66%) patients had a partial response of at least a 20% reduction in tumor volume. There were no complete responses, according to a press release.
The most common adverse events were vomiting, rash, abdominal pain, diarrhea, nausea, dry skin, fatigue, musculoskeletal pain, pyrexia, rash acneiform, stomatitis, headache, paronychia, and pruritus. Dose interruptions, dose reductions, and permanent drug discontinuation occurred in 80%, 24%, and 12% of patients, respectively.
Serious adverse reactions included cardiomyopathy, ocular toxicity, gastrointestinal toxicity, increased creatinine phosphokinase, and increased vitamin E levels and risk of bleeding, according to the press release.
“Previously, there were no medicines approved for this disease. This approval has the potential to change how symptomatic, inoperable NF1 plexiform neurofibromas are treated and provides new hope to these patients,” Roy Baynes, MD, PhD, senior vice president, head of global clinical development, and chief medical officer of Merck Research Laboratories, said in the press release.
The Food and Drug Administration has approved selumetinib (Koselugo) for the treatment of pediatric patients aged 2 years and older with type 1 neurofibromatosis (NF1) with symptomatic, inoperable plexiform neurofibromas.
FDA approval was based on results from the phase 2 SPRINT Stratum 1 trial, in which 50 patients with NF1 received selumetinib as twice-daily oral monotherapy. Of this group, 33 (66%) patients had a partial response of at least a 20% reduction in tumor volume. There were no complete responses, according to a press release.
The most common adverse events were vomiting, rash, abdominal pain, diarrhea, nausea, dry skin, fatigue, musculoskeletal pain, pyrexia, rash acneiform, stomatitis, headache, paronychia, and pruritus. Dose interruptions, dose reductions, and permanent drug discontinuation occurred in 80%, 24%, and 12% of patients, respectively.
Serious adverse reactions included cardiomyopathy, ocular toxicity, gastrointestinal toxicity, increased creatinine phosphokinase, and increased vitamin E levels and risk of bleeding, according to the press release.
“Previously, there were no medicines approved for this disease. This approval has the potential to change how symptomatic, inoperable NF1 plexiform neurofibromas are treated and provides new hope to these patients,” Roy Baynes, MD, PhD, senior vice president, head of global clinical development, and chief medical officer of Merck Research Laboratories, said in the press release.
CDC issues new return-to-work guidelines
The Centers for Disease Control and Prevention is releasing new guidance on return-to-work rules for critical workers exposed to a COVID-19 case, or a suspected case, replacing previous guidance to stay home for 14 days.
“One of the most important things we can do is keep our critical workforce working,” CDC Director Robert Redfield said at a White House briefing on April 8. “In certain circumstances they can go back to work,” he said.
Neither Redfield nor the other governmental officials specified what counts as an essential worker, although it has generally referred to food-service and health care workers.
They must take their temperature before work, wear a facial mask at all times and practice social distancing when at work, the new guidance says. They cannot share headsets or other objects used near the face.
Employers must take the worker’s temperature and assess each one for symptoms before work starts, sending a worker home if he or she is sick. Employers must increase the cleaning of frequently used surfaces, increase air exchange in the building and test the use of face masks to be sure they do not interfere with workflow.
Pressed on whether he would reopen the country at the end of the 30-day Stop the Spread effort on April 30 — since one model has revised the U.S. death toll down from 100,000-240,000 to 61,000 — President Donald Trump said meetings will take place soon to discuss the decision and that he will ‘’rely very heavily” on health experts.
“We know now for sure that the mitigation we have been doing is having a positive effect,” said Anthony Fauci, MD, a coronavirus task force member and director of the National Institute of Allergy and Infectious Diseases.
This article first appeared on WebMD.
The Centers for Disease Control and Prevention is releasing new guidance on return-to-work rules for critical workers exposed to a COVID-19 case, or a suspected case, replacing previous guidance to stay home for 14 days.
“One of the most important things we can do is keep our critical workforce working,” CDC Director Robert Redfield said at a White House briefing on April 8. “In certain circumstances they can go back to work,” he said.
Neither Redfield nor the other governmental officials specified what counts as an essential worker, although it has generally referred to food-service and health care workers.
