User login
Acute Generalized Exanthematous Pustulosis Caused by Pantoprazole
To the Editor:
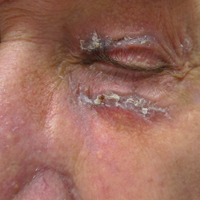
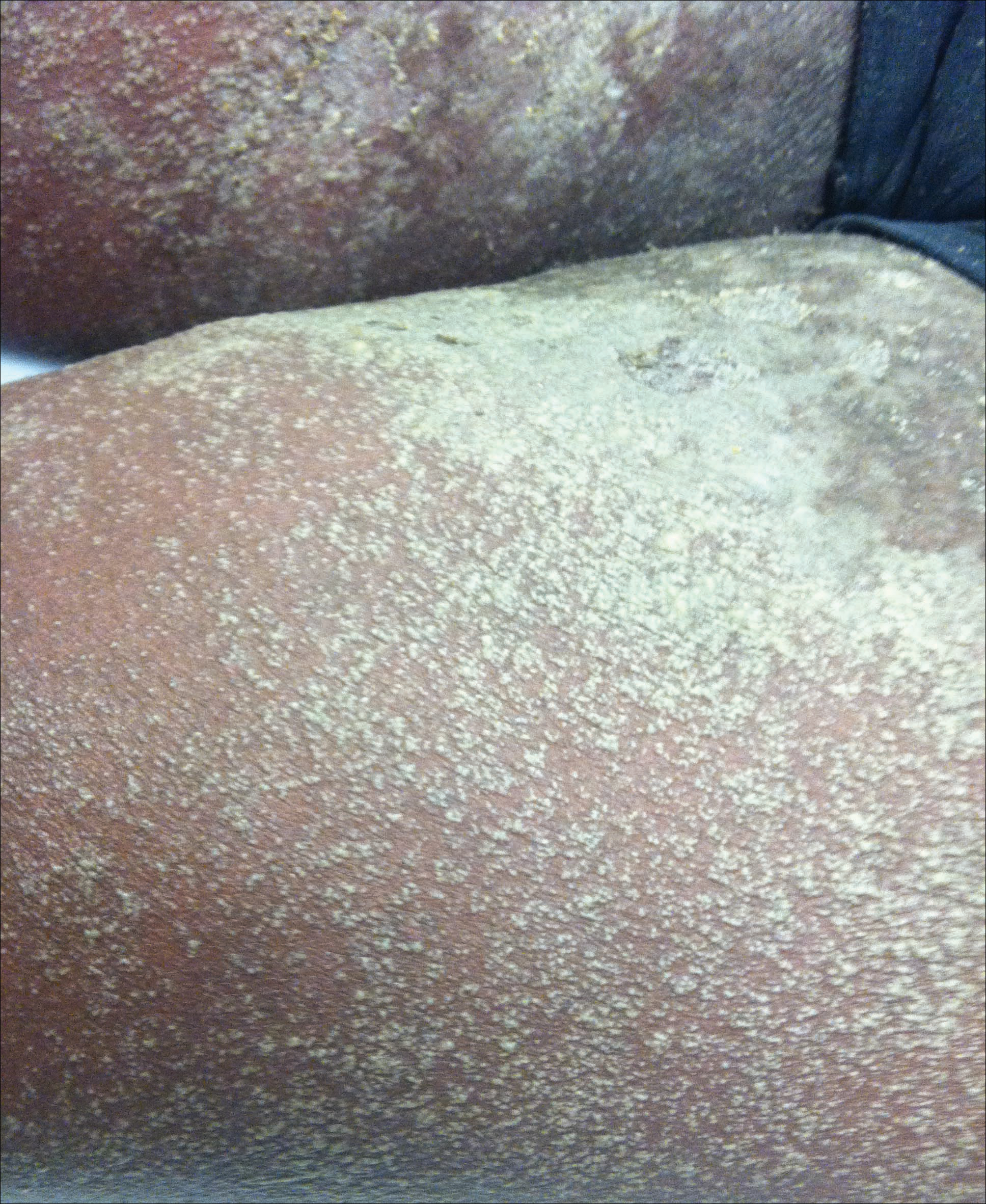
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.

Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
Interstitial Granulomatous Dermatitis and Palisaded Neutrophilic Granulomatous Dermatitis
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.

.")
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.

.")
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
Practice Points
- The clinical features of interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis exist on a spectrum, and these is considerable overlap between the features of these 2 clinicopathologic entities.
- Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis may respond to systemic steroids or treatment of the underlying systemic disease. Some cases spontaneously resolve.
Tinea Incognito in a Tattoo
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.

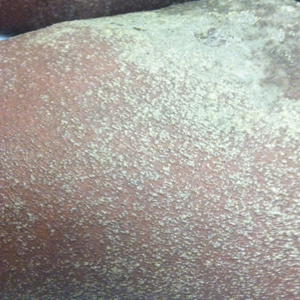
Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.
Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.
Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
Practice Points
- Health care providers should have a low threshold to perform a potassium hydroxide preparation when the possibility of a superficial fungal infection is considered.
- Tinea incognito occurs when a superficial fungal infection has unusual clinical features in the setting of local immune suppression.

Secondary Syphilis: An Atypical Presentation Complicated by a False Negative Rapid Plasma Reagin Test
To the Editor:
According to the Centers for Disease Control and Prevention, the number of syphilis cases in the United States decreased 95% from 1945 to 2000.1 Since 2000, the number of cases of syphilis in the United States has increased from 2.1 cases per 100,000 to 8.7 cases per 100,000.1 We report the case of an atypical presentation of secondary syphilis with a false negative rapid plasma reagin (RPR) test, which resulted in delayed diagnosis and treatment. The goal of this report is to raise awareness of the increasing prevalence of syphilis in the United States, draw attention to atypical presentations of syphilis, and inform physicians of some of the pitfalls in current syphilis screening and testing modalities.
A 37-year-old man presented with cutaneous ulcers on the forehead, thighs, and forearms of 3 months’ duration. The lesions started as a scarlet fever–like rash consisting of diffuse boils that would burst and become ulcerated. He reported arthralgias and drenching night sweats and had unintentionally lost 20 pounds over the last 3 months. He also had pharyngitis 8 months prior to presentation and sinusitis 4 months prior to presentation. These symptoms were present during his initial evaluation. One month prior to the current presentation, a nurse practitioner from an outside clinic had prescribed sulfamethoxazole/trimethoprim and ordered an RPR test, which was nonreactive. The lesions did not resolve, and the patient was referred to our dermatology department.
On physical examination, multiple 1- to 3-cm erythematous, well-defined papules were noted on the thighs and forearms. Some of the papules were covered with crusts, some were ulcerated with yellow discharge, and all were nontender. The differential diagnoses included dermatomyositis, polyarteritis nodosa, deep fungal infection, mycobacterial infection, leishmaniasis, and cutaneous anthrax. Secondary syphilis was a possible differential but was discounted due to the nonreactive RPR 1 month prior to presentation.
Punch biopsies were collected from lesions on the forehead, forearms, and thighs and sent to multiple institutions for pathology evaluation, which revealed dermal and pannicular necrosis and acute suppurative and granulomatous inflammation focally involving vessels (Figure 1). The biopsies were negative for acid-fast and fungal organisms, Mycobacterium tuberculosis, Leishmania, and anthrax. A work-up for Wegener granulomatosis was recommended by the pathology department.
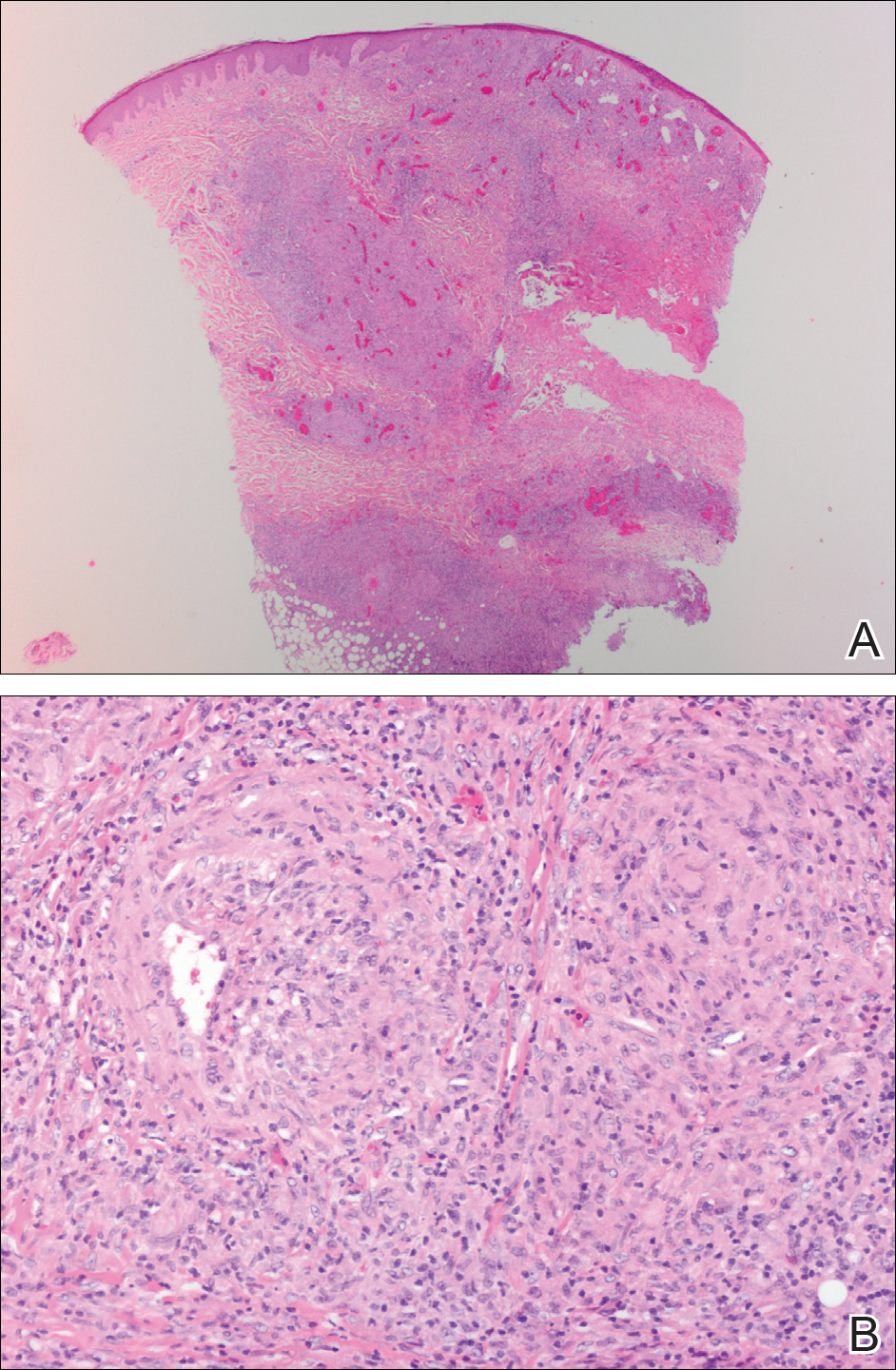
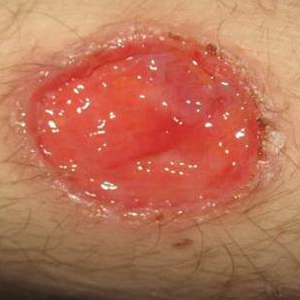
Three days later, the patient was admitted to the hospital for syncope. The hospitalist noted the cutaneous lesions and reordered the RPR test, which was now reactive. The ulcers had worsened since the original presentation (Figure 2). A fluorescent treponemal antibody absorption (FTA-ABS) test confirmed the reactive RPR, and a diagnosis of secondary syphilis was made. He was allergic to penicillin G, so the patient was prescribed doxycycline 100 mg twice daily for 28 days. His cutaneous ulcers have since healed with no recurrence of symptoms.
Secondary syphilis often is preceded by a prodrome of fever, malaise, sore throat, adenopathy, unintentional weight loss, myalgias, and headaches. It usually presents as a nonpruritic papulosquamous eruption with painless mucosal ulcers but rarely presents as cutaneous ulcers.2-4 Cutaneous ulcers are typical of lues maligna, which usually occurs in immunosuppressed patients.5,6 Our patient was human immunodeficiency virus–negative and was not otherwise immunocompromised.

Rapid plasma reagin is a common screening test for syphilis. In this case, it was initially negative, which may be attributed to the prozone phenomenon, a false negative result due to a high antibody titer that prevents the flocculation reaction from occurring. The prozone phenomenon can occur with a titer as low as 1:8.7 A 50% dilution of the negative sample should overcome the prozone phenomenon and yield a positive result7; unfortunately, this is not standard practice in all hospital laboratories.
The standard method of diagnosing syphilis in the United States is to screen with nontreponemal tests (eg, RPR) followed by treponemal tests (eg, FTA-ABS) to confirm a positive screen. According to the United States Preventive Services Task Force, the sensitivity of the RPR test is approximately 78% to 86%, while FTA-ABS has a sensitivity of 84% for detecting primary syphilis and 100% for secondary and tertiary syphilis.8 Seña et al4 suggest that FTA-ABS should be used as the screening test for syphilis. Fluorescent treponemal antibody absorption testing more accurately detects syphilis, while RPR testing is more useful in monitoring serum response once treatment has been initiated.
In conclusion, our patient could have benefited from earlier diagnosis and treatment if a treponemal test had been performed earlier or if the initial nonreactive RPR test was diluted and retested.
Acknowledgments
We would like to acknowledge Dr. Timothy Weiland (Pathology Department, Altru Health System, Grand Forks, North Dakota), and Dr. Mark Koponen (University of North Dakota, Grand Forks).
- Syphilis—CDC fact sheet. Centers for Disease Control and Prevention website. http://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed May 18, 2018.
- Stary A, Stary G. Sexually transmitted infections. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1368-1426.
- Habif TP. Sexually transmitted bacterial infections. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. China: Elsevier; 2016:377-417.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Bayramgürler D, Bilen N, Yıldız K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
- Bhate C, Tajirian AL, Kapila R, et al. Secondary syphilis resembling erythema multiforme. Int J Dermatol. 2010;49:1321-1324.
- Liu LL, Lin LR, Tong ML, et al. Incidence and risk factors for the prozone phenomenon in serologic testing for syphilis in a large cohort. Clin Infect Dis. 2014;59:384-389.
- Archived final recommendation statement. syphilis infection: screening. US Preventive Services Task Force website. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/syphilis-infection-screening. Published December 30, 2013. Accessed May 22, 2018.
To the Editor:
According to the Centers for Disease Control and Prevention, the number of syphilis cases in the United States decreased 95% from 1945 to 2000.1 Since 2000, the number of cases of syphilis in the United States has increased from 2.1 cases per 100,000 to 8.7 cases per 100,000.1 We report the case of an atypical presentation of secondary syphilis with a false negative rapid plasma reagin (RPR) test, which resulted in delayed diagnosis and treatment. The goal of this report is to raise awareness of the increasing prevalence of syphilis in the United States, draw attention to atypical presentations of syphilis, and inform physicians of some of the pitfalls in current syphilis screening and testing modalities.
A 37-year-old man presented with cutaneous ulcers on the forehead, thighs, and forearms of 3 months’ duration. The lesions started as a scarlet fever–like rash consisting of diffuse boils that would burst and become ulcerated. He reported arthralgias and drenching night sweats and had unintentionally lost 20 pounds over the last 3 months. He also had pharyngitis 8 months prior to presentation and sinusitis 4 months prior to presentation. These symptoms were present during his initial evaluation. One month prior to the current presentation, a nurse practitioner from an outside clinic had prescribed sulfamethoxazole/trimethoprim and ordered an RPR test, which was nonreactive. The lesions did not resolve, and the patient was referred to our dermatology department.
On physical examination, multiple 1- to 3-cm erythematous, well-defined papules were noted on the thighs and forearms. Some of the papules were covered with crusts, some were ulcerated with yellow discharge, and all were nontender. The differential diagnoses included dermatomyositis, polyarteritis nodosa, deep fungal infection, mycobacterial infection, leishmaniasis, and cutaneous anthrax. Secondary syphilis was a possible differential but was discounted due to the nonreactive RPR 1 month prior to presentation.
Punch biopsies were collected from lesions on the forehead, forearms, and thighs and sent to multiple institutions for pathology evaluation, which revealed dermal and pannicular necrosis and acute suppurative and granulomatous inflammation focally involving vessels (Figure 1). The biopsies were negative for acid-fast and fungal organisms, Mycobacterium tuberculosis, Leishmania, and anthrax. A work-up for Wegener granulomatosis was recommended by the pathology department.
Three days later, the patient was admitted to the hospital for syncope. The hospitalist noted the cutaneous lesions and reordered the RPR test, which was now reactive. The ulcers had worsened since the original presentation (Figure 2). A fluorescent treponemal antibody absorption (FTA-ABS) test confirmed the reactive RPR, and a diagnosis of secondary syphilis was made. He was allergic to penicillin G, so the patient was prescribed doxycycline 100 mg twice daily for 28 days. His cutaneous ulcers have since healed with no recurrence of symptoms.
Secondary syphilis often is preceded by a prodrome of fever, malaise, sore throat, adenopathy, unintentional weight loss, myalgias, and headaches. It usually presents as a nonpruritic papulosquamous eruption with painless mucosal ulcers but rarely presents as cutaneous ulcers.2-4 Cutaneous ulcers are typical of lues maligna, which usually occurs in immunosuppressed patients.5,6 Our patient was human immunodeficiency virus–negative and was not otherwise immunocompromised.
Rapid plasma reagin is a common screening test for syphilis. In this case, it was initially negative, which may be attributed to the prozone phenomenon, a false negative result due to a high antibody titer that prevents the flocculation reaction from occurring. The prozone phenomenon can occur with a titer as low as 1:8.7 A 50% dilution of the negative sample should overcome the prozone phenomenon and yield a positive result7; unfortunately, this is not standard practice in all hospital laboratories.
The standard method of diagnosing syphilis in the United States is to screen with nontreponemal tests (eg, RPR) followed by treponemal tests (eg, FTA-ABS) to confirm a positive screen. According to the United States Preventive Services Task Force, the sensitivity of the RPR test is approximately 78% to 86%, while FTA-ABS has a sensitivity of 84% for detecting primary syphilis and 100% for secondary and tertiary syphilis.8 Seña et al4 suggest that FTA-ABS should be used as the screening test for syphilis. Fluorescent treponemal antibody absorption testing more accurately detects syphilis, while RPR testing is more useful in monitoring serum response once treatment has been initiated.
