User login
MDedge conference coverage features onsite reporting of the latest study results and expert perspectives from leading researchers.
Patients given NSAIDs over antiemetics for headaches spend less time in the ED
based on data from approximately 7,000 patients.
Headache is the fourth-most common chief complaint in the ED, accounting for approximately 3% of all ED visits, said Philip Wang, a medical student at the Cleveland Clinic, in a presentation at the annual meeting of the American College of Emergency Physicians.
A variety of pharmacotherapies are used to manage headache, which leads to a range of resource use, he said.
To understand the association between route of drug administration and length of ED stay, Mr. Wang and colleagues reviewed data from 7,233 visits by 6,715 patients at any of the 21 Cleveland Clinic Health System EDs in 2018 with headache as the primary discharge diagnosis. Patients admitted to the hospital were excluded; those treated with opioids, antiemetics, and/or NSAIDs were included. The average age of the study population was 31 years, 57% were White, and approximately half were Medicaid or Medicare patients.
Approximately 68% of patients received antiemetics, 66.8% received NSAIDs, and 9.8% received opioids. Approximately 42% of patients received parenteral-only treatment and 42% received oral-only treatment; 15% received mixed treatment. The average length of ED stay was 202 minutes.
In a multivariate analysis adjusted for sex, age, income, race, insurance status, ED type, and arrival time, treatment with oral drugs only was associated with an 11% reduction of length of stay, compared with treatment with parenteral medication only (P < .001). However, the length of stay for patients treated with mixed route of administration was 10% longer, compared with parenteral only (P < .001).
In terms of drug class (a secondary outcome), patients treated with opioids had a 10% increase in length of stay (P < .01) and those treated with antiemetics had a 14% increase in length of stay; however, patients treated with NSAIDs had a 7% decrease in length of stay.
The study findings were limited in part by the challenge of isolating patients presenting with a primary headache diagnosis, Mr. Wang noted in the presentation.
The challenge of controlling for all the potential factors impacting length of stay, which is “provider, resource, and situation dependent,” is an additional limitation, he said.
However, the results show that route of administration has a significant impact on length of ED stay in patients presenting with headache, he concluded.
The study received no outside funding. The researchers disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
based on data from approximately 7,000 patients.
Headache is the fourth-most common chief complaint in the ED, accounting for approximately 3% of all ED visits, said Philip Wang, a medical student at the Cleveland Clinic, in a presentation at the annual meeting of the American College of Emergency Physicians.
A variety of pharmacotherapies are used to manage headache, which leads to a range of resource use, he said.
To understand the association between route of drug administration and length of ED stay, Mr. Wang and colleagues reviewed data from 7,233 visits by 6,715 patients at any of the 21 Cleveland Clinic Health System EDs in 2018 with headache as the primary discharge diagnosis. Patients admitted to the hospital were excluded; those treated with opioids, antiemetics, and/or NSAIDs were included. The average age of the study population was 31 years, 57% were White, and approximately half were Medicaid or Medicare patients.
Approximately 68% of patients received antiemetics, 66.8% received NSAIDs, and 9.8% received opioids. Approximately 42% of patients received parenteral-only treatment and 42% received oral-only treatment; 15% received mixed treatment. The average length of ED stay was 202 minutes.
In a multivariate analysis adjusted for sex, age, income, race, insurance status, ED type, and arrival time, treatment with oral drugs only was associated with an 11% reduction of length of stay, compared with treatment with parenteral medication only (P < .001). However, the length of stay for patients treated with mixed route of administration was 10% longer, compared with parenteral only (P < .001).
In terms of drug class (a secondary outcome), patients treated with opioids had a 10% increase in length of stay (P < .01) and those treated with antiemetics had a 14% increase in length of stay; however, patients treated with NSAIDs had a 7% decrease in length of stay.
The study findings were limited in part by the challenge of isolating patients presenting with a primary headache diagnosis, Mr. Wang noted in the presentation.
The challenge of controlling for all the potential factors impacting length of stay, which is “provider, resource, and situation dependent,” is an additional limitation, he said.
However, the results show that route of administration has a significant impact on length of ED stay in patients presenting with headache, he concluded.
The study received no outside funding. The researchers disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
based on data from approximately 7,000 patients.
Headache is the fourth-most common chief complaint in the ED, accounting for approximately 3% of all ED visits, said Philip Wang, a medical student at the Cleveland Clinic, in a presentation at the annual meeting of the American College of Emergency Physicians.
A variety of pharmacotherapies are used to manage headache, which leads to a range of resource use, he said.
To understand the association between route of drug administration and length of ED stay, Mr. Wang and colleagues reviewed data from 7,233 visits by 6,715 patients at any of the 21 Cleveland Clinic Health System EDs in 2018 with headache as the primary discharge diagnosis. Patients admitted to the hospital were excluded; those treated with opioids, antiemetics, and/or NSAIDs were included. The average age of the study population was 31 years, 57% were White, and approximately half were Medicaid or Medicare patients.
Approximately 68% of patients received antiemetics, 66.8% received NSAIDs, and 9.8% received opioids. Approximately 42% of patients received parenteral-only treatment and 42% received oral-only treatment; 15% received mixed treatment. The average length of ED stay was 202 minutes.
In a multivariate analysis adjusted for sex, age, income, race, insurance status, ED type, and arrival time, treatment with oral drugs only was associated with an 11% reduction of length of stay, compared with treatment with parenteral medication only (P < .001). However, the length of stay for patients treated with mixed route of administration was 10% longer, compared with parenteral only (P < .001).
In terms of drug class (a secondary outcome), patients treated with opioids had a 10% increase in length of stay (P < .01) and those treated with antiemetics had a 14% increase in length of stay; however, patients treated with NSAIDs had a 7% decrease in length of stay.
The study findings were limited in part by the challenge of isolating patients presenting with a primary headache diagnosis, Mr. Wang noted in the presentation.
The challenge of controlling for all the potential factors impacting length of stay, which is “provider, resource, and situation dependent,” is an additional limitation, he said.
However, the results show that route of administration has a significant impact on length of ED stay in patients presenting with headache, he concluded.
The study received no outside funding. The researchers disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Without PrEP, a third of new HIV cases occur in MSM at low risk
Nearly one in three gay and bisexual men who were diagnosed with HIV at U.K. sexual health clinics didn’t meet the criteria for “high risk” that would signal to a clinician that they would be good candidates for pre-exposure prophylaxis (PrEP).
And that means that people who appear lower risk may still be good candidates for the HIV prevention pills, said Ann Sullivan, MD, consulting physician at Chelsea and Westminster Hospital, London.
“If people are coming forward for PrEP, they have self-identified that they need PrEP, [and] we should be allowing them to take PrEP,” said Dr. Sullivan at the 18th European AIDS Society Conference (EACS 2021). “We just need to trust patients. People know their risk, and we just have to accept that they know what they need best.”
And while this trial was made up of 95% gay and bisexual men, that ethos applies to every other group that could benefit from PrEP, including cisgender and transgender women and other gender-diverse people, Latinos, and Black Americans. In the United States, these groups make up nearly half of those who could benefit from PrEP under older guidelines but account for just 8% of people currently taking PrEP.
The finding also reinforces growing calls from health care providers to reduce gatekeeping around PrEP. For instance, there’s a move underway by the U.S. Centers for Disease Control and Prevention, where drafts of updated PrEP guidelines call for clinicians to talk to any sexually active teenager and adult about PrEP.
For the PrEP Impact trial, gay and bisexual men who received sexual health care at UK National Health Service sexual health clinics were invited to enroll in the study based on national PrEP guidelines. Those guidelines included being a cisgender man who had had sex with men not currently living with HIV and reporting condomless anal sex in the last 3 months; having a male partner whose HIV status they don’t know or who doesn’t have an undetectable viral load and with whom they’ve had condomless anal sex; or someone who doesn’t reach those criteria but whom the clinician thinks would be a good candidate.
Between Oct. 2017 and Feb. 2020, a total of 17,770 gay and bisexual men and 503 transgender or nonbinary people enrolled in the trial and were paired with 97,098 gay and bisexual men who didn’t use PrEP. (Data from the transgender participants were reported in a separate presentation.) The median age was 27 years, with 14.4% of the cisgender gay men between the ages of 16 and 24. Three out of four cis men were White, most lived in London, and more than half came from very-low-income neighborhoods.
Participants and controls were assessed for whether they were at particularly high risk for acquiring HIV, such as having used PrEP, having had two or more HIV tests, having had a rectal bacterial sexually transmitted infection (STI), or having had contact with someone with HIV or syphilis.
At the end of Feb. 2020, 24 cisgender men on PrEP had acquired HIV compared with 670 in the control group – an 87% reduction in HIV acquisition. Only one of those 24 cis men had lab-confirmed high adherence to PrEP. However, because the hair samples used to judge drug concentration weren’t long enough, Dr. Sullivan and colleagues were unable to assess whether the person really was fully adherent to treatment for the length of the trial.
But when they looked at the assessed behavior of people who acquired HIV, the two groups diverged. While a full 92% of people using PrEP had had STI diagnoses and other markers of increased risk, that was true for only 71% of people not taking PrEP. That meant, Dr. Sullivan said in an interview, that screening guidelines for PrEP were missing 29% of people with low assessed risk for HIV who nevertheless acquired the virus.
The findings led Antonio Urbina, MD, who both prescribes PrEP and manages Mount Sinai Medical Center’s PrEP program in New York, to the same conclusion that Dr. Sullivan and her team came to: that no screener is going to account for everything, and that there may be things that patients don’t want to tell their clinicians about their risk, either because of their own internalized stigma or their calculation that they aren’t comfortable enough with their providers to be honest.
“It reinforces to me that I need to ask more open-ended questions regarding risk and then just talk more about PrEP,” said Dr. Urbina, professor of medicine at Icahn School of Medicine. “Risk is dynamic and changes. And the great thing about PrEP is that if the risk goes up or down, if you have PrEP on board, you maintain this protection against HIV.”
An accompanying presentation on the transgender and nonbinary participants in the Impact Trial found that just one of 503 PrEP users acquired HIV. But here, too, there were people who could have benefited from PrEP but didn’t take it: Of the 477 trans and nonbinary participants who acted as controls, 97 were eligible by current guidelines but didn’t take PrEP. One in four of those declined the offer to take PrEP; the rest weren’t able to take it because they lived outside the treatment area. That, combined with a significantly lower likelihood that Black trans and nonbinary people took PrEP, indicated that work needs to be done to address the needs of people geographically and ethnically.
The data on gay men also raised the “who’s left out” issue for Gina Simoncini, MD, medical director for the Philadelphia AIDS Healthcare Foundation Healthcare Center. Dr. Simoncini previously taught attending physicians at Temple University how to prescribe PrEP and has done many grand rounds for primary care providers on how to manage PrEP.
“My biggest issue with this data is: What about the people who aren’t going to sexual health clinics?” she said. “What about the kid who’s 16 and maybe just barely putting his feet into the waters of sex and doesn’t feel quite comfortable going to a sexual health clinic? What about the trans Indian girl who can’t get to sexual health clinics because of family stigma and cultural stigma? The more we move toward primary care, the more people need to get on board with this.”
Dr. Sullivan reports no relevant financial relationships. Dr. Simoncini is an employee of AIDS Healthcare Foundation and has received advisory board fees from ViiV Healthcare. Dr. Urbina sits on the scientific advisory councils for Gilead Sciences, ViiV Healthcare, and Merck.
A version of this article first appeared on Medscape.com.
Nearly one in three gay and bisexual men who were diagnosed with HIV at U.K. sexual health clinics didn’t meet the criteria for “high risk” that would signal to a clinician that they would be good candidates for pre-exposure prophylaxis (PrEP).
And that means that people who appear lower risk may still be good candidates for the HIV prevention pills, said Ann Sullivan, MD, consulting physician at Chelsea and Westminster Hospital, London.
“If people are coming forward for PrEP, they have self-identified that they need PrEP, [and] we should be allowing them to take PrEP,” said Dr. Sullivan at the 18th European AIDS Society Conference (EACS 2021). “We just need to trust patients. People know their risk, and we just have to accept that they know what they need best.”
And while this trial was made up of 95% gay and bisexual men, that ethos applies to every other group that could benefit from PrEP, including cisgender and transgender women and other gender-diverse people, Latinos, and Black Americans. In the United States, these groups make up nearly half of those who could benefit from PrEP under older guidelines but account for just 8% of people currently taking PrEP.
The finding also reinforces growing calls from health care providers to reduce gatekeeping around PrEP. For instance, there’s a move underway by the U.S. Centers for Disease Control and Prevention, where drafts of updated PrEP guidelines call for clinicians to talk to any sexually active teenager and adult about PrEP.
For the PrEP Impact trial, gay and bisexual men who received sexual health care at UK National Health Service sexual health clinics were invited to enroll in the study based on national PrEP guidelines. Those guidelines included being a cisgender man who had had sex with men not currently living with HIV and reporting condomless anal sex in the last 3 months; having a male partner whose HIV status they don’t know or who doesn’t have an undetectable viral load and with whom they’ve had condomless anal sex; or someone who doesn’t reach those criteria but whom the clinician thinks would be a good candidate.
Between Oct. 2017 and Feb. 2020, a total of 17,770 gay and bisexual men and 503 transgender or nonbinary people enrolled in the trial and were paired with 97,098 gay and bisexual men who didn’t use PrEP. (Data from the transgender participants were reported in a separate presentation.) The median age was 27 years, with 14.4% of the cisgender gay men between the ages of 16 and 24. Three out of four cis men were White, most lived in London, and more than half came from very-low-income neighborhoods.
Participants and controls were assessed for whether they were at particularly high risk for acquiring HIV, such as having used PrEP, having had two or more HIV tests, having had a rectal bacterial sexually transmitted infection (STI), or having had contact with someone with HIV or syphilis.
At the end of Feb. 2020, 24 cisgender men on PrEP had acquired HIV compared with 670 in the control group – an 87% reduction in HIV acquisition. Only one of those 24 cis men had lab-confirmed high adherence to PrEP. However, because the hair samples used to judge drug concentration weren’t long enough, Dr. Sullivan and colleagues were unable to assess whether the person really was fully adherent to treatment for the length of the trial.
But when they looked at the assessed behavior of people who acquired HIV, the two groups diverged. While a full 92% of people using PrEP had had STI diagnoses and other markers of increased risk, that was true for only 71% of people not taking PrEP. That meant, Dr. Sullivan said in an interview, that screening guidelines for PrEP were missing 29% of people with low assessed risk for HIV who nevertheless acquired the virus.
The findings led Antonio Urbina, MD, who both prescribes PrEP and manages Mount Sinai Medical Center’s PrEP program in New York, to the same conclusion that Dr. Sullivan and her team came to: that no screener is going to account for everything, and that there may be things that patients don’t want to tell their clinicians about their risk, either because of their own internalized stigma or their calculation that they aren’t comfortable enough with their providers to be honest.
“It reinforces to me that I need to ask more open-ended questions regarding risk and then just talk more about PrEP,” said Dr. Urbina, professor of medicine at Icahn School of Medicine. “Risk is dynamic and changes. And the great thing about PrEP is that if the risk goes up or down, if you have PrEP on board, you maintain this protection against HIV.”
An accompanying presentation on the transgender and nonbinary participants in the Impact Trial found that just one of 503 PrEP users acquired HIV. But here, too, there were people who could have benefited from PrEP but didn’t take it: Of the 477 trans and nonbinary participants who acted as controls, 97 were eligible by current guidelines but didn’t take PrEP. One in four of those declined the offer to take PrEP; the rest weren’t able to take it because they lived outside the treatment area. That, combined with a significantly lower likelihood that Black trans and nonbinary people took PrEP, indicated that work needs to be done to address the needs of people geographically and ethnically.
The data on gay men also raised the “who’s left out” issue for Gina Simoncini, MD, medical director for the Philadelphia AIDS Healthcare Foundation Healthcare Center. Dr. Simoncini previously taught attending physicians at Temple University how to prescribe PrEP and has done many grand rounds for primary care providers on how to manage PrEP.
“My biggest issue with this data is: What about the people who aren’t going to sexual health clinics?” she said. “What about the kid who’s 16 and maybe just barely putting his feet into the waters of sex and doesn’t feel quite comfortable going to a sexual health clinic? What about the trans Indian girl who can’t get to sexual health clinics because of family stigma and cultural stigma? The more we move toward primary care, the more people need to get on board with this.”
Dr. Sullivan reports no relevant financial relationships. Dr. Simoncini is an employee of AIDS Healthcare Foundation and has received advisory board fees from ViiV Healthcare. Dr. Urbina sits on the scientific advisory councils for Gilead Sciences, ViiV Healthcare, and Merck.
A version of this article first appeared on Medscape.com.
Nearly one in three gay and bisexual men who were diagnosed with HIV at U.K. sexual health clinics didn’t meet the criteria for “high risk” that would signal to a clinician that they would be good candidates for pre-exposure prophylaxis (PrEP).
And that means that people who appear lower risk may still be good candidates for the HIV prevention pills, said Ann Sullivan, MD, consulting physician at Chelsea and Westminster Hospital, London.
“If people are coming forward for PrEP, they have self-identified that they need PrEP, [and] we should be allowing them to take PrEP,” said Dr. Sullivan at the 18th European AIDS Society Conference (EACS 2021). “We just need to trust patients. People know their risk, and we just have to accept that they know what they need best.”
And while this trial was made up of 95% gay and bisexual men, that ethos applies to every other group that could benefit from PrEP, including cisgender and transgender women and other gender-diverse people, Latinos, and Black Americans. In the United States, these groups make up nearly half of those who could benefit from PrEP under older guidelines but account for just 8% of people currently taking PrEP.
The finding also reinforces growing calls from health care providers to reduce gatekeeping around PrEP. For instance, there’s a move underway by the U.S. Centers for Disease Control and Prevention, where drafts of updated PrEP guidelines call for clinicians to talk to any sexually active teenager and adult about PrEP.
For the PrEP Impact trial, gay and bisexual men who received sexual health care at UK National Health Service sexual health clinics were invited to enroll in the study based on national PrEP guidelines. Those guidelines included being a cisgender man who had had sex with men not currently living with HIV and reporting condomless anal sex in the last 3 months; having a male partner whose HIV status they don’t know or who doesn’t have an undetectable viral load and with whom they’ve had condomless anal sex; or someone who doesn’t reach those criteria but whom the clinician thinks would be a good candidate.
Between Oct. 2017 and Feb. 2020, a total of 17,770 gay and bisexual men and 503 transgender or nonbinary people enrolled in the trial and were paired with 97,098 gay and bisexual men who didn’t use PrEP. (Data from the transgender participants were reported in a separate presentation.) The median age was 27 years, with 14.4% of the cisgender gay men between the ages of 16 and 24. Three out of four cis men were White, most lived in London, and more than half came from very-low-income neighborhoods.
Participants and controls were assessed for whether they were at particularly high risk for acquiring HIV, such as having used PrEP, having had two or more HIV tests, having had a rectal bacterial sexually transmitted infection (STI), or having had contact with someone with HIV or syphilis.
