User login
FDA allows marketing of TMS to treat OCD
The Food and Drug Administration has cleared the way for marketing of the BrainsWay deep transcranial magnetic stimulation system for treating patients with treatment-resistant obsessive-compulsive disorder (OCD).
“With today’s marketing authorization, patients with OCD who have not responded to traditional treatments now have another option,” said Carlos Peña, PhD, director of the division of neurological and physical medicine devices in the FDA’s Center for Devices and Radiological Health, in an Aug. 17 news release.
49 of whom were treated with the BrainsWay device and 51 of whom with a sham device. Of the patients treated with the BrainsWay device, 38% had a 30% or greater reduction in their Yale-Brown Obsessive Compulsive Scale scores, which measures severity of OCD symptoms, whereas only 11% treated with the sham device experienced such a reduction.
The most common adverse reaction was headache, experienced by 37.5% of patients treated with the BrainsWay device and 35.0% of those treated with the sham device. No serious adverse events were reported. Other common reactions included application site pain or discomfort, spasm or twitching, and jaw, facial, muscle, or neck pain; all of those were reported as mild or moderate and resolved quickly after each treatment was completed.
The device is contraindicated in patients with any sort of metal in or near their heads, such as cochlear implants or vagus nerve stimulators. Patients with a history of seizure should discuss it with their health care clinician.
Transcranial magnetic stimulation also has been approved to treat major depressive disorder and migraine with aura.
The full press release can be found on the FDA website.
The Food and Drug Administration has cleared the way for marketing of the BrainsWay deep transcranial magnetic stimulation system for treating patients with treatment-resistant obsessive-compulsive disorder (OCD).
“With today’s marketing authorization, patients with OCD who have not responded to traditional treatments now have another option,” said Carlos Peña, PhD, director of the division of neurological and physical medicine devices in the FDA’s Center for Devices and Radiological Health, in an Aug. 17 news release.
49 of whom were treated with the BrainsWay device and 51 of whom with a sham device. Of the patients treated with the BrainsWay device, 38% had a 30% or greater reduction in their Yale-Brown Obsessive Compulsive Scale scores, which measures severity of OCD symptoms, whereas only 11% treated with the sham device experienced such a reduction.
The most common adverse reaction was headache, experienced by 37.5% of patients treated with the BrainsWay device and 35.0% of those treated with the sham device. No serious adverse events were reported. Other common reactions included application site pain or discomfort, spasm or twitching, and jaw, facial, muscle, or neck pain; all of those were reported as mild or moderate and resolved quickly after each treatment was completed.
The device is contraindicated in patients with any sort of metal in or near their heads, such as cochlear implants or vagus nerve stimulators. Patients with a history of seizure should discuss it with their health care clinician.
Transcranial magnetic stimulation also has been approved to treat major depressive disorder and migraine with aura.
The full press release can be found on the FDA website.
The Food and Drug Administration has cleared the way for marketing of the BrainsWay deep transcranial magnetic stimulation system for treating patients with treatment-resistant obsessive-compulsive disorder (OCD).
“With today’s marketing authorization, patients with OCD who have not responded to traditional treatments now have another option,” said Carlos Peña, PhD, director of the division of neurological and physical medicine devices in the FDA’s Center for Devices and Radiological Health, in an Aug. 17 news release.
49 of whom were treated with the BrainsWay device and 51 of whom with a sham device. Of the patients treated with the BrainsWay device, 38% had a 30% or greater reduction in their Yale-Brown Obsessive Compulsive Scale scores, which measures severity of OCD symptoms, whereas only 11% treated with the sham device experienced such a reduction.
The most common adverse reaction was headache, experienced by 37.5% of patients treated with the BrainsWay device and 35.0% of those treated with the sham device. No serious adverse events were reported. Other common reactions included application site pain or discomfort, spasm or twitching, and jaw, facial, muscle, or neck pain; all of those were reported as mild or moderate and resolved quickly after each treatment was completed.
The device is contraindicated in patients with any sort of metal in or near their heads, such as cochlear implants or vagus nerve stimulators. Patients with a history of seizure should discuss it with their health care clinician.
Transcranial magnetic stimulation also has been approved to treat major depressive disorder and migraine with aura.
The full press release can be found on the FDA website.
FDA requires companion diagnostics for checkpoint inhibitors in urothelial cancer
Two separate FDA-approved companion diagnostic tests are now required to determine PD-L1 levels in tumor tissue for those who are cisplatin-ineligible.
Pembrolizumab is now indicated for the treatment of patients who are not eligible for cisplatin-containing chemotherapy and whose tumors have a Combined Positive Score (CPS) for PD-L1 expression of greater than or equal to 10, or in patients who are not eligible for any platinum-containing chemotherapy regardless of PD-L1 status. The FDA approved the Dako PD-L1 IHC 22C3 PharmDx Assay for determining PD-L1 expression, through staining in tumor and immune cells, before prescribing pembrolizumab.
Atezolizumab is now indicated for the treatment of patients who are not eligible for cisplatin-containing chemotherapy, and whose tumors show PD-L1 expression through stained tumor-infiltrating immune cells covering greater than or equal to 5% of the tumor area, or patients who are not eligible for any platinum-containing therapy regardless of level of tumor PD-L1 expression. The FDA approved the Ventana PD-L1 (SP142) Assay as a companion diagnostic test to determine PD L1 expression in immune cells before prescribing atezolizumab.
PI is updated for both drugs to require use of an FDA-approved test for selection of patients being treated in the first-line setting who are cisplatin-ineligible, but second-line indications in urothelial carcinoma for both drugs remain unchanged, according to an FDA statement.
Two separate FDA-approved companion diagnostic tests are now required to determine PD-L1 levels in tumor tissue for those who are cisplatin-ineligible.
Pembrolizumab is now indicated for the treatment of patients who are not eligible for cisplatin-containing chemotherapy and whose tumors have a Combined Positive Score (CPS) for PD-L1 expression of greater than or equal to 10, or in patients who are not eligible for any platinum-containing chemotherapy regardless of PD-L1 status. The FDA approved the Dako PD-L1 IHC 22C3 PharmDx Assay for determining PD-L1 expression, through staining in tumor and immune cells, before prescribing pembrolizumab.
