User login
FDA approves tofacitinib for psoriatic arthritis
(PsA) who have had an inadequate response or intolerance to methotrexate or other disease-modifying antirheumatic drugs (DMARDs), according to a Dec. 14 announcement from its manufacturer, Pfizer.
The approvals of tofacitinib (Xeljanz) at 5 mg twice daily and extended-release tofacitinib (Xeljanz XR) at 11 mg once daily are based on data from the phase 3 Oral Psoriatic Arthritis Trial (OPAL) clinical development program, which consisted of two pivotal studies, OPAL Broaden and OPAL Beyond.
In the OPAL Broaden study, 50% of patients who received tofacitinib 5 mg twice daily in combination with a nonbiologic DMARD achieved an ACR20 response at 3 months, compared with 33% of those treated with placebo (P equal to or less than .05). In the OPAL Beyond study, 50% of patients achieved an ACR20 response with tofacitinib 5 mg twice daily at 3 months, when compared with 24% of patients taking placebo (P equal to or less than .05).
Tofacitinib is the first and only Janus kinase inhibitor approved by the FDA to treat both moderate to severe rheumatoid arthritis and active PsA, Pfizer said in its announcement. It is noted that the recommended dose of tofacitinib is in combination with nonbiologic DMARDs, and use in combination with biologic DMARDs or with potent immunosuppressants such as azathioprine and cyclosporine is not recommended.
The safety of tofacitinib in these trials was consistent with the safety profile observed in rheumatoid arthritis patients. The most common adverse events that occurred in greater than 3% of patients on tofacitinib 5 mg twice daily were nasopharyngitis, upper respiratory tract infection, headache, and diarrhea.
(PsA) who have had an inadequate response or intolerance to methotrexate or other disease-modifying antirheumatic drugs (DMARDs), according to a Dec. 14 announcement from its manufacturer, Pfizer.
The approvals of tofacitinib (Xeljanz) at 5 mg twice daily and extended-release tofacitinib (Xeljanz XR) at 11 mg once daily are based on data from the phase 3 Oral Psoriatic Arthritis Trial (OPAL) clinical development program, which consisted of two pivotal studies, OPAL Broaden and OPAL Beyond.
In the OPAL Broaden study, 50% of patients who received tofacitinib 5 mg twice daily in combination with a nonbiologic DMARD achieved an ACR20 response at 3 months, compared with 33% of those treated with placebo (P equal to or less than .05). In the OPAL Beyond study, 50% of patients achieved an ACR20 response with tofacitinib 5 mg twice daily at 3 months, when compared with 24% of patients taking placebo (P equal to or less than .05).
Tofacitinib is the first and only Janus kinase inhibitor approved by the FDA to treat both moderate to severe rheumatoid arthritis and active PsA, Pfizer said in its announcement. It is noted that the recommended dose of tofacitinib is in combination with nonbiologic DMARDs, and use in combination with biologic DMARDs or with potent immunosuppressants such as azathioprine and cyclosporine is not recommended.
The safety of tofacitinib in these trials was consistent with the safety profile observed in rheumatoid arthritis patients. The most common adverse events that occurred in greater than 3% of patients on tofacitinib 5 mg twice daily were nasopharyngitis, upper respiratory tract infection, headache, and diarrhea.
(PsA) who have had an inadequate response or intolerance to methotrexate or other disease-modifying antirheumatic drugs (DMARDs), according to a Dec. 14 announcement from its manufacturer, Pfizer.
The approvals of tofacitinib (Xeljanz) at 5 mg twice daily and extended-release tofacitinib (Xeljanz XR) at 11 mg once daily are based on data from the phase 3 Oral Psoriatic Arthritis Trial (OPAL) clinical development program, which consisted of two pivotal studies, OPAL Broaden and OPAL Beyond.
In the OPAL Broaden study, 50% of patients who received tofacitinib 5 mg twice daily in combination with a nonbiologic DMARD achieved an ACR20 response at 3 months, compared with 33% of those treated with placebo (P equal to or less than .05). In the OPAL Beyond study, 50% of patients achieved an ACR20 response with tofacitinib 5 mg twice daily at 3 months, when compared with 24% of patients taking placebo (P equal to or less than .05).
Tofacitinib is the first and only Janus kinase inhibitor approved by the FDA to treat both moderate to severe rheumatoid arthritis and active PsA, Pfizer said in its announcement. It is noted that the recommended dose of tofacitinib is in combination with nonbiologic DMARDs, and use in combination with biologic DMARDs or with potent immunosuppressants such as azathioprine and cyclosporine is not recommended.
The safety of tofacitinib in these trials was consistent with the safety profile observed in rheumatoid arthritis patients. The most common adverse events that occurred in greater than 3% of patients on tofacitinib 5 mg twice daily were nasopharyngitis, upper respiratory tract infection, headache, and diarrhea.
FDA approves infliximab biosimilar Ixifi for all of Remicade’s indications
The Food and Drug Administration has approved Ixifi (infliximab-qbtx), a biosimilar of Remicade, the original infliximab product. Ixifi is the third infliximab biosimilar to be approved by the FDA, and it is approved for all the same indications as Remicade, according to an announcement from its manufacturer, Pfizer.
Ixifi and Remicade are approved for the treatment of rheumatoid arthritis in combination with methotrexate, Crohn’s disease, pediatric Crohn’s disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis.![]()
The most common adverse events associated with Ixifi are upper respiratory infections, sinusitis, pharyngitis, infusion-related reactions, headache, and abdominal pain.
The Food and Drug Administration has approved Ixifi (infliximab-qbtx), a biosimilar of Remicade, the original infliximab product. Ixifi is the third infliximab biosimilar to be approved by the FDA, and it is approved for all the same indications as Remicade, according to an announcement from its manufacturer, Pfizer.
Ixifi and Remicade are approved for the treatment of rheumatoid arthritis in combination with methotrexate, Crohn’s disease, pediatric Crohn’s disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis.![]()
The most common adverse events associated with Ixifi are upper respiratory infections, sinusitis, pharyngitis, infusion-related reactions, headache, and abdominal pain.
The Food and Drug Administration has approved Ixifi (infliximab-qbtx), a biosimilar of Remicade, the original infliximab product. Ixifi is the third infliximab biosimilar to be approved by the FDA, and it is approved for all the same indications as Remicade, according to an announcement from its manufacturer, Pfizer.