At the end of Feb. 2020, 24 cisgender men on PrEP had acquired HIV compared with 670 in the control group – an 87% reduction in HIV acquisition. Only one of those 24 cis men had lab-confirmed high adherence to PrEP. However, because the hair samples used to judge drug concentration weren’t long enough, Dr. Sullivan and colleagues were unable to assess whether the person really was fully adherent to treatment for the length of the trial.
But when they looked at the assessed behavior of people who acquired HIV, the two groups diverged. While a full 92% of people using PrEP had had STI diagnoses and other markers of increased risk, that was true for only 71% of people not taking PrEP. That meant, Dr. Sullivan said in an interview, that screening guidelines for PrEP were missing 29% of people with low assessed risk for HIV who nevertheless acquired the virus.
The findings led Antonio Urbina, MD, who both prescribes PrEP and manages Mount Sinai Medical Center’s PrEP program in New York, to the same conclusion that Dr. Sullivan and her team came to: that no screener is going to account for everything, and that there may be things that patients don’t want to tell their clinicians about their risk, either because of their own internalized stigma or their calculation that they aren’t comfortable enough with their providers to be honest.
“It reinforces to me that I need to ask more open-ended questions regarding risk and then just talk more about PrEP,” said Dr. Urbina, professor of medicine at Icahn School of Medicine. “Risk is dynamic and changes. And the great thing about PrEP is that if the risk goes up or down, if you have PrEP on board, you maintain this protection against HIV.”
An accompanying presentation on the transgender and nonbinary participants in the Impact Trial found that just one of 503 PrEP users acquired HIV. But here, too, there were people who could have benefited from PrEP but didn’t take it: Of the 477 trans and nonbinary participants who acted as controls, 97 were eligible by current guidelines but didn’t take PrEP. One in four of those declined the offer to take PrEP; the rest weren’t able to take it because they lived outside the treatment area. That, combined with a significantly lower likelihood that Black trans and nonbinary people took PrEP, indicated that work needs to be done to address the needs of people geographically and ethnically.
The data on gay men also raised the “who’s left out” issue for Gina Simoncini, MD, medical director for the Philadelphia AIDS Healthcare Foundation Healthcare Center. Dr. Simoncini previously taught attending physicians at Temple University how to prescribe PrEP and has done many grand rounds for primary care providers on how to manage PrEP.
“My biggest issue with this data is: What about the people who aren’t going to sexual health clinics?” she said. “What about the kid who’s 16 and maybe just barely putting his feet into the waters of sex and doesn’t feel quite comfortable going to a sexual health clinic? What about the trans Indian girl who can’t get to sexual health clinics because of family stigma and cultural stigma? The more we move toward primary care, the more people need to get on board with this.”
Dr. Sullivan reports no relevant financial relationships. Dr. Simoncini is an employee of AIDS Healthcare Foundation and has received advisory board fees from ViiV Healthcare. Dr. Urbina sits on the scientific advisory councils for Gilead Sciences, ViiV Healthcare, and Merck.
A version of this article first appeared on Medscape.com.
Long-acting HIV ART: Lessons from a year of Cabenuva
One year into offering the first long-acting injectable HIV treatment to his patients, Jonathan Angel, MD, head of the division of infectious diseases at the University of Ottawa, reported that 15 of the 21 of patients who started on the regimen are still taking it, all with viral suppression. Those who weren’t cited a combination of inconvenience, injection site pain, and “injection fatigue.”
These are just a few things HIV providers are learning as they begin what Chloe Orkin, MD, professor of HIV medicine at Queen Mary University of London, called a paradigm shift to long-acting treatment, which may soon include not just shots but rings, implants, and microarray patches.
“It’s a paradigm shift, and we are at the very beginning of this paradigm shift,” said Dr. Orkin, commenting during the discussion session of the European AIDS Clinical Society 2021 annual meeting. “We’re having to change our model, and it’s challenging.”
In the United States, the Food and Drug Administration approved the first long-acting injectable, a combination of cabotegravir and rilpivirine (CAB/RIL; Cabenuva, ViiV Healthcare) in January 2021. But it has been approved in Canada since March 2020 and available at Dr. Angel’s clinic since November 2020. It’s also available in Canada as an every-other-month shot. Injected into the buttocks, the shot was found to be noninferior to standard daily oral treatment in many studies, including the ATLAS, the ATLAS-2M – which tested the every-other-month approach – and FLAIR trials.
Dr. Angel’s clinic was part of all three of those trials, so his clinic has had 5 years’ experience preparing for the change in workflow and the new approach the shots require.
Of the 21 people Dr. Angel has treated, 11 were white Canadians, nine were Black African, and one was Indigenous Canadian, with women making up a third of the participants. Median age was 51 years, and all patients had had undetectable viral loads before beginning the regimen. (Studies of the drug’s effectiveness in people who struggle to take daily pills are still ongoing.)
Most of those 21 patients had had undetectable viral loads for more than 5 years, but a few had been undetectable for only 6 months before beginning the shots. Their immune systems were also healthy, with a median CD4 count of 618 cells/mcL. As in the clinical trials, none of the participants had experienced antiretroviral treatment failure. Because public health insurers in Canada have yet to approve the shots, Dr. Angel’s patients receiving Cabenuva also have private health insurance. Up to 90% of people in Canada receive pharmaceutical coverage through public insurance; therefore, the shot is not yet widely available.
Twenty patients switched from integrase-inhibitor regimens, and one had been receiving a nonnucleoside reverse transcriptase inhibitor–based regimen before starting Cabenuva.
And although the drug has not been approved for shot initiation this way, two patients requested – and Dr. Angel agreed – to start them on the shots without first doing a month of daily pills to check for safety.
“This is my conclusion from these data: the oral lead-in period is not necessary,” Dr. Angel said in his presentation at the meeting. “It can provide some comfort to either a physician or a patient, but it does not seem to be medically necessary.”
That approach is not without data to back it up. Research presented at HIV Glasgow 2020 showed that people who switched from daily oral dolutegravir/abacavir/lamivudine straight to the injections did so without problems.
At last clinic visit, 15 of those 21 were still receiving the shots. None have experienced treatment failure, and all were still virally suppressed. Four participants left the trials and one more person opted to return to daily pills, citing some level of what Dr. Angel called “injection fatigue.”
“Just as we use the term ‘pill fatigue’ for patients who are tired of taking pills, patients do get tired of coming in monthly for their visits and injections,” he said. They find the trip to the clinic for the intramuscular injections “inconvenient,” he said.
Unlike in the United States, where Cabenuva is approved for only monthly injections, Health Canada has already approved the shot for every-other-month injections, which Dr. Angel said may reduce the odds of injection fatigue.
Dr. Angel’s presentation drew comments, questions, and excitement from the crowd. Annemarie Wensing, MD, assistant professor of medicine at University Medical Center Utrecht (the Netherlands), asked whether dispensing with the oral lead-in period could mean that these shots could be useful for people going on longer trips, people having surgeries where they can’t swallow pills, or in other scenarios.
“These are not hypothetical conversations,” Dr. Angel said. “I’m having these conversations with patients now – temporary use, they travel for 3 months and come back, can they go from injectable to oral to injectable.”
For now, he said, the answer is, “We’ll figure it out.”
Meanwhile, there’s another big question when it comes to injectables, said Marta Vas ylyev, MD, from Lviv (Ukraine) Regional AIDS Center: When will they be available to the people who might benefit most from them – people in resource-limited settings, people who so far have struggled to remember to take their pills every day?
For now, Dr. Angel replied, injectables continue to be a treatment only for those who are already doing well while receiving HIV treatment: those with already suppressed viral load, who are good at taking daily pills, and who are being treated at well-resourced clinics.
“There are huge obstacles to overcome if this is ever to be available [in resource-limited settings], and way more obstacles than there are with any oral therapies,” he said. “There’s not been much discussion here about the necessity of cold-chain requirements of pharmacies either centrally or locally, [or] the requirements of additional nurses or health care staff to administer the medication. So you’re looking at a very resource-intensive therapy, which now is fairly restrictive [as to] who will have access to it.”
Dr. Angel reports serving on advisory boards for ViiV Healthcare and Gilead Sciences and has done contract research for ViiV Healthcare, Gilead, and Merck. Dr. Orkin has received research grants, fees as a consultant, travel sponsorship, and speaker fees from ViiV, Merck, and GlaxoSmithKline. Dr. Vasylyev reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
One year into offering the first long-acting injectable HIV treatment to his patients, Jonathan Angel, MD, head of the division of infectious diseases at the University of Ottawa, reported that 15 of the 21 of patients who started on the regimen are still taking it, all with viral suppression. Those who weren’t cited a combination of inconvenience, injection site pain, and “injection fatigue.”
These are just a few things HIV providers are learning as they begin what Chloe Orkin, MD, professor of HIV medicine at Queen Mary University of London, called a paradigm shift to long-acting treatment, which may soon include not just shots but rings, implants, and microarray patches.
“It’s a paradigm shift, and we are at the very beginning of this paradigm shift,” said Dr. Orkin, commenting during the discussion session of the European AIDS Clinical Society 2021 annual meeting. “We’re having to change our model, and it’s challenging.”
In the United States, the Food and Drug Administration approved the first long-acting injectable, a combination of cabotegravir and rilpivirine (CAB/RIL; Cabenuva, ViiV Healthcare) in January 2021. But it has been approved in Canada since March 2020 and available at Dr. Angel’s clinic since November 2020. It’s also available in Canada as an every-other-month shot. Injected into the buttocks, the shot was found to be noninferior to standard daily oral treatment in many studies, including the ATLAS, the ATLAS-2M – which tested the every-other-month approach – and FLAIR trials.
Dr. Angel’s clinic was part of all three of those trials, so his clinic has had 5 years’ experience preparing for the change in workflow and the new approach the shots require.
Of the 21 people Dr. Angel has treated, 11 were white Canadians, nine were Black African, and one was Indigenous Canadian, with women making up a third of the participants. Median age was 51 years, and all patients had had undetectable viral loads before beginning the regimen. (Studies of the drug’s effectiveness in people who struggle to take daily pills are still ongoing.)
Most of those 21 patients had had undetectable viral loads for more than 5 years, but a few had been undetectable for only 6 months before beginning the shots. Their immune systems were also healthy, with a median CD4 count of 618 cells/mcL. As in the clinical trials, none of the participants had experienced antiretroviral treatment failure. Because public health insurers in Canada have yet to approve the shots, Dr. Angel’s patients receiving Cabenuva also have private health insurance. Up to 90% of people in Canada receive pharmaceutical coverage through public insurance; therefore, the shot is not yet widely available.
Twenty patients switched from integrase-inhibitor regimens, and one had been receiving a nonnucleoside reverse transcriptase inhibitor–based regimen before starting Cabenuva.
And although the drug has not been approved for shot initiation this way, two patients requested – and Dr. Angel agreed – to start them on the shots without first doing a month of daily pills to check for safety.
“This is my conclusion from these data: the oral lead-in period is not necessary,” Dr. Angel said in his presentation at the meeting. “It can provide some comfort to either a physician or a patient, but it does not seem to be medically necessary.”
That approach is not without data to back it up. Research presented at HIV Glasgow 2020 showed that people who switched from daily oral dolutegravir/abacavir/lamivudine straight to the injections did so without problems.
At last clinic visit, 15 of those 21 were still receiving the shots. None have experienced treatment failure, and all were still virally suppressed. Four participants left the trials and one more person opted to return to daily pills, citing some level of what Dr. Angel called “injection fatigue.”
“Just as we use the term ‘pill fatigue’ for patients who are tired of taking pills, patients do get tired of coming in monthly for their visits and injections,” he said. They find the trip to the clinic for the intramuscular injections “inconvenient,” he said.
Unlike in the United States, where Cabenuva is approved for only monthly injections, Health Canada has already approved the shot for every-other-month injections, which Dr. Angel said may reduce the odds of injection fatigue.
Dr. Angel’s presentation drew comments, questions, and excitement from the crowd. Annemarie Wensing, MD, assistant professor of medicine at University Medical Center Utrecht (the Netherlands), asked whether dispensing with the oral lead-in period could mean that these shots could be useful for people going on longer trips, people having surgeries where they can’t swallow pills, or in other scenarios.
“These are not hypothetical conversations,” Dr. Angel said. “I’m having these conversations with patients now – temporary use, they travel for 3 months and come back, can they go from injectable to oral to injectable.”
For now, he said, the answer is, “We’ll figure it out.”
Meanwhile, there’s another big question when it comes to injectables, said Marta Vas ylyev, MD, from Lviv (Ukraine) Regional AIDS Center: When will they be available to the people who might benefit most from them – people in resource-limited settings, people who so far have struggled to remember to take their pills every day?
For now, Dr. Angel replied, injectables continue to be a treatment only for those who are already doing well while receiving HIV treatment: those with already suppressed viral load, who are good at taking daily pills, and who are being treated at well-resourced clinics.
“There are huge obstacles to overcome if this is ever to be available [in resource-limited settings], and way more obstacles than there are with any oral therapies,” he said. “There’s not been much discussion here about the necessity of cold-chain requirements of pharmacies either centrally or locally, [or] the requirements of additional nurses or health care staff to administer the medication. So you’re looking at a very resource-intensive therapy, which now is fairly restrictive [as to] who will have access to it.”
Dr. Angel reports serving on advisory boards for ViiV Healthcare and Gilead Sciences and has done contract research for ViiV Healthcare, Gilead, and Merck. Dr. Orkin has received research grants, fees as a consultant, travel sponsorship, and speaker fees from ViiV, Merck, and GlaxoSmithKline. Dr. Vasylyev reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
One year into offering the first long-acting injectable HIV treatment to his patients, Jonathan Angel, MD, head of the division of infectious diseases at the University of Ottawa, reported that 15 of the 21 of patients who started on the regimen are still taking it, all with viral suppression. Those who weren’t cited a combination of inconvenience, injection site pain, and “injection fatigue.”
These are just a few things HIV providers are learning as they begin what Chloe Orkin, MD, professor of HIV medicine at Queen Mary University of London, called a paradigm shift to long-acting treatment, which may soon include not just shots but rings, implants, and microarray patches.
“It’s a paradigm shift, and we are at the very beginning of this paradigm shift,” said Dr. Orkin, commenting during the discussion session of the European AIDS Clinical Society 2021 annual meeting. “We’re having to change our model, and it’s challenging.”
In the United States, the Food and Drug Administration approved the first long-acting injectable, a combination of cabotegravir and rilpivirine (CAB/RIL; Cabenuva, ViiV Healthcare) in January 2021. But it has been approved in Canada since March 2020 and available at Dr. Angel’s clinic since November 2020. It’s also available in Canada as an every-other-month shot. Injected into the buttocks, the shot was found to be noninferior to standard daily oral treatment in many studies, including the ATLAS, the ATLAS-2M – which tested the every-other-month approach – and FLAIR trials.
Dr. Angel’s clinic was part of all three of those trials, so his clinic has had 5 years’ experience preparing for the change in workflow and the new approach the shots require.
Of the 21 people Dr. Angel has treated, 11 were white Canadians, nine were Black African, and one was Indigenous Canadian, with women making up a third of the participants. Median age was 51 years, and all patients had had undetectable viral loads before beginning the regimen. (Studies of the drug’s effectiveness in people who struggle to take daily pills are still ongoing.)
Most of those 21 patients had had undetectable viral loads for more than 5 years, but a few had been undetectable for only 6 months before beginning the shots. Their immune systems were also healthy, with a median CD4 count of 618 cells/mcL. As in the clinical trials, none of the participants had experienced antiretroviral treatment failure. Because public health insurers in Canada have yet to approve the shots, Dr. Angel’s patients receiving Cabenuva also have private health insurance. Up to 90% of people in Canada receive pharmaceutical coverage through public insurance; therefore, the shot is not yet widely available.
Twenty patients switched from integrase-inhibitor regimens, and one had been receiving a nonnucleoside reverse transcriptase inhibitor–based regimen before starting Cabenuva.
And although the drug has not been approved for shot initiation this way, two patients requested – and Dr. Angel agreed – to start them on the shots without first doing a month of daily pills to check for safety.
“This is my conclusion from these data: the oral lead-in period is not necessary,” Dr. Angel said in his presentation at the meeting. “It can provide some comfort to either a physician or a patient, but it does not seem to be medically necessary.”
That approach is not without data to back it up. Research presented at HIV Glasgow 2020 showed that people who switched from daily oral dolutegravir/abacavir/lamivudine straight to the injections did so without problems.
At last clinic visit, 15 of those 21 were still receiving the shots. None have experienced treatment failure, and all were still virally suppressed. Four participants left the trials and one more person opted to return to daily pills, citing some level of what Dr. Angel called “injection fatigue.”
“Just as we use the term ‘pill fatigue’ for patients who are tired of taking pills, patients do get tired of coming in monthly for their visits and injections,” he said. They find the trip to the clinic for the intramuscular injections “inconvenient,” he said.
Unlike in the United States, where Cabenuva is approved for only monthly injections, Health Canada has already approved the shot for every-other-month injections, which Dr. Angel said may reduce the odds of injection fatigue.
Dr. Angel’s presentation drew comments, questions, and excitement from the crowd. Annemarie Wensing, MD, assistant professor of medicine at University Medical Center Utrecht (the Netherlands), asked whether dispensing with the oral lead-in period could mean that these shots could be useful for people going on longer trips, people having surgeries where they can’t swallow pills, or in other scenarios.
“These are not hypothetical conversations,” Dr. Angel said. “I’m having these conversations with patients now – temporary use, they travel for 3 months and come back, can they go from injectable to oral to injectable.”
For now, he said, the answer is, “We’ll figure it out.”
Meanwhile, there’s another big question when it comes to injectables, said Marta Vas ylyev, MD, from Lviv (Ukraine) Regional AIDS Center: When will they be available to the people who might benefit most from them – people in resource-limited settings, people who so far have struggled to remember to take their pills every day?
For now, Dr. Angel replied, injectables continue to be a treatment only for those who are already doing well while receiving HIV treatment: those with already suppressed viral load, who are good at taking daily pills, and who are being treated at well-resourced clinics.
“There are huge obstacles to overcome if this is ever to be available [in resource-limited settings], and way more obstacles than there are with any oral therapies,” he said. “There’s not been much discussion here about the necessity of cold-chain requirements of pharmacies either centrally or locally, [or] the requirements of additional nurses or health care staff to administer the medication. So you’re looking at a very resource-intensive therapy, which now is fairly restrictive [as to] who will have access to it.”
Dr. Angel reports serving on advisory boards for ViiV Healthcare and Gilead Sciences and has done contract research for ViiV Healthcare, Gilead, and Merck. Dr. Orkin has received research grants, fees as a consultant, travel sponsorship, and speaker fees from ViiV, Merck, and GlaxoSmithKline. Dr. Vasylyev reported no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Multiple DMTs linked to alopecia, especially in women
a new study finds.
