User login
2023 Update on menopause
This year’s menopause Update highlights a highly effective nonhormonal medication that recently received approval by the US Food and Drug Administration (FDA) for the treatment of bothersome menopausal vasomotor symptoms. In addition, the Update provides guidance regarding how ObGyns should respond when an endometrial biopsy for postmenopausal bleeding reveals proliferative changes.
Breakthrough in women’s health: A new nonhormone therapy for vasomotor symptoms
Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate-to-severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. 2023;dgad058. doi:10.1210/clinem/dgad058.
Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401:1091-1102. doi:10.1016/S0140-6736(23)00085-5.
A new oral nonestrogen-containing medication for relief of moderate to severe hot flashes, fezolinetant (Veozah) 45 mg daily, has been approved by the FDA and was expected to be available by the end of May 2023. Fezolinetant is a selective neurokinin 3 (NK3) receptor antagonistthat offers a targeted nonhormonal approach to menopausal vasomotor symptoms (VMS), and it is the first in its class to make it to market.
The decline in estrogen at menopause appears to result in increased signaling at kisspeptin/neurokinin B/dynorphin (KNDy) neurons in the thermoregulatory center within the hypothalamus with resultant increases in hot flashes.1,2 Fezolinetant works by binding to and blocking the activities of the NK3 receptor.3-5
Key study findings
Selective NK3 receptor antagonists, including fezolinetant, effectively reduce the frequency and severity of VMS comparable to that of hormone therapy (HT). Two phase 3 clinical trials, Skylight 1 and 2, confirmed the efficacy and safety of fezolinetant 45 mg in treating VMS,6,7 and an additional 52-week placebo-controlled study, Skylight 4, confirmed long-term safety.8 Onset of action occurs within a week. Reported adverse events occurred in 1% to 2% of healthy menopausal women participating in clinical trials; these included headaches, abdominal pain, diarrhea, insomnia, back pain, hot flushes, and reversible elevated hepatic transaminase levels.6-9
The published phase 2 trials9 and the international randomized controlled trial (RCT) 12-week studies, Skylight 1 and 2,6,7 found that once-daily 30-mg and 45-mg doses of fezolinetant significantly reduced VMS frequency and severity at 12 weeks among women aged 40 to 60 years who reported an average of 7 moderate to severe VMS/day; the reduction in reported VMS was sustained at 40 weeks. Phase 3 data from Skylight 1 and 2 demonstrated fezolinetant’s efficacy in reducing the frequency and severity of VMS and provided information on the safety profile of fezolinetant compared with placebo over 12 weeks and a noncontrolled extension for an additional 40 weeks.6,7
Oral fezolinetant was associated with improved quality of life, including reduced VMS-related interference with daily life.10 Johnson and colleagues, reporting for Skylight 2, found VMS frequency and severity improvement by week 1, which achieved statistical significance at weeks 4 and 12, with this improvement maintained through week 52.6 A 64.3% reduction in mean daily VMS from baseline was seen at 12 weeks for fezolinetant 45 mg compared with a 45.4% reduction for placebo. VMS severity significantly decreased compared with placebo at 4 and 12 weeks.6
Serious treatment-emergent adverse events were infrequent, reported by 2%, 1%, and 0% of those receiving fezolinetant 30 mg, fezolinetant 45 mg, and placebo.6 Increases in levels of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) were noted and were described as asymptomatic, isolated, intermittent, or transient, and these levels returned to baseline during treatment or after discontinuation.6
Of the 5 participants taking fezolinetant in Skyline 1 with ALT or AST levels greater than 3 times the upper limit of normal in the 12-week randomized trial, levels returned to normal range while continuing treatment in 2 participants, with treatment interruption in 2, and with discontinuation in 1. No new safety signals were seen in the 40-week extension trial.6
Fezolinetant offers a much-needed effective and safe selective nonhormone NK3 receptor antagonist therapy that reduces the frequency and severity of menopausal VMS and has been shown to be safe through 52 weeks of treatment.
To read more about how fezolinetant specifically targets the hormone receptor that triggers hot flashes as well as on prescribing hormone therapy for women with menopausal symptoms, see “Focus on menopause: Q&A with Jan Shifren, MD, and Genevieve NealPerry, MD, PhD,” in the December 2022 issue of OBG Management at https://www.mdedge.com/obgyn/article/260380/menopause
Continue to: Endometrial and bone safety...
Endometrial and bone safety
Results from Skylight 4, a phase 3, randomized, double-blind, 52-week safety study, provided additional evidence that confirmed the longer-term safety of fezolinetant over a 52-week treatment period.8
Endometrial safety was assessed in postmenopausal women with normal baseline endometrium (n = 599).8 For fezolinetant 45 mg, 1 of 203 participants had endometrial hyperplasia (EH) (0.5%; upper limit of one-sided 95% confidence interval [CI], 2.3%); no cases of EH were noted in the placebo (0 of 186) or fezolinetant 30-mg (0 of 210) groups. The incidence of EH or malignancy in fezolinetant-treated participants was within prespecified limits, as assessed by blinded, centrally read endometrial biopsies. Endometrial malignancy occurred in 1 of 210 in the fezolinetant 30-mg group (0.5%; 95% CI, 2.2%) with no cases in the other groups, thus meeting FDA requirements for endometrial safety.8
In addition, no significant differences were noted in change from baseline endometrial thickness on transvaginal ultrasonography between fezolinetant-treated and placebo groups. Likewise, no loss of bone density was found on dual-energy x-ray absorptiometry (DEXA) scans or trabecular bone scores.8
Liver safety
Although no cases of severe liver injury were noted, elevations in serum transaminase concentrations greater than 3 times the upper limit of normal were observed in the clinical trials. In Skylight 4, liver enzyme elevations more than 3 times the upper limit of normal occurred in 6 of 583 participants taking placebo, 8 of 590 taking fezolinetant 30 mg, and 12 of 589 taking fezolinetant 45 mg.8
The prescribing information for fezolinetant includes a warning for elevated hepatic transaminases: Fezolinetant should not be started if baseline serum transaminase concentration is equal to or exceeds 2 times the upper limit of normal. Liver tests should be obtained at baseline and repeated every 3 months for the first 9 months and then if symptoms suggest liver injury.11,12
Unmet need for nonhormone treatment of VMS
Vasomotor symptoms affect up to 80% of women, with approximately 25% bothersome enough to warrant treatment. Vasomotor symptoms persist for a median of 7 years, with duration and severity differing by race and ethnicity. Black, Hispanic, and possibly Native American women experience the highest burden of VMS.2 Although VMS, including hot flashes, night sweats, and mood and sleep disturbances, often are considered an annoyance to those with mild symptoms, moderate to severe VMS impact women’s lives, including functioning at home or work, affecting relationships, and decreasing perceived quality of life, and they have been associated with workplace absenteeism and increased health care costs, both direct from medical care and testing and indirect costs from lost work.13-15
Women with 7 or more daily moderate to severe VMS (defined as with sweating or affecting function) reported interference with sleep (94%), concentration (84%), mood (85%), energy (77%), and sexual activity (61%).16 Moderately to severely bothersome VMS have been associated with impaired psychological and general well-being, affecting work performance.17 Based on a Mayo Clinic workplace survey, Faubion and colleagues estimated an annual loss of $1.8 billion in the United States for menopause-related missed work and a $28 billion loss when medical expenses were added.15
Menopausal HT has been the primary treatment for VMS and has been shown to reduce the frequency and severity of hot flashes, with additional benefits on sleep, mood, fatigue, bone loss and reduction of fracture, and genitourinary syndrome of menopause (GSM), and with potential improvement in cardiovascular health with decreased type 2 diabetes.18,19 For healthy women with early menopause and no contraindications, HT has been recommended until at least the age of natural menopause, as observational data suggest that HT prevents osteoporosis, cardiovascular disease, neurodegenerative changes, and sexual dysfunction for these women.19,20 Similarly, for healthy women younger than age 60 or within 10 years of menopause, initiating HT has been shown to be safe and effective in treating bothersome VMS and preventing osteoporotic fractures and genitourinary changes.19,21
Most systemic HT formulations are inexpensive (for example, available as generics), with multiple dosing and formulations available for use alone or combined as oral, transdermal, or vaginal therapies. Despite the fear that arose for clinicians and women from the initial 2002 findings of the Women’s Health Initiative regarding increased risk of breast cancer, stroke, venous thrombosis, cardiovascular disease, and dementia, major medical societies agree that when initiated at or soon after menopause, HT is a safe and effective therapy to relieve VMS, protect against bone loss, and treat genitourinary changes.19,21
Many women, however, cannot take HT, including those with estrogen-sensitive cancers, such as breast or uterine cancers; prior cardiovascular disease, stroke, or venous thrombotic events; severe endometriosis; or migraine headaches with visual auras.2 In addition, many symptomatic menopausal women without health contraindications choose not to take HT.2 Until now, the only FDA-approved VMS nonhormone therapy has been a low-dose 7.5-mg paroxetine salt. Unfortunately, this formulation, along with the off-label use of other antidepressants (selective serotonin reuptake inhibitors and serotonin and norepinephrine reuptake inhibitors), gabapentinoids, oxybutynin, and clonidine, are substantially less effective than HT in treating moderate to severe VMS.
Bottom line
A substantial unmet need remains for effective therapy for moderate to severe VMS for women who cannot or choose not to take menopausal HT to relieve VMS.2,16 Effective, safe nonhormone treatment options such as the new NK3 receptor antagonist fezolinetant will address this clinically important need.
One concern is that the cost of developing and bringing to market the first of a new type of medication will be passed on to consumers, which may put it out of the price range for the many women who need it. However, the development and FDA approval of fezolinetant as the first NK3 receptor antagonist to treat menopausal VMS is potentially a practice changer. It provides a novel, effective, and safe FDA-approved nonhormonal treatment for menopausal women with moderate to severe VMS, particularly for women who cannot or will not take hormone therapy.
Continue to: When endometrial biopsy for postmenopausal bleeding reveals proliferative changes, how should we respond?...
When endometrial biopsy for postmenopausal bleeding reveals proliferative changes, how should we respond?
Abraham C. Proliferative endometrium in menopause: to treat or not to treat? Obstet Gynecol. 2023;141:265-267. doi:10.1097/AOG.0000000000005054.
The following case represents a common scenario for ObGyns.
CASE Patient with proliferative endometrial changes
A menopausal patient with a body mass index (BMI) > 30 kg/m2 presents with uterine bleeding. She does not use systemic menopausal hormone therapy. Endometrial biopsy indicates proliferative changes.
When endometrial biopsy performed for bleeding reveals proliferative changes in menopausal women, we traditionally have responded by reassuring the patient that the findings are benign and advising that she should let us know if future spotting or bleeding occurs.
However, a recent review by Abraham published in Obstetrics and Gynecology details the implications of proliferative endometrial changes in menopausal patients, advising that treatment, as well as monitoring, may be appropriate.22
Endometrial changes and what they suggest
In premenopausal women, proliferative endometrial changes are physiologic and result from ovarian estrogen production early in each cycle, during what is called the proliferative (referring to the endometrium) or follicular (referring to the dominant follicle that synthesizes estrogen) phase. In menopausal women who are not using HT, however, proliferative endometrial changes, with orderly uniform glands seen on histologic evaluation, reflect aromatization of androgens by adipose and other tissues into estrogen.
The next step on the continuum to hyperplasia (benign or atypical) after proliferative endometrium is disordered proliferative endometrium. At this stage, histologic evaluation reveals scattered cystic and dilated glands that have a normal gland-to-stroma ratio with a low gland density overall and without any atypia. Randomly distributed glands may have tubal metaplasia or fibrin thrombi associated with microinfarcts, often presenting with irregular bleeding. This is a noncancerous change that occurs with excess estrogen (endogenous or exogenous).23
Progestins reverse endometrial hyperplasia by activating progesterone receptors, which leads to stromal decidualization with thinning of the endometrium. They have a pronounced effect on the histologic appearance of the endometrium. By contrast, endometrial intraepithelial neoplasia (EIN, previously known as endometrial hyperplasiawith atypia) shows underlying molecular mutations and histologic alterations and represents a sharp transition to true neoplasia, which greatly increases the risk of endometrioid endometrial adenocarcinoma.24
For decades, we have been aware that if women diagnosed with endometrial hyperplasia are not treated with progestational therapy, their future risk of endometrial cancer is elevated. More recently, we also recognize that menopausal women found to have proliferative endometrial changes, if not treated, have an increased risk of endometrial cancer.
In a retrospective cohort study of almost 300 menopausal women who were not treated after endometrial biopsy revealed proliferative changes, investigators followed participants for an average of 11 years.25 These women had a mean BMI of 34 kg/m2. During follow-up, almost 12% of these women were diagnosed with endometrial hyperplasia or cancer. This incidence of endometrial neoplasia was some 4 times higher than for women initially found to have atrophic endometrial changes.25
Progestin treatment
Oral progestin therapy with follow-up endometrial biopsy constitutes traditional management for endometrial hyperplasia. Such therapy minimizes the likelihood that hyperplasia will progress to endometrial cancer.
We now recognize that the convenience, as well as the high endometrial progestin levels achieved, with levonorgestrel-releasing intrauterine devices (LNG-IUDs) have advantages over oral progestin therapy in treating endometrial hyperplasia. Indeed, a recent US report found that among women with EIN managed medically, use of progestin-releasing IUDs has grown from 7.7% in 2008 to 35.6% in 2020.26
Although both oral and intrauterine progestin are highly effective in treating simple hyperplasia, progestin IUDs are substantially more effective than oral progestins in treating EIN.27 Progestin concentrations in the endometrium have been shown to be 100-fold higher after LNG-IUD placement compared with oral progestin use.22 In addition, adverse effects, including bloating, unpleasant mood changes, and increased appetite, are more common with oral than intrauterine progestin therapy.28
Unfortunately, data from randomized trials addressing progestational treatment of proliferative endometrium in menopausal women are not available to support the treatment of proliferative endometrium with either oral progestins or the LNG-IUD.22
Role of ultrasonography
Another concern is relying on a finding of thin endometrial thickness on vaginal ultrasonography. In a simulated retrospective cohort study, use of transvaginal ultrasonography to determine the appropriateness of a biopsy was found not to be sufficiently accurate or racially equitable with regard to Black women.29 In simulated data, transvaginal ultrasonography missed almost 5 times more cases of endometrial cancer among Black women compared with White women due to higher fibroid prevalence and nonendometrioid histologic type malignancies in Black women.29
Assessing risk
If proliferative endometrium is found, Abraham suggests assessing risk using22:
- age
- comorbidities (including obesity)
- endometrial echo thickness on vaginal ultrasonography.
Consider the patient’s risk and tolerance of recurrent bleeding as well as her tolerance for progestational adverse effects if medical therapy is chosen. Discussion about next steps should include reviewing the histologic findings with the patient and discussing the difference in risk of progression to endometrial cancer of a finding of proliferative endometrium compared with a histologic finding of endometrial hyperplasia.
Using this patient-centered approach, observation over time with follow-up endometrial biopsies remains a management option. Although some women may tolerate micronized progesterone over synthetic progestins, there is concern that it may be less effective in suppressing the endometrium than synthetic progestins.30 Accordingly, synthetic progestins represent first-line options in this setting.
In her review, Abraham suggests that when endometrial biopsy reveals proliferative changes in a menopausal woman, we should initiate progestin treatment and perform surveillance endometrial sampling every 3 to 6 months. If such sampling reveals benign but not proliferative endometrium, progestin therapy can be stopped and endometrial biopsy repeated if bleeding recurs.22 ●
ObGyns may choose to adopt Abraham’s approach or to hold off on progestin therapy while performing follow-up endometrial sampling. Either way, the take-home message is that the finding of proliferative endometrial changes on biopsy for postmenopausal bleeding requires proactive management.
- Modi M, Dhillo WS. Neurokinin 3 receptor antagonism: a novel treatment for menopausal hot flushes. Neuroendocrinology. 2019;109:242-248. doi:10.1159/000495889
- Pinkerton JV, Redick DL, Homewood LN, et al. Neurokinin receptor antagonist, fezolinetant, for treatment of menopausal vasomotor symptoms. J Clin Endocrinol Metab. 2023;dgad209. doi:10.1210/clinem/dgad209
- Rance NE, Dacks PA, Mittelman-Smith MA, et al. Modulation of body temperature and LH secretion by hypothalamic KNDy (kisspeptin, neurokinin B and dynorphin) neurons: a novel hypothesis on the mechanism of hot flushes. Front Neuroendocrinol. 2013;34:211-227. doi:10.1016 /j.yfrne.2013.07.003
- Mittelman-Smith MA, Williams H, Krajewski-Hall SJ, et al. Role for kisspeptin/neurokinin B/dynorphin (KNDy) neurons in cutaneous vasodilatation and the estrogen modulation of body temperature. Proc Natl Acad Sci USA. 2012;109:1984619851. doi:10.1073/pnas.1211517109
- Astellas Pharma. Astellas’ Veozah (fezolinetant) approved by US FDA for treatment of vasomotor symptoms due to menopause. May 12, 2023. PR Newswire. Accessed May 15, 2023. https://www.prnewswire.com/news-releases/astellas-veozah-fezolinetant-approved-by-us-fda-for -treatment-of-vasomotor-symptoms-due-to-menopause -301823639.html
- Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate-to-severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. 2023;dgad058. doi:10.1210/clinem/dgad058
- Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401:1091-1102. doi:10.1016 /S0140-6736(23)00085-5
- Neal-Perry G, Cano A, Lederman S, et al. Safety of fezolinetant for vasomotor symptoms associated with menopause: a randomized controlled trial. Obstet Gynecol. 2023;141:737-747. doi:10.1097/AOG.0000000000005114
- Depypere H, Timmerman D, Donders G, et al. Treatment of menopausal vasomotor symptoms with fezolinetant, a neurokinin 3 receptor antagonist: a phase 2a trial. J Clin Endocrinol Metab. 2019;104:5893-5905. doi: 10.1210/jc .2019-00677
- Santoro N, Waldbaum A, Lederman S, et al. Effect of the neurokinin 3 receptor antagonist fezolinetant on patientreported outcomes in postmenopausal women with vasomotor symptoms: results of a randomized, placebo-controlled, double-blind, dose-ranging study (VESTA). Menopause. 2020;27:1350-1356. doi:10.1097/GME.0000000000001621
- FDA approves novel drug to treat moderate to severe hot flashes caused by menopause. May 12, 2023. US Food and Drug Administration. Accessed May 15, 2023. https://www .fda.gov/news-events/press-announcements/fda-approves -novel-drug-treat-moderate-severe-hot-flashes-caused -menopause
- Veozah. Prescribing information. Astellas; 2023. Accessed May 16, 2023. https://www.astellas.com/us/system/files /veozah_uspi.pdf
- Pinkerton JV. Money talks: untreated hot flashes cost women, the workplace, and society. Menopause. 2015;22:254-255. doi:10.1097/GME.0000000000000427
- Sarrel P, Portman D, Lefebvre P, et al. Incremental direct and indirect costs of untreated vasomotor symptoms. Menopause. 2015;22(3):260-266. doi:10.1097/GME.0000000000000320
- Faubion SS, Enders F, Hedges MS, et al. Impact of menopause symptoms on women in the workplace. Mayo Clin Proc. 2023;98:833-845. doi:10.1016/j.mayocp.2023.02.025
- Williams RE, Levine KB, Kalilani L, et al. Menopause- specific questionnaire assessment in US populationbased study shows negative impact on health-related quality of life. Maturitas. 2009;62:153-159. doi:10.1016 /j.maturitas.2008.12.006
- Gartoulla P, Bell RJ, Worsley R, et al. Moderate-severely bothersome vasomotor symptoms are associated with lowered psychological general wellbeing in women at midlife. Maturitas. 2015;81:487-492. doi:10.1016 /j.maturitas.2015.06.004
- Manson JE, Kaunitz AM. Menopause management—getting clinical care back on track. N Engl J Med. 2016;374:803-806. doi:10.1056/NEJMp1514242
- 2022 Hormone Therapy Position Statement of the North American Menopause Society Advisory Panel. The 2022 hormone therapy position statement of the North American Menopause Society. Menopause. 2022;29:767-794. doi:10.1097/GME.0000000000002028
- Kaunitz AM, Kapoor E, Faubion S. Treatment of women after bilateral salpingo-oophorectomy performed prior to natural menopause. JAMA. 2021;12;326:1429-1430. doi:10.1001 /jama.2021.3305
- Pinkerton JV. Hormone therapy for postmenopausal women. N Engl J Med. 2020;382:446-455. doi:10.1056 /NEJMcp1714787
- Abraham C. Proliferative endometrium in menopause: to treat or not to treat? Obstet Gynecol. 2023;141:265-267. doi:10.1097/AOG.0000000000005054
- Chandra V, Kim JJ, Benbrook DM, et al. Therapeutic options for management of endometrial hyperplasia. J Gynecol Oncol. 2016;27:e8. doi:10.3802/jgo.2016.27.e8
- Owings RA, Quick CM. Endometrial intraepithelial neoplasia. Arch Pathol Lab Med. 2014;138:484-491. doi:10.5858 /arpa.2012-0709-RA
- Rotenberg O, Doulaveris G, Fridman D, et al. Long-term outcome of postmenopausal women with proliferative endometrium on endometrial sampling. Am J Obstet Gynecol. 2020;223:896.e1-896.e7. doi:10.1016/j.ajog.2020.06.045
- Suzuki Y, Chen L, Hou JY, et al. Systemic progestins and progestin-releasing intrauterine device therapy for premenopausal patients with endometrial intraepithelial neoplasia. Obstet Gynecol. 2023;141:979-987. doi:10.1097 /AOG.0000000000005124
- Mandelbaum RS, Ciccone MA, Nusbaum DJ, et al. Progestin therapy for obese women with complex atypical hyperplasia: levonorgestrel-releasing intrauterine device vs systemic therapy. Am J Obstet Gynecol. 2020;223:103.e1-103.e13. doi:10.1016/j.ajog.2019.12.273
- Liu S, Kciuk O, Frank M, et al. Progestins of today and tomorrow. Curr Opin Obstet Gynecol. 2022;34:344-350. doi:10.1097 /GCO.0000000000000819
- Doll KM, Romano SS, Marsh EE, et al. Estimated performance of transvaginal ultrasonography for evaluation of postmenopausal bleeding in a simulated cohort of black and white women in the US. JAMA Oncol. 2021;7:1158-1165. doi:10.1001/jamaoncol.2021.1700
- Gompel A. Progesterone and endometrial cancer. Best Pract Res Clin Obstet Gynaecol. 2020;69:95-107. doi:10.1016 /j.bpobgyn.2020.05.003

Andrew M. Kaunitz, MD, NCMP
Dr. Kaunitz is Tenured Professor and Associate Chair, Department of Obstetrics and Gynecology, University of Florida College of Medicine–Jacksonville; and Medical Director and Director of Menopause and Gynecologic Ultrasound Services, University of Florida Health Women’s Specialist Services–Emerson, Jacksonville. He serves on the OBG Management Board of Editors.

JoAnn V. Pinkerton, MD, NCMP
Dr. Pinkerton is Division Director, Midlife Health, and Professor, Department of Obstetrics and Gynecology, University of Virginia Health, Charlottesville; Virginia; Executive Director Emeritus, The North American Menopause Society. She serves on the OBG Management Board of Editors.
Dr. Kaunitz reports that the University of Florida receives research support from Bayer. Dr. Pinkerton reports participating in a multicenter clinical trial on nonhormone therapy for hot flashes, for which the University of Virginia received financial support from Bayer.
Andrew M. Kaunitz, MD, NCMP
Dr. Kaunitz is Tenured Professor and Associate Chair, Department of Obstetrics and Gynecology, University of Florida College of Medicine–Jacksonville; and Medical Director and Director of Menopause and Gynecologic Ultrasound Services, University of Florida Health Women’s Specialist Services–Emerson, Jacksonville. He serves on the OBG Management Board of Editors.
JoAnn V. Pinkerton, MD, NCMP
Dr. Pinkerton is Division Director, Midlife Health, and Professor, Department of Obstetrics and Gynecology, University of Virginia Health, Charlottesville; Virginia; Executive Director Emeritus, The North American Menopause Society. She serves on the OBG Management Board of Editors.
Dr. Kaunitz reports that the University of Florida receives research support from Bayer. Dr. Pinkerton reports participating in a multicenter clinical trial on nonhormone therapy for hot flashes, for which the University of Virginia received financial support from Bayer.
Andrew M. Kaunitz, MD, NCMP
Dr. Kaunitz is Tenured Professor and Associate Chair, Department of Obstetrics and Gynecology, University of Florida College of Medicine–Jacksonville; and Medical Director and Director of Menopause and Gynecologic Ultrasound Services, University of Florida Health Women’s Specialist Services–Emerson, Jacksonville. He serves on the OBG Management Board of Editors.
JoAnn V. Pinkerton, MD, NCMP
Dr. Pinkerton is Division Director, Midlife Health, and Professor, Department of Obstetrics and Gynecology, University of Virginia Health, Charlottesville; Virginia; Executive Director Emeritus, The North American Menopause Society. She serves on the OBG Management Board of Editors.
Dr. Kaunitz reports that the University of Florida receives research support from Bayer. Dr. Pinkerton reports participating in a multicenter clinical trial on nonhormone therapy for hot flashes, for which the University of Virginia received financial support from Bayer.
