User login
Coming to a Hospital Near You: E-prescribe

Coming to a Hospital Near You: E-prescribe
Will hospitalists and hospital medicine groups get to participate in the Centers for Medicare and Medicaid e-prescribe program?
Jettie Eddleman, BSN, RN, Quality Initiatives Program Director North Texas Specialty Physicians (NTSP), Fort Worth, Texas
Dr. Hospitalist responds: According to a December 2007 study by SureScripts, only 6% of U.S. physicians prescribe medications electronically. Medicare would like more physicians to electronically prescribe prescriptions because “e-prescribing is more efficient and convenient for consumers, improves the quality of care, lowers administrative costs, and its widespread use would eliminate thousands of medication errors every year.”
Medicare is not the only organization encouraging e-prescribing. Blue Cross Blue Shield of Massachusetts (BCBSMA) recently announced e-prescribing will be required for any physician to participate in any of the BCBSMA physician incentive programs, effective January, 2011. To speed the adoption of e-prescribing, Medicare will provide financial incentives to physicians who e-prescribe. Starting in this year, Medicare will pay a 2% bonus to physicians who prescribe under Part D. This incentive bonus will decrease to 1% in 2011 and 0.5% in 2013. Starting in 2012, physicians who are not e-prescribing will lose 1% of their Medicare payment. This penalty will increase to 1.5% in 2013 and 2% in 2014. In other words, you can e-prescribe sooner or later, but hospital medicine groups will increase revenue if they start sooner.
Do you have a problem or concern that you’d like Dr. Hospitalist to address? E-mail your questions to drhospit@wiley.com.
E-prescribing is, however, not without barriers. For example, the Drug Enforcement Agency (DEA) presently prohibits e-prescription of controlled substances. Medicare is working with the DEA to address this issue. In the meantime, providers who implement e-prescription will need to continue a separate system for prescription of controlled substances.
Some physicians have electronic medical record (EMR) systems, which send prescriptions to pharmacies via facsimile machines. For clarification, this is not considered e-prescribing. In fact, under Medicare statute, most EMR-faxed prescriptions no longer are allowed for Part D. As the plan currently stands, this incentive is tied to specific Current Procedural Terminology (CPT) codes, used primarily by primary care providers and not by hospitalists. Unless the hospitalists in your group also provide outpatient care, it is unlikely any will be able to participate in this Medicare e-prescribing incentive plan.
Test Results Post-Discharge
Some of my patients have laboratory test results pending at the time of discharge from the hospital. Most of my patients do not have a routine outpatient provider. At the time of discharge, I always set them up with outpatient follow-up with a new primary care provider, but I have no way of knowing if the patients are showing up at their appointments. My concern is if they don’t show up, they won’t know about their test results. Is this something I need to address? If so, how do you suggest I go about doing it?
Z. Taylor, Durant, Okla.
Dr. Hospitalist responds: This definitely is an issue that needs to be addressed. It is not only a quality of care issue, but also potentially a medical/legal issue. Transitions in care are risky for patients because these are periods with increased risk for medical error. For example, this is why the Joint Commission mandates medication reconciliation each time a patient sees a provider.
When patients are discharged from the hospital, which provider is responsible for notifying the patient of pending laboratory test results? Is it the primary care provider (PCP) or the hospitalist? As you described in your question, what if the patient does not have a regular PCP? Does the responsibility then rest with the hospitalist who discharged the patient? One could argue the physician who ordered the test is responsible. What if the hospitalist who ordered the test is not the same hospitalist who discharged the patient? It would seem under that circumstance, the hospitalist who discharged the patient bears more responsibility than the hospitalist who ordered the study.
To further complicate matters, what if the physician who ordered the test was a consultant? I am not aware of any rules specifying provider responsibility for notifying the patient. I typically recommend the hospitalist in charge of discharging the patient from the hospital make a practice of looking for studies whose results are pending at the time of discharge. The hospitalist should inform the patient the results are pending and discuss a plan of action for the patient to get the study results. Then I suggest the hospitalist document this discussion and plan/forward this documentation to the provider who is scheduled to see the patient in follow up. It is typically easier for hospitalists to include this information as part of the discharge summary sent to the PCP.
As you suggested, these steps may be insufficient when the patient does not follow up with the designated PCP. For that reason, it is necessary for the hospitalist who discharged the patient to follow up on these pending results. The hospitalist must notify the patient if the results are abnormal. To do this, prior to hospital discharge, one needs to know how to contact a patient post-discharge. Always document the fact you have notified the patient of the abnormal result. I recognize this type of follow up is not easy after a patient is discharged, especially when most results will return as normal studies. The volume-to-noise ratio is not great. But it is that one out of 100 abnormal result that will end up hurting the patient and potentially result in litigation.
One important piece of advice: Only order necessary tests. The fewer tests you order, the less it is likely you will have test results pending at discharge. If a test is not likely to change how you manage a patient during their inpatient stay, consider not ordering the test. Such practice is not only more cost-effective care, but also simplifies the system and minimizes the risk of error associated with notifying patients of abnormal test results.
A Little Common Courtesy, Please
I find it incredibly annoying when we are holding a staff meeting and some of my colleagues are checking e-mail on their Blackberry. At the risk of sounding like a codger, is it too much to ask for some common courtesy?
K. Moore, Austin, Texas
Dr. Hospitalist responds: You are, of course, correct at pointing out it is rude for people to check messages during meetings, not to mention anytime a supervisor or colleague is speaking. Do I condone the behavior? No. Do I understand the behavior? Yes. (In the spirit of full disclosure, I am addicted to my Blackberry and, occasionally, am guilty of checking for messages when I should be paying attention).
We live in an information age and the expectation for communication is greater than ever. As hospitalists, we know this all too well. For many of us, the Blackberry affords us the opportunity to multitask, shaving minutes or hours off our workday. I agree it is not an unreasonable request to ask everyone to turn off their cell phones and put down their Blackberrys during meetings.
That said, doing without the Blackberry for much longer than an hour or two is not an option for many of us.
Please note President-elect Obama will have to ditch his Blackberry this month, if not sooner, due to concerns surrounding e-mail privacy. He also is subject to the Presidential Records Act, which eliminates any privacy regarding this correspondence. (Memo to self, another reason not to run for president.) TH
Coming to a Hospital Near You: E-prescribe
Will hospitalists and hospital medicine groups get to participate in the Centers for Medicare and Medicaid e-prescribe program?
Jettie Eddleman, BSN, RN, Quality Initiatives Program Director North Texas Specialty Physicians (NTSP), Fort Worth, Texas
Dr. Hospitalist responds: According to a December 2007 study by SureScripts, only 6% of U.S. physicians prescribe medications electronically. Medicare would like more physicians to electronically prescribe prescriptions because “e-prescribing is more efficient and convenient for consumers, improves the quality of care, lowers administrative costs, and its widespread use would eliminate thousands of medication errors every year.”
Medicare is not the only organization encouraging e-prescribing. Blue Cross Blue Shield of Massachusetts (BCBSMA) recently announced e-prescribing will be required for any physician to participate in any of the BCBSMA physician incentive programs, effective January, 2011. To speed the adoption of e-prescribing, Medicare will provide financial incentives to physicians who e-prescribe. Starting in this year, Medicare will pay a 2% bonus to physicians who prescribe under Part D. This incentive bonus will decrease to 1% in 2011 and 0.5% in 2013. Starting in 2012, physicians who are not e-prescribing will lose 1% of their Medicare payment. This penalty will increase to 1.5% in 2013 and 2% in 2014. In other words, you can e-prescribe sooner or later, but hospital medicine groups will increase revenue if they start sooner.
Do you have a problem or concern that you’d like Dr. Hospitalist to address? E-mail your questions to drhospit@wiley.com.
E-prescribing is, however, not without barriers. For example, the Drug Enforcement Agency (DEA) presently prohibits e-prescription of controlled substances. Medicare is working with the DEA to address this issue. In the meantime, providers who implement e-prescription will need to continue a separate system for prescription of controlled substances.
Some physicians have electronic medical record (EMR) systems, which send prescriptions to pharmacies via facsimile machines. For clarification, this is not considered e-prescribing. In fact, under Medicare statute, most EMR-faxed prescriptions no longer are allowed for Part D. As the plan currently stands, this incentive is tied to specific Current Procedural Terminology (CPT) codes, used primarily by primary care providers and not by hospitalists. Unless the hospitalists in your group also provide outpatient care, it is unlikely any will be able to participate in this Medicare e-prescribing incentive plan.
Test Results Post-Discharge
Some of my patients have laboratory test results pending at the time of discharge from the hospital. Most of my patients do not have a routine outpatient provider. At the time of discharge, I always set them up with outpatient follow-up with a new primary care provider, but I have no way of knowing if the patients are showing up at their appointments. My concern is if they don’t show up, they won’t know about their test results. Is this something I need to address? If so, how do you suggest I go about doing it?
Z. Taylor, Durant, Okla.
Dr. Hospitalist responds: This definitely is an issue that needs to be addressed. It is not only a quality of care issue, but also potentially a medical/legal issue. Transitions in care are risky for patients because these are periods with increased risk for medical error. For example, this is why the Joint Commission mandates medication reconciliation each time a patient sees a provider.
When patients are discharged from the hospital, which provider is responsible for notifying the patient of pending laboratory test results? Is it the primary care provider (PCP) or the hospitalist? As you described in your question, what if the patient does not have a regular PCP? Does the responsibility then rest with the hospitalist who discharged the patient? One could argue the physician who ordered the test is responsible. What if the hospitalist who ordered the test is not the same hospitalist who discharged the patient? It would seem under that circumstance, the hospitalist who discharged the patient bears more responsibility than the hospitalist who ordered the study.
To further complicate matters, what if the physician who ordered the test was a consultant? I am not aware of any rules specifying provider responsibility for notifying the patient. I typically recommend the hospitalist in charge of discharging the patient from the hospital make a practice of looking for studies whose results are pending at the time of discharge. The hospitalist should inform the patient the results are pending and discuss a plan of action for the patient to get the study results. Then I suggest the hospitalist document this discussion and plan/forward this documentation to the provider who is scheduled to see the patient in follow up. It is typically easier for hospitalists to include this information as part of the discharge summary sent to the PCP.
As you suggested, these steps may be insufficient when the patient does not follow up with the designated PCP. For that reason, it is necessary for the hospitalist who discharged the patient to follow up on these pending results. The hospitalist must notify the patient if the results are abnormal. To do this, prior to hospital discharge, one needs to know how to contact a patient post-discharge. Always document the fact you have notified the patient of the abnormal result. I recognize this type of follow up is not easy after a patient is discharged, especially when most results will return as normal studies. The volume-to-noise ratio is not great. But it is that one out of 100 abnormal result that will end up hurting the patient and potentially result in litigation.
One important piece of advice: Only order necessary tests. The fewer tests you order, the less it is likely you will have test results pending at discharge. If a test is not likely to change how you manage a patient during their inpatient stay, consider not ordering the test. Such practice is not only more cost-effective care, but also simplifies the system and minimizes the risk of error associated with notifying patients of abnormal test results.
A Little Common Courtesy, Please
I find it incredibly annoying when we are holding a staff meeting and some of my colleagues are checking e-mail on their Blackberry. At the risk of sounding like a codger, is it too much to ask for some common courtesy?
K. Moore, Austin, Texas
Dr. Hospitalist responds: You are, of course, correct at pointing out it is rude for people to check messages during meetings, not to mention anytime a supervisor or colleague is speaking. Do I condone the behavior? No. Do I understand the behavior? Yes. (In the spirit of full disclosure, I am addicted to my Blackberry and, occasionally, am guilty of checking for messages when I should be paying attention).
We live in an information age and the expectation for communication is greater than ever. As hospitalists, we know this all too well. For many of us, the Blackberry affords us the opportunity to multitask, shaving minutes or hours off our workday. I agree it is not an unreasonable request to ask everyone to turn off their cell phones and put down their Blackberrys during meetings.
That said, doing without the Blackberry for much longer than an hour or two is not an option for many of us.
Please note President-elect Obama will have to ditch his Blackberry this month, if not sooner, due to concerns surrounding e-mail privacy. He also is subject to the Presidential Records Act, which eliminates any privacy regarding this correspondence. (Memo to self, another reason not to run for president.) TH
Coming to a Hospital Near You: E-prescribe
Will hospitalists and hospital medicine groups get to participate in the Centers for Medicare and Medicaid e-prescribe program?
Jettie Eddleman, BSN, RN, Quality Initiatives Program Director North Texas Specialty Physicians (NTSP), Fort Worth, Texas
Dr. Hospitalist responds: According to a December 2007 study by SureScripts, only 6% of U.S. physicians prescribe medications electronically. Medicare would like more physicians to electronically prescribe prescriptions because “e-prescribing is more efficient and convenient for consumers, improves the quality of care, lowers administrative costs, and its widespread use would eliminate thousands of medication errors every year.”
Medicare is not the only organization encouraging e-prescribing. Blue Cross Blue Shield of Massachusetts (BCBSMA) recently announced e-prescribing will be required for any physician to participate in any of the BCBSMA physician incentive programs, effective January, 2011. To speed the adoption of e-prescribing, Medicare will provide financial incentives to physicians who e-prescribe. Starting in this year, Medicare will pay a 2% bonus to physicians who prescribe under Part D. This incentive bonus will decrease to 1% in 2011 and 0.5% in 2013. Starting in 2012, physicians who are not e-prescribing will lose 1% of their Medicare payment. This penalty will increase to 1.5% in 2013 and 2% in 2014. In other words, you can e-prescribe sooner or later, but hospital medicine groups will increase revenue if they start sooner.
Do you have a problem or concern that you’d like Dr. Hospitalist to address? E-mail your questions to drhospit@wiley.com.
E-prescribing is, however, not without barriers. For example, the Drug Enforcement Agency (DEA) presently prohibits e-prescription of controlled substances. Medicare is working with the DEA to address this issue. In the meantime, providers who implement e-prescription will need to continue a separate system for prescription of controlled substances.
Some physicians have electronic medical record (EMR) systems, which send prescriptions to pharmacies via facsimile machines. For clarification, this is not considered e-prescribing. In fact, under Medicare statute, most EMR-faxed prescriptions no longer are allowed for Part D. As the plan currently stands, this incentive is tied to specific Current Procedural Terminology (CPT) codes, used primarily by primary care providers and not by hospitalists. Unless the hospitalists in your group also provide outpatient care, it is unlikely any will be able to participate in this Medicare e-prescribing incentive plan.
Test Results Post-Discharge
Some of my patients have laboratory test results pending at the time of discharge from the hospital. Most of my patients do not have a routine outpatient provider. At the time of discharge, I always set them up with outpatient follow-up with a new primary care provider, but I have no way of knowing if the patients are showing up at their appointments. My concern is if they don’t show up, they won’t know about their test results. Is this something I need to address? If so, how do you suggest I go about doing it?
Z. Taylor, Durant, Okla.
Dr. Hospitalist responds: This definitely is an issue that needs to be addressed. It is not only a quality of care issue, but also potentially a medical/legal issue. Transitions in care are risky for patients because these are periods with increased risk for medical error. For example, this is why the Joint Commission mandates medication reconciliation each time a patient sees a provider.
When patients are discharged from the hospital, which provider is responsible for notifying the patient of pending laboratory test results? Is it the primary care provider (PCP) or the hospitalist? As you described in your question, what if the patient does not have a regular PCP? Does the responsibility then rest with the hospitalist who discharged the patient? One could argue the physician who ordered the test is responsible. What if the hospitalist who ordered the test is not the same hospitalist who discharged the patient? It would seem under that circumstance, the hospitalist who discharged the patient bears more responsibility than the hospitalist who ordered the study.
To further complicate matters, what if the physician who ordered the test was a consultant? I am not aware of any rules specifying provider responsibility for notifying the patient. I typically recommend the hospitalist in charge of discharging the patient from the hospital make a practice of looking for studies whose results are pending at the time of discharge. The hospitalist should inform the patient the results are pending and discuss a plan of action for the patient to get the study results. Then I suggest the hospitalist document this discussion and plan/forward this documentation to the provider who is scheduled to see the patient in follow up. It is typically easier for hospitalists to include this information as part of the discharge summary sent to the PCP.
As you suggested, these steps may be insufficient when the patient does not follow up with the designated PCP. For that reason, it is necessary for the hospitalist who discharged the patient to follow up on these pending results. The hospitalist must notify the patient if the results are abnormal. To do this, prior to hospital discharge, one needs to know how to contact a patient post-discharge. Always document the fact you have notified the patient of the abnormal result. I recognize this type of follow up is not easy after a patient is discharged, especially when most results will return as normal studies. The volume-to-noise ratio is not great. But it is that one out of 100 abnormal result that will end up hurting the patient and potentially result in litigation.
One important piece of advice: Only order necessary tests. The fewer tests you order, the less it is likely you will have test results pending at discharge. If a test is not likely to change how you manage a patient during their inpatient stay, consider not ordering the test. Such practice is not only more cost-effective care, but also simplifies the system and minimizes the risk of error associated with notifying patients of abnormal test results.
A Little Common Courtesy, Please
I find it incredibly annoying when we are holding a staff meeting and some of my colleagues are checking e-mail on their Blackberry. At the risk of sounding like a codger, is it too much to ask for some common courtesy?
K. Moore, Austin, Texas
Dr. Hospitalist responds: You are, of course, correct at pointing out it is rude for people to check messages during meetings, not to mention anytime a supervisor or colleague is speaking. Do I condone the behavior? No. Do I understand the behavior? Yes. (In the spirit of full disclosure, I am addicted to my Blackberry and, occasionally, am guilty of checking for messages when I should be paying attention).
We live in an information age and the expectation for communication is greater than ever. As hospitalists, we know this all too well. For many of us, the Blackberry affords us the opportunity to multitask, shaving minutes or hours off our workday. I agree it is not an unreasonable request to ask everyone to turn off their cell phones and put down their Blackberrys during meetings.
That said, doing without the Blackberry for much longer than an hour or two is not an option for many of us.
Please note President-elect Obama will have to ditch his Blackberry this month, if not sooner, due to concerns surrounding e-mail privacy. He also is subject to the Presidential Records Act, which eliminates any privacy regarding this correspondence. (Memo to self, another reason not to run for president.) TH
Satisfaction Scorecard

Patient satisfaction became a much more important issue earlier in 2008 when hospitals began reporting their performance on the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey. The 18-item survey was developed by the Centers for Medicare and Medicaid Services (CMS) and the Agency for Healthcare Research and Quality, and is completed by patients to record their level of satisfaction with the hospital and the care they received.
If you don’t know your hospital’s publicly reported HCAHPS scores, you should make it your business to become familiar with them. Survey results from participating hospitals are updated periodically and available at www.hospitalcompare.hhs.gov.
I regularly hear hospitalists say they could more effectively direct their energy and resources in ways consistent with their hospital CEO’s goals, if they only knew what those goals were. If you find yourself in the same category, then you should know there is a really good chance the upper-level hospital leadership has a salary bonus in place for achieving certain patient safety and satisfaction goals. A recent survey in the Journal of Patient Safety showed this is the case for more than half of the nation’s hospital executives.1 Therefore, learning more about your hospital’s patient satisfaction scores, and your group’s contribution to the score, is worth the effort.
The publicly reported HCAHPS data does not address hospitalists’ effect on patient satisfaction separately from other doctors and hospital attributes. As a result, some hospitalist groups conduct their own survey. This may range from a very brief series of written questions, or a more-involved survey administered via phone. Since it is difficult to drill down to patient satisfaction with individual hospitalists, data from these surveys often times are collected for the entire hospitalist practice in aggregate, rather than by the individual doctor.
Incorporate Information
Investigate both internal and external resources, if you believe your group could benefit from an intervention (e.g., education or training to improve patient satisfaction). Many hospitals have someone on staff to provide employee training in this area; the trainer probably would be impressed and pleased if you initiate contact to learn more about available training. Resources external to your hospital include survey companies (e.g., Press Ganey, NCR Picker, among others). These companies can provide training and guidance, or put you in contact with firms with whom they work. I have seen something as simple as a one- or two-hour online or DVD course be a valuable tool for some practices.
I know some very efficient hospitalists who maintain incredibly high levels of patient satisfaction despite very high workloads. I think these doctors probably are outliers, and most of us will see satisfaction scores decline as workloads become unreasonably high. Each group should challenge itself continually to find the optimal point between patient volume and important outcomes like patient satisfaction.
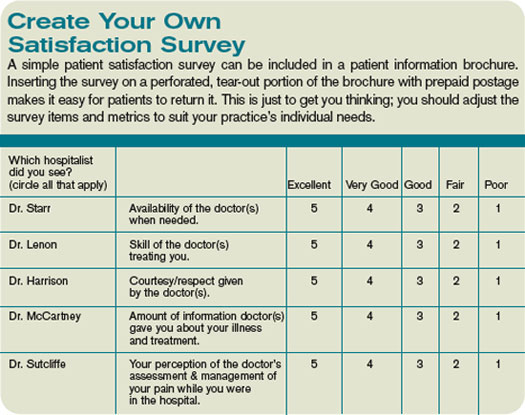
Here are some strategies hospitalist groups could use to improve patient satisfaction:
- Call patients after discharge. This is a potent way to improve patient satisfaction. It is a valuable clinical encounter, since it offers a chance to reinforce discharge instructions (e.g, the value of smoking cessation, or the need to have ongoing INR monitoring), and to address any issues arising in the interim. The calls are most effective if done by the doctor who discharged the patient, but can be of some value if made by a nurse or other person connected to the practice. Note, it is best not to inform patients you will be making this call, since it will reduce the surprise and pleasure when the call comes, and could lead to frustration if a patient never gets the call they were told to expect.
- Provide referring doctors, emergency room doctors, and nurses at the hospital with a “script” to use when introducing or describing the hospitalist practice to patients. Without coaching, most of these people likely will say to the patient something like “your doctor [primary care physician] doesn’t come to the hospital anymore, so the hospitalist will see you.” Such a description may make the patient feel like they’re getting a second-class doctor. Instead, encourage these workers to say something like “Your doctor has decided to focus her practice on the office, to be more available to you there. As a result, she has decided to refer you to Dr. McCartney, a doctor who specializes in the care of hospitalized patients with problems like yours. Dr. McCartney will communicate with your primary doctor, and you should plan to follow up with her when you are discharged.”
- Communicate regularly, even daily, with family members of patients who have dementia or other cognitive impairment. This usually means calling the family.
- During initial and subsequent visits with a patient, shake hands or gently touch the patient in some way. Sit down and, while still sitting, conclude the conversation by asking if the patient has any questions and asking “Is there anything special I can do for you today?”
- Provide patients with a copy of their discharge summary. It can serve as a valuable education tool for the patient, their loved ones, visiting nurses, etc. Ideally, the summary should be transcribed on a stat basis, so it can be available for the nurse to give to the patient on the way out of the hospital. Alternatively, it could be mailed to the patient later.
- Track your ongoing satisfaction performance by regularly including available data from HCAHPS and/or other sources in the practice dashboard or report card.
- Ensure patients and their families are provided a copy of your group’s brochure. Brochures delivered to primary care offices rarely find their way into the hands of the patients, and often times are forgotten or misplaced by the time hospital care is needed. It usually is more effective to ensure hospital staff provides the brochure. This could be done via a standing protocol stating that a clerical person will provide all patients with a hospitalist as attending or consultant will get a copy. Or, ensure an order to this effect is written on every one of your patients. It’s best to do this via pre-printed order sets.
If you don’t have a group brochure, develop one. It should include the name of the group with a one- or two-line biography and photograph for each hospitalist (e.g., where they attended medical school and residency), and other key information about the practice. If you’d like a sample brochure, visit www.hospitalmedicine.org and search “sample brochure for hospitalist program.”
Each hospitalist in your group could carry business cards with the doctor’s picture next to their name and key info. One small study showed this enhanced patients’ ability to correctly identify their hospitalist and presumably increased their satisfaction, as well.
- Maximize hospitalist-patient continuity. Each group should adjust their work schedule to maximize continuity between patient and hospitalist while still providing a sustainable lifestyle. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm. He is part of the faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official SHM position..
Reference
1. Vaughn T, Koepke M, Kroch E, et al. Engagement of leadership in quality improvement initiatives: executive quality improvement survey results. J Patient Saf. 2006;2(1):2-9.
Patient satisfaction became a much more important issue earlier in 2008 when hospitals began reporting their performance on the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey. The 18-item survey was developed by the Centers for Medicare and Medicaid Services (CMS) and the Agency for Healthcare Research and Quality, and is completed by patients to record their level of satisfaction with the hospital and the care they received.
If you don’t know your hospital’s publicly reported HCAHPS scores, you should make it your business to become familiar with them. Survey results from participating hospitals are updated periodically and available at www.hospitalcompare.hhs.gov.
I regularly hear hospitalists say they could more effectively direct their energy and resources in ways consistent with their hospital CEO’s goals, if they only knew what those goals were. If you find yourself in the same category, then you should know there is a really good chance the upper-level hospital leadership has a salary bonus in place for achieving certain patient safety and satisfaction goals. A recent survey in the Journal of Patient Safety showed this is the case for more than half of the nation’s hospital executives.1 Therefore, learning more about your hospital’s patient satisfaction scores, and your group’s contribution to the score, is worth the effort.
The publicly reported HCAHPS data does not address hospitalists’ effect on patient satisfaction separately from other doctors and hospital attributes. As a result, some hospitalist groups conduct their own survey. This may range from a very brief series of written questions, or a more-involved survey administered via phone. Since it is difficult to drill down to patient satisfaction with individual hospitalists, data from these surveys often times are collected for the entire hospitalist practice in aggregate, rather than by the individual doctor.
