Azithromycin is prescribed for a variety of acute respiratory and nonrespiratory infections. However, it is also used in several chronic respiratory diseases.
MECHANISM OF ACTION
Macrolide antibiotics like azithromycin inhibit bacterial growth and replication by interrupting protein synthesis. But azithromycin also has immunomodulatory properties.1
In the acute phase of inflammation, azithromycin exerts an initial neutrophil degranulation effect and enhances the oxidative response that is primed by particulate stimulus, which could facilitate its antibacterial effects. In the late phase, it down-regulates the oxidative burst and increases apoptosis of neutrophils to promote healing without compromising immunity. Azithromycin also attenuates airway mucus hypersecretion, improves ciliary function, and promotes pulmonary epithelial cell healing.2,3
Collectively, these effects make the drug effective in many chronic inflammatory lung conditions (Table 1).
CYSTIC FIBROSIS
Cystic fibrosis is a genetic disease affecting many organs, but its effect on the upper and lower airways has the greatest impact on quality of life and survival. Impaired mucociliary clearance and repeated respiratory infections contribute to chronic inflammation and a progressive decline in lung function.4,5
A 2012 Cochrane review of 5 studies in 549 patients found that, compared with those taking placebo, patients taking azithromycin 250–500 mg 3 times a week had improvement in forced expiratory volume in 1 second (FEV1). The mean difference at 6 months was 3.97% (95% cofidence interval [CI] 1.74– 6.19). Patients on azithromycin were free from pulmonary exacerbation approximately twice as long as patients on placebo (odds ratio 1.96, 95% CI 1.15–3.33).6,7
The Cystic Fibrosis Foundation recommends long-term azithromycin therapy to improve lung function and reduce exacerbations in patients age 6 or older who have persistent Pseudomonas aeruginosa airway cultures (level of evidence: fair).8
DIFFUSE PANBRONCHIOLITIS
Diffuse panbronchiolitis, or diffuse chronic inflammatory bronchiolitis and sinusitis, is seen mainly in patients of Asian descent.9 In the past, the mortality rate was greater than 90%, but between 1970 and 1979 the 10-year survival rate increased by more than 40% with chronic macrolide therapy, ie, with erythromycin.10,11
Later retrospective studies of azithromycin 500 mg 3 times a week showed results comparable to those with erythromycin, with improvement in symptoms, lung function, arterial partial pressure of oxygen, and radiologic findings, as well as fewer adverse effects.12 These benefits justify the current recommendation for azithromycin as the mainstay of therapy in diffuse panbronchiolitis.
BRONCHIOLITIS OBLITERANS SYNDROME
Bronchiolitis obliterans syndrome is an airflow limitation that arises without infection or imaging evidence of bronchiolitis in patients who received allogeneic hematopoietic stem cell or lung transplant. It occurs in 50% of lung transplant recipients as a form of chronic graft rejection and in 6% to 20% of allogeneic stem cell transplant recipients as a manifestation of chronic graft-vs-host disease.13,14
Azithromycin has been used in its management. A meta-analysis of lung transplant recipients found a significant improvement in the survival rate and overall lung function after an average of 7 months of treatment with azithromycin, with a mean increase in FEV1 of 8.8% (95% CI 5.1–12.47, P < .001).14 The evidence currently supports long-term azithromycin 250 mg 3 times a week after lung transplant to reduce any decline in lung function and to lower the mortality rate.14,15
In allogeneic stem cell transplant recipients, the evidence for long-term azithromycin treatment is sparse. A recent prospective multicenter study evaluated the effect of an azithromycin-based regimen (fluticasone, azithromycin, and montelukast, plus a steroid pulse) in stem cell recipients with bronchiolitis obliterans syndrome during the first 3 months after diagnosis. In the treated group, 6% had a drop in FEV1 of more than 10% at 3-month follow-up compared with 40% of historical controls (95% CI 1%–19%, P < .001). Also, treatment resulted in a 50% reduction in the dose of systemic steroids and a substantial improvement in functional status.16
Given the limited options in the management of these patients and until further studies are available, azithromycin 3 times weekly is suggested.
