User login
Secondary Syphilis
Syphilis often is referred to as the “great imitator” due to the protean presentations of secondary-stage disease, the most common of which are skin manifestations.1 Secondary syphilis typically begins 3 to 10 weeks after initial exposure due to systemic dissemination of Treponema pallidum, and although presentations can vary widely, the classic presentation includes nonspecific generalized symptoms (eg, fever, malaise, lymphadenopathy), variable skin findings (eg, nonpruritic papulosquamous eruption), and mucosal ulcerations or plaques.1 Early and accurate diagnosis of syphilis is critical to avoid the morbidity associated with advanced disease.
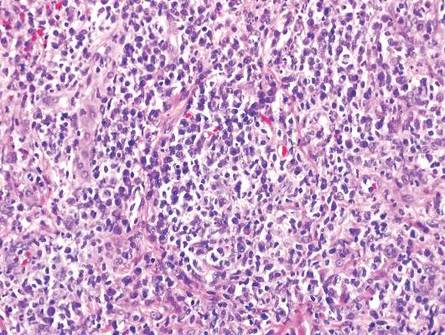
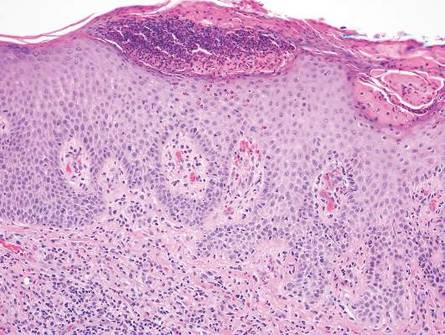
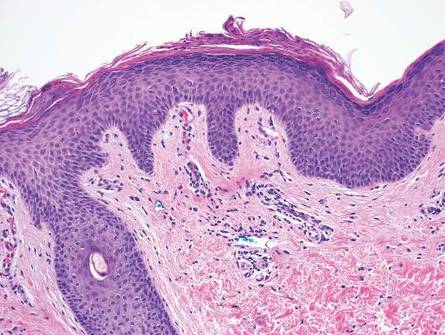
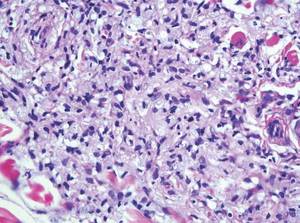
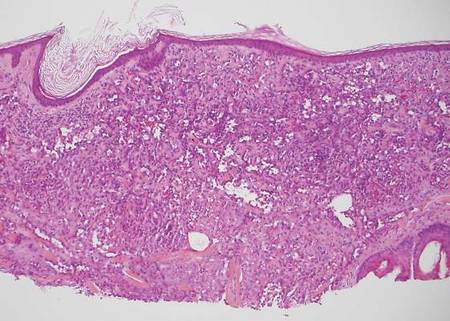
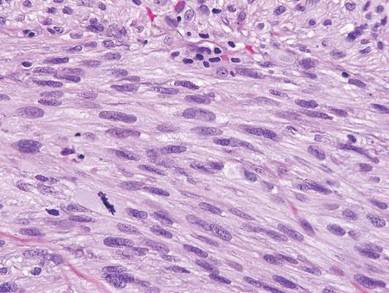
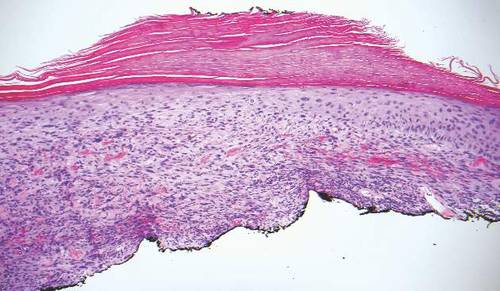
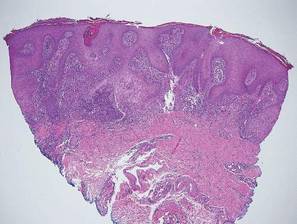
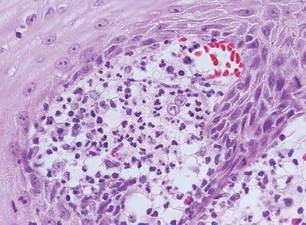
The classic histopathologic appearance of secondary syphilis is characterized by psoriasiform epidermal changes; a dermal inflammatory infiltrate of lymphocytes, histiocytes, and plasma cells in a lichenoid and/or superficial and deep perivascular distribution (Figure 1); and endothelial swelling of dermal blood vessels.1 The presence of plasma cells in the infiltrate (Figure 2) is particularly useful for differentiating secondary syphilis from other clinicopathological mimickers, but this finding is not always present. Silver-based histochemical stains (eg, Warthin-Starry silver stain) can be used to high-light T pallidum organisms; however, histochemical staining is plagued by low diagnostic sensitivity for identifying the causative organism, making immunohistochemical and/or serologic testing the preferred method for confirming the diagnosis.1

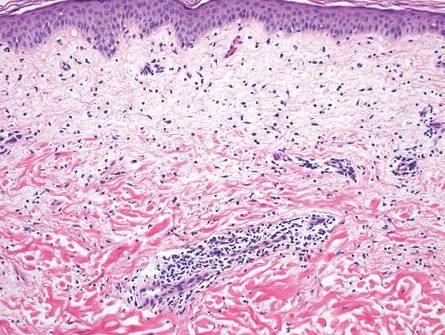
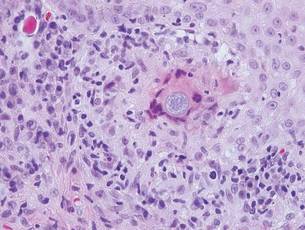
Arthropod assault is characterized by a superficial and deep perivascular lymphocytic inflammatory infiltrate with a variable number of polymorphonuclear cells.2 Overlying spongiosis or focal epidermal necrosis and increased eosinophils are typical of arthropod assault (Figure 3).2 The infiltrate seen following insect bites is classically described as wedge-shaped, although recent literature has disputed the sensitivity of this finding, identifying adnexal structure involvement as an alternative sensitive marker for identifying insect bites.2
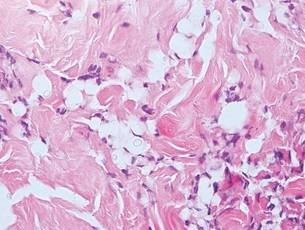
Chronic cutaneous lupus erythematosus demonstrates a spectrum of histopathologic changes depending on the age of the lesion biopsied; however, characteristic histopathologic features typically include variable epidermal atrophy or acanthosis with basal layer vacuolar degeneration, basement membrane thickening, follicular plugging, superficial and deep perivascular and periappendageal lymphocytic inflammation, and dermal mucin deposition (Figure 4).4
Fixed drug eruption histopathologically presents as an interface tissue reaction–associated single-cell necrosis to broader areas of epidermal necrosis, as well as superficial to mid-dermal lymphocytic infiltrate. Unlike secondary syphilis, a fixed drug eruption is characterized by prominent melanin pigment incontinence and eosinophils (Figure 5).5

Similar to secondary syphilis, pityriasis lichenoides et varioliformis acuta (PLEVA) demonstrates variable psoriasiform epidermal hyperplasia with a lichenoid and perivascular lymphocytic infiltrate. Other findings in PLEVA include parakeratosis, variable epidermal necrosis, and prominent exocytosis of lymphocytes. Unlike typical secondary syphilis, PLEVA often is associated with lymphocytic vasculitis, consisting of the invasion of vessel walls by lymphocytes with extravasation of erythrocytes and an absence of conspicuous plasma cells (Figure 6).6
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;3:595-599.
- Miteva M, Elsner P, Ziemer M. A histopathologic study of arthropod bite reactions in 20 patients highlights relevant adnexal involvement. J Cutan Pathol. 2009;36:26-33.
- Winkelmann RK, Reizner GT. Diffuse dermal neutrophilia in urticarial. Human Pathol. 1988;19:389-393.
- Sepehr A, Wenson S, Tahan SR. Histopathologic manifestations of systemic diseases: the example of cutaneous lupus erythematosus. J Cutan Pathol. 2010;37 (suppl 1):112-124.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727.
- Fernandes NF, Rozdeba PJ, Schwartz RA, et al. Pityriasis lichenoides et varioliformis acuta: a disease spectrum. Int J Dermatol. 2010;49:257-261.
Syphilis often is referred to as the “great imitator” due to the protean presentations of secondary-stage disease, the most common of which are skin manifestations.1 Secondary syphilis typically begins 3 to 10 weeks after initial exposure due to systemic dissemination of Treponema pallidum, and although presentations can vary widely, the classic presentation includes nonspecific generalized symptoms (eg, fever, malaise, lymphadenopathy), variable skin findings (eg, nonpruritic papulosquamous eruption), and mucosal ulcerations or plaques.1 Early and accurate diagnosis of syphilis is critical to avoid the morbidity associated with advanced disease.
The classic histopathologic appearance of secondary syphilis is characterized by psoriasiform epidermal changes; a dermal inflammatory infiltrate of lymphocytes, histiocytes, and plasma cells in a lichenoid and/or superficial and deep perivascular distribution (Figure 1); and endothelial swelling of dermal blood vessels.1 The presence of plasma cells in the infiltrate (Figure 2) is particularly useful for differentiating secondary syphilis from other clinicopathological mimickers, but this finding is not always present. Silver-based histochemical stains (eg, Warthin-Starry silver stain) can be used to high-light T pallidum organisms; however, histochemical staining is plagued by low diagnostic sensitivity for identifying the causative organism, making immunohistochemical and/or serologic testing the preferred method for confirming the diagnosis.1
Arthropod assault is characterized by a superficial and deep perivascular lymphocytic inflammatory infiltrate with a variable number of polymorphonuclear cells.2 Overlying spongiosis or focal epidermal necrosis and increased eosinophils are typical of arthropod assault (Figure 3).2 The infiltrate seen following insect bites is classically described as wedge-shaped, although recent literature has disputed the sensitivity of this finding, identifying adnexal structure involvement as an alternative sensitive marker for identifying insect bites.2
Chronic cutaneous lupus erythematosus demonstrates a spectrum of histopathologic changes depending on the age of the lesion biopsied; however, characteristic histopathologic features typically include variable epidermal atrophy or acanthosis with basal layer vacuolar degeneration, basement membrane thickening, follicular plugging, superficial and deep perivascular and periappendageal lymphocytic inflammation, and dermal mucin deposition (Figure 4).4
Fixed drug eruption histopathologically presents as an interface tissue reaction–associated single-cell necrosis to broader areas of epidermal necrosis, as well as superficial to mid-dermal lymphocytic infiltrate. Unlike secondary syphilis, a fixed drug eruption is characterized by prominent melanin pigment incontinence and eosinophils (Figure 5).5
Similar to secondary syphilis, pityriasis lichenoides et varioliformis acuta (PLEVA) demonstrates variable psoriasiform epidermal hyperplasia with a lichenoid and perivascular lymphocytic infiltrate. Other findings in PLEVA include parakeratosis, variable epidermal necrosis, and prominent exocytosis of lymphocytes. Unlike typical secondary syphilis, PLEVA often is associated with lymphocytic vasculitis, consisting of the invasion of vessel walls by lymphocytes with extravasation of erythrocytes and an absence of conspicuous plasma cells (Figure 6).6
Syphilis often is referred to as the “great imitator” due to the protean presentations of secondary-stage disease, the most common of which are skin manifestations.1 Secondary syphilis typically begins 3 to 10 weeks after initial exposure due to systemic dissemination of Treponema pallidum, and although presentations can vary widely, the classic presentation includes nonspecific generalized symptoms (eg, fever, malaise, lymphadenopathy), variable skin findings (eg, nonpruritic papulosquamous eruption), and mucosal ulcerations or plaques.1 Early and accurate diagnosis of syphilis is critical to avoid the morbidity associated with advanced disease.
The classic histopathologic appearance of secondary syphilis is characterized by psoriasiform epidermal changes; a dermal inflammatory infiltrate of lymphocytes, histiocytes, and plasma cells in a lichenoid and/or superficial and deep perivascular distribution (Figure 1); and endothelial swelling of dermal blood vessels.1 The presence of plasma cells in the infiltrate (Figure 2) is particularly useful for differentiating secondary syphilis from other clinicopathological mimickers, but this finding is not always present. Silver-based histochemical stains (eg, Warthin-Starry silver stain) can be used to high-light T pallidum organisms; however, histochemical staining is plagued by low diagnostic sensitivity for identifying the causative organism, making immunohistochemical and/or serologic testing the preferred method for confirming the diagnosis.1
Arthropod assault is characterized by a superficial and deep perivascular lymphocytic inflammatory infiltrate with a variable number of polymorphonuclear cells.2 Overlying spongiosis or focal epidermal necrosis and increased eosinophils are typical of arthropod assault (Figure 3).2 The infiltrate seen following insect bites is classically described as wedge-shaped, although recent literature has disputed the sensitivity of this finding, identifying adnexal structure involvement as an alternative sensitive marker for identifying insect bites.2
Chronic cutaneous lupus erythematosus demonstrates a spectrum of histopathologic changes depending on the age of the lesion biopsied; however, characteristic histopathologic features typically include variable epidermal atrophy or acanthosis with basal layer vacuolar degeneration, basement membrane thickening, follicular plugging, superficial and deep perivascular and periappendageal lymphocytic inflammation, and dermal mucin deposition (Figure 4).4
Fixed drug eruption histopathologically presents as an interface tissue reaction–associated single-cell necrosis to broader areas of epidermal necrosis, as well as superficial to mid-dermal lymphocytic infiltrate. Unlike secondary syphilis, a fixed drug eruption is characterized by prominent melanin pigment incontinence and eosinophils (Figure 5).5
Similar to secondary syphilis, pityriasis lichenoides et varioliformis acuta (PLEVA) demonstrates variable psoriasiform epidermal hyperplasia with a lichenoid and perivascular lymphocytic infiltrate. Other findings in PLEVA include parakeratosis, variable epidermal necrosis, and prominent exocytosis of lymphocytes. Unlike typical secondary syphilis, PLEVA often is associated with lymphocytic vasculitis, consisting of the invasion of vessel walls by lymphocytes with extravasation of erythrocytes and an absence of conspicuous plasma cells (Figure 6).6
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;3:595-599.
- Miteva M, Elsner P, Ziemer M. A histopathologic study of arthropod bite reactions in 20 patients highlights relevant adnexal involvement. J Cutan Pathol. 2009;36:26-33.
- Winkelmann RK, Reizner GT. Diffuse dermal neutrophilia in urticarial. Human Pathol. 1988;19:389-393.
- Sepehr A, Wenson S, Tahan SR. Histopathologic manifestations of systemic diseases: the example of cutaneous lupus erythematosus. J Cutan Pathol. 2010;37 (suppl 1):112-124.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727.
- Fernandes NF, Rozdeba PJ, Schwartz RA, et al. Pityriasis lichenoides et varioliformis acuta: a disease spectrum. Int J Dermatol. 2010;49:257-261.
- Hoang MP, High WA, Molberg KH. Secondary syphilis: a histologic and immunohistochemical evaluation. J Cutan Pathol. 2004;3:595-599.
- Miteva M, Elsner P, Ziemer M. A histopathologic study of arthropod bite reactions in 20 patients highlights relevant adnexal involvement. J Cutan Pathol. 2009;36:26-33.
- Winkelmann RK, Reizner GT. Diffuse dermal neutrophilia in urticarial. Human Pathol. 1988;19:389-393.
- Sepehr A, Wenson S, Tahan SR. Histopathologic manifestations of systemic diseases: the example of cutaneous lupus erythematosus. J Cutan Pathol. 2010;37 (suppl 1):112-124.
- Flowers H, Brodell R, Brents M, et al. Fixed drug eruptions: presentation, diagnosis, and management. South Med J. 2014;107:724-727.
- Fernandes NF, Rozdeba PJ, Schwartz RA, et al. Pityriasis lichenoides et varioliformis acuta: a disease spectrum. Int J Dermatol. 2010;49:257-261.
Cutaneous Leishmaniasis
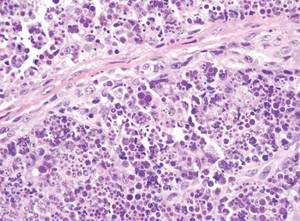
Cutaneous leishmaniasis is a parasitic infection caused by intracellular organisms found in tropical climates. Old World leishmaniasis is endemic to Asia, Africa, and parts of Europe, while New World leishmaniasis is native to Central and South Americas.1 Depending upon a host’s immune status and the specific Leishmania species, clinical presentations vary in appearance and severity, ranging from self-limited, localized cutaneous disease to potentially fatal visceral and mucocutaneous involvement. Most cutaneous manifestations of leishmaniasis begin as distinct, painless papules that may progress to nodules or become ulcerated over time.1 Histologically, leishmaniasis is diagnosed by the identification of intracellular organisms that characteristically align along the peripheral rim inside the vacuole of a histiocyte.2 This unique finding is called the “marquee sign” due to its resemblance to light bulbs arranged around a dressing room mirror (Figure 1).2Leishmania amastigotes (also known as Leishman-Donovan bodies) have kinetoplasts that are helpful in diagnosis but also may be difficult to detect.2 Along with the Leishmania parasites, there typically is a mixed inflammatory infiltrate of plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 2).1,2 There also may be varying degrees of pseudoepitheliomatous hyperplasia and overlying epidermal ulceration.1

Cutaneous botryomycosis can present clinically as a number of various primary lesions, including papules, nodules, or ulcers that may resemble leishmaniasis.3 Botryomycosis represents a specific histologic collection of bacterial granules, most commonly caused by Staphylococcus aureus.3 The dermal granulomatous infiltrate seen in botryomycosis often is similar to that seen in chronic leishmaniasis; however, one histologic feature unique to botryomycosis is the presence of characteristic basophilic staphylococcal grains that are arranged in clusters resembling bunches of grapes (the term botryo means “bunch of grapes” in Greek).3 A thin, eosinophilic rim consisting of antibodies, bacterial debris, and complement proteins and glycoproteins may encircle the basophilic grains but does not need to be present for diagnosis (Figure 3).3
Lepromatous leprosy presents as a symmetric, widespread eruption of macules, patches, plaques, or papules that are most prominent in acral areas.4 Perivascular infiltration of lymphocytes and histiocytes is characteristic of lepromatous leprosy.2 Mycobacteria bacilli also are seen within histiocytic vacuoles, similarly to leishmaniasis; however, collections of these bacilli congregate within the center of a foamy histiocyte to form a distinctive histologic finding known as a globus. These individual histiocytes containing central globi are called Virchow cells (Figure 4).2 However, lepromatous leprosy can be distinguished from leishmaniasis histologically by carefully observing the intracellular location of the infectious organism. Mycobacteria bacilli are located in the center of a histiocyte vacuole whereas Leishmania parasites demonstrate a peripheral alignment along a histiocyte vacuole. If any uncertainty remains between a diagnosis of leishmaniasis and lepromatous leprosy, positive Fite staining for mycobacteria easily differentiates between the 2 conditions.2,4
Cutaneous lobomycosis, a rare fungal infection transmitted by dolphins, manifests clinically as an asymptomatic nodule that is similar in appearance to a keloid. Histologic similarities to leishmaniasis include pseudoepitheliomatous hyperplasia and dermal granulomatous inflammation.4 The most distinguishing characteristic of lobomycosis is the presence of round, thick-walled, white organisms connected in a “string of beads” or chainlike configuration (Figure 5).2 Unlike leishmaniasis, lobomycosis fungal organisms would stain positive on periodic acid–Schiff staining.4

Cutaneous protothecosis is a rare clinical entity that presents as an isolated nodule or plaque or bursitis.4 It occurs following minor trauma and inoculation with Prototheca organisms, a genus of algae found in contaminated water.2,4 In its morula form, Prototheca adopts a characteristic arrangement within histiocytes that strikingly resembles a soccer ball (Figure 6).2 Conversely, nonmorulating forms of protothecosis can also be seen; these exhibit a central basophilic, dotlike structure within the histiocytes surrounded by a white halo.2 Definitive diagnosis of protothecosis can only be made upon successful culture of the algae.5
- Kevric I, Cappel MA, Keeling JH. New World and Old World leishmania infections: a practical review. Dermatol Clin. 2015;33:579-593.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 2nd ed. London, England: Elsevier Saunders; 2013.