This year’s menopause Update highlights a highly effective nonhormonal medication that recently received approval by the US Food and Drug Administration (FDA) for the treatment of bothersome menopausal vasomotor symptoms. In addition, the Update provides guidance regarding how ObGyns should respond when an endometrial biopsy for postmenopausal bleeding reveals proliferative changes.
Breakthrough in women’s health: A new nonhormone therapy for vasomotor symptoms
Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate-to-severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. 2023;dgad058. doi:10.1210/clinem/dgad058.
Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401:1091-1102. doi:10.1016/S0140-6736(23)00085-5.
A new oral nonestrogen-containing medication for relief of moderate to severe hot flashes, fezolinetant (Veozah) 45 mg daily, has been approved by the FDA and was expected to be available by the end of May 2023. Fezolinetant is a selective neurokinin 3 (NK3) receptor antagonistthat offers a targeted nonhormonal approach to menopausal vasomotor symptoms (VMS), and it is the first in its class to make it to market.
The decline in estrogen at menopause appears to result in increased signaling at kisspeptin/neurokinin B/dynorphin (KNDy) neurons in the thermoregulatory center within the hypothalamus with resultant increases in hot flashes.1,2 Fezolinetant works by binding to and blocking the activities of the NK3 receptor.3-5
Key study findings
Selective NK3 receptor antagonists, including fezolinetant, effectively reduce the frequency and severity of VMS comparable to that of hormone therapy (HT). Two phase 3 clinical trials, Skylight 1 and 2, confirmed the efficacy and safety of fezolinetant 45 mg in treating VMS,6,7 and an additional 52-week placebo-controlled study, Skylight 4, confirmed long-term safety.8 Onset of action occurs within a week. Reported adverse events occurred in 1% to 2% of healthy menopausal women participating in clinical trials; these included headaches, abdominal pain, diarrhea, insomnia, back pain, hot flushes, and reversible elevated hepatic transaminase levels.6-9
The published phase 2 trials9 and the international randomized controlled trial (RCT) 12-week studies, Skylight 1 and 2,6,7 found that once-daily 30-mg and 45-mg doses of fezolinetant significantly reduced VMS frequency and severity at 12 weeks among women aged 40 to 60 years who reported an average of 7 moderate to severe VMS/day; the reduction in reported VMS was sustained at 40 weeks. Phase 3 data from Skylight 1 and 2 demonstrated fezolinetant’s efficacy in reducing the frequency and severity of VMS and provided information on the safety profile of fezolinetant compared with placebo over 12 weeks and a noncontrolled extension for an additional 40 weeks.6,7
Oral fezolinetant was associated with improved quality of life, including reduced VMS-related interference with daily life.10 Johnson and colleagues, reporting for Skylight 2, found VMS frequency and severity improvement by week 1, which achieved statistical significance at weeks 4 and 12, with this improvement maintained through week 52.6 A 64.3% reduction in mean daily VMS from baseline was seen at 12 weeks for fezolinetant 45 mg compared with a 45.4% reduction for placebo. VMS severity significantly decreased compared with placebo at 4 and 12 weeks.6
Serious treatment-emergent adverse events were infrequent, reported by 2%, 1%, and 0% of those receiving fezolinetant 30 mg, fezolinetant 45 mg, and placebo.6 Increases in levels of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) were noted and were described as asymptomatic, isolated, intermittent, or transient, and these levels returned to baseline during treatment or after discontinuation.6
Of the 5 participants taking fezolinetant in Skyline 1 with ALT or AST levels greater than 3 times the upper limit of normal in the 12-week randomized trial, levels returned to normal range while continuing treatment in 2 participants, with treatment interruption in 2, and with discontinuation in 1. No new safety signals were seen in the 40-week extension trial.6
Fezolinetant offers a much-needed effective and safe selective nonhormone NK3 receptor antagonist therapy that reduces the frequency and severity of menopausal VMS and has been shown to be safe through 52 weeks of treatment.
To read more about how fezolinetant specifically targets the hormone receptor that triggers hot flashes as well as on prescribing hormone therapy for women with menopausal symptoms, see “Focus on menopause: Q&A with Jan Shifren, MD, and Genevieve NealPerry, MD, PhD,” in the December 2022 issue of OBG Management at https://www.mdedge.com/obgyn/article/260380/menopause
Continue to: Endometrial and bone safety...
Endometrial and bone safety
Results from Skylight 4, a phase 3, randomized, double-blind, 52-week safety study, provided additional evidence that confirmed the longer-term safety of fezolinetant over a 52-week treatment period.8
Endometrial safety was assessed in postmenopausal women with normal baseline endometrium (n = 599).8 For fezolinetant 45 mg, 1 of 203 participants had endometrial hyperplasia (EH) (0.5%; upper limit of one-sided 95% confidence interval [CI], 2.3%); no cases of EH were noted in the placebo (0 of 186) or fezolinetant 30-mg (0 of 210) groups. The incidence of EH or malignancy in fezolinetant-treated participants was within prespecified limits, as assessed by blinded, centrally read endometrial biopsies. Endometrial malignancy occurred in 1 of 210 in the fezolinetant 30-mg group (0.5%; 95% CI, 2.2%) with no cases in the other groups, thus meeting FDA requirements for endometrial safety.8
In addition, no significant differences were noted in change from baseline endometrial thickness on transvaginal ultrasonography between fezolinetant-treated and placebo groups. Likewise, no loss of bone density was found on dual-energy x-ray absorptiometry (DEXA) scans or trabecular bone scores.8
Liver safety
Although no cases of severe liver injury were noted, elevations in serum transaminase concentrations greater than 3 times the upper limit of normal were observed in the clinical trials. In Skylight 4, liver enzyme elevations more than 3 times the upper limit of normal occurred in 6 of 583 participants taking placebo, 8 of 590 taking fezolinetant 30 mg, and 12 of 589 taking fezolinetant 45 mg.8
The prescribing information for fezolinetant includes a warning for elevated hepatic transaminases: Fezolinetant should not be started if baseline serum transaminase concentration is equal to or exceeds 2 times the upper limit of normal. Liver tests should be obtained at baseline and repeated every 3 months for the first 9 months and then if symptoms suggest liver injury.11,12
Unmet need for nonhormone treatment of VMS
Vasomotor symptoms affect up to 80% of women, with approximately 25% bothersome enough to warrant treatment. Vasomotor symptoms persist for a median of 7 years, with duration and severity differing by race and ethnicity. Black, Hispanic, and possibly Native American women experience the highest burden of VMS.2 Although VMS, including hot flashes, night sweats, and mood and sleep disturbances, often are considered an annoyance to those with mild symptoms, moderate to severe VMS impact women’s lives, including functioning at home or work, affecting relationships, and decreasing perceived quality of life, and they have been associated with workplace absenteeism and increased health care costs, both direct from medical care and testing and indirect costs from lost work.13-15
Women with 7 or more daily moderate to severe VMS (defined as with sweating or affecting function) reported interference with sleep (94%), concentration (84%), mood (85%), energy (77%), and sexual activity (61%).16 Moderately to severely bothersome VMS have been associated with impaired psychological and general well-being, affecting work performance.17 Based on a Mayo Clinic workplace survey, Faubion and colleagues estimated an annual loss of $1.8 billion in the United States for menopause-related missed work and a $28 billion loss when medical expenses were added.15
Menopausal HT has been the primary treatment for VMS and has been shown to reduce the frequency and severity of hot flashes, with additional benefits on sleep, mood, fatigue, bone loss and reduction of fracture, and genitourinary syndrome of menopause (GSM), and with potential improvement in cardiovascular health with decreased type 2 diabetes.18,19 For healthy women with early menopause and no contraindications, HT has been recommended until at least the age of natural menopause, as observational data suggest that HT prevents osteoporosis, cardiovascular disease, neurodegenerative changes, and sexual dysfunction for these women.19,20 Similarly, for healthy women younger than age 60 or within 10 years of menopause, initiating HT has been shown to be safe and effective in treating bothersome VMS and preventing osteoporotic fractures and genitourinary changes.19,21
Most systemic HT formulations are inexpensive (for example, available as generics), with multiple dosing and formulations available for use alone or combined as oral, transdermal, or vaginal therapies. Despite the fear that arose for clinicians and women from the initial 2002 findings of the Women’s Health Initiative regarding increased risk of breast cancer, stroke, venous thrombosis, cardiovascular disease, and dementia, major medical societies agree that when initiated at or soon after menopause, HT is a safe and effective therapy to relieve VMS, protect against bone loss, and treat genitourinary changes.19,21
Many women, however, cannot take HT, including those with estrogen-sensitive cancers, such as breast or uterine cancers; prior cardiovascular disease, stroke, or venous thrombotic events; severe endometriosis; or migraine headaches with visual auras.2 In addition, many symptomatic menopausal women without health contraindications choose not to take HT.2 Until now, the only FDA-approved VMS nonhormone therapy has been a low-dose 7.5-mg paroxetine salt. Unfortunately, this formulation, along with the off-label use of other antidepressants (selective serotonin reuptake inhibitors and serotonin and norepinephrine reuptake inhibitors), gabapentinoids, oxybutynin, and clonidine, are substantially less effective than HT in treating moderate to severe VMS.
Bottom line
A substantial unmet need remains for effective therapy for moderate to severe VMS for women who cannot or choose not to take menopausal HT to relieve VMS.2,16 Effective, safe nonhormone treatment options such as the new NK3 receptor antagonist fezolinetant will address this clinically important need.
One concern is that the cost of developing and bringing to market the first of a new type of medication will be passed on to consumers, which may put it out of the price range for the many women who need it. However, the development and FDA approval of fezolinetant as the first NK3 receptor antagonist to treat menopausal VMS is potentially a practice changer. It provides a novel, effective, and safe FDA-approved nonhormonal treatment for menopausal women with moderate to severe VMS, particularly for women who cannot or will not take hormone therapy.
Continue to: When endometrial biopsy for postmenopausal bleeding reveals proliferative changes, how should we respond?...
When endometrial biopsy for postmenopausal bleeding reveals proliferative changes, how should we respond?
Abraham C. Proliferative endometrium in menopause: to treat or not to treat? Obstet Gynecol. 2023;141:265-267. doi:10.1097/AOG.0000000000005054.
The following case represents a common scenario for ObGyns.
CASE Patient with proliferative endometrial changes
A menopausal patient with a body mass index (BMI) > 30 kg/m2 presents with uterine bleeding. She does not use systemic menopausal hormone therapy. Endometrial biopsy indicates proliferative changes.
When endometrial biopsy performed for bleeding reveals proliferative changes in menopausal women, we traditionally have responded by reassuring the patient that the findings are benign and advising that she should let us know if future spotting or bleeding occurs.
However, a recent review by Abraham published in Obstetrics and Gynecology details the implications of proliferative endometrial changes in menopausal patients, advising that treatment, as well as monitoring, may be appropriate.22
Endometrial changes and what they suggest
In premenopausal women, proliferative endometrial changes are physiologic and result from ovarian estrogen production early in each cycle, during what is called the proliferative (referring to the endometrium) or follicular (referring to the dominant follicle that synthesizes estrogen) phase. In menopausal women who are not using HT, however, proliferative endometrial changes, with orderly uniform glands seen on histologic evaluation, reflect aromatization of androgens by adipose and other tissues into estrogen.
The next step on the continuum to hyperplasia (benign or atypical) after proliferative endometrium is disordered proliferative endometrium. At this stage, histologic evaluation reveals scattered cystic and dilated glands that have a normal gland-to-stroma ratio with a low gland density overall and without any atypia. Randomly distributed glands may have tubal metaplasia or fibrin thrombi associated with microinfarcts, often presenting with irregular bleeding. This is a noncancerous change that occurs with excess estrogen (endogenous or exogenous).23
Progestins reverse endometrial hyperplasia by activating progesterone receptors, which leads to stromal decidualization with thinning of the endometrium. They have a pronounced effect on the histologic appearance of the endometrium. By contrast, endometrial intraepithelial neoplasia (EIN, previously known as endometrial hyperplasiawith atypia) shows underlying molecular mutations and histologic alterations and represents a sharp transition to true neoplasia, which greatly increases the risk of endometrioid endometrial adenocarcinoma.24
For decades, we have been aware that if women diagnosed with endometrial hyperplasia are not treated with progestational therapy, their future risk of endometrial cancer is elevated. More recently, we also recognize that menopausal women found to have proliferative endometrial changes, if not treated, have an increased risk of endometrial cancer.
In a retrospective cohort study of almost 300 menopausal women who were not treated after endometrial biopsy revealed proliferative changes, investigators followed participants for an average of 11 years.25 These women had a mean BMI of 34 kg/m2. During follow-up, almost 12% of these women were diagnosed with endometrial hyperplasia or cancer. This incidence of endometrial neoplasia was some 4 times higher than for women initially found to have atrophic endometrial changes.25
Progestin treatment
Oral progestin therapy with follow-up endometrial biopsy constitutes traditional management for endometrial hyperplasia. Such therapy minimizes the likelihood that hyperplasia will progress to endometrial cancer.
We now recognize that the convenience, as well as the high endometrial progestin levels achieved, with levonorgestrel-releasing intrauterine devices (LNG-IUDs) have advantages over oral progestin therapy in treating endometrial hyperplasia. Indeed, a recent US report found that among women with EIN managed medically, use of progestin-releasing IUDs has grown from 7.7% in 2008 to 35.6% in 2020.26
Although both oral and intrauterine progestin are highly effective in treating simple hyperplasia, progestin IUDs are substantially more effective than oral progestins in treating EIN.27 Progestin concentrations in the endometrium have been shown to be 100-fold higher after LNG-IUD placement compared with oral progestin use.22 In addition, adverse effects, including bloating, unpleasant mood changes, and increased appetite, are more common with oral than intrauterine progestin therapy.28
Unfortunately, data from randomized trials addressing progestational treatment of proliferative endometrium in menopausal women are not available to support the treatment of proliferative endometrium with either oral progestins or the LNG-IUD.22
Role of ultrasonography
Another concern is relying on a finding of thin endometrial thickness on vaginal ultrasonography. In a simulated retrospective cohort study, use of transvaginal ultrasonography to determine the appropriateness of a biopsy was found not to be sufficiently accurate or racially equitable with regard to Black women.29 In simulated data, transvaginal ultrasonography missed almost 5 times more cases of endometrial cancer among Black women compared with White women due to higher fibroid prevalence and nonendometrioid histologic type malignancies in Black women.29
Assessing risk
If proliferative endometrium is found, Abraham suggests assessing risk using22:
- age
- comorbidities (including obesity)
- endometrial echo thickness on vaginal ultrasonography.
Consider the patient’s risk and tolerance of recurrent bleeding as well as her tolerance for progestational adverse effects if medical therapy is chosen. Discussion about next steps should include reviewing the histologic findings with the patient and discussing the difference in risk of progression to endometrial cancer of a finding of proliferative endometrium compared with a histologic finding of endometrial hyperplasia.
Using this patient-centered approach, observation over time with follow-up endometrial biopsies remains a management option. Although some women may tolerate micronized progesterone over synthetic progestins, there is concern that it may be less effective in suppressing the endometrium than synthetic progestins.30 Accordingly, synthetic progestins represent first-line options in this setting.
In her review, Abraham suggests that when endometrial biopsy reveals proliferative changes in a menopausal woman, we should initiate progestin treatment and perform surveillance endometrial sampling every 3 to 6 months. If such sampling reveals benign but not proliferative endometrium, progestin therapy can be stopped and endometrial biopsy repeated if bleeding recurs.22 ●
ObGyns may choose to adopt Abraham’s approach or to hold off on progestin therapy while performing follow-up endometrial sampling. Either way, the take-home message is that the finding of proliferative endometrial changes on biopsy for postmenopausal bleeding requires proactive management.
This year’s menopause Update highlights a highly effective nonhormonal medication that recently received approval by the US Food and Drug Administration (FDA) for the treatment of bothersome menopausal vasomotor symptoms. In addition, the Update provides guidance regarding how ObGyns should respond when an endometrial biopsy for postmenopausal bleeding reveals proliferative changes.
Breakthrough in women’s health: A new nonhormone therapy for vasomotor symptoms
Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate-to-severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. 2023;dgad058. doi:10.1210/clinem/dgad058.
Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401:1091-1102. doi:10.1016/S0140-6736(23)00085-5.
A new oral nonestrogen-containing medication for relief of moderate to severe hot flashes, fezolinetant (Veozah) 45 mg daily, has been approved by the FDA and was expected to be available by the end of May 2023. Fezolinetant is a selective neurokinin 3 (NK3) receptor antagonistthat offers a targeted nonhormonal approach to menopausal vasomotor symptoms (VMS), and it is the first in its class to make it to market.
The decline in estrogen at menopause appears to result in increased signaling at kisspeptin/neurokinin B/dynorphin (KNDy) neurons in the thermoregulatory center within the hypothalamus with resultant increases in hot flashes.1,2 Fezolinetant works by binding to and blocking the activities of the NK3 receptor.3-5
Key study findings
Selective NK3 receptor antagonists, including fezolinetant, effectively reduce the frequency and severity of VMS comparable to that of hormone therapy (HT). Two phase 3 clinical trials, Skylight 1 and 2, confirmed the efficacy and safety of fezolinetant 45 mg in treating VMS,6,7 and an additional 52-week placebo-controlled study, Skylight 4, confirmed long-term safety.8 Onset of action occurs within a week. Reported adverse events occurred in 1% to 2% of healthy menopausal women participating in clinical trials; these included headaches, abdominal pain, diarrhea, insomnia, back pain, hot flushes, and reversible elevated hepatic transaminase levels.6-9
The published phase 2 trials9 and the international randomized controlled trial (RCT) 12-week studies, Skylight 1 and 2,6,7 found that once-daily 30-mg and 45-mg doses of fezolinetant significantly reduced VMS frequency and severity at 12 weeks among women aged 40 to 60 years who reported an average of 7 moderate to severe VMS/day; the reduction in reported VMS was sustained at 40 weeks. Phase 3 data from Skylight 1 and 2 demonstrated fezolinetant’s efficacy in reducing the frequency and severity of VMS and provided information on the safety profile of fezolinetant compared with placebo over 12 weeks and a noncontrolled extension for an additional 40 weeks.6,7
Oral fezolinetant was associated with improved quality of life, including reduced VMS-related interference with daily life.10 Johnson and colleagues, reporting for Skylight 2, found VMS frequency and severity improvement by week 1, which achieved statistical significance at weeks 4 and 12, with this improvement maintained through week 52.6 A 64.3% reduction in mean daily VMS from baseline was seen at 12 weeks for fezolinetant 45 mg compared with a 45.4% reduction for placebo. VMS severity significantly decreased compared with placebo at 4 and 12 weeks.6
Serious treatment-emergent adverse events were infrequent, reported by 2%, 1%, and 0% of those receiving fezolinetant 30 mg, fezolinetant 45 mg, and placebo.6 Increases in levels of alanine aminotransferase (ALT) or aspartate aminotransferase (AST) were noted and were described as asymptomatic, isolated, intermittent, or transient, and these levels returned to baseline during treatment or after discontinuation.6
Of the 5 participants taking fezolinetant in Skyline 1 with ALT or AST levels greater than 3 times the upper limit of normal in the 12-week randomized trial, levels returned to normal range while continuing treatment in 2 participants, with treatment interruption in 2, and with discontinuation in 1. No new safety signals were seen in the 40-week extension trial.6
Fezolinetant offers a much-needed effective and safe selective nonhormone NK3 receptor antagonist therapy that reduces the frequency and severity of menopausal VMS and has been shown to be safe through 52 weeks of treatment.
To read more about how fezolinetant specifically targets the hormone receptor that triggers hot flashes as well as on prescribing hormone therapy for women with menopausal symptoms, see “Focus on menopause: Q&A with Jan Shifren, MD, and Genevieve NealPerry, MD, PhD,” in the December 2022 issue of OBG Management at https://www.mdedge.com/obgyn/article/260380/menopause
Continue to: Endometrial and bone safety...
Endometrial and bone safety
Results from Skylight 4, a phase 3, randomized, double-blind, 52-week safety study, provided additional evidence that confirmed the longer-term safety of fezolinetant over a 52-week treatment period.8
Endometrial safety was assessed in postmenopausal women with normal baseline endometrium (n = 599).8 For fezolinetant 45 mg, 1 of 203 participants had endometrial hyperplasia (EH) (0.5%; upper limit of one-sided 95% confidence interval [CI], 2.3%); no cases of EH were noted in the placebo (0 of 186) or fezolinetant 30-mg (0 of 210) groups. The incidence of EH or malignancy in fezolinetant-treated participants was within prespecified limits, as assessed by blinded, centrally read endometrial biopsies. Endometrial malignancy occurred in 1 of 210 in the fezolinetant 30-mg group (0.5%; 95% CI, 2.2%) with no cases in the other groups, thus meeting FDA requirements for endometrial safety.8
In addition, no significant differences were noted in change from baseline endometrial thickness on transvaginal ultrasonography between fezolinetant-treated and placebo groups. Likewise, no loss of bone density was found on dual-energy x-ray absorptiometry (DEXA) scans or trabecular bone scores.8
Liver safety
Although no cases of severe liver injury were noted, elevations in serum transaminase concentrations greater than 3 times the upper limit of normal were observed in the clinical trials. In Skylight 4, liver enzyme elevations more than 3 times the upper limit of normal occurred in 6 of 583 participants taking placebo, 8 of 590 taking fezolinetant 30 mg, and 12 of 589 taking fezolinetant 45 mg.8
The prescribing information for fezolinetant includes a warning for elevated hepatic transaminases: Fezolinetant should not be started if baseline serum transaminase concentration is equal to or exceeds 2 times the upper limit of normal. Liver tests should be obtained at baseline and repeated every 3 months for the first 9 months and then if symptoms suggest liver injury.11,12
Unmet need for nonhormone treatment of VMS
Vasomotor symptoms affect up to 80% of women, with approximately 25% bothersome enough to warrant treatment. Vasomotor symptoms persist for a median of 7 years, with duration and severity differing by race and ethnicity. Black, Hispanic, and possibly Native American women experience the highest burden of VMS.2 Although VMS, including hot flashes, night sweats, and mood and sleep disturbances, often are considered an annoyance to those with mild symptoms, moderate to severe VMS impact women’s lives, including functioning at home or work, affecting relationships, and decreasing perceived quality of life, and they have been associated with workplace absenteeism and increased health care costs, both direct from medical care and testing and indirect costs from lost work.13-15
Women with 7 or more daily moderate to severe VMS (defined as with sweating or affecting function) reported interference with sleep (94%), concentration (84%), mood (85%), energy (77%), and sexual activity (61%).16 Moderately to severely bothersome VMS have been associated with impaired psychological and general well-being, affecting work performance.17 Based on a Mayo Clinic workplace survey, Faubion and colleagues estimated an annual loss of $1.8 billion in the United States for menopause-related missed work and a $28 billion loss when medical expenses were added.15
Menopausal HT has been the primary treatment for VMS and has been shown to reduce the frequency and severity of hot flashes, with additional benefits on sleep, mood, fatigue, bone loss and reduction of fracture, and genitourinary syndrome of menopause (GSM), and with potential improvement in cardiovascular health with decreased type 2 diabetes.18,19 For healthy women with early menopause and no contraindications, HT has been recommended until at least the age of natural menopause, as observational data suggest that HT prevents osteoporosis, cardiovascular disease, neurodegenerative changes, and sexual dysfunction for these women.19,20 Similarly, for healthy women younger than age 60 or within 10 years of menopause, initiating HT has been shown to be safe and effective in treating bothersome VMS and preventing osteoporotic fractures and genitourinary changes.19,21
Most systemic HT formulations are inexpensive (for example, available as generics), with multiple dosing and formulations available for use alone or combined as oral, transdermal, or vaginal therapies. Despite the fear that arose for clinicians and women from the initial 2002 findings of the Women’s Health Initiative regarding increased risk of breast cancer, stroke, venous thrombosis, cardiovascular disease, and dementia, major medical societies agree that when initiated at or soon after menopause, HT is a safe and effective therapy to relieve VMS, protect against bone loss, and treat genitourinary changes.19,21
Many women, however, cannot take HT, including those with estrogen-sensitive cancers, such as breast or uterine cancers; prior cardiovascular disease, stroke, or venous thrombotic events; severe endometriosis; or migraine headaches with visual auras.2 In addition, many symptomatic menopausal women without health contraindications choose not to take HT.2 Until now, the only FDA-approved VMS nonhormone therapy has been a low-dose 7.5-mg paroxetine salt. Unfortunately, this formulation, along with the off-label use of other antidepressants (selective serotonin reuptake inhibitors and serotonin and norepinephrine reuptake inhibitors), gabapentinoids, oxybutynin, and clonidine, are substantially less effective than HT in treating moderate to severe VMS.
Bottom line
A substantial unmet need remains for effective therapy for moderate to severe VMS for women who cannot or choose not to take menopausal HT to relieve VMS.2,16 Effective, safe nonhormone treatment options such as the new NK3 receptor antagonist fezolinetant will address this clinically important need.
One concern is that the cost of developing and bringing to market the first of a new type of medication will be passed on to consumers, which may put it out of the price range for the many women who need it. However, the development and FDA approval of fezolinetant as the first NK3 receptor antagonist to treat menopausal VMS is potentially a practice changer. It provides a novel, effective, and safe FDA-approved nonhormonal treatment for menopausal women with moderate to severe VMS, particularly for women who cannot or will not take hormone therapy.