Ixifi and Remicade are approved for the treatment of rheumatoid arthritis in combination with methotrexate, Crohn’s disease, pediatric Crohn’s disease, ulcerative colitis, ankylosing spondylitis, psoriatic arthritis, and plaque psoriasis.![]()
The most common adverse events associated with Ixifi are upper respiratory infections, sinusitis, pharyngitis, infusion-related reactions, headache, and abdominal pain.
FDA approves premixed, low-volume colon-cleansing solution
in adults preparing to undergo colonoscopy, according to Ferring Pharmaceuticals.
The sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution is a relatively low-volume, premixed, cranberry-flavored solution, making it easier to use and more palatable for patients.
The oral solution is approved with two dosing options: split dose, one dose the evening prior and one dose the morning of the procedure, or the day before dose, which involves taking both doses the day prior to the procedure. Day before dosing is an alternative and should be used when split dosing is not appropriate. After each dose of sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution, clear liquids should be consumed based on the dosing recommendation. The American College of Gastroenterology recommends the split-dose regimen because of its improved cleansing quality of the colon and better tolerability of the liquid volume by patients.
Patients with impaired renal function should exercise caution if using sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution as it may effect renal function. A more comprehensive list of safety information is available at www.clenpiq.com.
in adults preparing to undergo colonoscopy, according to Ferring Pharmaceuticals.
The sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution is a relatively low-volume, premixed, cranberry-flavored solution, making it easier to use and more palatable for patients.
The oral solution is approved with two dosing options: split dose, one dose the evening prior and one dose the morning of the procedure, or the day before dose, which involves taking both doses the day prior to the procedure. Day before dosing is an alternative and should be used when split dosing is not appropriate. After each dose of sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution, clear liquids should be consumed based on the dosing recommendation. The American College of Gastroenterology recommends the split-dose regimen because of its improved cleansing quality of the colon and better tolerability of the liquid volume by patients.
Patients with impaired renal function should exercise caution if using sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution as it may effect renal function. A more comprehensive list of safety information is available at www.clenpiq.com.
in adults preparing to undergo colonoscopy, according to Ferring Pharmaceuticals.
The sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution is a relatively low-volume, premixed, cranberry-flavored solution, making it easier to use and more palatable for patients.
The oral solution is approved with two dosing options: split dose, one dose the evening prior and one dose the morning of the procedure, or the day before dose, which involves taking both doses the day prior to the procedure. Day before dosing is an alternative and should be used when split dosing is not appropriate. After each dose of sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution, clear liquids should be consumed based on the dosing recommendation. The American College of Gastroenterology recommends the split-dose regimen because of its improved cleansing quality of the colon and better tolerability of the liquid volume by patients.
Patients with impaired renal function should exercise caution if using sodium picosulfate, magnesium oxide, and anhydrous citric acid oral solution as it may effect renal function. A more comprehensive list of safety information is available at www.clenpiq.com.
FDA approves first therapy treatment for EGPA
The Food and Drug Administration announced Dec. 12 the approval of mepolizumab (Nucala) to treat adult patients with eosinophilic granulomatosis with polyangiitis (EGPA), making it the first FDA-approved therapy intended to treat this rare disease.
Approval was based on data from a 52-week clinical trial that compared mepolizumab with placebo, according to the FDA. Patients received 300 mg of mepolizumab once every 4 weeks while continuing stable daily oral corticosteroid therapy. Those patients receiving mepolizumab “achieved a significantly greater accrued time in remission compared with placebo,” and a significantly higher proportion of patients receiving 300 mg of mepolizumab had achieved remission at week 36 and week 48, the statement said. Additionally, significantly more patients treated with mepolizumab achieved remission within the first 24 weeks and remained in remission for the remainder of the 52-week study treatment period.
“The expanded indication of Nucala meets a critical, unmet need for EGPA patients. It’s notable that patients taking Nucala in clinical trials reported a significant improvement in their symptoms,” said Badrul Chowdhury, MD, PhD, director of the division of pulmonary, allergy, and rheumatology products in the FDA’s Center for Drug Evaluation and Research in the press release announcing the approval. EGPA was formerly known as Churg-Strauss syndrome, the statement pointed out.
Read the full press release on the FDA’s website.
SOURCE: FDA.gov
The Food and Drug Administration announced Dec. 12 the approval of mepolizumab (Nucala) to treat adult patients with eosinophilic granulomatosis with polyangiitis (EGPA), making it the first FDA-approved therapy intended to treat this rare disease.
Approval was based on data from a 52-week clinical trial that compared mepolizumab with placebo, according to the FDA. Patients received 300 mg of mepolizumab once every 4 weeks while continuing stable daily oral corticosteroid therapy. Those patients receiving mepolizumab “achieved a significantly greater accrued time in remission compared with placebo,” and a significantly higher proportion of patients receiving 300 mg of mepolizumab had achieved remission at week 36 and week 48, the statement said. Additionally, significantly more patients treated with mepolizumab achieved remission within the first 24 weeks and remained in remission for the remainder of the 52-week study treatment period.
“The expanded indication of Nucala meets a critical, unmet need for EGPA patients. It’s notable that patients taking Nucala in clinical trials reported a significant improvement in their symptoms,” said Badrul Chowdhury, MD, PhD, director of the division of pulmonary, allergy, and rheumatology products in the FDA’s Center for Drug Evaluation and Research in the press release announcing the approval. EGPA was formerly known as Churg-Strauss syndrome, the statement pointed out.
Read the full press release on the FDA’s website.
SOURCE: FDA.gov
The Food and Drug Administration announced Dec. 12 the approval of mepolizumab (Nucala) to treat adult patients with eosinophilic granulomatosis with polyangiitis (EGPA), making it the first FDA-approved therapy intended to treat this rare disease.
Approval was based on data from a 52-week clinical trial that compared mepolizumab with placebo, according to the FDA. Patients received 300 mg of mepolizumab once every 4 weeks while continuing stable daily oral corticosteroid therapy. Those patients receiving mepolizumab “achieved a significantly greater accrued time in remission compared with placebo,” and a significantly higher proportion of patients receiving 300 mg of mepolizumab had achieved remission at week 36 and week 48, the statement said. Additionally, significantly more patients treated with mepolizumab achieved remission within the first 24 weeks and remained in remission for the remainder of the 52-week study treatment period.