They must take their temperature before work, wear a facial mask at all times and practice social distancing when at work, the new guidance says. They cannot share headsets or other objects used near the face.
Employers must take the worker’s temperature and assess each one for symptoms before work starts, sending a worker home if he or she is sick. Employers must increase the cleaning of frequently used surfaces, increase air exchange in the building and test the use of face masks to be sure they do not interfere with workflow.
Pressed on whether he would reopen the country at the end of the 30-day Stop the Spread effort on April 30 — since one model has revised the U.S. death toll down from 100,000-240,000 to 61,000 — President Donald Trump said meetings will take place soon to discuss the decision and that he will ‘’rely very heavily” on health experts.
“We know now for sure that the mitigation we have been doing is having a positive effect,” said Anthony Fauci, MD, a coronavirus task force member and director of the National Institute of Allergy and Infectious Diseases.
This article first appeared on WebMD.
The Centers for Disease Control and Prevention is releasing new guidance on return-to-work rules for critical workers exposed to a COVID-19 case, or a suspected case, replacing previous guidance to stay home for 14 days.
“One of the most important things we can do is keep our critical workforce working,” CDC Director Robert Redfield said at a White House briefing on April 8. “In certain circumstances they can go back to work,” he said.
Neither Redfield nor the other governmental officials specified what counts as an essential worker, although it has generally referred to food-service and health care workers.
They must take their temperature before work, wear a facial mask at all times and practice social distancing when at work, the new guidance says. They cannot share headsets or other objects used near the face.
Employers must take the worker’s temperature and assess each one for symptoms before work starts, sending a worker home if he or she is sick. Employers must increase the cleaning of frequently used surfaces, increase air exchange in the building and test the use of face masks to be sure they do not interfere with workflow.
Pressed on whether he would reopen the country at the end of the 30-day Stop the Spread effort on April 30 — since one model has revised the U.S. death toll down from 100,000-240,000 to 61,000 — President Donald Trump said meetings will take place soon to discuss the decision and that he will ‘’rely very heavily” on health experts.
“We know now for sure that the mitigation we have been doing is having a positive effect,” said Anthony Fauci, MD, a coronavirus task force member and director of the National Institute of Allergy and Infectious Diseases.
This article first appeared on WebMD.
CDC: Screen nearly all adults, including pregnant women, for HCV
In the latest issue of the Morbidity and Mortality Weekly Report, the Centers for Disease Control and Prevention recommended hepatitis C virus screening for all adults and all pregnant women – during each of their pregnancies – in areas where prevalence of the infection is 0.1% or greater.
![]()
That’s essentially the entire United States; there’s no state with a statewide adult prevalence below 0.1%, and “few settings are known to exist” otherwise, the CDC noted (MMWR Recomm Rep. 2020 Apr 10;69(2):1-17).
The agency encouraged providers to consult state or local health departments or the CDC directly to determine local HCV prevalence. “As a general guide ... approximately 59% of anti-HCV positive persons are HCV RNA positive,” indicating active infection, the agency noted.
The advice was an expansion from the CDC’s last universal screening recommendation in 2012, which was limited to people born from 1945 to 1965; the incidence of acute infections has climbed since then and is highest now among younger people, so the guideline needed to be revisited, explained authors led by Sarah Schillie, MD, of the CDC’s Division of Viral Hepatitis, Atlanta.
The U.S. Preventive Services Task Force also recently recommended universal adult screening after previously limiting it to baby boomers.
As for pregnancy, the CDC’s past advice was to screen pregnant women with known risk factors, but that needed to be revisited as well. For one thing, the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America have since recommended testing all pregnant women.
But also, the CDC said, it’s an opportune time for screening because “many women only have access to health care during pregnancy and the immediate postpartum period,” when treatment, if needed, can be started. Plus, HCV status is important for management decisions, such as using amniocentesis in positive women instead of chorionic villus sampling.
The rest of CDC’s 2012 recommendations stand, including screening all people with risk factors and repeating screening while they persist. Also, “any person who requests hepatitis C testing should receive it, regardless of disclosure of risk,” because people might be reluctant to report things like IV drug use, the authors said.