From 2009 to 2019, the Food and Drug Administration received 7,978 reports of new-onset alopecia in patients taking DMTs, particularly teriflunomide (3,255, 40.8%; 90% female), dimethyl fumarate (1,641, 20.6%; 89% female), natalizumab (955, 12.0%; 92% female), and fingolimod (776, 9.7% of the total reports; 93% female), several researchers reported at the 2021 Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC). Of these, only teriflunomide had previously been linked to alopecia, study coauthor Ahmed Obeidat, MD, PhD, a neurologist at the Medical College of Wisconsin, Milwaukee, said in an interview.
“Our finding of frequent reports of alopecia on DMTs studied calls for further investigation into the subject,” Dr. Obeidat said. “Alopecia can cause deep personal impacts and can be a source of significant psychological concern for some patients.”
According to Dr. Obeidat, alopecia has been linked to the only a few DMTs – cladribine and the interferons – in addition to teriflunomide. “To our surprise, we received anecdotal reports of hair thinning from several of our MS patients treated with various other [DMTs]. Upon further investigation, we could not find substantial literature to explain this phenomenon which led us to conduct our investigation.”
Dr. Obeidat and colleagues identified DMT-related alopecia cases (18.3%) among 43,655 reports in the skin and subcutaneous tissue disorder category in the FDA Adverse Event Reporting System. Other DMTs with more than 1 case report were interferon beta-1a (635, 8.0%; 92% female), glatiramer acetate (332, 4.2%; 87% female), ocrelizumab (142, 1.8%; 94% female), interferon beta-1b (126, 1.6%; 95% female), alemtuzumab (86, 1.1%; 88% female), cladribine (17, 0.2%; 65% female), and rituximab (10, 0.1%; 90% female).
The average age for the case reports varied from 42 to 51 years for most of the drugs except alemtuzumab (mean age, 40 years) and cladribine (average age, 38 years), which had low numbers of cases.
Siponimod (three cases) and ozanimod (no cases) were not included in the age and gender analyses.
Why do so many women seem to be affected, well beyond their percentage of MS cases overall? The answer is unclear, said medical student Mokshal H. Porwal, the study’s lead author. “There could be a biological explanation,” Mr. Porwal said, “or women may report cases more often: “Earlier studies suggested that alopecia may affect women more adversely in terms of body image as well as overall psychological well-being, compared to males.”
The researchers also noted that patients – not medical professionals – provided most of the case reports in the FDA database. “We believe this indicates that alopecia is a patient-centered concern that may have a larger impact on their lives than what the health care teams may perceive,” Mr. Porwal said. “Oftentimes, we as health care providers, look for the more acute and apparent adverse events, which can overshadow issues such as hair thinning/alopecia that could have even greater psychological impacts on our patients.”
Dr. Obeidat said there are still multiple mysteries about DMT and alopecia risk: the true incidence of cases per DMT or DMT class, the mechanism(s) behind a link, the permanent or transient nature of the alopecia cases, and the risk factors in individual patients.
Going forward, he said, “we advise clinicians to discuss hair thinning or alopecia as a possible side effect that has been reported in association with all DMTs in the real-world, postmarketing era.”
No study funding was reported. Dr. Obeidat reported various disclosures; the other authors reported no disclosures.
a new study finds.
From 2009 to 2019, the Food and Drug Administration received 7,978 reports of new-onset alopecia in patients taking DMTs, particularly teriflunomide (3,255, 40.8%; 90% female), dimethyl fumarate (1,641, 20.6%; 89% female), natalizumab (955, 12.0%; 92% female), and fingolimod (776, 9.7% of the total reports; 93% female), several researchers reported at the 2021 Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC). Of these, only teriflunomide had previously been linked to alopecia, study coauthor Ahmed Obeidat, MD, PhD, a neurologist at the Medical College of Wisconsin, Milwaukee, said in an interview.
“Our finding of frequent reports of alopecia on DMTs studied calls for further investigation into the subject,” Dr. Obeidat said. “Alopecia can cause deep personal impacts and can be a source of significant psychological concern for some patients.”
According to Dr. Obeidat, alopecia has been linked to the only a few DMTs – cladribine and the interferons – in addition to teriflunomide. “To our surprise, we received anecdotal reports of hair thinning from several of our MS patients treated with various other [DMTs]. Upon further investigation, we could not find substantial literature to explain this phenomenon which led us to conduct our investigation.”
Dr. Obeidat and colleagues identified DMT-related alopecia cases (18.3%) among 43,655 reports in the skin and subcutaneous tissue disorder category in the FDA Adverse Event Reporting System. Other DMTs with more than 1 case report were interferon beta-1a (635, 8.0%; 92% female), glatiramer acetate (332, 4.2%; 87% female), ocrelizumab (142, 1.8%; 94% female), interferon beta-1b (126, 1.6%; 95% female), alemtuzumab (86, 1.1%; 88% female), cladribine (17, 0.2%; 65% female), and rituximab (10, 0.1%; 90% female).
The average age for the case reports varied from 42 to 51 years for most of the drugs except alemtuzumab (mean age, 40 years) and cladribine (average age, 38 years), which had low numbers of cases.
Siponimod (three cases) and ozanimod (no cases) were not included in the age and gender analyses.
Why do so many women seem to be affected, well beyond their percentage of MS cases overall? The answer is unclear, said medical student Mokshal H. Porwal, the study’s lead author. “There could be a biological explanation,” Mr. Porwal said, “or women may report cases more often: “Earlier studies suggested that alopecia may affect women more adversely in terms of body image as well as overall psychological well-being, compared to males.”
The researchers also noted that patients – not medical professionals – provided most of the case reports in the FDA database. “We believe this indicates that alopecia is a patient-centered concern that may have a larger impact on their lives than what the health care teams may perceive,” Mr. Porwal said. “Oftentimes, we as health care providers, look for the more acute and apparent adverse events, which can overshadow issues such as hair thinning/alopecia that could have even greater psychological impacts on our patients.”
Dr. Obeidat said there are still multiple mysteries about DMT and alopecia risk: the true incidence of cases per DMT or DMT class, the mechanism(s) behind a link, the permanent or transient nature of the alopecia cases, and the risk factors in individual patients.
Going forward, he said, “we advise clinicians to discuss hair thinning or alopecia as a possible side effect that has been reported in association with all DMTs in the real-world, postmarketing era.”
No study funding was reported. Dr. Obeidat reported various disclosures; the other authors reported no disclosures.
a new study finds.
From 2009 to 2019, the Food and Drug Administration received 7,978 reports of new-onset alopecia in patients taking DMTs, particularly teriflunomide (3,255, 40.8%; 90% female), dimethyl fumarate (1,641, 20.6%; 89% female), natalizumab (955, 12.0%; 92% female), and fingolimod (776, 9.7% of the total reports; 93% female), several researchers reported at the 2021 Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC). Of these, only teriflunomide had previously been linked to alopecia, study coauthor Ahmed Obeidat, MD, PhD, a neurologist at the Medical College of Wisconsin, Milwaukee, said in an interview.
“Our finding of frequent reports of alopecia on DMTs studied calls for further investigation into the subject,” Dr. Obeidat said. “Alopecia can cause deep personal impacts and can be a source of significant psychological concern for some patients.”
According to Dr. Obeidat, alopecia has been linked to the only a few DMTs – cladribine and the interferons – in addition to teriflunomide. “To our surprise, we received anecdotal reports of hair thinning from several of our MS patients treated with various other [DMTs]. Upon further investigation, we could not find substantial literature to explain this phenomenon which led us to conduct our investigation.”
Dr. Obeidat and colleagues identified DMT-related alopecia cases (18.3%) among 43,655 reports in the skin and subcutaneous tissue disorder category in the FDA Adverse Event Reporting System. Other DMTs with more than 1 case report were interferon beta-1a (635, 8.0%; 92% female), glatiramer acetate (332, 4.2%; 87% female), ocrelizumab (142, 1.8%; 94% female), interferon beta-1b (126, 1.6%; 95% female), alemtuzumab (86, 1.1%; 88% female), cladribine (17, 0.2%; 65% female), and rituximab (10, 0.1%; 90% female).
The average age for the case reports varied from 42 to 51 years for most of the drugs except alemtuzumab (mean age, 40 years) and cladribine (average age, 38 years), which had low numbers of cases.
Siponimod (three cases) and ozanimod (no cases) were not included in the age and gender analyses.
Why do so many women seem to be affected, well beyond their percentage of MS cases overall? The answer is unclear, said medical student Mokshal H. Porwal, the study’s lead author. “There could be a biological explanation,” Mr. Porwal said, “or women may report cases more often: “Earlier studies suggested that alopecia may affect women more adversely in terms of body image as well as overall psychological well-being, compared to males.”
The researchers also noted that patients – not medical professionals – provided most of the case reports in the FDA database. “We believe this indicates that alopecia is a patient-centered concern that may have a larger impact on their lives than what the health care teams may perceive,” Mr. Porwal said. “Oftentimes, we as health care providers, look for the more acute and apparent adverse events, which can overshadow issues such as hair thinning/alopecia that could have even greater psychological impacts on our patients.”
Dr. Obeidat said there are still multiple mysteries about DMT and alopecia risk: the true incidence of cases per DMT or DMT class, the mechanism(s) behind a link, the permanent or transient nature of the alopecia cases, and the risk factors in individual patients.
Going forward, he said, “we advise clinicians to discuss hair thinning or alopecia as a possible side effect that has been reported in association with all DMTs in the real-world, postmarketing era.”
No study funding was reported. Dr. Obeidat reported various disclosures; the other authors reported no disclosures.
FROM CMSC 2021
Clinicians may overprescribe clarithromycin for H. pylori
Clinicians are prescribing clarithromycin at high rates for Helicobacter pylori infections, despite increasing resistance to this antibiotic, researchers say.
In an analysis of 1 million U.S. prescriptions for H. pylori infections, 80% contained clarithromycin, said Carol Rockett, PharmD, associate vice president of RedHill Biopharma in Raleigh, N.C.
Dr. Rockett presented the findings at the annual meeting of the American College of Gastroenterology.
“Multiple talks [at the meeting] have suggested that the use of clarithromycin in H. pylori is obsolete,” she told this news organization. “Clarithromycin is particularly ineffective in people with a genetic variant that causes rapid metabolism.”
According to the 2017 ACG clinical guideline for treating H. pylori, patients diagnosed with this infection should be asked about their previous antibiotic exposure prior to treatment.
Additionally, clinicians should prescribe clarithromycin triple therapy with a proton pump inhibitor (PPI) and amoxicillin or metronidazole as a first-line treatment only in “regions where H. pylori clarithromycin resistance is known to be less than 15%” and in patients with no previous history of macrolide exposure.
The guideline puts bismuth quadruple therapy, consisting of a PPI, bismuth, tetracycline, and a nitroimidazole, at the top of its list of six alternative first-line therapies. However, three of the six alternatives include clarithromycin.
ERADICATE Hp and ERADICATE Hp2
To understand how U.S. physicians are treating patients with H. pylori, Dr. Rockett’s colleagues analyzed data from two phase 3 clinical trials of RedHill’s RHB-105 (Talicia): ERADICATE Hp and ERADICATE Hp2.
RHB-105 is an all-in‐one combination of omeprazole (40 mg), amoxicillin (1,000 mg), and rifabutin (50 mg) that the Food and Drug Administration approved for treatment of H pylori in 2019.
The researchers followed 38 subjects from ERADICATE Hp who remained positive for H. pylori after the study’s completion. A total of 33 had received a placebo in that trial, while the other 5 had received RHB-105.
The researchers obtained data on 31 of these patients. The overall cure rate was 61.3%. Of the 31 patients, 27 received a regimen including clarithromycin. Their cure rate was 59.3%.
Turning to ERADICATE Hp2, the researchers obtained data on 94 patients whose H. pylori infections persisted after the trial. Of those, 67 had received an active comparator (amoxicillin 250 mg and omeprazole 10 mg) and 27 had received RHB-105.
The overall cure rate was 56.2%. For the 48 subjects who received therapies including clarithromycin, the cure rate was 60.4%. For the 22 subjects who received a bismuth-based quadruple regimen, the cure rate was 45.4%.
In another analysis, the researchers crunched 12 months of numbers from IQVIA PharMetrics Plus medical and prescription claim database of over 1 million prescriptions for H. pylori. They found that 80% of the prescriptions made by gastroenterologists were for regimens containing clarithromycin. That proportion increased to 84% for physician assistants and internists, 85% for nurse practitioners, 86% for family practitioners, and 89% for general practitioners.
Finally, the researchers also analyzed patients for CYP2C19 gene status. They tested 65 subjects who received RHB-105 in ERADICATE Hp and all 445 subjects in ERADICATE Hp2. They found that 58.5% in ERADICATE Hp and 48.6% in ERADICATE Hp2 were normal metabolizers.
In 20 normal metabolizers who received clarithromycin, the drug eradicated the infection in 16 (80%). Out of 11 rapid metabolizers, clarithromycin eradicated the bacterium in 2 (18.2%). The difference was statistically significant (P = .0017).
“With clarithromycin, you can see that the efficacy is reduced in those patients who are rapid metabolizers,” Dr. Rockett said. “We didn’t see that with rifabutin [one of the drugs in RHB-105].”
Jared Magee, DO, MPH, a gastroenterology fellow at the Walter Reed National Military Medical Center in Bethesda, Md., said in treating H. pylori infections, he checks the patients’ medical records to see what antibiotics they have received in the past and generally begins treatment with the bismuth quadruple therapy.
“There is education needed to get the data out there that clarithromycin-based therapies may not be the right choice for patients,” he said. “There is a subset who will do well with it, but I think where we’re at now, with the frequency of macrolide prescriptions for other conditions, that clarithromycin is going to be a difficult therapy for a lot of people.”
Clinicians who are not gastroenterologists may not be aware of the guideline promulgated by the ACG, he pointed out.
Dr. Rockett is an employee of RedHill Biopharma. Dr. Magee has disclosed no relevant financial relationships. The study was funded by RedHill Biopharma.
A version of this article first appeared on Medscape.com.
Clinicians are prescribing clarithromycin at high rates for Helicobacter pylori infections, despite increasing resistance to this antibiotic, researchers say.
In an analysis of 1 million U.S. prescriptions for H. pylori infections, 80% contained clarithromycin, said Carol Rockett, PharmD, associate vice president of RedHill Biopharma in Raleigh, N.C.
Dr. Rockett presented the findings at the annual meeting of the American College of Gastroenterology.
“Multiple talks [at the meeting] have suggested that the use of clarithromycin in H. pylori is obsolete,” she told this news organization. “Clarithromycin is particularly ineffective in people with a genetic variant that causes rapid metabolism.”
According to the 2017 ACG clinical guideline for treating H. pylori, patients diagnosed with this infection should be asked about their previous antibiotic exposure prior to treatment.
Additionally, clinicians should prescribe clarithromycin triple therapy with a proton pump inhibitor (PPI) and amoxicillin or metronidazole as a first-line treatment only in “regions where H. pylori clarithromycin resistance is known to be less than 15%” and in patients with no previous history of macrolide exposure.
The guideline puts bismuth quadruple therapy, consisting of a PPI, bismuth, tetracycline, and a nitroimidazole, at the top of its list of six alternative first-line therapies. However, three of the six alternatives include clarithromycin.
ERADICATE Hp and ERADICATE Hp2
To understand how U.S. physicians are treating patients with H. pylori, Dr. Rockett’s colleagues analyzed data from two phase 3 clinical trials of RedHill’s RHB-105 (Talicia): ERADICATE Hp and ERADICATE Hp2.
RHB-105 is an all-in‐one combination of omeprazole (40 mg), amoxicillin (1,000 mg), and rifabutin (50 mg) that the Food and Drug Administration approved for treatment of H pylori in 2019.
The researchers followed 38 subjects from ERADICATE Hp who remained positive for H. pylori after the study’s completion. A total of 33 had received a placebo in that trial, while the other 5 had received RHB-105.
The researchers obtained data on 31 of these patients. The overall cure rate was 61.3%. Of the 31 patients, 27 received a regimen including clarithromycin. Their cure rate was 59.3%.
Turning to ERADICATE Hp2, the researchers obtained data on 94 patients whose H. pylori infections persisted after the trial. Of those, 67 had received an active comparator (amoxicillin 250 mg and omeprazole 10 mg) and 27 had received RHB-105.
The overall cure rate was 56.2%. For the 48 subjects who received therapies including clarithromycin, the cure rate was 60.4%. For the 22 subjects who received a bismuth-based quadruple regimen, the cure rate was 45.4%.
In another analysis, the researchers crunched 12 months of numbers from IQVIA PharMetrics Plus medical and prescription claim database of over 1 million prescriptions for H. pylori. They found that 80% of the prescriptions made by gastroenterologists were for regimens containing clarithromycin. That proportion increased to 84% for physician assistants and internists, 85% for nurse practitioners, 86% for family practitioners, and 89% for general practitioners.
Finally, the researchers also analyzed patients for CYP2C19 gene status. They tested 65 subjects who received RHB-105 in ERADICATE Hp and all 445 subjects in ERADICATE Hp2. They found that 58.5% in ERADICATE Hp and 48.6% in ERADICATE Hp2 were normal metabolizers.
In 20 normal metabolizers who received clarithromycin, the drug eradicated the infection in 16 (80%). Out of 11 rapid metabolizers, clarithromycin eradicated the bacterium in 2 (18.2%). The difference was statistically significant (P = .0017).
“With clarithromycin, you can see that the efficacy is reduced in those patients who are rapid metabolizers,” Dr. Rockett said. “We didn’t see that with rifabutin [one of the drugs in RHB-105].”
Jared Magee, DO, MPH, a gastroenterology fellow at the Walter Reed National Military Medical Center in Bethesda, Md., said in treating H. pylori infections, he checks the patients’ medical records to see what antibiotics they have received in the past and generally begins treatment with the bismuth quadruple therapy.
“There is education needed to get the data out there that clarithromycin-based therapies may not be the right choice for patients,” he said. “There is a subset who will do well with it, but I think where we’re at now, with the frequency of macrolide prescriptions for other conditions, that clarithromycin is going to be a difficult therapy for a lot of people.”
Clinicians who are not gastroenterologists may not be aware of the guideline promulgated by the ACG, he pointed out.
Dr. Rockett is an employee of RedHill Biopharma. Dr. Magee has disclosed no relevant financial relationships. The study was funded by RedHill Biopharma.
A version of this article first appeared on Medscape.com.
Clinicians are prescribing clarithromycin at high rates for Helicobacter pylori infections, despite increasing resistance to this antibiotic, researchers say.
In an analysis of 1 million U.S. prescriptions for H. pylori infections, 80% contained clarithromycin, said Carol Rockett, PharmD, associate vice president of RedHill Biopharma in Raleigh, N.C.
Dr. Rockett presented the findings at the annual meeting of the American College of Gastroenterology.
“Multiple talks [at the meeting] have suggested that the use of clarithromycin in H. pylori is obsolete,” she told this news organization. “Clarithromycin is particularly ineffective in people with a genetic variant that causes rapid metabolism.”