Continue to: When endometrial biopsy for postmenopausal bleeding reveals proliferative changes, how should we respond?...
When endometrial biopsy for postmenopausal bleeding reveals proliferative changes, how should we respond?
Abraham C. Proliferative endometrium in menopause: to treat or not to treat? Obstet Gynecol. 2023;141:265-267. doi:10.1097/AOG.0000000000005054.
The following case represents a common scenario for ObGyns.
CASE Patient with proliferative endometrial changes
A menopausal patient with a body mass index (BMI) > 30 kg/m2 presents with uterine bleeding. She does not use systemic menopausal hormone therapy. Endometrial biopsy indicates proliferative changes.
When endometrial biopsy performed for bleeding reveals proliferative changes in menopausal women, we traditionally have responded by reassuring the patient that the findings are benign and advising that she should let us know if future spotting or bleeding occurs.
However, a recent review by Abraham published in Obstetrics and Gynecology details the implications of proliferative endometrial changes in menopausal patients, advising that treatment, as well as monitoring, may be appropriate.22
Endometrial changes and what they suggest
In premenopausal women, proliferative endometrial changes are physiologic and result from ovarian estrogen production early in each cycle, during what is called the proliferative (referring to the endometrium) or follicular (referring to the dominant follicle that synthesizes estrogen) phase. In menopausal women who are not using HT, however, proliferative endometrial changes, with orderly uniform glands seen on histologic evaluation, reflect aromatization of androgens by adipose and other tissues into estrogen.
The next step on the continuum to hyperplasia (benign or atypical) after proliferative endometrium is disordered proliferative endometrium. At this stage, histologic evaluation reveals scattered cystic and dilated glands that have a normal gland-to-stroma ratio with a low gland density overall and without any atypia. Randomly distributed glands may have tubal metaplasia or fibrin thrombi associated with microinfarcts, often presenting with irregular bleeding. This is a noncancerous change that occurs with excess estrogen (endogenous or exogenous).23
Progestins reverse endometrial hyperplasia by activating progesterone receptors, which leads to stromal decidualization with thinning of the endometrium. They have a pronounced effect on the histologic appearance of the endometrium. By contrast, endometrial intraepithelial neoplasia (EIN, previously known as endometrial hyperplasiawith atypia) shows underlying molecular mutations and histologic alterations and represents a sharp transition to true neoplasia, which greatly increases the risk of endometrioid endometrial adenocarcinoma.24
For decades, we have been aware that if women diagnosed with endometrial hyperplasia are not treated with progestational therapy, their future risk of endometrial cancer is elevated. More recently, we also recognize that menopausal women found to have proliferative endometrial changes, if not treated, have an increased risk of endometrial cancer.
In a retrospective cohort study of almost 300 menopausal women who were not treated after endometrial biopsy revealed proliferative changes, investigators followed participants for an average of 11 years.25 These women had a mean BMI of 34 kg/m2. During follow-up, almost 12% of these women were diagnosed with endometrial hyperplasia or cancer. This incidence of endometrial neoplasia was some 4 times higher than for women initially found to have atrophic endometrial changes.25
Progestin treatment
Oral progestin therapy with follow-up endometrial biopsy constitutes traditional management for endometrial hyperplasia. Such therapy minimizes the likelihood that hyperplasia will progress to endometrial cancer.
We now recognize that the convenience, as well as the high endometrial progestin levels achieved, with levonorgestrel-releasing intrauterine devices (LNG-IUDs) have advantages over oral progestin therapy in treating endometrial hyperplasia. Indeed, a recent US report found that among women with EIN managed medically, use of progestin-releasing IUDs has grown from 7.7% in 2008 to 35.6% in 2020.26
Although both oral and intrauterine progestin are highly effective in treating simple hyperplasia, progestin IUDs are substantially more effective than oral progestins in treating EIN.27 Progestin concentrations in the endometrium have been shown to be 100-fold higher after LNG-IUD placement compared with oral progestin use.22 In addition, adverse effects, including bloating, unpleasant mood changes, and increased appetite, are more common with oral than intrauterine progestin therapy.28
Unfortunately, data from randomized trials addressing progestational treatment of proliferative endometrium in menopausal women are not available to support the treatment of proliferative endometrium with either oral progestins or the LNG-IUD.22
Role of ultrasonography
Another concern is relying on a finding of thin endometrial thickness on vaginal ultrasonography. In a simulated retrospective cohort study, use of transvaginal ultrasonography to determine the appropriateness of a biopsy was found not to be sufficiently accurate or racially equitable with regard to Black women.29 In simulated data, transvaginal ultrasonography missed almost 5 times more cases of endometrial cancer among Black women compared with White women due to higher fibroid prevalence and nonendometrioid histologic type malignancies in Black women.29
Assessing risk
If proliferative endometrium is found, Abraham suggests assessing risk using22:
- age
- comorbidities (including obesity)
- endometrial echo thickness on vaginal ultrasonography.
Consider the patient’s risk and tolerance of recurrent bleeding as well as her tolerance for progestational adverse effects if medical therapy is chosen. Discussion about next steps should include reviewing the histologic findings with the patient and discussing the difference in risk of progression to endometrial cancer of a finding of proliferative endometrium compared with a histologic finding of endometrial hyperplasia.
Using this patient-centered approach, observation over time with follow-up endometrial biopsies remains a management option. Although some women may tolerate micronized progesterone over synthetic progestins, there is concern that it may be less effective in suppressing the endometrium than synthetic progestins.30 Accordingly, synthetic progestins represent first-line options in this setting.
In her review, Abraham suggests that when endometrial biopsy reveals proliferative changes in a menopausal woman, we should initiate progestin treatment and perform surveillance endometrial sampling every 3 to 6 months. If such sampling reveals benign but not proliferative endometrium, progestin therapy can be stopped and endometrial biopsy repeated if bleeding recurs.22 ●
ObGyns may choose to adopt Abraham’s approach or to hold off on progestin therapy while performing follow-up endometrial sampling. Either way, the take-home message is that the finding of proliferative endometrial changes on biopsy for postmenopausal bleeding requires proactive management.
- Modi M, Dhillo WS. Neurokinin 3 receptor antagonism: a novel treatment for menopausal hot flushes. Neuroendocrinology. 2019;109:242-248. doi:10.1159/000495889
- Pinkerton JV, Redick DL, Homewood LN, et al. Neurokinin receptor antagonist, fezolinetant, for treatment of menopausal vasomotor symptoms. J Clin Endocrinol Metab. 2023;dgad209. doi:10.1210/clinem/dgad209
- Rance NE, Dacks PA, Mittelman-Smith MA, et al. Modulation of body temperature and LH secretion by hypothalamic KNDy (kisspeptin, neurokinin B and dynorphin) neurons: a novel hypothesis on the mechanism of hot flushes. Front Neuroendocrinol. 2013;34:211-227. doi:10.1016 /j.yfrne.2013.07.003
- Mittelman-Smith MA, Williams H, Krajewski-Hall SJ, et al. Role for kisspeptin/neurokinin B/dynorphin (KNDy) neurons in cutaneous vasodilatation and the estrogen modulation of body temperature. Proc Natl Acad Sci USA. 2012;109:1984619851. doi:10.1073/pnas.1211517109
- Astellas Pharma. Astellas’ Veozah (fezolinetant) approved by US FDA for treatment of vasomotor symptoms due to menopause. May 12, 2023. PR Newswire. Accessed May 15, 2023. https://www.prnewswire.com/news-releases/astellas-veozah-fezolinetant-approved-by-us-fda-for -treatment-of-vasomotor-symptoms-due-to-menopause -301823639.html
- Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate-to-severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. 2023;dgad058. doi:10.1210/clinem/dgad058
- Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401:1091-1102. doi:10.1016 /S0140-6736(23)00085-5
- Neal-Perry G, Cano A, Lederman S, et al. Safety of fezolinetant for vasomotor symptoms associated with menopause: a randomized controlled trial. Obstet Gynecol. 2023;141:737-747. doi:10.1097/AOG.0000000000005114
- Depypere H, Timmerman D, Donders G, et al. Treatment of menopausal vasomotor symptoms with fezolinetant, a neurokinin 3 receptor antagonist: a phase 2a trial. J Clin Endocrinol Metab. 2019;104:5893-5905. doi: 10.1210/jc .2019-00677
- Santoro N, Waldbaum A, Lederman S, et al. Effect of the neurokinin 3 receptor antagonist fezolinetant on patientreported outcomes in postmenopausal women with vasomotor symptoms: results of a randomized, placebo-controlled, double-blind, dose-ranging study (VESTA). Menopause. 2020;27:1350-1356. doi:10.1097/GME.0000000000001621
- FDA approves novel drug to treat moderate to severe hot flashes caused by menopause. May 12, 2023. US Food and Drug Administration. Accessed May 15, 2023. https://www .fda.gov/news-events/press-announcements/fda-approves -novel-drug-treat-moderate-severe-hot-flashes-caused -menopause
- Veozah. Prescribing information. Astellas; 2023. Accessed May 16, 2023. https://www.astellas.com/us/system/files /veozah_uspi.pdf
- Pinkerton JV. Money talks: untreated hot flashes cost women, the workplace, and society. Menopause. 2015;22:254-255. doi:10.1097/GME.0000000000000427
- Sarrel P, Portman D, Lefebvre P, et al. Incremental direct and indirect costs of untreated vasomotor symptoms. Menopause. 2015;22(3):260-266. doi:10.1097/GME.0000000000000320
- Faubion SS, Enders F, Hedges MS, et al. Impact of menopause symptoms on women in the workplace. Mayo Clin Proc. 2023;98:833-845. doi:10.1016/j.mayocp.2023.02.025
- Williams RE, Levine KB, Kalilani L, et al. Menopause- specific questionnaire assessment in US populationbased study shows negative impact on health-related quality of life. Maturitas. 2009;62:153-159. doi:10.1016 /j.maturitas.2008.12.006
- Gartoulla P, Bell RJ, Worsley R, et al. Moderate-severely bothersome vasomotor symptoms are associated with lowered psychological general wellbeing in women at midlife. Maturitas. 2015;81:487-492. doi:10.1016 /j.maturitas.2015.06.004
- Manson JE, Kaunitz AM. Menopause management—getting clinical care back on track. N Engl J Med. 2016;374:803-806. doi:10.1056/NEJMp1514242
- 2022 Hormone Therapy Position Statement of the North American Menopause Society Advisory Panel. The 2022 hormone therapy position statement of the North American Menopause Society. Menopause. 2022;29:767-794. doi:10.1097/GME.0000000000002028
- Kaunitz AM, Kapoor E, Faubion S. Treatment of women after bilateral salpingo-oophorectomy performed prior to natural menopause. JAMA. 2021;12;326:1429-1430. doi:10.1001 /jama.2021.3305
- Pinkerton JV. Hormone therapy for postmenopausal women. N Engl J Med. 2020;382:446-455. doi:10.1056 /NEJMcp1714787
- Abraham C. Proliferative endometrium in menopause: to treat or not to treat? Obstet Gynecol. 2023;141:265-267. doi:10.1097/AOG.0000000000005054
- Chandra V, Kim JJ, Benbrook DM, et al. Therapeutic options for management of endometrial hyperplasia. J Gynecol Oncol. 2016;27:e8. doi:10.3802/jgo.2016.27.e8
- Owings RA, Quick CM. Endometrial intraepithelial neoplasia. Arch Pathol Lab Med. 2014;138:484-491. doi:10.5858 /arpa.2012-0709-RA
- Rotenberg O, Doulaveris G, Fridman D, et al. Long-term outcome of postmenopausal women with proliferative endometrium on endometrial sampling. Am J Obstet Gynecol. 2020;223:896.e1-896.e7. doi:10.1016/j.ajog.2020.06.045
- Suzuki Y, Chen L, Hou JY, et al. Systemic progestins and progestin-releasing intrauterine device therapy for premenopausal patients with endometrial intraepithelial neoplasia. Obstet Gynecol. 2023;141:979-987. doi:10.1097 /AOG.0000000000005124
- Mandelbaum RS, Ciccone MA, Nusbaum DJ, et al. Progestin therapy for obese women with complex atypical hyperplasia: levonorgestrel-releasing intrauterine device vs systemic therapy. Am J Obstet Gynecol. 2020;223:103.e1-103.e13. doi:10.1016/j.ajog.2019.12.273
- Liu S, Kciuk O, Frank M, et al. Progestins of today and tomorrow. Curr Opin Obstet Gynecol. 2022;34:344-350. doi:10.1097 /GCO.0000000000000819
- Doll KM, Romano SS, Marsh EE, et al. Estimated performance of transvaginal ultrasonography for evaluation of postmenopausal bleeding in a simulated cohort of black and white women in the US. JAMA Oncol. 2021;7:1158-1165. doi:10.1001/jamaoncol.2021.1700
- Gompel A. Progesterone and endometrial cancer. Best Pract Res Clin Obstet Gynaecol. 2020;69:95-107. doi:10.1016 /j.bpobgyn.2020.05.003
- Modi M, Dhillo WS. Neurokinin 3 receptor antagonism: a novel treatment for menopausal hot flushes. Neuroendocrinology. 2019;109:242-248. doi:10.1159/000495889
- Pinkerton JV, Redick DL, Homewood LN, et al. Neurokinin receptor antagonist, fezolinetant, for treatment of menopausal vasomotor symptoms. J Clin Endocrinol Metab. 2023;dgad209. doi:10.1210/clinem/dgad209
- Rance NE, Dacks PA, Mittelman-Smith MA, et al. Modulation of body temperature and LH secretion by hypothalamic KNDy (kisspeptin, neurokinin B and dynorphin) neurons: a novel hypothesis on the mechanism of hot flushes. Front Neuroendocrinol. 2013;34:211-227. doi:10.1016 /j.yfrne.2013.07.003
- Mittelman-Smith MA, Williams H, Krajewski-Hall SJ, et al. Role for kisspeptin/neurokinin B/dynorphin (KNDy) neurons in cutaneous vasodilatation and the estrogen modulation of body temperature. Proc Natl Acad Sci USA. 2012;109:1984619851. doi:10.1073/pnas.1211517109
- Astellas Pharma. Astellas’ Veozah (fezolinetant) approved by US FDA for treatment of vasomotor symptoms due to menopause. May 12, 2023. PR Newswire. Accessed May 15, 2023. https://www.prnewswire.com/news-releases/astellas-veozah-fezolinetant-approved-by-us-fda-for -treatment-of-vasomotor-symptoms-due-to-menopause -301823639.html
- Johnson KA, Martin N, Nappi RE, et al. Efficacy and safety of fezolinetant in moderate-to-severe vasomotor symptoms associated with menopause: a phase 3 RCT. J Clin Endocrinol Metab. 2023;dgad058. doi:10.1210/clinem/dgad058
- Lederman S, Ottery FD, Cano A, et al. Fezolinetant for treatment of moderate-to-severe vasomotor symptoms associated with menopause (SKYLIGHT 1): a phase 3 randomised controlled study. Lancet. 2023;401:1091-1102. doi:10.1016 /S0140-6736(23)00085-5
- Neal-Perry G, Cano A, Lederman S, et al. Safety of fezolinetant for vasomotor symptoms associated with menopause: a randomized controlled trial. Obstet Gynecol. 2023;141:737-747. doi:10.1097/AOG.0000000000005114
- Depypere H, Timmerman D, Donders G, et al. Treatment of menopausal vasomotor symptoms with fezolinetant, a neurokinin 3 receptor antagonist: a phase 2a trial. J Clin Endocrinol Metab. 2019;104:5893-5905. doi: 10.1210/jc .2019-00677
- Santoro N, Waldbaum A, Lederman S, et al. Effect of the neurokinin 3 receptor antagonist fezolinetant on patientreported outcomes in postmenopausal women with vasomotor symptoms: results of a randomized, placebo-controlled, double-blind, dose-ranging study (VESTA). Menopause. 2020;27:1350-1356. doi:10.1097/GME.0000000000001621
- FDA approves novel drug to treat moderate to severe hot flashes caused by menopause. May 12, 2023. US Food and Drug Administration. Accessed May 15, 2023. https://www .fda.gov/news-events/press-announcements/fda-approves -novel-drug-treat-moderate-severe-hot-flashes-caused -menopause
- Veozah. Prescribing information. Astellas; 2023. Accessed May 16, 2023. https://www.astellas.com/us/system/files /veozah_uspi.pdf
- Pinkerton JV. Money talks: untreated hot flashes cost women, the workplace, and society. Menopause. 2015;22:254-255. doi:10.1097/GME.0000000000000427
- Sarrel P, Portman D, Lefebvre P, et al. Incremental direct and indirect costs of untreated vasomotor symptoms. Menopause. 2015;22(3):260-266. doi:10.1097/GME.0000000000000320
- Faubion SS, Enders F, Hedges MS, et al. Impact of menopause symptoms on women in the workplace. Mayo Clin Proc. 2023;98:833-845. doi:10.1016/j.mayocp.2023.02.025
- Williams RE, Levine KB, Kalilani L, et al. Menopause- specific questionnaire assessment in US populationbased study shows negative impact on health-related quality of life. Maturitas. 2009;62:153-159. doi:10.1016 /j.maturitas.2008.12.006
- Gartoulla P, Bell RJ, Worsley R, et al. Moderate-severely bothersome vasomotor symptoms are associated with lowered psychological general wellbeing in women at midlife. Maturitas. 2015;81:487-492. doi:10.1016 /j.maturitas.2015.06.004
- Manson JE, Kaunitz AM. Menopause management—getting clinical care back on track. N Engl J Med. 2016;374:803-806. doi:10.1056/NEJMp1514242
- 2022 Hormone Therapy Position Statement of the North American Menopause Society Advisory Panel. The 2022 hormone therapy position statement of the North American Menopause Society. Menopause. 2022;29:767-794. doi:10.1097/GME.0000000000002028
- Kaunitz AM, Kapoor E, Faubion S. Treatment of women after bilateral salpingo-oophorectomy performed prior to natural menopause. JAMA. 2021;12;326:1429-1430. doi:10.1001 /jama.2021.3305
- Pinkerton JV. Hormone therapy for postmenopausal women. N Engl J Med. 2020;382:446-455. doi:10.1056 /NEJMcp1714787
- Abraham C. Proliferative endometrium in menopause: to treat or not to treat? Obstet Gynecol. 2023;141:265-267. doi:10.1097/AOG.0000000000005054
- Chandra V, Kim JJ, Benbrook DM, et al. Therapeutic options for management of endometrial hyperplasia. J Gynecol Oncol. 2016;27:e8. doi:10.3802/jgo.2016.27.e8
- Owings RA, Quick CM. Endometrial intraepithelial neoplasia. Arch Pathol Lab Med. 2014;138:484-491. doi:10.5858 /arpa.2012-0709-RA
- Rotenberg O, Doulaveris G, Fridman D, et al. Long-term outcome of postmenopausal women with proliferative endometrium on endometrial sampling. Am J Obstet Gynecol. 2020;223:896.e1-896.e7. doi:10.1016/j.ajog.2020.06.045
- Suzuki Y, Chen L, Hou JY, et al. Systemic progestins and progestin-releasing intrauterine device therapy for premenopausal patients with endometrial intraepithelial neoplasia. Obstet Gynecol. 2023;141:979-987. doi:10.1097 /AOG.0000000000005124
- Mandelbaum RS, Ciccone MA, Nusbaum DJ, et al. Progestin therapy for obese women with complex atypical hyperplasia: levonorgestrel-releasing intrauterine device vs systemic therapy. Am J Obstet Gynecol. 2020;223:103.e1-103.e13. doi:10.1016/j.ajog.2019.12.273
- Liu S, Kciuk O, Frank M, et al. Progestins of today and tomorrow. Curr Opin Obstet Gynecol. 2022;34:344-350. doi:10.1097 /GCO.0000000000000819
- Doll KM, Romano SS, Marsh EE, et al. Estimated performance of transvaginal ultrasonography for evaluation of postmenopausal bleeding in a simulated cohort of black and white women in the US. JAMA Oncol. 2021;7:1158-1165. doi:10.1001/jamaoncol.2021.1700
- Gompel A. Progesterone and endometrial cancer. Best Pract Res Clin Obstet Gynaecol. 2020;69:95-107. doi:10.1016 /j.bpobgyn.2020.05.003

End of the road for transcranial brain stimulation as an adjunct in major depression?
The study showed no difference in mean improvement in Montgomery-Åsberg Depression Rating Scale (MADRS) scores at 6 weeks between active and sham tDCS.
“Our trial does not support the efficacy of tDCS as an additional treatment to SSRIs in adults with MDD,” the investigators, led by Frank Padberg, MD, department of psychiatry and psychotherapy, Ludwig-Maximilians-University Munich, write.
The study was published online in The Lancet.
Rigorous trial
Because it neurophysiologically modulates prefrontal cortex connectivity, tDCS has been proposed as a potential treatment for MDD.
Yet evidence for its efficacy has been inconsistent, and there is a scarcity of multicenter trial data, the researchers note.
The DepressionDC trial assessed the efficacy of tDCS in combination with SSRIs in 160 adults with MDD. Participants had a score of at least 15 on the Hamilton Depression Rating Scale (21-item version); their conditions had not responded to at least one antidepressant trial in their current depressive episode; and they had received treatment with an SSRI at a stable dose for at least 4 weeks. The SSRI was continued at the same dose during stimulation.
Eighty-three patients were allocated to undergo 30 min of 2-mA bifrontal tDCS every weekday for 4 weeks, then two tDCS sessions per week for 2 weeks; 77 patients were assigned to receive matching sham stimulation. Randomization was stratified by baseline MADRS score of less than 31 or 31 and higher.
In intention-to-treat analysis, there was no between-group difference in mean improvement on the MADRS at week 6 (–8.2 with active and –8.0 with sham tDCS; difference, 0.3; 95% confidence interval, –2.4 to 2.9).
There was also no significant difference for all secondary outcomes, including response and remission rates, patient-reported depression, and functioning, as well as at 18-week and 30-week follow-up visits.
There were significantly more mild adverse events reported in the active tDCS group than in the sham group (60% vs. 43%; P = .028). The most common adverse events were headaches, local skin reactions, and sleep-related problems.
Still reason for optimism
These findings call into question the efficacy of tDCS as add-on therapy to SSRI treatment for individuals with MDD and highlight the need for supportive evidence from multicenter studies, the investigators write.
Yet Dr. Padberg said it’s not the end of the road for tDCS for depression.
tDCS exerts a “rather subtle and nonfocal effect on neuronal activity. Thus, tDCS may need to be combined with specific behavioral or cognitive interventions which functionally involve the brain region where tDCS is targeted at,” he said.
Another “promising avenue” is personalization of tDCS by “individual MRI-based computational modeling of tDCS-induced electric fields,” he noted.
The coauthors of an accompanying commentary note that the DepressionDC trial was “carefully designed” and “well executed.”
And while the results did not show the superiority of active tDCS over sham stimulation as an additional treatment to SSRI therapy, “clinicians and researchers should not disregard this treatment for people with MDD,” write Daphne Voineskos, MD, PhD, and Daniel Blumberger, MD, with the University of Toronto.
“Specifically, further exploration of placebo response in less heterogeneous MDD samples and the evaluation of tDCS as an earlier treatment option for people with MDD are important areas of future research,” they suggest.
“Moreover, elucidating the effects of interindividual anatomical variability on electrical current distribution might lead to tDCS protocols that individualize treatment to optimize therapeutic effects as opposed to a so-called one-size-fits-all approach.
“Overall, there is reason for optimism about the potential to individualize tDCS and deliver it outside of the clinical setting,” Dr. Voineskos and Dr. Blumberger conclude.
Funding for the study was provided by the German Federal Ministry of Education and Research. Several authors disclosed relationships with the pharmaceutical industry. A complete list of disclosures of authors and comment writers is available with the original article.
A version of this article first appeared on Medscape.com.
The study showed no difference in mean improvement in Montgomery-Åsberg Depression Rating Scale (MADRS) scores at 6 weeks between active and sham tDCS.
“Our trial does not support the efficacy of tDCS as an additional treatment to SSRIs in adults with MDD,” the investigators, led by Frank Padberg, MD, department of psychiatry and psychotherapy, Ludwig-Maximilians-University Munich, write.
The study was published online in The Lancet.
Rigorous trial
Because it neurophysiologically modulates prefrontal cortex connectivity, tDCS has been proposed as a potential treatment for MDD.
Yet evidence for its efficacy has been inconsistent, and there is a scarcity of multicenter trial data, the researchers note.
The DepressionDC trial assessed the efficacy of tDCS in combination with SSRIs in 160 adults with MDD. Participants had a score of at least 15 on the Hamilton Depression Rating Scale (21-item version); their conditions had not responded to at least one antidepressant trial in their current depressive episode; and they had received treatment with an SSRI at a stable dose for at least 4 weeks. The SSRI was continued at the same dose during stimulation.
Eighty-three patients were allocated to undergo 30 min of 2-mA bifrontal tDCS every weekday for 4 weeks, then two tDCS sessions per week for 2 weeks; 77 patients were assigned to receive matching sham stimulation. Randomization was stratified by baseline MADRS score of less than 31 or 31 and higher.
In intention-to-treat analysis, there was no between-group difference in mean improvement on the MADRS at week 6 (–8.2 with active and –8.0 with sham tDCS; difference, 0.3; 95% confidence interval, –2.4 to 2.9).