Screening in the guidelines means an HCV antibody test, followed by a nucleic acid test to check for active infection. The CDC encouraged automatic reflex testing, meaning immediately checking antibody positive samples for HCV RNA. RNA in the blood indicates active, replicating virus.
The new recommendations penciled out in modeling, with an incremental cost-effectiveness ratio (ICER) for universal adult screening of approximately $36,000 per quality-adjusted life year (QALY) gained, and an ICER of approximately $15,000 per QALY gained for pregnancy screening, where HCV prevalence is 0.1%; the 0.1% cost/benefit cutpoint was one of the reasons it was chosen as the prevalence threshold. An ICER under $50,000 is the conservative benchmark for cost-effectiveness, the authors noted.
There was no external funding, and the authors had no disclosures.
SOURCE: Schillie S et al. MMWR Recomm Rep. 2020 Apr 10;69[2]:1-17).
In the latest issue of the Morbidity and Mortality Weekly Report, the Centers for Disease Control and Prevention recommended hepatitis C virus screening for all adults and all pregnant women – during each of their pregnancies – in areas where prevalence of the infection is 0.1% or greater.
![]()
That’s essentially the entire United States; there’s no state with a statewide adult prevalence below 0.1%, and “few settings are known to exist” otherwise, the CDC noted (MMWR Recomm Rep. 2020 Apr 10;69(2):1-17).
The agency encouraged providers to consult state or local health departments or the CDC directly to determine local HCV prevalence. “As a general guide ... approximately 59% of anti-HCV positive persons are HCV RNA positive,” indicating active infection, the agency noted.
The advice was an expansion from the CDC’s last universal screening recommendation in 2012, which was limited to people born from 1945 to 1965; the incidence of acute infections has climbed since then and is highest now among younger people, so the guideline needed to be revisited, explained authors led by Sarah Schillie, MD, of the CDC’s Division of Viral Hepatitis, Atlanta.
The U.S. Preventive Services Task Force also recently recommended universal adult screening after previously limiting it to baby boomers.
As for pregnancy, the CDC’s past advice was to screen pregnant women with known risk factors, but that needed to be revisited as well. For one thing, the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America have since recommended testing all pregnant women.
But also, the CDC said, it’s an opportune time for screening because “many women only have access to health care during pregnancy and the immediate postpartum period,” when treatment, if needed, can be started. Plus, HCV status is important for management decisions, such as using amniocentesis in positive women instead of chorionic villus sampling.
The rest of CDC’s 2012 recommendations stand, including screening all people with risk factors and repeating screening while they persist. Also, “any person who requests hepatitis C testing should receive it, regardless of disclosure of risk,” because people might be reluctant to report things like IV drug use, the authors said.
Screening in the guidelines means an HCV antibody test, followed by a nucleic acid test to check for active infection. The CDC encouraged automatic reflex testing, meaning immediately checking antibody positive samples for HCV RNA. RNA in the blood indicates active, replicating virus.
The new recommendations penciled out in modeling, with an incremental cost-effectiveness ratio (ICER) for universal adult screening of approximately $36,000 per quality-adjusted life year (QALY) gained, and an ICER of approximately $15,000 per QALY gained for pregnancy screening, where HCV prevalence is 0.1%; the 0.1% cost/benefit cutpoint was one of the reasons it was chosen as the prevalence threshold. An ICER under $50,000 is the conservative benchmark for cost-effectiveness, the authors noted.
There was no external funding, and the authors had no disclosures.
SOURCE: Schillie S et al. MMWR Recomm Rep. 2020 Apr 10;69[2]:1-17).
In the latest issue of the Morbidity and Mortality Weekly Report, the Centers for Disease Control and Prevention recommended hepatitis C virus screening for all adults and all pregnant women – during each of their pregnancies – in areas where prevalence of the infection is 0.1% or greater.
![]()
That’s essentially the entire United States; there’s no state with a statewide adult prevalence below 0.1%, and “few settings are known to exist” otherwise, the CDC noted (MMWR Recomm Rep. 2020 Apr 10;69(2):1-17).