In conclusion, our patient could have benefited from earlier diagnosis and treatment if a treponemal test had been performed earlier or if the initial nonreactive RPR test was diluted and retested.
Acknowledgments
We would like to acknowledge Dr. Timothy Weiland (Pathology Department, Altru Health System, Grand Forks, North Dakota), and Dr. Mark Koponen (University of North Dakota, Grand Forks).
To the Editor:
According to the Centers for Disease Control and Prevention, the number of syphilis cases in the United States decreased 95% from 1945 to 2000.1 Since 2000, the number of cases of syphilis in the United States has increased from 2.1 cases per 100,000 to 8.7 cases per 100,000.1 We report the case of an atypical presentation of secondary syphilis with a false negative rapid plasma reagin (RPR) test, which resulted in delayed diagnosis and treatment. The goal of this report is to raise awareness of the increasing prevalence of syphilis in the United States, draw attention to atypical presentations of syphilis, and inform physicians of some of the pitfalls in current syphilis screening and testing modalities.
A 37-year-old man presented with cutaneous ulcers on the forehead, thighs, and forearms of 3 months’ duration. The lesions started as a scarlet fever–like rash consisting of diffuse boils that would burst and become ulcerated. He reported arthralgias and drenching night sweats and had unintentionally lost 20 pounds over the last 3 months. He also had pharyngitis 8 months prior to presentation and sinusitis 4 months prior to presentation. These symptoms were present during his initial evaluation. One month prior to the current presentation, a nurse practitioner from an outside clinic had prescribed sulfamethoxazole/trimethoprim and ordered an RPR test, which was nonreactive. The lesions did not resolve, and the patient was referred to our dermatology department.
On physical examination, multiple 1- to 3-cm erythematous, well-defined papules were noted on the thighs and forearms. Some of the papules were covered with crusts, some were ulcerated with yellow discharge, and all were nontender. The differential diagnoses included dermatomyositis, polyarteritis nodosa, deep fungal infection, mycobacterial infection, leishmaniasis, and cutaneous anthrax. Secondary syphilis was a possible differential but was discounted due to the nonreactive RPR 1 month prior to presentation.
Punch biopsies were collected from lesions on the forehead, forearms, and thighs and sent to multiple institutions for pathology evaluation, which revealed dermal and pannicular necrosis and acute suppurative and granulomatous inflammation focally involving vessels (Figure 1). The biopsies were negative for acid-fast and fungal organisms, Mycobacterium tuberculosis, Leishmania, and anthrax. A work-up for Wegener granulomatosis was recommended by the pathology department.
Three days later, the patient was admitted to the hospital for syncope. The hospitalist noted the cutaneous lesions and reordered the RPR test, which was now reactive. The ulcers had worsened since the original presentation (Figure 2). A fluorescent treponemal antibody absorption (FTA-ABS) test confirmed the reactive RPR, and a diagnosis of secondary syphilis was made. He was allergic to penicillin G, so the patient was prescribed doxycycline 100 mg twice daily for 28 days. His cutaneous ulcers have since healed with no recurrence of symptoms.
Secondary syphilis often is preceded by a prodrome of fever, malaise, sore throat, adenopathy, unintentional weight loss, myalgias, and headaches. It usually presents as a nonpruritic papulosquamous eruption with painless mucosal ulcers but rarely presents as cutaneous ulcers.2-4 Cutaneous ulcers are typical of lues maligna, which usually occurs in immunosuppressed patients.5,6 Our patient was human immunodeficiency virus–negative and was not otherwise immunocompromised.
Rapid plasma reagin is a common screening test for syphilis. In this case, it was initially negative, which may be attributed to the prozone phenomenon, a false negative result due to a high antibody titer that prevents the flocculation reaction from occurring. The prozone phenomenon can occur with a titer as low as 1:8.7 A 50% dilution of the negative sample should overcome the prozone phenomenon and yield a positive result7; unfortunately, this is not standard practice in all hospital laboratories.
The standard method of diagnosing syphilis in the United States is to screen with nontreponemal tests (eg, RPR) followed by treponemal tests (eg, FTA-ABS) to confirm a positive screen. According to the United States Preventive Services Task Force, the sensitivity of the RPR test is approximately 78% to 86%, while FTA-ABS has a sensitivity of 84% for detecting primary syphilis and 100% for secondary and tertiary syphilis.8 Seña et al4 suggest that FTA-ABS should be used as the screening test for syphilis. Fluorescent treponemal antibody absorption testing more accurately detects syphilis, while RPR testing is more useful in monitoring serum response once treatment has been initiated.
In conclusion, our patient could have benefited from earlier diagnosis and treatment if a treponemal test had been performed earlier or if the initial nonreactive RPR test was diluted and retested.
Acknowledgments
We would like to acknowledge Dr. Timothy Weiland (Pathology Department, Altru Health System, Grand Forks, North Dakota), and Dr. Mark Koponen (University of North Dakota, Grand Forks).
- Syphilis—CDC fact sheet. Centers for Disease Control and Prevention website. http://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed May 18, 2018.
- Stary A, Stary G. Sexually transmitted infections. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1368-1426.
- Habif TP. Sexually transmitted bacterial infections. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. China: Elsevier; 2016:377-417.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Bayramgürler D, Bilen N, Yıldız K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
- Bhate C, Tajirian AL, Kapila R, et al. Secondary syphilis resembling erythema multiforme. Int J Dermatol. 2010;49:1321-1324.
- Liu LL, Lin LR, Tong ML, et al. Incidence and risk factors for the prozone phenomenon in serologic testing for syphilis in a large cohort. Clin Infect Dis. 2014;59:384-389.
- Archived final recommendation statement. syphilis infection: screening. US Preventive Services Task Force website. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/syphilis-infection-screening. Published December 30, 2013. Accessed May 22, 2018.
- Syphilis—CDC fact sheet. Centers for Disease Control and Prevention website. http://www.cdc.gov/std/syphilis/stdfact-syphilis.htm. Updated June 13, 2017. Accessed May 18, 2018.
- Stary A, Stary G. Sexually transmitted infections. In: Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. China: Elsevier Saunders; 2012:1368-1426.
- Habif TP. Sexually transmitted bacterial infections. Clinical Dermatology: A Color Guide to Diagnosis and Therapy. 6th ed. China: Elsevier; 2016:377-417.
- Seña AC, White BL, Sparling PF. Novel Treponema pallidum serologic tests: a paradigm shift in syphilis screening for the 21st century. Clin Infect Dis. 2010;51:700-708.
- Bayramgürler D, Bilen N, Yıldız K, et al. Lues maligna in a chronic alcoholic patient. J Dermatol. 2005;32:217-219.
- Bhate C, Tajirian AL, Kapila R, et al. Secondary syphilis resembling erythema multiforme. Int J Dermatol. 2010;49:1321-1324.
- Liu LL, Lin LR, Tong ML, et al. Incidence and risk factors for the prozone phenomenon in serologic testing for syphilis in a large cohort. Clin Infect Dis. 2014;59:384-389.
- Archived final recommendation statement. syphilis infection: screening. US Preventive Services Task Force website. https://www.uspreventiveservicestaskforce.org/Page/Document/RecommendationStatementFinal/syphilis-infection-screening. Published December 30, 2013. Accessed May 22, 2018.
Practice Points
- Fluorescent treponemal antibody absorption testing more accurately detects syphilis than rapid plasma reagin (RPR).
- Rapid plasma reagin testing is more useful in monitoring serum response once treatment has been initiated.
- If only RPR is being performed at your institution, ensure the laboratory is performing serial dilutions to negate the prozone phenomenon.
Solitary Angiokeratoma of the Vulva Mimicking Malignant Melanoma
To the Editor:
Angiokeratoma is a benign vascular tumor characterized by several dilated vessels in the superficial dermis accompanied by epidermal hyperplasia and hyperkeratosis.1 Angiokeratoma of the vulva is a rare clinical finding, usually involving multiple lesions as part of the Fordyce type.2 Solitary angiokeratoma occurs predominantly on the lower legs,3 and although other locations have been described, the presence of a solitary angiokeratoma on the vulva is rare.4 We report 2 cases of solitary angiokeratoma on the vulva that was misdiagnosed as malignant melanoma. Both patients were referred to our center for evaluation and excision.
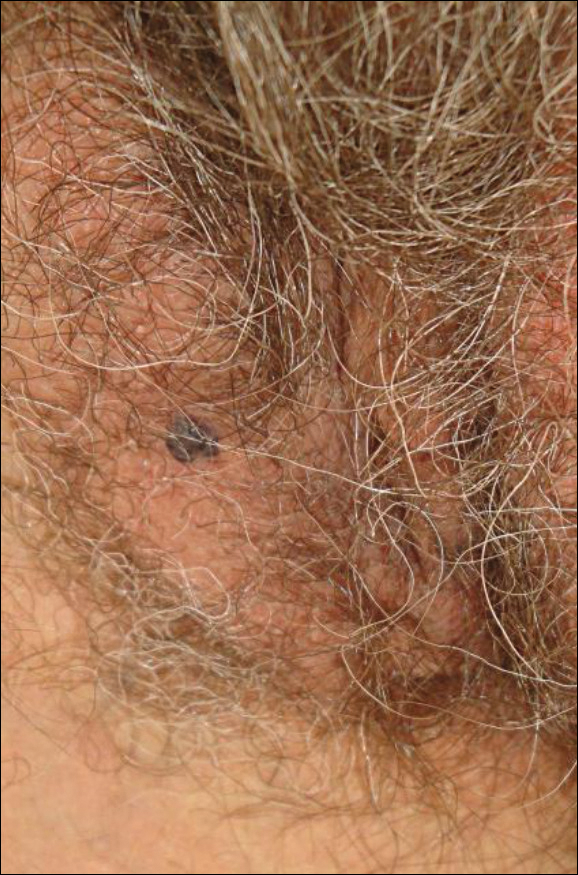
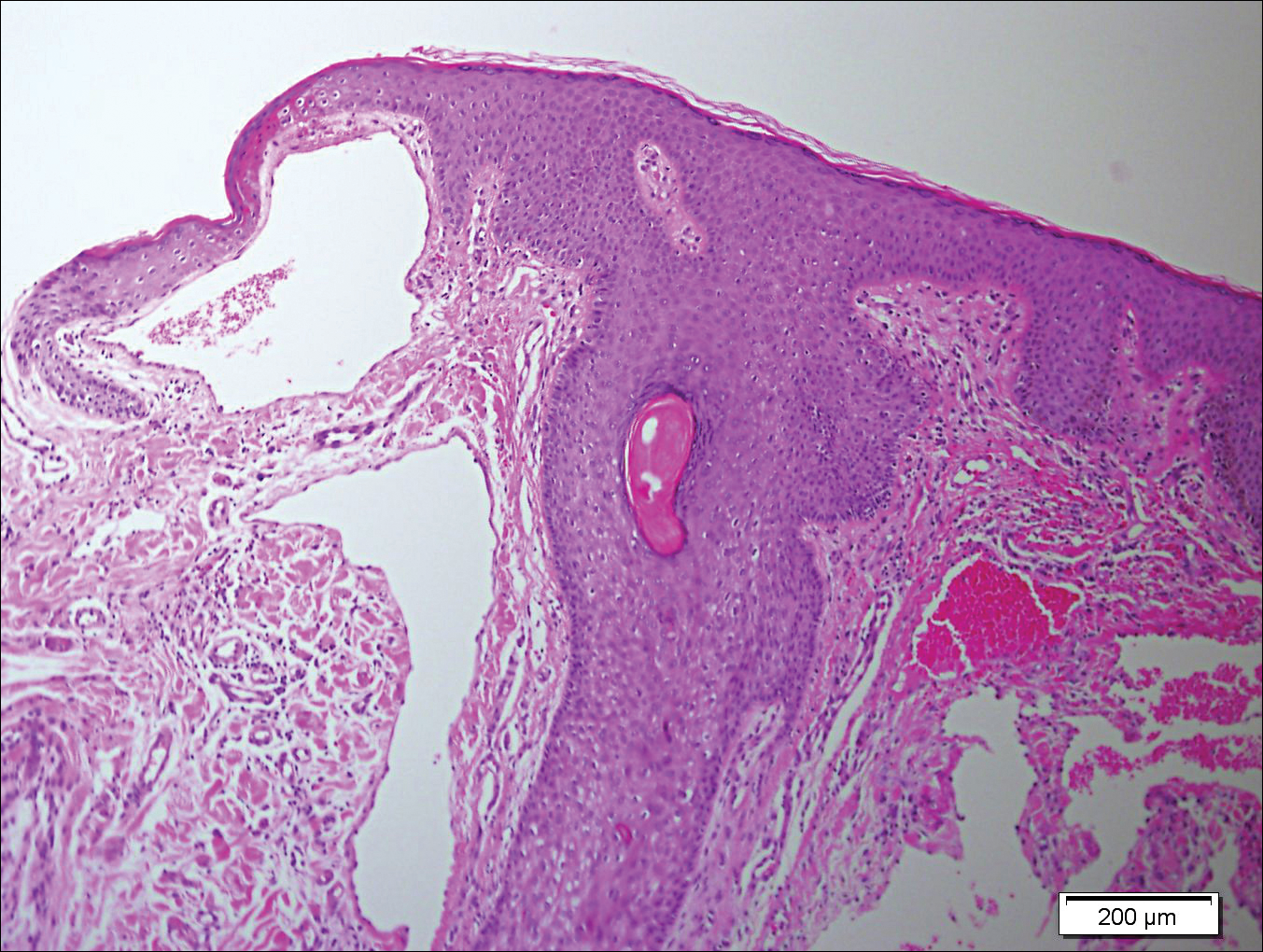
A 65-year-old woman (patient 1) and a 67-year-old woman (patient 2) presented with a bluish black, growing, asymptomatic lesion on the right (Figure 1) and left labia majora, respectively. Both patients were referred by outside physicians for excision because of suspected malignant melanoma. Physical examinations revealed bluish black globular nodules that measured 0.5 and 0.3 cm in diameter, respectively. Dermoscopy (patient 1) revealed dark lacunae. Histopathologic examination of the vulvar lesion (patient 2) showed dilated, blood-filled, vascular spaces in the papillary dermis, accompanied by overlying acanthosis, hyperkeratosis, and papillomatosis that was consistent with angiokeratoma (Figure 2).
Angiokeratoma, particularly the solitary type, often is misdiagnosed. Clinical differential diagnoses may include a wide range of pathologic conditions, including condyloma acuminata, basal cell carcinoma, pyogenic granuloma, lymphangioma, nevi, condyloma lata, nodular prurigo, seborrheic keratosis, granuloma inguinale, and deep fungal infection.2,5 However, due to its quickly growing nature and its dark complexion, malignant melanoma often is initially diagnosed. Because patients affected by angiokeratoma of the vulva usually are aged 20 to 40 years,5 and vulvar melanoma is typical for middle-aged women (median age, 68 years),6 this misdiagnosis is more likely in older patients. It should be noted that a high index of suspicion for melanoma often is present when examining the vulva, considering that this area is difficult to monitor, and there is an especially poor prognosis of vulvar melanoma due to its late detection.6,7
In the past, biopsy was considered mandatory for confirming the diagnosis of vulvar angiokeratoma.5,8,9 However, dermoscopy has emerged as a valuable tool for diagnosis of angiokeratoma10 and also was helpful as a diagnostic aid in one of our patients (patient 1). Therefore, we believe that dermoscopy should be performed prior to a biopsy of angiokeratomas of the vulva.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. part I. hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol. 1997;37:523-549.
- Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-282.
- Gomi H, Eriyama Y, Horikawa E, et al. Solitary angiokeratoma. J Dermatol. 1988;15:349-350.
- Yamazaki M, Hiruma M, Irie H, et al. Angiokeratoma of the clitoris: a subtype of angiokeratoma vulvae. J Dermatol. 1992;19:553-555.
- Cohen PR, Young AW Jr, Tovell HM. Angiokeratoma of the vulva: diagnosis and review of the literature. Obstet Gynecol Surv. 1989;44:339-346.
- Sugiyama VE, Chan JK, Shin JY, et al. Vulvar melanoma: a multivariable analysis of 644 patients. Obstet Gynecol. 2007;110:296-301.
- De Simone P, Silipo V, Buccini P, et al. Vulvar melanoma: a report of 10 cases and review of the literature. Melanoma Res. 2008;18:127-133.
- Novick NL. Angiokeratoma vulvae. J Am Acad Dermatol. 1985;12:561-563.
- Yigiter M, Arda IS, Tosun E, et al. Angiokeratoma of clitoris: a rare lesion in an adolescent girl. Urology. 2008;71:604-606.
- Zaballos P, Daufi C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325.
To the Editor:
Angiokeratoma is a benign vascular tumor characterized by several dilated vessels in the superficial dermis accompanied by epidermal hyperplasia and hyperkeratosis.1 Angiokeratoma of the vulva is a rare clinical finding, usually involving multiple lesions as part of the Fordyce type.2 Solitary angiokeratoma occurs predominantly on the lower legs,3 and although other locations have been described, the presence of a solitary angiokeratoma on the vulva is rare.4 We report 2 cases of solitary angiokeratoma on the vulva that was misdiagnosed as malignant melanoma. Both patients were referred to our center for evaluation and excision.
A 65-year-old woman (patient 1) and a 67-year-old woman (patient 2) presented with a bluish black, growing, asymptomatic lesion on the right (Figure 1) and left labia majora, respectively. Both patients were referred by outside physicians for excision because of suspected malignant melanoma. Physical examinations revealed bluish black globular nodules that measured 0.5 and 0.3 cm in diameter, respectively. Dermoscopy (patient 1) revealed dark lacunae. Histopathologic examination of the vulvar lesion (patient 2) showed dilated, blood-filled, vascular spaces in the papillary dermis, accompanied by overlying acanthosis, hyperkeratosis, and papillomatosis that was consistent with angiokeratoma (Figure 2).
Angiokeratoma, particularly the solitary type, often is misdiagnosed. Clinical differential diagnoses may include a wide range of pathologic conditions, including condyloma acuminata, basal cell carcinoma, pyogenic granuloma, lymphangioma, nevi, condyloma lata, nodular prurigo, seborrheic keratosis, granuloma inguinale, and deep fungal infection.2,5 However, due to its quickly growing nature and its dark complexion, malignant melanoma often is initially diagnosed. Because patients affected by angiokeratoma of the vulva usually are aged 20 to 40 years,5 and vulvar melanoma is typical for middle-aged women (median age, 68 years),6 this misdiagnosis is more likely in older patients. It should be noted that a high index of suspicion for melanoma often is present when examining the vulva, considering that this area is difficult to monitor, and there is an especially poor prognosis of vulvar melanoma due to its late detection.6,7
In the past, biopsy was considered mandatory for confirming the diagnosis of vulvar angiokeratoma.5,8,9 However, dermoscopy has emerged as a valuable tool for diagnosis of angiokeratoma10 and also was helpful as a diagnostic aid in one of our patients (patient 1). Therefore, we believe that dermoscopy should be performed prior to a biopsy of angiokeratomas of the vulva.