There was also no significant difference for all secondary outcomes, including response and remission rates, patient-reported depression, and functioning, as well as at 18-week and 30-week follow-up visits.
There were significantly more mild adverse events reported in the active tDCS group than in the sham group (60% vs. 43%; P = .028). The most common adverse events were headaches, local skin reactions, and sleep-related problems.
Still reason for optimism
These findings call into question the efficacy of tDCS as add-on therapy to SSRI treatment for individuals with MDD and highlight the need for supportive evidence from multicenter studies, the investigators write.
Yet Dr. Padberg said it’s not the end of the road for tDCS for depression.
tDCS exerts a “rather subtle and nonfocal effect on neuronal activity. Thus, tDCS may need to be combined with specific behavioral or cognitive interventions which functionally involve the brain region where tDCS is targeted at,” he said.
Another “promising avenue” is personalization of tDCS by “individual MRI-based computational modeling of tDCS-induced electric fields,” he noted.
The coauthors of an accompanying commentary note that the DepressionDC trial was “carefully designed” and “well executed.”
And while the results did not show the superiority of active tDCS over sham stimulation as an additional treatment to SSRI therapy, “clinicians and researchers should not disregard this treatment for people with MDD,” write Daphne Voineskos, MD, PhD, and Daniel Blumberger, MD, with the University of Toronto.
“Specifically, further exploration of placebo response in less heterogeneous MDD samples and the evaluation of tDCS as an earlier treatment option for people with MDD are important areas of future research,” they suggest.
“Moreover, elucidating the effects of interindividual anatomical variability on electrical current distribution might lead to tDCS protocols that individualize treatment to optimize therapeutic effects as opposed to a so-called one-size-fits-all approach.
“Overall, there is reason for optimism about the potential to individualize tDCS and deliver it outside of the clinical setting,” Dr. Voineskos and Dr. Blumberger conclude.
Funding for the study was provided by the German Federal Ministry of Education and Research. Several authors disclosed relationships with the pharmaceutical industry. A complete list of disclosures of authors and comment writers is available with the original article.
A version of this article first appeared on Medscape.com.
The study showed no difference in mean improvement in Montgomery-Åsberg Depression Rating Scale (MADRS) scores at 6 weeks between active and sham tDCS.
“Our trial does not support the efficacy of tDCS as an additional treatment to SSRIs in adults with MDD,” the investigators, led by Frank Padberg, MD, department of psychiatry and psychotherapy, Ludwig-Maximilians-University Munich, write.
The study was published online in The Lancet.
Rigorous trial
Because it neurophysiologically modulates prefrontal cortex connectivity, tDCS has been proposed as a potential treatment for MDD.
Yet evidence for its efficacy has been inconsistent, and there is a scarcity of multicenter trial data, the researchers note.
The DepressionDC trial assessed the efficacy of tDCS in combination with SSRIs in 160 adults with MDD. Participants had a score of at least 15 on the Hamilton Depression Rating Scale (21-item version); their conditions had not responded to at least one antidepressant trial in their current depressive episode; and they had received treatment with an SSRI at a stable dose for at least 4 weeks. The SSRI was continued at the same dose during stimulation.
Eighty-three patients were allocated to undergo 30 min of 2-mA bifrontal tDCS every weekday for 4 weeks, then two tDCS sessions per week for 2 weeks; 77 patients were assigned to receive matching sham stimulation. Randomization was stratified by baseline MADRS score of less than 31 or 31 and higher.
In intention-to-treat analysis, there was no between-group difference in mean improvement on the MADRS at week 6 (–8.2 with active and –8.0 with sham tDCS; difference, 0.3; 95% confidence interval, –2.4 to 2.9).
There was also no significant difference for all secondary outcomes, including response and remission rates, patient-reported depression, and functioning, as well as at 18-week and 30-week follow-up visits.
There were significantly more mild adverse events reported in the active tDCS group than in the sham group (60% vs. 43%; P = .028). The most common adverse events were headaches, local skin reactions, and sleep-related problems.
Still reason for optimism
These findings call into question the efficacy of tDCS as add-on therapy to SSRI treatment for individuals with MDD and highlight the need for supportive evidence from multicenter studies, the investigators write.
Yet Dr. Padberg said it’s not the end of the road for tDCS for depression.
tDCS exerts a “rather subtle and nonfocal effect on neuronal activity. Thus, tDCS may need to be combined with specific behavioral or cognitive interventions which functionally involve the brain region where tDCS is targeted at,” he said.
Another “promising avenue” is personalization of tDCS by “individual MRI-based computational modeling of tDCS-induced electric fields,” he noted.
The coauthors of an accompanying commentary note that the DepressionDC trial was “carefully designed” and “well executed.”
And while the results did not show the superiority of active tDCS over sham stimulation as an additional treatment to SSRI therapy, “clinicians and researchers should not disregard this treatment for people with MDD,” write Daphne Voineskos, MD, PhD, and Daniel Blumberger, MD, with the University of Toronto.
“Specifically, further exploration of placebo response in less heterogeneous MDD samples and the evaluation of tDCS as an earlier treatment option for people with MDD are important areas of future research,” they suggest.
“Moreover, elucidating the effects of interindividual anatomical variability on electrical current distribution might lead to tDCS protocols that individualize treatment to optimize therapeutic effects as opposed to a so-called one-size-fits-all approach.
“Overall, there is reason for optimism about the potential to individualize tDCS and deliver it outside of the clinical setting,” Dr. Voineskos and Dr. Blumberger conclude.
Funding for the study was provided by the German Federal Ministry of Education and Research. Several authors disclosed relationships with the pharmaceutical industry. A complete list of disclosures of authors and comment writers is available with the original article.
A version of this article first appeared on Medscape.com.
FROM THE LANCET
Affordable IVF – Are we there yet?
The price for an in vitro fertilization (IVF) cycle continues to increase annually by many clinics, particularly because of “add-ons” of dubious value.

The initial application of IVF was for tubal factor infertility. Over the decades since 1981, the year of the first successful live birth in the United States, indications for IVF have dramatically expanded – ovulation dysfunction, unexplained infertility, male factor, advanced stage endometriosis, unexplained infertility, embryo testing to avoid an inherited genetic disease from the intended parents carrying the same mutation, and family balancing for gender, along with fertility preservation, including before potentially gonadotoxic treatment and “elective” planned oocyte cryopreservation.
From RESOLVE.org, the National Infertility Association: “As of June 2022, 20 states have passed fertility insurance coverage laws, 14 of those laws include IVF coverage, and 12 states have fertility preservation laws for iatrogenic (medically induced) infertility.” Consequently, “affordable IVF” is paramount to providing equal access for patients.

I spoke with the past president of The Society for Assisted Reproductive Technology (SART.org), Kevin Doody, MD, HCLD, to discuss current IVF treatment options for couples that may decrease their financial burden, particularly by applying a novel approach – called INVOcell – that involves using the woman’s vagina as the embryo “incubator.” Dr. Doody is director of CARE Fertility in Bedford, Tex., and clinical professor at UT Southwestern Medical Center, Dallas.
How does limiting the dosage of gonadotropins in IVF cycles, known as “minimal stimulation,” affect pregnancy outcomes?
IVF medications are often costly, so it is logical to try and minimize expenses by using them judiciously. “Minimal stimulation” generally is not the best approach, as having more eggs usually leads to better pregnancy rates. High egg yield increases short-term success and provides additional embryos for future attempts.
However, extremely high gonadotropin doses do not necessarily yield more eggs or successful pregnancies. The dose response to gonadotropins follows a sigmoid curve, and typically doses beyond 225-300 IU per day do not offer additional benefits, except for women with an elevated body weight. Yet, some physicians continue to use higher doses in women with low ovarian reserve, which is often not beneficial and can add unnecessary costs.
Is “natural cycle” IVF cost-effective with acceptable pregnancy success rates?
Although the first-ever IVF baby was conceived through a natural cycle, this approach has very low success rates. Even with advancements in IVF laboratory technologies, the outcomes of natural cycle IVF have remained disappointingly low and are generally considered unacceptable.
Are there other cost-saving alternatives for IVF that still maintain reasonable success rates?
Some patients can undergo a more simplified ovarian stimulation protocol that reduces the number of monitoring visits, thus reducing costs. In couples without a severe male factor, the application and additional expense of intracytoplasmic sperm injection (ICSI) is unnecessary. Pre-implantation genetic testing for embryo aneuploidy, another “add-on” procedure, has specific indications and medical evidence does not support its use in all patient cycles.
How can the cost of a standard IVF cycle be reduced, especially in areas without mandated infertility insurance coverage?
Addressing this issue involves considering principles of justice in medical ethics, which emphasize equal health care access for all individuals. Infertility is a medical condition and IVF is expensive, so lack of insurance coverage often restricts access. Our clinic offers a more affordable option called “effortless IVF” using an intravaginal culture system (INVOcell), which minimizes the monitoring process while maintaining satisfactory success rates and reducing the risks associated with ovarian hyperstimulation syndrome.
What is INVOcell, and how successful is it in terms of live birth rates?
INVOcell is an innovative approach to IVF, where an intravaginal culture system is used as an “embryo incubator whereby freshly harvested eggs along with sperm are immediately added to a small chamber device that is placed in the woman’s vagina for up to 5 days to allow for fertilization and embryo development.” The woman, typically, has no discomfort from the device. For appropriately selected patients, the literature has shown live birth rates are comparable to those achieved using conventional laboratory incubation systems.
As an early participant in INVOcell research, can you share insights on the ideal candidates for this procedure and any contraindications?
The INVOcell system is best suited for straightforward cases. It is not recommended for severe male factor infertility requiring ICSI, since this will delay application of the chamber device and increase cost. Further, cases involving preimplantation genetic testing are not recommended because the embryos may not develop synchronously within the device to the embryo stage needed for a biopsy.
What training is required for embryologists and physicians to use INVOcell?
Embryologists require training for a few hours to learn the basics of INVOcell. They must master loading eggs into and retrieving embryos from the device. Practicing on discarded eggs and embryos, embryologists can accelerate the acquisition of the proper technique needed for INVOcell. Physicians find the training easier; they mainly need to learn the correct placement and removal of the device in the vagina.
Is INVOcell gaining acceptance among patients and IVF centers?
Acceptance varies. In our practice, INVOcell has largely replaced superovulation and intrauterine insemination treatments. However, some clinics still need to determine how this tool fits within their practice.
Have IVF success rates plateaued as affordable options increase?
IVF success rates grew substantially in the 1980s and 1990s, fostered by improved embryo culture systems and higher numbers of embryos transferred, the latter at the expense of a multiple gestation. While the rate of improvement has slowed, coinciding with the increasing use of single embryo transfer, advancements in IVF continue toward the goal of improving the singleton live birth rate per IVF cycle. There is still room for enhancement in success rates alongside cost reduction. Continued innovation is needed, especially for patients with challenging underlying biological issues.
Can you provide insight into the next potential breakthrough in IVF that may reduce costs, be less invasive, and maintain optimal pregnancy rates?
I am very excited about recent breakthroughs in in vitro maturation (IVM) of oocytes. The bottleneck in IVF clinics (and significant expense) primarily relates to the need to stimulate the ovaries to get mature and competent eggs. The technology of IVM has existed for decades but has yet to be fully embraced by clinics because of the poor competency of oocytes matured in the laboratory.
Immature eggs resume meiosis immediately upon removal from the ovary. Nuclear maturation of eggs in the lab is easy. In fact, it happens too quickly, thereby not allowing for the maturation of the egg cytoplasm. This has previously led to poor development of embryos following fertilization and low success rates.
Recently, a new laboratory strategy has resulted in a significant improvement in success. This improved culture system uses a peptide that prevents the resumption of meiosis for the initial culture time frame. Substances, including follicle stimulating hormone, can be added to the media to promote oocyte cytoplasmic maturation. Following this, the eggs are placed in a media without the meiosis inhibitor to allow for nuclear maturation. This results in a significantly higher proportion of competent mature eggs.
Dr. Trolice is director of The IVF Center in Winter Park, Fla., and professor of obstetrics and gynecology at the University of Central Florida, Orlando.
The price for an in vitro fertilization (IVF) cycle continues to increase annually by many clinics, particularly because of “add-ons” of dubious value.
The initial application of IVF was for tubal factor infertility. Over the decades since 1981, the year of the first successful live birth in the United States, indications for IVF have dramatically expanded – ovulation dysfunction, unexplained infertility, male factor, advanced stage endometriosis, unexplained infertility, embryo testing to avoid an inherited genetic disease from the intended parents carrying the same mutation, and family balancing for gender, along with fertility preservation, including before potentially gonadotoxic treatment and “elective” planned oocyte cryopreservation.
From RESOLVE.org, the National Infertility Association: “As of June 2022, 20 states have passed fertility insurance coverage laws, 14 of those laws include IVF coverage, and 12 states have fertility preservation laws for iatrogenic (medically induced) infertility.” Consequently, “affordable IVF” is paramount to providing equal access for patients.
I spoke with the past president of The Society for Assisted Reproductive Technology (SART.org), Kevin Doody, MD, HCLD, to discuss current IVF treatment options for couples that may decrease their financial burden, particularly by applying a novel approach – called INVOcell – that involves using the woman’s vagina as the embryo “incubator.” Dr. Doody is director of CARE Fertility in Bedford, Tex., and clinical professor at UT Southwestern Medical Center, Dallas.
How does limiting the dosage of gonadotropins in IVF cycles, known as “minimal stimulation,” affect pregnancy outcomes?
IVF medications are often costly, so it is logical to try and minimize expenses by using them judiciously. “Minimal stimulation” generally is not the best approach, as having more eggs usually leads to better pregnancy rates. High egg yield increases short-term success and provides additional embryos for future attempts.
However, extremely high gonadotropin doses do not necessarily yield more eggs or successful pregnancies. The dose response to gonadotropins follows a sigmoid curve, and typically doses beyond 225-300 IU per day do not offer additional benefits, except for women with an elevated body weight. Yet, some physicians continue to use higher doses in women with low ovarian reserve, which is often not beneficial and can add unnecessary costs.
Is “natural cycle” IVF cost-effective with acceptable pregnancy success rates?
Although the first-ever IVF baby was conceived through a natural cycle, this approach has very low success rates. Even with advancements in IVF laboratory technologies, the outcomes of natural cycle IVF have remained disappointingly low and are generally considered unacceptable.
Are there other cost-saving alternatives for IVF that still maintain reasonable success rates?
Some patients can undergo a more simplified ovarian stimulation protocol that reduces the number of monitoring visits, thus reducing costs. In couples without a severe male factor, the application and additional expense of intracytoplasmic sperm injection (ICSI) is unnecessary. Pre-implantation genetic testing for embryo aneuploidy, another “add-on” procedure, has specific indications and medical evidence does not support its use in all patient cycles.
How can the cost of a standard IVF cycle be reduced, especially in areas without mandated infertility insurance coverage?
Addressing this issue involves considering principles of justice in medical ethics, which emphasize equal health care access for all individuals. Infertility is a medical condition and IVF is expensive, so lack of insurance coverage often restricts access. Our clinic offers a more affordable option called “effortless IVF” using an intravaginal culture system (INVOcell), which minimizes the monitoring process while maintaining satisfactory success rates and reducing the risks associated with ovarian hyperstimulation syndrome.
What is INVOcell, and how successful is it in terms of live birth rates?
INVOcell is an innovative approach to IVF, where an intravaginal culture system is used as an “embryo incubator whereby freshly harvested eggs along with sperm are immediately added to a small chamber device that is placed in the woman’s vagina for up to 5 days to allow for fertilization and embryo development.” The woman, typically, has no discomfort from the device. For appropriately selected patients, the literature has shown live birth rates are comparable to those achieved using conventional laboratory incubation systems.
As an early participant in INVOcell research, can you share insights on the ideal candidates for this procedure and any contraindications?
The INVOcell system is best suited for straightforward cases. It is not recommended for severe male factor infertility requiring ICSI, since this will delay application of the chamber device and increase cost. Further, cases involving preimplantation genetic testing are not recommended because the embryos may not develop synchronously within the device to the embryo stage needed for a biopsy.
What training is required for embryologists and physicians to use INVOcell?
Embryologists require training for a few hours to learn the basics of INVOcell. They must master loading eggs into and retrieving embryos from the device. Practicing on discarded eggs and embryos, embryologists can accelerate the acquisition of the proper technique needed for INVOcell. Physicians find the training easier; they mainly need to learn the correct placement and removal of the device in the vagina.
Is INVOcell gaining acceptance among patients and IVF centers?
Acceptance varies. In our practice, INVOcell has largely replaced superovulation and intrauterine insemination treatments. However, some clinics still need to determine how this tool fits within their practice.
Have IVF success rates plateaued as affordable options increase?
IVF success rates grew substantially in the 1980s and 1990s, fostered by improved embryo culture systems and higher numbers of embryos transferred, the latter at the expense of a multiple gestation. While the rate of improvement has slowed, coinciding with the increasing use of single embryo transfer, advancements in IVF continue toward the goal of improving the singleton live birth rate per IVF cycle. There is still room for enhancement in success rates alongside cost reduction. Continued innovation is needed, especially for patients with challenging underlying biological issues.
Can you provide insight into the next potential breakthrough in IVF that may reduce costs, be less invasive, and maintain optimal pregnancy rates?
I am very excited about recent breakthroughs in in vitro maturation (IVM) of oocytes. The bottleneck in IVF clinics (and significant expense) primarily relates to the need to stimulate the ovaries to get mature and competent eggs. The technology of IVM has existed for decades but has yet to be fully embraced by clinics because of the poor competency of oocytes matured in the laboratory.
Immature eggs resume meiosis immediately upon removal from the ovary. Nuclear maturation of eggs in the lab is easy. In fact, it happens too quickly, thereby not allowing for the maturation of the egg cytoplasm. This has previously led to poor development of embryos following fertilization and low success rates.
Recently, a new laboratory strategy has resulted in a significant improvement in success. This improved culture system uses a peptide that prevents the resumption of meiosis for the initial culture time frame. Substances, including follicle stimulating hormone, can be added to the media to promote oocyte cytoplasmic maturation. Following this, the eggs are placed in a media without the meiosis inhibitor to allow for nuclear maturation. This results in a significantly higher proportion of competent mature eggs.
Dr. Trolice is director of The IVF Center in Winter Park, Fla., and professor of obstetrics and gynecology at the University of Central Florida, Orlando.
The price for an in vitro fertilization (IVF) cycle continues to increase annually by many clinics, particularly because of “add-ons” of dubious value.
The initial application of IVF was for tubal factor infertility. Over the decades since 1981, the year of the first successful live birth in the United States, indications for IVF have dramatically expanded – ovulation dysfunction, unexplained infertility, male factor, advanced stage endometriosis, unexplained infertility, embryo testing to avoid an inherited genetic disease from the intended parents carrying the same mutation, and family balancing for gender, along with fertility preservation, including before potentially gonadotoxic treatment and “elective” planned oocyte cryopreservation.
From RESOLVE.org, the National Infertility Association: “As of June 2022, 20 states have passed fertility insurance coverage laws, 14 of those laws include IVF coverage, and 12 states have fertility preservation laws for iatrogenic (medically induced) infertility.” Consequently, “affordable IVF” is paramount to providing equal access for patients.
I spoke with the past president of The Society for Assisted Reproductive Technology (SART.org), Kevin Doody, MD, HCLD, to discuss current IVF treatment options for couples that may decrease their financial burden, particularly by applying a novel approach – called INVOcell – that involves using the woman’s vagina as the embryo “incubator.” Dr. Doody is director of CARE Fertility in Bedford, Tex., and clinical professor at UT Southwestern Medical Center, Dallas.
How does limiting the dosage of gonadotropins in IVF cycles, known as “minimal stimulation,” affect pregnancy outcomes?
IVF medications are often costly, so it is logical to try and minimize expenses by using them judiciously. “Minimal stimulation” generally is not the best approach, as having more eggs usually leads to better pregnancy rates. High egg yield increases short-term success and provides additional embryos for future attempts.
However, extremely high gonadotropin doses do not necessarily yield more eggs or successful pregnancies. The dose response to gonadotropins follows a sigmoid curve, and typically doses beyond 225-300 IU per day do not offer additional benefits, except for women with an elevated body weight. Yet, some physicians continue to use higher doses in women with low ovarian reserve, which is often not beneficial and can add unnecessary costs.
Is “natural cycle” IVF cost-effective with acceptable pregnancy success rates?
Although the first-ever IVF baby was conceived through a natural cycle, this approach has very low success rates. Even with advancements in IVF laboratory technologies, the outcomes of natural cycle IVF have remained disappointingly low and are generally considered unacceptable.
Are there other cost-saving alternatives for IVF that still maintain reasonable success rates?
Some patients can undergo a more simplified ovarian stimulation protocol that reduces the number of monitoring visits, thus reducing costs. In couples without a severe male factor, the application and additional expense of intracytoplasmic sperm injection (ICSI) is unnecessary. Pre-implantation genetic testing for embryo aneuploidy, another “add-on” procedure, has specific indications and medical evidence does not support its use in all patient cycles.
How can the cost of a standard IVF cycle be reduced, especially in areas without mandated infertility insurance coverage?
Addressing this issue involves considering principles of justice in medical ethics, which emphasize equal health care access for all individuals. Infertility is a medical condition and IVF is expensive, so lack of insurance coverage often restricts access. Our clinic offers a more affordable option called “effortless IVF” using an intravaginal culture system (INVOcell), which minimizes the monitoring process while maintaining satisfactory success rates and reducing the risks associated with ovarian hyperstimulation syndrome.
What is INVOcell, and how successful is it in terms of live birth rates?
INVOcell is an innovative approach to IVF, where an intravaginal culture system is used as an “embryo incubator whereby freshly harvested eggs along with sperm are immediately added to a small chamber device that is placed in the woman’s vagina for up to 5 days to allow for fertilization and embryo development.” The woman, typically, has no discomfort from the device. For appropriately selected patients, the literature has shown live birth rates are comparable to those achieved using conventional laboratory incubation systems.
As an early participant in INVOcell research, can you share insights on the ideal candidates for this procedure and any contraindications?
The INVOcell system is best suited for straightforward cases. It is not recommended for severe male factor infertility requiring ICSI, since this will delay application of the chamber device and increase cost. Further, cases involving preimplantation genetic testing are not recommended because the embryos may not develop synchronously within the device to the embryo stage needed for a biopsy.
What training is required for embryologists and physicians to use INVOcell?
Embryologists require training for a few hours to learn the basics of INVOcell. They must master loading eggs into and retrieving embryos from the device. Practicing on discarded eggs and embryos, embryologists can accelerate the acquisition of the proper technique needed for INVOcell. Physicians find the training easier; they mainly need to learn the correct placement and removal of the device in the vagina.
Is INVOcell gaining acceptance among patients and IVF centers?
Acceptance varies. In our practice, INVOcell has largely replaced superovulation and intrauterine insemination treatments. However, some clinics still need to determine how this tool fits within their practice.
Have IVF success rates plateaued as affordable options increase?
IVF success rates grew substantially in the 1980s and 1990s, fostered by improved embryo culture systems and higher numbers of embryos transferred, the latter at the expense of a multiple gestation. While the rate of improvement has slowed, coinciding with the increasing use of single embryo transfer, advancements in IVF continue toward the goal of improving the singleton live birth rate per IVF cycle. There is still room for enhancement in success rates alongside cost reduction. Continued innovation is needed, especially for patients with challenging underlying biological issues.
Can you provide insight into the next potential breakthrough in IVF that may reduce costs, be less invasive, and maintain optimal pregnancy rates?
I am very excited about recent breakthroughs in in vitro maturation (IVM) of oocytes. The bottleneck in IVF clinics (and significant expense) primarily relates to the need to stimulate the ovaries to get mature and competent eggs. The technology of IVM has existed for decades but has yet to be fully embraced by clinics because of the poor competency of oocytes matured in the laboratory.
Immature eggs resume meiosis immediately upon removal from the ovary. Nuclear maturation of eggs in the lab is easy. In fact, it happens too quickly, thereby not allowing for the maturation of the egg cytoplasm. This has previously led to poor development of embryos following fertilization and low success rates.
Recently, a new laboratory strategy has resulted in a significant improvement in success. This improved culture system uses a peptide that prevents the resumption of meiosis for the initial culture time frame. Substances, including follicle stimulating hormone, can be added to the media to promote oocyte cytoplasmic maturation. Following this, the eggs are placed in a media without the meiosis inhibitor to allow for nuclear maturation. This results in a significantly higher proportion of competent mature eggs.
Dr. Trolice is director of The IVF Center in Winter Park, Fla., and professor of obstetrics and gynecology at the University of Central Florida, Orlando.

Does rapid weight loss from GLP-1s decrease muscle mass?
Recently, the glucagonlike peptide 1 (GLP-1) receptor agonist semaglutide has changed the obesity treatment landscape. This and other similar medications approaching the market are in high demand because of their ease of use, effectiveness, and lack of interactions with other medications.
Semaglutide is a weekly subcutaneous injection approved by the U.S. Food and Drug Administration for weight loss in conjunction with lifestyle change. It elicits an average weight loss of 15%-18% from baseline in adults with overweight or obesity (body mass index ≥ 27 with at least one obesity-related comorbidity or BMI ≥ 30) in a period of 52-68 weeks (Wilding et al; Rubino et al). Liraglutide is a daily GLP-1 agonist, which is FDA approved for treatment of overweight, with an average weight loss of 8% from treatment start.
