User login
What's your diagnosis?
The diagnosis
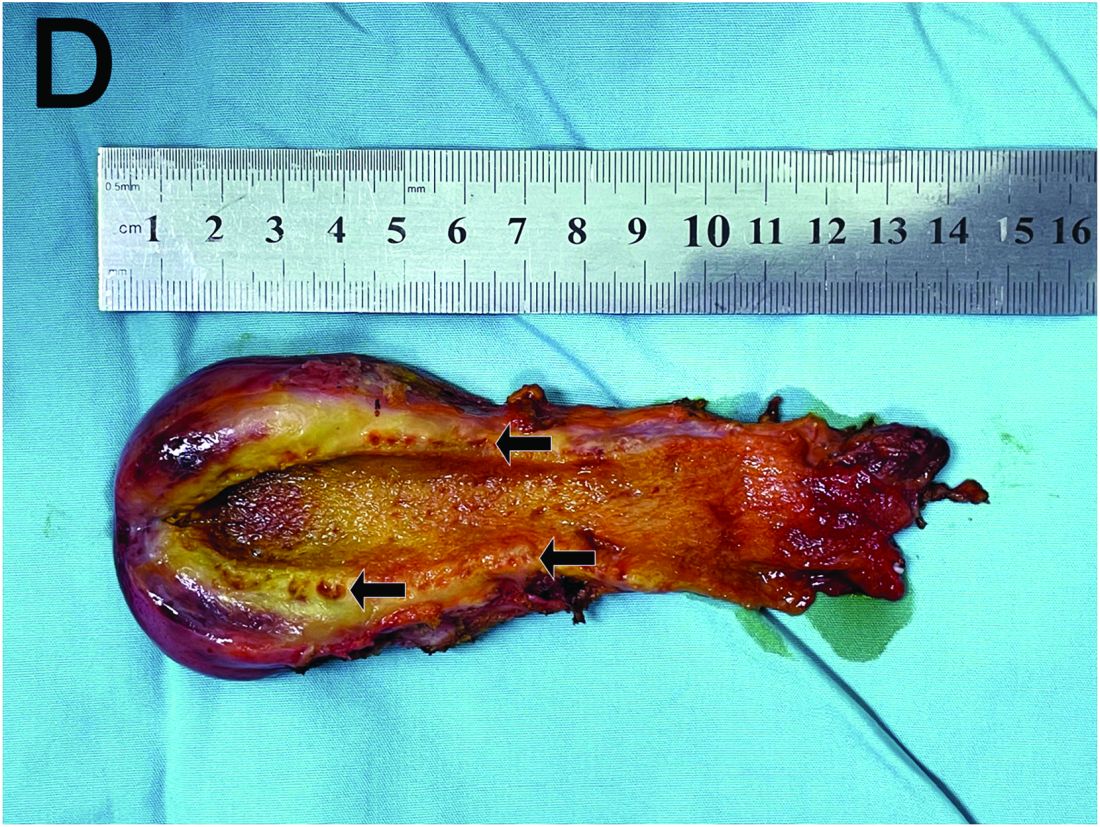
Based on the clinical and imaging findings, a diagnosis of gallbladder adenomyomatosis was made. GA is a benign and usually asymptomatic condition that occurs mainly beyond the age of 50-60 years and is very rare in childhood.1 Symptomatic gallbladder adenomyomatosis indicates cholecystectomy, considering the presence of inflammation or gallbladder stones.2 Therefore, a laparoscopic cholecystectomy was performed on our patient. Rokitansky-Aschoff sinuses were seen in the entire thickened gallbladder wall on gross pathologic examination (Figure D). Histopathologic examination confirmed the diagnosis of GA with cholecystitis. The patient was eventually diagnosed with diffuse GA. She was successfully discharged from the hospital 4 days after surgery, and 3 months of follow-up were uneventful.
References
Eroglu N et al. Diffuse adenomyomatosis of the gallbladder in a child. J Pediatr Hematol Oncol. 2016;38:e307-9.
Bonatti M. et al. Gallbladder adenomyomatosis: imaging findings, tricks and pitfalls. Insights Imaging. 2017;8:243-53.
Hammad AY et al. A literature review of radiological findings to guide the diagnosis of gallbladder adenomyomatosis. HPB (Oxford). 2016;18:129-35.
The diagnosis
Based on the clinical and imaging findings, a diagnosis of gallbladder adenomyomatosis was made. GA is a benign and usually asymptomatic condition that occurs mainly beyond the age of 50-60 years and is very rare in childhood.1 Symptomatic gallbladder adenomyomatosis indicates cholecystectomy, considering the presence of inflammation or gallbladder stones.2 Therefore, a laparoscopic cholecystectomy was performed on our patient. Rokitansky-Aschoff sinuses were seen in the entire thickened gallbladder wall on gross pathologic examination (Figure D). Histopathologic examination confirmed the diagnosis of GA with cholecystitis. The patient was eventually diagnosed with diffuse GA. She was successfully discharged from the hospital 4 days after surgery, and 3 months of follow-up were uneventful.
References
Eroglu N et al. Diffuse adenomyomatosis of the gallbladder in a child. J Pediatr Hematol Oncol. 2016;38:e307-9.
Bonatti M. et al. Gallbladder adenomyomatosis: imaging findings, tricks and pitfalls. Insights Imaging. 2017;8:243-53.
Hammad AY et al. A literature review of radiological findings to guide the diagnosis of gallbladder adenomyomatosis. HPB (Oxford). 2016;18:129-35.
The diagnosis
Based on the clinical and imaging findings, a diagnosis of gallbladder adenomyomatosis was made. GA is a benign and usually asymptomatic condition that occurs mainly beyond the age of 50-60 years and is very rare in childhood.1 Symptomatic gallbladder adenomyomatosis indicates cholecystectomy, considering the presence of inflammation or gallbladder stones.2 Therefore, a laparoscopic cholecystectomy was performed on our patient. Rokitansky-Aschoff sinuses were seen in the entire thickened gallbladder wall on gross pathologic examination (Figure D). Histopathologic examination confirmed the diagnosis of GA with cholecystitis. The patient was eventually diagnosed with diffuse GA. She was successfully discharged from the hospital 4 days after surgery, and 3 months of follow-up were uneventful.
References
Eroglu N et al. Diffuse adenomyomatosis of the gallbladder in a child. J Pediatr Hematol Oncol. 2016;38:e307-9.
Bonatti M. et al. Gallbladder adenomyomatosis: imaging findings, tricks and pitfalls. Insights Imaging. 2017;8:243-53.
Hammad AY et al. A literature review of radiological findings to guide the diagnosis of gallbladder adenomyomatosis. HPB (Oxford). 2016;18:129-35.
A 15-year-old girl presented with an 18-month history of intermittent right upper quadrant pain that appeared after meals and was relieved after rest. She denied any nausea, vomiting, chills, diarrhea, or constipation. The patient reported no trauma. At admission, physical examination showed tenderness in the right upper abdomen without rebound or guarding. Murphy's sign was also present. The laboratory tests were unremarkable.
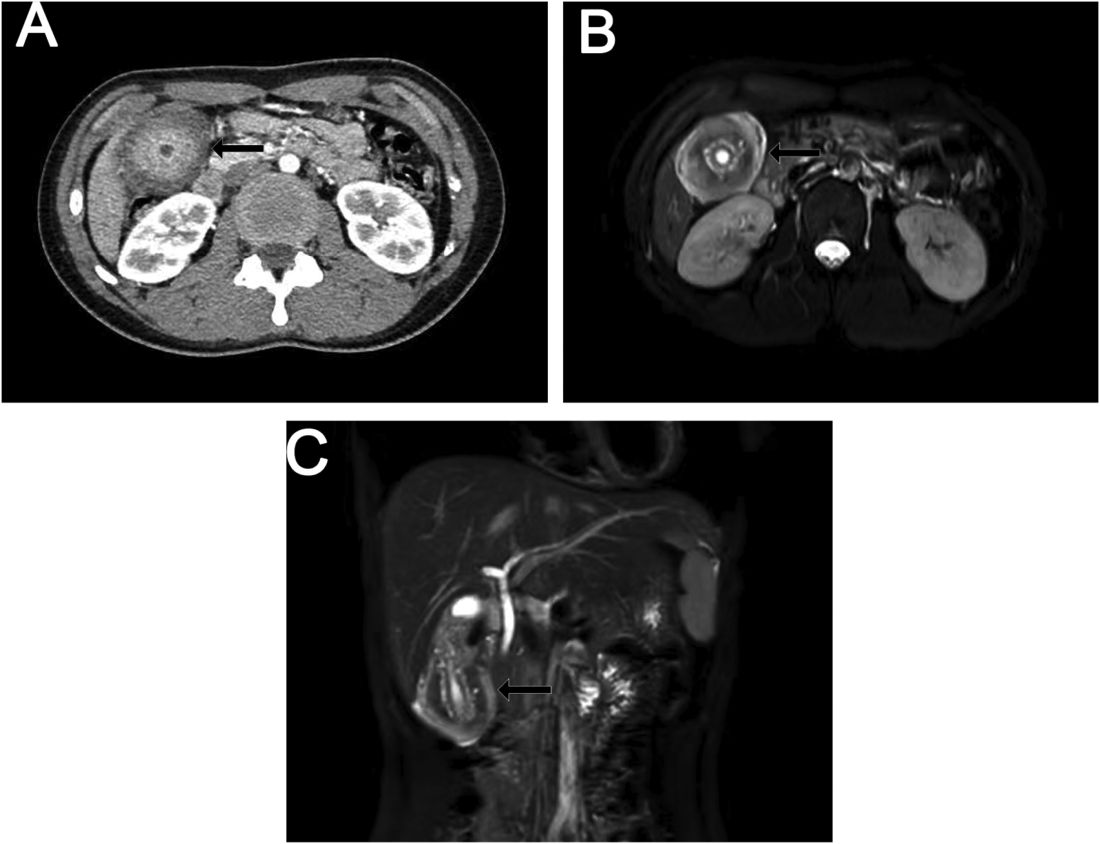
Ultrasound examination indicated gallbladder wall thickening. Furthermore, a contrast-enhanced computed tomographic scan showed marked gallbladder wall thickening with an annular unenhanced proliferative muscularis layer surrounding enhanced proliferative mucosal epithelium (Figure A), and magnetic resonance imaging showed multiple cyst-like spaces in the gallbladder wall (Figures B and C).
What is the diagnosis, and how should it be managed?
Previously published in Gastroenterology
What's your diagnosis?
Answer to ‘What’s your diagnosis?’: Gastric adenocarcinoma and proximal polyposis of the stomach syndrome.
Fundic gland polyps (FGPs) are the most common gastric polyps and when occurring in the sporadic setting are typically benign; however, FGPs that occur in gastrointestinal polyposis syndromes such as familial adenomatosis polyposis can progress to adenocarcinoma and require surveillance. Therefore, it is important to distinguish sporadic versus syndromic fundic gland polyposis. Gastric adenocarcinoma and proximal polyposis of the stomach is a recently described condition that significantly increases the risk of developing invasive gastric adenocarcinoma from FGPs. Diagnostic criteria include (1) gastric polyposis restricted to the body and fundus with no small bowel or colonic involvement, (2) >100 gastric polyps or >30 polyps in a first-degree relative, (3) histology consistent with FGP with areas of dysplasia, (4) a family history consistent with an autosomal-dominant pattern of inheritance, and (5) exclusion of other syndromes and proton pump inhibitor use.1 Unlike familial adenomatosis polyposis, the polyposis is restricted to the oxyntic mucosa of the gastric body and fundus with sparing of the gastric antrum, small bowel, and colon. The genetic basis of the disease has been attributed to a point mutation in the APC gene promotor IB region leading to a loss of tumor suppressor function.2 Typical histology shows large FGPs with areas of low-grade and high-grade dysplasia, as seen in our patient.
There are few data on the natural history of gastric adenocarcinoma and proximal polyposis of the stomach, but effective surveillance is limited by the degree of polyposis. There are multiple reports of hidden adenocarcinoma on surgically resected specimens, as well as rapid progression to metastatic adenocarcinoma despite adequate diagnosis and surveillance.1,3 Therefore, total gastrectomy should be offered to patients who are surgical candidates. Our patient underwent genetic testing that revealed a point mutation in the APC promotor IB. He declined surgical intervention and opted for surveillance endoscopy every 6 months.
References
1. Worthley D.L. et al. Gut. 2012;61:774-9
2. Li J et al. Am J Hum Genet. 2016;98:830-42
3. Rudloff U. Clin Exp Gastroenterol. 2018;11:447-59
Answer to ‘What’s your diagnosis?’: Gastric adenocarcinoma and proximal polyposis of the stomach syndrome.
Fundic gland polyps (FGPs) are the most common gastric polyps and when occurring in the sporadic setting are typically benign; however, FGPs that occur in gastrointestinal polyposis syndromes such as familial adenomatosis polyposis can progress to adenocarcinoma and require surveillance. Therefore, it is important to distinguish sporadic versus syndromic fundic gland polyposis. Gastric adenocarcinoma and proximal polyposis of the stomach is a recently described condition that significantly increases the risk of developing invasive gastric adenocarcinoma from FGPs. Diagnostic criteria include (1) gastric polyposis restricted to the body and fundus with no small bowel or colonic involvement, (2) >100 gastric polyps or >30 polyps in a first-degree relative, (3) histology consistent with FGP with areas of dysplasia, (4) a family history consistent with an autosomal-dominant pattern of inheritance, and (5) exclusion of other syndromes and proton pump inhibitor use.1 Unlike familial adenomatosis polyposis, the polyposis is restricted to the oxyntic mucosa of the gastric body and fundus with sparing of the gastric antrum, small bowel, and colon. The genetic basis of the disease has been attributed to a point mutation in the APC gene promotor IB region leading to a loss of tumor suppressor function.2 Typical histology shows large FGPs with areas of low-grade and high-grade dysplasia, as seen in our patient.
There are few data on the natural history of gastric adenocarcinoma and proximal polyposis of the stomach, but effective surveillance is limited by the degree of polyposis. There are multiple reports of hidden adenocarcinoma on surgically resected specimens, as well as rapid progression to metastatic adenocarcinoma despite adequate diagnosis and surveillance.1,3 Therefore, total gastrectomy should be offered to patients who are surgical candidates. Our patient underwent genetic testing that revealed a point mutation in the APC promotor IB. He declined surgical intervention and opted for surveillance endoscopy every 6 months.
References
1. Worthley D.L. et al. Gut. 2012;61:774-9
2. Li J et al. Am J Hum Genet. 2016;98:830-42
3. Rudloff U. Clin Exp Gastroenterol. 2018;11:447-59
Answer to ‘What’s your diagnosis?’: Gastric adenocarcinoma and proximal polyposis of the stomach syndrome.