“The expanded indication of Nucala meets a critical, unmet need for EGPA patients. It’s notable that patients taking Nucala in clinical trials reported a significant improvement in their symptoms,” said Badrul Chowdhury, MD, PhD, director of the division of pulmonary, allergy, and rheumatology products in the FDA’s Center for Drug Evaluation and Research in the press release announcing the approval. EGPA was formerly known as Churg-Strauss syndrome, the statement pointed out.
Read the full press release on the FDA’s website.
SOURCE: FDA.gov
Hawaii experiencing a statewide outbreak of mumps
As of Nov. 30, 2017, 636 cases of mumps had been confirmed in Hawaii, according to the state’s Department of Health (DOH).
The Hawaii DOH originally reported 14 confirmed mumps cases statewide in April 2017, but the number of confirmed cases has increased by more than 4,000% since that time.
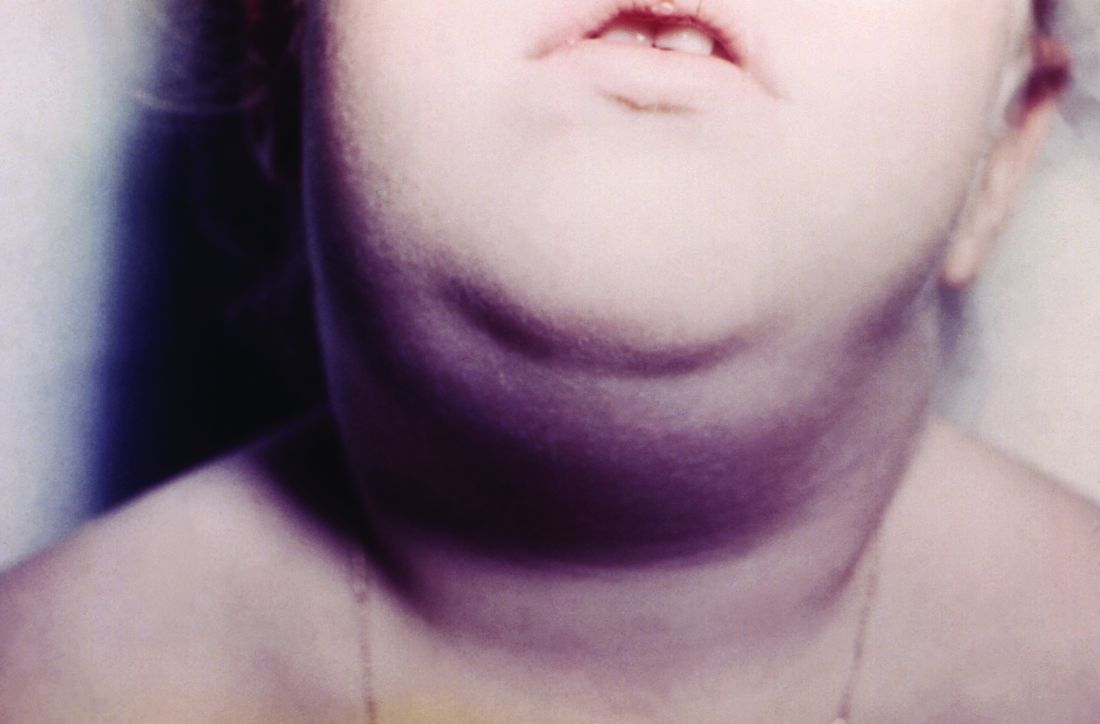
In the midst of the outbreak, the Hawaii DOH recommends that all adolescents between the aged 10-19 years old, and adults born in or after 1957, should receive an additional MMR vaccine dose as soon as possible. The outbreak dose is recommended regardless of previous vaccination or documented immunity to mumps. Administering additional doses of vaccine is not an ideal situation, the DOH noted, but said it should not cause any medical complications.
The Hawaii DOH will investigate mumps cases statewide as the outbreak continues.
As of Nov. 30, 2017, 636 cases of mumps had been confirmed in Hawaii, according to the state’s Department of Health (DOH).
The Hawaii DOH originally reported 14 confirmed mumps cases statewide in April 2017, but the number of confirmed cases has increased by more than 4,000% since that time.
In the midst of the outbreak, the Hawaii DOH recommends that all adolescents between the aged 10-19 years old, and adults born in or after 1957, should receive an additional MMR vaccine dose as soon as possible. The outbreak dose is recommended regardless of previous vaccination or documented immunity to mumps. Administering additional doses of vaccine is not an ideal situation, the DOH noted, but said it should not cause any medical complications.
The Hawaii DOH will investigate mumps cases statewide as the outbreak continues.
As of Nov. 30, 2017, 636 cases of mumps had been confirmed in Hawaii, according to the state’s Department of Health (DOH).
The Hawaii DOH originally reported 14 confirmed mumps cases statewide in April 2017, but the number of confirmed cases has increased by more than 4,000% since that time.
In the midst of the outbreak, the Hawaii DOH recommends that all adolescents between the aged 10-19 years old, and adults born in or after 1957, should receive an additional MMR vaccine dose as soon as possible. The outbreak dose is recommended regardless of previous vaccination or documented immunity to mumps. Administering additional doses of vaccine is not an ideal situation, the DOH noted, but said it should not cause any medical complications.
The Hawaii DOH will investigate mumps cases statewide as the outbreak continues.
FDA approves injectable diabetes drug that improves A1c scores
The Food and Drug Administration has approved semaglutide (OZEMPIC) injections for treatment of type 2 diabetes in adults, according to a press release from Novo Nordisk.
Semaglutide is a once-weekly injection of glucagon-like peptide (GLP-1) receptor agonist that, combined with diet and exercise, can improve glycemic control in adults with type 2 diabetes. Weekly injections are administered by health care providers in a prefilled pen subcutaneously in the stomach, abdomen, thigh, or upper arm as a 0.5-mg or 1-mg formulation. It is important that all doses be administered on the same day each week, according to the OZEMPIC package insert.
“The OZEMPIC (semaglutide) approval builds on Novo Nordisk’s commitment to offering health care professionals a range of treatments that effectively addresses the complex needs of diabetes management and fits their patients’ lifestyles,” said Todd Hobbs, vice president and U.S. chief medical officer of Novo Nordisk.