According to the 2017 ACG clinical guideline for treating H. pylori, patients diagnosed with this infection should be asked about their previous antibiotic exposure prior to treatment.
Additionally, clinicians should prescribe clarithromycin triple therapy with a proton pump inhibitor (PPI) and amoxicillin or metronidazole as a first-line treatment only in “regions where H. pylori clarithromycin resistance is known to be less than 15%” and in patients with no previous history of macrolide exposure.
The guideline puts bismuth quadruple therapy, consisting of a PPI, bismuth, tetracycline, and a nitroimidazole, at the top of its list of six alternative first-line therapies. However, three of the six alternatives include clarithromycin.
ERADICATE Hp and ERADICATE Hp2
To understand how U.S. physicians are treating patients with H. pylori, Dr. Rockett’s colleagues analyzed data from two phase 3 clinical trials of RedHill’s RHB-105 (Talicia): ERADICATE Hp and ERADICATE Hp2.
RHB-105 is an all-in‐one combination of omeprazole (40 mg), amoxicillin (1,000 mg), and rifabutin (50 mg) that the Food and Drug Administration approved for treatment of H pylori in 2019.
The researchers followed 38 subjects from ERADICATE Hp who remained positive for H. pylori after the study’s completion. A total of 33 had received a placebo in that trial, while the other 5 had received RHB-105.
The researchers obtained data on 31 of these patients. The overall cure rate was 61.3%. Of the 31 patients, 27 received a regimen including clarithromycin. Their cure rate was 59.3%.
Turning to ERADICATE Hp2, the researchers obtained data on 94 patients whose H. pylori infections persisted after the trial. Of those, 67 had received an active comparator (amoxicillin 250 mg and omeprazole 10 mg) and 27 had received RHB-105.
The overall cure rate was 56.2%. For the 48 subjects who received therapies including clarithromycin, the cure rate was 60.4%. For the 22 subjects who received a bismuth-based quadruple regimen, the cure rate was 45.4%.
In another analysis, the researchers crunched 12 months of numbers from IQVIA PharMetrics Plus medical and prescription claim database of over 1 million prescriptions for H. pylori. They found that 80% of the prescriptions made by gastroenterologists were for regimens containing clarithromycin. That proportion increased to 84% for physician assistants and internists, 85% for nurse practitioners, 86% for family practitioners, and 89% for general practitioners.
Finally, the researchers also analyzed patients for CYP2C19 gene status. They tested 65 subjects who received RHB-105 in ERADICATE Hp and all 445 subjects in ERADICATE Hp2. They found that 58.5% in ERADICATE Hp and 48.6% in ERADICATE Hp2 were normal metabolizers.
In 20 normal metabolizers who received clarithromycin, the drug eradicated the infection in 16 (80%). Out of 11 rapid metabolizers, clarithromycin eradicated the bacterium in 2 (18.2%). The difference was statistically significant (P = .0017).
“With clarithromycin, you can see that the efficacy is reduced in those patients who are rapid metabolizers,” Dr. Rockett said. “We didn’t see that with rifabutin [one of the drugs in RHB-105].”
Jared Magee, DO, MPH, a gastroenterology fellow at the Walter Reed National Military Medical Center in Bethesda, Md., said in treating H. pylori infections, he checks the patients’ medical records to see what antibiotics they have received in the past and generally begins treatment with the bismuth quadruple therapy.
“There is education needed to get the data out there that clarithromycin-based therapies may not be the right choice for patients,” he said. “There is a subset who will do well with it, but I think where we’re at now, with the frequency of macrolide prescriptions for other conditions, that clarithromycin is going to be a difficult therapy for a lot of people.”
Clinicians who are not gastroenterologists may not be aware of the guideline promulgated by the ACG, he pointed out.
Dr. Rockett is an employee of RedHill Biopharma. Dr. Magee has disclosed no relevant financial relationships. The study was funded by RedHill Biopharma.
A version of this article first appeared on Medscape.com.
Certain DMTs in MS linked to more psoriasis
, a new study finds. However, overall rates of reported disease are very low, and there’s no confirmation of a connection.
“People with MS and comorbid psoriasis – or those at a known high-risk for developing psoriasis – may benefit from a careful consideration of disease-modifying therapy (DMT), specifically when B cell-depleting therapies are considered,” study coauthor and Medical College of Wisconsin neurologist Ahmed Obeidat, MD, PhD, said in an interview. The findings were presented at the 2021 Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC).
Dr. Obeidat and colleagues launched the study after noticing cases of psoriasis that developed months to years after patients started taking ocrelizumab, a B cell-depleting therapy. “We referred to the published literature and only found very scant reports of MS, psoriasis, and B cell-depleting therapy use,” he said. “Thus we decided to pursue an investigation of a large [Food and Drug Administration] database to examine for possible out-of-proportion reports for psoriasis in patients with MS who were receiving B cell-depleting therapy.”
The researchers tracked case reports of psoriasis in patients with MS on DMTs from 2009 to 2020 via the FDA Adverse Event Reporting System. They found 517 psoriasis reports among 45,547 reports of skin/cutaneous conditions. The reports were linked to interferon beta 1a (136 reports, 26% of total), natalizumab (107, 21%), fingolimod (75, 15%), dimethyl fumarate (64, 12%), ocrelizumab (49, 10%), teriflunomide (28, 5%), interferon beta 1b (22, 4%), glatiramer acetate (12, 2%), rituximab (10, 2%), and alemtuzumab (9, 2%).
The total numbers of cases is low, but this may reflect underreporting due to the assumption that “autoimmunity begets autoimmunity” and therefore cases of psoriasis in MS are not alarming, medical student Mokshal H. Porwal, the study lead author, said in an interview.
The average age of patients – 48-51 – was similar for all of the drugs except alemtuzumab (mean age 41), which had a very small number of cases. The percentage of cases in females was 71%-77% for most of the drugs, with a few exceptions: rituximab (60%), ocrelizumab (63%), and alemtuzumab (33%).
Other drugs – cladribine, siponimod, and ozanimod – had 1, 1, and 0 reports, respectively, and were not included in the age and gender analyses.
The researchers also found that psoriasis made up about 65% of all skin/cutaneous adverse reports for rituximab, the highest number among DMTs. By comparison, that number was about 30% for ocrelizumab and under 1% for dimethyl fumarate and alemtuzumab.
Links between psoriasis and MS are murky, Dr. Obeidat said. “Some studies consider the presence of psoriasis as a possible indicator of increased future risk for MS, but there’s no clear association between the two conditions,” he said.
As for DMTs, “a few case reports of psoriasis in association with interferon-beta and rare case reports in association with ocrelizumab therapy have been published. However, the possible association between certain DMTs and psoriasis remains unclear,” he said.
Going forward, “we advise that patients with psoriasis on B cell-depleting agents are monitored more closely,” Dr. Obeidat said. “If the psoriasis worsens, it may be beneficial to think about potential alternative therapies.”
No study funding is reported. Dr. Obeidat reports various disclosures; the other authors report no disclosures.
, a new study finds. However, overall rates of reported disease are very low, and there’s no confirmation of a connection.
“People with MS and comorbid psoriasis – or those at a known high-risk for developing psoriasis – may benefit from a careful consideration of disease-modifying therapy (DMT), specifically when B cell-depleting therapies are considered,” study coauthor and Medical College of Wisconsin neurologist Ahmed Obeidat, MD, PhD, said in an interview. The findings were presented at the 2021 Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC).
Dr. Obeidat and colleagues launched the study after noticing cases of psoriasis that developed months to years after patients started taking ocrelizumab, a B cell-depleting therapy. “We referred to the published literature and only found very scant reports of MS, psoriasis, and B cell-depleting therapy use,” he said. “Thus we decided to pursue an investigation of a large [Food and Drug Administration] database to examine for possible out-of-proportion reports for psoriasis in patients with MS who were receiving B cell-depleting therapy.”
The researchers tracked case reports of psoriasis in patients with MS on DMTs from 2009 to 2020 via the FDA Adverse Event Reporting System. They found 517 psoriasis reports among 45,547 reports of skin/cutaneous conditions. The reports were linked to interferon beta 1a (136 reports, 26% of total), natalizumab (107, 21%), fingolimod (75, 15%), dimethyl fumarate (64, 12%), ocrelizumab (49, 10%), teriflunomide (28, 5%), interferon beta 1b (22, 4%), glatiramer acetate (12, 2%), rituximab (10, 2%), and alemtuzumab (9, 2%).
The total numbers of cases is low, but this may reflect underreporting due to the assumption that “autoimmunity begets autoimmunity” and therefore cases of psoriasis in MS are not alarming, medical student Mokshal H. Porwal, the study lead author, said in an interview.
The average age of patients – 48-51 – was similar for all of the drugs except alemtuzumab (mean age 41), which had a very small number of cases. The percentage of cases in females was 71%-77% for most of the drugs, with a few exceptions: rituximab (60%), ocrelizumab (63%), and alemtuzumab (33%).
Other drugs – cladribine, siponimod, and ozanimod – had 1, 1, and 0 reports, respectively, and were not included in the age and gender analyses.
The researchers also found that psoriasis made up about 65% of all skin/cutaneous adverse reports for rituximab, the highest number among DMTs. By comparison, that number was about 30% for ocrelizumab and under 1% for dimethyl fumarate and alemtuzumab.
Links between psoriasis and MS are murky, Dr. Obeidat said. “Some studies consider the presence of psoriasis as a possible indicator of increased future risk for MS, but there’s no clear association between the two conditions,” he said.
As for DMTs, “a few case reports of psoriasis in association with interferon-beta and rare case reports in association with ocrelizumab therapy have been published. However, the possible association between certain DMTs and psoriasis remains unclear,” he said.
Going forward, “we advise that patients with psoriasis on B cell-depleting agents are monitored more closely,” Dr. Obeidat said. “If the psoriasis worsens, it may be beneficial to think about potential alternative therapies.”
No study funding is reported. Dr. Obeidat reports various disclosures; the other authors report no disclosures.
, a new study finds. However, overall rates of reported disease are very low, and there’s no confirmation of a connection.
“People with MS and comorbid psoriasis – or those at a known high-risk for developing psoriasis – may benefit from a careful consideration of disease-modifying therapy (DMT), specifically when B cell-depleting therapies are considered,” study coauthor and Medical College of Wisconsin neurologist Ahmed Obeidat, MD, PhD, said in an interview. The findings were presented at the 2021 Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC).
Dr. Obeidat and colleagues launched the study after noticing cases of psoriasis that developed months to years after patients started taking ocrelizumab, a B cell-depleting therapy. “We referred to the published literature and only found very scant reports of MS, psoriasis, and B cell-depleting therapy use,” he said. “Thus we decided to pursue an investigation of a large [Food and Drug Administration] database to examine for possible out-of-proportion reports for psoriasis in patients with MS who were receiving B cell-depleting therapy.”
The researchers tracked case reports of psoriasis in patients with MS on DMTs from 2009 to 2020 via the FDA Adverse Event Reporting System. They found 517 psoriasis reports among 45,547 reports of skin/cutaneous conditions. The reports were linked to interferon beta 1a (136 reports, 26% of total), natalizumab (107, 21%), fingolimod (75, 15%), dimethyl fumarate (64, 12%), ocrelizumab (49, 10%), teriflunomide (28, 5%), interferon beta 1b (22, 4%), glatiramer acetate (12, 2%), rituximab (10, 2%), and alemtuzumab (9, 2%).
The total numbers of cases is low, but this may reflect underreporting due to the assumption that “autoimmunity begets autoimmunity” and therefore cases of psoriasis in MS are not alarming, medical student Mokshal H. Porwal, the study lead author, said in an interview.
The average age of patients – 48-51 – was similar for all of the drugs except alemtuzumab (mean age 41), which had a very small number of cases. The percentage of cases in females was 71%-77% for most of the drugs, with a few exceptions: rituximab (60%), ocrelizumab (63%), and alemtuzumab (33%).
Other drugs – cladribine, siponimod, and ozanimod – had 1, 1, and 0 reports, respectively, and were not included in the age and gender analyses.
The researchers also found that psoriasis made up about 65% of all skin/cutaneous adverse reports for rituximab, the highest number among DMTs. By comparison, that number was about 30% for ocrelizumab and under 1% for dimethyl fumarate and alemtuzumab.
Links between psoriasis and MS are murky, Dr. Obeidat said. “Some studies consider the presence of psoriasis as a possible indicator of increased future risk for MS, but there’s no clear association between the two conditions,” he said.
As for DMTs, “a few case reports of psoriasis in association with interferon-beta and rare case reports in association with ocrelizumab therapy have been published. However, the possible association between certain DMTs and psoriasis remains unclear,” he said.
Going forward, “we advise that patients with psoriasis on B cell-depleting agents are monitored more closely,” Dr. Obeidat said. “If the psoriasis worsens, it may be beneficial to think about potential alternative therapies.”
No study funding is reported. Dr. Obeidat reports various disclosures; the other authors report no disclosures.
FROM CMSC 2021
Upadacitinib shows potential for ulcerative colitis
LAS VEGAS – An oral Janus kinase 1 inhibitor upadacitinib (Rinvoq, AbbVie) showed high efficacy and good safety as a treatment for ulcerative colitis in a phase 3 trial.
The finding could provide some reassurance after the Food and Drug Administration recently warned of an increased risk of cancer and heart disease associated with medications in the same class as upadacitinib.
“Serious adverse events were numerically lower in patients on upadacitinib, and discontinuations from the study due to adverse events were also lower” than in patients taking a placebo, said Edward Loftus, MD, a gastroenterologist at the Mayo Clinic in Rochester, Minn.
Dr. Loftus presented the findings from the U-ACCOMPLISH study at the annual meeting of the American College of Gastroenterology.
Although other medications are approved for the treatment of ulcerative colitis, including biologics, many patients do not respond. In 2019, tofacitinib (Xeljanz) became the first JAK inhibitor approved for this condition. It works by blocking the JAK1 and JAK3 inflammation pathways, and at high concentrations, it also blocks the tyrosine kinase 2 and JAK2 pathways.
However, adverse events seen in clinical trials of tofacitinib include pneumonia, herpes zoster, anal abscess, and Clostridioides difficile infections. And, as reported by this news organization in September, the FDA required its manufacturer, Pfizer, to add a boxed warning that includes information about the risks of stroke, cancer, blood clots, and death.
Upadacitinib may be more selective and reversible because it preferentially blocks JAK1 or JAK1/3. In August 2019, it received FDA approval at a dose of 15 mg for adult patients with moderately to severely active RA who have had an inadequate response or intolerance to methotrexate.
But the FDA applied the same warnings to upadacitinib – and to a third related drug, baricitinib (Olumiant) – that it required for tofacitinib, even though they are not as well studied.
The FDA also limited approved uses of these three medications to patients who have not responded well to tumor necrosis factor blockers to ensure their benefits outweigh their risks.
A well-tolerated treatment
U-ACCOMPLISH is one of two phase 3 trials induction trials completed on upadacitinib.
Investigators randomized 522 people with moderately to severely active ulcerative colitis, defined as Adapted Mayo Score 5-9 with a centrally read endoscopic score of 2-3. Of those patients, the intent to treat population included 341 in the upadacitinib group (45 mg once daily) and 174 in the placebo group.
The baseline demographics and disease characteristics were similar between groups. More than two-thirds of patients in both groups were White, and more than two-thirds were men. In the upadacitinib group, 50.7% had responded inadequately to biologic treatments, compared with 51.1% in the placebo group.
After 8 weeks, a significantly higher proportion of patients receiving upadacitinib achieved clinical remission as defined by the adapted Mayo Score (stool frequency subscore ≤1 and not greater than baseline, rectal bleeding subscore of 0, and Mayo endoscopic subscore ≤1).
“In terms of the efficacy, I think it’s very, very promising,” said Derrick Eichele, MD, an assistant professor of gastroenterology-hepatology at the University of Nebraska Medical Center in Omaha, who was not involved in the trial.
The efficacy data were similar to those reported for tofacitinib in clinical trials, he said in an interview. “But I think again, what we’re waiting to see is how is this going to be positioned in relation to tofacitinib in terms of safety profile.”
More patients in the upadacitinib group reported adverse events, including those deemed related to the drug. However, the proportion that were severe, serious, or led to discontinuation was higher in the placebo group. No one in the study died, and no one in the upadacitinib group had an adjudicated major adverse cardiovascular event, tuberculosis, or malignancy.
The most common adverse events were acne, blood creatine phosphokinase elevation, and anemia, which were all more common in the upadacitinib group, and headache and worsening of ulcerative colitis, which were more common in the placebo group.
Among adverse events of special interest, anemia, neutropenia, hepatic disorder, lymphopenia, serious infection, and opportunistic infection were more common in the upadacitinib group than in the placebo group. The four opportunistic infections in the upadacitinib group included two cases of herpes zoster.
In reviewing the poster presented at this meeting, the cases of neutropenia and hepatic disorder in the upadacitinib group stood out for Dr. Eichele. But he said it’s hard to pass judgment based on this amount of data. He is looking forward to a peer-reviewed publication. “I’ll be interested to see what it shows in terms of the details.”
Phase 3 trials of upadacitinib are underway in atopic dermatitis, RA, psoriatic arthritis, axial spondyloarthritis, Crohn’s disease, giant cell arteritis, and Takayasu arteritis as well as ulcerative colitis.
In a 52-week maintenance trial, according to a press release, malignancies (excluding nonmelanoma skin cancer) included one event among 148 people taking a 15-mg dose of upadacitinib 15, two events among 154 people taking a 30-mg dose of upadacitinib, and one event among 149 people in the placebo group.
Two cases of pulmonary embolism were reported in the 15-mg group and two cases of deep vein thrombosis were reported in the 30-mg group, compared with one event of ovarian vein thrombosis in the placebo group. One adjudicated major cardiovascular event each were reported in the upadacitinib 30-mg group and the placebo group. No one died.
The study was funded by AbbVie. Dr. Loftus reported that he is a consultant for AbbVie as well as multiple other gastroenterology drug companies. Dr. Eichele disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – An oral Janus kinase 1 inhibitor upadacitinib (Rinvoq, AbbVie) showed high efficacy and good safety as a treatment for ulcerative colitis in a phase 3 trial.
The finding could provide some reassurance after the Food and Drug Administration recently warned of an increased risk of cancer and heart disease associated with medications in the same class as upadacitinib.
“Serious adverse events were numerically lower in patients on upadacitinib, and discontinuations from the study due to adverse events were also lower” than in patients taking a placebo, said Edward Loftus, MD, a gastroenterologist at the Mayo Clinic in Rochester, Minn.
Dr. Loftus presented the findings from the U-ACCOMPLISH study at the annual meeting of the American College of Gastroenterology.
Although other medications are approved for the treatment of ulcerative colitis, including biologics, many patients do not respond. In 2019, tofacitinib (Xeljanz) became the first JAK inhibitor approved for this condition. It works by blocking the JAK1 and JAK3 inflammation pathways, and at high concentrations, it also blocks the tyrosine kinase 2 and JAK2 pathways.