- De Vries HJ, Van Noesel CJ, Hoekzema R, et al. Botryomycosis in an HIV-positive subject. J Eur Acad Dermatol Venereol. 2003;17:87-90.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences UK; 2012.
- Hillesheim PB, Bahrami S. Cutaneous protothecosis. Arch Pathol Lab Med. 2011;135:941-944.
Cutaneous leishmaniasis is a parasitic infection caused by intracellular organisms found in tropical climates. Old World leishmaniasis is endemic to Asia, Africa, and parts of Europe, while New World leishmaniasis is native to Central and South Americas.1 Depending upon a host’s immune status and the specific Leishmania species, clinical presentations vary in appearance and severity, ranging from self-limited, localized cutaneous disease to potentially fatal visceral and mucocutaneous involvement. Most cutaneous manifestations of leishmaniasis begin as distinct, painless papules that may progress to nodules or become ulcerated over time.1 Histologically, leishmaniasis is diagnosed by the identification of intracellular organisms that characteristically align along the peripheral rim inside the vacuole of a histiocyte.2 This unique finding is called the “marquee sign” due to its resemblance to light bulbs arranged around a dressing room mirror (Figure 1).2Leishmania amastigotes (also known as Leishman-Donovan bodies) have kinetoplasts that are helpful in diagnosis but also may be difficult to detect.2 Along with the Leishmania parasites, there typically is a mixed inflammatory infiltrate of plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 2).1,2 There also may be varying degrees of pseudoepitheliomatous hyperplasia and overlying epidermal ulceration.1
Cutaneous botryomycosis can present clinically as a number of various primary lesions, including papules, nodules, or ulcers that may resemble leishmaniasis.3 Botryomycosis represents a specific histologic collection of bacterial granules, most commonly caused by Staphylococcus aureus.3 The dermal granulomatous infiltrate seen in botryomycosis often is similar to that seen in chronic leishmaniasis; however, one histologic feature unique to botryomycosis is the presence of characteristic basophilic staphylococcal grains that are arranged in clusters resembling bunches of grapes (the term botryo means “bunch of grapes” in Greek).3 A thin, eosinophilic rim consisting of antibodies, bacterial debris, and complement proteins and glycoproteins may encircle the basophilic grains but does not need to be present for diagnosis (Figure 3).3
Lepromatous leprosy presents as a symmetric, widespread eruption of macules, patches, plaques, or papules that are most prominent in acral areas.4 Perivascular infiltration of lymphocytes and histiocytes is characteristic of lepromatous leprosy.2 Mycobacteria bacilli also are seen within histiocytic vacuoles, similarly to leishmaniasis; however, collections of these bacilli congregate within the center of a foamy histiocyte to form a distinctive histologic finding known as a globus. These individual histiocytes containing central globi are called Virchow cells (Figure 4).2 However, lepromatous leprosy can be distinguished from leishmaniasis histologically by carefully observing the intracellular location of the infectious organism. Mycobacteria bacilli are located in the center of a histiocyte vacuole whereas Leishmania parasites demonstrate a peripheral alignment along a histiocyte vacuole. If any uncertainty remains between a diagnosis of leishmaniasis and lepromatous leprosy, positive Fite staining for mycobacteria easily differentiates between the 2 conditions.2,4
Cutaneous lobomycosis, a rare fungal infection transmitted by dolphins, manifests clinically as an asymptomatic nodule that is similar in appearance to a keloid. Histologic similarities to leishmaniasis include pseudoepitheliomatous hyperplasia and dermal granulomatous inflammation.4 The most distinguishing characteristic of lobomycosis is the presence of round, thick-walled, white organisms connected in a “string of beads” or chainlike configuration (Figure 5).2 Unlike leishmaniasis, lobomycosis fungal organisms would stain positive on periodic acid–Schiff staining.4
Cutaneous protothecosis is a rare clinical entity that presents as an isolated nodule or plaque or bursitis.4 It occurs following minor trauma and inoculation with Prototheca organisms, a genus of algae found in contaminated water.2,4 In its morula form, Prototheca adopts a characteristic arrangement within histiocytes that strikingly resembles a soccer ball (Figure 6).2 Conversely, nonmorulating forms of protothecosis can also be seen; these exhibit a central basophilic, dotlike structure within the histiocytes surrounded by a white halo.2 Definitive diagnosis of protothecosis can only be made upon successful culture of the algae.5
Cutaneous leishmaniasis is a parasitic infection caused by intracellular organisms found in tropical climates. Old World leishmaniasis is endemic to Asia, Africa, and parts of Europe, while New World leishmaniasis is native to Central and South Americas.1 Depending upon a host’s immune status and the specific Leishmania species, clinical presentations vary in appearance and severity, ranging from self-limited, localized cutaneous disease to potentially fatal visceral and mucocutaneous involvement. Most cutaneous manifestations of leishmaniasis begin as distinct, painless papules that may progress to nodules or become ulcerated over time.1 Histologically, leishmaniasis is diagnosed by the identification of intracellular organisms that characteristically align along the peripheral rim inside the vacuole of a histiocyte.2 This unique finding is called the “marquee sign” due to its resemblance to light bulbs arranged around a dressing room mirror (Figure 1).2Leishmania amastigotes (also known as Leishman-Donovan bodies) have kinetoplasts that are helpful in diagnosis but also may be difficult to detect.2 Along with the Leishmania parasites, there typically is a mixed inflammatory infiltrate of plasma cells, lymphocytes, histiocytes, and neutrophils (Figure 2).1,2 There also may be varying degrees of pseudoepitheliomatous hyperplasia and overlying epidermal ulceration.1
Cutaneous botryomycosis can present clinically as a number of various primary lesions, including papules, nodules, or ulcers that may resemble leishmaniasis.3 Botryomycosis represents a specific histologic collection of bacterial granules, most commonly caused by Staphylococcus aureus.3 The dermal granulomatous infiltrate seen in botryomycosis often is similar to that seen in chronic leishmaniasis; however, one histologic feature unique to botryomycosis is the presence of characteristic basophilic staphylococcal grains that are arranged in clusters resembling bunches of grapes (the term botryo means “bunch of grapes” in Greek).3 A thin, eosinophilic rim consisting of antibodies, bacterial debris, and complement proteins and glycoproteins may encircle the basophilic grains but does not need to be present for diagnosis (Figure 3).3
Lepromatous leprosy presents as a symmetric, widespread eruption of macules, patches, plaques, or papules that are most prominent in acral areas.4 Perivascular infiltration of lymphocytes and histiocytes is characteristic of lepromatous leprosy.2 Mycobacteria bacilli also are seen within histiocytic vacuoles, similarly to leishmaniasis; however, collections of these bacilli congregate within the center of a foamy histiocyte to form a distinctive histologic finding known as a globus. These individual histiocytes containing central globi are called Virchow cells (Figure 4).2 However, lepromatous leprosy can be distinguished from leishmaniasis histologically by carefully observing the intracellular location of the infectious organism. Mycobacteria bacilli are located in the center of a histiocyte vacuole whereas Leishmania parasites demonstrate a peripheral alignment along a histiocyte vacuole. If any uncertainty remains between a diagnosis of leishmaniasis and lepromatous leprosy, positive Fite staining for mycobacteria easily differentiates between the 2 conditions.2,4
Cutaneous lobomycosis, a rare fungal infection transmitted by dolphins, manifests clinically as an asymptomatic nodule that is similar in appearance to a keloid. Histologic similarities to leishmaniasis include pseudoepitheliomatous hyperplasia and dermal granulomatous inflammation.4 The most distinguishing characteristic of lobomycosis is the presence of round, thick-walled, white organisms connected in a “string of beads” or chainlike configuration (Figure 5).2 Unlike leishmaniasis, lobomycosis fungal organisms would stain positive on periodic acid–Schiff staining.4
Cutaneous protothecosis is a rare clinical entity that presents as an isolated nodule or plaque or bursitis.4 It occurs following minor trauma and inoculation with Prototheca organisms, a genus of algae found in contaminated water.2,4 In its morula form, Prototheca adopts a characteristic arrangement within histiocytes that strikingly resembles a soccer ball (Figure 6).2 Conversely, nonmorulating forms of protothecosis can also be seen; these exhibit a central basophilic, dotlike structure within the histiocytes surrounded by a white halo.2 Definitive diagnosis of protothecosis can only be made upon successful culture of the algae.5
- Kevric I, Cappel MA, Keeling JH. New World and Old World leishmania infections: a practical review. Dermatol Clin. 2015;33:579-593.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 2nd ed. London, England: Elsevier Saunders; 2013.
- De Vries HJ, Van Noesel CJ, Hoekzema R, et al. Botryomycosis in an HIV-positive subject. J Eur Acad Dermatol Venereol. 2003;17:87-90.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences UK; 2012.
- Hillesheim PB, Bahrami S. Cutaneous protothecosis. Arch Pathol Lab Med. 2011;135:941-944.
- Kevric I, Cappel MA, Keeling JH. New World and Old World leishmania infections: a practical review. Dermatol Clin. 2015;33:579-593.
- Elston DM, Ferringer T, Ko CJ, et al. Dermatopathology. 2nd ed. London, England: Elsevier Saunders; 2013.
- De Vries HJ, Van Noesel CJ, Hoekzema R, et al. Botryomycosis in an HIV-positive subject. J Eur Acad Dermatol Venereol. 2003;17:87-90.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier Health Sciences UK; 2012.
- Hillesheim PB, Bahrami S. Cutaneous protothecosis. Arch Pathol Lab Med. 2011;135:941-944.
Desmoplastic Melanoma
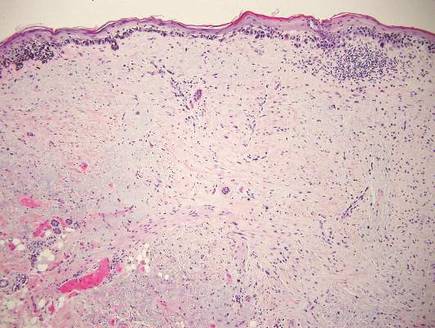
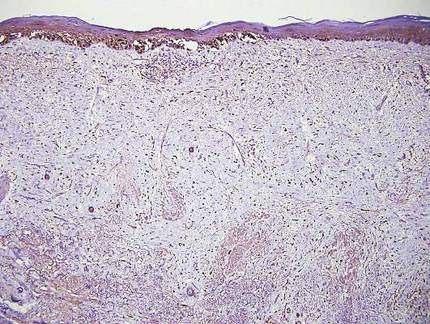
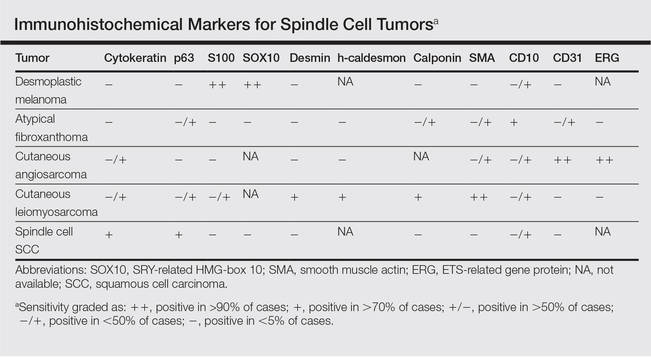
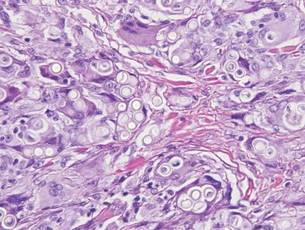
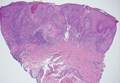
Desmoplastic melanoma, an uncommon variant of melanoma, poses a diagnostic challenge to the clinician because the tumors frequently appear as nonspecific flesh-colored or amelanotic plaques or nodules. They are more common in men than in women and are frequently found on the head and neck.1,2 Their innocuous appearance may lead to a delay in diagnosis and may explain why desmoplastic melanomas often are deeply infiltrative at the time of biopsy. Desmoplastic melanoma arises de novo in approximately one-third of cases.1 In the remainder of cases, it is seen in conjunction with overlying melanoma in situ, most commonly lentigo maligna melanoma.1 Histologically, desmoplastic melanomas are characterized by malignant spindle cells within a densely fibrotic stroma (Figure 1). Adjacent lymphoid aggregates and perineural involvement are common features,2 while pigment and atypical mitoses can be infrequent. Desmoplastic melanoma can be classified as mixed or pure based on the degree of desmoplasia and cellularity. Within mixed desmoplastic melanomas, there are areas that have histologic features of conventional melanomas while others demonstrate more typical desmoplastic characteristics. Pure desmoplastic melanoma has a higher degree of desmoplasia and fewer tumor cells than the mixed type.1 The pure subtype tends to be less aggressive and is less likely to metastasize to the lymph nodes.1 In the absence of an in situ component (Figure 2), desmoplastic melanoma may be indistinguishable from other spindle cell tumors on routine hematoxylin and eosin staining; thus, immunohistochemical staining generally is required. The most reliable stains in confirming a diagnosis of desmoplastic melanoma are S100 and SOX10 (SRY-related HMG-box 10)(Figure 3)(eTable).3
Atypical fibroxathoma typically presents as a nodule in the head and neck region or other sun-exposed areas in elderly individuals and is more commonly seen in men than in women.4 Histologically, atypical fibroxanthomas are composed of pleomorphic spindle, epithelioid, and multinucleated giant cells with numerous and atypical mitoses (Figure 4).5 Atypical fibroxanthoma is considered a diagnosis of exclusion; therefore, other dermal spindle cell tumors need to be ruled out before diagnosis can be made. Atypical fibroxanthomas generally stain negative for cytokeratin, S100, SOX10, and desmin, but in some cases there is positive focal staining for smooth muscle actin.4 Multiple immunohistochemical markers, including CD10, have shown reactivity in atypical fibroxanthomas,4 but none of these markers has a high specificity for this tumor; thus, it remains a diagnosis of exclusion.
Cutaneous angiosarcomas are aggressive tumors associated with a high mortality rate despite appropriate treatment with surgical resection and postoperative radiation treatment. They typically present as ecchymotic macules or nodules on the face or scalp of elderly patients.6,7 Ionizing radiation and chronic lymphedema are risk factors for cutaneous angiosarcoma.6 Histologically, well-differentiated cutaneous angiosarcomas are composed of irregular, anastomosing vascular channels that dissect through the dermis (Figure 5).6,7 Less well-differentiated tumors may contain spindle cells and lack obvious vascular structures; thus immunohistochemistry is essential for making the correct diagnosis in these cases. Cutaneous angiosarcomas typically stain positive for ERG (ETS-related gene) protein, CD31, CD34, and factor VIII.6,8 Unfortunately these tumors may also occasionally stain with cytokeratin, which may lead to the erroneous diagnosis of a carcinoma.6
| 
| |
| Figure 4. Pleomorphic spindle, epithelioid, and multinucleate giant cells with atypical mitoses filling the dermis in atypical fibroxanthoma (H&E, original magnification ×200). | Figure 5. Anastamosing vascular channels dissecting through collagen bundles and consuming the epidermis in cutaneous angiosarcoma (H&E, original magnification ×100). |
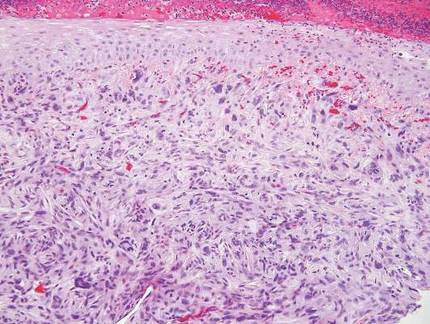
Cutaneous leiomyosarcoma is a smooth muscle neoplasm that arises from arrector pili muscles, genital smooth muscles, or vascular smooth muscles. It typically presents as a single plaque or nodule on the arms and legs of individuals older than 50 years of age.9 Cutaneous leiomyosarcomas can be classified as either dermal, in which at least 90% of the tumor is confined to the dermis, or subcutaneous; this distinction is important because the latter type has a higher rate of metastasis and a poorer prognosis.9 Because of this tumor’s smooth muscle derivation, well-differentiated tumors may retain features of typical smooth muscle cells, including cigar-shaped nuclei with adjacent glycogen vacuoles (Figure 6). If fascicle formation is observed, this may be an additional clue to the diagnosis. In poorly differentiated tumors, immunohistochemistry is invaluable. Leiomyosarcoma often stains positive for smooth muscle actin, muscle specific actin, h-caldesmon, desmin, and calponin.9-11
Spindle cell squamous cell carcinomas often present as ulcerated nodules on sun-exposed skin or on sites of prior ionizing radiation.2,12 Like desmoplastic melanoma, spindle cell squamous cell carcinomas are characterized by spindle cells in the dermis. Helpful diagnostic clues may include evidence of squamous differentiation, including keratin pearls or overlying actinic keratosis (Figure 7). However, actinic keratosis is common on sun-damaged skin and cannot be used to definitively confirm this diagnosis. There also may be areas of the tumor with more typical epithelioid cells that are easily identified as squamous cell carcinoma.2 Spindle cell squamous cell carcinoma stains positive for high–molecular weight cytokeratin antibodies and p63,2 which can help to differentiate it from the other spindle cell tumors in the differential.
| 
| |
| Figure 6. Spindle cells of leiomyosarcoma with cigar-shaped nuclei and adjacent glycogen vacuoles (H&E, original magnification ×600). | Figure 7. Spindle cell squamous cell carcinoma with overlying epidermal atypia that blends with the underlying dermal spindle cells (H&E, original magnification ×100). |
1. Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
3. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
4. Luzar B, Calonje E. Morphological and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
5. Iorizzo LJ III, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
6. Luca DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
7. Mendenhall WM, Mendenhall CM, Werning JW, et al. Cutaneous angiosarcoma. Am J Oncol. 2006;29:524-528.
8. Thum C, Husain EA, Mulholland K, et al. Atypical fibroxanthoma with pseudoangiomatous features: a histological and immunohistochemical mimic of cutaneous angiosarcoma. Ann Diagn Pathol. 2013;17:502-507.
9. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
10. Hall BJ, Grossmann AH, Webber NP, et al. Atypical intradermal smooth muscle neoplasms (formerly cutaneous leiomyosarcomas): case series, immunohistochemical profile and review of the literature. Appl Immunohistochem Mol Morphol. 2013;21:132-138.
11. Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
12. Cassarino DS, DeRienzo DP, Barr RJ. Cutaneous squamous cell carcinoma: a comprehensive clinicopathologic classification. part one. J Cutan Pathol. 2006;33:191-205.
Desmoplastic melanoma, an uncommon variant of melanoma, poses a diagnostic challenge to the clinician because the tumors frequently appear as nonspecific flesh-colored or amelanotic plaques or nodules. They are more common in men than in women and are frequently found on the head and neck.1,2 Their innocuous appearance may lead to a delay in diagnosis and may explain why desmoplastic melanomas often are deeply infiltrative at the time of biopsy. Desmoplastic melanoma arises de novo in approximately one-third of cases.1 In the remainder of cases, it is seen in conjunction with overlying melanoma in situ, most commonly lentigo maligna melanoma.1 Histologically, desmoplastic melanomas are characterized by malignant spindle cells within a densely fibrotic stroma (Figure 1). Adjacent lymphoid aggregates and perineural involvement are common features,2 while pigment and atypical mitoses can be infrequent. Desmoplastic melanoma can be classified as mixed or pure based on the degree of desmoplasia and cellularity. Within mixed desmoplastic melanomas, there are areas that have histologic features of conventional melanomas while others demonstrate more typical desmoplastic characteristics. Pure desmoplastic melanoma has a higher degree of desmoplasia and fewer tumor cells than the mixed type.1 The pure subtype tends to be less aggressive and is less likely to metastasize to the lymph nodes.1 In the absence of an in situ component (Figure 2), desmoplastic melanoma may be indistinguishable from other spindle cell tumors on routine hematoxylin and eosin staining; thus, immunohistochemical staining generally is required. The most reliable stains in confirming a diagnosis of desmoplastic melanoma are S100 and SOX10 (SRY-related HMG-box 10)(Figure 3)(eTable).3
Atypical fibroxathoma typically presents as a nodule in the head and neck region or other sun-exposed areas in elderly individuals and is more commonly seen in men than in women.4 Histologically, atypical fibroxanthomas are composed of pleomorphic spindle, epithelioid, and multinucleated giant cells with numerous and atypical mitoses (Figure 4).5 Atypical fibroxanthoma is considered a diagnosis of exclusion; therefore, other dermal spindle cell tumors need to be ruled out before diagnosis can be made. Atypical fibroxanthomas generally stain negative for cytokeratin, S100, SOX10, and desmin, but in some cases there is positive focal staining for smooth muscle actin.4 Multiple immunohistochemical markers, including CD10, have shown reactivity in atypical fibroxanthomas,4 but none of these markers has a high specificity for this tumor; thus, it remains a diagnosis of exclusion.
Cutaneous angiosarcomas are aggressive tumors associated with a high mortality rate despite appropriate treatment with surgical resection and postoperative radiation treatment. They typically present as ecchymotic macules or nodules on the face or scalp of elderly patients.6,7 Ionizing radiation and chronic lymphedema are risk factors for cutaneous angiosarcoma.6 Histologically, well-differentiated cutaneous angiosarcomas are composed of irregular, anastomosing vascular channels that dissect through the dermis (Figure 5).6,7 Less well-differentiated tumors may contain spindle cells and lack obvious vascular structures; thus immunohistochemistry is essential for making the correct diagnosis in these cases. Cutaneous angiosarcomas typically stain positive for ERG (ETS-related gene) protein, CD31, CD34, and factor VIII.6,8 Unfortunately these tumors may also occasionally stain with cytokeratin, which may lead to the erroneous diagnosis of a carcinoma.6
|
|
| |
| Figure 4. Pleomorphic spindle, epithelioid, and multinucleate giant cells with atypical mitoses filling the dermis in atypical fibroxanthoma (H&E, original magnification ×200). | Figure 5. Anastamosing vascular channels dissecting through collagen bundles and consuming the epidermis in cutaneous angiosarcoma (H&E, original magnification ×100). |
Cutaneous leiomyosarcoma is a smooth muscle neoplasm that arises from arrector pili muscles, genital smooth muscles, or vascular smooth muscles. It typically presents as a single plaque or nodule on the arms and legs of individuals older than 50 years of age.9 Cutaneous leiomyosarcomas can be classified as either dermal, in which at least 90% of the tumor is confined to the dermis, or subcutaneous; this distinction is important because the latter type has a higher rate of metastasis and a poorer prognosis.9 Because of this tumor’s smooth muscle derivation, well-differentiated tumors may retain features of typical smooth muscle cells, including cigar-shaped nuclei with adjacent glycogen vacuoles (Figure 6). If fascicle formation is observed, this may be an additional clue to the diagnosis. In poorly differentiated tumors, immunohistochemistry is invaluable. Leiomyosarcoma often stains positive for smooth muscle actin, muscle specific actin, h-caldesmon, desmin, and calponin.9-11
Spindle cell squamous cell carcinomas often present as ulcerated nodules on sun-exposed skin or on sites of prior ionizing radiation.2,12 Like desmoplastic melanoma, spindle cell squamous cell carcinomas are characterized by spindle cells in the dermis. Helpful diagnostic clues may include evidence of squamous differentiation, including keratin pearls or overlying actinic keratosis (Figure 7). However, actinic keratosis is common on sun-damaged skin and cannot be used to definitively confirm this diagnosis. There also may be areas of the tumor with more typical epithelioid cells that are easily identified as squamous cell carcinoma.2 Spindle cell squamous cell carcinoma stains positive for high–molecular weight cytokeratin antibodies and p63,2 which can help to differentiate it from the other spindle cell tumors in the differential.
|
|
| |
| Figure 6. Spindle cells of leiomyosarcoma with cigar-shaped nuclei and adjacent glycogen vacuoles (H&E, original magnification ×600). | Figure 7. Spindle cell squamous cell carcinoma with overlying epidermal atypia that blends with the underlying dermal spindle cells (H&E, original magnification ×100). |
Desmoplastic melanoma, an uncommon variant of melanoma, poses a diagnostic challenge to the clinician because the tumors frequently appear as nonspecific flesh-colored or amelanotic plaques or nodules. They are more common in men than in women and are frequently found on the head and neck.1,2 Their innocuous appearance may lead to a delay in diagnosis and may explain why desmoplastic melanomas often are deeply infiltrative at the time of biopsy. Desmoplastic melanoma arises de novo in approximately one-third of cases.1 In the remainder of cases, it is seen in conjunction with overlying melanoma in situ, most commonly lentigo maligna melanoma.1 Histologically, desmoplastic melanomas are characterized by malignant spindle cells within a densely fibrotic stroma (Figure 1). Adjacent lymphoid aggregates and perineural involvement are common features,2 while pigment and atypical mitoses can be infrequent. Desmoplastic melanoma can be classified as mixed or pure based on the degree of desmoplasia and cellularity. Within mixed desmoplastic melanomas, there are areas that have histologic features of conventional melanomas while others demonstrate more typical desmoplastic characteristics. Pure desmoplastic melanoma has a higher degree of desmoplasia and fewer tumor cells than the mixed type.1 The pure subtype tends to be less aggressive and is less likely to metastasize to the lymph nodes.1 In the absence of an in situ component (Figure 2), desmoplastic melanoma may be indistinguishable from other spindle cell tumors on routine hematoxylin and eosin staining; thus, immunohistochemical staining generally is required. The most reliable stains in confirming a diagnosis of desmoplastic melanoma are S100 and SOX10 (SRY-related HMG-box 10)(Figure 3)(eTable).3
Atypical fibroxathoma typically presents as a nodule in the head and neck region or other sun-exposed areas in elderly individuals and is more commonly seen in men than in women.4 Histologically, atypical fibroxanthomas are composed of pleomorphic spindle, epithelioid, and multinucleated giant cells with numerous and atypical mitoses (Figure 4).5 Atypical fibroxanthoma is considered a diagnosis of exclusion; therefore, other dermal spindle cell tumors need to be ruled out before diagnosis can be made. Atypical fibroxanthomas generally stain negative for cytokeratin, S100, SOX10, and desmin, but in some cases there is positive focal staining for smooth muscle actin.4 Multiple immunohistochemical markers, including CD10, have shown reactivity in atypical fibroxanthomas,4 but none of these markers has a high specificity for this tumor; thus, it remains a diagnosis of exclusion.
Cutaneous angiosarcomas are aggressive tumors associated with a high mortality rate despite appropriate treatment with surgical resection and postoperative radiation treatment. They typically present as ecchymotic macules or nodules on the face or scalp of elderly patients.6,7 Ionizing radiation and chronic lymphedema are risk factors for cutaneous angiosarcoma.6 Histologically, well-differentiated cutaneous angiosarcomas are composed of irregular, anastomosing vascular channels that dissect through the dermis (Figure 5).6,7 Less well-differentiated tumors may contain spindle cells and lack obvious vascular structures; thus immunohistochemistry is essential for making the correct diagnosis in these cases. Cutaneous angiosarcomas typically stain positive for ERG (ETS-related gene) protein, CD31, CD34, and factor VIII.6,8 Unfortunately these tumors may also occasionally stain with cytokeratin, which may lead to the erroneous diagnosis of a carcinoma.6
|
|
| |
| Figure 4. Pleomorphic spindle, epithelioid, and multinucleate giant cells with atypical mitoses filling the dermis in atypical fibroxanthoma (H&E, original magnification ×200). | Figure 5. Anastamosing vascular channels dissecting through collagen bundles and consuming the epidermis in cutaneous angiosarcoma (H&E, original magnification ×100). |
Cutaneous leiomyosarcoma is a smooth muscle neoplasm that arises from arrector pili muscles, genital smooth muscles, or vascular smooth muscles. It typically presents as a single plaque or nodule on the arms and legs of individuals older than 50 years of age.9 Cutaneous leiomyosarcomas can be classified as either dermal, in which at least 90% of the tumor is confined to the dermis, or subcutaneous; this distinction is important because the latter type has a higher rate of metastasis and a poorer prognosis.9 Because of this tumor’s smooth muscle derivation, well-differentiated tumors may retain features of typical smooth muscle cells, including cigar-shaped nuclei with adjacent glycogen vacuoles (Figure 6). If fascicle formation is observed, this may be an additional clue to the diagnosis. In poorly differentiated tumors, immunohistochemistry is invaluable. Leiomyosarcoma often stains positive for smooth muscle actin, muscle specific actin, h-caldesmon, desmin, and calponin.9-11
Spindle cell squamous cell carcinomas often present as ulcerated nodules on sun-exposed skin or on sites of prior ionizing radiation.2,12 Like desmoplastic melanoma, spindle cell squamous cell carcinomas are characterized by spindle cells in the dermis. Helpful diagnostic clues may include evidence of squamous differentiation, including keratin pearls or overlying actinic keratosis (Figure 7). However, actinic keratosis is common on sun-damaged skin and cannot be used to definitively confirm this diagnosis. There also may be areas of the tumor with more typical epithelioid cells that are easily identified as squamous cell carcinoma.2 Spindle cell squamous cell carcinoma stains positive for high–molecular weight cytokeratin antibodies and p63,2 which can help to differentiate it from the other spindle cell tumors in the differential.
|
|
| |
| Figure 6. Spindle cells of leiomyosarcoma with cigar-shaped nuclei and adjacent glycogen vacuoles (H&E, original magnification ×600). | Figure 7. Spindle cell squamous cell carcinoma with overlying epidermal atypia that blends with the underlying dermal spindle cells (H&E, original magnification ×100). |
1. Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
3. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
4. Luzar B, Calonje E. Morphological and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
5. Iorizzo LJ III, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
6. Luca DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
7. Mendenhall WM, Mendenhall CM, Werning JW, et al. Cutaneous angiosarcoma. Am J Oncol. 2006;29:524-528.
8. Thum C, Husain EA, Mulholland K, et al. Atypical fibroxanthoma with pseudoangiomatous features: a histological and immunohistochemical mimic of cutaneous angiosarcoma. Ann Diagn Pathol. 2013;17:502-507.
9. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
10. Hall BJ, Grossmann AH, Webber NP, et al. Atypical intradermal smooth muscle neoplasms (formerly cutaneous leiomyosarcomas): case series, immunohistochemical profile and review of the literature. Appl Immunohistochem Mol Morphol. 2013;21:132-138.
11. Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
12. Cassarino DS, DeRienzo DP, Barr RJ. Cutaneous squamous cell carcinoma: a comprehensive clinicopathologic classification. part one. J Cutan Pathol. 2006;33:191-205.
1. Chen LL, Jaimes N, Barker CA, et al. Desmoplastic melanoma: a review. J Am Acad Dermatol. 2013;68:825-833.
2. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
3. Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
4. Luzar B, Calonje E. Morphological and immunohistochemical characteristics of atypical fibroxanthoma with a special emphasis on potential diagnostic pitfalls: a review. J Cutan Pathol. 2010;37:301-309.
5. Iorizzo LJ III, Brown MD. Atypical fibroxanthoma: a review of the literature. Dermatol Surg. 2011;37:146-157.
6. Luca DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
7. Mendenhall WM, Mendenhall CM, Werning JW, et al. Cutaneous angiosarcoma. Am J Oncol. 2006;29:524-528.
8. Thum C, Husain EA, Mulholland K, et al. Atypical fibroxanthoma with pseudoangiomatous features: a histological and immunohistochemical mimic of cutaneous angiosarcoma. Ann Diagn Pathol. 2013;17:502-507.
9. Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
10. Hall BJ, Grossmann AH, Webber NP, et al. Atypical intradermal smooth muscle neoplasms (formerly cutaneous leiomyosarcomas): case series, immunohistochemical profile and review of the literature. Appl Immunohistochem Mol Morphol. 2013;21:132-138.
11. Perez-Montiel MD, Plaza JA, Dominguez-Malagon H, et al. Differential expression of smooth muscle myosin, smooth muscle actin, h-caldesmon, and calponin in the diagnosis of myofibroblastic and smooth muscle lesions of skin and soft tissue. Am J Dermatopathol. 2006;28:105-111.
12. Cassarino DS, DeRienzo DP, Barr RJ. Cutaneous squamous cell carcinoma: a comprehensive clinicopathologic classification. part one. J Cutan Pathol. 2006;33:191-205.