Though GLP-1 agonists are very effective for weight loss, questions about side effects have arisen.
Current modalities of weight loss don’t specifically target fat mass (FM), so it is expected that, to a degree, fat-free mass (FFM), including muscle mass, will also be lost along with fat mass.
Loss of muscle mass is associated with an increased risk for lower bone density, fatigue, injuries, and decreased strength. In addition, sarcopenic obesity, a combination of high body fat percentage and low skeletal muscle mass, is concerning in patients older than 65 years and/or postmenopausal patients. Because GLP-1 agonists cause more rapid and sustainable weight loss, compared with intensive behavioral lifestyle therapy, there has been more media attention recently about possible muscle mass loss with GLP-1–agonist use.
However, proper well-rounded approaches to obesity treatment can mitigate the issue of muscle mass loss even when rapid weight loss occurs. When weight loss is achieved with very-low-calorie dietary changes alone (without exercise), it is also associated with significant reductions in lean muscle mass; however, incorporating exercise, preferably resistance training, can mitigate the muscle mass loss. The muscle-preserving effect of exercise is especially prominent in older populations where it is needed most and should be incorporated (Armanento-Villareal et al.; Winter et al.; Batsis and Zagaria; Mason et al.).
Furthermore, studies in rat models demonstrate liraglutide induces myogenesis in myoblasts and protects against muscular atrophy. In human studies, GLP-1 infusion was associated with an improved skeletal and cardiac muscle microvasculature, suggesting that GLP-1 agonists may have some positive effects on the muscle. A 2020 systematic review examined the effect of gradual vs. rapid weight loss and demonstrated no significant difference in muscle loss between the rapid weight-loss group and gradual weight-loss group. Even after gastric bypass surgery, most of the muscle mass loss occurred during the first year, when weight loss is happening. However, after the first year, skeletal muscle was maintained even without introducing additional dietary or exercise interventions.
Age, although a consideration, should not be a discriminating factor against treating obesity. Sarcopenic obesity is a serious risk especially in patients aged 65 years or older, but GLP-1–agonist therapy can be beneficial to prevent muscle atrophy and increase blood flow to skeletal and cardiac muscle. In addition, patients must be encouraged to maintain an appropriate dietary and exercise regimen to treat their obesity. Management of obesity is complex and multifaceted, and patients should understand their responsibility to follow clinician recommendations during this journey to decrease the associated side effects.
Overall, with any level of weight loss achieved with current strategies, a certain amount of muscle mass loss is expected. All efforts to actively preserve muscle mass can prevent too much muscle loss.
Therefore, providers prescribing medications like GLP-1 agonists to treat obesity must also counsel patients about incorporating aerobic exercise and resistance training as part of the treatment plan as well as ensuring they eat a high-protein diet. Generally, resistance training is preferred over aerobic exercise for muscle mass preservation and increased strength, but studies also demonstrate benefit with aerobic exercise.
In the first few visits of initiating obesity treatment, patients should be encouraged to start to incorporate light physical activity as tolerable while starting to make dietary changes to include at least 0.8g/kg/day of protein (Fappi et al.). These initial visits are also an important opportunity for clinicians to ingrain the importance of exercise as part of healthy weight loss. At every visit, physical activity level should be assessed.
Dr. Ahn is a clinical fellow in obesity medicine, Weight Management Center, at Massachusetts General Hospital, Boston. Dr. Singhal is an assistant professor of pediatrics, Harvard Medical School, Boston, and director, Pediatric Program, MGH Weight Center, Massachusetts General Hospital. Dr. Singhal reported that his spouse consults with AstraZeneca, Dilachi Pharma, Eli Lilly, Genetech, Immunomedics, Pfizer, Sanofi, and Novartis.
A version of this article first appeared on Medscape.com.
Recently, the glucagonlike peptide 1 (GLP-1) receptor agonist semaglutide has changed the obesity treatment landscape. This and other similar medications approaching the market are in high demand because of their ease of use, effectiveness, and lack of interactions with other medications.
Semaglutide is a weekly subcutaneous injection approved by the U.S. Food and Drug Administration for weight loss in conjunction with lifestyle change. It elicits an average weight loss of 15%-18% from baseline in adults with overweight or obesity (body mass index ≥ 27 with at least one obesity-related comorbidity or BMI ≥ 30) in a period of 52-68 weeks (Wilding et al; Rubino et al). Liraglutide is a daily GLP-1 agonist, which is FDA approved for treatment of overweight, with an average weight loss of 8% from treatment start.
Though GLP-1 agonists are very effective for weight loss, questions about side effects have arisen.
Current modalities of weight loss don’t specifically target fat mass (FM), so it is expected that, to a degree, fat-free mass (FFM), including muscle mass, will also be lost along with fat mass.
Loss of muscle mass is associated with an increased risk for lower bone density, fatigue, injuries, and decreased strength. In addition, sarcopenic obesity, a combination of high body fat percentage and low skeletal muscle mass, is concerning in patients older than 65 years and/or postmenopausal patients. Because GLP-1 agonists cause more rapid and sustainable weight loss, compared with intensive behavioral lifestyle therapy, there has been more media attention recently about possible muscle mass loss with GLP-1–agonist use.
However, proper well-rounded approaches to obesity treatment can mitigate the issue of muscle mass loss even when rapid weight loss occurs. When weight loss is achieved with very-low-calorie dietary changes alone (without exercise), it is also associated with significant reductions in lean muscle mass; however, incorporating exercise, preferably resistance training, can mitigate the muscle mass loss. The muscle-preserving effect of exercise is especially prominent in older populations where it is needed most and should be incorporated (Armanento-Villareal et al.; Winter et al.; Batsis and Zagaria; Mason et al.).
Furthermore, studies in rat models demonstrate liraglutide induces myogenesis in myoblasts and protects against muscular atrophy. In human studies, GLP-1 infusion was associated with an improved skeletal and cardiac muscle microvasculature, suggesting that GLP-1 agonists may have some positive effects on the muscle. A 2020 systematic review examined the effect of gradual vs. rapid weight loss and demonstrated no significant difference in muscle loss between the rapid weight-loss group and gradual weight-loss group. Even after gastric bypass surgery, most of the muscle mass loss occurred during the first year, when weight loss is happening. However, after the first year, skeletal muscle was maintained even without introducing additional dietary or exercise interventions.
Age, although a consideration, should not be a discriminating factor against treating obesity. Sarcopenic obesity is a serious risk especially in patients aged 65 years or older, but GLP-1–agonist therapy can be beneficial to prevent muscle atrophy and increase blood flow to skeletal and cardiac muscle. In addition, patients must be encouraged to maintain an appropriate dietary and exercise regimen to treat their obesity. Management of obesity is complex and multifaceted, and patients should understand their responsibility to follow clinician recommendations during this journey to decrease the associated side effects.
Overall, with any level of weight loss achieved with current strategies, a certain amount of muscle mass loss is expected. All efforts to actively preserve muscle mass can prevent too much muscle loss.
Therefore, providers prescribing medications like GLP-1 agonists to treat obesity must also counsel patients about incorporating aerobic exercise and resistance training as part of the treatment plan as well as ensuring they eat a high-protein diet. Generally, resistance training is preferred over aerobic exercise for muscle mass preservation and increased strength, but studies also demonstrate benefit with aerobic exercise.
In the first few visits of initiating obesity treatment, patients should be encouraged to start to incorporate light physical activity as tolerable while starting to make dietary changes to include at least 0.8g/kg/day of protein (Fappi et al.). These initial visits are also an important opportunity for clinicians to ingrain the importance of exercise as part of healthy weight loss. At every visit, physical activity level should be assessed.
Dr. Ahn is a clinical fellow in obesity medicine, Weight Management Center, at Massachusetts General Hospital, Boston. Dr. Singhal is an assistant professor of pediatrics, Harvard Medical School, Boston, and director, Pediatric Program, MGH Weight Center, Massachusetts General Hospital. Dr. Singhal reported that his spouse consults with AstraZeneca, Dilachi Pharma, Eli Lilly, Genetech, Immunomedics, Pfizer, Sanofi, and Novartis.
A version of this article first appeared on Medscape.com.
Recently, the glucagonlike peptide 1 (GLP-1) receptor agonist semaglutide has changed the obesity treatment landscape. This and other similar medications approaching the market are in high demand because of their ease of use, effectiveness, and lack of interactions with other medications.
Semaglutide is a weekly subcutaneous injection approved by the U.S. Food and Drug Administration for weight loss in conjunction with lifestyle change. It elicits an average weight loss of 15%-18% from baseline in adults with overweight or obesity (body mass index ≥ 27 with at least one obesity-related comorbidity or BMI ≥ 30) in a period of 52-68 weeks (Wilding et al; Rubino et al). Liraglutide is a daily GLP-1 agonist, which is FDA approved for treatment of overweight, with an average weight loss of 8% from treatment start.
Though GLP-1 agonists are very effective for weight loss, questions about side effects have arisen.
Current modalities of weight loss don’t specifically target fat mass (FM), so it is expected that, to a degree, fat-free mass (FFM), including muscle mass, will also be lost along with fat mass.
Loss of muscle mass is associated with an increased risk for lower bone density, fatigue, injuries, and decreased strength. In addition, sarcopenic obesity, a combination of high body fat percentage and low skeletal muscle mass, is concerning in patients older than 65 years and/or postmenopausal patients. Because GLP-1 agonists cause more rapid and sustainable weight loss, compared with intensive behavioral lifestyle therapy, there has been more media attention recently about possible muscle mass loss with GLP-1–agonist use.
However, proper well-rounded approaches to obesity treatment can mitigate the issue of muscle mass loss even when rapid weight loss occurs. When weight loss is achieved with very-low-calorie dietary changes alone (without exercise), it is also associated with significant reductions in lean muscle mass; however, incorporating exercise, preferably resistance training, can mitigate the muscle mass loss. The muscle-preserving effect of exercise is especially prominent in older populations where it is needed most and should be incorporated (Armanento-Villareal et al.; Winter et al.; Batsis and Zagaria; Mason et al.).
Furthermore, studies in rat models demonstrate liraglutide induces myogenesis in myoblasts and protects against muscular atrophy. In human studies, GLP-1 infusion was associated with an improved skeletal and cardiac muscle microvasculature, suggesting that GLP-1 agonists may have some positive effects on the muscle. A 2020 systematic review examined the effect of gradual vs. rapid weight loss and demonstrated no significant difference in muscle loss between the rapid weight-loss group and gradual weight-loss group. Even after gastric bypass surgery, most of the muscle mass loss occurred during the first year, when weight loss is happening. However, after the first year, skeletal muscle was maintained even without introducing additional dietary or exercise interventions.
Age, although a consideration, should not be a discriminating factor against treating obesity. Sarcopenic obesity is a serious risk especially in patients aged 65 years or older, but GLP-1–agonist therapy can be beneficial to prevent muscle atrophy and increase blood flow to skeletal and cardiac muscle. In addition, patients must be encouraged to maintain an appropriate dietary and exercise regimen to treat their obesity. Management of obesity is complex and multifaceted, and patients should understand their responsibility to follow clinician recommendations during this journey to decrease the associated side effects.
Overall, with any level of weight loss achieved with current strategies, a certain amount of muscle mass loss is expected. All efforts to actively preserve muscle mass can prevent too much muscle loss.
Therefore, providers prescribing medications like GLP-1 agonists to treat obesity must also counsel patients about incorporating aerobic exercise and resistance training as part of the treatment plan as well as ensuring they eat a high-protein diet. Generally, resistance training is preferred over aerobic exercise for muscle mass preservation and increased strength, but studies also demonstrate benefit with aerobic exercise.
In the first few visits of initiating obesity treatment, patients should be encouraged to start to incorporate light physical activity as tolerable while starting to make dietary changes to include at least 0.8g/kg/day of protein (Fappi et al.). These initial visits are also an important opportunity for clinicians to ingrain the importance of exercise as part of healthy weight loss. At every visit, physical activity level should be assessed.
Dr. Ahn is a clinical fellow in obesity medicine, Weight Management Center, at Massachusetts General Hospital, Boston. Dr. Singhal is an assistant professor of pediatrics, Harvard Medical School, Boston, and director, Pediatric Program, MGH Weight Center, Massachusetts General Hospital. Dr. Singhal reported that his spouse consults with AstraZeneca, Dilachi Pharma, Eli Lilly, Genetech, Immunomedics, Pfizer, Sanofi, and Novartis.
A version of this article first appeared on Medscape.com.
Camp Discovery: A place for children to be comfortable in their own skin
The talent show, the grand finale of the 1-week camp, was nearly 7 years ago, but Emily Haygood of Houston, now 17 and about to start her senior year, remembers it in detail. She sang “Death of a Bachelor,” an R&B pop song and Billboard No. 1 hit at the time about a former bachelor who had happily married. These days, she said, if she watched the video of her 10-year-old singing self, “I would probably throw up.” But she still treasures the audience response, “having all those people I’d gotten close to cheer for me.”
Emily was at , but share one feature: they are the kind of dermatologic issues that can make doing everyday kid or teen activities like swimming difficult and can elicit mean comments from classmates and other would-be friends.

Emily was first diagnosed with atopic dermatitis at age 4, her mother, Amber Haygood, says. By age 9, it had become severe. Emily remembers being teased some in elementary school. “I did feel bad a lot of the time, when asked insensitive questions.” Her mother still bristles that adults often could be cruel, too.
But at Camp Discovery, those issues were nonexistent. “Camp was so cool,” Emily said. Besides the usual camp activities, it had things that “normal” camp didn’t, like other kids who didn’t stare at your skin condition or make fun of it.
30th anniversary season begins
This year is the 30th anniversary of Camp Discovery. Sessions began July 23 and continue through Aug. 18, with locations in Crosslake, Minn.; Hebron, Conn.; and Millville, Pa., in addition to Burton, Tex. About 300 campers will attend this year, according to the AAD, and 6,151 campers have attended from 1993 to 2022.

The 1-week camp accepts youth with conditions ranging from eczema and psoriasis to vitiligo, alopecia, epidermolysis bullosa, and ichthyosis, according to the academy. A dermatologist first refers a child, downloading and completing the referral form and sending it to the academy.
The 1-week session, including travel, is free for the campers, thanks to donors. As a nonprofit and membership-based organization, the AAD does not release the detailed financial information about the operating budget for the camp. Dermatologists, nurses, and counselors volunteer their time.
In his presidential address at the AAD’s annual meeting in March, outgoing president Mark D. Kaufmann, MD, of the department of dermatology at the Icahn School of Medicine at Mount Sinai in New York, referred to camp volunteering as an antidote to professional burnout. Remembering why as a dermatologist one entered the profession can be one solution, he said, and described his own recent 3-day volunteer stint at the camp.
“Those 3 magical days, being with kids as they discovered they weren’t alone in the world, sharing their experiences and ideas, reminded me why I became a physician in the first place,” he told the audience of meeting attendees. He vowed to expand the program, with a goal of having every dermatology resident attend Camp Discovery.
Mental health effects of skin conditions
Much research has focused on the mental health fallout from living with chronic skin conditions, and even young children can be adversely affected. In one review of the literature, researchers concluded that pediatric skin disease, including acne, atopic dermatitis, and psoriasis, can affect quality of life, carry stigma, and lead to bullying and eventually even suicidal behavior. Another study, published earlier this year, found that atopic dermatitis affected children’s quality of life, impacting sleep and leading to feelings of being ashamed.

“It’s not necessarily about what their skin condition is and more about the psychosocial impact,’’ said Samantha Hill, MD, a pediatric and general dermatologist in Lynchburg, Va., who is the medical director of Camp Discovery in Minnesota this year.
Camp activities, reactions
The overriding theme of camp is allowing all the youth to be “just one of the kids at camp,” Dr. Hill said in an interview. “They come to do all kinds of things they don’t do in normal life because people don’t give them the credit to [be able to] do it.”
Every year, she said, “I tell my staff we are in the business of making things happen, so if there is a kid bandaged head to toe [because of a skin condition] and they want to go tubing and get in the lake, we figure out how to make it happen. We have done that multiple times.”
Newcomers are initially nervous, Dr. Hill acknowledged, but in time let their guard down. Returnees are a different story. “When kids who have been at camp before arrive, you can see them start breathing again, looking for their friends. You can see them relax right before your eyes.”
“The single most empowering thing is the realization you are not alone,” said Meena Julapalli, MD, a Houston dermatologist who is a medical team member and long-time volunteer at Camp Discovery. That, she said, and “You get to be a kid, and you don’t have to have people staring at you.”

Dr. Julapalli remembers one of her patients with keratitis-ichthyosis-deafness (KID) syndrome. “She needed more than what I could offer,” she said. “She needed camp.” At camp, the organizers found a counselor who knew sign language to accompany her. At first, she was quiet and didn’t smile much. By the end of the week, as she was about to observe her birthday, things changed. After breakfast, she was led to the stage, where fellow campers began singing – and signing the song they had just learned.
Camp staff gets it
Allyson Garin, who was diagnosed with vitiligo at age 6 months, is a camp program director at Camp Discovery in Crosslake, Minn. She first went to camp in 1990 at age 11, returning until she “aged out” at 16, then worked as a counselor. She gets it when campers tell her they hear rude comments about their skin conditions.

“I remember being in swimming pools, in lines at fairgrounds or amusement parks,” she said in an interview, “and hearing people say, ‘Don’t touch her,’ ’’ fearing contagion, perhaps. “People would make jokes about cows, since they are spotted,” she said, or people would simply step back.
All those years ago, her mother found out about the camp and decided to figure out how to get her there. She got there, and she met a fellow camper with vitiligo, and they became pen pals. “We still talk,” she said.
Meeting someone with the same skin condition, she said, isn’t just about commiserating. “There is a lot of information sharing,” on topics such as best treatments, strategies, and other conversations.
Other lessons
While campers can feel comfortable around others who also have skin conditions, and understand, the lesson extends beyond that, Ms. Garin said. “It gave me a perspective,” she said of her camp experience. “I always felt, ‘Woe is me.’ ” But when she met others with, as she said, conditions “way worse than vitiligo, it really grounds you.”
Dr. Hill agreed. Campers get the benefit of others accepting and including them, but also practicing that same attitude toward fellow campers, she said. “It insures that we are providing this environment of inclusion, but that they are practicing it as well. They need to practice it like everyone else.”
Getting parents on board
The idea of camp, especially for those at the younger end of the 8- to 16-years age range accepted for Camp Discovery, can take some getting used to for some parents. Ms. Haygood, Emily’s mother, relates to that. Her daughter’s dermatologist at the time, who is now retired, had first suggested the camp. Her first reaction? “I am not sending my chronically ill child to camp with strangers.” She also acknowledged that she, like other parents of children with a chronic illness, can be a helicopter parent.

Then, she noticed that Emily seemed interested, so she got more information, finding out that it was staffed by doctors. It all sounded good, she said, and the social interaction, she knew, would be beneficial. “Then my husband was a no,” she said, concerned about their daughter being with strangers. “Eventually he came around,” Ms. Haygood said. All along, Emily said, “it seemed fun. I was probably trying to talk them into it.” She admits she was very nervous at first, but calmed down when she realized her own dermatologist was going to be there.
Vanessa Hadley of Spring, Tex., was on board the moment she heard about Camp Discovery. “I just thought it was amazing,” she said. Her daughter Isabelle, 13, has been to the camp. “She has alopecia areata and severe eczema,” Ms. Hadley said. Now, Isabelle is returning to camp and coaching her sister Penelope, 8, who has eczema and mild alopecia and is a first-timer this summer.
One tip the 8-year-old has learned so far: Turn to your counselor for support if you’re nervous. That worked, Isabelle said, the first year when she was wary of the zipline – then surprised herself and conquered it.
Dr. Hill and Dr. Julapalli have no disclosures.
The talent show, the grand finale of the 1-week camp, was nearly 7 years ago, but Emily Haygood of Houston, now 17 and about to start her senior year, remembers it in detail. She sang “Death of a Bachelor,” an R&B pop song and Billboard No. 1 hit at the time about a former bachelor who had happily married. These days, she said, if she watched the video of her 10-year-old singing self, “I would probably throw up.” But she still treasures the audience response, “having all those people I’d gotten close to cheer for me.”
Emily was at , but share one feature: they are the kind of dermatologic issues that can make doing everyday kid or teen activities like swimming difficult and can elicit mean comments from classmates and other would-be friends.
Emily was first diagnosed with atopic dermatitis at age 4, her mother, Amber Haygood, says. By age 9, it had become severe. Emily remembers being teased some in elementary school. “I did feel bad a lot of the time, when asked insensitive questions.” Her mother still bristles that adults often could be cruel, too.
But at Camp Discovery, those issues were nonexistent. “Camp was so cool,” Emily said. Besides the usual camp activities, it had things that “normal” camp didn’t, like other kids who didn’t stare at your skin condition or make fun of it.
30th anniversary season begins
This year is the 30th anniversary of Camp Discovery. Sessions began July 23 and continue through Aug. 18, with locations in Crosslake, Minn.; Hebron, Conn.; and Millville, Pa., in addition to Burton, Tex. About 300 campers will attend this year, according to the AAD, and 6,151 campers have attended from 1993 to 2022.
The 1-week camp accepts youth with conditions ranging from eczema and psoriasis to vitiligo, alopecia, epidermolysis bullosa, and ichthyosis, according to the academy. A dermatologist first refers a child, downloading and completing the referral form and sending it to the academy.
The 1-week session, including travel, is free for the campers, thanks to donors. As a nonprofit and membership-based organization, the AAD does not release the detailed financial information about the operating budget for the camp. Dermatologists, nurses, and counselors volunteer their time.
In his presidential address at the AAD’s annual meeting in March, outgoing president Mark D. Kaufmann, MD, of the department of dermatology at the Icahn School of Medicine at Mount Sinai in New York, referred to camp volunteering as an antidote to professional burnout. Remembering why as a dermatologist one entered the profession can be one solution, he said, and described his own recent 3-day volunteer stint at the camp.
“Those 3 magical days, being with kids as they discovered they weren’t alone in the world, sharing their experiences and ideas, reminded me why I became a physician in the first place,” he told the audience of meeting attendees. He vowed to expand the program, with a goal of having every dermatology resident attend Camp Discovery.
Mental health effects of skin conditions
Much research has focused on the mental health fallout from living with chronic skin conditions, and even young children can be adversely affected. In one review of the literature, researchers concluded that pediatric skin disease, including acne, atopic dermatitis, and psoriasis, can affect quality of life, carry stigma, and lead to bullying and eventually even suicidal behavior. Another study, published earlier this year, found that atopic dermatitis affected children’s quality of life, impacting sleep and leading to feelings of being ashamed.
“It’s not necessarily about what their skin condition is and more about the psychosocial impact,’’ said Samantha Hill, MD, a pediatric and general dermatologist in Lynchburg, Va., who is the medical director of Camp Discovery in Minnesota this year.
Camp activities, reactions
The overriding theme of camp is allowing all the youth to be “just one of the kids at camp,” Dr. Hill said in an interview. “They come to do all kinds of things they don’t do in normal life because people don’t give them the credit to [be able to] do it.”
Every year, she said, “I tell my staff we are in the business of making things happen, so if there is a kid bandaged head to toe [because of a skin condition] and they want to go tubing and get in the lake, we figure out how to make it happen. We have done that multiple times.”
Newcomers are initially nervous, Dr. Hill acknowledged, but in time let their guard down. Returnees are a different story. “When kids who have been at camp before arrive, you can see them start breathing again, looking for their friends. You can see them relax right before your eyes.”
“The single most empowering thing is the realization you are not alone,” said Meena Julapalli, MD, a Houston dermatologist who is a medical team member and long-time volunteer at Camp Discovery. That, she said, and “You get to be a kid, and you don’t have to have people staring at you.”
Dr. Julapalli remembers one of her patients with keratitis-ichthyosis-deafness (KID) syndrome. “She needed more than what I could offer,” she said. “She needed camp.” At camp, the organizers found a counselor who knew sign language to accompany her. At first, she was quiet and didn’t smile much. By the end of the week, as she was about to observe her birthday, things changed. After breakfast, she was led to the stage, where fellow campers began singing – and signing the song they had just learned.
Camp staff gets it
Allyson Garin, who was diagnosed with vitiligo at age 6 months, is a camp program director at Camp Discovery in Crosslake, Minn. She first went to camp in 1990 at age 11, returning until she “aged out” at 16, then worked as a counselor. She gets it when campers tell her they hear rude comments about their skin conditions.
“I remember being in swimming pools, in lines at fairgrounds or amusement parks,” she said in an interview, “and hearing people say, ‘Don’t touch her,’ ’’ fearing contagion, perhaps. “People would make jokes about cows, since they are spotted,” she said, or people would simply step back.
All those years ago, her mother found out about the camp and decided to figure out how to get her there. She got there, and she met a fellow camper with vitiligo, and they became pen pals. “We still talk,” she said.
Meeting someone with the same skin condition, she said, isn’t just about commiserating. “There is a lot of information sharing,” on topics such as best treatments, strategies, and other conversations.
Other lessons
While campers can feel comfortable around others who also have skin conditions, and understand, the lesson extends beyond that, Ms. Garin said. “It gave me a perspective,” she said of her camp experience. “I always felt, ‘Woe is me.’ ” But when she met others with, as she said, conditions “way worse than vitiligo, it really grounds you.”
