User login
In youth, hours of screen viewing is associated with severity of depression
Spending time in front of a screen may increase adolescents’ risks of suffering from depression and anxiety, according to a study of 2,482 Canadian middle and high schoolers.
To assess the mental status of the participants, the researchers used self-report questionnaires, the Children’s Depression Inventory, and the Multidimensional Anxiety Scale for Children-10. The more time a student spent viewing a screen was significantly associated with depressive symptoms and the severity of anxiety symptoms, after controlling for the covariates of age, sex, ethnicity, parental education, body mass index, and physical activity. More severe depressive symptoms were significantly associated with the increased amounts of time a student played video games and used a computer, but not with the hours spent watching television. The duration of video game playing also was significantly associated with more severe symptoms of anxiety.
The study’s findings suggest that “screen time may represent a risk factor for, or a marker” of depression and anxiety disorders in adolescents, according to Danijela Maras of Carleton University, Ottawa, and her colleagues. The researchers recommended that future studies determine if “reducing screen time can have a significant impact on the prevention and treatment of anxiety and depression in adolescents.”
Read the full study in Preventive Medicine (doi:10.1016/j.ypmed2015.01.029).
Spending time in front of a screen may increase adolescents’ risks of suffering from depression and anxiety, according to a study of 2,482 Canadian middle and high schoolers.
To assess the mental status of the participants, the researchers used self-report questionnaires, the Children’s Depression Inventory, and the Multidimensional Anxiety Scale for Children-10. The more time a student spent viewing a screen was significantly associated with depressive symptoms and the severity of anxiety symptoms, after controlling for the covariates of age, sex, ethnicity, parental education, body mass index, and physical activity. More severe depressive symptoms were significantly associated with the increased amounts of time a student played video games and used a computer, but not with the hours spent watching television. The duration of video game playing also was significantly associated with more severe symptoms of anxiety.
The study’s findings suggest that “screen time may represent a risk factor for, or a marker” of depression and anxiety disorders in adolescents, according to Danijela Maras of Carleton University, Ottawa, and her colleagues. The researchers recommended that future studies determine if “reducing screen time can have a significant impact on the prevention and treatment of anxiety and depression in adolescents.”
Read the full study in Preventive Medicine (doi:10.1016/j.ypmed2015.01.029).
Spending time in front of a screen may increase adolescents’ risks of suffering from depression and anxiety, according to a study of 2,482 Canadian middle and high schoolers.
To assess the mental status of the participants, the researchers used self-report questionnaires, the Children’s Depression Inventory, and the Multidimensional Anxiety Scale for Children-10. The more time a student spent viewing a screen was significantly associated with depressive symptoms and the severity of anxiety symptoms, after controlling for the covariates of age, sex, ethnicity, parental education, body mass index, and physical activity. More severe depressive symptoms were significantly associated with the increased amounts of time a student played video games and used a computer, but not with the hours spent watching television. The duration of video game playing also was significantly associated with more severe symptoms of anxiety.
The study’s findings suggest that “screen time may represent a risk factor for, or a marker” of depression and anxiety disorders in adolescents, according to Danijela Maras of Carleton University, Ottawa, and her colleagues. The researchers recommended that future studies determine if “reducing screen time can have a significant impact on the prevention and treatment of anxiety and depression in adolescents.”
Read the full study in Preventive Medicine (doi:10.1016/j.ypmed2015.01.029).
FROM PREVENTIVE MEDICINE
ACOG, SMFM, and others address safety concerns in labor and delivery
At least half of all cases of maternal morbidity and mortality could be prevented, or so studies suggest.1,2
The main stumbling block?
Faulty communication.
That’s the word from the American College of Obstetricians and Gynecologists, the Society for Maternal-Fetal Medicine, the American College of Nurse-Midwives, and the Association of Women’s Health, Obstetric and Neonatal Nurses.3
In a joint “blueprint” to transform communication and enhance the safety culture in intrapartum care, these organizations, led by Audrey Lyndon, PhD, RN, from the University of California, San Francisco, School of Nursing, describe the extent of the problem, steps that various team members can take to improve safety, notable success stories, and communication strategies.3 In this article, the joint blueprint is summarized, with a focus on steps obstetricians can take to improve the intrapartum safety culture.
Scope of the problem
A study of more than 3,282 physicians, midwives, and registered nurses produced a troubling statistic: More than 90% of respondents said that they had “witnessed shortcuts, missing competencies, disrespect, or performance problems” during the preceding year of practice.4 Few of these clinicians reported that they had discussed their concerns with the parties involved.
A second study of 1,932 clinicians found that 34% of physicians, 40% of midwives, and 56% of registered nurses had witnessed patients being put at risk within the preceding 2 years by other team members’ inattentiveness or lack of responsiveness.5
These findings suggest that health care providers often witness weak links in intrapartum safety but do not always address or report them. Among the reasons team members may be hesitant to speak up when they perceive a potential problem:
- feelings of resignation or inability to change the situation
- fear of retribution or ridicule
- fear of interpersonal or intrateam conflict.
Although Lyndon and colleagues acknowledge that it is impossible to eliminate adverse outcomes entirely or completely eradicate human error, they argue that significant improvements can be made by adopting a number of manageable strategies.3
Recommended strategies
Lyndon and colleagues describe some of the challenges of effective communication in a health care setting:
Lyndon and colleagues go on to mention a number of strategies to improve communication, boost safety, and reduce medical errors.3
1. Remember that the patient is part of the team
The patient and her family play a key role in identifying the potential for harm during labor and delivery, Lyndon and colleagues assert. Patients should be considered members of the intrapartum team, care should be patient-focused, and any communications from the patient should not only be heard but fully considered. In fact, explicit elicitation of her experience and concerns is recommended.3
2. Consider that you might be part of the problem
It is human nature to attribute a communication problem to the other people involved, rather than take responsibility for it oneself. One potential solution to this mindset is team training, where all members are encouraged to communicate clearly and listen attentively. Organizations that have been successful at improving their culture of safety have implemented such training, as well as the use of checklists, training in fetal heart-rate monitoring, formation of a patient safety committee, external review of safety practices, and designation of a key clinicianto lead the safety program and oversee team training.
3. Structure handoffs
The team should standardize handoffs so that they occur smoothly and all channels of communication remain open and clear.
“Having structured formats for debriefing and handoffs are steps in the right direction, but solving the problem of communication breakdowns is more complicated than standardizing the flow and format of information transfer,” Lyndon and colleagues assert. “Indeed, solving communication breakdowns is a matter of individual, group, organizational, and professional responsibility for creating and sustaining an environment of mutual respect, curiosity, and accountability for behavior and performance.”3
4. Learn to communicate responsibly
“Differences of opinion about clinical assessments, goals of care, and the pathway to optimal outcomes are bound to occur with some regularity in the dynamic environment of labor and delivery,” note Lyndon and colleagues. “Every person has the responsibility to contribute to improving how we relate to and communicate with each other. Collectively, we must create environments in which every team member (woman, family member, physician, midwife, nurse, unit clerk, patient care assistant, or scrub tech) is comfortable expressing and discussing concerns about safety or performance, is encouraged to do so, and has the support of the team to articulate the rationale for and urgency of the concern without fear of put-downs, retribution, or receiving poor-quality care.”3
5. Be persistent and proactive
When team members have differing expectations and communication styles, useful approaches include structured communication tools such as situation, background, assessment, recommendation (SBAR); structured handoffs; board rounds; huddles; attentive listening; and explicit elicitation of the patient’s concerns and desires.3
If someone fails to pay attention to a concern you raise, be persistent about restating that concern until you elicit a response.
If someone exhibits disruptive behavior, point to or establish a code of conduct that clearly describes professional behavior.
If there is a difference of opinion on patient management, such as fetal monitoring and interpretation, conduct regular case reviews and standardize a plan for notification of complications.
6. If you’re a team leader, set clear goals
Then ask team members what will be needed to achieve the outcomes desired.
“Team leaders need to develop outstanding skills for listening and eliciting feedback and cross-monitoring (being aware of each other’s actions and performance) from other team members,” note Lyndon and colleagues.3
7. Increase public awareness of safety concepts
When these concepts and best practices are made known to the public, women and families become “empowered” to speak up when they have concerns about care.
And when they do speak up, it pays to listen.
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
1. Geller SE, Rosenberg D, Cox SM, et al. The continuum of maternal morbidity and mortality: factors associated with severity. Am J Obstet Gynecol. 2004;191(3):939–944.
2. Mitchell C, Lawton E, Morton C, McCain C, Holtby S, Main E. California Pregnancy-Associated Mortality Review: mixed methods approach for improved case identification, cause of death analyses and translation of findings. Matern Child Health J. 2014;18(3):518–526.
3. Lyndon A, Johnson MC, Bingham D, et al. Transforming communication and safety culture in intrapartum care: a multi-organization blueprint. Obstet Gynecol. 2015;125(5):1049–1055.
4. Maxfield DG, Lyndon A, Kennedy HP, O’Keeffe DF, Ziatnik MG. Confronting safety gaps across labor and delivery teams. Am J Obstet Gynecol. 2013;209(5):402–408.e3.
5. Lyndon A, Zlatnik MG, Maxfield DG, Lewis A, McMillan C, Kennedy HP. Contributions of clinical disconnections and unresolved conflict to failures in intrapartum safety. J Obstet Gynecol Neonatal Nurs. 2014;43(1):2–12.
At least half of all cases of maternal morbidity and mortality could be prevented, or so studies suggest.1,2
The main stumbling block?
Faulty communication.
That’s the word from the American College of Obstetricians and Gynecologists, the Society for Maternal-Fetal Medicine, the American College of Nurse-Midwives, and the Association of Women’s Health, Obstetric and Neonatal Nurses.3
In a joint “blueprint” to transform communication and enhance the safety culture in intrapartum care, these organizations, led by Audrey Lyndon, PhD, RN, from the University of California, San Francisco, School of Nursing, describe the extent of the problem, steps that various team members can take to improve safety, notable success stories, and communication strategies.3 In this article, the joint blueprint is summarized, with a focus on steps obstetricians can take to improve the intrapartum safety culture.
Scope of the problem
A study of more than 3,282 physicians, midwives, and registered nurses produced a troubling statistic: More than 90% of respondents said that they had “witnessed shortcuts, missing competencies, disrespect, or performance problems” during the preceding year of practice.4 Few of these clinicians reported that they had discussed their concerns with the parties involved.
A second study of 1,932 clinicians found that 34% of physicians, 40% of midwives, and 56% of registered nurses had witnessed patients being put at risk within the preceding 2 years by other team members’ inattentiveness or lack of responsiveness.5
These findings suggest that health care providers often witness weak links in intrapartum safety but do not always address or report them. Among the reasons team members may be hesitant to speak up when they perceive a potential problem:
- feelings of resignation or inability to change the situation
- fear of retribution or ridicule
- fear of interpersonal or intrateam conflict.
Although Lyndon and colleagues acknowledge that it is impossible to eliminate adverse outcomes entirely or completely eradicate human error, they argue that significant improvements can be made by adopting a number of manageable strategies.3
Recommended strategies
Lyndon and colleagues describe some of the challenges of effective communication in a health care setting:
Lyndon and colleagues go on to mention a number of strategies to improve communication, boost safety, and reduce medical errors.3
1. Remember that the patient is part of the team
The patient and her family play a key role in identifying the potential for harm during labor and delivery, Lyndon and colleagues assert. Patients should be considered members of the intrapartum team, care should be patient-focused, and any communications from the patient should not only be heard but fully considered. In fact, explicit elicitation of her experience and concerns is recommended.3
2. Consider that you might be part of the problem
It is human nature to attribute a communication problem to the other people involved, rather than take responsibility for it oneself. One potential solution to this mindset is team training, where all members are encouraged to communicate clearly and listen attentively. Organizations that have been successful at improving their culture of safety have implemented such training, as well as the use of checklists, training in fetal heart-rate monitoring, formation of a patient safety committee, external review of safety practices, and designation of a key clinicianto lead the safety program and oversee team training.
3. Structure handoffs
The team should standardize handoffs so that they occur smoothly and all channels of communication remain open and clear.
“Having structured formats for debriefing and handoffs are steps in the right direction, but solving the problem of communication breakdowns is more complicated than standardizing the flow and format of information transfer,” Lyndon and colleagues assert. “Indeed, solving communication breakdowns is a matter of individual, group, organizational, and professional responsibility for creating and sustaining an environment of mutual respect, curiosity, and accountability for behavior and performance.”3
4. Learn to communicate responsibly
“Differences of opinion about clinical assessments, goals of care, and the pathway to optimal outcomes are bound to occur with some regularity in the dynamic environment of labor and delivery,” note Lyndon and colleagues. “Every person has the responsibility to contribute to improving how we relate to and communicate with each other. Collectively, we must create environments in which every team member (woman, family member, physician, midwife, nurse, unit clerk, patient care assistant, or scrub tech) is comfortable expressing and discussing concerns about safety or performance, is encouraged to do so, and has the support of the team to articulate the rationale for and urgency of the concern without fear of put-downs, retribution, or receiving poor-quality care.”3
5. Be persistent and proactive
When team members have differing expectations and communication styles, useful approaches include structured communication tools such as situation, background, assessment, recommendation (SBAR); structured handoffs; board rounds; huddles; attentive listening; and explicit elicitation of the patient’s concerns and desires.3
If someone fails to pay attention to a concern you raise, be persistent about restating that concern until you elicit a response.
If someone exhibits disruptive behavior, point to or establish a code of conduct that clearly describes professional behavior.
If there is a difference of opinion on patient management, such as fetal monitoring and interpretation, conduct regular case reviews and standardize a plan for notification of complications.
6. If you’re a team leader, set clear goals
Then ask team members what will be needed to achieve the outcomes desired.
“Team leaders need to develop outstanding skills for listening and eliciting feedback and cross-monitoring (being aware of each other’s actions and performance) from other team members,” note Lyndon and colleagues.3
7. Increase public awareness of safety concepts
When these concepts and best practices are made known to the public, women and families become “empowered” to speak up when they have concerns about care.
And when they do speak up, it pays to listen.
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
At least half of all cases of maternal morbidity and mortality could be prevented, or so studies suggest.1,2
The main stumbling block?
Faulty communication.
That’s the word from the American College of Obstetricians and Gynecologists, the Society for Maternal-Fetal Medicine, the American College of Nurse-Midwives, and the Association of Women’s Health, Obstetric and Neonatal Nurses.3
In a joint “blueprint” to transform communication and enhance the safety culture in intrapartum care, these organizations, led by Audrey Lyndon, PhD, RN, from the University of California, San Francisco, School of Nursing, describe the extent of the problem, steps that various team members can take to improve safety, notable success stories, and communication strategies.3 In this article, the joint blueprint is summarized, with a focus on steps obstetricians can take to improve the intrapartum safety culture.
Scope of the problem
A study of more than 3,282 physicians, midwives, and registered nurses produced a troubling statistic: More than 90% of respondents said that they had “witnessed shortcuts, missing competencies, disrespect, or performance problems” during the preceding year of practice.4 Few of these clinicians reported that they had discussed their concerns with the parties involved.
A second study of 1,932 clinicians found that 34% of physicians, 40% of midwives, and 56% of registered nurses had witnessed patients being put at risk within the preceding 2 years by other team members’ inattentiveness or lack of responsiveness.5
These findings suggest that health care providers often witness weak links in intrapartum safety but do not always address or report them. Among the reasons team members may be hesitant to speak up when they perceive a potential problem:
- feelings of resignation or inability to change the situation
- fear of retribution or ridicule
- fear of interpersonal or intrateam conflict.
Although Lyndon and colleagues acknowledge that it is impossible to eliminate adverse outcomes entirely or completely eradicate human error, they argue that significant improvements can be made by adopting a number of manageable strategies.3
Recommended strategies
Lyndon and colleagues describe some of the challenges of effective communication in a health care setting:
Lyndon and colleagues go on to mention a number of strategies to improve communication, boost safety, and reduce medical errors.3
1. Remember that the patient is part of the team
The patient and her family play a key role in identifying the potential for harm during labor and delivery, Lyndon and colleagues assert. Patients should be considered members of the intrapartum team, care should be patient-focused, and any communications from the patient should not only be heard but fully considered. In fact, explicit elicitation of her experience and concerns is recommended.3
2. Consider that you might be part of the problem
It is human nature to attribute a communication problem to the other people involved, rather than take responsibility for it oneself. One potential solution to this mindset is team training, where all members are encouraged to communicate clearly and listen attentively. Organizations that have been successful at improving their culture of safety have implemented such training, as well as the use of checklists, training in fetal heart-rate monitoring, formation of a patient safety committee, external review of safety practices, and designation of a key clinicianto lead the safety program and oversee team training.
3. Structure handoffs
The team should standardize handoffs so that they occur smoothly and all channels of communication remain open and clear.
“Having structured formats for debriefing and handoffs are steps in the right direction, but solving the problem of communication breakdowns is more complicated than standardizing the flow and format of information transfer,” Lyndon and colleagues assert. “Indeed, solving communication breakdowns is a matter of individual, group, organizational, and professional responsibility for creating and sustaining an environment of mutual respect, curiosity, and accountability for behavior and performance.”3
4. Learn to communicate responsibly
“Differences of opinion about clinical assessments, goals of care, and the pathway to optimal outcomes are bound to occur with some regularity in the dynamic environment of labor and delivery,” note Lyndon and colleagues. “Every person has the responsibility to contribute to improving how we relate to and communicate with each other. Collectively, we must create environments in which every team member (woman, family member, physician, midwife, nurse, unit clerk, patient care assistant, or scrub tech) is comfortable expressing and discussing concerns about safety or performance, is encouraged to do so, and has the support of the team to articulate the rationale for and urgency of the concern without fear of put-downs, retribution, or receiving poor-quality care.”3
5. Be persistent and proactive
When team members have differing expectations and communication styles, useful approaches include structured communication tools such as situation, background, assessment, recommendation (SBAR); structured handoffs; board rounds; huddles; attentive listening; and explicit elicitation of the patient’s concerns and desires.3
If someone fails to pay attention to a concern you raise, be persistent about restating that concern until you elicit a response.
If someone exhibits disruptive behavior, point to or establish a code of conduct that clearly describes professional behavior.
If there is a difference of opinion on patient management, such as fetal monitoring and interpretation, conduct regular case reviews and standardize a plan for notification of complications.
6. If you’re a team leader, set clear goals
Then ask team members what will be needed to achieve the outcomes desired.
“Team leaders need to develop outstanding skills for listening and eliciting feedback and cross-monitoring (being aware of each other’s actions and performance) from other team members,” note Lyndon and colleagues.3
7. Increase public awareness of safety concepts
When these concepts and best practices are made known to the public, women and families become “empowered” to speak up when they have concerns about care.
And when they do speak up, it pays to listen.
Share your thoughts on this article! Send your Letter to the Editor to rbarbieri@frontlinemedcom.com. Please include your name and the city and state in which you practice.
1. Geller SE, Rosenberg D, Cox SM, et al. The continuum of maternal morbidity and mortality: factors associated with severity. Am J Obstet Gynecol. 2004;191(3):939–944.
2. Mitchell C, Lawton E, Morton C, McCain C, Holtby S, Main E. California Pregnancy-Associated Mortality Review: mixed methods approach for improved case identification, cause of death analyses and translation of findings. Matern Child Health J. 2014;18(3):518–526.
3. Lyndon A, Johnson MC, Bingham D, et al. Transforming communication and safety culture in intrapartum care: a multi-organization blueprint. Obstet Gynecol. 2015;125(5):1049–1055.
4. Maxfield DG, Lyndon A, Kennedy HP, O’Keeffe DF, Ziatnik MG. Confronting safety gaps across labor and delivery teams. Am J Obstet Gynecol. 2013;209(5):402–408.e3.
5. Lyndon A, Zlatnik MG, Maxfield DG, Lewis A, McMillan C, Kennedy HP. Contributions of clinical disconnections and unresolved conflict to failures in intrapartum safety. J Obstet Gynecol Neonatal Nurs. 2014;43(1):2–12.
1. Geller SE, Rosenberg D, Cox SM, et al. The continuum of maternal morbidity and mortality: factors associated with severity. Am J Obstet Gynecol. 2004;191(3):939–944.
2. Mitchell C, Lawton E, Morton C, McCain C, Holtby S, Main E. California Pregnancy-Associated Mortality Review: mixed methods approach for improved case identification, cause of death analyses and translation of findings. Matern Child Health J. 2014;18(3):518–526.
3. Lyndon A, Johnson MC, Bingham D, et al. Transforming communication and safety culture in intrapartum care: a multi-organization blueprint. Obstet Gynecol. 2015;125(5):1049–1055.
4. Maxfield DG, Lyndon A, Kennedy HP, O’Keeffe DF, Ziatnik MG. Confronting safety gaps across labor and delivery teams. Am J Obstet Gynecol. 2013;209(5):402–408.e3.
5. Lyndon A, Zlatnik MG, Maxfield DG, Lewis A, McMillan C, Kennedy HP. Contributions of clinical disconnections and unresolved conflict to failures in intrapartum safety. J Obstet Gynecol Neonatal Nurs. 2014;43(1):2–12.
In this article
- Why handoffs should be structured
- Goals for teamleaders
Asthma Self-Management in Women
From the Department of Health Behavior and Health Education, School of Public Health (Dr. Janevic) and the Medical School (Dr. Sanders), University of Michigan Ann Arbor, MI.
Abstract
- Objective: Asthma prevalence, morbidity, and mortality are all greater among adult women compared to men. Appropriate asthma self-management can improve asthma control. We reviewed published literature about sex- and gender-related factors that influence asthma self-management among women, as well as evidence-based interventions to promote effective asthma self-management in this population.
- Design: Based on evidence from the published literature, factors influencing women’s asthma self-management were categorized as follows: social roles and socioeconomic status, comorbidities, obesity, hormonal factors, and aging-related changes.
- Results: A number of factors were identified that affect women’s asthma self-management. These include: exposure to asthma triggers associated with gender roles, such as cleaning products; financial barriers to asthma management; comorbidities that divert attention or otherwise interfere with asthma management; a link between obesity and poor asthma outcomes; the effects of hormonal shifts associated with menstrual cycles and menopause on asthma control; and aging-associated barriers to effective self-management such as functional limitations and caregiving. Certain groups, such as African-American women, are at higher risk for poor asthma outcomes linked to many of the above factors. At least 1 health coaching intervention designed for women with asthma has been shown in a randomized trial to reduce symptoms and health care use.
- Conclusion: Future research on women and asthma self-management should include a focus on the relationship between hormonal changes and asthma symptoms. Interventions are also needed that address the separate and interacting effects of risk factors for poor asthma control that tend to cluster in women, such as obesity, depression, and gastroesophageal reflux disease.
In childhood, asthma is more prevalent in boys than in girls. In adolescence and adulthood, however, asthma becomes a predominantly female disease, with hormonal factors likely playing a role in this shift [1,2]. Fu et al [3] reviewed daily asthma symptom diaries of 418 children. From age 5 to 7, boys had more severe symptoms, but by age 10 girls’ symptoms were becoming more severe. By age 14, the girls’ symptoms continued increasing while the boys’ symptoms began to decline. A meta-analysis by Lieberoth et al [4] found a 37% increased risk of post-menarchal asthma in girls with onset of menarche < 12 years. Together, these studies implicate female sex hormones in both the increased incidence and severity of asthma after puberty. In 2012, nearly 10% of adult women reported current asthma, compared to only 6% of men [5]. Among adults with asthma, women have a 30% higher mortality rate than men [6]. Disparities that disadvantage women are also evident across a range of other asthma-related outcomes, including disease severity, rescue inhaler use, activity limitations, asthma-related quality of life, and health care utilization [7–12].

Factors Influencing Asthma Self-Management in Women
Social Roles and Socioeconomic Status
Traditional gender roles involve various responsibilities, such as household cleaning, cooking, and care of young children, that are associated with exposures to precipitants of asthma symptoms [17]. Gender norms also promote the use of personal care products, like fragrances and hair sprays, which are potential asthma triggers [17]. Recent observational studies in Europe have examined the link between women’s use of cleaning products and asthma. Bédard and colleagues [18] found an association between weekly use of cleaning sprays at home and asthma among women, and Dumas and colleagues [19] found that workplace exposure to cleaning products among women with asthma was related to increased symptoms and severity of asthma. These researchers conclude that “while domestic exposure is much more frequent in the general population, exposure levels are probably higher at the workplace” and therefore both contribute to asthma disease burden [19]. Although little-discussed in the literature, sexual activity is another common trigger of asthma symptoms in women. Clark et al [15] found that more than one-third of women taking part in a randomized controlled trial (RCT) of an asthma self-management intervention reported being bothered by symptoms of asthma during sexual activity. This topic was rarely discussed, however, by their health care providers [20].
Socioeconomic factors also play a significant role in asthma management. There is a well-recognized and persistent gender gap in income in the U.S. population such that women who work full-time only earn three-quarters of what their male counterparts earn [21]. Challenges related to low socioeconomic status (SES) may contribute to poor medication adherence among asthma patients [22]. Although a comprehensive review of the impact of SES-related factors on asthma prevalence, severity, and disease-management behaviors is beyond the scope of this article, recent research demonstrates the impact of financial stress on women’s asthma self-management. Patel et al (2014) studied health-related financial burden among African-American women with asthma [23]. Despite the fact that the majority of women in this qualitative study had health insurance, they felt greatly burdened by out-of-pocket expenses such as high co-pays for medications or ambulance use, lost wages due to sick time, and gaps in insurance coverage. These financial concerns—and related issues such as time spent navigating health care insurance and cycling through private and public insurance programs—were described as a significant source of ongoing stress by this group of vulnerable asthma patients [23]. Focus group participants reported several strategies for dealing with asthma-related financial challenges, including stockpiling medications when feasible (eg, when covered by current insurance plan) for future use by the patient or a family member, seeking out and using community assistance programs, and foregoing medications altogether during periods when they could not afford them [23].
Comorbidities
The 2010 publication of Multiple chronic conditions: a strategic framework by the US Department of Health and Human Services [24] brought the attention of the medical and research communities to the scope and significance of multimorbidity in the US population, including the challenges that individuals face in managing multiple chronic health conditions. Although the prevalence of specific comorbidities with asthma differs by age, some that are most commonly associated with asthma and that may complicate asthma control are obstructive sleep apnea, gastroesophageal reflux disease (GERD), rhinitis, and sinusitis [25,26]. Among women with asthma, multimorbidity appears to be the rule, not the exception. Using nationally-representative data from the US National Health and Nutrition Examination Survey (NHANES), Patel et al [27] found that more than half of adults with asthma reported also being diagnosed with at least 1 additional major chronic condition. A recent study found that asthma/arthritis and asthma/hypertension were the second and third most prevalent disease dyads among all US women aged 18–44 years [28]. Studies have found that comorbidities among asthma patients are associated with worse asthma outcomes, including increased symptoms, activity limitations and sleep disturbance due to asthma [27], and ED use for asthma [15,27].
Qualitative research yields insight into the patient perspective of multimorbidity, that is, how women with asthma and coexisting chronic diseases perceive the effect of their health conditions on their ability to engage in self-management. Janevic and colleagues [29] conducted face-to-face interviews with African-American women participating in a randomized controlled trial of a culturally and gender-tailored asthma-management intervention to learn about their experiences managing asthma and concurrent health conditions. Interviewees had an average of 5.7 chronic conditions in addition to asthma. Women reported that managing their asthma often “took a backseat” to other chronic conditions. Participants also discussed reduced motivation or capacity for asthma self-management due to depression, chronic pain, mobility limitations or combinations of these, and reduced adherence to asthma medications due to the psychological and logistical burdens of polypharmacy.
Depression and anxiety are common comorbidities that are associated with worse asthma outcomes [26,30–32] and reduced asthma medication adherence [33,34]. In general population studies as well as among asthma patients, women are more likely than men to report depression and anxiety [30,35–37]. Screening for and treating depression and anxiety are indicated in women with asthma and may lead to improved adherence and outcomes [30].
Obesity
Adults with asthma are at increased risk of obesity [38]. Obesity is a possible risk factor for development of asthma in women [2] and for resting dyspnea in women with asthma [39]. It is associated with poor asthma-related QOL and use of emergency/urgent services [40]. Evidence is mixed regarding the link between BMI and asthma control [41–43], but the following studies suggest that women who are overweight/obese face unique asthma management challenges. Valerio and colleagues found that in a sample of 808 women enrolled in a randomized trial of an asthma-education intervention, nearly 7 out of 10 were overweight (BMI ≥ 25) or obese (BMI ≥ 30), and nearly a quarter were “extremely obese” (BMI > 35) [44]. This subgroup of women was more likely to have persistent asthma, comorbid GERD and urinary incontinence, to be non-white, and to have lower levels of education and income. Being overweight was also associated with greater use of health care services and having greater psychosocial challenges (ie, a higher need for asthma-related social support and lower asthma-related quality of life). These authors suggest the need to design communications for overweight women with asthma that recognize “the specific cultural and social influences on their asthma management behaviors” [44] with a focus on psychosocial needs, while incorporating existing social support networks. In the previously discussed study by Janevic and colleagues [29] the average BMI of the interview participants was 36.0, and a number of respondents identified weight loss as the self-care behavior that they thought would benefit them the most across multimorbid conditions. Therefore, health care providers should provide appropriate counseling and/or referrals to help women with asthma achieve weight loss goals. Given trends over time showing increasing prevalence of asthma and obesity [45,46], interest is growing in the asthma research community about the interaction of the 2 conditions.
Hormonal Factors
Hormones exert a significant effect on asthma in women, and must be considered in clinical and self-management of the disease. Hormone levels fluctuate during the menstrual cycle, with a surge of estradiol (a type of estrogen) at the time of ovulation around day 14, accompanied by low levels of progesterone. During the luteal phase (day 14–28 of the menstrual cycle), estrogens decrease while progesterone levels increase then decrease again before onset of menstruation [47]. During pregnancy, levels of estrogens and progesterone increase and are the highest during the third trimester, when women usually experience good asthma control. Then, during menopause both estradiol and progesterone levels drop to lower levels than those during any phase of menstruation. In addition to the role in the menstrual cycle, there are estrogen receptors (ER-α and ER-β) which are expressed in the human lung and have a role in both airway responsiveness (relaxation) and inflammation [48]. Estrogen also acts directly on cells of the immune system to stimulate airway inflammation, particularly when allergens are present [48]. Further discussion about these contrasting actions of estrogen can be found in a recent review [48].
During the reproductive years, 30% to 40% of women with asthma report perimenstrual symptoms. Forced expiratory volume in 1 second and forced vital capacity are lowest in the periovulatory period, when estrogen levels are high. In contrast, during the luteal phase, studies have shown increased airway hyperreactivity, especially in the premenstrual period when estrogen levels are low [49]. However, when asthma patients with and without perimenstrual symptoms are evaluated, there is no significant difference in their perimenstrual estrogen and progesterone levels [50]. Clark et al [15] found women participating in a self-management intervention, which included checking daily peak flow rates, reported significantly more menstrual and perimenstrual asthma symptomatology than the control group. This suggests that some women with asthma have may have, but do not recognize, perimenstrual symptoms. Further elucidation of the incidence of symptomatology related to the menstrual cycle as well as the role of hormonal variation is an area for future research efforts.
At the time of menopause and continuing to postmenopause, levels of both estrogen and progesterone drop to below those during the reproductive years, leading to uncomfortable symptoms in many women. Hormone replacement therapy (HRT) with either estrogen alone or estrogen-progesterone combination effectively improves these, but there is concern for potential effects on asthma prevalence and severity. Two recent large studies support this concern. Postmenopausal women followed for 10 years in the Nurses’ Health Study with a history of HRT had an increased risk of new onset asthma when compared to postmenopausal women with no history of estrogen use (RR = 2.30, 95% CI 1.69–3.14) [51]. This persisted in estrogen-progesterone users. A large French cohort confirmed the increased onset of new asthma in users of estrogen-alone replacement therapy (HR = 1.54, 95% CI 1.13–2.09). However, this effect decreased with time if estrogen had been discontinued, and they did not find a similar increase in users of estrogen-progesterone combination therapy [52]. In contrast, Bonelykke et al [53] found an association between ever using HRT and first-ever hospital admission for asthma, in postmenopausal women (HR 1.46, CI 1.21–1.76), and this risk increased with duration of HRT use. It is clear that physicians need to be aware of these potential respiratory complications, inform their patients, and consider new-onset asthma when women on HRT bring complaints of dyspnea, cough, or wheeze. Future randomized trials are needed to clarify the relationship between HRT and asthma, and to test ways to optimize asthma self-management in women experiencing these transitions.
Older Women and Asthma
Although the bulk of research on asthma focuses on children and young adults, asthma in the elderly is receiving increased attention [54], in part because this demographic group has the highest asthma mortality rate and the most frequent hospitalizations [6,55]. In a sample of midlife and older women from the Nurses’ Health Study who had been diagnosed with persistent asthma, Barr et al found that adherence to asthma medication guidelines decreased with age [54]. In this study, women with more severe asthma and those with multimorbidity were less adherent than those without comorbidities, as were women who spent more hours caregiving for an ill spouse. The authors concluded that asthma is undertreated among older women.
Baptist et al (2014) describe several challenges to asthma management of older women by clinicians and by the women themselves [55]. For example, elderly women may be at increased risk for adverse effects of inhaled corticosteroids. Certain medications used to treat comorbidities, such as beta-blockers and aspirin, may also exacerbate asthma symptoms. In terms of self-management, older women may have a decreased ability to perceive breathlessness, requiring monitoring with a peak flow meter to detect reductions in airflow. Comorbidities are particularly prevalent in this age group, and asthma symptoms may be confused with symptoms of other conditions, such as heart disease [56]. Baptist and colleagues note factors common among elderly women that pose potential barriers to successful self-management of asthma, including limited income, poverty, depression, and caregiving [55]. They also mention that functional limitations such as those due to arthritis, visual difficulties, or weakened inspiratory strength can make inhaler use more difficult. It should also be noted that some behaviors may promote asthma self-management in this group; for example, Valerio and colleagues [57] found that women over age 50 were more likely than younger women to keep a daily asthma diary when asked to do so as part of a self-management intervention [57].
Evidence-Based Asthma Self-Management Interventions for Women
For women to achieve optimal asthma control, the unique factors as described above that influence their symptoms and management need to be addressed [58]. Several examples can be found in the literature of behavioral interventions that focus on the particular self-management challenges faced by women. Clark and colleagues reported the results of an RCT of the Women Breathe Free (WBF) program [15,16]. This intervention consisted of asthma self-management education delivered over 5 telephone sessions by a health educator. WBF content was based on self-regulation theory, which involves observing one’s behavior and making judgments on the observations, testing strategies to improve asthma management, and reacting to positive results of these strategies with enhanced self-efficacy and outcome expectations, ie, the belief that a given strategy will produce the desired results [59]. In WBF, participants used a problem-solving process based on this framework to carry out recommendations in their physician’s therapeutic plan. WBF also incorporated special attention to sex- and gender-based factors in asthma management.