Fundic gland polyps (FGPs) are the most common gastric polyps and when occurring in the sporadic setting are typically benign; however, FGPs that occur in gastrointestinal polyposis syndromes such as familial adenomatosis polyposis can progress to adenocarcinoma and require surveillance. Therefore, it is important to distinguish sporadic versus syndromic fundic gland polyposis. Gastric adenocarcinoma and proximal polyposis of the stomach is a recently described condition that significantly increases the risk of developing invasive gastric adenocarcinoma from FGPs. Diagnostic criteria include (1) gastric polyposis restricted to the body and fundus with no small bowel or colonic involvement, (2) >100 gastric polyps or >30 polyps in a first-degree relative, (3) histology consistent with FGP with areas of dysplasia, (4) a family history consistent with an autosomal-dominant pattern of inheritance, and (5) exclusion of other syndromes and proton pump inhibitor use.1 Unlike familial adenomatosis polyposis, the polyposis is restricted to the oxyntic mucosa of the gastric body and fundus with sparing of the gastric antrum, small bowel, and colon. The genetic basis of the disease has been attributed to a point mutation in the APC gene promotor IB region leading to a loss of tumor suppressor function.2 Typical histology shows large FGPs with areas of low-grade and high-grade dysplasia, as seen in our patient.
There are few data on the natural history of gastric adenocarcinoma and proximal polyposis of the stomach, but effective surveillance is limited by the degree of polyposis. There are multiple reports of hidden adenocarcinoma on surgically resected specimens, as well as rapid progression to metastatic adenocarcinoma despite adequate diagnosis and surveillance.1,3 Therefore, total gastrectomy should be offered to patients who are surgical candidates. Our patient underwent genetic testing that revealed a point mutation in the APC promotor IB. He declined surgical intervention and opted for surveillance endoscopy every 6 months.
References
1. Worthley D.L. et al. Gut. 2012;61:774-9
2. Li J et al. Am J Hum Genet. 2016;98:830-42
3. Rudloff U. Clin Exp Gastroenterol. 2018;11:447-59
A 72-year-old man with compensated cirrhosis owing to autoimmune hepatitis presented for evaluation of an indeterminate gastric lesion found during an otherwise normal endoscopic retrograde cholangiopancreatography performed for incidental ductal dilation seen on cross-sectional imaging. He did not endorse any abdominal pain, dyspepsia, or weight loss and was not on a proton pump inhibitor. Family history was notable for a daughter diagnosed with metastatic gastric adenocarcinoma at the age of 44 years.
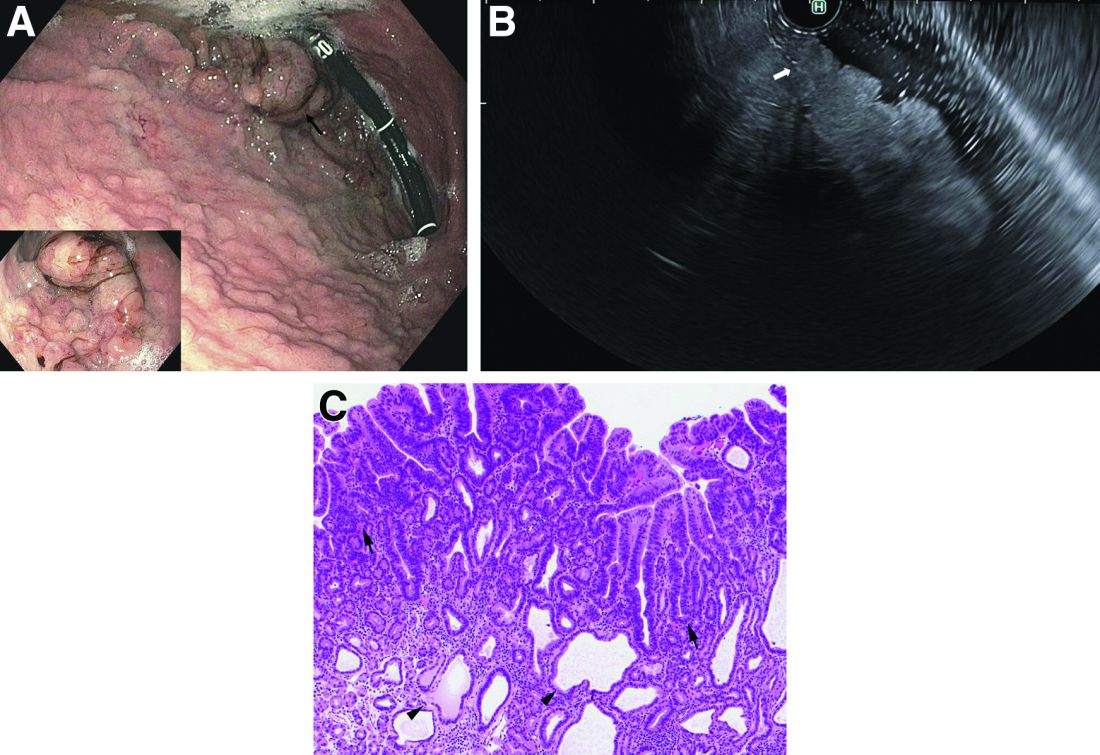
Upper endoscopy showed innumerable sessile polyps of variable size carpeting the gastric body and fundus (Figure A) with a large, mound-like mass lesion in the fundus (Figure A, arrow and inset). Echoendoscopy revealed a hypoechoic, noncircumferential mass restricted to the mucosal surface with well-defined borders (Figure B, arrow). A technically challenging, piecemeal endoscopic mucosal resection was performed. The patient also underwent a colonoscopy that was unremarkable. Pathology of the gastric lesion was consistent with a fundic gland polyp (Figure C, arrowheads) containing low-grade and high-grade dysplasia (Figure C, arrows).
What is the most likely diagnosis?
What's your diagnosis?
Answer: Blue rubber bleb nevus syndrome.
According to an American College of Gastroenterology Clinical Guideline,1 for patients with recurrence of small bowel bleeding, endoscopic management could be considered depending on the patient’s clinical course and response to prior therapy. Consequently, injections of lauromacrogol with SBE (single-balloon enteroscopy) were given (Figure D). Lesions that ranged from 1 to 2 cm were injected with 1-2 mL lauromacrogol until the mucosa turned white. Three SBEs had been performed in a 5-month period. A total of 20 lesions were successfully treated with lauromacrogol. The treated hemangiomas became small, and the site healed 5 months after treatment (Figures E and F). The patient has been followed for 1 year, and he remains in good clinical condition with his latest hemoglobin level at 110 g/L. No further blood transfusion is needed.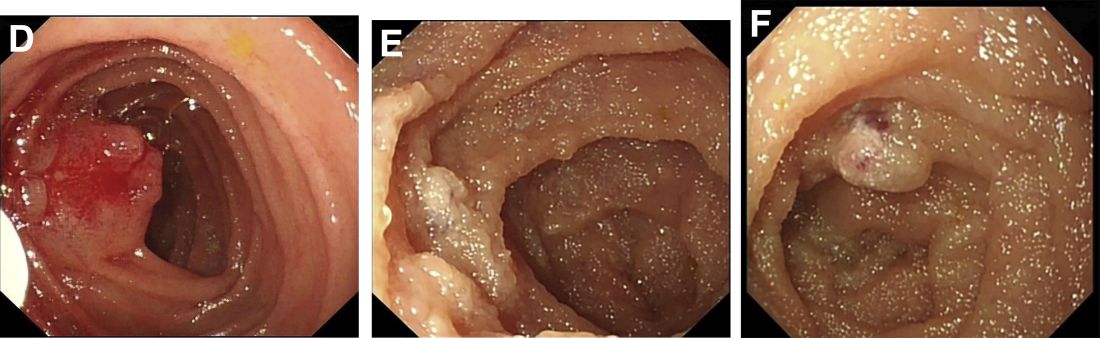
BRBNS is a rare disorder characterized by discrete venous malformations of varying size and appearance that are present on the skin and within the gastrointestinal tract.2With wider application of video capsule endoscopy (VCE) and the increase of image resolution, the detection rate and diagnostic accuracy of BRBNS are significantly improved. Treatment of BRBNS varies depending on the site, size, and number of lesions. Medication, surgery, and endoscopic therapy are currently clinically applied. The successful use of sirolimus was recently reported in the treatment of vascular lesions.3Sirolimus has potential adverse effects on renal function, bone marrow, and cholesterol metabolism, however. In consideration of the patient’s young age, we did not adopt this method. Surgical resection is more suitable for limited or life-threatening lesions. The lesions in this patient were mild and sporadic. Consequently, in this case, endoscopic injection of lauromacrogol was performed. This was the most complicated case of endoscopic treatment of BRBNS in our center and proved lauromacrogol injection was a feasible approach. According to a literature review, lauromacrogol has been used to treat vascular lesions for decades, but there is still no standard instruction for the dosage of lauromacrogol. We hope that our experience can be a reference for the endoscopic treatment of BRBNS.
References (add links)
1. Gerson LB et al. ACG clinical guideline: Diagnosis and management of small bowel bleeding. Am J Gastroenterol.
2. Felton SJ and Ferguson JE. Multiple cutaneous swellings associated with sudden collapse. JAMA.
3. Yuksekkaya H et al. Blue rubber bleb nevus syndrome: Successful treatment with sirolimus. Pediatrics.
Answer: Blue rubber bleb nevus syndrome.
According to an American College of Gastroenterology Clinical Guideline,1 for patients with recurrence of small bowel bleeding, endoscopic management could be considered depending on the patient’s clinical course and response to prior therapy. Consequently, injections of lauromacrogol with SBE (single-balloon enteroscopy) were given (Figure D). Lesions that ranged from 1 to 2 cm were injected with 1-2 mL lauromacrogol until the mucosa turned white. Three SBEs had been performed in a 5-month period. A total of 20 lesions were successfully treated with lauromacrogol. The treated hemangiomas became small, and the site healed 5 months after treatment (Figures E and F). The patient has been followed for 1 year, and he remains in good clinical condition with his latest hemoglobin level at 110 g/L. No further blood transfusion is needed.
BRBNS is a rare disorder characterized by discrete venous malformations of varying size and appearance that are present on the skin and within the gastrointestinal tract.2With wider application of video capsule endoscopy (VCE) and the increase of image resolution, the detection rate and diagnostic accuracy of BRBNS are significantly improved. Treatment of BRBNS varies depending on the site, size, and number of lesions. Medication, surgery, and endoscopic therapy are currently clinically applied. The successful use of sirolimus was recently reported in the treatment of vascular lesions.3Sirolimus has potential adverse effects on renal function, bone marrow, and cholesterol metabolism, however. In consideration of the patient’s young age, we did not adopt this method. Surgical resection is more suitable for limited or life-threatening lesions. The lesions in this patient were mild and sporadic. Consequently, in this case, endoscopic injection of lauromacrogol was performed. This was the most complicated case of endoscopic treatment of BRBNS in our center and proved lauromacrogol injection was a feasible approach. According to a literature review, lauromacrogol has been used to treat vascular lesions for decades, but there is still no standard instruction for the dosage of lauromacrogol. We hope that our experience can be a reference for the endoscopic treatment of BRBNS.
References (add links)
1. Gerson LB et al. ACG clinical guideline: Diagnosis and management of small bowel bleeding. Am J Gastroenterol.
2. Felton SJ and Ferguson JE. Multiple cutaneous swellings associated with sudden collapse. JAMA.
3. Yuksekkaya H et al. Blue rubber bleb nevus syndrome: Successful treatment with sirolimus. Pediatrics.
Answer: Blue rubber bleb nevus syndrome.
According to an American College of Gastroenterology Clinical Guideline,1 for patients with recurrence of small bowel bleeding, endoscopic management could be considered depending on the patient’s clinical course and response to prior therapy. Consequently, injections of lauromacrogol with SBE (single-balloon enteroscopy) were given (Figure D). Lesions that ranged from 1 to 2 cm were injected with 1-2 mL lauromacrogol until the mucosa turned white. Three SBEs had been performed in a 5-month period. A total of 20 lesions were successfully treated with lauromacrogol. The treated hemangiomas became small, and the site healed 5 months after treatment (Figures E and F). The patient has been followed for 1 year, and he remains in good clinical condition with his latest hemoglobin level at 110 g/L. No further blood transfusion is needed.
BRBNS is a rare disorder characterized by discrete venous malformations of varying size and appearance that are present on the skin and within the gastrointestinal tract.2With wider application of video capsule endoscopy (VCE) and the increase of image resolution, the detection rate and diagnostic accuracy of BRBNS are significantly improved. Treatment of BRBNS varies depending on the site, size, and number of lesions. Medication, surgery, and endoscopic therapy are currently clinically applied. The successful use of sirolimus was recently reported in the treatment of vascular lesions.3Sirolimus has potential adverse effects on renal function, bone marrow, and cholesterol metabolism, however. In consideration of the patient’s young age, we did not adopt this method. Surgical resection is more suitable for limited or life-threatening lesions. The lesions in this patient were mild and sporadic. Consequently, in this case, endoscopic injection of lauromacrogol was performed. This was the most complicated case of endoscopic treatment of BRBNS in our center and proved lauromacrogol injection was a feasible approach. According to a literature review, lauromacrogol has been used to treat vascular lesions for decades, but there is still no standard instruction for the dosage of lauromacrogol. We hope that our experience can be a reference for the endoscopic treatment of BRBNS.
References (add links)
1. Gerson LB et al. ACG clinical guideline: Diagnosis and management of small bowel bleeding. Am J Gastroenterol.
2. Felton SJ and Ferguson JE. Multiple cutaneous swellings associated with sudden collapse. JAMA.
3. Yuksekkaya H et al. Blue rubber bleb nevus syndrome: Successful treatment with sirolimus. Pediatrics.
A 13-year-old boy presented with recurrent melena for 10 years accompanied with dizziness and fatigue. This patient had no history of nonsteroidal anti-inflammatory drug use, peptic ulcer, or chronic liver disease, and no family history of gastrointestinal bleeding. He was born with a right foot hemangioma that was resected when he was 2 years old. Additionally, he had received multiple blood transfusions for iron deficiency anemia since childhood. The body mass index was 16.5 kg/m2 and physical examination revealed active bowel sounds.
Laboratory examinations showed severe iron deficiency anemia (the lowest hemoglobin available was 36 g/L) and positive stool occult blood. Gastroscopy unveiled superficial gastritis and colonoscopy was normal. Second-look examinations showed the same results. No clinically important signs were observed on computed tomography scan. Given these results, small intestinal bleeding was considered. Therefore, a video capsule endoscopy (VCE) was carried out and revealed multifocal hemangioma-like purplish blue lesions in jejunum and ileum (Figure A). Then a single-balloon enteroscopy (SBE) was performed, which showed multifocal vascular lesions ranging between 1.0 and 2.0 cm in the jejunum and ileum (Figure B, C).