Dr. Hill agreed. Campers get the benefit of others accepting and including them, but also practicing that same attitude toward fellow campers, she said. “It insures that we are providing this environment of inclusion, but that they are practicing it as well. They need to practice it like everyone else.”
Getting parents on board
The idea of camp, especially for those at the younger end of the 8- to 16-years age range accepted for Camp Discovery, can take some getting used to for some parents. Ms. Haygood, Emily’s mother, relates to that. Her daughter’s dermatologist at the time, who is now retired, had first suggested the camp. Her first reaction? “I am not sending my chronically ill child to camp with strangers.” She also acknowledged that she, like other parents of children with a chronic illness, can be a helicopter parent.
Then, she noticed that Emily seemed interested, so she got more information, finding out that it was staffed by doctors. It all sounded good, she said, and the social interaction, she knew, would be beneficial. “Then my husband was a no,” she said, concerned about their daughter being with strangers. “Eventually he came around,” Ms. Haygood said. All along, Emily said, “it seemed fun. I was probably trying to talk them into it.” She admits she was very nervous at first, but calmed down when she realized her own dermatologist was going to be there.
Vanessa Hadley of Spring, Tex., was on board the moment she heard about Camp Discovery. “I just thought it was amazing,” she said. Her daughter Isabelle, 13, has been to the camp. “She has alopecia areata and severe eczema,” Ms. Hadley said. Now, Isabelle is returning to camp and coaching her sister Penelope, 8, who has eczema and mild alopecia and is a first-timer this summer.
One tip the 8-year-old has learned so far: Turn to your counselor for support if you’re nervous. That worked, Isabelle said, the first year when she was wary of the zipline – then surprised herself and conquered it.
Dr. Hill and Dr. Julapalli have no disclosures.
The talent show, the grand finale of the 1-week camp, was nearly 7 years ago, but Emily Haygood of Houston, now 17 and about to start her senior year, remembers it in detail. She sang “Death of a Bachelor,” an R&B pop song and Billboard No. 1 hit at the time about a former bachelor who had happily married. These days, she said, if she watched the video of her 10-year-old singing self, “I would probably throw up.” But she still treasures the audience response, “having all those people I’d gotten close to cheer for me.”
Emily was at , but share one feature: they are the kind of dermatologic issues that can make doing everyday kid or teen activities like swimming difficult and can elicit mean comments from classmates and other would-be friends.
Emily was first diagnosed with atopic dermatitis at age 4, her mother, Amber Haygood, says. By age 9, it had become severe. Emily remembers being teased some in elementary school. “I did feel bad a lot of the time, when asked insensitive questions.” Her mother still bristles that adults often could be cruel, too.
But at Camp Discovery, those issues were nonexistent. “Camp was so cool,” Emily said. Besides the usual camp activities, it had things that “normal” camp didn’t, like other kids who didn’t stare at your skin condition or make fun of it.
30th anniversary season begins
This year is the 30th anniversary of Camp Discovery. Sessions began July 23 and continue through Aug. 18, with locations in Crosslake, Minn.; Hebron, Conn.; and Millville, Pa., in addition to Burton, Tex. About 300 campers will attend this year, according to the AAD, and 6,151 campers have attended from 1993 to 2022.
The 1-week camp accepts youth with conditions ranging from eczema and psoriasis to vitiligo, alopecia, epidermolysis bullosa, and ichthyosis, according to the academy. A dermatologist first refers a child, downloading and completing the referral form and sending it to the academy.
The 1-week session, including travel, is free for the campers, thanks to donors. As a nonprofit and membership-based organization, the AAD does not release the detailed financial information about the operating budget for the camp. Dermatologists, nurses, and counselors volunteer their time.
In his presidential address at the AAD’s annual meeting in March, outgoing president Mark D. Kaufmann, MD, of the department of dermatology at the Icahn School of Medicine at Mount Sinai in New York, referred to camp volunteering as an antidote to professional burnout. Remembering why as a dermatologist one entered the profession can be one solution, he said, and described his own recent 3-day volunteer stint at the camp.
“Those 3 magical days, being with kids as they discovered they weren’t alone in the world, sharing their experiences and ideas, reminded me why I became a physician in the first place,” he told the audience of meeting attendees. He vowed to expand the program, with a goal of having every dermatology resident attend Camp Discovery.
Mental health effects of skin conditions
Much research has focused on the mental health fallout from living with chronic skin conditions, and even young children can be adversely affected. In one review of the literature, researchers concluded that pediatric skin disease, including acne, atopic dermatitis, and psoriasis, can affect quality of life, carry stigma, and lead to bullying and eventually even suicidal behavior. Another study, published earlier this year, found that atopic dermatitis affected children’s quality of life, impacting sleep and leading to feelings of being ashamed.
“It’s not necessarily about what their skin condition is and more about the psychosocial impact,’’ said Samantha Hill, MD, a pediatric and general dermatologist in Lynchburg, Va., who is the medical director of Camp Discovery in Minnesota this year.
Camp activities, reactions
The overriding theme of camp is allowing all the youth to be “just one of the kids at camp,” Dr. Hill said in an interview. “They come to do all kinds of things they don’t do in normal life because people don’t give them the credit to [be able to] do it.”
Every year, she said, “I tell my staff we are in the business of making things happen, so if there is a kid bandaged head to toe [because of a skin condition] and they want to go tubing and get in the lake, we figure out how to make it happen. We have done that multiple times.”
Newcomers are initially nervous, Dr. Hill acknowledged, but in time let their guard down. Returnees are a different story. “When kids who have been at camp before arrive, you can see them start breathing again, looking for their friends. You can see them relax right before your eyes.”
“The single most empowering thing is the realization you are not alone,” said Meena Julapalli, MD, a Houston dermatologist who is a medical team member and long-time volunteer at Camp Discovery. That, she said, and “You get to be a kid, and you don’t have to have people staring at you.”
Dr. Julapalli remembers one of her patients with keratitis-ichthyosis-deafness (KID) syndrome. “She needed more than what I could offer,” she said. “She needed camp.” At camp, the organizers found a counselor who knew sign language to accompany her. At first, she was quiet and didn’t smile much. By the end of the week, as she was about to observe her birthday, things changed. After breakfast, she was led to the stage, where fellow campers began singing – and signing the song they had just learned.
Camp staff gets it
Allyson Garin, who was diagnosed with vitiligo at age 6 months, is a camp program director at Camp Discovery in Crosslake, Minn. She first went to camp in 1990 at age 11, returning until she “aged out” at 16, then worked as a counselor. She gets it when campers tell her they hear rude comments about their skin conditions.
“I remember being in swimming pools, in lines at fairgrounds or amusement parks,” she said in an interview, “and hearing people say, ‘Don’t touch her,’ ’’ fearing contagion, perhaps. “People would make jokes about cows, since they are spotted,” she said, or people would simply step back.
All those years ago, her mother found out about the camp and decided to figure out how to get her there. She got there, and she met a fellow camper with vitiligo, and they became pen pals. “We still talk,” she said.
Meeting someone with the same skin condition, she said, isn’t just about commiserating. “There is a lot of information sharing,” on topics such as best treatments, strategies, and other conversations.
Other lessons
While campers can feel comfortable around others who also have skin conditions, and understand, the lesson extends beyond that, Ms. Garin said. “It gave me a perspective,” she said of her camp experience. “I always felt, ‘Woe is me.’ ” But when she met others with, as she said, conditions “way worse than vitiligo, it really grounds you.”
Dr. Hill agreed. Campers get the benefit of others accepting and including them, but also practicing that same attitude toward fellow campers, she said. “It insures that we are providing this environment of inclusion, but that they are practicing it as well. They need to practice it like everyone else.”
Getting parents on board
The idea of camp, especially for those at the younger end of the 8- to 16-years age range accepted for Camp Discovery, can take some getting used to for some parents. Ms. Haygood, Emily’s mother, relates to that. Her daughter’s dermatologist at the time, who is now retired, had first suggested the camp. Her first reaction? “I am not sending my chronically ill child to camp with strangers.” She also acknowledged that she, like other parents of children with a chronic illness, can be a helicopter parent.
Then, she noticed that Emily seemed interested, so she got more information, finding out that it was staffed by doctors. It all sounded good, she said, and the social interaction, she knew, would be beneficial. “Then my husband was a no,” she said, concerned about their daughter being with strangers. “Eventually he came around,” Ms. Haygood said. All along, Emily said, “it seemed fun. I was probably trying to talk them into it.” She admits she was very nervous at first, but calmed down when she realized her own dermatologist was going to be there.
Vanessa Hadley of Spring, Tex., was on board the moment she heard about Camp Discovery. “I just thought it was amazing,” she said. Her daughter Isabelle, 13, has been to the camp. “She has alopecia areata and severe eczema,” Ms. Hadley said. Now, Isabelle is returning to camp and coaching her sister Penelope, 8, who has eczema and mild alopecia and is a first-timer this summer.
One tip the 8-year-old has learned so far: Turn to your counselor for support if you’re nervous. That worked, Isabelle said, the first year when she was wary of the zipline – then surprised herself and conquered it.
Dr. Hill and Dr. Julapalli have no disclosures.
Naltrexone is safe and beneficial in AUD with cirrhosis
VIENNA – , results of the first randomized controlled trial (RCT) show.
After 3 months, 64% of patients who received naltrexone were abstinent from alcohol, compared with 22% of patients who received placebo, Manasa Alla, MD, a hepatologist from the Institute of Liver and Biliary Sciences (ILBS), New Delhi, said at the European Association for the Study of the Liver (EASL) 2023, where she presented the study findings.
Importantly, naltrexone was found to be safe for patients with compensated cirrhosis. “This fragile population of patients has limited drugs to help them quit alcohol. Naltrexone can be a valuable addition to their measures to reduce craving and on their journey to reach de-addiction and abstinence,” Dr. Alla said.
Hepatotoxicity with naltrexone is rare and data are limited. The Food and Drug Administration previously placed a warning on its use for patients with alcoholic liver disease and underlying cirrhosis.
As a clinician constantly challenged with treating patients with AUD and cirrhosis, Dr. Alla wanted to explore the safety of naltrexone and to test its suitability for these patients who struggle to quit alcohol.
“Here we aimed to primarily test the safety of naltrexone in achieving abstinence and reducing alcohol cravings in patients with alcohol-related cirrhosis,” she said, adding, “The FDA black box warning has been removed, but it has never been tested in an RCT in patients with cirrhosis, so this is exactly what we did here.
“Naltrexone is a very good anti-alcohol-craving drug. If we can establish its safety in cirrhotic patients, it may have very good potential in reducing AUD and reducing the related complications of continued alcohol intake,” Dr. Alla said.
Safety, abstinence, lapse, and relapse assessed
The prospective, double-blind, single-center study at the ILBS in New Delhi, enrolled 100 patients with alcohol dependence and cirrhosis between 2020 and 2022. Participants were randomly assigned in a 1:1 ratio to receive naltrexone (50 mg/d) or placebo for 12 weeks. All participants attended regular counseling sessions with the resident psychiatrist. At baseline, the biochemical and drinking-related assessment scores between active and placebo groups of patients with compensated cirrhosis were matched.
Abstinence from alcohol was assessed through self-reported mean number of standard drinks (12 g alcohol per day). Findings were corroborated through an interview with a family member. Serum ethyl glucuronide levels were measured in cases of discrepancy. A relapse was considered to be consumption of over four standard alcoholic drinks/month; a lapse was considered any other alcohol drinking event not classified as relapse.
The primary outcome was the proportion of patients who achieved and maintained alcohol abstinence at 12 weeks; secondary outcomes were the proportion of patients who took naltrexone without a liver-related adverse effect compared with placebo at 12 weeks, the number of relapses and lapses, the difference in craving scores on the Obsessive Compulsive Drinking Scale (OCDS) between groups at 4, 8, and 12 weeks and at 6 months and 12 months, and the proportion of patients who achieved and maintained alcohol abstinence at 6 months.
Abstinence at 3 months
After 3 months, abstinence was noted in 64% of the study population who received naltrexone, compared to 22% of those who received placebo (P < .001). At 6 months, a higher proportion of patients in the naltrexone group achieved abstinence (22% vs. 8% with placebo; P = .09).
“We still need to look at the longer-term effects of naltrexone,” Dr. Alla said. “Here we gave the drug plus counseling for 3 months only, so despite encouraging findings, we need further studies to understand more.”
The researchers analyzed the predictors of abstinence at 3 months. They found that patients who consumed fewer than 17 drinks per month at baseline were more likely to achieve abstinence (sensitivity, 81%).
“Our study showed that patients who are consuming less alcohol at baseline can quit alcohol if adequately motivated. We need the motivation, as well as the drug,” she said.
Patient counseling was also very important and was provided for the 3 months of the study. “Even in the placebo arm, we had some patients who became abstinent [11/50 patients], but this dropped at 6 months [to 4/50],” Dr. Alla said.
At 12 weeks, 28% in the naltrexone group experienced relapse, vs. 72% in the placebo group (P < .001). Regarding the secondary outcome of craving scores and how they were affected by naltrexone, the mean OCDS-O (obsessive element) scores were 6.63, compared with 9.29 in naltrexone and placebo, respectively (P < .01). The mean OCDS-C (compulsive element) scores were 6.34 and 9.02, respectively (P < .01).
“Most important, was the safety of naltrexone in this study,” she said. There were no significant adverse events in either arm, and only one patient discontinued the drug in the naltrexone arm. Three patients in the naltrexone group who continued alcohol consumption developed jaundice, “so the jaundice can be attributed to continuous alcohol intake and may not be secondary to the naltrexone per se. We concluded that naltrexone is safe in a compensated cirrhotic patient,” Dr. Alla said.
Regarding other adverse events, 13.7% of patients experienced gastritis with naltrexone, vs. 3.7% among patients who received placebo. Nausea was more common in the placebo group, at 11.1% compared with 6.8% among patients who received naltrexone. Vomiting was more common in the naltrexone arm, at 10.3% vs. 7.4% with placebo. None of these differences reached statistical significance.
A longer-term study and comparisons to other drugs would provide valuable insights going forward.
Moderator Aleksander Krag, MD, professor and head of hepatology at the University of Southern Denmark and Odense University Hospital, Denmark, said: “Any intervention that can reduce or stop alcohol use in patients with cirrhosis and more advanced cirrhosis will improve outcome as well as reduce complications and mortality.
“In some cases, alcohol rehabilitation can completely revert the damaged liver. We have lots of data that show that continuous alcohol use at the more advanced stages can be devastating and reduction [in alcohol use] improves outcome. Therefore, any intervention that can help us to achieve this on behalf of all patients is most welcome,” he said.
Naltrexone (ADDTREX) and identical placebos were supplied by Rusan Pharma. Dr. Alla has disclosed no relevant financial relationships. Dr. Krag has served as speaker for Norgine, Siemens, and Nordic Bioscience and has participated in advisory boards for Norgine and Siemens outside the submitted work. He receives royalties from Gyldendal and Echosens.
A version of this article appeared on Medscape.com.
VIENNA – , results of the first randomized controlled trial (RCT) show.
After 3 months, 64% of patients who received naltrexone were abstinent from alcohol, compared with 22% of patients who received placebo, Manasa Alla, MD, a hepatologist from the Institute of Liver and Biliary Sciences (ILBS), New Delhi, said at the European Association for the Study of the Liver (EASL) 2023, where she presented the study findings.
Importantly, naltrexone was found to be safe for patients with compensated cirrhosis. “This fragile population of patients has limited drugs to help them quit alcohol. Naltrexone can be a valuable addition to their measures to reduce craving and on their journey to reach de-addiction and abstinence,” Dr. Alla said.
Hepatotoxicity with naltrexone is rare and data are limited. The Food and Drug Administration previously placed a warning on its use for patients with alcoholic liver disease and underlying cirrhosis.
As a clinician constantly challenged with treating patients with AUD and cirrhosis, Dr. Alla wanted to explore the safety of naltrexone and to test its suitability for these patients who struggle to quit alcohol.
“Here we aimed to primarily test the safety of naltrexone in achieving abstinence and reducing alcohol cravings in patients with alcohol-related cirrhosis,” she said, adding, “The FDA black box warning has been removed, but it has never been tested in an RCT in patients with cirrhosis, so this is exactly what we did here.
“Naltrexone is a very good anti-alcohol-craving drug. If we can establish its safety in cirrhotic patients, it may have very good potential in reducing AUD and reducing the related complications of continued alcohol intake,” Dr. Alla said.
Safety, abstinence, lapse, and relapse assessed
The prospective, double-blind, single-center study at the ILBS in New Delhi, enrolled 100 patients with alcohol dependence and cirrhosis between 2020 and 2022. Participants were randomly assigned in a 1:1 ratio to receive naltrexone (50 mg/d) or placebo for 12 weeks. All participants attended regular counseling sessions with the resident psychiatrist. At baseline, the biochemical and drinking-related assessment scores between active and placebo groups of patients with compensated cirrhosis were matched.
Abstinence from alcohol was assessed through self-reported mean number of standard drinks (12 g alcohol per day). Findings were corroborated through an interview with a family member. Serum ethyl glucuronide levels were measured in cases of discrepancy. A relapse was considered to be consumption of over four standard alcoholic drinks/month; a lapse was considered any other alcohol drinking event not classified as relapse.
The primary outcome was the proportion of patients who achieved and maintained alcohol abstinence at 12 weeks; secondary outcomes were the proportion of patients who took naltrexone without a liver-related adverse effect compared with placebo at 12 weeks, the number of relapses and lapses, the difference in craving scores on the Obsessive Compulsive Drinking Scale (OCDS) between groups at 4, 8, and 12 weeks and at 6 months and 12 months, and the proportion of patients who achieved and maintained alcohol abstinence at 6 months.
Abstinence at 3 months
After 3 months, abstinence was noted in 64% of the study population who received naltrexone, compared to 22% of those who received placebo (P < .001). At 6 months, a higher proportion of patients in the naltrexone group achieved abstinence (22% vs. 8% with placebo; P = .09).
“We still need to look at the longer-term effects of naltrexone,” Dr. Alla said. “Here we gave the drug plus counseling for 3 months only, so despite encouraging findings, we need further studies to understand more.”
The researchers analyzed the predictors of abstinence at 3 months. They found that patients who consumed fewer than 17 drinks per month at baseline were more likely to achieve abstinence (sensitivity, 81%).
“Our study showed that patients who are consuming less alcohol at baseline can quit alcohol if adequately motivated. We need the motivation, as well as the drug,” she said.
Patient counseling was also very important and was provided for the 3 months of the study. “Even in the placebo arm, we had some patients who became abstinent [11/50 patients], but this dropped at 6 months [to 4/50],” Dr. Alla said.
At 12 weeks, 28% in the naltrexone group experienced relapse, vs. 72% in the placebo group (P < .001). Regarding the secondary outcome of craving scores and how they were affected by naltrexone, the mean OCDS-O (obsessive element) scores were 6.63, compared with 9.29 in naltrexone and placebo, respectively (P < .01). The mean OCDS-C (compulsive element) scores were 6.34 and 9.02, respectively (P < .01).
“Most important, was the safety of naltrexone in this study,” she said. There were no significant adverse events in either arm, and only one patient discontinued the drug in the naltrexone arm. Three patients in the naltrexone group who continued alcohol consumption developed jaundice, “so the jaundice can be attributed to continuous alcohol intake and may not be secondary to the naltrexone per se. We concluded that naltrexone is safe in a compensated cirrhotic patient,” Dr. Alla said.
Regarding other adverse events, 13.7% of patients experienced gastritis with naltrexone, vs. 3.7% among patients who received placebo. Nausea was more common in the placebo group, at 11.1% compared with 6.8% among patients who received naltrexone. Vomiting was more common in the naltrexone arm, at 10.3% vs. 7.4% with placebo. None of these differences reached statistical significance.
A longer-term study and comparisons to other drugs would provide valuable insights going forward.
Moderator Aleksander Krag, MD, professor and head of hepatology at the University of Southern Denmark and Odense University Hospital, Denmark, said: “Any intervention that can reduce or stop alcohol use in patients with cirrhosis and more advanced cirrhosis will improve outcome as well as reduce complications and mortality.
“In some cases, alcohol rehabilitation can completely revert the damaged liver. We have lots of data that show that continuous alcohol use at the more advanced stages can be devastating and reduction [in alcohol use] improves outcome. Therefore, any intervention that can help us to achieve this on behalf of all patients is most welcome,” he said.
Naltrexone (ADDTREX) and identical placebos were supplied by Rusan Pharma. Dr. Alla has disclosed no relevant financial relationships. Dr. Krag has served as speaker for Norgine, Siemens, and Nordic Bioscience and has participated in advisory boards for Norgine and Siemens outside the submitted work. He receives royalties from Gyldendal and Echosens.
A version of this article appeared on Medscape.com.
VIENNA – , results of the first randomized controlled trial (RCT) show.
After 3 months, 64% of patients who received naltrexone were abstinent from alcohol, compared with 22% of patients who received placebo, Manasa Alla, MD, a hepatologist from the Institute of Liver and Biliary Sciences (ILBS), New Delhi, said at the European Association for the Study of the Liver (EASL) 2023, where she presented the study findings.
Importantly, naltrexone was found to be safe for patients with compensated cirrhosis. “This fragile population of patients has limited drugs to help them quit alcohol. Naltrexone can be a valuable addition to their measures to reduce craving and on their journey to reach de-addiction and abstinence,” Dr. Alla said.
Hepatotoxicity with naltrexone is rare and data are limited. The Food and Drug Administration previously placed a warning on its use for patients with alcoholic liver disease and underlying cirrhosis.
As a clinician constantly challenged with treating patients with AUD and cirrhosis, Dr. Alla wanted to explore the safety of naltrexone and to test its suitability for these patients who struggle to quit alcohol.
“Here we aimed to primarily test the safety of naltrexone in achieving abstinence and reducing alcohol cravings in patients with alcohol-related cirrhosis,” she said, adding, “The FDA black box warning has been removed, but it has never been tested in an RCT in patients with cirrhosis, so this is exactly what we did here.
“Naltrexone is a very good anti-alcohol-craving drug. If we can establish its safety in cirrhotic patients, it may have very good potential in reducing AUD and reducing the related complications of continued alcohol intake,” Dr. Alla said.
Safety, abstinence, lapse, and relapse assessed
The prospective, double-blind, single-center study at the ILBS in New Delhi, enrolled 100 patients with alcohol dependence and cirrhosis between 2020 and 2022. Participants were randomly assigned in a 1:1 ratio to receive naltrexone (50 mg/d) or placebo for 12 weeks. All participants attended regular counseling sessions with the resident psychiatrist. At baseline, the biochemical and drinking-related assessment scores between active and placebo groups of patients with compensated cirrhosis were matched.
Abstinence from alcohol was assessed through self-reported mean number of standard drinks (12 g alcohol per day). Findings were corroborated through an interview with a family member. Serum ethyl glucuronide levels were measured in cases of discrepancy. A relapse was considered to be consumption of over four standard alcoholic drinks/month; a lapse was considered any other alcohol drinking event not classified as relapse.
The primary outcome was the proportion of patients who achieved and maintained alcohol abstinence at 12 weeks; secondary outcomes were the proportion of patients who took naltrexone without a liver-related adverse effect compared with placebo at 12 weeks, the number of relapses and lapses, the difference in craving scores on the Obsessive Compulsive Drinking Scale (OCDS) between groups at 4, 8, and 12 weeks and at 6 months and 12 months, and the proportion of patients who achieved and maintained alcohol abstinence at 6 months.
Abstinence at 3 months
After 3 months, abstinence was noted in 64% of the study population who received naltrexone, compared to 22% of those who received placebo (P < .001). At 6 months, a higher proportion of patients in the naltrexone group achieved abstinence (22% vs. 8% with placebo; P = .09).
“We still need to look at the longer-term effects of naltrexone,” Dr. Alla said. “Here we gave the drug plus counseling for 3 months only, so despite encouraging findings, we need further studies to understand more.”
The researchers analyzed the predictors of abstinence at 3 months. They found that patients who consumed fewer than 17 drinks per month at baseline were more likely to achieve abstinence (sensitivity, 81%).
“Our study showed that patients who are consuming less alcohol at baseline can quit alcohol if adequately motivated. We need the motivation, as well as the drug,” she said.
Patient counseling was also very important and was provided for the 3 months of the study. “Even in the placebo arm, we had some patients who became abstinent [11/50 patients], but this dropped at 6 months [to 4/50],” Dr. Alla said.
At 12 weeks, 28% in the naltrexone group experienced relapse, vs. 72% in the placebo group (P < .001). Regarding the secondary outcome of craving scores and how they were affected by naltrexone, the mean OCDS-O (obsessive element) scores were 6.63, compared with 9.29 in naltrexone and placebo, respectively (P < .01). The mean OCDS-C (compulsive element) scores were 6.34 and 9.02, respectively (P < .01).
“Most important, was the safety of naltrexone in this study,” she said. There were no significant adverse events in either arm, and only one patient discontinued the drug in the naltrexone arm. Three patients in the naltrexone group who continued alcohol consumption developed jaundice, “so the jaundice can be attributed to continuous alcohol intake and may not be secondary to the naltrexone per se. We concluded that naltrexone is safe in a compensated cirrhotic patient,” Dr. Alla said.