Over a 12-month period, women who participated in the intervention relative to controls experienced significant reductions in nighttime symptoms, days of missed work/school, emergency department visits, and both scheduled and urgent office visits. Intervention group women also reported decreased asthma symptoms during sexual activity, improved asthma-related quality of life, and increased confidence to manage asthma. At long-term follow-up (2 years from baseline), persistent positive effects of the intervention were found on outpatient visits for asthma symptom level during sexual activity, days of missed work/school, asthma-related quality of life, and confidence to manage asthma [60].
In a follow-up study, Clark and colleagues [61] developed the “Women of Color and Asthma Control” (WCAC) program. WCAC incorporates the theoretical orientation and many of the program elements of Women Breathe Free, but has been adapted to be responsive to the needs and preferences of African-American women. Poverty and race are associated with greater asthma morbidity and mortality [5,62,63]. African-American women and women of low socioeconomic status are particularly vulnerable to asthma and associated morbidity and mortality, making this an important group for intervention. Culturally responsive components in the WCAC intervention include use of culturally relevant activities and beliefs when discussing triggers and barriers to asthma management, as well as culturally appropriate visuals. This ongoing trial will test WCAC’s effect on ED visits, hospitalizations, and urgent care; asthma symptoms; and asthma-related quality of life at 1 year and 18 months from baseline.
In a small RCT among women with asthma, Bidwell and colleagues tested a program consisting of 10 weeks of yoga instruction (including breathing practices, poses, and meditation/relaxation skills) in a group setting followed by 10 weeks of home practice [64]. Women in the intervention group reported improved quality of life, as measured by the St. George’s Respiratory Quality of Life questionnaire [65], and participants also had decreased parasympathetic modulation in response to an isometric forearm exercise. They conclude that yoga is a promising modality for improving quality of life among asthma patients and that these changes may be linked to better autonomic modulation. Although this program was not designed specifically for women, yoga is practiced significantly more frequently among women compared to men [66,67], and thus has the potential to be widely used in this group.
Based on our experience conducting self-management research among women with asthma, and unpublished process data from these studies, we observe that the following elements appear to contribute to high participant engagement in these programs and successful outcomes. First, in participant feedback questionnaires from the Women Breathe Free and Women of Color and Asthma Control studies, women have singled out the importance of their relationship with their assigned telephone asthma educator as motivating them to make positive changes in their asthma self-management behaviors. The popularity of health and wellness coaching, including for chronic disease management, is rapidly growing [68]. This is a patient-centered approach that guides patients in setting their own goals for disease management and devising their own strategies for achieving them [68]. Strong interpersonal relationships are thought to enhance the coaching process and this may be especially important for women [68]. Participants have also indicated that they are able to apply the goal-setting and problem-solving skills they have learned as part of the intervention to management of other health or psychosocial issues in their lives; therefore this component seems especially critical for women with asthma who are typically managing multiple health issues as well as those of others. Finally, maximizing the flexibility of interventions is important for working-age women who typically are engaged in part- or full-time employment and also have significant responsibilities caring for others. This flexibility can come in the form of telephone-based or “mHealth” interventions that use mobile technologies such as text messaging [69], as well as internet-based or smartphone/tablet “apps” that can be completed at a pace and schedule that is convenient for the participant [70]. Such interventions could be easily tailored to address sex- and gender-specific issues in asthma management.
Future Research and Practice Directions
This review points to several promising directions for research and practice in the area of supporting women’s asthma self-management. The first is a systematic exploration of the added value of gender-tailoring asthma self-management support interventions to determine which subgroups of women benefit from which type of sex- and gender-specific information, and in which form. More research is needed on the relationship between hormone levels and changes and asthma symptoms, and how this affects women’s self-management. This includes recognition of new or worsening asthma with the use of hormone replacement therapy in menopausal and post-menopausal women, a group that is rapidly increasing in number in the US population. Another direction for research is a family-systems approach to asthma education and supporting asthma management. Asthma in one or more first-degree relatives has been shown across diverse populations to be a risk factor for asthma [71]. Women with asthma are therefore more likely to have children with asthma, and vice-versa; however, no prior research was identified that addresses asthma self-management in mother/child dyads. For example, it is possible that teaching women to better manage their own asthma could have “trickle down” effects to how they help a child manage asthma. Last, as the above discussion of factors affecting women’s asthma makes clear, many risk factors for poor asthma management and control in women cluster together, such as obesity, depression, and GERD. Interventions that attempt to address the separate and interacting effects of these factors and comorbidities, may yield better outcomes among the most vulnerable asthma patients.
Corresponding author: Mary R. Janevic, PhD, Center for Managing Chronic Disease, University of Michigan School of Public Health, 1425 Washington Heights, Ann Arbor, MI 48109, mjanevic@umich.edu.
Financial disclosures: None.
1. Postma DS. Gender differences in asthma development and progression. Gend Med 2007;4 Suppl B:S133–46.
2. Melgert BN, Ray A, Hylkema MN, et al. Are there reasons why adult asthma is more common in females? Curr Allergy Asthma Rep 2007;7:143–50.
3. Fu L, Freishtat RJ, Gordish-Dressman H, et al. Natural progression of childhood asthma symptoms and strong influence of sex and puberty. Ann Am Thor Soc 2014;11:939–44.
4. Lieberoth S, Gade EJ, Brok J, et al. Age at menarche and risk of asthma: systematic review and meta-analysis. J Asthma 2014;51:559–65.
5. Blackwell D, Lucas J, Clarke T. Summary health statistics for U.S. adults: national health interview survey, 2012. Vital Health Stat 10 2014;(260):1–161.
6. Akinbami LJ, Moorman JE, Bailey C, et al. Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010. NCHS Data Brief 2012 May;(94):1–8.
7. Sinclair AH, Tolsma DD. Gender differences in asthma experience and disease care in a managed care organization. J Asthma 2006;43:363–7.
8. Naleway AL, Vollmer WM, Frazier EA, et al. Gender differences in asthma management and quality of life. J Asthma 2006;43:549–52.
9. Lisspers K, Ställberg B, Janson C, et al. Sex-differences in quality of life and asthma control in Swedish asthma patients. J Asthma 2013;50:1090–5.
10. Ostrom NK. Women with asthma: a review of potential variables and preferred medical management. Ann Allergy Asthma Immunol 2006;96:655–65.
11. Kynyk JA, Mastronarde JG, McCallister JW. Asthma, the sex difference. Curr Opin Pulm Med 2011;17:6–11.
12. Schiller JS, Lucas JW, Ward BW, Peregoy JA. Summary health statistics for U.S. adults: National Health Interview Survey, 2010. Vital Health Stat 10 2012;(252):1–207.
13. Clark NM, Becker MH, Janz NK, et al. Self-management of chronic disease by older adults: A review and directions for research. J Aging Health 1991;3:3–27.
14. Sundberg R, Torén K, Franklin KA, et al. Asthma in men and women: Treatment adherence, anxiety, and quality of sleep. Resp Med 2010;104:337–44.
15. Clark NM, Gong ZM, Wang SJ, et al. A randomized trial of a self-regulation intervention for women with asthma. Chest 2007;132:88–97.
16. Clark NM, Gong ZM, Wang SJ, et al. From the female perspective: Long-term effects on quality of life of a program for women with asthma. Gend Med 2010;7:125–36.
17. Thomas LJ, Janevic MR, Sanders G, Clark NM. Gender-related asthma challenges in a sample of African American women. Poster presented at 2012 Women’s Health Congress.
18. Bedard A, Varraso R, Sanchez M, et al. Cleaning sprays, household help and asthma among elderly women. Respir Med 2014;108:171–80.
19. Dumas O, Siroux V, Luu F, et al. Cleaning and asthma characteristics in women. Am J Ind Med 2014;57:303–11.
20. Clark NM, Valerio MA, Gong ZM. Self-regulation and women with asthma. Curr Opin Allergy Clin Immunol 2008;8:222–7.
21. Lips HM. The gender pay gap: Challenging the rationalizations. Perceived equity, discrimination, and the limits of human capital models. Sex Roles 2013;68:169–85.
22. Apter AJ, Reisine ST, Affleck G, et al. Adherence with twice-daily dosing of inhaled steroids. Socioeconomic and health-belief differences. Am J Respir Crit Care Med 1998;157(6 Pt 1):1810–7.
23. Patel MR, Nelson BW, Id-Deen E, Caldwell CH. Beyond co-pays and out-of-pocket costs: perceptions of health-related financial burden in managing asthma among African American women. J Asthma 2014;51:1083–8.
24. U.S. Department of Health & Human Services. Multiple chronic conditions—a strategic framework: optimum health and quality of life for individuals with multiple chronic conditions. Washington, DC; 2010.
25. Boulet LP, Boulay ME. Asthma-related comorbidities. Expert Rev Respir Med 2011;5:377–93.
26. Sumino K, O’Brian K, Bartle B, et al. Coexisting chronic conditions associated with mortality and morbidity in adult patients with asthma. J Asthma 2014;51:306–14.
27. Patel MR, Janevic MR, Heeringa SG, et al. An examination of adverse asthma outcomes in U.S. Adults with multiple morbidities. Ann Am Thorac Soc 2013;10:426–31.
28. Ward BW, Schiller JS. Prevalence of multiple chronic conditions among US adults: estimates from the National Health Interview Survey, 2010. Prev Chronic Dis 2013;10:E65.
29. Janevic MR, Ellis KR, Sanders GM, et al. Self-management of multiple chronic conditions among African American women with asthma: a qualitative study. J Asthma 2014;51:243–52.
30. Eisner MD, Katz PP, Lactao G, Iribarren C. Impact of depressive symptoms on adult asthma outcomes. Ann Allergy Asthma Immunol 2005;94:566–74.
31. Strine TW, Mokdad AH, Balluz LS, et al. Impact of depression and anxiety on quality of life, health behaviors, and asthma control among adults in the United States with asthma, 2006. J Asthma 2008;45:123–33.
32. Di Marco F, Verga M, Santus P, et al. Close correlation between anxiety, depression, and asthma control. Resp Med 2010;104:22–8.
33. Smith A, Krishnan JA, Bilderback A, et al. Depressive symptoms and adherence to asthma therapy after hospital discharge. Chest 2006;130:1034–8.
34. Krauskopf KA, Sofianou A, Goel MS, et al. Depressive symptoms, low adherence, and poor asthma outcomes in the elderly. J Asthma 2013;50:260–6.
35. Current depression among adults---United States, 2006 and 2008. MMWR 2010;59:1229–35.
36. McLean CP, Asnaani A, Litz BT, Hofmann SG. Gender differences in anxiety disorders: prevalence, course of illness, comorbidity and burden of illness. J Psychiatr Res 2011;45:1027–35.
37. Sundberg R, Toren K, Franklin KA, et al. Asthma in men and women: treatment adherence, anxiety, and quality of sleep. Respir Med 2010;104:337–44.
38. Ford ES. The epidemiology of obesity and asthma. J Allergy Clin Immunol 2005;115:897–909.
39. Essalhi M, Gillaizeau F, Chevallier JM, et al. Cross-sectional assessment of the roles of comorbidities in resting and activity-related dyspnea in severely obese women. J Asthma 2013;50:565–72.
40. Grammer LC, Weiss KB, Pedicano JB, et al. Obesity and asthma morbidity in a community-based adult cohort in a large urban area: the Chicago Initiative to Raise Asthma Health Equity (CHIRAH). J Asthma 2010;47:491–5.
41. Clerisme-Beaty EM, Karam S, Rand C, et al. Does higher body mass index contribute to worse asthma control in an urban population? J Allergy Clin Immunol 2009;124:207–12.
42. Boudreau M, Bacon SL, Ouellet K, et al. Mediator effect of depressive symptoms on the association between BMI and asthma control in adults. Chest 2014;146:348–54.
43. Camargo CA Jr, Sutherland ER, Bailey W, et al. Effect of increased body mass index on asthma risk, impairment and response to asthma controller therapy in African Americans. Curr Med Res Opin 2010;26:1629–35.
44. Valerio MA, Gong ZM, Wang S, et al. Overweight women and management of asthma. Women Health Issues 2009;19:
300–5.
45. Fryar CD, Carroll MD, Ogden CL. Prevalence of overweight, obesity, and extreme obesity among adults: United States, trends 1960–1962 through 2009–2010. Hyattsville, MD: National Center for Health Statistics; 2012.
46. Manion AB. Asthma and obesity: the dose effect. Nurs Clin North Am 2013;48:151–8.
47. Tam A, Morrish D, Wadsworth S, et al. The role of female hormones on lung function in chronic lung diseases. BMC Women Health 2011;11:24.
48. Ticconi C, Pietropolli A, Piccione E. Estrogen replacement therapy and asthma. Pulm Pharmacol Ther 2013;26:617–23.
49. Bonds RS, Midoro-Horiuti T. Estrogen effects in allergy and asthma. Curr Opin Allergy Clin Immunol 2013;13:92–9.
50. Pereira-Vega A, Sanchez Ramos JL, Vazquez Oliva R, et al. Premenstrual asthma and female sex hormones. J Investig Allergol Clin Immunol 2012;22:437–9.
51. Barr RG, Wentowski CC, Grodstein F, et al. Prospective study of postmenopausal hormone use and newly diagnosed asthma and chronic obstructive pulmonary disease. Arch Intern Med 2004;164:379–86.
52. Romieu I, Fabre A, Fournier A, et al. Postmenopausal hormone therapy and asthma onset in the E3N cohort. Thorax 2010;65:292–7.
53. Bonnelykke K, Raaschou-Nielsen O, Tjonneland A, et al. Postmenopausal hormone therapy and asthma-related hospital admission. J Allergy Clin Immunol 2015.
54. Barr RG, Somers SC, Speizer FE, Camargo CA, Jr. Patient factors and medication guideline adherence among older women with asthma. Arch Intern Med 2002;162:1761–8.
55. Baptist AP, Hamad A, Patel MR. Special challenges in treatment and self-management of older women with asthma. Ann Allergy Asthma Immunol 2014;113:125–30.
56. Baptist AP, Deol BB, Reddy RC, et al. Age-specific factors influencing asthma management by older adults. Qual Health Res 2010;20:117–24.
57. Valerio MA, Parker EA, Couper MP, et al. Demographic and clinical characteristics predictive of asthma diary use among women. J Asthma 2008;45:357–61.
58. Ostrom NK. Women with asthma: a review of potential variables and preferred medical management. Ann Allergy Asthma Immunol 2006;96:655–65.
59. Clark NM, Valerio MA, Gong ZM. Self-regulation and women with asthma. Curr Opin Allergy Clin Immunol 2008;8:222.
60. Clark NM, Gong ZM, Wang SJ, et al. From the female perspective: Long-term effects on quality of life of a program for women with asthma. Gend Med 2010;7:125–36.
61. Janevic MR, Sanders GM, Thomas LJ, et al. Study protocol for Women of Color and Asthma Control: a randomized controlled trial of an asthma-management intervention for African American women. BMC Public Health 2012;12:76.
62. Rand CS, Apter AJ. Mind the widening gap: have improvements in asthma care increased asthma disparities? J Allergy Clin Immunol 2008;2:319–21.
63. Moorman JE, Mannino DM. Increasing U.S. asthma mortality rates: who is really dying? J Asthma 2001;38:65–71.
64. Bidwell AJ, Yazel B, Davin D, et al. Yoga training improves quality of life in women with asthma. J Altern Complement Med 2012;18:749–55.
65. Jones PW. St. George’s Respiratory Questionnaire: MCID. COPD 2005;2:75–9.
66. Ding D, Stamatakis E. Yoga practice in England 1997-2008: prevalence, temporal trends, and correlates of participation. BMC Res Notes 2014;7:172.
67. Birdee GS, Legedza AT, Saper RB, et al. Characteristics of yoga users: results of a national survey. J Gen Intern Med 2008;23:1653–8.
68. Wolever RQ, Simmons LA, Sforzo GA, et al. A systematic review of the literature on health and wellness coaching: defining a key behavioral intervention in healthcare. Glob Adv Health Med 2013;2:38–57.
69. Free C, Phillips G, Galli L, et al. The effectiveness of mobile-health technology-based health behaviour change or disease management interventions for health care consumers: a systematic review. PLoS Med 2013;10:e1001362.
70. Marcano Belisario JS, Huckvale K, Greenfield G, et al. Smartphone and tablet self management apps for asthma. Cochrane Database Syst Rev 2013;11:CD010013.
71. Burke W, Fesinmeyer M, Reed K, et al. Family history as a predictor of asthma risk. Am J Prev Med 2003;24:160–9.
From the Department of Health Behavior and Health Education, School of Public Health (Dr. Janevic) and the Medical School (Dr. Sanders), University of Michigan Ann Arbor, MI.
Abstract
- Objective: Asthma prevalence, morbidity, and mortality are all greater among adult women compared to men. Appropriate asthma self-management can improve asthma control. We reviewed published literature about sex- and gender-related factors that influence asthma self-management among women, as well as evidence-based interventions to promote effective asthma self-management in this population.
- Design: Based on evidence from the published literature, factors influencing women’s asthma self-management were categorized as follows: social roles and socioeconomic status, comorbidities, obesity, hormonal factors, and aging-related changes.
- Results: A number of factors were identified that affect women’s asthma self-management. These include: exposure to asthma triggers associated with gender roles, such as cleaning products; financial barriers to asthma management; comorbidities that divert attention or otherwise interfere with asthma management; a link between obesity and poor asthma outcomes; the effects of hormonal shifts associated with menstrual cycles and menopause on asthma control; and aging-associated barriers to effective self-management such as functional limitations and caregiving. Certain groups, such as African-American women, are at higher risk for poor asthma outcomes linked to many of the above factors. At least 1 health coaching intervention designed for women with asthma has been shown in a randomized trial to reduce symptoms and health care use.
- Conclusion: Future research on women and asthma self-management should include a focus on the relationship between hormonal changes and asthma symptoms. Interventions are also needed that address the separate and interacting effects of risk factors for poor asthma control that tend to cluster in women, such as obesity, depression, and gastroesophageal reflux disease.
In childhood, asthma is more prevalent in boys than in girls. In adolescence and adulthood, however, asthma becomes a predominantly female disease, with hormonal factors likely playing a role in this shift [1,2]. Fu et al [3] reviewed daily asthma symptom diaries of 418 children. From age 5 to 7, boys had more severe symptoms, but by age 10 girls’ symptoms were becoming more severe. By age 14, the girls’ symptoms continued increasing while the boys’ symptoms began to decline. A meta-analysis by Lieberoth et al [4] found a 37% increased risk of post-menarchal asthma in girls with onset of menarche < 12 years. Together, these studies implicate female sex hormones in both the increased incidence and severity of asthma after puberty. In 2012, nearly 10% of adult women reported current asthma, compared to only 6% of men [5]. Among adults with asthma, women have a 30% higher mortality rate than men [6]. Disparities that disadvantage women are also evident across a range of other asthma-related outcomes, including disease severity, rescue inhaler use, activity limitations, asthma-related quality of life, and health care utilization [7–12].
Factors Influencing Asthma Self-Management in Women
Social Roles and Socioeconomic Status
Traditional gender roles involve various responsibilities, such as household cleaning, cooking, and care of young children, that are associated with exposures to precipitants of asthma symptoms [17]. Gender norms also promote the use of personal care products, like fragrances and hair sprays, which are potential asthma triggers [17]. Recent observational studies in Europe have examined the link between women’s use of cleaning products and asthma. Bédard and colleagues [18] found an association between weekly use of cleaning sprays at home and asthma among women, and Dumas and colleagues [19] found that workplace exposure to cleaning products among women with asthma was related to increased symptoms and severity of asthma. These researchers conclude that “while domestic exposure is much more frequent in the general population, exposure levels are probably higher at the workplace” and therefore both contribute to asthma disease burden [19]. Although little-discussed in the literature, sexual activity is another common trigger of asthma symptoms in women. Clark et al [15] found that more than one-third of women taking part in a randomized controlled trial (RCT) of an asthma self-management intervention reported being bothered by symptoms of asthma during sexual activity. This topic was rarely discussed, however, by their health care providers [20].
Socioeconomic factors also play a significant role in asthma management. There is a well-recognized and persistent gender gap in income in the U.S. population such that women who work full-time only earn three-quarters of what their male counterparts earn [21]. Challenges related to low socioeconomic status (SES) may contribute to poor medication adherence among asthma patients [22]. Although a comprehensive review of the impact of SES-related factors on asthma prevalence, severity, and disease-management behaviors is beyond the scope of this article, recent research demonstrates the impact of financial stress on women’s asthma self-management. Patel et al (2014) studied health-related financial burden among African-American women with asthma [23]. Despite the fact that the majority of women in this qualitative study had health insurance, they felt greatly burdened by out-of-pocket expenses such as high co-pays for medications or ambulance use, lost wages due to sick time, and gaps in insurance coverage. These financial concerns—and related issues such as time spent navigating health care insurance and cycling through private and public insurance programs—were described as a significant source of ongoing stress by this group of vulnerable asthma patients [23]. Focus group participants reported several strategies for dealing with asthma-related financial challenges, including stockpiling medications when feasible (eg, when covered by current insurance plan) for future use by the patient or a family member, seeking out and using community assistance programs, and foregoing medications altogether during periods when they could not afford them [23].
Comorbidities
The 2010 publication of Multiple chronic conditions: a strategic framework by the US Department of Health and Human Services [24] brought the attention of the medical and research communities to the scope and significance of multimorbidity in the US population, including the challenges that individuals face in managing multiple chronic health conditions. Although the prevalence of specific comorbidities with asthma differs by age, some that are most commonly associated with asthma and that may complicate asthma control are obstructive sleep apnea, gastroesophageal reflux disease (GERD), rhinitis, and sinusitis [25,26]. Among women with asthma, multimorbidity appears to be the rule, not the exception. Using nationally-representative data from the US National Health and Nutrition Examination Survey (NHANES), Patel et al [27] found that more than half of adults with asthma reported also being diagnosed with at least 1 additional major chronic condition. A recent study found that asthma/arthritis and asthma/hypertension were the second and third most prevalent disease dyads among all US women aged 18–44 years [28]. Studies have found that comorbidities among asthma patients are associated with worse asthma outcomes, including increased symptoms, activity limitations and sleep disturbance due to asthma [27], and ED use for asthma [15,27].
Qualitative research yields insight into the patient perspective of multimorbidity, that is, how women with asthma and coexisting chronic diseases perceive the effect of their health conditions on their ability to engage in self-management. Janevic and colleagues [29] conducted face-to-face interviews with African-American women participating in a randomized controlled trial of a culturally and gender-tailored asthma-management intervention to learn about their experiences managing asthma and concurrent health conditions. Interviewees had an average of 5.7 chronic conditions in addition to asthma. Women reported that managing their asthma often “took a backseat” to other chronic conditions. Participants also discussed reduced motivation or capacity for asthma self-management due to depression, chronic pain, mobility limitations or combinations of these, and reduced adherence to asthma medications due to the psychological and logistical burdens of polypharmacy.
Depression and anxiety are common comorbidities that are associated with worse asthma outcomes [26,30–32] and reduced asthma medication adherence [33,34]. In general population studies as well as among asthma patients, women are more likely than men to report depression and anxiety [30,35–37]. Screening for and treating depression and anxiety are indicated in women with asthma and may lead to improved adherence and outcomes [30].
Obesity
Adults with asthma are at increased risk of obesity [38]. Obesity is a possible risk factor for development of asthma in women [2] and for resting dyspnea in women with asthma [39]. It is associated with poor asthma-related QOL and use of emergency/urgent services [40]. Evidence is mixed regarding the link between BMI and asthma control [41–43], but the following studies suggest that women who are overweight/obese face unique asthma management challenges. Valerio and colleagues found that in a sample of 808 women enrolled in a randomized trial of an asthma-education intervention, nearly 7 out of 10 were overweight (BMI ≥ 25) or obese (BMI ≥ 30), and nearly a quarter were “extremely obese” (BMI > 35) [44]. This subgroup of women was more likely to have persistent asthma, comorbid GERD and urinary incontinence, to be non-white, and to have lower levels of education and income. Being overweight was also associated with greater use of health care services and having greater psychosocial challenges (ie, a higher need for asthma-related social support and lower asthma-related quality of life). These authors suggest the need to design communications for overweight women with asthma that recognize “the specific cultural and social influences on their asthma management behaviors” [44] with a focus on psychosocial needs, while incorporating existing social support networks. In the previously discussed study by Janevic and colleagues [29] the average BMI of the interview participants was 36.0, and a number of respondents identified weight loss as the self-care behavior that they thought would benefit them the most across multimorbid conditions. Therefore, health care providers should provide appropriate counseling and/or referrals to help women with asthma achieve weight loss goals. Given trends over time showing increasing prevalence of asthma and obesity [45,46], interest is growing in the asthma research community about the interaction of the 2 conditions.
Hormonal Factors
Hormones exert a significant effect on asthma in women, and must be considered in clinical and self-management of the disease. Hormone levels fluctuate during the menstrual cycle, with a surge of estradiol (a type of estrogen) at the time of ovulation around day 14, accompanied by low levels of progesterone. During the luteal phase (day 14–28 of the menstrual cycle), estrogens decrease while progesterone levels increase then decrease again before onset of menstruation [47]. During pregnancy, levels of estrogens and progesterone increase and are the highest during the third trimester, when women usually experience good asthma control. Then, during menopause both estradiol and progesterone levels drop to lower levels than those during any phase of menstruation. In addition to the role in the menstrual cycle, there are estrogen receptors (ER-α and ER-β) which are expressed in the human lung and have a role in both airway responsiveness (relaxation) and inflammation [48]. Estrogen also acts directly on cells of the immune system to stimulate airway inflammation, particularly when allergens are present [48]. Further discussion about these contrasting actions of estrogen can be found in a recent review [48].
During the reproductive years, 30% to 40% of women with asthma report perimenstrual symptoms. Forced expiratory volume in 1 second and forced vital capacity are lowest in the periovulatory period, when estrogen levels are high. In contrast, during the luteal phase, studies have shown increased airway hyperreactivity, especially in the premenstrual period when estrogen levels are low [49]. However, when asthma patients with and without perimenstrual symptoms are evaluated, there is no significant difference in their perimenstrual estrogen and progesterone levels [50]. Clark et al [15] found women participating in a self-management intervention, which included checking daily peak flow rates, reported significantly more menstrual and perimenstrual asthma symptomatology than the control group. This suggests that some women with asthma have may have, but do not recognize, perimenstrual symptoms. Further elucidation of the incidence of symptomatology related to the menstrual cycle as well as the role of hormonal variation is an area for future research efforts.
At the time of menopause and continuing to postmenopause, levels of both estrogen and progesterone drop to below those during the reproductive years, leading to uncomfortable symptoms in many women. Hormone replacement therapy (HRT) with either estrogen alone or estrogen-progesterone combination effectively improves these, but there is concern for potential effects on asthma prevalence and severity. Two recent large studies support this concern. Postmenopausal women followed for 10 years in the Nurses’ Health Study with a history of HRT had an increased risk of new onset asthma when compared to postmenopausal women with no history of estrogen use (RR = 2.30, 95% CI 1.69–3.14) [51]. This persisted in estrogen-progesterone users. A large French cohort confirmed the increased onset of new asthma in users of estrogen-alone replacement therapy (HR = 1.54, 95% CI 1.13–2.09). However, this effect decreased with time if estrogen had been discontinued, and they did not find a similar increase in users of estrogen-progesterone combination therapy [52]. In contrast, Bonelykke et al [53] found an association between ever using HRT and first-ever hospital admission for asthma, in postmenopausal women (HR 1.46, CI 1.21–1.76), and this risk increased with duration of HRT use. It is clear that physicians need to be aware of these potential respiratory complications, inform their patients, and consider new-onset asthma when women on HRT bring complaints of dyspnea, cough, or wheeze. Future randomized trials are needed to clarify the relationship between HRT and asthma, and to test ways to optimize asthma self-management in women experiencing these transitions.
Older Women and Asthma
Although the bulk of research on asthma focuses on children and young adults, asthma in the elderly is receiving increased attention [54], in part because this demographic group has the highest asthma mortality rate and the most frequent hospitalizations [6,55]. In a sample of midlife and older women from the Nurses’ Health Study who had been diagnosed with persistent asthma, Barr et al found that adherence to asthma medication guidelines decreased with age [54]. In this study, women with more severe asthma and those with multimorbidity were less adherent than those without comorbidities, as were women who spent more hours caregiving for an ill spouse. The authors concluded that asthma is undertreated among older women.
Baptist et al (2014) describe several challenges to asthma management of older women by clinicians and by the women themselves [55]. For example, elderly women may be at increased risk for adverse effects of inhaled corticosteroids. Certain medications used to treat comorbidities, such as beta-blockers and aspirin, may also exacerbate asthma symptoms. In terms of self-management, older women may have a decreased ability to perceive breathlessness, requiring monitoring with a peak flow meter to detect reductions in airflow. Comorbidities are particularly prevalent in this age group, and asthma symptoms may be confused with symptoms of other conditions, such as heart disease [56]. Baptist and colleagues note factors common among elderly women that pose potential barriers to successful self-management of asthma, including limited income, poverty, depression, and caregiving [55]. They also mention that functional limitations such as those due to arthritis, visual difficulties, or weakened inspiratory strength can make inhaler use more difficult. It should also be noted that some behaviors may promote asthma self-management in this group; for example, Valerio and colleagues [57] found that women over age 50 were more likely than younger women to keep a daily asthma diary when asked to do so as part of a self-management intervention [57].
Evidence-Based Asthma Self-Management Interventions for Women
For women to achieve optimal asthma control, the unique factors as described above that influence their symptoms and management need to be addressed [58]. Several examples can be found in the literature of behavioral interventions that focus on the particular self-management challenges faced by women. Clark and colleagues reported the results of an RCT of the Women Breathe Free (WBF) program [15,16]. This intervention consisted of asthma self-management education delivered over 5 telephone sessions by a health educator. WBF content was based on self-regulation theory, which involves observing one’s behavior and making judgments on the observations, testing strategies to improve asthma management, and reacting to positive results of these strategies with enhanced self-efficacy and outcome expectations, ie, the belief that a given strategy will produce the desired results [59]. In WBF, participants used a problem-solving process based on this framework to carry out recommendations in their physician’s therapeutic plan. WBF also incorporated special attention to sex- and gender-based factors in asthma management.
Over a 12-month period, women who participated in the intervention relative to controls experienced significant reductions in nighttime symptoms, days of missed work/school, emergency department visits, and both scheduled and urgent office visits. Intervention group women also reported decreased asthma symptoms during sexual activity, improved asthma-related quality of life, and increased confidence to manage asthma. At long-term follow-up (2 years from baseline), persistent positive effects of the intervention were found on outpatient visits for asthma symptom level during sexual activity, days of missed work/school, asthma-related quality of life, and confidence to manage asthma [60].
In a follow-up study, Clark and colleagues [61] developed the “Women of Color and Asthma Control” (WCAC) program. WCAC incorporates the theoretical orientation and many of the program elements of Women Breathe Free, but has been adapted to be responsive to the needs and preferences of African-American women. Poverty and race are associated with greater asthma morbidity and mortality [5,62,63]. African-American women and women of low socioeconomic status are particularly vulnerable to asthma and associated morbidity and mortality, making this an important group for intervention. Culturally responsive components in the WCAC intervention include use of culturally relevant activities and beliefs when discussing triggers and barriers to asthma management, as well as culturally appropriate visuals. This ongoing trial will test WCAC’s effect on ED visits, hospitalizations, and urgent care; asthma symptoms; and asthma-related quality of life at 1 year and 18 months from baseline.
In a small RCT among women with asthma, Bidwell and colleagues tested a program consisting of 10 weeks of yoga instruction (including breathing practices, poses, and meditation/relaxation skills) in a group setting followed by 10 weeks of home practice [64]. Women in the intervention group reported improved quality of life, as measured by the St. George’s Respiratory Quality of Life questionnaire [65], and participants also had decreased parasympathetic modulation in response to an isometric forearm exercise. They conclude that yoga is a promising modality for improving quality of life among asthma patients and that these changes may be linked to better autonomic modulation. Although this program was not designed specifically for women, yoga is practiced significantly more frequently among women compared to men [66,67], and thus has the potential to be widely used in this group.
Based on our experience conducting self-management research among women with asthma, and unpublished process data from these studies, we observe that the following elements appear to contribute to high participant engagement in these programs and successful outcomes. First, in participant feedback questionnaires from the Women Breathe Free and Women of Color and Asthma Control studies, women have singled out the importance of their relationship with their assigned telephone asthma educator as motivating them to make positive changes in their asthma self-management behaviors. The popularity of health and wellness coaching, including for chronic disease management, is rapidly growing [68]. This is a patient-centered approach that guides patients in setting their own goals for disease management and devising their own strategies for achieving them [68]. Strong interpersonal relationships are thought to enhance the coaching process and this may be especially important for women [68]. Participants have also indicated that they are able to apply the goal-setting and problem-solving skills they have learned as part of the intervention to management of other health or psychosocial issues in their lives; therefore this component seems especially critical for women with asthma who are typically managing multiple health issues as well as those of others. Finally, maximizing the flexibility of interventions is important for working-age women who typically are engaged in part- or full-time employment and also have significant responsibilities caring for others. This flexibility can come in the form of telephone-based or “mHealth” interventions that use mobile technologies such as text messaging [69], as well as internet-based or smartphone/tablet “apps” that can be completed at a pace and schedule that is convenient for the participant [70]. Such interventions could be easily tailored to address sex- and gender-specific issues in asthma management.
Future Research and Practice Directions
This review points to several promising directions for research and practice in the area of supporting women’s asthma self-management. The first is a systematic exploration of the added value of gender-tailoring asthma self-management support interventions to determine which subgroups of women benefit from which type of sex- and gender-specific information, and in which form. More research is needed on the relationship between hormone levels and changes and asthma symptoms, and how this affects women’s self-management. This includes recognition of new or worsening asthma with the use of hormone replacement therapy in menopausal and post-menopausal women, a group that is rapidly increasing in number in the US population. Another direction for research is a family-systems approach to asthma education and supporting asthma management. Asthma in one or more first-degree relatives has been shown across diverse populations to be a risk factor for asthma [71]. Women with asthma are therefore more likely to have children with asthma, and vice-versa; however, no prior research was identified that addresses asthma self-management in mother/child dyads. For example, it is possible that teaching women to better manage their own asthma could have “trickle down” effects to how they help a child manage asthma. Last, as the above discussion of factors affecting women’s asthma makes clear, many risk factors for poor asthma management and control in women cluster together, such as obesity, depression, and GERD. Interventions that attempt to address the separate and interacting effects of these factors and comorbidities, may yield better outcomes among the most vulnerable asthma patients.