Chromoblastomycosis
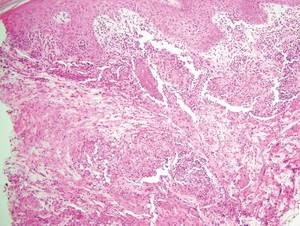
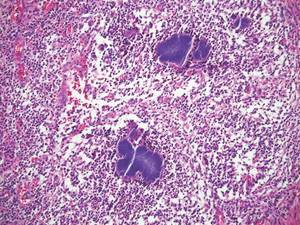
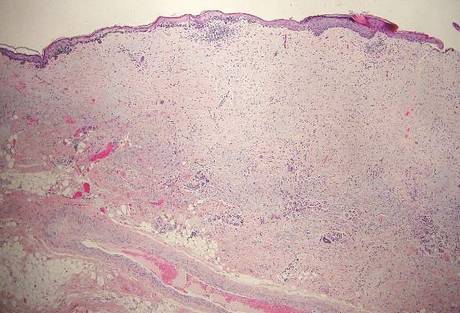
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7

Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7
Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
Chromoblastomycosis is a chronic fungal infection of the skin and subcutaneous tissues that demonstrates characteristic Medlar or sclerotic bodies that resemble copper pennies on histopathology.1 Cutaneous infection often results from direct inoculation, such as from a wood splinter. Clinically, the lesion typically is a pink papule that progresses to a verrucous plaque on the legs of farmers or rural workers in the tropics or subtropics. There usually are no associated constitutional symptoms. Several dematiaceous (darkly pigmented) fungi cause chromoblastomycosis, including Fonsecaea compacta, Cladophialophora carrionii, Rhinocladiella aquaspersa, Phialophora verrucosa, and Fonsecaea pedrosoi. Cellular division occurs by internal septation rather than budding. Skin biopsy can confirm the diagnosis.1 Chromoblastomycosis is histopathologically characterized by pseudoepitheli- omatous hyperplasia (Figure 1) with histiocytes and neutrophils surrounding distinct copper-colored Medlar bodies (6–12 μm)(Figure 2), which are fungal spores.1-3 Several conditions demonstrate pseudoepitheliomatous hyperplasia with intraepidermal pustules and can be remembered by the mnemonic “here come big green leafy vegetables”: halogenoderma, chromoblastomycosis, blastomycosis, granuloma inguinale, leishmaniasis, and pemphigus vegetans.2 Treatment of chromoblastomycosis can be challenging, as no standard treatment has been established and therapy can be complicated by low cure rates and high relapse rates, especially in chronic and extensive disease. Treatment can include cryotherapy or surgical excision for small lesions in combination with systemic antifungals.4 Itraconazole (200–400 mg daily) for at least 6 months has been reported to have up to a 90% cure rate with mild to moderate disease and 44% with severe disease.5 Combination oral antifungal treatment with itraconazole and terbinafine has been recommended.6 There are reports of progression of chromoblastomycosis to squamous cell carcinoma, which is rare and occurred after long-standing, inadequately treated lesions.7
Blastomycosis also presents with pseudoepitheliomatous hyperplasia, as seen in chromoblastomycosis, but organisms typically are few in number and demonstrate a thick, asymmetrical, refractile wall and a dark nucleus. Although chromoblastomycosis and blastomycosis are similar in size (8–15 μm), the broad-based budding of blastomycosis (Figure 3) is a key feature and the yeast are not pigmented.1-3 Blastomycosis is caused by Blastomyces dermatitidis and is endemic to the Mississippi and Ohio River valleys, Great Lakes region, and Southeastern United States. Cutaneous infection typically occurs from inhalation of the dimorphic fungi into the lungs and occasional dissemination involving the skin, causing papulopustules and thick, crusted, warty plaques with central ulceration. Rarely, primary cutaneous blastomycosis can occur from direct inoculation, typically in a laboratory. Treatment of disseminated blastomycosis includes systemic antifungals.1
Coccidioidomycosis is characterized by large spherules (10–80 μm) with refractile walls and granular gray cytoplasm.2,3 Coccidioidomycosis spherules occasionally contain endospores2 and often are noticeably larger than surrounding histiocyte nuclei (Figure 4), whereas chromoblastomycosis, blastomycosis, cryptococcosis, and lobomycosis are more similar in size to histiocyte nuclei. Coccidioidomycosis is caused by Coccidioides immitis, a highly virulent dimorphic fungus found in the Southwestern United States, northern Mexico, and Central and South America. Pulmonary infection occurs by inhalation of arthroconidia, often from soil, and is asymptomatic in most patients; however, immunocompromised patients are predisposed to disseminated cutaneous infection. Facial lesions are most common and can present as papules, pustules, plaques, abscesses, sinus tracts, and/or ulcerations. Treatment of disseminated infection requires systemic antifungals; amphotericin B has proven most effective.1
Cryptococcosis is characterized by vacuoles with small (2–20 μm), central, pleomorphic yeast (Figure 5). The vacuole is due to a gelati- nous capsule that stains red with mucicarmine and blue with Alcian blue.2,3 Cryptococcosis is caused by Cryptococcus neoformans and is associated with pigeon droppings. Disseminated infection in patients with human immunodefi- ciency virus often presents as umbilicated molluscumlike lesions and portends a poor prognosis with a mortality rate of up to 80%.8 Disseminated infection necessitates aggressive treatment with systemic antifungals.1
Lobomycosis demonstrates thick-walled, refractile spherules with surrounding histiocytes and multinucleated giant cells. The yeast of lobomycosis (6–12 μm) is of similar size to chromoblastomycosis and blastomycosis, but linear chains resembling a child’s pop beads are characteristic of this condition (Figure 6).2,3 Lobomycosis is caused by Lacazia loboi and is acquired most frequently through contact with dolphins in Central and South America. Clinically, lesions present as slow-growing, keloidlike nodules, often on the face, ears, and distal extremities. Surgical treatment may be required given that oral antifungals typically are ineffective.1
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
- Bolognia JL, Jorizzo JL, Shaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier; 2012.
- Elston DM, Ferringer TC, Ko C, et al. Dermatopathology: Requisites in Dermatology. 2nd ed. Philadelphia, PA: Saunders Elsevier; 2014.
- Fernandez-Flores A, Saeb-Lima M, Arenas-Guzman R. Morphological findings of deep cutaneous fungal infections. Am J Dermatopathol. 2014;36:531-556.
- Ameen M. Chromoblastomycosis: clinical presentation and management. Clin Exp Dermatol. 2009;34:849-854.
- Queiroz-Telles F, McGinnis MR, Salkin I, et al. Subcutaneous mycoses. Infect Dis Clin North Am. 2003;17:59-85.
- Bonifaz A, Paredes-Solís, Saúl A. Treating chromoblastomycosis with systemic antifungals. Expert Opin Pharmacother. 2004;5:247-254.
- Rojas OC, González GM, Moreno-Treviño M, et al. Chromoblastomycosis by Cladophialophora carrionii associated with squamous cell carcinoma and review of published reports. Mycopathologia. 2015;179:153-157.
- Durden FM, Elewski B. Cutaneous involvement with Cryptococcus neoformans in AIDS. J Am Acad Dermatol. 1994;30:844-848.
Syringoid Eccrine Carcinoma
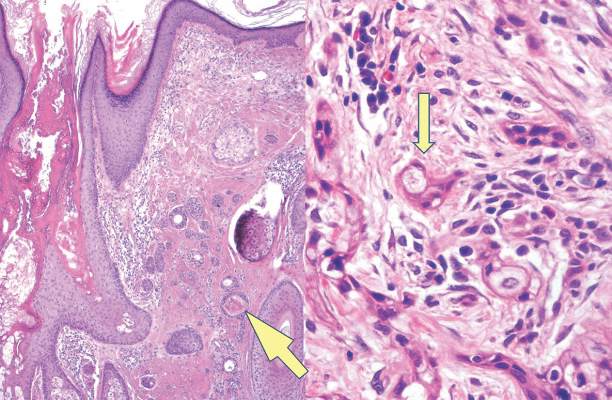
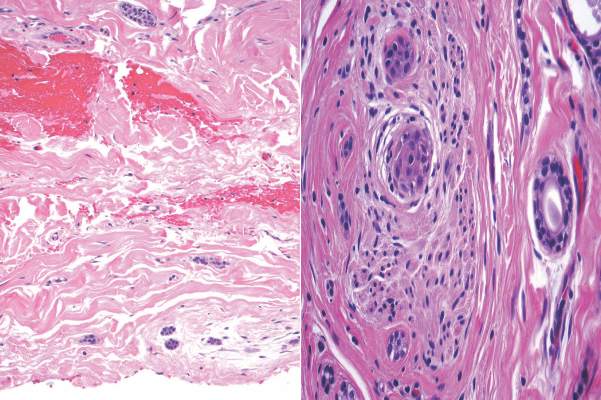
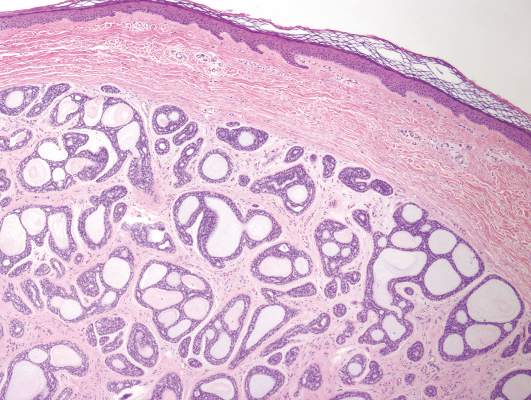
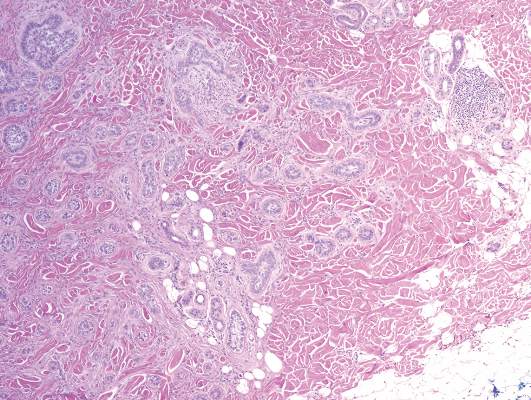
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
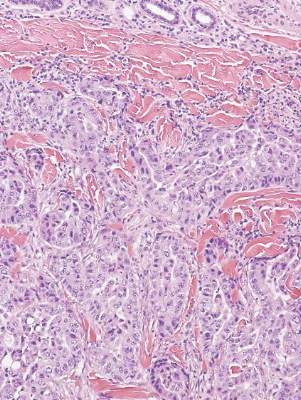
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
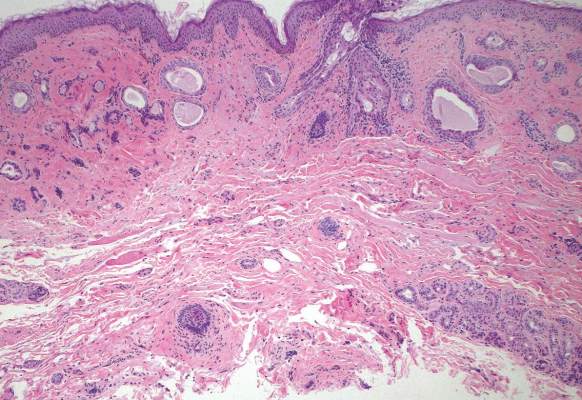
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
Syringoid eccrine carcinoma is a rare malignant adnexal tumor with eccrine differentiation that histologically resembles a syringoma.1 Originally described as eccrine epithelioma by Freeman and Winklemann2 in 1969, syringoid eccrine carcinoma has been reported in the literature as eccrine carcinoma, eccrine syringomatous carcinoma, and sclerosing sweat duct carcinoma.3 Clinically, syringoid eccrine carcinoma most commonly presents as a tender plaque or nodule on the scalp, and histologic examination generally reveals a dermal-based lesion that rarely shows epidermal connection. It demonstrates syringomalike tadpole morphology (epithelial strands with lumen formation) composed of basaloid epithelium with uniform hyperchromatic nuclei (Figure 1). There usually is an infiltrative growth pattern to the subcutis (Figure 2 [left]) or skeletal muscle as well as remarkable perineural invasion (Figure 2 [right]). Mitotic activity is minimal to absent. The tumor cells of syringoid eccrine carcinoma typically show positive immuno-staining for high- and low-molecular-weight cytokeratin, while the lumina are highlighted by epithelial membrane antigen and carcinoembryonic antigen.4 However, immunohistochemistry often is not contributory in diagnosing primary eccrine carcinomas.
The differential diagnosis of syringoid eccrine carcinoma includes cutaneous adenoid cystic carcinoma, metastatic adenocarcinoma, sclerosing basal cell carcinoma, and syringoma. Cutaneous adenoid cystic carcinoma is a rare, slow-growing, flesh-colored tumor that consists of lobules, islands, and cords of basaloid cells with prominent cystic cribriforming (Figure 3). The tumor cells typically are small, cuboidal, and monomorphic. Metastatic adenoid cystic carcinoma, such as from a primary tumor of the salivary glands or breasts, must be excluded before rendering a diagnosis of primary cutaneous disease.
Metastatic adenocarcinoma of the skin usually presents in patients with a clinical history of preexisting disease. The breasts, colon, stomach, and ovaries are common origins of metastases. The histopathologic and immunohistochemical findings depend on the particular site of origin of the metastasis. Compared with primary eccrine carcinomas, metastatic adenocarcinomas of the skin generally are high-grade lesions with prominent atypia, mitosis, and necrosis (Figure 4).
Sclerosing basal cell carcinoma shows basaloid tumor cells with deep infiltration. Unlike syringoid eccrine carcinoma, basal cell carcinoma is an epidermal tumor that does not have true lumen formation. Furthermore, other variants of basal cell carcinoma, including nodular, micronodular, or superficial multicentric tumors, often coexist with the sclerosing variant in the same lesion and constitute a useful diagnostic clue (Figure 5). Staining for epithelial membrane antigen may be useful in identifying the absence of lumen formation, and Ber-EP4 highlights the epidermal origin of the lesion.5
Syringomas most commonly present as multiple small flesh-colored papules on the eyelids. On histology, syringomas present as small superficial dermal lesions composed of small ducts that may form tadpolelike structures in a fibrotic stroma (Figure 6). The ducts are lined by benign cuboidal cells. In contrast to syringoid eccrine carcinomas, syringomas usually present as multiple lesions that are microscopically superficial without perineural involvement.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
1. Sidiropoulos M, Sade S, Al-Habeeb A, et al. Syringoid eccrine carcinoma: a clinicopathological and immunohistochemical study of four cases. J Clin Pathol. 2011;64:788-792.
2. Freeman RG, Winklemann RK. Basal cell tumor with eccrine differentiations (eccrine epithelioma). Arch Dermatol. 1969;100:234-242.
3. Nishizawa A, Nakanishi Y, Sasajima Y, et al. Syringoid carcinoma with apparently aggressive transformation: case report and review of the literature. Int J Dermatol. 2006;45:1218-1221.
4. Urso C, Bondi R, Paglierani M, et al. Carcinomas of sweat glands: report of 60 cases. Arch Pathol Lab Med. 2001;125:498-505.
5. Cassarino D. Diagnostic Pathology: Neoplastic Dermatopathology. Salt Lake City, UT: Amirsys Publishing Inc; 2012.
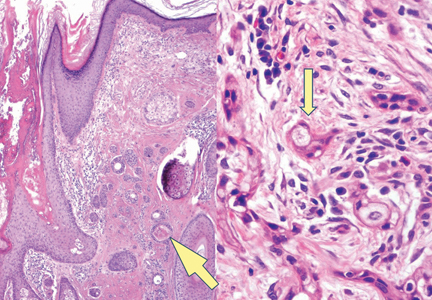
Trichilemmoma
Trichilemmomas are benign follicular neoplasms that exhibit differentiation toward the outer root sheath of the pilosebaceous follicular epithelium.1 Trichilemmomas clinically present as individual or multiple, slowly growing, verrucous papules appearing most commonly on the face or neck. The lesions may coalesce to form small plaques. Although trichilemmomas typically are isolated, patients with multiple trichilemmomas require a cancer screening workup due to their association with Cowden disease, which results from a mutation in the phosphatase and tensin homolog tumor suppressor gene, PTEN.2 An easy way to remember the association between trichilemmomas and Cowden disease is to alter the spelling to “trichile-moo-moo,” using the “moo moo” sound of an animal cow as a clue linking the tumor to Cowden disease.
Histologically, trichilemmomas exhibit a lobular epidermal downgrowth into the dermis (Figure 1). The surface of the lesion may be hyperkeratotic and somewhat papillomatous. Cells toward the center of the lobule are pale staining, periodic acid–Schiff positive, and diastase labile due to high levels of intracellular glycogen (Figure 2). Cells toward the periphery of the lobule usually appear basophilic with a palisading arrangement of the peripheral cells. The entire lobule is enclosed within an eosinophilic basement membrane that stains positively with periodic acid–Schiff (Figure 2).1 Consistent with the tumor’s differentiation toward the outer root sheath of the hair follicle, trichilemmomas have been reported to express CD34 focally or diffusely.3
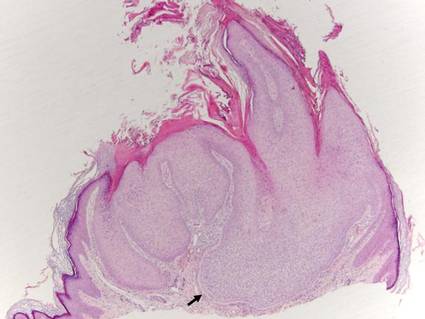
| 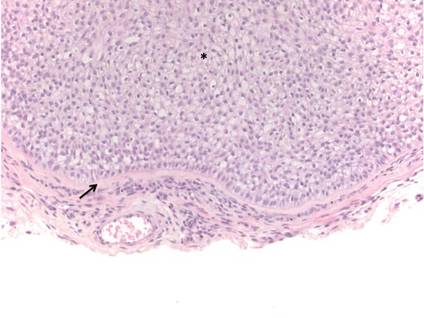
|
Similar to trichilemmoma, inverted follicular keratosis (IFK) commonly presents as a solitary asymptomatic papule on the face. Inverted follicular keratosis is a somewhat controversial entity, with some authorities arguing IFK is a variant of verruca vulgaris or seborrheic keratosis. Histologically, IFKs can be differentiated by the presence of squamous eddies (concentric layers of squamous cells in a whorled pattern), which are diagnostic, and central longitudinal crypts that contain keratin and are lined by squamous epithelium.4 Basaloid cells can be seen at the periphery of the tumors; however, IFKs lack an eosinophilic basement membrane surrounding the tumor (Figure 3).

Squamous cell carcinoma in situ classically appears as an erythematous hyperkeratotic papule or plaque on sun-exposed sites that can become crusted or ulcerated. Microscopically, squamous cell carcinoma in situ displays full-thickness disorderly maturation of keratinocytes. The keratinocytes exhibit nuclear pleomorphism. Atypical mitotic figures and dyskeratotic keratinocytes also can be seen throughout the full thickness of the epidermis (Figure 4).5
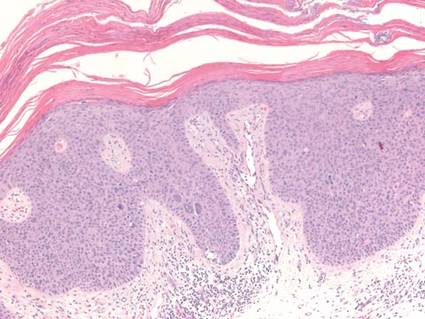
Verruca vulgaris (Figure 5) histologically demonstrates hyperkeratosis with tiers of parakeratosis, digitated epidermal hyperplasia, and dilated tortuous capillaries within the dermal papillae. At the edges of the lesion there often is inward turning of elongated rete ridges,6,7 which can be thought of as the rete reaching out for a hug of sorts to spread the human papillomavirus infection. Although the surface of a trichilemmoma can bear resemblance to a verruca vulgaris, the remainder of the histologic features can be used to help differentiate these tumors. Additionally, there has been no evidence suggestive of a viral etiology for trichilemmomas.8

Warty dyskeratoma features an umbilicated papule, usually on the face, head, or neck, that is associated with a follicular unit. The papule shows a cup-shaped, keratin-filled invagination; suprabasilar clefting; and acantholytic dyskeratotic cells, which are features that are not seen in trichilemmomas (Figure 6).9

Acknowledgment—The authors would like to thank Brandon Litzner, MD, St Louis, Missouri, for proofreading the manuscript.
1. Brownstein MH, Shapiro L. Trichilemmoma: analysis of 40 new cases. Arch Dermatol. 1973;107:866-869.
2. Al-Zaid T, Ditelberg J, Prieto V, et al. Trichilemmomas show loss of PTEN in Cowden syndrome but only rarely in sporadic tumors. J Cutan Pathol. 2012;39:493-499.
3. Tardío JC. CD34-reactive tumors of the skin. an updated review of an ever-growing list of lesions. J Cutan Pathol. 2009;36:89-102.