Incorporate Information
Investigate both internal and external resources, if you believe your group could benefit from an intervention (e.g., education or training to improve patient satisfaction). Many hospitals have someone on staff to provide employee training in this area; the trainer probably would be impressed and pleased if you initiate contact to learn more about available training. Resources external to your hospital include survey companies (e.g., Press Ganey, NCR Picker, among others). These companies can provide training and guidance, or put you in contact with firms with whom they work. I have seen something as simple as a one- or two-hour online or DVD course be a valuable tool for some practices.
I know some very efficient hospitalists who maintain incredibly high levels of patient satisfaction despite very high workloads. I think these doctors probably are outliers, and most of us will see satisfaction scores decline as workloads become unreasonably high. Each group should challenge itself continually to find the optimal point between patient volume and important outcomes like patient satisfaction.
Here are some strategies hospitalist groups could use to improve patient satisfaction:
- Call patients after discharge. This is a potent way to improve patient satisfaction. It is a valuable clinical encounter, since it offers a chance to reinforce discharge instructions (e.g, the value of smoking cessation, or the need to have ongoing INR monitoring), and to address any issues arising in the interim. The calls are most effective if done by the doctor who discharged the patient, but can be of some value if made by a nurse or other person connected to the practice. Note, it is best not to inform patients you will be making this call, since it will reduce the surprise and pleasure when the call comes, and could lead to frustration if a patient never gets the call they were told to expect.
- Provide referring doctors, emergency room doctors, and nurses at the hospital with a “script” to use when introducing or describing the hospitalist practice to patients. Without coaching, most of these people likely will say to the patient something like “your doctor [primary care physician] doesn’t come to the hospital anymore, so the hospitalist will see you.” Such a description may make the patient feel like they’re getting a second-class doctor. Instead, encourage these workers to say something like “Your doctor has decided to focus her practice on the office, to be more available to you there. As a result, she has decided to refer you to Dr. McCartney, a doctor who specializes in the care of hospitalized patients with problems like yours. Dr. McCartney will communicate with your primary doctor, and you should plan to follow up with her when you are discharged.”
- Communicate regularly, even daily, with family members of patients who have dementia or other cognitive impairment. This usually means calling the family.
- During initial and subsequent visits with a patient, shake hands or gently touch the patient in some way. Sit down and, while still sitting, conclude the conversation by asking if the patient has any questions and asking “Is there anything special I can do for you today?”
- Provide patients with a copy of their discharge summary. It can serve as a valuable education tool for the patient, their loved ones, visiting nurses, etc. Ideally, the summary should be transcribed on a stat basis, so it can be available for the nurse to give to the patient on the way out of the hospital. Alternatively, it could be mailed to the patient later.
- Track your ongoing satisfaction performance by regularly including available data from HCAHPS and/or other sources in the practice dashboard or report card.
- Ensure patients and their families are provided a copy of your group’s brochure. Brochures delivered to primary care offices rarely find their way into the hands of the patients, and often times are forgotten or misplaced by the time hospital care is needed. It usually is more effective to ensure hospital staff provides the brochure. This could be done via a standing protocol stating that a clerical person will provide all patients with a hospitalist as attending or consultant will get a copy. Or, ensure an order to this effect is written on every one of your patients. It’s best to do this via pre-printed order sets.
If you don’t have a group brochure, develop one. It should include the name of the group with a one- or two-line biography and photograph for each hospitalist (e.g., where they attended medical school and residency), and other key information about the practice. If you’d like a sample brochure, visit www.hospitalmedicine.org and search “sample brochure for hospitalist program.”
Each hospitalist in your group could carry business cards with the doctor’s picture next to their name and key info. One small study showed this enhanced patients’ ability to correctly identify their hospitalist and presumably increased their satisfaction, as well.
- Maximize hospitalist-patient continuity. Each group should adjust their work schedule to maximize continuity between patient and hospitalist while still providing a sustainable lifestyle. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm. He is part of the faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official SHM position..
Reference
1. Vaughn T, Koepke M, Kroch E, et al. Engagement of leadership in quality improvement initiatives: executive quality improvement survey results. J Patient Saf. 2006;2(1):2-9.
Patient satisfaction became a much more important issue earlier in 2008 when hospitals began reporting their performance on the Hospital Consumer Assessment of Healthcare Providers and Systems (HCAHPS) survey. The 18-item survey was developed by the Centers for Medicare and Medicaid Services (CMS) and the Agency for Healthcare Research and Quality, and is completed by patients to record their level of satisfaction with the hospital and the care they received.
If you don’t know your hospital’s publicly reported HCAHPS scores, you should make it your business to become familiar with them. Survey results from participating hospitals are updated periodically and available at www.hospitalcompare.hhs.gov.
I regularly hear hospitalists say they could more effectively direct their energy and resources in ways consistent with their hospital CEO’s goals, if they only knew what those goals were. If you find yourself in the same category, then you should know there is a really good chance the upper-level hospital leadership has a salary bonus in place for achieving certain patient safety and satisfaction goals. A recent survey in the Journal of Patient Safety showed this is the case for more than half of the nation’s hospital executives.1 Therefore, learning more about your hospital’s patient satisfaction scores, and your group’s contribution to the score, is worth the effort.
The publicly reported HCAHPS data does not address hospitalists’ effect on patient satisfaction separately from other doctors and hospital attributes. As a result, some hospitalist groups conduct their own survey. This may range from a very brief series of written questions, or a more-involved survey administered via phone. Since it is difficult to drill down to patient satisfaction with individual hospitalists, data from these surveys often times are collected for the entire hospitalist practice in aggregate, rather than by the individual doctor.
Incorporate Information
Investigate both internal and external resources, if you believe your group could benefit from an intervention (e.g., education or training to improve patient satisfaction). Many hospitals have someone on staff to provide employee training in this area; the trainer probably would be impressed and pleased if you initiate contact to learn more about available training. Resources external to your hospital include survey companies (e.g., Press Ganey, NCR Picker, among others). These companies can provide training and guidance, or put you in contact with firms with whom they work. I have seen something as simple as a one- or two-hour online or DVD course be a valuable tool for some practices.
I know some very efficient hospitalists who maintain incredibly high levels of patient satisfaction despite very high workloads. I think these doctors probably are outliers, and most of us will see satisfaction scores decline as workloads become unreasonably high. Each group should challenge itself continually to find the optimal point between patient volume and important outcomes like patient satisfaction.
Here are some strategies hospitalist groups could use to improve patient satisfaction:
- Call patients after discharge. This is a potent way to improve patient satisfaction. It is a valuable clinical encounter, since it offers a chance to reinforce discharge instructions (e.g, the value of smoking cessation, or the need to have ongoing INR monitoring), and to address any issues arising in the interim. The calls are most effective if done by the doctor who discharged the patient, but can be of some value if made by a nurse or other person connected to the practice. Note, it is best not to inform patients you will be making this call, since it will reduce the surprise and pleasure when the call comes, and could lead to frustration if a patient never gets the call they were told to expect.
- Provide referring doctors, emergency room doctors, and nurses at the hospital with a “script” to use when introducing or describing the hospitalist practice to patients. Without coaching, most of these people likely will say to the patient something like “your doctor [primary care physician] doesn’t come to the hospital anymore, so the hospitalist will see you.” Such a description may make the patient feel like they’re getting a second-class doctor. Instead, encourage these workers to say something like “Your doctor has decided to focus her practice on the office, to be more available to you there. As a result, she has decided to refer you to Dr. McCartney, a doctor who specializes in the care of hospitalized patients with problems like yours. Dr. McCartney will communicate with your primary doctor, and you should plan to follow up with her when you are discharged.”
- Communicate regularly, even daily, with family members of patients who have dementia or other cognitive impairment. This usually means calling the family.
- During initial and subsequent visits with a patient, shake hands or gently touch the patient in some way. Sit down and, while still sitting, conclude the conversation by asking if the patient has any questions and asking “Is there anything special I can do for you today?”
- Provide patients with a copy of their discharge summary. It can serve as a valuable education tool for the patient, their loved ones, visiting nurses, etc. Ideally, the summary should be transcribed on a stat basis, so it can be available for the nurse to give to the patient on the way out of the hospital. Alternatively, it could be mailed to the patient later.
- Track your ongoing satisfaction performance by regularly including available data from HCAHPS and/or other sources in the practice dashboard or report card.
- Ensure patients and their families are provided a copy of your group’s brochure. Brochures delivered to primary care offices rarely find their way into the hands of the patients, and often times are forgotten or misplaced by the time hospital care is needed. It usually is more effective to ensure hospital staff provides the brochure. This could be done via a standing protocol stating that a clerical person will provide all patients with a hospitalist as attending or consultant will get a copy. Or, ensure an order to this effect is written on every one of your patients. It’s best to do this via pre-printed order sets.
If you don’t have a group brochure, develop one. It should include the name of the group with a one- or two-line biography and photograph for each hospitalist (e.g., where they attended medical school and residency), and other key information about the practice. If you’d like a sample brochure, visit www.hospitalmedicine.org and search “sample brochure for hospitalist program.”
Each hospitalist in your group could carry business cards with the doctor’s picture next to their name and key info. One small study showed this enhanced patients’ ability to correctly identify their hospitalist and presumably increased their satisfaction, as well.
- Maximize hospitalist-patient continuity. Each group should adjust their work schedule to maximize continuity between patient and hospitalist while still providing a sustainable lifestyle. TH
Dr. Nelson has been a practicing hospitalist since 1988 and is co-founder and past president of SHM. He is a principal in Nelson/Flores Associates, a national hospitalist practice management consulting firm. He is part of the faculty for SHM’s “Best Practices in Managing a Hospital Medicine Program” course. This column represents his views and is not intended to reflect an official SHM position..
Reference
1. Vaughn T, Koepke M, Kroch E, et al. Engagement of leadership in quality improvement initiatives: executive quality improvement survey results. J Patient Saf. 2006;2(1):2-9.
The Fast, Furious Future

I read the ads.” “The what?” I replied, dumbfounded.
“The ads.”
“Not ‘In the Literature,’ ‘Key Clinical Questions,’ the cover stories, John Nelson’s practice management column?”
“No,” “no,” “no” and “no” were the responses.
Encasing my slightly bruised ego behind a facade of curiosity, I inquired of my friend who directs a large community hospitalist practice why the first thing he reads in The Hospitalist are the advertisements.
“Because I need to know what the competition is offering, so that I can keep my docs,” my friend explained.
“With that it was clearer than ever that we have a shortage of hospitalists. Let’s take a look at some numbers.”
Numbers Part I
The good news is there are close to 20,000 hospitalists in the U.S. This meteoric rise occurred in 11 years; something emergency medicine took 20 years to accomplish. It is commonly assumed the mature hospital medicine field will grow to 30,000 providers. More grandiose estimates place the top estimate as high as 70,000.
Numbers Part II
The concerning news is the number of adults 65 and older is expected to double by 2030. This demographic accounts for nearly 50% of all hospital admissions, and thus heralds an onslaught of growth in hospital medicine. Workforce models predict a physician shortfall of 200,000 by the year 2020.1
Numbers Part III
The frightening news is the number of U.S. medical school graduates matching into internal medicine (IM) residency programs has dropped from 3,884 in 1985 to 2,660 in 2007.1 Only 22% of graduating U.S. medical school students chooses to match in IM and only 55% of IM residency positions are filled by U.S. medical school graduates.2 Nearly 60% of IM graduates choose subspecialist careers, with only 8% choosing hospitalist careers.3
Runaway Train
The U.S. is likely to witness an astronomic growth in the supply of hospitalized patients. Fewer students are choosing IM, with the majority of these opting for subspecialty careers. Hospital medicine faces an ever growing workforce shortage that has the potential to cripple the field.
I often wonder if hospital medicine is developing too fast. Our haste to grow and take on new business can, at times, lead us astray. To be sure, some of this growth is thrust on us by external forces, such as the mass exodus of primary care doctors out of the hospital and hospital executives’ eagerness to tap the resource efficiency of the hospitalist model. However, we cannot allow our foremost mission—improving patient experiences and outcomes—to be lost in a landslide of growth that outstrips our staffing.
Limit Growth
To that end, I think one very logical solution to this pending workforce shortage is to limit growth. Take a breather and let the supply catch up with the demand before growing further. The skeptic will say this is not possible, but recall that hospitalized patients received care for hundreds of years prior to hospitalists. Ask yourself how much of the work you do could be done by another physician group. A recent study I was part of reported more than 22% of a community hospitalist’s workload consists of orthopedic, neurological, and general surgical diagnoses.4 Our study design was unable to account for patients previously cared for by medical sub-specialists, such as cardiologists and gastroenterologists, but I suspect the number is large, as well.
Although a 22% decrease in volume across the board would not solve all of our problems, it would be a solid start. Consider this the next time a surgical or medical subspecialty group requests you care for their patients. It may be a poor business move to refuse this work, but perhaps you can build a collaborative, long-term plan that allows you to better care for the patient obligations you have today while incrementally caring for their patients as your group matures.
Higher, Faster, Farther
Another method to tackle the hospitalist shortage is to see more patients with the same amount of providers. Doing this requires more than just increasing the patient numerator as you hold the provider denominator steady. Rather, it requires real systems changes to enhance provider efficiency. A significant amount of a hospitalists’ daily workload consists of non-patient care activity, such as searching for charts, waiting for consultants to call back and scheduling follow-up appointments. The challenge to future hospitalists will be to tame these inefficiencies by developing more streamlined hospital systems and care processes.
This may include hiring assistants, rounding staff, and/or mid-level providers to incrementally increase the number of patients the group can see per physician. These are not without risks and barriers, and many groups currently are wrestling with how to best utilize and integrate these providers in a cost-effective manner. However, I think it is likely these components will provide the future scaffolding to allow groups to care for ever increasing numbers of patients.
Prime the Pump
Perhaps, most importantly, we need to develop ways to attract more medical students and residents to hospitalist careers. This will be challenging and falls squarely to my academic hospital medicine colleagues, as our learners see us as the yardstick for a hospitalist career. Unfortunately, the growth trend in academic hospital medicine has been in non-teaching or uncovered services. These jobs often are an academic necropolis, with the providers routinely being overworked and devalued by their institution.
As a result, academic hospitalist positions often times are filled by recent residency graduates awaiting a fellowship. These “R4” or “pretending” positions provide very rickety underpinnings from which to build the foundation of hospital medicine. Don’t for an instant think this goes unnoticed by our student and resident colleagues who choose their career based on the role models they see early in their training.
It is essential hospital medicine develops truly sustainable academic careers replete with opportunities to fulfill the inimitable tenets of academic medicine—teaching and scholarly work. For these reasons, SHM has partnered with the Society of General Internal Medicine and the Association of Chiefs in General Internal Medicine to develop the Academic Hospitalist Academy. The four-day academy premiers next fall. Its goal is to enable academic hospitalists to become exceptional educators, institutional leaders, and successful scholars—the exact type of role models that will attract the best and the brightest to the field of hospital medicine for generations to come. TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the hospital medicine program and the hospitalist training program, and as associate program director of the Internal Medicine Residency Program.
References
1. Hauer KE, Durning SJ, Kernan WN, et al. Factors associated with medical students’ career choices regarding internal medicine. JAMA. 2008;300:1154-1156,1164.
2. Hauer KE, Fagan MJ, Kernan W, Mintz M, Durning SJ. Internal medicine clerkship directors’ perceptions about student interest in internal medicine careers. J Gen Intern Med. 2008;1101-1104.
3. McDonald FS, West CP, Popkave C, Kolars JC. Educational debt and reported career plans among internal medicine residents. Ann Inter Med. 2008;149:416-420.
4. Glasheen JJ, Epstein KR, Siegal E, Kutner JS, Prochazka AV. The spectrum of community based hospitalist practice: A call to tailor internal medicine residency training. Arch Intern Med. 2007;167:727-729.
I read the ads.” “The what?” I replied, dumbfounded.
“The ads.”
“Not ‘In the Literature,’ ‘Key Clinical Questions,’ the cover stories, John Nelson’s practice management column?”
“No,” “no,” “no” and “no” were the responses.
Encasing my slightly bruised ego behind a facade of curiosity, I inquired of my friend who directs a large community hospitalist practice why the first thing he reads in The Hospitalist are the advertisements.
“Because I need to know what the competition is offering, so that I can keep my docs,” my friend explained.
“With that it was clearer than ever that we have a shortage of hospitalists. Let’s take a look at some numbers.”
Numbers Part I
The good news is there are close to 20,000 hospitalists in the U.S. This meteoric rise occurred in 11 years; something emergency medicine took 20 years to accomplish. It is commonly assumed the mature hospital medicine field will grow to 30,000 providers. More grandiose estimates place the top estimate as high as 70,000.
Numbers Part II
The concerning news is the number of adults 65 and older is expected to double by 2030. This demographic accounts for nearly 50% of all hospital admissions, and thus heralds an onslaught of growth in hospital medicine. Workforce models predict a physician shortfall of 200,000 by the year 2020.1
Numbers Part III
The frightening news is the number of U.S. medical school graduates matching into internal medicine (IM) residency programs has dropped from 3,884 in 1985 to 2,660 in 2007.1 Only 22% of graduating U.S. medical school students chooses to match in IM and only 55% of IM residency positions are filled by U.S. medical school graduates.2 Nearly 60% of IM graduates choose subspecialist careers, with only 8% choosing hospitalist careers.3
Runaway Train
The U.S. is likely to witness an astronomic growth in the supply of hospitalized patients. Fewer students are choosing IM, with the majority of these opting for subspecialty careers. Hospital medicine faces an ever growing workforce shortage that has the potential to cripple the field.
I often wonder if hospital medicine is developing too fast. Our haste to grow and take on new business can, at times, lead us astray. To be sure, some of this growth is thrust on us by external forces, such as the mass exodus of primary care doctors out of the hospital and hospital executives’ eagerness to tap the resource efficiency of the hospitalist model. However, we cannot allow our foremost mission—improving patient experiences and outcomes—to be lost in a landslide of growth that outstrips our staffing.
Limit Growth
To that end, I think one very logical solution to this pending workforce shortage is to limit growth. Take a breather and let the supply catch up with the demand before growing further. The skeptic will say this is not possible, but recall that hospitalized patients received care for hundreds of years prior to hospitalists. Ask yourself how much of the work you do could be done by another physician group. A recent study I was part of reported more than 22% of a community hospitalist’s workload consists of orthopedic, neurological, and general surgical diagnoses.4 Our study design was unable to account for patients previously cared for by medical sub-specialists, such as cardiologists and gastroenterologists, but I suspect the number is large, as well.
Although a 22% decrease in volume across the board would not solve all of our problems, it would be a solid start. Consider this the next time a surgical or medical subspecialty group requests you care for their patients. It may be a poor business move to refuse this work, but perhaps you can build a collaborative, long-term plan that allows you to better care for the patient obligations you have today while incrementally caring for their patients as your group matures.
Higher, Faster, Farther
Another method to tackle the hospitalist shortage is to see more patients with the same amount of providers. Doing this requires more than just increasing the patient numerator as you hold the provider denominator steady. Rather, it requires real systems changes to enhance provider efficiency. A significant amount of a hospitalists’ daily workload consists of non-patient care activity, such as searching for charts, waiting for consultants to call back and scheduling follow-up appointments. The challenge to future hospitalists will be to tame these inefficiencies by developing more streamlined hospital systems and care processes.
This may include hiring assistants, rounding staff, and/or mid-level providers to incrementally increase the number of patients the group can see per physician. These are not without risks and barriers, and many groups currently are wrestling with how to best utilize and integrate these providers in a cost-effective manner. However, I think it is likely these components will provide the future scaffolding to allow groups to care for ever increasing numbers of patients.
Prime the Pump
Perhaps, most importantly, we need to develop ways to attract more medical students and residents to hospitalist careers. This will be challenging and falls squarely to my academic hospital medicine colleagues, as our learners see us as the yardstick for a hospitalist career. Unfortunately, the growth trend in academic hospital medicine has been in non-teaching or uncovered services. These jobs often are an academic necropolis, with the providers routinely being overworked and devalued by their institution.
As a result, academic hospitalist positions often times are filled by recent residency graduates awaiting a fellowship. These “R4” or “pretending” positions provide very rickety underpinnings from which to build the foundation of hospital medicine. Don’t for an instant think this goes unnoticed by our student and resident colleagues who choose their career based on the role models they see early in their training.
It is essential hospital medicine develops truly sustainable academic careers replete with opportunities to fulfill the inimitable tenets of academic medicine—teaching and scholarly work. For these reasons, SHM has partnered with the Society of General Internal Medicine and the Association of Chiefs in General Internal Medicine to develop the Academic Hospitalist Academy. The four-day academy premiers next fall. Its goal is to enable academic hospitalists to become exceptional educators, institutional leaders, and successful scholars—the exact type of role models that will attract the best and the brightest to the field of hospital medicine for generations to come. TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the hospital medicine program and the hospitalist training program, and as associate program director of the Internal Medicine Residency Program.
References
1. Hauer KE, Durning SJ, Kernan WN, et al. Factors associated with medical students’ career choices regarding internal medicine. JAMA. 2008;300:1154-1156,1164.
2. Hauer KE, Fagan MJ, Kernan W, Mintz M, Durning SJ. Internal medicine clerkship directors’ perceptions about student interest in internal medicine careers. J Gen Intern Med. 2008;1101-1104.
3. McDonald FS, West CP, Popkave C, Kolars JC. Educational debt and reported career plans among internal medicine residents. Ann Inter Med. 2008;149:416-420.
4. Glasheen JJ, Epstein KR, Siegal E, Kutner JS, Prochazka AV. The spectrum of community based hospitalist practice: A call to tailor internal medicine residency training. Arch Intern Med. 2007;167:727-729.
I read the ads.” “The what?” I replied, dumbfounded.
“The ads.”
“Not ‘In the Literature,’ ‘Key Clinical Questions,’ the cover stories, John Nelson’s practice management column?”
“No,” “no,” “no” and “no” were the responses.
Encasing my slightly bruised ego behind a facade of curiosity, I inquired of my friend who directs a large community hospitalist practice why the first thing he reads in The Hospitalist are the advertisements.
“Because I need to know what the competition is offering, so that I can keep my docs,” my friend explained.
“With that it was clearer than ever that we have a shortage of hospitalists. Let’s take a look at some numbers.”
Numbers Part I
The good news is there are close to 20,000 hospitalists in the U.S. This meteoric rise occurred in 11 years; something emergency medicine took 20 years to accomplish. It is commonly assumed the mature hospital medicine field will grow to 30,000 providers. More grandiose estimates place the top estimate as high as 70,000.
Numbers Part II
The concerning news is the number of adults 65 and older is expected to double by 2030. This demographic accounts for nearly 50% of all hospital admissions, and thus heralds an onslaught of growth in hospital medicine. Workforce models predict a physician shortfall of 200,000 by the year 2020.1
Numbers Part III
The frightening news is the number of U.S. medical school graduates matching into internal medicine (IM) residency programs has dropped from 3,884 in 1985 to 2,660 in 2007.1 Only 22% of graduating U.S. medical school students chooses to match in IM and only 55% of IM residency positions are filled by U.S. medical school graduates.2 Nearly 60% of IM graduates choose subspecialist careers, with only 8% choosing hospitalist careers.3
Runaway Train
The U.S. is likely to witness an astronomic growth in the supply of hospitalized patients. Fewer students are choosing IM, with the majority of these opting for subspecialty careers. Hospital medicine faces an ever growing workforce shortage that has the potential to cripple the field.
I often wonder if hospital medicine is developing too fast. Our haste to grow and take on new business can, at times, lead us astray. To be sure, some of this growth is thrust on us by external forces, such as the mass exodus of primary care doctors out of the hospital and hospital executives’ eagerness to tap the resource efficiency of the hospitalist model. However, we cannot allow our foremost mission—improving patient experiences and outcomes—to be lost in a landslide of growth that outstrips our staffing.
Limit Growth
To that end, I think one very logical solution to this pending workforce shortage is to limit growth. Take a breather and let the supply catch up with the demand before growing further. The skeptic will say this is not possible, but recall that hospitalized patients received care for hundreds of years prior to hospitalists. Ask yourself how much of the work you do could be done by another physician group. A recent study I was part of reported more than 22% of a community hospitalist’s workload consists of orthopedic, neurological, and general surgical diagnoses.4 Our study design was unable to account for patients previously cared for by medical sub-specialists, such as cardiologists and gastroenterologists, but I suspect the number is large, as well.
Although a 22% decrease in volume across the board would not solve all of our problems, it would be a solid start. Consider this the next time a surgical or medical subspecialty group requests you care for their patients. It may be a poor business move to refuse this work, but perhaps you can build a collaborative, long-term plan that allows you to better care for the patient obligations you have today while incrementally caring for their patients as your group matures.
Higher, Faster, Farther
Another method to tackle the hospitalist shortage is to see more patients with the same amount of providers. Doing this requires more than just increasing the patient numerator as you hold the provider denominator steady. Rather, it requires real systems changes to enhance provider efficiency. A significant amount of a hospitalists’ daily workload consists of non-patient care activity, such as searching for charts, waiting for consultants to call back and scheduling follow-up appointments. The challenge to future hospitalists will be to tame these inefficiencies by developing more streamlined hospital systems and care processes.
This may include hiring assistants, rounding staff, and/or mid-level providers to incrementally increase the number of patients the group can see per physician. These are not without risks and barriers, and many groups currently are wrestling with how to best utilize and integrate these providers in a cost-effective manner. However, I think it is likely these components will provide the future scaffolding to allow groups to care for ever increasing numbers of patients.
Prime the Pump
Perhaps, most importantly, we need to develop ways to attract more medical students and residents to hospitalist careers. This will be challenging and falls squarely to my academic hospital medicine colleagues, as our learners see us as the yardstick for a hospitalist career. Unfortunately, the growth trend in academic hospital medicine has been in non-teaching or uncovered services. These jobs often are an academic necropolis, with the providers routinely being overworked and devalued by their institution.