Atezolizumab is now indicated for the treatment of patients who are not eligible for cisplatin-containing chemotherapy, and whose tumors show PD-L1 expression through stained tumor-infiltrating immune cells covering greater than or equal to 5% of the tumor area, or patients who are not eligible for any platinum-containing therapy regardless of level of tumor PD-L1 expression. The FDA approved the Ventana PD-L1 (SP142) Assay as a companion diagnostic test to determine PD L1 expression in immune cells before prescribing atezolizumab.
PI is updated for both drugs to require use of an FDA-approved test for selection of patients being treated in the first-line setting who are cisplatin-ineligible, but second-line indications in urothelial carcinoma for both drugs remain unchanged, according to an FDA statement.
Two separate FDA-approved companion diagnostic tests are now required to determine PD-L1 levels in tumor tissue for those who are cisplatin-ineligible.
Pembrolizumab is now indicated for the treatment of patients who are not eligible for cisplatin-containing chemotherapy and whose tumors have a Combined Positive Score (CPS) for PD-L1 expression of greater than or equal to 10, or in patients who are not eligible for any platinum-containing chemotherapy regardless of PD-L1 status. The FDA approved the Dako PD-L1 IHC 22C3 PharmDx Assay for determining PD-L1 expression, through staining in tumor and immune cells, before prescribing pembrolizumab.
Atezolizumab is now indicated for the treatment of patients who are not eligible for cisplatin-containing chemotherapy, and whose tumors show PD-L1 expression through stained tumor-infiltrating immune cells covering greater than or equal to 5% of the tumor area, or patients who are not eligible for any platinum-containing therapy regardless of level of tumor PD-L1 expression. The FDA approved the Ventana PD-L1 (SP142) Assay as a companion diagnostic test to determine PD L1 expression in immune cells before prescribing atezolizumab.
PI is updated for both drugs to require use of an FDA-approved test for selection of patients being treated in the first-line setting who are cisplatin-ineligible, but second-line indications in urothelial carcinoma for both drugs remain unchanged, according to an FDA statement.
FDA alert: Artificial heart driver linked to higher mortality
Postapproval results for SynCardia Systems’ Companion 2 (C2) driver system for temporary total artificial hearts (TAH-t) have shown higher mortality and stroke rates than were seen with the previous system, the circulatory support system. As a result, the Food and Drug Administration has issued a safety alert cautioning them to weigh the risks and benefits carefully. The alert, issued on August 17, is based on a postapproval study conducted by SynCardia Systems.

Furthermore, patients and health care professionals are encouraged to report any adverse events using the FDA’s Medwatch reporting form, as well as return any devices associated with adverse events to the SynCardia Systems to help them and the FDA better understand the issue.
The C2 driver system is an external pneumatic system that activates an implanted TAH-t in eligible heart failure patients who have severe biventricular failure and are waiting for transplant. It is smaller than its predecessor, but per the device’s approved use, patients must still remain in the hospital while on the device. Since its approval in 2012, the Freedom driver system was approved in 2014, which allows patients to return home.
The full safety alert can be found on the FDA website.
Postapproval results for SynCardia Systems’ Companion 2 (C2) driver system for temporary total artificial hearts (TAH-t) have shown higher mortality and stroke rates than were seen with the previous system, the circulatory support system. As a result, the Food and Drug Administration has issued a safety alert cautioning them to weigh the risks and benefits carefully. The alert, issued on August 17, is based on a postapproval study conducted by SynCardia Systems.
Furthermore, patients and health care professionals are encouraged to report any adverse events using the FDA’s Medwatch reporting form, as well as return any devices associated with adverse events to the SynCardia Systems to help them and the FDA better understand the issue.
The C2 driver system is an external pneumatic system that activates an implanted TAH-t in eligible heart failure patients who have severe biventricular failure and are waiting for transplant. It is smaller than its predecessor, but per the device’s approved use, patients must still remain in the hospital while on the device. Since its approval in 2012, the Freedom driver system was approved in 2014, which allows patients to return home.
The full safety alert can be found on the FDA website.
Postapproval results for SynCardia Systems’ Companion 2 (C2) driver system for temporary total artificial hearts (TAH-t) have shown higher mortality and stroke rates than were seen with the previous system, the circulatory support system. As a result, the Food and Drug Administration has issued a safety alert cautioning them to weigh the risks and benefits carefully. The alert, issued on August 17, is based on a postapproval study conducted by SynCardia Systems.
Furthermore, patients and health care professionals are encouraged to report any adverse events using the FDA’s Medwatch reporting form, as well as return any devices associated with adverse events to the SynCardia Systems to help them and the FDA better understand the issue.
The C2 driver system is an external pneumatic system that activates an implanted TAH-t in eligible heart failure patients who have severe biventricular failure and are waiting for transplant. It is smaller than its predecessor, but per the device’s approved use, patients must still remain in the hospital while on the device. Since its approval in 2012, the Freedom driver system was approved in 2014, which allows patients to return home.
The full safety alert can be found on the FDA website.
CDC: 2017 worst year yet for drug overdoses
An estimated 72,000 drug overdose deaths occurred in 2017 in the United States, making it the worst year on record, according to preliminary data released by the Centers for Disease Control and Prevention.
The record high was driven by a sharp increase in deaths attributed to synthetic opioids, such as fentanyl and tramadol, data published on the agency’s website show.
The provisional counts are based on death records sent to the CDC’s National Center for Health Statistics from state vital registration offices in all 50 states and the District of Columbia, reported lead author Farida B. Ahmad, MPH, of the division of vital statistics at the NCHS. Overall, the predicted number of drug overdose deaths has climbed steadily, from 54,207 in November 2015 to 66,012 in November 2016, and to 72,287 in November 2017, according to an interactive chart accessible on the website.
Deaths attributable to synthetic opioids have climbed faster than any other drug class, soaring from just 9,983 in 2015 to 20,310 in 2016, and to 29,418 in 2017.
The next-largest category, heroin-related deaths, increased from 13,407 in 2015 to 16,012 in 2016, but appeared to plateau at 15,959 in 2017. However, the CDC cautioned that flat or declining numbers could be attributable to incomplete data, true decreases in deaths, or some combination of the two. “True declines or plateaus in the numbers of drug overdose deaths across the U.S. cannot be ascertained until final data become available.”