4. Mehregan A. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
5. Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. 2000;42(1, pt 2):11-17.
6. Jabłonska S, Majewski S, Obalek S, et al. Cutaneous warts. Clin Dermatol. 1997;15:309-319.
7. Hardin J, Gardner J, Colome M, et al. Verrucous cyst with melanocytic and sebaceous differentiation. Arch Path Lab Med. 2013;137:576-579.
8. Johnson BL, Kramer EM, Lavker RM. The keratotic tumors of Cowden’s disease: an electron microscopy study. J Cutan Pathol. 1987;14:291-298.
9. Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma—“follicular dyskeratoma”: analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
Trichilemmomas are benign follicular neoplasms that exhibit differentiation toward the outer root sheath of the pilosebaceous follicular epithelium.1 Trichilemmomas clinically present as individual or multiple, slowly growing, verrucous papules appearing most commonly on the face or neck. The lesions may coalesce to form small plaques. Although trichilemmomas typically are isolated, patients with multiple trichilemmomas require a cancer screening workup due to their association with Cowden disease, which results from a mutation in the phosphatase and tensin homolog tumor suppressor gene, PTEN.2 An easy way to remember the association between trichilemmomas and Cowden disease is to alter the spelling to “trichile-moo-moo,” using the “moo moo” sound of an animal cow as a clue linking the tumor to Cowden disease.
Histologically, trichilemmomas exhibit a lobular epidermal downgrowth into the dermis (Figure 1). The surface of the lesion may be hyperkeratotic and somewhat papillomatous. Cells toward the center of the lobule are pale staining, periodic acid–Schiff positive, and diastase labile due to high levels of intracellular glycogen (Figure 2). Cells toward the periphery of the lobule usually appear basophilic with a palisading arrangement of the peripheral cells. The entire lobule is enclosed within an eosinophilic basement membrane that stains positively with periodic acid–Schiff (Figure 2).1 Consistent with the tumor’s differentiation toward the outer root sheath of the hair follicle, trichilemmomas have been reported to express CD34 focally or diffusely.3
|
|
|
Similar to trichilemmoma, inverted follicular keratosis (IFK) commonly presents as a solitary asymptomatic papule on the face. Inverted follicular keratosis is a somewhat controversial entity, with some authorities arguing IFK is a variant of verruca vulgaris or seborrheic keratosis. Histologically, IFKs can be differentiated by the presence of squamous eddies (concentric layers of squamous cells in a whorled pattern), which are diagnostic, and central longitudinal crypts that contain keratin and are lined by squamous epithelium.4 Basaloid cells can be seen at the periphery of the tumors; however, IFKs lack an eosinophilic basement membrane surrounding the tumor (Figure 3).
Squamous cell carcinoma in situ classically appears as an erythematous hyperkeratotic papule or plaque on sun-exposed sites that can become crusted or ulcerated. Microscopically, squamous cell carcinoma in situ displays full-thickness disorderly maturation of keratinocytes. The keratinocytes exhibit nuclear pleomorphism. Atypical mitotic figures and dyskeratotic keratinocytes also can be seen throughout the full thickness of the epidermis (Figure 4).5
Verruca vulgaris (Figure 5) histologically demonstrates hyperkeratosis with tiers of parakeratosis, digitated epidermal hyperplasia, and dilated tortuous capillaries within the dermal papillae. At the edges of the lesion there often is inward turning of elongated rete ridges,6,7 which can be thought of as the rete reaching out for a hug of sorts to spread the human papillomavirus infection. Although the surface of a trichilemmoma can bear resemblance to a verruca vulgaris, the remainder of the histologic features can be used to help differentiate these tumors. Additionally, there has been no evidence suggestive of a viral etiology for trichilemmomas.8
Warty dyskeratoma features an umbilicated papule, usually on the face, head, or neck, that is associated with a follicular unit. The papule shows a cup-shaped, keratin-filled invagination; suprabasilar clefting; and acantholytic dyskeratotic cells, which are features that are not seen in trichilemmomas (Figure 6).9
Acknowledgment—The authors would like to thank Brandon Litzner, MD, St Louis, Missouri, for proofreading the manuscript.
Trichilemmomas are benign follicular neoplasms that exhibit differentiation toward the outer root sheath of the pilosebaceous follicular epithelium.1 Trichilemmomas clinically present as individual or multiple, slowly growing, verrucous papules appearing most commonly on the face or neck. The lesions may coalesce to form small plaques. Although trichilemmomas typically are isolated, patients with multiple trichilemmomas require a cancer screening workup due to their association with Cowden disease, which results from a mutation in the phosphatase and tensin homolog tumor suppressor gene, PTEN.2 An easy way to remember the association between trichilemmomas and Cowden disease is to alter the spelling to “trichile-moo-moo,” using the “moo moo” sound of an animal cow as a clue linking the tumor to Cowden disease.
Histologically, trichilemmomas exhibit a lobular epidermal downgrowth into the dermis (Figure 1). The surface of the lesion may be hyperkeratotic and somewhat papillomatous. Cells toward the center of the lobule are pale staining, periodic acid–Schiff positive, and diastase labile due to high levels of intracellular glycogen (Figure 2). Cells toward the periphery of the lobule usually appear basophilic with a palisading arrangement of the peripheral cells. The entire lobule is enclosed within an eosinophilic basement membrane that stains positively with periodic acid–Schiff (Figure 2).1 Consistent with the tumor’s differentiation toward the outer root sheath of the hair follicle, trichilemmomas have been reported to express CD34 focally or diffusely.3
|
|
|
Similar to trichilemmoma, inverted follicular keratosis (IFK) commonly presents as a solitary asymptomatic papule on the face. Inverted follicular keratosis is a somewhat controversial entity, with some authorities arguing IFK is a variant of verruca vulgaris or seborrheic keratosis. Histologically, IFKs can be differentiated by the presence of squamous eddies (concentric layers of squamous cells in a whorled pattern), which are diagnostic, and central longitudinal crypts that contain keratin and are lined by squamous epithelium.4 Basaloid cells can be seen at the periphery of the tumors; however, IFKs lack an eosinophilic basement membrane surrounding the tumor (Figure 3).
Squamous cell carcinoma in situ classically appears as an erythematous hyperkeratotic papule or plaque on sun-exposed sites that can become crusted or ulcerated. Microscopically, squamous cell carcinoma in situ displays full-thickness disorderly maturation of keratinocytes. The keratinocytes exhibit nuclear pleomorphism. Atypical mitotic figures and dyskeratotic keratinocytes also can be seen throughout the full thickness of the epidermis (Figure 4).5
Verruca vulgaris (Figure 5) histologically demonstrates hyperkeratosis with tiers of parakeratosis, digitated epidermal hyperplasia, and dilated tortuous capillaries within the dermal papillae. At the edges of the lesion there often is inward turning of elongated rete ridges,6,7 which can be thought of as the rete reaching out for a hug of sorts to spread the human papillomavirus infection. Although the surface of a trichilemmoma can bear resemblance to a verruca vulgaris, the remainder of the histologic features can be used to help differentiate these tumors. Additionally, there has been no evidence suggestive of a viral etiology for trichilemmomas.8
Warty dyskeratoma features an umbilicated papule, usually on the face, head, or neck, that is associated with a follicular unit. The papule shows a cup-shaped, keratin-filled invagination; suprabasilar clefting; and acantholytic dyskeratotic cells, which are features that are not seen in trichilemmomas (Figure 6).9
Acknowledgment—The authors would like to thank Brandon Litzner, MD, St Louis, Missouri, for proofreading the manuscript.
1. Brownstein MH, Shapiro L. Trichilemmoma: analysis of 40 new cases. Arch Dermatol. 1973;107:866-869.
2. Al-Zaid T, Ditelberg J, Prieto V, et al. Trichilemmomas show loss of PTEN in Cowden syndrome but only rarely in sporadic tumors. J Cutan Pathol. 2012;39:493-499.
3. Tardío JC. CD34-reactive tumors of the skin. an updated review of an ever-growing list of lesions. J Cutan Pathol. 2009;36:89-102.
4. Mehregan A. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
5. Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. 2000;42(1, pt 2):11-17.
6. Jabłonska S, Majewski S, Obalek S, et al. Cutaneous warts. Clin Dermatol. 1997;15:309-319.
7. Hardin J, Gardner J, Colome M, et al. Verrucous cyst with melanocytic and sebaceous differentiation. Arch Path Lab Med. 2013;137:576-579.
8. Johnson BL, Kramer EM, Lavker RM. The keratotic tumors of Cowden’s disease: an electron microscopy study. J Cutan Pathol. 1987;14:291-298.
9. Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma—“follicular dyskeratoma”: analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
1. Brownstein MH, Shapiro L. Trichilemmoma: analysis of 40 new cases. Arch Dermatol. 1973;107:866-869.
2. Al-Zaid T, Ditelberg J, Prieto V, et al. Trichilemmomas show loss of PTEN in Cowden syndrome but only rarely in sporadic tumors. J Cutan Pathol. 2012;39:493-499.
3. Tardío JC. CD34-reactive tumors of the skin. an updated review of an ever-growing list of lesions. J Cutan Pathol. 2009;36:89-102.
4. Mehregan A. Inverted follicular keratosis is a distinct follicular tumor. Am J Dermatopathol. 1983;5:467-470.
5. Cockerell CJ. Histopathology of incipient intraepidermal squamous cell carcinoma (“actinic keratosis”). J Am Acad Dermatol. 2000;42(1, pt 2):11-17.
6. Jabłonska S, Majewski S, Obalek S, et al. Cutaneous warts. Clin Dermatol. 1997;15:309-319.
7. Hardin J, Gardner J, Colome M, et al. Verrucous cyst with melanocytic and sebaceous differentiation. Arch Path Lab Med. 2013;137:576-579.
8. Johnson BL, Kramer EM, Lavker RM. The keratotic tumors of Cowden’s disease: an electron microscopy study. J Cutan Pathol. 1987;14:291-298.
9. Kaddu S, Dong H, Mayer G, et al. Warty dyskeratoma—“follicular dyskeratoma”: analysis of clinicopathologic features of a distinctive follicular adnexal neoplasm. J Am Acad Dermatol. 2002;47:423-428.
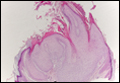
Rosai-Dorfman Disease
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare benign histioproliferative disorder of unknown etiology.1 Clinically, it is most frequently characterized by massive painless cervical lymphadenopathy with other systemic manifestations, including fever, night sweats, and weight loss. Accompanying laboratory findings include leukocytosis with neutrophilia, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. Extranodal involvement has been noted in more than 40% of cases, and cutaneous lesions represent the most common form of extranodal disease.2 Cutaneous RDD is a distinct and rare entity limited to the skin without lymphadenopathy or other extracutaneous involvement.3 Patients with cutaneous RDD typically present with papules and plaques that can grow to form nodules with satellite lesions that resolve into fibrotic plaques before spontaneous regression.4
Histologic examination of cutaneous lesions of RDD reveals a dense nodular dermal and often subcutaneous infiltrate of characteristic large polygonal histiocytes termed Rosai-Dorfman cells, which feature abundant pale to eosinophilic cytoplasm, indistinct borders, and large vesicular nuclei with prominent nucleoli (Figure 1).4,5 Some multinucleate forms may be seen. These Rosai-Dorfman cells display positive staining for CD68 and S-100, and negative staining for CD1a on immunohistochemistry. Lymphocytes and plasma cells often are admixed with the Rosai-Dorfman cells, and neutrophils and eosinophils also may be present in the infiltrate.4 The histologic hallmark of RDD is emperipolesis, a phenomenon whereby inflammatory cells such as lymphocytes and plasma cells reside intact within the cytoplasm of histiocytes (Figure 2).5
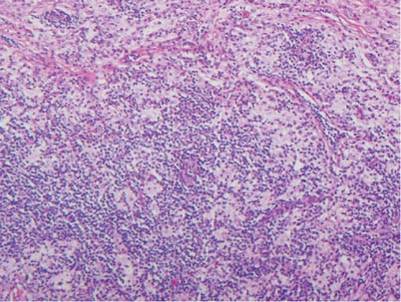
| 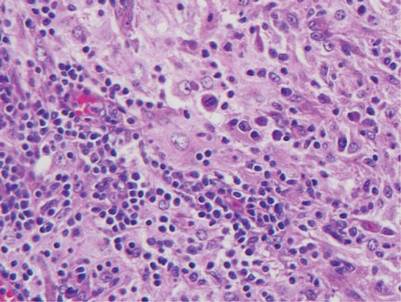
|
The histologic differential diagnosis of cutaneous lesions of RDD includes other histiocytic and xanthomatous diseases, including eruptive xanthoma, juvenile xanthogranuloma, Langerhans cell histiocytosis, and solitary reticulohistiocytoma, which should not display emperipolesis. Eruptive xanthomas display collections of foamy histiocytes in the dermis and typically contain extracellular lipid. They may contain infiltrates of lymphocytes (Figure 3). Juvenile xanthogranuloma also features a dense infiltrate of histiocytes in the papillary and reticular dermis but distinctly shows Touton giant cells and lipidization of histiocytes (Figure 4). Both eruptive xanthomas and juvenile xanthogranulomas typically stain negatively for S-100. Langerhans cell histiocytosis is histologically characterized by a dermal infiltrate of Langerhans cells that have their own distinctive morphologic features. They are uniformly ovoid with abundant eosinophilic cytoplasm. Their nuclei are smaller than those of Rosai-Dorfman cells and have a kidney bean shape with inconspicuous nucleoli (Figure 5). Epidermotropism of these cells can be observed. Immunohistochemically, Langerhans cell histiocytosis typically is S-100 positive, CD1a positive, and langerin positive. Reticulohistiocytoma features histiocytes that have a characteristic dusty rose or ground glass cytoplasm with two-toned darker and lighter areas (Figure 6). Reticulohistiocytoma cells stain positively for CD68 but typically stain negatively for both CD1a and S-100.
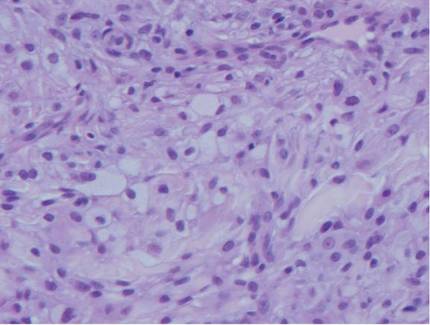
| 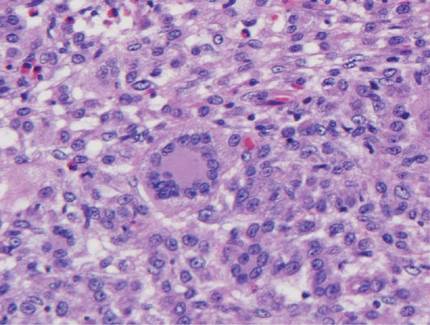
| ||
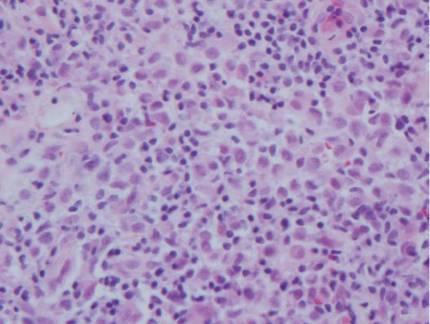
| 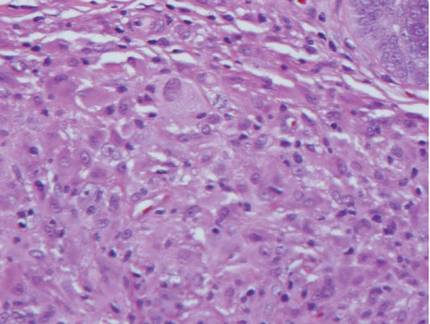
|
1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
2. Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): a review of the entity. Semin Diagn Pathol. 1990;7:19-73.
3. Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385-391.
4. Wang KH, Chen WY, Lie HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
5. Chu P, LeBoit PE. Histologic features of cutaneous sinus histiocytosis (Rosai-Dorfman disease): study of cases both with and without systemic involvement. J Cutan Pathol. 1992;19:201-206.