To ensure access to semaglutide, Novo Nordisk is pricing the drug competitively with other GLP-1 receptor agonists and will offer an associated savings card program to reduce copays for insured patients, the company said. Novo Nordisk expects to launch OZEMPIC in the United States in the first quarter of 2018, and is working on contracting solutions with health insurance providers to increase patient access to the drug.
According to the Novo Nordisk statement, clinicians should not consider semaglutide as a first choice option for treating diabetes or as a substitute for insulin in patients with type 1 diabetes and diabetic ketoacidosis. Whether semaglutide can be used by people who have had pancreatitis or is safe in patients under the age of 18 years old remains to be seen.
“Type 2 diabetes is a serious condition that affects more than 28 million people in the U.S., and despite advancements in treatment, some people with type 2 diabetes do not achieve their A1c goals,” said Helena Rodbard, MD, past president of the American Association of Clinical Endocrinologists. “The approval of semaglutide offers health care professionals an important new treatment option to help adults with type 2 diabetes meet their A1c goals.”
The Food and Drug Administration has approved semaglutide (OZEMPIC) injections for treatment of type 2 diabetes in adults, according to a press release from Novo Nordisk.
Semaglutide is a once-weekly injection of glucagon-like peptide (GLP-1) receptor agonist that, combined with diet and exercise, can improve glycemic control in adults with type 2 diabetes. Weekly injections are administered by health care providers in a prefilled pen subcutaneously in the stomach, abdomen, thigh, or upper arm as a 0.5-mg or 1-mg formulation. It is important that all doses be administered on the same day each week, according to the OZEMPIC package insert.
“The OZEMPIC (semaglutide) approval builds on Novo Nordisk’s commitment to offering health care professionals a range of treatments that effectively addresses the complex needs of diabetes management and fits their patients’ lifestyles,” said Todd Hobbs, vice president and U.S. chief medical officer of Novo Nordisk.
To ensure access to semaglutide, Novo Nordisk is pricing the drug competitively with other GLP-1 receptor agonists and will offer an associated savings card program to reduce copays for insured patients, the company said. Novo Nordisk expects to launch OZEMPIC in the United States in the first quarter of 2018, and is working on contracting solutions with health insurance providers to increase patient access to the drug.
According to the Novo Nordisk statement, clinicians should not consider semaglutide as a first choice option for treating diabetes or as a substitute for insulin in patients with type 1 diabetes and diabetic ketoacidosis. Whether semaglutide can be used by people who have had pancreatitis or is safe in patients under the age of 18 years old remains to be seen.
“Type 2 diabetes is a serious condition that affects more than 28 million people in the U.S., and despite advancements in treatment, some people with type 2 diabetes do not achieve their A1c goals,” said Helena Rodbard, MD, past president of the American Association of Clinical Endocrinologists. “The approval of semaglutide offers health care professionals an important new treatment option to help adults with type 2 diabetes meet their A1c goals.”
The Food and Drug Administration has approved semaglutide (OZEMPIC) injections for treatment of type 2 diabetes in adults, according to a press release from Novo Nordisk.
Semaglutide is a once-weekly injection of glucagon-like peptide (GLP-1) receptor agonist that, combined with diet and exercise, can improve glycemic control in adults with type 2 diabetes. Weekly injections are administered by health care providers in a prefilled pen subcutaneously in the stomach, abdomen, thigh, or upper arm as a 0.5-mg or 1-mg formulation. It is important that all doses be administered on the same day each week, according to the OZEMPIC package insert.
“The OZEMPIC (semaglutide) approval builds on Novo Nordisk’s commitment to offering health care professionals a range of treatments that effectively addresses the complex needs of diabetes management and fits their patients’ lifestyles,” said Todd Hobbs, vice president and U.S. chief medical officer of Novo Nordisk.
To ensure access to semaglutide, Novo Nordisk is pricing the drug competitively with other GLP-1 receptor agonists and will offer an associated savings card program to reduce copays for insured patients, the company said. Novo Nordisk expects to launch OZEMPIC in the United States in the first quarter of 2018, and is working on contracting solutions with health insurance providers to increase patient access to the drug.
According to the Novo Nordisk statement, clinicians should not consider semaglutide as a first choice option for treating diabetes or as a substitute for insulin in patients with type 1 diabetes and diabetic ketoacidosis. Whether semaglutide can be used by people who have had pancreatitis or is safe in patients under the age of 18 years old remains to be seen.
“Type 2 diabetes is a serious condition that affects more than 28 million people in the U.S., and despite advancements in treatment, some people with type 2 diabetes do not achieve their A1c goals,” said Helena Rodbard, MD, past president of the American Association of Clinical Endocrinologists. “The approval of semaglutide offers health care professionals an important new treatment option to help adults with type 2 diabetes meet their A1c goals.”
FDA recommends voluntary recall of Limbrel
The Food and Drug Administration announced on Dec. 4 that it recommends the voluntary recall of Limbrel, a medical food product in capsule form that is currently marketed to “manage the metabolic processes associated with osteoarthritis.”
![]()
The FDA’s ongoing investigation at this point considers the product to be an unapproved new drug rather than a medical food product. However, the agency does not have mandatory recall authority. It has recommended the recall to the product’s manufacturer, Primus Pharmaceuticals, on the basis of the risk of liver injury and hypersensitivity pneumonitis associated with continued use of the product.
The agency had received 194 adverse event reports as of Nov. 21, of which it found a likely association of the events with Limbrel in at least 30 cases, and continues to evaluate reports, which consumers can submit through MedWatch. The FDA is currently testing samples of the product and has advised consumers to cease taking it, though the manufacturer has declined thus far to recall it.
The safety alert advises that “health care providers who are aware that their patients are taking Limbrel should advise them to immediately stop taking the product.”
The Food and Drug Administration announced on Dec. 4 that it recommends the voluntary recall of Limbrel, a medical food product in capsule form that is currently marketed to “manage the metabolic processes associated with osteoarthritis.”
![]()
The FDA’s ongoing investigation at this point considers the product to be an unapproved new drug rather than a medical food product. However, the agency does not have mandatory recall authority. It has recommended the recall to the product’s manufacturer, Primus Pharmaceuticals, on the basis of the risk of liver injury and hypersensitivity pneumonitis associated with continued use of the product.