As a result, academic hospitalist positions often times are filled by recent residency graduates awaiting a fellowship. These “R4” or “pretending” positions provide very rickety underpinnings from which to build the foundation of hospital medicine. Don’t for an instant think this goes unnoticed by our student and resident colleagues who choose their career based on the role models they see early in their training.
It is essential hospital medicine develops truly sustainable academic careers replete with opportunities to fulfill the inimitable tenets of academic medicine—teaching and scholarly work. For these reasons, SHM has partnered with the Society of General Internal Medicine and the Association of Chiefs in General Internal Medicine to develop the Academic Hospitalist Academy. The four-day academy premiers next fall. Its goal is to enable academic hospitalists to become exceptional educators, institutional leaders, and successful scholars—the exact type of role models that will attract the best and the brightest to the field of hospital medicine for generations to come. TH
Dr. Glasheen is associate professor of medicine at the University of Colorado Denver, where he serves as director of the hospital medicine program and the hospitalist training program, and as associate program director of the Internal Medicine Residency Program.
References
1. Hauer KE, Durning SJ, Kernan WN, et al. Factors associated with medical students’ career choices regarding internal medicine. JAMA. 2008;300:1154-1156,1164.
2. Hauer KE, Fagan MJ, Kernan W, Mintz M, Durning SJ. Internal medicine clerkship directors’ perceptions about student interest in internal medicine careers. J Gen Intern Med. 2008;1101-1104.
3. McDonald FS, West CP, Popkave C, Kolars JC. Educational debt and reported career plans among internal medicine residents. Ann Inter Med. 2008;149:416-420.
4. Glasheen JJ, Epstein KR, Siegal E, Kutner JS, Prochazka AV. The spectrum of community based hospitalist practice: A call to tailor internal medicine residency training. Arch Intern Med. 2007;167:727-729.
Welcome, President Obama

On Jan. 20, Barack Obama will become the 44th President of the United States against the backdrop of two foreign wars and one of the worst economic crisis since the Great Depression. U.S. business icons are failing; unemployment is at its highest rate in decades; housing values are plummeting as foreclosures and credit tightening make the American dream of home ownership more of a nightmare than a reality. Personal net worth is shrinking and the financial ability for some to even consider retirement is fading away.
Yet, at the same time there is dire need to structure and re-invent many institutions. Our infrastructure of roads, buildings, and bridges has been neglected and are in disrepair. Our education system is not preparing our young people for a global market, a place where the best and brightest may now be found in India, Japan, Europe, and China, as much as in the U.S.
And healthcare, my oh my healthcare, needs more than just a face lift; it needs an overhaul, a righting of the ship, and a truing up of its direction for the future.
Yet, fixing healthcare is very much intermingled with the rest of our economic woes. With company failures and layoffs comes the loss of health benefits, and, ironically, more time available to seek healthcare. Even those with jobs may find themselves with no insurance or inadequate coverage. It is not unlikely the current 47 million uninsured will soon be joined by another 15 million uninsured or underinsured, made up mostly of middle-class workers who have never before been faced with the prospect of financial ruin if they or their family members take ill. Never before has the middle class been faced with the choice between the right care they need and losing all of their net worth, including their homes.
President Obama could be another Jimmy Carter, an intelligent, well-meaning man whose presidency was disabled by 13% inflation, gas lines, and being held hostage in Iran. Or he will be the next FDR, a president who remade America for generations to come, with Social Security, work programs, and a new economy.
Fortunately, Obama’s team is loaded with thought leaders who come with a strong interest in reforming and rebuilding healthcare. Tom Daschle, the new secretary of Health and Human Services has a long history of healthcare policy and can work well to move things through a Democratic Congress. Obama’s Director of the Office of Management and Budget, Peter Orszag, is a well-regarded health policy expert. Melody Barnes, his domestic policy advisor, was the executive director of a think tank, the Center for American Progress, which has developed a detailed road map for change in our healthcare system, involving some of the best minds in American healthcare, such as Don Berwick, David Blumenthal and Paul Ginsburg. You can read the center’s 120-page report at http://www.americanprogress.org/issues/2008/10/health_care_delivery.html.
While this group appears primed for a revolution, rather than just rearranging the deck chairs on the Titanic, at this point Obama and his team have been more visionary than specific. Here is my best guess as to what some of the aspects of a new healthcare approach might be. (And most of these changes are of more than a passing interest to hospital medicine.)
Less Uninsured
There is no doubt it is unsustainable for a first-class society to have so many citizens without access and payment for healthcare. Although this may start with covering all children and offering people affordable insurance not specifically tied to their employment, the U.S. must move closer to something that looks like Medicare for all. Along the way, this will lead to more regulation of insurance companies to raise the percentage of the premium dollar that actually goes for medical care (75% is just not tolerable). Do not expect the $2.1 trillion healthcare pie to expand, so doctors and hospitals will need to be more efficient and effective as they supply better, more accountable healthcare to a larger patient population. Some may perceive this as more work for less pay.
This expansion of coverage, however, cannot be a broadening of Medicaid and its dysfunctional payment system. It cannot be a single-payer, Canadian system, which creates irrational rationing and does nothing to address the need to bolster primary care. We need a new health paradigm where performance and access mean just as much as new technology.
More Primary Care
What good is insurance if you have no access? Just ask the citizens of Massachusetts, where the newly insured can’t find a primary care physician (PCP). One “benefit” of the economic downturn and stock market tumble is late-career PCPs can’t afford to retire. But primary care is in shambles, and throwing a few more dollars at PCPs or creating a “home” won’t make being a PCP more attractive to medical students. For a more revolutionary approach, check out the New England Journal of Medicine video roundtable (www.nejm.org/perspective/primary-care-video/?query=TOC) to hear of a “new” primary care model, which is more centered on population management than a series of 10-minute visits. Hospitalists, as much as anyone, need a strong, sustainable primary care partnership, if we are to tackle the difficult problems inside the hospital.
Value-Based Purchasing
This new payment model is being pushed by Sen. Max Baucas (D-Mont.), the powerful chairman of the Senate Finance Committee. Value-based purchasing (VBP) basically moves us away from just paying for care by the unit of the visit or the procedure, regardless of medical necessity or outcome. This plays into the strengths of hospital medicine where performance and communication are valued. Paying more when the customer gets more is an American value, which, at times, has been overlooked in American medicine. It is time we brought VBP into the healthcare equation.
Bundled Payment
All politics is local, and in many ways all healthcare is local. By changing the payment for hospital care to a composite fee for the facility and all the health professionals, an opportunity exists for the physicians and the hospital at a local level to creatively reward work, performance, outcomes, and patient satisfaction. This is not giving the hospital the entire fee, but more relying on a physician-hospital organization (as currently exists in many places in the country) deciding how to allocate resources. Once again, hospitalists are managing up to 80% of inpatients at some hospitals, so we are right in the middle of a new distribution of compensation for inpatient care.
Transitions of Care
It is time to look at our healthcare system from the patient’s point of view. It is not enough to perform the surgery perfectly or order the correct treatment. Patients need to be involved in their care, to clearly understand what medications they should be taking, to know who is responsible to answer their questions, and what their expectations for recovery should be. It also is an opportunity to prevent unnecessary visits back to the emergency room or readmissions to the hospital. The current, 15% readmission rate within 30 days for Medicare patients points to how broken the system is. Patients deserve accountability, transparency and clarity on their terms.
Once again, SHM and hospitalists have taken the lead in this issue. With a grant from the Hartford Foundation, SHM already has demonstrated practical strategies to improve the discharge process.
What It Means to You
In calmer, less-chaotic times, I suspect there would be calls for tinkering around the edges. But these are dangerous times that call for decisive, some might say, disruptive change. A new, patient-centered healthcare system based on access, inclusiveness, performance, communication, and safety is coming. There will be those who feel less well-off in the new order—insurance companies, some physicians and some hospitals—but there will be many who feel, for the first time, that the system is equitable, open, and responsive to their needs. The latter group includes U.S. business, some physicians (e.g. hospitalists), some hospitals, and, most importantly, the American people.
Hospitalists are uniquely positioned to shoulder the full force of this change. Hospitalists now practice in most hospitals throughout the country, and they are right at the intersection of the patient and the illness, thrown into the caldron of change along with allied health and our institutions. We must embrace change and we mold it into a new system of care, a system that benefits our patients based on data, evidence, and compassion. There is an outcome where hospitalists and our patients both win; it is the future, and now it seems closer to reality. TH
Dr. Wellikson is the CEO of SHM
On Jan. 20, Barack Obama will become the 44th President of the United States against the backdrop of two foreign wars and one of the worst economic crisis since the Great Depression. U.S. business icons are failing; unemployment is at its highest rate in decades; housing values are plummeting as foreclosures and credit tightening make the American dream of home ownership more of a nightmare than a reality. Personal net worth is shrinking and the financial ability for some to even consider retirement is fading away.
Yet, at the same time there is dire need to structure and re-invent many institutions. Our infrastructure of roads, buildings, and bridges has been neglected and are in disrepair. Our education system is not preparing our young people for a global market, a place where the best and brightest may now be found in India, Japan, Europe, and China, as much as in the U.S.
And healthcare, my oh my healthcare, needs more than just a face lift; it needs an overhaul, a righting of the ship, and a truing up of its direction for the future.
Yet, fixing healthcare is very much intermingled with the rest of our economic woes. With company failures and layoffs comes the loss of health benefits, and, ironically, more time available to seek healthcare. Even those with jobs may find themselves with no insurance or inadequate coverage. It is not unlikely the current 47 million uninsured will soon be joined by another 15 million uninsured or underinsured, made up mostly of middle-class workers who have never before been faced with the prospect of financial ruin if they or their family members take ill. Never before has the middle class been faced with the choice between the right care they need and losing all of their net worth, including their homes.
President Obama could be another Jimmy Carter, an intelligent, well-meaning man whose presidency was disabled by 13% inflation, gas lines, and being held hostage in Iran. Or he will be the next FDR, a president who remade America for generations to come, with Social Security, work programs, and a new economy.
Fortunately, Obama’s team is loaded with thought leaders who come with a strong interest in reforming and rebuilding healthcare. Tom Daschle, the new secretary of Health and Human Services has a long history of healthcare policy and can work well to move things through a Democratic Congress. Obama’s Director of the Office of Management and Budget, Peter Orszag, is a well-regarded health policy expert. Melody Barnes, his domestic policy advisor, was the executive director of a think tank, the Center for American Progress, which has developed a detailed road map for change in our healthcare system, involving some of the best minds in American healthcare, such as Don Berwick, David Blumenthal and Paul Ginsburg. You can read the center’s 120-page report at http://www.americanprogress.org/issues/2008/10/health_care_delivery.html.
While this group appears primed for a revolution, rather than just rearranging the deck chairs on the Titanic, at this point Obama and his team have been more visionary than specific. Here is my best guess as to what some of the aspects of a new healthcare approach might be. (And most of these changes are of more than a passing interest to hospital medicine.)
Less Uninsured
There is no doubt it is unsustainable for a first-class society to have so many citizens without access and payment for healthcare. Although this may start with covering all children and offering people affordable insurance not specifically tied to their employment, the U.S. must move closer to something that looks like Medicare for all. Along the way, this will lead to more regulation of insurance companies to raise the percentage of the premium dollar that actually goes for medical care (75% is just not tolerable). Do not expect the $2.1 trillion healthcare pie to expand, so doctors and hospitals will need to be more efficient and effective as they supply better, more accountable healthcare to a larger patient population. Some may perceive this as more work for less pay.
This expansion of coverage, however, cannot be a broadening of Medicaid and its dysfunctional payment system. It cannot be a single-payer, Canadian system, which creates irrational rationing and does nothing to address the need to bolster primary care. We need a new health paradigm where performance and access mean just as much as new technology.
More Primary Care
What good is insurance if you have no access? Just ask the citizens of Massachusetts, where the newly insured can’t find a primary care physician (PCP). One “benefit” of the economic downturn and stock market tumble is late-career PCPs can’t afford to retire. But primary care is in shambles, and throwing a few more dollars at PCPs or creating a “home” won’t make being a PCP more attractive to medical students. For a more revolutionary approach, check out the New England Journal of Medicine video roundtable (www.nejm.org/perspective/primary-care-video/?query=TOC) to hear of a “new” primary care model, which is more centered on population management than a series of 10-minute visits. Hospitalists, as much as anyone, need a strong, sustainable primary care partnership, if we are to tackle the difficult problems inside the hospital.
Value-Based Purchasing
This new payment model is being pushed by Sen. Max Baucas (D-Mont.), the powerful chairman of the Senate Finance Committee. Value-based purchasing (VBP) basically moves us away from just paying for care by the unit of the visit or the procedure, regardless of medical necessity or outcome. This plays into the strengths of hospital medicine where performance and communication are valued. Paying more when the customer gets more is an American value, which, at times, has been overlooked in American medicine. It is time we brought VBP into the healthcare equation.
Bundled Payment
All politics is local, and in many ways all healthcare is local. By changing the payment for hospital care to a composite fee for the facility and all the health professionals, an opportunity exists for the physicians and the hospital at a local level to creatively reward work, performance, outcomes, and patient satisfaction. This is not giving the hospital the entire fee, but more relying on a physician-hospital organization (as currently exists in many places in the country) deciding how to allocate resources. Once again, hospitalists are managing up to 80% of inpatients at some hospitals, so we are right in the middle of a new distribution of compensation for inpatient care.
Transitions of Care
It is time to look at our healthcare system from the patient’s point of view. It is not enough to perform the surgery perfectly or order the correct treatment. Patients need to be involved in their care, to clearly understand what medications they should be taking, to know who is responsible to answer their questions, and what their expectations for recovery should be. It also is an opportunity to prevent unnecessary visits back to the emergency room or readmissions to the hospital. The current, 15% readmission rate within 30 days for Medicare patients points to how broken the system is. Patients deserve accountability, transparency and clarity on their terms.
Once again, SHM and hospitalists have taken the lead in this issue. With a grant from the Hartford Foundation, SHM already has demonstrated practical strategies to improve the discharge process.
What It Means to You
In calmer, less-chaotic times, I suspect there would be calls for tinkering around the edges. But these are dangerous times that call for decisive, some might say, disruptive change. A new, patient-centered healthcare system based on access, inclusiveness, performance, communication, and safety is coming. There will be those who feel less well-off in the new order—insurance companies, some physicians and some hospitals—but there will be many who feel, for the first time, that the system is equitable, open, and responsive to their needs. The latter group includes U.S. business, some physicians (e.g. hospitalists), some hospitals, and, most importantly, the American people.
Hospitalists are uniquely positioned to shoulder the full force of this change. Hospitalists now practice in most hospitals throughout the country, and they are right at the intersection of the patient and the illness, thrown into the caldron of change along with allied health and our institutions. We must embrace change and we mold it into a new system of care, a system that benefits our patients based on data, evidence, and compassion. There is an outcome where hospitalists and our patients both win; it is the future, and now it seems closer to reality. TH
Dr. Wellikson is the CEO of SHM
On Jan. 20, Barack Obama will become the 44th President of the United States against the backdrop of two foreign wars and one of the worst economic crisis since the Great Depression. U.S. business icons are failing; unemployment is at its highest rate in decades; housing values are plummeting as foreclosures and credit tightening make the American dream of home ownership more of a nightmare than a reality. Personal net worth is shrinking and the financial ability for some to even consider retirement is fading away.
Yet, at the same time there is dire need to structure and re-invent many institutions. Our infrastructure of roads, buildings, and bridges has been neglected and are in disrepair. Our education system is not preparing our young people for a global market, a place where the best and brightest may now be found in India, Japan, Europe, and China, as much as in the U.S.
And healthcare, my oh my healthcare, needs more than just a face lift; it needs an overhaul, a righting of the ship, and a truing up of its direction for the future.
Yet, fixing healthcare is very much intermingled with the rest of our economic woes. With company failures and layoffs comes the loss of health benefits, and, ironically, more time available to seek healthcare. Even those with jobs may find themselves with no insurance or inadequate coverage. It is not unlikely the current 47 million uninsured will soon be joined by another 15 million uninsured or underinsured, made up mostly of middle-class workers who have never before been faced with the prospect of financial ruin if they or their family members take ill. Never before has the middle class been faced with the choice between the right care they need and losing all of their net worth, including their homes.
President Obama could be another Jimmy Carter, an intelligent, well-meaning man whose presidency was disabled by 13% inflation, gas lines, and being held hostage in Iran. Or he will be the next FDR, a president who remade America for generations to come, with Social Security, work programs, and a new economy.
Fortunately, Obama’s team is loaded with thought leaders who come with a strong interest in reforming and rebuilding healthcare. Tom Daschle, the new secretary of Health and Human Services has a long history of healthcare policy and can work well to move things through a Democratic Congress. Obama’s Director of the Office of Management and Budget, Peter Orszag, is a well-regarded health policy expert. Melody Barnes, his domestic policy advisor, was the executive director of a think tank, the Center for American Progress, which has developed a detailed road map for change in our healthcare system, involving some of the best minds in American healthcare, such as Don Berwick, David Blumenthal and Paul Ginsburg. You can read the center’s 120-page report at http://www.americanprogress.org/issues/2008/10/health_care_delivery.html.
While this group appears primed for a revolution, rather than just rearranging the deck chairs on the Titanic, at this point Obama and his team have been more visionary than specific. Here is my best guess as to what some of the aspects of a new healthcare approach might be. (And most of these changes are of more than a passing interest to hospital medicine.)
Less Uninsured
There is no doubt it is unsustainable for a first-class society to have so many citizens without access and payment for healthcare. Although this may start with covering all children and offering people affordable insurance not specifically tied to their employment, the U.S. must move closer to something that looks like Medicare for all. Along the way, this will lead to more regulation of insurance companies to raise the percentage of the premium dollar that actually goes for medical care (75% is just not tolerable). Do not expect the $2.1 trillion healthcare pie to expand, so doctors and hospitals will need to be more efficient and effective as they supply better, more accountable healthcare to a larger patient population. Some may perceive this as more work for less pay.
This expansion of coverage, however, cannot be a broadening of Medicaid and its dysfunctional payment system. It cannot be a single-payer, Canadian system, which creates irrational rationing and does nothing to address the need to bolster primary care. We need a new health paradigm where performance and access mean just as much as new technology.
More Primary Care
What good is insurance if you have no access? Just ask the citizens of Massachusetts, where the newly insured can’t find a primary care physician (PCP). One “benefit” of the economic downturn and stock market tumble is late-career PCPs can’t afford to retire. But primary care is in shambles, and throwing a few more dollars at PCPs or creating a “home” won’t make being a PCP more attractive to medical students. For a more revolutionary approach, check out the New England Journal of Medicine video roundtable (www.nejm.org/perspective/primary-care-video/?query=TOC) to hear of a “new” primary care model, which is more centered on population management than a series of 10-minute visits. Hospitalists, as much as anyone, need a strong, sustainable primary care partnership, if we are to tackle the difficult problems inside the hospital.
Value-Based Purchasing
This new payment model is being pushed by Sen. Max Baucas (D-Mont.), the powerful chairman of the Senate Finance Committee. Value-based purchasing (VBP) basically moves us away from just paying for care by the unit of the visit or the procedure, regardless of medical necessity or outcome. This plays into the strengths of hospital medicine where performance and communication are valued. Paying more when the customer gets more is an American value, which, at times, has been overlooked in American medicine. It is time we brought VBP into the healthcare equation.
Bundled Payment
All politics is local, and in many ways all healthcare is local. By changing the payment for hospital care to a composite fee for the facility and all the health professionals, an opportunity exists for the physicians and the hospital at a local level to creatively reward work, performance, outcomes, and patient satisfaction. This is not giving the hospital the entire fee, but more relying on a physician-hospital organization (as currently exists in many places in the country) deciding how to allocate resources. Once again, hospitalists are managing up to 80% of inpatients at some hospitals, so we are right in the middle of a new distribution of compensation for inpatient care.
Transitions of Care
It is time to look at our healthcare system from the patient’s point of view. It is not enough to perform the surgery perfectly or order the correct treatment. Patients need to be involved in their care, to clearly understand what medications they should be taking, to know who is responsible to answer their questions, and what their expectations for recovery should be. It also is an opportunity to prevent unnecessary visits back to the emergency room or readmissions to the hospital. The current, 15% readmission rate within 30 days for Medicare patients points to how broken the system is. Patients deserve accountability, transparency and clarity on their terms.
Once again, SHM and hospitalists have taken the lead in this issue. With a grant from the Hartford Foundation, SHM already has demonstrated practical strategies to improve the discharge process.
What It Means to You
In calmer, less-chaotic times, I suspect there would be calls for tinkering around the edges. But these are dangerous times that call for decisive, some might say, disruptive change. A new, patient-centered healthcare system based on access, inclusiveness, performance, communication, and safety is coming. There will be those who feel less well-off in the new order—insurance companies, some physicians and some hospitals—but there will be many who feel, for the first time, that the system is equitable, open, and responsive to their needs. The latter group includes U.S. business, some physicians (e.g. hospitalists), some hospitals, and, most importantly, the American people.
Hospitalists are uniquely positioned to shoulder the full force of this change. Hospitalists now practice in most hospitals throughout the country, and they are right at the intersection of the patient and the illness, thrown into the caldron of change along with allied health and our institutions. We must embrace change and we mold it into a new system of care, a system that benefits our patients based on data, evidence, and compassion. There is an outcome where hospitalists and our patients both win; it is the future, and now it seems closer to reality. TH
Dr. Wellikson is the CEO of SHM
It’s Good to Be Country

Think a big hospital is where it’s at? Not according to Randy Ferrance, DC, MD, a hospitalist at Riverside Tappahanock Hospital, a 67-bed facility in rural Tappahannock, Va. The community is home to 2,172 residents and located about an hour east of Richmond, just up river from the Chesapeake Bay. Dr. Ferrance, a former chiropractor who has been practicing as a hospitalist at Riverside since 2002, recently spoke with The Hospitalist about why he enjoys the rural setting.
How is Riverside Tappahanock different from other hospitalist groups you’ve worked at?
Answer: The thing I like about it is I get to wear a lot of different hats. We don’t have intensivists; we manage our own ventilators and do our own critical care. And we’re also limited by the number of specialists we have, so of course, anything that is too difficult for us to do we transfer out. I don’t manage consultants, which seems like what hospitalists at a lot of big hospitals do. I’m often wondering what those hospitalists are left doing. Here we have cardiologists available to us, and as far as other specialists go, we have one gastroenterologist and a part-time nephrologist. I like the fact that I’m actually treating and not just stepping back and watching others treat. I especially like the ICU. This way I get to do critical care, and I think do it fairly well.
What are the challenges at a rural hospital?
A: A number of people just assume that, since we are just a small hospital, we can’t be giving good care. They come through the doors and they immediately want us to transfer them to a bigger hospital.
Is there a need for more rural hospitalists?
A: There have been times in the past when we’ve had trouble getting people [recruits] to look at us just because of the location, although I think we’re in a great location. We’re not far from Richmond, not far from good things to do.
Is there a solution to the recruitment problem?
A: The bottom line is we need more primary care physicians. We’re pretty selective and we’ve managed to do well despite that.
How many patients, on average, do you see?
A: We average about five admissions a day. We tend to follow about eight patients at a time. We don’t really do shifts. We take call a quarter of the time, doing admissions for a 24-hour stretch, averaging seven or eight calls in a month. Then we round on our post call days, as well, and the days in between. On average, we take every third day call, with a week off each month. We work 90 hours a week—pretty awful hours. So this is clearly a drawback. There are only four of us here, so if one of us were taken ill, we’d either have to get a [temporary doctor] or pick up the slack.
What are the other drawbacks to a rural hospital?
A: Our denominators are so small that, if we miss aspirin on arrival for one patient, it can pull us from first to the fourth in quality ratings. Everything has to be perfect. We can’t make any omissions. I think it certainly adds to perception. People in small towns talk a lot, and what people talk about are things that did not go well. They don’t talk about things that did go well.
What advice do you have for those considering a position at a rural hospital?
A: You have to be willing to work more than you would at a larger hospital, but I think you get to do more, which is more rewarding from my point of view.
What can rural hospitalists teach other hospitalists?
A: We probably can teach workload management a bit better. I think we can also talk about quality referral patterns. The things we need to do to make sure our quality numbers are good are probably a lot more stringent because our capture needs to be better. TH
Think a big hospital is where it’s at? Not according to Randy Ferrance, DC, MD, a hospitalist at Riverside Tappahanock Hospital, a 67-bed facility in rural Tappahannock, Va. The community is home to 2,172 residents and located about an hour east of Richmond, just up river from the Chesapeake Bay. Dr. Ferrance, a former chiropractor who has been practicing as a hospitalist at Riverside since 2002, recently spoke with The Hospitalist about why he enjoys the rural setting.
How is Riverside Tappahanock different from other hospitalist groups you’ve worked at?
Answer: The thing I like about it is I get to wear a lot of different hats. We don’t have intensivists; we manage our own ventilators and do our own critical care. And we’re also limited by the number of specialists we have, so of course, anything that is too difficult for us to do we transfer out. I don’t manage consultants, which seems like what hospitalists at a lot of big hospitals do. I’m often wondering what those hospitalists are left doing. Here we have cardiologists available to us, and as far as other specialists go, we have one gastroenterologist and a part-time nephrologist. I like the fact that I’m actually treating and not just stepping back and watching others treat. I especially like the ICU. This way I get to do critical care, and I think do it fairly well.
What are the challenges at a rural hospital?
A: A number of people just assume that, since we are just a small hospital, we can’t be giving good care. They come through the doors and they immediately want us to transfer them to a bigger hospital.
Is there a need for more rural hospitalists?
A: There have been times in the past when we’ve had trouble getting people [recruits] to look at us just because of the location, although I think we’re in a great location. We’re not far from Richmond, not far from good things to do.