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare benign histioproliferative disorder of unknown etiology.1 Clinically, it is most frequently characterized by massive painless cervical lymphadenopathy with other systemic manifestations, including fever, night sweats, and weight loss. Accompanying laboratory findings include leukocytosis with neutrophilia, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. Extranodal involvement has been noted in more than 40% of cases, and cutaneous lesions represent the most common form of extranodal disease.2 Cutaneous RDD is a distinct and rare entity limited to the skin without lymphadenopathy or other extracutaneous involvement.3 Patients with cutaneous RDD typically present with papules and plaques that can grow to form nodules with satellite lesions that resolve into fibrotic plaques before spontaneous regression.4
Histologic examination of cutaneous lesions of RDD reveals a dense nodular dermal and often subcutaneous infiltrate of characteristic large polygonal histiocytes termed Rosai-Dorfman cells, which feature abundant pale to eosinophilic cytoplasm, indistinct borders, and large vesicular nuclei with prominent nucleoli (Figure 1).4,5 Some multinucleate forms may be seen. These Rosai-Dorfman cells display positive staining for CD68 and S-100, and negative staining for CD1a on immunohistochemistry. Lymphocytes and plasma cells often are admixed with the Rosai-Dorfman cells, and neutrophils and eosinophils also may be present in the infiltrate.4 The histologic hallmark of RDD is emperipolesis, a phenomenon whereby inflammatory cells such as lymphocytes and plasma cells reside intact within the cytoplasm of histiocytes (Figure 2).5
|
|
|
The histologic differential diagnosis of cutaneous lesions of RDD includes other histiocytic and xanthomatous diseases, including eruptive xanthoma, juvenile xanthogranuloma, Langerhans cell histiocytosis, and solitary reticulohistiocytoma, which should not display emperipolesis. Eruptive xanthomas display collections of foamy histiocytes in the dermis and typically contain extracellular lipid. They may contain infiltrates of lymphocytes (Figure 3). Juvenile xanthogranuloma also features a dense infiltrate of histiocytes in the papillary and reticular dermis but distinctly shows Touton giant cells and lipidization of histiocytes (Figure 4). Both eruptive xanthomas and juvenile xanthogranulomas typically stain negatively for S-100. Langerhans cell histiocytosis is histologically characterized by a dermal infiltrate of Langerhans cells that have their own distinctive morphologic features. They are uniformly ovoid with abundant eosinophilic cytoplasm. Their nuclei are smaller than those of Rosai-Dorfman cells and have a kidney bean shape with inconspicuous nucleoli (Figure 5). Epidermotropism of these cells can be observed. Immunohistochemically, Langerhans cell histiocytosis typically is S-100 positive, CD1a positive, and langerin positive. Reticulohistiocytoma features histiocytes that have a characteristic dusty rose or ground glass cytoplasm with two-toned darker and lighter areas (Figure 6). Reticulohistiocytoma cells stain positively for CD68 but typically stain negatively for both CD1a and S-100.
|
|
| ||
|
|
|
Rosai-Dorfman disease (RDD), also known as sinus histiocytosis with massive lymphadenopathy, is a rare benign histioproliferative disorder of unknown etiology.1 Clinically, it is most frequently characterized by massive painless cervical lymphadenopathy with other systemic manifestations, including fever, night sweats, and weight loss. Accompanying laboratory findings include leukocytosis with neutrophilia, elevated erythrocyte sedimentation rate, and polyclonal hypergammaglobulinemia. Extranodal involvement has been noted in more than 40% of cases, and cutaneous lesions represent the most common form of extranodal disease.2 Cutaneous RDD is a distinct and rare entity limited to the skin without lymphadenopathy or other extracutaneous involvement.3 Patients with cutaneous RDD typically present with papules and plaques that can grow to form nodules with satellite lesions that resolve into fibrotic plaques before spontaneous regression.4
Histologic examination of cutaneous lesions of RDD reveals a dense nodular dermal and often subcutaneous infiltrate of characteristic large polygonal histiocytes termed Rosai-Dorfman cells, which feature abundant pale to eosinophilic cytoplasm, indistinct borders, and large vesicular nuclei with prominent nucleoli (Figure 1).4,5 Some multinucleate forms may be seen. These Rosai-Dorfman cells display positive staining for CD68 and S-100, and negative staining for CD1a on immunohistochemistry. Lymphocytes and plasma cells often are admixed with the Rosai-Dorfman cells, and neutrophils and eosinophils also may be present in the infiltrate.4 The histologic hallmark of RDD is emperipolesis, a phenomenon whereby inflammatory cells such as lymphocytes and plasma cells reside intact within the cytoplasm of histiocytes (Figure 2).5
|
|
|
The histologic differential diagnosis of cutaneous lesions of RDD includes other histiocytic and xanthomatous diseases, including eruptive xanthoma, juvenile xanthogranuloma, Langerhans cell histiocytosis, and solitary reticulohistiocytoma, which should not display emperipolesis. Eruptive xanthomas display collections of foamy histiocytes in the dermis and typically contain extracellular lipid. They may contain infiltrates of lymphocytes (Figure 3). Juvenile xanthogranuloma also features a dense infiltrate of histiocytes in the papillary and reticular dermis but distinctly shows Touton giant cells and lipidization of histiocytes (Figure 4). Both eruptive xanthomas and juvenile xanthogranulomas typically stain negatively for S-100. Langerhans cell histiocytosis is histologically characterized by a dermal infiltrate of Langerhans cells that have their own distinctive morphologic features. They are uniformly ovoid with abundant eosinophilic cytoplasm. Their nuclei are smaller than those of Rosai-Dorfman cells and have a kidney bean shape with inconspicuous nucleoli (Figure 5). Epidermotropism of these cells can be observed. Immunohistochemically, Langerhans cell histiocytosis typically is S-100 positive, CD1a positive, and langerin positive. Reticulohistiocytoma features histiocytes that have a characteristic dusty rose or ground glass cytoplasm with two-toned darker and lighter areas (Figure 6). Reticulohistiocytoma cells stain positively for CD68 but typically stain negatively for both CD1a and S-100.
|
|
| ||
|
|
|
1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
2. Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): a review of the entity. Semin Diagn Pathol. 1990;7:19-73.
3. Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385-391.
4. Wang KH, Chen WY, Lie HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
5. Chu P, LeBoit PE. Histologic features of cutaneous sinus histiocytosis (Rosai-Dorfman disease): study of cases both with and without systemic involvement. J Cutan Pathol. 1992;19:201-206.
1. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. a newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
2. Foucar E, Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease): a review of the entity. Semin Diagn Pathol. 1990;7:19-73.
3. Brenn T, Calonje E, Granter SR, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385-391.
4. Wang KH, Chen WY, Lie HN, et al. Cutaneous Rosai-Dorfman disease: clinicopathological profiles, spectrum and evolution of 21 lesions in six patients. Br J Dermatol. 2006;154:277-286.
5. Chu P, LeBoit PE. Histologic features of cutaneous sinus histiocytosis (Rosai-Dorfman disease): study of cases both with and without systemic involvement. J Cutan Pathol. 1992;19:201-206.
Extramammary Paget Disease
Extramammary Paget disease (EMPD) is an uncommon condition that usually presents in apocrine sweat gland–rich areas, most commonly the vulva followed by the perianal region. Lesions clinically present as erythematous, well-demarcated plaques that may become ulcerated, erosive, scaly, or eczematous. Extramammary Paget disease has a female predominance and usually occurs in the sixth to eighth decades of life.1 Histologically, EMPD displays intraepidermal spread of large cells with plentiful amphophilic cytoplasm and large nuclei (Figure 1). These atypical cells may be seen “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells (Figure 2). Frequently, the cytoplasm of these tumor cells is positive on mucicarmine staining, which indicates the presence of mucin, giving the cytoplasm a bluish gray color on hematoxylin and eosin–stained sections. Typically, EMPD cells can be found alone or in nests throughout the epithelium. The basal layer of the epithelium will appear crushed but not infiltrated by these atypical cells in some areas.2 Extramammary Paget disease is epithelial membrane antigen and cytokeratin 7 positive, unlike other conditions in the differential diagnosis such as benign acral nevus, Bowen disease, mycosis fungoides, and superficial spreading melanoma in situ, with the rare exception of cytokeratin 7 positivity in Bowen disease.3
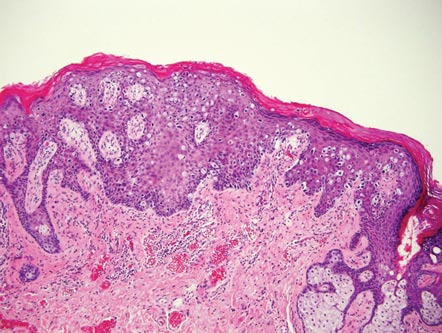
| 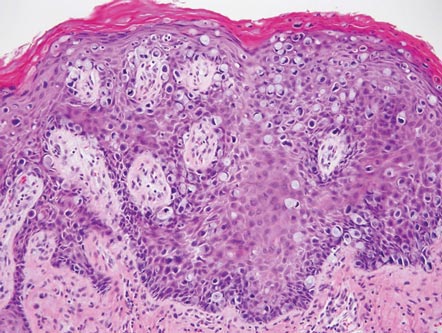
|
Benign acral nevi, similar to melanoma in situ, can have melanocytes scattered above the basal layer, but they usually appear in the lower half of the epidermis without cytologic atypia.4 When present, these pagetoid cells are most often limited to the center of a well-delineated lesion. The compact thick stratum corneum characteristic of acral skin also is helpful in distinguishing a benign acral nevus from EMPD, which does not involve acral sites (Figure 3).2
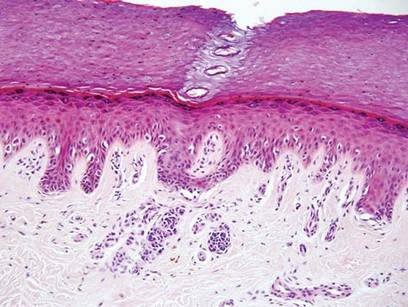
Bowen disease (squamous cell carcinoma in situ) may have pagetoid spread (or buckshot scatter) through the epidermis similar to EMPD and melanoma in situ. However, in Bowen disease the malignant cells are keratinocytes that keratinize and become incorporated into the stratum corneum as parakeratotic nuclei rather than intact “spit out” cells, as seen in melanoma in situ and EMPD. Usually the pagetoid spread is only focal in Bowen disease with other areas of more characteristic full-thickness keratinocyte atypia (Figure 4).2
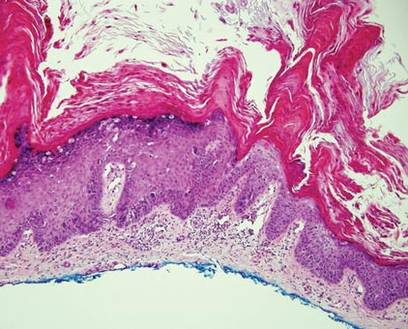
Mycosis fungoides displays atypical lymphocytes with large dark nuclei and minimal to no cytoplasm scattered throughout the epidermis. The atypical cells have irregular nuclear contours and often a clear perinuclear space (Figure 5). These cells tend to line up along the dermoepidermal junction and form intraepidermal clusters known as Pautrier microabscesses. Papillary dermal fibroplasia also is usually present in mycosis fungoides.2
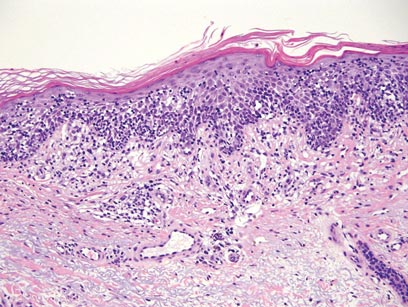
Similar to EMPD, superficial spreading melanoma in situ shows single or nested atypical cells scattered throughout all levels of the epithelium and may be “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells. However, in melanoma, nests of atypical melanocytes predominate and involve the basal layer (Figure 6), whereas clusters of cells in EMPD typically are located superficial to the basal layer. The cells of melanoma also lack the amphophilic mucinous cytoplasm of EMPD.1
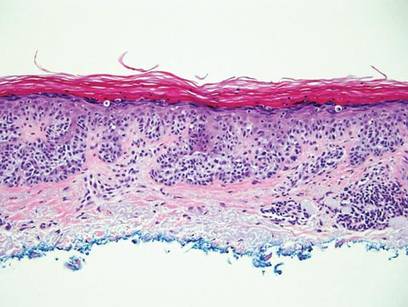
1. Calonje E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier Saunders; 2011.
2. Ferringer T, Elston D, eds. Dermatopathology. 2nd ed. London, England: Elsevier; 2014.
3. Sah SP, Kelly PJ, McManus DT, et al. Diffuse CK7, CAM5.2 and BerEP4 positivity in pagetoid squamous cell carcinoma in situ (pagetoid Bowen’s disease) of the perianal region: a mimic of extramammary Paget’s disease. Histopathology. 2013;62:511-514.
4. LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol. 2000;22:556-558.
Extramammary Paget disease (EMPD) is an uncommon condition that usually presents in apocrine sweat gland–rich areas, most commonly the vulva followed by the perianal region. Lesions clinically present as erythematous, well-demarcated plaques that may become ulcerated, erosive, scaly, or eczematous. Extramammary Paget disease has a female predominance and usually occurs in the sixth to eighth decades of life.1 Histologically, EMPD displays intraepidermal spread of large cells with plentiful amphophilic cytoplasm and large nuclei (Figure 1). These atypical cells may be seen “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells (Figure 2). Frequently, the cytoplasm of these tumor cells is positive on mucicarmine staining, which indicates the presence of mucin, giving the cytoplasm a bluish gray color on hematoxylin and eosin–stained sections. Typically, EMPD cells can be found alone or in nests throughout the epithelium. The basal layer of the epithelium will appear crushed but not infiltrated by these atypical cells in some areas.2 Extramammary Paget disease is epithelial membrane antigen and cytokeratin 7 positive, unlike other conditions in the differential diagnosis such as benign acral nevus, Bowen disease, mycosis fungoides, and superficial spreading melanoma in situ, with the rare exception of cytokeratin 7 positivity in Bowen disease.3
|
|
|
Benign acral nevi, similar to melanoma in situ, can have melanocytes scattered above the basal layer, but they usually appear in the lower half of the epidermis without cytologic atypia.4 When present, these pagetoid cells are most often limited to the center of a well-delineated lesion. The compact thick stratum corneum characteristic of acral skin also is helpful in distinguishing a benign acral nevus from EMPD, which does not involve acral sites (Figure 3).2
Bowen disease (squamous cell carcinoma in situ) may have pagetoid spread (or buckshot scatter) through the epidermis similar to EMPD and melanoma in situ. However, in Bowen disease the malignant cells are keratinocytes that keratinize and become incorporated into the stratum corneum as parakeratotic nuclei rather than intact “spit out” cells, as seen in melanoma in situ and EMPD. Usually the pagetoid spread is only focal in Bowen disease with other areas of more characteristic full-thickness keratinocyte atypia (Figure 4).2
Mycosis fungoides displays atypical lymphocytes with large dark nuclei and minimal to no cytoplasm scattered throughout the epidermis. The atypical cells have irregular nuclear contours and often a clear perinuclear space (Figure 5). These cells tend to line up along the dermoepidermal junction and form intraepidermal clusters known as Pautrier microabscesses. Papillary dermal fibroplasia also is usually present in mycosis fungoides.2
Similar to EMPD, superficial spreading melanoma in situ shows single or nested atypical cells scattered throughout all levels of the epithelium and may be “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells. However, in melanoma, nests of atypical melanocytes predominate and involve the basal layer (Figure 6), whereas clusters of cells in EMPD typically are located superficial to the basal layer. The cells of melanoma also lack the amphophilic mucinous cytoplasm of EMPD.1
Extramammary Paget disease (EMPD) is an uncommon condition that usually presents in apocrine sweat gland–rich areas, most commonly the vulva followed by the perianal region. Lesions clinically present as erythematous, well-demarcated plaques that may become ulcerated, erosive, scaly, or eczematous. Extramammary Paget disease has a female predominance and usually occurs in the sixth to eighth decades of life.1 Histologically, EMPD displays intraepidermal spread of large cells with plentiful amphophilic cytoplasm and large nuclei (Figure 1). These atypical cells may be seen “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells (Figure 2). Frequently, the cytoplasm of these tumor cells is positive on mucicarmine staining, which indicates the presence of mucin, giving the cytoplasm a bluish gray color on hematoxylin and eosin–stained sections. Typically, EMPD cells can be found alone or in nests throughout the epithelium. The basal layer of the epithelium will appear crushed but not infiltrated by these atypical cells in some areas.2 Extramammary Paget disease is epithelial membrane antigen and cytokeratin 7 positive, unlike other conditions in the differential diagnosis such as benign acral nevus, Bowen disease, mycosis fungoides, and superficial spreading melanoma in situ, with the rare exception of cytokeratin 7 positivity in Bowen disease.3
|
|
|
Benign acral nevi, similar to melanoma in situ, can have melanocytes scattered above the basal layer, but they usually appear in the lower half of the epidermis without cytologic atypia.4 When present, these pagetoid cells are most often limited to the center of a well-delineated lesion. The compact thick stratum corneum characteristic of acral skin also is helpful in distinguishing a benign acral nevus from EMPD, which does not involve acral sites (Figure 3).2
Bowen disease (squamous cell carcinoma in situ) may have pagetoid spread (or buckshot scatter) through the epidermis similar to EMPD and melanoma in situ. However, in Bowen disease the malignant cells are keratinocytes that keratinize and become incorporated into the stratum corneum as parakeratotic nuclei rather than intact “spit out” cells, as seen in melanoma in situ and EMPD. Usually the pagetoid spread is only focal in Bowen disease with other areas of more characteristic full-thickness keratinocyte atypia (Figure 4).2
Mycosis fungoides displays atypical lymphocytes with large dark nuclei and minimal to no cytoplasm scattered throughout the epidermis. The atypical cells have irregular nuclear contours and often a clear perinuclear space (Figure 5). These cells tend to line up along the dermoepidermal junction and form intraepidermal clusters known as Pautrier microabscesses. Papillary dermal fibroplasia also is usually present in mycosis fungoides.2
Similar to EMPD, superficial spreading melanoma in situ shows single or nested atypical cells scattered throughout all levels of the epithelium and may be “spit out” whole into the stratum corneum rather than keratinizing into parakeratotic cells. However, in melanoma, nests of atypical melanocytes predominate and involve the basal layer (Figure 6), whereas clusters of cells in EMPD typically are located superficial to the basal layer. The cells of melanoma also lack the amphophilic mucinous cytoplasm of EMPD.1
1. Calonje E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier Saunders; 2011.
2. Ferringer T, Elston D, eds. Dermatopathology. 2nd ed. London, England: Elsevier; 2014.
3. Sah SP, Kelly PJ, McManus DT, et al. Diffuse CK7, CAM5.2 and BerEP4 positivity in pagetoid squamous cell carcinoma in situ (pagetoid Bowen’s disease) of the perianal region: a mimic of extramammary Paget’s disease. Histopathology. 2013;62:511-514.
4. LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol. 2000;22:556-558.
1. Calonje E, Brenn T, Lazar A, et al. McKee’s Pathology of the Skin. 4th ed. London, England: Elsevier Saunders; 2011.
2. Ferringer T, Elston D, eds. Dermatopathology. 2nd ed. London, England: Elsevier; 2014.
3. Sah SP, Kelly PJ, McManus DT, et al. Diffuse CK7, CAM5.2 and BerEP4 positivity in pagetoid squamous cell carcinoma in situ (pagetoid Bowen’s disease) of the perianal region: a mimic of extramammary Paget’s disease. Histopathology. 2013;62:511-514.