The agency encouraged providers to consult state or local health departments or the CDC directly to determine local HCV prevalence. “As a general guide ... approximately 59% of anti-HCV positive persons are HCV RNA positive,” indicating active infection, the agency noted.
The advice was an expansion from the CDC’s last universal screening recommendation in 2012, which was limited to people born from 1945 to 1965; the incidence of acute infections has climbed since then and is highest now among younger people, so the guideline needed to be revisited, explained authors led by Sarah Schillie, MD, of the CDC’s Division of Viral Hepatitis, Atlanta.
The U.S. Preventive Services Task Force also recently recommended universal adult screening after previously limiting it to baby boomers.
As for pregnancy, the CDC’s past advice was to screen pregnant women with known risk factors, but that needed to be revisited as well. For one thing, the American Association for the Study of Liver Diseases and the Infectious Diseases Society of America have since recommended testing all pregnant women.
But also, the CDC said, it’s an opportune time for screening because “many women only have access to health care during pregnancy and the immediate postpartum period,” when treatment, if needed, can be started. Plus, HCV status is important for management decisions, such as using amniocentesis in positive women instead of chorionic villus sampling.
The rest of CDC’s 2012 recommendations stand, including screening all people with risk factors and repeating screening while they persist. Also, “any person who requests hepatitis C testing should receive it, regardless of disclosure of risk,” because people might be reluctant to report things like IV drug use, the authors said.
Screening in the guidelines means an HCV antibody test, followed by a nucleic acid test to check for active infection. The CDC encouraged automatic reflex testing, meaning immediately checking antibody positive samples for HCV RNA. RNA in the blood indicates active, replicating virus.
The new recommendations penciled out in modeling, with an incremental cost-effectiveness ratio (ICER) for universal adult screening of approximately $36,000 per quality-adjusted life year (QALY) gained, and an ICER of approximately $15,000 per QALY gained for pregnancy screening, where HCV prevalence is 0.1%; the 0.1% cost/benefit cutpoint was one of the reasons it was chosen as the prevalence threshold. An ICER under $50,000 is the conservative benchmark for cost-effectiveness, the authors noted.
There was no external funding, and the authors had no disclosures.
SOURCE: Schillie S et al. MMWR Recomm Rep. 2020 Apr 10;69[2]:1-17).
First advance in MDS for decade: Luspatercept for anemia
The US Food and Drug Administration has approved luspatercept (Reblozyl, Bristol-Myers Squibb/Acceleron) for the treatment of anemia in patients with myelodysplastic syndromes (MDS).
The green light represents the first treatment advancement in MDS in more than a decade, says an expert in the field.
Luspatercept is the first and so far only erythroid maturation agent (EMA), and was launched last year when it was approved for the treatment of anemia in adults with beta thalassemia, who require regular red blood cell transfusions.
The new approval is for the treatment of anemia in adult patients with very low- to intermediate-risk MDS with ring sideroblasts and patients with myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis, after they have progressed on treatment with an erythropoiesis-stimulating agent and who require two or more red blood cell (RBC) units over 8 weeks.
Luspatercept is not a substitute for RBC transfusions in patients who require immediate correction of anemia.
The FDA approval in MDS is based on results from the pivotal, placebo-controlled, phase 3 MEDALIST trial, conducted in 229 patients with very-low–, low- and intermediate-risk non-del(5q) MDS with ring sideroblasts. All patients were RBC transfusion-dependent and had disease that was refractory to, or unlikely to respond to, erythropoiesis-stimulating agents. Results were published in January in the New England Journal of Medicine. The study was funded by Acceleron Pharma and Celgene, which was later acquired by Bristol-Myers Squibb.
These results were first presented at the 2018 annual meeting of the American Society of Hematology (ASH), as reported by Medscape Medical News. At the time, ASH President Alexis Thompson, MD, said it appears that luspatercept can improve the production of endogenous RBCs by enhancing the maturation of these cells in the bone marrow. The drug significantly reduced the need for RBC transfusions, and “this is a very exciting advance for patients who would have few other treatment options,” she said.
“Anemia and the chronic need for transfusions is a very big issue for these patients,” commented lead study author Pierre Fenaux, MD, PhD, from Hôpital Saint-Louis in Paris, France. “With low hemoglobin levels, patients are tired all the time and have an increased risk of falls and cardiovascular events. When you can improve hemoglobin levels, you really see a difference in quality of life.”