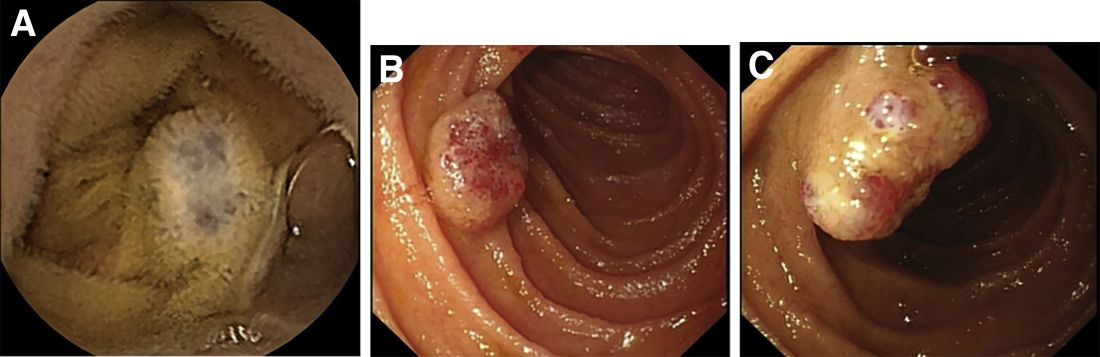
Based on these findings, what is your diagnosis? What is the next step in management for this patient?
What is the diagnosis?
Syphilis
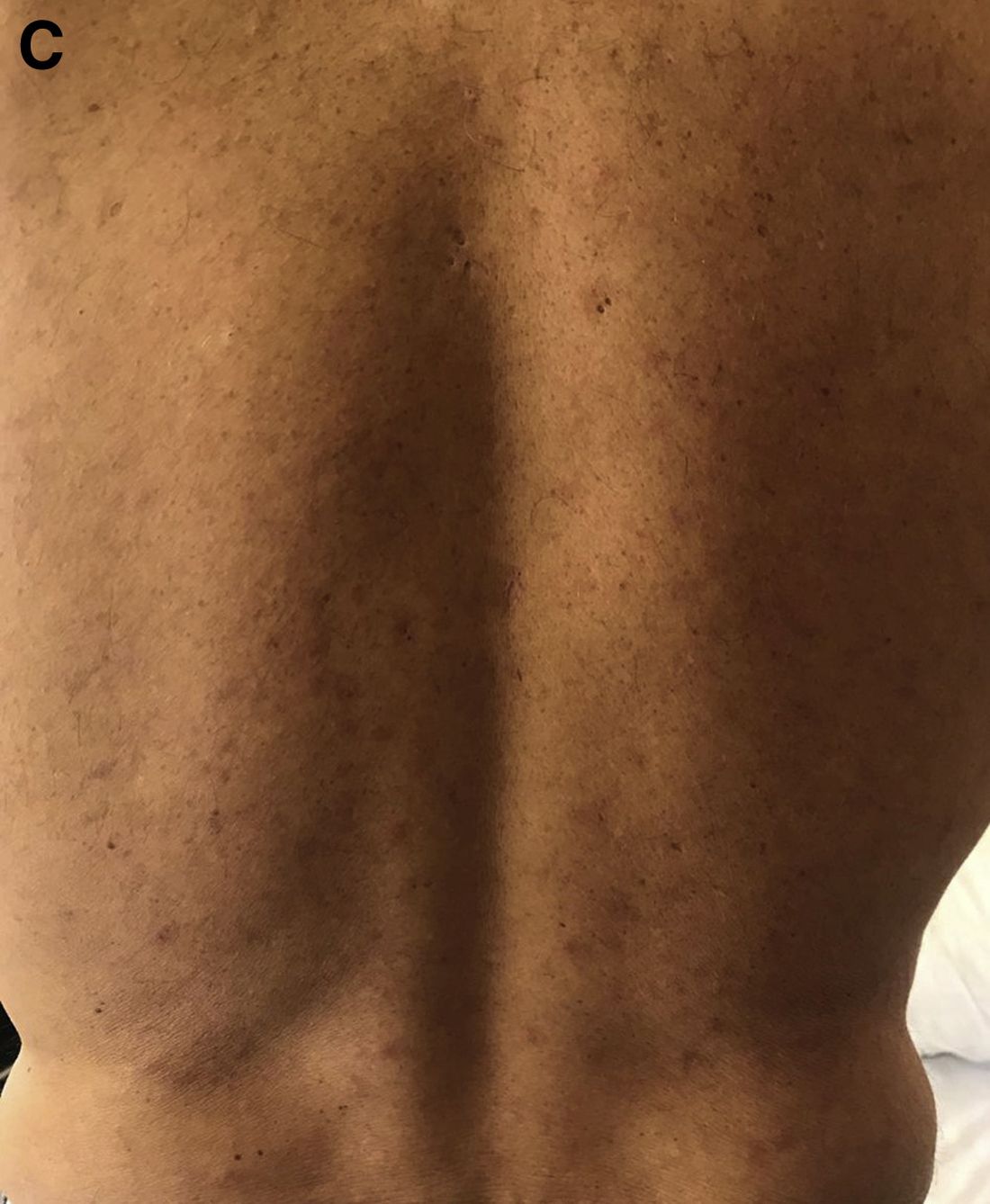
Although extremely rare, we checked a Venereal Disease Research Laboratory PCR, the results of which came back positive. A Treponema pallidum hemagglutination assay also returned positive with titers 1:5120 (ULN, < 1:80), confirming the diagnosis of syphilis. During his hospitalization, the patient developed a syphilitic skin rash on his back, chest, palms, feet, and soles (Figure C). The patient was started on penicillin G, 4 million units intravenously every 4 hours. The fever broke 36 hours after antibiotic initiation and 48 hours later, his bilirubin started to downtrend, followed by the alkaline phosphatase and GGT 3 days later. His rash completely disappeared 5 days after antibiotic initiation. He received a total of 2 weeks of penicillin G intravenously at 24 million units a day and his liver enzymes normalized 7 weeks later.
Syphilitic hepatitis is extremely rare and occurs in 0.2% of patients with secondary syphilis.1 There are few cases of syphilitic hepatitis in HIV carriers reported in the literature. Of the described cases, only two patients had an undetectable viral load.2,3 The clinical presentation of syphilitic hepatitis includes jaundice, pruritus, nausea, and vomiting, in addition to generalized symptoms of fatigue, malaise, and weight loss. Biochemically, alkaline phosphatase and GGT are predominantly elevated with mild elevation in the transaminases. Few cases describe an elevation in the bilirubin. Diagnosis is made based on treponemal testing and/or evaluation of tissue for spirochetes on liver biopsy. The majority of cases used penicillin G with excellent response. Doxycycline was also used in one case and ceftriaxone was used in another.
In our case, the patient had several other possible reasons for his liver enzyme elevation, including drug-induced liver injury, cocaine, and alcohol use, which could have contributed to his disturbed liver enzymes. The steady improvement in his cholestatic liver enzymes, fever, and rash, shortly after the initiation of penicillin G indicates that syphilis was the cause of his hepatitis. Given the improvement in his symptoms and biochemical markers, we refrained from obtaining a liver biopsy.
References
1. Lee M., Wang C., Dorer R. et al. A great masquerader: Acute syphilitic hepatitis. Dig Dis Sci. 2013;58:923-5.
2. Mullick CJ. Liappis A.P. Benator D.A. et al. Syphilitic hepatitis in HIV-infected patients: A report of 7 cases and review of the literature. Clin Infect Dis. 2004;39:e100-e105.
3. German MN. Matkowskyj K.A. Hoffman R.J. et al. A case of syphilitic hepatitis in an HIV-infected patient. Hum Pathol. 2018;79:184-7.
Syphilis
Although extremely rare, we checked a Venereal Disease Research Laboratory PCR, the results of which came back positive. A Treponema pallidum hemagglutination assay also returned positive with titers 1:5120 (ULN, < 1:80), confirming the diagnosis of syphilis. During his hospitalization, the patient developed a syphilitic skin rash on his back, chest, palms, feet, and soles (Figure C). The patient was started on penicillin G, 4 million units intravenously every 4 hours. The fever broke 36 hours after antibiotic initiation and 48 hours later, his bilirubin started to downtrend, followed by the alkaline phosphatase and GGT 3 days later. His rash completely disappeared 5 days after antibiotic initiation. He received a total of 2 weeks of penicillin G intravenously at 24 million units a day and his liver enzymes normalized 7 weeks later.
Syphilitic hepatitis is extremely rare and occurs in 0.2% of patients with secondary syphilis.1 There are few cases of syphilitic hepatitis in HIV carriers reported in the literature. Of the described cases, only two patients had an undetectable viral load.2,3 The clinical presentation of syphilitic hepatitis includes jaundice, pruritus, nausea, and vomiting, in addition to generalized symptoms of fatigue, malaise, and weight loss. Biochemically, alkaline phosphatase and GGT are predominantly elevated with mild elevation in the transaminases. Few cases describe an elevation in the bilirubin. Diagnosis is made based on treponemal testing and/or evaluation of tissue for spirochetes on liver biopsy. The majority of cases used penicillin G with excellent response. Doxycycline was also used in one case and ceftriaxone was used in another.
In our case, the patient had several other possible reasons for his liver enzyme elevation, including drug-induced liver injury, cocaine, and alcohol use, which could have contributed to his disturbed liver enzymes. The steady improvement in his cholestatic liver enzymes, fever, and rash, shortly after the initiation of penicillin G indicates that syphilis was the cause of his hepatitis. Given the improvement in his symptoms and biochemical markers, we refrained from obtaining a liver biopsy.
References
1. Lee M., Wang C., Dorer R. et al. A great masquerader: Acute syphilitic hepatitis. Dig Dis Sci. 2013;58:923-5.
2. Mullick CJ. Liappis A.P. Benator D.A. et al. Syphilitic hepatitis in HIV-infected patients: A report of 7 cases and review of the literature. Clin Infect Dis. 2004;39:e100-e105.
3. German MN. Matkowskyj K.A. Hoffman R.J. et al. A case of syphilitic hepatitis in an HIV-infected patient. Hum Pathol. 2018;79:184-7.
Syphilis
Although extremely rare, we checked a Venereal Disease Research Laboratory PCR, the results of which came back positive. A Treponema pallidum hemagglutination assay also returned positive with titers 1:5120 (ULN, < 1:80), confirming the diagnosis of syphilis. During his hospitalization, the patient developed a syphilitic skin rash on his back, chest, palms, feet, and soles (Figure C). The patient was started on penicillin G, 4 million units intravenously every 4 hours. The fever broke 36 hours after antibiotic initiation and 48 hours later, his bilirubin started to downtrend, followed by the alkaline phosphatase and GGT 3 days later. His rash completely disappeared 5 days after antibiotic initiation. He received a total of 2 weeks of penicillin G intravenously at 24 million units a day and his liver enzymes normalized 7 weeks later.
Syphilitic hepatitis is extremely rare and occurs in 0.2% of patients with secondary syphilis.1 There are few cases of syphilitic hepatitis in HIV carriers reported in the literature. Of the described cases, only two patients had an undetectable viral load.2,3 The clinical presentation of syphilitic hepatitis includes jaundice, pruritus, nausea, and vomiting, in addition to generalized symptoms of fatigue, malaise, and weight loss. Biochemically, alkaline phosphatase and GGT are predominantly elevated with mild elevation in the transaminases. Few cases describe an elevation in the bilirubin. Diagnosis is made based on treponemal testing and/or evaluation of tissue for spirochetes on liver biopsy. The majority of cases used penicillin G with excellent response. Doxycycline was also used in one case and ceftriaxone was used in another.
In our case, the patient had several other possible reasons for his liver enzyme elevation, including drug-induced liver injury, cocaine, and alcohol use, which could have contributed to his disturbed liver enzymes. The steady improvement in his cholestatic liver enzymes, fever, and rash, shortly after the initiation of penicillin G indicates that syphilis was the cause of his hepatitis. Given the improvement in his symptoms and biochemical markers, we refrained from obtaining a liver biopsy.
References
1. Lee M., Wang C., Dorer R. et al. A great masquerader: Acute syphilitic hepatitis. Dig Dis Sci. 2013;58:923-5.
2. Mullick CJ. Liappis A.P. Benator D.A. et al. Syphilitic hepatitis in HIV-infected patients: A report of 7 cases and review of the literature. Clin Infect Dis. 2004;39:e100-e105.
3. German MN. Matkowskyj K.A. Hoffman R.J. et al. A case of syphilitic hepatitis in an HIV-infected patient. Hum Pathol. 2018;79:184-7.
A 48-year-old man with HIV infection (PCR undetectable CD4 483) who used cocaine and was a heavy user of alcohol presented with jaundice, fever, and acute-onset left-upper quadrant abdominal pain. The pain was exacerbated by breathing. He had associated intermittent fevers and weight loss starting 3 weeks before presentation. He denied chest pain, nausea, vomiting, or a change in bowel habits. Home medications included dolutegravir, emtricitabine, tenofovir disoproxil fumarate, and recent intake of tamoxifen, clomiphene, and chorionic gonadotropin to counteract the effects of anabolic steroids that were used 4 months before presentation.
On examination, his temperature was 39°C, he was jaundiced, and he had icteric sclera. The abdomen was soft and nondistended with minimal left-upper quadrant tenderness. Blood work showed a white blood cell count of 7,800/mcL, hemoglobin of 12.2 g/dL, platelets of 378,000/mcL, alanine aminotransferase 236 IU/L (upper limit of normal [ULN], 65 IU/L), aspartate aminotransferase 166 IU/L (ULN, 50 IU/L), total bilirubin 3.4 mg/dL (ULN, 1.2 mg/dL), direct bilirubin 2.6 mg/dL (ULN, 0.3 mg/dL), alkaline phosphatase 1,064 IU/L (ULN, 120 IU/L), gamma-glutamyl transferase (GGT) of 655 (ULN, 50 IU/L), protein of 73 g/L, and albumin of 34 g/L. Lipase, lactate dehydrogenase, and international normalized ratio were normal. Blood smear was unrevealing. A contrasted computed tomography scan showed multiple subcentimetric mesenteric and multiple retroperitoneal lymph nodes, the largest of which was 1.3 cm in the aortocaval area. All medications were discontinued. Hepatitis A, B, and C serologies were negative, including hepatitis B and C PCR. Epstein-Barr virus IgM was negative and cytomegalovirus IgM was equivocal.
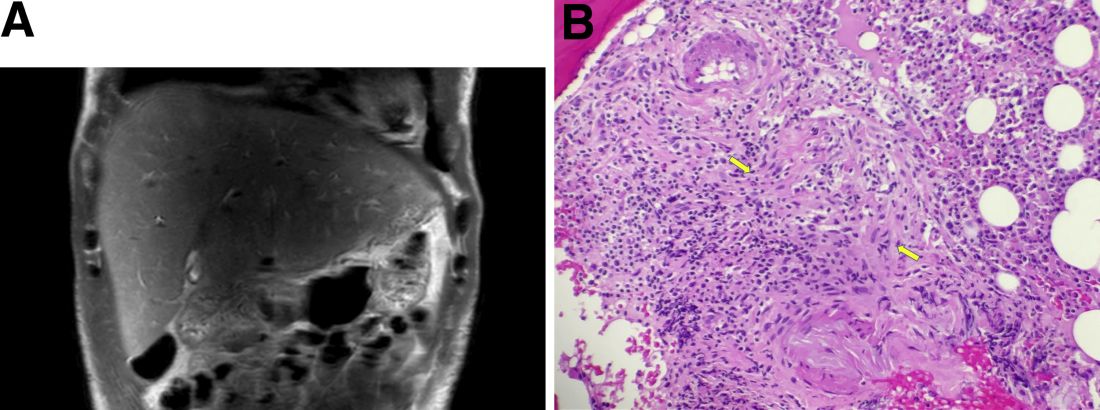
During this hospitalization, his cholestatic liver enzymes continued to rise, reaching a maximum value of total bilirubin of 7.8 mg/dL, direct bilirubin of 6.5 mg/dL, and 3 days later, alkaline phosphatase of 1,637 IU/L and GGT of 1,171 IU/L. Alanine aminotransferase and aspartate aminotransferase slowly downtrended during the hospitalization. Magnetic resonance cholangiopancreatography showed an edematous enlarged liver with minimal peripheral intrahepatic dilatation without an obstructing mass or extrahepatic biliary ductal dilatation (Figure A). Comprehensive autoimmune hepatic serology, iron studies, ceruloplasmin, and alpha-1 antitrypsin labs were negative. The patient remained febrile, so a positron emission tomography computed tomography scan was done and it showed active and enlarged (2.8-cm) portocaval and porta hepatis lymph nodes. Bone marrow biopsy showed no lymphoproliferative disorder, but there was a small poorly formed granuloma (Figure B, between the arrows).