4. LeBoit PE. A diagnosis for maniacs. Am J Dermatopathol. 2000;22:556-558.
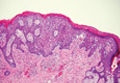
Necrobiosis Lipoidica Diabeticorum
Necrobiosis lipoidica diabeticorum (NLD) is a rare granulomatous skin manifestation that is strongly associated with diabetes mellitus. Necrobiosis lipoidica diabeticorum is more common among females and occurs primarily in the pretibial area.1 Necrobiosis lipoidica diabeticorum may clinically manifest as single or multiple lesions that begin as small red papules and progress into patches or plaques. Lesions ultimately develop into areas of yellowish brown atrophic tissue with central depression and telangiectasia. The etiology of NLD is not completely understood, but it is thought to be a presentation of diabetic microangiopathy.1 Histologically, NLD demonstrates broad horizontal zones of necrobiosis with a surrounding inflammatory infiltrate that is principally composed of histiocytes but also may contain multinucleated giant cells, lymphocytes, and plasma cells (Figures 1 and 2). Occasionally, sarcoidal granulomas are seen in NLD. There also may be thickening of vessel walls and edema of the endothelial cells.1
| 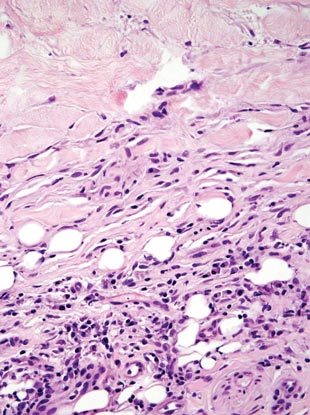
|
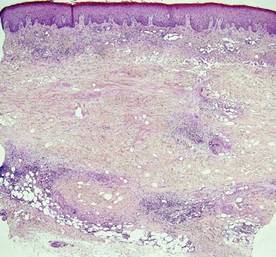
Cutaneous Rosai-Dorfman disease (RDD) is characterized by the presence of diffuse, large, pale histiocytes (commonly known as Rosai-Dorfman cells) with an admixed infiltrate of lymphocytes and plasma cells (Figure 3).2 Additionally, Rosai-Dorfman cells display emperipolesis. They stain positively for S-100 protein and CD68 and negatively for CD1a.2 Clinically, cutaneous RDD has a myriad of manifestations but most commonly presents as cutaneous nodules that can be tender or pruritic. It also may be associated with systemic symptoms. Patients with cutaneous RDD often have an elevated erythrocyte sedimentation rate and concomitant anemia.2
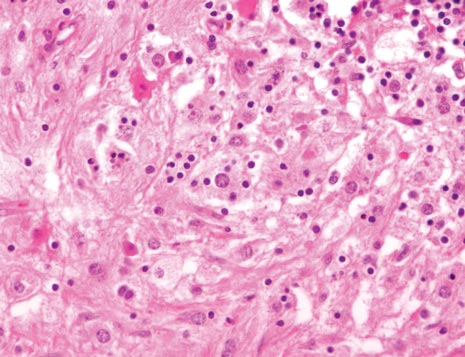
Granuloma annulare demonstrates necrobiosis and palisaded granulomatous dermatitis similar to NLD; however, the necrobiotic foci in granuloma annulare usually are more focal than in NLD and typically are surrounded by well-formed palisaded granulomas. There also is an increase in dermal mucin (Figure 4), which can be highlighted on colloidal iron or Alcian blue staining.1 Granuloma annulare also typically has scattered eosinophils rather than plasma cells as seen in NLD. Granuloma annulare also may present in an interstitial pattern, with scattered histiocytes, mucin, and eosinophils between collagen bundles. Granuloma annulare clinically presents as variably colored papules arranged in an annular pattern on the distal extremities but also can present as widespread papules or plaques.
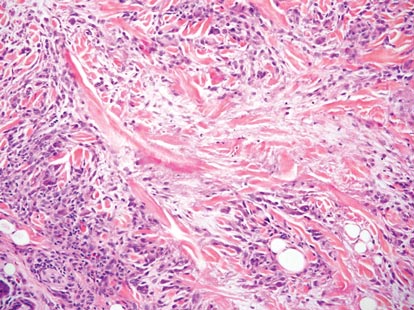
Juvenile xanthogranuloma (JXG) is a benign condition typically seen in children that is characterized by the presence of 1 or more pink or yellow nodules, most commonly presenting on the head and neck. Histologically, JXG demonstrates a dermal collection of histiocytes, lymphocytes, eosinophils, and characteristic Touton giant cells, which contain nuclei that are arranged in a wreathlike pattern and exhibit peripheral xanthomatization (Figure 5).3 The histiocytes in JXG typically stain positive for CD68 and negative for S-100 protein, though occasional S-100–positive cases are reported.3
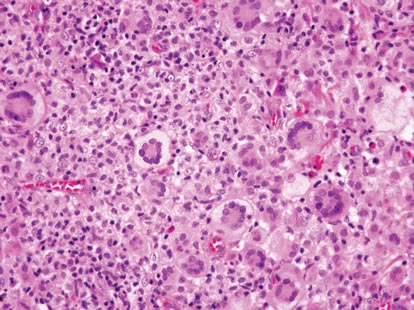
Necrobiotic xanthogranuloma presents as yellowish to brown plaques and nodules most commonly in the periorbital area. Necrobiotic xanthogranuloma is strongly associated with monoclonal gammopathy, typically IgGk monoclonal gammopathy. Necrobiotic xanthogranuloma is histologically similar to NLD but is distinguished by a nodular pattern of inflammation and the frequent presence of cholesterol clefts (Figure 6).4
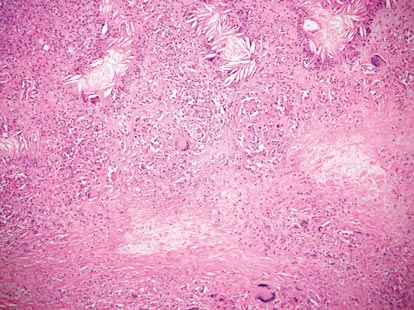
Figure 6. Necrobiotic xanthogranuloma is distinguished by the nodularity
of the infiltrate, with necrobiotic collagen, palisaded histiocytes and giant
cells, and the presence of cholesterol clefts (H&E, original
magnification ×40).
1. Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
2. Khoo JJ, Rahmat BO. Cutaneous Rosai-Dorfman disease. Malays J Pathol. 2007;29:49-52.
3. Cypel TK, Zuker RM. Juvenile xanthogranuloma: case report and review of the literature. Can J Plast Surg. 2008;16:175-177.
4. Inthasotti S, Wanitphakdeedecha R, Manonukul J. A 7-year history of necrobiotic xanthogranuloma following asymptomatic multiple myeloma: a case report. Dermatol Res Pract. 2011;2011:927852.
Necrobiosis lipoidica diabeticorum (NLD) is a rare granulomatous skin manifestation that is strongly associated with diabetes mellitus. Necrobiosis lipoidica diabeticorum is more common among females and occurs primarily in the pretibial area.1 Necrobiosis lipoidica diabeticorum may clinically manifest as single or multiple lesions that begin as small red papules and progress into patches or plaques. Lesions ultimately develop into areas of yellowish brown atrophic tissue with central depression and telangiectasia. The etiology of NLD is not completely understood, but it is thought to be a presentation of diabetic microangiopathy.1 Histologically, NLD demonstrates broad horizontal zones of necrobiosis with a surrounding inflammatory infiltrate that is principally composed of histiocytes but also may contain multinucleated giant cells, lymphocytes, and plasma cells (Figures 1 and 2). Occasionally, sarcoidal granulomas are seen in NLD. There also may be thickening of vessel walls and edema of the endothelial cells.1
|
|
Cutaneous Rosai-Dorfman disease (RDD) is characterized by the presence of diffuse, large, pale histiocytes (commonly known as Rosai-Dorfman cells) with an admixed infiltrate of lymphocytes and plasma cells (Figure 3).2 Additionally, Rosai-Dorfman cells display emperipolesis. They stain positively for S-100 protein and CD68 and negatively for CD1a.2 Clinically, cutaneous RDD has a myriad of manifestations but most commonly presents as cutaneous nodules that can be tender or pruritic. It also may be associated with systemic symptoms. Patients with cutaneous RDD often have an elevated erythrocyte sedimentation rate and concomitant anemia.2
Granuloma annulare demonstrates necrobiosis and palisaded granulomatous dermatitis similar to NLD; however, the necrobiotic foci in granuloma annulare usually are more focal than in NLD and typically are surrounded by well-formed palisaded granulomas. There also is an increase in dermal mucin (Figure 4), which can be highlighted on colloidal iron or Alcian blue staining.1 Granuloma annulare also typically has scattered eosinophils rather than plasma cells as seen in NLD. Granuloma annulare also may present in an interstitial pattern, with scattered histiocytes, mucin, and eosinophils between collagen bundles. Granuloma annulare clinically presents as variably colored papules arranged in an annular pattern on the distal extremities but also can present as widespread papules or plaques.
Juvenile xanthogranuloma (JXG) is a benign condition typically seen in children that is characterized by the presence of 1 or more pink or yellow nodules, most commonly presenting on the head and neck. Histologically, JXG demonstrates a dermal collection of histiocytes, lymphocytes, eosinophils, and characteristic Touton giant cells, which contain nuclei that are arranged in a wreathlike pattern and exhibit peripheral xanthomatization (Figure 5).3 The histiocytes in JXG typically stain positive for CD68 and negative for S-100 protein, though occasional S-100–positive cases are reported.3
Necrobiotic xanthogranuloma presents as yellowish to brown plaques and nodules most commonly in the periorbital area. Necrobiotic xanthogranuloma is strongly associated with monoclonal gammopathy, typically IgGk monoclonal gammopathy. Necrobiotic xanthogranuloma is histologically similar to NLD but is distinguished by a nodular pattern of inflammation and the frequent presence of cholesterol clefts (Figure 6).4
Figure 6. Necrobiotic xanthogranuloma is distinguished by the nodularity
of the infiltrate, with necrobiotic collagen, palisaded histiocytes and giant
cells, and the presence of cholesterol clefts (H&E, original
magnification ×40).
Necrobiosis lipoidica diabeticorum (NLD) is a rare granulomatous skin manifestation that is strongly associated with diabetes mellitus. Necrobiosis lipoidica diabeticorum is more common among females and occurs primarily in the pretibial area.1 Necrobiosis lipoidica diabeticorum may clinically manifest as single or multiple lesions that begin as small red papules and progress into patches or plaques. Lesions ultimately develop into areas of yellowish brown atrophic tissue with central depression and telangiectasia. The etiology of NLD is not completely understood, but it is thought to be a presentation of diabetic microangiopathy.1 Histologically, NLD demonstrates broad horizontal zones of necrobiosis with a surrounding inflammatory infiltrate that is principally composed of histiocytes but also may contain multinucleated giant cells, lymphocytes, and plasma cells (Figures 1 and 2). Occasionally, sarcoidal granulomas are seen in NLD. There also may be thickening of vessel walls and edema of the endothelial cells.1
|
|
Cutaneous Rosai-Dorfman disease (RDD) is characterized by the presence of diffuse, large, pale histiocytes (commonly known as Rosai-Dorfman cells) with an admixed infiltrate of lymphocytes and plasma cells (Figure 3).2 Additionally, Rosai-Dorfman cells display emperipolesis. They stain positively for S-100 protein and CD68 and negatively for CD1a.2 Clinically, cutaneous RDD has a myriad of manifestations but most commonly presents as cutaneous nodules that can be tender or pruritic. It also may be associated with systemic symptoms. Patients with cutaneous RDD often have an elevated erythrocyte sedimentation rate and concomitant anemia.2
Granuloma annulare demonstrates necrobiosis and palisaded granulomatous dermatitis similar to NLD; however, the necrobiotic foci in granuloma annulare usually are more focal than in NLD and typically are surrounded by well-formed palisaded granulomas. There also is an increase in dermal mucin (Figure 4), which can be highlighted on colloidal iron or Alcian blue staining.1 Granuloma annulare also typically has scattered eosinophils rather than plasma cells as seen in NLD. Granuloma annulare also may present in an interstitial pattern, with scattered histiocytes, mucin, and eosinophils between collagen bundles. Granuloma annulare clinically presents as variably colored papules arranged in an annular pattern on the distal extremities but also can present as widespread papules or plaques.
Juvenile xanthogranuloma (JXG) is a benign condition typically seen in children that is characterized by the presence of 1 or more pink or yellow nodules, most commonly presenting on the head and neck. Histologically, JXG demonstrates a dermal collection of histiocytes, lymphocytes, eosinophils, and characteristic Touton giant cells, which contain nuclei that are arranged in a wreathlike pattern and exhibit peripheral xanthomatization (Figure 5).3 The histiocytes in JXG typically stain positive for CD68 and negative for S-100 protein, though occasional S-100–positive cases are reported.3
Necrobiotic xanthogranuloma presents as yellowish to brown plaques and nodules most commonly in the periorbital area. Necrobiotic xanthogranuloma is strongly associated with monoclonal gammopathy, typically IgGk monoclonal gammopathy. Necrobiotic xanthogranuloma is histologically similar to NLD but is distinguished by a nodular pattern of inflammation and the frequent presence of cholesterol clefts (Figure 6).4
Figure 6. Necrobiotic xanthogranuloma is distinguished by the nodularity
of the infiltrate, with necrobiotic collagen, palisaded histiocytes and giant
cells, and the presence of cholesterol clefts (H&E, original
magnification ×40).
1. Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
2. Khoo JJ, Rahmat BO. Cutaneous Rosai-Dorfman disease. Malays J Pathol. 2007;29:49-52.
3. Cypel TK, Zuker RM. Juvenile xanthogranuloma: case report and review of the literature. Can J Plast Surg. 2008;16:175-177.
4. Inthasotti S, Wanitphakdeedecha R, Manonukul J. A 7-year history of necrobiotic xanthogranuloma following asymptomatic multiple myeloma: a case report. Dermatol Res Pract. 2011;2011:927852.
1. Kota SK, Jammula S, Kota SK, et al. Necrobiosis lipoidica diabeticorum: a case-based review of literature. Indian J Endocrinol Metab. 2012;16:614-620.
2. Khoo JJ, Rahmat BO. Cutaneous Rosai-Dorfman disease. Malays J Pathol. 2007;29:49-52.
3. Cypel TK, Zuker RM. Juvenile xanthogranuloma: case report and review of the literature. Can J Plast Surg. 2008;16:175-177.
4. Inthasotti S, Wanitphakdeedecha R, Manonukul J. A 7-year history of necrobiotic xanthogranuloma following asymptomatic multiple myeloma: a case report. Dermatol Res Pract. 2011;2011:927852.

Trichoepithelioma and Spiradenoma Collision Tumor
The coexistence of more than one cutaneous adnexal neoplasm in a single biopsy specimen is unusual and is most frequently recognized in the context of a nevus sebaceous or Brooke-Spiegler syndrome, an autosomal-dominant inherited disease characterized by cutaneous adnexal neoplasms, most commonly cylindromas and trichoepitheliomas.1-3 Brooke-Spiegler syndrome is caused by germline mutations in the cylindromatosis gene, CYLD, located on band 16q12; it functions as a tumor suppressor gene and has regulatory roles in development, immunity, and inflammation.1 Weyers et al3 first recognized the tendency for adnexal collision tumors to present in patients with Brooke-Spiegler syndrome; they reported a patient with Brooke-Spiegler syndrome with spiradenomas found in the immediate vicinity of trichoepitheliomas and in continuity with hair follicles.
Spiradenomas are composed of large, sharply demarcated, rounded nodules of basaloid cells with little cytoplasm (Figure 1).4 The basaloid nodules may demonstrate a trabecular architecture, and on close inspection 2 cell types—paler cells with more cytoplasm and darker cells with less cytoplasm—are distinguishable (Figure 2A). Lymphocytes often are scattered within the tumor nodules and/or stroma. In Brooke-Spiegler syndrome, collision tumors containing a spiradenomatous component in collision with trichoepithelioma are not uncommon.1 Spiradenomas in Brooke-Spiegler syndrome have been reported to contain sebaceous differentiation or foci with an adenoid cystic carcinoma (ACC)–like pattern and are known to occur as hybrid lesions of spiradenoma and cylindroma or trichoepithelioma (as in this case).
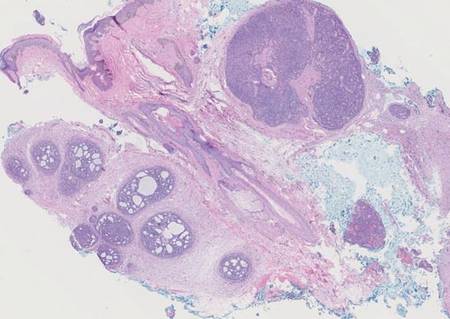
In this case, 2 distinct neoplasms (spiradenoma and trichoepithelioma) are apparent, side by side, with an intervening hair follicle (Figure 1). Trichoepitheliomas, also known as cribriform trichoblastomas,5 are characterized by lobules of basaloid cells resembling basal cell carcinoma surrounded by a fibroblast-rich stroma. They often contain fingerlike projections and adopt a cribriform morphology within the tumor lobules (Figure 2B).4 Numerous horn cysts may be present, but their absence does not preclude the diagnosis. Mucin may be present within the cribriform tumor islands (Figure 2B) but not in the stroma. Characteristically, trichoepitheliomas are distinctly negative for CK7 (Figure 3), and unlike spiradenomas, they lack a myoepithelial component.6 This staining pattern in combination with the tumor’s proximity to an adjacent hair follicle makes a diagnosis of trichoepithelioma and spiradenoma collision tumor most likely and supports a clinical suspicion for Brooke-Spiegler syndrome.
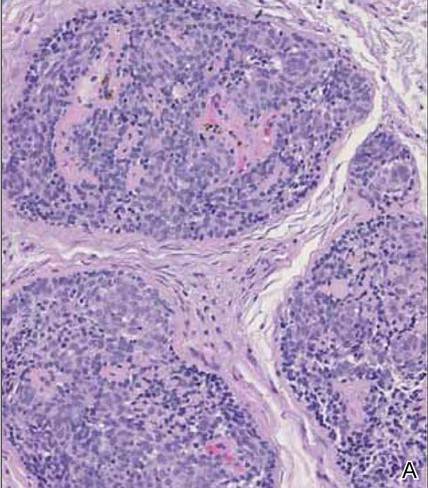
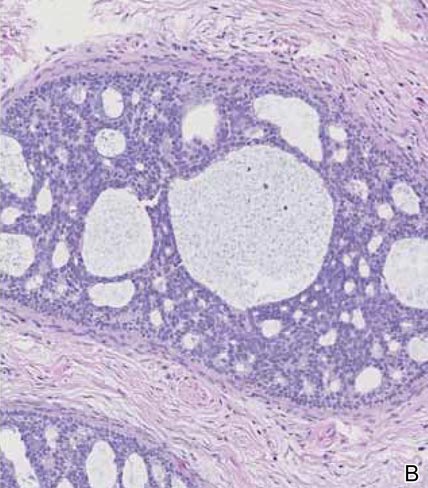
|
Although spiradenomas sometimes contain cystic cavities (microcystic change), they typically are filled with finely granular eosinophilic material, not mucin, that is diastase resistant and periodic acid–Schiff positive (Figure 4).7 Spiradenomas classically stain positive with CK7 (Figure 3), epithelial membrane antigen, and carcinoembryonic antigen, and have a substantial myoepithelial component, as evidenced by the myoepithelial component staining with p63, S-100, and smooth muscle actin (SMA).7-9 The distinct lack of staining with CK7 and SMA in the tumor on the left in Figure 3 confirms that these tumors are of different lineage, rather than representing cystic change within a spiradenoma.