However, adverse events seen in clinical trials of tofacitinib include pneumonia, herpes zoster, anal abscess, and Clostridioides difficile infections. And, as reported by this news organization in September, the FDA required its manufacturer, Pfizer, to add a boxed warning that includes information about the risks of stroke, cancer, blood clots, and death.
Upadacitinib may be more selective and reversible because it preferentially blocks JAK1 or JAK1/3. In August 2019, it received FDA approval at a dose of 15 mg for adult patients with moderately to severely active RA who have had an inadequate response or intolerance to methotrexate.
But the FDA applied the same warnings to upadacitinib – and to a third related drug, baricitinib (Olumiant) – that it required for tofacitinib, even though they are not as well studied.
The FDA also limited approved uses of these three medications to patients who have not responded well to tumor necrosis factor blockers to ensure their benefits outweigh their risks.
A well-tolerated treatment
U-ACCOMPLISH is one of two phase 3 trials induction trials completed on upadacitinib.
Investigators randomized 522 people with moderately to severely active ulcerative colitis, defined as Adapted Mayo Score 5-9 with a centrally read endoscopic score of 2-3. Of those patients, the intent to treat population included 341 in the upadacitinib group (45 mg once daily) and 174 in the placebo group.
The baseline demographics and disease characteristics were similar between groups. More than two-thirds of patients in both groups were White, and more than two-thirds were men. In the upadacitinib group, 50.7% had responded inadequately to biologic treatments, compared with 51.1% in the placebo group.
After 8 weeks, a significantly higher proportion of patients receiving upadacitinib achieved clinical remission as defined by the adapted Mayo Score (stool frequency subscore ≤1 and not greater than baseline, rectal bleeding subscore of 0, and Mayo endoscopic subscore ≤1).
“In terms of the efficacy, I think it’s very, very promising,” said Derrick Eichele, MD, an assistant professor of gastroenterology-hepatology at the University of Nebraska Medical Center in Omaha, who was not involved in the trial.
The efficacy data were similar to those reported for tofacitinib in clinical trials, he said in an interview. “But I think again, what we’re waiting to see is how is this going to be positioned in relation to tofacitinib in terms of safety profile.”
More patients in the upadacitinib group reported adverse events, including those deemed related to the drug. However, the proportion that were severe, serious, or led to discontinuation was higher in the placebo group. No one in the study died, and no one in the upadacitinib group had an adjudicated major adverse cardiovascular event, tuberculosis, or malignancy.
The most common adverse events were acne, blood creatine phosphokinase elevation, and anemia, which were all more common in the upadacitinib group, and headache and worsening of ulcerative colitis, which were more common in the placebo group.
Among adverse events of special interest, anemia, neutropenia, hepatic disorder, lymphopenia, serious infection, and opportunistic infection were more common in the upadacitinib group than in the placebo group. The four opportunistic infections in the upadacitinib group included two cases of herpes zoster.
In reviewing the poster presented at this meeting, the cases of neutropenia and hepatic disorder in the upadacitinib group stood out for Dr. Eichele. But he said it’s hard to pass judgment based on this amount of data. He is looking forward to a peer-reviewed publication. “I’ll be interested to see what it shows in terms of the details.”
Phase 3 trials of upadacitinib are underway in atopic dermatitis, RA, psoriatic arthritis, axial spondyloarthritis, Crohn’s disease, giant cell arteritis, and Takayasu arteritis as well as ulcerative colitis.
In a 52-week maintenance trial, according to a press release, malignancies (excluding nonmelanoma skin cancer) included one event among 148 people taking a 15-mg dose of upadacitinib 15, two events among 154 people taking a 30-mg dose of upadacitinib, and one event among 149 people in the placebo group.
Two cases of pulmonary embolism were reported in the 15-mg group and two cases of deep vein thrombosis were reported in the 30-mg group, compared with one event of ovarian vein thrombosis in the placebo group. One adjudicated major cardiovascular event each were reported in the upadacitinib 30-mg group and the placebo group. No one died.
The study was funded by AbbVie. Dr. Loftus reported that he is a consultant for AbbVie as well as multiple other gastroenterology drug companies. Dr. Eichele disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
LAS VEGAS – An oral Janus kinase 1 inhibitor upadacitinib (Rinvoq, AbbVie) showed high efficacy and good safety as a treatment for ulcerative colitis in a phase 3 trial.
The finding could provide some reassurance after the Food and Drug Administration recently warned of an increased risk of cancer and heart disease associated with medications in the same class as upadacitinib.
“Serious adverse events were numerically lower in patients on upadacitinib, and discontinuations from the study due to adverse events were also lower” than in patients taking a placebo, said Edward Loftus, MD, a gastroenterologist at the Mayo Clinic in Rochester, Minn.
Dr. Loftus presented the findings from the U-ACCOMPLISH study at the annual meeting of the American College of Gastroenterology.
Although other medications are approved for the treatment of ulcerative colitis, including biologics, many patients do not respond. In 2019, tofacitinib (Xeljanz) became the first JAK inhibitor approved for this condition. It works by blocking the JAK1 and JAK3 inflammation pathways, and at high concentrations, it also blocks the tyrosine kinase 2 and JAK2 pathways.
However, adverse events seen in clinical trials of tofacitinib include pneumonia, herpes zoster, anal abscess, and Clostridioides difficile infections. And, as reported by this news organization in September, the FDA required its manufacturer, Pfizer, to add a boxed warning that includes information about the risks of stroke, cancer, blood clots, and death.
Upadacitinib may be more selective and reversible because it preferentially blocks JAK1 or JAK1/3. In August 2019, it received FDA approval at a dose of 15 mg for adult patients with moderately to severely active RA who have had an inadequate response or intolerance to methotrexate.
But the FDA applied the same warnings to upadacitinib – and to a third related drug, baricitinib (Olumiant) – that it required for tofacitinib, even though they are not as well studied.
The FDA also limited approved uses of these three medications to patients who have not responded well to tumor necrosis factor blockers to ensure their benefits outweigh their risks.
A well-tolerated treatment
U-ACCOMPLISH is one of two phase 3 trials induction trials completed on upadacitinib.
Investigators randomized 522 people with moderately to severely active ulcerative colitis, defined as Adapted Mayo Score 5-9 with a centrally read endoscopic score of 2-3. Of those patients, the intent to treat population included 341 in the upadacitinib group (45 mg once daily) and 174 in the placebo group.
The baseline demographics and disease characteristics were similar between groups. More than two-thirds of patients in both groups were White, and more than two-thirds were men. In the upadacitinib group, 50.7% had responded inadequately to biologic treatments, compared with 51.1% in the placebo group.
After 8 weeks, a significantly higher proportion of patients receiving upadacitinib achieved clinical remission as defined by the adapted Mayo Score (stool frequency subscore ≤1 and not greater than baseline, rectal bleeding subscore of 0, and Mayo endoscopic subscore ≤1).
“In terms of the efficacy, I think it’s very, very promising,” said Derrick Eichele, MD, an assistant professor of gastroenterology-hepatology at the University of Nebraska Medical Center in Omaha, who was not involved in the trial.
The efficacy data were similar to those reported for tofacitinib in clinical trials, he said in an interview. “But I think again, what we’re waiting to see is how is this going to be positioned in relation to tofacitinib in terms of safety profile.”
More patients in the upadacitinib group reported adverse events, including those deemed related to the drug. However, the proportion that were severe, serious, or led to discontinuation was higher in the placebo group. No one in the study died, and no one in the upadacitinib group had an adjudicated major adverse cardiovascular event, tuberculosis, or malignancy.
The most common adverse events were acne, blood creatine phosphokinase elevation, and anemia, which were all more common in the upadacitinib group, and headache and worsening of ulcerative colitis, which were more common in the placebo group.
Among adverse events of special interest, anemia, neutropenia, hepatic disorder, lymphopenia, serious infection, and opportunistic infection were more common in the upadacitinib group than in the placebo group. The four opportunistic infections in the upadacitinib group included two cases of herpes zoster.
In reviewing the poster presented at this meeting, the cases of neutropenia and hepatic disorder in the upadacitinib group stood out for Dr. Eichele. But he said it’s hard to pass judgment based on this amount of data. He is looking forward to a peer-reviewed publication. “I’ll be interested to see what it shows in terms of the details.”
Phase 3 trials of upadacitinib are underway in atopic dermatitis, RA, psoriatic arthritis, axial spondyloarthritis, Crohn’s disease, giant cell arteritis, and Takayasu arteritis as well as ulcerative colitis.
In a 52-week maintenance trial, according to a press release, malignancies (excluding nonmelanoma skin cancer) included one event among 148 people taking a 15-mg dose of upadacitinib 15, two events among 154 people taking a 30-mg dose of upadacitinib, and one event among 149 people in the placebo group.
Two cases of pulmonary embolism were reported in the 15-mg group and two cases of deep vein thrombosis were reported in the 30-mg group, compared with one event of ovarian vein thrombosis in the placebo group. One adjudicated major cardiovascular event each were reported in the upadacitinib 30-mg group and the placebo group. No one died.
The study was funded by AbbVie. Dr. Loftus reported that he is a consultant for AbbVie as well as multiple other gastroenterology drug companies. Dr. Eichele disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
AT ACG 2021
Which agent is best for neuromyelitis optica?
The Alexion-sponsored study was presented at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) by Dean Wingerchuk, MD, of the Mayo Clinic in Scottsdale, Ariz.
Other experts in the field have highlighted limitations to the analysis and pointed out that all three agents are very effective in treating AQP4+ NMOSD, and many other considerations need to be taken into account as well as time to first relapse when selecting a therapy, leaving the door open for all three agents.
Dr. Wingerchuk explained that NMOSD is a rare severely disabling complement-mediated autoimmune neuroinflammatory disease of the central nervous system, characterized by devastating and unpredictable attacks (relapses) that can cause immediate and irreversible damage.
There are three recently approved monoclonal antibody treatment options in the United States for adults with AQP4+ NMOSD: eculizumab (Soliris, Alexion), inebilizumab (Uplizna, Horizon), and satralizumab (Enspryng, Genentech). A comparison of the relative treatment effects of these drugs would facilitate the treatment selection process, Dr. Wingerchuk said.
The objective of this study was to perform an indirect treatment comparison on the efficacy of these three FDA-approved treatment options for adults with AQP4+ NMOSD, in the absence of any head-to-head studies.
Using published data from randomized controlled trials, which were identified by a systematic literature review in September 2020, the researchers performed a Bayesian network meta-analysis to estimate the relative effects between eculizumab, inebilizumab, and satralizumab.
Network meta-analyses were performed for clinically relevant subpopulations based on three treatment networks: (1) patients who received monotherapy with one of the monoclonal antibodies or in combination with an immunosuppressant therapy; (2) patients who received monotherapy with the monoclonal antibody alone; and (3) patients who received a combination of both the monoclonal antibody and immunosuppressant therapy.
Time to first relapse was the primary efficacy outcome assessed. Relative treatment effects were expressed as hazard ratios and the probability that a treatment was the best at delaying time to first relapse was also evaluated.
In the systematic literature review, 29 publications from four unique clinical trials were identified and include in the network meta-analysis. These included publications from congress proceedings and peer-reviewed journals.
The four clinical trials were the N-MOmentum trial of inebilizumab versus placebo; the PREVENT trial of eculizumab with or without immunosuppressant therapy versus placebo with or without immunosuppressant therapy; the SAkuraSky trial of satralizumab plus immunosuppressant therapy versus placebo plus immunosuppressant therapy; and the SAkuraStar trial of satralizumab versus placebo.
Results showed that for the first analysis of mono or combination therapy, patients treated with eculizumab with or without immunosuppressant therapy were 76% less likely to experience a first relapse when compared with patients treated with satralizumab with or without immunosuppressant therapy.
In the monotherapy network, patients on eculizumab were 90% less likely to experience a first relapse when compared with patients treated with satralizumab, and patients on eculizumab were 89% less likely to experience a first relapse when compared with patients treated with inebilizumab.
In the third network analysis – a comparison of eculizumab plus immunosuppressant therapy with inebilizumab plus immunosuppressant therapy (Table 1) – the point estimate appeared to favor eculizumab but the confidence intervals were wide and not significant.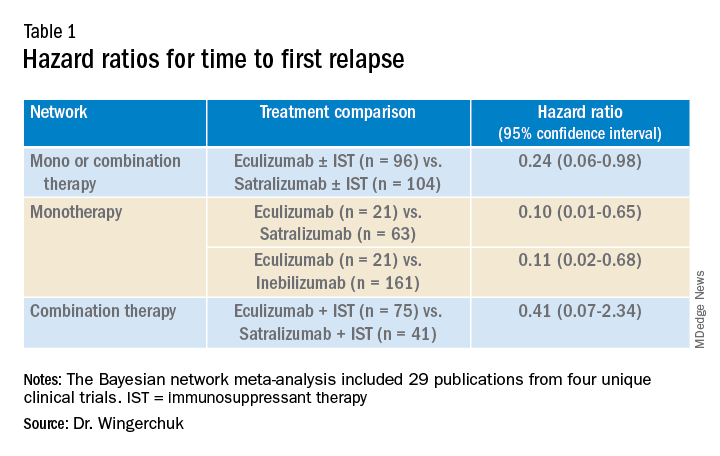
A subsequent analysis looked at the rank order of the best treatment option, with eculizumab coming out first in all three networks (Table 2).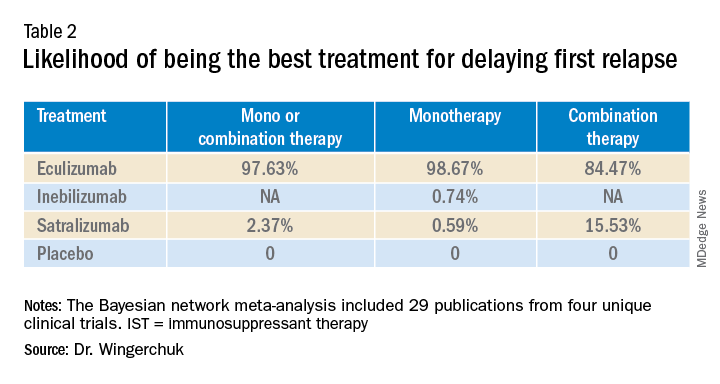
Dr. Wingerchuk acknowledged that there were many limitations to this study, including that analyses for annualized relapse rate, disability, and quality of life were not included because of a lack of consistent outcome reporting by AQP4+ status in the randomized trials.
Safety outcomes were excluded because of a lack of standardized baseline risks and inconsistent reporting by AQP4+ status across trials.
Because this study focused on drugs approved in the United States in a rare disease area, there were a limited number of studies with intervention effects.
There were differences in follow-up durations across the different trials, with N-MOmentum having a follow-up of 197 days compared with 144 weeks for other trials.
“In the absence of head-to-head trials, this network meta-analysis provides important evidence on the relative efficacy of eculizumab versus satralizumab or inebilizumab for the treatment of patients with AQP4+ NMOSD, with significant differences in two out of the three treatment comparison scenarios observed,” Dr. Wingerchuk concluded.
“Based on current evidence, monotherapy and mono-combination therapy with eculizumab appear to more efficacious at preventing relapses than satralizumab or inebilizumab for the treatment of adults with AQP4+ NMOSD. These findings appear to suggest that C5 complement inhibition with treatments such as eculizumab appear to prevent relapses more effectively than other mechanisms involving IL-6 receptor or CD19 inhibition among adults with AQP4+ NMOSD,” he added.
Experts respond
Commenting on the study, several experts in the field provided some balancing views.
Bruce Cree, MD, University of California San Francisco, who was the chief investigator of the N-MOmentum study with inebilizumab, said he was skeptical about this new indirect comparison. “The results of this study seem too good to be true; a 90% difference between agents has to be an overestimate,” he said.
“We know from independent studies that all three drugs are very effective. If we take each trial separately, eculizumab reduced attack risk by 90% versus placebo; and the other two drugs by 77% to 78% versus placebo. For eculizumab to be 90% better than inebilizumab or satralizumab its basically like saying these drugs perform like placebo, but we know that is not the case,” Dr. Cree argued.
He pointed out that when comparing results across studies there are many factors that have to be considered, including the different patient populations included in the different studies, with the characteristic of each population in each trial being unique to that dataset.
In addition, Dr. Cree suggested that all the studies included in the comparison were relatively small for this type of analysis. “Normally this type of analysis is done with much larger studies, so the resulting database is closer to a representation of the disease state itself,” he said.
Dr. Cree also questioned the role of the sponsor in this meta-analysis. “The analysis was sponsored by Alexion and several coauthors were employees of Alexion. There was not much description available of how the statistics were done. I am concerned that the company was involved in the analysis, which could introduce bias. I look forward to seeing details of the statistical methodology,” he said.
“This is definitely a provocative study. They have thrown down the gauntlet. If they are so confident in the results they should now do a head-to-head study to back this result up. If they don’t do that, then I think physicians should ignore it as there are just too many problems with this analysis,” Dr. Cree stated.
Dr. Cree acknowledged that when looking at the four trials separately, eculizumab does look a little better than the other two agents in delaying time to first relapse. “But there are some caveats. Despite a larger reduction in relapse rate there was no reduction in disability in the eculizumab trial. Whereas the inebilizumab trial did show a reduction in disability. And while the PREVENT trial with eculizumab was a good study, during the course of the trial the definition of clinical relapse was changed, and as a consequence that increased the product’s performance – that’s a little bit curious,” he added.
How to choose?
On how to choose between the three agents, Dr. Cree said they are all “extraordinarily effective” at reducing relapse activity. “They are all ‘home run’ products, but they have differences in safety,” he said.
“Inebilizumab is linked to hypogammaglobulinemia over time – we haven’t seen an increase in infection risk linked to this, but with enough time, I would expect that there probably will be. But inebilizumab is a B-cell-depleting agent like the agents used in MS, and we now have a lot of experiences with this type of product, which gives us more confidence on the safety profile,” Dr. Cree noted.
“Eculizumab was linked to a risk of meningococcal meningitis and other bacterial infections, and satralizumab seems to [be] overall well tolerated with no obvious safety concerns to date, but the studies have been quite small,” he added.
On routes of administration and frequency of dosing, Dr. Cree pointed out that while all three drugs have an intensive loading schedule, for maintenance, eculizumab needs to be given as an IV infusion every 2 weeks. Inebilizumab needs just two infusions per year for maintenance, while satralizumab is given by subcutaneous injection once per month.
“It may be that eculizumab could be used at the time of an acute attack but then treatment could be switched to one of the other two for long-term maintenance,” he suggested.
But Dr. Cree pointed out that the biggest challenge for all three agents is access. “The costs are astronomically high ($200,000-$770,000). They are prohibitively expensive and very few insurance companies are covering them.”
Also commenting, Brian Weinshenker, MD, from the Mayo Clinic in Rochester, Minn., who was a member of the attack adjudication committee for both PREVENT and N-MOmentum studies, pointed out that as well as differences in the populations enrolled, and study designs, the studies with the three different drugs also had differences in attack adjudication criteria.