Regarding other adverse events, 13.7% of patients experienced gastritis with naltrexone, vs. 3.7% among patients who received placebo. Nausea was more common in the placebo group, at 11.1% compared with 6.8% among patients who received naltrexone. Vomiting was more common in the naltrexone arm, at 10.3% vs. 7.4% with placebo. None of these differences reached statistical significance.
A longer-term study and comparisons to other drugs would provide valuable insights going forward.
Moderator Aleksander Krag, MD, professor and head of hepatology at the University of Southern Denmark and Odense University Hospital, Denmark, said: “Any intervention that can reduce or stop alcohol use in patients with cirrhosis and more advanced cirrhosis will improve outcome as well as reduce complications and mortality.
“In some cases, alcohol rehabilitation can completely revert the damaged liver. We have lots of data that show that continuous alcohol use at the more advanced stages can be devastating and reduction [in alcohol use] improves outcome. Therefore, any intervention that can help us to achieve this on behalf of all patients is most welcome,” he said.
Naltrexone (ADDTREX) and identical placebos were supplied by Rusan Pharma. Dr. Alla has disclosed no relevant financial relationships. Dr. Krag has served as speaker for Norgine, Siemens, and Nordic Bioscience and has participated in advisory boards for Norgine and Siemens outside the submitted work. He receives royalties from Gyldendal and Echosens.
A version of this article appeared on Medscape.com.
AT EASL CONGRESS 2023
Does timing of surgery affect rectal cancer outcomes?
TOPLINE:
METHODOLOGY:
- A total of 1,506 patients with locally advanced rectal cancer who underwent neoadjuvant therapy followed by total mesorectal excision were divided into three groups based on the time interval between therapy and surgery: short (8 weeks), intermediate (> 8 to 12 weeks), and long (> 12 weeks).
- The primary outcome was pathologic complete response, and secondary outcomes included other histopathologic results, perioperative events, and survival outcomes.
- Median follow-up was 33 months.
TAKEAWAY:
- Overall, a pathologic complete response was observed in 255 patients (17.2%).
- Compared with the intermediate interval (reference) group, investigators found no association between time interval and pathologic complete response in the short-interval (odds ratio, 0.74; 95% CI, 0.55-1.01) or long-interval groups (OR, 1.07; P = .70).
- A long interval was significantly associated with a lower risk of a bad response as measured by tumor regression grade 2-3, compared with the reference category (OR, 0.47), but a higher risk of minor postoperative complications (OR, 1.43), conversion to open surgery (OR, 3.14), and longer operative time.
- The long-interval group was associated with a significantly reduced risk of systemic recurrence, compared with the reference group (hazard ratio, 0.59; P = .04), but not improved overall survival (HR, 1.38; P = .11) or locoregional recurrence (HR, 0.53; P = .18); no significant findings occurred for the short versus intermediate group.
IN PRACTICE:
“Findings suggest that delaying surgery may improve tumor regression and decrease risk of distant metastasis but increase surgical complexity,” the authors conclude. “Nonetheless, the reported improvements in tumor regression and systemic recurrence in the long-interval group were unexpectedly not followed by improved [overall survival].”
SOURCE:
F. Borja de Lacy, MD, PhD, Hospital Clinic of Barcelona, University of Barcelona, led the study, published online in JAMA Surgery, with an accompanying editorial.
LIMITATIONS:
- The study’s main limitation was its retrospective design, which could have resulted in missing or inconsistent data, as well as the short follow-up time.
- Decisions about time interval were based more on professional preference rather than specific tumor characteristics.
DISCLOSURES:
Dr. de Lacy has reported no relevant financial relationships. No outside funding source was disclosed.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- A total of 1,506 patients with locally advanced rectal cancer who underwent neoadjuvant therapy followed by total mesorectal excision were divided into three groups based on the time interval between therapy and surgery: short (8 weeks), intermediate (> 8 to 12 weeks), and long (> 12 weeks).
- The primary outcome was pathologic complete response, and secondary outcomes included other histopathologic results, perioperative events, and survival outcomes.
- Median follow-up was 33 months.
TAKEAWAY:
- Overall, a pathologic complete response was observed in 255 patients (17.2%).
- Compared with the intermediate interval (reference) group, investigators found no association between time interval and pathologic complete response in the short-interval (odds ratio, 0.74; 95% CI, 0.55-1.01) or long-interval groups (OR, 1.07; P = .70).
- A long interval was significantly associated with a lower risk of a bad response as measured by tumor regression grade 2-3, compared with the reference category (OR, 0.47), but a higher risk of minor postoperative complications (OR, 1.43), conversion to open surgery (OR, 3.14), and longer operative time.
- The long-interval group was associated with a significantly reduced risk of systemic recurrence, compared with the reference group (hazard ratio, 0.59; P = .04), but not improved overall survival (HR, 1.38; P = .11) or locoregional recurrence (HR, 0.53; P = .18); no significant findings occurred for the short versus intermediate group.
IN PRACTICE:
“Findings suggest that delaying surgery may improve tumor regression and decrease risk of distant metastasis but increase surgical complexity,” the authors conclude. “Nonetheless, the reported improvements in tumor regression and systemic recurrence in the long-interval group were unexpectedly not followed by improved [overall survival].”
SOURCE:
F. Borja de Lacy, MD, PhD, Hospital Clinic of Barcelona, University of Barcelona, led the study, published online in JAMA Surgery, with an accompanying editorial.
LIMITATIONS:
- The study’s main limitation was its retrospective design, which could have resulted in missing or inconsistent data, as well as the short follow-up time.
- Decisions about time interval were based more on professional preference rather than specific tumor characteristics.
DISCLOSURES:
Dr. de Lacy has reported no relevant financial relationships. No outside funding source was disclosed.
A version of this article first appeared on Medscape.com.
TOPLINE:
METHODOLOGY:
- A total of 1,506 patients with locally advanced rectal cancer who underwent neoadjuvant therapy followed by total mesorectal excision were divided into three groups based on the time interval between therapy and surgery: short (8 weeks), intermediate (> 8 to 12 weeks), and long (> 12 weeks).
- The primary outcome was pathologic complete response, and secondary outcomes included other histopathologic results, perioperative events, and survival outcomes.
- Median follow-up was 33 months.
TAKEAWAY:
- Overall, a pathologic complete response was observed in 255 patients (17.2%).
- Compared with the intermediate interval (reference) group, investigators found no association between time interval and pathologic complete response in the short-interval (odds ratio, 0.74; 95% CI, 0.55-1.01) or long-interval groups (OR, 1.07; P = .70).
- A long interval was significantly associated with a lower risk of a bad response as measured by tumor regression grade 2-3, compared with the reference category (OR, 0.47), but a higher risk of minor postoperative complications (OR, 1.43), conversion to open surgery (OR, 3.14), and longer operative time.
- The long-interval group was associated with a significantly reduced risk of systemic recurrence, compared with the reference group (hazard ratio, 0.59; P = .04), but not improved overall survival (HR, 1.38; P = .11) or locoregional recurrence (HR, 0.53; P = .18); no significant findings occurred for the short versus intermediate group.
IN PRACTICE:
“Findings suggest that delaying surgery may improve tumor regression and decrease risk of distant metastasis but increase surgical complexity,” the authors conclude. “Nonetheless, the reported improvements in tumor regression and systemic recurrence in the long-interval group were unexpectedly not followed by improved [overall survival].”
SOURCE:
F. Borja de Lacy, MD, PhD, Hospital Clinic of Barcelona, University of Barcelona, led the study, published online in JAMA Surgery, with an accompanying editorial.
LIMITATIONS:
- The study’s main limitation was its retrospective design, which could have resulted in missing or inconsistent data, as well as the short follow-up time.
- Decisions about time interval were based more on professional preference rather than specific tumor characteristics.
DISCLOSURES:
Dr. de Lacy has reported no relevant financial relationships. No outside funding source was disclosed.
A version of this article first appeared on Medscape.com.
Postmenopausal screening mammogram
The findings in this case are suggestive of invasive lobular carcinoma (ILC).
Globally, breast cancer remains the most common life-threatening cancer diagnosed and the second leading cause of cancer-related deaths in women. In the United States, approximately 287,850 new cases of invasive breast cancer were diagnosed in 2022 and 43,250 deaths were attributed to breast cancer in the same year. Worldwide, approximately 2.3 million new diagnoses and 685,000 breast cancer-related deaths were reported in 2020.
ILC is one of the leading histologic types of invasive carcinoma, second in incidence only to invasive carcinoma of no special type. ILC accounts for 5%-15% of all invasive breast cancers, and its incidence has been steadily increasing — particularly among postmenopausal women — over the past two decades. ILC has distinct molecular and histopathologic features, including the loss of cell-cell adhesion molecule E-cadherin, resulting in small, discohesive cells proliferating in single-file strands; positivity for both the estrogen and progesterone receptor; and human epidermal growth factor receptor 2 negativity.
The diagnosis of ILC can be challenging, as it is difficult to detect both on physical examination and with standard imaging techniques. Patients are often diagnosed with late-stage disease, characterized by large tumors and lymph node involvement. The signs of ILC are often vague, such as skin thickening or dimpling. In addition, measuring the extent of ILC can be challenging, as traditional screening methods (eg, mammography and ultrasonography) have a low sensitivity for detecting ILC compared with other invasive breast tumors. This difficulty is usually ascribed to the diffuse infiltrative growth pattern of ILC. MRI has a greater sensitivity for detecting ILC.
Risk factors for the development of ILC have been identified and include:
• Alcohol consumption
• Use of combined hormone replacement therapy
• Early menarche (menarche before the age of 12 years)
• Late-onset menopause (menopause after the age of 55 years)
• Nulliparity/low parity (defined by World Health Organization as fewer than five pregnancies with gestation periods of ≥ 20 weeks)
• Late age at birth (> 30 years)
• Family history (eg, hereditary diffuse gastric cancer syndrome)
• Genetics (eg, CDH1 mutations)
Treatment protocols for ILC align with those used in other breast cancer subtypes and typically involve a multidisciplinary approach comprising surgery, radiotherapy, and systemic therapies. Cancers that are deemed resectable will typically be managed surgically upfront, although some patients may require neoadjuvant therapy to reduce tumor burden and facilitate surgical intervention. Breast-conserving surgery using a wide local excision can frequently be performed; however, in up to 65% of cases, a second surgery will be required (re-excision or mastectomy). Axillary lymph node status is a crucial factor in the prognosis of all breast cancers and affects surgical planning. Sentinel node biopsy is the standard method of assessing the axilla.
Systemic therapy is an integral part of the multidisciplinary approach to treating breast cancer and usually involves the use of chemotherapy. However, because of the unique molecular biology of ILC, treatment response to chemotherapy is often poor, resulting in lower rates of complete pathologic response and higher rates of mastectomy. Conversely, ILC has been shown to respond well to endocrine therapy, making it the optimal treatment choice. Novel therapeutic approaches are under investigation.
Detailed guidance on the treatment of ILC is available from the National Comprehensive Cancer Network.
Avan J. Armaghani, MD, Assistant Member, Department of Breast Oncology, Moffitt Cancer Center, University of South Florida, Tampa, FL.
Avan J. Armaghani, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The findings in this case are suggestive of invasive lobular carcinoma (ILC).
Globally, breast cancer remains the most common life-threatening cancer diagnosed and the second leading cause of cancer-related deaths in women. In the United States, approximately 287,850 new cases of invasive breast cancer were diagnosed in 2022 and 43,250 deaths were attributed to breast cancer in the same year. Worldwide, approximately 2.3 million new diagnoses and 685,000 breast cancer-related deaths were reported in 2020.
ILC is one of the leading histologic types of invasive carcinoma, second in incidence only to invasive carcinoma of no special type. ILC accounts for 5%-15% of all invasive breast cancers, and its incidence has been steadily increasing — particularly among postmenopausal women — over the past two decades. ILC has distinct molecular and histopathologic features, including the loss of cell-cell adhesion molecule E-cadherin, resulting in small, discohesive cells proliferating in single-file strands; positivity for both the estrogen and progesterone receptor; and human epidermal growth factor receptor 2 negativity.
The diagnosis of ILC can be challenging, as it is difficult to detect both on physical examination and with standard imaging techniques. Patients are often diagnosed with late-stage disease, characterized by large tumors and lymph node involvement. The signs of ILC are often vague, such as skin thickening or dimpling. In addition, measuring the extent of ILC can be challenging, as traditional screening methods (eg, mammography and ultrasonography) have a low sensitivity for detecting ILC compared with other invasive breast tumors. This difficulty is usually ascribed to the diffuse infiltrative growth pattern of ILC. MRI has a greater sensitivity for detecting ILC.
Risk factors for the development of ILC have been identified and include:
• Alcohol consumption
• Use of combined hormone replacement therapy
• Early menarche (menarche before the age of 12 years)
• Late-onset menopause (menopause after the age of 55 years)
• Nulliparity/low parity (defined by World Health Organization as fewer than five pregnancies with gestation periods of ≥ 20 weeks)
• Late age at birth (> 30 years)
• Family history (eg, hereditary diffuse gastric cancer syndrome)
• Genetics (eg, CDH1 mutations)
Treatment protocols for ILC align with those used in other breast cancer subtypes and typically involve a multidisciplinary approach comprising surgery, radiotherapy, and systemic therapies. Cancers that are deemed resectable will typically be managed surgically upfront, although some patients may require neoadjuvant therapy to reduce tumor burden and facilitate surgical intervention. Breast-conserving surgery using a wide local excision can frequently be performed; however, in up to 65% of cases, a second surgery will be required (re-excision or mastectomy). Axillary lymph node status is a crucial factor in the prognosis of all breast cancers and affects surgical planning. Sentinel node biopsy is the standard method of assessing the axilla.
Systemic therapy is an integral part of the multidisciplinary approach to treating breast cancer and usually involves the use of chemotherapy. However, because of the unique molecular biology of ILC, treatment response to chemotherapy is often poor, resulting in lower rates of complete pathologic response and higher rates of mastectomy. Conversely, ILC has been shown to respond well to endocrine therapy, making it the optimal treatment choice. Novel therapeutic approaches are under investigation.
Detailed guidance on the treatment of ILC is available from the National Comprehensive Cancer Network.
Avan J. Armaghani, MD, Assistant Member, Department of Breast Oncology, Moffitt Cancer Center, University of South Florida, Tampa, FL.
Avan J. Armaghani, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
The findings in this case are suggestive of invasive lobular carcinoma (ILC).
Globally, breast cancer remains the most common life-threatening cancer diagnosed and the second leading cause of cancer-related deaths in women. In the United States, approximately 287,850 new cases of invasive breast cancer were diagnosed in 2022 and 43,250 deaths were attributed to breast cancer in the same year. Worldwide, approximately 2.3 million new diagnoses and 685,000 breast cancer-related deaths were reported in 2020.
ILC is one of the leading histologic types of invasive carcinoma, second in incidence only to invasive carcinoma of no special type. ILC accounts for 5%-15% of all invasive breast cancers, and its incidence has been steadily increasing — particularly among postmenopausal women — over the past two decades. ILC has distinct molecular and histopathologic features, including the loss of cell-cell adhesion molecule E-cadherin, resulting in small, discohesive cells proliferating in single-file strands; positivity for both the estrogen and progesterone receptor; and human epidermal growth factor receptor 2 negativity.
The diagnosis of ILC can be challenging, as it is difficult to detect both on physical examination and with standard imaging techniques. Patients are often diagnosed with late-stage disease, characterized by large tumors and lymph node involvement. The signs of ILC are often vague, such as skin thickening or dimpling. In addition, measuring the extent of ILC can be challenging, as traditional screening methods (eg, mammography and ultrasonography) have a low sensitivity for detecting ILC compared with other invasive breast tumors. This difficulty is usually ascribed to the diffuse infiltrative growth pattern of ILC. MRI has a greater sensitivity for detecting ILC.
Risk factors for the development of ILC have been identified and include:
• Alcohol consumption
• Use of combined hormone replacement therapy
• Early menarche (menarche before the age of 12 years)
• Late-onset menopause (menopause after the age of 55 years)
• Nulliparity/low parity (defined by World Health Organization as fewer than five pregnancies with gestation periods of ≥ 20 weeks)
• Late age at birth (> 30 years)
• Family history (eg, hereditary diffuse gastric cancer syndrome)
• Genetics (eg, CDH1 mutations)
Treatment protocols for ILC align with those used in other breast cancer subtypes and typically involve a multidisciplinary approach comprising surgery, radiotherapy, and systemic therapies. Cancers that are deemed resectable will typically be managed surgically upfront, although some patients may require neoadjuvant therapy to reduce tumor burden and facilitate surgical intervention. Breast-conserving surgery using a wide local excision can frequently be performed; however, in up to 65% of cases, a second surgery will be required (re-excision or mastectomy). Axillary lymph node status is a crucial factor in the prognosis of all breast cancers and affects surgical planning. Sentinel node biopsy is the standard method of assessing the axilla.
Systemic therapy is an integral part of the multidisciplinary approach to treating breast cancer and usually involves the use of chemotherapy. However, because of the unique molecular biology of ILC, treatment response to chemotherapy is often poor, resulting in lower rates of complete pathologic response and higher rates of mastectomy. Conversely, ILC has been shown to respond well to endocrine therapy, making it the optimal treatment choice. Novel therapeutic approaches are under investigation.
Detailed guidance on the treatment of ILC is available from the National Comprehensive Cancer Network.
Avan J. Armaghani, MD, Assistant Member, Department of Breast Oncology, Moffitt Cancer Center, University of South Florida, Tampa, FL.
Avan J. Armaghani, MD, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
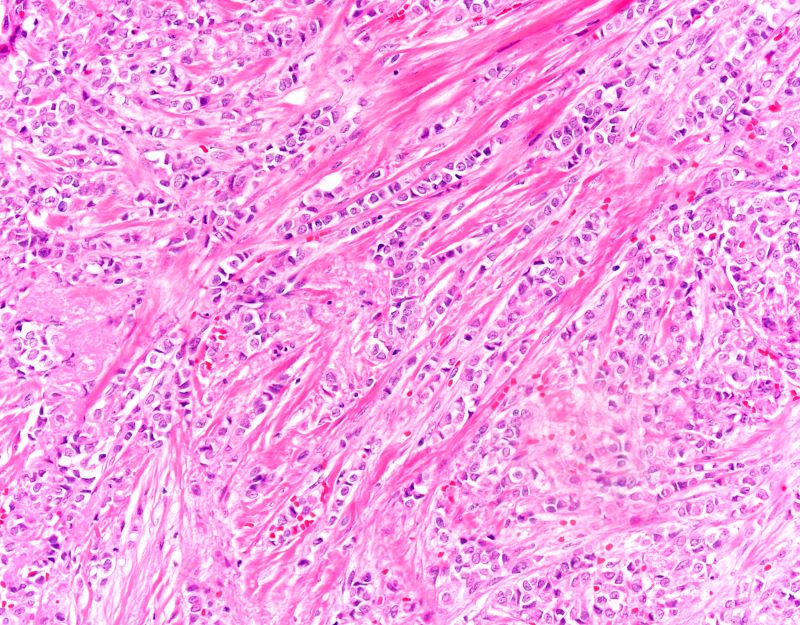
A 58-year-old postmenopausal woman presents for screening mammography. The patient's last mammogram was 18 months ago and showed dense breast tissue with no abnormalities. The patient states that she has no breast symptoms. She is 5 ft 3 in and weighs 196 lb (BMI 34.7). Her previous medical history is unremarkable. There is a family history of breast cancer (two maternal cousins) and colon cancer (paternal grandmother). Bilateral mammography reveals an irregular mass that is approximately 2.4 cm and calcifications in the upper outer quadrant of the right breast. Physical examination reveals no palpable abnormalities. The patient undergoes a stereotactic breast biopsy. Pathology findings include malignant monomorphic cells that form loosely dispersed linear columns encircling the mammary ducts and infiltrating breast tissue and fat.
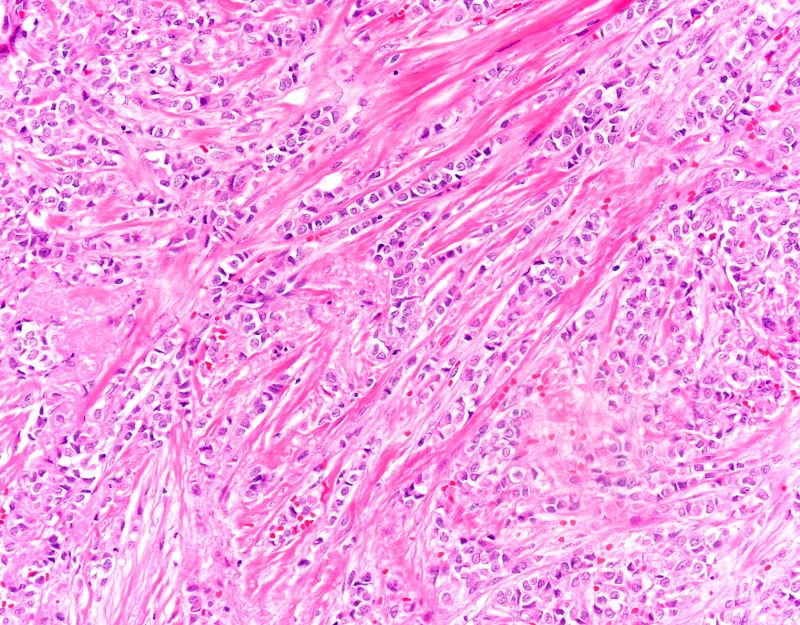
WHO declares aspartame as possible carcinogen
Although this means aspartame might cause cancer in humans, the Group 2B classification from the IARC means the evidence is “limited.” A summary of the working group’s evaluation, also published in The Lancet Oncology, explained that the classification was based on data from three studies assessing the link between aspartame intake and primary liver cancer.
Using that evidence, the World Health Organization and Food and Agriculture Organization Joint Expert Committee on Food Additives confirmed its existing stance that aspartame consumption of up to 40 mg/kg of body weight per day – the amount found in 9-14 diet soft drinks – is safe.
The decision, which was anticipated by Reuters in late June, drew praise from an array of experts who weighed in on the study results via the U.K.-based Science Media Centre. Many emphasized the lack of data showing a causal relationship between the low-calorie artificial sweetener and sought to temper any alarmism related to the decision.
“In short the evidence that aspartame causes primary liver cancer, or any other cancer in humans, is very weak,” said Paul Pharoah, MD, PhD, a professor of cancer epidemiology at Cedars-Sinai Medical Center, Los Angeles. “Group 2B is a very conservative classification in that almost any evidence of carcinogenicity, however flawed, will put a chemical in that category or above.”
Other examples of substances classified as Group 2B are extract of aloe vera, diesel oil, and caffeic acid found in coffee and tea, Dr. Pharoah explained, adding that “this is reflected in the view of the [JECFA] who concluded that there was no convincing evidence from experimental animal or human data that aspartame has adverse effects after ingestion.”
“The general public should not be worried about the risk of cancer associated with a chemical classed as Group 2B by IARC,” he stressed.
Alan Boobis, OBE, PhD, similarly noted that the Group 2B classification “reflects a lack of confidence that the data from experimental animals or from humans is sufficiently convincing to reach a clear conclusion that aspartame is carcinogenic.”
“Hence, exposure at current levels would not be anticipated to have any detrimental effects,” added Dr. Boobis, emeritus professor of toxicology, Imperial College London.
The IARC/JECFA opinion is “very welcome” and “ends the speculation about the safety of aspartame,” added Gunter Kuhnle, Dr. rer. nat., a professor of nutrition and food science at the University of Reading (England).
“It is unfortunate that leaking some information might have created unnecessary uncertainty and concern as consumers might be rightfully worried if they are told that something that is in many foods could cause cancer,” Dr. Kuhnle said. “The published opinion puts this into perspective and makes it very clear that there is no cause for concern when consumed at the current amounts.”
The data reviewed by the IARC Working Group included three studies, comprising four prospective cohorts, that “assessed the association of artificially sweetened beverage consumption with liver cancer risk,” the group reported.
The cohort studies – including one conducted within 10 European countries, one that pooled data from two large U.S. cohorts, and a prospective study also conducted in the United States – each “showed positive associations between artificially sweetened beverage consumption and cancer incidence or cancer mortality” in the overall study population or in relevant subgroups.
Although the studies were of “high quality and controlled for many potential confounders,” the Working Group concluded that “chance, bias, or confounding could not be ruled out with reasonable confidence.” Thus, the evidence for cancer in humans was deemed “limited” for hepatocellular carcinoma and “inadequate” for other cancer types.”
Dr. Pharoah and Dr. Kuhnle disclosed no relevant financial relationships. Dr. Boobis is a member of a number of advisory committees in the public and private sectors, including the International Life Science Institute and The Center for Research on Ingredient Safety at Michigan State University.
A version of this article first appeared on Medscape.com.
Although this means aspartame might cause cancer in humans, the Group 2B classification from the IARC means the evidence is “limited.” A summary of the working group’s evaluation, also published in The Lancet Oncology, explained that the classification was based on data from three studies assessing the link between aspartame intake and primary liver cancer.
Using that evidence, the World Health Organization and Food and Agriculture Organization Joint Expert Committee on Food Additives confirmed its existing stance that aspartame consumption of up to 40 mg/kg of body weight per day – the amount found in 9-14 diet soft drinks – is safe.