The agency had received 194 adverse event reports as of Nov. 21, of which it found a likely association of the events with Limbrel in at least 30 cases, and continues to evaluate reports, which consumers can submit through MedWatch. The FDA is currently testing samples of the product and has advised consumers to cease taking it, though the manufacturer has declined thus far to recall it.
The safety alert advises that “health care providers who are aware that their patients are taking Limbrel should advise them to immediately stop taking the product.”
The Food and Drug Administration announced on Dec. 4 that it recommends the voluntary recall of Limbrel, a medical food product in capsule form that is currently marketed to “manage the metabolic processes associated with osteoarthritis.”
![]()
The FDA’s ongoing investigation at this point considers the product to be an unapproved new drug rather than a medical food product. However, the agency does not have mandatory recall authority. It has recommended the recall to the product’s manufacturer, Primus Pharmaceuticals, on the basis of the risk of liver injury and hypersensitivity pneumonitis associated with continued use of the product.
The agency had received 194 adverse event reports as of Nov. 21, of which it found a likely association of the events with Limbrel in at least 30 cases, and continues to evaluate reports, which consumers can submit through MedWatch. The FDA is currently testing samples of the product and has advised consumers to cease taking it, though the manufacturer has declined thus far to recall it.
The safety alert advises that “health care providers who are aware that their patients are taking Limbrel should advise them to immediately stop taking the product.”
Anti-BCMA CAR T-cell therapy being fast tracked at FDA
The Food and Drug Administration has granted breakthrough therapy designation to bb2121, a chimeric antigen receptor T-cell (CAR T) therapy that targets b-cell maturation antigen (BCMA) in patients with relapsed/refractory multiple myeloma.
The therapy, being developed jointly by Celgene and bluebird bio, will be given expedited review by the FDA under the program. Meanwhile, European drug officials have granted it Priority Medicines eligibility, which also provides accelerated review.
The decision to fast track the review of bb2121 is based on preliminary data from the ongoing phase I CRB-401 trial. As of May 2017, there was 1-month clinical response data from 18 patients with multiple myeloma who were infused with bb2121. The overall response rate was 89%, but was 100% for patients who had been treated with doses of 150 × 106 CAR+ T cells or higher, according to an abstract from the annual meeting of the American Society of Hematology. Five months of follow-up data on these patients, plus initial data on additional patients, will be presented at ASH 2017 on Dec. 11.
The Food and Drug Administration has granted breakthrough therapy designation to bb2121, a chimeric antigen receptor T-cell (CAR T) therapy that targets b-cell maturation antigen (BCMA) in patients with relapsed/refractory multiple myeloma.
The therapy, being developed jointly by Celgene and bluebird bio, will be given expedited review by the FDA under the program. Meanwhile, European drug officials have granted it Priority Medicines eligibility, which also provides accelerated review.
The decision to fast track the review of bb2121 is based on preliminary data from the ongoing phase I CRB-401 trial. As of May 2017, there was 1-month clinical response data from 18 patients with multiple myeloma who were infused with bb2121. The overall response rate was 89%, but was 100% for patients who had been treated with doses of 150 × 106 CAR+ T cells or higher, according to an abstract from the annual meeting of the American Society of Hematology. Five months of follow-up data on these patients, plus initial data on additional patients, will be presented at ASH 2017 on Dec. 11.
The Food and Drug Administration has granted breakthrough therapy designation to bb2121, a chimeric antigen receptor T-cell (CAR T) therapy that targets b-cell maturation antigen (BCMA) in patients with relapsed/refractory multiple myeloma.
The therapy, being developed jointly by Celgene and bluebird bio, will be given expedited review by the FDA under the program. Meanwhile, European drug officials have granted it Priority Medicines eligibility, which also provides accelerated review.
The decision to fast track the review of bb2121 is based on preliminary data from the ongoing phase I CRB-401 trial. As of May 2017, there was 1-month clinical response data from 18 patients with multiple myeloma who were infused with bb2121. The overall response rate was 89%, but was 100% for patients who had been treated with doses of 150 × 106 CAR+ T cells or higher, according to an abstract from the annual meeting of the American Society of Hematology. Five months of follow-up data on these patients, plus initial data on additional patients, will be presented at ASH 2017 on Dec. 11.
Panel votes against universal blood donor screens for Zika virus
SILVER SPRING, MD – Universal testing of individual blood donations for the presence of Zika virus was unanimously rejected by voting members of the Food and Drug Administration’s Blood Products Advisory Committee at a December 1 meeting.
Universal individual donor testing, while comprehensive, is resource intensive and places a burden on the blood system that is not outweighed by the benefits, 10 of the 11 committee members concluded. The other committee member could not be reached by phone for this vote.
The committee instead recommended by a vote of 10 to 1 that mini-pool nucleic acid testing (MP-NAT) be performed in all states and territories with known cases of Zika virus infection and the presence of A. aegypti mosquitoes, as well as in states and territories with a high number of travelers from areas with Zika virus infections. Also, the committee members agreed that a trigger needs to be defined for when to undertake universal individual donor nucleic acid testing (ID-NAT) in those areas.
Additionally, the committee agreed that it was not necessary to maintain a Zika virus-negative blood inventory for at-risk patients, such as pregnant women and newborns. Zika virus, a vector-borne disease carried by the Aedes aegypti and Aedes albopictus mosquitoes, has also been transmitted via sexual contact and blood transfusion. Infection has been linked to fetal loss and microcephaly in the offspring of infected pregnant women. Other neurological disorders, including Guillain-Barré Syndrome, also have been linked to Zika virus infection.
Noting the complexity of managing an inventory of tested and non-tested blood, the committee rejected the separate inventory approach by a vote of 9 to 2.*
The panel was clearly divided on the possibility of eliminating all Zika virus testing in some states and territories; 5 members supported this measure, 4 opposed it, and 2 abstained from voting.
The panel unanimously rejected using screening questionnaires to determine whether to selectively test individual at-risk donors in areas with active vector-borne Zika virus infections. This option was considered particularly troublesome, they agreed, because it relies on the use of nonspecific, insensitive, and error-prone questionnaires.
Some level of Zika virus testing is needed to safeguard blood products, the committee said. Eliminating all Zika virus testing of blood products would open the door for infections via transfusion and would diminish preparedness against a potential epidemic, they unanimously determined.
Prior to voting, the committee listened to presentations on the epidemiology of Zika virus infections, the effectiveness of screening tests, and the risk for transmission via transfusion.