Corresponding author: Mary R. Janevic, PhD, Center for Managing Chronic Disease, University of Michigan School of Public Health, 1425 Washington Heights, Ann Arbor, MI 48109, mjanevic@umich.edu.
Financial disclosures: None.
From the Department of Health Behavior and Health Education, School of Public Health (Dr. Janevic) and the Medical School (Dr. Sanders), University of Michigan Ann Arbor, MI.
Abstract
- Objective: Asthma prevalence, morbidity, and mortality are all greater among adult women compared to men. Appropriate asthma self-management can improve asthma control. We reviewed published literature about sex- and gender-related factors that influence asthma self-management among women, as well as evidence-based interventions to promote effective asthma self-management in this population.
- Design: Based on evidence from the published literature, factors influencing women’s asthma self-management were categorized as follows: social roles and socioeconomic status, comorbidities, obesity, hormonal factors, and aging-related changes.
- Results: A number of factors were identified that affect women’s asthma self-management. These include: exposure to asthma triggers associated with gender roles, such as cleaning products; financial barriers to asthma management; comorbidities that divert attention or otherwise interfere with asthma management; a link between obesity and poor asthma outcomes; the effects of hormonal shifts associated with menstrual cycles and menopause on asthma control; and aging-associated barriers to effective self-management such as functional limitations and caregiving. Certain groups, such as African-American women, are at higher risk for poor asthma outcomes linked to many of the above factors. At least 1 health coaching intervention designed for women with asthma has been shown in a randomized trial to reduce symptoms and health care use.
- Conclusion: Future research on women and asthma self-management should include a focus on the relationship between hormonal changes and asthma symptoms. Interventions are also needed that address the separate and interacting effects of risk factors for poor asthma control that tend to cluster in women, such as obesity, depression, and gastroesophageal reflux disease.
In childhood, asthma is more prevalent in boys than in girls. In adolescence and adulthood, however, asthma becomes a predominantly female disease, with hormonal factors likely playing a role in this shift [1,2]. Fu et al [3] reviewed daily asthma symptom diaries of 418 children. From age 5 to 7, boys had more severe symptoms, but by age 10 girls’ symptoms were becoming more severe. By age 14, the girls’ symptoms continued increasing while the boys’ symptoms began to decline. A meta-analysis by Lieberoth et al [4] found a 37% increased risk of post-menarchal asthma in girls with onset of menarche < 12 years. Together, these studies implicate female sex hormones in both the increased incidence and severity of asthma after puberty. In 2012, nearly 10% of adult women reported current asthma, compared to only 6% of men [5]. Among adults with asthma, women have a 30% higher mortality rate than men [6]. Disparities that disadvantage women are also evident across a range of other asthma-related outcomes, including disease severity, rescue inhaler use, activity limitations, asthma-related quality of life, and health care utilization [7–12].
Factors Influencing Asthma Self-Management in Women
Social Roles and Socioeconomic Status
Traditional gender roles involve various responsibilities, such as household cleaning, cooking, and care of young children, that are associated with exposures to precipitants of asthma symptoms [17]. Gender norms also promote the use of personal care products, like fragrances and hair sprays, which are potential asthma triggers [17]. Recent observational studies in Europe have examined the link between women’s use of cleaning products and asthma. Bédard and colleagues [18] found an association between weekly use of cleaning sprays at home and asthma among women, and Dumas and colleagues [19] found that workplace exposure to cleaning products among women with asthma was related to increased symptoms and severity of asthma. These researchers conclude that “while domestic exposure is much more frequent in the general population, exposure levels are probably higher at the workplace” and therefore both contribute to asthma disease burden [19]. Although little-discussed in the literature, sexual activity is another common trigger of asthma symptoms in women. Clark et al [15] found that more than one-third of women taking part in a randomized controlled trial (RCT) of an asthma self-management intervention reported being bothered by symptoms of asthma during sexual activity. This topic was rarely discussed, however, by their health care providers [20].
Socioeconomic factors also play a significant role in asthma management. There is a well-recognized and persistent gender gap in income in the U.S. population such that women who work full-time only earn three-quarters of what their male counterparts earn [21]. Challenges related to low socioeconomic status (SES) may contribute to poor medication adherence among asthma patients [22]. Although a comprehensive review of the impact of SES-related factors on asthma prevalence, severity, and disease-management behaviors is beyond the scope of this article, recent research demonstrates the impact of financial stress on women’s asthma self-management. Patel et al (2014) studied health-related financial burden among African-American women with asthma [23]. Despite the fact that the majority of women in this qualitative study had health insurance, they felt greatly burdened by out-of-pocket expenses such as high co-pays for medications or ambulance use, lost wages due to sick time, and gaps in insurance coverage. These financial concerns—and related issues such as time spent navigating health care insurance and cycling through private and public insurance programs—were described as a significant source of ongoing stress by this group of vulnerable asthma patients [23]. Focus group participants reported several strategies for dealing with asthma-related financial challenges, including stockpiling medications when feasible (eg, when covered by current insurance plan) for future use by the patient or a family member, seeking out and using community assistance programs, and foregoing medications altogether during periods when they could not afford them [23].
Comorbidities
The 2010 publication of Multiple chronic conditions: a strategic framework by the US Department of Health and Human Services [24] brought the attention of the medical and research communities to the scope and significance of multimorbidity in the US population, including the challenges that individuals face in managing multiple chronic health conditions. Although the prevalence of specific comorbidities with asthma differs by age, some that are most commonly associated with asthma and that may complicate asthma control are obstructive sleep apnea, gastroesophageal reflux disease (GERD), rhinitis, and sinusitis [25,26]. Among women with asthma, multimorbidity appears to be the rule, not the exception. Using nationally-representative data from the US National Health and Nutrition Examination Survey (NHANES), Patel et al [27] found that more than half of adults with asthma reported also being diagnosed with at least 1 additional major chronic condition. A recent study found that asthma/arthritis and asthma/hypertension were the second and third most prevalent disease dyads among all US women aged 18–44 years [28]. Studies have found that comorbidities among asthma patients are associated with worse asthma outcomes, including increased symptoms, activity limitations and sleep disturbance due to asthma [27], and ED use for asthma [15,27].
Qualitative research yields insight into the patient perspective of multimorbidity, that is, how women with asthma and coexisting chronic diseases perceive the effect of their health conditions on their ability to engage in self-management. Janevic and colleagues [29] conducted face-to-face interviews with African-American women participating in a randomized controlled trial of a culturally and gender-tailored asthma-management intervention to learn about their experiences managing asthma and concurrent health conditions. Interviewees had an average of 5.7 chronic conditions in addition to asthma. Women reported that managing their asthma often “took a backseat” to other chronic conditions. Participants also discussed reduced motivation or capacity for asthma self-management due to depression, chronic pain, mobility limitations or combinations of these, and reduced adherence to asthma medications due to the psychological and logistical burdens of polypharmacy.
Depression and anxiety are common comorbidities that are associated with worse asthma outcomes [26,30–32] and reduced asthma medication adherence [33,34]. In general population studies as well as among asthma patients, women are more likely than men to report depression and anxiety [30,35–37]. Screening for and treating depression and anxiety are indicated in women with asthma and may lead to improved adherence and outcomes [30].
Obesity
Adults with asthma are at increased risk of obesity [38]. Obesity is a possible risk factor for development of asthma in women [2] and for resting dyspnea in women with asthma [39]. It is associated with poor asthma-related QOL and use of emergency/urgent services [40]. Evidence is mixed regarding the link between BMI and asthma control [41–43], but the following studies suggest that women who are overweight/obese face unique asthma management challenges. Valerio and colleagues found that in a sample of 808 women enrolled in a randomized trial of an asthma-education intervention, nearly 7 out of 10 were overweight (BMI ≥ 25) or obese (BMI ≥ 30), and nearly a quarter were “extremely obese” (BMI > 35) [44]. This subgroup of women was more likely to have persistent asthma, comorbid GERD and urinary incontinence, to be non-white, and to have lower levels of education and income. Being overweight was also associated with greater use of health care services and having greater psychosocial challenges (ie, a higher need for asthma-related social support and lower asthma-related quality of life). These authors suggest the need to design communications for overweight women with asthma that recognize “the specific cultural and social influences on their asthma management behaviors” [44] with a focus on psychosocial needs, while incorporating existing social support networks. In the previously discussed study by Janevic and colleagues [29] the average BMI of the interview participants was 36.0, and a number of respondents identified weight loss as the self-care behavior that they thought would benefit them the most across multimorbid conditions. Therefore, health care providers should provide appropriate counseling and/or referrals to help women with asthma achieve weight loss goals. Given trends over time showing increasing prevalence of asthma and obesity [45,46], interest is growing in the asthma research community about the interaction of the 2 conditions.
Hormonal Factors
Hormones exert a significant effect on asthma in women, and must be considered in clinical and self-management of the disease. Hormone levels fluctuate during the menstrual cycle, with a surge of estradiol (a type of estrogen) at the time of ovulation around day 14, accompanied by low levels of progesterone. During the luteal phase (day 14–28 of the menstrual cycle), estrogens decrease while progesterone levels increase then decrease again before onset of menstruation [47]. During pregnancy, levels of estrogens and progesterone increase and are the highest during the third trimester, when women usually experience good asthma control. Then, during menopause both estradiol and progesterone levels drop to lower levels than those during any phase of menstruation. In addition to the role in the menstrual cycle, there are estrogen receptors (ER-α and ER-β) which are expressed in the human lung and have a role in both airway responsiveness (relaxation) and inflammation [48]. Estrogen also acts directly on cells of the immune system to stimulate airway inflammation, particularly when allergens are present [48]. Further discussion about these contrasting actions of estrogen can be found in a recent review [48].
During the reproductive years, 30% to 40% of women with asthma report perimenstrual symptoms. Forced expiratory volume in 1 second and forced vital capacity are lowest in the periovulatory period, when estrogen levels are high. In contrast, during the luteal phase, studies have shown increased airway hyperreactivity, especially in the premenstrual period when estrogen levels are low [49]. However, when asthma patients with and without perimenstrual symptoms are evaluated, there is no significant difference in their perimenstrual estrogen and progesterone levels [50]. Clark et al [15] found women participating in a self-management intervention, which included checking daily peak flow rates, reported significantly more menstrual and perimenstrual asthma symptomatology than the control group. This suggests that some women with asthma have may have, but do not recognize, perimenstrual symptoms. Further elucidation of the incidence of symptomatology related to the menstrual cycle as well as the role of hormonal variation is an area for future research efforts.
At the time of menopause and continuing to postmenopause, levels of both estrogen and progesterone drop to below those during the reproductive years, leading to uncomfortable symptoms in many women. Hormone replacement therapy (HRT) with either estrogen alone or estrogen-progesterone combination effectively improves these, but there is concern for potential effects on asthma prevalence and severity. Two recent large studies support this concern. Postmenopausal women followed for 10 years in the Nurses’ Health Study with a history of HRT had an increased risk of new onset asthma when compared to postmenopausal women with no history of estrogen use (RR = 2.30, 95% CI 1.69–3.14) [51]. This persisted in estrogen-progesterone users. A large French cohort confirmed the increased onset of new asthma in users of estrogen-alone replacement therapy (HR = 1.54, 95% CI 1.13–2.09). However, this effect decreased with time if estrogen had been discontinued, and they did not find a similar increase in users of estrogen-progesterone combination therapy [52]. In contrast, Bonelykke et al [53] found an association between ever using HRT and first-ever hospital admission for asthma, in postmenopausal women (HR 1.46, CI 1.21–1.76), and this risk increased with duration of HRT use. It is clear that physicians need to be aware of these potential respiratory complications, inform their patients, and consider new-onset asthma when women on HRT bring complaints of dyspnea, cough, or wheeze. Future randomized trials are needed to clarify the relationship between HRT and asthma, and to test ways to optimize asthma self-management in women experiencing these transitions.
Older Women and Asthma
Although the bulk of research on asthma focuses on children and young adults, asthma in the elderly is receiving increased attention [54], in part because this demographic group has the highest asthma mortality rate and the most frequent hospitalizations [6,55]. In a sample of midlife and older women from the Nurses’ Health Study who had been diagnosed with persistent asthma, Barr et al found that adherence to asthma medication guidelines decreased with age [54]. In this study, women with more severe asthma and those with multimorbidity were less adherent than those without comorbidities, as were women who spent more hours caregiving for an ill spouse. The authors concluded that asthma is undertreated among older women.
Baptist et al (2014) describe several challenges to asthma management of older women by clinicians and by the women themselves [55]. For example, elderly women may be at increased risk for adverse effects of inhaled corticosteroids. Certain medications used to treat comorbidities, such as beta-blockers and aspirin, may also exacerbate asthma symptoms. In terms of self-management, older women may have a decreased ability to perceive breathlessness, requiring monitoring with a peak flow meter to detect reductions in airflow. Comorbidities are particularly prevalent in this age group, and asthma symptoms may be confused with symptoms of other conditions, such as heart disease [56]. Baptist and colleagues note factors common among elderly women that pose potential barriers to successful self-management of asthma, including limited income, poverty, depression, and caregiving [55]. They also mention that functional limitations such as those due to arthritis, visual difficulties, or weakened inspiratory strength can make inhaler use more difficult. It should also be noted that some behaviors may promote asthma self-management in this group; for example, Valerio and colleagues [57] found that women over age 50 were more likely than younger women to keep a daily asthma diary when asked to do so as part of a self-management intervention [57].
Evidence-Based Asthma Self-Management Interventions for Women
For women to achieve optimal asthma control, the unique factors as described above that influence their symptoms and management need to be addressed [58]. Several examples can be found in the literature of behavioral interventions that focus on the particular self-management challenges faced by women. Clark and colleagues reported the results of an RCT of the Women Breathe Free (WBF) program [15,16]. This intervention consisted of asthma self-management education delivered over 5 telephone sessions by a health educator. WBF content was based on self-regulation theory, which involves observing one’s behavior and making judgments on the observations, testing strategies to improve asthma management, and reacting to positive results of these strategies with enhanced self-efficacy and outcome expectations, ie, the belief that a given strategy will produce the desired results [59]. In WBF, participants used a problem-solving process based on this framework to carry out recommendations in their physician’s therapeutic plan. WBF also incorporated special attention to sex- and gender-based factors in asthma management.
Over a 12-month period, women who participated in the intervention relative to controls experienced significant reductions in nighttime symptoms, days of missed work/school, emergency department visits, and both scheduled and urgent office visits. Intervention group women also reported decreased asthma symptoms during sexual activity, improved asthma-related quality of life, and increased confidence to manage asthma. At long-term follow-up (2 years from baseline), persistent positive effects of the intervention were found on outpatient visits for asthma symptom level during sexual activity, days of missed work/school, asthma-related quality of life, and confidence to manage asthma [60].
In a follow-up study, Clark and colleagues [61] developed the “Women of Color and Asthma Control” (WCAC) program. WCAC incorporates the theoretical orientation and many of the program elements of Women Breathe Free, but has been adapted to be responsive to the needs and preferences of African-American women. Poverty and race are associated with greater asthma morbidity and mortality [5,62,63]. African-American women and women of low socioeconomic status are particularly vulnerable to asthma and associated morbidity and mortality, making this an important group for intervention. Culturally responsive components in the WCAC intervention include use of culturally relevant activities and beliefs when discussing triggers and barriers to asthma management, as well as culturally appropriate visuals. This ongoing trial will test WCAC’s effect on ED visits, hospitalizations, and urgent care; asthma symptoms; and asthma-related quality of life at 1 year and 18 months from baseline.
In a small RCT among women with asthma, Bidwell and colleagues tested a program consisting of 10 weeks of yoga instruction (including breathing practices, poses, and meditation/relaxation skills) in a group setting followed by 10 weeks of home practice [64]. Women in the intervention group reported improved quality of life, as measured by the St. George’s Respiratory Quality of Life questionnaire [65], and participants also had decreased parasympathetic modulation in response to an isometric forearm exercise. They conclude that yoga is a promising modality for improving quality of life among asthma patients and that these changes may be linked to better autonomic modulation. Although this program was not designed specifically for women, yoga is practiced significantly more frequently among women compared to men [66,67], and thus has the potential to be widely used in this group.
Based on our experience conducting self-management research among women with asthma, and unpublished process data from these studies, we observe that the following elements appear to contribute to high participant engagement in these programs and successful outcomes. First, in participant feedback questionnaires from the Women Breathe Free and Women of Color and Asthma Control studies, women have singled out the importance of their relationship with their assigned telephone asthma educator as motivating them to make positive changes in their asthma self-management behaviors. The popularity of health and wellness coaching, including for chronic disease management, is rapidly growing [68]. This is a patient-centered approach that guides patients in setting their own goals for disease management and devising their own strategies for achieving them [68]. Strong interpersonal relationships are thought to enhance the coaching process and this may be especially important for women [68]. Participants have also indicated that they are able to apply the goal-setting and problem-solving skills they have learned as part of the intervention to management of other health or psychosocial issues in their lives; therefore this component seems especially critical for women with asthma who are typically managing multiple health issues as well as those of others. Finally, maximizing the flexibility of interventions is important for working-age women who typically are engaged in part- or full-time employment and also have significant responsibilities caring for others. This flexibility can come in the form of telephone-based or “mHealth” interventions that use mobile technologies such as text messaging [69], as well as internet-based or smartphone/tablet “apps” that can be completed at a pace and schedule that is convenient for the participant [70]. Such interventions could be easily tailored to address sex- and gender-specific issues in asthma management.
Future Research and Practice Directions
This review points to several promising directions for research and practice in the area of supporting women’s asthma self-management. The first is a systematic exploration of the added value of gender-tailoring asthma self-management support interventions to determine which subgroups of women benefit from which type of sex- and gender-specific information, and in which form. More research is needed on the relationship between hormone levels and changes and asthma symptoms, and how this affects women’s self-management. This includes recognition of new or worsening asthma with the use of hormone replacement therapy in menopausal and post-menopausal women, a group that is rapidly increasing in number in the US population. Another direction for research is a family-systems approach to asthma education and supporting asthma management. Asthma in one or more first-degree relatives has been shown across diverse populations to be a risk factor for asthma [71]. Women with asthma are therefore more likely to have children with asthma, and vice-versa; however, no prior research was identified that addresses asthma self-management in mother/child dyads. For example, it is possible that teaching women to better manage their own asthma could have “trickle down” effects to how they help a child manage asthma. Last, as the above discussion of factors affecting women’s asthma makes clear, many risk factors for poor asthma management and control in women cluster together, such as obesity, depression, and GERD. Interventions that attempt to address the separate and interacting effects of these factors and comorbidities, may yield better outcomes among the most vulnerable asthma patients.
Corresponding author: Mary R. Janevic, PhD, Center for Managing Chronic Disease, University of Michigan School of Public Health, 1425 Washington Heights, Ann Arbor, MI 48109, mjanevic@umich.edu.
Financial disclosures: None.
1. Postma DS. Gender differences in asthma development and progression. Gend Med 2007;4 Suppl B:S133–46.
2. Melgert BN, Ray A, Hylkema MN, et al. Are there reasons why adult asthma is more common in females? Curr Allergy Asthma Rep 2007;7:143–50.
3. Fu L, Freishtat RJ, Gordish-Dressman H, et al. Natural progression of childhood asthma symptoms and strong influence of sex and puberty. Ann Am Thor Soc 2014;11:939–44.
4. Lieberoth S, Gade EJ, Brok J, et al. Age at menarche and risk of asthma: systematic review and meta-analysis. J Asthma 2014;51:559–65.
5. Blackwell D, Lucas J, Clarke T. Summary health statistics for U.S. adults: national health interview survey, 2012. Vital Health Stat 10 2014;(260):1–161.
6. Akinbami LJ, Moorman JE, Bailey C, et al. Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010. NCHS Data Brief 2012 May;(94):1–8.
7. Sinclair AH, Tolsma DD. Gender differences in asthma experience and disease care in a managed care organization. J Asthma 2006;43:363–7.
8. Naleway AL, Vollmer WM, Frazier EA, et al. Gender differences in asthma management and quality of life. J Asthma 2006;43:549–52.
9. Lisspers K, Ställberg B, Janson C, et al. Sex-differences in quality of life and asthma control in Swedish asthma patients. J Asthma 2013;50:1090–5.
10. Ostrom NK. Women with asthma: a review of potential variables and preferred medical management. Ann Allergy Asthma Immunol 2006;96:655–65.
11. Kynyk JA, Mastronarde JG, McCallister JW. Asthma, the sex difference. Curr Opin Pulm Med 2011;17:6–11.
12. Schiller JS, Lucas JW, Ward BW, Peregoy JA. Summary health statistics for U.S. adults: National Health Interview Survey, 2010. Vital Health Stat 10 2012;(252):1–207.
13. Clark NM, Becker MH, Janz NK, et al. Self-management of chronic disease by older adults: A review and directions for research. J Aging Health 1991;3:3–27.
14. Sundberg R, Torén K, Franklin KA, et al. Asthma in men and women: Treatment adherence, anxiety, and quality of sleep. Resp Med 2010;104:337–44.
15. Clark NM, Gong ZM, Wang SJ, et al. A randomized trial of a self-regulation intervention for women with asthma. Chest 2007;132:88–97.
16. Clark NM, Gong ZM, Wang SJ, et al. From the female perspective: Long-term effects on quality of life of a program for women with asthma. Gend Med 2010;7:125–36.
17. Thomas LJ, Janevic MR, Sanders G, Clark NM. Gender-related asthma challenges in a sample of African American women. Poster presented at 2012 Women’s Health Congress.
18. Bedard A, Varraso R, Sanchez M, et al. Cleaning sprays, household help and asthma among elderly women. Respir Med 2014;108:171–80.
19. Dumas O, Siroux V, Luu F, et al. Cleaning and asthma characteristics in women. Am J Ind Med 2014;57:303–11.
20. Clark NM, Valerio MA, Gong ZM. Self-regulation and women with asthma. Curr Opin Allergy Clin Immunol 2008;8:222–7.
21. Lips HM. The gender pay gap: Challenging the rationalizations. Perceived equity, discrimination, and the limits of human capital models. Sex Roles 2013;68:169–85.
22. Apter AJ, Reisine ST, Affleck G, et al. Adherence with twice-daily dosing of inhaled steroids. Socioeconomic and health-belief differences. Am J Respir Crit Care Med 1998;157(6 Pt 1):1810–7.
23. Patel MR, Nelson BW, Id-Deen E, Caldwell CH. Beyond co-pays and out-of-pocket costs: perceptions of health-related financial burden in managing asthma among African American women. J Asthma 2014;51:1083–8.
24. U.S. Department of Health & Human Services. Multiple chronic conditions—a strategic framework: optimum health and quality of life for individuals with multiple chronic conditions. Washington, DC; 2010.
25. Boulet LP, Boulay ME. Asthma-related comorbidities. Expert Rev Respir Med 2011;5:377–93.
26. Sumino K, O’Brian K, Bartle B, et al. Coexisting chronic conditions associated with mortality and morbidity in adult patients with asthma. J Asthma 2014;51:306–14.
27. Patel MR, Janevic MR, Heeringa SG, et al. An examination of adverse asthma outcomes in U.S. Adults with multiple morbidities. Ann Am Thorac Soc 2013;10:426–31.
28. Ward BW, Schiller JS. Prevalence of multiple chronic conditions among US adults: estimates from the National Health Interview Survey, 2010. Prev Chronic Dis 2013;10:E65.
29. Janevic MR, Ellis KR, Sanders GM, et al. Self-management of multiple chronic conditions among African American women with asthma: a qualitative study. J Asthma 2014;51:243–52.
30. Eisner MD, Katz PP, Lactao G, Iribarren C. Impact of depressive symptoms on adult asthma outcomes. Ann Allergy Asthma Immunol 2005;94:566–74.
31. Strine TW, Mokdad AH, Balluz LS, et al. Impact of depression and anxiety on quality of life, health behaviors, and asthma control among adults in the United States with asthma, 2006. J Asthma 2008;45:123–33.
32. Di Marco F, Verga M, Santus P, et al. Close correlation between anxiety, depression, and asthma control. Resp Med 2010;104:22–8.
33. Smith A, Krishnan JA, Bilderback A, et al. Depressive symptoms and adherence to asthma therapy after hospital discharge. Chest 2006;130:1034–8.
34. Krauskopf KA, Sofianou A, Goel MS, et al. Depressive symptoms, low adherence, and poor asthma outcomes in the elderly. J Asthma 2013;50:260–6.
35. Current depression among adults---United States, 2006 and 2008. MMWR 2010;59:1229–35.
36. McLean CP, Asnaani A, Litz BT, Hofmann SG. Gender differences in anxiety disorders: prevalence, course of illness, comorbidity and burden of illness. J Psychiatr Res 2011;45:1027–35.
37. Sundberg R, Toren K, Franklin KA, et al. Asthma in men and women: treatment adherence, anxiety, and quality of sleep. Respir Med 2010;104:337–44.
38. Ford ES. The epidemiology of obesity and asthma. J Allergy Clin Immunol 2005;115:897–909.
39. Essalhi M, Gillaizeau F, Chevallier JM, et al. Cross-sectional assessment of the roles of comorbidities in resting and activity-related dyspnea in severely obese women. J Asthma 2013;50:565–72.
40. Grammer LC, Weiss KB, Pedicano JB, et al. Obesity and asthma morbidity in a community-based adult cohort in a large urban area: the Chicago Initiative to Raise Asthma Health Equity (CHIRAH). J Asthma 2010;47:491–5.
41. Clerisme-Beaty EM, Karam S, Rand C, et al. Does higher body mass index contribute to worse asthma control in an urban population? J Allergy Clin Immunol 2009;124:207–12.
42. Boudreau M, Bacon SL, Ouellet K, et al. Mediator effect of depressive symptoms on the association between BMI and asthma control in adults. Chest 2014;146:348–54.
43. Camargo CA Jr, Sutherland ER, Bailey W, et al. Effect of increased body mass index on asthma risk, impairment and response to asthma controller therapy in African Americans. Curr Med Res Opin 2010;26:1629–35.
44. Valerio MA, Gong ZM, Wang S, et al. Overweight women and management of asthma. Women Health Issues 2009;19:
300–5.
45. Fryar CD, Carroll MD, Ogden CL. Prevalence of overweight, obesity, and extreme obesity among adults: United States, trends 1960–1962 through 2009–2010. Hyattsville, MD: National Center for Health Statistics; 2012.
46. Manion AB. Asthma and obesity: the dose effect. Nurs Clin North Am 2013;48:151–8.
47. Tam A, Morrish D, Wadsworth S, et al. The role of female hormones on lung function in chronic lung diseases. BMC Women Health 2011;11:24.
48. Ticconi C, Pietropolli A, Piccione E. Estrogen replacement therapy and asthma. Pulm Pharmacol Ther 2013;26:617–23.
49. Bonds RS, Midoro-Horiuti T. Estrogen effects in allergy and asthma. Curr Opin Allergy Clin Immunol 2013;13:92–9.
50. Pereira-Vega A, Sanchez Ramos JL, Vazquez Oliva R, et al. Premenstrual asthma and female sex hormones. J Investig Allergol Clin Immunol 2012;22:437–9.
51. Barr RG, Wentowski CC, Grodstein F, et al. Prospective study of postmenopausal hormone use and newly diagnosed asthma and chronic obstructive pulmonary disease. Arch Intern Med 2004;164:379–86.
52. Romieu I, Fabre A, Fournier A, et al. Postmenopausal hormone therapy and asthma onset in the E3N cohort. Thorax 2010;65:292–7.
53. Bonnelykke K, Raaschou-Nielsen O, Tjonneland A, et al. Postmenopausal hormone therapy and asthma-related hospital admission. J Allergy Clin Immunol 2015.
54. Barr RG, Somers SC, Speizer FE, Camargo CA, Jr. Patient factors and medication guideline adherence among older women with asthma. Arch Intern Med 2002;162:1761–8.
55. Baptist AP, Hamad A, Patel MR. Special challenges in treatment and self-management of older women with asthma. Ann Allergy Asthma Immunol 2014;113:125–30.
56. Baptist AP, Deol BB, Reddy RC, et al. Age-specific factors influencing asthma management by older adults. Qual Health Res 2010;20:117–24.
57. Valerio MA, Parker EA, Couper MP, et al. Demographic and clinical characteristics predictive of asthma diary use among women. J Asthma 2008;45:357–61.
58. Ostrom NK. Women with asthma: a review of potential variables and preferred medical management. Ann Allergy Asthma Immunol 2006;96:655–65.
59. Clark NM, Valerio MA, Gong ZM. Self-regulation and women with asthma. Curr Opin Allergy Clin Immunol 2008;8:222.
60. Clark NM, Gong ZM, Wang SJ, et al. From the female perspective: Long-term effects on quality of life of a program for women with asthma. Gend Med 2010;7:125–36.
61. Janevic MR, Sanders GM, Thomas LJ, et al. Study protocol for Women of Color and Asthma Control: a randomized controlled trial of an asthma-management intervention for African American women. BMC Public Health 2012;12:76.
62. Rand CS, Apter AJ. Mind the widening gap: have improvements in asthma care increased asthma disparities? J Allergy Clin Immunol 2008;2:319–21.
63. Moorman JE, Mannino DM. Increasing U.S. asthma mortality rates: who is really dying? J Asthma 2001;38:65–71.
64. Bidwell AJ, Yazel B, Davin D, et al. Yoga training improves quality of life in women with asthma. J Altern Complement Med 2012;18:749–55.
65. Jones PW. St. George’s Respiratory Questionnaire: MCID. COPD 2005;2:75–9.
66. Ding D, Stamatakis E. Yoga practice in England 1997-2008: prevalence, temporal trends, and correlates of participation. BMC Res Notes 2014;7:172.
67. Birdee GS, Legedza AT, Saper RB, et al. Characteristics of yoga users: results of a national survey. J Gen Intern Med 2008;23:1653–8.
68. Wolever RQ, Simmons LA, Sforzo GA, et al. A systematic review of the literature on health and wellness coaching: defining a key behavioral intervention in healthcare. Glob Adv Health Med 2013;2:38–57.
69. Free C, Phillips G, Galli L, et al. The effectiveness of mobile-health technology-based health behaviour change or disease management interventions for health care consumers: a systematic review. PLoS Med 2013;10:e1001362.
70. Marcano Belisario JS, Huckvale K, Greenfield G, et al. Smartphone and tablet self management apps for asthma. Cochrane Database Syst Rev 2013;11:CD010013.
71. Burke W, Fesinmeyer M, Reed K, et al. Family history as a predictor of asthma risk. Am J Prev Med 2003;24:160–9.
1. Postma DS. Gender differences in asthma development and progression. Gend Med 2007;4 Suppl B:S133–46.
2. Melgert BN, Ray A, Hylkema MN, et al. Are there reasons why adult asthma is more common in females? Curr Allergy Asthma Rep 2007;7:143–50.
3. Fu L, Freishtat RJ, Gordish-Dressman H, et al. Natural progression of childhood asthma symptoms and strong influence of sex and puberty. Ann Am Thor Soc 2014;11:939–44.
4. Lieberoth S, Gade EJ, Brok J, et al. Age at menarche and risk of asthma: systematic review and meta-analysis. J Asthma 2014;51:559–65.
5. Blackwell D, Lucas J, Clarke T. Summary health statistics for U.S. adults: national health interview survey, 2012. Vital Health Stat 10 2014;(260):1–161.
6. Akinbami LJ, Moorman JE, Bailey C, et al. Trends in asthma prevalence, health care use, and mortality in the United States, 2001–2010. NCHS Data Brief 2012 May;(94):1–8.
7. Sinclair AH, Tolsma DD. Gender differences in asthma experience and disease care in a managed care organization. J Asthma 2006;43:363–7.
8. Naleway AL, Vollmer WM, Frazier EA, et al. Gender differences in asthma management and quality of life. J Asthma 2006;43:549–52.
9. Lisspers K, Ställberg B, Janson C, et al. Sex-differences in quality of life and asthma control in Swedish asthma patients. J Asthma 2013;50:1090–5.
10. Ostrom NK. Women with asthma: a review of potential variables and preferred medical management. Ann Allergy Asthma Immunol 2006;96:655–65.
11. Kynyk JA, Mastronarde JG, McCallister JW. Asthma, the sex difference. Curr Opin Pulm Med 2011;17:6–11.
12. Schiller JS, Lucas JW, Ward BW, Peregoy JA. Summary health statistics for U.S. adults: National Health Interview Survey, 2010. Vital Health Stat 10 2012;(252):1–207.
13. Clark NM, Becker MH, Janz NK, et al. Self-management of chronic disease by older adults: A review and directions for research. J Aging Health 1991;3:3–27.
14. Sundberg R, Torén K, Franklin KA, et al. Asthma in men and women: Treatment adherence, anxiety, and quality of sleep. Resp Med 2010;104:337–44.
15. Clark NM, Gong ZM, Wang SJ, et al. A randomized trial of a self-regulation intervention for women with asthma. Chest 2007;132:88–97.
16. Clark NM, Gong ZM, Wang SJ, et al. From the female perspective: Long-term effects on quality of life of a program for women with asthma. Gend Med 2010;7:125–36.
17. Thomas LJ, Janevic MR, Sanders G, Clark NM. Gender-related asthma challenges in a sample of African American women. Poster presented at 2012 Women’s Health Congress.
18. Bedard A, Varraso R, Sanchez M, et al. Cleaning sprays, household help and asthma among elderly women. Respir Med 2014;108:171–80.
19. Dumas O, Siroux V, Luu F, et al. Cleaning and asthma characteristics in women. Am J Ind Med 2014;57:303–11.
20. Clark NM, Valerio MA, Gong ZM. Self-regulation and women with asthma. Curr Opin Allergy Clin Immunol 2008;8:222–7.
21. Lips HM. The gender pay gap: Challenging the rationalizations. Perceived equity, discrimination, and the limits of human capital models. Sex Roles 2013;68:169–85.
22. Apter AJ, Reisine ST, Affleck G, et al. Adherence with twice-daily dosing of inhaled steroids. Socioeconomic and health-belief differences. Am J Respir Crit Care Med 1998;157(6 Pt 1):1810–7.
23. Patel MR, Nelson BW, Id-Deen E, Caldwell CH. Beyond co-pays and out-of-pocket costs: perceptions of health-related financial burden in managing asthma among African American women. J Asthma 2014;51:1083–8.
24. U.S. Department of Health & Human Services. Multiple chronic conditions—a strategic framework: optimum health and quality of life for individuals with multiple chronic conditions. Washington, DC; 2010.
25. Boulet LP, Boulay ME. Asthma-related comorbidities. Expert Rev Respir Med 2011;5:377–93.
26. Sumino K, O’Brian K, Bartle B, et al. Coexisting chronic conditions associated with mortality and morbidity in adult patients with asthma. J Asthma 2014;51:306–14.