To the Editor:
Angiokeratoma is a benign vascular tumor characterized by several dilated vessels in the superficial dermis accompanied by epidermal hyperplasia and hyperkeratosis.1 Angiokeratoma of the vulva is a rare clinical finding, usually involving multiple lesions as part of the Fordyce type.2 Solitary angiokeratoma occurs predominantly on the lower legs,3 and although other locations have been described, the presence of a solitary angiokeratoma on the vulva is rare.4 We report 2 cases of solitary angiokeratoma on the vulva that was misdiagnosed as malignant melanoma. Both patients were referred to our center for evaluation and excision.
A 65-year-old woman (patient 1) and a 67-year-old woman (patient 2) presented with a bluish black, growing, asymptomatic lesion on the right (Figure 1) and left labia majora, respectively. Both patients were referred by outside physicians for excision because of suspected malignant melanoma. Physical examinations revealed bluish black globular nodules that measured 0.5 and 0.3 cm in diameter, respectively. Dermoscopy (patient 1) revealed dark lacunae. Histopathologic examination of the vulvar lesion (patient 2) showed dilated, blood-filled, vascular spaces in the papillary dermis, accompanied by overlying acanthosis, hyperkeratosis, and papillomatosis that was consistent with angiokeratoma (Figure 2).
Angiokeratoma, particularly the solitary type, often is misdiagnosed. Clinical differential diagnoses may include a wide range of pathologic conditions, including condyloma acuminata, basal cell carcinoma, pyogenic granuloma, lymphangioma, nevi, condyloma lata, nodular prurigo, seborrheic keratosis, granuloma inguinale, and deep fungal infection.2,5 However, due to its quickly growing nature and its dark complexion, malignant melanoma often is initially diagnosed. Because patients affected by angiokeratoma of the vulva usually are aged 20 to 40 years,5 and vulvar melanoma is typical for middle-aged women (median age, 68 years),6 this misdiagnosis is more likely in older patients. It should be noted that a high index of suspicion for melanoma often is present when examining the vulva, considering that this area is difficult to monitor, and there is an especially poor prognosis of vulvar melanoma due to its late detection.6,7
In the past, biopsy was considered mandatory for confirming the diagnosis of vulvar angiokeratoma.5,8,9 However, dermoscopy has emerged as a valuable tool for diagnosis of angiokeratoma10 and also was helpful as a diagnostic aid in one of our patients (patient 1). Therefore, we believe that dermoscopy should be performed prior to a biopsy of angiokeratomas of the vulva.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. part I. hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol. 1997;37:523-549.
- Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-282.
- Gomi H, Eriyama Y, Horikawa E, et al. Solitary angiokeratoma. J Dermatol. 1988;15:349-350.
- Yamazaki M, Hiruma M, Irie H, et al. Angiokeratoma of the clitoris: a subtype of angiokeratoma vulvae. J Dermatol. 1992;19:553-555.
- Cohen PR, Young AW Jr, Tovell HM. Angiokeratoma of the vulva: diagnosis and review of the literature. Obstet Gynecol Surv. 1989;44:339-346.
- Sugiyama VE, Chan JK, Shin JY, et al. Vulvar melanoma: a multivariable analysis of 644 patients. Obstet Gynecol. 2007;110:296-301.
- De Simone P, Silipo V, Buccini P, et al. Vulvar melanoma: a report of 10 cases and review of the literature. Melanoma Res. 2008;18:127-133.
- Novick NL. Angiokeratoma vulvae. J Am Acad Dermatol. 1985;12:561-563.
- Yigiter M, Arda IS, Tosun E, et al. Angiokeratoma of clitoris: a rare lesion in an adolescent girl. Urology. 2008;71:604-606.
- Zaballos P, Daufi C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325.
- Requena L, Sangueza OP. Cutaneous vascular anomalies. part I. hamartomas, malformations, and dilation of preexisting vessels. J Am Acad Dermatol. 1997;37:523-549.
- Schiller PI, Itin PH. Angiokeratomas: an update. Dermatology. 1996;193:275-282.
- Gomi H, Eriyama Y, Horikawa E, et al. Solitary angiokeratoma. J Dermatol. 1988;15:349-350.
- Yamazaki M, Hiruma M, Irie H, et al. Angiokeratoma of the clitoris: a subtype of angiokeratoma vulvae. J Dermatol. 1992;19:553-555.
- Cohen PR, Young AW Jr, Tovell HM. Angiokeratoma of the vulva: diagnosis and review of the literature. Obstet Gynecol Surv. 1989;44:339-346.
- Sugiyama VE, Chan JK, Shin JY, et al. Vulvar melanoma: a multivariable analysis of 644 patients. Obstet Gynecol. 2007;110:296-301.
- De Simone P, Silipo V, Buccini P, et al. Vulvar melanoma: a report of 10 cases and review of the literature. Melanoma Res. 2008;18:127-133.
- Novick NL. Angiokeratoma vulvae. J Am Acad Dermatol. 1985;12:561-563.
- Yigiter M, Arda IS, Tosun E, et al. Angiokeratoma of clitoris: a rare lesion in an adolescent girl. Urology. 2008;71:604-606.
- Zaballos P, Daufi C, Puig S, et al. Dermoscopy of solitary angiokeratomas: a morphological study. Arch Dermatol. 2007;143:318-325.
Practice Points
- Solitary angiokeratoma of the vulva often is misdiagnosed as malignant melanoma due to its rapid growth and dark color.
- Dermoscopy is a valuable tool for diagnosing vulvar angiokeratoma to avoid unnecessary excisions.

Carotenoderma Associated With a Diet Rich in Red Palm Oil
To the Editor:
Carotenoderma is a cutaneous manifestation of elevated serum β-carotene levels and classically localizes to fatty tissues and areas rich in sweat glands. We present a case of carotenoderma associated with a diet rich in red palm oil, a common food additive in parts of the world outside of the United States.
A previously healthy 8-year-old boy who recently immigrated to the United States from Liberia was hospitalized for treatment of a febrile illness that subsequently was attributed to a viral syndrome. On physical examination by the dermatology department, the patient was noted to have marked orange discoloration on the palms and soles (Figure). Laboratory workup revealed elevated serum β-carotene levels of 809 μg/dL (reference range, 10–85 μg/dL). Testing of hemoglobin/hematocrit levels and liver, thyroid, and kidney function was normal, and systemic examination revealed no further abnormalities. Upon further inquiry by the dermatology department, the patient’s family reported frequent addition of red palm oil to all of the child’s meals. The patient subsequently was diagnosed with carotenoderma and was instructed to limit inclusion of red palm oil in his diet.

Red palm oil is a rich source of β-carotene and is commonly used outside the United States as a dietary supplement or food flavoring. Excessive consumption of red palm oil or other sources rich in carotenes can result in elevated serum carotene levels or hypercarotenemia. An elevation in serum β-carotene levels may be recognized from 4 to 7 weeks after starting a β-carotene–rich diet.1
While dietary consumption of carotenes is the most common cause of carotenoderma, others include kidney or liver disease, hyperlipidemia, porphyria, diabetes mellitus, hypothyroidism, and anorexia nervosa.2-4 Moreover, since carotenoids are enzymatically converted to vitamin A in the small intestine, a mutation of the gene of the conversion enzyme β-carotene 15,15’-monooxygenase 1 (BCMO1) also can cause be a rare cause of hypercarotenemia.3
Carotenoderma, the clinical cutaneous manifestation of hypercarotenemia, occurs as a result of β-carotene deposits in the skin when serum concentration exceeds 250 μg/dL. More specifically, β-carotene accumulates mainly in the lipid-rich stratum corneum as well as in sweat and sebum, which explains the localized discoloration in fatty tissues and areas rich in sweat glands (eg, nasolabial folds, palms, soles).3,4 The sclerae of the eyes are not affected by the surplus of β-carotene in carotenoderma, which helps distinguish it from jaundice.5
The differential diagnosis of yellow discoloration of the skin includes jaundice, encompassing the prehepatic, hepatocellular, and posthepatic categories.4 Also noteworthy in the differential diagnosis is lycopenemia, which occurs as a result of eating lycopene-rich foods (eg, tomatoes), resulting in a deeper orange-yellow pigmentation when compared to the cutaneous manifestation of hypercarotenemia.2,4,6 Several drugs also have been reported to induce yellow discoloration of the skin, including sunitinib,7 sorafenib,8 quinacrine, saffron supplements, santonin, fluorescein, 2,4-dinitrophenol, canthaxanthin, tetryl and picric acids, and acriflavine.2,4
Carotenoderma caused by a diet rich in β-carotene is a benign condition in which a diet low in β-carotene is implicated for treatment. Contrary to popular belief, vitamin A toxicity does not occur in the presence of a surplus of β-carotenes because the enzymatic conversion of β-carotene to vitamin A is strictly regulated.9 Although acknowledging the various causes of carotenoderma is important, a simple history and laboratory testing for elevated serum β-carotene levels can eliminate further unnecessary testing and allow for prompt recognition of the condition. Appropriate dietary modifications also may be warranted.
- Roe DA. Assessment of risk factors for carotenodermia and cutaneous signs of hypervitaminosis A in college-aged populations. Semin Dermatol. 1991;10:303-308.
- Manolios N, Samaras K. Hypercarotenaemia. Intern Med J. 2006;36:534.
- Wageesha ND, Ekanayake S, Jansz ER, et al. Studies on hypercarotenemia due to excessive ingestion of carrot, pumpkin and papaw [published online September 27, 2010]. Int J Food Sci Nutr. 2011;62:20-25.
- Maharshak N, Shapiro J, Trau H. Carotenoderma—a review of the current literature. Int J Dermatol. 2003;42:178-181.
- Maruani A, Labarthe F, Dupré T, et al. Hypercarotenaemia in an infant [in French]. Ann Dermatol Venereol. 2010;137:32-35.
- Shaw JA, Koti M. Clinical images. CMAJ. 2009;180:895.
- Vignand-Courtin C, Martin C, Le Beller C, et al. Cutaneous side effects associated with sunitinib: an analysis of 8 cases. Int J Clin Pharm. 2012;34:286-289.
- Dasanu CA, Alexandrescu DT, Dutcher J. Yellow skin discoloration associated with sorafenib use for treatment of metastatic renal cell carcinoma. South Med J. 2007;100:328-330.
- Lascari AD. Carotenemia. a review. Clin Pediatr (Phila). 1981;20:25-29.
To the Editor:
Carotenoderma is a cutaneous manifestation of elevated serum β-carotene levels and classically localizes to fatty tissues and areas rich in sweat glands. We present a case of carotenoderma associated with a diet rich in red palm oil, a common food additive in parts of the world outside of the United States.
A previously healthy 8-year-old boy who recently immigrated to the United States from Liberia was hospitalized for treatment of a febrile illness that subsequently was attributed to a viral syndrome. On physical examination by the dermatology department, the patient was noted to have marked orange discoloration on the palms and soles (Figure). Laboratory workup revealed elevated serum β-carotene levels of 809 μg/dL (reference range, 10–85 μg/dL). Testing of hemoglobin/hematocrit levels and liver, thyroid, and kidney function was normal, and systemic examination revealed no further abnormalities. Upon further inquiry by the dermatology department, the patient’s family reported frequent addition of red palm oil to all of the child’s meals. The patient subsequently was diagnosed with carotenoderma and was instructed to limit inclusion of red palm oil in his diet.
Red palm oil is a rich source of β-carotene and is commonly used outside the United States as a dietary supplement or food flavoring. Excessive consumption of red palm oil or other sources rich in carotenes can result in elevated serum carotene levels or hypercarotenemia. An elevation in serum β-carotene levels may be recognized from 4 to 7 weeks after starting a β-carotene–rich diet.1
While dietary consumption of carotenes is the most common cause of carotenoderma, others include kidney or liver disease, hyperlipidemia, porphyria, diabetes mellitus, hypothyroidism, and anorexia nervosa.2-4 Moreover, since carotenoids are enzymatically converted to vitamin A in the small intestine, a mutation of the gene of the conversion enzyme β-carotene 15,15’-monooxygenase 1 (BCMO1) also can cause be a rare cause of hypercarotenemia.3
Carotenoderma, the clinical cutaneous manifestation of hypercarotenemia, occurs as a result of β-carotene deposits in the skin when serum concentration exceeds 250 μg/dL. More specifically, β-carotene accumulates mainly in the lipid-rich stratum corneum as well as in sweat and sebum, which explains the localized discoloration in fatty tissues and areas rich in sweat glands (eg, nasolabial folds, palms, soles).3,4 The sclerae of the eyes are not affected by the surplus of β-carotene in carotenoderma, which helps distinguish it from jaundice.5
The differential diagnosis of yellow discoloration of the skin includes jaundice, encompassing the prehepatic, hepatocellular, and posthepatic categories.4 Also noteworthy in the differential diagnosis is lycopenemia, which occurs as a result of eating lycopene-rich foods (eg, tomatoes), resulting in a deeper orange-yellow pigmentation when compared to the cutaneous manifestation of hypercarotenemia.2,4,6 Several drugs also have been reported to induce yellow discoloration of the skin, including sunitinib,7 sorafenib,8 quinacrine, saffron supplements, santonin, fluorescein, 2,4-dinitrophenol, canthaxanthin, tetryl and picric acids, and acriflavine.2,4
Carotenoderma caused by a diet rich in β-carotene is a benign condition in which a diet low in β-carotene is implicated for treatment. Contrary to popular belief, vitamin A toxicity does not occur in the presence of a surplus of β-carotenes because the enzymatic conversion of β-carotene to vitamin A is strictly regulated.9 Although acknowledging the various causes of carotenoderma is important, a simple history and laboratory testing for elevated serum β-carotene levels can eliminate further unnecessary testing and allow for prompt recognition of the condition. Appropriate dietary modifications also may be warranted.
To the Editor:
Carotenoderma is a cutaneous manifestation of elevated serum β-carotene levels and classically localizes to fatty tissues and areas rich in sweat glands. We present a case of carotenoderma associated with a diet rich in red palm oil, a common food additive in parts of the world outside of the United States.
A previously healthy 8-year-old boy who recently immigrated to the United States from Liberia was hospitalized for treatment of a febrile illness that subsequently was attributed to a viral syndrome. On physical examination by the dermatology department, the patient was noted to have marked orange discoloration on the palms and soles (Figure). Laboratory workup revealed elevated serum β-carotene levels of 809 μg/dL (reference range, 10–85 μg/dL). Testing of hemoglobin/hematocrit levels and liver, thyroid, and kidney function was normal, and systemic examination revealed no further abnormalities. Upon further inquiry by the dermatology department, the patient’s family reported frequent addition of red palm oil to all of the child’s meals. The patient subsequently was diagnosed with carotenoderma and was instructed to limit inclusion of red palm oil in his diet.
Red palm oil is a rich source of β-carotene and is commonly used outside the United States as a dietary supplement or food flavoring. Excessive consumption of red palm oil or other sources rich in carotenes can result in elevated serum carotene levels or hypercarotenemia. An elevation in serum β-carotene levels may be recognized from 4 to 7 weeks after starting a β-carotene–rich diet.1
While dietary consumption of carotenes is the most common cause of carotenoderma, others include kidney or liver disease, hyperlipidemia, porphyria, diabetes mellitus, hypothyroidism, and anorexia nervosa.2-4 Moreover, since carotenoids are enzymatically converted to vitamin A in the small intestine, a mutation of the gene of the conversion enzyme β-carotene 15,15’-monooxygenase 1 (BCMO1) also can cause be a rare cause of hypercarotenemia.3
Carotenoderma, the clinical cutaneous manifestation of hypercarotenemia, occurs as a result of β-carotene deposits in the skin when serum concentration exceeds 250 μg/dL. More specifically, β-carotene accumulates mainly in the lipid-rich stratum corneum as well as in sweat and sebum, which explains the localized discoloration in fatty tissues and areas rich in sweat glands (eg, nasolabial folds, palms, soles).3,4 The sclerae of the eyes are not affected by the surplus of β-carotene in carotenoderma, which helps distinguish it from jaundice.5
The differential diagnosis of yellow discoloration of the skin includes jaundice, encompassing the prehepatic, hepatocellular, and posthepatic categories.4 Also noteworthy in the differential diagnosis is lycopenemia, which occurs as a result of eating lycopene-rich foods (eg, tomatoes), resulting in a deeper orange-yellow pigmentation when compared to the cutaneous manifestation of hypercarotenemia.2,4,6 Several drugs also have been reported to induce yellow discoloration of the skin, including sunitinib,7 sorafenib,8 quinacrine, saffron supplements, santonin, fluorescein, 2,4-dinitrophenol, canthaxanthin, tetryl and picric acids, and acriflavine.2,4
Carotenoderma caused by a diet rich in β-carotene is a benign condition in which a diet low in β-carotene is implicated for treatment. Contrary to popular belief, vitamin A toxicity does not occur in the presence of a surplus of β-carotenes because the enzymatic conversion of β-carotene to vitamin A is strictly regulated.9 Although acknowledging the various causes of carotenoderma is important, a simple history and laboratory testing for elevated serum β-carotene levels can eliminate further unnecessary testing and allow for prompt recognition of the condition. Appropriate dietary modifications also may be warranted.
- Roe DA. Assessment of risk factors for carotenodermia and cutaneous signs of hypervitaminosis A in college-aged populations. Semin Dermatol. 1991;10:303-308.
- Manolios N, Samaras K. Hypercarotenaemia. Intern Med J. 2006;36:534.
- Wageesha ND, Ekanayake S, Jansz ER, et al. Studies on hypercarotenemia due to excessive ingestion of carrot, pumpkin and papaw [published online September 27, 2010]. Int J Food Sci Nutr. 2011;62:20-25.
- Maharshak N, Shapiro J, Trau H. Carotenoderma—a review of the current literature. Int J Dermatol. 2003;42:178-181.
- Maruani A, Labarthe F, Dupré T, et al. Hypercarotenaemia in an infant [in French]. Ann Dermatol Venereol. 2010;137:32-35.
- Shaw JA, Koti M. Clinical images. CMAJ. 2009;180:895.