“These factors make it very difficult to compare across studies, which is what was done in this analysis, so I would be reluctant to reach many conclusions about differences.”
Dr. Weinshenker added: “All three treatments provided strong benefit. We are still learning about long-term benefits, but emerging data have suggested that all three seem to provide persistent benefits for the length of the open-label extension study. We don’t have much evidence about the severity of the attacks that did occur, although some limited data suggest that both eculizumab and inebilizumab reduce attack severity.”
Dennis Bourdette, MD, professor emeritus, department of neurology, Oregon Health & Science University, Portland, who was not involved in any of the studies, said he thought the new analysis was “a worthwhile effort to determine the relative effectiveness of the three different drugs in treating AQP4+ NMOSD.
“Given the rarity of APQ4+ NMOSD, it will be difficult to perform randomized head-to-head clinical trials of the agents, so this type of comparison is the best we can do at this time,” he said.
While Dr. Bourdette feels this study supports the notion that eculizumab is more effective at delaying time to first relapse than inebilizumab and satralizumab, he does not believe the results should have a major impact on decisions about which agent to use in clinical practice.
“A difference in delaying time to first relapse tells us little about the relative effectiveness of the long-term benefit of these [agents], particularly with regards to permanent disability or frequency of relapses. However, it is possible that the difference reflects the efficacy kinetics of the agents with eculizumab working faster than the other two agents, which would be useful in making a decision about a patient with very active NMOSD where one wants to get the disease under control as quickly as possible,” Dr. Bourdette noted.
But he added that neurologists should also consider safety profile, convenience, and contraindications. “Eculizumab is clearly less convenient in terms of dosing schedule than the other two agents, and patient convenience is important for long-term compliance.”
Dr. Bourdette pointed out that another consideration is prior treatment. “Many patients with NMOSD will receive the anti-CD20 monoclonal antibody, rituximab – which depletes B cells – off label. Inebilizumab also depletes B cells, so a patient who has had continued NMOSD disease activity on rituximab probably should not be treated with inebilizumab, making eculizumab or satralizumab preferable,” he suggested.
Finally, Dr. Bourdette highlighted the sponsorship of the current study by the manufacturer of eculizumab, Alexion, and that all of the authors have some financial relationship with Alexion as described in their disclosures. “Whether this resulted in any biases about the design, conduct, or interpretation of the study is uncertain but is always a concern,” he said.
Company statements
The companies selling inebilizumab and satralizumab sent statements on the new analysis and repeated many of the above points.
Genentech noted that new longer-term data presented at ECTRIMS show that satralizumab is effective in significantly reducing relapses over 4 years of treatment in people with AQP4+ NMOSD, with a favorable safety profile both as a monotherapy and in conjunction with immunosuppressive therapy. More than 70% of people treated with satralizumab remained relapse free after 4 years in the SAkuraStar (73%) and SAkuraSky (71%) open-label extension studies, and 90% and 91%, respectively, were free from severe relapse, the company reported.
Horizon said: “We are confident in the efficacy and safety of Uplizna (inebilizumab) – a convenient, twice-annual monotherapy – that was studied in the largest randomized, placebo-controlled, global trial of a monotherapy in NMOSD. The endpoints in this trial were prospectively defined and assessed by an adjudication committee as published in The Lancet, with long-term follow-up data now published in the Multiple Sclerosis Journal that further support the efficacy and safety.”
The current study was funded by Alexion–AstraZeneca Rare Disease. Dr. Wingerchuk has participated on data safety monitoring or advisory boards for Roche, Viela Bio, Genentech, Biogen, Reistone, TG Therapeutics, Celgene, Novartis, and Alexion–AstraZeneca Rare Disease. He has received grants for clinical trials through Alexion–AstraZeneca Rare Disease and Terumo BCT, and has been paid consulting fees by Mitsubishi Tanabe. Several coauthors of this study are employees of Alexion Pharmaceutics. Dr. Cree was principal investigator on the N-MOmentum study with inebilizumab. He has a grant from Genentech for MS research, and has consulted for Alexion in the past. Dr. Weinshenker has served as a member of the attack adjudication committee for both PREVENT and N-MOmentum studies and has financial relationships with the manufacturers of all three drugs. Dr. Bourdette has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The Alexion-sponsored study was presented at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) by Dean Wingerchuk, MD, of the Mayo Clinic in Scottsdale, Ariz.
Other experts in the field have highlighted limitations to the analysis and pointed out that all three agents are very effective in treating AQP4+ NMOSD, and many other considerations need to be taken into account as well as time to first relapse when selecting a therapy, leaving the door open for all three agents.
Dr. Wingerchuk explained that NMOSD is a rare severely disabling complement-mediated autoimmune neuroinflammatory disease of the central nervous system, characterized by devastating and unpredictable attacks (relapses) that can cause immediate and irreversible damage.
There are three recently approved monoclonal antibody treatment options in the United States for adults with AQP4+ NMOSD: eculizumab (Soliris, Alexion), inebilizumab (Uplizna, Horizon), and satralizumab (Enspryng, Genentech). A comparison of the relative treatment effects of these drugs would facilitate the treatment selection process, Dr. Wingerchuk said.
The objective of this study was to perform an indirect treatment comparison on the efficacy of these three FDA-approved treatment options for adults with AQP4+ NMOSD, in the absence of any head-to-head studies.
Using published data from randomized controlled trials, which were identified by a systematic literature review in September 2020, the researchers performed a Bayesian network meta-analysis to estimate the relative effects between eculizumab, inebilizumab, and satralizumab.
Network meta-analyses were performed for clinically relevant subpopulations based on three treatment networks: (1) patients who received monotherapy with one of the monoclonal antibodies or in combination with an immunosuppressant therapy; (2) patients who received monotherapy with the monoclonal antibody alone; and (3) patients who received a combination of both the monoclonal antibody and immunosuppressant therapy.
Time to first relapse was the primary efficacy outcome assessed. Relative treatment effects were expressed as hazard ratios and the probability that a treatment was the best at delaying time to first relapse was also evaluated.
In the systematic literature review, 29 publications from four unique clinical trials were identified and include in the network meta-analysis. These included publications from congress proceedings and peer-reviewed journals.
The four clinical trials were the N-MOmentum trial of inebilizumab versus placebo; the PREVENT trial of eculizumab with or without immunosuppressant therapy versus placebo with or without immunosuppressant therapy; the SAkuraSky trial of satralizumab plus immunosuppressant therapy versus placebo plus immunosuppressant therapy; and the SAkuraStar trial of satralizumab versus placebo.
Results showed that for the first analysis of mono or combination therapy, patients treated with eculizumab with or without immunosuppressant therapy were 76% less likely to experience a first relapse when compared with patients treated with satralizumab with or without immunosuppressant therapy.
In the monotherapy network, patients on eculizumab were 90% less likely to experience a first relapse when compared with patients treated with satralizumab, and patients on eculizumab were 89% less likely to experience a first relapse when compared with patients treated with inebilizumab.
In the third network analysis – a comparison of eculizumab plus immunosuppressant therapy with inebilizumab plus immunosuppressant therapy (Table 1) – the point estimate appeared to favor eculizumab but the confidence intervals were wide and not significant.
A subsequent analysis looked at the rank order of the best treatment option, with eculizumab coming out first in all three networks (Table 2).
Dr. Wingerchuk acknowledged that there were many limitations to this study, including that analyses for annualized relapse rate, disability, and quality of life were not included because of a lack of consistent outcome reporting by AQP4+ status in the randomized trials.
Safety outcomes were excluded because of a lack of standardized baseline risks and inconsistent reporting by AQP4+ status across trials.
Because this study focused on drugs approved in the United States in a rare disease area, there were a limited number of studies with intervention effects.
There were differences in follow-up durations across the different trials, with N-MOmentum having a follow-up of 197 days compared with 144 weeks for other trials.
“In the absence of head-to-head trials, this network meta-analysis provides important evidence on the relative efficacy of eculizumab versus satralizumab or inebilizumab for the treatment of patients with AQP4+ NMOSD, with significant differences in two out of the three treatment comparison scenarios observed,” Dr. Wingerchuk concluded.
“Based on current evidence, monotherapy and mono-combination therapy with eculizumab appear to more efficacious at preventing relapses than satralizumab or inebilizumab for the treatment of adults with AQP4+ NMOSD. These findings appear to suggest that C5 complement inhibition with treatments such as eculizumab appear to prevent relapses more effectively than other mechanisms involving IL-6 receptor or CD19 inhibition among adults with AQP4+ NMOSD,” he added.
Experts respond
Commenting on the study, several experts in the field provided some balancing views.
Bruce Cree, MD, University of California San Francisco, who was the chief investigator of the N-MOmentum study with inebilizumab, said he was skeptical about this new indirect comparison. “The results of this study seem too good to be true; a 90% difference between agents has to be an overestimate,” he said.
“We know from independent studies that all three drugs are very effective. If we take each trial separately, eculizumab reduced attack risk by 90% versus placebo; and the other two drugs by 77% to 78% versus placebo. For eculizumab to be 90% better than inebilizumab or satralizumab its basically like saying these drugs perform like placebo, but we know that is not the case,” Dr. Cree argued.
He pointed out that when comparing results across studies there are many factors that have to be considered, including the different patient populations included in the different studies, with the characteristic of each population in each trial being unique to that dataset.
In addition, Dr. Cree suggested that all the studies included in the comparison were relatively small for this type of analysis. “Normally this type of analysis is done with much larger studies, so the resulting database is closer to a representation of the disease state itself,” he said.
Dr. Cree also questioned the role of the sponsor in this meta-analysis. “The analysis was sponsored by Alexion and several coauthors were employees of Alexion. There was not much description available of how the statistics were done. I am concerned that the company was involved in the analysis, which could introduce bias. I look forward to seeing details of the statistical methodology,” he said.
“This is definitely a provocative study. They have thrown down the gauntlet. If they are so confident in the results they should now do a head-to-head study to back this result up. If they don’t do that, then I think physicians should ignore it as there are just too many problems with this analysis,” Dr. Cree stated.
Dr. Cree acknowledged that when looking at the four trials separately, eculizumab does look a little better than the other two agents in delaying time to first relapse. “But there are some caveats. Despite a larger reduction in relapse rate there was no reduction in disability in the eculizumab trial. Whereas the inebilizumab trial did show a reduction in disability. And while the PREVENT trial with eculizumab was a good study, during the course of the trial the definition of clinical relapse was changed, and as a consequence that increased the product’s performance – that’s a little bit curious,” he added.
How to choose?
On how to choose between the three agents, Dr. Cree said they are all “extraordinarily effective” at reducing relapse activity. “They are all ‘home run’ products, but they have differences in safety,” he said.
“Inebilizumab is linked to hypogammaglobulinemia over time – we haven’t seen an increase in infection risk linked to this, but with enough time, I would expect that there probably will be. But inebilizumab is a B-cell-depleting agent like the agents used in MS, and we now have a lot of experiences with this type of product, which gives us more confidence on the safety profile,” Dr. Cree noted.
“Eculizumab was linked to a risk of meningococcal meningitis and other bacterial infections, and satralizumab seems to [be] overall well tolerated with no obvious safety concerns to date, but the studies have been quite small,” he added.
On routes of administration and frequency of dosing, Dr. Cree pointed out that while all three drugs have an intensive loading schedule, for maintenance, eculizumab needs to be given as an IV infusion every 2 weeks. Inebilizumab needs just two infusions per year for maintenance, while satralizumab is given by subcutaneous injection once per month.
“It may be that eculizumab could be used at the time of an acute attack but then treatment could be switched to one of the other two for long-term maintenance,” he suggested.
But Dr. Cree pointed out that the biggest challenge for all three agents is access. “The costs are astronomically high ($200,000-$770,000). They are prohibitively expensive and very few insurance companies are covering them.”
Also commenting, Brian Weinshenker, MD, from the Mayo Clinic in Rochester, Minn., who was a member of the attack adjudication committee for both PREVENT and N-MOmentum studies, pointed out that as well as differences in the populations enrolled, and study designs, the studies with the three different drugs also had differences in attack adjudication criteria.
“These factors make it very difficult to compare across studies, which is what was done in this analysis, so I would be reluctant to reach many conclusions about differences.”
Dr. Weinshenker added: “All three treatments provided strong benefit. We are still learning about long-term benefits, but emerging data have suggested that all three seem to provide persistent benefits for the length of the open-label extension study. We don’t have much evidence about the severity of the attacks that did occur, although some limited data suggest that both eculizumab and inebilizumab reduce attack severity.”
Dennis Bourdette, MD, professor emeritus, department of neurology, Oregon Health & Science University, Portland, who was not involved in any of the studies, said he thought the new analysis was “a worthwhile effort to determine the relative effectiveness of the three different drugs in treating AQP4+ NMOSD.
“Given the rarity of APQ4+ NMOSD, it will be difficult to perform randomized head-to-head clinical trials of the agents, so this type of comparison is the best we can do at this time,” he said.
While Dr. Bourdette feels this study supports the notion that eculizumab is more effective at delaying time to first relapse than inebilizumab and satralizumab, he does not believe the results should have a major impact on decisions about which agent to use in clinical practice.
“A difference in delaying time to first relapse tells us little about the relative effectiveness of the long-term benefit of these [agents], particularly with regards to permanent disability or frequency of relapses. However, it is possible that the difference reflects the efficacy kinetics of the agents with eculizumab working faster than the other two agents, which would be useful in making a decision about a patient with very active NMOSD where one wants to get the disease under control as quickly as possible,” Dr. Bourdette noted.
But he added that neurologists should also consider safety profile, convenience, and contraindications. “Eculizumab is clearly less convenient in terms of dosing schedule than the other two agents, and patient convenience is important for long-term compliance.”
Dr. Bourdette pointed out that another consideration is prior treatment. “Many patients with NMOSD will receive the anti-CD20 monoclonal antibody, rituximab – which depletes B cells – off label. Inebilizumab also depletes B cells, so a patient who has had continued NMOSD disease activity on rituximab probably should not be treated with inebilizumab, making eculizumab or satralizumab preferable,” he suggested.
Finally, Dr. Bourdette highlighted the sponsorship of the current study by the manufacturer of eculizumab, Alexion, and that all of the authors have some financial relationship with Alexion as described in their disclosures. “Whether this resulted in any biases about the design, conduct, or interpretation of the study is uncertain but is always a concern,” he said.
Company statements
The companies selling inebilizumab and satralizumab sent statements on the new analysis and repeated many of the above points.
Genentech noted that new longer-term data presented at ECTRIMS show that satralizumab is effective in significantly reducing relapses over 4 years of treatment in people with AQP4+ NMOSD, with a favorable safety profile both as a monotherapy and in conjunction with immunosuppressive therapy. More than 70% of people treated with satralizumab remained relapse free after 4 years in the SAkuraStar (73%) and SAkuraSky (71%) open-label extension studies, and 90% and 91%, respectively, were free from severe relapse, the company reported.
Horizon said: “We are confident in the efficacy and safety of Uplizna (inebilizumab) – a convenient, twice-annual monotherapy – that was studied in the largest randomized, placebo-controlled, global trial of a monotherapy in NMOSD. The endpoints in this trial were prospectively defined and assessed by an adjudication committee as published in The Lancet, with long-term follow-up data now published in the Multiple Sclerosis Journal that further support the efficacy and safety.”
The current study was funded by Alexion–AstraZeneca Rare Disease. Dr. Wingerchuk has participated on data safety monitoring or advisory boards for Roche, Viela Bio, Genentech, Biogen, Reistone, TG Therapeutics, Celgene, Novartis, and Alexion–AstraZeneca Rare Disease. He has received grants for clinical trials through Alexion–AstraZeneca Rare Disease and Terumo BCT, and has been paid consulting fees by Mitsubishi Tanabe. Several coauthors of this study are employees of Alexion Pharmaceutics. Dr. Cree was principal investigator on the N-MOmentum study with inebilizumab. He has a grant from Genentech for MS research, and has consulted for Alexion in the past. Dr. Weinshenker has served as a member of the attack adjudication committee for both PREVENT and N-MOmentum studies and has financial relationships with the manufacturers of all three drugs. Dr. Bourdette has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The Alexion-sponsored study was presented at the annual meeting of the European Committee for Treatment and Research in Multiple Sclerosis (ECTRIMS) by Dean Wingerchuk, MD, of the Mayo Clinic in Scottsdale, Ariz.
Other experts in the field have highlighted limitations to the analysis and pointed out that all three agents are very effective in treating AQP4+ NMOSD, and many other considerations need to be taken into account as well as time to first relapse when selecting a therapy, leaving the door open for all three agents.
Dr. Wingerchuk explained that NMOSD is a rare severely disabling complement-mediated autoimmune neuroinflammatory disease of the central nervous system, characterized by devastating and unpredictable attacks (relapses) that can cause immediate and irreversible damage.
There are three recently approved monoclonal antibody treatment options in the United States for adults with AQP4+ NMOSD: eculizumab (Soliris, Alexion), inebilizumab (Uplizna, Horizon), and satralizumab (Enspryng, Genentech). A comparison of the relative treatment effects of these drugs would facilitate the treatment selection process, Dr. Wingerchuk said.
The objective of this study was to perform an indirect treatment comparison on the efficacy of these three FDA-approved treatment options for adults with AQP4+ NMOSD, in the absence of any head-to-head studies.
Using published data from randomized controlled trials, which were identified by a systematic literature review in September 2020, the researchers performed a Bayesian network meta-analysis to estimate the relative effects between eculizumab, inebilizumab, and satralizumab.
Network meta-analyses were performed for clinically relevant subpopulations based on three treatment networks: (1) patients who received monotherapy with one of the monoclonal antibodies or in combination with an immunosuppressant therapy; (2) patients who received monotherapy with the monoclonal antibody alone; and (3) patients who received a combination of both the monoclonal antibody and immunosuppressant therapy.
Time to first relapse was the primary efficacy outcome assessed. Relative treatment effects were expressed as hazard ratios and the probability that a treatment was the best at delaying time to first relapse was also evaluated.
In the systematic literature review, 29 publications from four unique clinical trials were identified and include in the network meta-analysis. These included publications from congress proceedings and peer-reviewed journals.
The four clinical trials were the N-MOmentum trial of inebilizumab versus placebo; the PREVENT trial of eculizumab with or without immunosuppressant therapy versus placebo with or without immunosuppressant therapy; the SAkuraSky trial of satralizumab plus immunosuppressant therapy versus placebo plus immunosuppressant therapy; and the SAkuraStar trial of satralizumab versus placebo.
Results showed that for the first analysis of mono or combination therapy, patients treated with eculizumab with or without immunosuppressant therapy were 76% less likely to experience a first relapse when compared with patients treated with satralizumab with or without immunosuppressant therapy.
In the monotherapy network, patients on eculizumab were 90% less likely to experience a first relapse when compared with patients treated with satralizumab, and patients on eculizumab were 89% less likely to experience a first relapse when compared with patients treated with inebilizumab.
In the third network analysis – a comparison of eculizumab plus immunosuppressant therapy with inebilizumab plus immunosuppressant therapy (Table 1) – the point estimate appeared to favor eculizumab but the confidence intervals were wide and not significant.