27. Patel MR, Janevic MR, Heeringa SG, et al. An examination of adverse asthma outcomes in U.S. Adults with multiple morbidities. Ann Am Thorac Soc 2013;10:426–31.
28. Ward BW, Schiller JS. Prevalence of multiple chronic conditions among US adults: estimates from the National Health Interview Survey, 2010. Prev Chronic Dis 2013;10:E65.
29. Janevic MR, Ellis KR, Sanders GM, et al. Self-management of multiple chronic conditions among African American women with asthma: a qualitative study. J Asthma 2014;51:243–52.
30. Eisner MD, Katz PP, Lactao G, Iribarren C. Impact of depressive symptoms on adult asthma outcomes. Ann Allergy Asthma Immunol 2005;94:566–74.
31. Strine TW, Mokdad AH, Balluz LS, et al. Impact of depression and anxiety on quality of life, health behaviors, and asthma control among adults in the United States with asthma, 2006. J Asthma 2008;45:123–33.
32. Di Marco F, Verga M, Santus P, et al. Close correlation between anxiety, depression, and asthma control. Resp Med 2010;104:22–8.
33. Smith A, Krishnan JA, Bilderback A, et al. Depressive symptoms and adherence to asthma therapy after hospital discharge. Chest 2006;130:1034–8.
34. Krauskopf KA, Sofianou A, Goel MS, et al. Depressive symptoms, low adherence, and poor asthma outcomes in the elderly. J Asthma 2013;50:260–6.
35. Current depression among adults---United States, 2006 and 2008. MMWR 2010;59:1229–35.
36. McLean CP, Asnaani A, Litz BT, Hofmann SG. Gender differences in anxiety disorders: prevalence, course of illness, comorbidity and burden of illness. J Psychiatr Res 2011;45:1027–35.
37. Sundberg R, Toren K, Franklin KA, et al. Asthma in men and women: treatment adherence, anxiety, and quality of sleep. Respir Med 2010;104:337–44.
38. Ford ES. The epidemiology of obesity and asthma. J Allergy Clin Immunol 2005;115:897–909.
39. Essalhi M, Gillaizeau F, Chevallier JM, et al. Cross-sectional assessment of the roles of comorbidities in resting and activity-related dyspnea in severely obese women. J Asthma 2013;50:565–72.
40. Grammer LC, Weiss KB, Pedicano JB, et al. Obesity and asthma morbidity in a community-based adult cohort in a large urban area: the Chicago Initiative to Raise Asthma Health Equity (CHIRAH). J Asthma 2010;47:491–5.
41. Clerisme-Beaty EM, Karam S, Rand C, et al. Does higher body mass index contribute to worse asthma control in an urban population? J Allergy Clin Immunol 2009;124:207–12.
42. Boudreau M, Bacon SL, Ouellet K, et al. Mediator effect of depressive symptoms on the association between BMI and asthma control in adults. Chest 2014;146:348–54.
43. Camargo CA Jr, Sutherland ER, Bailey W, et al. Effect of increased body mass index on asthma risk, impairment and response to asthma controller therapy in African Americans. Curr Med Res Opin 2010;26:1629–35.
44. Valerio MA, Gong ZM, Wang S, et al. Overweight women and management of asthma. Women Health Issues 2009;19:
300–5.
45. Fryar CD, Carroll MD, Ogden CL. Prevalence of overweight, obesity, and extreme obesity among adults: United States, trends 1960–1962 through 2009–2010. Hyattsville, MD: National Center for Health Statistics; 2012.
46. Manion AB. Asthma and obesity: the dose effect. Nurs Clin North Am 2013;48:151–8.
47. Tam A, Morrish D, Wadsworth S, et al. The role of female hormones on lung function in chronic lung diseases. BMC Women Health 2011;11:24.
48. Ticconi C, Pietropolli A, Piccione E. Estrogen replacement therapy and asthma. Pulm Pharmacol Ther 2013;26:617–23.
49. Bonds RS, Midoro-Horiuti T. Estrogen effects in allergy and asthma. Curr Opin Allergy Clin Immunol 2013;13:92–9.
50. Pereira-Vega A, Sanchez Ramos JL, Vazquez Oliva R, et al. Premenstrual asthma and female sex hormones. J Investig Allergol Clin Immunol 2012;22:437–9.
51. Barr RG, Wentowski CC, Grodstein F, et al. Prospective study of postmenopausal hormone use and newly diagnosed asthma and chronic obstructive pulmonary disease. Arch Intern Med 2004;164:379–86.
52. Romieu I, Fabre A, Fournier A, et al. Postmenopausal hormone therapy and asthma onset in the E3N cohort. Thorax 2010;65:292–7.
53. Bonnelykke K, Raaschou-Nielsen O, Tjonneland A, et al. Postmenopausal hormone therapy and asthma-related hospital admission. J Allergy Clin Immunol 2015.
54. Barr RG, Somers SC, Speizer FE, Camargo CA, Jr. Patient factors and medication guideline adherence among older women with asthma. Arch Intern Med 2002;162:1761–8.
55. Baptist AP, Hamad A, Patel MR. Special challenges in treatment and self-management of older women with asthma. Ann Allergy Asthma Immunol 2014;113:125–30.
56. Baptist AP, Deol BB, Reddy RC, et al. Age-specific factors influencing asthma management by older adults. Qual Health Res 2010;20:117–24.
57. Valerio MA, Parker EA, Couper MP, et al. Demographic and clinical characteristics predictive of asthma diary use among women. J Asthma 2008;45:357–61.
58. Ostrom NK. Women with asthma: a review of potential variables and preferred medical management. Ann Allergy Asthma Immunol 2006;96:655–65.
59. Clark NM, Valerio MA, Gong ZM. Self-regulation and women with asthma. Curr Opin Allergy Clin Immunol 2008;8:222.
60. Clark NM, Gong ZM, Wang SJ, et al. From the female perspective: Long-term effects on quality of life of a program for women with asthma. Gend Med 2010;7:125–36.
61. Janevic MR, Sanders GM, Thomas LJ, et al. Study protocol for Women of Color and Asthma Control: a randomized controlled trial of an asthma-management intervention for African American women. BMC Public Health 2012;12:76.
62. Rand CS, Apter AJ. Mind the widening gap: have improvements in asthma care increased asthma disparities? J Allergy Clin Immunol 2008;2:319–21.
63. Moorman JE, Mannino DM. Increasing U.S. asthma mortality rates: who is really dying? J Asthma 2001;38:65–71.
64. Bidwell AJ, Yazel B, Davin D, et al. Yoga training improves quality of life in women with asthma. J Altern Complement Med 2012;18:749–55.
65. Jones PW. St. George’s Respiratory Questionnaire: MCID. COPD 2005;2:75–9.
66. Ding D, Stamatakis E. Yoga practice in England 1997-2008: prevalence, temporal trends, and correlates of participation. BMC Res Notes 2014;7:172.
67. Birdee GS, Legedza AT, Saper RB, et al. Characteristics of yoga users: results of a national survey. J Gen Intern Med 2008;23:1653–8.
68. Wolever RQ, Simmons LA, Sforzo GA, et al. A systematic review of the literature on health and wellness coaching: defining a key behavioral intervention in healthcare. Glob Adv Health Med 2013;2:38–57.
69. Free C, Phillips G, Galli L, et al. The effectiveness of mobile-health technology-based health behaviour change or disease management interventions for health care consumers: a systematic review. PLoS Med 2013;10:e1001362.
70. Marcano Belisario JS, Huckvale K, Greenfield G, et al. Smartphone and tablet self management apps for asthma. Cochrane Database Syst Rev 2013;11:CD010013.
71. Burke W, Fesinmeyer M, Reed K, et al. Family history as a predictor of asthma risk. Am J Prev Med 2003;24:160–9.
Unplanned Exubations in the ICU: Risk Factors and Strategies for Reducing Adverse Events
From the MetroHealth System, Cleveland, OH.
Abstract
- Objective: To describe risk factors for unplanned extubation (UE) among critically ill adults requiring mechanical ventilation and to identify strategies to reduce the occurrence of this adverse event.
- Methods: Review of the literature.
- Results: Inadvertent removal of an endotracheal tube, or a UE, occurs in 7% to 22.5% of mechanically ventilated adult patients and is often due to deliberate patient removal. Despite the multitude of research examining risk factors and predictors of UE, rates have remained unchanged for the past 2 decades. Risk factors can be classified by intensive care unit (ICU) type, including medical ICUs, surgical ICUs, and mixed medical-surgical ICUs. The majority of risk factors for UEs across ICUs may be amenable to changes in unit processes, such as programs for agitation management, use of weaning protocols, increased surveillance of patients, and ongoing education for patients and health care staff.
- Conclusion: Prevention of UE remains an elusive target. Changes in unit processes that target identified risk factors may be an effective method to decrease prevalence of UE.
Unplanned extubation (UE) is the inadvertent removal of an endotracheal tube, either by a patient (deliberate self-extubation), or by a member of the health care team providing routine care such as repositioning, suctioning, or procedures (accidental extubation). Approximately 7% to 22.5% of mechanically ventilated patients in the intensive care unit (ICU) experience UE [1–7]. Estimates are likely higher, as current regulatory and accreditation standards do not include mandatory reporting of this event. Despite numerous studies investigating risk factors associated with UE, it remains a prevalent problem with adverse outcomes for patients and hospitals. The purpose of this review is to provide a summary of the literature on risk factors for UE, review effects on patient and organizational outcomes, and identify evidence-based strategies for reducing occurrence of UE among mechanically ventilated patients.
Prevalence of Unplanned Exubation
There is substantial heterogeneity in how UE is calculated and reported in the research literature. UE is calculated as the number of UE events per 100 or 1000 patient days, or the number of UE per total ventilator days. Rates of UE are also reported as the proportion of patients who experience UE out of all intubated patients over a set time period [8]. Despite efforts aimed at mitigating risk factors for UE, rates have remained static over the past 2 decades. Reported UE rates from 1994–2002 were 2.6% to 14% [3,6,9–11], while rates from 2004–2014 ranged from 1% to 22% [3–5,8,12–15]. Interventions utilizing a multidisciplinary approach have been implemented with the aim of decreasing UE, yet few have proven successful on improving rates nationally.
Unplanned self-extubation by the patient (deliberate self-extubation) is the most common type of UE [3,10,12,16–18]. A multicenter trial of 426 patients from 11 medical centers indicates that 46 patients experienced UE, with 36 of these (78.2%) caused by patient self-extubation [6]. Prospective single-site studies report similar or higher estimates of patient self-extubation, ranging from 75.8% to 91.7% [3,5], while a multisite study of 10,112 patients revealed 32 of 35 UE (91.4%) were due to patient self-extubation [12]. Similarly, a 4-year analysis of 85 UEs reported 82 incidences (96.5%) were a result of deliberate patient removal [13]. Patients either physically pull out the endotracheal tube or use their tongue or coughing/gagging maneuvers to displace or intentionally remove the endotracheal tube [5]. Only 3% to 8% of UEs are caused by inadvertent removal by health care staff [3,5,12,13].
Effects on Patient and Organizational Outcomes
Regardless of the cause of the UE, there are adverse consequences for both patients and hospitals. Some patients who experience UE have higher rates of in-hospital mortality; however, this is often due to contributing factors associated with severity of injury, the need for reintubation, and underlying chronic diseases [13]. Patients who experience accidental UE have higher incidence of nosocomial pneumonia (27.6% vs. 138%, P = 0.002) [11], longer duration of mechanical ventilation, and increased length of stay (LOS) [7,13]. While some studies report UE can result in serious consequences such as respiratory distress, hypoxia [13], and even death [6,12], others report lower mortality and length of stay when UE occurs, likely due to the fact that many patients are ready for liberation from mechanical ventilation at the time of UE [5,15].
Despite the emergent nature of UE, not all patients experience immediate reintubation. Many instances of UE occur during patient weaning trials or in preparation for planned extubations [5,11], which explains why only 10% to 60% of patients require reintubation [3,5,10,11,15,19,20]. When reintubation is necessary, it results in increased number of ventilator days [10,11], and increased ICU and hospital LOS [1,11]. There is little evidence directly linking reintubation with in-hospital mortality; however, it can cause serious complications such as hypotension, hypertension, arrhythmias, and airway trauma [21]. For hospitals and health care organizations, the need for reintubation results in increased hospital costs, estimated to be $1000 per reintubation event [17,22]. This estimate does not take into account additional costs incurred with increased ICU care, longer periods of mechanical ventilation, and increased LOS. Estimates of these additional costs in pediatric patients are approximately $36,000 [23]. Costs are likely higher in adult patients, due to multiple comorbidities that often accompany the need for mechanical ventilation, as well as increased pharmacy, lab, and diagnostic charges [1].
Risk Factors for Unplanned Extubation
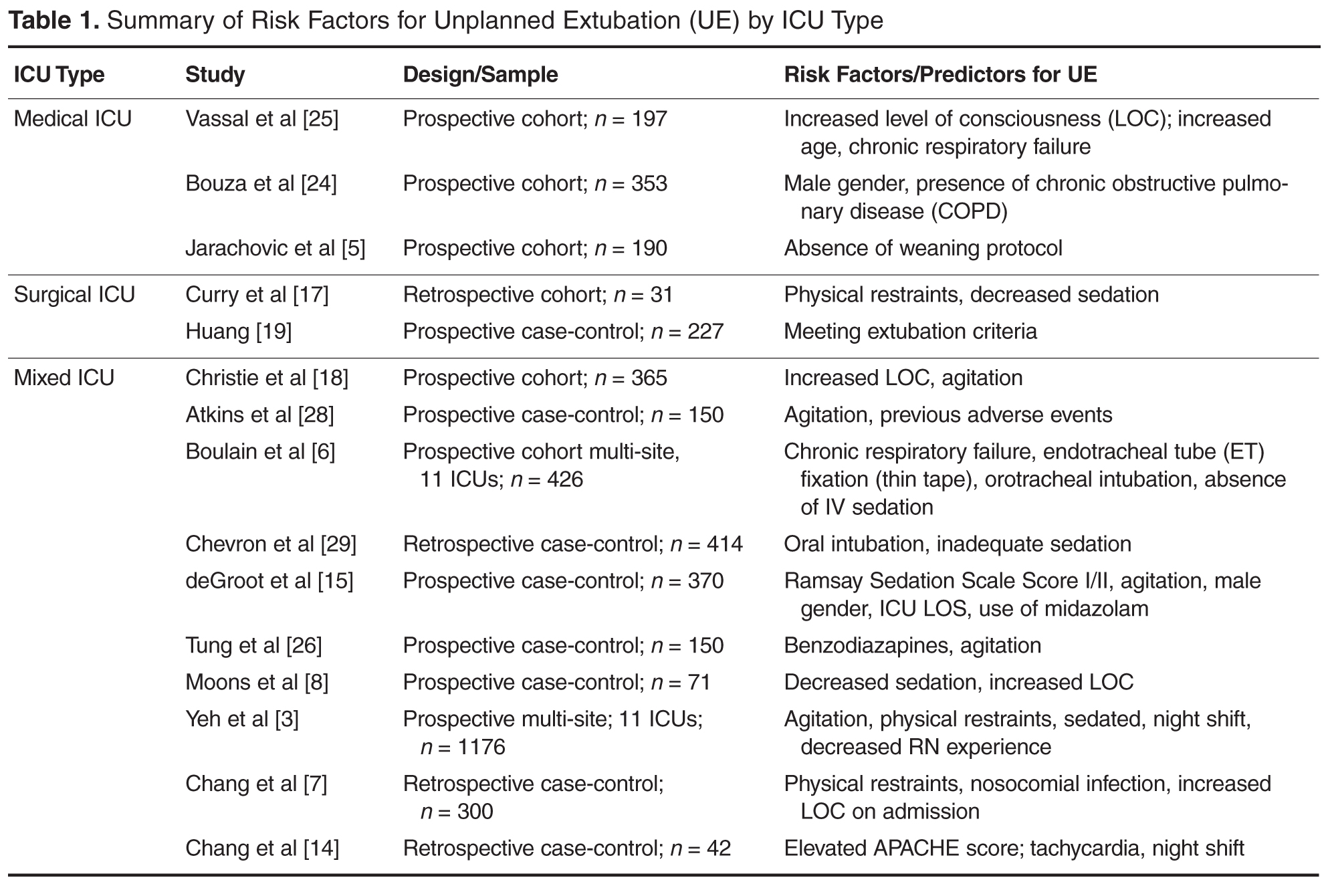
Medical ICU Risk Factors
MICUs traditionally have the highest rates of UE [4,8]. Data from a national prevalence study indicated that there were 23.4 episodes of UE in MICUs per 1000 ventilator days [4]. Approximately 9.5% to 15% of all ventilated patients in the MICU experience UE [4,5,8]. Patients in the MICU who require mechanical ventilation often have complex chronic illness with underlying respiratory disease, which can result in prolonged periods of ventilation and increased risk of UE. Specific risk factors investigated in UE research include patient specific factors (age, gender, diagnosis, comorbidities, agitation, level of consciousness, laboratory values), ventilatory factors (ventilator type and setting, type of tracheal tube, method of tube fixation), as well as type of sedation and use of protocols [5,6,24]. Surprisingly, few variables emerge as significant risk factors for UE among MICU patients. Risk factors associated with UE have included male gender [24], presence of chronic obstructive pulmonary disease (COPD) [24], increased level of consciousness [25], and use of weaning protocols [5]. While gender, COPD, and level of consciousness increase risk of UE, the presence of weaning protocols is shown to decrease risk of UE [5]. Although UE are reported most often in MICUs, few risk factors consistently emerge for this specific cohort, making definitive recommendations for prevention of UE difficult.
Surgical ICU Risk Factors
The prevalence of UE for mechanically ventilated patients in the SICU tend to be lower than those for MICU cohorts. Prevalence of UE in the SICU is reported at 1.41 episodes per 100 ventilator days [13], or 6.8 episodes per 1000 ventilator days [4]. Percentages of UE in the SICU range from 2% to 6% [4,8,19]. Similar to MICU patients, critically ill patients in the SICU often have specific risk factors placing them at risk for UE. Causative factors examined in research studies with this population include gender, age, sedation scale scores, need for reintubation, time from intubation to extubation, use of sedatives/analgesics, restraints, ICU nurse experience, location of staff at time of UE, and criteria for extubation [17,19]. Similar to MICU cohorts, few variables are identified as predictors of UE. Significant predictors include use of restraints, decreased sedation [17], and meeting criteria for extubation [19]. Among patients who experienced an UE, 87% were restrained at the time of the UE [17], and most had low levels of sedation (mean Ramsay sedation scale score = 2.42 in the hour preceding the UE). Approximately 64% of patients who experienced UE met criteria for planned extubation and did not require re-intubation [19], suggesting many patients were essentially ready for planned extubation.
Mixed ICU Risk Factors
The majority of research investigating risk factors for UE is conducted within medical-surgical or mixed/general ICUs. The prevalence of UE within this type of unit is reported at 1.59 episodes per 100 patient days [6], or approximately 2% to 10% [4,6,7]. Among this population, potential risk factors are similar to those included in solely MICU or SICU studies. Because of the high number of studies investigating UE in a mixed ICU setting, there are significantly more variables included in as potential risk factors. Variables include patient age, gender, admission diagnosis, injury severity using Acute Physiological and Chronic Health Evaluation (APACHE II), ICU and hospital LOS, patient level of consciousness, agitation, days of mechanical ventilation, ventilator settings, nosocomial infection, sedation, physical restraints, vital signs [7,14,26], laboratory values, medication types, and body mass index [15,26]. One study also included time of UE and ICU nurse level of experience [3]. Among all factors, several were significant predictors of UE: male gender [15], decreased sedation and increased level of consciousness [8], agitation [3,19,26], use of restraints [3,7], sedation practices (particularly use of benzodiazapines) [3,7,15,26,27], lack of strong tube fixation, absence of IV sedation, and orotracheal intubation [6]. UE were more likely to occur on the night shift and among staff that included nurses with fewer years of experience [3]. Many episodes of UE occurred during weaning [10] or among patients who could communicate and were alert [3]. One study reports 57% of patients who intentionally self-extubated explained they simply removed the tube because it was uncomfortable [3].
Strategies for Reducing Adverse Events
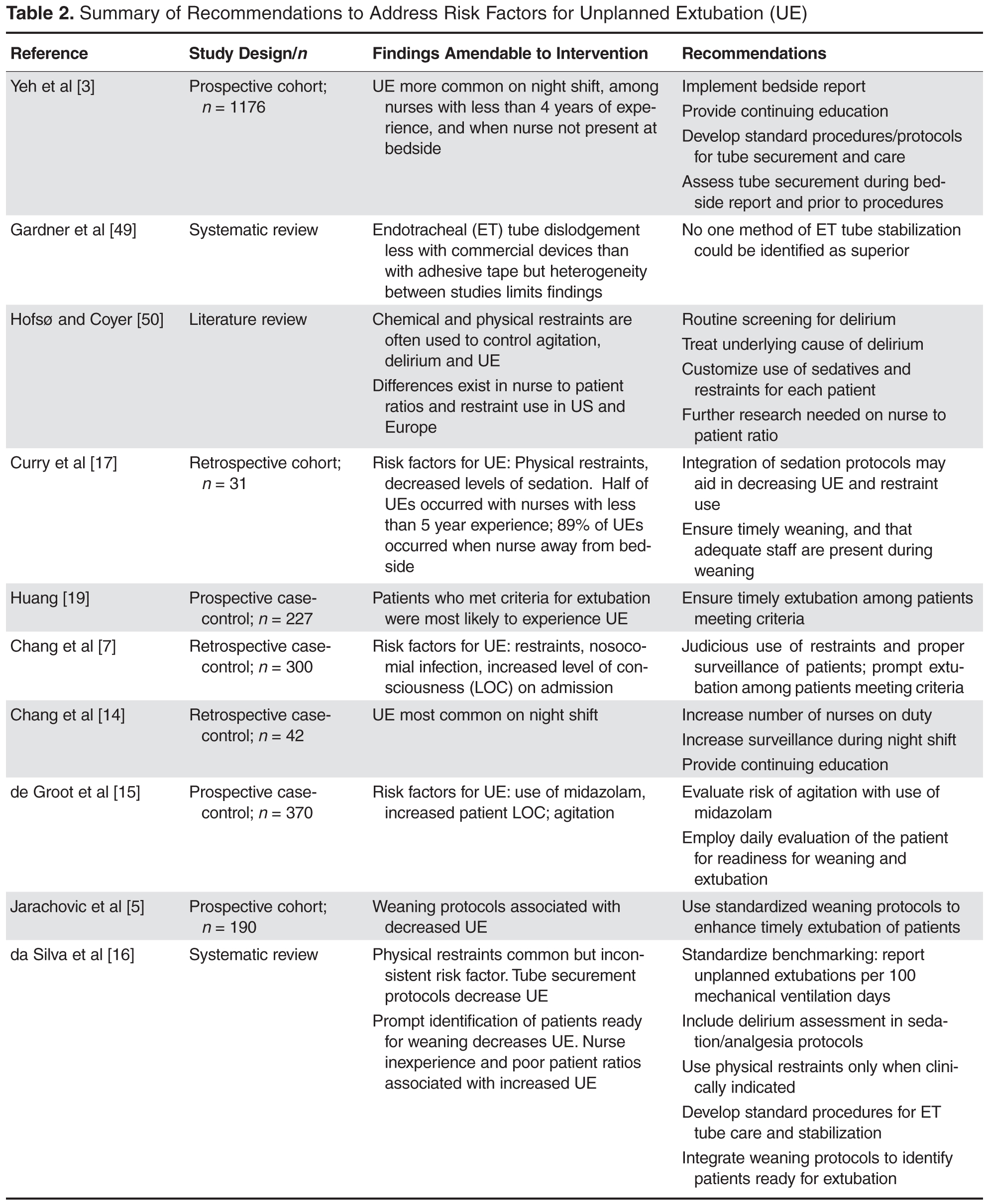
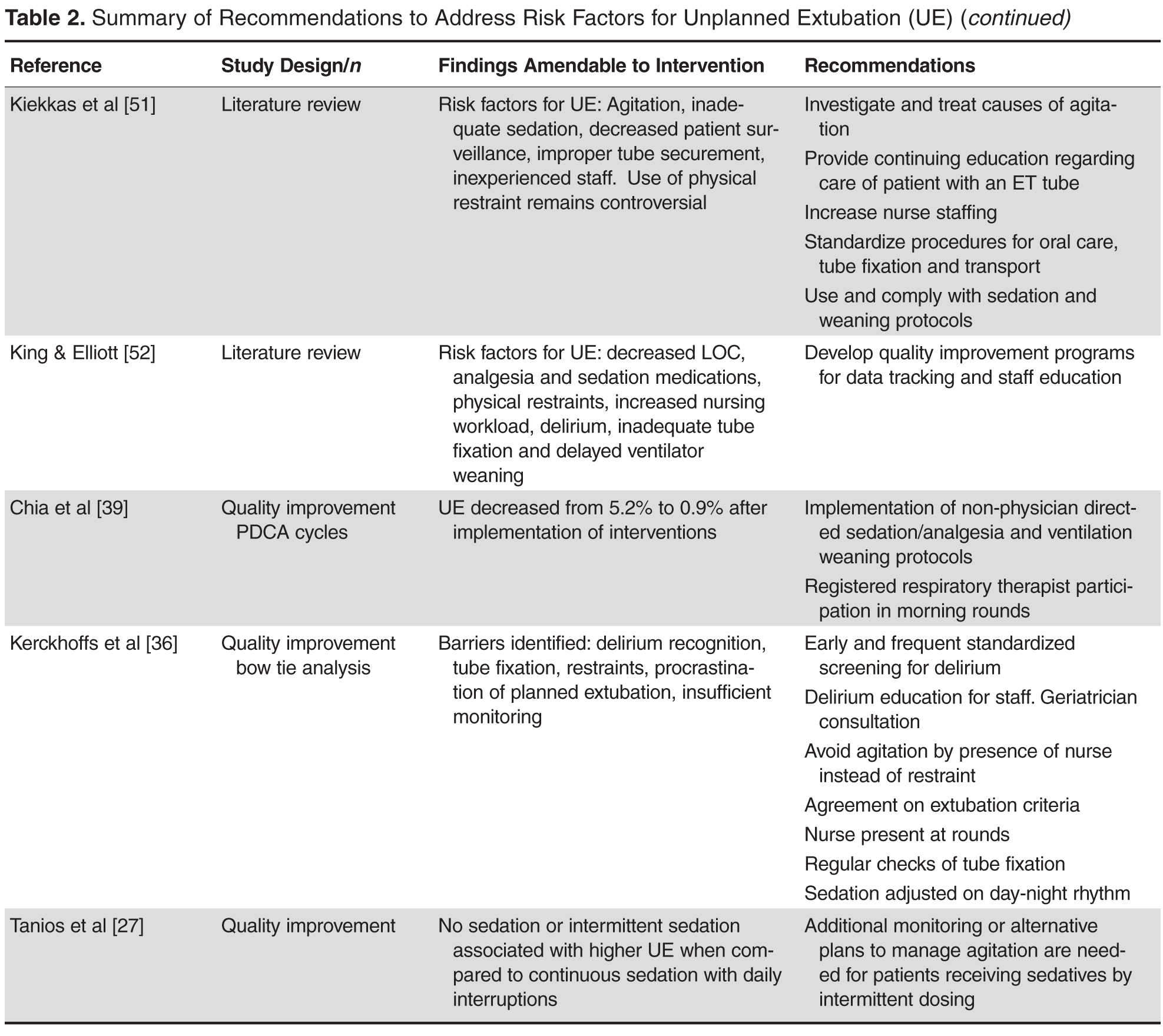
Agitation Management
The majority of studies cited agitation, altered level of consciousness, or inadequate sedation as risk factors for UE [3,6–8,15,17,18,25,26,28,29]. These factors directly impact restraint use, another common risk factor for UE [3,7,17]. A key recommendation for agitation management is to identify the source of agitation, which is often caused by delirium onset in the ICU [30–32]. Prevalence of delirium in the ICU ranges from 20% to 80% [33–35]. ICU patients are at high risk for delirium due to sleep deprivation, older age, restraints, abnormal lab values, medications, infection, and respiratory complications [31]. Treatment for delirium centers on prevention, early recognition, interdisciplinary and pharmacologic protocols, increased nursing presence, and use of short-acting sedation when necessary [30–32,36]. While there is no research specifically linking delirium to UE, a quality analysis of risk factors present at the time of UE using bow-tie analysis methods identified delirium as a key factor present in the majority of UE cases [36]. It is possible that agitation reported in other studies investigating risk factors for UE may actually be reflective of underlying delirium. Routine screening using validated tools, such as the Confusion Assessment Method-ICU (CAM-ICU) [37] would aid in early detection and management of delirium, and would provide a standardized method for exploring the relationship of delirium and UE in future trials.
Integration of Weaning Protocols
Protocol-directed weaning is beneficial for decreasing ventilator days, time to wean from mechanical ventilation, and ICU LOS [38]. A systematic review including 7 trials (2434 patients) comparing protocol/non-protocol for weaning from mechanical ventilation reported a 26% decrease in the mean duration of mechanical ventilation for the protocol groups (95% CI 13%–37%, P < 0.001), a 70% reduction in time to wean, (95% CI 27%–88%, P = 0.009), and a decrease in ICU LOS by 11% (95% CI 3%–19%, P = 0.01). Weaning protocols are also an important risk factor for UE [5]. Findings from a prospective cohort study specifically identify the presence of weaning protocols as an important factor for reducing UE; patients who had weaning protocols ordered and followed were least likely to experience UE (P = 0.02) [5]. A separate quality improvement initiative demonstrated an overall decrease in the number of UEs (from 5.2% to 0.9%) after implementing weaning protocols as standard of care [39]. Considering many UEs occur during weaning [10], integration of weaning protocols aids in expediting the process and ensuring timely extubation.
Increased Surveillance
Increasing surveillance and monitoring of ventilated patients is a recommendation based on risk factors presented at the time of UE. Specifically, staffing levels and shifts and the use of physical restraints are variables associated with UE that are amendable to changes in unit processes based on increased surveillance. It is reported that 40% to 76% of UEs occurred during the night shift [14,17,24,40]; many more occur during change of shift or when there is not a nurse present at the bedside [3,17]. Recent trends towards mandatory bedside reporting is a specific intervention that may positively impact UE among patients in the ICU [41]. Meta-analyses of observational studies investigating the effect of nurse staffing on hospital outcomes indicate that increasing the number of RNs is associated with decreased risk of adverse patient outcomes, including UE [42,43]. The addition of 1 additional nurse per patient day can result in a 51% decrease in UE, while a decrease in nursing workload could result in a 45% decrease in UE [42]. Data from a national prevalence study reports ICUs with fewer available resources, including staff, experienced a higher number of UEs [4].
Increasing surveillance by nursing and health care staff may also impact prevalence of physical restraint use. A significant number of patients who experience UE are physically restrained at the time of the incident, ranging from 40% to 90% of intubated patients [5–7,14,17,40]. It is well documented that UE continue to occur despite the use of restraints [5,7,28,29,44] Patients who are physically restrained often experience higher rates of unplanned extubation (42.9% vs. 16.5% , P < 0.001 in Chang et al’s study [7]), and longer ICU LOS (20.3 days vs. 15.8 days, P = 0.009) [7]. Soft wrist restraints are commonly used to prevent pulling of the endotracheal tube; however, research evidence on UE demonstrates this is not always an effective intervention. Increasing surveillance of ventilated patients, treating their agitation and screening for underlying delirium, and integration of weaning protocols are all interventions that may decrease UE and the need for routine use of physical restraints.
Ongoing Education for Patients and Health Care Staff
Initial and ongoing education about UE, risk factors, and effective interventions is beneficial for patients and health care staff. Although there are no trials investigating effects of educational interventions for patients on UE outcomes, pre-education of surgical patients regarding what to expect while intubated may aid in decreasing delirium risk, agitation, physical restraint use, and possibly UE. Verbal and written educational information during pre-admission testing is a feasible method easily integrated into pre-operative programs.
Because UEs often occur more frequently among less experienced staff, initial education about risk factors for UE is crucial to include in ICU staff orientation programs [3,7]. Educational initiatives should incorporate training on routine delirium screening and avoidance of agitation, use of protocols, and increased surveillance of patients receiving mechanical ventilation [5,15,17,39,45]. Ongoing education of staff regarding ventilatory equipment and risk factors for UE can be particularly effective in decreasing UE [46]. Initial educational efforts should be followed by routine updates for all members of the healthcare team about ongoing quality improvement efforts to monitor UE. Associated factors for UE that may be unit- or process-specific, including methods for endotracheal tube securement and intra-hospital transport, should be communicated with all individuals involved in patient care. Integration of continuous quality improvement programs can decrease UE rates by 22% to 53% [16]. Quality efforts typically focus on standardization of reporting and tracking tools, protocol implementations, and ongoing monitoring, auditing, and recording of UE.
Current Trends and Future Directions
Recent trends in critical care recommendations may mitigate potential risk factors identified in UE research. Integration of lightened sedation and daily wake up periods for intubated patients may decrease prevalence of risk factors for UE, specifically agitation, physical restraint use, and altered level of consciousness [30], while routine weaning protocols may improve ventilatory outcomes, including UE [5,38,40]. Nursing bedside report and purposeful hourly rounding are quickly emerging as mainstays of professional nursing care [41]. Inherent in these 2 initiatives are increased surveillance and vigilance by health care staff, which can result in timely extubation of those who indicate readiness, as well as decreased incidence of adverse events. Delirium remains a key factor that may be a likely cause for UE; recent trends towards early detection and proper management of delirium among ICU staff may result in improved ventilatory outcomes, including weaning, planned extubation, and the prevalence of UE.
Another important trend in critical care is the emergence of a neurocritical care specialty and routine admission of neurocritically ill patients to neuroscience ICUs [47,48]. However, there are no studies investigating prevalence of UE among these patients, who often have higher rates of agitation or restlessness due to cognitive impairment. Among general ICUs, patients with a primary respiratory diagnosis accounted for 23% of all UE in one study, while those with a neurological diagnosis accounted for the second highest percentage (12%) among the study population [15]. A separate study concluded that presence of neurological injury with a concomitant nosocomial infection increased risk of UE among patients in a mixed ICU [7]. A recent systematic review of weaning protocols highlights positive effects on ventilatory outcomes but cites lack of evidence for effectiveness of protocols among those with neurological injury [38]. Areas for future UE research should include factors specific to this patient population, as they may be at higher risk for adverse ventilatory outcomes due to the nature of the neurological injury.
Conclusion
Prevention of UE remains an elusive target, evidenced by little change in reported rates over 2 decades. Research provides data on risk factors that may be patient, unit, or process related. Structuring prevention efforts around modifiable risk factors for UE is a feasible approach amenable to ongoing monitoring for effectiveness. Integration of current trends in health care safety and quality may produce an added benefit of reducing the occurrence of UE in critical care units. Future research evaluating these trends and the prevalence of UE in subspecialty populations is warranted.