- Vignand-Courtin C, Martin C, Le Beller C, et al. Cutaneous side effects associated with sunitinib: an analysis of 8 cases. Int J Clin Pharm. 2012;34:286-289.
- Dasanu CA, Alexandrescu DT, Dutcher J. Yellow skin discoloration associated with sorafenib use for treatment of metastatic renal cell carcinoma. South Med J. 2007;100:328-330.
- Lascari AD. Carotenemia. a review. Clin Pediatr (Phila). 1981;20:25-29.
- Roe DA. Assessment of risk factors for carotenodermia and cutaneous signs of hypervitaminosis A in college-aged populations. Semin Dermatol. 1991;10:303-308.
- Manolios N, Samaras K. Hypercarotenaemia. Intern Med J. 2006;36:534.
- Wageesha ND, Ekanayake S, Jansz ER, et al. Studies on hypercarotenemia due to excessive ingestion of carrot, pumpkin and papaw [published online September 27, 2010]. Int J Food Sci Nutr. 2011;62:20-25.
- Maharshak N, Shapiro J, Trau H. Carotenoderma—a review of the current literature. Int J Dermatol. 2003;42:178-181.
- Maruani A, Labarthe F, Dupré T, et al. Hypercarotenaemia in an infant [in French]. Ann Dermatol Venereol. 2010;137:32-35.
- Shaw JA, Koti M. Clinical images. CMAJ. 2009;180:895.
- Vignand-Courtin C, Martin C, Le Beller C, et al. Cutaneous side effects associated with sunitinib: an analysis of 8 cases. Int J Clin Pharm. 2012;34:286-289.
- Dasanu CA, Alexandrescu DT, Dutcher J. Yellow skin discoloration associated with sorafenib use for treatment of metastatic renal cell carcinoma. South Med J. 2007;100:328-330.
- Lascari AD. Carotenemia. a review. Clin Pediatr (Phila). 1981;20:25-29.
Practice Points
- Carotenoderma is a cutaneous manifestation of elevated serum β-carotene levels and classically localizes to fatty tissues and areas rich in sweat glands.
- Carotenoderma caused by a diet rich in β-carotene is a benign condition in which a diet low in β-carotene is implicated for treatment.
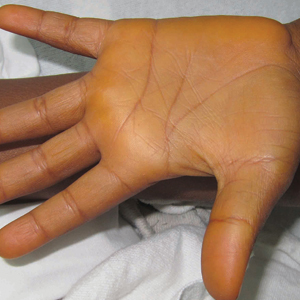
Atypical Presentation of Acquired Angioedema
To the Editor:
A 65-year-old woman with B-cell marginal zone lymphoma presented with asymptomatic swelling and redness of the upper and lower eyelids of 1 week’s duration that was unresponsive to topical corticosteroids for presumptive allergic contact dermatitis. She denied any lip or tongue swelling, abdominal pain, or difficulty breathing or swallowing. Diagnosis of acquired angioedema (AAE) was confirmed on laboratory analysis, which showed C1q levels less than 3.6 mg/dL (reference range, 5.0–8.6 mg/dL), complement component 4 levels less than 8 mg/dL (reference range, 14–44 mg/dL), and C1 esterase inhibitor (C1-INH) levels of 3 mg/dL (reference range, 12–30 mg/dL).
A review of the patient’s medical record showed chronic thrombocytopenia secondary to previous chemotherapy. It was determined that the patient’s ecchymosis and purpura of the eyelids was secondary to a low platelet count resulting in bleeding into the area of angioedema (Figure). Serum protein electrophoresis did not demonstrate a monoclonal spike, and flow cytometry showed persistent B-cell leukemia without evidence of an aberrant T-cell antigenic profile. The edema and purpura of the eyelids spontaneously resolved over days, and the patient has had no recurrences to date. She was prescribed icatibant for treatment of future acute AAE attacks.

The common pathway of AAE involves the inability of C1-INH to stop activation of the complement, fibrinolytic, and contact systems. Failure to control the contact system leads to increased bradykinin production resulting in vasodilation and edema. Diagnosis of hereditary angioedema (HAE) types 1 and 2 can be confirmed in the setting of low complement component 4 and C1-INH functional levels and normal C1q levels; in AAE, C1q levels also are low.1,2
The malignancies most frequently associated with AAE are non-Hodgkin lymphomas (eg, nodal marginal zone lymphoma, splenic marginal zone lymphoma), such as in our patient, as well as monoclonal gammopathies.2 Triggers of AAE include trauma (eg, surgery, strenuous exercise), infection, and use of certain medications such as angiotensin-converting enzyme inhibitors and estrogen, but most episodes are spontaneous. Swelling of any cutaneous surface can occur in the setting of AAE. Mucosal involvement appears to be limited to the upper airway and gastrointestinal tract. Edema of the upper airway mucosa can lead to asphyxiation. In these cases, asphyxia can occur rapidly, and therefore all patients with upper airway involvement should present to the emergency room or call 911. Pain from swelling in the gastrointestinal tract can mimic an acute abdomen.3
Newly developed targeted therapies for HAE also appear to be effective in treating AAE. A summary of available treatments for angioedema is provided in the Table. Human plasma C1-INH can be used intravenously to treat acute attacks or can be given prophylactically to prevent attacks, but large doses may be necessary due to consumption of the protein.1,3 The risk of bloodborne disease as a result of treatment exists, but screening and processing during production of the plasma makes this unlikely. Ecallantide is a reversible inhibitor of plasma kallikrein.1,3 Rapid onset and subcutaneous dosing make it useful for treatment of acute AAE attacks. Because anaphylaxis has been reported in up to 3% of patients, ecallantide includes a boxed warning indicating that it must be administered by a health care professional with appropriate medical support to manage anaphylaxis and HAE.4 Icatibant is a selective competitive antagonist of bradykinin receptor B2. It can be administered subcutaneously by the patient, making it ideal for rapid treatment of angioedema.1,3 Adverse events include pain and irritation at the injection site.
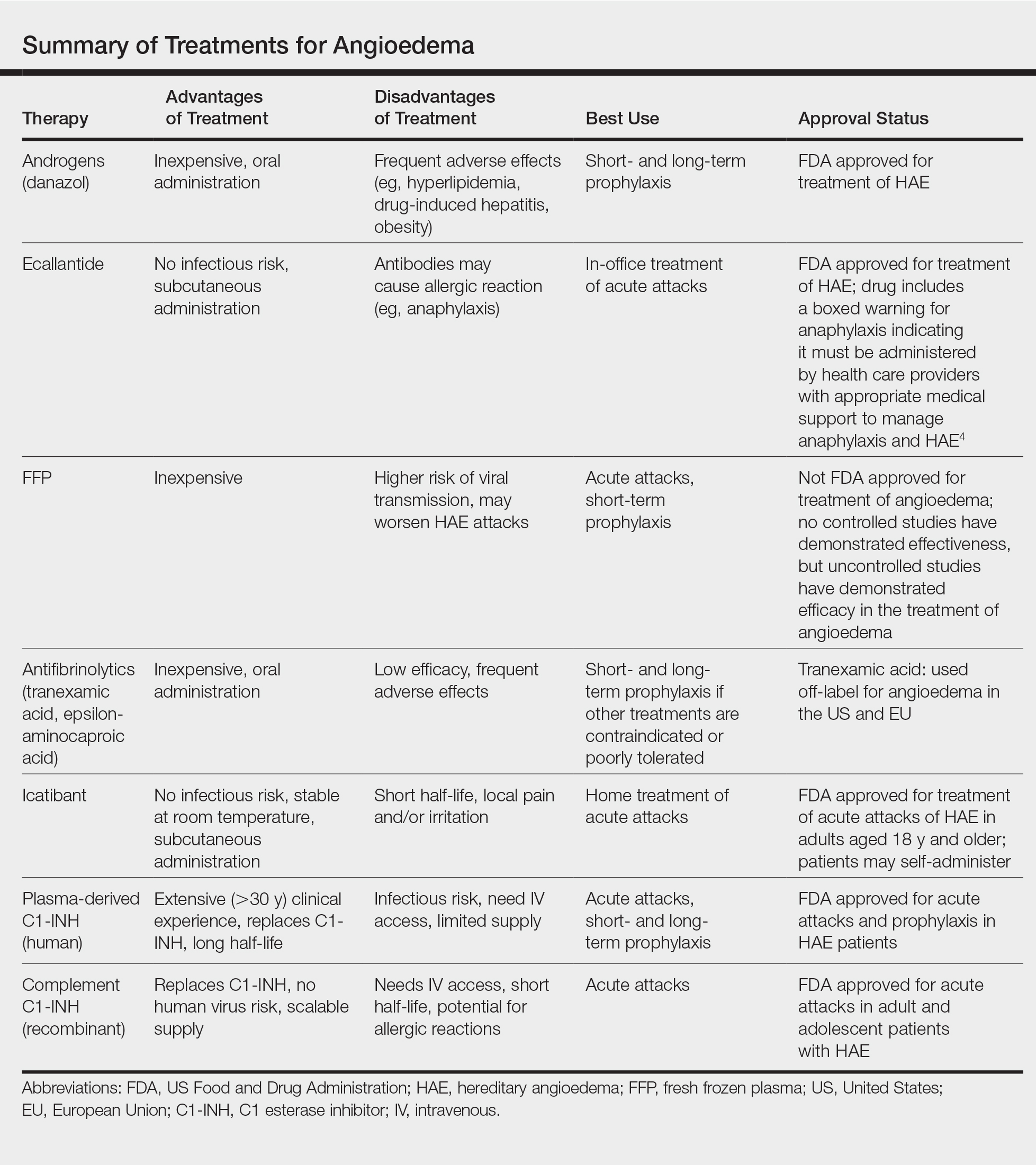
The most appropriate therapy for AAE is treatment of the underlying malignancy. Recognition and proper treatment of AAE is essential, as bradykinin-induced angioedema (AAE, HAE and angiotensin-converting enzyme inhibitor induced angioedema) does not respond to antihistamines and corticosteroids and instead requires therapy as discussed above.
- Craig T, Riedl M, Dykewicz MS, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009;102:366-372.
- Cugno M, Castelli R, Cicardi M. Angioedema due to acquired C1-inhibitor deficiency: a bridging connection between autoimmunity and lymphoproliferation. Autoimmun Rev. 2008;8:156-159.
- Buyantseva LV, Sardana N, Craig TJ. Update on treatment of hereditary angioedema. Asian Pac J Allergy Immunol. 2012;30:89-98.
- Kalbitor [package insert]. Burlington, MA: Dyax Corp; 2015.
To the Editor:
A 65-year-old woman with B-cell marginal zone lymphoma presented with asymptomatic swelling and redness of the upper and lower eyelids of 1 week’s duration that was unresponsive to topical corticosteroids for presumptive allergic contact dermatitis. She denied any lip or tongue swelling, abdominal pain, or difficulty breathing or swallowing. Diagnosis of acquired angioedema (AAE) was confirmed on laboratory analysis, which showed C1q levels less than 3.6 mg/dL (reference range, 5.0–8.6 mg/dL), complement component 4 levels less than 8 mg/dL (reference range, 14–44 mg/dL), and C1 esterase inhibitor (C1-INH) levels of 3 mg/dL (reference range, 12–30 mg/dL).
A review of the patient’s medical record showed chronic thrombocytopenia secondary to previous chemotherapy. It was determined that the patient’s ecchymosis and purpura of the eyelids was secondary to a low platelet count resulting in bleeding into the area of angioedema (Figure). Serum protein electrophoresis did not demonstrate a monoclonal spike, and flow cytometry showed persistent B-cell leukemia without evidence of an aberrant T-cell antigenic profile. The edema and purpura of the eyelids spontaneously resolved over days, and the patient has had no recurrences to date. She was prescribed icatibant for treatment of future acute AAE attacks.
The common pathway of AAE involves the inability of C1-INH to stop activation of the complement, fibrinolytic, and contact systems. Failure to control the contact system leads to increased bradykinin production resulting in vasodilation and edema. Diagnosis of hereditary angioedema (HAE) types 1 and 2 can be confirmed in the setting of low complement component 4 and C1-INH functional levels and normal C1q levels; in AAE, C1q levels also are low.1,2
The malignancies most frequently associated with AAE are non-Hodgkin lymphomas (eg, nodal marginal zone lymphoma, splenic marginal zone lymphoma), such as in our patient, as well as monoclonal gammopathies.2 Triggers of AAE include trauma (eg, surgery, strenuous exercise), infection, and use of certain medications such as angiotensin-converting enzyme inhibitors and estrogen, but most episodes are spontaneous. Swelling of any cutaneous surface can occur in the setting of AAE. Mucosal involvement appears to be limited to the upper airway and gastrointestinal tract. Edema of the upper airway mucosa can lead to asphyxiation. In these cases, asphyxia can occur rapidly, and therefore all patients with upper airway involvement should present to the emergency room or call 911. Pain from swelling in the gastrointestinal tract can mimic an acute abdomen.3
Newly developed targeted therapies for HAE also appear to be effective in treating AAE. A summary of available treatments for angioedema is provided in the Table. Human plasma C1-INH can be used intravenously to treat acute attacks or can be given prophylactically to prevent attacks, but large doses may be necessary due to consumption of the protein.1,3 The risk of bloodborne disease as a result of treatment exists, but screening and processing during production of the plasma makes this unlikely. Ecallantide is a reversible inhibitor of plasma kallikrein.1,3 Rapid onset and subcutaneous dosing make it useful for treatment of acute AAE attacks. Because anaphylaxis has been reported in up to 3% of patients, ecallantide includes a boxed warning indicating that it must be administered by a health care professional with appropriate medical support to manage anaphylaxis and HAE.4 Icatibant is a selective competitive antagonist of bradykinin receptor B2. It can be administered subcutaneously by the patient, making it ideal for rapid treatment of angioedema.1,3 Adverse events include pain and irritation at the injection site.
The most appropriate therapy for AAE is treatment of the underlying malignancy. Recognition and proper treatment of AAE is essential, as bradykinin-induced angioedema (AAE, HAE and angiotensin-converting enzyme inhibitor induced angioedema) does not respond to antihistamines and corticosteroids and instead requires therapy as discussed above.
To the Editor:
A 65-year-old woman with B-cell marginal zone lymphoma presented with asymptomatic swelling and redness of the upper and lower eyelids of 1 week’s duration that was unresponsive to topical corticosteroids for presumptive allergic contact dermatitis. She denied any lip or tongue swelling, abdominal pain, or difficulty breathing or swallowing. Diagnosis of acquired angioedema (AAE) was confirmed on laboratory analysis, which showed C1q levels less than 3.6 mg/dL (reference range, 5.0–8.6 mg/dL), complement component 4 levels less than 8 mg/dL (reference range, 14–44 mg/dL), and C1 esterase inhibitor (C1-INH) levels of 3 mg/dL (reference range, 12–30 mg/dL).
A review of the patient’s medical record showed chronic thrombocytopenia secondary to previous chemotherapy. It was determined that the patient’s ecchymosis and purpura of the eyelids was secondary to a low platelet count resulting in bleeding into the area of angioedema (Figure). Serum protein electrophoresis did not demonstrate a monoclonal spike, and flow cytometry showed persistent B-cell leukemia without evidence of an aberrant T-cell antigenic profile. The edema and purpura of the eyelids spontaneously resolved over days, and the patient has had no recurrences to date. She was prescribed icatibant for treatment of future acute AAE attacks.
The common pathway of AAE involves the inability of C1-INH to stop activation of the complement, fibrinolytic, and contact systems. Failure to control the contact system leads to increased bradykinin production resulting in vasodilation and edema. Diagnosis of hereditary angioedema (HAE) types 1 and 2 can be confirmed in the setting of low complement component 4 and C1-INH functional levels and normal C1q levels; in AAE, C1q levels also are low.1,2
The malignancies most frequently associated with AAE are non-Hodgkin lymphomas (eg, nodal marginal zone lymphoma, splenic marginal zone lymphoma), such as in our patient, as well as monoclonal gammopathies.2 Triggers of AAE include trauma (eg, surgery, strenuous exercise), infection, and use of certain medications such as angiotensin-converting enzyme inhibitors and estrogen, but most episodes are spontaneous. Swelling of any cutaneous surface can occur in the setting of AAE. Mucosal involvement appears to be limited to the upper airway and gastrointestinal tract. Edema of the upper airway mucosa can lead to asphyxiation. In these cases, asphyxia can occur rapidly, and therefore all patients with upper airway involvement should present to the emergency room or call 911. Pain from swelling in the gastrointestinal tract can mimic an acute abdomen.3
Newly developed targeted therapies for HAE also appear to be effective in treating AAE. A summary of available treatments for angioedema is provided in the Table. Human plasma C1-INH can be used intravenously to treat acute attacks or can be given prophylactically to prevent attacks, but large doses may be necessary due to consumption of the protein.1,3 The risk of bloodborne disease as a result of treatment exists, but screening and processing during production of the plasma makes this unlikely. Ecallantide is a reversible inhibitor of plasma kallikrein.1,3 Rapid onset and subcutaneous dosing make it useful for treatment of acute AAE attacks. Because anaphylaxis has been reported in up to 3% of patients, ecallantide includes a boxed warning indicating that it must be administered by a health care professional with appropriate medical support to manage anaphylaxis and HAE.4 Icatibant is a selective competitive antagonist of bradykinin receptor B2. It can be administered subcutaneously by the patient, making it ideal for rapid treatment of angioedema.1,3 Adverse events include pain and irritation at the injection site.
The most appropriate therapy for AAE is treatment of the underlying malignancy. Recognition and proper treatment of AAE is essential, as bradykinin-induced angioedema (AAE, HAE and angiotensin-converting enzyme inhibitor induced angioedema) does not respond to antihistamines and corticosteroids and instead requires therapy as discussed above.
- Craig T, Riedl M, Dykewicz MS, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009;102:366-372.
- Cugno M, Castelli R, Cicardi M. Angioedema due to acquired C1-inhibitor deficiency: a bridging connection between autoimmunity and lymphoproliferation. Autoimmun Rev. 2008;8:156-159.
- Buyantseva LV, Sardana N, Craig TJ. Update on treatment of hereditary angioedema. Asian Pac J Allergy Immunol. 2012;30:89-98.
- Kalbitor [package insert]. Burlington, MA: Dyax Corp; 2015.
- Craig T, Riedl M, Dykewicz MS, et al. When is prophylaxis for hereditary angioedema necessary? Ann Allergy Asthma Immunol. 2009;102:366-372.
- Cugno M, Castelli R, Cicardi M. Angioedema due to acquired C1-inhibitor deficiency: a bridging connection between autoimmunity and lymphoproliferation. Autoimmun Rev. 2008;8:156-159.