What other testing would you obtain to evaluate this patient's fever and abnormal liver enzymes?
Previously published in Gastroenterology
What's your diagnosis?
Whipple's disease
The ultrasound features were highly suggestive of malabsorption, a hypothesis that was supported by the laboratory findings. Celiac disease, one of the most common causes of malabsorption, was excluded by serology tests. Esophagogastroduodenoscopy was therefore repeated: The mucosa of the distal first part and second part of the duodenum appeared completely covered with tiny white spots (Figure C). Histologic examination revealed that the mucosal architecture of the villi was altered by the presence of infiltrates of macrophages with wide cytoplasm filled with round periodic acid-Schiff (PAS)-positive inclusions, associated to aggregates of neutrophils attacking the epithelium (Figure D). These histologic findings are consistent with Whipple's disease.
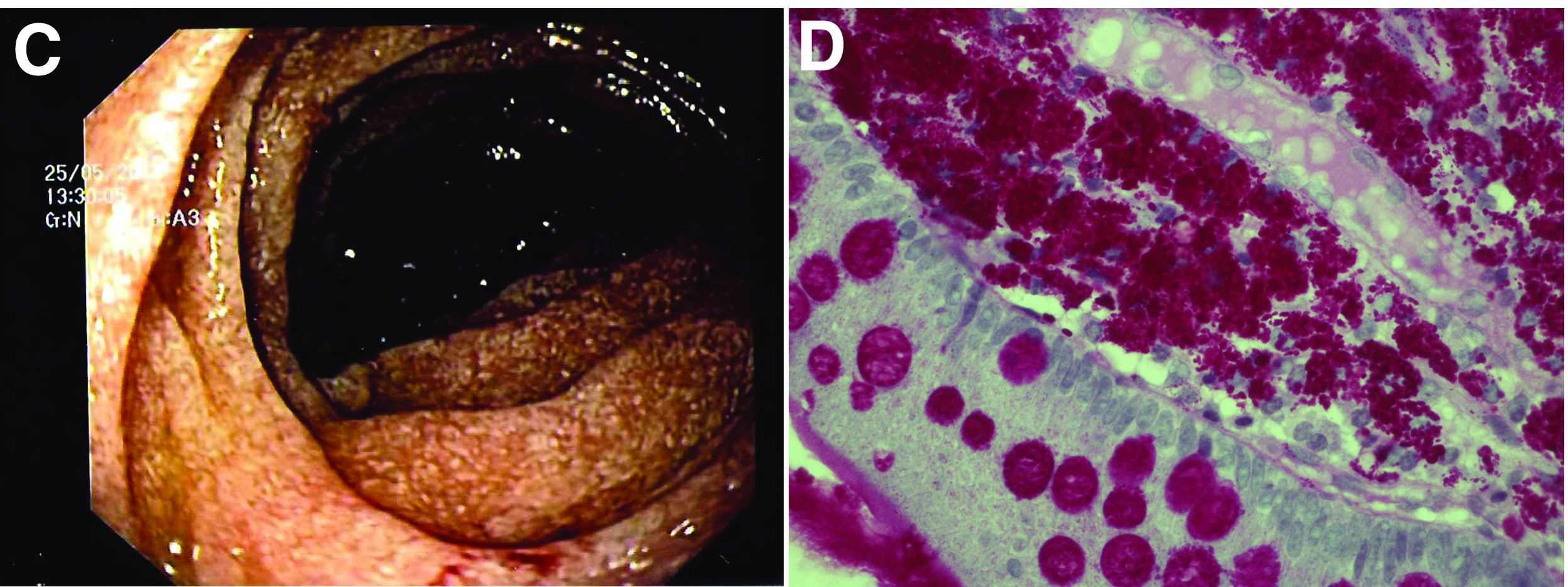
Whipple's disease is a chronic infectious disease caused by a gram-positive ubiquitous bacterium named Tropheryma whipplei. In predisposed subjects with an insufficient T-helper response, for example, those undergoing treatment with tumor necrosis factor-alpha inhibitors as in our patient, T. whipplei is able to survive and replicate inside the macrophages of the intestinal mucosa and to spread to other organs.1 Whipple's disease can thus manifest as a multisystemic disease or as a single-organ disease with extraintestinal involvement (e.g., central nervous system, eyes, heart, or lung). The classic form is characterized by weight loss, diarrhea, abdominal pain, and signs of malabsorption, typically preceded by a history of arthralgia. The arthralgia is often misdiagnosed as a form of rheumatoid arthritis and therefore treated with immunosuppressant therapy, which favors the onset of the classic intestinal symptoms.
In the literature, few case reports describe the ultrasound findings in patients with Whipple's disease. The most frequent sonographic features include small-bowel dilatation with wall thickening, the presence of peri-intestinal fluid effusion and mesenteric and retroperitoneal lymphadenopathy.2,3
The final diagnosis relies on intestinal biopsy and the histologic finding of foamy macrophages containing large amounts of diastase-resistant PAS-positive particles in the lamina propria of the duodenum, jejunum, ileum, or gastric antral region.
The diagnosis, particularly in cases of extraintestinal involvement, can be confirmed by polymerase chain reaction positivity for T. whipplei in the examined tissue.
Therapy consists of the administration of ceftriaxone (2 g IV once daily) for 2 weeks followed by oral therapy with trimethoprim-sulfamethoxazole for 1 year.
References
1. Schneider T et al. Whipple's disease: New aspects of pathogenesis and treatment. Lancet Infect Dis. 2008;8:179-90.
2. Brindicci D et al. Ultrasonic findings in Whipple's disease. J Clin Ultrasound. 1984;12:286-8.
3. Neye H et al. Der Morbus Whipple's Disease - A rare intestinal disease and its sonographic characteristics. Ultraschall Med. 2012;33(04):314-5.
Whipple's disease
The ultrasound features were highly suggestive of malabsorption, a hypothesis that was supported by the laboratory findings. Celiac disease, one of the most common causes of malabsorption, was excluded by serology tests. Esophagogastroduodenoscopy was therefore repeated: The mucosa of the distal first part and second part of the duodenum appeared completely covered with tiny white spots (Figure C). Histologic examination revealed that the mucosal architecture of the villi was altered by the presence of infiltrates of macrophages with wide cytoplasm filled with round periodic acid-Schiff (PAS)-positive inclusions, associated to aggregates of neutrophils attacking the epithelium (Figure D). These histologic findings are consistent with Whipple's disease.
Whipple's disease is a chronic infectious disease caused by a gram-positive ubiquitous bacterium named Tropheryma whipplei. In predisposed subjects with an insufficient T-helper response, for example, those undergoing treatment with tumor necrosis factor-alpha inhibitors as in our patient, T. whipplei is able to survive and replicate inside the macrophages of the intestinal mucosa and to spread to other organs.1 Whipple's disease can thus manifest as a multisystemic disease or as a single-organ disease with extraintestinal involvement (e.g., central nervous system, eyes, heart, or lung). The classic form is characterized by weight loss, diarrhea, abdominal pain, and signs of malabsorption, typically preceded by a history of arthralgia. The arthralgia is often misdiagnosed as a form of rheumatoid arthritis and therefore treated with immunosuppressant therapy, which favors the onset of the classic intestinal symptoms.
In the literature, few case reports describe the ultrasound findings in patients with Whipple's disease. The most frequent sonographic features include small-bowel dilatation with wall thickening, the presence of peri-intestinal fluid effusion and mesenteric and retroperitoneal lymphadenopathy.2,3
The final diagnosis relies on intestinal biopsy and the histologic finding of foamy macrophages containing large amounts of diastase-resistant PAS-positive particles in the lamina propria of the duodenum, jejunum, ileum, or gastric antral region.
The diagnosis, particularly in cases of extraintestinal involvement, can be confirmed by polymerase chain reaction positivity for T. whipplei in the examined tissue.
Therapy consists of the administration of ceftriaxone (2 g IV once daily) for 2 weeks followed by oral therapy with trimethoprim-sulfamethoxazole for 1 year.
References
1. Schneider T et al. Whipple's disease: New aspects of pathogenesis and treatment. Lancet Infect Dis. 2008;8:179-90.
2. Brindicci D et al. Ultrasonic findings in Whipple's disease. J Clin Ultrasound. 1984;12:286-8.
3. Neye H et al. Der Morbus Whipple's Disease - A rare intestinal disease and its sonographic characteristics. Ultraschall Med. 2012;33(04):314-5.
Whipple's disease
The ultrasound features were highly suggestive of malabsorption, a hypothesis that was supported by the laboratory findings. Celiac disease, one of the most common causes of malabsorption, was excluded by serology tests. Esophagogastroduodenoscopy was therefore repeated: The mucosa of the distal first part and second part of the duodenum appeared completely covered with tiny white spots (Figure C). Histologic examination revealed that the mucosal architecture of the villi was altered by the presence of infiltrates of macrophages with wide cytoplasm filled with round periodic acid-Schiff (PAS)-positive inclusions, associated to aggregates of neutrophils attacking the epithelium (Figure D). These histologic findings are consistent with Whipple's disease.
Whipple's disease is a chronic infectious disease caused by a gram-positive ubiquitous bacterium named Tropheryma whipplei. In predisposed subjects with an insufficient T-helper response, for example, those undergoing treatment with tumor necrosis factor-alpha inhibitors as in our patient, T. whipplei is able to survive and replicate inside the macrophages of the intestinal mucosa and to spread to other organs.1 Whipple's disease can thus manifest as a multisystemic disease or as a single-organ disease with extraintestinal involvement (e.g., central nervous system, eyes, heart, or lung). The classic form is characterized by weight loss, diarrhea, abdominal pain, and signs of malabsorption, typically preceded by a history of arthralgia. The arthralgia is often misdiagnosed as a form of rheumatoid arthritis and therefore treated with immunosuppressant therapy, which favors the onset of the classic intestinal symptoms.
In the literature, few case reports describe the ultrasound findings in patients with Whipple's disease. The most frequent sonographic features include small-bowel dilatation with wall thickening, the presence of peri-intestinal fluid effusion and mesenteric and retroperitoneal lymphadenopathy.2,3
The final diagnosis relies on intestinal biopsy and the histologic finding of foamy macrophages containing large amounts of diastase-resistant PAS-positive particles in the lamina propria of the duodenum, jejunum, ileum, or gastric antral region.
The diagnosis, particularly in cases of extraintestinal involvement, can be confirmed by polymerase chain reaction positivity for T. whipplei in the examined tissue.
Therapy consists of the administration of ceftriaxone (2 g IV once daily) for 2 weeks followed by oral therapy with trimethoprim-sulfamethoxazole for 1 year.
References
1. Schneider T et al. Whipple's disease: New aspects of pathogenesis and treatment. Lancet Infect Dis. 2008;8:179-90.
2. Brindicci D et al. Ultrasonic findings in Whipple's disease. J Clin Ultrasound. 1984;12:286-8.
3. Neye H et al. Der Morbus Whipple's Disease - A rare intestinal disease and its sonographic characteristics. Ultraschall Med. 2012;33(04):314-5.
67-year-old woman presented with a year-long history of general malaise, low-grade fever, diarrhea, and a 20-kg weight loss. She had a history of hypertension and depressive disorder. In the previous 4 years, she had undergone several rheumatologic examinations for polyarthritis and, having been diagnosed with seronegative rheumatoid arthritis, she had been treated with steroids, methotrexate, and etanercept, with little benefit.
Recent laboratory tests showed: hemoglobin, 8.3 g/dL; mean corpuscular volume, 70 fL; erythrocyte sedimentation rate, 78; and C-reactive protein, 6.4 mg/dL. To evaluate the microcytic anemia and the diarrhea, endoscopic investigations had been performed a few months earlier. Esophagogastroduodenoscopy showed villous atrophy at the level of DII; histology was compatible with intramucosal xanthoma. There were no pathologic findings at colonoscopy. The situation had not been further investigated.
At presentation, the physical examination revealed lower-limb edema, skin and mucosal pallor, and a body mass index of 17.4 kg/m2. Laboratory tests showed microcytic anemia (hemoglobin, 10.0 g/dL; mean corpuscular volume, 74 fL), increased acute-phase proteins (erythrocyte sedimentation rate, 59; C-reactive protein, 8.53 mg/dL), and malabsorption (albumin, 2.5 g/dL; multiple electrolytes deficiencies including iron, vitamin A, and vitamin D deficiency).
Abdominal ultrasound examination revealed three small lymph nodes in the periaortic region (maximum diameter, 10 mm), marked mesenteric and ileal wall thickening, mild jejunal wall thickening, an increased number of connivent valves, and a mild amount of peri-intestinal fluid effusion (Figure A, B).

What is the likely diagnosis and the appropriate treatment?
What's your diagnosis?
Pancreatic adenocarcinoma arising from main duct intraductal papillary mucinous neoplasm with inadvertent main pancreatic duct stenting.
The FNA was positive for carcinoma with abundant mucin, which, taken together with the imaging findings, was indicative of pancreatic adenocarcinoma arising from main duct intraductal papillary mucinous neoplasm (M-IPMN).
The post-endoscopic retrograde cholangiopancreatography (ERCP) CT revealed inadvertent placement of the fully covered self-expanding metallic stent (fcSEMS) within the main pancreatic duct (MPD) stricture and persistent common bile duct (CBD) obstruction. On post hoc review of the fluoroscopic and cross-sectional imaging, it became evident that the massively dilated MPD was mistaken during ERCP for the CBD and left hepatic duct (Figure F). In addition, the patient also had several cysts within the liver (compatible with incidental polycystic liver disease), which further complicated real-time image interpretation.