Cocaine-related deaths were fewer in number but appear to have risen substantially to the point where the number of deaths now nearly rival that of heroin. The number of deaths was 7,106 in 2015, 10,868 in 2016, and 14,614 in 2017.
The count of drug overdose deaths varied by state. coming in at 33.3%, though the absolute numbers of cases were low (114 through January 2017 and 152 through January 2018). North Carolina also showed substantial increases (from 2,053 to 2,515 cases, 22.5%), as did New Jersey (2,219 to 2,687 cases, 21.1%), the CDC data showed.
Provisional data will be updated on a monthly basis as additional records are received, the CDC said.
An estimated 72,000 drug overdose deaths occurred in 2017 in the United States, making it the worst year on record, according to preliminary data released by the Centers for Disease Control and Prevention.
The record high was driven by a sharp increase in deaths attributed to synthetic opioids, such as fentanyl and tramadol, data published on the agency’s website show.
The provisional counts are based on death records sent to the CDC’s National Center for Health Statistics from state vital registration offices in all 50 states and the District of Columbia, reported lead author Farida B. Ahmad, MPH, of the division of vital statistics at the NCHS. Overall, the predicted number of drug overdose deaths has climbed steadily, from 54,207 in November 2015 to 66,012 in November 2016, and to 72,287 in November 2017, according to an interactive chart accessible on the website.
Deaths attributable to synthetic opioids have climbed faster than any other drug class, soaring from just 9,983 in 2015 to 20,310 in 2016, and to 29,418 in 2017.
The next-largest category, heroin-related deaths, increased from 13,407 in 2015 to 16,012 in 2016, but appeared to plateau at 15,959 in 2017. However, the CDC cautioned that flat or declining numbers could be attributable to incomplete data, true decreases in deaths, or some combination of the two. “True declines or plateaus in the numbers of drug overdose deaths across the U.S. cannot be ascertained until final data become available.”
Cocaine-related deaths were fewer in number but appear to have risen substantially to the point where the number of deaths now nearly rival that of heroin. The number of deaths was 7,106 in 2015, 10,868 in 2016, and 14,614 in 2017.
The count of drug overdose deaths varied by state. coming in at 33.3%, though the absolute numbers of cases were low (114 through January 2017 and 152 through January 2018). North Carolina also showed substantial increases (from 2,053 to 2,515 cases, 22.5%), as did New Jersey (2,219 to 2,687 cases, 21.1%), the CDC data showed.
Provisional data will be updated on a monthly basis as additional records are received, the CDC said.
An estimated 72,000 drug overdose deaths occurred in 2017 in the United States, making it the worst year on record, according to preliminary data released by the Centers for Disease Control and Prevention.
The record high was driven by a sharp increase in deaths attributed to synthetic opioids, such as fentanyl and tramadol, data published on the agency’s website show.
The provisional counts are based on death records sent to the CDC’s National Center for Health Statistics from state vital registration offices in all 50 states and the District of Columbia, reported lead author Farida B. Ahmad, MPH, of the division of vital statistics at the NCHS. Overall, the predicted number of drug overdose deaths has climbed steadily, from 54,207 in November 2015 to 66,012 in November 2016, and to 72,287 in November 2017, according to an interactive chart accessible on the website.
Deaths attributable to synthetic opioids have climbed faster than any other drug class, soaring from just 9,983 in 2015 to 20,310 in 2016, and to 29,418 in 2017.
The next-largest category, heroin-related deaths, increased from 13,407 in 2015 to 16,012 in 2016, but appeared to plateau at 15,959 in 2017. However, the CDC cautioned that flat or declining numbers could be attributable to incomplete data, true decreases in deaths, or some combination of the two. “True declines or plateaus in the numbers of drug overdose deaths across the U.S. cannot be ascertained until final data become available.”
Cocaine-related deaths were fewer in number but appear to have risen substantially to the point where the number of deaths now nearly rival that of heroin. The number of deaths was 7,106 in 2015, 10,868 in 2016, and 14,614 in 2017.
The count of drug overdose deaths varied by state. coming in at 33.3%, though the absolute numbers of cases were low (114 through January 2017 and 152 through January 2018). North Carolina also showed substantial increases (from 2,053 to 2,515 cases, 22.5%), as did New Jersey (2,219 to 2,687 cases, 21.1%), the CDC data showed.
Provisional data will be updated on a monthly basis as additional records are received, the CDC said.
CPI-613 receives orphan designation for PTCL
The Food and Drug Administration has granted orphan drug designation to CPI-613 for the treatment of peripheral T-cell lymphoma (PTCL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
Rafael Pharmaceuticals is developing CPI-613 as a treatment for hematologic malignancies and solid tumors.
CPI-613 is currently under investigation in combination with bendamustine in a phase 1 study of patients with relapsed or refractory T-cell lymphoma or classical Hodgkin lymphoma, according to the press release from the company.
Results from this trial were presented at the 2016 annual meeting of the American Society of Hematology.
CPI-613 was given at escalating doses starting at 2,000 mg/m2 over 2 hours on days 1-4 as well as on days 8, 11, 15, and 18. Bendamustine was infused at 90 mg/m2 on days 4 and 5 of each 4-week treatment cycle. The treatment cycles were repeated for up to six cycles. There was no intrapatient dose escalation.
The ASH presentation included safety data on eight patients. The most common grade 3 or higher toxicities – lymphopenia and neutropenia – occurred in four patients.
A patient dosed at 2,750 mg/m2 had a dose-limiting toxicity of grade 3 acute kidney injury and grade 4 lactic acidosis. Because of this, the study protocol was amended to discontinue dose escalation at doses of 2,750 mg/m2 or higher and to expand the 2,500 mg/m2 cohort.
Six patients were evaluable for efficacy, and the overall response rate was 83% (5/6).
There were three complete responses in patients with PTCL not otherwise specified, a partial response in a patient with mycosis fungoides, and a partial response in a patient with angioimmunoblastic T-cell lymphoma.