- Buyantseva LV, Sardana N, Craig TJ. Update on treatment of hereditary angioedema. Asian Pac J Allergy Immunol. 2012;30:89-98.
- Kalbitor [package insert]. Burlington, MA: Dyax Corp; 2015.
Practice Points
- Late-onset angioedema without urticaria can be secondary to acquired angioedema with C1 esterase inhibitor deficiency (C1-INH).
- Most patients with angioedema with C1-INH inhibitor deficiency will have either a monoclonal gammopathy or a lymphoma.
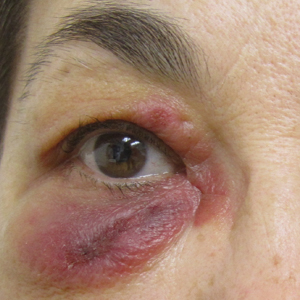
Carcinoma Erysipeloides of Papillary Serous Ovarian Cancer Mimicking Cellulitis of the Abdominal Wall
To the Editor:
A 40-year-old woman with a history of stage IIIC ovarian cancer presented with progressing abdominal erythema and pain of 1 month’s duration. She had been diagnosed 4 years prior with grade 3, poorly differentiated papillary serous carcinoma involving the bilateral ovaries, uterine tubes, uterus, and omentum with lymphovascular invasion. She underwent tumor resection and debulking followed by paclitaxel plus platinum-based chemotherapy. The cancer recurred 2 years later with carcinomatous ascites. She declined chemotherapy but underwent therapeutic paracentesis.
One month prior to presentation, the patient developed a small, tender, erythematous patch on the abdomen. Her primary physician started her on cephalexin for presumed cellulitis without improvement. The erythema continued to spread on the abdomen with worsening pain, which prompted her presentation to the emergency department. She was admitted and started on intravenous vancomycin.
On admission to the hospital, the patient was cachexic and afebrile with a white blood cell count of 10,400/µL (reference range, 4500–11,000/µL). Physical examination revealed a well-demarcated, 15×20-cm, erythematous, blanchable, indurated plaque in the periumbilical region (Figure 1). The plaque was tender to palpation with guarding but no increased warmth. Punch biopsies of the abdominal skin revealed carcinoma within the lymphatic channels in the deep dermis and dilated lymphatics throughout the overlying dermis (Figure 2). These findings were diagnostic for carcinoma erysipeloides. Tissue and blood cultures were negative for bacterial, fungal, or mycobacterial growth. Vancomycin was discontinued, and she was discharged with pain medication. She declined chemotherapy due to the potential side effects and elected to continue symptomatic management with palliative paracentesis. After she was discharged, she underwent a tunneled pleural catheterization for recurrent malignant pleural effusions.
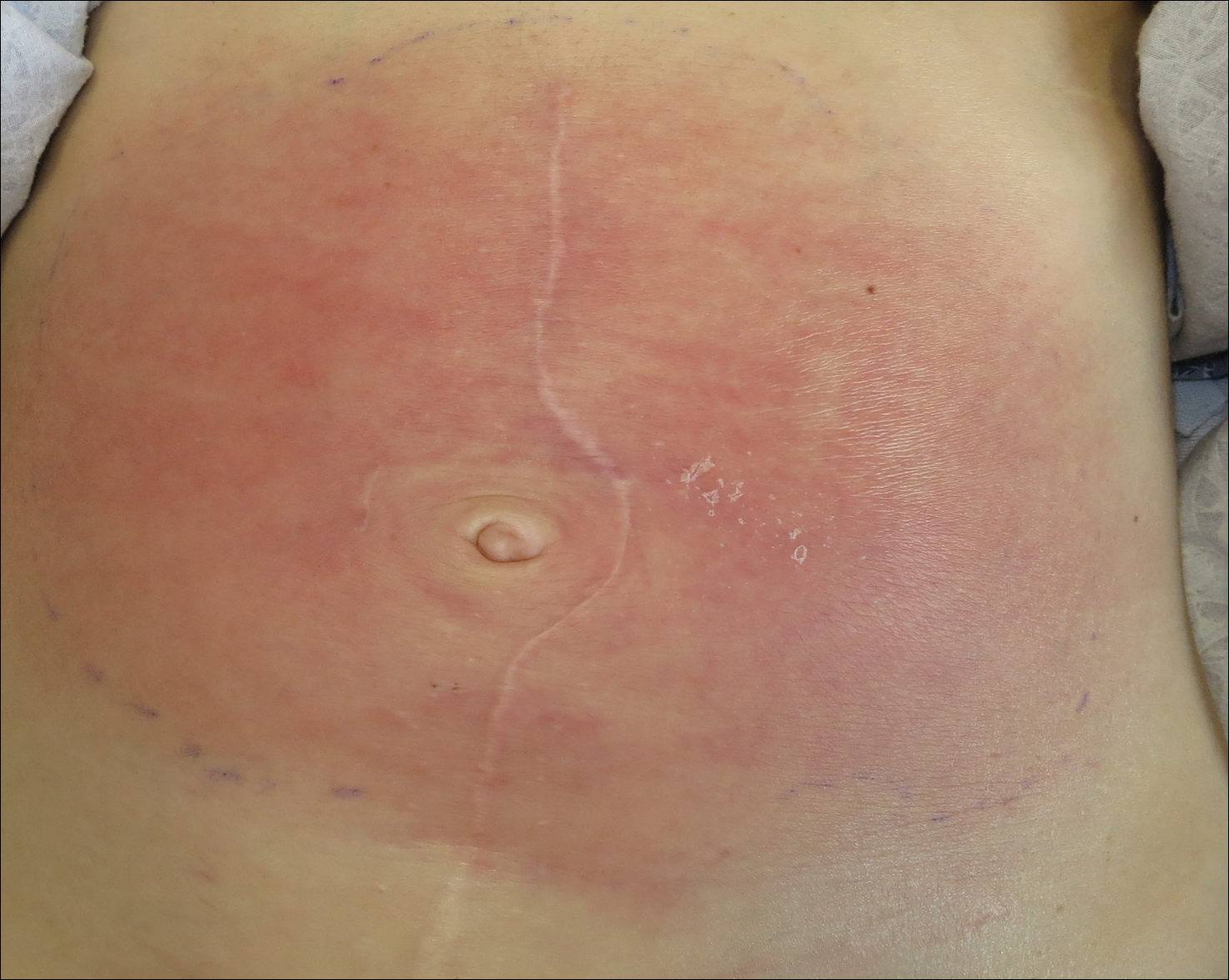
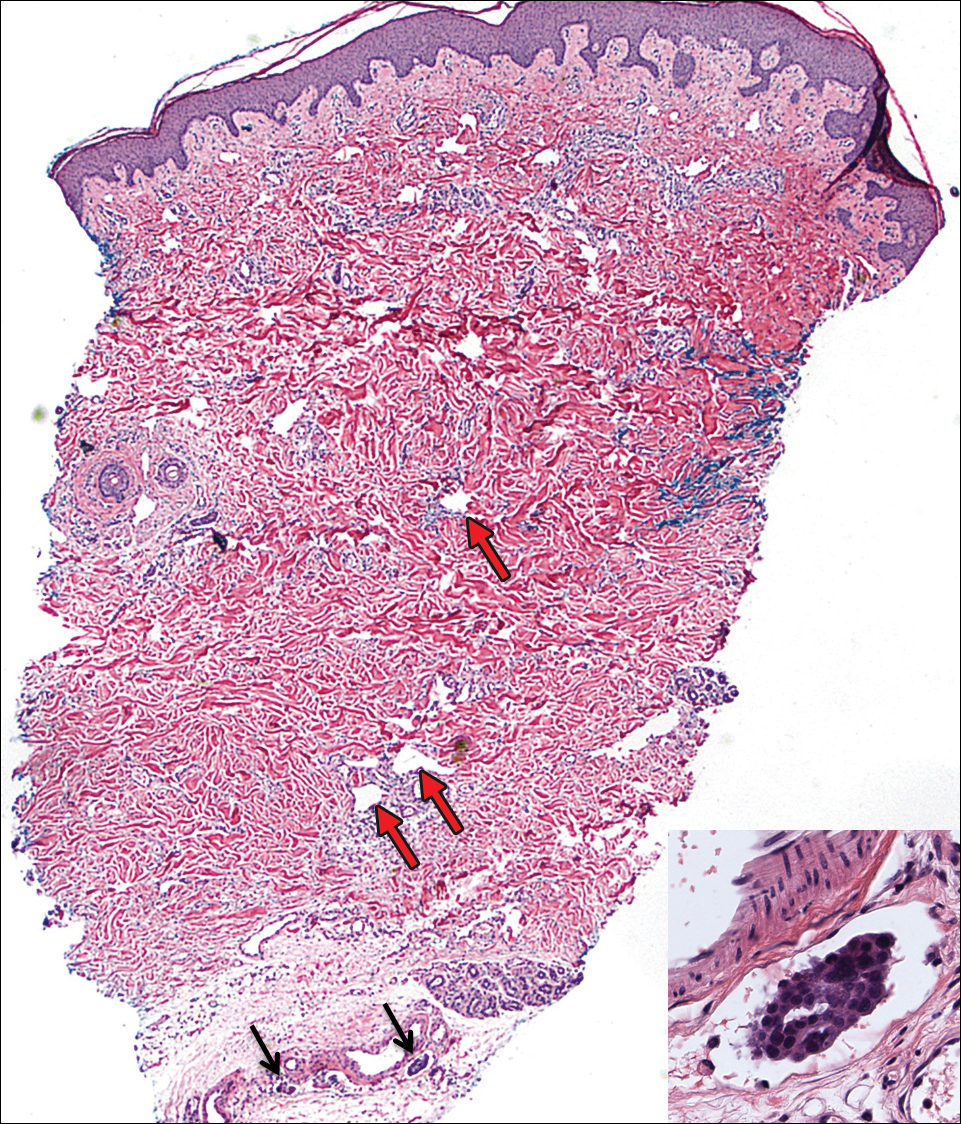
Carcinoma erysipeloides is a rare cutaneous metastasis secondary to internal malignancy that presents as well-demarcated areas of erythema and is sometimes misdiagnosed as cellulitis or erysipelas. Histology is notable for lymphovascular congestion without inflammation. Carcinoma erysipeloides most commonly is associated with breast cancer, but it also has been described in cancers of the prostate, larynx, stomach, lungs, thyroid, parotid gland, fallopian tubes, cervix, pancreas, and metastatic melanoma.1-5 While the pathogenesis of carcinoma erysipeloides is poorly understood, it is thought to occur by direct spread of tumor cells from the lymph nodes to the cutaneous lymphatics, causing obstruction and edema.
Ovarian cancer has the highest mortality of all gynecologic cancers and often is associated with delayed diagnosis. Cutaneous metastasis is a late manifestation often presenting as subcutaneous nodules.6,7 Carcinoma erysipeloides is an even rarer presentation of ovarian cancer, with a poor prognosis and a median survival of 18 months.8 A PubMed search of articles indexed for MEDLINE using the term carcinoma erysipeloides revealed 9 cases of carcinoma erysipeloides from ovarian cancer: 1 describing erythematous papules, plaques, and zosteriform vesicles on the upper thighs to the lower abdomen,9 and 8 describing erythematous plaques on the breasts.8,10 We report a case of carcinoma erysipeloides associated with stage IIIc ovarian cancer localized to the abdominal wall mimicking cellulitis. Our report reminds clinicians of this important diagnosis in ovarian cancer and of the importance of a skin biopsy to expedite a definitive diagnosis. Immunohistochemistry using ovarian tumor markers (eg, paired-box gene 8, cancer antigen 125) is an additional tool to accurately identify malignant cells in skin biopsy.8,10 Once diagnosed, primary treatment for carcinoma erysipeloides is treatment of the underlying malignancy.
- Cormio G, Capotorto M, Di Vagno G, et al. Skin metastases in ovarian carcinoma: a report of nine cases and a review of the literature. Gynecol Oncol. 2003;90:682-685.
- Kim MK, Kim SH, Lee YY, et al. Metastatic skin lesions on lower extremities in a patient with recurrent serous papillary ovarian carcinoma: a case report and literature review. Cancer Res Treat. 2012;44:142-145.
- Karmali S, Rudmik L, Temple W, et al. Melanoma erysipeloides. Can J Surg. 2005;48:159-160.
- Godinez-Puig V, Frangos J, Hollmann TJ, et al. Carcinoma erysipeloides of the breast in a patient with advanced ovarian carcinoma. Clin Infect Dis. 2012;54:575-576.
- Hazelrigg DE, Rudolph AH. Inflammatory metastic carcinoma. carcinoma erysipelatoides. Arch Dermatol. 1977;113:69-70.
- Cowan LJ, Roller JI, Connelly PJ, et al. Extraovarian stage IV peritoneal serous papillary carcinoma presenting as an asymptomatic skin lesion—a case report and literature review. Gynecol Oncol. 1995;57:433-435.
- Schonmann R, Altaras M, Biron T, et al. Inflammatory skin metastases from ovarian carcinoma—a case report and review of the literature. Gynecol Oncol. 2003;90:670-672.
- Klein RL, Brown AR, Gomez-Castro CM, et al. Ovarian cancer metastatic to the breast presenting as inflammatory breast cancer: a case report and literature review. J Cancer. 2010;1:27-31.
- Lee HC, Chu CY, Hsiao CH. Carcinoma erysipeloides from ovarian clear-cell carcinoma. J Clin Oncol. 2007;25:5828-5830.
- Godinez-Puig V, Frangos J, Hollmann TJ, et al. Photo quiz. rash in a patient with ovarian cancer. Clin Infect Dis. 2012;54:538, 575-576.
To the Editor:
A 40-year-old woman with a history of stage IIIC ovarian cancer presented with progressing abdominal erythema and pain of 1 month’s duration. She had been diagnosed 4 years prior with grade 3, poorly differentiated papillary serous carcinoma involving the bilateral ovaries, uterine tubes, uterus, and omentum with lymphovascular invasion. She underwent tumor resection and debulking followed by paclitaxel plus platinum-based chemotherapy. The cancer recurred 2 years later with carcinomatous ascites. She declined chemotherapy but underwent therapeutic paracentesis.
One month prior to presentation, the patient developed a small, tender, erythematous patch on the abdomen. Her primary physician started her on cephalexin for presumed cellulitis without improvement. The erythema continued to spread on the abdomen with worsening pain, which prompted her presentation to the emergency department. She was admitted and started on intravenous vancomycin.
On admission to the hospital, the patient was cachexic and afebrile with a white blood cell count of 10,400/µL (reference range, 4500–11,000/µL). Physical examination revealed a well-demarcated, 15×20-cm, erythematous, blanchable, indurated plaque in the periumbilical region (Figure 1). The plaque was tender to palpation with guarding but no increased warmth. Punch biopsies of the abdominal skin revealed carcinoma within the lymphatic channels in the deep dermis and dilated lymphatics throughout the overlying dermis (Figure 2). These findings were diagnostic for carcinoma erysipeloides. Tissue and blood cultures were negative for bacterial, fungal, or mycobacterial growth. Vancomycin was discontinued, and she was discharged with pain medication. She declined chemotherapy due to the potential side effects and elected to continue symptomatic management with palliative paracentesis. After she was discharged, she underwent a tunneled pleural catheterization for recurrent malignant pleural effusions.
Carcinoma erysipeloides is a rare cutaneous metastasis secondary to internal malignancy that presents as well-demarcated areas of erythema and is sometimes misdiagnosed as cellulitis or erysipelas. Histology is notable for lymphovascular congestion without inflammation. Carcinoma erysipeloides most commonly is associated with breast cancer, but it also has been described in cancers of the prostate, larynx, stomach, lungs, thyroid, parotid gland, fallopian tubes, cervix, pancreas, and metastatic melanoma.1-5 While the pathogenesis of carcinoma erysipeloides is poorly understood, it is thought to occur by direct spread of tumor cells from the lymph nodes to the cutaneous lymphatics, causing obstruction and edema.
Ovarian cancer has the highest mortality of all gynecologic cancers and often is associated with delayed diagnosis. Cutaneous metastasis is a late manifestation often presenting as subcutaneous nodules.6,7 Carcinoma erysipeloides is an even rarer presentation of ovarian cancer, with a poor prognosis and a median survival of 18 months.8 A PubMed search of articles indexed for MEDLINE using the term carcinoma erysipeloides revealed 9 cases of carcinoma erysipeloides from ovarian cancer: 1 describing erythematous papules, plaques, and zosteriform vesicles on the upper thighs to the lower abdomen,9 and 8 describing erythematous plaques on the breasts.8,10 We report a case of carcinoma erysipeloides associated with stage IIIc ovarian cancer localized to the abdominal wall mimicking cellulitis. Our report reminds clinicians of this important diagnosis in ovarian cancer and of the importance of a skin biopsy to expedite a definitive diagnosis. Immunohistochemistry using ovarian tumor markers (eg, paired-box gene 8, cancer antigen 125) is an additional tool to accurately identify malignant cells in skin biopsy.8,10 Once diagnosed, primary treatment for carcinoma erysipeloides is treatment of the underlying malignancy.
To the Editor:
A 40-year-old woman with a history of stage IIIC ovarian cancer presented with progressing abdominal erythema and pain of 1 month’s duration. She had been diagnosed 4 years prior with grade 3, poorly differentiated papillary serous carcinoma involving the bilateral ovaries, uterine tubes, uterus, and omentum with lymphovascular invasion. She underwent tumor resection and debulking followed by paclitaxel plus platinum-based chemotherapy. The cancer recurred 2 years later with carcinomatous ascites. She declined chemotherapy but underwent therapeutic paracentesis.
One month prior to presentation, the patient developed a small, tender, erythematous patch on the abdomen. Her primary physician started her on cephalexin for presumed cellulitis without improvement. The erythema continued to spread on the abdomen with worsening pain, which prompted her presentation to the emergency department. She was admitted and started on intravenous vancomycin.
On admission to the hospital, the patient was cachexic and afebrile with a white blood cell count of 10,400/µL (reference range, 4500–11,000/µL). Physical examination revealed a well-demarcated, 15×20-cm, erythematous, blanchable, indurated plaque in the periumbilical region (Figure 1). The plaque was tender to palpation with guarding but no increased warmth. Punch biopsies of the abdominal skin revealed carcinoma within the lymphatic channels in the deep dermis and dilated lymphatics throughout the overlying dermis (Figure 2). These findings were diagnostic for carcinoma erysipeloides. Tissue and blood cultures were negative for bacterial, fungal, or mycobacterial growth. Vancomycin was discontinued, and she was discharged with pain medication. She declined chemotherapy due to the potential side effects and elected to continue symptomatic management with palliative paracentesis. After she was discharged, she underwent a tunneled pleural catheterization for recurrent malignant pleural effusions.