Is there a solution to the recruitment problem?
A: The bottom line is we need more primary care physicians. We’re pretty selective and we’ve managed to do well despite that.
How many patients, on average, do you see?
A: We average about five admissions a day. We tend to follow about eight patients at a time. We don’t really do shifts. We take call a quarter of the time, doing admissions for a 24-hour stretch, averaging seven or eight calls in a month. Then we round on our post call days, as well, and the days in between. On average, we take every third day call, with a week off each month. We work 90 hours a week—pretty awful hours. So this is clearly a drawback. There are only four of us here, so if one of us were taken ill, we’d either have to get a [temporary doctor] or pick up the slack.
What are the other drawbacks to a rural hospital?
A: Our denominators are so small that, if we miss aspirin on arrival for one patient, it can pull us from first to the fourth in quality ratings. Everything has to be perfect. We can’t make any omissions. I think it certainly adds to perception. People in small towns talk a lot, and what people talk about are things that did not go well. They don’t talk about things that did go well.
What advice do you have for those considering a position at a rural hospital?
A: You have to be willing to work more than you would at a larger hospital, but I think you get to do more, which is more rewarding from my point of view.
What can rural hospitalists teach other hospitalists?
A: We probably can teach workload management a bit better. I think we can also talk about quality referral patterns. The things we need to do to make sure our quality numbers are good are probably a lot more stringent because our capture needs to be better. TH
Think a big hospital is where it’s at? Not according to Randy Ferrance, DC, MD, a hospitalist at Riverside Tappahanock Hospital, a 67-bed facility in rural Tappahannock, Va. The community is home to 2,172 residents and located about an hour east of Richmond, just up river from the Chesapeake Bay. Dr. Ferrance, a former chiropractor who has been practicing as a hospitalist at Riverside since 2002, recently spoke with The Hospitalist about why he enjoys the rural setting.
How is Riverside Tappahanock different from other hospitalist groups you’ve worked at?
Answer: The thing I like about it is I get to wear a lot of different hats. We don’t have intensivists; we manage our own ventilators and do our own critical care. And we’re also limited by the number of specialists we have, so of course, anything that is too difficult for us to do we transfer out. I don’t manage consultants, which seems like what hospitalists at a lot of big hospitals do. I’m often wondering what those hospitalists are left doing. Here we have cardiologists available to us, and as far as other specialists go, we have one gastroenterologist and a part-time nephrologist. I like the fact that I’m actually treating and not just stepping back and watching others treat. I especially like the ICU. This way I get to do critical care, and I think do it fairly well.
What are the challenges at a rural hospital?
A: A number of people just assume that, since we are just a small hospital, we can’t be giving good care. They come through the doors and they immediately want us to transfer them to a bigger hospital.
Is there a need for more rural hospitalists?
A: There have been times in the past when we’ve had trouble getting people [recruits] to look at us just because of the location, although I think we’re in a great location. We’re not far from Richmond, not far from good things to do.
Is there a solution to the recruitment problem?
A: The bottom line is we need more primary care physicians. We’re pretty selective and we’ve managed to do well despite that.
How many patients, on average, do you see?
A: We average about five admissions a day. We tend to follow about eight patients at a time. We don’t really do shifts. We take call a quarter of the time, doing admissions for a 24-hour stretch, averaging seven or eight calls in a month. Then we round on our post call days, as well, and the days in between. On average, we take every third day call, with a week off each month. We work 90 hours a week—pretty awful hours. So this is clearly a drawback. There are only four of us here, so if one of us were taken ill, we’d either have to get a [temporary doctor] or pick up the slack.
What are the other drawbacks to a rural hospital?
A: Our denominators are so small that, if we miss aspirin on arrival for one patient, it can pull us from first to the fourth in quality ratings. Everything has to be perfect. We can’t make any omissions. I think it certainly adds to perception. People in small towns talk a lot, and what people talk about are things that did not go well. They don’t talk about things that did go well.
What advice do you have for those considering a position at a rural hospital?
A: You have to be willing to work more than you would at a larger hospital, but I think you get to do more, which is more rewarding from my point of view.
What can rural hospitalists teach other hospitalists?
A: We probably can teach workload management a bit better. I think we can also talk about quality referral patterns. The things we need to do to make sure our quality numbers are good are probably a lot more stringent because our capture needs to be better. TH
When should a hospitalized patient be transfused?
Case
A 65-year-old male nursing home resident is sent to the emergency room with a productive cough, fever, and low blood pressure, and is diagnosed with community-acquired pneumonia. He has a history of tobacco abuse, hypertension, and a right middle cerebral artery stroke. His admission labs show a hemoglobin level of 9.0 g/dL. The day after admission his hypotension has resolved and he reports feeling much better after two liters of intravenous fluids and antibiotics. However, his hemoglobin level is 7.9 g/dL. There is no evidence of bleeding. Should this hospitalized patient be transfused?
Overview
When to give a red blood cell transfusion is a clinical question commonly encountered by hospitalists. Individuals with acute blood loss, chronic blood loss, anemia of chronic disease, and hemolytic anemia often are given transfusions. Hospitalists serving as consultants may be asked when to transfuse patients perioperatively.
It is estimated up to 25% of the red blood cells transfused in the U.S. are inappropriate.1-4 Many physicians transfuse based on a number, rather than on objective findings. Overuse is common because of the wide availability of red blood cells, the belief complications are infrequent, and an unfounded fear of adverse outcomes if a patient is not transfused.
Tachycardia, low blood pressure, and declining oxygen saturations are signs clinicians can use when making the decision to transfuse. Electrocardiographic changes associated with tissue hypoxia can occur at a hemoglobin level <5 g/dL in healthy adults. Studies show mortality and morbidity increase rapidly at levels <5.0 to 6.0 g/dL.5 Currently, no diagnostic serological test exists for tissue hypoxia, which is the physiologic reason to give red blood cells.
Red blood cell transfusion can be a life-saving therapy; however, it is not a benign intervention. It is estimated 10% of transfusion reactions will have some adverse event.6 Red blood cell use exposes patients to hemolytic transfusion reactions, infections, and transfusion related acute lung injury.7,8 Additionally, unnecessary economic expenses are incurred and a scarce resource is diverted from other patients.
Hospitalists should be able to describe the indications for red blood cell transfusion and understand the evidence for and against its use. Physicians who appreciate the risks and benefits of red blood cell use tend to transfuse less blood that those who less informed. 9, 10
Review of the Data
General outcomes: Despite the long history of red blood cell transfusion, which dates back to 1818, when James Blundell successfully saved a woman exsanguinating from a postpartum hemorrhage, little evidence has been accumulated for its appropriate use. In the 1980s, the discovery of the human immunodeficiency virus sparked blood product safety concerns. This stimulated research and a debate over red blood cell transfusion practices, with a growing body of literature unsupportive of transfusion for an arbitrary trigger, for example the “10/30 rule,” which referred to 10 g/dL hemoglobin or hematocrit of 30%.9
Observational studies have raised concerns by linking morbidity and mortality to red blood cell use. Among 1,958 surgical patients who refused blood transfusion on religious grounds, there was an increase in mortality when hemoglobin levels were <6.0 g/dL. Hemoglobin levels higher than 7.0 g/dL showed no increased mortality.11 A recent comprehensive review included 272,596 surgical, trauma, and ICU patients in 45 observational studies. The review included studies with end points, including mortality, infections, multiorgan dysfunction syndrome, and acute respiratory distress syndrome, and concluded transfusions are associated with a higher risk of morbidity and mortality.12 (see Figure 1, p. 20)
Higher rates of infection associated with transfusions occurred in patients with post-operative trauma, acute injuries, gastrointestinal cancer undergoing surgery, coronary bypass surgery, hip surgery, burns, critical illness, and patients requiring ventilation. (see Figure 2, p. 21)12 The increased infection risk likely is due to the transient depression of the immune system induced by red blood cell transfusion. Prolonged hospital stays in postoperative colorectal surgery patients and ICU patients have been associated with transfusions.13
A meta-analysis of the few randomized controlled trials favors the restrictive use of red blood cells. The preponderance of the evidence comes from the Transfusion Requirements in Critical Care (TRICC) trial.14 This randomized control trial in critically ill medical and surgical patients demonstrated a restrictive strategy (transfusion trigger of <7.0 g/dL) and was as effective as a liberal transfusion strategy (transfusion trigger <10.0 g/dL). (see Figure 3, p. 22) Indeed, patients in the restrictive arm of the trial, who were less ill and under age 55 had a lower mortality rate than those who were transfused liberally.15 To date, there are no hospital-based randomized control trials that evaluate outcomes of anemic non-ICU medical patients.
This evidence has created a growing consensus that a restrictive use of blood results in improved patient outcomes. In patients without cardiovascular disease the evidence suggests most patients tolerate a hemoglobin level of 7.0 g/dL.5
Cardiac Patients
Experimental and clinical evidence suggests patients with cardiovascular disease are less tolerant of anemia. Patients with coronary disease are more likely to have adverse outcomes than those without coronary disease, if they do not have a red blood cell transfusion.11,16
The myocardium has a higher oxygen extraction ratio compared to the tissue oxygen extraction ratio, making it more sensitive to anemia.17,18 The presence of cardiac disease may require a higher threshold to transfuse blood; however, the precise recommended threshold remains controversial. A restrictive red blood cell transfusion strategy (maintaining the hemoglobin between 7.0 g/dL and 9.0 g/dL) appeared to be safe in most critically ill patients with cardiovascular disease.14
The data is more conflicting for patients with an acute coronary syndrome (ACS). Some studies have found increased mortality and another concluded ACS decreased with red blood cell use.19-21 Further research is needed to determine when red blood cells should be given to patients with coronary disease.
Gastrointestinal Bleeding
The decision to transfuse for gastrointestinal (GI) bleeding takes into account the site and etiology of the bleeding, availability of treatments, and risk of continued bleeding. Once the blood loss is controlled, a decision must be made on how to treat the anemia. Currently, no studies have looked at outcomes for patients who did and did not receive blood for an acute or chronic GI bleed.
Additionally, no studies have been conducted to delineate when to transfuse patients with chronic GI blood loss. Studies of patients with an acute GI bleed and cardiovascular disease have shown an increase in mortality, but it is unknown if the use of specific transfusion triggers affects outcomes in this group.

In patients with GI bleeding, experts feel the use of red blood cells should be guided by available evidence. For patients without cardiac disease, red blood cell transfusion is rarely required following definitive treatment and cessation of blood loss unless the hemoglobin is <7.0 g/dL.22
Back to the Case
The patient described in our case should not be transfused unless he has clinical signs or symptoms of tissue hypoxemia. An appropriate workup for his anemia should be initiated and, if an etiology identified, definitive treatment or intervention applied.
Bottom Line
Unless there are clinical signs of tissue hypoxia, symptomatic anemia, or a hemoglobin of <7.0 g/dL, red blood cell transfusion is not recommended, unless the patient has active ACS or significant underlying coronary disease. TH
Dr. Dressler is associate program director, assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta. Dr. VanderEnde is assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta.
References
1. Welch HG, Meehan KR, Goodnough LT. Prudent strategies for elective red blood cell transfusion. Ann Intern Med. 1992;116(5):393-402.
2. Tartter PI, Barron DM. Unnecessary blood transfusions in elective colorectal cancer surgery. Transfusion. 1985;25(2):113-115.
3. Saxena S, Weiner JM, Rabinowitz A, Fridey J, Shulman IA, Carmel R. Transfusion practice in medical patients. Arch Int Med. 1993;153(22):2575-80.
4. Palermo G, Bove J, Katz AJ. Patterns of blood use in Connecticut. Transfusion. 1980;20(6):704-710.
5. Carson JL, Reynolds RC. In search of the transfusion threshold. Hematology. 2005;10(Suppl 1):86-88.
6. Walker RH. Special report: transfusion risks. Am J Clin Pathol. 1987;88(3):374-378.
7. Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infections. N Engl J Med. 2006;355(13):1303-1305.
8. Spiess BD. Risks of transfusion: outcome focus. Transfusion. 2004;44(Suppl 12):4S-14S.
9. Salem-Schatz SR, Avorn J, Soumerai SB. Influence of clinical knowledge, organizational context, and practice style on transfusion decision-making. JAMA. 1990;264(4):476-483.
10. Wilson K, MacDougall L, Fergusson D, Graham I, Tinmouth A, Hebert PC. The effectiveness of interventions to reduce physician’s levels of inappropriate transfusion: what can be learned from a systematic review of the literature. Transfusion. 2002;42(9):1224-1229.
11. Carson JL, Duff A, Poses RM, et al. Effect of anemia and cardiovascular disease on surgical mortality and morbidity. Lancet. 1996;348(9034):1055-1060.
12. Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008;36(9):2667-2674.
13. Raghavan M, Marik PE. Anemia, allogenic blood transfusion, and immunomodulation in the critically ill. Chest. 2005;127(1):295-307.
14. Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion requirements in critical care investigators, Canadian critical care trials group. N Engl J Med. 1999;340(6):409-417.
15. Carson JL, Hill S, Carless P, Hebert P, Henry D. Transfusion triggers: a systematic review of the literature. Transfus Med Rev. 2002;16(3):187-199.
16. Sabatine MS, Morrow DA, Giugliano RP, et al. Association of hemoglobin levels with clinical outcomes in acute coronary syndromes. Circulation. 2005; 111(16):2042-2049.
17. Jan KM, Chien S. Effect of hematocrit variations on coronary hemodynamics and oxygen utilization. Am J Physiol. 1977;233(1):H106-H113.
18. Wilderson DK RASL, Gould SA, Sehgal HL, Moss GS. Limits of cardiac compensation in anemic baboons. Surgery. 1988;103(6):665-670.
19. Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA. 2004; 292(13):1555-1562.
20. Wu WC, Rathore SS, Wang Y, Radford MJ, Krumholz HM. Blood transfusion in elderly patients with acute myocardial infarction. N Engl J Med. 2001; 345(17):1230-1236.
21. Hebert PC, Fergusson DA. Do transfusions get to the heart of the matter? JAMA. 2004;292(13):1610-1612.
22. Hearnshaw S, Travis S, Murphy M. The role of blood transfusion in the management of upper and lower intestinal tract bleeding. Best Pract Res Clin Gastroenterology. 2008;22(2):355-371.
Case
A 65-year-old male nursing home resident is sent to the emergency room with a productive cough, fever, and low blood pressure, and is diagnosed with community-acquired pneumonia. He has a history of tobacco abuse, hypertension, and a right middle cerebral artery stroke. His admission labs show a hemoglobin level of 9.0 g/dL. The day after admission his hypotension has resolved and he reports feeling much better after two liters of intravenous fluids and antibiotics. However, his hemoglobin level is 7.9 g/dL. There is no evidence of bleeding. Should this hospitalized patient be transfused?
Overview
When to give a red blood cell transfusion is a clinical question commonly encountered by hospitalists. Individuals with acute blood loss, chronic blood loss, anemia of chronic disease, and hemolytic anemia often are given transfusions. Hospitalists serving as consultants may be asked when to transfuse patients perioperatively.
It is estimated up to 25% of the red blood cells transfused in the U.S. are inappropriate.1-4 Many physicians transfuse based on a number, rather than on objective findings. Overuse is common because of the wide availability of red blood cells, the belief complications are infrequent, and an unfounded fear of adverse outcomes if a patient is not transfused.
Tachycardia, low blood pressure, and declining oxygen saturations are signs clinicians can use when making the decision to transfuse. Electrocardiographic changes associated with tissue hypoxia can occur at a hemoglobin level <5 g/dL in healthy adults. Studies show mortality and morbidity increase rapidly at levels <5.0 to 6.0 g/dL.5 Currently, no diagnostic serological test exists for tissue hypoxia, which is the physiologic reason to give red blood cells.
Red blood cell transfusion can be a life-saving therapy; however, it is not a benign intervention. It is estimated 10% of transfusion reactions will have some adverse event.6 Red blood cell use exposes patients to hemolytic transfusion reactions, infections, and transfusion related acute lung injury.7,8 Additionally, unnecessary economic expenses are incurred and a scarce resource is diverted from other patients.
Hospitalists should be able to describe the indications for red blood cell transfusion and understand the evidence for and against its use. Physicians who appreciate the risks and benefits of red blood cell use tend to transfuse less blood that those who less informed. 9, 10
Review of the Data
General outcomes: Despite the long history of red blood cell transfusion, which dates back to 1818, when James Blundell successfully saved a woman exsanguinating from a postpartum hemorrhage, little evidence has been accumulated for its appropriate use. In the 1980s, the discovery of the human immunodeficiency virus sparked blood product safety concerns. This stimulated research and a debate over red blood cell transfusion practices, with a growing body of literature unsupportive of transfusion for an arbitrary trigger, for example the “10/30 rule,” which referred to 10 g/dL hemoglobin or hematocrit of 30%.9
Observational studies have raised concerns by linking morbidity and mortality to red blood cell use. Among 1,958 surgical patients who refused blood transfusion on religious grounds, there was an increase in mortality when hemoglobin levels were <6.0 g/dL. Hemoglobin levels higher than 7.0 g/dL showed no increased mortality.11 A recent comprehensive review included 272,596 surgical, trauma, and ICU patients in 45 observational studies. The review included studies with end points, including mortality, infections, multiorgan dysfunction syndrome, and acute respiratory distress syndrome, and concluded transfusions are associated with a higher risk of morbidity and mortality.12 (see Figure 1, p. 20)
Higher rates of infection associated with transfusions occurred in patients with post-operative trauma, acute injuries, gastrointestinal cancer undergoing surgery, coronary bypass surgery, hip surgery, burns, critical illness, and patients requiring ventilation. (see Figure 2, p. 21)12 The increased infection risk likely is due to the transient depression of the immune system induced by red blood cell transfusion. Prolonged hospital stays in postoperative colorectal surgery patients and ICU patients have been associated with transfusions.13
A meta-analysis of the few randomized controlled trials favors the restrictive use of red blood cells. The preponderance of the evidence comes from the Transfusion Requirements in Critical Care (TRICC) trial.14 This randomized control trial in critically ill medical and surgical patients demonstrated a restrictive strategy (transfusion trigger of <7.0 g/dL) and was as effective as a liberal transfusion strategy (transfusion trigger <10.0 g/dL). (see Figure 3, p. 22) Indeed, patients in the restrictive arm of the trial, who were less ill and under age 55 had a lower mortality rate than those who were transfused liberally.15 To date, there are no hospital-based randomized control trials that evaluate outcomes of anemic non-ICU medical patients.
This evidence has created a growing consensus that a restrictive use of blood results in improved patient outcomes. In patients without cardiovascular disease the evidence suggests most patients tolerate a hemoglobin level of 7.0 g/dL.5
Cardiac Patients
Experimental and clinical evidence suggests patients with cardiovascular disease are less tolerant of anemia. Patients with coronary disease are more likely to have adverse outcomes than those without coronary disease, if they do not have a red blood cell transfusion.11,16
The myocardium has a higher oxygen extraction ratio compared to the tissue oxygen extraction ratio, making it more sensitive to anemia.17,18 The presence of cardiac disease may require a higher threshold to transfuse blood; however, the precise recommended threshold remains controversial. A restrictive red blood cell transfusion strategy (maintaining the hemoglobin between 7.0 g/dL and 9.0 g/dL) appeared to be safe in most critically ill patients with cardiovascular disease.14
The data is more conflicting for patients with an acute coronary syndrome (ACS). Some studies have found increased mortality and another concluded ACS decreased with red blood cell use.19-21 Further research is needed to determine when red blood cells should be given to patients with coronary disease.
Gastrointestinal Bleeding
The decision to transfuse for gastrointestinal (GI) bleeding takes into account the site and etiology of the bleeding, availability of treatments, and risk of continued bleeding. Once the blood loss is controlled, a decision must be made on how to treat the anemia. Currently, no studies have looked at outcomes for patients who did and did not receive blood for an acute or chronic GI bleed.
Additionally, no studies have been conducted to delineate when to transfuse patients with chronic GI blood loss. Studies of patients with an acute GI bleed and cardiovascular disease have shown an increase in mortality, but it is unknown if the use of specific transfusion triggers affects outcomes in this group.
In patients with GI bleeding, experts feel the use of red blood cells should be guided by available evidence. For patients without cardiac disease, red blood cell transfusion is rarely required following definitive treatment and cessation of blood loss unless the hemoglobin is <7.0 g/dL.22
Back to the Case
The patient described in our case should not be transfused unless he has clinical signs or symptoms of tissue hypoxemia. An appropriate workup for his anemia should be initiated and, if an etiology identified, definitive treatment or intervention applied.
Bottom Line
Unless there are clinical signs of tissue hypoxia, symptomatic anemia, or a hemoglobin of <7.0 g/dL, red blood cell transfusion is not recommended, unless the patient has active ACS or significant underlying coronary disease. TH
Dr. Dressler is associate program director, assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta. Dr. VanderEnde is assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta.
References
1. Welch HG, Meehan KR, Goodnough LT. Prudent strategies for elective red blood cell transfusion. Ann Intern Med. 1992;116(5):393-402.
2. Tartter PI, Barron DM. Unnecessary blood transfusions in elective colorectal cancer surgery. Transfusion. 1985;25(2):113-115.
3. Saxena S, Weiner JM, Rabinowitz A, Fridey J, Shulman IA, Carmel R. Transfusion practice in medical patients. Arch Int Med. 1993;153(22):2575-80.
4. Palermo G, Bove J, Katz AJ. Patterns of blood use in Connecticut. Transfusion. 1980;20(6):704-710.
5. Carson JL, Reynolds RC. In search of the transfusion threshold. Hematology. 2005;10(Suppl 1):86-88.
6. Walker RH. Special report: transfusion risks. Am J Clin Pathol. 1987;88(3):374-378.
7. Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infections. N Engl J Med. 2006;355(13):1303-1305.
8. Spiess BD. Risks of transfusion: outcome focus. Transfusion. 2004;44(Suppl 12):4S-14S.
9. Salem-Schatz SR, Avorn J, Soumerai SB. Influence of clinical knowledge, organizational context, and practice style on transfusion decision-making. JAMA. 1990;264(4):476-483.
10. Wilson K, MacDougall L, Fergusson D, Graham I, Tinmouth A, Hebert PC. The effectiveness of interventions to reduce physician’s levels of inappropriate transfusion: what can be learned from a systematic review of the literature. Transfusion. 2002;42(9):1224-1229.
11. Carson JL, Duff A, Poses RM, et al. Effect of anemia and cardiovascular disease on surgical mortality and morbidity. Lancet. 1996;348(9034):1055-1060.
12. Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008;36(9):2667-2674.
13. Raghavan M, Marik PE. Anemia, allogenic blood transfusion, and immunomodulation in the critically ill. Chest. 2005;127(1):295-307.
14. Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion requirements in critical care investigators, Canadian critical care trials group. N Engl J Med. 1999;340(6):409-417.
15. Carson JL, Hill S, Carless P, Hebert P, Henry D. Transfusion triggers: a systematic review of the literature. Transfus Med Rev. 2002;16(3):187-199.
16. Sabatine MS, Morrow DA, Giugliano RP, et al. Association of hemoglobin levels with clinical outcomes in acute coronary syndromes. Circulation. 2005; 111(16):2042-2049.
17. Jan KM, Chien S. Effect of hematocrit variations on coronary hemodynamics and oxygen utilization. Am J Physiol. 1977;233(1):H106-H113.
18. Wilderson DK RASL, Gould SA, Sehgal HL, Moss GS. Limits of cardiac compensation in anemic baboons. Surgery. 1988;103(6):665-670.
19. Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA. 2004; 292(13):1555-1562.
20. Wu WC, Rathore SS, Wang Y, Radford MJ, Krumholz HM. Blood transfusion in elderly patients with acute myocardial infarction. N Engl J Med. 2001; 345(17):1230-1236.
21. Hebert PC, Fergusson DA. Do transfusions get to the heart of the matter? JAMA. 2004;292(13):1610-1612.
22. Hearnshaw S, Travis S, Murphy M. The role of blood transfusion in the management of upper and lower intestinal tract bleeding. Best Pract Res Clin Gastroenterology. 2008;22(2):355-371.
Case
A 65-year-old male nursing home resident is sent to the emergency room with a productive cough, fever, and low blood pressure, and is diagnosed with community-acquired pneumonia. He has a history of tobacco abuse, hypertension, and a right middle cerebral artery stroke. His admission labs show a hemoglobin level of 9.0 g/dL. The day after admission his hypotension has resolved and he reports feeling much better after two liters of intravenous fluids and antibiotics. However, his hemoglobin level is 7.9 g/dL. There is no evidence of bleeding. Should this hospitalized patient be transfused?
Overview
When to give a red blood cell transfusion is a clinical question commonly encountered by hospitalists. Individuals with acute blood loss, chronic blood loss, anemia of chronic disease, and hemolytic anemia often are given transfusions. Hospitalists serving as consultants may be asked when to transfuse patients perioperatively.
It is estimated up to 25% of the red blood cells transfused in the U.S. are inappropriate.1-4 Many physicians transfuse based on a number, rather than on objective findings. Overuse is common because of the wide availability of red blood cells, the belief complications are infrequent, and an unfounded fear of adverse outcomes if a patient is not transfused.
Tachycardia, low blood pressure, and declining oxygen saturations are signs clinicians can use when making the decision to transfuse. Electrocardiographic changes associated with tissue hypoxia can occur at a hemoglobin level <5 g/dL in healthy adults. Studies show mortality and morbidity increase rapidly at levels <5.0 to 6.0 g/dL.5 Currently, no diagnostic serological test exists for tissue hypoxia, which is the physiologic reason to give red blood cells.
Red blood cell transfusion can be a life-saving therapy; however, it is not a benign intervention. It is estimated 10% of transfusion reactions will have some adverse event.6 Red blood cell use exposes patients to hemolytic transfusion reactions, infections, and transfusion related acute lung injury.7,8 Additionally, unnecessary economic expenses are incurred and a scarce resource is diverted from other patients.