Corresponding author: Molly McNett, PhD, RN, CNRN, Attn: NBO, MetroHealth Medical Center, 2500 MetroHealth Drive; Cleveland, OH 44109, mmcnett@metrohealth.org.
Financial disclosures: None.
1. Krinsley JS, Barone JE. The drive to survive: unplanned extubation in the ICU. Chest 2005;128:560–6.
2. Coppolo DP, May JJ. Self-extubations. A 12-month experience. Chest 1990;98:165–9.
3. Yeh SH, Lee LN, Ho TH, et al. Implications of nursing care in the occurrence and consequences of unplanned extubation in adult intensive care units. Int J Nurs Stud 2004;41:255–62.
4. Mion LC, Minnick AF, Leipzig R, et al. Patient-initiated device removal in intensive care units: a national prevalence study. Crit Care Med 2007;35:2714–20.
5. Jarachovic M, Mason M, Kerber K. The role of standardized protocols in unplanned extubations in a medical intensive care unit. Am J Crit Care 2011;20:304–11.
6. Boulain T. Unplanned extubations in the adult intensive care unit: a prospective multicenter study. Association des Reanimateurs du Centre-Ouest. Am J Resp Crit Care Med 1998;157(4 Pt 1):1131–7.
7. Chang LY, Wang KW, Chao YF. Influence of physical restraint on unplanned extubation of adult intensive care patients: a case-control study. Am J Crit Care 2008;17:408–15.
8. Moons P, Sels K, De Becker W, et al. Development of a risk assessment tool for deliberate self-extubation in intensive care patients. Intensive Care Med 2004;30:1348–55.
9. Chiang AA, Lee KC, Lee JC, Wei CH. Effectiveness of a continuous quality improvement program aiming to reduce unplanned extubation: a prospective study. Intensive Care Med 1996;22:1269–71.
10. Betbese AJ, Perez M, Bak E, et al. A prospective study of unplanned endotracheal extubation in intensive care unit patients. Crit Care Med 1998;26:1180–6.
11. de Lassence A, Alberti C, Azoulay E, et al. Impact of unplanned extubation and reintubation after weaning on nosocomial pneumonia risk in the intensive care unit: a prospective multicenter study. Anesthesiology 2002;97:148–56.
12. Kapadia FN, Tekawade PC, Nath SS, et al. A prolonged observational study of tracheal tube displacements: Benchmarking an incidence <0.5-1% in a medical-surgical adult intensive care unit. Ind J Crit Care Med 2014;18:273–7.
13. Lee JH, Lee HC, Jeon YT, et al. Clinical outcomes after unplanned extubation in a surgical intensive care population. World J Surg 2014;38:203–10.
14. Chang LC, Liu PF, Huang YL, et al. Risk factors associated with unplanned endotracheal self-extubation of hospitalized intubated patients: a 3-year retrospective case-control study. Appl Nurs Res 2011;24:188–92.
15. de Groot RI, Dekkers OM, Herold IH, et al. Risk factors and outcomes after unplanned extubations on the ICU: a case-control study. Crit Care 2011;15:R19.
16. da Silva PS, Fonseca MC. Unplanned endotracheal extubations in the intensive care unit: systematic review, critical appraisal, and evidence-based recommendations. Anesth Analg 2012;114:1003–14.
17. Curry K, Cobb S, Kutash M, Diggs C. Characteristics associated with unplanned extubations in a surgical intensive care unit. Am J Crit Care 2008;17:45–51.
18. Christie JM, Dethlefsen M, Cane RD. Unplanned endotracheal extubation in the intensive care unit. J Clin Anesth 1996;8:289–93.
19. Huang YT. Factors leading to self-extubation of endotracheal tubes in the intensive care unit. Nurs Crit Care 2009;14:68–74.
20. Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet 2008;371:126–34.
21. Mort TC. Unplanned tracheal extubation outside the operating room: a quality improvement audit of hemodynamic and tracheal airway complications associated with emergency tracheal reintubation. Anesth Analg 1998;86:1171–6.
22. Jaber S, Chanques G, Altairac C, et al. A prospective study of agitation in a medical-surgical ICU: incidence, risk factors, and outcomes. Chest 2005;128:2749–57.
23. Roddy DJ, Spaeder MC, Pastor W, Stockwell DC, Klugman D. Unplanned extubations in children: impact on hospital cost and length of stay. Ped Crit Care Med 2015.
24. Bouza C, Garcia E, Diaz M, et al. Unplanned extubation in orally intubated medical patients in the intensive care unit: a prospective cohort study. Heart Lung 2007;36:270–6.
25. Vassal T, Anh NG, Gabillet JM, et al. Prospective evaluation of self-extubations in a medical intensive care unit. Intensive Care Med 1993;19:340-342.
26. Tung A, Tadimeti L, Caruana-Montaldo B, et al. The relationship of sedation to deliberate self-extubation. J Clin Anesth 2001;13:24–9.
27. Tanios M, Epstein S, Grzeskowiak M, et al. Influence of sedation strategies on unplanned extubation in a mixed intensive care unit. Am J Crit Care 2014;23:306–14.
28. Atkins PM, Mion LC, Mendelson W, et al. Characteristics and outcomes of patients who self-extubate from ventilatory support: a case-control study. Chest 1997;112:1317–23.
29. Chevron V, Menard JF, Richard JC, et al. Unplanned extubation: risk factors of development and predictive criteria for reintubation. Crit Care Med 1998;26:1049–53.
30. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med 2013;41:263–306.
31. Morandi A, Jackson JC. Delirium in the intensive care unit: a review. Neurol Clin 2011;29:749–63.
32. Banerjee A, Vasilevskis, EE, Pandharipande, P. Strategies to improve delirium assessment practices in the intensive care unit. J Clin Outcomes Manag 2010;17:459–68.
33. Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001;286:2703–10.
34. Ely EW, Stephens RK, Jackson JC, et al. Current opinions regarding the importance, diagnosis, and management of delirium in the intensive care unit: a survey of 912 healthcare professionals. Crit Care Med 2004;32:106–12.
35. McNicoll L, Pisani MA, Zhang Y, et al. Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc 2003;51:591–8.
36. Kerckhoffs MC, van der Sluijs AF, Binnekade JM, Dongelmans DA. Improving patient safety in the ICU by prospective identification of missing safety barriers using the bow-tie prospective risk analysis model. J Patient Safe 2013;9:154–9.
37. Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med 1990;113:941–8.
38. Blackwood B, Burns KE, Cardwell CR, O’Halloran P. Protocolized versus non-protocolized weaning for reducing the duration of mechanical ventilation in critically ill adult patients. Cochrane Database Syst Rev 2014;11:CD006904.
39. Chia PL, Santos DR, Tan TC, et al. Clinical quality improvement: eliminating unplanned extubation in the CCU. Int J Health Care Qual Ass 2013;26:642–52.
40. Balon JA. Common factors of spontaneous self-extubation in a critical care setting. Int J Trauma Nurs 2001;7:93–9.
41. Gregory S, Tan D, Tilrico M, et al. Bedside shift reports: what does the evidence say? J Nurs Admin 2014;44:541–5.
42. Kane RL, Shamliyan TA, Mueller C, et al. The association of registered nurse staffing levels and patient outcomes: systematic review and meta-analysis. Med Care 2007;45:1195–204.
43. Penoyer DA. Nurse staffing and patient outcomes in critical care: a concise review. Crit Care Med 2010;38:1521–8; quiz 1529.
44. Tindol GA, Jr., DiBenedetto RJ, Kosciuk L. Unplanned extubations. Chest 1994;105:1804–7.
45. Chen CM CK, Fong Y, Hsing SC, et al. Age is an important predictor of failed unplanned extubation. Int J Gerontol 2010;4:120–9.
46. Richmond AL, Jarog DL, Hanson VM. Unplanned extubation in adult critical care. Quality improvement and education payoff. Crit Care Nurs 2004;24:32–7.
47. Kurtz P, Fitts V, Sumer Z, et al. How does care differ for neurological patients admitted to a neurocritical care unit versus a general ICU? Neurocrit Care 2011;15:477–80.
48. McNett MM, Horowitz DA. International multidisciplinary consensus conference on multimodality monitoring: ICU processes of care. Neurocrit Care 2014;21 Suppl 2:S215–28.
49. Gardner A, Hughes, D, Cook R, et al. Best practice in stabilisation of oral endotracheal tubes: a systematic review. Database of abstracts of reivews of effects (DARE): Quality-assessed reviews. 2005. York: Center for Reviews and Dissemination.
50. Hofso K, Coyer FM. Part 1: Chemical and physical restraints in the management of mechanically ventliated patients in the ICU: Contributing factors. Intensive Crit Care Nurs 2007; 23:249–55.
51. Kiekkas P, Diamanto A, Panteli E, et al. Unplanned extubation in critially ill adults: Clinical reviews. Nurs Crit Care 2012;18:123–34.
52. King JN, Elliiot VA. Self/unplanned extubation: Safety, surveillance, and monitoring of the mechanically ventilated patient. Crit Care Nurs Clin North Am 2012;24:469–79.
From the MetroHealth System, Cleveland, OH.
Abstract
- Objective: To describe risk factors for unplanned extubation (UE) among critically ill adults requiring mechanical ventilation and to identify strategies to reduce the occurrence of this adverse event.
- Methods: Review of the literature.
- Results: Inadvertent removal of an endotracheal tube, or a UE, occurs in 7% to 22.5% of mechanically ventilated adult patients and is often due to deliberate patient removal. Despite the multitude of research examining risk factors and predictors of UE, rates have remained unchanged for the past 2 decades. Risk factors can be classified by intensive care unit (ICU) type, including medical ICUs, surgical ICUs, and mixed medical-surgical ICUs. The majority of risk factors for UEs across ICUs may be amenable to changes in unit processes, such as programs for agitation management, use of weaning protocols, increased surveillance of patients, and ongoing education for patients and health care staff.
- Conclusion: Prevention of UE remains an elusive target. Changes in unit processes that target identified risk factors may be an effective method to decrease prevalence of UE.
Unplanned extubation (UE) is the inadvertent removal of an endotracheal tube, either by a patient (deliberate self-extubation), or by a member of the health care team providing routine care such as repositioning, suctioning, or procedures (accidental extubation). Approximately 7% to 22.5% of mechanically ventilated patients in the intensive care unit (ICU) experience UE [1–7]. Estimates are likely higher, as current regulatory and accreditation standards do not include mandatory reporting of this event. Despite numerous studies investigating risk factors associated with UE, it remains a prevalent problem with adverse outcomes for patients and hospitals. The purpose of this review is to provide a summary of the literature on risk factors for UE, review effects on patient and organizational outcomes, and identify evidence-based strategies for reducing occurrence of UE among mechanically ventilated patients.
Prevalence of Unplanned Exubation
There is substantial heterogeneity in how UE is calculated and reported in the research literature. UE is calculated as the number of UE events per 100 or 1000 patient days, or the number of UE per total ventilator days. Rates of UE are also reported as the proportion of patients who experience UE out of all intubated patients over a set time period [8]. Despite efforts aimed at mitigating risk factors for UE, rates have remained static over the past 2 decades. Reported UE rates from 1994–2002 were 2.6% to 14% [3,6,9–11], while rates from 2004–2014 ranged from 1% to 22% [3–5,8,12–15]. Interventions utilizing a multidisciplinary approach have been implemented with the aim of decreasing UE, yet few have proven successful on improving rates nationally.
Unplanned self-extubation by the patient (deliberate self-extubation) is the most common type of UE [3,10,12,16–18]. A multicenter trial of 426 patients from 11 medical centers indicates that 46 patients experienced UE, with 36 of these (78.2%) caused by patient self-extubation [6]. Prospective single-site studies report similar or higher estimates of patient self-extubation, ranging from 75.8% to 91.7% [3,5], while a multisite study of 10,112 patients revealed 32 of 35 UE (91.4%) were due to patient self-extubation [12]. Similarly, a 4-year analysis of 85 UEs reported 82 incidences (96.5%) were a result of deliberate patient removal [13]. Patients either physically pull out the endotracheal tube or use their tongue or coughing/gagging maneuvers to displace or intentionally remove the endotracheal tube [5]. Only 3% to 8% of UEs are caused by inadvertent removal by health care staff [3,5,12,13].
Effects on Patient and Organizational Outcomes
Regardless of the cause of the UE, there are adverse consequences for both patients and hospitals. Some patients who experience UE have higher rates of in-hospital mortality; however, this is often due to contributing factors associated with severity of injury, the need for reintubation, and underlying chronic diseases [13]. Patients who experience accidental UE have higher incidence of nosocomial pneumonia (27.6% vs. 138%, P = 0.002) [11], longer duration of mechanical ventilation, and increased length of stay (LOS) [7,13]. While some studies report UE can result in serious consequences such as respiratory distress, hypoxia [13], and even death [6,12], others report lower mortality and length of stay when UE occurs, likely due to the fact that many patients are ready for liberation from mechanical ventilation at the time of UE [5,15].
Despite the emergent nature of UE, not all patients experience immediate reintubation. Many instances of UE occur during patient weaning trials or in preparation for planned extubations [5,11], which explains why only 10% to 60% of patients require reintubation [3,5,10,11,15,19,20]. When reintubation is necessary, it results in increased number of ventilator days [10,11], and increased ICU and hospital LOS [1,11]. There is little evidence directly linking reintubation with in-hospital mortality; however, it can cause serious complications such as hypotension, hypertension, arrhythmias, and airway trauma [21]. For hospitals and health care organizations, the need for reintubation results in increased hospital costs, estimated to be $1000 per reintubation event [17,22]. This estimate does not take into account additional costs incurred with increased ICU care, longer periods of mechanical ventilation, and increased LOS. Estimates of these additional costs in pediatric patients are approximately $36,000 [23]. Costs are likely higher in adult patients, due to multiple comorbidities that often accompany the need for mechanical ventilation, as well as increased pharmacy, lab, and diagnostic charges [1].
Risk Factors for Unplanned Extubation
Medical ICU Risk Factors
MICUs traditionally have the highest rates of UE [4,8]. Data from a national prevalence study indicated that there were 23.4 episodes of UE in MICUs per 1000 ventilator days [4]. Approximately 9.5% to 15% of all ventilated patients in the MICU experience UE [4,5,8]. Patients in the MICU who require mechanical ventilation often have complex chronic illness with underlying respiratory disease, which can result in prolonged periods of ventilation and increased risk of UE. Specific risk factors investigated in UE research include patient specific factors (age, gender, diagnosis, comorbidities, agitation, level of consciousness, laboratory values), ventilatory factors (ventilator type and setting, type of tracheal tube, method of tube fixation), as well as type of sedation and use of protocols [5,6,24]. Surprisingly, few variables emerge as significant risk factors for UE among MICU patients. Risk factors associated with UE have included male gender [24], presence of chronic obstructive pulmonary disease (COPD) [24], increased level of consciousness [25], and use of weaning protocols [5]. While gender, COPD, and level of consciousness increase risk of UE, the presence of weaning protocols is shown to decrease risk of UE [5]. Although UE are reported most often in MICUs, few risk factors consistently emerge for this specific cohort, making definitive recommendations for prevention of UE difficult.
Surgical ICU Risk Factors
The prevalence of UE for mechanically ventilated patients in the SICU tend to be lower than those for MICU cohorts. Prevalence of UE in the SICU is reported at 1.41 episodes per 100 ventilator days [13], or 6.8 episodes per 1000 ventilator days [4]. Percentages of UE in the SICU range from 2% to 6% [4,8,19]. Similar to MICU patients, critically ill patients in the SICU often have specific risk factors placing them at risk for UE. Causative factors examined in research studies with this population include gender, age, sedation scale scores, need for reintubation, time from intubation to extubation, use of sedatives/analgesics, restraints, ICU nurse experience, location of staff at time of UE, and criteria for extubation [17,19]. Similar to MICU cohorts, few variables are identified as predictors of UE. Significant predictors include use of restraints, decreased sedation [17], and meeting criteria for extubation [19]. Among patients who experienced an UE, 87% were restrained at the time of the UE [17], and most had low levels of sedation (mean Ramsay sedation scale score = 2.42 in the hour preceding the UE). Approximately 64% of patients who experienced UE met criteria for planned extubation and did not require re-intubation [19], suggesting many patients were essentially ready for planned extubation.
Mixed ICU Risk Factors
The majority of research investigating risk factors for UE is conducted within medical-surgical or mixed/general ICUs. The prevalence of UE within this type of unit is reported at 1.59 episodes per 100 patient days [6], or approximately 2% to 10% [4,6,7]. Among this population, potential risk factors are similar to those included in solely MICU or SICU studies. Because of the high number of studies investigating UE in a mixed ICU setting, there are significantly more variables included in as potential risk factors. Variables include patient age, gender, admission diagnosis, injury severity using Acute Physiological and Chronic Health Evaluation (APACHE II), ICU and hospital LOS, patient level of consciousness, agitation, days of mechanical ventilation, ventilator settings, nosocomial infection, sedation, physical restraints, vital signs [7,14,26], laboratory values, medication types, and body mass index [15,26]. One study also included time of UE and ICU nurse level of experience [3]. Among all factors, several were significant predictors of UE: male gender [15], decreased sedation and increased level of consciousness [8], agitation [3,19,26], use of restraints [3,7], sedation practices (particularly use of benzodiazapines) [3,7,15,26,27], lack of strong tube fixation, absence of IV sedation, and orotracheal intubation [6]. UE were more likely to occur on the night shift and among staff that included nurses with fewer years of experience [3]. Many episodes of UE occurred during weaning [10] or among patients who could communicate and were alert [3]. One study reports 57% of patients who intentionally self-extubated explained they simply removed the tube because it was uncomfortable [3].
Strategies for Reducing Adverse Events
Agitation Management
The majority of studies cited agitation, altered level of consciousness, or inadequate sedation as risk factors for UE [3,6–8,15,17,18,25,26,28,29]. These factors directly impact restraint use, another common risk factor for UE [3,7,17]. A key recommendation for agitation management is to identify the source of agitation, which is often caused by delirium onset in the ICU [30–32]. Prevalence of delirium in the ICU ranges from 20% to 80% [33–35]. ICU patients are at high risk for delirium due to sleep deprivation, older age, restraints, abnormal lab values, medications, infection, and respiratory complications [31]. Treatment for delirium centers on prevention, early recognition, interdisciplinary and pharmacologic protocols, increased nursing presence, and use of short-acting sedation when necessary [30–32,36]. While there is no research specifically linking delirium to UE, a quality analysis of risk factors present at the time of UE using bow-tie analysis methods identified delirium as a key factor present in the majority of UE cases [36]. It is possible that agitation reported in other studies investigating risk factors for UE may actually be reflective of underlying delirium. Routine screening using validated tools, such as the Confusion Assessment Method-ICU (CAM-ICU) [37] would aid in early detection and management of delirium, and would provide a standardized method for exploring the relationship of delirium and UE in future trials.
Integration of Weaning Protocols
Protocol-directed weaning is beneficial for decreasing ventilator days, time to wean from mechanical ventilation, and ICU LOS [38]. A systematic review including 7 trials (2434 patients) comparing protocol/non-protocol for weaning from mechanical ventilation reported a 26% decrease in the mean duration of mechanical ventilation for the protocol groups (95% CI 13%–37%, P < 0.001), a 70% reduction in time to wean, (95% CI 27%–88%, P = 0.009), and a decrease in ICU LOS by 11% (95% CI 3%–19%, P = 0.01). Weaning protocols are also an important risk factor for UE [5]. Findings from a prospective cohort study specifically identify the presence of weaning protocols as an important factor for reducing UE; patients who had weaning protocols ordered and followed were least likely to experience UE (P = 0.02) [5]. A separate quality improvement initiative demonstrated an overall decrease in the number of UEs (from 5.2% to 0.9%) after implementing weaning protocols as standard of care [39]. Considering many UEs occur during weaning [10], integration of weaning protocols aids in expediting the process and ensuring timely extubation.
Increased Surveillance
Increasing surveillance and monitoring of ventilated patients is a recommendation based on risk factors presented at the time of UE. Specifically, staffing levels and shifts and the use of physical restraints are variables associated with UE that are amendable to changes in unit processes based on increased surveillance. It is reported that 40% to 76% of UEs occurred during the night shift [14,17,24,40]; many more occur during change of shift or when there is not a nurse present at the bedside [3,17]. Recent trends towards mandatory bedside reporting is a specific intervention that may positively impact UE among patients in the ICU [41]. Meta-analyses of observational studies investigating the effect of nurse staffing on hospital outcomes indicate that increasing the number of RNs is associated with decreased risk of adverse patient outcomes, including UE [42,43]. The addition of 1 additional nurse per patient day can result in a 51% decrease in UE, while a decrease in nursing workload could result in a 45% decrease in UE [42]. Data from a national prevalence study reports ICUs with fewer available resources, including staff, experienced a higher number of UEs [4].
Increasing surveillance by nursing and health care staff may also impact prevalence of physical restraint use. A significant number of patients who experience UE are physically restrained at the time of the incident, ranging from 40% to 90% of intubated patients [5–7,14,17,40]. It is well documented that UE continue to occur despite the use of restraints [5,7,28,29,44] Patients who are physically restrained often experience higher rates of unplanned extubation (42.9% vs. 16.5% , P < 0.001 in Chang et al’s study [7]), and longer ICU LOS (20.3 days vs. 15.8 days, P = 0.009) [7]. Soft wrist restraints are commonly used to prevent pulling of the endotracheal tube; however, research evidence on UE demonstrates this is not always an effective intervention. Increasing surveillance of ventilated patients, treating their agitation and screening for underlying delirium, and integration of weaning protocols are all interventions that may decrease UE and the need for routine use of physical restraints.
Ongoing Education for Patients and Health Care Staff
Initial and ongoing education about UE, risk factors, and effective interventions is beneficial for patients and health care staff. Although there are no trials investigating effects of educational interventions for patients on UE outcomes, pre-education of surgical patients regarding what to expect while intubated may aid in decreasing delirium risk, agitation, physical restraint use, and possibly UE. Verbal and written educational information during pre-admission testing is a feasible method easily integrated into pre-operative programs.
Because UEs often occur more frequently among less experienced staff, initial education about risk factors for UE is crucial to include in ICU staff orientation programs [3,7]. Educational initiatives should incorporate training on routine delirium screening and avoidance of agitation, use of protocols, and increased surveillance of patients receiving mechanical ventilation [5,15,17,39,45]. Ongoing education of staff regarding ventilatory equipment and risk factors for UE can be particularly effective in decreasing UE [46]. Initial educational efforts should be followed by routine updates for all members of the healthcare team about ongoing quality improvement efforts to monitor UE. Associated factors for UE that may be unit- or process-specific, including methods for endotracheal tube securement and intra-hospital transport, should be communicated with all individuals involved in patient care. Integration of continuous quality improvement programs can decrease UE rates by 22% to 53% [16]. Quality efforts typically focus on standardization of reporting and tracking tools, protocol implementations, and ongoing monitoring, auditing, and recording of UE.
Current Trends and Future Directions
Recent trends in critical care recommendations may mitigate potential risk factors identified in UE research. Integration of lightened sedation and daily wake up periods for intubated patients may decrease prevalence of risk factors for UE, specifically agitation, physical restraint use, and altered level of consciousness [30], while routine weaning protocols may improve ventilatory outcomes, including UE [5,38,40]. Nursing bedside report and purposeful hourly rounding are quickly emerging as mainstays of professional nursing care [41]. Inherent in these 2 initiatives are increased surveillance and vigilance by health care staff, which can result in timely extubation of those who indicate readiness, as well as decreased incidence of adverse events. Delirium remains a key factor that may be a likely cause for UE; recent trends towards early detection and proper management of delirium among ICU staff may result in improved ventilatory outcomes, including weaning, planned extubation, and the prevalence of UE.
Another important trend in critical care is the emergence of a neurocritical care specialty and routine admission of neurocritically ill patients to neuroscience ICUs [47,48]. However, there are no studies investigating prevalence of UE among these patients, who often have higher rates of agitation or restlessness due to cognitive impairment. Among general ICUs, patients with a primary respiratory diagnosis accounted for 23% of all UE in one study, while those with a neurological diagnosis accounted for the second highest percentage (12%) among the study population [15]. A separate study concluded that presence of neurological injury with a concomitant nosocomial infection increased risk of UE among patients in a mixed ICU [7]. A recent systematic review of weaning protocols highlights positive effects on ventilatory outcomes but cites lack of evidence for effectiveness of protocols among those with neurological injury [38]. Areas for future UE research should include factors specific to this patient population, as they may be at higher risk for adverse ventilatory outcomes due to the nature of the neurological injury.
Conclusion
Prevention of UE remains an elusive target, evidenced by little change in reported rates over 2 decades. Research provides data on risk factors that may be patient, unit, or process related. Structuring prevention efforts around modifiable risk factors for UE is a feasible approach amenable to ongoing monitoring for effectiveness. Integration of current trends in health care safety and quality may produce an added benefit of reducing the occurrence of UE in critical care units. Future research evaluating these trends and the prevalence of UE in subspecialty populations is warranted.
Corresponding author: Molly McNett, PhD, RN, CNRN, Attn: NBO, MetroHealth Medical Center, 2500 MetroHealth Drive; Cleveland, OH 44109, mmcnett@metrohealth.org.
Financial disclosures: None.
From the MetroHealth System, Cleveland, OH.
Abstract
- Objective: To describe risk factors for unplanned extubation (UE) among critically ill adults requiring mechanical ventilation and to identify strategies to reduce the occurrence of this adverse event.
- Methods: Review of the literature.
- Results: Inadvertent removal of an endotracheal tube, or a UE, occurs in 7% to 22.5% of mechanically ventilated adult patients and is often due to deliberate patient removal. Despite the multitude of research examining risk factors and predictors of UE, rates have remained unchanged for the past 2 decades. Risk factors can be classified by intensive care unit (ICU) type, including medical ICUs, surgical ICUs, and mixed medical-surgical ICUs. The majority of risk factors for UEs across ICUs may be amenable to changes in unit processes, such as programs for agitation management, use of weaning protocols, increased surveillance of patients, and ongoing education for patients and health care staff.
- Conclusion: Prevention of UE remains an elusive target. Changes in unit processes that target identified risk factors may be an effective method to decrease prevalence of UE.
Unplanned extubation (UE) is the inadvertent removal of an endotracheal tube, either by a patient (deliberate self-extubation), or by a member of the health care team providing routine care such as repositioning, suctioning, or procedures (accidental extubation). Approximately 7% to 22.5% of mechanically ventilated patients in the intensive care unit (ICU) experience UE [1–7]. Estimates are likely higher, as current regulatory and accreditation standards do not include mandatory reporting of this event. Despite numerous studies investigating risk factors associated with UE, it remains a prevalent problem with adverse outcomes for patients and hospitals. The purpose of this review is to provide a summary of the literature on risk factors for UE, review effects on patient and organizational outcomes, and identify evidence-based strategies for reducing occurrence of UE among mechanically ventilated patients.
Prevalence of Unplanned Exubation
There is substantial heterogeneity in how UE is calculated and reported in the research literature. UE is calculated as the number of UE events per 100 or 1000 patient days, or the number of UE per total ventilator days. Rates of UE are also reported as the proportion of patients who experience UE out of all intubated patients over a set time period [8]. Despite efforts aimed at mitigating risk factors for UE, rates have remained static over the past 2 decades. Reported UE rates from 1994–2002 were 2.6% to 14% [3,6,9–11], while rates from 2004–2014 ranged from 1% to 22% [3–5,8,12–15]. Interventions utilizing a multidisciplinary approach have been implemented with the aim of decreasing UE, yet few have proven successful on improving rates nationally.
Unplanned self-extubation by the patient (deliberate self-extubation) is the most common type of UE [3,10,12,16–18]. A multicenter trial of 426 patients from 11 medical centers indicates that 46 patients experienced UE, with 36 of these (78.2%) caused by patient self-extubation [6]. Prospective single-site studies report similar or higher estimates of patient self-extubation, ranging from 75.8% to 91.7% [3,5], while a multisite study of 10,112 patients revealed 32 of 35 UE (91.4%) were due to patient self-extubation [12]. Similarly, a 4-year analysis of 85 UEs reported 82 incidences (96.5%) were a result of deliberate patient removal [13]. Patients either physically pull out the endotracheal tube or use their tongue or coughing/gagging maneuvers to displace or intentionally remove the endotracheal tube [5]. Only 3% to 8% of UEs are caused by inadvertent removal by health care staff [3,5,12,13].
Effects on Patient and Organizational Outcomes
Regardless of the cause of the UE, there are adverse consequences for both patients and hospitals. Some patients who experience UE have higher rates of in-hospital mortality; however, this is often due to contributing factors associated with severity of injury, the need for reintubation, and underlying chronic diseases [13]. Patients who experience accidental UE have higher incidence of nosocomial pneumonia (27.6% vs. 138%, P = 0.002) [11], longer duration of mechanical ventilation, and increased length of stay (LOS) [7,13]. While some studies report UE can result in serious consequences such as respiratory distress, hypoxia [13], and even death [6,12], others report lower mortality and length of stay when UE occurs, likely due to the fact that many patients are ready for liberation from mechanical ventilation at the time of UE [5,15].
Despite the emergent nature of UE, not all patients experience immediate reintubation. Many instances of UE occur during patient weaning trials or in preparation for planned extubations [5,11], which explains why only 10% to 60% of patients require reintubation [3,5,10,11,15,19,20]. When reintubation is necessary, it results in increased number of ventilator days [10,11], and increased ICU and hospital LOS [1,11]. There is little evidence directly linking reintubation with in-hospital mortality; however, it can cause serious complications such as hypotension, hypertension, arrhythmias, and airway trauma [21]. For hospitals and health care organizations, the need for reintubation results in increased hospital costs, estimated to be $1000 per reintubation event [17,22]. This estimate does not take into account additional costs incurred with increased ICU care, longer periods of mechanical ventilation, and increased LOS. Estimates of these additional costs in pediatric patients are approximately $36,000 [23]. Costs are likely higher in adult patients, due to multiple comorbidities that often accompany the need for mechanical ventilation, as well as increased pharmacy, lab, and diagnostic charges [1].
Risk Factors for Unplanned Extubation
Medical ICU Risk Factors
MICUs traditionally have the highest rates of UE [4,8]. Data from a national prevalence study indicated that there were 23.4 episodes of UE in MICUs per 1000 ventilator days [4]. Approximately 9.5% to 15% of all ventilated patients in the MICU experience UE [4,5,8]. Patients in the MICU who require mechanical ventilation often have complex chronic illness with underlying respiratory disease, which can result in prolonged periods of ventilation and increased risk of UE. Specific risk factors investigated in UE research include patient specific factors (age, gender, diagnosis, comorbidities, agitation, level of consciousness, laboratory values), ventilatory factors (ventilator type and setting, type of tracheal tube, method of tube fixation), as well as type of sedation and use of protocols [5,6,24]. Surprisingly, few variables emerge as significant risk factors for UE among MICU patients. Risk factors associated with UE have included male gender [24], presence of chronic obstructive pulmonary disease (COPD) [24], increased level of consciousness [25], and use of weaning protocols [5]. While gender, COPD, and level of consciousness increase risk of UE, the presence of weaning protocols is shown to decrease risk of UE [5]. Although UE are reported most often in MICUs, few risk factors consistently emerge for this specific cohort, making definitive recommendations for prevention of UE difficult.
Surgical ICU Risk Factors
The prevalence of UE for mechanically ventilated patients in the SICU tend to be lower than those for MICU cohorts. Prevalence of UE in the SICU is reported at 1.41 episodes per 100 ventilator days [13], or 6.8 episodes per 1000 ventilator days [4]. Percentages of UE in the SICU range from 2% to 6% [4,8,19]. Similar to MICU patients, critically ill patients in the SICU often have specific risk factors placing them at risk for UE. Causative factors examined in research studies with this population include gender, age, sedation scale scores, need for reintubation, time from intubation to extubation, use of sedatives/analgesics, restraints, ICU nurse experience, location of staff at time of UE, and criteria for extubation [17,19]. Similar to MICU cohorts, few variables are identified as predictors of UE. Significant predictors include use of restraints, decreased sedation [17], and meeting criteria for extubation [19]. Among patients who experienced an UE, 87% were restrained at the time of the UE [17], and most had low levels of sedation (mean Ramsay sedation scale score = 2.42 in the hour preceding the UE). Approximately 64% of patients who experienced UE met criteria for planned extubation and did not require re-intubation [19], suggesting many patients were essentially ready for planned extubation.
Mixed ICU Risk Factors
The majority of research investigating risk factors for UE is conducted within medical-surgical or mixed/general ICUs. The prevalence of UE within this type of unit is reported at 1.59 episodes per 100 patient days [6], or approximately 2% to 10% [4,6,7]. Among this population, potential risk factors are similar to those included in solely MICU or SICU studies. Because of the high number of studies investigating UE in a mixed ICU setting, there are significantly more variables included in as potential risk factors. Variables include patient age, gender, admission diagnosis, injury severity using Acute Physiological and Chronic Health Evaluation (APACHE II), ICU and hospital LOS, patient level of consciousness, agitation, days of mechanical ventilation, ventilator settings, nosocomial infection, sedation, physical restraints, vital signs [7,14,26], laboratory values, medication types, and body mass index [15,26]. One study also included time of UE and ICU nurse level of experience [3]. Among all factors, several were significant predictors of UE: male gender [15], decreased sedation and increased level of consciousness [8], agitation [3,19,26], use of restraints [3,7], sedation practices (particularly use of benzodiazapines) [3,7,15,26,27], lack of strong tube fixation, absence of IV sedation, and orotracheal intubation [6]. UE were more likely to occur on the night shift and among staff that included nurses with fewer years of experience [3]. Many episodes of UE occurred during weaning [10] or among patients who could communicate and were alert [3]. One study reports 57% of patients who intentionally self-extubated explained they simply removed the tube because it was uncomfortable [3].
Strategies for Reducing Adverse Events
Agitation Management
The majority of studies cited agitation, altered level of consciousness, or inadequate sedation as risk factors for UE [3,6–8,15,17,18,25,26,28,29]. These factors directly impact restraint use, another common risk factor for UE [3,7,17]. A key recommendation for agitation management is to identify the source of agitation, which is often caused by delirium onset in the ICU [30–32]. Prevalence of delirium in the ICU ranges from 20% to 80% [33–35]. ICU patients are at high risk for delirium due to sleep deprivation, older age, restraints, abnormal lab values, medications, infection, and respiratory complications [31]. Treatment for delirium centers on prevention, early recognition, interdisciplinary and pharmacologic protocols, increased nursing presence, and use of short-acting sedation when necessary [30–32,36]. While there is no research specifically linking delirium to UE, a quality analysis of risk factors present at the time of UE using bow-tie analysis methods identified delirium as a key factor present in the majority of UE cases [36]. It is possible that agitation reported in other studies investigating risk factors for UE may actually be reflective of underlying delirium. Routine screening using validated tools, such as the Confusion Assessment Method-ICU (CAM-ICU) [37] would aid in early detection and management of delirium, and would provide a standardized method for exploring the relationship of delirium and UE in future trials.