Carcinoma erysipeloides is a rare cutaneous metastasis secondary to internal malignancy that presents as well-demarcated areas of erythema and is sometimes misdiagnosed as cellulitis or erysipelas. Histology is notable for lymphovascular congestion without inflammation. Carcinoma erysipeloides most commonly is associated with breast cancer, but it also has been described in cancers of the prostate, larynx, stomach, lungs, thyroid, parotid gland, fallopian tubes, cervix, pancreas, and metastatic melanoma.1-5 While the pathogenesis of carcinoma erysipeloides is poorly understood, it is thought to occur by direct spread of tumor cells from the lymph nodes to the cutaneous lymphatics, causing obstruction and edema.
Ovarian cancer has the highest mortality of all gynecologic cancers and often is associated with delayed diagnosis. Cutaneous metastasis is a late manifestation often presenting as subcutaneous nodules.6,7 Carcinoma erysipeloides is an even rarer presentation of ovarian cancer, with a poor prognosis and a median survival of 18 months.8 A PubMed search of articles indexed for MEDLINE using the term carcinoma erysipeloides revealed 9 cases of carcinoma erysipeloides from ovarian cancer: 1 describing erythematous papules, plaques, and zosteriform vesicles on the upper thighs to the lower abdomen,9 and 8 describing erythematous plaques on the breasts.8,10 We report a case of carcinoma erysipeloides associated with stage IIIc ovarian cancer localized to the abdominal wall mimicking cellulitis. Our report reminds clinicians of this important diagnosis in ovarian cancer and of the importance of a skin biopsy to expedite a definitive diagnosis. Immunohistochemistry using ovarian tumor markers (eg, paired-box gene 8, cancer antigen 125) is an additional tool to accurately identify malignant cells in skin biopsy.8,10 Once diagnosed, primary treatment for carcinoma erysipeloides is treatment of the underlying malignancy.
- Cormio G, Capotorto M, Di Vagno G, et al. Skin metastases in ovarian carcinoma: a report of nine cases and a review of the literature. Gynecol Oncol. 2003;90:682-685.
- Kim MK, Kim SH, Lee YY, et al. Metastatic skin lesions on lower extremities in a patient with recurrent serous papillary ovarian carcinoma: a case report and literature review. Cancer Res Treat. 2012;44:142-145.
- Karmali S, Rudmik L, Temple W, et al. Melanoma erysipeloides. Can J Surg. 2005;48:159-160.
- Godinez-Puig V, Frangos J, Hollmann TJ, et al. Carcinoma erysipeloides of the breast in a patient with advanced ovarian carcinoma. Clin Infect Dis. 2012;54:575-576.
- Hazelrigg DE, Rudolph AH. Inflammatory metastic carcinoma. carcinoma erysipelatoides. Arch Dermatol. 1977;113:69-70.
- Cowan LJ, Roller JI, Connelly PJ, et al. Extraovarian stage IV peritoneal serous papillary carcinoma presenting as an asymptomatic skin lesion—a case report and literature review. Gynecol Oncol. 1995;57:433-435.
- Schonmann R, Altaras M, Biron T, et al. Inflammatory skin metastases from ovarian carcinoma—a case report and review of the literature. Gynecol Oncol. 2003;90:670-672.
- Klein RL, Brown AR, Gomez-Castro CM, et al. Ovarian cancer metastatic to the breast presenting as inflammatory breast cancer: a case report and literature review. J Cancer. 2010;1:27-31.
- Lee HC, Chu CY, Hsiao CH. Carcinoma erysipeloides from ovarian clear-cell carcinoma. J Clin Oncol. 2007;25:5828-5830.
- Godinez-Puig V, Frangos J, Hollmann TJ, et al. Photo quiz. rash in a patient with ovarian cancer. Clin Infect Dis. 2012;54:538, 575-576.
- Cormio G, Capotorto M, Di Vagno G, et al. Skin metastases in ovarian carcinoma: a report of nine cases and a review of the literature. Gynecol Oncol. 2003;90:682-685.
- Kim MK, Kim SH, Lee YY, et al. Metastatic skin lesions on lower extremities in a patient with recurrent serous papillary ovarian carcinoma: a case report and literature review. Cancer Res Treat. 2012;44:142-145.
- Karmali S, Rudmik L, Temple W, et al. Melanoma erysipeloides. Can J Surg. 2005;48:159-160.
- Godinez-Puig V, Frangos J, Hollmann TJ, et al. Carcinoma erysipeloides of the breast in a patient with advanced ovarian carcinoma. Clin Infect Dis. 2012;54:575-576.
- Hazelrigg DE, Rudolph AH. Inflammatory metastic carcinoma. carcinoma erysipelatoides. Arch Dermatol. 1977;113:69-70.
- Cowan LJ, Roller JI, Connelly PJ, et al. Extraovarian stage IV peritoneal serous papillary carcinoma presenting as an asymptomatic skin lesion—a case report and literature review. Gynecol Oncol. 1995;57:433-435.
- Schonmann R, Altaras M, Biron T, et al. Inflammatory skin metastases from ovarian carcinoma—a case report and review of the literature. Gynecol Oncol. 2003;90:670-672.
- Klein RL, Brown AR, Gomez-Castro CM, et al. Ovarian cancer metastatic to the breast presenting as inflammatory breast cancer: a case report and literature review. J Cancer. 2010;1:27-31.
- Lee HC, Chu CY, Hsiao CH. Carcinoma erysipeloides from ovarian clear-cell carcinoma. J Clin Oncol. 2007;25:5828-5830.
- Godinez-Puig V, Frangos J, Hollmann TJ, et al. Photo quiz. rash in a patient with ovarian cancer. Clin Infect Dis. 2012;54:538, 575-576.
Practice Points
- Carcinoma erysipeloides is a rare cutaneous marker of metastatic ovarian cancer.
- Clinicians should be aware of carcinoma erysipeloides in ovarian cancer and maintain a low threshold for biopsy for accurate diagnosis and management planning.
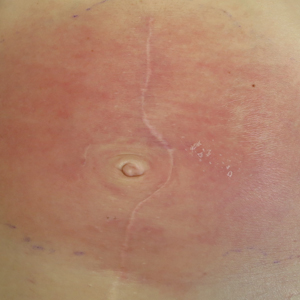
Metastatic Melanoma and Prostatic Adenocarcinoma in the Same Sentinel Lymph Node
To the Editor:
Sentinel lymph node (SLN) biopsies routinely are performed to detect regional metastases in a variety of malignancies, including breast cancer, squamous cell carcinoma, Merkel cell carcinoma, and melanoma. Histologic examination of an SLN occasionally enables detection of other unsuspected underlying diseases that typically are inflammatory in nature. Although concomitant hematolymphoid malignancy, particularly chronic lymphocytic leukemia, has been reported in SLNs, collision of 2 different solid tumors in the same SLN is rare.1,2 We report a unique case documenting collision of both metastatic melanoma and prostatic adenocarcinoma detected in an SLN to raise awareness of the diagnostic challenges occurring in patients with coexisting malignancies.
A 71-year-old man with a history of metastatic prostatic adenocarcinoma to the bone presented for treatment of a melanoma that was newly diagnosed by an outside dermatologist. The patient’s medical history was notable for radical prostatectomy performed 15 years prior for treatment of a prostatic adenocarcinoma (Gleason score unknown) followed by bilateral orchiectomy performed 7 years later after his serum prostate-specific antigen (PSA) level began to rise, with no response to goserelin (a gonadotropin-releasing hormone agonist) therapy. Two years prior to the diagnosis of metastatic disease, his PSA level started to rise again and the patient received bicalutamide with little improvement, followed by 8 cycles of docetaxel. His PSA level improved and he most recently was being treated with abiraterone acetate. The patient’s latest computed tomography scan showed that the bony metastases secondary to prostatic adenocarcinoma had progressed. His serum PSA level was 105 ng/mL (reference range, <4.0 ng/mL) at the current presentation, elevated from 64 ng/mL one year prior.
Recently, the patient had noted a changing pigmented skin lesion on the left side of the flank. The patient described the lesion as a “black mole” first appearing 2 years prior, which had begun to ooze, change shape, and become darker and more nodular. A shave biopsy revealed a primary cutaneous malignant melanoma at least 3.4 mm in depth with ulceration and a mitotic rate of 15/mm2. No molecular studies were performed on the melanoma. Standard treatment via wide local excision and sentinel lymphadenectomy was planned.
Lymphoscintigraphy revealed 3 left draining axillary lymph nodes. The patient was treated with wide local excision and left axillary SLN biopsy. Five SLNs and 3 non-SLNs were excised. Per protocol, all SLNs were examined pathologically with serial sections: 2 hematoxylin and eosin–stained levels, S-100, and melan-A immunohistochemical stains. No residual melanoma was identified in the wide-excision specimen. Examination of the left axillary SLNs revealed metastatic melanoma in 3 of 5 SLNs. Two SLNs demonstrated total replacement by metastatic melanoma. A third SLN revealed a metastatic malignant neoplasm occupying 75% of the nodal area (Figure, A). S-100 and melan-A immunohistochemical staining were negative in this nodule but revealed small aggregates and isolated tumor cells distinct from this nodule that were diagnostic of micrometastatic melanoma (Figures, B and C). The tumor cells in the large nodule were histologically distinct from the melanoma and were instead composed of nests of epithelioid cells with clear cytoplasm (Figure, D). Upon further immunohistochemical staining, this tumor was strongly positive for AE1/AE3 keratin and PIN4 cocktail (cytokeratin 5, cytokeratin 15, p63, and p504s/alpha-methylacyl-CoA-racemase)(Figure, E) with focal positivity for PSA and prostatic acid phosphatase, diagnostic of metastatic adenocarcinoma of prostate origin.
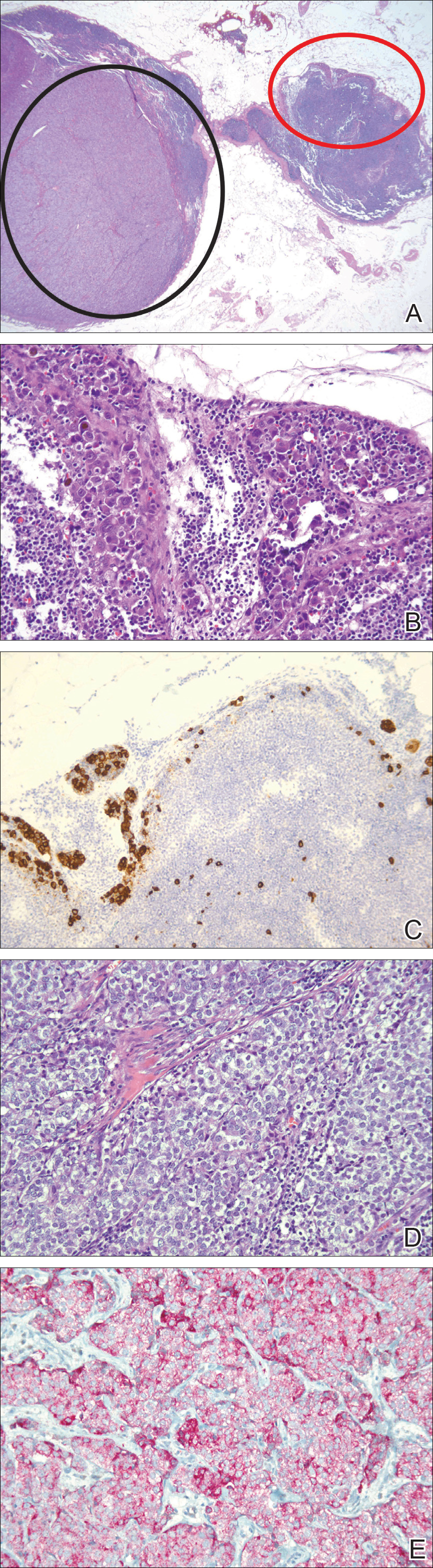
A positron emission tomography scan performed a few days after the discovery of metastatic prostatic adenocarcinoma in the SLNs showed expected postoperative changes (eg, increased activity from procedure-related inflammation) in the left side of the flank and axilla as well as moderately hypermetabolic left supraclavicular lymph nodes suspicious for viable metastatic disease. Subsequent fine-needle aspiration of the aforementioned lymph nodes revealed metastatic prostatic adenocarcinoma. The preoperative lymphoscintigraphy at the time of SLN biopsy did not show drainage to the left supraclavicular nodal basin.
Based on a discussion of the patient’s case during a multidisciplinary tumor board consultation, the benefit of performing completion lymph node dissection for melanoma management did not outweigh the risks. Accordingly, the patient received adjuvant radiation therapy to the axillary nodal basin. He was started on ketoconazole and zoledronic acid therapy for metastatic prostate adenocarcinoma and was alive with disease at 6-month follow-up. The finding of both metastatic melanoma and prostate adenocarcinoma detected in an SLN after wide excision and SLN biopsy for cutaneous melanoma is a unique report of collision of these 2 tumors. Rare cases of collision between 2 solid tumors occurring in the same lymph node have involved prostate adenocarcinoma as one of the solid tumor components.1,3 Detection of tumor collision on lymph node biopsy between prostatic adenocarcinoma and urothelial carcinoma has been documented in 2 separate cases.1 Three additional cases of concurrent prostatic adenocarcinoma and colorectal adenocarcinoma identified on lymph node biopsy have been reported.1,3 Although never proven statistically, it is likely that these concurrent diagnoses are due to the high incidences of prostate and colorectal adenocarcinomas in the general US population; they are ranked first and third, respectively, for cancer incidence in US males.4
As demonstrated in the current case and the available literature, immunohistochemical stains play a vital role in the detection of tumor collision phenomena as well as identification of histologic source of the metastases. Furthermore, thorough histopathologic examination of biopsy specimens in the context of a patient’s clinical history remains paramount in obtaining an accurate diagnosis. Earlier identification of second malignancies in SLNs can alert the clinician to the presence of relapse of a known concurrent malignancy before it is clinically apparent, enhancing the possibility of more effective treatment of earlier disease. As has been demonstrated for lymphoma and melanoma, in rare cases awareness of the possibility of a second malignancy in the SLN can result in earlier initial diagnosis of undiscovered malignancy.2
- Sughayer MA, Zakarneh L, Abu-Shakra R. Collision metastasis of breast and ovarian adenocarcinoma in axillary lymph nodes: a case report and review of the literature. Pathol Oncol Res. 2009;15:423-427.
- Farma JM, Zager JS, Barnica-Elvir V, et al. A collision of diseases: chronic lymphocytic leukemia discovered during lymph node biopsy for melanoma. Ann Surg Oncol. 2013;20:1360-1364.
- Wade ZK, Shippey JE, Hamon GA, et al. Collision metastasis of prostatic and colonic adenocarcinoma: report of 2 cases. Arch Pathol Lab Med. 2004;128:318-320.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30.
To the Editor:
Sentinel lymph node (SLN) biopsies routinely are performed to detect regional metastases in a variety of malignancies, including breast cancer, squamous cell carcinoma, Merkel cell carcinoma, and melanoma. Histologic examination of an SLN occasionally enables detection of other unsuspected underlying diseases that typically are inflammatory in nature. Although concomitant hematolymphoid malignancy, particularly chronic lymphocytic leukemia, has been reported in SLNs, collision of 2 different solid tumors in the same SLN is rare.1,2 We report a unique case documenting collision of both metastatic melanoma and prostatic adenocarcinoma detected in an SLN to raise awareness of the diagnostic challenges occurring in patients with coexisting malignancies.
A 71-year-old man with a history of metastatic prostatic adenocarcinoma to the bone presented for treatment of a melanoma that was newly diagnosed by an outside dermatologist. The patient’s medical history was notable for radical prostatectomy performed 15 years prior for treatment of a prostatic adenocarcinoma (Gleason score unknown) followed by bilateral orchiectomy performed 7 years later after his serum prostate-specific antigen (PSA) level began to rise, with no response to goserelin (a gonadotropin-releasing hormone agonist) therapy. Two years prior to the diagnosis of metastatic disease, his PSA level started to rise again and the patient received bicalutamide with little improvement, followed by 8 cycles of docetaxel. His PSA level improved and he most recently was being treated with abiraterone acetate. The patient’s latest computed tomography scan showed that the bony metastases secondary to prostatic adenocarcinoma had progressed. His serum PSA level was 105 ng/mL (reference range, <4.0 ng/mL) at the current presentation, elevated from 64 ng/mL one year prior.
Recently, the patient had noted a changing pigmented skin lesion on the left side of the flank. The patient described the lesion as a “black mole” first appearing 2 years prior, which had begun to ooze, change shape, and become darker and more nodular. A shave biopsy revealed a primary cutaneous malignant melanoma at least 3.4 mm in depth with ulceration and a mitotic rate of 15/mm2. No molecular studies were performed on the melanoma. Standard treatment via wide local excision and sentinel lymphadenectomy was planned.
Lymphoscintigraphy revealed 3 left draining axillary lymph nodes. The patient was treated with wide local excision and left axillary SLN biopsy. Five SLNs and 3 non-SLNs were excised. Per protocol, all SLNs were examined pathologically with serial sections: 2 hematoxylin and eosin–stained levels, S-100, and melan-A immunohistochemical stains. No residual melanoma was identified in the wide-excision specimen. Examination of the left axillary SLNs revealed metastatic melanoma in 3 of 5 SLNs. Two SLNs demonstrated total replacement by metastatic melanoma. A third SLN revealed a metastatic malignant neoplasm occupying 75% of the nodal area (Figure, A). S-100 and melan-A immunohistochemical staining were negative in this nodule but revealed small aggregates and isolated tumor cells distinct from this nodule that were diagnostic of micrometastatic melanoma (Figures, B and C). The tumor cells in the large nodule were histologically distinct from the melanoma and were instead composed of nests of epithelioid cells with clear cytoplasm (Figure, D). Upon further immunohistochemical staining, this tumor was strongly positive for AE1/AE3 keratin and PIN4 cocktail (cytokeratin 5, cytokeratin 15, p63, and p504s/alpha-methylacyl-CoA-racemase)(Figure, E) with focal positivity for PSA and prostatic acid phosphatase, diagnostic of metastatic adenocarcinoma of prostate origin.