NON-CYSTIC FIBROSIS BRONCHIECTASIS
Non-cystic fibrosis bronchiectasis is a chronic inflammatory lung condition characterized by irreversible dilation of the bronchi and bronchioles due to a variety of causes including recurrent or old infection, immunodeficiency, autoimmune conditions, and connective tissue disease; it can also be idiopathic.17
Altenburg et al,18 in a randomized, double-blind, placebo-controlled trial, found that azithromycin 250 mg 3 times a week for 12 months reduced the number of exacerbations from a median number of 2 per patient with placebo to 0 per patient with azithromycin (P < .001). At 3 months, the FEV1 as a percent of predicted had increased by 1.03% in the azithromycin group and decreased by 0.10% in the placebo group (P = .047). The number needed to treat with azithromycin to maintain clinical stability was 3.0.
Wong et al19 randomized patients to receive azithromycin 500 mg 3 times a week or placebo for 6 months. The rate of exacerbations was 0.59 per patient in the azithromycin group and 1.57 per patient in the placebo group (P < .0001). The FEV1 remained unchanged from baseline in the azithromycin group while decreasing in the placebo group, but the difference was not significant.
EXACERBATIONS OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE
Acute exacerbations of chronic obstructive pulmonary disease (COPD) are a major cause of death, poor quality of life, and healthcare expenditures.20 Prevention is therefore of the utmost importance.
Several studies have shown that azithromycin prophylaxis can reduce acute exacerbations of COPD. A recent meta-analysis showed that long-term macrolide prophylaxis significantly reduced exacerbations compared with rates in controls (risk ratio = 0.70, 95% CI 0.56–0.87, P < .01) and increased the median time to first COPD exacerbation by more than 90 days (P < .01).21 Long-term azithromycin therapy may be considered in selected patients who have frequent exacerbations despite optimal maintenance inhaler therapy.
PROPHYLAXIS IN IMMUNODEFICIENCY
Disseminated Mycobacterium avium complex (MAC) is an opportunistic infection most commonly occurring in patients with acquired immunodeficiency syndrome with CD4 counts below 50 cells/µL.22,23
In a double-blinded, randomized trial, patients who received azithromycin had a 47% reduction in the incidence of MAC infection.
Given the long half-life of azithromycin, it is effective with once-weekly dosing of 1,200 mg.23 Ideally, patients are placed on a prophylactic agent for disseminated MAC infection until the CD4 count reaches 100 cells/µL and remains at or above this level for 3 consecutive months.24
ADVERSE EFFECTS AND PRECAUTIONS
Long-term azithromycin therapy may produce bacterial resistance; the risk has been estimated at 2.7 times greater in patients who are on long-term azithromycin treatment.25 Also, patients at risk for MAC infection, such as those with cystic fibrosis, should be screened for it before starting treatment in order to prevent resistance to azithromycin.
The US Food and Drug Administration warns that azithromycin can lead to a prolonged corrected QT interval and potential fatal arrhythmias such as torsades de pointes. Major reviews have largely agreed that arrhythmias are more pronounced in patients with a coexisting cardiac risk factor such as existing QT-interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or arrhythmias, or who are on class IA and III antiarrhythmic drugs.26–28
Other potential adverse effects of long-term azithromycin treatment are gastrointestinal symptoms and hearing impairment.29,30 A review of potential drug interactions is advised when patients are placed on long-term azithromycin therapy.
Although azithromycin is generally well tolerated, long-term treatment should be individualized and the benefits weighed against the risks. Patients should be monitored during treatment for any of the above adverse effects.
References
Bailly S, Pocidalo JJ, Fay M, Gougerot-Pocidalo MA. Differential modulation of cytokine production by macrolides: interleukin-6 production is increased by spiramycin and erythromycin. Antimicrob Agents Chemother 1991; 35:2016–2019.
Kanoh S, Rubin BK. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin Microbiol Rev 2010; 23:590–615.
Culić O, Eraković V, Cepelak I, et al. Azithromycin modulates neutrophil function and circulating inflammatory mediators in healthy human subjects. Eur J Pharmacol 2002; 450:277–289.
Cohen-Cymberknoh M, Kerem E, Ferkol T, Elizur A. Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax 2013; 68:1157–1162.
Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med 2012; 186:857–865.
Saiman L, Anstead M, Mayer-Hamblett N, et al; AZ0004 Azithromycin Study Group. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2010; 303:1707–1715.
Southern KW, Barker PM, Solis-Moya A, Patel L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2012;11:CD002203.
Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007; 176:957–969.
Yanagihara K, Kadoto J, Kohno S. Diffuse panbronchiolitis—pathophysiology and treatment mechanisms. Int J Antimicrob Agents 2001; 18(suppl 1):S83–S87.
Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
Schultz MJ. Macrolide activities beyond their antimicrobial effects: macrolides in diffuse panbronchiolitis and cystic fibrosis. J Antimicrob Chemother 2004; 54:21–28.
Hui D, Yan F, Chen RH. The effects of azithromycin on patients with diffuse panbronchiolitis: a retrospective study of 29 cases. J Thorac Dis 2013; 5:613–617.
Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005; 25:490–493.
Kingah PL, Muma G, Soubani A. Azithromycin improves lung function in patients with post-lung transplant bronchiolitis obliterans syndrome: a meta-analysis. Clin Transplant 2014; 28:906–910.
Corris PA, Ryan VA, Small T, et al. A randomised controlled trial of azithromycin therapy in bronchiolitis obliterans syndrome (BOS) post lung transplantation. Thorax 2015; 70:442–450.
Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016; 22:710–716.
Haworth CS, Bilton D, Elborn JS. Long-term macrolide maintenance therapy in non-CF bronchiectasis: evidence and questions. Respir Med 2014; 108:1397–1408.
Altenburg J, de Graaff CS, Stienstra Y, et al. Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: the BAT randomized controlled trial. JAMA 2013; 309:1251–1259.
Wong C, Jayaram L, Karalus N, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet 2012; 380:660–667.
Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax 2012; 67:957–963.
Ni W, Shao X, Cai X, et al. Prophylactic use of macrolide antibiotics for the prevention of chronic obstructive pulmonary disease exacerbation: a meta-analysis. PLoS One 2015; 10:e0121257.
Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416.
Havlir DV, Dubé MP, Sattler FR, et al. Prophylaxis against disseminated Mycobacterium avium complex with weekly azithromycin, daily rifabutin, or both. California Collaborative Treatment Group. N Engl J Med 1996; 335:392–398.
Uthman MM, Uthman OA, Yahaya I. Interventions for the prevention of Mycobacterium avium complex in adults and children with HIV. Cochrane Database Syst Rev 2013; 4:CD007191.
Li H, Liu DH, Chen LL, et al. Meta-analysis of the adverse effects of long-term azithromycin use in patients with chronic lung diseases. Antimicrob Agents Chemother 2014; 58:511–517.
Svanström H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013; 368:1704–1712.
Albert RK, Schuller JL; COPD Clinical Research Network. Macrolide antibiotics and the risk of cardiac arrhythmias. Am J Respir Crit Care Med 2014; 189:1173–1180.
Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366:1881–1890.
Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
Broad J, Sanger GJ. The antibiotic azithromycin is a motilin receptor agonist in human stomach: comparison with erythromycin. Br J Pharmacol 2013; 168:1859–1867.
Abdulrazak Alchakaki, MD Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Cassondra Cramer Division of Pulmonary, Critical Care and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Allie Patterson Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Ayman O. Soubani Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Address: Abdulrazak Alchakaki, MD, Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, 3990 John R, 3 Hudson, Detroit, MI 48201; aalchaka@med.wayne.edu
Abdulrazak Alchakaki, MD Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Cassondra Cramer Division of Pulmonary, Critical Care and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Allie Patterson Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Ayman O. Soubani Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Address: Abdulrazak Alchakaki, MD, Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, 3990 John R, 3 Hudson, Detroit, MI 48201; aalchaka@med.wayne.edu
Author and Disclosure Information
Abdulrazak Alchakaki, MD Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Cassondra Cramer Division of Pulmonary, Critical Care and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Allie Patterson Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Ayman O. Soubani Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, Detroit, MI
Address: Abdulrazak Alchakaki, MD, Division of Pulmonary, Critical Care, and Sleep Medicine, Wayne State University School of Medicine, 3990 John R, 3 Hudson, Detroit, MI 48201; aalchaka@med.wayne.edu
Azithromycin is prescribed for a variety of acute respiratory and nonrespiratory infections. However, it is also used in several chronic respiratory diseases.