Hospitalists should be able to describe the indications for red blood cell transfusion and understand the evidence for and against its use. Physicians who appreciate the risks and benefits of red blood cell use tend to transfuse less blood that those who less informed. 9, 10
Review of the Data
General outcomes: Despite the long history of red blood cell transfusion, which dates back to 1818, when James Blundell successfully saved a woman exsanguinating from a postpartum hemorrhage, little evidence has been accumulated for its appropriate use. In the 1980s, the discovery of the human immunodeficiency virus sparked blood product safety concerns. This stimulated research and a debate over red blood cell transfusion practices, with a growing body of literature unsupportive of transfusion for an arbitrary trigger, for example the “10/30 rule,” which referred to 10 g/dL hemoglobin or hematocrit of 30%.9
Observational studies have raised concerns by linking morbidity and mortality to red blood cell use. Among 1,958 surgical patients who refused blood transfusion on religious grounds, there was an increase in mortality when hemoglobin levels were <6.0 g/dL. Hemoglobin levels higher than 7.0 g/dL showed no increased mortality.11 A recent comprehensive review included 272,596 surgical, trauma, and ICU patients in 45 observational studies. The review included studies with end points, including mortality, infections, multiorgan dysfunction syndrome, and acute respiratory distress syndrome, and concluded transfusions are associated with a higher risk of morbidity and mortality.12 (see Figure 1, p. 20)
Higher rates of infection associated with transfusions occurred in patients with post-operative trauma, acute injuries, gastrointestinal cancer undergoing surgery, coronary bypass surgery, hip surgery, burns, critical illness, and patients requiring ventilation. (see Figure 2, p. 21)12 The increased infection risk likely is due to the transient depression of the immune system induced by red blood cell transfusion. Prolonged hospital stays in postoperative colorectal surgery patients and ICU patients have been associated with transfusions.13
A meta-analysis of the few randomized controlled trials favors the restrictive use of red blood cells. The preponderance of the evidence comes from the Transfusion Requirements in Critical Care (TRICC) trial.14 This randomized control trial in critically ill medical and surgical patients demonstrated a restrictive strategy (transfusion trigger of <7.0 g/dL) and was as effective as a liberal transfusion strategy (transfusion trigger <10.0 g/dL). (see Figure 3, p. 22) Indeed, patients in the restrictive arm of the trial, who were less ill and under age 55 had a lower mortality rate than those who were transfused liberally.15 To date, there are no hospital-based randomized control trials that evaluate outcomes of anemic non-ICU medical patients.
This evidence has created a growing consensus that a restrictive use of blood results in improved patient outcomes. In patients without cardiovascular disease the evidence suggests most patients tolerate a hemoglobin level of 7.0 g/dL.5
Cardiac Patients
Experimental and clinical evidence suggests patients with cardiovascular disease are less tolerant of anemia. Patients with coronary disease are more likely to have adverse outcomes than those without coronary disease, if they do not have a red blood cell transfusion.11,16
The myocardium has a higher oxygen extraction ratio compared to the tissue oxygen extraction ratio, making it more sensitive to anemia.17,18 The presence of cardiac disease may require a higher threshold to transfuse blood; however, the precise recommended threshold remains controversial. A restrictive red blood cell transfusion strategy (maintaining the hemoglobin between 7.0 g/dL and 9.0 g/dL) appeared to be safe in most critically ill patients with cardiovascular disease.14
The data is more conflicting for patients with an acute coronary syndrome (ACS). Some studies have found increased mortality and another concluded ACS decreased with red blood cell use.19-21 Further research is needed to determine when red blood cells should be given to patients with coronary disease.
Gastrointestinal Bleeding
The decision to transfuse for gastrointestinal (GI) bleeding takes into account the site and etiology of the bleeding, availability of treatments, and risk of continued bleeding. Once the blood loss is controlled, a decision must be made on how to treat the anemia. Currently, no studies have looked at outcomes for patients who did and did not receive blood for an acute or chronic GI bleed.
Additionally, no studies have been conducted to delineate when to transfuse patients with chronic GI blood loss. Studies of patients with an acute GI bleed and cardiovascular disease have shown an increase in mortality, but it is unknown if the use of specific transfusion triggers affects outcomes in this group.
In patients with GI bleeding, experts feel the use of red blood cells should be guided by available evidence. For patients without cardiac disease, red blood cell transfusion is rarely required following definitive treatment and cessation of blood loss unless the hemoglobin is <7.0 g/dL.22
Back to the Case
The patient described in our case should not be transfused unless he has clinical signs or symptoms of tissue hypoxemia. An appropriate workup for his anemia should be initiated and, if an etiology identified, definitive treatment or intervention applied.
Bottom Line
Unless there are clinical signs of tissue hypoxia, symptomatic anemia, or a hemoglobin of <7.0 g/dL, red blood cell transfusion is not recommended, unless the patient has active ACS or significant underlying coronary disease. TH
Dr. Dressler is associate program director, assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta. Dr. VanderEnde is assistant professor of medicine, Division of General Internal Medicine, Emory University Hospital, Atlanta.
References
1. Welch HG, Meehan KR, Goodnough LT. Prudent strategies for elective red blood cell transfusion. Ann Intern Med. 1992;116(5):393-402.
2. Tartter PI, Barron DM. Unnecessary blood transfusions in elective colorectal cancer surgery. Transfusion. 1985;25(2):113-115.
3. Saxena S, Weiner JM, Rabinowitz A, Fridey J, Shulman IA, Carmel R. Transfusion practice in medical patients. Arch Int Med. 1993;153(22):2575-80.
4. Palermo G, Bove J, Katz AJ. Patterns of blood use in Connecticut. Transfusion. 1980;20(6):704-710.
5. Carson JL, Reynolds RC. In search of the transfusion threshold. Hematology. 2005;10(Suppl 1):86-88.
6. Walker RH. Special report: transfusion risks. Am J Clin Pathol. 1987;88(3):374-378.
7. Blajchman MA, Vamvakas EC. The continuing risk of transfusion-transmitted infections. N Engl J Med. 2006;355(13):1303-1305.
8. Spiess BD. Risks of transfusion: outcome focus. Transfusion. 2004;44(Suppl 12):4S-14S.
9. Salem-Schatz SR, Avorn J, Soumerai SB. Influence of clinical knowledge, organizational context, and practice style on transfusion decision-making. JAMA. 1990;264(4):476-483.
10. Wilson K, MacDougall L, Fergusson D, Graham I, Tinmouth A, Hebert PC. The effectiveness of interventions to reduce physician’s levels of inappropriate transfusion: what can be learned from a systematic review of the literature. Transfusion. 2002;42(9):1224-1229.
11. Carson JL, Duff A, Poses RM, et al. Effect of anemia and cardiovascular disease on surgical mortality and morbidity. Lancet. 1996;348(9034):1055-1060.
12. Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008;36(9):2667-2674.
13. Raghavan M, Marik PE. Anemia, allogenic blood transfusion, and immunomodulation in the critically ill. Chest. 2005;127(1):295-307.
14. Hebert PC, Wells G, Blajchman MA, et al. A multicenter, randomized, controlled clinical trial of transfusion requirements in critical care. Transfusion requirements in critical care investigators, Canadian critical care trials group. N Engl J Med. 1999;340(6):409-417.
15. Carson JL, Hill S, Carless P, Hebert P, Henry D. Transfusion triggers: a systematic review of the literature. Transfus Med Rev. 2002;16(3):187-199.
16. Sabatine MS, Morrow DA, Giugliano RP, et al. Association of hemoglobin levels with clinical outcomes in acute coronary syndromes. Circulation. 2005; 111(16):2042-2049.
17. Jan KM, Chien S. Effect of hematocrit variations on coronary hemodynamics and oxygen utilization. Am J Physiol. 1977;233(1):H106-H113.
18. Wilderson DK RASL, Gould SA, Sehgal HL, Moss GS. Limits of cardiac compensation in anemic baboons. Surgery. 1988;103(6):665-670.
19. Rao SV, Jollis JG, Harrington RA, et al. Relationship of blood transfusion and clinical outcomes in patients with acute coronary syndromes. JAMA. 2004; 292(13):1555-1562.
20. Wu WC, Rathore SS, Wang Y, Radford MJ, Krumholz HM. Blood transfusion in elderly patients with acute myocardial infarction. N Engl J Med. 2001; 345(17):1230-1236.
21. Hebert PC, Fergusson DA. Do transfusions get to the heart of the matter? JAMA. 2004;292(13):1610-1612.
22. Hearnshaw S, Travis S, Murphy M. The role of blood transfusion in the management of upper and lower intestinal tract bleeding. Best Pract Res Clin Gastroenterology. 2008;22(2):355-371.
Face-to-Face Improvement
The American Medical Association recently released Current Procedural Terminology (CPT) 2009. New, deleted, and revised codes went into effect Jan. 1. The biggest change to hospitalist billing involves prolonged care codes (99354-99357). CPT 2009 descriptor revisions make it possible for physicians to contribute non-face-to-face time toward prolonged care services.
Inpatient Prolonged Care
Previous versions of CPT defined code 99356 as the first hour of prolonged physician [inpatient] services requiring direct (face-to-face) patient contact beyond the usual services (reportable after the initial 30 minutes); and 99357 for each additional 30 minutes of prolonged [inpatient] care beyond the first hour (reportable after the first 15 minutes of each additional segment). CPT 2009 has changed prolonged care guidelines to be more consistent with other time-based services: all unit/floor time spent by the physician is considered when reporting 99356 and 99357.1
As with most other evaluation and management services, a face-to-face encounter still must occur. In addition to the time associated with the face-to-face encounter, count the time associated with all other physician activities occurring on the unit/floor (e.g., reviewing images, obtaining information involving overnight events, discussing management options with the family) directed toward an individual patient. The cumulative time spent by the billing provider on a single calendar day is considered for billing. Time spent by someone other than the billing provider cannot be credited toward prolonged care.
As example, a physician cares for a 65-year-old male with uncontrolled diabetes, diabetic nephropathy, and congestive heart failure. Early in the day, the physician rounds, spending a total of 20 minutes reviewing the overnight course of events on the unit, re-confirming the patient history, and performing an exam with the patient. Anticipating the patient’s needs, the physician discusses post-discharge options and care with the patient and his family for 45 minutes. After the discussion, the physician spends an additional 30 minutes relaying information to the team and coordinating care. Merely reporting the highest-level subsequent hospital care service (99233), does not capture the physician’s cumulative effort. It only would account for 40 of the 95 minutes spent throughout the day. In order to capture the remaining 55 minutes, the physician reports 99356 on the same claim form as 99233.
Do not report prolonged care codes on a separate claim form. Prolonged care codes do not represent an independent service. These codes are reported along with a primary service. They must appear as a separate line item on the claim form, which includes a code representing the primary service. For prolonged care in the inpatient setting, the primary service must be initial hospital care (99221-99223), subsequent hospital care (99231-99233), inpatient consultations (99251-99255), or nursing facility services (99304-99318). Additional examples of billable prolonged care services are in Section 30.6.15.1I of the Medicare manual, available at www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf.
Threshold Time
Prolonged care guidelines refer to “threshold” time. Threshold time requires the physician to exceed the time requirements associated with the “primary” codes before reporting prolonged care. Table 1 identifies the typical times associated with inpatient services qualifying for prolonged care. The physician must exceed the typical time by a minimum of 30 minutes. (For example, 99232 + 99356 = 25 minutes + 30 minutes = 55 total minutes). Additionally, the physician must document the total time spent during the face-to-face portion of the encounter, and the additional unit or floor time in one cumulative note or in separate notes representing the physician services provided to the patient throughout the day.
Prolonged Outpatient Services
Prolonged care (99354-99355) provided to outpatients remains unchanged. Physicians only report personally provided face-to-face time with the patient. Time spent by other staff members does not count toward prolonged care.
As with prolonged inpatient care, report 99354 and 99355 in addition to a primary service code. The companion outpatient codes are outpatient/office visits (99201-99205 or 99212–99215), outpatient consultation (99241–99245), domiciliary/custodial care (99324–99328 or 99334–99337), and home services (99341-99350). Hospitalists more often use outpatient prolonged care with office consultation codes for services provided in the emergency department, as appropriate.
Do not report 99354 or 99355 with observation care (99217-99220) or emergency department visits (99281-99288), since these service categories typically require prolonged periods of physician monitoring, thereby prohibiting use of prolonged care codes. As with inpatient-prolonged care, the concept of threshold time exists. Refer to Table 2 (pg. 25) for the typical threshold times associated with office consultation codes.

Medicare Consideration
Although CPT has offered revisions to this code, Medicare guidelines remain unchanged. The Medicare Claims Processing Manual still states: “In the case of prolonged hospital services, time spent reviewing charts or discussion of a patient with house medical staff and not with direct face-to-face contact with the patient, or waiting for test results, for changes in the patient’s condition, for end of a therapy, or for use of facilities, cannot be billed as prolonged services.”4 It is yet to be determined if the Centers for Medicare and Medicaid Services (CMS) will issue a transmittal to revise the current description in the processing manual. Physicians and staff may access past and present transmittal information at www.cms.hhs.gov/ Transmittals/.
As always, be sure to query payers about prolonged care services, since some non-Medicare insurers may not recognize these codes.
Modifier 21
Modifier 21 has been deleted from the CPT. Modifier 21 was appended to an appropriate visit code (e.g., 99232-21) when the face-to-face or floor/unit service(s) provided is prolonged or otherwise greater than usually required for the highest level of evaluation and management service within a given category.5 Since the descriptors for codes 99354-99357 have been revised to more consistently reflect the description formerly associated with modifier 21, there is no need to maintain its existence. Additionally, Medicare and most other payers did not recognize this modifier.
Code This Case
Question: A newly diagnosed diabetic requires extensive counseling regarding lifestyle changes, medication regime, the disease process, as well as coordination of care for outpatient programs and services. The hospitalist reviews some of the pertinent information with the patient (15 minutes), and performs an abbreviated service (problem-focused history and exam). The attending physician asks the resident to assist him with the remaining counseling efforts and coordination of care (30 minutes).
Each physician documents his or her portion of the service. What visit level can the hospitalist report?
Answer: When two billing providers (i.e., two attending physicians) from the same group practice split the threshold time (e.g., physician A provided morning rounds, and physician B spoke with the family in the afternoon), only one physician can report the cumulative service, since 99356 must be reported on the same invoice as the primary visit code (e.g., 99231).6
The example above involves the resident’s time as well as the attending physician’s time. Documentation must be very clear to demonstrate the attending physician actively participated in the entire 45-minute service. Otherwise, only the attending may report the amount of time he actually spent providing the service.
Billing options for this scenario can vary. When the physician performs and documents the key components of history, exam, and decision making for the primary encounter, report 99231 (0.76 physician work relative value units; $33.90) and 99356 (1.71 physician work relative value units; $76.46) for the cumulative service. Alternatively, in those evaluation and management services for which the [primary] code level is selected based on time alone (i.e., history and exam was not performed or required), prolonged services may only be reported with the highest code level in that family of codes as the companion code.7
Therefore, this 45-minute service may be reported as 99233 (2.0 physician work relative value units; $86.92) since more than half of the total visit time was dedicated to counseling/coordi-nation of care (see Section 30.6.1B-C available at www. cms.hhs.gov/manuals/ downloads/clm104c12.pdf for additional information on billing for counseling/coordination of care time).
If a payer does not recognize prolonged care codes, only the latter billing option is possible. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References
1. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 25-26.
2. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1G. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
3. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1F. www.cms.hhs.gov/manuals/dowloads/ clm104c12.pdf. Accessed November 19, 2008.
4. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1C. www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf. Accessed November 19, 2008.
5. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 457.
6. Pohlig, C. Bill by time spent on case. The Hospitalist. Jul 2008;19.
7. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1H. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
The American Medical Association recently released Current Procedural Terminology (CPT) 2009. New, deleted, and revised codes went into effect Jan. 1. The biggest change to hospitalist billing involves prolonged care codes (99354-99357). CPT 2009 descriptor revisions make it possible for physicians to contribute non-face-to-face time toward prolonged care services.
Inpatient Prolonged Care
Previous versions of CPT defined code 99356 as the first hour of prolonged physician [inpatient] services requiring direct (face-to-face) patient contact beyond the usual services (reportable after the initial 30 minutes); and 99357 for each additional 30 minutes of prolonged [inpatient] care beyond the first hour (reportable after the first 15 minutes of each additional segment). CPT 2009 has changed prolonged care guidelines to be more consistent with other time-based services: all unit/floor time spent by the physician is considered when reporting 99356 and 99357.1
As with most other evaluation and management services, a face-to-face encounter still must occur. In addition to the time associated with the face-to-face encounter, count the time associated with all other physician activities occurring on the unit/floor (e.g., reviewing images, obtaining information involving overnight events, discussing management options with the family) directed toward an individual patient. The cumulative time spent by the billing provider on a single calendar day is considered for billing. Time spent by someone other than the billing provider cannot be credited toward prolonged care.
As example, a physician cares for a 65-year-old male with uncontrolled diabetes, diabetic nephropathy, and congestive heart failure. Early in the day, the physician rounds, spending a total of 20 minutes reviewing the overnight course of events on the unit, re-confirming the patient history, and performing an exam with the patient. Anticipating the patient’s needs, the physician discusses post-discharge options and care with the patient and his family for 45 minutes. After the discussion, the physician spends an additional 30 minutes relaying information to the team and coordinating care. Merely reporting the highest-level subsequent hospital care service (99233), does not capture the physician’s cumulative effort. It only would account for 40 of the 95 minutes spent throughout the day. In order to capture the remaining 55 minutes, the physician reports 99356 on the same claim form as 99233.
Do not report prolonged care codes on a separate claim form. Prolonged care codes do not represent an independent service. These codes are reported along with a primary service. They must appear as a separate line item on the claim form, which includes a code representing the primary service. For prolonged care in the inpatient setting, the primary service must be initial hospital care (99221-99223), subsequent hospital care (99231-99233), inpatient consultations (99251-99255), or nursing facility services (99304-99318). Additional examples of billable prolonged care services are in Section 30.6.15.1I of the Medicare manual, available at www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf.
Threshold Time
Prolonged care guidelines refer to “threshold” time. Threshold time requires the physician to exceed the time requirements associated with the “primary” codes before reporting prolonged care. Table 1 identifies the typical times associated with inpatient services qualifying for prolonged care. The physician must exceed the typical time by a minimum of 30 minutes. (For example, 99232 + 99356 = 25 minutes + 30 minutes = 55 total minutes). Additionally, the physician must document the total time spent during the face-to-face portion of the encounter, and the additional unit or floor time in one cumulative note or in separate notes representing the physician services provided to the patient throughout the day.
Prolonged Outpatient Services
Prolonged care (99354-99355) provided to outpatients remains unchanged. Physicians only report personally provided face-to-face time with the patient. Time spent by other staff members does not count toward prolonged care.
As with prolonged inpatient care, report 99354 and 99355 in addition to a primary service code. The companion outpatient codes are outpatient/office visits (99201-99205 or 99212–99215), outpatient consultation (99241–99245), domiciliary/custodial care (99324–99328 or 99334–99337), and home services (99341-99350). Hospitalists more often use outpatient prolonged care with office consultation codes for services provided in the emergency department, as appropriate.
Do not report 99354 or 99355 with observation care (99217-99220) or emergency department visits (99281-99288), since these service categories typically require prolonged periods of physician monitoring, thereby prohibiting use of prolonged care codes. As with inpatient-prolonged care, the concept of threshold time exists. Refer to Table 2 (pg. 25) for the typical threshold times associated with office consultation codes.
Medicare Consideration
Although CPT has offered revisions to this code, Medicare guidelines remain unchanged. The Medicare Claims Processing Manual still states: “In the case of prolonged hospital services, time spent reviewing charts or discussion of a patient with house medical staff and not with direct face-to-face contact with the patient, or waiting for test results, for changes in the patient’s condition, for end of a therapy, or for use of facilities, cannot be billed as prolonged services.”4 It is yet to be determined if the Centers for Medicare and Medicaid Services (CMS) will issue a transmittal to revise the current description in the processing manual. Physicians and staff may access past and present transmittal information at www.cms.hhs.gov/ Transmittals/.
As always, be sure to query payers about prolonged care services, since some non-Medicare insurers may not recognize these codes.
Modifier 21
Modifier 21 has been deleted from the CPT. Modifier 21 was appended to an appropriate visit code (e.g., 99232-21) when the face-to-face or floor/unit service(s) provided is prolonged or otherwise greater than usually required for the highest level of evaluation and management service within a given category.5 Since the descriptors for codes 99354-99357 have been revised to more consistently reflect the description formerly associated with modifier 21, there is no need to maintain its existence. Additionally, Medicare and most other payers did not recognize this modifier.
Code This Case
Question: A newly diagnosed diabetic requires extensive counseling regarding lifestyle changes, medication regime, the disease process, as well as coordination of care for outpatient programs and services. The hospitalist reviews some of the pertinent information with the patient (15 minutes), and performs an abbreviated service (problem-focused history and exam). The attending physician asks the resident to assist him with the remaining counseling efforts and coordination of care (30 minutes).
Each physician documents his or her portion of the service. What visit level can the hospitalist report?
Answer: When two billing providers (i.e., two attending physicians) from the same group practice split the threshold time (e.g., physician A provided morning rounds, and physician B spoke with the family in the afternoon), only one physician can report the cumulative service, since 99356 must be reported on the same invoice as the primary visit code (e.g., 99231).6
The example above involves the resident’s time as well as the attending physician’s time. Documentation must be very clear to demonstrate the attending physician actively participated in the entire 45-minute service. Otherwise, only the attending may report the amount of time he actually spent providing the service.
Billing options for this scenario can vary. When the physician performs and documents the key components of history, exam, and decision making for the primary encounter, report 99231 (0.76 physician work relative value units; $33.90) and 99356 (1.71 physician work relative value units; $76.46) for the cumulative service. Alternatively, in those evaluation and management services for which the [primary] code level is selected based on time alone (i.e., history and exam was not performed or required), prolonged services may only be reported with the highest code level in that family of codes as the companion code.7
Therefore, this 45-minute service may be reported as 99233 (2.0 physician work relative value units; $86.92) since more than half of the total visit time was dedicated to counseling/coordi-nation of care (see Section 30.6.1B-C available at www. cms.hhs.gov/manuals/ downloads/clm104c12.pdf for additional information on billing for counseling/coordination of care time).
If a payer does not recognize prolonged care codes, only the latter billing option is possible. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References
1. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 25-26.
2. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1G. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
3. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1F. www.cms.hhs.gov/manuals/dowloads/ clm104c12.pdf. Accessed November 19, 2008.
4. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1C. www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf. Accessed November 19, 2008.
5. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 457.
6. Pohlig, C. Bill by time spent on case. The Hospitalist. Jul 2008;19.
7. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1H. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
The American Medical Association recently released Current Procedural Terminology (CPT) 2009. New, deleted, and revised codes went into effect Jan. 1. The biggest change to hospitalist billing involves prolonged care codes (99354-99357). CPT 2009 descriptor revisions make it possible for physicians to contribute non-face-to-face time toward prolonged care services.
Inpatient Prolonged Care
Previous versions of CPT defined code 99356 as the first hour of prolonged physician [inpatient] services requiring direct (face-to-face) patient contact beyond the usual services (reportable after the initial 30 minutes); and 99357 for each additional 30 minutes of prolonged [inpatient] care beyond the first hour (reportable after the first 15 minutes of each additional segment). CPT 2009 has changed prolonged care guidelines to be more consistent with other time-based services: all unit/floor time spent by the physician is considered when reporting 99356 and 99357.1
As with most other evaluation and management services, a face-to-face encounter still must occur. In addition to the time associated with the face-to-face encounter, count the time associated with all other physician activities occurring on the unit/floor (e.g., reviewing images, obtaining information involving overnight events, discussing management options with the family) directed toward an individual patient. The cumulative time spent by the billing provider on a single calendar day is considered for billing. Time spent by someone other than the billing provider cannot be credited toward prolonged care.
As example, a physician cares for a 65-year-old male with uncontrolled diabetes, diabetic nephropathy, and congestive heart failure. Early in the day, the physician rounds, spending a total of 20 minutes reviewing the overnight course of events on the unit, re-confirming the patient history, and performing an exam with the patient. Anticipating the patient’s needs, the physician discusses post-discharge options and care with the patient and his family for 45 minutes. After the discussion, the physician spends an additional 30 minutes relaying information to the team and coordinating care. Merely reporting the highest-level subsequent hospital care service (99233), does not capture the physician’s cumulative effort. It only would account for 40 of the 95 minutes spent throughout the day. In order to capture the remaining 55 minutes, the physician reports 99356 on the same claim form as 99233.
Do not report prolonged care codes on a separate claim form. Prolonged care codes do not represent an independent service. These codes are reported along with a primary service. They must appear as a separate line item on the claim form, which includes a code representing the primary service. For prolonged care in the inpatient setting, the primary service must be initial hospital care (99221-99223), subsequent hospital care (99231-99233), inpatient consultations (99251-99255), or nursing facility services (99304-99318). Additional examples of billable prolonged care services are in Section 30.6.15.1I of the Medicare manual, available at www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf.
Threshold Time
Prolonged care guidelines refer to “threshold” time. Threshold time requires the physician to exceed the time requirements associated with the “primary” codes before reporting prolonged care. Table 1 identifies the typical times associated with inpatient services qualifying for prolonged care. The physician must exceed the typical time by a minimum of 30 minutes. (For example, 99232 + 99356 = 25 minutes + 30 minutes = 55 total minutes). Additionally, the physician must document the total time spent during the face-to-face portion of the encounter, and the additional unit or floor time in one cumulative note or in separate notes representing the physician services provided to the patient throughout the day.