A positron emission tomography scan performed a few days after the discovery of metastatic prostatic adenocarcinoma in the SLNs showed expected postoperative changes (eg, increased activity from procedure-related inflammation) in the left side of the flank and axilla as well as moderately hypermetabolic left supraclavicular lymph nodes suspicious for viable metastatic disease. Subsequent fine-needle aspiration of the aforementioned lymph nodes revealed metastatic prostatic adenocarcinoma. The preoperative lymphoscintigraphy at the time of SLN biopsy did not show drainage to the left supraclavicular nodal basin.
Based on a discussion of the patient’s case during a multidisciplinary tumor board consultation, the benefit of performing completion lymph node dissection for melanoma management did not outweigh the risks. Accordingly, the patient received adjuvant radiation therapy to the axillary nodal basin. He was started on ketoconazole and zoledronic acid therapy for metastatic prostate adenocarcinoma and was alive with disease at 6-month follow-up. The finding of both metastatic melanoma and prostate adenocarcinoma detected in an SLN after wide excision and SLN biopsy for cutaneous melanoma is a unique report of collision of these 2 tumors. Rare cases of collision between 2 solid tumors occurring in the same lymph node have involved prostate adenocarcinoma as one of the solid tumor components.1,3 Detection of tumor collision on lymph node biopsy between prostatic adenocarcinoma and urothelial carcinoma has been documented in 2 separate cases.1 Three additional cases of concurrent prostatic adenocarcinoma and colorectal adenocarcinoma identified on lymph node biopsy have been reported.1,3 Although never proven statistically, it is likely that these concurrent diagnoses are due to the high incidences of prostate and colorectal adenocarcinomas in the general US population; they are ranked first and third, respectively, for cancer incidence in US males.4
As demonstrated in the current case and the available literature, immunohistochemical stains play a vital role in the detection of tumor collision phenomena as well as identification of histologic source of the metastases. Furthermore, thorough histopathologic examination of biopsy specimens in the context of a patient’s clinical history remains paramount in obtaining an accurate diagnosis. Earlier identification of second malignancies in SLNs can alert the clinician to the presence of relapse of a known concurrent malignancy before it is clinically apparent, enhancing the possibility of more effective treatment of earlier disease. As has been demonstrated for lymphoma and melanoma, in rare cases awareness of the possibility of a second malignancy in the SLN can result in earlier initial diagnosis of undiscovered malignancy.2
To the Editor:
Sentinel lymph node (SLN) biopsies routinely are performed to detect regional metastases in a variety of malignancies, including breast cancer, squamous cell carcinoma, Merkel cell carcinoma, and melanoma. Histologic examination of an SLN occasionally enables detection of other unsuspected underlying diseases that typically are inflammatory in nature. Although concomitant hematolymphoid malignancy, particularly chronic lymphocytic leukemia, has been reported in SLNs, collision of 2 different solid tumors in the same SLN is rare.1,2 We report a unique case documenting collision of both metastatic melanoma and prostatic adenocarcinoma detected in an SLN to raise awareness of the diagnostic challenges occurring in patients with coexisting malignancies.
A 71-year-old man with a history of metastatic prostatic adenocarcinoma to the bone presented for treatment of a melanoma that was newly diagnosed by an outside dermatologist. The patient’s medical history was notable for radical prostatectomy performed 15 years prior for treatment of a prostatic adenocarcinoma (Gleason score unknown) followed by bilateral orchiectomy performed 7 years later after his serum prostate-specific antigen (PSA) level began to rise, with no response to goserelin (a gonadotropin-releasing hormone agonist) therapy. Two years prior to the diagnosis of metastatic disease, his PSA level started to rise again and the patient received bicalutamide with little improvement, followed by 8 cycles of docetaxel. His PSA level improved and he most recently was being treated with abiraterone acetate. The patient’s latest computed tomography scan showed that the bony metastases secondary to prostatic adenocarcinoma had progressed. His serum PSA level was 105 ng/mL (reference range, <4.0 ng/mL) at the current presentation, elevated from 64 ng/mL one year prior.
Recently, the patient had noted a changing pigmented skin lesion on the left side of the flank. The patient described the lesion as a “black mole” first appearing 2 years prior, which had begun to ooze, change shape, and become darker and more nodular. A shave biopsy revealed a primary cutaneous malignant melanoma at least 3.4 mm in depth with ulceration and a mitotic rate of 15/mm2. No molecular studies were performed on the melanoma. Standard treatment via wide local excision and sentinel lymphadenectomy was planned.
Lymphoscintigraphy revealed 3 left draining axillary lymph nodes. The patient was treated with wide local excision and left axillary SLN biopsy. Five SLNs and 3 non-SLNs were excised. Per protocol, all SLNs were examined pathologically with serial sections: 2 hematoxylin and eosin–stained levels, S-100, and melan-A immunohistochemical stains. No residual melanoma was identified in the wide-excision specimen. Examination of the left axillary SLNs revealed metastatic melanoma in 3 of 5 SLNs. Two SLNs demonstrated total replacement by metastatic melanoma. A third SLN revealed a metastatic malignant neoplasm occupying 75% of the nodal area (Figure, A). S-100 and melan-A immunohistochemical staining were negative in this nodule but revealed small aggregates and isolated tumor cells distinct from this nodule that were diagnostic of micrometastatic melanoma (Figures, B and C). The tumor cells in the large nodule were histologically distinct from the melanoma and were instead composed of nests of epithelioid cells with clear cytoplasm (Figure, D). Upon further immunohistochemical staining, this tumor was strongly positive for AE1/AE3 keratin and PIN4 cocktail (cytokeratin 5, cytokeratin 15, p63, and p504s/alpha-methylacyl-CoA-racemase)(Figure, E) with focal positivity for PSA and prostatic acid phosphatase, diagnostic of metastatic adenocarcinoma of prostate origin.
A positron emission tomography scan performed a few days after the discovery of metastatic prostatic adenocarcinoma in the SLNs showed expected postoperative changes (eg, increased activity from procedure-related inflammation) in the left side of the flank and axilla as well as moderately hypermetabolic left supraclavicular lymph nodes suspicious for viable metastatic disease. Subsequent fine-needle aspiration of the aforementioned lymph nodes revealed metastatic prostatic adenocarcinoma. The preoperative lymphoscintigraphy at the time of SLN biopsy did not show drainage to the left supraclavicular nodal basin.
Based on a discussion of the patient’s case during a multidisciplinary tumor board consultation, the benefit of performing completion lymph node dissection for melanoma management did not outweigh the risks. Accordingly, the patient received adjuvant radiation therapy to the axillary nodal basin. He was started on ketoconazole and zoledronic acid therapy for metastatic prostate adenocarcinoma and was alive with disease at 6-month follow-up. The finding of both metastatic melanoma and prostate adenocarcinoma detected in an SLN after wide excision and SLN biopsy for cutaneous melanoma is a unique report of collision of these 2 tumors. Rare cases of collision between 2 solid tumors occurring in the same lymph node have involved prostate adenocarcinoma as one of the solid tumor components.1,3 Detection of tumor collision on lymph node biopsy between prostatic adenocarcinoma and urothelial carcinoma has been documented in 2 separate cases.1 Three additional cases of concurrent prostatic adenocarcinoma and colorectal adenocarcinoma identified on lymph node biopsy have been reported.1,3 Although never proven statistically, it is likely that these concurrent diagnoses are due to the high incidences of prostate and colorectal adenocarcinomas in the general US population; they are ranked first and third, respectively, for cancer incidence in US males.4
As demonstrated in the current case and the available literature, immunohistochemical stains play a vital role in the detection of tumor collision phenomena as well as identification of histologic source of the metastases. Furthermore, thorough histopathologic examination of biopsy specimens in the context of a patient’s clinical history remains paramount in obtaining an accurate diagnosis. Earlier identification of second malignancies in SLNs can alert the clinician to the presence of relapse of a known concurrent malignancy before it is clinically apparent, enhancing the possibility of more effective treatment of earlier disease. As has been demonstrated for lymphoma and melanoma, in rare cases awareness of the possibility of a second malignancy in the SLN can result in earlier initial diagnosis of undiscovered malignancy.2
- Sughayer MA, Zakarneh L, Abu-Shakra R. Collision metastasis of breast and ovarian adenocarcinoma in axillary lymph nodes: a case report and review of the literature. Pathol Oncol Res. 2009;15:423-427.
- Farma JM, Zager JS, Barnica-Elvir V, et al. A collision of diseases: chronic lymphocytic leukemia discovered during lymph node biopsy for melanoma. Ann Surg Oncol. 2013;20:1360-1364.
- Wade ZK, Shippey JE, Hamon GA, et al. Collision metastasis of prostatic and colonic adenocarcinoma: report of 2 cases. Arch Pathol Lab Med. 2004;128:318-320.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30.
- Sughayer MA, Zakarneh L, Abu-Shakra R. Collision metastasis of breast and ovarian adenocarcinoma in axillary lymph nodes: a case report and review of the literature. Pathol Oncol Res. 2009;15:423-427.
- Farma JM, Zager JS, Barnica-Elvir V, et al. A collision of diseases: chronic lymphocytic leukemia discovered during lymph node biopsy for melanoma. Ann Surg Oncol. 2013;20:1360-1364.
- Wade ZK, Shippey JE, Hamon GA, et al. Collision metastasis of prostatic and colonic adenocarcinoma: report of 2 cases. Arch Pathol Lab Med. 2004;128:318-320.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30.
Practice Points
- Immunohistochemical stains play a vital role in the detection of tumor collision phenomena as well as identification of histologic sources of metastases.
- Thorough histopathologic examination of biopsy specimens in the context of a patient’s clinical history remains paramount in obtaining an accurate diagnosis, enhancing the possibility of more effective treatment of earlier disease.
Periorbital Lupuslike Presentation of Graft-versus-host Disease
To the Editor:
A 79-year-old man presented with a scaling eruption in the periorbital area, on the bilateral forearms, and on the chest of 4 weeks’ duration. The patient denied systemic symptoms including lethargy, muscle weakness, and fevers. His medical history was notable for blastic plasmacytoid dendritic cell neoplasm, a form of acute myeloid leukemia, diagnosed 3 years prior to presentation. The patient received an allogeneic hematopoietic stem cell transplant 8 months later. His posttransplant course was complicated by gastrointestinal graft-versus-host disease (GVHD); progressive graft loss requiring a donor lymphocyte infusion after 1 month; and leukemia cutis, which spontaneously resolved after 1 month. The patient was taken off all immunosuppressive therapy 5 months after the transplant and had been doing well for 2 years with only mild mucosal GVHD affecting the oral mucosa and the head of the penis.
Physical examination at the current presentation revealed linear, atrophic, scaling, purplish plaques with adherent white scale on the upper and lower eyelids (Figure 1). The patient also had scattered purple scaling patches on the bilateral forearms and chest. Laboratory tests including complete blood cell count, comprehensive metabolic panel, and lactate dehydrogenase demonstrated no gross abnormalities. Two shave biopsies of the right lower eyelid (Figure 2) and left arm (Figure 3) were performed for histologic examination and revealed basket weave hyperkeratosis, irregular acanthosis, sawtooth rete ridges, and scattered dyskeratotic cells. Vacuolar changes and smudging of the basement membrane zone along with a bandlike lymphocytic infiltrate in the upper dermis also were noted in both biopsies. A diagnosis of lupuslike grade 1 GVHD was made.
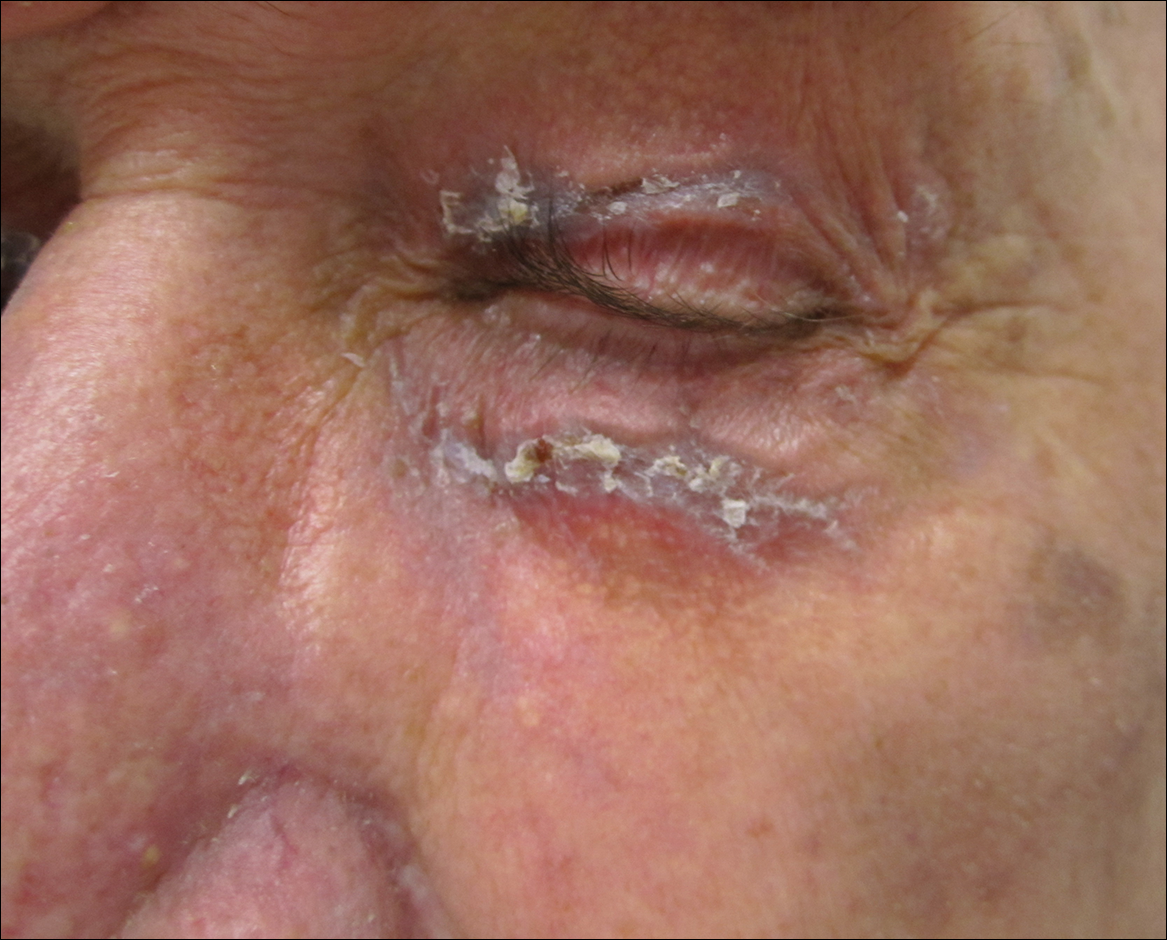
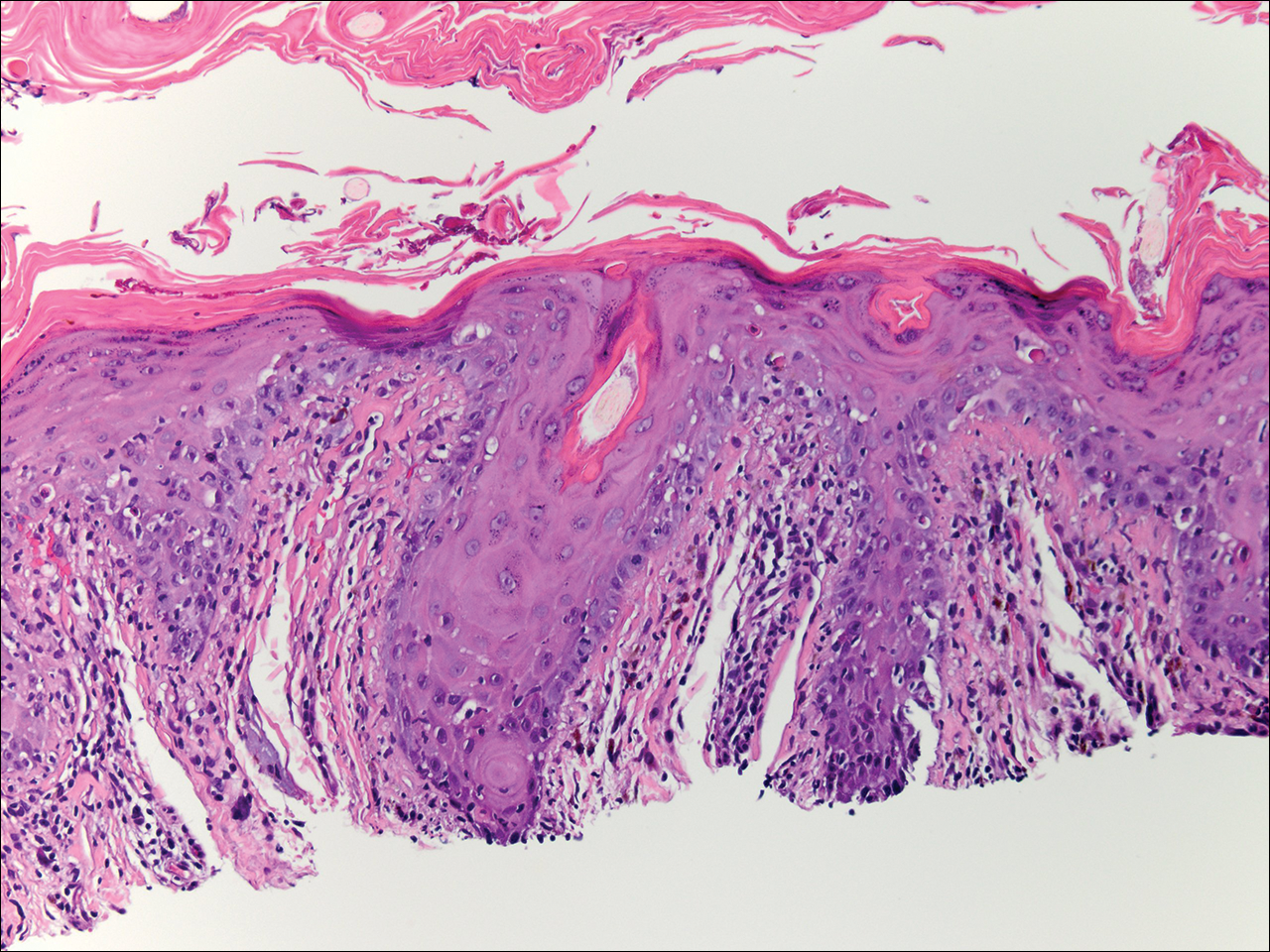
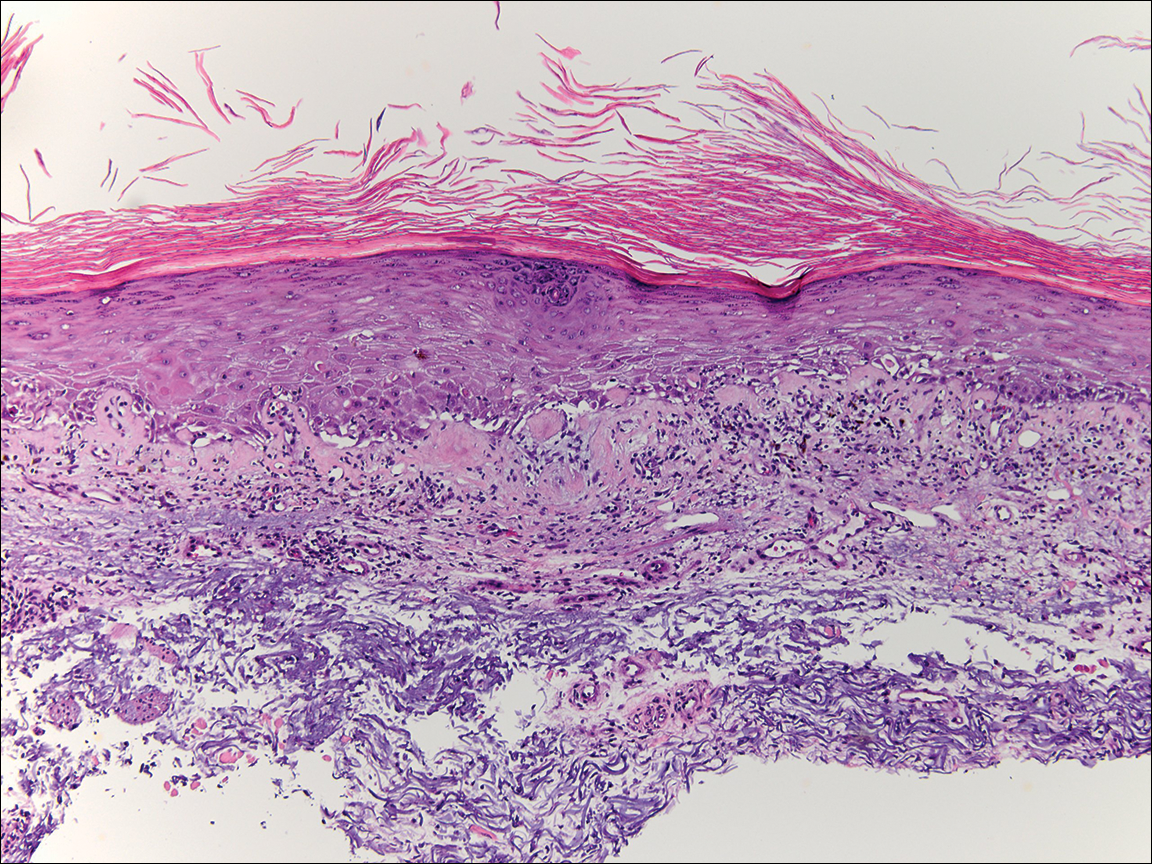
Graft-versus-host disease remains a notable cause of morbidity and mortality in allogenic hematopoietic stem cell transplant patients.1 Skin manifestations represent the most common manifestation of GVHD and have been reclassified as acute or chronic disease based on clinical and histologic findings rather than time of onset. Although acute GVHD classically presents as diffuse morbilliform papules and macules, chronic GVHD has a large range of clinical presentations most commonly mimicking the skin findings of lichen planus, morphea, scleroderma, or lichen sclerosus.1
Lupuslike GVHD is a rarely reported manifestation of chronic GVHD that predominantly affects the lower eyelids and malar regions.2,3 Our case documents extensive involvement of both the upper and lower eyelids. A lupuslike manifestation of GVHD may portend a poor prognosis. In a case series of 5 patients with chronic GVHD presenting as facial lupuslike plaques, 1 patient died from a relapse of leukemia and 3 patients developed sclerodermatous GVHD. The fifth patient was lost to follow-up.2 In another case series, a retrospective analysis discovered that 3 of 7 patients with sclerodermatous GVHD initially presented with hyperpigmented periorbital plaques.4 Resolution of skin findings with topical steroids and oral tacrolimus was reported in a case of GVHD presenting with periorbital lupuslike plaques.3 Although further reports are needed to validate the relationship, a lupuslike presentation of chronic GVHD may be an important harbinger for the development of extensive sclerodermatous GVHD.