MECHANISM OF ACTION
Macrolide antibiotics like azithromycin inhibit bacterial growth and replication by interrupting protein synthesis. But azithromycin also has immunomodulatory properties.1
In the acute phase of inflammation, azithromycin exerts an initial neutrophil degranulation effect and enhances the oxidative response that is primed by particulate stimulus, which could facilitate its antibacterial effects. In the late phase, it down-regulates the oxidative burst and increases apoptosis of neutrophils to promote healing without compromising immunity. Azithromycin also attenuates airway mucus hypersecretion, improves ciliary function, and promotes pulmonary epithelial cell healing.2,3
Collectively, these effects make the drug effective in many chronic inflammatory lung conditions (Table 1).
CYSTIC FIBROSIS
Cystic fibrosis is a genetic disease affecting many organs, but its effect on the upper and lower airways has the greatest impact on quality of life and survival. Impaired mucociliary clearance and repeated respiratory infections contribute to chronic inflammation and a progressive decline in lung function.4,5
A 2012 Cochrane review of 5 studies in 549 patients found that, compared with those taking placebo, patients taking azithromycin 250–500 mg 3 times a week had improvement in forced expiratory volume in 1 second (FEV1). The mean difference at 6 months was 3.97% (95% cofidence interval [CI] 1.74– 6.19). Patients on azithromycin were free from pulmonary exacerbation approximately twice as long as patients on placebo (odds ratio 1.96, 95% CI 1.15–3.33).6,7
The Cystic Fibrosis Foundation recommends long-term azithromycin therapy to improve lung function and reduce exacerbations in patients age 6 or older who have persistent Pseudomonas aeruginosa airway cultures (level of evidence: fair).8
DIFFUSE PANBRONCHIOLITIS
Diffuse panbronchiolitis, or diffuse chronic inflammatory bronchiolitis and sinusitis, is seen mainly in patients of Asian descent.9 In the past, the mortality rate was greater than 90%, but between 1970 and 1979 the 10-year survival rate increased by more than 40% with chronic macrolide therapy, ie, with erythromycin.10,11
Later retrospective studies of azithromycin 500 mg 3 times a week showed results comparable to those with erythromycin, with improvement in symptoms, lung function, arterial partial pressure of oxygen, and radiologic findings, as well as fewer adverse effects.12 These benefits justify the current recommendation for azithromycin as the mainstay of therapy in diffuse panbronchiolitis.
BRONCHIOLITIS OBLITERANS SYNDROME
Bronchiolitis obliterans syndrome is an airflow limitation that arises without infection or imaging evidence of bronchiolitis in patients who received allogeneic hematopoietic stem cell or lung transplant. It occurs in 50% of lung transplant recipients as a form of chronic graft rejection and in 6% to 20% of allogeneic stem cell transplant recipients as a manifestation of chronic graft-vs-host disease.13,14
Azithromycin has been used in its management. A meta-analysis of lung transplant recipients found a significant improvement in the survival rate and overall lung function after an average of 7 months of treatment with azithromycin, with a mean increase in FEV1 of 8.8% (95% CI 5.1–12.47, P < .001).14 The evidence currently supports long-term azithromycin 250 mg 3 times a week after lung transplant to reduce any decline in lung function and to lower the mortality rate.14,15
In allogeneic stem cell transplant recipients, the evidence for long-term azithromycin treatment is sparse. A recent prospective multicenter study evaluated the effect of an azithromycin-based regimen (fluticasone, azithromycin, and montelukast, plus a steroid pulse) in stem cell recipients with bronchiolitis obliterans syndrome during the first 3 months after diagnosis. In the treated group, 6% had a drop in FEV1 of more than 10% at 3-month follow-up compared with 40% of historical controls (95% CI 1%–19%, P < .001). Also, treatment resulted in a 50% reduction in the dose of systemic steroids and a substantial improvement in functional status.16
Given the limited options in the management of these patients and until further studies are available, azithromycin 3 times weekly is suggested.