A subsequent analysis looked at the rank order of the best treatment option, with eculizumab coming out first in all three networks (Table 2).
Dr. Wingerchuk acknowledged that there were many limitations to this study, including that analyses for annualized relapse rate, disability, and quality of life were not included because of a lack of consistent outcome reporting by AQP4+ status in the randomized trials.
Safety outcomes were excluded because of a lack of standardized baseline risks and inconsistent reporting by AQP4+ status across trials.
Because this study focused on drugs approved in the United States in a rare disease area, there were a limited number of studies with intervention effects.
There were differences in follow-up durations across the different trials, with N-MOmentum having a follow-up of 197 days compared with 144 weeks for other trials.
“In the absence of head-to-head trials, this network meta-analysis provides important evidence on the relative efficacy of eculizumab versus satralizumab or inebilizumab for the treatment of patients with AQP4+ NMOSD, with significant differences in two out of the three treatment comparison scenarios observed,” Dr. Wingerchuk concluded.
“Based on current evidence, monotherapy and mono-combination therapy with eculizumab appear to more efficacious at preventing relapses than satralizumab or inebilizumab for the treatment of adults with AQP4+ NMOSD. These findings appear to suggest that C5 complement inhibition with treatments such as eculizumab appear to prevent relapses more effectively than other mechanisms involving IL-6 receptor or CD19 inhibition among adults with AQP4+ NMOSD,” he added.
Experts respond
Commenting on the study, several experts in the field provided some balancing views.
Bruce Cree, MD, University of California San Francisco, who was the chief investigator of the N-MOmentum study with inebilizumab, said he was skeptical about this new indirect comparison. “The results of this study seem too good to be true; a 90% difference between agents has to be an overestimate,” he said.
“We know from independent studies that all three drugs are very effective. If we take each trial separately, eculizumab reduced attack risk by 90% versus placebo; and the other two drugs by 77% to 78% versus placebo. For eculizumab to be 90% better than inebilizumab or satralizumab its basically like saying these drugs perform like placebo, but we know that is not the case,” Dr. Cree argued.
He pointed out that when comparing results across studies there are many factors that have to be considered, including the different patient populations included in the different studies, with the characteristic of each population in each trial being unique to that dataset.
In addition, Dr. Cree suggested that all the studies included in the comparison were relatively small for this type of analysis. “Normally this type of analysis is done with much larger studies, so the resulting database is closer to a representation of the disease state itself,” he said.
Dr. Cree also questioned the role of the sponsor in this meta-analysis. “The analysis was sponsored by Alexion and several coauthors were employees of Alexion. There was not much description available of how the statistics were done. I am concerned that the company was involved in the analysis, which could introduce bias. I look forward to seeing details of the statistical methodology,” he said.
“This is definitely a provocative study. They have thrown down the gauntlet. If they are so confident in the results they should now do a head-to-head study to back this result up. If they don’t do that, then I think physicians should ignore it as there are just too many problems with this analysis,” Dr. Cree stated.
Dr. Cree acknowledged that when looking at the four trials separately, eculizumab does look a little better than the other two agents in delaying time to first relapse. “But there are some caveats. Despite a larger reduction in relapse rate there was no reduction in disability in the eculizumab trial. Whereas the inebilizumab trial did show a reduction in disability. And while the PREVENT trial with eculizumab was a good study, during the course of the trial the definition of clinical relapse was changed, and as a consequence that increased the product’s performance – that’s a little bit curious,” he added.
How to choose?
On how to choose between the three agents, Dr. Cree said they are all “extraordinarily effective” at reducing relapse activity. “They are all ‘home run’ products, but they have differences in safety,” he said.
“Inebilizumab is linked to hypogammaglobulinemia over time – we haven’t seen an increase in infection risk linked to this, but with enough time, I would expect that there probably will be. But inebilizumab is a B-cell-depleting agent like the agents used in MS, and we now have a lot of experiences with this type of product, which gives us more confidence on the safety profile,” Dr. Cree noted.
“Eculizumab was linked to a risk of meningococcal meningitis and other bacterial infections, and satralizumab seems to [be] overall well tolerated with no obvious safety concerns to date, but the studies have been quite small,” he added.
On routes of administration and frequency of dosing, Dr. Cree pointed out that while all three drugs have an intensive loading schedule, for maintenance, eculizumab needs to be given as an IV infusion every 2 weeks. Inebilizumab needs just two infusions per year for maintenance, while satralizumab is given by subcutaneous injection once per month.
“It may be that eculizumab could be used at the time of an acute attack but then treatment could be switched to one of the other two for long-term maintenance,” he suggested.
But Dr. Cree pointed out that the biggest challenge for all three agents is access. “The costs are astronomically high ($200,000-$770,000). They are prohibitively expensive and very few insurance companies are covering them.”
Also commenting, Brian Weinshenker, MD, from the Mayo Clinic in Rochester, Minn., who was a member of the attack adjudication committee for both PREVENT and N-MOmentum studies, pointed out that as well as differences in the populations enrolled, and study designs, the studies with the three different drugs also had differences in attack adjudication criteria.
“These factors make it very difficult to compare across studies, which is what was done in this analysis, so I would be reluctant to reach many conclusions about differences.”
Dr. Weinshenker added: “All three treatments provided strong benefit. We are still learning about long-term benefits, but emerging data have suggested that all three seem to provide persistent benefits for the length of the open-label extension study. We don’t have much evidence about the severity of the attacks that did occur, although some limited data suggest that both eculizumab and inebilizumab reduce attack severity.”
Dennis Bourdette, MD, professor emeritus, department of neurology, Oregon Health & Science University, Portland, who was not involved in any of the studies, said he thought the new analysis was “a worthwhile effort to determine the relative effectiveness of the three different drugs in treating AQP4+ NMOSD.
“Given the rarity of APQ4+ NMOSD, it will be difficult to perform randomized head-to-head clinical trials of the agents, so this type of comparison is the best we can do at this time,” he said.
While Dr. Bourdette feels this study supports the notion that eculizumab is more effective at delaying time to first relapse than inebilizumab and satralizumab, he does not believe the results should have a major impact on decisions about which agent to use in clinical practice.
“A difference in delaying time to first relapse tells us little about the relative effectiveness of the long-term benefit of these [agents], particularly with regards to permanent disability or frequency of relapses. However, it is possible that the difference reflects the efficacy kinetics of the agents with eculizumab working faster than the other two agents, which would be useful in making a decision about a patient with very active NMOSD where one wants to get the disease under control as quickly as possible,” Dr. Bourdette noted.
But he added that neurologists should also consider safety profile, convenience, and contraindications. “Eculizumab is clearly less convenient in terms of dosing schedule than the other two agents, and patient convenience is important for long-term compliance.”
Dr. Bourdette pointed out that another consideration is prior treatment. “Many patients with NMOSD will receive the anti-CD20 monoclonal antibody, rituximab – which depletes B cells – off label. Inebilizumab also depletes B cells, so a patient who has had continued NMOSD disease activity on rituximab probably should not be treated with inebilizumab, making eculizumab or satralizumab preferable,” he suggested.
Finally, Dr. Bourdette highlighted the sponsorship of the current study by the manufacturer of eculizumab, Alexion, and that all of the authors have some financial relationship with Alexion as described in their disclosures. “Whether this resulted in any biases about the design, conduct, or interpretation of the study is uncertain but is always a concern,” he said.
Company statements
The companies selling inebilizumab and satralizumab sent statements on the new analysis and repeated many of the above points.
Genentech noted that new longer-term data presented at ECTRIMS show that satralizumab is effective in significantly reducing relapses over 4 years of treatment in people with AQP4+ NMOSD, with a favorable safety profile both as a monotherapy and in conjunction with immunosuppressive therapy. More than 70% of people treated with satralizumab remained relapse free after 4 years in the SAkuraStar (73%) and SAkuraSky (71%) open-label extension studies, and 90% and 91%, respectively, were free from severe relapse, the company reported.
Horizon said: “We are confident in the efficacy and safety of Uplizna (inebilizumab) – a convenient, twice-annual monotherapy – that was studied in the largest randomized, placebo-controlled, global trial of a monotherapy in NMOSD. The endpoints in this trial were prospectively defined and assessed by an adjudication committee as published in The Lancet, with long-term follow-up data now published in the Multiple Sclerosis Journal that further support the efficacy and safety.”
The current study was funded by Alexion–AstraZeneca Rare Disease. Dr. Wingerchuk has participated on data safety monitoring or advisory boards for Roche, Viela Bio, Genentech, Biogen, Reistone, TG Therapeutics, Celgene, Novartis, and Alexion–AstraZeneca Rare Disease. He has received grants for clinical trials through Alexion–AstraZeneca Rare Disease and Terumo BCT, and has been paid consulting fees by Mitsubishi Tanabe. Several coauthors of this study are employees of Alexion Pharmaceutics. Dr. Cree was principal investigator on the N-MOmentum study with inebilizumab. He has a grant from Genentech for MS research, and has consulted for Alexion in the past. Dr. Weinshenker has served as a member of the attack adjudication committee for both PREVENT and N-MOmentum studies and has financial relationships with the manufacturers of all three drugs. Dr. Bourdette has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ECTRIMS 2021
Evaluations of novel approaches to treating NF-1 tumors are underway
In the clinical experience of R. Rox Anderson, MD, currently available treatment options for benign tumors caused by neurofibromatosis type 1 (NF-1) are not acceptable.
“Simply removing the tumors with surgery is not the answer,” Dr. Anderson, a dermatologist who is the director of the Wellman Center for Photomedicine at Massachusetts General Hospital, Boston, said during a virtual course on laser and aesthetic skin therapy. “We need a way to inhibit the cutaneous neurofibromatosis early in life and prevent disfigurement that occurs when kids become adults.
“Kids with NF-1 are born looking normal,” he said. “They have café au lait macules and Lisch nodules in their eye, but they’re normal-looking kids. By early adulthood, many will grow hundreds of tumors that are disfiguring.”
In patients with NF-1, surgical excision works for cutaneous tumors but is expensive and not widely available, and is usually not covered by health insurance. “Plus, you have these adults who have already been through a lot of trauma, with the disfigurement in their lives, who have to be put under general anesthesia to remove a large number of tumors,” Dr. Anderson said at the meeting, which was named What’s the Truth and was sponsored by Harvard Medical School, Massachusetts General Hospital, and the Wellman Center for Photomedicine. Cryotherapy is a minimally invasive way to treat cutaneous neurofibroma tumors, “but this destroys the overlying skin, so you get unwanted destruction,” he said. “I like the idea of selecting heating, but we don’t know yet by what method.”
Dr. Anderson and his colleagues just launched a comparative clinical They plan to perform one or more treatment methods per patient in a single treatment session, then follow up at least 6 months later. Baseline and untreated cutaneous NF lesions will serve as controls. The researchers plan to conduct three-dimensional imaging, clinical assessments, and evaluate pain and other subjective measures.
Use of deoxycholate in a pilot trial was well tolerated and induced tumor regression in adults with cutaneous NF, he said.
Dr. Anderson noted that other researchers are studying the potential role of topical or local mitogen-activated protein kinase (MEK) inhibitors for these tumors. “Systemic MEK inhibitors are effective for plexiform neuromas, but cause significant cutaneous side effects,” he said. A “soft” MEK inhibitor, NFX-179 is rapidly metabolized such that high drug levels are achieved in skin without systemic drug levels. However, Dr. Anderson said that it remains unclear if this approach will prevent cutaneous NF tumors from forming, arrest their growth, or induce their regression.
Dr. Anderson reported having received research funding and/or consulting fees from numerous device and pharmaceutical companies.
Commentary by Lawrence F. Eichenfield, MD
Neurofibromatosis type 1 (NF1) is a common genodermatosis, associated with the development of neurofibromas derived from nerves, soft tissue, and skin. Cutaneous NFs often develop in later childhood onward and may be deforming, associated with pruritus, pain, and significant effect on quality of life. Dr. Anderson is a world leader in laser treatment, having developed the theories behind laser development for medical usage, as well as the laser technology used for vascular birthmarks and hair removal, laser and cooling techniques targeting fat, and “fractionating” laser energy, which has revolutionized scar management. We look forward to his group’s insights into better management of NF1 lesions!
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children's Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. He disclosed that he has served as an investigator and/or consultant to AbbVie, Lilly, Pfizer, Regeneron, Sanofi-Genzyme, and Verrica.
A version of this article first appeared on Medscape.com.
This article was updated 6/18/22.
In the clinical experience of R. Rox Anderson, MD, currently available treatment options for benign tumors caused by neurofibromatosis type 1 (NF-1) are not acceptable.
“Simply removing the tumors with surgery is not the answer,” Dr. Anderson, a dermatologist who is the director of the Wellman Center for Photomedicine at Massachusetts General Hospital, Boston, said during a virtual course on laser and aesthetic skin therapy. “We need a way to inhibit the cutaneous neurofibromatosis early in life and prevent disfigurement that occurs when kids become adults.
“Kids with NF-1 are born looking normal,” he said. “They have café au lait macules and Lisch nodules in their eye, but they’re normal-looking kids. By early adulthood, many will grow hundreds of tumors that are disfiguring.”
In patients with NF-1, surgical excision works for cutaneous tumors but is expensive and not widely available, and is usually not covered by health insurance. “Plus, you have these adults who have already been through a lot of trauma, with the disfigurement in their lives, who have to be put under general anesthesia to remove a large number of tumors,” Dr. Anderson said at the meeting, which was named What’s the Truth and was sponsored by Harvard Medical School, Massachusetts General Hospital, and the Wellman Center for Photomedicine. Cryotherapy is a minimally invasive way to treat cutaneous neurofibroma tumors, “but this destroys the overlying skin, so you get unwanted destruction,” he said. “I like the idea of selecting heating, but we don’t know yet by what method.”
Dr. Anderson and his colleagues just launched a comparative clinical They plan to perform one or more treatment methods per patient in a single treatment session, then follow up at least 6 months later. Baseline and untreated cutaneous NF lesions will serve as controls. The researchers plan to conduct three-dimensional imaging, clinical assessments, and evaluate pain and other subjective measures.
Use of deoxycholate in a pilot trial was well tolerated and induced tumor regression in adults with cutaneous NF, he said.
Dr. Anderson noted that other researchers are studying the potential role of topical or local mitogen-activated protein kinase (MEK) inhibitors for these tumors. “Systemic MEK inhibitors are effective for plexiform neuromas, but cause significant cutaneous side effects,” he said. A “soft” MEK inhibitor, NFX-179 is rapidly metabolized such that high drug levels are achieved in skin without systemic drug levels. However, Dr. Anderson said that it remains unclear if this approach will prevent cutaneous NF tumors from forming, arrest their growth, or induce their regression.
Dr. Anderson reported having received research funding and/or consulting fees from numerous device and pharmaceutical companies.
Commentary by Lawrence F. Eichenfield, MD
Neurofibromatosis type 1 (NF1) is a common genodermatosis, associated with the development of neurofibromas derived from nerves, soft tissue, and skin. Cutaneous NFs often develop in later childhood onward and may be deforming, associated with pruritus, pain, and significant effect on quality of life. Dr. Anderson is a world leader in laser treatment, having developed the theories behind laser development for medical usage, as well as the laser technology used for vascular birthmarks and hair removal, laser and cooling techniques targeting fat, and “fractionating” laser energy, which has revolutionized scar management. We look forward to his group’s insights into better management of NF1 lesions!
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children's Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. He disclosed that he has served as an investigator and/or consultant to AbbVie, Lilly, Pfizer, Regeneron, Sanofi-Genzyme, and Verrica.
A version of this article first appeared on Medscape.com.
This article was updated 6/18/22.
In the clinical experience of R. Rox Anderson, MD, currently available treatment options for benign tumors caused by neurofibromatosis type 1 (NF-1) are not acceptable.
“Simply removing the tumors with surgery is not the answer,” Dr. Anderson, a dermatologist who is the director of the Wellman Center for Photomedicine at Massachusetts General Hospital, Boston, said during a virtual course on laser and aesthetic skin therapy. “We need a way to inhibit the cutaneous neurofibromatosis early in life and prevent disfigurement that occurs when kids become adults.
“Kids with NF-1 are born looking normal,” he said. “They have café au lait macules and Lisch nodules in their eye, but they’re normal-looking kids. By early adulthood, many will grow hundreds of tumors that are disfiguring.”
In patients with NF-1, surgical excision works for cutaneous tumors but is expensive and not widely available, and is usually not covered by health insurance. “Plus, you have these adults who have already been through a lot of trauma, with the disfigurement in their lives, who have to be put under general anesthesia to remove a large number of tumors,” Dr. Anderson said at the meeting, which was named What’s the Truth and was sponsored by Harvard Medical School, Massachusetts General Hospital, and the Wellman Center for Photomedicine. Cryotherapy is a minimally invasive way to treat cutaneous neurofibroma tumors, “but this destroys the overlying skin, so you get unwanted destruction,” he said. “I like the idea of selecting heating, but we don’t know yet by what method.”
Dr. Anderson and his colleagues just launched a comparative clinical They plan to perform one or more treatment methods per patient in a single treatment session, then follow up at least 6 months later. Baseline and untreated cutaneous NF lesions will serve as controls. The researchers plan to conduct three-dimensional imaging, clinical assessments, and evaluate pain and other subjective measures.
Use of deoxycholate in a pilot trial was well tolerated and induced tumor regression in adults with cutaneous NF, he said.
Dr. Anderson noted that other researchers are studying the potential role of topical or local mitogen-activated protein kinase (MEK) inhibitors for these tumors. “Systemic MEK inhibitors are effective for plexiform neuromas, but cause significant cutaneous side effects,” he said. A “soft” MEK inhibitor, NFX-179 is rapidly metabolized such that high drug levels are achieved in skin without systemic drug levels. However, Dr. Anderson said that it remains unclear if this approach will prevent cutaneous NF tumors from forming, arrest their growth, or induce their regression.
Dr. Anderson reported having received research funding and/or consulting fees from numerous device and pharmaceutical companies.
Commentary by Lawrence F. Eichenfield, MD
Neurofibromatosis type 1 (NF1) is a common genodermatosis, associated with the development of neurofibromas derived from nerves, soft tissue, and skin. Cutaneous NFs often develop in later childhood onward and may be deforming, associated with pruritus, pain, and significant effect on quality of life. Dr. Anderson is a world leader in laser treatment, having developed the theories behind laser development for medical usage, as well as the laser technology used for vascular birthmarks and hair removal, laser and cooling techniques targeting fat, and “fractionating” laser energy, which has revolutionized scar management. We look forward to his group’s insights into better management of NF1 lesions!
Dr. Eichenfield is chief of pediatric and adolescent dermatology at Rady Children's Hospital-San Diego. He is vice chair of the department of dermatology and professor of dermatology and pediatrics at the University of California, San Diego. He disclosed that he has served as an investigator and/or consultant to AbbVie, Lilly, Pfizer, Regeneron, Sanofi-Genzyme, and Verrica.