Carolyn Gould, MD, of the Centers for Disease Control and Prevention, Atlanta, reported that the number of laboratory-confirmed Zika virus infections in 2016 was 4,830 for travelers to endemic areas and 224 for locally-acquired cases. In 2017, those numbers dropped to 344 confirmed cases for travelers and 2 for locally-acquired cases.
More than 4 million blood donations in the United States and Puerto Rico have been screened for Zika virus RNA using the cobas assay, which is now FDA approved. The overall confirmed positive rate of Zika virus is 0.0007% in donations from the continental United States (29 positive results in 4,341,770 donations) and 0.326% in donations from Puerto Rico (356 out of 111,808 donations) based on data obtained from May 23, 2016 to October 7, 2017, according to Tony Hardiman, Blood Screening, Life Cycle Leader at Roche Molecular Systems.
Of the Zika virus-positive donors who were available for follow up, 23 of 27 had traveled to Zika-active areas, including 3 cases associated with domestic travel to Florida. “I was surprised that 4 of the 29 were from Cuba, but it does seem, as we just saw, (that) an increasing number are coming out from Cuba, from travel to Cuba,” Mr. Hardiman said during his presentation to the committee.
Findings concerning viral RNA duration in blood and other body fluids were presented by Michael Busch, MD of the University of California, San Francisco, who spoke during the public hearing portion of the meeting.**
According to Dr. Busch, blood is likely not infectious to others once donors develop Zika virus-neutralizing antibodies and their viral load levels become very low.
Based on his review of various studies, Dr. Busch concluded that mini-pool testing options with triggers for individual testing are appropriate and effective for detecting Zika virus in endemic areas.
“In Puerto Rico, within a day of picking up a mini-pool positive [result}, we would have ID-NAT in place,” Dr. Busch said. “The mini-pool testing is picking up 90% of those at highest risk.”
The committee recommendations serve as guidance to the FDA, which is not obligated to follow the committee’s recommendations.
*Correction 12/14/17: An earlier version of this story misstated the vote on maintaining a Zika virus-negative blood inventory for at-risk patients. The advisory committee voted against that approach.
**Correction 12/14/17: Dr. Michael Busch's name was misstated in an earlier version of this article.
SILVER SPRING, MD – Universal testing of individual blood donations for the presence of Zika virus was unanimously rejected by voting members of the Food and Drug Administration’s Blood Products Advisory Committee at a December 1 meeting.
Universal individual donor testing, while comprehensive, is resource intensive and places a burden on the blood system that is not outweighed by the benefits, 10 of the 11 committee members concluded. The other committee member could not be reached by phone for this vote.
The committee instead recommended by a vote of 10 to 1 that mini-pool nucleic acid testing (MP-NAT) be performed in all states and territories with known cases of Zika virus infection and the presence of A. aegypti mosquitoes, as well as in states and territories with a high number of travelers from areas with Zika virus infections. Also, the committee members agreed that a trigger needs to be defined for when to undertake universal individual donor nucleic acid testing (ID-NAT) in those areas.
Additionally, the committee agreed that it was not necessary to maintain a Zika virus-negative blood inventory for at-risk patients, such as pregnant women and newborns. Zika virus, a vector-borne disease carried by the Aedes aegypti and Aedes albopictus mosquitoes, has also been transmitted via sexual contact and blood transfusion. Infection has been linked to fetal loss and microcephaly in the offspring of infected pregnant women. Other neurological disorders, including Guillain-Barré Syndrome, also have been linked to Zika virus infection.
Noting the complexity of managing an inventory of tested and non-tested blood, the committee rejected the separate inventory approach by a vote of 9 to 2.*
The panel was clearly divided on the possibility of eliminating all Zika virus testing in some states and territories; 5 members supported this measure, 4 opposed it, and 2 abstained from voting.
The panel unanimously rejected using screening questionnaires to determine whether to selectively test individual at-risk donors in areas with active vector-borne Zika virus infections. This option was considered particularly troublesome, they agreed, because it relies on the use of nonspecific, insensitive, and error-prone questionnaires.
Some level of Zika virus testing is needed to safeguard blood products, the committee said. Eliminating all Zika virus testing of blood products would open the door for infections via transfusion and would diminish preparedness against a potential epidemic, they unanimously determined.
Prior to voting, the committee listened to presentations on the epidemiology of Zika virus infections, the effectiveness of screening tests, and the risk for transmission via transfusion.
Carolyn Gould, MD, of the Centers for Disease Control and Prevention, Atlanta, reported that the number of laboratory-confirmed Zika virus infections in 2016 was 4,830 for travelers to endemic areas and 224 for locally-acquired cases. In 2017, those numbers dropped to 344 confirmed cases for travelers and 2 for locally-acquired cases.
More than 4 million blood donations in the United States and Puerto Rico have been screened for Zika virus RNA using the cobas assay, which is now FDA approved. The overall confirmed positive rate of Zika virus is 0.0007% in donations from the continental United States (29 positive results in 4,341,770 donations) and 0.326% in donations from Puerto Rico (356 out of 111,808 donations) based on data obtained from May 23, 2016 to October 7, 2017, according to Tony Hardiman, Blood Screening, Life Cycle Leader at Roche Molecular Systems.
Of the Zika virus-positive donors who were available for follow up, 23 of 27 had traveled to Zika-active areas, including 3 cases associated with domestic travel to Florida. “I was surprised that 4 of the 29 were from Cuba, but it does seem, as we just saw, (that) an increasing number are coming out from Cuba, from travel to Cuba,” Mr. Hardiman said during his presentation to the committee.
Findings concerning viral RNA duration in blood and other body fluids were presented by Michael Busch, MD of the University of California, San Francisco, who spoke during the public hearing portion of the meeting.**
According to Dr. Busch, blood is likely not infectious to others once donors develop Zika virus-neutralizing antibodies and their viral load levels become very low.
Based on his review of various studies, Dr. Busch concluded that mini-pool testing options with triggers for individual testing are appropriate and effective for detecting Zika virus in endemic areas.
“In Puerto Rico, within a day of picking up a mini-pool positive [result}, we would have ID-NAT in place,” Dr. Busch said. “The mini-pool testing is picking up 90% of those at highest risk.”
The committee recommendations serve as guidance to the FDA, which is not obligated to follow the committee’s recommendations.