The MEDALIST trial is an important milestone for patients with lower-risk, transfusion-dependent MDS, commented Elizabeth Griffiths, MD, associate professor of oncology and director of MDS, Roswell Park Comprehensive Cancer Center, Buffalo, New York.
“No new agents have been approved for MDS in the last 10 years, highlighting this development as a substantial step forward for the MDS community,” she told Medscape Medical News. “Current therapies are time-intensive and only modestly beneficial.”
“The availability of a new, effective drug — particularly relevant to those harboring SF3B1 mutations — is an exciting development and is likely to offer meaningful improvements in quality of life,” Griffiths said. “Since these patients tend to live longer than others with MDS, there are many patients in my clinical practice who would have fit the enrollment criteria for this study. Such patients are eagerly awaiting the opportunity for a decrease in transfusion burden.”
Study Details
In the trial, luspatercept reduced the severity of anemia — 38% of the 153 patients who received luspatercept achieved transfusion independence for 8 weeks or longer compared with 13% of the 76 patients receiving placebo (P < .001).
In the study, patients received luspatercept at a starting dose of 1.0 mg/kg with titration up to 1.75 mg/kg, if needed, or placebo, subcutaneously every 3 weeks for at least 24 weeks.
During the 16 weeks before the initiation of treatment, study patients had received a median of 5 RBC units transfusions during an 8-week period (43.2% of patients had ≥ 6 RBC units, 27.9% had ≥ 4 to < 6 RBC units, and 28.8% had < 4 RBC units). At baseline, 138 (60.3%), 58 (25.3%), and 32 (14%) patients had serum erythropoietin levels less than 200 IU/L, 200-500 IU/L, and greater than 500 IU/L, respectively.
The most common luspatercept-associated adverse events (any grade) in the trial were fatigue, diarrhea, asthenia, nausea, and dizziness. Grade 3 or 4 treatment-emergent adverse events were reported in 42.5% of patients who received luspatercept and 44.7% of patients who received placebo. The incidence of adverse events decreased over time, according to the study authors.
This article first appeared on Medscape.com.
The US Food and Drug Administration has approved luspatercept (Reblozyl, Bristol-Myers Squibb/Acceleron) for the treatment of anemia in patients with myelodysplastic syndromes (MDS).
The green light represents the first treatment advancement in MDS in more than a decade, says an expert in the field.
Luspatercept is the first and so far only erythroid maturation agent (EMA), and was launched last year when it was approved for the treatment of anemia in adults with beta thalassemia, who require regular red blood cell transfusions.
The new approval is for the treatment of anemia in adult patients with very low- to intermediate-risk MDS with ring sideroblasts and patients with myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis, after they have progressed on treatment with an erythropoiesis-stimulating agent and who require two or more red blood cell (RBC) units over 8 weeks.
Luspatercept is not a substitute for RBC transfusions in patients who require immediate correction of anemia.
The FDA approval in MDS is based on results from the pivotal, placebo-controlled, phase 3 MEDALIST trial, conducted in 229 patients with very-low–, low- and intermediate-risk non-del(5q) MDS with ring sideroblasts. All patients were RBC transfusion-dependent and had disease that was refractory to, or unlikely to respond to, erythropoiesis-stimulating agents. Results were published in January in the New England Journal of Medicine. The study was funded by Acceleron Pharma and Celgene, which was later acquired by Bristol-Myers Squibb.
These results were first presented at the 2018 annual meeting of the American Society of Hematology (ASH), as reported by Medscape Medical News. At the time, ASH President Alexis Thompson, MD, said it appears that luspatercept can improve the production of endogenous RBCs by enhancing the maturation of these cells in the bone marrow. The drug significantly reduced the need for RBC transfusions, and “this is a very exciting advance for patients who would have few other treatment options,” she said.
“Anemia and the chronic need for transfusions is a very big issue for these patients,” commented lead study author Pierre Fenaux, MD, PhD, from Hôpital Saint-Louis in Paris, France. “With low hemoglobin levels, patients are tired all the time and have an increased risk of falls and cardiovascular events. When you can improve hemoglobin levels, you really see a difference in quality of life.”