One patient with T-cell acute lymphoblastic leukemia experienced progressive disease.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the United States.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The Food and Drug Administration has granted orphan drug designation to CPI-613 for the treatment of peripheral T-cell lymphoma (PTCL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
Rafael Pharmaceuticals is developing CPI-613 as a treatment for hematologic malignancies and solid tumors.
CPI-613 is currently under investigation in combination with bendamustine in a phase 1 study of patients with relapsed or refractory T-cell lymphoma or classical Hodgkin lymphoma, according to the press release from the company.
Results from this trial were presented at the 2016 annual meeting of the American Society of Hematology.
CPI-613 was given at escalating doses starting at 2,000 mg/m2 over 2 hours on days 1-4 as well as on days 8, 11, 15, and 18. Bendamustine was infused at 90 mg/m2 on days 4 and 5 of each 4-week treatment cycle. The treatment cycles were repeated for up to six cycles. There was no intrapatient dose escalation.
The ASH presentation included safety data on eight patients. The most common grade 3 or higher toxicities – lymphopenia and neutropenia – occurred in four patients.
A patient dosed at 2,750 mg/m2 had a dose-limiting toxicity of grade 3 acute kidney injury and grade 4 lactic acidosis. Because of this, the study protocol was amended to discontinue dose escalation at doses of 2,750 mg/m2 or higher and to expand the 2,500 mg/m2 cohort.
Six patients were evaluable for efficacy, and the overall response rate was 83% (5/6).
There were three complete responses in patients with PTCL not otherwise specified, a partial response in a patient with mycosis fungoides, and a partial response in a patient with angioimmunoblastic T-cell lymphoma.
One patient with T-cell acute lymphoblastic leukemia experienced progressive disease.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the United States.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
The Food and Drug Administration has granted orphan drug designation to CPI-613 for the treatment of peripheral T-cell lymphoma (PTCL).
CPI-613 is a novel lipoic acid analogue that inhibits multiple enzyme targets within the tricarboxylic acid cycle.
Rafael Pharmaceuticals is developing CPI-613 as a treatment for hematologic malignancies and solid tumors.
CPI-613 is currently under investigation in combination with bendamustine in a phase 1 study of patients with relapsed or refractory T-cell lymphoma or classical Hodgkin lymphoma, according to the press release from the company.
Results from this trial were presented at the 2016 annual meeting of the American Society of Hematology.
CPI-613 was given at escalating doses starting at 2,000 mg/m2 over 2 hours on days 1-4 as well as on days 8, 11, 15, and 18. Bendamustine was infused at 90 mg/m2 on days 4 and 5 of each 4-week treatment cycle. The treatment cycles were repeated for up to six cycles. There was no intrapatient dose escalation.
The ASH presentation included safety data on eight patients. The most common grade 3 or higher toxicities – lymphopenia and neutropenia – occurred in four patients.
A patient dosed at 2,750 mg/m2 had a dose-limiting toxicity of grade 3 acute kidney injury and grade 4 lactic acidosis. Because of this, the study protocol was amended to discontinue dose escalation at doses of 2,750 mg/m2 or higher and to expand the 2,500 mg/m2 cohort.
Six patients were evaluable for efficacy, and the overall response rate was 83% (5/6).
There were three complete responses in patients with PTCL not otherwise specified, a partial response in a patient with mycosis fungoides, and a partial response in a patient with angioimmunoblastic T-cell lymphoma.
One patient with T-cell acute lymphoblastic leukemia experienced progressive disease.
The FDA grants orphan designation to products intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the United States.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity if the product is approved.
FDA approves lenvatinib for HCC
Approval was based on a noninferiority trial of 954 patients with previously untreated, metastatic or unresectable HCC, comparing treatment with lenvatinib to sorafenib, according to an FDA statement.
Lenvatinib was found noninferior but not statistically superior to sorafenib for overall survival (hazard ratio, 0.92; 95% confidence interval, 0.79-1.06). Median overall survival was 13.6 months for patients in the lenvatinib arm, compared with 12.3 months for patients in the sorafenib arm.
The most common adverse reactions with lenvatinib were hypertension, fatigue, diarrhea, decreased appetite, arthralgia/myalgia, decreased weight, abdominal pain, palmar-plantar erythrodysesthesia syndrome, proteinuria, dysphonia, hemorrhagic events, hypothyroidism, and nausea.
The recommended lenvatinib dosages are 12 mg orally once daily in patients weighing 60 kg or greater actual body weight or 8 mg orally once daily in patients weighing less than 60 kg actual body weight, the FDA said.
Lenvatinib is marketed as Lenvima by Eisai.
Approval was based on a noninferiority trial of 954 patients with previously untreated, metastatic or unresectable HCC, comparing treatment with lenvatinib to sorafenib, according to an FDA statement.
Lenvatinib was found noninferior but not statistically superior to sorafenib for overall survival (hazard ratio, 0.92; 95% confidence interval, 0.79-1.06). Median overall survival was 13.6 months for patients in the lenvatinib arm, compared with 12.3 months for patients in the sorafenib arm.
The most common adverse reactions with lenvatinib were hypertension, fatigue, diarrhea, decreased appetite, arthralgia/myalgia, decreased weight, abdominal pain, palmar-plantar erythrodysesthesia syndrome, proteinuria, dysphonia, hemorrhagic events, hypothyroidism, and nausea.
The recommended lenvatinib dosages are 12 mg orally once daily in patients weighing 60 kg or greater actual body weight or 8 mg orally once daily in patients weighing less than 60 kg actual body weight, the FDA said.
Lenvatinib is marketed as Lenvima by Eisai.
Approval was based on a noninferiority trial of 954 patients with previously untreated, metastatic or unresectable HCC, comparing treatment with lenvatinib to sorafenib, according to an FDA statement.
Lenvatinib was found noninferior but not statistically superior to sorafenib for overall survival (hazard ratio, 0.92; 95% confidence interval, 0.79-1.06). Median overall survival was 13.6 months for patients in the lenvatinib arm, compared with 12.3 months for patients in the sorafenib arm.
The most common adverse reactions with lenvatinib were hypertension, fatigue, diarrhea, decreased appetite, arthralgia/myalgia, decreased weight, abdominal pain, palmar-plantar erythrodysesthesia syndrome, proteinuria, dysphonia, hemorrhagic events, hypothyroidism, and nausea.