Prolonged Outpatient Services
Prolonged care (99354-99355) provided to outpatients remains unchanged. Physicians only report personally provided face-to-face time with the patient. Time spent by other staff members does not count toward prolonged care.
As with prolonged inpatient care, report 99354 and 99355 in addition to a primary service code. The companion outpatient codes are outpatient/office visits (99201-99205 or 99212–99215), outpatient consultation (99241–99245), domiciliary/custodial care (99324–99328 or 99334–99337), and home services (99341-99350). Hospitalists more often use outpatient prolonged care with office consultation codes for services provided in the emergency department, as appropriate.
Do not report 99354 or 99355 with observation care (99217-99220) or emergency department visits (99281-99288), since these service categories typically require prolonged periods of physician monitoring, thereby prohibiting use of prolonged care codes. As with inpatient-prolonged care, the concept of threshold time exists. Refer to Table 2 (pg. 25) for the typical threshold times associated with office consultation codes.
Medicare Consideration
Although CPT has offered revisions to this code, Medicare guidelines remain unchanged. The Medicare Claims Processing Manual still states: “In the case of prolonged hospital services, time spent reviewing charts or discussion of a patient with house medical staff and not with direct face-to-face contact with the patient, or waiting for test results, for changes in the patient’s condition, for end of a therapy, or for use of facilities, cannot be billed as prolonged services.”4 It is yet to be determined if the Centers for Medicare and Medicaid Services (CMS) will issue a transmittal to revise the current description in the processing manual. Physicians and staff may access past and present transmittal information at www.cms.hhs.gov/ Transmittals/.
As always, be sure to query payers about prolonged care services, since some non-Medicare insurers may not recognize these codes.
Modifier 21
Modifier 21 has been deleted from the CPT. Modifier 21 was appended to an appropriate visit code (e.g., 99232-21) when the face-to-face or floor/unit service(s) provided is prolonged or otherwise greater than usually required for the highest level of evaluation and management service within a given category.5 Since the descriptors for codes 99354-99357 have been revised to more consistently reflect the description formerly associated with modifier 21, there is no need to maintain its existence. Additionally, Medicare and most other payers did not recognize this modifier.
Code This Case
Question: A newly diagnosed diabetic requires extensive counseling regarding lifestyle changes, medication regime, the disease process, as well as coordination of care for outpatient programs and services. The hospitalist reviews some of the pertinent information with the patient (15 minutes), and performs an abbreviated service (problem-focused history and exam). The attending physician asks the resident to assist him with the remaining counseling efforts and coordination of care (30 minutes).
Each physician documents his or her portion of the service. What visit level can the hospitalist report?
Answer: When two billing providers (i.e., two attending physicians) from the same group practice split the threshold time (e.g., physician A provided morning rounds, and physician B spoke with the family in the afternoon), only one physician can report the cumulative service, since 99356 must be reported on the same invoice as the primary visit code (e.g., 99231).6
The example above involves the resident’s time as well as the attending physician’s time. Documentation must be very clear to demonstrate the attending physician actively participated in the entire 45-minute service. Otherwise, only the attending may report the amount of time he actually spent providing the service.
Billing options for this scenario can vary. When the physician performs and documents the key components of history, exam, and decision making for the primary encounter, report 99231 (0.76 physician work relative value units; $33.90) and 99356 (1.71 physician work relative value units; $76.46) for the cumulative service. Alternatively, in those evaluation and management services for which the [primary] code level is selected based on time alone (i.e., history and exam was not performed or required), prolonged services may only be reported with the highest code level in that family of codes as the companion code.7
Therefore, this 45-minute service may be reported as 99233 (2.0 physician work relative value units; $86.92) since more than half of the total visit time was dedicated to counseling/coordi-nation of care (see Section 30.6.1B-C available at www. cms.hhs.gov/manuals/ downloads/clm104c12.pdf for additional information on billing for counseling/coordination of care time).
If a payer does not recognize prolonged care codes, only the latter billing option is possible. TH
Carol Pohlig is a billing and coding expert with the University of Pennsylvania Medical Center, Philadelphia. She is on the faculty of SHM’s inpatient coding course.
References
1. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 25-26.
2. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1G. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
3. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1F. www.cms.hhs.gov/manuals/dowloads/ clm104c12.pdf. Accessed November 19, 2008.
4. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1C. www.cms.hhs.gov/manuals/ downloads/clm104c12.pdf. Accessed November 19, 2008.
5. Beebe M, Dalton J, Espronceda M, Evans D, Glenn R. Current Procedural Terminology Professional Edition. Chicago, IL: American Medical Association, 2008; 457.
6. Pohlig, C. Bill by time spent on case. The Hospitalist. Jul 2008;19.
7. Centers for Medicare and Medicaid Services. Medicare Claims Processing Manual: Chapter 12, Section 30.6.15.1H. www.cms.hhs.gov/manuals/downloads/ clm104c12.pdf. Accessed November 19, 2008.
Medicare Modifications
Physicians who count Medicare among their payers already know the government green-lighted a 1.1% increase in Medicare Part B payments to physicians last summer. The increase was made official by the Centers for Medicare and Medicaid Services (CMS) on Oct. 30, with the release of the Medicare Physician Fee Schedule Final Rule for fiscal year 2009. The Final Rule governs what services are reimbursed by Medicare, the reimbursement levels for those services, and other rules pertaining to Medicare. Many of these changes, additions, and deletions were dictated by the Medicare Improvements for Patients and Providers Act, or MIPPA. (See “MIPPA Matters,” December 2008, p. 18.)
The 2009 Final Rule not only makes official the short-term, 1.1% payment increase, it also marks significant increases in payments for inpatient evaluation and management services, higher bonuses for participation in the Physician Quality Reporting Initiative (PQRI), and new policies to help direct the future of healthcare.
Here is a look at a few of the key aspects of the Final Rule, of which you may not be aware:
Transparent Physicians
In a continued effort to make healthcare transparent, CMS will begin posting the names of physicians who successfully report through the 2009 PQRI on a physician compare Web site in 2010. (2007 and 2008 PQRI participants will not be included.) Just as the Hospital Compare site enables consumers to view data on facilities, this site will allow consumers to view data reported by individual doctors.
Although consumers may be interested in checking for information on their primary care physician, it is unlikely inpatients will check the site before agreeing to see a specific hospitalist. However, the Physician Compare site will have some impact on hospital medicine. “I think this is the beginning of physicians’ commitment to greater transparency,” says Eric Siegal, MD, chair of SHM’s Public Policy Committee. “In a very broad sense, physicians who agree to be listed on the Physician Compare site very clearly value transparency and quality of care. Their inclusion could be seen as a differentiator, though a small one.”
Another factor to consider regarding transparency: “Physician Compare is not just about patients,” Dr. Siegal points out. “Third-party payers will look at this, as well. If they’re looking for someone to help take care of their patients, this data might sway them in their decision.”
Telehealth and Inpatients
Medicare already reimburses for certain exchanges of medical information from off-site physicians or vendors via interactive electronic communications, also known as telehealth or telemedicine services. Under the 2009 Final Rule, CMS will create a new series of Healthcare Common Procedure Coding System (HCPCS) codes for follow-up inpatient telehealth consultations, allowing practitioners to bill for follow-up inpatient consultations delivered via telehealth.
These codes are intended for use by physicians or non-physician providers when an inpatient consultation is requested from an appropriate source, such as the patient’s attending physician. CMS emphasizes the codes are not intended for use in billing for the ongoing evaluation and management of a hospital inpatient.
E-prescribe Out of Reach
Much attention has been given to a new Medicare program, which promotes the widespread adoption of electronic prescribing (e-prescribing). Physicians who successfully participate in CMS’ Electronic Prescribing Incentive Program will earn an extra bonus; however, the program was designed for primary care programs and hospitalists are unlikely to be able to take advantage of this.
“We don’t even know if hospitalists will be able to participate,” Dr. Siegal explains. The only way a hospitalist can take part in the e-prescribing initiative is if the hospital already has an acceptable system. However, Dr. Siegal warns, “If you create a mandate requiring a system for medication reconciliation at discharge, and then require another, separate system for e-prescribing, you’ve got problems. The primary driver should be that the hospital’s system supports both. And as far as we can tell, most hospital systems don’t do this.”
In August, SHM and the American College of Emergency Physicians conducted a teleconference with CMS to voice concerns with the e-prescribe initiative. “What we wanted was an exception,” Dr. Siegal says. SHM’s concern: When CMS stops rewarding physicians for e-prescribing and begins to penalize those who don’t—currently scheduled for 2013—hospitalists who can’t participate will be penalized through their Medicare payments. The outcome of the meeting, Dr. Siegal says, is “CMS turned around and said ‘either you can participate or you can’t.’ But at least they are considering our points; they seem to understand them.”
The good news is there is time to work the problem out, “At the moment, while e-prescribing is all bonus and no penalty, there’s no urgency to address it,” Dr. Siegal says.
Patient Safety
The Final Rule also includes improvements to PQRI, which allows eligible professionals to report on 153 quality measures. Physicians who successfully report on cases during 2009 will be able to earn an incentive payment, which has been increased to 2% (up from 1.5% in 2008), of their total allowed charges for covered professional services.
“I hope that more hospitalists will get on board with this,” Dr. Siegal says. He believes PQRI will be around for a while, and any hospital medicine group waiting to see if it is worth investing in the program can safely do so. “My feeling is that there’s growing bi-partisan support for something like this. I think it’s here to stay,” Dr. Siegal says.
SHM’s Opinion Counts
One reason the Final Rule is especially hospitalist-friendly is because SHM submitted extensive comment on CMS’s proposals in August. “SHM had a fair amount to say, and there are things in the rule that dovetail with our comments,” Dr. Siegal explains. “Part of the challenge is picking which battles to fight; there is a lot covered in this rule. We ended up focusing on areas that were really important to us, and on items where we thought we had a unique voice where nobody else was going to articulate.”
The Final Rule is available at www.cms.hhs.gov/center/physician.asp under “CMS-1403-FC.” Fact sheets covering major provisions of the Final Rule are available at www.cms.hhs.gov/apps/media/ fact_sheets.asp. TH
Jane Jerrard is a medical writer based in Chicago.
Physicians who count Medicare among their payers already know the government green-lighted a 1.1% increase in Medicare Part B payments to physicians last summer. The increase was made official by the Centers for Medicare and Medicaid Services (CMS) on Oct. 30, with the release of the Medicare Physician Fee Schedule Final Rule for fiscal year 2009. The Final Rule governs what services are reimbursed by Medicare, the reimbursement levels for those services, and other rules pertaining to Medicare. Many of these changes, additions, and deletions were dictated by the Medicare Improvements for Patients and Providers Act, or MIPPA. (See “MIPPA Matters,” December 2008, p. 18.)
The 2009 Final Rule not only makes official the short-term, 1.1% payment increase, it also marks significant increases in payments for inpatient evaluation and management services, higher bonuses for participation in the Physician Quality Reporting Initiative (PQRI), and new policies to help direct the future of healthcare.
Here is a look at a few of the key aspects of the Final Rule, of which you may not be aware:
Transparent Physicians
In a continued effort to make healthcare transparent, CMS will begin posting the names of physicians who successfully report through the 2009 PQRI on a physician compare Web site in 2010. (2007 and 2008 PQRI participants will not be included.) Just as the Hospital Compare site enables consumers to view data on facilities, this site will allow consumers to view data reported by individual doctors.
Although consumers may be interested in checking for information on their primary care physician, it is unlikely inpatients will check the site before agreeing to see a specific hospitalist. However, the Physician Compare site will have some impact on hospital medicine. “I think this is the beginning of physicians’ commitment to greater transparency,” says Eric Siegal, MD, chair of SHM’s Public Policy Committee. “In a very broad sense, physicians who agree to be listed on the Physician Compare site very clearly value transparency and quality of care. Their inclusion could be seen as a differentiator, though a small one.”
Another factor to consider regarding transparency: “Physician Compare is not just about patients,” Dr. Siegal points out. “Third-party payers will look at this, as well. If they’re looking for someone to help take care of their patients, this data might sway them in their decision.”
Telehealth and Inpatients
Medicare already reimburses for certain exchanges of medical information from off-site physicians or vendors via interactive electronic communications, also known as telehealth or telemedicine services. Under the 2009 Final Rule, CMS will create a new series of Healthcare Common Procedure Coding System (HCPCS) codes for follow-up inpatient telehealth consultations, allowing practitioners to bill for follow-up inpatient consultations delivered via telehealth.
These codes are intended for use by physicians or non-physician providers when an inpatient consultation is requested from an appropriate source, such as the patient’s attending physician. CMS emphasizes the codes are not intended for use in billing for the ongoing evaluation and management of a hospital inpatient.
E-prescribe Out of Reach
Much attention has been given to a new Medicare program, which promotes the widespread adoption of electronic prescribing (e-prescribing). Physicians who successfully participate in CMS’ Electronic Prescribing Incentive Program will earn an extra bonus; however, the program was designed for primary care programs and hospitalists are unlikely to be able to take advantage of this.
“We don’t even know if hospitalists will be able to participate,” Dr. Siegal explains. The only way a hospitalist can take part in the e-prescribing initiative is if the hospital already has an acceptable system. However, Dr. Siegal warns, “If you create a mandate requiring a system for medication reconciliation at discharge, and then require another, separate system for e-prescribing, you’ve got problems. The primary driver should be that the hospital’s system supports both. And as far as we can tell, most hospital systems don’t do this.”
In August, SHM and the American College of Emergency Physicians conducted a teleconference with CMS to voice concerns with the e-prescribe initiative. “What we wanted was an exception,” Dr. Siegal says. SHM’s concern: When CMS stops rewarding physicians for e-prescribing and begins to penalize those who don’t—currently scheduled for 2013—hospitalists who can’t participate will be penalized through their Medicare payments. The outcome of the meeting, Dr. Siegal says, is “CMS turned around and said ‘either you can participate or you can’t.’ But at least they are considering our points; they seem to understand them.”
The good news is there is time to work the problem out, “At the moment, while e-prescribing is all bonus and no penalty, there’s no urgency to address it,” Dr. Siegal says.
Patient Safety
The Final Rule also includes improvements to PQRI, which allows eligible professionals to report on 153 quality measures. Physicians who successfully report on cases during 2009 will be able to earn an incentive payment, which has been increased to 2% (up from 1.5% in 2008), of their total allowed charges for covered professional services.
“I hope that more hospitalists will get on board with this,” Dr. Siegal says. He believes PQRI will be around for a while, and any hospital medicine group waiting to see if it is worth investing in the program can safely do so. “My feeling is that there’s growing bi-partisan support for something like this. I think it’s here to stay,” Dr. Siegal says.
SHM’s Opinion Counts
One reason the Final Rule is especially hospitalist-friendly is because SHM submitted extensive comment on CMS’s proposals in August. “SHM had a fair amount to say, and there are things in the rule that dovetail with our comments,” Dr. Siegal explains. “Part of the challenge is picking which battles to fight; there is a lot covered in this rule. We ended up focusing on areas that were really important to us, and on items where we thought we had a unique voice where nobody else was going to articulate.”
The Final Rule is available at www.cms.hhs.gov/center/physician.asp under “CMS-1403-FC.” Fact sheets covering major provisions of the Final Rule are available at www.cms.hhs.gov/apps/media/ fact_sheets.asp. TH
Jane Jerrard is a medical writer based in Chicago.
Physicians who count Medicare among their payers already know the government green-lighted a 1.1% increase in Medicare Part B payments to physicians last summer. The increase was made official by the Centers for Medicare and Medicaid Services (CMS) on Oct. 30, with the release of the Medicare Physician Fee Schedule Final Rule for fiscal year 2009. The Final Rule governs what services are reimbursed by Medicare, the reimbursement levels for those services, and other rules pertaining to Medicare. Many of these changes, additions, and deletions were dictated by the Medicare Improvements for Patients and Providers Act, or MIPPA. (See “MIPPA Matters,” December 2008, p. 18.)
The 2009 Final Rule not only makes official the short-term, 1.1% payment increase, it also marks significant increases in payments for inpatient evaluation and management services, higher bonuses for participation in the Physician Quality Reporting Initiative (PQRI), and new policies to help direct the future of healthcare.
Here is a look at a few of the key aspects of the Final Rule, of which you may not be aware:
Transparent Physicians
In a continued effort to make healthcare transparent, CMS will begin posting the names of physicians who successfully report through the 2009 PQRI on a physician compare Web site in 2010. (2007 and 2008 PQRI participants will not be included.) Just as the Hospital Compare site enables consumers to view data on facilities, this site will allow consumers to view data reported by individual doctors.
Although consumers may be interested in checking for information on their primary care physician, it is unlikely inpatients will check the site before agreeing to see a specific hospitalist. However, the Physician Compare site will have some impact on hospital medicine. “I think this is the beginning of physicians’ commitment to greater transparency,” says Eric Siegal, MD, chair of SHM’s Public Policy Committee. “In a very broad sense, physicians who agree to be listed on the Physician Compare site very clearly value transparency and quality of care. Their inclusion could be seen as a differentiator, though a small one.”
Another factor to consider regarding transparency: “Physician Compare is not just about patients,” Dr. Siegal points out. “Third-party payers will look at this, as well. If they’re looking for someone to help take care of their patients, this data might sway them in their decision.”
Telehealth and Inpatients
Medicare already reimburses for certain exchanges of medical information from off-site physicians or vendors via interactive electronic communications, also known as telehealth or telemedicine services. Under the 2009 Final Rule, CMS will create a new series of Healthcare Common Procedure Coding System (HCPCS) codes for follow-up inpatient telehealth consultations, allowing practitioners to bill for follow-up inpatient consultations delivered via telehealth.
These codes are intended for use by physicians or non-physician providers when an inpatient consultation is requested from an appropriate source, such as the patient’s attending physician. CMS emphasizes the codes are not intended for use in billing for the ongoing evaluation and management of a hospital inpatient.
E-prescribe Out of Reach
Much attention has been given to a new Medicare program, which promotes the widespread adoption of electronic prescribing (e-prescribing). Physicians who successfully participate in CMS’ Electronic Prescribing Incentive Program will earn an extra bonus; however, the program was designed for primary care programs and hospitalists are unlikely to be able to take advantage of this.
“We don’t even know if hospitalists will be able to participate,” Dr. Siegal explains. The only way a hospitalist can take part in the e-prescribing initiative is if the hospital already has an acceptable system. However, Dr. Siegal warns, “If you create a mandate requiring a system for medication reconciliation at discharge, and then require another, separate system for e-prescribing, you’ve got problems. The primary driver should be that the hospital’s system supports both. And as far as we can tell, most hospital systems don’t do this.”
In August, SHM and the American College of Emergency Physicians conducted a teleconference with CMS to voice concerns with the e-prescribe initiative. “What we wanted was an exception,” Dr. Siegal says. SHM’s concern: When CMS stops rewarding physicians for e-prescribing and begins to penalize those who don’t—currently scheduled for 2013—hospitalists who can’t participate will be penalized through their Medicare payments. The outcome of the meeting, Dr. Siegal says, is “CMS turned around and said ‘either you can participate or you can’t.’ But at least they are considering our points; they seem to understand them.”
The good news is there is time to work the problem out, “At the moment, while e-prescribing is all bonus and no penalty, there’s no urgency to address it,” Dr. Siegal says.
Patient Safety
The Final Rule also includes improvements to PQRI, which allows eligible professionals to report on 153 quality measures. Physicians who successfully report on cases during 2009 will be able to earn an incentive payment, which has been increased to 2% (up from 1.5% in 2008), of their total allowed charges for covered professional services.
“I hope that more hospitalists will get on board with this,” Dr. Siegal says. He believes PQRI will be around for a while, and any hospital medicine group waiting to see if it is worth investing in the program can safely do so. “My feeling is that there’s growing bi-partisan support for something like this. I think it’s here to stay,” Dr. Siegal says.
SHM’s Opinion Counts
One reason the Final Rule is especially hospitalist-friendly is because SHM submitted extensive comment on CMS’s proposals in August. “SHM had a fair amount to say, and there are things in the rule that dovetail with our comments,” Dr. Siegal explains. “Part of the challenge is picking which battles to fight; there is a lot covered in this rule. We ended up focusing on areas that were really important to us, and on items where we thought we had a unique voice where nobody else was going to articulate.”
The Final Rule is available at www.cms.hhs.gov/center/physician.asp under “CMS-1403-FC.” Fact sheets covering major provisions of the Final Rule are available at www.cms.hhs.gov/apps/media/ fact_sheets.asp. TH
Jane Jerrard is a medical writer based in Chicago.
Beware Office Politics
Hospitalists routinely confront clinical, administrative, and ethical issues. Sometimes they face less-identifiable issues, such as office politics. Webster’s Dictionary defines office politics as “factional scheming for power and status within a group.” Wikipedia describes office politics as “the use of one’s individual or assigned power within an employing organization for the purpose of obtaining advantages beyond one’s legitimate authority.”
How much does office politics affect hospital medicine?
“Of course there is office politics in any work environment,” says Heather A. Harris, MD, former director of Eden Inpatient Services in Castro Valley, Calif., and currently splitting time as a hospitalist at the University of California San Francisco and the Palo Alto Medical Foundation. Dr. Harris, however, believes office politics is rare within hospital medicine because, “It is a young field and a growing field; everyone is growing together, so things tend to be pretty democratic. This is especially true of newer groups.”
Then again, there are times hospitalists find themselves embroiled in office politics. When this happens, what should you do?
Take the High Ground
Although she’s encountered few cases of office politics in her career, Dr. Harris’ general advice for hospitalists is, “First, recognize it, and then try to be a good team player.” Stay above the fray and try to tread carefully around political situations, especially if you’re a manager or informal leader.
Mary Jo Gorman, MD, MBA, CEO of Advanced ICU Care in St. Louis, and former SHM president, advises hospitalists and group directors to “take the high ground, no matter how frustrated you become.” She stresses discretion: “You can talk about it to your spouse, but if you’re a leader, you can’t even [comment on someone’s behavior] in front of your group. You never know, especially if you’re in a relatively small community, when you’re going to need someone’s support. You need to stay on good terms with people.” Dr. Gorman’s advice for leaders holds true for individuals hospitalists caught up in office politics.
Power Struggles
The role hospital medicine groups play as change agents probably is the main reason office politics may develop. “Any time you’re introducing a new concept that somebody feels threatened by, you’re going to incur some defensive maneuvers,” Dr. Gorman warns. “Whether you’re introducing a new hospital medicine group, or trying to change something, like the admissions process in the emergency room, you’re going to disrupt someone’s actions. Then you’ll find a whole broad range of reactions. And the more a person feels threatened, the more aggressive they’ll become.”
Based on her experiences establishing Eden Inpatient Services in 2003, Dr. Harris knows bringing a hospital medicine group into a hospital for the first time can be “a very political situation.” You can be stepping on personal, professional, and financial toes. “When you’re part of a new hospital medicine group … you’re potentially poised to take a lot of business away from people,” Dr. Harris explains. “It’s difficult to navigate those waters and build relationships” with physicians you’re consulting with and with primary care physicians. “In a way, this even extends to nurses,” she says. “You’re suddenly going to be working with them on patient care, and changing the way they work.” Dr. Harris encourages hospitalists to be aware of touchy situations, so as not to inadvertently fuel the fire of office politics. “Especially for young physicians just starting out, there can be a lack of recognition of other people’s feelings and turfs,” she cautions.
Hospitalists faced with an office issue should combine the cautionary approach with a willingness to work with people, even those who are engaging in office politics. “When you’re implementing a change, regardless of what it is, you need to identify who will think it’s a good thing and who will not,” Dr. Gorman advises. “You need to speak with individuals in the latter group, or choose others to speak to them, to garner their support.”
Take, for example, proposing a new project for your hospital’s Quality Improvement committee. A cautionary approach and team building will go a long way. “You’ve got to get to the people on the committee ahead of time, explain what you want to do, and get their feedback and support,” Dr. Gorman says. “If you find someone who opposes it, make sure you have enough support to override them. Or, better yet, find someone who can approach them on the topic, maybe their partner or another member of their group. This is a very practical approach.”
Identify Informal Leadership
When considering this inclusive approach, don’t forget the indirect leadership. “You may have a member of the medical staff who has some informal authority or power, maybe they have the most years of experience, or bring a lot of patients to the hospital, or maybe they are a member of the same group as someone in power,” Dr. Gorman says. “These informal leaders can create a lot of disturbance.”
To avoid problems, either direct or indirect, with these types of people, identify them early and make it a point to include them in the plan. “Usually, you know who holds informal power within your organization or the hospital,” Dr. Gorman says. “All you have to do is talk to them and explain what you’re doing. No one likes to be surprised. You might have to make some changes to accommodate their concerns.”
If this tactic fails and you still face opposition, you might have to weigh how important the opposition is. “You may decide to move ahead, even if you have to make changes and the project takes more time,” she says. “For physicians working in hospitals, we’re all used to instant results. You have to understand that process change takes time and you may have to take something bit by bit, and not get immediate results or responses.”
Be tactful about other professionals’ territories and feelings. Keep communication open and avoid springing surprises on stakeholders. Most importantly, stick to the high ground. These simple steps can help you stay far from the minefield known as office politics. TH
Jane Jerrard is a medical writer based in Chicago. She also writes “Public Policy” for The Hospitalist.
Hospitalists routinely confront clinical, administrative, and ethical issues. Sometimes they face less-identifiable issues, such as office politics. Webster’s Dictionary defines office politics as “factional scheming for power and status within a group.” Wikipedia describes office politics as “the use of one’s individual or assigned power within an employing organization for the purpose of obtaining advantages beyond one’s legitimate authority.”
How much does office politics affect hospital medicine?