| 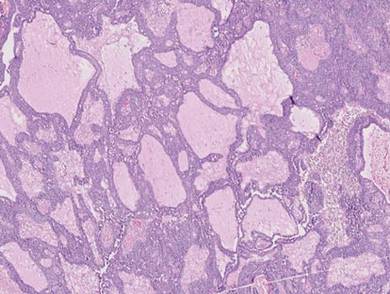
|
Adenoid cystic carcinoma is a rare neoplasm that may occur in a primary cutaneous form, as a direct extension from an underlying salivary gland neoplasm, or rarely as a focal pattern within spiradenomas occurring both sporadically or in the context of Brooke-Spiegler syndrome.2,7 The tumor is composed of variably sized cribriform islands of basaloid to pink cells concentrically arranged around glandlike spaces filled with mucin (Figure 5A). In contrast to trichoepithelioma, ACC occurs in the mid to deep dermis, often extending into subcutaneous fat with an infiltrative border, and is not often found in close proximity to hair follicles.7 Characteristically, hyaline basement membrane–like material that is periodic acid–Schiff positive is found between the tumor cells and also surrounding the individual lobules. Immunohistochemically, ACC has a myoepithelial component that stains positive with SMA, S-100, and p63; additionally, the tumor cells express low- and high-molecular-weight keratin and demonstrate variable epithelial membrane antigen positivity.10 In the current case, the superficial location, close association with a hair follicle, and lack of staining with both CK7 (Figure 3) and SMA (not shown) make ACC arising within a spiradenoma a less likely diagnosis.
Cylindromas are composed of basaloid islands interconnected in a jigsaw puzzle configuration (Figure 5B).4 Similar to spiradenomas, they also are composed of 2 cell populations. Characteristically, the tumor islands are outlined by a hyalinized eosinophilic basement membrane. Hyalinized droplets of basement membrane zone material also may be noted in the islands. Unlike spiradenomas, they lack both intratumoral lymphocytes and a trabecular growth pattern. Although spiradenocylindromas (cylindroma and spiradenoma collision tumors) are perhaps the most common collision tumor associated with Brooke-Spiegler syndrome, there is no evidence suggesting the presence of a cylindroma in the current case.
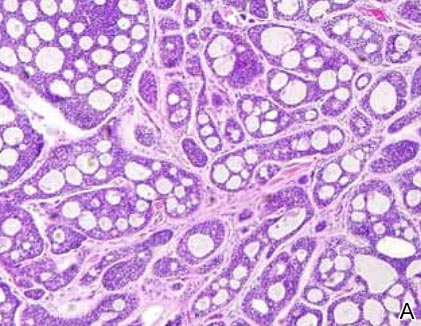
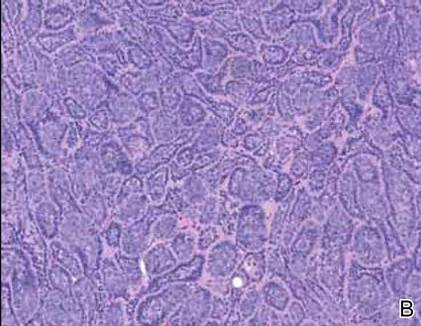
|
Primary cutaneous mucinous carcinoma is a rare neoplasm with a predilection for the eyelids; lesions occurring outside of this facial distribution, particularly of the breast, warrant a workup for metastatic disease.7 It typically occurs in the deeper dermis with involvement of the subcutaneous fat and is characterized by delicate fibrous septa enveloping large lakes of mucin, which contain islands of tumor cells (Figure 6). It has not been reported in association with spiradenomas. In addition, the tumor cells typically are CK7 positive.
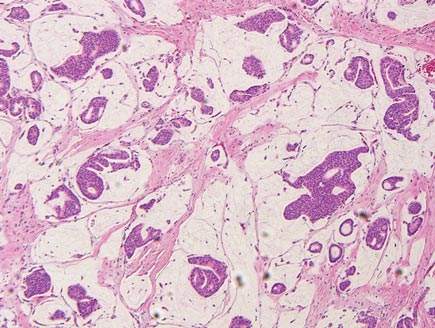
1. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
2. Petersson F, Kutzner H, Spagnolo DV, et al. Adenoid cystic carcinoma-like pattern in spiradenoma and spiradenocylindroma: a rare feature in sporadic neoplasms and those associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2009;31:642-648.
3. Weyers W, Nilles M, Eckert F, et al. Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol. 1993;15:156-161.
4. Elston DM, Ferringer T. Dermatopathology. Edinburgh, Scotland: Elsevier Saunders; 2009.
5. Ackerman AB, de Viragh PA, Chongchitnant N. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
6. Yamamoto O, Asahi M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140:8-16.
7. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
8. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19:154-161.
9. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrinespiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21:121-123.
10. Thompson LD, Penner C, Ho NJ, et al. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014;8:88-109.
The coexistence of more than one cutaneous adnexal neoplasm in a single biopsy specimen is unusual and is most frequently recognized in the context of a nevus sebaceous or Brooke-Spiegler syndrome, an autosomal-dominant inherited disease characterized by cutaneous adnexal neoplasms, most commonly cylindromas and trichoepitheliomas.1-3 Brooke-Spiegler syndrome is caused by germline mutations in the cylindromatosis gene, CYLD, located on band 16q12; it functions as a tumor suppressor gene and has regulatory roles in development, immunity, and inflammation.1 Weyers et al3 first recognized the tendency for adnexal collision tumors to present in patients with Brooke-Spiegler syndrome; they reported a patient with Brooke-Spiegler syndrome with spiradenomas found in the immediate vicinity of trichoepitheliomas and in continuity with hair follicles.
Spiradenomas are composed of large, sharply demarcated, rounded nodules of basaloid cells with little cytoplasm (Figure 1).4 The basaloid nodules may demonstrate a trabecular architecture, and on close inspection 2 cell types—paler cells with more cytoplasm and darker cells with less cytoplasm—are distinguishable (Figure 2A). Lymphocytes often are scattered within the tumor nodules and/or stroma. In Brooke-Spiegler syndrome, collision tumors containing a spiradenomatous component in collision with trichoepithelioma are not uncommon.1 Spiradenomas in Brooke-Spiegler syndrome have been reported to contain sebaceous differentiation or foci with an adenoid cystic carcinoma (ACC)–like pattern and are known to occur as hybrid lesions of spiradenoma and cylindroma or trichoepithelioma (as in this case).
In this case, 2 distinct neoplasms (spiradenoma and trichoepithelioma) are apparent, side by side, with an intervening hair follicle (Figure 1). Trichoepitheliomas, also known as cribriform trichoblastomas,5 are characterized by lobules of basaloid cells resembling basal cell carcinoma surrounded by a fibroblast-rich stroma. They often contain fingerlike projections and adopt a cribriform morphology within the tumor lobules (Figure 2B).4 Numerous horn cysts may be present, but their absence does not preclude the diagnosis. Mucin may be present within the cribriform tumor islands (Figure 2B) but not in the stroma. Characteristically, trichoepitheliomas are distinctly negative for CK7 (Figure 3), and unlike spiradenomas, they lack a myoepithelial component.6 This staining pattern in combination with the tumor’s proximity to an adjacent hair follicle makes a diagnosis of trichoepithelioma and spiradenoma collision tumor most likely and supports a clinical suspicion for Brooke-Spiegler syndrome.
|
|
Although spiradenomas sometimes contain cystic cavities (microcystic change), they typically are filled with finely granular eosinophilic material, not mucin, that is diastase resistant and periodic acid–Schiff positive (Figure 4).7 Spiradenomas classically stain positive with CK7 (Figure 3), epithelial membrane antigen, and carcinoembryonic antigen, and have a substantial myoepithelial component, as evidenced by the myoepithelial component staining with p63, S-100, and smooth muscle actin (SMA).7-9 The distinct lack of staining with CK7 and SMA in the tumor on the left in Figure 3 confirms that these tumors are of different lineage, rather than representing cystic change within a spiradenoma.
|
|
|
Adenoid cystic carcinoma is a rare neoplasm that may occur in a primary cutaneous form, as a direct extension from an underlying salivary gland neoplasm, or rarely as a focal pattern within spiradenomas occurring both sporadically or in the context of Brooke-Spiegler syndrome.2,7 The tumor is composed of variably sized cribriform islands of basaloid to pink cells concentrically arranged around glandlike spaces filled with mucin (Figure 5A). In contrast to trichoepithelioma, ACC occurs in the mid to deep dermis, often extending into subcutaneous fat with an infiltrative border, and is not often found in close proximity to hair follicles.7 Characteristically, hyaline basement membrane–like material that is periodic acid–Schiff positive is found between the tumor cells and also surrounding the individual lobules. Immunohistochemically, ACC has a myoepithelial component that stains positive with SMA, S-100, and p63; additionally, the tumor cells express low- and high-molecular-weight keratin and demonstrate variable epithelial membrane antigen positivity.10 In the current case, the superficial location, close association with a hair follicle, and lack of staining with both CK7 (Figure 3) and SMA (not shown) make ACC arising within a spiradenoma a less likely diagnosis.
Cylindromas are composed of basaloid islands interconnected in a jigsaw puzzle configuration (Figure 5B).4 Similar to spiradenomas, they also are composed of 2 cell populations. Characteristically, the tumor islands are outlined by a hyalinized eosinophilic basement membrane. Hyalinized droplets of basement membrane zone material also may be noted in the islands. Unlike spiradenomas, they lack both intratumoral lymphocytes and a trabecular growth pattern. Although spiradenocylindromas (cylindroma and spiradenoma collision tumors) are perhaps the most common collision tumor associated with Brooke-Spiegler syndrome, there is no evidence suggesting the presence of a cylindroma in the current case.
|
|
Primary cutaneous mucinous carcinoma is a rare neoplasm with a predilection for the eyelids; lesions occurring outside of this facial distribution, particularly of the breast, warrant a workup for metastatic disease.7 It typically occurs in the deeper dermis with involvement of the subcutaneous fat and is characterized by delicate fibrous septa enveloping large lakes of mucin, which contain islands of tumor cells (Figure 6). It has not been reported in association with spiradenomas. In addition, the tumor cells typically are CK7 positive.
The coexistence of more than one cutaneous adnexal neoplasm in a single biopsy specimen is unusual and is most frequently recognized in the context of a nevus sebaceous or Brooke-Spiegler syndrome, an autosomal-dominant inherited disease characterized by cutaneous adnexal neoplasms, most commonly cylindromas and trichoepitheliomas.1-3 Brooke-Spiegler syndrome is caused by germline mutations in the cylindromatosis gene, CYLD, located on band 16q12; it functions as a tumor suppressor gene and has regulatory roles in development, immunity, and inflammation.1 Weyers et al3 first recognized the tendency for adnexal collision tumors to present in patients with Brooke-Spiegler syndrome; they reported a patient with Brooke-Spiegler syndrome with spiradenomas found in the immediate vicinity of trichoepitheliomas and in continuity with hair follicles.
Spiradenomas are composed of large, sharply demarcated, rounded nodules of basaloid cells with little cytoplasm (Figure 1).4 The basaloid nodules may demonstrate a trabecular architecture, and on close inspection 2 cell types—paler cells with more cytoplasm and darker cells with less cytoplasm—are distinguishable (Figure 2A). Lymphocytes often are scattered within the tumor nodules and/or stroma. In Brooke-Spiegler syndrome, collision tumors containing a spiradenomatous component in collision with trichoepithelioma are not uncommon.1 Spiradenomas in Brooke-Spiegler syndrome have been reported to contain sebaceous differentiation or foci with an adenoid cystic carcinoma (ACC)–like pattern and are known to occur as hybrid lesions of spiradenoma and cylindroma or trichoepithelioma (as in this case).
In this case, 2 distinct neoplasms (spiradenoma and trichoepithelioma) are apparent, side by side, with an intervening hair follicle (Figure 1). Trichoepitheliomas, also known as cribriform trichoblastomas,5 are characterized by lobules of basaloid cells resembling basal cell carcinoma surrounded by a fibroblast-rich stroma. They often contain fingerlike projections and adopt a cribriform morphology within the tumor lobules (Figure 2B).4 Numerous horn cysts may be present, but their absence does not preclude the diagnosis. Mucin may be present within the cribriform tumor islands (Figure 2B) but not in the stroma. Characteristically, trichoepitheliomas are distinctly negative for CK7 (Figure 3), and unlike spiradenomas, they lack a myoepithelial component.6 This staining pattern in combination with the tumor’s proximity to an adjacent hair follicle makes a diagnosis of trichoepithelioma and spiradenoma collision tumor most likely and supports a clinical suspicion for Brooke-Spiegler syndrome.
|
|
Although spiradenomas sometimes contain cystic cavities (microcystic change), they typically are filled with finely granular eosinophilic material, not mucin, that is diastase resistant and periodic acid–Schiff positive (Figure 4).7 Spiradenomas classically stain positive with CK7 (Figure 3), epithelial membrane antigen, and carcinoembryonic antigen, and have a substantial myoepithelial component, as evidenced by the myoepithelial component staining with p63, S-100, and smooth muscle actin (SMA).7-9 The distinct lack of staining with CK7 and SMA in the tumor on the left in Figure 3 confirms that these tumors are of different lineage, rather than representing cystic change within a spiradenoma.
|
|
|
Adenoid cystic carcinoma is a rare neoplasm that may occur in a primary cutaneous form, as a direct extension from an underlying salivary gland neoplasm, or rarely as a focal pattern within spiradenomas occurring both sporadically or in the context of Brooke-Spiegler syndrome.2,7 The tumor is composed of variably sized cribriform islands of basaloid to pink cells concentrically arranged around glandlike spaces filled with mucin (Figure 5A). In contrast to trichoepithelioma, ACC occurs in the mid to deep dermis, often extending into subcutaneous fat with an infiltrative border, and is not often found in close proximity to hair follicles.7 Characteristically, hyaline basement membrane–like material that is periodic acid–Schiff positive is found between the tumor cells and also surrounding the individual lobules. Immunohistochemically, ACC has a myoepithelial component that stains positive with SMA, S-100, and p63; additionally, the tumor cells express low- and high-molecular-weight keratin and demonstrate variable epithelial membrane antigen positivity.10 In the current case, the superficial location, close association with a hair follicle, and lack of staining with both CK7 (Figure 3) and SMA (not shown) make ACC arising within a spiradenoma a less likely diagnosis.
Cylindromas are composed of basaloid islands interconnected in a jigsaw puzzle configuration (Figure 5B).4 Similar to spiradenomas, they also are composed of 2 cell populations. Characteristically, the tumor islands are outlined by a hyalinized eosinophilic basement membrane. Hyalinized droplets of basement membrane zone material also may be noted in the islands. Unlike spiradenomas, they lack both intratumoral lymphocytes and a trabecular growth pattern. Although spiradenocylindromas (cylindroma and spiradenoma collision tumors) are perhaps the most common collision tumor associated with Brooke-Spiegler syndrome, there is no evidence suggesting the presence of a cylindroma in the current case.
|
|
Primary cutaneous mucinous carcinoma is a rare neoplasm with a predilection for the eyelids; lesions occurring outside of this facial distribution, particularly of the breast, warrant a workup for metastatic disease.7 It typically occurs in the deeper dermis with involvement of the subcutaneous fat and is characterized by delicate fibrous septa enveloping large lakes of mucin, which contain islands of tumor cells (Figure 6). It has not been reported in association with spiradenomas. In addition, the tumor cells typically are CK7 positive.
1. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
2. Petersson F, Kutzner H, Spagnolo DV, et al. Adenoid cystic carcinoma-like pattern in spiradenoma and spiradenocylindroma: a rare feature in sporadic neoplasms and those associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2009;31:642-648.
3. Weyers W, Nilles M, Eckert F, et al. Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol. 1993;15:156-161.
4. Elston DM, Ferringer T. Dermatopathology. Edinburgh, Scotland: Elsevier Saunders; 2009.
5. Ackerman AB, de Viragh PA, Chongchitnant N. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
6. Yamamoto O, Asahi M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140:8-16.
7. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
8. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19:154-161.
9. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrinespiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21:121-123.
10. Thompson LD, Penner C, Ho NJ, et al. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014;8:88-109.
1. Kazakov DV, Soukup R, Mukensnabl P, et al. Brooke-Spiegler syndrome: report of a case with combined lesions containing cylindromatous, spiradenomatous, trichoblastomatous, and sebaceous differentiation. Am J Dermatopathol. 2005;27:27-33.
2. Petersson F, Kutzner H, Spagnolo DV, et al. Adenoid cystic carcinoma-like pattern in spiradenoma and spiradenocylindroma: a rare feature in sporadic neoplasms and those associated with Brooke-Spiegler syndrome. Am J Dermatopathol. 2009;31:642-648.
3. Weyers W, Nilles M, Eckert F, et al. Spiradenomas in Brooke-Spiegler syndrome. Am J Dermatopathol. 1993;15:156-161.
4. Elston DM, Ferringer T. Dermatopathology. Edinburgh, Scotland: Elsevier Saunders; 2009.
5. Ackerman AB, de Viragh PA, Chongchitnant N. Neoplasms with Follicular Differentiation. Philadelphia, PA: Lea & Febiger; 1993.
6. Yamamoto O, Asahi M. Cytokeratin expression in trichoblastic fibroma (small nodular type trichoblastoma), trichoepithelioma and basal cell carcinoma. Br J Dermatol. 1999;140:8-16.
7. Calonje JE, Brenn T, Lazar AJ, et al. McKee’s Pathology of the Skin with Clinical Correlations. 4th ed. St Louis, MO: Elsevier Saunders; 2012.
8. Meybehm M, Fischer HP. Spiradenoma and dermal cylindroma: comparative immunohistochemical analysis and histogenetic considerations. Am J Dermatopathol. 1997;19:154-161.
9. Kurokawa I, Nishimura K, Tarumi C, et al. Eccrinespiradenoma: co-expression of cytokeratin and smooth muscle actin suggesting differentiation toward myoepithelial cells. J Eur Acad Dermatol Venereol. 2007;21:121-123.
10. Thompson LD, Penner C, Ho NJ, et al. Sinonasal tract and nasopharyngeal adenoid cystic carcinoma: a clinicopathologic and immunophenotypic study of 86 cases. Head Neck Pathol. 2014;8:88-109.