The recommended lenvatinib dosages are 12 mg orally once daily in patients weighing 60 kg or greater actual body weight or 8 mg orally once daily in patients weighing less than 60 kg actual body weight, the FDA said.
Lenvatinib is marketed as Lenvima by Eisai.
FDA approves first EpiPen generic
The , including anaphylaxis, for adults and children weighing more than 33 pounds, according to an announcement from the agency.
“Today’s approval of the first generic version of the most widely prescribed epinephrine autoinjector in the U.S. is part of our longstanding commitment to advance access to lower cost, safe, and effective generic alternatives once patents and other exclusivities no longer prevent approval,” FDA commissioner Scott Gottlieb, MD, said in the release.
Manufactured by Teva Pharmaceuticals USA, the two strengths of the generic versions are 0.3 mg and 0.15 mg.
The FDA has previously approved other epinephrine autoinjectors, which include brand-name products and so-called “authorized generic” versions of Epi-Pen and Adrenaclick. An authorized generic “is made under the brand name’s existing drug application using the same formulation, process, and manufacturing facilities that are used by the brand name manufacturer. The labeling or packaging is, however, changed to remove the brand name or other trade dress. In some cases, a company may choose to sell an authorized generic at a lower cost than the brand-name drug product,” according to the FDA statement.
“Complex” generics – those that, as with this generic, include both a drug and a delivery device – face a tougher path to approval because the FDA has to evaluate and approve both components. “We remain committed to doing our part to provide scientific and regulatory clarity for sponsors seeking to develop complex generics, as well as prioritize the approval of medicines with little or no generic competition, as part of our overarching effort to remove barriers to generic development and market entry of critically important medicines,” Dr. Gottlieb explained.
Side effects of epinephrine autoinjectors include anxiety, restlessness, palpitations, nausea, and weakness; rarely, serious skin and soft-tissue infections after use of epinephrine autoinjectors have been reported.
Find more information about this approval in the FDA press announcement.
The , including anaphylaxis, for adults and children weighing more than 33 pounds, according to an announcement from the agency.
“Today’s approval of the first generic version of the most widely prescribed epinephrine autoinjector in the U.S. is part of our longstanding commitment to advance access to lower cost, safe, and effective generic alternatives once patents and other exclusivities no longer prevent approval,” FDA commissioner Scott Gottlieb, MD, said in the release.
Manufactured by Teva Pharmaceuticals USA, the two strengths of the generic versions are 0.3 mg and 0.15 mg.
The FDA has previously approved other epinephrine autoinjectors, which include brand-name products and so-called “authorized generic” versions of Epi-Pen and Adrenaclick. An authorized generic “is made under the brand name’s existing drug application using the same formulation, process, and manufacturing facilities that are used by the brand name manufacturer. The labeling or packaging is, however, changed to remove the brand name or other trade dress. In some cases, a company may choose to sell an authorized generic at a lower cost than the brand-name drug product,” according to the FDA statement.
“Complex” generics – those that, as with this generic, include both a drug and a delivery device – face a tougher path to approval because the FDA has to evaluate and approve both components. “We remain committed to doing our part to provide scientific and regulatory clarity for sponsors seeking to develop complex generics, as well as prioritize the approval of medicines with little or no generic competition, as part of our overarching effort to remove barriers to generic development and market entry of critically important medicines,” Dr. Gottlieb explained.
Side effects of epinephrine autoinjectors include anxiety, restlessness, palpitations, nausea, and weakness; rarely, serious skin and soft-tissue infections after use of epinephrine autoinjectors have been reported.
Find more information about this approval in the FDA press announcement.
The , including anaphylaxis, for adults and children weighing more than 33 pounds, according to an announcement from the agency.
“Today’s approval of the first generic version of the most widely prescribed epinephrine autoinjector in the U.S. is part of our longstanding commitment to advance access to lower cost, safe, and effective generic alternatives once patents and other exclusivities no longer prevent approval,” FDA commissioner Scott Gottlieb, MD, said in the release.
Manufactured by Teva Pharmaceuticals USA, the two strengths of the generic versions are 0.3 mg and 0.15 mg.
The FDA has previously approved other epinephrine autoinjectors, which include brand-name products and so-called “authorized generic” versions of Epi-Pen and Adrenaclick. An authorized generic “is made under the brand name’s existing drug application using the same formulation, process, and manufacturing facilities that are used by the brand name manufacturer. The labeling or packaging is, however, changed to remove the brand name or other trade dress. In some cases, a company may choose to sell an authorized generic at a lower cost than the brand-name drug product,” according to the FDA statement.
“Complex” generics – those that, as with this generic, include both a drug and a delivery device – face a tougher path to approval because the FDA has to evaluate and approve both components. “We remain committed to doing our part to provide scientific and regulatory clarity for sponsors seeking to develop complex generics, as well as prioritize the approval of medicines with little or no generic competition, as part of our overarching effort to remove barriers to generic development and market entry of critically important medicines,” Dr. Gottlieb explained.
Side effects of epinephrine autoinjectors include anxiety, restlessness, palpitations, nausea, and weakness; rarely, serious skin and soft-tissue infections after use of epinephrine autoinjectors have been reported.
Find more information about this approval in the FDA press announcement.
CDC supports Ebola response in DRC
The Centers for Disease Control and Prevention has been working with the Ministry of Health of the Democratic Republic of the Congo (DRC) on a new Ebola outbreak reported on Aug, 1, 2018, in North Kivu province.
![]()
“For the current outbreak, CDC has deployed experienced Ebola experts to DRC and the World Health Organization [WHO] to provide guidance on coordination of outbreak response, laboratory testing, disease contact tracing, infection control, and health communication,” according to a CDC press release.
The CDC’s online response also provides a Traveler’s Health notice of Watch Level 1 for the DRC, which advises standard precautions and avoiding infected individuals, but not an advisory against travel.