*Correction 12/14/17: An earlier version of this story misstated the vote on maintaining a Zika virus-negative blood inventory for at-risk patients. The advisory committee voted against that approach.
**Correction 12/14/17: Dr. Michael Busch's name was misstated in an earlier version of this article.
SILVER SPRING, MD – Universal testing of individual blood donations for the presence of Zika virus was unanimously rejected by voting members of the Food and Drug Administration’s Blood Products Advisory Committee at a December 1 meeting.
Universal individual donor testing, while comprehensive, is resource intensive and places a burden on the blood system that is not outweighed by the benefits, 10 of the 11 committee members concluded. The other committee member could not be reached by phone for this vote.
The committee instead recommended by a vote of 10 to 1 that mini-pool nucleic acid testing (MP-NAT) be performed in all states and territories with known cases of Zika virus infection and the presence of A. aegypti mosquitoes, as well as in states and territories with a high number of travelers from areas with Zika virus infections. Also, the committee members agreed that a trigger needs to be defined for when to undertake universal individual donor nucleic acid testing (ID-NAT) in those areas.
Additionally, the committee agreed that it was not necessary to maintain a Zika virus-negative blood inventory for at-risk patients, such as pregnant women and newborns. Zika virus, a vector-borne disease carried by the Aedes aegypti and Aedes albopictus mosquitoes, has also been transmitted via sexual contact and blood transfusion. Infection has been linked to fetal loss and microcephaly in the offspring of infected pregnant women. Other neurological disorders, including Guillain-Barré Syndrome, also have been linked to Zika virus infection.
Noting the complexity of managing an inventory of tested and non-tested blood, the committee rejected the separate inventory approach by a vote of 9 to 2.*
The panel was clearly divided on the possibility of eliminating all Zika virus testing in some states and territories; 5 members supported this measure, 4 opposed it, and 2 abstained from voting.
The panel unanimously rejected using screening questionnaires to determine whether to selectively test individual at-risk donors in areas with active vector-borne Zika virus infections. This option was considered particularly troublesome, they agreed, because it relies on the use of nonspecific, insensitive, and error-prone questionnaires.
Some level of Zika virus testing is needed to safeguard blood products, the committee said. Eliminating all Zika virus testing of blood products would open the door for infections via transfusion and would diminish preparedness against a potential epidemic, they unanimously determined.
Prior to voting, the committee listened to presentations on the epidemiology of Zika virus infections, the effectiveness of screening tests, and the risk for transmission via transfusion.
Carolyn Gould, MD, of the Centers for Disease Control and Prevention, Atlanta, reported that the number of laboratory-confirmed Zika virus infections in 2016 was 4,830 for travelers to endemic areas and 224 for locally-acquired cases. In 2017, those numbers dropped to 344 confirmed cases for travelers and 2 for locally-acquired cases.
More than 4 million blood donations in the United States and Puerto Rico have been screened for Zika virus RNA using the cobas assay, which is now FDA approved. The overall confirmed positive rate of Zika virus is 0.0007% in donations from the continental United States (29 positive results in 4,341,770 donations) and 0.326% in donations from Puerto Rico (356 out of 111,808 donations) based on data obtained from May 23, 2016 to October 7, 2017, according to Tony Hardiman, Blood Screening, Life Cycle Leader at Roche Molecular Systems.
Of the Zika virus-positive donors who were available for follow up, 23 of 27 had traveled to Zika-active areas, including 3 cases associated with domestic travel to Florida. “I was surprised that 4 of the 29 were from Cuba, but it does seem, as we just saw, (that) an increasing number are coming out from Cuba, from travel to Cuba,” Mr. Hardiman said during his presentation to the committee.
Findings concerning viral RNA duration in blood and other body fluids were presented by Michael Busch, MD of the University of California, San Francisco, who spoke during the public hearing portion of the meeting.**
According to Dr. Busch, blood is likely not infectious to others once donors develop Zika virus-neutralizing antibodies and their viral load levels become very low.
Based on his review of various studies, Dr. Busch concluded that mini-pool testing options with triggers for individual testing are appropriate and effective for detecting Zika virus in endemic areas.
“In Puerto Rico, within a day of picking up a mini-pool positive [result}, we would have ID-NAT in place,” Dr. Busch said. “The mini-pool testing is picking up 90% of those at highest risk.”
The committee recommendations serve as guidance to the FDA, which is not obligated to follow the committee’s recommendations.
*Correction 12/14/17: An earlier version of this story misstated the vote on maintaining a Zika virus-negative blood inventory for at-risk patients. The advisory committee voted against that approach.
**Correction 12/14/17: Dr. Michael Busch's name was misstated in an earlier version of this article.
AT AN FDA ADVISORY COMMITTEE MEETING
New buprenorphine formulation approved for medication-assisted treatment
The Food and Drug Administration has approved an extended-release, subcutaneous injection formulation of buprenorphine for use in treating moderate to severe opioid use disorder (OUD), the manufacturer of the drug announced Nov. 30.
The new product, called Sublocade, is a monthly injection intended for use in patients who have already begun treatment of OUD with transmucosal buprenorphine products, followed by a dose adjustment for a minimum of 7 days. Sublocade contains the partial mu-opioid agonist buprenorphine. By administering a consistent level of buprenorphine into the body, it ensures that levels of buprenorphine are delivered to the mu-opioid receptors, diminishing the effects of opioids, including the euphoric sensations associated with opioid use. During the clinical trial program, buprenorphine plasma concentrations of 2-3 ng/mL were found to bind to greater than 70% of mu-opioid receptors.
According to a statement from the FDA, Sublocade will be distributed only to health care providers as part of a Risk Evaluation and Mitigation Strategy to ensure that the product is not distributed directly to patients. Sublocade should be administered only by a health care professional. Self-injection of Sublocade into the blood stream instead of subcutaneously could lead to occlusion of blood vessels and embolism, according to one of the drug’s boxed warnings. It also should be used as part of a complete treatment program that includes counseling and psychosocial support.
The FDA is also requiring the manufacturer to conduct postmarketing studies to assess which patients would benefit from a higher dosing regimen, to determine whether Sublocade can be safely initiated without a dose stabilization period of sublingual buprenorphine, to assess the feasibility of administering Sublocade at a longer inter-dose interval than once monthly, and to determine a process for transitioning patients with long-term stability on a transmucosal buprenorphine dose to a monthly dose of Sublocade without the use of a higher dose for the first 2 months of treatment.