The MEDALIST trial is an important milestone for patients with lower-risk, transfusion-dependent MDS, commented Elizabeth Griffiths, MD, associate professor of oncology and director of MDS, Roswell Park Comprehensive Cancer Center, Buffalo, New York.
“No new agents have been approved for MDS in the last 10 years, highlighting this development as a substantial step forward for the MDS community,” she told Medscape Medical News. “Current therapies are time-intensive and only modestly beneficial.”
“The availability of a new, effective drug — particularly relevant to those harboring SF3B1 mutations — is an exciting development and is likely to offer meaningful improvements in quality of life,” Griffiths said. “Since these patients tend to live longer than others with MDS, there are many patients in my clinical practice who would have fit the enrollment criteria for this study. Such patients are eagerly awaiting the opportunity for a decrease in transfusion burden.”
Study Details
In the trial, luspatercept reduced the severity of anemia — 38% of the 153 patients who received luspatercept achieved transfusion independence for 8 weeks or longer compared with 13% of the 76 patients receiving placebo (P < .001).
In the study, patients received luspatercept at a starting dose of 1.0 mg/kg with titration up to 1.75 mg/kg, if needed, or placebo, subcutaneously every 3 weeks for at least 24 weeks.
During the 16 weeks before the initiation of treatment, study patients had received a median of 5 RBC units transfusions during an 8-week period (43.2% of patients had ≥ 6 RBC units, 27.9% had ≥ 4 to < 6 RBC units, and 28.8% had < 4 RBC units). At baseline, 138 (60.3%), 58 (25.3%), and 32 (14%) patients had serum erythropoietin levels less than 200 IU/L, 200-500 IU/L, and greater than 500 IU/L, respectively.
The most common luspatercept-associated adverse events (any grade) in the trial were fatigue, diarrhea, asthenia, nausea, and dizziness. Grade 3 or 4 treatment-emergent adverse events were reported in 42.5% of patients who received luspatercept and 44.7% of patients who received placebo. The incidence of adverse events decreased over time, according to the study authors.
This article first appeared on Medscape.com.
The US Food and Drug Administration has approved luspatercept (Reblozyl, Bristol-Myers Squibb/Acceleron) for the treatment of anemia in patients with myelodysplastic syndromes (MDS).
The green light represents the first treatment advancement in MDS in more than a decade, says an expert in the field.
Luspatercept is the first and so far only erythroid maturation agent (EMA), and was launched last year when it was approved for the treatment of anemia in adults with beta thalassemia, who require regular red blood cell transfusions.
The new approval is for the treatment of anemia in adult patients with very low- to intermediate-risk MDS with ring sideroblasts and patients with myelodysplastic/myeloproliferative neoplasm with ring sideroblasts and thrombocytosis, after they have progressed on treatment with an erythropoiesis-stimulating agent and who require two or more red blood cell (RBC) units over 8 weeks.
Luspatercept is not a substitute for RBC transfusions in patients who require immediate correction of anemia.
The FDA approval in MDS is based on results from the pivotal, placebo-controlled, phase 3 MEDALIST trial, conducted in 229 patients with very-low–, low- and intermediate-risk non-del(5q) MDS with ring sideroblasts. All patients were RBC transfusion-dependent and had disease that was refractory to, or unlikely to respond to, erythropoiesis-stimulating agents. Results were published in January in the New England Journal of Medicine. The study was funded by Acceleron Pharma and Celgene, which was later acquired by Bristol-Myers Squibb.
These results were first presented at the 2018 annual meeting of the American Society of Hematology (ASH), as reported by Medscape Medical News. At the time, ASH President Alexis Thompson, MD, said it appears that luspatercept can improve the production of endogenous RBCs by enhancing the maturation of these cells in the bone marrow. The drug significantly reduced the need for RBC transfusions, and “this is a very exciting advance for patients who would have few other treatment options,” she said.