A version of this article first appeared on Medscape.com.
This article was updated 6/18/22.
FROM A LASER & AESTHETIC SKIN THERAPY COURSE
Two diets linked to improved cognition, fatigue in MS
A Paleolithic elimination diet (Wahls diet) or a low-saturated fat diet (Swank diet) are associated with improved cognition, among other clinical outcomes, in relapsing-remitting multiple sclerosis (RRMS), new research suggests.
In a randomized study of patients with RRMS, the group that followed a Wahls diet and the group that followed a Swank diet both showed significant, unique improvement in measures of cognitive dysfunction, fatigue, and quality of life.
“Several dietary intervention studies have demonstrated favorable results on MS-related fatigue and quality of life. However, these results are among the first to show favorable reductions in cognitive dysfunction,” said co-investigator Tyler Titcomb, PhD, department of internal medicine, University of Iowa, Iowa City.
The results were presented at the 2021 Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC).
Similar diets
The CMSC findings came from a secondary analysis of a randomized trial published online in July in the Multiple Sclerosis Journal Experimental, Translational, and Clinical (MSJ-ETC).
The primary analysis of the single-blind, parallel group, randomized trial showed the Wahls and Swank diets were linked to significant improvement in outcomes on the Fatigue Severity Scale (FSS), the Modified Fatigue Impact Scale (MFIS), and other measures among participants with RRMS. There were no significant differences between the two dietary regimens.
The Swank diet restricts saturated fat to a maximum of 15 g per day while providing 20 g to 50 g (4 to 10 teaspoons) of unsaturated fat per day, with four servings each of whole grains, fruits, and vegetables.
The Wahls diet recommends six to nine servings of fruits and vegetables per day, in addition to 6 to 12 ounces of meat per day, according to gender. Grains, legumes, eggs, and dairy, with the exception of clarified butter or ghee, are not permitted on this diet. Both diets eschew processed foods.
To further evaluate the diets’ effects on perceived fatigue and cognitive dysfunction, the researchers returned to the trial, which enrolled 95 adults with stable RRMS at the University of Iowa Prevention Intervention Center between August 2016 and May 2019.
After a 12-week run-in period with support and education from registered dietitians, participants were randomly assigned to either the Swank or Wahls diets in a 24-week intervention that did not include dietitian support.
Inclusion criteria included having moderate to severe fatigue, as shown by an FSS score of at least 4.0, while not having severe mental impairment, an eating disorder, or liver or kidney disease. There were no significant differences in baseline demographic or clinical characteristics between the groups.
Of the patients, 77 completed the 12-week run-in (38 in the Swank diet group and 39 in the Wahls group). A total of 72 participants completed the 24-week follow-up (37 and 35, respectively).
Reduction in fatigue, cognitive dysfunction
After the researchers controlled for smoking, alcohol consumption, age, sex, baseline distance 6-minute walk test, body mass index, serum vitamin D, and years with MS, results at 12 and 24 weeks showed significant improvements from baseline in the key outcomes of fatigue and cognitive function, as measured by the Fatigue Scale for Motor and Cognitive Function (FSMC).
Scores were −5.7 and −9.0, respectively, for the Swank diet group and −9.3 and −14.9 for the Wahls group (P ≤ .001 for all comparisons).
In addition, there was a significant reduction in both groups on the total Perceived Deficits Questionnaire (PDQ) at 12 and 24 weeks (Swank, −7.4 and −6.3, respectively; Wahls, −6.8 and −10.8; P ≤ .001 for all).
There were similar improvements for both diets in an analysis of the mental and physical scores on FSMC and on the subscales on PDQ of attention, retrospective memory, prospective memory, and planning.
As observed in the primary analysis, there were no significant differences between the two groups in absolute mean scores on FSMC, PDQ, or their subscales at any timepoint.
“Both diets led to significant reductions in fatigue and cognitive dysfunction,” Dr. Titcomb said.
Of note, the primary analysis further showed statistically and clinically significant increases in the 6-minute walk test at 24-weeks of 6% in the Wahls group (P = .007). After removal of nonadherent participants, the improvement was still significant at 24 weeks in the Wahls group (P = .02), as well as in the Swank group (P = .001).
Dr. Titcomb noted that the majority of study participants were taking disease-modifying therapies (DMTs). However, there were no interactions between any specific DMTs and dietary benefits.
Potential mechanisms
Although the similar outcomes between the diets point to a common mechanism, there are also various other possibilities, said Dr. Titcomb. These include modulation of the microbiome, inflammation, immune system, or micronutrient optimization, he said.
Previous research has shown reduced mass and diversity in the gut microbiota among patients with MS compared with those without MS, potentially promoting inflammation. Other research has shown improvements in those factors with dietary modification.
While there is no evidence of gut microbiota changes with the Wahls and Swank diets, each is rich in fiber and plant-derived phytochemicals, which are known to be associated with improvements in gut microbiota and neuroinflammation, the investigators noted.
Dr. Titcomb reported that research into the diets is continuing as they evaluate longer-term and other effects. “This trial was a short-term parallel arm trial that did not include MRI or a control group,” he said, adding that the investigators will soon start recruiting for a follow-up study that will include a control group, long-term follow-up, and MRIs.
That upcoming study “has the potential to answer several of the unknown questions regarding the effect of diet on MS,” Dr. Titcomb said.
Notable research with limitations
Commenting on the study, Rebecca Spain, MD, MSPH, associate professor of neurology at the Oregon Health & Science University, and associate director of clinical care at VA MS Center of Excellence West in Portland, said there were several notable findings.
This includes that most people with MS “were able to adhere to the protocols for significant lengths of time, even without the support of dietitians for the final 12 weeks of the study,” said Spain, who was not involved with the research.
A significant limitation was the lack of a control group. Without that, “it’s hard to know for sure if the improvements in fatigue and cognition were from the diets or were simply from the social support of participating in a research study,” she said
Nevertheless, trials reporting on dietary effects in MS such as the current study are important, Dr. Spain noted. They demonstrate “that it is feasible and safe to conduct dietary studies and suggest which key MS symptoms may benefit and should be evaluated in future studies.”
“Critically, diet studies address one of the most frequent concerns of people with MS, promoting self-management and empowerment,” Dr. Spain concluded.
General guidelines for common dietary elements with evidence of improving fatigue, cognition, and mood are available on the National MS Society’s website.
The study received no outside funding. Dr. Titcomb and Dr. Spain have disclosed no relevant financial relationships. Terry L. Wahls, MD, who developed the Wahls diet, was a senior author of the study.
A version of this article first appeared on Medscape.com.
A Paleolithic elimination diet (Wahls diet) or a low-saturated fat diet (Swank diet) are associated with improved cognition, among other clinical outcomes, in relapsing-remitting multiple sclerosis (RRMS), new research suggests.
In a randomized study of patients with RRMS, the group that followed a Wahls diet and the group that followed a Swank diet both showed significant, unique improvement in measures of cognitive dysfunction, fatigue, and quality of life.
“Several dietary intervention studies have demonstrated favorable results on MS-related fatigue and quality of life. However, these results are among the first to show favorable reductions in cognitive dysfunction,” said co-investigator Tyler Titcomb, PhD, department of internal medicine, University of Iowa, Iowa City.
The results were presented at the 2021 Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC).
Similar diets
The CMSC findings came from a secondary analysis of a randomized trial published online in July in the Multiple Sclerosis Journal Experimental, Translational, and Clinical (MSJ-ETC).
The primary analysis of the single-blind, parallel group, randomized trial showed the Wahls and Swank diets were linked to significant improvement in outcomes on the Fatigue Severity Scale (FSS), the Modified Fatigue Impact Scale (MFIS), and other measures among participants with RRMS. There were no significant differences between the two dietary regimens.
The Swank diet restricts saturated fat to a maximum of 15 g per day while providing 20 g to 50 g (4 to 10 teaspoons) of unsaturated fat per day, with four servings each of whole grains, fruits, and vegetables.
The Wahls diet recommends six to nine servings of fruits and vegetables per day, in addition to 6 to 12 ounces of meat per day, according to gender. Grains, legumes, eggs, and dairy, with the exception of clarified butter or ghee, are not permitted on this diet. Both diets eschew processed foods.
To further evaluate the diets’ effects on perceived fatigue and cognitive dysfunction, the researchers returned to the trial, which enrolled 95 adults with stable RRMS at the University of Iowa Prevention Intervention Center between August 2016 and May 2019.
After a 12-week run-in period with support and education from registered dietitians, participants were randomly assigned to either the Swank or Wahls diets in a 24-week intervention that did not include dietitian support.
Inclusion criteria included having moderate to severe fatigue, as shown by an FSS score of at least 4.0, while not having severe mental impairment, an eating disorder, or liver or kidney disease. There were no significant differences in baseline demographic or clinical characteristics between the groups.
Of the patients, 77 completed the 12-week run-in (38 in the Swank diet group and 39 in the Wahls group). A total of 72 participants completed the 24-week follow-up (37 and 35, respectively).
Reduction in fatigue, cognitive dysfunction
After the researchers controlled for smoking, alcohol consumption, age, sex, baseline distance 6-minute walk test, body mass index, serum vitamin D, and years with MS, results at 12 and 24 weeks showed significant improvements from baseline in the key outcomes of fatigue and cognitive function, as measured by the Fatigue Scale for Motor and Cognitive Function (FSMC).
Scores were −5.7 and −9.0, respectively, for the Swank diet group and −9.3 and −14.9 for the Wahls group (P ≤ .001 for all comparisons).
In addition, there was a significant reduction in both groups on the total Perceived Deficits Questionnaire (PDQ) at 12 and 24 weeks (Swank, −7.4 and −6.3, respectively; Wahls, −6.8 and −10.8; P ≤ .001 for all).
There were similar improvements for both diets in an analysis of the mental and physical scores on FSMC and on the subscales on PDQ of attention, retrospective memory, prospective memory, and planning.
As observed in the primary analysis, there were no significant differences between the two groups in absolute mean scores on FSMC, PDQ, or their subscales at any timepoint.
“Both diets led to significant reductions in fatigue and cognitive dysfunction,” Dr. Titcomb said.
Of note, the primary analysis further showed statistically and clinically significant increases in the 6-minute walk test at 24-weeks of 6% in the Wahls group (P = .007). After removal of nonadherent participants, the improvement was still significant at 24 weeks in the Wahls group (P = .02), as well as in the Swank group (P = .001).
Dr. Titcomb noted that the majority of study participants were taking disease-modifying therapies (DMTs). However, there were no interactions between any specific DMTs and dietary benefits.
Potential mechanisms
Although the similar outcomes between the diets point to a common mechanism, there are also various other possibilities, said Dr. Titcomb. These include modulation of the microbiome, inflammation, immune system, or micronutrient optimization, he said.
Previous research has shown reduced mass and diversity in the gut microbiota among patients with MS compared with those without MS, potentially promoting inflammation. Other research has shown improvements in those factors with dietary modification.
While there is no evidence of gut microbiota changes with the Wahls and Swank diets, each is rich in fiber and plant-derived phytochemicals, which are known to be associated with improvements in gut microbiota and neuroinflammation, the investigators noted.
Dr. Titcomb reported that research into the diets is continuing as they evaluate longer-term and other effects. “This trial was a short-term parallel arm trial that did not include MRI or a control group,” he said, adding that the investigators will soon start recruiting for a follow-up study that will include a control group, long-term follow-up, and MRIs.
That upcoming study “has the potential to answer several of the unknown questions regarding the effect of diet on MS,” Dr. Titcomb said.
Notable research with limitations
Commenting on the study, Rebecca Spain, MD, MSPH, associate professor of neurology at the Oregon Health & Science University, and associate director of clinical care at VA MS Center of Excellence West in Portland, said there were several notable findings.
This includes that most people with MS “were able to adhere to the protocols for significant lengths of time, even without the support of dietitians for the final 12 weeks of the study,” said Spain, who was not involved with the research.
A significant limitation was the lack of a control group. Without that, “it’s hard to know for sure if the improvements in fatigue and cognition were from the diets or were simply from the social support of participating in a research study,” she said
Nevertheless, trials reporting on dietary effects in MS such as the current study are important, Dr. Spain noted. They demonstrate “that it is feasible and safe to conduct dietary studies and suggest which key MS symptoms may benefit and should be evaluated in future studies.”
“Critically, diet studies address one of the most frequent concerns of people with MS, promoting self-management and empowerment,” Dr. Spain concluded.
General guidelines for common dietary elements with evidence of improving fatigue, cognition, and mood are available on the National MS Society’s website.
The study received no outside funding. Dr. Titcomb and Dr. Spain have disclosed no relevant financial relationships. Terry L. Wahls, MD, who developed the Wahls diet, was a senior author of the study.
A version of this article first appeared on Medscape.com.
A Paleolithic elimination diet (Wahls diet) or a low-saturated fat diet (Swank diet) are associated with improved cognition, among other clinical outcomes, in relapsing-remitting multiple sclerosis (RRMS), new research suggests.
In a randomized study of patients with RRMS, the group that followed a Wahls diet and the group that followed a Swank diet both showed significant, unique improvement in measures of cognitive dysfunction, fatigue, and quality of life.
“Several dietary intervention studies have demonstrated favorable results on MS-related fatigue and quality of life. However, these results are among the first to show favorable reductions in cognitive dysfunction,” said co-investigator Tyler Titcomb, PhD, department of internal medicine, University of Iowa, Iowa City.
The results were presented at the 2021 Annual Meeting of the Consortium of Multiple Sclerosis Centers (CMSC).
Similar diets
The CMSC findings came from a secondary analysis of a randomized trial published online in July in the Multiple Sclerosis Journal Experimental, Translational, and Clinical (MSJ-ETC).
The primary analysis of the single-blind, parallel group, randomized trial showed the Wahls and Swank diets were linked to significant improvement in outcomes on the Fatigue Severity Scale (FSS), the Modified Fatigue Impact Scale (MFIS), and other measures among participants with RRMS. There were no significant differences between the two dietary regimens.
The Swank diet restricts saturated fat to a maximum of 15 g per day while providing 20 g to 50 g (4 to 10 teaspoons) of unsaturated fat per day, with four servings each of whole grains, fruits, and vegetables.
The Wahls diet recommends six to nine servings of fruits and vegetables per day, in addition to 6 to 12 ounces of meat per day, according to gender. Grains, legumes, eggs, and dairy, with the exception of clarified butter or ghee, are not permitted on this diet. Both diets eschew processed foods.
To further evaluate the diets’ effects on perceived fatigue and cognitive dysfunction, the researchers returned to the trial, which enrolled 95 adults with stable RRMS at the University of Iowa Prevention Intervention Center between August 2016 and May 2019.
After a 12-week run-in period with support and education from registered dietitians, participants were randomly assigned to either the Swank or Wahls diets in a 24-week intervention that did not include dietitian support.
Inclusion criteria included having moderate to severe fatigue, as shown by an FSS score of at least 4.0, while not having severe mental impairment, an eating disorder, or liver or kidney disease. There were no significant differences in baseline demographic or clinical characteristics between the groups.
Of the patients, 77 completed the 12-week run-in (38 in the Swank diet group and 39 in the Wahls group). A total of 72 participants completed the 24-week follow-up (37 and 35, respectively).
Reduction in fatigue, cognitive dysfunction
After the researchers controlled for smoking, alcohol consumption, age, sex, baseline distance 6-minute walk test, body mass index, serum vitamin D, and years with MS, results at 12 and 24 weeks showed significant improvements from baseline in the key outcomes of fatigue and cognitive function, as measured by the Fatigue Scale for Motor and Cognitive Function (FSMC).
Scores were −5.7 and −9.0, respectively, for the Swank diet group and −9.3 and −14.9 for the Wahls group (P ≤ .001 for all comparisons).
In addition, there was a significant reduction in both groups on the total Perceived Deficits Questionnaire (PDQ) at 12 and 24 weeks (Swank, −7.4 and −6.3, respectively; Wahls, −6.8 and −10.8; P ≤ .001 for all).
There were similar improvements for both diets in an analysis of the mental and physical scores on FSMC and on the subscales on PDQ of attention, retrospective memory, prospective memory, and planning.
As observed in the primary analysis, there were no significant differences between the two groups in absolute mean scores on FSMC, PDQ, or their subscales at any timepoint.
“Both diets led to significant reductions in fatigue and cognitive dysfunction,” Dr. Titcomb said.
Of note, the primary analysis further showed statistically and clinically significant increases in the 6-minute walk test at 24-weeks of 6% in the Wahls group (P = .007). After removal of nonadherent participants, the improvement was still significant at 24 weeks in the Wahls group (P = .02), as well as in the Swank group (P = .001).
Dr. Titcomb noted that the majority of study participants were taking disease-modifying therapies (DMTs). However, there were no interactions between any specific DMTs and dietary benefits.
Potential mechanisms
Although the similar outcomes between the diets point to a common mechanism, there are also various other possibilities, said Dr. Titcomb. These include modulation of the microbiome, inflammation, immune system, or micronutrient optimization, he said.
Previous research has shown reduced mass and diversity in the gut microbiota among patients with MS compared with those without MS, potentially promoting inflammation. Other research has shown improvements in those factors with dietary modification.
While there is no evidence of gut microbiota changes with the Wahls and Swank diets, each is rich in fiber and plant-derived phytochemicals, which are known to be associated with improvements in gut microbiota and neuroinflammation, the investigators noted.
Dr. Titcomb reported that research into the diets is continuing as they evaluate longer-term and other effects. “This trial was a short-term parallel arm trial that did not include MRI or a control group,” he said, adding that the investigators will soon start recruiting for a follow-up study that will include a control group, long-term follow-up, and MRIs.
That upcoming study “has the potential to answer several of the unknown questions regarding the effect of diet on MS,” Dr. Titcomb said.
Notable research with limitations
Commenting on the study, Rebecca Spain, MD, MSPH, associate professor of neurology at the Oregon Health & Science University, and associate director of clinical care at VA MS Center of Excellence West in Portland, said there were several notable findings.
This includes that most people with MS “were able to adhere to the protocols for significant lengths of time, even without the support of dietitians for the final 12 weeks of the study,” said Spain, who was not involved with the research.
A significant limitation was the lack of a control group. Without that, “it’s hard to know for sure if the improvements in fatigue and cognition were from the diets or were simply from the social support of participating in a research study,” she said
Nevertheless, trials reporting on dietary effects in MS such as the current study are important, Dr. Spain noted. They demonstrate “that it is feasible and safe to conduct dietary studies and suggest which key MS symptoms may benefit and should be evaluated in future studies.”
“Critically, diet studies address one of the most frequent concerns of people with MS, promoting self-management and empowerment,” Dr. Spain concluded.
General guidelines for common dietary elements with evidence of improving fatigue, cognition, and mood are available on the National MS Society’s website.
The study received no outside funding. Dr. Titcomb and Dr. Spain have disclosed no relevant financial relationships. Terry L. Wahls, MD, who developed the Wahls diet, was a senior author of the study.
A version of this article first appeared on Medscape.com.
FROM CMSC 2021