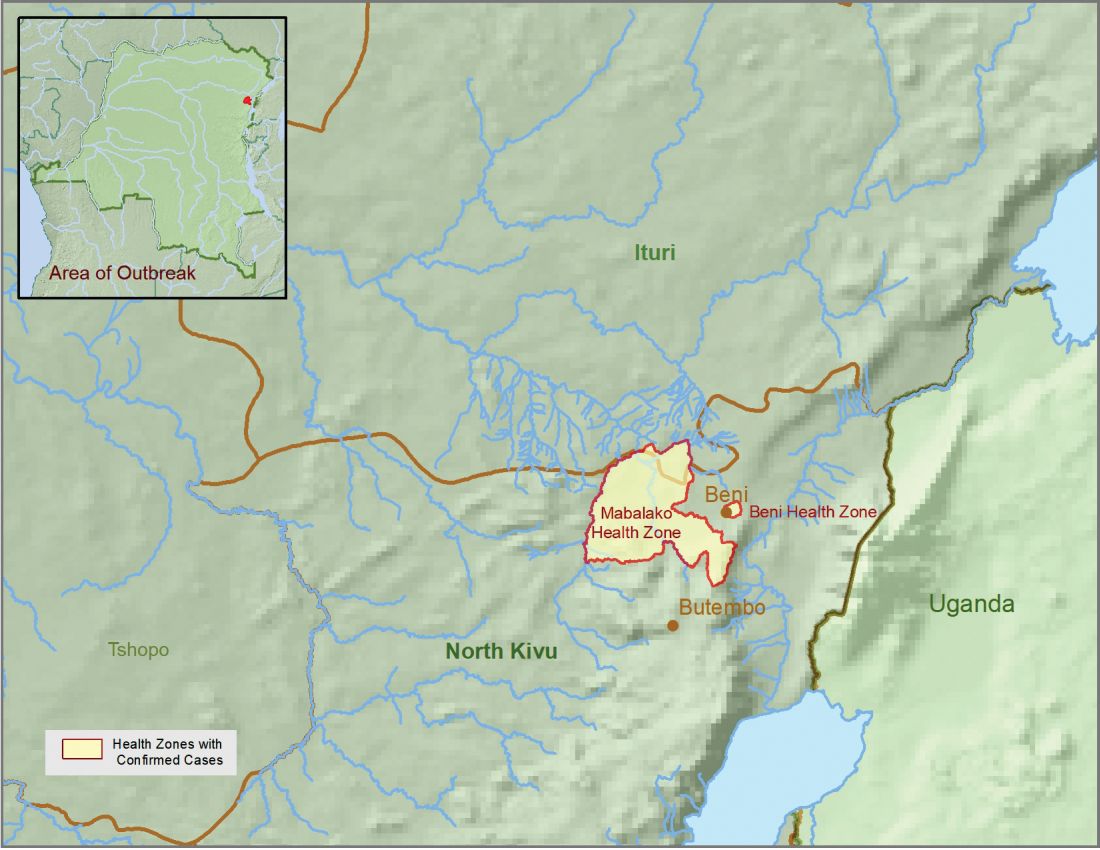
The Ebola virus associated with the current outbreak is Zaire ebolavirus, according to genetic testing by scientists in the DRC. This is the same species that caused an outbreak earlier this year in Equateur province in northwestern DRC, although differences between the genes of the viruses suggest the two outbreaks are not linked, according to the CDC media announcement.
As of Aug. 12, 2018, the following statistics were reported by the WHO on the outbreak:
- Confirmed cases: 30
- Probable cases: 27
- Total cases: 57
- Deaths: 41 (14 confirmed, 27 probable)
The outbreak is of particular concern because of the instability of the area, which hampers relief and quarantine efforts. The outbreak is in a part of the country identified by the U.S. State Department as a “restricted travel” zone due to armed conflict and violence targeting civilians, according to the CDC.
The Centers for Disease Control and Prevention has been working with the Ministry of Health of the Democratic Republic of the Congo (DRC) on a new Ebola outbreak reported on Aug, 1, 2018, in North Kivu province.
![]()
“For the current outbreak, CDC has deployed experienced Ebola experts to DRC and the World Health Organization [WHO] to provide guidance on coordination of outbreak response, laboratory testing, disease contact tracing, infection control, and health communication,” according to a CDC press release.
The CDC’s online response also provides a Traveler’s Health notice of Watch Level 1 for the DRC, which advises standard precautions and avoiding infected individuals, but not an advisory against travel.
The Ebola virus associated with the current outbreak is Zaire ebolavirus, according to genetic testing by scientists in the DRC. This is the same species that caused an outbreak earlier this year in Equateur province in northwestern DRC, although differences between the genes of the viruses suggest the two outbreaks are not linked, according to the CDC media announcement.
As of Aug. 12, 2018, the following statistics were reported by the WHO on the outbreak:
- Confirmed cases: 30
- Probable cases: 27
- Total cases: 57
- Deaths: 41 (14 confirmed, 27 probable)
The outbreak is of particular concern because of the instability of the area, which hampers relief and quarantine efforts. The outbreak is in a part of the country identified by the U.S. State Department as a “restricted travel” zone due to armed conflict and violence targeting civilians, according to the CDC.
The Centers for Disease Control and Prevention has been working with the Ministry of Health of the Democratic Republic of the Congo (DRC) on a new Ebola outbreak reported on Aug, 1, 2018, in North Kivu province.
![]()
“For the current outbreak, CDC has deployed experienced Ebola experts to DRC and the World Health Organization [WHO] to provide guidance on coordination of outbreak response, laboratory testing, disease contact tracing, infection control, and health communication,” according to a CDC press release.
The CDC’s online response also provides a Traveler’s Health notice of Watch Level 1 for the DRC, which advises standard precautions and avoiding infected individuals, but not an advisory against travel.
The Ebola virus associated with the current outbreak is Zaire ebolavirus, according to genetic testing by scientists in the DRC. This is the same species that caused an outbreak earlier this year in Equateur province in northwestern DRC, although differences between the genes of the viruses suggest the two outbreaks are not linked, according to the CDC media announcement.
As of Aug. 12, 2018, the following statistics were reported by the WHO on the outbreak:
- Confirmed cases: 30
- Probable cases: 27
- Total cases: 57
- Deaths: 41 (14 confirmed, 27 probable)
The outbreak is of particular concern because of the instability of the area, which hampers relief and quarantine efforts. The outbreak is in a part of the country identified by the U.S. State Department as a “restricted travel” zone due to armed conflict and violence targeting civilians, according to the CDC.