“Anemia and the chronic need for transfusions is a very big issue for these patients,” commented lead study author Pierre Fenaux, MD, PhD, from Hôpital Saint-Louis in Paris, France. “With low hemoglobin levels, patients are tired all the time and have an increased risk of falls and cardiovascular events. When you can improve hemoglobin levels, you really see a difference in quality of life.”
The MEDALIST trial is an important milestone for patients with lower-risk, transfusion-dependent MDS, commented Elizabeth Griffiths, MD, associate professor of oncology and director of MDS, Roswell Park Comprehensive Cancer Center, Buffalo, New York.
“No new agents have been approved for MDS in the last 10 years, highlighting this development as a substantial step forward for the MDS community,” she told Medscape Medical News. “Current therapies are time-intensive and only modestly beneficial.”
“The availability of a new, effective drug — particularly relevant to those harboring SF3B1 mutations — is an exciting development and is likely to offer meaningful improvements in quality of life,” Griffiths said. “Since these patients tend to live longer than others with MDS, there are many patients in my clinical practice who would have fit the enrollment criteria for this study. Such patients are eagerly awaiting the opportunity for a decrease in transfusion burden.”
Study Details
In the trial, luspatercept reduced the severity of anemia — 38% of the 153 patients who received luspatercept achieved transfusion independence for 8 weeks or longer compared with 13% of the 76 patients receiving placebo (P < .001).
In the study, patients received luspatercept at a starting dose of 1.0 mg/kg with titration up to 1.75 mg/kg, if needed, or placebo, subcutaneously every 3 weeks for at least 24 weeks.
During the 16 weeks before the initiation of treatment, study patients had received a median of 5 RBC units transfusions during an 8-week period (43.2% of patients had ≥ 6 RBC units, 27.9% had ≥ 4 to < 6 RBC units, and 28.8% had < 4 RBC units). At baseline, 138 (60.3%), 58 (25.3%), and 32 (14%) patients had serum erythropoietin levels less than 200 IU/L, 200-500 IU/L, and greater than 500 IU/L, respectively.
The most common luspatercept-associated adverse events (any grade) in the trial were fatigue, diarrhea, asthenia, nausea, and dizziness. Grade 3 or 4 treatment-emergent adverse events were reported in 42.5% of patients who received luspatercept and 44.7% of patients who received placebo. The incidence of adverse events decreased over time, according to the study authors.
This article first appeared on Medscape.com.
FDA grants emergency authorization for first rapid antibody test for COVID-19
The U.S. Food and Drug Administration has granted Cellex an emergency use authorization to market a rapid antibody test for COVID-19, the first antibody test released amidst the pandemic.

“It is reasonable to believe that your product may be effective in diagnosing COVID-19,” and “there is no adequate, approved, and available alternative,” the agency said in a letter to Cellex.
A drop of serum, plasma, or whole blood is placed into a well on a small cartridge, and the results are read 15-20 minutes later; lines indicate the presence of IgM, IgG, or both antibodies against the SARS-CoV-2 virus.
Of 128 samples confirmed positive by reverse transcription polymerase chain reaction in premarket testing, 120 tested positive by IgG, IgM, or both. Of 250 confirmed negative, 239 were negative by the rapid test.
The numbers translated to a positive percent agreement with RT-PCR of 93.8% (95% CI: 88.06-97.26%) and a negative percent agreement of 96.4% (95% CI: 92.26-97.78%), according to labeling.
“Results from antibody testing should not be used as the sole basis to diagnose or exclude SARS-CoV-2 infection,” the labeling states.
Negative results do not rule out infection; antibodies might not have had enough time to form or the virus could have had a minor amino acid mutation in the epitope recognized by the antibodies screened for in the test. False positives can occur due to cross-reactivity with antibodies from previous infections, such as from other coronaviruses.
Labeling suggests that people who test negative should be checked again in a few days, and positive results should be confirmed by other methods. Also, the intensity of the test lines do not necessarily correlate with SARS-CoV-2 antibody titers.
As part of its authorization, the FDA waived good manufacturing practice requirements, but stipulated that advertising must state that the test has not been formally approved by the agency.
Testing is limited to Clinical Laboratory Improvement Amendments-certified labs. Positive results are required to be reported to public health authorities. The test can be ordered through Cellex distributors or directly from the company.