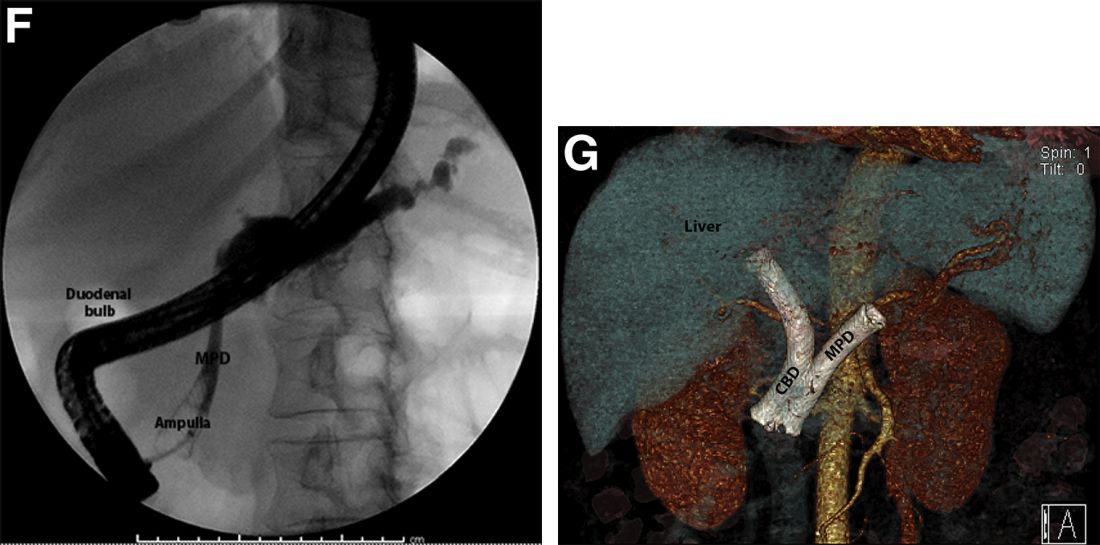
Based on multidisciplinary discussion, the precedent of a prior series of successful palliative MPD stenting in the setting of adenocarcinoma,1 and the notable improvement in the patient's steatorrhea and abdominal pain, the initially placed fcSEMS was left in situ across the MPD stricture, and a second fcSEMS was successfully deployed across the CBD stricture (Figure G), resulting in prompt improvement in serum liver tests. The patient was thereafter initiated on palliative chemotherapy with gemcitabine and abraxane and has maintained clinically stable disease for the last 9 months.
M-IPMN is a premalignant condition in which endoscopy plays an important role. In our patient, because of anatomic and morphologic abnormalities, including the massive dilation of the MPD and severe distal biliary compression in the context of an obstructing pancreatic head mass arising from M-IPMN, initial deployment of the fcSEMS occurred unwittingly into the MPD. Little is known about the impact of fcSEMS in the MPD in patients with pancreatic adenocarcinoma, although in select cases, alleviation of pain caused by MPD obstruction and improvement in quality of life have been reported.2,3 In the case of our patient, fcSEMS placement in the MPD indeed led to symptomatic relief as manifested by a decrease in both diarrhea and pain and an increase in appetite; the addition of a fcSEMS in the CBD led to serum liver test normalization and permitted the initiation of chemotherapy. Further studies are needed to examine the outcomes of palliative MPD stenting in patients with obstructing pancreatic malignancies as well as the epidemiology and biology of M-IPMN and associated pancreatic adenocarcinoma in minority populations.
References
1. Tham TC et al. Am J Gastroenterol. 2000 Apr;95(4):956-60.
2. Grimm IS, Baron TH. Gastroenterology. 2015 Jul;149(1):20-2.
3. Wehrmann T et al. Eur J Gastroenterol Hepatol. 2005 Dec;17(12):1395-400.
Pancreatic adenocarcinoma arising from main duct intraductal papillary mucinous neoplasm with inadvertent main pancreatic duct stenting.
The FNA was positive for carcinoma with abundant mucin, which, taken together with the imaging findings, was indicative of pancreatic adenocarcinoma arising from main duct intraductal papillary mucinous neoplasm (M-IPMN).
The post-endoscopic retrograde cholangiopancreatography (ERCP) CT revealed inadvertent placement of the fully covered self-expanding metallic stent (fcSEMS) within the main pancreatic duct (MPD) stricture and persistent common bile duct (CBD) obstruction. On post hoc review of the fluoroscopic and cross-sectional imaging, it became evident that the massively dilated MPD was mistaken during ERCP for the CBD and left hepatic duct (Figure F). In addition, the patient also had several cysts within the liver (compatible with incidental polycystic liver disease), which further complicated real-time image interpretation.
Based on multidisciplinary discussion, the precedent of a prior series of successful palliative MPD stenting in the setting of adenocarcinoma,1 and the notable improvement in the patient's steatorrhea and abdominal pain, the initially placed fcSEMS was left in situ across the MPD stricture, and a second fcSEMS was successfully deployed across the CBD stricture (Figure G), resulting in prompt improvement in serum liver tests. The patient was thereafter initiated on palliative chemotherapy with gemcitabine and abraxane and has maintained clinically stable disease for the last 9 months.
M-IPMN is a premalignant condition in which endoscopy plays an important role. In our patient, because of anatomic and morphologic abnormalities, including the massive dilation of the MPD and severe distal biliary compression in the context of an obstructing pancreatic head mass arising from M-IPMN, initial deployment of the fcSEMS occurred unwittingly into the MPD. Little is known about the impact of fcSEMS in the MPD in patients with pancreatic adenocarcinoma, although in select cases, alleviation of pain caused by MPD obstruction and improvement in quality of life have been reported.2,3 In the case of our patient, fcSEMS placement in the MPD indeed led to symptomatic relief as manifested by a decrease in both diarrhea and pain and an increase in appetite; the addition of a fcSEMS in the CBD led to serum liver test normalization and permitted the initiation of chemotherapy. Further studies are needed to examine the outcomes of palliative MPD stenting in patients with obstructing pancreatic malignancies as well as the epidemiology and biology of M-IPMN and associated pancreatic adenocarcinoma in minority populations.
References
1. Tham TC et al. Am J Gastroenterol. 2000 Apr;95(4):956-60.
2. Grimm IS, Baron TH. Gastroenterology. 2015 Jul;149(1):20-2.
3. Wehrmann T et al. Eur J Gastroenterol Hepatol. 2005 Dec;17(12):1395-400.
Pancreatic adenocarcinoma arising from main duct intraductal papillary mucinous neoplasm with inadvertent main pancreatic duct stenting.
The FNA was positive for carcinoma with abundant mucin, which, taken together with the imaging findings, was indicative of pancreatic adenocarcinoma arising from main duct intraductal papillary mucinous neoplasm (M-IPMN).
The post-endoscopic retrograde cholangiopancreatography (ERCP) CT revealed inadvertent placement of the fully covered self-expanding metallic stent (fcSEMS) within the main pancreatic duct (MPD) stricture and persistent common bile duct (CBD) obstruction. On post hoc review of the fluoroscopic and cross-sectional imaging, it became evident that the massively dilated MPD was mistaken during ERCP for the CBD and left hepatic duct (Figure F). In addition, the patient also had several cysts within the liver (compatible with incidental polycystic liver disease), which further complicated real-time image interpretation.
Based on multidisciplinary discussion, the precedent of a prior series of successful palliative MPD stenting in the setting of adenocarcinoma,1 and the notable improvement in the patient's steatorrhea and abdominal pain, the initially placed fcSEMS was left in situ across the MPD stricture, and a second fcSEMS was successfully deployed across the CBD stricture (Figure G), resulting in prompt improvement in serum liver tests. The patient was thereafter initiated on palliative chemotherapy with gemcitabine and abraxane and has maintained clinically stable disease for the last 9 months.
M-IPMN is a premalignant condition in which endoscopy plays an important role. In our patient, because of anatomic and morphologic abnormalities, including the massive dilation of the MPD and severe distal biliary compression in the context of an obstructing pancreatic head mass arising from M-IPMN, initial deployment of the fcSEMS occurred unwittingly into the MPD. Little is known about the impact of fcSEMS in the MPD in patients with pancreatic adenocarcinoma, although in select cases, alleviation of pain caused by MPD obstruction and improvement in quality of life have been reported.2,3 In the case of our patient, fcSEMS placement in the MPD indeed led to symptomatic relief as manifested by a decrease in both diarrhea and pain and an increase in appetite; the addition of a fcSEMS in the CBD led to serum liver test normalization and permitted the initiation of chemotherapy. Further studies are needed to examine the outcomes of palliative MPD stenting in patients with obstructing pancreatic malignancies as well as the epidemiology and biology of M-IPMN and associated pancreatic adenocarcinoma in minority populations.
References
1. Tham TC et al. Am J Gastroenterol. 2000 Apr;95(4):956-60.
2. Grimm IS, Baron TH. Gastroenterology. 2015 Jul;149(1):20-2.
3. Wehrmann T et al. Eur J Gastroenterol Hepatol. 2005 Dec;17(12):1395-400.
A 69-year-old Filipino American woman presented with increasing epigastralgia, worsening appetite, jaundice, and oily diarrhea over the course of 3 months. Her past medical history consisted of diabetes, hypertension, hyperlipidemia, and osteopenia being managed with metformin, losartan, and atorvastatin, respectively.
Physical examination revealed she was thin (body mass index, 22 kg/m2) and jaundiced with moderate tenderness to epigastric palpation and 1+ peripheral pitting edema. Laboratory tests were significant for normal complete blood count and elevated alanine aminotransferase (113 U/L), alkaline phosphatase (235 U/L), bilirubin (7.3 mg/dL), international normalized ratio (1.3), and carbohydrate antigen 19-9 (7886 U/L). A CT scan of the abdomen revealed severe extrahepatic and intrahepatic ductal dilation, with a common bile duct (CBD) and main pancreatic duct (MPD) diameter of 2.5 and 1.7 cm, respectively, as well an infiltrating, malignant-appearing, 4.5-cm spheroid mass in the head of the pancreas (Figure A). The mass involved the superior mesenteric vein at the portal confluence and encased >50% of the superior mesenteric artery.

To further characterize these findings, magnetic resonance cholangiopancreatography was performed, which additionally revealed multifocal cysts throughout the liver ranging from 0.5 to 5.0 cm in greatest diameter, as seen on maximal intensity projection algorithm (Figure B).
The patient was referred for same-session endoscopic ultrasound examination with fine needle aspiration (FNA) and endoscopic retrograde cholangiopancreatography (ERCP) for further diagnosis and treatment. Endoscopic ultrasound demonstrated a large, hypoechoic mass in the pancreatic head with severe CBD and MPD dilation proximally, corresponding with the cross-sectional imaging findings; FNA was performed. ERCP demonstrated a long, distal CBD stricture and what appeared to be nonopacification of the right hepatic ductal system; a 10 × 60-mm fully covered self-expanding metallic stent (fcSEMS) was placed across the stricture (Figure C, D). Over the subsequent 3 days, the patient's diarrhea resolved and epigastralgia improved; however, serum liver tests did not downtrend, thus prompting repeat imaging (Figure E).
Based on the patient's clinical history, cross-sectional imaging findings, and only partial response to therapeutic ERCP, what are the patient's likely diagnoses?
What's your diagnosis?
Answer: Colonic Malakoplakia.
Histopathologic examination of the biopsy specimens revealed nodular mixed inflammatory cells and infiltration of the epithelioid histiocytes in lamina propria (Figure D; stain: hematoxylin and eosin; original magnification 40×). The histiocytes showed foamy and eosinophilic cytoplasm (Figure E, arrow) and some of them had a targetoid appearance (Figure E, arrow head; stain: hematoxylin and eosin; original magnification 200×). Von Kossa stains highlighted the targetoid structures in the histiocytes (Figure F, Michaelis-Gutmann bodies). The granular cytoplasm of the histiocytes was positive on periodic acid-Schiff stain (Figure G). Based on these findings, the patient was diagnosed with colonic malakoplakia.
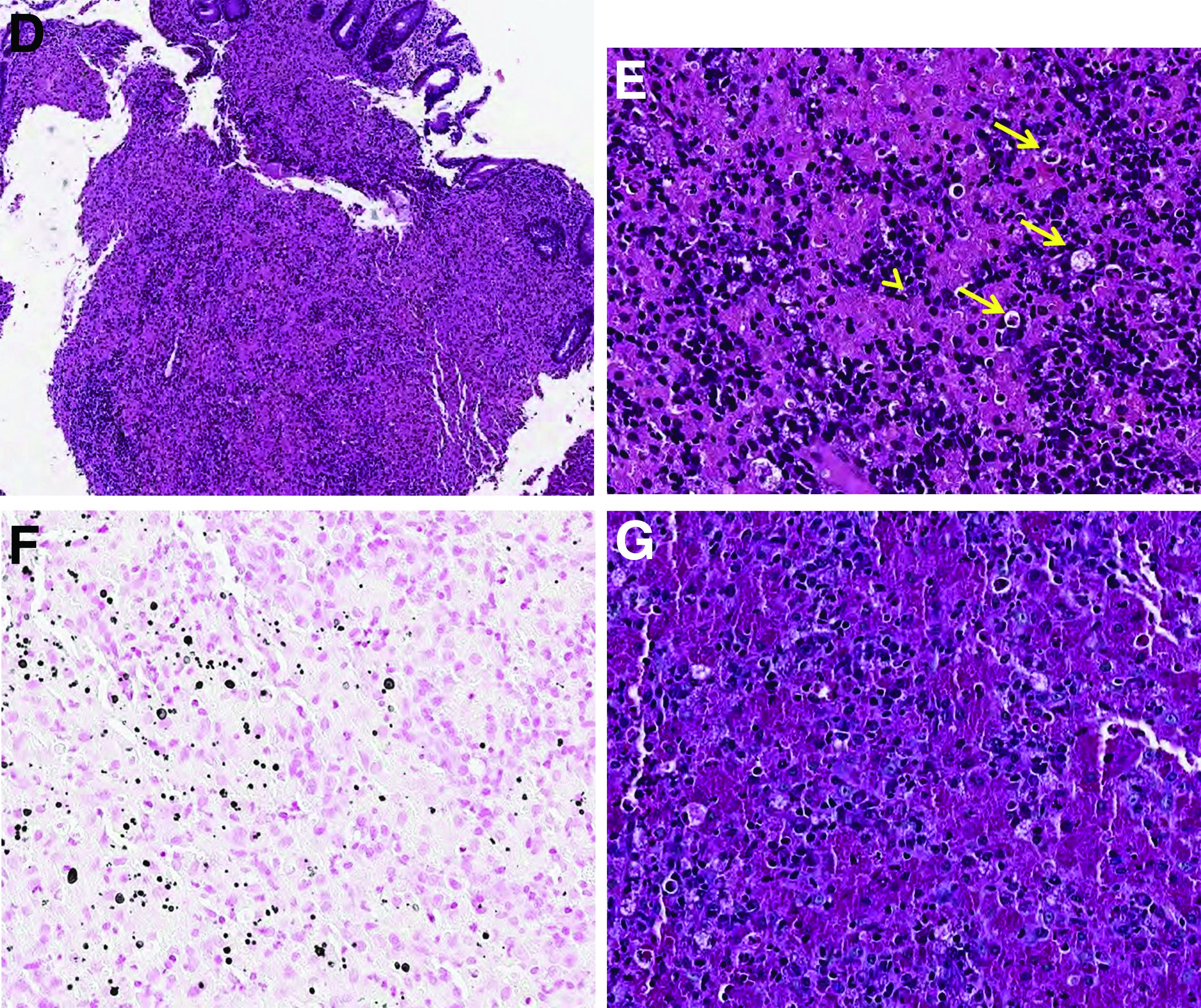
Malakoplakia is an uncommon, chronic, granulomatous inflammatory disease. It most commonly affects the urinary tract and gastrointestinal tract, but may occur at any anatomic site. Malakoplakia of the gastrointestinal tract are seen most frequently in the rectum, sigmoid, and right colon.1 It is diagnosed by the characteristic histologic feature of accumulated histiocytes with abundant eosinophilic granular cytoplasm containing basophilic inclusions, consistent with Michaelis-Gutmann bodies. Although the exact etiology and pathogenesis of malakoplakia are unclear, it seems to originate from an acquired defect in the intracellular destruction of phagocytosed bacteria, usually associated with Escherichia coli, Klebsiella, and Mycobacterium.2 It can have various causes, such as immunosuppression, malignant neoplasms, systemic diseases, and genetic diseases. Clinical manifestation of colonic malakoplakia is diverse, ranging from asymptomatic to malaise, fever, abdominal pain, diarrhea, hematochezia, and intestinal obstruction. Granulomatous reaction of malakoplakia generates the endoscopic appearance of lesions, which ranges from plaques to nodules and yellow-brown masses. In the early stage, malakoplakia commonly presents as soft yellow to tan mucosal plaques endoscopically, as seen in our case (Figure A). As the disease progresses in the later stage, malakoplakia presents as raised, grey to tan polypoid lesions of various sizes with peripheral hyperemia and a central depressed area, as seen in our case (Figure B).3 Owing to this endoscopic morphology, colonic malakoplakia may be misdiagnosed as atypical lymphoma, familial adenomatous polyposis, and metastatic carcinoma. To date, the natural course of malakoplakia of the colon is unclear, and no guidelines for treatment, treatment methods, duration of treatment, or surveillance are currently available. However, treatment of malakoplakia is essential to reduce immunosuppression and includes antibiotics with intracellular action and choline agonists that replenish the decreased cyclic 3’, 5’-guanosine monophosphate levels. In summary, although malakoplakia of the colon is very rare, it should be considered in the differential diagnosis of polypoid colonic lesions, especially in immunocompromised or malnourished patients.