A diagnosis of lupuslike GVHD is made based on the correlation of a comprehensive medical history, clinical examination, and histopathologic findings. Although other cases of chronic GVHD resembling dermatomyositis presented with purple periorbital plaques, these patients demonstrated dermatomyositislike systemic symptoms including muscle weakness and fatigue, which were not present in our patient.5,6 Antinuclear antibody (ANA) testing is unlikely to be helpful in the diagnosis of this uncommon presentation, as 65% (41/63) of chronic GVHD patients developed ANA antibodies in one study.7 Also, other patients with lupuslike GVHD who progressed to sclerodermatous GVHD have had both positive and negative ANA serology.2 The histopathology of GVHD and lupus erythematosus can exhibit overlapping features, such as lymphocytic infiltrate with interface changes; however, in lupus erythematosus, mucin usually is present, the infiltrate usually is denser and deeper, and a thickened basement membrane zone may be present. Necrotic keratinocytes also usually are not seen in lupus erythematosus unless the patient’s photosensitivity has led to a sunburn reaction.
After his initial presentation, our patient’s mucosal GVHD flared in the mouth and on the penis, and he was started on prednisone 50 mg once daily and mycophenolate mofetil 1 g twice daily. With this treatment, our patient’s periorbital scaling plaques resolved to residual hyperpigmentation along with remarkable improvement of the mucosal GVHD. He has not manifested any signs of leukemia relapse or sclerodermatous GVHD; however, he remains under close clinical evaluation.
This case highlights an unusual presentation of GVHD with periorbital plaques mimicking hypertrophic lupus erythematous. A greater recognition of this rare entity is essential to further elucidate its prognosis and its relationship with sclerodermatous GVHD.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-5.15e18; quiz 533-534.
- Goiriz R, Peñas PF, Delgado-Jiménez Y, et al. Cutaneous lichenoid graft-versus-host disease mimicking lupus erythematosus. Lupus. 2008;17:591-595.
- Hu SW, Myskowski PL, Papadopoulos EB, et al. Chronic cutaneous graft-versus host disease simulating hypertrophic lupus erythematosus—a case report of a new morphologic variant of graft-versus-host disease. Am J Dermatopathol. 2012;34:E81-E83.
- Chosidow O, Bagot M, Vernant JP, et al. Sclerodermatous chronic graft-versus-host disease. J Am Acad Dermatol. 1992;26:49-55.
- Ollivier I, Wolkenstein P, Gherardi R, et al. Dermatomyositis-like graft-versus-host disease. Br J Dermatol. 1998;138:558-559.
- Arin MJ, Scheid C, Hübel K, et al. Chronic graft-versus-host disease with skin signs suggestive of dermatomyositis. Clin Exp Dermatol. 2006;31:141-143.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
To the Editor:
A 79-year-old man presented with a scaling eruption in the periorbital area, on the bilateral forearms, and on the chest of 4 weeks’ duration. The patient denied systemic symptoms including lethargy, muscle weakness, and fevers. His medical history was notable for blastic plasmacytoid dendritic cell neoplasm, a form of acute myeloid leukemia, diagnosed 3 years prior to presentation. The patient received an allogeneic hematopoietic stem cell transplant 8 months later. His posttransplant course was complicated by gastrointestinal graft-versus-host disease (GVHD); progressive graft loss requiring a donor lymphocyte infusion after 1 month; and leukemia cutis, which spontaneously resolved after 1 month. The patient was taken off all immunosuppressive therapy 5 months after the transplant and had been doing well for 2 years with only mild mucosal GVHD affecting the oral mucosa and the head of the penis.
Physical examination at the current presentation revealed linear, atrophic, scaling, purplish plaques with adherent white scale on the upper and lower eyelids (Figure 1). The patient also had scattered purple scaling patches on the bilateral forearms and chest. Laboratory tests including complete blood cell count, comprehensive metabolic panel, and lactate dehydrogenase demonstrated no gross abnormalities. Two shave biopsies of the right lower eyelid (Figure 2) and left arm (Figure 3) were performed for histologic examination and revealed basket weave hyperkeratosis, irregular acanthosis, sawtooth rete ridges, and scattered dyskeratotic cells. Vacuolar changes and smudging of the basement membrane zone along with a bandlike lymphocytic infiltrate in the upper dermis also were noted in both biopsies. A diagnosis of lupuslike grade 1 GVHD was made.
Graft-versus-host disease remains a notable cause of morbidity and mortality in allogenic hematopoietic stem cell transplant patients.1 Skin manifestations represent the most common manifestation of GVHD and have been reclassified as acute or chronic disease based on clinical and histologic findings rather than time of onset. Although acute GVHD classically presents as diffuse morbilliform papules and macules, chronic GVHD has a large range of clinical presentations most commonly mimicking the skin findings of lichen planus, morphea, scleroderma, or lichen sclerosus.1
Lupuslike GVHD is a rarely reported manifestation of chronic GVHD that predominantly affects the lower eyelids and malar regions.2,3 Our case documents extensive involvement of both the upper and lower eyelids. A lupuslike manifestation of GVHD may portend a poor prognosis. In a case series of 5 patients with chronic GVHD presenting as facial lupuslike plaques, 1 patient died from a relapse of leukemia and 3 patients developed sclerodermatous GVHD. The fifth patient was lost to follow-up.2 In another case series, a retrospective analysis discovered that 3 of 7 patients with sclerodermatous GVHD initially presented with hyperpigmented periorbital plaques.4 Resolution of skin findings with topical steroids and oral tacrolimus was reported in a case of GVHD presenting with periorbital lupuslike plaques.3 Although further reports are needed to validate the relationship, a lupuslike presentation of chronic GVHD may be an important harbinger for the development of extensive sclerodermatous GVHD.
A diagnosis of lupuslike GVHD is made based on the correlation of a comprehensive medical history, clinical examination, and histopathologic findings. Although other cases of chronic GVHD resembling dermatomyositis presented with purple periorbital plaques, these patients demonstrated dermatomyositislike systemic symptoms including muscle weakness and fatigue, which were not present in our patient.5,6 Antinuclear antibody (ANA) testing is unlikely to be helpful in the diagnosis of this uncommon presentation, as 65% (41/63) of chronic GVHD patients developed ANA antibodies in one study.7 Also, other patients with lupuslike GVHD who progressed to sclerodermatous GVHD have had both positive and negative ANA serology.2 The histopathology of GVHD and lupus erythematosus can exhibit overlapping features, such as lymphocytic infiltrate with interface changes; however, in lupus erythematosus, mucin usually is present, the infiltrate usually is denser and deeper, and a thickened basement membrane zone may be present. Necrotic keratinocytes also usually are not seen in lupus erythematosus unless the patient’s photosensitivity has led to a sunburn reaction.
After his initial presentation, our patient’s mucosal GVHD flared in the mouth and on the penis, and he was started on prednisone 50 mg once daily and mycophenolate mofetil 1 g twice daily. With this treatment, our patient’s periorbital scaling plaques resolved to residual hyperpigmentation along with remarkable improvement of the mucosal GVHD. He has not manifested any signs of leukemia relapse or sclerodermatous GVHD; however, he remains under close clinical evaluation.
This case highlights an unusual presentation of GVHD with periorbital plaques mimicking hypertrophic lupus erythematous. A greater recognition of this rare entity is essential to further elucidate its prognosis and its relationship with sclerodermatous GVHD.
To the Editor:
A 79-year-old man presented with a scaling eruption in the periorbital area, on the bilateral forearms, and on the chest of 4 weeks’ duration. The patient denied systemic symptoms including lethargy, muscle weakness, and fevers. His medical history was notable for blastic plasmacytoid dendritic cell neoplasm, a form of acute myeloid leukemia, diagnosed 3 years prior to presentation. The patient received an allogeneic hematopoietic stem cell transplant 8 months later. His posttransplant course was complicated by gastrointestinal graft-versus-host disease (GVHD); progressive graft loss requiring a donor lymphocyte infusion after 1 month; and leukemia cutis, which spontaneously resolved after 1 month. The patient was taken off all immunosuppressive therapy 5 months after the transplant and had been doing well for 2 years with only mild mucosal GVHD affecting the oral mucosa and the head of the penis.
Physical examination at the current presentation revealed linear, atrophic, scaling, purplish plaques with adherent white scale on the upper and lower eyelids (Figure 1). The patient also had scattered purple scaling patches on the bilateral forearms and chest. Laboratory tests including complete blood cell count, comprehensive metabolic panel, and lactate dehydrogenase demonstrated no gross abnormalities. Two shave biopsies of the right lower eyelid (Figure 2) and left arm (Figure 3) were performed for histologic examination and revealed basket weave hyperkeratosis, irregular acanthosis, sawtooth rete ridges, and scattered dyskeratotic cells. Vacuolar changes and smudging of the basement membrane zone along with a bandlike lymphocytic infiltrate in the upper dermis also were noted in both biopsies. A diagnosis of lupuslike grade 1 GVHD was made.
Graft-versus-host disease remains a notable cause of morbidity and mortality in allogenic hematopoietic stem cell transplant patients.1 Skin manifestations represent the most common manifestation of GVHD and have been reclassified as acute or chronic disease based on clinical and histologic findings rather than time of onset. Although acute GVHD classically presents as diffuse morbilliform papules and macules, chronic GVHD has a large range of clinical presentations most commonly mimicking the skin findings of lichen planus, morphea, scleroderma, or lichen sclerosus.1
Lupuslike GVHD is a rarely reported manifestation of chronic GVHD that predominantly affects the lower eyelids and malar regions.2,3 Our case documents extensive involvement of both the upper and lower eyelids. A lupuslike manifestation of GVHD may portend a poor prognosis. In a case series of 5 patients with chronic GVHD presenting as facial lupuslike plaques, 1 patient died from a relapse of leukemia and 3 patients developed sclerodermatous GVHD. The fifth patient was lost to follow-up.2 In another case series, a retrospective analysis discovered that 3 of 7 patients with sclerodermatous GVHD initially presented with hyperpigmented periorbital plaques.4 Resolution of skin findings with topical steroids and oral tacrolimus was reported in a case of GVHD presenting with periorbital lupuslike plaques.3 Although further reports are needed to validate the relationship, a lupuslike presentation of chronic GVHD may be an important harbinger for the development of extensive sclerodermatous GVHD.
A diagnosis of lupuslike GVHD is made based on the correlation of a comprehensive medical history, clinical examination, and histopathologic findings. Although other cases of chronic GVHD resembling dermatomyositis presented with purple periorbital plaques, these patients demonstrated dermatomyositislike systemic symptoms including muscle weakness and fatigue, which were not present in our patient.5,6 Antinuclear antibody (ANA) testing is unlikely to be helpful in the diagnosis of this uncommon presentation, as 65% (41/63) of chronic GVHD patients developed ANA antibodies in one study.7 Also, other patients with lupuslike GVHD who progressed to sclerodermatous GVHD have had both positive and negative ANA serology.2 The histopathology of GVHD and lupus erythematosus can exhibit overlapping features, such as lymphocytic infiltrate with interface changes; however, in lupus erythematosus, mucin usually is present, the infiltrate usually is denser and deeper, and a thickened basement membrane zone may be present. Necrotic keratinocytes also usually are not seen in lupus erythematosus unless the patient’s photosensitivity has led to a sunburn reaction.
After his initial presentation, our patient’s mucosal GVHD flared in the mouth and on the penis, and he was started on prednisone 50 mg once daily and mycophenolate mofetil 1 g twice daily. With this treatment, our patient’s periorbital scaling plaques resolved to residual hyperpigmentation along with remarkable improvement of the mucosal GVHD. He has not manifested any signs of leukemia relapse or sclerodermatous GVHD; however, he remains under close clinical evaluation.
This case highlights an unusual presentation of GVHD with periorbital plaques mimicking hypertrophic lupus erythematous. A greater recognition of this rare entity is essential to further elucidate its prognosis and its relationship with sclerodermatous GVHD.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-5.15e18; quiz 533-534.
- Goiriz R, Peñas PF, Delgado-Jiménez Y, et al. Cutaneous lichenoid graft-versus-host disease mimicking lupus erythematosus. Lupus. 2008;17:591-595.
- Hu SW, Myskowski PL, Papadopoulos EB, et al. Chronic cutaneous graft-versus host disease simulating hypertrophic lupus erythematosus—a case report of a new morphologic variant of graft-versus-host disease. Am J Dermatopathol. 2012;34:E81-E83.
- Chosidow O, Bagot M, Vernant JP, et al. Sclerodermatous chronic graft-versus-host disease. J Am Acad Dermatol. 1992;26:49-55.
- Ollivier I, Wolkenstein P, Gherardi R, et al. Dermatomyositis-like graft-versus-host disease. Br J Dermatol. 1998;138:558-559.
- Arin MJ, Scheid C, Hübel K, et al. Chronic graft-versus-host disease with skin signs suggestive of dermatomyositis. Clin Exp Dermatol. 2006;31:141-143.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.
- Hymes SR, Alousi AM, Cowen EW. Graft-versus-host disease: part I. pathogenesis and clinical manifestations of graft-versus-host disease. J Am Acad Dermatol. 2012;66:515.e1-5.15e18; quiz 533-534.
- Goiriz R, Peñas PF, Delgado-Jiménez Y, et al. Cutaneous lichenoid graft-versus-host disease mimicking lupus erythematosus. Lupus. 2008;17:591-595.
- Hu SW, Myskowski PL, Papadopoulos EB, et al. Chronic cutaneous graft-versus host disease simulating hypertrophic lupus erythematosus—a case report of a new morphologic variant of graft-versus-host disease. Am J Dermatopathol. 2012;34:E81-E83.
- Chosidow O, Bagot M, Vernant JP, et al. Sclerodermatous chronic graft-versus-host disease. J Am Acad Dermatol. 1992;26:49-55.
- Ollivier I, Wolkenstein P, Gherardi R, et al. Dermatomyositis-like graft-versus-host disease. Br J Dermatol. 1998;138:558-559.
- Arin MJ, Scheid C, Hübel K, et al. Chronic graft-versus-host disease with skin signs suggestive of dermatomyositis. Clin Exp Dermatol. 2006;31:141-143.
- Patriarca F, Skert C, Sperotto A, et al. The development of autoantibodies after allogeneic stem cell transplantation is related with chronic graft-vs-host disease and immune recovery. Exp Hematol. 2006;34:389-396.