FDA approves vaginal ring contraceptive Annovera
The first vaginal ring contraceptive that can be used for 1 year has been approved by the Food and Drug Administration.
The FDA granted approval of Annovera (segesterone acetate and ethinyl estradiol vaginal system), a reusable donut-shaped ring, to the Population Council. The nonbiodegradable, flexible vaginal system is placed in the vagina for 3 weeks followed by 1 week out of the vagina, at which time women may experience a menstrual period. This schedule is repeated every 4 weeks for 13 28-day menstrual cycles.
The contraceptive ring is washed and stored in a compact case for the 7 days when it is not in use. Annovera does not require refrigeration prior to dispensing and can withstand storage temperatures up to 30° C (86° F).
“Today’s approval builds on available birth control options,” said Victor Crentsil, MD, acting deputy director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research.
The efficacy and safety of Annovera were studied in three, open-label clinical trials with healthy women ranging from 18 to 40 years of age. Based on the results, about 2%-4% of women may get pregnant during the first year they use Annovera.
Annovera carries a boxed warning regarding cigarette smoking and serious cardiovascular events. Women over age 35 who smoke should not use Annovera. Cigarette smoking increases the risk of serious cardiovascular events from combination hormonal contraceptive use.
Annovera is contraindicated for women with a high risk of arterial or venous thrombotic diseases; a history of breast cancer or another estrogen- or progestin-sensitive cancer; liver tumors, acute hepatitis, or severe (decompensated) cirrhosis; undiagnosed abnormal uterine bleeding; hypersensitivity to any of the components of Annovera; and use of hepatitis C drug combinations containing ombitasvir/paritaprevir/ritonavir, with or without dasabuvir, according to an FDA press release.
The most common side effects of Annovera are similar to those of other combined hormonal contraceptive products and include headache, nausea and vomiting, yeast infections, abdominal pain, dysmenorrhea, breast tenderness, irregular bleeding, diarrhea, and genital itching.
The FDA is requiring postmarketing studies to further evaluate the risks of venous thromboembolism and the effects of CYP3A-modulating drugs and tampon use on the pharmacokinetics of Annovera.
It is very exciting to see ongoing research and development for new contraceptives and approval of these new methods! We know that each person’s contraceptive needs are unique and having more options from which to choose will help us as providers connect our patients to methods that meet their goals and individual needs. These two methods fill important gaps in our current contraceptive portfolio: a patient-controlled, long-acting method and a facilitated non-hormonal, non-prescription method (the Natural Cycles app). I am looking forward to hearing feedback from patients about their thoughts and experiences with these new methods.

Melissa Kottke, MD, MPH, MBA, is an associate professor of obstetrics and gynecology and director of the Jane Fonda Center for Adolescent Reproductive Health, Emory University, Atlanta.
It is very exciting to see ongoing research and development for new contraceptives and approval of these new methods! We know that each person’s contraceptive needs are unique and having more options from which to choose will help us as providers connect our patients to methods that meet their goals and individual needs. These two methods fill important gaps in our current contraceptive portfolio: a patient-controlled, long-acting method and a facilitated non-hormonal, non-prescription method (the Natural Cycles app). I am looking forward to hearing feedback from patients about their thoughts and experiences with these new methods.
Melissa Kottke, MD, MPH, MBA, is an associate professor of obstetrics and gynecology and director of the Jane Fonda Center for Adolescent Reproductive Health, Emory University, Atlanta.
It is very exciting to see ongoing research and development for new contraceptives and approval of these new methods! We know that each person’s contraceptive needs are unique and having more options from which to choose will help us as providers connect our patients to methods that meet their goals and individual needs. These two methods fill important gaps in our current contraceptive portfolio: a patient-controlled, long-acting method and a facilitated non-hormonal, non-prescription method (the Natural Cycles app). I am looking forward to hearing feedback from patients about their thoughts and experiences with these new methods.
Melissa Kottke, MD, MPH, MBA, is an associate professor of obstetrics and gynecology and director of the Jane Fonda Center for Adolescent Reproductive Health, Emory University, Atlanta.
The first vaginal ring contraceptive that can be used for 1 year has been approved by the Food and Drug Administration.
The FDA granted approval of Annovera (segesterone acetate and ethinyl estradiol vaginal system), a reusable donut-shaped ring, to the Population Council. The nonbiodegradable, flexible vaginal system is placed in the vagina for 3 weeks followed by 1 week out of the vagina, at which time women may experience a menstrual period. This schedule is repeated every 4 weeks for 13 28-day menstrual cycles.
The contraceptive ring is washed and stored in a compact case for the 7 days when it is not in use. Annovera does not require refrigeration prior to dispensing and can withstand storage temperatures up to 30° C (86° F).
“Today’s approval builds on available birth control options,” said Victor Crentsil, MD, acting deputy director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research.
The efficacy and safety of Annovera were studied in three, open-label clinical trials with healthy women ranging from 18 to 40 years of age. Based on the results, about 2%-4% of women may get pregnant during the first year they use Annovera.
Annovera carries a boxed warning regarding cigarette smoking and serious cardiovascular events. Women over age 35 who smoke should not use Annovera. Cigarette smoking increases the risk of serious cardiovascular events from combination hormonal contraceptive use.
Annovera is contraindicated for women with a high risk of arterial or venous thrombotic diseases; a history of breast cancer or another estrogen- or progestin-sensitive cancer; liver tumors, acute hepatitis, or severe (decompensated) cirrhosis; undiagnosed abnormal uterine bleeding; hypersensitivity to any of the components of Annovera; and use of hepatitis C drug combinations containing ombitasvir/paritaprevir/ritonavir, with or without dasabuvir, according to an FDA press release.
The most common side effects of Annovera are similar to those of other combined hormonal contraceptive products and include headache, nausea and vomiting, yeast infections, abdominal pain, dysmenorrhea, breast tenderness, irregular bleeding, diarrhea, and genital itching.
The FDA is requiring postmarketing studies to further evaluate the risks of venous thromboembolism and the effects of CYP3A-modulating drugs and tampon use on the pharmacokinetics of Annovera.
The first vaginal ring contraceptive that can be used for 1 year has been approved by the Food and Drug Administration.