References
1. Cipolletta L et al. Gastrointest Endosc. 1995 Mar;41(3):255-8.
2. Berney T et al. Transpl Int. 1999;12(4):293-6.
3. Weinrach DM et al. Arch Pathol Lab Med. 2004 Oct;128(10):e133-4.
Answer: Colonic Malakoplakia.
Histopathologic examination of the biopsy specimens revealed nodular mixed inflammatory cells and infiltration of the epithelioid histiocytes in lamina propria (Figure D; stain: hematoxylin and eosin; original magnification 40×). The histiocytes showed foamy and eosinophilic cytoplasm (Figure E, arrow) and some of them had a targetoid appearance (Figure E, arrow head; stain: hematoxylin and eosin; original magnification 200×). Von Kossa stains highlighted the targetoid structures in the histiocytes (Figure F, Michaelis-Gutmann bodies). The granular cytoplasm of the histiocytes was positive on periodic acid-Schiff stain (Figure G). Based on these findings, the patient was diagnosed with colonic malakoplakia.
Malakoplakia is an uncommon, chronic, granulomatous inflammatory disease. It most commonly affects the urinary tract and gastrointestinal tract, but may occur at any anatomic site. Malakoplakia of the gastrointestinal tract are seen most frequently in the rectum, sigmoid, and right colon.1 It is diagnosed by the characteristic histologic feature of accumulated histiocytes with abundant eosinophilic granular cytoplasm containing basophilic inclusions, consistent with Michaelis-Gutmann bodies. Although the exact etiology and pathogenesis of malakoplakia are unclear, it seems to originate from an acquired defect in the intracellular destruction of phagocytosed bacteria, usually associated with Escherichia coli, Klebsiella, and Mycobacterium.2 It can have various causes, such as immunosuppression, malignant neoplasms, systemic diseases, and genetic diseases. Clinical manifestation of colonic malakoplakia is diverse, ranging from asymptomatic to malaise, fever, abdominal pain, diarrhea, hematochezia, and intestinal obstruction. Granulomatous reaction of malakoplakia generates the endoscopic appearance of lesions, which ranges from plaques to nodules and yellow-brown masses. In the early stage, malakoplakia commonly presents as soft yellow to tan mucosal plaques endoscopically, as seen in our case (Figure A). As the disease progresses in the later stage, malakoplakia presents as raised, grey to tan polypoid lesions of various sizes with peripheral hyperemia and a central depressed area, as seen in our case (Figure B).3 Owing to this endoscopic morphology, colonic malakoplakia may be misdiagnosed as atypical lymphoma, familial adenomatous polyposis, and metastatic carcinoma. To date, the natural course of malakoplakia of the colon is unclear, and no guidelines for treatment, treatment methods, duration of treatment, or surveillance are currently available. However, treatment of malakoplakia is essential to reduce immunosuppression and includes antibiotics with intracellular action and choline agonists that replenish the decreased cyclic 3’, 5’-guanosine monophosphate levels. In summary, although malakoplakia of the colon is very rare, it should be considered in the differential diagnosis of polypoid colonic lesions, especially in immunocompromised or malnourished patients.
References
1. Cipolletta L et al. Gastrointest Endosc. 1995 Mar;41(3):255-8.
2. Berney T et al. Transpl Int. 1999;12(4):293-6.
3. Weinrach DM et al. Arch Pathol Lab Med. 2004 Oct;128(10):e133-4.
Answer: Colonic Malakoplakia.
Histopathologic examination of the biopsy specimens revealed nodular mixed inflammatory cells and infiltration of the epithelioid histiocytes in lamina propria (Figure D; stain: hematoxylin and eosin; original magnification 40×). The histiocytes showed foamy and eosinophilic cytoplasm (Figure E, arrow) and some of them had a targetoid appearance (Figure E, arrow head; stain: hematoxylin and eosin; original magnification 200×). Von Kossa stains highlighted the targetoid structures in the histiocytes (Figure F, Michaelis-Gutmann bodies). The granular cytoplasm of the histiocytes was positive on periodic acid-Schiff stain (Figure G). Based on these findings, the patient was diagnosed with colonic malakoplakia.
Malakoplakia is an uncommon, chronic, granulomatous inflammatory disease. It most commonly affects the urinary tract and gastrointestinal tract, but may occur at any anatomic site. Malakoplakia of the gastrointestinal tract are seen most frequently in the rectum, sigmoid, and right colon.1 It is diagnosed by the characteristic histologic feature of accumulated histiocytes with abundant eosinophilic granular cytoplasm containing basophilic inclusions, consistent with Michaelis-Gutmann bodies. Although the exact etiology and pathogenesis of malakoplakia are unclear, it seems to originate from an acquired defect in the intracellular destruction of phagocytosed bacteria, usually associated with Escherichia coli, Klebsiella, and Mycobacterium.2 It can have various causes, such as immunosuppression, malignant neoplasms, systemic diseases, and genetic diseases. Clinical manifestation of colonic malakoplakia is diverse, ranging from asymptomatic to malaise, fever, abdominal pain, diarrhea, hematochezia, and intestinal obstruction. Granulomatous reaction of malakoplakia generates the endoscopic appearance of lesions, which ranges from plaques to nodules and yellow-brown masses. In the early stage, malakoplakia commonly presents as soft yellow to tan mucosal plaques endoscopically, as seen in our case (Figure A). As the disease progresses in the later stage, malakoplakia presents as raised, grey to tan polypoid lesions of various sizes with peripheral hyperemia and a central depressed area, as seen in our case (Figure B).3 Owing to this endoscopic morphology, colonic malakoplakia may be misdiagnosed as atypical lymphoma, familial adenomatous polyposis, and metastatic carcinoma. To date, the natural course of malakoplakia of the colon is unclear, and no guidelines for treatment, treatment methods, duration of treatment, or surveillance are currently available. However, treatment of malakoplakia is essential to reduce immunosuppression and includes antibiotics with intracellular action and choline agonists that replenish the decreased cyclic 3’, 5’-guanosine monophosphate levels. In summary, although malakoplakia of the colon is very rare, it should be considered in the differential diagnosis of polypoid colonic lesions, especially in immunocompromised or malnourished patients.
References
1. Cipolletta L et al. Gastrointest Endosc. 1995 Mar;41(3):255-8.
2. Berney T et al. Transpl Int. 1999;12(4):293-6.
3. Weinrach DM et al. Arch Pathol Lab Med. 2004 Oct;128(10):e133-4.
A 60-year-old man with C3 tetraplegia was referred to our department for evaluation of abdominal pain and hematochezia. He was diagnosed with adrenal insufficiency 5 years prior and has been taking low-dose prednisolone (7.5 mg) once a day. One year before presentation, he complained of intermittent loose, mucoid stool and abdominal pain. Sigmoidoscopy revealed multiple small yellowish plaques in the sigmoid colon (Figure A). However, symptoms improved without any treatment, and he was discharged from the rehabilitation department. He was readmitted for respiratory rehabilitation owing to dyspnea. On hospital day 4, he complained of abdominal pain and passing loose stool with foul odor 4-5 times a day. On hospital day 7, the abdominal pain worsened, and hematochezia occurred.
On physical examination, he was hemodynamically stable and afebrile. The abdomen was soft with mild tenderness on palpation in the periumbilical area without peritoneal signs. Laboratory studies were notable with a hemoglobin level of 10.7 g/dL, total protein of 4.09 g/dL, and albumin of 2.21 g/dL. Inflammatory marker (C-reactive protein) was mildly elevated to 1.83 mg/dL. Serology for human immunodeficiency virus was negative. Tumor markers, such as carcinoembryonic antigen, carbohydrate antigenic determinant, and alpha-fetoprotein, were within the normal range. Antineutrophil cytoplasmic antibody was negative, and rheumatic factor was within the normal range. Findings from stool for acid-fast bacillus and Clostridioides difficile toxin were negative; no pathogens were cultured, and no parasites were identified.
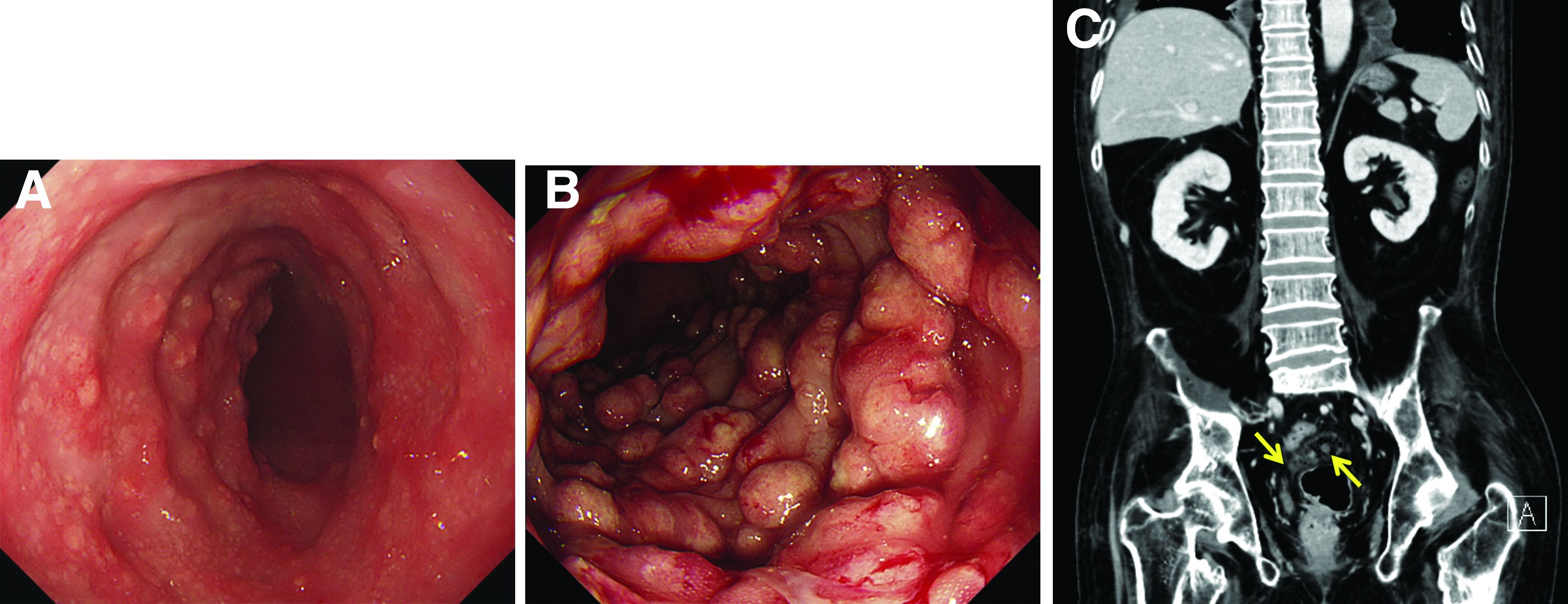
Sigmoidoscopy revealed diverse, multiple polypoid lesions (3-10 mm) with erythema, edema, and friability surrounding the entire lumen on the sigmoid colon (Figure B). The number and size of the polypoid lesions increased compared with the endoscopic findings obtained 1 year prior. The lesions easily bled on contact. Multiple biopsies of different sites were taken. An abdominal computed tomography scan showed multiple polyps of <1 cm that were confined to the sigmoid colon (Figure C, arrow).
Based on this information, what is the most likely diagnosis?
Previously published in Gastroenterology (2020 Feb;158[3]:482-4).
What's your diagnosis?
Mucous membrane pemphigoid with esophageal web stricture.
Additional laboratory examination showed that his serum anti-BP180 antibody level was high (11.7 U/mL; normal range, <9.0 U/mL). Biopsy specimens taken from the laryngopharyngeal erosion showed subepithelial blister formation and it was consistent with pemphigoid pathologically (Figure D). He did not have cutaneous lesions and was diagnosed with mucous membrane pemphigoid (MMP). After performing endoscopic dilation, prednisolone (20 mg/d) was administered orally. Three months after starting the prednisolone treatment, follow-up endoscopy showed improvements of the laryngopharyngeal erosions (Figure E) and esophageal blister on the web. However, esophageal narrowing remained, and thus endoscopic balloon dilation was performed (Figure F-H). Three months after the dilation, the narrowing improved (Figure I).