Integration of Weaning Protocols
Protocol-directed weaning is beneficial for decreasing ventilator days, time to wean from mechanical ventilation, and ICU LOS [38]. A systematic review including 7 trials (2434 patients) comparing protocol/non-protocol for weaning from mechanical ventilation reported a 26% decrease in the mean duration of mechanical ventilation for the protocol groups (95% CI 13%–37%, P < 0.001), a 70% reduction in time to wean, (95% CI 27%–88%, P = 0.009), and a decrease in ICU LOS by 11% (95% CI 3%–19%, P = 0.01). Weaning protocols are also an important risk factor for UE [5]. Findings from a prospective cohort study specifically identify the presence of weaning protocols as an important factor for reducing UE; patients who had weaning protocols ordered and followed were least likely to experience UE (P = 0.02) [5]. A separate quality improvement initiative demonstrated an overall decrease in the number of UEs (from 5.2% to 0.9%) after implementing weaning protocols as standard of care [39]. Considering many UEs occur during weaning [10], integration of weaning protocols aids in expediting the process and ensuring timely extubation.
Increased Surveillance
Increasing surveillance and monitoring of ventilated patients is a recommendation based on risk factors presented at the time of UE. Specifically, staffing levels and shifts and the use of physical restraints are variables associated with UE that are amendable to changes in unit processes based on increased surveillance. It is reported that 40% to 76% of UEs occurred during the night shift [14,17,24,40]; many more occur during change of shift or when there is not a nurse present at the bedside [3,17]. Recent trends towards mandatory bedside reporting is a specific intervention that may positively impact UE among patients in the ICU [41]. Meta-analyses of observational studies investigating the effect of nurse staffing on hospital outcomes indicate that increasing the number of RNs is associated with decreased risk of adverse patient outcomes, including UE [42,43]. The addition of 1 additional nurse per patient day can result in a 51% decrease in UE, while a decrease in nursing workload could result in a 45% decrease in UE [42]. Data from a national prevalence study reports ICUs with fewer available resources, including staff, experienced a higher number of UEs [4].
Increasing surveillance by nursing and health care staff may also impact prevalence of physical restraint use. A significant number of patients who experience UE are physically restrained at the time of the incident, ranging from 40% to 90% of intubated patients [5–7,14,17,40]. It is well documented that UE continue to occur despite the use of restraints [5,7,28,29,44] Patients who are physically restrained often experience higher rates of unplanned extubation (42.9% vs. 16.5% , P < 0.001 in Chang et al’s study [7]), and longer ICU LOS (20.3 days vs. 15.8 days, P = 0.009) [7]. Soft wrist restraints are commonly used to prevent pulling of the endotracheal tube; however, research evidence on UE demonstrates this is not always an effective intervention. Increasing surveillance of ventilated patients, treating their agitation and screening for underlying delirium, and integration of weaning protocols are all interventions that may decrease UE and the need for routine use of physical restraints.
Ongoing Education for Patients and Health Care Staff
Initial and ongoing education about UE, risk factors, and effective interventions is beneficial for patients and health care staff. Although there are no trials investigating effects of educational interventions for patients on UE outcomes, pre-education of surgical patients regarding what to expect while intubated may aid in decreasing delirium risk, agitation, physical restraint use, and possibly UE. Verbal and written educational information during pre-admission testing is a feasible method easily integrated into pre-operative programs.
Because UEs often occur more frequently among less experienced staff, initial education about risk factors for UE is crucial to include in ICU staff orientation programs [3,7]. Educational initiatives should incorporate training on routine delirium screening and avoidance of agitation, use of protocols, and increased surveillance of patients receiving mechanical ventilation [5,15,17,39,45]. Ongoing education of staff regarding ventilatory equipment and risk factors for UE can be particularly effective in decreasing UE [46]. Initial educational efforts should be followed by routine updates for all members of the healthcare team about ongoing quality improvement efforts to monitor UE. Associated factors for UE that may be unit- or process-specific, including methods for endotracheal tube securement and intra-hospital transport, should be communicated with all individuals involved in patient care. Integration of continuous quality improvement programs can decrease UE rates by 22% to 53% [16]. Quality efforts typically focus on standardization of reporting and tracking tools, protocol implementations, and ongoing monitoring, auditing, and recording of UE.
Current Trends and Future Directions
Recent trends in critical care recommendations may mitigate potential risk factors identified in UE research. Integration of lightened sedation and daily wake up periods for intubated patients may decrease prevalence of risk factors for UE, specifically agitation, physical restraint use, and altered level of consciousness [30], while routine weaning protocols may improve ventilatory outcomes, including UE [5,38,40]. Nursing bedside report and purposeful hourly rounding are quickly emerging as mainstays of professional nursing care [41]. Inherent in these 2 initiatives are increased surveillance and vigilance by health care staff, which can result in timely extubation of those who indicate readiness, as well as decreased incidence of adverse events. Delirium remains a key factor that may be a likely cause for UE; recent trends towards early detection and proper management of delirium among ICU staff may result in improved ventilatory outcomes, including weaning, planned extubation, and the prevalence of UE.
Another important trend in critical care is the emergence of a neurocritical care specialty and routine admission of neurocritically ill patients to neuroscience ICUs [47,48]. However, there are no studies investigating prevalence of UE among these patients, who often have higher rates of agitation or restlessness due to cognitive impairment. Among general ICUs, patients with a primary respiratory diagnosis accounted for 23% of all UE in one study, while those with a neurological diagnosis accounted for the second highest percentage (12%) among the study population [15]. A separate study concluded that presence of neurological injury with a concomitant nosocomial infection increased risk of UE among patients in a mixed ICU [7]. A recent systematic review of weaning protocols highlights positive effects on ventilatory outcomes but cites lack of evidence for effectiveness of protocols among those with neurological injury [38]. Areas for future UE research should include factors specific to this patient population, as they may be at higher risk for adverse ventilatory outcomes due to the nature of the neurological injury.
Conclusion
Prevention of UE remains an elusive target, evidenced by little change in reported rates over 2 decades. Research provides data on risk factors that may be patient, unit, or process related. Structuring prevention efforts around modifiable risk factors for UE is a feasible approach amenable to ongoing monitoring for effectiveness. Integration of current trends in health care safety and quality may produce an added benefit of reducing the occurrence of UE in critical care units. Future research evaluating these trends and the prevalence of UE in subspecialty populations is warranted.
Corresponding author: Molly McNett, PhD, RN, CNRN, Attn: NBO, MetroHealth Medical Center, 2500 MetroHealth Drive; Cleveland, OH 44109, mmcnett@metrohealth.org.
Financial disclosures: None.
1. Krinsley JS, Barone JE. The drive to survive: unplanned extubation in the ICU. Chest 2005;128:560–6.
2. Coppolo DP, May JJ. Self-extubations. A 12-month experience. Chest 1990;98:165–9.
3. Yeh SH, Lee LN, Ho TH, et al. Implications of nursing care in the occurrence and consequences of unplanned extubation in adult intensive care units. Int J Nurs Stud 2004;41:255–62.
4. Mion LC, Minnick AF, Leipzig R, et al. Patient-initiated device removal in intensive care units: a national prevalence study. Crit Care Med 2007;35:2714–20.
5. Jarachovic M, Mason M, Kerber K. The role of standardized protocols in unplanned extubations in a medical intensive care unit. Am J Crit Care 2011;20:304–11.
6. Boulain T. Unplanned extubations in the adult intensive care unit: a prospective multicenter study. Association des Reanimateurs du Centre-Ouest. Am J Resp Crit Care Med 1998;157(4 Pt 1):1131–7.
7. Chang LY, Wang KW, Chao YF. Influence of physical restraint on unplanned extubation of adult intensive care patients: a case-control study. Am J Crit Care 2008;17:408–15.
8. Moons P, Sels K, De Becker W, et al. Development of a risk assessment tool for deliberate self-extubation in intensive care patients. Intensive Care Med 2004;30:1348–55.
9. Chiang AA, Lee KC, Lee JC, Wei CH. Effectiveness of a continuous quality improvement program aiming to reduce unplanned extubation: a prospective study. Intensive Care Med 1996;22:1269–71.
10. Betbese AJ, Perez M, Bak E, et al. A prospective study of unplanned endotracheal extubation in intensive care unit patients. Crit Care Med 1998;26:1180–6.
11. de Lassence A, Alberti C, Azoulay E, et al. Impact of unplanned extubation and reintubation after weaning on nosocomial pneumonia risk in the intensive care unit: a prospective multicenter study. Anesthesiology 2002;97:148–56.
12. Kapadia FN, Tekawade PC, Nath SS, et al. A prolonged observational study of tracheal tube displacements: Benchmarking an incidence <0.5-1% in a medical-surgical adult intensive care unit. Ind J Crit Care Med 2014;18:273–7.
13. Lee JH, Lee HC, Jeon YT, et al. Clinical outcomes after unplanned extubation in a surgical intensive care population. World J Surg 2014;38:203–10.
14. Chang LC, Liu PF, Huang YL, et al. Risk factors associated with unplanned endotracheal self-extubation of hospitalized intubated patients: a 3-year retrospective case-control study. Appl Nurs Res 2011;24:188–92.
15. de Groot RI, Dekkers OM, Herold IH, et al. Risk factors and outcomes after unplanned extubations on the ICU: a case-control study. Crit Care 2011;15:R19.
16. da Silva PS, Fonseca MC. Unplanned endotracheal extubations in the intensive care unit: systematic review, critical appraisal, and evidence-based recommendations. Anesth Analg 2012;114:1003–14.
17. Curry K, Cobb S, Kutash M, Diggs C. Characteristics associated with unplanned extubations in a surgical intensive care unit. Am J Crit Care 2008;17:45–51.
18. Christie JM, Dethlefsen M, Cane RD. Unplanned endotracheal extubation in the intensive care unit. J Clin Anesth 1996;8:289–93.
19. Huang YT. Factors leading to self-extubation of endotracheal tubes in the intensive care unit. Nurs Crit Care 2009;14:68–74.
20. Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet 2008;371:126–34.
21. Mort TC. Unplanned tracheal extubation outside the operating room: a quality improvement audit of hemodynamic and tracheal airway complications associated with emergency tracheal reintubation. Anesth Analg 1998;86:1171–6.
22. Jaber S, Chanques G, Altairac C, et al. A prospective study of agitation in a medical-surgical ICU: incidence, risk factors, and outcomes. Chest 2005;128:2749–57.
23. Roddy DJ, Spaeder MC, Pastor W, Stockwell DC, Klugman D. Unplanned extubations in children: impact on hospital cost and length of stay. Ped Crit Care Med 2015.
24. Bouza C, Garcia E, Diaz M, et al. Unplanned extubation in orally intubated medical patients in the intensive care unit: a prospective cohort study. Heart Lung 2007;36:270–6.
25. Vassal T, Anh NG, Gabillet JM, et al. Prospective evaluation of self-extubations in a medical intensive care unit. Intensive Care Med 1993;19:340-342.
26. Tung A, Tadimeti L, Caruana-Montaldo B, et al. The relationship of sedation to deliberate self-extubation. J Clin Anesth 2001;13:24–9.
27. Tanios M, Epstein S, Grzeskowiak M, et al. Influence of sedation strategies on unplanned extubation in a mixed intensive care unit. Am J Crit Care 2014;23:306–14.
28. Atkins PM, Mion LC, Mendelson W, et al. Characteristics and outcomes of patients who self-extubate from ventilatory support: a case-control study. Chest 1997;112:1317–23.
29. Chevron V, Menard JF, Richard JC, et al. Unplanned extubation: risk factors of development and predictive criteria for reintubation. Crit Care Med 1998;26:1049–53.
30. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med 2013;41:263–306.
31. Morandi A, Jackson JC. Delirium in the intensive care unit: a review. Neurol Clin 2011;29:749–63.
32. Banerjee A, Vasilevskis, EE, Pandharipande, P. Strategies to improve delirium assessment practices in the intensive care unit. J Clin Outcomes Manag 2010;17:459–68.
33. Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001;286:2703–10.
34. Ely EW, Stephens RK, Jackson JC, et al. Current opinions regarding the importance, diagnosis, and management of delirium in the intensive care unit: a survey of 912 healthcare professionals. Crit Care Med 2004;32:106–12.
35. McNicoll L, Pisani MA, Zhang Y, et al. Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc 2003;51:591–8.
36. Kerckhoffs MC, van der Sluijs AF, Binnekade JM, Dongelmans DA. Improving patient safety in the ICU by prospective identification of missing safety barriers using the bow-tie prospective risk analysis model. J Patient Safe 2013;9:154–9.
37. Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med 1990;113:941–8.
38. Blackwood B, Burns KE, Cardwell CR, O’Halloran P. Protocolized versus non-protocolized weaning for reducing the duration of mechanical ventilation in critically ill adult patients. Cochrane Database Syst Rev 2014;11:CD006904.
39. Chia PL, Santos DR, Tan TC, et al. Clinical quality improvement: eliminating unplanned extubation in the CCU. Int J Health Care Qual Ass 2013;26:642–52.
40. Balon JA. Common factors of spontaneous self-extubation in a critical care setting. Int J Trauma Nurs 2001;7:93–9.
41. Gregory S, Tan D, Tilrico M, et al. Bedside shift reports: what does the evidence say? J Nurs Admin 2014;44:541–5.
42. Kane RL, Shamliyan TA, Mueller C, et al. The association of registered nurse staffing levels and patient outcomes: systematic review and meta-analysis. Med Care 2007;45:1195–204.
43. Penoyer DA. Nurse staffing and patient outcomes in critical care: a concise review. Crit Care Med 2010;38:1521–8; quiz 1529.
44. Tindol GA, Jr., DiBenedetto RJ, Kosciuk L. Unplanned extubations. Chest 1994;105:1804–7.
45. Chen CM CK, Fong Y, Hsing SC, et al. Age is an important predictor of failed unplanned extubation. Int J Gerontol 2010;4:120–9.
46. Richmond AL, Jarog DL, Hanson VM. Unplanned extubation in adult critical care. Quality improvement and education payoff. Crit Care Nurs 2004;24:32–7.
47. Kurtz P, Fitts V, Sumer Z, et al. How does care differ for neurological patients admitted to a neurocritical care unit versus a general ICU? Neurocrit Care 2011;15:477–80.
48. McNett MM, Horowitz DA. International multidisciplinary consensus conference on multimodality monitoring: ICU processes of care. Neurocrit Care 2014;21 Suppl 2:S215–28.
49. Gardner A, Hughes, D, Cook R, et al. Best practice in stabilisation of oral endotracheal tubes: a systematic review. Database of abstracts of reivews of effects (DARE): Quality-assessed reviews. 2005. York: Center for Reviews and Dissemination.
50. Hofso K, Coyer FM. Part 1: Chemical and physical restraints in the management of mechanically ventliated patients in the ICU: Contributing factors. Intensive Crit Care Nurs 2007; 23:249–55.
51. Kiekkas P, Diamanto A, Panteli E, et al. Unplanned extubation in critially ill adults: Clinical reviews. Nurs Crit Care 2012;18:123–34.
52. King JN, Elliiot VA. Self/unplanned extubation: Safety, surveillance, and monitoring of the mechanically ventilated patient. Crit Care Nurs Clin North Am 2012;24:469–79.
1. Krinsley JS, Barone JE. The drive to survive: unplanned extubation in the ICU. Chest 2005;128:560–6.
2. Coppolo DP, May JJ. Self-extubations. A 12-month experience. Chest 1990;98:165–9.
3. Yeh SH, Lee LN, Ho TH, et al. Implications of nursing care in the occurrence and consequences of unplanned extubation in adult intensive care units. Int J Nurs Stud 2004;41:255–62.
4. Mion LC, Minnick AF, Leipzig R, et al. Patient-initiated device removal in intensive care units: a national prevalence study. Crit Care Med 2007;35:2714–20.
5. Jarachovic M, Mason M, Kerber K. The role of standardized protocols in unplanned extubations in a medical intensive care unit. Am J Crit Care 2011;20:304–11.
6. Boulain T. Unplanned extubations in the adult intensive care unit: a prospective multicenter study. Association des Reanimateurs du Centre-Ouest. Am J Resp Crit Care Med 1998;157(4 Pt 1):1131–7.
7. Chang LY, Wang KW, Chao YF. Influence of physical restraint on unplanned extubation of adult intensive care patients: a case-control study. Am J Crit Care 2008;17:408–15.
8. Moons P, Sels K, De Becker W, et al. Development of a risk assessment tool for deliberate self-extubation in intensive care patients. Intensive Care Med 2004;30:1348–55.
9. Chiang AA, Lee KC, Lee JC, Wei CH. Effectiveness of a continuous quality improvement program aiming to reduce unplanned extubation: a prospective study. Intensive Care Med 1996;22:1269–71.
10. Betbese AJ, Perez M, Bak E, et al. A prospective study of unplanned endotracheal extubation in intensive care unit patients. Crit Care Med 1998;26:1180–6.
11. de Lassence A, Alberti C, Azoulay E, et al. Impact of unplanned extubation and reintubation after weaning on nosocomial pneumonia risk in the intensive care unit: a prospective multicenter study. Anesthesiology 2002;97:148–56.
12. Kapadia FN, Tekawade PC, Nath SS, et al. A prolonged observational study of tracheal tube displacements: Benchmarking an incidence <0.5-1% in a medical-surgical adult intensive care unit. Ind J Crit Care Med 2014;18:273–7.
13. Lee JH, Lee HC, Jeon YT, et al. Clinical outcomes after unplanned extubation in a surgical intensive care population. World J Surg 2014;38:203–10.
14. Chang LC, Liu PF, Huang YL, et al. Risk factors associated with unplanned endotracheal self-extubation of hospitalized intubated patients: a 3-year retrospective case-control study. Appl Nurs Res 2011;24:188–92.
15. de Groot RI, Dekkers OM, Herold IH, et al. Risk factors and outcomes after unplanned extubations on the ICU: a case-control study. Crit Care 2011;15:R19.
16. da Silva PS, Fonseca MC. Unplanned endotracheal extubations in the intensive care unit: systematic review, critical appraisal, and evidence-based recommendations. Anesth Analg 2012;114:1003–14.
17. Curry K, Cobb S, Kutash M, Diggs C. Characteristics associated with unplanned extubations in a surgical intensive care unit. Am J Crit Care 2008;17:45–51.
18. Christie JM, Dethlefsen M, Cane RD. Unplanned endotracheal extubation in the intensive care unit. J Clin Anesth 1996;8:289–93.
19. Huang YT. Factors leading to self-extubation of endotracheal tubes in the intensive care unit. Nurs Crit Care 2009;14:68–74.
20. Girard TD, Kress JP, Fuchs BD, et al. Efficacy and safety of a paired sedation and ventilator weaning protocol for mechanically ventilated patients in intensive care (Awakening and Breathing Controlled trial): a randomised controlled trial. Lancet 2008;371:126–34.
21. Mort TC. Unplanned tracheal extubation outside the operating room: a quality improvement audit of hemodynamic and tracheal airway complications associated with emergency tracheal reintubation. Anesth Analg 1998;86:1171–6.
22. Jaber S, Chanques G, Altairac C, et al. A prospective study of agitation in a medical-surgical ICU: incidence, risk factors, and outcomes. Chest 2005;128:2749–57.
23. Roddy DJ, Spaeder MC, Pastor W, Stockwell DC, Klugman D. Unplanned extubations in children: impact on hospital cost and length of stay. Ped Crit Care Med 2015.
24. Bouza C, Garcia E, Diaz M, et al. Unplanned extubation in orally intubated medical patients in the intensive care unit: a prospective cohort study. Heart Lung 2007;36:270–6.
25. Vassal T, Anh NG, Gabillet JM, et al. Prospective evaluation of self-extubations in a medical intensive care unit. Intensive Care Med 1993;19:340-342.
26. Tung A, Tadimeti L, Caruana-Montaldo B, et al. The relationship of sedation to deliberate self-extubation. J Clin Anesth 2001;13:24–9.
27. Tanios M, Epstein S, Grzeskowiak M, et al. Influence of sedation strategies on unplanned extubation in a mixed intensive care unit. Am J Crit Care 2014;23:306–14.
28. Atkins PM, Mion LC, Mendelson W, et al. Characteristics and outcomes of patients who self-extubate from ventilatory support: a case-control study. Chest 1997;112:1317–23.
29. Chevron V, Menard JF, Richard JC, et al. Unplanned extubation: risk factors of development and predictive criteria for reintubation. Crit Care Med 1998;26:1049–53.
30. Barr J, Fraser GL, Puntillo K, et al. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med 2013;41:263–306.
31. Morandi A, Jackson JC. Delirium in the intensive care unit: a review. Neurol Clin 2011;29:749–63.
32. Banerjee A, Vasilevskis, EE, Pandharipande, P. Strategies to improve delirium assessment practices in the intensive care unit. J Clin Outcomes Manag 2010;17:459–68.
33. Ely EW, Inouye SK, Bernard GR, et al. Delirium in mechanically ventilated patients: validity and reliability of the confusion assessment method for the intensive care unit (CAM-ICU). JAMA 2001;286:2703–10.
34. Ely EW, Stephens RK, Jackson JC, et al. Current opinions regarding the importance, diagnosis, and management of delirium in the intensive care unit: a survey of 912 healthcare professionals. Crit Care Med 2004;32:106–12.
35. McNicoll L, Pisani MA, Zhang Y, et al. Delirium in the intensive care unit: occurrence and clinical course in older patients. J Am Geriatr Soc 2003;51:591–8.
36. Kerckhoffs MC, van der Sluijs AF, Binnekade JM, Dongelmans DA. Improving patient safety in the ICU by prospective identification of missing safety barriers using the bow-tie prospective risk analysis model. J Patient Safe 2013;9:154–9.
37. Inouye SK, van Dyck CH, Alessi CA, et al. Clarifying confusion: the confusion assessment method. A new method for detection of delirium. Ann Intern Med 1990;113:941–8.
38. Blackwood B, Burns KE, Cardwell CR, O’Halloran P. Protocolized versus non-protocolized weaning for reducing the duration of mechanical ventilation in critically ill adult patients. Cochrane Database Syst Rev 2014;11:CD006904.
39. Chia PL, Santos DR, Tan TC, et al. Clinical quality improvement: eliminating unplanned extubation in the CCU. Int J Health Care Qual Ass 2013;26:642–52.
40. Balon JA. Common factors of spontaneous self-extubation in a critical care setting. Int J Trauma Nurs 2001;7:93–9.
41. Gregory S, Tan D, Tilrico M, et al. Bedside shift reports: what does the evidence say? J Nurs Admin 2014;44:541–5.
42. Kane RL, Shamliyan TA, Mueller C, et al. The association of registered nurse staffing levels and patient outcomes: systematic review and meta-analysis. Med Care 2007;45:1195–204.
43. Penoyer DA. Nurse staffing and patient outcomes in critical care: a concise review. Crit Care Med 2010;38:1521–8; quiz 1529.
44. Tindol GA, Jr., DiBenedetto RJ, Kosciuk L. Unplanned extubations. Chest 1994;105:1804–7.
45. Chen CM CK, Fong Y, Hsing SC, et al. Age is an important predictor of failed unplanned extubation. Int J Gerontol 2010;4:120–9.
46. Richmond AL, Jarog DL, Hanson VM. Unplanned extubation in adult critical care. Quality improvement and education payoff. Crit Care Nurs 2004;24:32–7.
47. Kurtz P, Fitts V, Sumer Z, et al. How does care differ for neurological patients admitted to a neurocritical care unit versus a general ICU? Neurocrit Care 2011;15:477–80.
48. McNett MM, Horowitz DA. International multidisciplinary consensus conference on multimodality monitoring: ICU processes of care. Neurocrit Care 2014;21 Suppl 2:S215–28.
49. Gardner A, Hughes, D, Cook R, et al. Best practice in stabilisation of oral endotracheal tubes: a systematic review. Database of abstracts of reivews of effects (DARE): Quality-assessed reviews. 2005. York: Center for Reviews and Dissemination.
50. Hofso K, Coyer FM. Part 1: Chemical and physical restraints in the management of mechanically ventliated patients in the ICU: Contributing factors. Intensive Crit Care Nurs 2007; 23:249–55.
51. Kiekkas P, Diamanto A, Panteli E, et al. Unplanned extubation in critially ill adults: Clinical reviews. Nurs Crit Care 2012;18:123–34.
52. King JN, Elliiot VA. Self/unplanned extubation: Safety, surveillance, and monitoring of the mechanically ventilated patient. Crit Care Nurs Clin North Am 2012;24:469–79.
Increase in the Incidence of Hypertensive Emergency Syndrome?
Study Overview
Objective. To investigate national trends in hospital admissions for malignant hypertension and hypertensive encephalopathy.
Study design. Retrospective cohort study. The Nation-wide Inpatient Sample [1] was used to identify all hospitalizations between January 2000 and December 2011 during which a primary diagnosis of malignant hypertension, hypertensive encephalopathy, or essential hypertension occurred. Time series models were estimated for these diagnoses and also for the combined series. A piecewise linear regression analysis was done to investigate whether there were changes in the trends of these series. In addition, the researchers compared patient characteristics.
Results. There was a gradual increase in the number of hypertension-related hospitalizations from 2000 to 2011. However, after 2007, the number of admissions for malignant hypertension and hypertensive encephalopathy increased dramatically, whereas diagnoses for essential hypertension fell (P < 0.001). Mortality for malignant hypertension significantly fell after the change point of 2007 (–36%, P = 0.02) but there was no significant difference in mortality for hypertensive encephalopathy or essential hypertension. The number of diagnoses and the adjusted average charges significantly increased after the change point for all hypertension series, although the increase in malignant hypertension and hypertensive encephalopathy was higher than in essential hypertension. Length of stay significantly decreased after 2007 for all series. Mean patient age and number of procedures for all series were similar before and after the change point.
Conclusion. Since the dramatic increase in the number of hospital admissions did not result in dramatic increases in morbidity, which would have been expected, the increase was most likely related to a change in coding practices that was implemented in 2007 and not actual changes in disease incidence.
Commentary
Hypertension is a major public health problem associated with significant morbidity and mortality [2]. In general, hypertension is asymptomatic; however, life-threatening manifestations of hypertension can develop. A hypertensive emergency is a situation in which uncontrolled hypertension is associated with acute end-organ damage. Most patients presenting with hypertensive emergency have chronic hypertension, although the disorder can present in previously normotensive individuals [3]. The 2 major emergency syndromes are malignant hypertension and hypertensive encephalopathy. They usually require hospitalization, and therefore monitoring trends in admissions for these conditions is a reasonable population-based indicator for failures related to hypertension management.
In this epidemiologic study by Polgreen et al, the authors found a increasing trend in admissions for malig-nant hypertension and hypertensive emergencies, with a substantial increase after 2007. Although the authors considered the possibility that their findings represented a true change in the epidemiology of hypertensive emergencies, they concluded that this appears unlikely, as the diagnoses of essential hypertension fell, and in addition, an expected associated increase in morbidity was not seen. They attribute the shift to a change in assignment of administrative billing codes. In 2007, DRG codes were changed to medical severity DRG codes [4]. The authors acknowledge that there was a recession from 2007 to 2009 that led to an increase in the number of uninsured Americans [5]. However, they noted that the uninsured were no more likely to be diagnosed with malignant hypertension or hypertensive encephalopathy than essential hypertension and there is no reason to think that a change provider’s management of hypertension could have been responsible.
Limitations to this study were the use of administrative data only and the lack of data on outpatient medication use.
Applications for Clinical Practice
As the authors suggest, the study raised questions regarding the use of administrative data for monitoring hypertension outcomes. Future studies are needed to examine whether the rise in diagnoses for malignant hypertension and hypertensive encephalopathy are related to coding practices or other variables.
—Paloma Cesar de Sales, BN, RN, MS
1. Nationwide Inpatient Sample overview. Available at www.hcup-us.ahrq.gov/nisoverview.jsp.
2. American Heart Association. High blood pressure: statistical fact sheet 2013 Update. Available at www.heart.org.
3. Vaughan CJ, Delanty N. Hypertensive emergencies. Lancet 2000;356:411–7.
4. Centers for Medicare and Medicaid Services. Acute care hospital inpatient prospective payment system. Payment system fact sheet series. April 2013. Available at www.cms.gov/outreach-and-education/.
5. Holahan J. The 2007-09 recession and health insurance coverage. Health Aff (Millwood) 2011;30:145–52.
Study Overview
Objective. To investigate national trends in hospital admissions for malignant hypertension and hypertensive encephalopathy.
Study design. Retrospective cohort study. The Nation-wide Inpatient Sample [1] was used to identify all hospitalizations between January 2000 and December 2011 during which a primary diagnosis of malignant hypertension, hypertensive encephalopathy, or essential hypertension occurred. Time series models were estimated for these diagnoses and also for the combined series. A piecewise linear regression analysis was done to investigate whether there were changes in the trends of these series. In addition, the researchers compared patient characteristics.
Results. There was a gradual increase in the number of hypertension-related hospitalizations from 2000 to 2011. However, after 2007, the number of admissions for malignant hypertension and hypertensive encephalopathy increased dramatically, whereas diagnoses for essential hypertension fell (P < 0.001). Mortality for malignant hypertension significantly fell after the change point of 2007 (–36%, P = 0.02) but there was no significant difference in mortality for hypertensive encephalopathy or essential hypertension. The number of diagnoses and the adjusted average charges significantly increased after the change point for all hypertension series, although the increase in malignant hypertension and hypertensive encephalopathy was higher than in essential hypertension. Length of stay significantly decreased after 2007 for all series. Mean patient age and number of procedures for all series were similar before and after the change point.
Conclusion. Since the dramatic increase in the number of hospital admissions did not result in dramatic increases in morbidity, which would have been expected, the increase was most likely related to a change in coding practices that was implemented in 2007 and not actual changes in disease incidence.
Commentary
Hypertension is a major public health problem associated with significant morbidity and mortality [2]. In general, hypertension is asymptomatic; however, life-threatening manifestations of hypertension can develop. A hypertensive emergency is a situation in which uncontrolled hypertension is associated with acute end-organ damage. Most patients presenting with hypertensive emergency have chronic hypertension, although the disorder can present in previously normotensive individuals [3]. The 2 major emergency syndromes are malignant hypertension and hypertensive encephalopathy. They usually require hospitalization, and therefore monitoring trends in admissions for these conditions is a reasonable population-based indicator for failures related to hypertension management.
In this epidemiologic study by Polgreen et al, the authors found a increasing trend in admissions for malig-nant hypertension and hypertensive emergencies, with a substantial increase after 2007. Although the authors considered the possibility that their findings represented a true change in the epidemiology of hypertensive emergencies, they concluded that this appears unlikely, as the diagnoses of essential hypertension fell, and in addition, an expected associated increase in morbidity was not seen. They attribute the shift to a change in assignment of administrative billing codes. In 2007, DRG codes were changed to medical severity DRG codes [4]. The authors acknowledge that there was a recession from 2007 to 2009 that led to an increase in the number of uninsured Americans [5]. However, they noted that the uninsured were no more likely to be diagnosed with malignant hypertension or hypertensive encephalopathy than essential hypertension and there is no reason to think that a change provider’s management of hypertension could have been responsible.
Limitations to this study were the use of administrative data only and the lack of data on outpatient medication use.
Applications for Clinical Practice
As the authors suggest, the study raised questions regarding the use of administrative data for monitoring hypertension outcomes. Future studies are needed to examine whether the rise in diagnoses for malignant hypertension and hypertensive encephalopathy are related to coding practices or other variables.
—Paloma Cesar de Sales, BN, RN, MS
Study Overview
Objective. To investigate national trends in hospital admissions for malignant hypertension and hypertensive encephalopathy.
Study design. Retrospective cohort study. The Nation-wide Inpatient Sample [1] was used to identify all hospitalizations between January 2000 and December 2011 during which a primary diagnosis of malignant hypertension, hypertensive encephalopathy, or essential hypertension occurred. Time series models were estimated for these diagnoses and also for the combined series. A piecewise linear regression analysis was done to investigate whether there were changes in the trends of these series. In addition, the researchers compared patient characteristics.
Results. There was a gradual increase in the number of hypertension-related hospitalizations from 2000 to 2011. However, after 2007, the number of admissions for malignant hypertension and hypertensive encephalopathy increased dramatically, whereas diagnoses for essential hypertension fell (P < 0.001). Mortality for malignant hypertension significantly fell after the change point of 2007 (–36%, P = 0.02) but there was no significant difference in mortality for hypertensive encephalopathy or essential hypertension. The number of diagnoses and the adjusted average charges significantly increased after the change point for all hypertension series, although the increase in malignant hypertension and hypertensive encephalopathy was higher than in essential hypertension. Length of stay significantly decreased after 2007 for all series. Mean patient age and number of procedures for all series were similar before and after the change point.
Conclusion. Since the dramatic increase in the number of hospital admissions did not result in dramatic increases in morbidity, which would have been expected, the increase was most likely related to a change in coding practices that was implemented in 2007 and not actual changes in disease incidence.
Commentary
Hypertension is a major public health problem associated with significant morbidity and mortality [2]. In general, hypertension is asymptomatic; however, life-threatening manifestations of hypertension can develop. A hypertensive emergency is a situation in which uncontrolled hypertension is associated with acute end-organ damage. Most patients presenting with hypertensive emergency have chronic hypertension, although the disorder can present in previously normotensive individuals [3]. The 2 major emergency syndromes are malignant hypertension and hypertensive encephalopathy. They usually require hospitalization, and therefore monitoring trends in admissions for these conditions is a reasonable population-based indicator for failures related to hypertension management.
In this epidemiologic study by Polgreen et al, the authors found a increasing trend in admissions for malig-nant hypertension and hypertensive emergencies, with a substantial increase after 2007. Although the authors considered the possibility that their findings represented a true change in the epidemiology of hypertensive emergencies, they concluded that this appears unlikely, as the diagnoses of essential hypertension fell, and in addition, an expected associated increase in morbidity was not seen. They attribute the shift to a change in assignment of administrative billing codes. In 2007, DRG codes were changed to medical severity DRG codes [4]. The authors acknowledge that there was a recession from 2007 to 2009 that led to an increase in the number of uninsured Americans [5]. However, they noted that the uninsured were no more likely to be diagnosed with malignant hypertension or hypertensive encephalopathy than essential hypertension and there is no reason to think that a change provider’s management of hypertension could have been responsible.
Limitations to this study were the use of administrative data only and the lack of data on outpatient medication use.