“Of course there is office politics in any work environment,” says Heather A. Harris, MD, former director of Eden Inpatient Services in Castro Valley, Calif., and currently splitting time as a hospitalist at the University of California San Francisco and the Palo Alto Medical Foundation. Dr. Harris, however, believes office politics is rare within hospital medicine because, “It is a young field and a growing field; everyone is growing together, so things tend to be pretty democratic. This is especially true of newer groups.”
Then again, there are times hospitalists find themselves embroiled in office politics. When this happens, what should you do?
Take the High Ground
Although she’s encountered few cases of office politics in her career, Dr. Harris’ general advice for hospitalists is, “First, recognize it, and then try to be a good team player.” Stay above the fray and try to tread carefully around political situations, especially if you’re a manager or informal leader.
Mary Jo Gorman, MD, MBA, CEO of Advanced ICU Care in St. Louis, and former SHM president, advises hospitalists and group directors to “take the high ground, no matter how frustrated you become.” She stresses discretion: “You can talk about it to your spouse, but if you’re a leader, you can’t even [comment on someone’s behavior] in front of your group. You never know, especially if you’re in a relatively small community, when you’re going to need someone’s support. You need to stay on good terms with people.” Dr. Gorman’s advice for leaders holds true for individuals hospitalists caught up in office politics.
Power Struggles
The role hospital medicine groups play as change agents probably is the main reason office politics may develop. “Any time you’re introducing a new concept that somebody feels threatened by, you’re going to incur some defensive maneuvers,” Dr. Gorman warns. “Whether you’re introducing a new hospital medicine group, or trying to change something, like the admissions process in the emergency room, you’re going to disrupt someone’s actions. Then you’ll find a whole broad range of reactions. And the more a person feels threatened, the more aggressive they’ll become.”
Based on her experiences establishing Eden Inpatient Services in 2003, Dr. Harris knows bringing a hospital medicine group into a hospital for the first time can be “a very political situation.” You can be stepping on personal, professional, and financial toes. “When you’re part of a new hospital medicine group … you’re potentially poised to take a lot of business away from people,” Dr. Harris explains. “It’s difficult to navigate those waters and build relationships” with physicians you’re consulting with and with primary care physicians. “In a way, this even extends to nurses,” she says. “You’re suddenly going to be working with them on patient care, and changing the way they work.” Dr. Harris encourages hospitalists to be aware of touchy situations, so as not to inadvertently fuel the fire of office politics. “Especially for young physicians just starting out, there can be a lack of recognition of other people’s feelings and turfs,” she cautions.
Hospitalists faced with an office issue should combine the cautionary approach with a willingness to work with people, even those who are engaging in office politics. “When you’re implementing a change, regardless of what it is, you need to identify who will think it’s a good thing and who will not,” Dr. Gorman advises. “You need to speak with individuals in the latter group, or choose others to speak to them, to garner their support.”
Take, for example, proposing a new project for your hospital’s Quality Improvement committee. A cautionary approach and team building will go a long way. “You’ve got to get to the people on the committee ahead of time, explain what you want to do, and get their feedback and support,” Dr. Gorman says. “If you find someone who opposes it, make sure you have enough support to override them. Or, better yet, find someone who can approach them on the topic, maybe their partner or another member of their group. This is a very practical approach.”
Identify Informal Leadership
When considering this inclusive approach, don’t forget the indirect leadership. “You may have a member of the medical staff who has some informal authority or power, maybe they have the most years of experience, or bring a lot of patients to the hospital, or maybe they are a member of the same group as someone in power,” Dr. Gorman says. “These informal leaders can create a lot of disturbance.”
To avoid problems, either direct or indirect, with these types of people, identify them early and make it a point to include them in the plan. “Usually, you know who holds informal power within your organization or the hospital,” Dr. Gorman says. “All you have to do is talk to them and explain what you’re doing. No one likes to be surprised. You might have to make some changes to accommodate their concerns.”
If this tactic fails and you still face opposition, you might have to weigh how important the opposition is. “You may decide to move ahead, even if you have to make changes and the project takes more time,” she says. “For physicians working in hospitals, we’re all used to instant results. You have to understand that process change takes time and you may have to take something bit by bit, and not get immediate results or responses.”
Be tactful about other professionals’ territories and feelings. Keep communication open and avoid springing surprises on stakeholders. Most importantly, stick to the high ground. These simple steps can help you stay far from the minefield known as office politics. TH
Jane Jerrard is a medical writer based in Chicago. She also writes “Public Policy” for The Hospitalist.
Hospitalists routinely confront clinical, administrative, and ethical issues. Sometimes they face less-identifiable issues, such as office politics. Webster’s Dictionary defines office politics as “factional scheming for power and status within a group.” Wikipedia describes office politics as “the use of one’s individual or assigned power within an employing organization for the purpose of obtaining advantages beyond one’s legitimate authority.”
How much does office politics affect hospital medicine?
“Of course there is office politics in any work environment,” says Heather A. Harris, MD, former director of Eden Inpatient Services in Castro Valley, Calif., and currently splitting time as a hospitalist at the University of California San Francisco and the Palo Alto Medical Foundation. Dr. Harris, however, believes office politics is rare within hospital medicine because, “It is a young field and a growing field; everyone is growing together, so things tend to be pretty democratic. This is especially true of newer groups.”
Then again, there are times hospitalists find themselves embroiled in office politics. When this happens, what should you do?
Take the High Ground
Although she’s encountered few cases of office politics in her career, Dr. Harris’ general advice for hospitalists is, “First, recognize it, and then try to be a good team player.” Stay above the fray and try to tread carefully around political situations, especially if you’re a manager or informal leader.
Mary Jo Gorman, MD, MBA, CEO of Advanced ICU Care in St. Louis, and former SHM president, advises hospitalists and group directors to “take the high ground, no matter how frustrated you become.” She stresses discretion: “You can talk about it to your spouse, but if you’re a leader, you can’t even [comment on someone’s behavior] in front of your group. You never know, especially if you’re in a relatively small community, when you’re going to need someone’s support. You need to stay on good terms with people.” Dr. Gorman’s advice for leaders holds true for individuals hospitalists caught up in office politics.
Power Struggles
The role hospital medicine groups play as change agents probably is the main reason office politics may develop. “Any time you’re introducing a new concept that somebody feels threatened by, you’re going to incur some defensive maneuvers,” Dr. Gorman warns. “Whether you’re introducing a new hospital medicine group, or trying to change something, like the admissions process in the emergency room, you’re going to disrupt someone’s actions. Then you’ll find a whole broad range of reactions. And the more a person feels threatened, the more aggressive they’ll become.”
Based on her experiences establishing Eden Inpatient Services in 2003, Dr. Harris knows bringing a hospital medicine group into a hospital for the first time can be “a very political situation.” You can be stepping on personal, professional, and financial toes. “When you’re part of a new hospital medicine group … you’re potentially poised to take a lot of business away from people,” Dr. Harris explains. “It’s difficult to navigate those waters and build relationships” with physicians you’re consulting with and with primary care physicians. “In a way, this even extends to nurses,” she says. “You’re suddenly going to be working with them on patient care, and changing the way they work.” Dr. Harris encourages hospitalists to be aware of touchy situations, so as not to inadvertently fuel the fire of office politics. “Especially for young physicians just starting out, there can be a lack of recognition of other people’s feelings and turfs,” she cautions.
Hospitalists faced with an office issue should combine the cautionary approach with a willingness to work with people, even those who are engaging in office politics. “When you’re implementing a change, regardless of what it is, you need to identify who will think it’s a good thing and who will not,” Dr. Gorman advises. “You need to speak with individuals in the latter group, or choose others to speak to them, to garner their support.”
Take, for example, proposing a new project for your hospital’s Quality Improvement committee. A cautionary approach and team building will go a long way. “You’ve got to get to the people on the committee ahead of time, explain what you want to do, and get their feedback and support,” Dr. Gorman says. “If you find someone who opposes it, make sure you have enough support to override them. Or, better yet, find someone who can approach them on the topic, maybe their partner or another member of their group. This is a very practical approach.”
Identify Informal Leadership
When considering this inclusive approach, don’t forget the indirect leadership. “You may have a member of the medical staff who has some informal authority or power, maybe they have the most years of experience, or bring a lot of patients to the hospital, or maybe they are a member of the same group as someone in power,” Dr. Gorman says. “These informal leaders can create a lot of disturbance.”
To avoid problems, either direct or indirect, with these types of people, identify them early and make it a point to include them in the plan. “Usually, you know who holds informal power within your organization or the hospital,” Dr. Gorman says. “All you have to do is talk to them and explain what you’re doing. No one likes to be surprised. You might have to make some changes to accommodate their concerns.”
If this tactic fails and you still face opposition, you might have to weigh how important the opposition is. “You may decide to move ahead, even if you have to make changes and the project takes more time,” she says. “For physicians working in hospitals, we’re all used to instant results. You have to understand that process change takes time and you may have to take something bit by bit, and not get immediate results or responses.”
Be tactful about other professionals’ territories and feelings. Keep communication open and avoid springing surprises on stakeholders. Most importantly, stick to the high ground. These simple steps can help you stay far from the minefield known as office politics. TH
Jane Jerrard is a medical writer based in Chicago. She also writes “Public Policy” for The Hospitalist.
The latest research you need to know
In This Edition
- Aspirin plus extended-release dipyridamole and clopidogrel provide similar outcomes in stroke.
- Traditional readings of bedside chest radiographs are insensitive in detecting intraatrial central venous catheter placement.
- Improved outcomes with bortezomib in myeloma treatment.
- ICD firings in cardiomyopathy patients are associated with worse outcomes.
- The clinical dehydration scale rapidly assesses the severity of dehydration in children.
- Liberal red blood cell transfusions may be harming patients.
- Five-year risk of colorectal neoplasia is low in patients with negative screening colonoscopy.
- Vital sign instability and oxygenation predict prognosis following hospitalization for community acquired pneumonia.
Is aspirin plus extended release dipyridamole more efficacious and safer than clopidogrel in preventing recurrent stroke?
Background: Recurrent stroke is a frequent (7% to 8% thrombotic stroke recurrence in first year) and disabling event after ischemic stroke. Multiple randomized trials demonstrate efficacy of anti-platelet agents for the prevention of recurrent stroke after non-cardioembolic stroke. However, direct comparisons and relative benefits of various antiplatelet agents are not well defined.
Study design: Randomized, double-blinded, two-by-two factorial design with intention-to-treat analysis.
Setting: A total of 20,333 patients from 695 centers in 35 countries, including the U.S.
Synopsis: This study directly compared aspirin plus extended-release dipyridamole to clopidogrel within the PRoFESS trial. A total of 20,333 patients were enrolled and followed up for a mean duration of 2.5 years. Eligible patients randomly were assigned to receive either 25 mg aspirin plus 200 mg extended-release twice a day, or clopidogrel 75 mg a day; and either telmisartan 80 mg once a day or placebo. Groups were similar at baseline.
The primary outcome of recurrent stroke was similar in both the aspirin plus extended-release dipyrimadole group and the clopidogrel group (9.0% vs. 8.8%). The composite secondary outcome of stroke, myocardial infarction or vascular death, and tertiary outcomes were similar in both groups. The trial showed statistical equivalence in the rates of recurrent stroke in the two groups.
Despite more frequent hemorrhagic strokes in the group receiving aspirin plus extended-release dipyridamole (4.1% vs. 3.6%), there was no significant difference in the risk of fatal or disabling stroke between both the groups.
Bottom line: Aspirin plus extended-release dipyridamole is equivalent to clopidogrel in the prevention of recurrent stroke, in terms of relevant efficacy and safety parameters.
Citation: Sacco RL, Diener H, Yusuf S, et.al. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008:359:1238-1251.
Is there a better method to judge the placement of central venous catheters?
Background: Placement of central venous catheters is common, particularly in critical care settings. Correct placement is usually confirmed by bedside chest radiography. The recommended location of the distal catheter tip is superior to the superior vena cava and right atria junction. However, determining this landmark on traditional bedside chest radiographs is frequently inaccurate.
Study design: Prospective, blinded study.
Setting: University hospital in Germany.
Synopsis: The researchers enrolled 212 patients scheduled for elective cardiac surgery. Either left or right internal jugular vein central lines were placed via EKG guidance, and more precisely evaluated by transesophageal echocardiography. Bedside chest radiographs were performed within three hours of admission to the ICU.
The radiologists were able to detect between 40% and 60% of incorrect central venous catheter placements when compared to transesophageal echocardiography. The researchers concluded TC-distance (tip of catheter to carina) of greater than 55 mm on chest X-ray performed better (98% accurate) in the detection of intra-atrial catheter placement, compared to conventional judgment by attending (93% accurate) or resident (53% accurate) radiologists. Limitations of the study include the use of only one attending radiologist. Secondly, the chest radiographs and echocardiograms were not done simultaneously, allowing for possible movement of the catheters between studies.
Bottom line: A TC distance of greater than 55 mm on chest X-ray should be further investigated as an accurate method to detect intra-atrial central venous catheters.
Citation: Wirsing M, Schummer C, Neumann R, et al. Is traditional reading of the bedside chest radiograph appropriate to detect intra-atrial central venous catheter position? Chest. 2008;134:527-533.
Does adding bortezomib to melphalan and prednisone improve outcomes in newly diagnosed myeloma?
Background: More than 50% of newly diagnosed myeloma patients are older than 65 and cannot receive optimal treatment with high-dose chemotherapy and stem-cell transplant. Previous trials have demonstrated patients with relapsed or refractory myeloma benefit from the administration of bortezomib, which sensitizes the myeloma cell lines to melphalan.
Study design: Randomized, open-label (unblinded) phase 3 study.
Setting: 151 centers, 22 countries in Asia, Europe, South and North America.
Synopsis: 682 patients with untreated multiple myeloma, who were ineligible for high-dose chemotherapy and stem cell transplant, were treated with bortezomib in combination with standard melphalan and prednisone, or melphalan and prednisone alone. The bortezomib group had improved partial or complete response (71% vs. 35%; NNT=3; p<0.001), increased median time to progression of disease (19.9 months vs. 13.1 months), and improved overall survival (87% vs. 78% over a 16-month median follow up; NNT=11; p=0.008). There were increased grade 3 adverse effects with the intervention, but no increase in grade 4 events or treatment related deaths compared to control. Limitations of the study include lack of blinding and involvement of the pharmaceutical company in data collection analysis, writing and editing of the manuscript.
Bottom line: Bortezomib is a valuable adjunct to standard treatment of multiple myeloma in patients over the age of 65 who may be ineligible for high-dose chemotherapy and stem cell transplant.
Citation: San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for the initial treatment of multiple myeloma. NEJM. 2008;359:906-917.
Does the occurrence of a shock increase the risk of death in cardiomyopathy patients with defibrillators?
Background: The SCD-HeFT trial, originally published in 2005, was instrumental in demonstrating the utility of defibrillators in the primary prevention of sudden cardiac deaths in patients with either ischemic or non-ischemic cardiomyopathy, NYHA class II or III, ejection fraction <35%, and no history of sustained ventricular tachycardia or fibrillation. This study re-examined the data derived from the SCD-HeFT trial to better understand the long-term prognosis of these patients with defibrillators who receive either appropriate shocks (ventricular fibrillation, sustained ventricular tachycardia), inappropriate shocks, or no shocks. Inappropriate shocks were defined as defibrillations due to supraventricular tachycardia, oversensing P or T waves as R waves, double counting of R waves, and artifact.
Study design: Retrospective cohort (analysis of patients randomized to implantable cardioverter-defibrillator (ICD) group in SCD-HeFT).
Setting: Multicenter trial.
Synopsis: The analysis demonstrated patients who received shocks were 11 times more likely to die compared with those who had no defibrillations (Hazard Ratio [HR]=11.3, p<0.001). These shocked patients were at more risk (HR=5.7, p<0.001) than those with inappropriate shocks (HR=2.0, p=0.002). Therefore, even inappropriate shocks themselves doubled the risk of death. Patients who received more than one shock, either appropriate or not, were at even higher odds of death (HR=8.3, p<0.001). The results highlight the higher mortality risk when patients with ICDs have received a shock (appropriate or inappropriate) and the need for further therapies to modify outcome in these patients.
Bottom Line: Appropriate or inappropriate defibrillations are associated with a poorer prognosis in patients with cardiomyopathy.
Citation: Poole JE, Johnson GW, Hellkamp AS, et al. Prognostic importance of defibrillator shocks in patients with heart failure. N Engl J Med. 2008(359):1009-1017.
Can a simple physical exam tool assess the degree of dehydration in children?
Background: Despite the frequency and the associated cost of acute gastroenteritis (AGE) in the pediatric population, there is no unified scale to assess the severity of dehydration. The authors of this paper previously reported a clinical dehydration scale (CDS) and applied it prospectively in a new cohort of patients ages 1 month to 5 years.
Study design: Prospective, observational study.
Setting: Single, tertiary care emergency department (ED) in Canada.
Synopsis: The CDS score is based purely on the physical findings of the child, including a) general appearance, b) eyes, c) mucous membranes, and d) amount of tears. On a point system, the patient is placed in one of three categories: no dehydration, some dehydration, moderate/severe dehydration. The trial enrolled 205 children and the CDS was applied by the triaging nurse. The attending ED pediatricians were blinded from this assessment. The CDS was able to predict the length of stay (mean + SD: no dehyrdation 245 + 181 mins; some dehydration, 397 + 302 mins; mod/severe dehydration, 501 + 389 mins), need for intravenous rehydration (none, 15%; some, 49%; mod/severe, 80%), and frequency of emesis/diarrhea as reported by the parents (none, 8.4 + 7.7; some, 13 + 10.7; mod/severe, 30.2 + 14.8). Only five children were categorized in the moderate/severe dehydration category, which may limit the ability to generalize the scoring system to that group of patients.
Bottom line: The CDS is an easy to use and promising tool to assess the severity of illness, expected ED length of stay, and need for intravenous rehydration in children with acute gastroenteritis.
Citation: Goldman RD, Friedman JN, Parkin PC. Validation of the clinical dehydration scale for children with acute gastroenteritis. Pediatrics. 2008;122(3):545-549.
Does maintaining a higher hemoglobin level benefit critical care patients?
Background: Historically, medical and surgical critical care patients liberally were transfused with little prospective evidence to support this approach. However, recent evidence has led to the use of a more-restricted transfusion threshold.
Study design: Systematic review and meta-analysis of cohort studies evaluating the effect of red blood cell (RBC) transfusion on patient outcomes.
Setting: Data sources include MEDLINE, Embase, and Cochrane databases.
Synopsis: The 45 cohort studies, including more than 272,000 patients, were selected due to focus on outcome measures, such as mortality, multiorgan dysfunction, acute respiratory distress syndrome, and infections. Primary studies were then placed into one of three categories: benefits outweigh the risk, neutral, or risks outweigh the benefit. Forty-two of these studies found the risks outweigh the benefits; two were neutral; and only one sub-study (in elderly patients with acute myocardial infarctions) suggested benefit outweighs the risk.
Although a systematic review of cohort studies has inherent limitations, the overwhelming direction of the results suggests statistically significant harm due to liberal transfusion practices (Summary Odd Ratios [OR] for a) death, OR = 1.69; b) infection, OR = 1.88; and c) Acute Respiratory Distress Syndrome, OR = 2.5). Due to the observational nature of the cohort studies, one might suspect RBC transfusions could simply reflect patient severity of illness. Thus, the harm suggested by the more liberal transfusion standards could just reflect the fact these patients carried a worse prognosis due to their respective illnesses.
Bottom Line: The preponderance of evidence suggests liberal transfusion practice is associated with increased morbidity and mortality of ICU patients. When considering RBC transfusions, the risks and benefits to each individual patient should be considered carefully.
Citation: Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008; 36(9);2667-2674.
What is the appropriate frequency of rescreening for patients with initial screening colonoscopies negative for adenomas?
Background: Colonoscopy is the preferred primary screening method for the detection of colorectal cancer and precancerous polyps. Data suggest colonoscopy may be performed too frequently and for inappropriate indications.
Study design: Retrospective cohort study.
Setting: Seven sites in central Indiana.
Synopsis: In this study of 2,436 persons with no adenomas on baseline screening colonoscopies, 1,256 of them (51.6%) were rescreened a mean of 5.34 + 1.34 years later. No cancers were found on rescreening. One or more adenomas were found in 201 persons (16.0%). Nineteen advanced adenomas were found in 16 persons (1.3%). Patients in this study were relatively young (mean age at baseline was 56.7 years). Men were more likely than women to have adenomas (RR 1.88; 95% CI 1.42-2.51) and to have advanced adenomas (RR 3.31; 95% CI 1.02-10.8).
Limitations included a small cohort sample size, as well as incomplete information on persons who did not follow up with the five-year examination. Also, there is uncertainty about the clinical significance of advanced adenomas.
Bottom Line: Among persons previously screened with colonoscopy who have no colorectal adenomas, the five-year risk of detecting an advanced adenoma is extremely low (1.3%), supporting a rescreening interval of more than five years after a normal colonoscopy. Men have greater risk, and may deserve a shorter interval screening.
Citation: Imperiale TF, Glowinski EA, Lin-Cooper C, et al. Five-year risk of colorectal neoplasia after negative screening colonoscopy. N Engl J Med. 2008;359:1218-1224.
Can validated discharge instability criteria predict mortality or readmission within 30 days of hospital discharge for community acquired pneumonia (CAP)?
Background: Prior prospective cohort data have delineated instability criteria utilizing vital sign criteria at hospital discharge for CAP. However, guidelines for determining patient readiness for discharge remain largely unstudied.
Study design: Prospective, observational cohort study.
Setting: A single, non-urban teaching hospital in Spain.
Synopsis: In this study, 870 adults with CAP were evaluated following discharge. Abnormal oxygenation and vital signs were utilized to calculate an instability score. Criteria for instability were defined as temperature >37.5° C, heart rate <100, respiratory rate >24, systolic blood pressure (SBP) <90 (or diastolic blood pressure, DBP <60), and oxygen saturation <90% (or PaO2 <60).
Of all the instability criteria, only low oxygenation predicted readmission at 30 days (Hazard Ratio [HR] 1.4, p=0.03). However, mortality was significantly increased when instability criteria of temperature (HR 4.5, p=0.04), blood pressure (HR 2.6, p=0.02), respiratory rate (HR 2.4, p=0.03), or oxygenation (HR 2.4, p=0.03) were met. Elevated heart rate was not found to predict death.
The authors assigned each of the significant instability criteria a score of one or two (based on the weight of its hazard ratio), with respiratory rate, low blood pressure, and low oxygenation each assigned one point, and temperature assigned two points. Patients with an instability score of two or more had a six-fold increased risk of death (HR 5.8; 95%, p=0.0001). The negative predictive value (NPV) of an instability score less than two was very helpful (NPV=98%) in identifying patients with low mortality risk; however, the positive predictive value (PPV) of an instability score >2 is not necessarily helpful (PPV=13%) clinically.
Bottom line: Patients with a temperature >37.5° C or any combination of RR >24, SBP <90 (or DBP <60), and SpO2 <90% (or Pa02 <60) are at increased risk of death. Identifying a low instability score is most helpful in clinical practice.
Citation: Capelastegui A, Espana P, Bilbao A, et al. Pneumonia: criteria for patient instability on hospital discharge. Chest. 2008;34:595-600.
In This Edition
- Aspirin plus extended-release dipyridamole and clopidogrel provide similar outcomes in stroke.
- Traditional readings of bedside chest radiographs are insensitive in detecting intraatrial central venous catheter placement.
- Improved outcomes with bortezomib in myeloma treatment.
- ICD firings in cardiomyopathy patients are associated with worse outcomes.
- The clinical dehydration scale rapidly assesses the severity of dehydration in children.
- Liberal red blood cell transfusions may be harming patients.
- Five-year risk of colorectal neoplasia is low in patients with negative screening colonoscopy.
- Vital sign instability and oxygenation predict prognosis following hospitalization for community acquired pneumonia.
Is aspirin plus extended release dipyridamole more efficacious and safer than clopidogrel in preventing recurrent stroke?
Background: Recurrent stroke is a frequent (7% to 8% thrombotic stroke recurrence in first year) and disabling event after ischemic stroke. Multiple randomized trials demonstrate efficacy of anti-platelet agents for the prevention of recurrent stroke after non-cardioembolic stroke. However, direct comparisons and relative benefits of various antiplatelet agents are not well defined.
Study design: Randomized, double-blinded, two-by-two factorial design with intention-to-treat analysis.
Setting: A total of 20,333 patients from 695 centers in 35 countries, including the U.S.
Synopsis: This study directly compared aspirin plus extended-release dipyridamole to clopidogrel within the PRoFESS trial. A total of 20,333 patients were enrolled and followed up for a mean duration of 2.5 years. Eligible patients randomly were assigned to receive either 25 mg aspirin plus 200 mg extended-release twice a day, or clopidogrel 75 mg a day; and either telmisartan 80 mg once a day or placebo. Groups were similar at baseline.
The primary outcome of recurrent stroke was similar in both the aspirin plus extended-release dipyrimadole group and the clopidogrel group (9.0% vs. 8.8%). The composite secondary outcome of stroke, myocardial infarction or vascular death, and tertiary outcomes were similar in both groups. The trial showed statistical equivalence in the rates of recurrent stroke in the two groups.
Despite more frequent hemorrhagic strokes in the group receiving aspirin plus extended-release dipyridamole (4.1% vs. 3.6%), there was no significant difference in the risk of fatal or disabling stroke between both the groups.
Bottom line: Aspirin plus extended-release dipyridamole is equivalent to clopidogrel in the prevention of recurrent stroke, in terms of relevant efficacy and safety parameters.
Citation: Sacco RL, Diener H, Yusuf S, et.al. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008:359:1238-1251.
Is there a better method to judge the placement of central venous catheters?