The FDA granted approval of Annovera (segesterone acetate and ethinyl estradiol vaginal system), a reusable donut-shaped ring, to the Population Council. The nonbiodegradable, flexible vaginal system is placed in the vagina for 3 weeks followed by 1 week out of the vagina, at which time women may experience a menstrual period. This schedule is repeated every 4 weeks for 13 28-day menstrual cycles.
The contraceptive ring is washed and stored in a compact case for the 7 days when it is not in use. Annovera does not require refrigeration prior to dispensing and can withstand storage temperatures up to 30° C (86° F).
“Today’s approval builds on available birth control options,” said Victor Crentsil, MD, acting deputy director of the Office of Drug Evaluation III in FDA’s Center for Drug Evaluation and Research.
The efficacy and safety of Annovera were studied in three, open-label clinical trials with healthy women ranging from 18 to 40 years of age. Based on the results, about 2%-4% of women may get pregnant during the first year they use Annovera.
Annovera carries a boxed warning regarding cigarette smoking and serious cardiovascular events. Women over age 35 who smoke should not use Annovera. Cigarette smoking increases the risk of serious cardiovascular events from combination hormonal contraceptive use.
Annovera is contraindicated for women with a high risk of arterial or venous thrombotic diseases; a history of breast cancer or another estrogen- or progestin-sensitive cancer; liver tumors, acute hepatitis, or severe (decompensated) cirrhosis; undiagnosed abnormal uterine bleeding; hypersensitivity to any of the components of Annovera; and use of hepatitis C drug combinations containing ombitasvir/paritaprevir/ritonavir, with or without dasabuvir, according to an FDA press release.
The most common side effects of Annovera are similar to those of other combined hormonal contraceptive products and include headache, nausea and vomiting, yeast infections, abdominal pain, dysmenorrhea, breast tenderness, irregular bleeding, diarrhea, and genital itching.
The FDA is requiring postmarketing studies to further evaluate the risks of venous thromboembolism and the effects of CYP3A-modulating drugs and tampon use on the pharmacokinetics of Annovera.
FDA approves first mobile app for contraceptive use
The Food and Drug Administration has approved marketing of the first medical mobile application that can be used as a contraceptive, the agency announced in a written statement.

The app, called Natural Cycles, contains an algorithm that calculates the days of the month women are likely to be fertile, based on daily body temperature readings and menstrual cycle information, and is intended for premenopausal women aged 18 years and older. App users are instructed to take their daily body temperatures with a basal body thermometer and enter the information into the app. The more-sensitive basal body thermometer can detect temperature elevations during ovulation, and the app will display a warning on days when users are most fertile. During these days, women should either abstain from sex or use protection.
In clinical studies comprising more than 15,000 women, Natural Cycles had a “perfect use” failure rate of 1.8% and a “normal use” failure rate of 6.5%. This compares favorably with other forms of contraception and birth control: Male condoms have a perfect use failure rate of 2.0% and a normal use failure rate of 18.0%, and combined oral contraceptives have a perfect use failure rate of 0.3% and a normal use failure rate of 9.0%, according to the Association of Reproductive Care Professionals.
“Consumers are increasingly using digital health technologies to inform their everyday health decisions, and this new app can provide an effective method of contraception if it’s used carefully and correctly,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in the statement.
The app should not be used by women with a condition in which a pregnancy would present risk to the mother or fetus or by women who are using a birth control or hormonal treatment that inhibits ovulation. Natural Cycles does not protect against sexually transmitted infections.
The Food and Drug Administration has approved marketing of the first medical mobile application that can be used as a contraceptive, the agency announced in a written statement.
The app, called Natural Cycles, contains an algorithm that calculates the days of the month women are likely to be fertile, based on daily body temperature readings and menstrual cycle information, and is intended for premenopausal women aged 18 years and older. App users are instructed to take their daily body temperatures with a basal body thermometer and enter the information into the app. The more-sensitive basal body thermometer can detect temperature elevations during ovulation, and the app will display a warning on days when users are most fertile. During these days, women should either abstain from sex or use protection.
In clinical studies comprising more than 15,000 women, Natural Cycles had a “perfect use” failure rate of 1.8% and a “normal use” failure rate of 6.5%. This compares favorably with other forms of contraception and birth control: Male condoms have a perfect use failure rate of 2.0% and a normal use failure rate of 18.0%, and combined oral contraceptives have a perfect use failure rate of 0.3% and a normal use failure rate of 9.0%, according to the Association of Reproductive Care Professionals.
“Consumers are increasingly using digital health technologies to inform their everyday health decisions, and this new app can provide an effective method of contraception if it’s used carefully and correctly,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in the statement.
The app should not be used by women with a condition in which a pregnancy would present risk to the mother or fetus or by women who are using a birth control or hormonal treatment that inhibits ovulation. Natural Cycles does not protect against sexually transmitted infections.
The Food and Drug Administration has approved marketing of the first medical mobile application that can be used as a contraceptive, the agency announced in a written statement.
The app, called Natural Cycles, contains an algorithm that calculates the days of the month women are likely to be fertile, based on daily body temperature readings and menstrual cycle information, and is intended for premenopausal women aged 18 years and older. App users are instructed to take their daily body temperatures with a basal body thermometer and enter the information into the app. The more-sensitive basal body thermometer can detect temperature elevations during ovulation, and the app will display a warning on days when users are most fertile. During these days, women should either abstain from sex or use protection.
In clinical studies comprising more than 15,000 women, Natural Cycles had a “perfect use” failure rate of 1.8% and a “normal use” failure rate of 6.5%. This compares favorably with other forms of contraception and birth control: Male condoms have a perfect use failure rate of 2.0% and a normal use failure rate of 18.0%, and combined oral contraceptives have a perfect use failure rate of 0.3% and a normal use failure rate of 9.0%, according to the Association of Reproductive Care Professionals.
“Consumers are increasingly using digital health technologies to inform their everyday health decisions, and this new app can provide an effective method of contraception if it’s used carefully and correctly,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in the statement.
The app should not be used by women with a condition in which a pregnancy would present risk to the mother or fetus or by women who are using a birth control or hormonal treatment that inhibits ovulation. Natural Cycles does not protect against sexually transmitted infections.