At recent joint meetings of the FDA’s Psychopharmacologic Drugs and Drug Safety and Risk Management advisory committees, panelists voted on Oct. 31 to recommend approval of Sublocade and on Nov. 1 for another subcutaneous buprenorphine injection formulation. These actions have not gone unnoticed by the American Medical Association.
“The AMA enthusiastically supports Food and Drug Administration Commissioner Scott Gottlieb’s efforts to advance policies and actions to treat those suffering from an opioid use disorder,” Patrice Harris, MD, immediate past chair of the American Medical Association Board of Trustees and a member of the AMA Opioid Task Force, said in a statement. “We also second his bold acknowledgment that criminal justice systems should offer [medication-assisted treatment] to those being detained. As he points out, ‘At the very moment when the criminal justice system could be dramatically lowering the risk of overdose, it is creating the conditions of reduced tolerance to opioids that substantially raises the risk of death upon release.’ With his clear explanation of the problem and solution, this situation can be remedied.”
The Food and Drug Administration has approved an extended-release, subcutaneous injection formulation of buprenorphine for use in treating moderate to severe opioid use disorder (OUD), the manufacturer of the drug announced Nov. 30.
The new product, called Sublocade, is a monthly injection intended for use in patients who have already begun treatment of OUD with transmucosal buprenorphine products, followed by a dose adjustment for a minimum of 7 days. Sublocade contains the partial mu-opioid agonist buprenorphine. By administering a consistent level of buprenorphine into the body, it ensures that levels of buprenorphine are delivered to the mu-opioid receptors, diminishing the effects of opioids, including the euphoric sensations associated with opioid use. During the clinical trial program, buprenorphine plasma concentrations of 2-3 ng/mL were found to bind to greater than 70% of mu-opioid receptors.
According to a statement from the FDA, Sublocade will be distributed only to health care providers as part of a Risk Evaluation and Mitigation Strategy to ensure that the product is not distributed directly to patients. Sublocade should be administered only by a health care professional. Self-injection of Sublocade into the blood stream instead of subcutaneously could lead to occlusion of blood vessels and embolism, according to one of the drug’s boxed warnings. It also should be used as part of a complete treatment program that includes counseling and psychosocial support.
The FDA is also requiring the manufacturer to conduct postmarketing studies to assess which patients would benefit from a higher dosing regimen, to determine whether Sublocade can be safely initiated without a dose stabilization period of sublingual buprenorphine, to assess the feasibility of administering Sublocade at a longer inter-dose interval than once monthly, and to determine a process for transitioning patients with long-term stability on a transmucosal buprenorphine dose to a monthly dose of Sublocade without the use of a higher dose for the first 2 months of treatment.
At recent joint meetings of the FDA’s Psychopharmacologic Drugs and Drug Safety and Risk Management advisory committees, panelists voted on Oct. 31 to recommend approval of Sublocade and on Nov. 1 for another subcutaneous buprenorphine injection formulation. These actions have not gone unnoticed by the American Medical Association.
“The AMA enthusiastically supports Food and Drug Administration Commissioner Scott Gottlieb’s efforts to advance policies and actions to treat those suffering from an opioid use disorder,” Patrice Harris, MD, immediate past chair of the American Medical Association Board of Trustees and a member of the AMA Opioid Task Force, said in a statement. “We also second his bold acknowledgment that criminal justice systems should offer [medication-assisted treatment] to those being detained. As he points out, ‘At the very moment when the criminal justice system could be dramatically lowering the risk of overdose, it is creating the conditions of reduced tolerance to opioids that substantially raises the risk of death upon release.’ With his clear explanation of the problem and solution, this situation can be remedied.”
The Food and Drug Administration has approved an extended-release, subcutaneous injection formulation of buprenorphine for use in treating moderate to severe opioid use disorder (OUD), the manufacturer of the drug announced Nov. 30.
The new product, called Sublocade, is a monthly injection intended for use in patients who have already begun treatment of OUD with transmucosal buprenorphine products, followed by a dose adjustment for a minimum of 7 days. Sublocade contains the partial mu-opioid agonist buprenorphine. By administering a consistent level of buprenorphine into the body, it ensures that levels of buprenorphine are delivered to the mu-opioid receptors, diminishing the effects of opioids, including the euphoric sensations associated with opioid use. During the clinical trial program, buprenorphine plasma concentrations of 2-3 ng/mL were found to bind to greater than 70% of mu-opioid receptors.
According to a statement from the FDA, Sublocade will be distributed only to health care providers as part of a Risk Evaluation and Mitigation Strategy to ensure that the product is not distributed directly to patients. Sublocade should be administered only by a health care professional. Self-injection of Sublocade into the blood stream instead of subcutaneously could lead to occlusion of blood vessels and embolism, according to one of the drug’s boxed warnings. It also should be used as part of a complete treatment program that includes counseling and psychosocial support.
The FDA is also requiring the manufacturer to conduct postmarketing studies to assess which patients would benefit from a higher dosing regimen, to determine whether Sublocade can be safely initiated without a dose stabilization period of sublingual buprenorphine, to assess the feasibility of administering Sublocade at a longer inter-dose interval than once monthly, and to determine a process for transitioning patients with long-term stability on a transmucosal buprenorphine dose to a monthly dose of Sublocade without the use of a higher dose for the first 2 months of treatment.
At recent joint meetings of the FDA’s Psychopharmacologic Drugs and Drug Safety and Risk Management advisory committees, panelists voted on Oct. 31 to recommend approval of Sublocade and on Nov. 1 for another subcutaneous buprenorphine injection formulation. These actions have not gone unnoticed by the American Medical Association.
“The AMA enthusiastically supports Food and Drug Administration Commissioner Scott Gottlieb’s efforts to advance policies and actions to treat those suffering from an opioid use disorder,” Patrice Harris, MD, immediate past chair of the American Medical Association Board of Trustees and a member of the AMA Opioid Task Force, said in a statement. “We also second his bold acknowledgment that criminal justice systems should offer [medication-assisted treatment] to those being detained. As he points out, ‘At the very moment when the criminal justice system could be dramatically lowering the risk of overdose, it is creating the conditions of reduced tolerance to opioids that substantially raises the risk of death upon release.’ With his clear explanation of the problem and solution, this situation can be remedied.”