Background: Placement of central venous catheters is common, particularly in critical care settings. Correct placement is usually confirmed by bedside chest radiography. The recommended location of the distal catheter tip is superior to the superior vena cava and right atria junction. However, determining this landmark on traditional bedside chest radiographs is frequently inaccurate.
Study design: Prospective, blinded study.
Setting: University hospital in Germany.
Synopsis: The researchers enrolled 212 patients scheduled for elective cardiac surgery. Either left or right internal jugular vein central lines were placed via EKG guidance, and more precisely evaluated by transesophageal echocardiography. Bedside chest radiographs were performed within three hours of admission to the ICU.
The radiologists were able to detect between 40% and 60% of incorrect central venous catheter placements when compared to transesophageal echocardiography. The researchers concluded TC-distance (tip of catheter to carina) of greater than 55 mm on chest X-ray performed better (98% accurate) in the detection of intra-atrial catheter placement, compared to conventional judgment by attending (93% accurate) or resident (53% accurate) radiologists. Limitations of the study include the use of only one attending radiologist. Secondly, the chest radiographs and echocardiograms were not done simultaneously, allowing for possible movement of the catheters between studies.
Bottom line: A TC distance of greater than 55 mm on chest X-ray should be further investigated as an accurate method to detect intra-atrial central venous catheters.
Citation: Wirsing M, Schummer C, Neumann R, et al. Is traditional reading of the bedside chest radiograph appropriate to detect intra-atrial central venous catheter position? Chest. 2008;134:527-533.
Does adding bortezomib to melphalan and prednisone improve outcomes in newly diagnosed myeloma?
Background: More than 50% of newly diagnosed myeloma patients are older than 65 and cannot receive optimal treatment with high-dose chemotherapy and stem-cell transplant. Previous trials have demonstrated patients with relapsed or refractory myeloma benefit from the administration of bortezomib, which sensitizes the myeloma cell lines to melphalan.
Study design: Randomized, open-label (unblinded) phase 3 study.
Setting: 151 centers, 22 countries in Asia, Europe, South and North America.
Synopsis: 682 patients with untreated multiple myeloma, who were ineligible for high-dose chemotherapy and stem cell transplant, were treated with bortezomib in combination with standard melphalan and prednisone, or melphalan and prednisone alone. The bortezomib group had improved partial or complete response (71% vs. 35%; NNT=3; p<0.001), increased median time to progression of disease (19.9 months vs. 13.1 months), and improved overall survival (87% vs. 78% over a 16-month median follow up; NNT=11; p=0.008). There were increased grade 3 adverse effects with the intervention, but no increase in grade 4 events or treatment related deaths compared to control. Limitations of the study include lack of blinding and involvement of the pharmaceutical company in data collection analysis, writing and editing of the manuscript.
Bottom line: Bortezomib is a valuable adjunct to standard treatment of multiple myeloma in patients over the age of 65 who may be ineligible for high-dose chemotherapy and stem cell transplant.
Citation: San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for the initial treatment of multiple myeloma. NEJM. 2008;359:906-917.
Does the occurrence of a shock increase the risk of death in cardiomyopathy patients with defibrillators?
Background: The SCD-HeFT trial, originally published in 2005, was instrumental in demonstrating the utility of defibrillators in the primary prevention of sudden cardiac deaths in patients with either ischemic or non-ischemic cardiomyopathy, NYHA class II or III, ejection fraction <35%, and no history of sustained ventricular tachycardia or fibrillation. This study re-examined the data derived from the SCD-HeFT trial to better understand the long-term prognosis of these patients with defibrillators who receive either appropriate shocks (ventricular fibrillation, sustained ventricular tachycardia), inappropriate shocks, or no shocks. Inappropriate shocks were defined as defibrillations due to supraventricular tachycardia, oversensing P or T waves as R waves, double counting of R waves, and artifact.
Study design: Retrospective cohort (analysis of patients randomized to implantable cardioverter-defibrillator (ICD) group in SCD-HeFT).
Setting: Multicenter trial.
Synopsis: The analysis demonstrated patients who received shocks were 11 times more likely to die compared with those who had no defibrillations (Hazard Ratio [HR]=11.3, p<0.001). These shocked patients were at more risk (HR=5.7, p<0.001) than those with inappropriate shocks (HR=2.0, p=0.002). Therefore, even inappropriate shocks themselves doubled the risk of death. Patients who received more than one shock, either appropriate or not, were at even higher odds of death (HR=8.3, p<0.001). The results highlight the higher mortality risk when patients with ICDs have received a shock (appropriate or inappropriate) and the need for further therapies to modify outcome in these patients.
Bottom Line: Appropriate or inappropriate defibrillations are associated with a poorer prognosis in patients with cardiomyopathy.
Citation: Poole JE, Johnson GW, Hellkamp AS, et al. Prognostic importance of defibrillator shocks in patients with heart failure. N Engl J Med. 2008(359):1009-1017.
Can a simple physical exam tool assess the degree of dehydration in children?
Background: Despite the frequency and the associated cost of acute gastroenteritis (AGE) in the pediatric population, there is no unified scale to assess the severity of dehydration. The authors of this paper previously reported a clinical dehydration scale (CDS) and applied it prospectively in a new cohort of patients ages 1 month to 5 years.
Study design: Prospective, observational study.
Setting: Single, tertiary care emergency department (ED) in Canada.
Synopsis: The CDS score is based purely on the physical findings of the child, including a) general appearance, b) eyes, c) mucous membranes, and d) amount of tears. On a point system, the patient is placed in one of three categories: no dehydration, some dehydration, moderate/severe dehydration. The trial enrolled 205 children and the CDS was applied by the triaging nurse. The attending ED pediatricians were blinded from this assessment. The CDS was able to predict the length of stay (mean + SD: no dehyrdation 245 + 181 mins; some dehydration, 397 + 302 mins; mod/severe dehydration, 501 + 389 mins), need for intravenous rehydration (none, 15%; some, 49%; mod/severe, 80%), and frequency of emesis/diarrhea as reported by the parents (none, 8.4 + 7.7; some, 13 + 10.7; mod/severe, 30.2 + 14.8). Only five children were categorized in the moderate/severe dehydration category, which may limit the ability to generalize the scoring system to that group of patients.
Bottom line: The CDS is an easy to use and promising tool to assess the severity of illness, expected ED length of stay, and need for intravenous rehydration in children with acute gastroenteritis.
Citation: Goldman RD, Friedman JN, Parkin PC. Validation of the clinical dehydration scale for children with acute gastroenteritis. Pediatrics. 2008;122(3):545-549.
Does maintaining a higher hemoglobin level benefit critical care patients?
Background: Historically, medical and surgical critical care patients liberally were transfused with little prospective evidence to support this approach. However, recent evidence has led to the use of a more-restricted transfusion threshold.
Study design: Systematic review and meta-analysis of cohort studies evaluating the effect of red blood cell (RBC) transfusion on patient outcomes.
Setting: Data sources include MEDLINE, Embase, and Cochrane databases.
Synopsis: The 45 cohort studies, including more than 272,000 patients, were selected due to focus on outcome measures, such as mortality, multiorgan dysfunction, acute respiratory distress syndrome, and infections. Primary studies were then placed into one of three categories: benefits outweigh the risk, neutral, or risks outweigh the benefit. Forty-two of these studies found the risks outweigh the benefits; two were neutral; and only one sub-study (in elderly patients with acute myocardial infarctions) suggested benefit outweighs the risk.
Although a systematic review of cohort studies has inherent limitations, the overwhelming direction of the results suggests statistically significant harm due to liberal transfusion practices (Summary Odd Ratios [OR] for a) death, OR = 1.69; b) infection, OR = 1.88; and c) Acute Respiratory Distress Syndrome, OR = 2.5). Due to the observational nature of the cohort studies, one might suspect RBC transfusions could simply reflect patient severity of illness. Thus, the harm suggested by the more liberal transfusion standards could just reflect the fact these patients carried a worse prognosis due to their respective illnesses.
Bottom Line: The preponderance of evidence suggests liberal transfusion practice is associated with increased morbidity and mortality of ICU patients. When considering RBC transfusions, the risks and benefits to each individual patient should be considered carefully.
Citation: Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008; 36(9);2667-2674.
What is the appropriate frequency of rescreening for patients with initial screening colonoscopies negative for adenomas?
Background: Colonoscopy is the preferred primary screening method for the detection of colorectal cancer and precancerous polyps. Data suggest colonoscopy may be performed too frequently and for inappropriate indications.
Study design: Retrospective cohort study.
Setting: Seven sites in central Indiana.
Synopsis: In this study of 2,436 persons with no adenomas on baseline screening colonoscopies, 1,256 of them (51.6%) were rescreened a mean of 5.34 + 1.34 years later. No cancers were found on rescreening. One or more adenomas were found in 201 persons (16.0%). Nineteen advanced adenomas were found in 16 persons (1.3%). Patients in this study were relatively young (mean age at baseline was 56.7 years). Men were more likely than women to have adenomas (RR 1.88; 95% CI 1.42-2.51) and to have advanced adenomas (RR 3.31; 95% CI 1.02-10.8).
Limitations included a small cohort sample size, as well as incomplete information on persons who did not follow up with the five-year examination. Also, there is uncertainty about the clinical significance of advanced adenomas.
Bottom Line: Among persons previously screened with colonoscopy who have no colorectal adenomas, the five-year risk of detecting an advanced adenoma is extremely low (1.3%), supporting a rescreening interval of more than five years after a normal colonoscopy. Men have greater risk, and may deserve a shorter interval screening.
Citation: Imperiale TF, Glowinski EA, Lin-Cooper C, et al. Five-year risk of colorectal neoplasia after negative screening colonoscopy. N Engl J Med. 2008;359:1218-1224.
Can validated discharge instability criteria predict mortality or readmission within 30 days of hospital discharge for community acquired pneumonia (CAP)?
Background: Prior prospective cohort data have delineated instability criteria utilizing vital sign criteria at hospital discharge for CAP. However, guidelines for determining patient readiness for discharge remain largely unstudied.
Study design: Prospective, observational cohort study.
Setting: A single, non-urban teaching hospital in Spain.
Synopsis: In this study, 870 adults with CAP were evaluated following discharge. Abnormal oxygenation and vital signs were utilized to calculate an instability score. Criteria for instability were defined as temperature >37.5° C, heart rate <100, respiratory rate >24, systolic blood pressure (SBP) <90 (or diastolic blood pressure, DBP <60), and oxygen saturation <90% (or PaO2 <60).
Of all the instability criteria, only low oxygenation predicted readmission at 30 days (Hazard Ratio [HR] 1.4, p=0.03). However, mortality was significantly increased when instability criteria of temperature (HR 4.5, p=0.04), blood pressure (HR 2.6, p=0.02), respiratory rate (HR 2.4, p=0.03), or oxygenation (HR 2.4, p=0.03) were met. Elevated heart rate was not found to predict death.
The authors assigned each of the significant instability criteria a score of one or two (based on the weight of its hazard ratio), with respiratory rate, low blood pressure, and low oxygenation each assigned one point, and temperature assigned two points. Patients with an instability score of two or more had a six-fold increased risk of death (HR 5.8; 95%, p=0.0001). The negative predictive value (NPV) of an instability score less than two was very helpful (NPV=98%) in identifying patients with low mortality risk; however, the positive predictive value (PPV) of an instability score >2 is not necessarily helpful (PPV=13%) clinically.
Bottom line: Patients with a temperature >37.5° C or any combination of RR >24, SBP <90 (or DBP <60), and SpO2 <90% (or Pa02 <60) are at increased risk of death. Identifying a low instability score is most helpful in clinical practice.
Citation: Capelastegui A, Espana P, Bilbao A, et al. Pneumonia: criteria for patient instability on hospital discharge. Chest. 2008;34:595-600.
In This Edition
- Aspirin plus extended-release dipyridamole and clopidogrel provide similar outcomes in stroke.
- Traditional readings of bedside chest radiographs are insensitive in detecting intraatrial central venous catheter placement.
- Improved outcomes with bortezomib in myeloma treatment.
- ICD firings in cardiomyopathy patients are associated with worse outcomes.
- The clinical dehydration scale rapidly assesses the severity of dehydration in children.
- Liberal red blood cell transfusions may be harming patients.
- Five-year risk of colorectal neoplasia is low in patients with negative screening colonoscopy.
- Vital sign instability and oxygenation predict prognosis following hospitalization for community acquired pneumonia.
Is aspirin plus extended release dipyridamole more efficacious and safer than clopidogrel in preventing recurrent stroke?
Background: Recurrent stroke is a frequent (7% to 8% thrombotic stroke recurrence in first year) and disabling event after ischemic stroke. Multiple randomized trials demonstrate efficacy of anti-platelet agents for the prevention of recurrent stroke after non-cardioembolic stroke. However, direct comparisons and relative benefits of various antiplatelet agents are not well defined.
Study design: Randomized, double-blinded, two-by-two factorial design with intention-to-treat analysis.
Setting: A total of 20,333 patients from 695 centers in 35 countries, including the U.S.
Synopsis: This study directly compared aspirin plus extended-release dipyridamole to clopidogrel within the PRoFESS trial. A total of 20,333 patients were enrolled and followed up for a mean duration of 2.5 years. Eligible patients randomly were assigned to receive either 25 mg aspirin plus 200 mg extended-release twice a day, or clopidogrel 75 mg a day; and either telmisartan 80 mg once a day or placebo. Groups were similar at baseline.
The primary outcome of recurrent stroke was similar in both the aspirin plus extended-release dipyrimadole group and the clopidogrel group (9.0% vs. 8.8%). The composite secondary outcome of stroke, myocardial infarction or vascular death, and tertiary outcomes were similar in both groups. The trial showed statistical equivalence in the rates of recurrent stroke in the two groups.
Despite more frequent hemorrhagic strokes in the group receiving aspirin plus extended-release dipyridamole (4.1% vs. 3.6%), there was no significant difference in the risk of fatal or disabling stroke between both the groups.
Bottom line: Aspirin plus extended-release dipyridamole is equivalent to clopidogrel in the prevention of recurrent stroke, in terms of relevant efficacy and safety parameters.
Citation: Sacco RL, Diener H, Yusuf S, et.al. Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med. 2008:359:1238-1251.
Is there a better method to judge the placement of central venous catheters?
Background: Placement of central venous catheters is common, particularly in critical care settings. Correct placement is usually confirmed by bedside chest radiography. The recommended location of the distal catheter tip is superior to the superior vena cava and right atria junction. However, determining this landmark on traditional bedside chest radiographs is frequently inaccurate.
Study design: Prospective, blinded study.
Setting: University hospital in Germany.
Synopsis: The researchers enrolled 212 patients scheduled for elective cardiac surgery. Either left or right internal jugular vein central lines were placed via EKG guidance, and more precisely evaluated by transesophageal echocardiography. Bedside chest radiographs were performed within three hours of admission to the ICU.
The radiologists were able to detect between 40% and 60% of incorrect central venous catheter placements when compared to transesophageal echocardiography. The researchers concluded TC-distance (tip of catheter to carina) of greater than 55 mm on chest X-ray performed better (98% accurate) in the detection of intra-atrial catheter placement, compared to conventional judgment by attending (93% accurate) or resident (53% accurate) radiologists. Limitations of the study include the use of only one attending radiologist. Secondly, the chest radiographs and echocardiograms were not done simultaneously, allowing for possible movement of the catheters between studies.
Bottom line: A TC distance of greater than 55 mm on chest X-ray should be further investigated as an accurate method to detect intra-atrial central venous catheters.
Citation: Wirsing M, Schummer C, Neumann R, et al. Is traditional reading of the bedside chest radiograph appropriate to detect intra-atrial central venous catheter position? Chest. 2008;134:527-533.
Does adding bortezomib to melphalan and prednisone improve outcomes in newly diagnosed myeloma?
Background: More than 50% of newly diagnosed myeloma patients are older than 65 and cannot receive optimal treatment with high-dose chemotherapy and stem-cell transplant. Previous trials have demonstrated patients with relapsed or refractory myeloma benefit from the administration of bortezomib, which sensitizes the myeloma cell lines to melphalan.
Study design: Randomized, open-label (unblinded) phase 3 study.
Setting: 151 centers, 22 countries in Asia, Europe, South and North America.
Synopsis: 682 patients with untreated multiple myeloma, who were ineligible for high-dose chemotherapy and stem cell transplant, were treated with bortezomib in combination with standard melphalan and prednisone, or melphalan and prednisone alone. The bortezomib group had improved partial or complete response (71% vs. 35%; NNT=3; p<0.001), increased median time to progression of disease (19.9 months vs. 13.1 months), and improved overall survival (87% vs. 78% over a 16-month median follow up; NNT=11; p=0.008). There were increased grade 3 adverse effects with the intervention, but no increase in grade 4 events or treatment related deaths compared to control. Limitations of the study include lack of blinding and involvement of the pharmaceutical company in data collection analysis, writing and editing of the manuscript.
Bottom line: Bortezomib is a valuable adjunct to standard treatment of multiple myeloma in patients over the age of 65 who may be ineligible for high-dose chemotherapy and stem cell transplant.
Citation: San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for the initial treatment of multiple myeloma. NEJM. 2008;359:906-917.
Does the occurrence of a shock increase the risk of death in cardiomyopathy patients with defibrillators?
Background: The SCD-HeFT trial, originally published in 2005, was instrumental in demonstrating the utility of defibrillators in the primary prevention of sudden cardiac deaths in patients with either ischemic or non-ischemic cardiomyopathy, NYHA class II or III, ejection fraction <35%, and no history of sustained ventricular tachycardia or fibrillation. This study re-examined the data derived from the SCD-HeFT trial to better understand the long-term prognosis of these patients with defibrillators who receive either appropriate shocks (ventricular fibrillation, sustained ventricular tachycardia), inappropriate shocks, or no shocks. Inappropriate shocks were defined as defibrillations due to supraventricular tachycardia, oversensing P or T waves as R waves, double counting of R waves, and artifact.
Study design: Retrospective cohort (analysis of patients randomized to implantable cardioverter-defibrillator (ICD) group in SCD-HeFT).
Setting: Multicenter trial.
Synopsis: The analysis demonstrated patients who received shocks were 11 times more likely to die compared with those who had no defibrillations (Hazard Ratio [HR]=11.3, p<0.001). These shocked patients were at more risk (HR=5.7, p<0.001) than those with inappropriate shocks (HR=2.0, p=0.002). Therefore, even inappropriate shocks themselves doubled the risk of death. Patients who received more than one shock, either appropriate or not, were at even higher odds of death (HR=8.3, p<0.001). The results highlight the higher mortality risk when patients with ICDs have received a shock (appropriate or inappropriate) and the need for further therapies to modify outcome in these patients.
Bottom Line: Appropriate or inappropriate defibrillations are associated with a poorer prognosis in patients with cardiomyopathy.
Citation: Poole JE, Johnson GW, Hellkamp AS, et al. Prognostic importance of defibrillator shocks in patients with heart failure. N Engl J Med. 2008(359):1009-1017.
Can a simple physical exam tool assess the degree of dehydration in children?
Background: Despite the frequency and the associated cost of acute gastroenteritis (AGE) in the pediatric population, there is no unified scale to assess the severity of dehydration. The authors of this paper previously reported a clinical dehydration scale (CDS) and applied it prospectively in a new cohort of patients ages 1 month to 5 years.
Study design: Prospective, observational study.
Setting: Single, tertiary care emergency department (ED) in Canada.
Synopsis: The CDS score is based purely on the physical findings of the child, including a) general appearance, b) eyes, c) mucous membranes, and d) amount of tears. On a point system, the patient is placed in one of three categories: no dehydration, some dehydration, moderate/severe dehydration. The trial enrolled 205 children and the CDS was applied by the triaging nurse. The attending ED pediatricians were blinded from this assessment. The CDS was able to predict the length of stay (mean + SD: no dehyrdation 245 + 181 mins; some dehydration, 397 + 302 mins; mod/severe dehydration, 501 + 389 mins), need for intravenous rehydration (none, 15%; some, 49%; mod/severe, 80%), and frequency of emesis/diarrhea as reported by the parents (none, 8.4 + 7.7; some, 13 + 10.7; mod/severe, 30.2 + 14.8). Only five children were categorized in the moderate/severe dehydration category, which may limit the ability to generalize the scoring system to that group of patients.
Bottom line: The CDS is an easy to use and promising tool to assess the severity of illness, expected ED length of stay, and need for intravenous rehydration in children with acute gastroenteritis.
Citation: Goldman RD, Friedman JN, Parkin PC. Validation of the clinical dehydration scale for children with acute gastroenteritis. Pediatrics. 2008;122(3):545-549.
Does maintaining a higher hemoglobin level benefit critical care patients?
Background: Historically, medical and surgical critical care patients liberally were transfused with little prospective evidence to support this approach. However, recent evidence has led to the use of a more-restricted transfusion threshold.
Study design: Systematic review and meta-analysis of cohort studies evaluating the effect of red blood cell (RBC) transfusion on patient outcomes.
Setting: Data sources include MEDLINE, Embase, and Cochrane databases.
Synopsis: The 45 cohort studies, including more than 272,000 patients, were selected due to focus on outcome measures, such as mortality, multiorgan dysfunction, acute respiratory distress syndrome, and infections. Primary studies were then placed into one of three categories: benefits outweigh the risk, neutral, or risks outweigh the benefit. Forty-two of these studies found the risks outweigh the benefits; two were neutral; and only one sub-study (in elderly patients with acute myocardial infarctions) suggested benefit outweighs the risk.
Although a systematic review of cohort studies has inherent limitations, the overwhelming direction of the results suggests statistically significant harm due to liberal transfusion practices (Summary Odd Ratios [OR] for a) death, OR = 1.69; b) infection, OR = 1.88; and c) Acute Respiratory Distress Syndrome, OR = 2.5). Due to the observational nature of the cohort studies, one might suspect RBC transfusions could simply reflect patient severity of illness. Thus, the harm suggested by the more liberal transfusion standards could just reflect the fact these patients carried a worse prognosis due to their respective illnesses.
Bottom Line: The preponderance of evidence suggests liberal transfusion practice is associated with increased morbidity and mortality of ICU patients. When considering RBC transfusions, the risks and benefits to each individual patient should be considered carefully.
Citation: Marik PE, Corwin HL. Efficacy of red blood cell transfusion in the critically ill: a systematic review of the literature. Crit Care Med. 2008; 36(9);2667-2674.
What is the appropriate frequency of rescreening for patients with initial screening colonoscopies negative for adenomas?
Background: Colonoscopy is the preferred primary screening method for the detection of colorectal cancer and precancerous polyps. Data suggest colonoscopy may be performed too frequently and for inappropriate indications.
Study design: Retrospective cohort study.
Setting: Seven sites in central Indiana.
Synopsis: In this study of 2,436 persons with no adenomas on baseline screening colonoscopies, 1,256 of them (51.6%) were rescreened a mean of 5.34 + 1.34 years later. No cancers were found on rescreening. One or more adenomas were found in 201 persons (16.0%). Nineteen advanced adenomas were found in 16 persons (1.3%). Patients in this study were relatively young (mean age at baseline was 56.7 years). Men were more likely than women to have adenomas (RR 1.88; 95% CI 1.42-2.51) and to have advanced adenomas (RR 3.31; 95% CI 1.02-10.8).
Limitations included a small cohort sample size, as well as incomplete information on persons who did not follow up with the five-year examination. Also, there is uncertainty about the clinical significance of advanced adenomas.
Bottom Line: Among persons previously screened with colonoscopy who have no colorectal adenomas, the five-year risk of detecting an advanced adenoma is extremely low (1.3%), supporting a rescreening interval of more than five years after a normal colonoscopy. Men have greater risk, and may deserve a shorter interval screening.
Citation: Imperiale TF, Glowinski EA, Lin-Cooper C, et al. Five-year risk of colorectal neoplasia after negative screening colonoscopy. N Engl J Med. 2008;359:1218-1224.
Can validated discharge instability criteria predict mortality or readmission within 30 days of hospital discharge for community acquired pneumonia (CAP)?
Background: Prior prospective cohort data have delineated instability criteria utilizing vital sign criteria at hospital discharge for CAP. However, guidelines for determining patient readiness for discharge remain largely unstudied.
Study design: Prospective, observational cohort study.
Setting: A single, non-urban teaching hospital in Spain.
Synopsis: In this study, 870 adults with CAP were evaluated following discharge. Abnormal oxygenation and vital signs were utilized to calculate an instability score. Criteria for instability were defined as temperature >37.5° C, heart rate <100, respiratory rate >24, systolic blood pressure (SBP) <90 (or diastolic blood pressure, DBP <60), and oxygen saturation <90% (or PaO2 <60).
Of all the instability criteria, only low oxygenation predicted readmission at 30 days (Hazard Ratio [HR] 1.4, p=0.03). However, mortality was significantly increased when instability criteria of temperature (HR 4.5, p=0.04), blood pressure (HR 2.6, p=0.02), respiratory rate (HR 2.4, p=0.03), or oxygenation (HR 2.4, p=0.03) were met. Elevated heart rate was not found to predict death.
The authors assigned each of the significant instability criteria a score of one or two (based on the weight of its hazard ratio), with respiratory rate, low blood pressure, and low oxygenation each assigned one point, and temperature assigned two points. Patients with an instability score of two or more had a six-fold increased risk of death (HR 5.8; 95%, p=0.0001). The negative predictive value (NPV) of an instability score less than two was very helpful (NPV=98%) in identifying patients with low mortality risk; however, the positive predictive value (PPV) of an instability score >2 is not necessarily helpful (PPV=13%) clinically.
Bottom line: Patients with a temperature >37.5° C or any combination of RR >24, SBP <90 (or DBP <60), and SpO2 <90% (or Pa02 <60) are at increased risk of death. Identifying a low instability score is most helpful in clinical practice.
Citation: Capelastegui A, Espana P, Bilbao A, et al. Pneumonia: criteria for patient instability on hospital discharge. Chest. 2008;34:595-600.