Applications for Clinical Practice
As the authors suggest, the study raised questions regarding the use of administrative data for monitoring hypertension outcomes. Future studies are needed to examine whether the rise in diagnoses for malignant hypertension and hypertensive encephalopathy are related to coding practices or other variables.
—Paloma Cesar de Sales, BN, RN, MS
1. Nationwide Inpatient Sample overview. Available at www.hcup-us.ahrq.gov/nisoverview.jsp.
2. American Heart Association. High blood pressure: statistical fact sheet 2013 Update. Available at www.heart.org.
3. Vaughan CJ, Delanty N. Hypertensive emergencies. Lancet 2000;356:411–7.
4. Centers for Medicare and Medicaid Services. Acute care hospital inpatient prospective payment system. Payment system fact sheet series. April 2013. Available at www.cms.gov/outreach-and-education/.
5. Holahan J. The 2007-09 recession and health insurance coverage. Health Aff (Millwood) 2011;30:145–52.
1. Nationwide Inpatient Sample overview. Available at www.hcup-us.ahrq.gov/nisoverview.jsp.
2. American Heart Association. High blood pressure: statistical fact sheet 2013 Update. Available at www.heart.org.
3. Vaughan CJ, Delanty N. Hypertensive emergencies. Lancet 2000;356:411–7.
4. Centers for Medicare and Medicaid Services. Acute care hospital inpatient prospective payment system. Payment system fact sheet series. April 2013. Available at www.cms.gov/outreach-and-education/.
5. Holahan J. The 2007-09 recession and health insurance coverage. Health Aff (Millwood) 2011;30:145–52.
More Evidence That a High-Fiber Diet May Prevent Type 2 Diabetes
Study Overview
Objective. To evaluate the association between intake of dietary fiber and type 2 diabetes.
Design. Case-cohort study (EPIC-InterAct Study), which is nested within the large prospective cohort study EPIC (European Prospective Investigation into Cancer & Nutrition) [1]. EPIC includes participants from 10 European countries and was designed to investigate the relationships between diet, nutritional status, lifestyle, and environmental factors and the incidence of cancer and other chronic disease [1].
Setting and participants. The EPIC-Interact study used data from 8 European countries (Denmark, France, Germany, Italy, the Netherlands, Spain, Sweden, and UK). The Interact sample includes 12,403 individuals who were identified as having developed type 2 diabetes, and a random subcohort of controls who were free of diabetes at baseline (n = 16,835, including 778 cases of incident diabetes) selected from 340,234 eligible EPIC participants. Of the 28,460 participants in the EPIC-InterAct study, excluded were those with prevalent diabetes, missing diabetes status information, post-censoring diabetes, extreme energy intake (top and bottom 1%), and missing values for education level, physical activity, smoking status, and BMI, leaving a final sample of 11,559 cases and 15,258 subcohort participants. No differences were observed in baseline characteristics between the included and excluded participants.
Analysis. Country-specific hazard ratios (HRs) were estimated using Prentice-weighted Cox proportional hazards models and were pooled using a random effects meta-analysis. Dietary intake over the previous 12 months before recruitment was assessed by country-specific or center-specific assessment methods (food-frequency questionnaire and dietary histories) that were developed and validated locally, and data were converted to nutrient intake.
Main outcome measure. Incident cases of diabetes.
Main results. During a median of 10.8 years of follow-up, total fiber intake was associated with a lower risk of diabetes after adjusting for lifestyle and dietary factors (hazard ratio for the highest quartile of fiber intake [> 26 g/day] vs the lowest [< 19 g/day], 0.82; 95% confidence interval, 0.69–0.97, P for trend = 0.02). When the researchers focused on specific types of fiber, they found that people who consumed the highest amounts of cereal and vegetable fiber were 19% and 16%, respectively, less likely to develop type 2 diabetes compared with those who consumed the lowest amounts (P < 0.001). Intake of fruit fiber was not associated with risk of diabetes. When the analyses were additionally adjusted for BMI, the inverse associations between intake of fiber and diabetes were attenuated and no longer statistically significant.
The researchers also conducted a meta-analysis that included 18 other cohorts in addition to the current EPIC-InterAct study. The summary relative risks per 10/g day increase in intake were 0.91 (95% CI, 0.87–0.96) for total fiber, 0.75 (95% CI, 0.65–0.86) for cereal fiber, 0.95 (95% CI, 0.87–1.03) for fruit fiber, and 0.93 (95% CI 0.82–1.05) for vegetable fiber.
Conclusion. Individuals with diets rich in fiber, in particular cereal fiber, may be at lower risk of type 2 diabetes.
Commentary
The current study by the European InterAct Consortium adds to the available evidence supporting the association of dietary fiber and risk of diabetes. Higher intake of dietary fiber, especially cereal fiber, has been consistently associated with a lower risk of diabetes [2,3].
This study showed that a high intake of total fiber (primarily cereal and vegetable fiber) was associated with an 18% lower risk of type 2 diabetes (adjusted for dietary and lifestyle factors). While there was no association after adjusting for BMI, their meta-analysis of 18 studies did support the an inverse association between total fiber and cereal fiber intake and risk of type 2 diabetes.
What is it about fiber that is protective? With regard to whole grains, a rich source of fiber, potential mechanisms have been identified [5], including the possible impact of improved postprandial glucose response. However, whole grains are rich in nutrients and phytochemicals, and new hypotheses for the health-protective mechanisms of whole grains beyond fiber are being proposed [6]. In addition, the beneficial effect of fiber seen in this and other studies may be partly mediated by a lower BMI. Dietary fiber may affect appetite and energy intake through a range of processes.
It should be noted that although the effect of whole-grain foods for the prevention of type 2 diabetes is strongly supported by numerous epidemiological studies, a 2008 systematic review by the Cochrane Collaboration [4], which included 1 low-quality RCT and 11 cohort studies, stated that the evidence is too weak to be able to draw a definite conclusion about the preventive effect of this dietary factor.
Strengths of the study included its prospective design and large sample size. Limitations of the study include that dietary intake was assessed only at baseline and measurement error through the use of a questionnaire may have occurred. Although food-frequency questionnaires are widely used, they are subjective estimates and subject to recall bias, and some researchers have questioned their value for use in epidemiologic studies [7]. In addition, the authors note that the inverse association for total fiber and cereal fiber intake in the meta-analysis could be due to residual confounding as fiber intake has been associated with healthier diet, lower BMI, and greater physical activity.
Applications for Clinical Practice
The prevalence of type 2 diabetes has increased rapidly during the past decades in the United States. Dietary guidelines recommend the consumption of whole grains to prevent chronic diseases. The results presented in the current study strengthen the evidence supporting cereal fiber as an important determinant of type 2 diabetes risk. Randomized controlled trials are needed and should elucidate this matter.
1. Riboli E, Hunt KJ, Slimani N, European Prospective Investigation into Cancer and Nutrition (EPIC): study populations and data collection. Public Health Nutr 2002;5(6B):1113–24.
2. Ye EQ, Chacko SA, Chou EL, et al. Greater whole-grain intake is associated with lower risk of type 2 diabetes, cardiovascular disease, and weight gain. J Nutr 2012;142:1304–13.
3. Huang T, Xu M, Lee A, et al. Consumption of whole grains and cereal fiber and total and cause-specific mortality: prospective analysis of 367,442 individuals. BMC Med 2015;13:59.
4. Priebe MG, van Binsbergen JJ, de Vos R, Vonk RJ. Whole grain foods for theprevention of type 2 diabetes mellitus. Cochrane Database Syst Rev 2008;(1):CD006061.
5. Slavin JL, Martini MC, Jacobs DR Jr, Marquart L. Plausible mechanisms for the protectiveness of whole grains. Am J Clin Nutr 1999;70(3 Suppl):459S–463S.
6. Fardet A. New hypotheses for the health-protective mechanisms of whole-grain cereals: what is beyond fibre? Nutr Res Rev 2010;23:65–134.
7. Shim J-S, Oh K, Kim HC. Dietary assessment methods in epidemiologic studies. Epidemiol Health 2014;36:e2014009.
Study Overview
Objective. To evaluate the association between intake of dietary fiber and type 2 diabetes.
Design. Case-cohort study (EPIC-InterAct Study), which is nested within the large prospective cohort study EPIC (European Prospective Investigation into Cancer & Nutrition) [1]. EPIC includes participants from 10 European countries and was designed to investigate the relationships between diet, nutritional status, lifestyle, and environmental factors and the incidence of cancer and other chronic disease [1].
Setting and participants. The EPIC-Interact study used data from 8 European countries (Denmark, France, Germany, Italy, the Netherlands, Spain, Sweden, and UK). The Interact sample includes 12,403 individuals who were identified as having developed type 2 diabetes, and a random subcohort of controls who were free of diabetes at baseline (n = 16,835, including 778 cases of incident diabetes) selected from 340,234 eligible EPIC participants. Of the 28,460 participants in the EPIC-InterAct study, excluded were those with prevalent diabetes, missing diabetes status information, post-censoring diabetes, extreme energy intake (top and bottom 1%), and missing values for education level, physical activity, smoking status, and BMI, leaving a final sample of 11,559 cases and 15,258 subcohort participants. No differences were observed in baseline characteristics between the included and excluded participants.
Analysis. Country-specific hazard ratios (HRs) were estimated using Prentice-weighted Cox proportional hazards models and were pooled using a random effects meta-analysis. Dietary intake over the previous 12 months before recruitment was assessed by country-specific or center-specific assessment methods (food-frequency questionnaire and dietary histories) that were developed and validated locally, and data were converted to nutrient intake.
Main outcome measure. Incident cases of diabetes.
Main results. During a median of 10.8 years of follow-up, total fiber intake was associated with a lower risk of diabetes after adjusting for lifestyle and dietary factors (hazard ratio for the highest quartile of fiber intake [> 26 g/day] vs the lowest [< 19 g/day], 0.82; 95% confidence interval, 0.69–0.97, P for trend = 0.02). When the researchers focused on specific types of fiber, they found that people who consumed the highest amounts of cereal and vegetable fiber were 19% and 16%, respectively, less likely to develop type 2 diabetes compared with those who consumed the lowest amounts (P < 0.001). Intake of fruit fiber was not associated with risk of diabetes. When the analyses were additionally adjusted for BMI, the inverse associations between intake of fiber and diabetes were attenuated and no longer statistically significant.
The researchers also conducted a meta-analysis that included 18 other cohorts in addition to the current EPIC-InterAct study. The summary relative risks per 10/g day increase in intake were 0.91 (95% CI, 0.87–0.96) for total fiber, 0.75 (95% CI, 0.65–0.86) for cereal fiber, 0.95 (95% CI, 0.87–1.03) for fruit fiber, and 0.93 (95% CI 0.82–1.05) for vegetable fiber.
Conclusion. Individuals with diets rich in fiber, in particular cereal fiber, may be at lower risk of type 2 diabetes.
Commentary
The current study by the European InterAct Consortium adds to the available evidence supporting the association of dietary fiber and risk of diabetes. Higher intake of dietary fiber, especially cereal fiber, has been consistently associated with a lower risk of diabetes [2,3].
This study showed that a high intake of total fiber (primarily cereal and vegetable fiber) was associated with an 18% lower risk of type 2 diabetes (adjusted for dietary and lifestyle factors). While there was no association after adjusting for BMI, their meta-analysis of 18 studies did support the an inverse association between total fiber and cereal fiber intake and risk of type 2 diabetes.
What is it about fiber that is protective? With regard to whole grains, a rich source of fiber, potential mechanisms have been identified [5], including the possible impact of improved postprandial glucose response. However, whole grains are rich in nutrients and phytochemicals, and new hypotheses for the health-protective mechanisms of whole grains beyond fiber are being proposed [6]. In addition, the beneficial effect of fiber seen in this and other studies may be partly mediated by a lower BMI. Dietary fiber may affect appetite and energy intake through a range of processes.
It should be noted that although the effect of whole-grain foods for the prevention of type 2 diabetes is strongly supported by numerous epidemiological studies, a 2008 systematic review by the Cochrane Collaboration [4], which included 1 low-quality RCT and 11 cohort studies, stated that the evidence is too weak to be able to draw a definite conclusion about the preventive effect of this dietary factor.
Strengths of the study included its prospective design and large sample size. Limitations of the study include that dietary intake was assessed only at baseline and measurement error through the use of a questionnaire may have occurred. Although food-frequency questionnaires are widely used, they are subjective estimates and subject to recall bias, and some researchers have questioned their value for use in epidemiologic studies [7]. In addition, the authors note that the inverse association for total fiber and cereal fiber intake in the meta-analysis could be due to residual confounding as fiber intake has been associated with healthier diet, lower BMI, and greater physical activity.
Applications for Clinical Practice
The prevalence of type 2 diabetes has increased rapidly during the past decades in the United States. Dietary guidelines recommend the consumption of whole grains to prevent chronic diseases. The results presented in the current study strengthen the evidence supporting cereal fiber as an important determinant of type 2 diabetes risk. Randomized controlled trials are needed and should elucidate this matter.
Study Overview
Objective. To evaluate the association between intake of dietary fiber and type 2 diabetes.
Design. Case-cohort study (EPIC-InterAct Study), which is nested within the large prospective cohort study EPIC (European Prospective Investigation into Cancer & Nutrition) [1]. EPIC includes participants from 10 European countries and was designed to investigate the relationships between diet, nutritional status, lifestyle, and environmental factors and the incidence of cancer and other chronic disease [1].
Setting and participants. The EPIC-Interact study used data from 8 European countries (Denmark, France, Germany, Italy, the Netherlands, Spain, Sweden, and UK). The Interact sample includes 12,403 individuals who were identified as having developed type 2 diabetes, and a random subcohort of controls who were free of diabetes at baseline (n = 16,835, including 778 cases of incident diabetes) selected from 340,234 eligible EPIC participants. Of the 28,460 participants in the EPIC-InterAct study, excluded were those with prevalent diabetes, missing diabetes status information, post-censoring diabetes, extreme energy intake (top and bottom 1%), and missing values for education level, physical activity, smoking status, and BMI, leaving a final sample of 11,559 cases and 15,258 subcohort participants. No differences were observed in baseline characteristics between the included and excluded participants.
Analysis. Country-specific hazard ratios (HRs) were estimated using Prentice-weighted Cox proportional hazards models and were pooled using a random effects meta-analysis. Dietary intake over the previous 12 months before recruitment was assessed by country-specific or center-specific assessment methods (food-frequency questionnaire and dietary histories) that were developed and validated locally, and data were converted to nutrient intake.
Main outcome measure. Incident cases of diabetes.
Main results. During a median of 10.8 years of follow-up, total fiber intake was associated with a lower risk of diabetes after adjusting for lifestyle and dietary factors (hazard ratio for the highest quartile of fiber intake [> 26 g/day] vs the lowest [< 19 g/day], 0.82; 95% confidence interval, 0.69–0.97, P for trend = 0.02). When the researchers focused on specific types of fiber, they found that people who consumed the highest amounts of cereal and vegetable fiber were 19% and 16%, respectively, less likely to develop type 2 diabetes compared with those who consumed the lowest amounts (P < 0.001). Intake of fruit fiber was not associated with risk of diabetes. When the analyses were additionally adjusted for BMI, the inverse associations between intake of fiber and diabetes were attenuated and no longer statistically significant.
The researchers also conducted a meta-analysis that included 18 other cohorts in addition to the current EPIC-InterAct study. The summary relative risks per 10/g day increase in intake were 0.91 (95% CI, 0.87–0.96) for total fiber, 0.75 (95% CI, 0.65–0.86) for cereal fiber, 0.95 (95% CI, 0.87–1.03) for fruit fiber, and 0.93 (95% CI 0.82–1.05) for vegetable fiber.
Conclusion. Individuals with diets rich in fiber, in particular cereal fiber, may be at lower risk of type 2 diabetes.
Commentary
The current study by the European InterAct Consortium adds to the available evidence supporting the association of dietary fiber and risk of diabetes. Higher intake of dietary fiber, especially cereal fiber, has been consistently associated with a lower risk of diabetes [2,3].
This study showed that a high intake of total fiber (primarily cereal and vegetable fiber) was associated with an 18% lower risk of type 2 diabetes (adjusted for dietary and lifestyle factors). While there was no association after adjusting for BMI, their meta-analysis of 18 studies did support the an inverse association between total fiber and cereal fiber intake and risk of type 2 diabetes.
What is it about fiber that is protective? With regard to whole grains, a rich source of fiber, potential mechanisms have been identified [5], including the possible impact of improved postprandial glucose response. However, whole grains are rich in nutrients and phytochemicals, and new hypotheses for the health-protective mechanisms of whole grains beyond fiber are being proposed [6]. In addition, the beneficial effect of fiber seen in this and other studies may be partly mediated by a lower BMI. Dietary fiber may affect appetite and energy intake through a range of processes.
It should be noted that although the effect of whole-grain foods for the prevention of type 2 diabetes is strongly supported by numerous epidemiological studies, a 2008 systematic review by the Cochrane Collaboration [4], which included 1 low-quality RCT and 11 cohort studies, stated that the evidence is too weak to be able to draw a definite conclusion about the preventive effect of this dietary factor.
Strengths of the study included its prospective design and large sample size. Limitations of the study include that dietary intake was assessed only at baseline and measurement error through the use of a questionnaire may have occurred. Although food-frequency questionnaires are widely used, they are subjective estimates and subject to recall bias, and some researchers have questioned their value for use in epidemiologic studies [7]. In addition, the authors note that the inverse association for total fiber and cereal fiber intake in the meta-analysis could be due to residual confounding as fiber intake has been associated with healthier diet, lower BMI, and greater physical activity.
Applications for Clinical Practice
The prevalence of type 2 diabetes has increased rapidly during the past decades in the United States. Dietary guidelines recommend the consumption of whole grains to prevent chronic diseases. The results presented in the current study strengthen the evidence supporting cereal fiber as an important determinant of type 2 diabetes risk. Randomized controlled trials are needed and should elucidate this matter.
1. Riboli E, Hunt KJ, Slimani N, European Prospective Investigation into Cancer and Nutrition (EPIC): study populations and data collection. Public Health Nutr 2002;5(6B):1113–24.
2. Ye EQ, Chacko SA, Chou EL, et al. Greater whole-grain intake is associated with lower risk of type 2 diabetes, cardiovascular disease, and weight gain. J Nutr 2012;142:1304–13.
3. Huang T, Xu M, Lee A, et al. Consumption of whole grains and cereal fiber and total and cause-specific mortality: prospective analysis of 367,442 individuals. BMC Med 2015;13:59.
4. Priebe MG, van Binsbergen JJ, de Vos R, Vonk RJ. Whole grain foods for theprevention of type 2 diabetes mellitus. Cochrane Database Syst Rev 2008;(1):CD006061.
5. Slavin JL, Martini MC, Jacobs DR Jr, Marquart L. Plausible mechanisms for the protectiveness of whole grains. Am J Clin Nutr 1999;70(3 Suppl):459S–463S.
6. Fardet A. New hypotheses for the health-protective mechanisms of whole-grain cereals: what is beyond fibre? Nutr Res Rev 2010;23:65–134.
7. Shim J-S, Oh K, Kim HC. Dietary assessment methods in epidemiologic studies. Epidemiol Health 2014;36:e2014009.
1. Riboli E, Hunt KJ, Slimani N, European Prospective Investigation into Cancer and Nutrition (EPIC): study populations and data collection. Public Health Nutr 2002;5(6B):1113–24.
2. Ye EQ, Chacko SA, Chou EL, et al. Greater whole-grain intake is associated with lower risk of type 2 diabetes, cardiovascular disease, and weight gain. J Nutr 2012;142:1304–13.
3. Huang T, Xu M, Lee A, et al. Consumption of whole grains and cereal fiber and total and cause-specific mortality: prospective analysis of 367,442 individuals. BMC Med 2015;13:59.
4. Priebe MG, van Binsbergen JJ, de Vos R, Vonk RJ. Whole grain foods for theprevention of type 2 diabetes mellitus. Cochrane Database Syst Rev 2008;(1):CD006061.
5. Slavin JL, Martini MC, Jacobs DR Jr, Marquart L. Plausible mechanisms for the protectiveness of whole grains. Am J Clin Nutr 1999;70(3 Suppl):459S–463S.
6. Fardet A. New hypotheses for the health-protective mechanisms of whole-grain cereals: what is beyond fibre? Nutr Res Rev 2010;23:65–134.
7. Shim J-S, Oh K, Kim HC. Dietary assessment methods in epidemiologic studies. Epidemiol Health 2014;36:e2014009.
Idarucizumab reverses dabigatran’s anticoagulant effects
TORONTO – Idarucizumab is a promising agent that quickly and safely reverses the anticoagulant effects of dabigatran whether the goal is to control serious bleeding or to permit urgent surgery, according to interim results of a multicenter trial.
Idarucizumab is a monoclonal antibody that binds to dabigatran to reverse its activity. The data, presented by Dr. V. Charles Pollack Jr. at the International Society on Thrombosis and Haemostasis congress, involved the first 90 patients of an ongoing trial with a planned enrollment of 300. The data from this trial, called REVERSE-AD, were published online simultaneously with the June 22 presentation at the congress (N. Engl. J. Med 2015 [doi:10.1056/NEJMoa1502000]).

“Non–vitamin K antagonist oral anticoagulants (NOACs) are generally safer than warfarin, and provide similar or improved efficacy in the prevention of stroke in patients with nonvalvular atrial fibrillation and in the prevention and treatment of venous thromboembolism,” Dr. Pollack said in an interview. “Nonetheless, serious bleeding events may occur with NOAC use, and patients taking one of these agents occasionally require urgent surgery or other intervention for which normal hemostasis is required,” added Dr. Pollack, chair of the department of emergency medicine at Pennsylvania Hospital in Philadelphia.
In RE-VERSE AD (a study of the reversal effects of idarucizumab on active dabigatran), the first 90 patients were divided into two distinct groups. Group A, with 51 patients, included those on dabigatran with serious bleeding. Group B, with 39 patients, required reversal of dabigatran for urgent or emergent procedures. In both, idarucizumab provided a median maximum reversal of 100% (95% confidence interval, 100-100) of the anticoagulation effect within 4 hours.
Clotting assays were normalized almost immediately in almost 90% of patients, and the effect was durable, with 80% having measured dabigatran levels reflecting no significant anticoagulation 24 hours later, Dr. Pollack said.
“Clinical outcomes were quite good in this multimorbid patient population, with restoration of hemostasis as reported by local investigators achieved in less than 12 hours when assessable, and with 92% of surgical patients being reported as having normal hemostasis at the time of the procedure,” he said.
Idarucizumab was generally well tolerated in the patient population. “There were no serious adverse events related to the reversal agent ... and only one patient experienced a thrombotic complication within 72 hours, and that patient had not been restarted on any antithrombotic medications,” Dr. Pollack said.
“The study is ongoing,” he added, “but these interim results show rather convincingly that idarucizumab completely and safely reverses the anticoagulant effects of dabigatran within minutes.”
In addition, Dr. Pollack said the availability of a specific reversal agent for dabigatran would enhance its safety margin, and thus alleviate the fears of providers who may hesitate to use a NOAC because of the lack of an “antidote.”
“In fact, most such cases can already be successfully and safely managed with general support and ‘tincture of time’ (the half-life of dabigatran is much shorter than that of warfarin), but having a specific ‘go-to’ option could streamline the care of the most significantly compromised patients,” he said.
Dr. Pollack emphasized, however, that idarucizumab is a specific reversal agent for dabigatran, not an antidote. “To me, the latter would imply that idarucizumab immediately stops bleeding associated with active use of dabigatran,” he said.
Providers should realize that while idarucizumab seems capable of removing dabigatran-induced coagulopathy from the list of concerns when managing a patient with serious bleeding or before a “sharp” procedure, bleeding is a multifaceted issue that also may be due to traumatized blood vessels, other causes of coagulopathy such as liver disease, or concurrent use of antiplatelet medications, he said.
“The patient with a serious or life-threatening bleed on dabigatran will likely need additional care to investigate and manage such concerns,” Dr. Pollack said. “But at least idarucizumab can specifically, safely, and rapidly address the primary consideration.
“The safety of anticoagulation therapy with dabigatran is further enhanced with idarucizumab, a specific reversal agent that won’t need to be used often, but the availability of which would be reassuring to prescribers,” he concluded.
Boehringer Ingelheim sponsored RE-VERSE AD. Idarucizumab was given a fast-track status by the Food and Drug Administration, and BI submitted a new drug application in March 2015, according to the company.
Dr. Pollack reported receiving personal fees from BI, Janssen, Daiichi-Sankyo, Bristol-Myers Squibb, and Pfizer. Disclosures for all the investigators are available at NEJM.org.
TORONTO – Idarucizumab is a promising agent that quickly and safely reverses the anticoagulant effects of dabigatran whether the goal is to control serious bleeding or to permit urgent surgery, according to interim results of a multicenter trial.
Idarucizumab is a monoclonal antibody that binds to dabigatran to reverse its activity. The data, presented by Dr. V. Charles Pollack Jr. at the International Society on Thrombosis and Haemostasis congress, involved the first 90 patients of an ongoing trial with a planned enrollment of 300. The data from this trial, called REVERSE-AD, were published online simultaneously with the June 22 presentation at the congress (N. Engl. J. Med 2015 [doi:10.1056/NEJMoa1502000]).
“Non–vitamin K antagonist oral anticoagulants (NOACs) are generally safer than warfarin, and provide similar or improved efficacy in the prevention of stroke in patients with nonvalvular atrial fibrillation and in the prevention and treatment of venous thromboembolism,” Dr. Pollack said in an interview. “Nonetheless, serious bleeding events may occur with NOAC use, and patients taking one of these agents occasionally require urgent surgery or other intervention for which normal hemostasis is required,” added Dr. Pollack, chair of the department of emergency medicine at Pennsylvania Hospital in Philadelphia.
In RE-VERSE AD (a study of the reversal effects of idarucizumab on active dabigatran), the first 90 patients were divided into two distinct groups. Group A, with 51 patients, included those on dabigatran with serious bleeding. Group B, with 39 patients, required reversal of dabigatran for urgent or emergent procedures. In both, idarucizumab provided a median maximum reversal of 100% (95% confidence interval, 100-100) of the anticoagulation effect within 4 hours.
Clotting assays were normalized almost immediately in almost 90% of patients, and the effect was durable, with 80% having measured dabigatran levels reflecting no significant anticoagulation 24 hours later, Dr. Pollack said.
“Clinical outcomes were quite good in this multimorbid patient population, with restoration of hemostasis as reported by local investigators achieved in less than 12 hours when assessable, and with 92% of surgical patients being reported as having normal hemostasis at the time of the procedure,” he said.
Idarucizumab was generally well tolerated in the patient population. “There were no serious adverse events related to the reversal agent ... and only one patient experienced a thrombotic complication within 72 hours, and that patient had not been restarted on any antithrombotic medications,” Dr. Pollack said.
“The study is ongoing,” he added, “but these interim results show rather convincingly that idarucizumab completely and safely reverses the anticoagulant effects of dabigatran within minutes.”
In addition, Dr. Pollack said the availability of a specific reversal agent for dabigatran would enhance its safety margin, and thus alleviate the fears of providers who may hesitate to use a NOAC because of the lack of an “antidote.”
“In fact, most such cases can already be successfully and safely managed with general support and ‘tincture of time’ (the half-life of dabigatran is much shorter than that of warfarin), but having a specific ‘go-to’ option could streamline the care of the most significantly compromised patients,” he said.
Dr. Pollack emphasized, however, that idarucizumab is a specific reversal agent for dabigatran, not an antidote. “To me, the latter would imply that idarucizumab immediately stops bleeding associated with active use of dabigatran,” he said.
Providers should realize that while idarucizumab seems capable of removing dabigatran-induced coagulopathy from the list of concerns when managing a patient with serious bleeding or before a “sharp” procedure, bleeding is a multifaceted issue that also may be due to traumatized blood vessels, other causes of coagulopathy such as liver disease, or concurrent use of antiplatelet medications, he said.
“The patient with a serious or life-threatening bleed on dabigatran will likely need additional care to investigate and manage such concerns,” Dr. Pollack said. “But at least idarucizumab can specifically, safely, and rapidly address the primary consideration.
“The safety of anticoagulation therapy with dabigatran is further enhanced with idarucizumab, a specific reversal agent that won’t need to be used often, but the availability of which would be reassuring to prescribers,” he concluded.
Boehringer Ingelheim sponsored RE-VERSE AD. Idarucizumab was given a fast-track status by the Food and Drug Administration, and BI submitted a new drug application in March 2015, according to the company.
Dr. Pollack reported receiving personal fees from BI, Janssen, Daiichi-Sankyo, Bristol-Myers Squibb, and Pfizer. Disclosures for all the investigators are available at NEJM.org.
TORONTO – Idarucizumab is a promising agent that quickly and safely reverses the anticoagulant effects of dabigatran whether the goal is to control serious bleeding or to permit urgent surgery, according to interim results of a multicenter trial.
Idarucizumab is a monoclonal antibody that binds to dabigatran to reverse its activity. The data, presented by Dr. V. Charles Pollack Jr. at the International Society on Thrombosis and Haemostasis congress, involved the first 90 patients of an ongoing trial with a planned enrollment of 300. The data from this trial, called REVERSE-AD, were published online simultaneously with the June 22 presentation at the congress (N. Engl. J. Med 2015 [doi:10.1056/NEJMoa1502000]).
“Non–vitamin K antagonist oral anticoagulants (NOACs) are generally safer than warfarin, and provide similar or improved efficacy in the prevention of stroke in patients with nonvalvular atrial fibrillation and in the prevention and treatment of venous thromboembolism,” Dr. Pollack said in an interview. “Nonetheless, serious bleeding events may occur with NOAC use, and patients taking one of these agents occasionally require urgent surgery or other intervention for which normal hemostasis is required,” added Dr. Pollack, chair of the department of emergency medicine at Pennsylvania Hospital in Philadelphia.
In RE-VERSE AD (a study of the reversal effects of idarucizumab on active dabigatran), the first 90 patients were divided into two distinct groups. Group A, with 51 patients, included those on dabigatran with serious bleeding. Group B, with 39 patients, required reversal of dabigatran for urgent or emergent procedures. In both, idarucizumab provided a median maximum reversal of 100% (95% confidence interval, 100-100) of the anticoagulation effect within 4 hours.
Clotting assays were normalized almost immediately in almost 90% of patients, and the effect was durable, with 80% having measured dabigatran levels reflecting no significant anticoagulation 24 hours later, Dr. Pollack said.
“Clinical outcomes were quite good in this multimorbid patient population, with restoration of hemostasis as reported by local investigators achieved in less than 12 hours when assessable, and with 92% of surgical patients being reported as having normal hemostasis at the time of the procedure,” he said.
Idarucizumab was generally well tolerated in the patient population. “There were no serious adverse events related to the reversal agent ... and only one patient experienced a thrombotic complication within 72 hours, and that patient had not been restarted on any antithrombotic medications,” Dr. Pollack said.
“The study is ongoing,” he added, “but these interim results show rather convincingly that idarucizumab completely and safely reverses the anticoagulant effects of dabigatran within minutes.”
In addition, Dr. Pollack said the availability of a specific reversal agent for dabigatran would enhance its safety margin, and thus alleviate the fears of providers who may hesitate to use a NOAC because of the lack of an “antidote.”
“In fact, most such cases can already be successfully and safely managed with general support and ‘tincture of time’ (the half-life of dabigatran is much shorter than that of warfarin), but having a specific ‘go-to’ option could streamline the care of the most significantly compromised patients,” he said.
Dr. Pollack emphasized, however, that idarucizumab is a specific reversal agent for dabigatran, not an antidote. “To me, the latter would imply that idarucizumab immediately stops bleeding associated with active use of dabigatran,” he said.
Providers should realize that while idarucizumab seems capable of removing dabigatran-induced coagulopathy from the list of concerns when managing a patient with serious bleeding or before a “sharp” procedure, bleeding is a multifaceted issue that also may be due to traumatized blood vessels, other causes of coagulopathy such as liver disease, or concurrent use of antiplatelet medications, he said.
“The patient with a serious or life-threatening bleed on dabigatran will likely need additional care to investigate and manage such concerns,” Dr. Pollack said. “But at least idarucizumab can specifically, safely, and rapidly address the primary consideration.
“The safety of anticoagulation therapy with dabigatran is further enhanced with idarucizumab, a specific reversal agent that won’t need to be used often, but the availability of which would be reassuring to prescribers,” he concluded.
Boehringer Ingelheim sponsored RE-VERSE AD. Idarucizumab was given a fast-track status by the Food and Drug Administration, and BI submitted a new drug application in March 2015, according to the company.
Dr. Pollack reported receiving personal fees from BI, Janssen, Daiichi-Sankyo, Bristol-Myers Squibb, and Pfizer. Disclosures for all the investigators are available at NEJM.org.
AT 2015 ISTH CONGRESS
Key clinical point: The investigational monoclonal antibody idarucizumab reversed the anticoagulant effects of dabigatran.
Major finding: Idarucizumab provided a median maximum dabigatran reversal of 100% (95% CI, 100-100) of the anticoagulation effect within 4 hours in an interim analysis.
Data source: RE-VERSE AD, a prospective cohort study in which 90 patients treated with dabigatran who had uncontrolled bleeding or required emergency surgery or procedures were given 5.0 g idarucizumab.
Disclosures: Boehringer Ingelheim sponsored RE-VERSE AD. Dr. Pollack reported receiving personal fees from Boehringer Ingelheim, Janssen, Daiichi-Sankyo, Bristol-Myers Squibb, and Pfizer. Disclosures for all the investigators are available at NEJM.org.

FDA approves cangrelor, an intravenous antiplatelet drug
Cangrelor became the first intravenous antiplatelet agent acting on ADP receptors for adult patients undergoing percutaneous coronary intervention to receive marketing approval from the Food and Drug Administration, The Medicines Company announced on June 22.
While cangrelor’s unique delivery route and rapid onset and off-set of action set it apart and may give it certain clinical advantages over the three approved oral drugs that target the same platelet receptor – clopidogrel, prasugrel (Effient), and ticagrelor (Brilinta) – cangrelor will also be distinguished by its much higher price. The standard dosage to treat one patient undergoing percutaneous coronary intervention (PCI) with cangrelor (Kengreal) will have a wholesale acquisition cost of $749, Raymond Russo, senior vice president of The Medicines Company, said at a June 23 press briefing. That prices cangrelor substantially above its brand-name competition, which costs roughly $10 for similar treatment, as well as generic clopidogrel, which costs about $3 for the same indication.

“I believe in the strength of the data that showed that cangrelor was superior to the comparator drug [clopidogrel], and if cost were not an issue I’d use cangrelor routinely, but I am not naive; cost is an issue,” said Dr. Deepak L. Bhatt, professor of medicine at Harvard University and executive director of interventional cardiology programs at Brigham and Women’s Hospital in Boston, and co–lead investigator for the CHAMPION PHOENIX pivotal trial that led to cangrelor’s approval (N. Engl. J. Med. 2013;368:1303-13).