The decision, which was anticipated by Reuters in late June, drew praise from an array of experts who weighed in on the study results via the U.K.-based Science Media Centre. Many emphasized the lack of data showing a causal relationship between the low-calorie artificial sweetener and sought to temper any alarmism related to the decision.
“In short the evidence that aspartame causes primary liver cancer, or any other cancer in humans, is very weak,” said Paul Pharoah, MD, PhD, a professor of cancer epidemiology at Cedars-Sinai Medical Center, Los Angeles. “Group 2B is a very conservative classification in that almost any evidence of carcinogenicity, however flawed, will put a chemical in that category or above.”
Other examples of substances classified as Group 2B are extract of aloe vera, diesel oil, and caffeic acid found in coffee and tea, Dr. Pharoah explained, adding that “this is reflected in the view of the [JECFA] who concluded that there was no convincing evidence from experimental animal or human data that aspartame has adverse effects after ingestion.”
“The general public should not be worried about the risk of cancer associated with a chemical classed as Group 2B by IARC,” he stressed.
Alan Boobis, OBE, PhD, similarly noted that the Group 2B classification “reflects a lack of confidence that the data from experimental animals or from humans is sufficiently convincing to reach a clear conclusion that aspartame is carcinogenic.”
“Hence, exposure at current levels would not be anticipated to have any detrimental effects,” added Dr. Boobis, emeritus professor of toxicology, Imperial College London.
The IARC/JECFA opinion is “very welcome” and “ends the speculation about the safety of aspartame,” added Gunter Kuhnle, Dr. rer. nat., a professor of nutrition and food science at the University of Reading (England).
“It is unfortunate that leaking some information might have created unnecessary uncertainty and concern as consumers might be rightfully worried if they are told that something that is in many foods could cause cancer,” Dr. Kuhnle said. “The published opinion puts this into perspective and makes it very clear that there is no cause for concern when consumed at the current amounts.”
The data reviewed by the IARC Working Group included three studies, comprising four prospective cohorts, that “assessed the association of artificially sweetened beverage consumption with liver cancer risk,” the group reported.
The cohort studies – including one conducted within 10 European countries, one that pooled data from two large U.S. cohorts, and a prospective study also conducted in the United States – each “showed positive associations between artificially sweetened beverage consumption and cancer incidence or cancer mortality” in the overall study population or in relevant subgroups.
Although the studies were of “high quality and controlled for many potential confounders,” the Working Group concluded that “chance, bias, or confounding could not be ruled out with reasonable confidence.” Thus, the evidence for cancer in humans was deemed “limited” for hepatocellular carcinoma and “inadequate” for other cancer types.”
Dr. Pharoah and Dr. Kuhnle disclosed no relevant financial relationships. Dr. Boobis is a member of a number of advisory committees in the public and private sectors, including the International Life Science Institute and The Center for Research on Ingredient Safety at Michigan State University.
A version of this article first appeared on Medscape.com.
Although this means aspartame might cause cancer in humans, the Group 2B classification from the IARC means the evidence is “limited.” A summary of the working group’s evaluation, also published in The Lancet Oncology, explained that the classification was based on data from three studies assessing the link between aspartame intake and primary liver cancer.
Using that evidence, the World Health Organization and Food and Agriculture Organization Joint Expert Committee on Food Additives confirmed its existing stance that aspartame consumption of up to 40 mg/kg of body weight per day – the amount found in 9-14 diet soft drinks – is safe.
The decision, which was anticipated by Reuters in late June, drew praise from an array of experts who weighed in on the study results via the U.K.-based Science Media Centre. Many emphasized the lack of data showing a causal relationship between the low-calorie artificial sweetener and sought to temper any alarmism related to the decision.
“In short the evidence that aspartame causes primary liver cancer, or any other cancer in humans, is very weak,” said Paul Pharoah, MD, PhD, a professor of cancer epidemiology at Cedars-Sinai Medical Center, Los Angeles. “Group 2B is a very conservative classification in that almost any evidence of carcinogenicity, however flawed, will put a chemical in that category or above.”
Other examples of substances classified as Group 2B are extract of aloe vera, diesel oil, and caffeic acid found in coffee and tea, Dr. Pharoah explained, adding that “this is reflected in the view of the [JECFA] who concluded that there was no convincing evidence from experimental animal or human data that aspartame has adverse effects after ingestion.”
“The general public should not be worried about the risk of cancer associated with a chemical classed as Group 2B by IARC,” he stressed.
Alan Boobis, OBE, PhD, similarly noted that the Group 2B classification “reflects a lack of confidence that the data from experimental animals or from humans is sufficiently convincing to reach a clear conclusion that aspartame is carcinogenic.”
“Hence, exposure at current levels would not be anticipated to have any detrimental effects,” added Dr. Boobis, emeritus professor of toxicology, Imperial College London.
The IARC/JECFA opinion is “very welcome” and “ends the speculation about the safety of aspartame,” added Gunter Kuhnle, Dr. rer. nat., a professor of nutrition and food science at the University of Reading (England).
“It is unfortunate that leaking some information might have created unnecessary uncertainty and concern as consumers might be rightfully worried if they are told that something that is in many foods could cause cancer,” Dr. Kuhnle said. “The published opinion puts this into perspective and makes it very clear that there is no cause for concern when consumed at the current amounts.”
The data reviewed by the IARC Working Group included three studies, comprising four prospective cohorts, that “assessed the association of artificially sweetened beverage consumption with liver cancer risk,” the group reported.
The cohort studies – including one conducted within 10 European countries, one that pooled data from two large U.S. cohorts, and a prospective study also conducted in the United States – each “showed positive associations between artificially sweetened beverage consumption and cancer incidence or cancer mortality” in the overall study population or in relevant subgroups.
Although the studies were of “high quality and controlled for many potential confounders,” the Working Group concluded that “chance, bias, or confounding could not be ruled out with reasonable confidence.” Thus, the evidence for cancer in humans was deemed “limited” for hepatocellular carcinoma and “inadequate” for other cancer types.”
Dr. Pharoah and Dr. Kuhnle disclosed no relevant financial relationships. Dr. Boobis is a member of a number of advisory committees in the public and private sectors, including the International Life Science Institute and The Center for Research on Ingredient Safety at Michigan State University.
A version of this article first appeared on Medscape.com.
FROM THE LANCET ONCOLOGY
Setmelanotide offers significant, long-lasting weight loss
MARSEILLE, FRANCE – , according to results presented at the latest French Pediatric Society conference. The treatment is effective for adults and children alike.
Setmelanotide is the culmination of two decades of research involving the identification of genes involved in early-onset obesity and the characterization of the melanocortin 4 receptor. It became available via early access in 2021.
Currently limited to use in treating obesity linked to a biallelic POMC/PCSK1 or LEPR deficiency, setmelanotide is being tested with respect to other mutations responsible for severe obesity, raising hopes that it will soon be indicated for use in a larger number of patients.
Fewer than 1% of patients who suffer from severe obesity have monogenic forms of obesity. In recent years, the hope for a targeted treatment for patients with these monogenic forms has become a reality.
Although only a small number of patients currently meet the criteria for setmelanotide treatment (namely, those with obesity linked to a biallelic POMC/PCSK1 enzyme or LEPR deficiency and patients with Bardet-Biedl syndrome), there is a real hope that patients with other forms of severe obesity will be able to benefit from this product, including those with heterozygous (not just homozygous) monoallelic variants, who account for more than 10% of patients with severe early-onset obesity.
Restoring satiety signaling
“Setmelanotide is a melanocortin 4 receptor agonist,” said Béatrice Dubern, MD, PhD, pediatrician at Trousseau Hospital, Paris, and member of the French medical research institute (INSERM)/Sorbonne University team on nutrition and obesity. “Its mode of action rests on activation of the leptin-melanocortin signaling pathway in the hypothalamus, which regulates hunger, satiety, energy expenditure, and, therefore, body weight. Rare genetic variants in the leptin-melanocortin pathway are associated with polyphagia and severe early-onset obesity. It is believed that more than 60 genes involved in this leptin-melanocortin pathway are currently associated with obesity.”
In July 2021, the European Medicines Agency approved setmelanotide for daily use via subcutaneous administration.
Weight loss maintained
In phase 3 studies, setmelanotide (melanocortin 4 receptor agonist) demonstrated its effectiveness in reducing weight and hunger for patients with obesity caused by a POMC/PCSK1 or LEPR deficiency.
Twenty-four patients aged 6 years and older showed a significant response to setmelanotide after 1 year of treatment and were included in the extension study. “Significant response” was defined as a reduction in body weight greater than or equal to 10% after 52 weeks for patients aged 18 years and older or a reduction in body mass index (BMI) z-score greater than or equal to 0.3 after 52 weeks for patients younger than 18 years.
Among all patients, the mean variation (standard deviation) in BMI was −24.8% (8.2%, n = 24), −21% (13%, n = 23), and −24% (17.9%, n = 15) at 12, 24, and 36 months, respectively.
For patients aged greater than or equal to 18 years (n = 11), the mean variation (standard deviation) in weight was −25.1% (7.7%, n = 11), −22.9% (12.5%, n = 11), and −24.4% (13.2%, n = 8) at 12, 24, and 36 months, respectively.
For children and adolescents (patients aged < 18 years, n = 13), the mean reduction (standard deviation) in BMI z-score was −1.31 ([0.66], n = 13), −1.10 ([0.79], n = 11), and −1.01 (1.22], n = 4) at 12, 24, and 36 months, respectively.
For patients younger than 18 years, the mean variation in BMI z-score was −1.01 SD after three years on setmelanotide (standard deviation, 1.22 [n = 4]). The mean BMI z-score was +2.42 SD (standard deviation, 1.22 [n = 4]) after 3 years of treatment with setmelanotide.
In sum, the patients who achieved a reduction in body weight of at least 10% or greater than or equal to 0.3 mean variation in BMI z-score after 1 year experienced long-lasting, clinically significant benefit after 3 years. The finding supports the long-term use of setmelanotide for this group.
“We feared that setmelanotide’s effectiveness would decrease over time, but after 3 years, this had not happened, and we are hopeful that this sustained efficacy will be long lasting. The first two patients who took the drug in 2016 have not noticed any loss of efficacy as it stands,” said Dr. Dubern.
This is all the more encouraging. In the study presented at the 2023 pediatric conference, setmelanotide’s safety profile was reassuring, and it was consistent with previous studies. Side effects reported in greater than or equal to 15% of patients include injection site reactions, skin hyperpigmentation, nausea, diarrhea, mood disturbances, abdominal pain, vomiting, gastroenteritis, and spontaneous erection.
In addition to the lack of control group, Dr. Dubern acknowledged one other constraint of this study. “Only the patients who responded to treatment with setmelanotide during early-phase trials (85.7%) were enrolled.”
Dr. Dubern summarized the clinical implications: “In patients with early-onset obesity, starting before 5 years of age, doctors should really be considering the possibility that genetics might be involved in such cases. For confirmation and to seek expert opinion, specialist obesity clinics can be found throughout France. Additionally, the INSERM NutriOmics research team headed by Prof Karine Clément, MD, PhD, Sorbonne University, in conjunction with Prof Christine Poitou, MD, PhD, has developed a diagnostic tool [called] ObsGen for practitioners faced with patients with potentially genetic causes of obesity. We can answer any questions they have about the likelihood of a particular patient having a genetic form of obesity and guide their next steps. Treating patients with genetic obesity early on helps limit the condition worsening during adolescence, prevents related complications, and can reduce the stigmatization and suffering experienced by these people. It’s a huge issue. A clinical trial with setmelanotide is currently being carried out in children over 2 years of age.”
Hypothalamic obesity
During the pediatric conference, another speaker presented the results of a phase 2 study that evaluated the efficacy and tolerability of setmelanotide as a new treatment for hypothalamic obesity. Lesions in the hypothalamus can alter melanocortin 4 receptor pathway signaling and thus lead to hypothalamic obesity. Eighteen patients aged 6-40 years with a BMI greater than or equal to the 95th percentile (for patients aged 6-18 years) or greater than or equal to 35 kg/m2 (for adults aged ≥ 18 years) and hypothalamic obesity (craniopharyngioma or other benign brain tumors, surgical removal, and/or chemotherapy) were enrolled. A significant proportion of patients achieved a reduction of greater than or equal to 5% of their BMI (n = 16, 88.9%, CI 90%, 69%-89%, P < .0001), and 72.2% achieved a reduction of greater than or equal to 10% by week 16. The mean change in BMI was −14.9% (9.6%, n = 17). These early results may justify further studies of setmelanotide in treating hypothalamic obesity.
Dr. Dubern has collaborated with Rhythm Pharmaceuticals and Novo Nordisk.
This article was translated from the Medscape French Edition and a version appeared on Medscape.com.
MARSEILLE, FRANCE – , according to results presented at the latest French Pediatric Society conference. The treatment is effective for adults and children alike.
Setmelanotide is the culmination of two decades of research involving the identification of genes involved in early-onset obesity and the characterization of the melanocortin 4 receptor. It became available via early access in 2021.
Currently limited to use in treating obesity linked to a biallelic POMC/PCSK1 or LEPR deficiency, setmelanotide is being tested with respect to other mutations responsible for severe obesity, raising hopes that it will soon be indicated for use in a larger number of patients.
Fewer than 1% of patients who suffer from severe obesity have monogenic forms of obesity. In recent years, the hope for a targeted treatment for patients with these monogenic forms has become a reality.
Although only a small number of patients currently meet the criteria for setmelanotide treatment (namely, those with obesity linked to a biallelic POMC/PCSK1 enzyme or LEPR deficiency and patients with Bardet-Biedl syndrome), there is a real hope that patients with other forms of severe obesity will be able to benefit from this product, including those with heterozygous (not just homozygous) monoallelic variants, who account for more than 10% of patients with severe early-onset obesity.
Restoring satiety signaling
“Setmelanotide is a melanocortin 4 receptor agonist,” said Béatrice Dubern, MD, PhD, pediatrician at Trousseau Hospital, Paris, and member of the French medical research institute (INSERM)/Sorbonne University team on nutrition and obesity. “Its mode of action rests on activation of the leptin-melanocortin signaling pathway in the hypothalamus, which regulates hunger, satiety, energy expenditure, and, therefore, body weight. Rare genetic variants in the leptin-melanocortin pathway are associated with polyphagia and severe early-onset obesity. It is believed that more than 60 genes involved in this leptin-melanocortin pathway are currently associated with obesity.”
In July 2021, the European Medicines Agency approved setmelanotide for daily use via subcutaneous administration.
Weight loss maintained
In phase 3 studies, setmelanotide (melanocortin 4 receptor agonist) demonstrated its effectiveness in reducing weight and hunger for patients with obesity caused by a POMC/PCSK1 or LEPR deficiency.
Twenty-four patients aged 6 years and older showed a significant response to setmelanotide after 1 year of treatment and were included in the extension study. “Significant response” was defined as a reduction in body weight greater than or equal to 10% after 52 weeks for patients aged 18 years and older or a reduction in body mass index (BMI) z-score greater than or equal to 0.3 after 52 weeks for patients younger than 18 years.
Among all patients, the mean variation (standard deviation) in BMI was −24.8% (8.2%, n = 24), −21% (13%, n = 23), and −24% (17.9%, n = 15) at 12, 24, and 36 months, respectively.
For patients aged greater than or equal to 18 years (n = 11), the mean variation (standard deviation) in weight was −25.1% (7.7%, n = 11), −22.9% (12.5%, n = 11), and −24.4% (13.2%, n = 8) at 12, 24, and 36 months, respectively.
For children and adolescents (patients aged < 18 years, n = 13), the mean reduction (standard deviation) in BMI z-score was −1.31 ([0.66], n = 13), −1.10 ([0.79], n = 11), and −1.01 (1.22], n = 4) at 12, 24, and 36 months, respectively.
For patients younger than 18 years, the mean variation in BMI z-score was −1.01 SD after three years on setmelanotide (standard deviation, 1.22 [n = 4]). The mean BMI z-score was +2.42 SD (standard deviation, 1.22 [n = 4]) after 3 years of treatment with setmelanotide.
In sum, the patients who achieved a reduction in body weight of at least 10% or greater than or equal to 0.3 mean variation in BMI z-score after 1 year experienced long-lasting, clinically significant benefit after 3 years. The finding supports the long-term use of setmelanotide for this group.
“We feared that setmelanotide’s effectiveness would decrease over time, but after 3 years, this had not happened, and we are hopeful that this sustained efficacy will be long lasting. The first two patients who took the drug in 2016 have not noticed any loss of efficacy as it stands,” said Dr. Dubern.
This is all the more encouraging. In the study presented at the 2023 pediatric conference, setmelanotide’s safety profile was reassuring, and it was consistent with previous studies. Side effects reported in greater than or equal to 15% of patients include injection site reactions, skin hyperpigmentation, nausea, diarrhea, mood disturbances, abdominal pain, vomiting, gastroenteritis, and spontaneous erection.
In addition to the lack of control group, Dr. Dubern acknowledged one other constraint of this study. “Only the patients who responded to treatment with setmelanotide during early-phase trials (85.7%) were enrolled.”
Dr. Dubern summarized the clinical implications: “In patients with early-onset obesity, starting before 5 years of age, doctors should really be considering the possibility that genetics might be involved in such cases. For confirmation and to seek expert opinion, specialist obesity clinics can be found throughout France. Additionally, the INSERM NutriOmics research team headed by Prof Karine Clément, MD, PhD, Sorbonne University, in conjunction with Prof Christine Poitou, MD, PhD, has developed a diagnostic tool [called] ObsGen for practitioners faced with patients with potentially genetic causes of obesity. We can answer any questions they have about the likelihood of a particular patient having a genetic form of obesity and guide their next steps. Treating patients with genetic obesity early on helps limit the condition worsening during adolescence, prevents related complications, and can reduce the stigmatization and suffering experienced by these people. It’s a huge issue. A clinical trial with setmelanotide is currently being carried out in children over 2 years of age.”
Hypothalamic obesity
During the pediatric conference, another speaker presented the results of a phase 2 study that evaluated the efficacy and tolerability of setmelanotide as a new treatment for hypothalamic obesity. Lesions in the hypothalamus can alter melanocortin 4 receptor pathway signaling and thus lead to hypothalamic obesity. Eighteen patients aged 6-40 years with a BMI greater than or equal to the 95th percentile (for patients aged 6-18 years) or greater than or equal to 35 kg/m2 (for adults aged ≥ 18 years) and hypothalamic obesity (craniopharyngioma or other benign brain tumors, surgical removal, and/or chemotherapy) were enrolled. A significant proportion of patients achieved a reduction of greater than or equal to 5% of their BMI (n = 16, 88.9%, CI 90%, 69%-89%, P < .0001), and 72.2% achieved a reduction of greater than or equal to 10% by week 16. The mean change in BMI was −14.9% (9.6%, n = 17). These early results may justify further studies of setmelanotide in treating hypothalamic obesity.
Dr. Dubern has collaborated with Rhythm Pharmaceuticals and Novo Nordisk.
This article was translated from the Medscape French Edition and a version appeared on Medscape.com.
MARSEILLE, FRANCE – , according to results presented at the latest French Pediatric Society conference. The treatment is effective for adults and children alike.
Setmelanotide is the culmination of two decades of research involving the identification of genes involved in early-onset obesity and the characterization of the melanocortin 4 receptor. It became available via early access in 2021.
Currently limited to use in treating obesity linked to a biallelic POMC/PCSK1 or LEPR deficiency, setmelanotide is being tested with respect to other mutations responsible for severe obesity, raising hopes that it will soon be indicated for use in a larger number of patients.
Fewer than 1% of patients who suffer from severe obesity have monogenic forms of obesity. In recent years, the hope for a targeted treatment for patients with these monogenic forms has become a reality.
Although only a small number of patients currently meet the criteria for setmelanotide treatment (namely, those with obesity linked to a biallelic POMC/PCSK1 enzyme or LEPR deficiency and patients with Bardet-Biedl syndrome), there is a real hope that patients with other forms of severe obesity will be able to benefit from this product, including those with heterozygous (not just homozygous) monoallelic variants, who account for more than 10% of patients with severe early-onset obesity.
Restoring satiety signaling
“Setmelanotide is a melanocortin 4 receptor agonist,” said Béatrice Dubern, MD, PhD, pediatrician at Trousseau Hospital, Paris, and member of the French medical research institute (INSERM)/Sorbonne University team on nutrition and obesity. “Its mode of action rests on activation of the leptin-melanocortin signaling pathway in the hypothalamus, which regulates hunger, satiety, energy expenditure, and, therefore, body weight. Rare genetic variants in the leptin-melanocortin pathway are associated with polyphagia and severe early-onset obesity. It is believed that more than 60 genes involved in this leptin-melanocortin pathway are currently associated with obesity.”
In July 2021, the European Medicines Agency approved setmelanotide for daily use via subcutaneous administration.
Weight loss maintained
In phase 3 studies, setmelanotide (melanocortin 4 receptor agonist) demonstrated its effectiveness in reducing weight and hunger for patients with obesity caused by a POMC/PCSK1 or LEPR deficiency.
Twenty-four patients aged 6 years and older showed a significant response to setmelanotide after 1 year of treatment and were included in the extension study. “Significant response” was defined as a reduction in body weight greater than or equal to 10% after 52 weeks for patients aged 18 years and older or a reduction in body mass index (BMI) z-score greater than or equal to 0.3 after 52 weeks for patients younger than 18 years.
Among all patients, the mean variation (standard deviation) in BMI was −24.8% (8.2%, n = 24), −21% (13%, n = 23), and −24% (17.9%, n = 15) at 12, 24, and 36 months, respectively.
For patients aged greater than or equal to 18 years (n = 11), the mean variation (standard deviation) in weight was −25.1% (7.7%, n = 11), −22.9% (12.5%, n = 11), and −24.4% (13.2%, n = 8) at 12, 24, and 36 months, respectively.
For children and adolescents (patients aged < 18 years, n = 13), the mean reduction (standard deviation) in BMI z-score was −1.31 ([0.66], n = 13), −1.10 ([0.79], n = 11), and −1.01 (1.22], n = 4) at 12, 24, and 36 months, respectively.
For patients younger than 18 years, the mean variation in BMI z-score was −1.01 SD after three years on setmelanotide (standard deviation, 1.22 [n = 4]). The mean BMI z-score was +2.42 SD (standard deviation, 1.22 [n = 4]) after 3 years of treatment with setmelanotide.
In sum, the patients who achieved a reduction in body weight of at least 10% or greater than or equal to 0.3 mean variation in BMI z-score after 1 year experienced long-lasting, clinically significant benefit after 3 years. The finding supports the long-term use of setmelanotide for this group.
“We feared that setmelanotide’s effectiveness would decrease over time, but after 3 years, this had not happened, and we are hopeful that this sustained efficacy will be long lasting. The first two patients who took the drug in 2016 have not noticed any loss of efficacy as it stands,” said Dr. Dubern.
This is all the more encouraging. In the study presented at the 2023 pediatric conference, setmelanotide’s safety profile was reassuring, and it was consistent with previous studies. Side effects reported in greater than or equal to 15% of patients include injection site reactions, skin hyperpigmentation, nausea, diarrhea, mood disturbances, abdominal pain, vomiting, gastroenteritis, and spontaneous erection.
In addition to the lack of control group, Dr. Dubern acknowledged one other constraint of this study. “Only the patients who responded to treatment with setmelanotide during early-phase trials (85.7%) were enrolled.”
Dr. Dubern summarized the clinical implications: “In patients with early-onset obesity, starting before 5 years of age, doctors should really be considering the possibility that genetics might be involved in such cases. For confirmation and to seek expert opinion, specialist obesity clinics can be found throughout France. Additionally, the INSERM NutriOmics research team headed by Prof Karine Clément, MD, PhD, Sorbonne University, in conjunction with Prof Christine Poitou, MD, PhD, has developed a diagnostic tool [called] ObsGen for practitioners faced with patients with potentially genetic causes of obesity. We can answer any questions they have about the likelihood of a particular patient having a genetic form of obesity and guide their next steps. Treating patients with genetic obesity early on helps limit the condition worsening during adolescence, prevents related complications, and can reduce the stigmatization and suffering experienced by these people. It’s a huge issue. A clinical trial with setmelanotide is currently being carried out in children over 2 years of age.”
Hypothalamic obesity
During the pediatric conference, another speaker presented the results of a phase 2 study that evaluated the efficacy and tolerability of setmelanotide as a new treatment for hypothalamic obesity. Lesions in the hypothalamus can alter melanocortin 4 receptor pathway signaling and thus lead to hypothalamic obesity. Eighteen patients aged 6-40 years with a BMI greater than or equal to the 95th percentile (for patients aged 6-18 years) or greater than or equal to 35 kg/m2 (for adults aged ≥ 18 years) and hypothalamic obesity (craniopharyngioma or other benign brain tumors, surgical removal, and/or chemotherapy) were enrolled. A significant proportion of patients achieved a reduction of greater than or equal to 5% of their BMI (n = 16, 88.9%, CI 90%, 69%-89%, P < .0001), and 72.2% achieved a reduction of greater than or equal to 10% by week 16. The mean change in BMI was −14.9% (9.6%, n = 17). These early results may justify further studies of setmelanotide in treating hypothalamic obesity.
Dr. Dubern has collaborated with Rhythm Pharmaceuticals and Novo Nordisk.
This article was translated from the Medscape French Edition and a version appeared on Medscape.com.