IgM antibodies are generally detectable several days after the initial infection, while IgG antibodies take longer. It’s not known how long COVID-19 antibodies persist after the infection has cleared, the agency said.
The U.S. Food and Drug Administration has granted Cellex an emergency use authorization to market a rapid antibody test for COVID-19, the first antibody test released amidst the pandemic.
“It is reasonable to believe that your product may be effective in diagnosing COVID-19,” and “there is no adequate, approved, and available alternative,” the agency said in a letter to Cellex.
A drop of serum, plasma, or whole blood is placed into a well on a small cartridge, and the results are read 15-20 minutes later; lines indicate the presence of IgM, IgG, or both antibodies against the SARS-CoV-2 virus.
Of 128 samples confirmed positive by reverse transcription polymerase chain reaction in premarket testing, 120 tested positive by IgG, IgM, or both. Of 250 confirmed negative, 239 were negative by the rapid test.
The numbers translated to a positive percent agreement with RT-PCR of 93.8% (95% CI: 88.06-97.26%) and a negative percent agreement of 96.4% (95% CI: 92.26-97.78%), according to labeling.
“Results from antibody testing should not be used as the sole basis to diagnose or exclude SARS-CoV-2 infection,” the labeling states.
Negative results do not rule out infection; antibodies might not have had enough time to form or the virus could have had a minor amino acid mutation in the epitope recognized by the antibodies screened for in the test. False positives can occur due to cross-reactivity with antibodies from previous infections, such as from other coronaviruses.
Labeling suggests that people who test negative should be checked again in a few days, and positive results should be confirmed by other methods. Also, the intensity of the test lines do not necessarily correlate with SARS-CoV-2 antibody titers.
As part of its authorization, the FDA waived good manufacturing practice requirements, but stipulated that advertising must state that the test has not been formally approved by the agency.
Testing is limited to Clinical Laboratory Improvement Amendments-certified labs. Positive results are required to be reported to public health authorities. The test can be ordered through Cellex distributors or directly from the company.
IgM antibodies are generally detectable several days after the initial infection, while IgG antibodies take longer. It’s not known how long COVID-19 antibodies persist after the infection has cleared, the agency said.
The U.S. Food and Drug Administration has granted Cellex an emergency use authorization to market a rapid antibody test for COVID-19, the first antibody test released amidst the pandemic.
“It is reasonable to believe that your product may be effective in diagnosing COVID-19,” and “there is no adequate, approved, and available alternative,” the agency said in a letter to Cellex.
A drop of serum, plasma, or whole blood is placed into a well on a small cartridge, and the results are read 15-20 minutes later; lines indicate the presence of IgM, IgG, or both antibodies against the SARS-CoV-2 virus.
Of 128 samples confirmed positive by reverse transcription polymerase chain reaction in premarket testing, 120 tested positive by IgG, IgM, or both. Of 250 confirmed negative, 239 were negative by the rapid test.
The numbers translated to a positive percent agreement with RT-PCR of 93.8% (95% CI: 88.06-97.26%) and a negative percent agreement of 96.4% (95% CI: 92.26-97.78%), according to labeling.
“Results from antibody testing should not be used as the sole basis to diagnose or exclude SARS-CoV-2 infection,” the labeling states.
Negative results do not rule out infection; antibodies might not have had enough time to form or the virus could have had a minor amino acid mutation in the epitope recognized by the antibodies screened for in the test. False positives can occur due to cross-reactivity with antibodies from previous infections, such as from other coronaviruses.
Labeling suggests that people who test negative should be checked again in a few days, and positive results should be confirmed by other methods. Also, the intensity of the test lines do not necessarily correlate with SARS-CoV-2 antibody titers.
As part of its authorization, the FDA waived good manufacturing practice requirements, but stipulated that advertising must state that the test has not been formally approved by the agency.
Testing is limited to Clinical Laboratory Improvement Amendments-certified labs. Positive results are required to be reported to public health authorities. The test can be ordered through Cellex distributors or directly from the company.
IgM antibodies are generally detectable several days after the initial infection, while IgG antibodies take longer. It’s not known how long COVID-19 antibodies persist after the infection has cleared, the agency said.