Whether or not interventional cardiologists and the centers where they work decide to use cangrelor or one of the oral antiplatelet drugs for coronary artery disease (CAD) patients undergoing PCI will likely depend on a series of considerations that will need to take into account not just drug cost but also practice strategies, a patient’s clinical state, and the potential for ancillary costs from following an entirely different management approach.
The first issue is whether the interventionalist decides to pretreat a patient scheduled for angioplasty and possible immediate PCI following angiography with an ADP-receptor antagonist (also known as a P2Y12-receptor inhibitor) prior to the start of angiography or opts to defer that treatment until the angiography results are available and a decision is made to proceed with PCI. Recent nationwide registry data suggest that roughly half of U.S. interventionalists treat their patients upfront with an ADP-receptor antagonist, usually clopidogrel for patients with stable angina or prasugrel or ticagrelor if they have either a non-ST-elevation MI or a ST-elevation MI, while the other 50% of interventionalists will wait to administer the ADP-receptor antagonist until angiography is complete, Dr. Bhatt explained in an interview.
The advantage to upfront treatment is that by the time the patient is ready for PCI an oral ADP-receptor antagonist is fully absorbed and on board. The disadvantage is that if the coronary anatomy demands a surgical approach to revascularization many surgeons would elect not to operate on a patient freshly dosed with an antiplatelet agent, and these patients often remain hospitalized for several days until the ADP-receptor antagonist clears and the patient’s platelet function returns to normal. Angiography generally identifies 10%-15% of these patients with a CAD distribution that necessitates surgical coronary bypass, and the potential hospitalization expense of waiting for their ADP-receptor antagonist to clear could be a major cost to counterbalance the price of cangrelor, which would obviate this expense if the quick-to-start-and-to-clear cangrelor were used instead of a more lumbering oral drug, he noted.
The other 50% of U.S. interventionalists, Dr. Bhatt included, take a different approach. Recognizing the potential downside of upfront oral antiplatelet therapy if the patient is pegged for bypass surgery following angiography, they elect to wait until the angiography results are in hand. If the angiography results show the patient is destined for surgery or for medical management, then the patient receives no ADP-receptor antagonist. The cardiologist administers an ADP-receptor antagonist only if the patient’s CAD is appropriate for PCI, the fate for most of these CAD patients following angiography. It’s under these circumstances that the advantages of cangrelor kick in, as shown in the results from CHAMPION PHOENIX.
This trial randomized patients to two different types of ADP-receptor antagonist treatment while they were in the coronary catheterization laboratory. The study results showed a statistically significant, 22% relative-risk reduction in the primary endpoint in favor of intravenous cangrelor compared with oral clopidogrel delivered while patients were “on the table” in the interval between angiography and PCI. That 22% relative improvement in outcomes, driven primarily by reductions in periprocedural MIs and stent thrombosis, improved to a 31% relative-risk reduction when The Medicines Company performed a new analysis of the study results at the FDA’s request using a more stringent and conventional definition of periprocedural MIs and stent thrombosis. The time needed to perform this and other FDA-requested analyses largely caused the greater than 2-year gap between the 2013 publication of the CHAMPION PHOENIX results and the FDA’s approval.
But the editorial that accompanied the 2013 publication highlighted what the editorialists perceived as flaws in the study’s design, such as an inadequate loading dose of clopidogrel delivered to a quarter of the patients randomized to that arm, inadequate time allowed for the clopidogrel to fully kick in before PCI began in a third of patients, and the use of clopidogrel as the comparator drug and not a more potent alternative drug, either prasugrel or ticagrelor (N. Engl. J. Med. 2013;368:1356-7).
“Cangrelor was never tested against prasugrel or ticagrelor, and it was compared with inadequate clopidogrel treatment. That was a problem,” reiterated Dr. Richard A. Lange, one of the 2013 editorialists, when interviewed following news of cangrelor’s FDA approval. CHAMPION PHOENIX “wasn’t really a comparison [of two drugs], it was a study of an intravenous strategy, and it’s not a strategy that is needed very often,” said Dr. Lange, an interventional cardiologist and president of the Texas Tech University Health Sciences Center in El Paso. In Dr. Lange’s opinion, the only real need for an intravenous ADP-receptor antagonist is for CAD patients undergoing PCI who are unable to take an oral agent, for example because they are on a ventilator, unable to hold down an oral pill, or unconscious, which collectively are “rare” situations, he said.
Dr. Bhatt noted that another clear indication for an intravenous agent is when MI patients receive morphine for their pain, a situation recently documented to interfere with absorption of oral ADP-receptor antagonists.
From Dr. Bhatt’s perspective, the major issue is practice patterns: “Do the interventionalists treat [with an ADP-receptor antagonist] upstream or not. If they do, then they should do the math,” and determine if the expense of holding a significant minority of patients in the hospital just to allow them to clear the ADP-receptor antagonist prior to coronary bypass surgery outweighs the cost for delaying this treatment and administering cangrelor later only to patients scheduled for PCI. At the center where he practices, Brigham and Women’s Hospital in Boston, he sees a roughly equal mix of interventionalists who prefer to treat patients with clopidogrel upfront, those who treat with ticagrelor upfront, and those who practice as he does and wait until the PCI is a go.
“For my personal practice, cangrelor will fit in quite nicely,” Dr. Bhatt said.
On Twitter@mitchelzoler
Cangrelor became the first intravenous antiplatelet agent acting on ADP receptors for adult patients undergoing percutaneous coronary intervention to receive marketing approval from the Food and Drug Administration, The Medicines Company announced on June 22.
While cangrelor’s unique delivery route and rapid onset and off-set of action set it apart and may give it certain clinical advantages over the three approved oral drugs that target the same platelet receptor – clopidogrel, prasugrel (Effient), and ticagrelor (Brilinta) – cangrelor will also be distinguished by its much higher price. The standard dosage to treat one patient undergoing percutaneous coronary intervention (PCI) with cangrelor (Kengreal) will have a wholesale acquisition cost of $749, Raymond Russo, senior vice president of The Medicines Company, said at a June 23 press briefing. That prices cangrelor substantially above its brand-name competition, which costs roughly $10 for similar treatment, as well as generic clopidogrel, which costs about $3 for the same indication.
“I believe in the strength of the data that showed that cangrelor was superior to the comparator drug [clopidogrel], and if cost were not an issue I’d use cangrelor routinely, but I am not naive; cost is an issue,” said Dr. Deepak L. Bhatt, professor of medicine at Harvard University and executive director of interventional cardiology programs at Brigham and Women’s Hospital in Boston, and co–lead investigator for the CHAMPION PHOENIX pivotal trial that led to cangrelor’s approval (N. Engl. J. Med. 2013;368:1303-13).
Whether or not interventional cardiologists and the centers where they work decide to use cangrelor or one of the oral antiplatelet drugs for coronary artery disease (CAD) patients undergoing PCI will likely depend on a series of considerations that will need to take into account not just drug cost but also practice strategies, a patient’s clinical state, and the potential for ancillary costs from following an entirely different management approach.
The first issue is whether the interventionalist decides to pretreat a patient scheduled for angioplasty and possible immediate PCI following angiography with an ADP-receptor antagonist (also known as a P2Y12-receptor inhibitor) prior to the start of angiography or opts to defer that treatment until the angiography results are available and a decision is made to proceed with PCI. Recent nationwide registry data suggest that roughly half of U.S. interventionalists treat their patients upfront with an ADP-receptor antagonist, usually clopidogrel for patients with stable angina or prasugrel or ticagrelor if they have either a non-ST-elevation MI or a ST-elevation MI, while the other 50% of interventionalists will wait to administer the ADP-receptor antagonist until angiography is complete, Dr. Bhatt explained in an interview.
The advantage to upfront treatment is that by the time the patient is ready for PCI an oral ADP-receptor antagonist is fully absorbed and on board. The disadvantage is that if the coronary anatomy demands a surgical approach to revascularization many surgeons would elect not to operate on a patient freshly dosed with an antiplatelet agent, and these patients often remain hospitalized for several days until the ADP-receptor antagonist clears and the patient’s platelet function returns to normal. Angiography generally identifies 10%-15% of these patients with a CAD distribution that necessitates surgical coronary bypass, and the potential hospitalization expense of waiting for their ADP-receptor antagonist to clear could be a major cost to counterbalance the price of cangrelor, which would obviate this expense if the quick-to-start-and-to-clear cangrelor were used instead of a more lumbering oral drug, he noted.
The other 50% of U.S. interventionalists, Dr. Bhatt included, take a different approach. Recognizing the potential downside of upfront oral antiplatelet therapy if the patient is pegged for bypass surgery following angiography, they elect to wait until the angiography results are in hand. If the angiography results show the patient is destined for surgery or for medical management, then the patient receives no ADP-receptor antagonist. The cardiologist administers an ADP-receptor antagonist only if the patient’s CAD is appropriate for PCI, the fate for most of these CAD patients following angiography. It’s under these circumstances that the advantages of cangrelor kick in, as shown in the results from CHAMPION PHOENIX.
This trial randomized patients to two different types of ADP-receptor antagonist treatment while they were in the coronary catheterization laboratory. The study results showed a statistically significant, 22% relative-risk reduction in the primary endpoint in favor of intravenous cangrelor compared with oral clopidogrel delivered while patients were “on the table” in the interval between angiography and PCI. That 22% relative improvement in outcomes, driven primarily by reductions in periprocedural MIs and stent thrombosis, improved to a 31% relative-risk reduction when The Medicines Company performed a new analysis of the study results at the FDA’s request using a more stringent and conventional definition of periprocedural MIs and stent thrombosis. The time needed to perform this and other FDA-requested analyses largely caused the greater than 2-year gap between the 2013 publication of the CHAMPION PHOENIX results and the FDA’s approval.
But the editorial that accompanied the 2013 publication highlighted what the editorialists perceived as flaws in the study’s design, such as an inadequate loading dose of clopidogrel delivered to a quarter of the patients randomized to that arm, inadequate time allowed for the clopidogrel to fully kick in before PCI began in a third of patients, and the use of clopidogrel as the comparator drug and not a more potent alternative drug, either prasugrel or ticagrelor (N. Engl. J. Med. 2013;368:1356-7).
“Cangrelor was never tested against prasugrel or ticagrelor, and it was compared with inadequate clopidogrel treatment. That was a problem,” reiterated Dr. Richard A. Lange, one of the 2013 editorialists, when interviewed following news of cangrelor’s FDA approval. CHAMPION PHOENIX “wasn’t really a comparison [of two drugs], it was a study of an intravenous strategy, and it’s not a strategy that is needed very often,” said Dr. Lange, an interventional cardiologist and president of the Texas Tech University Health Sciences Center in El Paso. In Dr. Lange’s opinion, the only real need for an intravenous ADP-receptor antagonist is for CAD patients undergoing PCI who are unable to take an oral agent, for example because they are on a ventilator, unable to hold down an oral pill, or unconscious, which collectively are “rare” situations, he said.
Dr. Bhatt noted that another clear indication for an intravenous agent is when MI patients receive morphine for their pain, a situation recently documented to interfere with absorption of oral ADP-receptor antagonists.
From Dr. Bhatt’s perspective, the major issue is practice patterns: “Do the interventionalists treat [with an ADP-receptor antagonist] upstream or not. If they do, then they should do the math,” and determine if the expense of holding a significant minority of patients in the hospital just to allow them to clear the ADP-receptor antagonist prior to coronary bypass surgery outweighs the cost for delaying this treatment and administering cangrelor later only to patients scheduled for PCI. At the center where he practices, Brigham and Women’s Hospital in Boston, he sees a roughly equal mix of interventionalists who prefer to treat patients with clopidogrel upfront, those who treat with ticagrelor upfront, and those who practice as he does and wait until the PCI is a go.
“For my personal practice, cangrelor will fit in quite nicely,” Dr. Bhatt said.
On Twitter@mitchelzoler
Cangrelor became the first intravenous antiplatelet agent acting on ADP receptors for adult patients undergoing percutaneous coronary intervention to receive marketing approval from the Food and Drug Administration, The Medicines Company announced on June 22.
While cangrelor’s unique delivery route and rapid onset and off-set of action set it apart and may give it certain clinical advantages over the three approved oral drugs that target the same platelet receptor – clopidogrel, prasugrel (Effient), and ticagrelor (Brilinta) – cangrelor will also be distinguished by its much higher price. The standard dosage to treat one patient undergoing percutaneous coronary intervention (PCI) with cangrelor (Kengreal) will have a wholesale acquisition cost of $749, Raymond Russo, senior vice president of The Medicines Company, said at a June 23 press briefing. That prices cangrelor substantially above its brand-name competition, which costs roughly $10 for similar treatment, as well as generic clopidogrel, which costs about $3 for the same indication.
“I believe in the strength of the data that showed that cangrelor was superior to the comparator drug [clopidogrel], and if cost were not an issue I’d use cangrelor routinely, but I am not naive; cost is an issue,” said Dr. Deepak L. Bhatt, professor of medicine at Harvard University and executive director of interventional cardiology programs at Brigham and Women’s Hospital in Boston, and co–lead investigator for the CHAMPION PHOENIX pivotal trial that led to cangrelor’s approval (N. Engl. J. Med. 2013;368:1303-13).
Whether or not interventional cardiologists and the centers where they work decide to use cangrelor or one of the oral antiplatelet drugs for coronary artery disease (CAD) patients undergoing PCI will likely depend on a series of considerations that will need to take into account not just drug cost but also practice strategies, a patient’s clinical state, and the potential for ancillary costs from following an entirely different management approach.
The first issue is whether the interventionalist decides to pretreat a patient scheduled for angioplasty and possible immediate PCI following angiography with an ADP-receptor antagonist (also known as a P2Y12-receptor inhibitor) prior to the start of angiography or opts to defer that treatment until the angiography results are available and a decision is made to proceed with PCI. Recent nationwide registry data suggest that roughly half of U.S. interventionalists treat their patients upfront with an ADP-receptor antagonist, usually clopidogrel for patients with stable angina or prasugrel or ticagrelor if they have either a non-ST-elevation MI or a ST-elevation MI, while the other 50% of interventionalists will wait to administer the ADP-receptor antagonist until angiography is complete, Dr. Bhatt explained in an interview.
The advantage to upfront treatment is that by the time the patient is ready for PCI an oral ADP-receptor antagonist is fully absorbed and on board. The disadvantage is that if the coronary anatomy demands a surgical approach to revascularization many surgeons would elect not to operate on a patient freshly dosed with an antiplatelet agent, and these patients often remain hospitalized for several days until the ADP-receptor antagonist clears and the patient’s platelet function returns to normal. Angiography generally identifies 10%-15% of these patients with a CAD distribution that necessitates surgical coronary bypass, and the potential hospitalization expense of waiting for their ADP-receptor antagonist to clear could be a major cost to counterbalance the price of cangrelor, which would obviate this expense if the quick-to-start-and-to-clear cangrelor were used instead of a more lumbering oral drug, he noted.
The other 50% of U.S. interventionalists, Dr. Bhatt included, take a different approach. Recognizing the potential downside of upfront oral antiplatelet therapy if the patient is pegged for bypass surgery following angiography, they elect to wait until the angiography results are in hand. If the angiography results show the patient is destined for surgery or for medical management, then the patient receives no ADP-receptor antagonist. The cardiologist administers an ADP-receptor antagonist only if the patient’s CAD is appropriate for PCI, the fate for most of these CAD patients following angiography. It’s under these circumstances that the advantages of cangrelor kick in, as shown in the results from CHAMPION PHOENIX.
This trial randomized patients to two different types of ADP-receptor antagonist treatment while they were in the coronary catheterization laboratory. The study results showed a statistically significant, 22% relative-risk reduction in the primary endpoint in favor of intravenous cangrelor compared with oral clopidogrel delivered while patients were “on the table” in the interval between angiography and PCI. That 22% relative improvement in outcomes, driven primarily by reductions in periprocedural MIs and stent thrombosis, improved to a 31% relative-risk reduction when The Medicines Company performed a new analysis of the study results at the FDA’s request using a more stringent and conventional definition of periprocedural MIs and stent thrombosis. The time needed to perform this and other FDA-requested analyses largely caused the greater than 2-year gap between the 2013 publication of the CHAMPION PHOENIX results and the FDA’s approval.
But the editorial that accompanied the 2013 publication highlighted what the editorialists perceived as flaws in the study’s design, such as an inadequate loading dose of clopidogrel delivered to a quarter of the patients randomized to that arm, inadequate time allowed for the clopidogrel to fully kick in before PCI began in a third of patients, and the use of clopidogrel as the comparator drug and not a more potent alternative drug, either prasugrel or ticagrelor (N. Engl. J. Med. 2013;368:1356-7).
“Cangrelor was never tested against prasugrel or ticagrelor, and it was compared with inadequate clopidogrel treatment. That was a problem,” reiterated Dr. Richard A. Lange, one of the 2013 editorialists, when interviewed following news of cangrelor’s FDA approval. CHAMPION PHOENIX “wasn’t really a comparison [of two drugs], it was a study of an intravenous strategy, and it’s not a strategy that is needed very often,” said Dr. Lange, an interventional cardiologist and president of the Texas Tech University Health Sciences Center in El Paso. In Dr. Lange’s opinion, the only real need for an intravenous ADP-receptor antagonist is for CAD patients undergoing PCI who are unable to take an oral agent, for example because they are on a ventilator, unable to hold down an oral pill, or unconscious, which collectively are “rare” situations, he said.
Dr. Bhatt noted that another clear indication for an intravenous agent is when MI patients receive morphine for their pain, a situation recently documented to interfere with absorption of oral ADP-receptor antagonists.
From Dr. Bhatt’s perspective, the major issue is practice patterns: “Do the interventionalists treat [with an ADP-receptor antagonist] upstream or not. If they do, then they should do the math,” and determine if the expense of holding a significant minority of patients in the hospital just to allow them to clear the ADP-receptor antagonist prior to coronary bypass surgery outweighs the cost for delaying this treatment and administering cangrelor later only to patients scheduled for PCI. At the center where he practices, Brigham and Women’s Hospital in Boston, he sees a roughly equal mix of interventionalists who prefer to treat patients with clopidogrel upfront, those who treat with ticagrelor upfront, and those who practice as he does and wait until the PCI is a go.
“For my personal practice, cangrelor will fit in quite nicely,” Dr. Bhatt said.
On Twitter@mitchelzoler

Physician suicide needs attention
Recently, there have been several news stories about physicians committing suicide. This is across all levels of the profession, including medical students, residents, and attendings.
Historically, doctors have had a higher rate of suicide than most professions. I’m not sure if that number has crept up recently, or if events are garnering more attention than before. They’re certainly mentioned prominently on various medical blogs.
Why do you see this in medicine? There are probably a number of factors that overlap:
• A high pressure job, where mistakes aren’t allowed (which isn’t humanly possible).
• A culture of litigation, where even minor mistakes are taken to court.
• Declining financial reimbursement, making it harder to support a practice and family, especially when you’re already six figures in debt coming out of medical school.
• Pressure to work longer hours and see far more patients than is possible, which increases the potential for mistakes. This further reduces the amount of family and recreational time available to balance ourselves.
• An increase in “empowered patients” demanding unnecessary tests and treatments because it said so on the Internet.
• A general lack of respect for the profession, to where we’re now “providers” who are vilified for political reasons by insurance companies, consumer groups, and both major parties.
• The need for us not to admit or seek treatment for human vulnerabilities. Our own health (mental and physical) is neglected because we can’t take time off to address it and a fear that doing so may result in us having our licenses penalized.
Any of the above makes life unpleasant, but when you combine them … it can be a perfect storm that tips a person over the edge.
In medicine, seeking help is often seen as a weakness, and even the most rational person under difficult circumstances can snap. None of the physicians who’ve ended their lives started out saying that was how they wanted their medical career to wind up. But when stressors pile up, it may appear to them to be the only way out. In that frame of mind, you think doing something so drastic is better for everyone around you. It isn’t true, but at that point you don’t believe it.
A physician’s suicide, even outside of its effects on their family, is a loss. A physician is a community resource, leaving behind relationships with patients in various stages of work-ups and treatments. There’s always another doctor, but it’s not easy, or immediate, to find someone who’s a good fit for the area.
I don’t know if this is a peculiarly American phenomenon or if my colleagues in Canada, Europe, and elsewhere face similar challenges. If the suicide rate elsewhere is lower, what can we learn from them to make things better here? If it’s the same, what can we do collectively to find an answer? Every country needs doctors and can’t afford to lose them.
Is there an easy solution? Probably not. Too many factors to fix. But it’s a serious problem and needs attention.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Recently, there have been several news stories about physicians committing suicide. This is across all levels of the profession, including medical students, residents, and attendings.
Historically, doctors have had a higher rate of suicide than most professions. I’m not sure if that number has crept up recently, or if events are garnering more attention than before. They’re certainly mentioned prominently on various medical blogs.
Why do you see this in medicine? There are probably a number of factors that overlap:
• A high pressure job, where mistakes aren’t allowed (which isn’t humanly possible).
• A culture of litigation, where even minor mistakes are taken to court.
• Declining financial reimbursement, making it harder to support a practice and family, especially when you’re already six figures in debt coming out of medical school.
• Pressure to work longer hours and see far more patients than is possible, which increases the potential for mistakes. This further reduces the amount of family and recreational time available to balance ourselves.
• An increase in “empowered patients” demanding unnecessary tests and treatments because it said so on the Internet.
• A general lack of respect for the profession, to where we’re now “providers” who are vilified for political reasons by insurance companies, consumer groups, and both major parties.
• The need for us not to admit or seek treatment for human vulnerabilities. Our own health (mental and physical) is neglected because we can’t take time off to address it and a fear that doing so may result in us having our licenses penalized.
Any of the above makes life unpleasant, but when you combine them … it can be a perfect storm that tips a person over the edge.
In medicine, seeking help is often seen as a weakness, and even the most rational person under difficult circumstances can snap. None of the physicians who’ve ended their lives started out saying that was how they wanted their medical career to wind up. But when stressors pile up, it may appear to them to be the only way out. In that frame of mind, you think doing something so drastic is better for everyone around you. It isn’t true, but at that point you don’t believe it.
A physician’s suicide, even outside of its effects on their family, is a loss. A physician is a community resource, leaving behind relationships with patients in various stages of work-ups and treatments. There’s always another doctor, but it’s not easy, or immediate, to find someone who’s a good fit for the area.
I don’t know if this is a peculiarly American phenomenon or if my colleagues in Canada, Europe, and elsewhere face similar challenges. If the suicide rate elsewhere is lower, what can we learn from them to make things better here? If it’s the same, what can we do collectively to find an answer? Every country needs doctors and can’t afford to lose them.
Is there an easy solution? Probably not. Too many factors to fix. But it’s a serious problem and needs attention.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Recently, there have been several news stories about physicians committing suicide. This is across all levels of the profession, including medical students, residents, and attendings.
Historically, doctors have had a higher rate of suicide than most professions. I’m not sure if that number has crept up recently, or if events are garnering more attention than before. They’re certainly mentioned prominently on various medical blogs.
Why do you see this in medicine? There are probably a number of factors that overlap:
• A high pressure job, where mistakes aren’t allowed (which isn’t humanly possible).
• A culture of litigation, where even minor mistakes are taken to court.
• Declining financial reimbursement, making it harder to support a practice and family, especially when you’re already six figures in debt coming out of medical school.
• Pressure to work longer hours and see far more patients than is possible, which increases the potential for mistakes. This further reduces the amount of family and recreational time available to balance ourselves.
• An increase in “empowered patients” demanding unnecessary tests and treatments because it said so on the Internet.
• A general lack of respect for the profession, to where we’re now “providers” who are vilified for political reasons by insurance companies, consumer groups, and both major parties.
• The need for us not to admit or seek treatment for human vulnerabilities. Our own health (mental and physical) is neglected because we can’t take time off to address it and a fear that doing so may result in us having our licenses penalized.
Any of the above makes life unpleasant, but when you combine them … it can be a perfect storm that tips a person over the edge.
In medicine, seeking help is often seen as a weakness, and even the most rational person under difficult circumstances can snap. None of the physicians who’ve ended their lives started out saying that was how they wanted their medical career to wind up. But when stressors pile up, it may appear to them to be the only way out. In that frame of mind, you think doing something so drastic is better for everyone around you. It isn’t true, but at that point you don’t believe it.
A physician’s suicide, even outside of its effects on their family, is a loss. A physician is a community resource, leaving behind relationships with patients in various stages of work-ups and treatments. There’s always another doctor, but it’s not easy, or immediate, to find someone who’s a good fit for the area.
I don’t know if this is a peculiarly American phenomenon or if my colleagues in Canada, Europe, and elsewhere face similar challenges. If the suicide rate elsewhere is lower, what can we learn from them to make things better here? If it’s the same, what can we do collectively to find an answer? Every country needs doctors and can’t afford to lose them.
Is there an easy solution? Probably not. Too many factors to fix. But it’s a serious problem and needs attention.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.

To FDA: Wait for clinical outcomes data
The recent endorsements by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee of the new genetically manufactured, subcutaneously administered cholesterol-lowering agents alirocumab (Sanofi-Regeneron) and evolocumab (Pfizer) have sent a jolt through the world of cholesterol therapy. Proposed for the treatment of patients with high-risk cardiovascular disease and homozygous familial hypercholesterolemia (HoFH), the decisions suggest that the committee has developed a severe case of scientific amnesia.
A case can be made for the approval for the very-high-risk and untreatable patients with HoFH, but the decision to treat millions of Americans based on the paucity of clinical data available would be disturbing, if not irresponsible.
It is clear that these proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors provide a powerful therapy for reducing LDL cholesterol, with a reported decrease of about 60% after only 12 or 52 weeks of treatment. Based on four small studies lasting 10-12 weeks and without any clinical outcome data, the committee recommended approving evolocumab for lifetime therapy without anything to support its clinical effectiveness or safety.
Dr. Robert J. Smith, who chaired the advisory committee meeting, faced with approving the drug or delaying a decision until a randomized clinical trial can be completed said, “I am unwilling to subject patients to the wait.” A publication of the American College of Cardiology suggested that these two monoclonal antibody drugs could replace generic statins in 70 million Americans at an estimated cost of $7,000-$12,000 a year. Talk about breaking the bank.
Only recently, we decided that the appropriate statin therapy for cholesterol control should use the dosing from the original clinical trials rather than chasing LDL levels to the lowest level possible. We now have a class of drugs that clearly can lower the cholesterol to a level never seen before, and we are about to discard that therapeutic advice.
We have experience in chasing blood levels with uncertain outcomes. Reducing blood sugar to a very low hemoglobin A1c level led to adverse clinical events and increased mortality in type 2 diabetes. Raising HDL with the cholesterol ester transfer protein (CETP) inhibitor torcetrapib in a study of 15,000 patients in an outcome trial carried out over a number of years led to increased blood pressure and mortality by 58%, which was associated with a 25% decrease in LDL cholesterol and a 72% increase in HDL, both effects presumed to be remarkably beneficial (N. Engl. J. Med. 2007;357:2109-22).
It is quite possible that these new agents can further decrease coronary vascular mortality. A healthy controversy has raged for some time in regard to the “LDL hypothesis,” compared with therapy based upon the clinical trial outcome. The recent IMPROVE-IT report (N. Engl. J. Med. 2015;372:2387-97) provides some data to support the LDL hypothesis. In that study, the addition of 10 mg of ezetimibe to 40 mg of simvastatin in 18,144 patients followed for up to 6 years resulted in a 2% decrease in mortality associated with a 15.8-mg decrease in serum LDL.
Would it not be prudent to have similar data with the new drug on the street? If it is as potent as it appears to be, a trial of much shorter duration might demonstrate its potency and safety before it is offered to 70 million Americans. The FDA has been reluctant to use surrogate endpoints for approving drugs, but its position has not always been consistent. For some time the FDA has, on occasion, approved drugs that can lower cholesterol with limited outcome data as it did with ezetimibe. However, the decision in regard to the PCSK9 inhibitors will have a much larger impact on care than an add-on drug like ezetimibe. Let’s hope that the FDA shows better judgment than its advisory committee.
Dr. Goldstein, medical editor of Cardiology News, is professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
The recent endorsements by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee of the new genetically manufactured, subcutaneously administered cholesterol-lowering agents alirocumab (Sanofi-Regeneron) and evolocumab (Pfizer) have sent a jolt through the world of cholesterol therapy. Proposed for the treatment of patients with high-risk cardiovascular disease and homozygous familial hypercholesterolemia (HoFH), the decisions suggest that the committee has developed a severe case of scientific amnesia.
A case can be made for the approval for the very-high-risk and untreatable patients with HoFH, but the decision to treat millions of Americans based on the paucity of clinical data available would be disturbing, if not irresponsible.
It is clear that these proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors provide a powerful therapy for reducing LDL cholesterol, with a reported decrease of about 60% after only 12 or 52 weeks of treatment. Based on four small studies lasting 10-12 weeks and without any clinical outcome data, the committee recommended approving evolocumab for lifetime therapy without anything to support its clinical effectiveness or safety.
Dr. Robert J. Smith, who chaired the advisory committee meeting, faced with approving the drug or delaying a decision until a randomized clinical trial can be completed said, “I am unwilling to subject patients to the wait.” A publication of the American College of Cardiology suggested that these two monoclonal antibody drugs could replace generic statins in 70 million Americans at an estimated cost of $7,000-$12,000 a year. Talk about breaking the bank.
Only recently, we decided that the appropriate statin therapy for cholesterol control should use the dosing from the original clinical trials rather than chasing LDL levels to the lowest level possible. We now have a class of drugs that clearly can lower the cholesterol to a level never seen before, and we are about to discard that therapeutic advice.
We have experience in chasing blood levels with uncertain outcomes. Reducing blood sugar to a very low hemoglobin A1c level led to adverse clinical events and increased mortality in type 2 diabetes. Raising HDL with the cholesterol ester transfer protein (CETP) inhibitor torcetrapib in a study of 15,000 patients in an outcome trial carried out over a number of years led to increased blood pressure and mortality by 58%, which was associated with a 25% decrease in LDL cholesterol and a 72% increase in HDL, both effects presumed to be remarkably beneficial (N. Engl. J. Med. 2007;357:2109-22).
It is quite possible that these new agents can further decrease coronary vascular mortality. A healthy controversy has raged for some time in regard to the “LDL hypothesis,” compared with therapy based upon the clinical trial outcome. The recent IMPROVE-IT report (N. Engl. J. Med. 2015;372:2387-97) provides some data to support the LDL hypothesis. In that study, the addition of 10 mg of ezetimibe to 40 mg of simvastatin in 18,144 patients followed for up to 6 years resulted in a 2% decrease in mortality associated with a 15.8-mg decrease in serum LDL.
Would it not be prudent to have similar data with the new drug on the street? If it is as potent as it appears to be, a trial of much shorter duration might demonstrate its potency and safety before it is offered to 70 million Americans. The FDA has been reluctant to use surrogate endpoints for approving drugs, but its position has not always been consistent. For some time the FDA has, on occasion, approved drugs that can lower cholesterol with limited outcome data as it did with ezetimibe. However, the decision in regard to the PCSK9 inhibitors will have a much larger impact on care than an add-on drug like ezetimibe. Let’s hope that the FDA shows better judgment than its advisory committee.
Dr. Goldstein, medical editor of Cardiology News, is professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
The recent endorsements by the FDA’s Endocrinologic and Metabolic Drugs Advisory Committee of the new genetically manufactured, subcutaneously administered cholesterol-lowering agents alirocumab (Sanofi-Regeneron) and evolocumab (Pfizer) have sent a jolt through the world of cholesterol therapy. Proposed for the treatment of patients with high-risk cardiovascular disease and homozygous familial hypercholesterolemia (HoFH), the decisions suggest that the committee has developed a severe case of scientific amnesia.
A case can be made for the approval for the very-high-risk and untreatable patients with HoFH, but the decision to treat millions of Americans based on the paucity of clinical data available would be disturbing, if not irresponsible.
It is clear that these proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors provide a powerful therapy for reducing LDL cholesterol, with a reported decrease of about 60% after only 12 or 52 weeks of treatment. Based on four small studies lasting 10-12 weeks and without any clinical outcome data, the committee recommended approving evolocumab for lifetime therapy without anything to support its clinical effectiveness or safety.
Dr. Robert J. Smith, who chaired the advisory committee meeting, faced with approving the drug or delaying a decision until a randomized clinical trial can be completed said, “I am unwilling to subject patients to the wait.” A publication of the American College of Cardiology suggested that these two monoclonal antibody drugs could replace generic statins in 70 million Americans at an estimated cost of $7,000-$12,000 a year. Talk about breaking the bank.
Only recently, we decided that the appropriate statin therapy for cholesterol control should use the dosing from the original clinical trials rather than chasing LDL levels to the lowest level possible. We now have a class of drugs that clearly can lower the cholesterol to a level never seen before, and we are about to discard that therapeutic advice.
We have experience in chasing blood levels with uncertain outcomes. Reducing blood sugar to a very low hemoglobin A1c level led to adverse clinical events and increased mortality in type 2 diabetes. Raising HDL with the cholesterol ester transfer protein (CETP) inhibitor torcetrapib in a study of 15,000 patients in an outcome trial carried out over a number of years led to increased blood pressure and mortality by 58%, which was associated with a 25% decrease in LDL cholesterol and a 72% increase in HDL, both effects presumed to be remarkably beneficial (N. Engl. J. Med. 2007;357:2109-22).
It is quite possible that these new agents can further decrease coronary vascular mortality. A healthy controversy has raged for some time in regard to the “LDL hypothesis,” compared with therapy based upon the clinical trial outcome. The recent IMPROVE-IT report (N. Engl. J. Med. 2015;372:2387-97) provides some data to support the LDL hypothesis. In that study, the addition of 10 mg of ezetimibe to 40 mg of simvastatin in 18,144 patients followed for up to 6 years resulted in a 2% decrease in mortality associated with a 15.8-mg decrease in serum LDL.
Would it not be prudent to have similar data with the new drug on the street? If it is as potent as it appears to be, a trial of much shorter duration might demonstrate its potency and safety before it is offered to 70 million Americans. The FDA has been reluctant to use surrogate endpoints for approving drugs, but its position has not always been consistent. For some time the FDA has, on occasion, approved drugs that can lower cholesterol with limited outcome data as it did with ezetimibe. However, the decision in regard to the PCSK9 inhibitors will have a much larger impact on care than an add-on drug like ezetimibe. Let’s hope that the FDA shows better judgment than its advisory committee.
Dr. Goldstein, medical editor of Cardiology News, is professor of medicine at Wayne State University and division head emeritus of cardiovascular medicine at Henry Ford Hospital, both in Detroit. He is on data safety monitoring committees for the National Institutes of Health and several pharmaceutical companies.