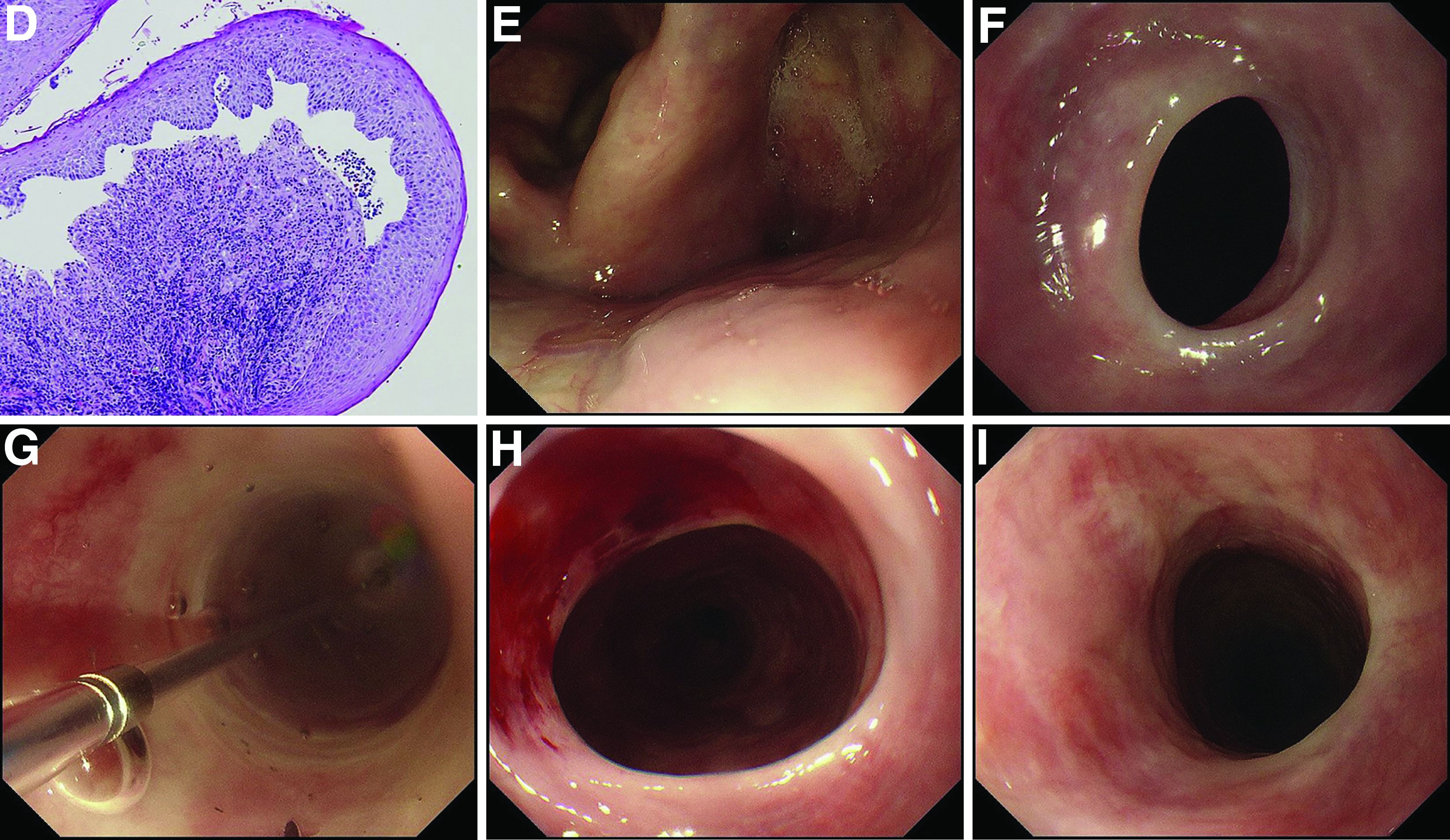
MMP is an autoimmune blistering disease that induces the formation of mucous membrane subepithelial bullae. Basement membrane zone components such as collagen XVII (also known as BP180) are targets of autoantibodies in MMP. Symptomatic esophageal involvement affects 5.4% of patients with MMP and dysphagia is the most frequent symptom.1 Endoscopic findings include erosion, web stricture, subepithelial hematomas, and scars.2,3 Endoscopic dilation is sometimes necessary for the treatment of severe esophageal strictures.1
References
1. Zehou O et al. Br J Dermatol. 2017 Oct;177(4):1074-85.
2. Sallout H et al. Gastrointest Endosc. 2000 Sep;52(3):429-33.
3. Gaspar R et al. Gastrointest Endosc. 2017 Aug;86(2):400-2.
Mucous membrane pemphigoid with esophageal web stricture.
Additional laboratory examination showed that his serum anti-BP180 antibody level was high (11.7 U/mL; normal range, <9.0 U/mL). Biopsy specimens taken from the laryngopharyngeal erosion showed subepithelial blister formation and it was consistent with pemphigoid pathologically (Figure D). He did not have cutaneous lesions and was diagnosed with mucous membrane pemphigoid (MMP). After performing endoscopic dilation, prednisolone (20 mg/d) was administered orally. Three months after starting the prednisolone treatment, follow-up endoscopy showed improvements of the laryngopharyngeal erosions (Figure E) and esophageal blister on the web. However, esophageal narrowing remained, and thus endoscopic balloon dilation was performed (Figure F-H). Three months after the dilation, the narrowing improved (Figure I).
MMP is an autoimmune blistering disease that induces the formation of mucous membrane subepithelial bullae. Basement membrane zone components such as collagen XVII (also known as BP180) are targets of autoantibodies in MMP. Symptomatic esophageal involvement affects 5.4% of patients with MMP and dysphagia is the most frequent symptom.1 Endoscopic findings include erosion, web stricture, subepithelial hematomas, and scars.2,3 Endoscopic dilation is sometimes necessary for the treatment of severe esophageal strictures.1
References
1. Zehou O et al. Br J Dermatol. 2017 Oct;177(4):1074-85.
2. Sallout H et al. Gastrointest Endosc. 2000 Sep;52(3):429-33.
3. Gaspar R et al. Gastrointest Endosc. 2017 Aug;86(2):400-2.
Mucous membrane pemphigoid with esophageal web stricture.
Additional laboratory examination showed that his serum anti-BP180 antibody level was high (11.7 U/mL; normal range, <9.0 U/mL). Biopsy specimens taken from the laryngopharyngeal erosion showed subepithelial blister formation and it was consistent with pemphigoid pathologically (Figure D). He did not have cutaneous lesions and was diagnosed with mucous membrane pemphigoid (MMP). After performing endoscopic dilation, prednisolone (20 mg/d) was administered orally. Three months after starting the prednisolone treatment, follow-up endoscopy showed improvements of the laryngopharyngeal erosions (Figure E) and esophageal blister on the web. However, esophageal narrowing remained, and thus endoscopic balloon dilation was performed (Figure F-H). Three months after the dilation, the narrowing improved (Figure I).
MMP is an autoimmune blistering disease that induces the formation of mucous membrane subepithelial bullae. Basement membrane zone components such as collagen XVII (also known as BP180) are targets of autoantibodies in MMP. Symptomatic esophageal involvement affects 5.4% of patients with MMP and dysphagia is the most frequent symptom.1 Endoscopic findings include erosion, web stricture, subepithelial hematomas, and scars.2,3 Endoscopic dilation is sometimes necessary for the treatment of severe esophageal strictures.1
References
1. Zehou O et al. Br J Dermatol. 2017 Oct;177(4):1074-85.
2. Sallout H et al. Gastrointest Endosc. 2000 Sep;52(3):429-33.
3. Gaspar R et al. Gastrointest Endosc. 2017 Aug;86(2):400-2.
A 70-year-old man with a history of rectal cancer was referred to our clinic for chronic dysphagia and odynophagia. He did not have fevers or an allergic history. Physical examination was unremarkable except for multiple erosions in the oral cavity. Upper gastrointestinal endoscopy revealed multiple erosions in the palate and laryngopharynx (Figure A), a web stricture in the cervical esophagus (Figure B), and multiple scars in the thoracic esophagus.
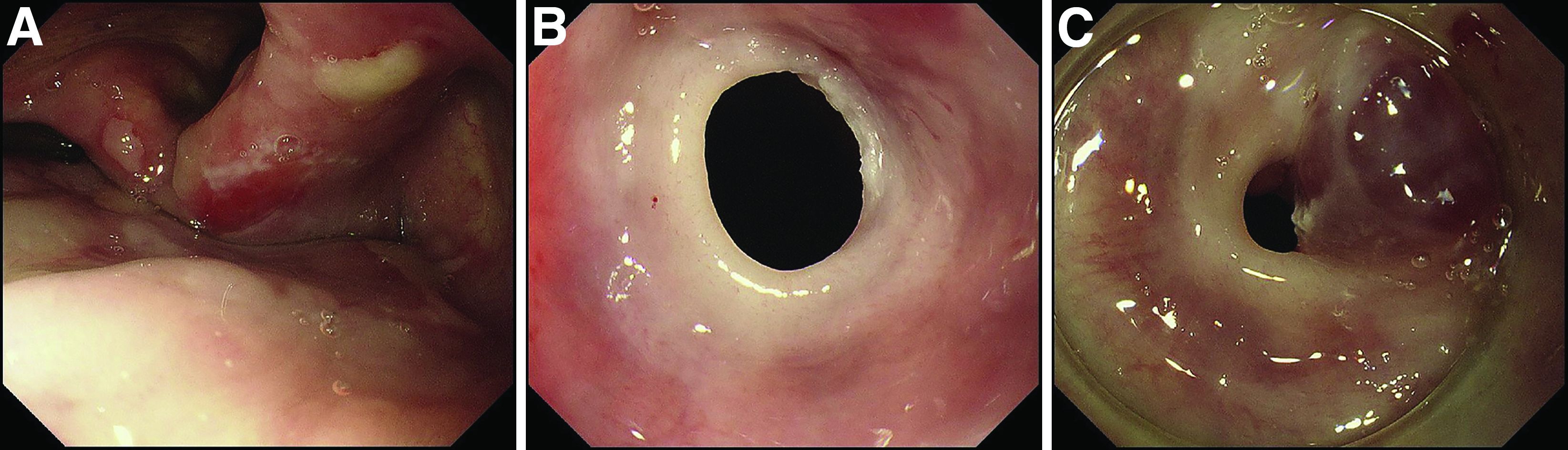
Laboratory examination showed normal results including a normal white blood cell count (8010/mcL; eosinophils 360/mcL), hemoglobin level (14.0 g/dL), mean corpuscular volume (97.8 fL), serum iron level (140 mcg/dL), and ferritin level (50.5 mg/L). His dysphagia gradually worsened and he finally could not take pills nor solid food. Two weeks after the first endoscopy, a second endoscopic examination was performed and it showed exacerbation of esophageal stricture and appearance of a bloody blister (Figure C).
What is the diagnosis?
Previously published in Gastroenterology
What is your diagnosis?
Answer: Infectious gastroparesis secondary to acute hepatitis A infection.
A computed tomography scan of the abdomen/pelvis demonstrated marked gastric distention without obvious obstructing mass and normal caliber small bowel and colon. Additional laboratory workup revealed a positive hepatitis A IgM antibody. Hepatitis B surface antigen and core IgM antibody were negative, as was the hepatitis C virus antibody. Human immunodeficiency virus antigen and antibody were negative. An esophagogastroduodenoscopy was performed that showed a large amount of food in a dilated and atonic stomach.
With conservative treatment, the patient’s liver enzymes trended down over the next 2 days to alanine aminotransferase 993 U/L, aspartate aminotransferase 244 U/L, and direct bilirubin 3.8 mg/dL. At the time of discharge, she was tolerating soft foods without any difficulty. She was educated on taking appropriate precautions to avoid transmitting the hepatitis A infection to others. Her risk factor for hepatitis A was recent incarceration.
Here we highlight a rare case of infectious gastroparesis secondary to hepatitis A infection. Hepatitis A virus is a small, nonenveloped, RNA-containing virus.1 It typically presents with a self-limited illness with liver failure occurring in rare cases. Common presenting symptoms including nausea, vomiting, jaundice, fever, diarrhea, and abdominal pain.Laboratory abnormalities include elevations in the serum aminotransferases, alkaline phosphatase, and total bilirubin.2 The diagnosis is confirmed with a positive hepatitis A IgM antibody. The most common route of transmission is the fecal-oral route such as through consumption of contaminated water and food or from person-to-person contact.1 Individuals can develop immunity to the virus either from prior infection or vaccination.
Gastroparesis refers to delayed emptying of gastric contents when mechanical obstruction has been ruled out. Common causes of gastroparesis include diabetes mellitus, medications, postoperative complications, and infections. Infectious gastroparesis may present acutely after a viral prodrome and symptoms may be severe and slow to resolve.3
References
1. Lemon SM. N Engl J Med. 1985 Oct 24;313(17):1059-67.
2. Tong MJ et al. J Infect Dis. 1995 Mar;171 Suppl 1:S15-8.
3. Bityutskiy LP. Am J Gastroenterol. 1997 Sep;92(9):1501-4.
Answer: Infectious gastroparesis secondary to acute hepatitis A infection.
A computed tomography scan of the abdomen/pelvis demonstrated marked gastric distention without obvious obstructing mass and normal caliber small bowel and colon. Additional laboratory workup revealed a positive hepatitis A IgM antibody. Hepatitis B surface antigen and core IgM antibody were negative, as was the hepatitis C virus antibody. Human immunodeficiency virus antigen and antibody were negative. An esophagogastroduodenoscopy was performed that showed a large amount of food in a dilated and atonic stomach.
With conservative treatment, the patient’s liver enzymes trended down over the next 2 days to alanine aminotransferase 993 U/L, aspartate aminotransferase 244 U/L, and direct bilirubin 3.8 mg/dL. At the time of discharge, she was tolerating soft foods without any difficulty. She was educated on taking appropriate precautions to avoid transmitting the hepatitis A infection to others. Her risk factor for hepatitis A was recent incarceration.
Here we highlight a rare case of infectious gastroparesis secondary to hepatitis A infection. Hepatitis A virus is a small, nonenveloped, RNA-containing virus.1 It typically presents with a self-limited illness with liver failure occurring in rare cases. Common presenting symptoms including nausea, vomiting, jaundice, fever, diarrhea, and abdominal pain.Laboratory abnormalities include elevations in the serum aminotransferases, alkaline phosphatase, and total bilirubin.2 The diagnosis is confirmed with a positive hepatitis A IgM antibody. The most common route of transmission is the fecal-oral route such as through consumption of contaminated water and food or from person-to-person contact.1 Individuals can develop immunity to the virus either from prior infection or vaccination.
Gastroparesis refers to delayed emptying of gastric contents when mechanical obstruction has been ruled out. Common causes of gastroparesis include diabetes mellitus, medications, postoperative complications, and infections. Infectious gastroparesis may present acutely after a viral prodrome and symptoms may be severe and slow to resolve.3
References
1. Lemon SM. N Engl J Med. 1985 Oct 24;313(17):1059-67.
2. Tong MJ et al. J Infect Dis. 1995 Mar;171 Suppl 1:S15-8.
3. Bityutskiy LP. Am J Gastroenterol. 1997 Sep;92(9):1501-4.
Answer: Infectious gastroparesis secondary to acute hepatitis A infection.
A computed tomography scan of the abdomen/pelvis demonstrated marked gastric distention without obvious obstructing mass and normal caliber small bowel and colon. Additional laboratory workup revealed a positive hepatitis A IgM antibody. Hepatitis B surface antigen and core IgM antibody were negative, as was the hepatitis C virus antibody. Human immunodeficiency virus antigen and antibody were negative. An esophagogastroduodenoscopy was performed that showed a large amount of food in a dilated and atonic stomach.
With conservative treatment, the patient’s liver enzymes trended down over the next 2 days to alanine aminotransferase 993 U/L, aspartate aminotransferase 244 U/L, and direct bilirubin 3.8 mg/dL. At the time of discharge, she was tolerating soft foods without any difficulty. She was educated on taking appropriate precautions to avoid transmitting the hepatitis A infection to others. Her risk factor for hepatitis A was recent incarceration.
Here we highlight a rare case of infectious gastroparesis secondary to hepatitis A infection. Hepatitis A virus is a small, nonenveloped, RNA-containing virus.1 It typically presents with a self-limited illness with liver failure occurring in rare cases. Common presenting symptoms including nausea, vomiting, jaundice, fever, diarrhea, and abdominal pain.Laboratory abnormalities include elevations in the serum aminotransferases, alkaline phosphatase, and total bilirubin.2 The diagnosis is confirmed with a positive hepatitis A IgM antibody. The most common route of transmission is the fecal-oral route such as through consumption of contaminated water and food or from person-to-person contact.1 Individuals can develop immunity to the virus either from prior infection or vaccination.
Gastroparesis refers to delayed emptying of gastric contents when mechanical obstruction has been ruled out. Common causes of gastroparesis include diabetes mellitus, medications, postoperative complications, and infections. Infectious gastroparesis may present acutely after a viral prodrome and symptoms may be severe and slow to resolve.3
References
1. Lemon SM. N Engl J Med. 1985 Oct 24;313(17):1059-67.
2. Tong MJ et al. J Infect Dis. 1995 Mar;171 Suppl 1:S15-8.
3. Bityutskiy LP. Am J Gastroenterol. 1997 Sep;92(9):1501-4.
A 33-year-old woman presented with a 10-day history of painless jaundice. During this time, she also noted decreased appetite, malaise, and pruritus. On occasion, she would have heartburn and belching that would improve with an antacid. She denied any right upper quadrant pain and weight loss. She was not currently taking any medications, including acetaminophen. She had a past medical history of methamphetamine use in recent remission. She had recently been incarcerated for about 1 month. 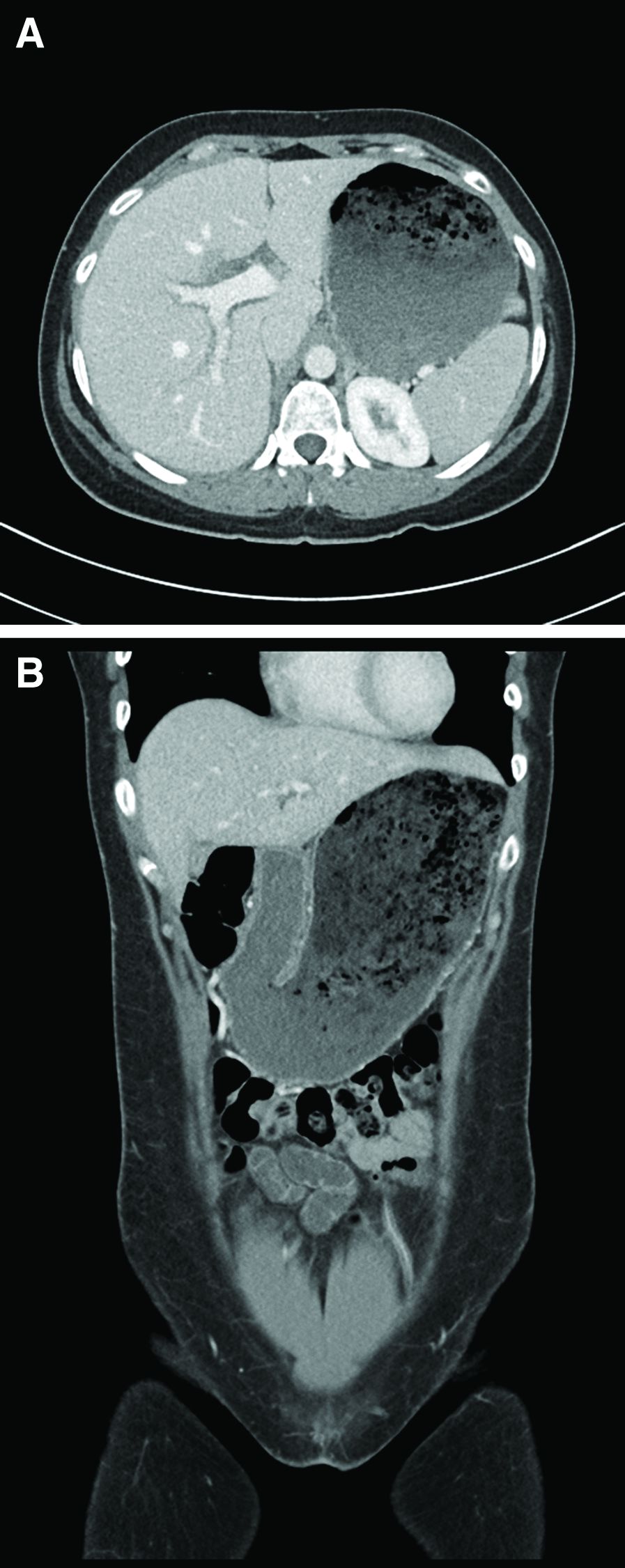
What is the most likely etiology of the patient's condition?
What's your diagnosis?
Sigmoid colon perforation secondary to transcutaneous epicardial pacer wires.
A plain film image (Figure A) shows diffusely dilated loops of bowel with subdiaphragmatic air concerning for GI viscous perforation. Dedicated cross-sectional imaging confirms intra-abdominal free air, and in representative cross section, the epicardial pacing wires can be visualized within the gastrointestinal lumen (Figure D, arrows). At the time of surgical consultation, the radiology report was notable for concern regarding possible disruption of peritoneum secondary to the difficult surgical chest tube placement in a patient with a high-riding left hemidiaphragm. Urgent laparoscopic exploration secondary to these findings unexpectedly revealed that the transcutaneous epicardial pacing wires had been inadvertently placed through the sigmoid colon (Figure C). The pacer wires were cut and removed intraoperatively. Unfortunately, 4 days after removal of pacer wires, the patient continued to have ongoing distension and was found to have sigmoid volvulus necessitating endoscopic decompression. After a prolonged hospitalization and recovery, he was discharged with a normal bowel pattern and tolerating oral intake to a skilled nursing facility.
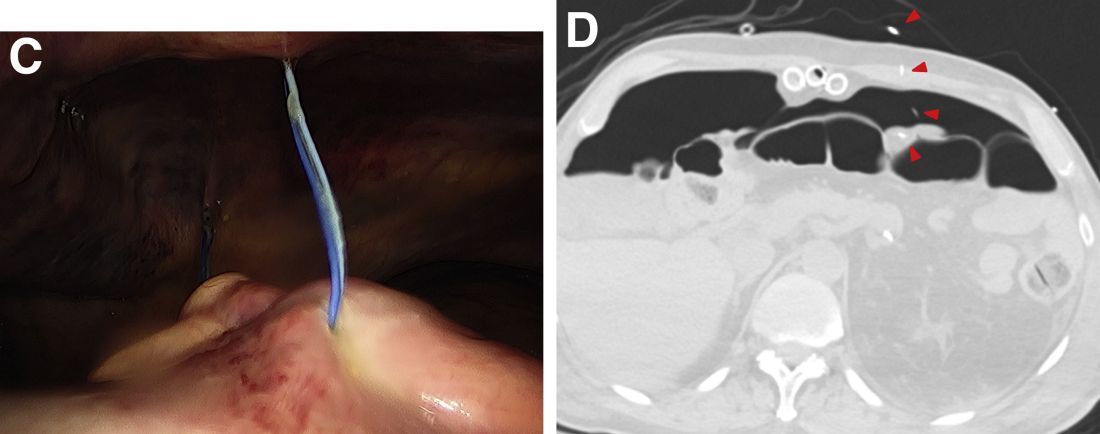
Temporary transcutaneous epicardial pacing wires are often placed after complex cardiovascular surgical procedures. Complications from wire placement are thought to be relatively rare and are typically associated with migration into local structures after wire placement and infectious complications secondary to retained wires.1,2 Perforation of local structures during placement is less common, and GI viscous perforation in particular is not a well-characterized cause of associated morbidity.3
Our case demonstrates that, in patients with hemidiaphragm elevation, epicardial wire placement risks GI viscous perforation. Furthermore, given the frequency of concomitant surgical hardware in this patient population, identification of malpositioned epicardial wires on plain film and even cross-sectional imaging can be difficult and can delay diagnosis.
References
1. Del Nido P, Goldman BS. J Card Surg. 1989 Mar;4(1):99-103.
2. Kapoor A et al. Interact Cardiovasc Thorac Surg. 2011 Jul;13(1):104-6.
3. Haba J et al. Can Assoc Radiol J. 2013 Feb;64(1):77-80.
Sigmoid colon perforation secondary to transcutaneous epicardial pacer wires.
A plain film image (Figure A) shows diffusely dilated loops of bowel with subdiaphragmatic air concerning for GI viscous perforation. Dedicated cross-sectional imaging confirms intra-abdominal free air, and in representative cross section, the epicardial pacing wires can be visualized within the gastrointestinal lumen (Figure D, arrows). At the time of surgical consultation, the radiology report was notable for concern regarding possible disruption of peritoneum secondary to the difficult surgical chest tube placement in a patient with a high-riding left hemidiaphragm. Urgent laparoscopic exploration secondary to these findings unexpectedly revealed that the transcutaneous epicardial pacing wires had been inadvertently placed through the sigmoid colon (Figure C). The pacer wires were cut and removed intraoperatively. Unfortunately, 4 days after removal of pacer wires, the patient continued to have ongoing distension and was found to have sigmoid volvulus necessitating endoscopic decompression. After a prolonged hospitalization and recovery, he was discharged with a normal bowel pattern and tolerating oral intake to a skilled nursing facility.
Temporary transcutaneous epicardial pacing wires are often placed after complex cardiovascular surgical procedures. Complications from wire placement are thought to be relatively rare and are typically associated with migration into local structures after wire placement and infectious complications secondary to retained wires.1,2 Perforation of local structures during placement is less common, and GI viscous perforation in particular is not a well-characterized cause of associated morbidity.3
Our case demonstrates that, in patients with hemidiaphragm elevation, epicardial wire placement risks GI viscous perforation. Furthermore, given the frequency of concomitant surgical hardware in this patient population, identification of malpositioned epicardial wires on plain film and even cross-sectional imaging can be difficult and can delay diagnosis.
References
1. Del Nido P, Goldman BS. J Card Surg. 1989 Mar;4(1):99-103.
2. Kapoor A et al. Interact Cardiovasc Thorac Surg. 2011 Jul;13(1):104-6.
3. Haba J et al. Can Assoc Radiol J. 2013 Feb;64(1):77-80.
Sigmoid colon perforation secondary to transcutaneous epicardial pacer wires.
A plain film image (Figure A) shows diffusely dilated loops of bowel with subdiaphragmatic air concerning for GI viscous perforation. Dedicated cross-sectional imaging confirms intra-abdominal free air, and in representative cross section, the epicardial pacing wires can be visualized within the gastrointestinal lumen (Figure D, arrows). At the time of surgical consultation, the radiology report was notable for concern regarding possible disruption of peritoneum secondary to the difficult surgical chest tube placement in a patient with a high-riding left hemidiaphragm. Urgent laparoscopic exploration secondary to these findings unexpectedly revealed that the transcutaneous epicardial pacing wires had been inadvertently placed through the sigmoid colon (Figure C). The pacer wires were cut and removed intraoperatively. Unfortunately, 4 days after removal of pacer wires, the patient continued to have ongoing distension and was found to have sigmoid volvulus necessitating endoscopic decompression. After a prolonged hospitalization and recovery, he was discharged with a normal bowel pattern and tolerating oral intake to a skilled nursing facility.
Temporary transcutaneous epicardial pacing wires are often placed after complex cardiovascular surgical procedures. Complications from wire placement are thought to be relatively rare and are typically associated with migration into local structures after wire placement and infectious complications secondary to retained wires.1,2 Perforation of local structures during placement is less common, and GI viscous perforation in particular is not a well-characterized cause of associated morbidity.3
Our case demonstrates that, in patients with hemidiaphragm elevation, epicardial wire placement risks GI viscous perforation. Furthermore, given the frequency of concomitant surgical hardware in this patient population, identification of malpositioned epicardial wires on plain film and even cross-sectional imaging can be difficult and can delay diagnosis.
References
1. Del Nido P, Goldman BS. J Card Surg. 1989 Mar;4(1):99-103.
2. Kapoor A et al. Interact Cardiovasc Thorac Surg. 2011 Jul;13(1):104-6.
3. Haba J et al. Can Assoc Radiol J. 2013 Feb;64(1):77-80.
Question: An 82-year-old man was admitted for urgent coronary artery bypass and concurrent mitral valve repair. Intraoperatively, he underwent cardiopulmonary bypass, epicardial pacing, and placement of two anterior mediastinal and one pleural chest tubes. After a relatively unremarkable initial postoperative course and nonnarcotic pain control, concern for ileus developed on postoperative day 4. A nasogastric tube was placed out of concern for worsening somnolence, nausea, and the inability to safely tolerate oral intake. The patient had been passing flatus but had yet to have a bowel movement since the operation. Physical examination at the time was notable for a soft abdomen with diffuse tenderness and voluntary guarding. Subsequent plain film imaging to confirm nasogastric tube placement (Figure A) and follow-up computed tomography imaging (Figure B) are shown.
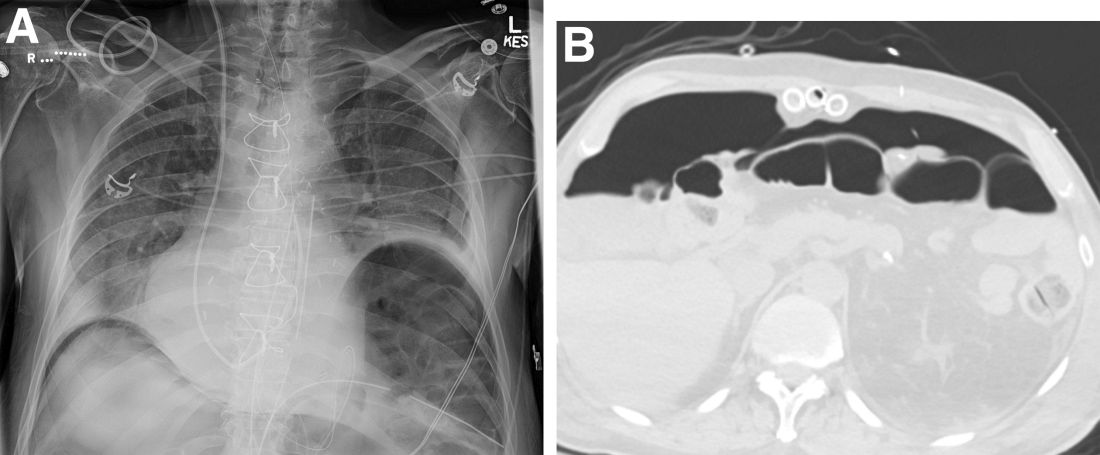
What's the diagnosis?