NON-CYSTIC FIBROSIS BRONCHIECTASIS
Non-cystic fibrosis bronchiectasis is a chronic inflammatory lung condition characterized by irreversible dilation of the bronchi and bronchioles due to a variety of causes including recurrent or old infection, immunodeficiency, autoimmune conditions, and connective tissue disease; it can also be idiopathic.17
Altenburg et al,18 in a randomized, double-blind, placebo-controlled trial, found that azithromycin 250 mg 3 times a week for 12 months reduced the number of exacerbations from a median number of 2 per patient with placebo to 0 per patient with azithromycin (P < .001). At 3 months, the FEV1 as a percent of predicted had increased by 1.03% in the azithromycin group and decreased by 0.10% in the placebo group (P = .047). The number needed to treat with azithromycin to maintain clinical stability was 3.0.
Wong et al19 randomized patients to receive azithromycin 500 mg 3 times a week or placebo for 6 months. The rate of exacerbations was 0.59 per patient in the azithromycin group and 1.57 per patient in the placebo group (P < .0001). The FEV1 remained unchanged from baseline in the azithromycin group while decreasing in the placebo group, but the difference was not significant.
EXACERBATIONS OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE
Acute exacerbations of chronic obstructive pulmonary disease (COPD) are a major cause of death, poor quality of life, and healthcare expenditures.20 Prevention is therefore of the utmost importance.
Several studies have shown that azithromycin prophylaxis can reduce acute exacerbations of COPD. A recent meta-analysis showed that long-term macrolide prophylaxis significantly reduced exacerbations compared with rates in controls (risk ratio = 0.70, 95% CI 0.56–0.87, P < .01) and increased the median time to first COPD exacerbation by more than 90 days (P < .01).21 Long-term azithromycin therapy may be considered in selected patients who have frequent exacerbations despite optimal maintenance inhaler therapy.
PROPHYLAXIS IN IMMUNODEFICIENCY
Disseminated Mycobacterium avium complex (MAC) is an opportunistic infection most commonly occurring in patients with acquired immunodeficiency syndrome with CD4 counts below 50 cells/µL.22,23
In a double-blinded, randomized trial, patients who received azithromycin had a 47% reduction in the incidence of MAC infection.
Given the long half-life of azithromycin, it is effective with once-weekly dosing of 1,200 mg.23 Ideally, patients are placed on a prophylactic agent for disseminated MAC infection until the CD4 count reaches 100 cells/µL and remains at or above this level for 3 consecutive months.24
ADVERSE EFFECTS AND PRECAUTIONS
Long-term azithromycin therapy may produce bacterial resistance; the risk has been estimated at 2.7 times greater in patients who are on long-term azithromycin treatment.25 Also, patients at risk for MAC infection, such as those with cystic fibrosis, should be screened for it before starting treatment in order to prevent resistance to azithromycin.
The US Food and Drug Administration warns that azithromycin can lead to a prolonged corrected QT interval and potential fatal arrhythmias such as torsades de pointes. Major reviews have largely agreed that arrhythmias are more pronounced in patients with a coexisting cardiac risk factor such as existing QT-interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or arrhythmias, or who are on class IA and III antiarrhythmic drugs.26–28
Other potential adverse effects of long-term azithromycin treatment are gastrointestinal symptoms and hearing impairment.29,30 A review of potential drug interactions is advised when patients are placed on long-term azithromycin therapy.
Although azithromycin is generally well tolerated, long-term treatment should be individualized and the benefits weighed against the risks. Patients should be monitored during treatment for any of the above adverse effects.
Azithromycin is prescribed for a variety of acute respiratory and nonrespiratory infections. However, it is also used in several chronic respiratory diseases.
MECHANISM OF ACTION
Macrolide antibiotics like azithromycin inhibit bacterial growth and replication by interrupting protein synthesis. But azithromycin also has immunomodulatory properties.1
In the acute phase of inflammation, azithromycin exerts an initial neutrophil degranulation effect and enhances the oxidative response that is primed by particulate stimulus, which could facilitate its antibacterial effects. In the late phase, it down-regulates the oxidative burst and increases apoptosis of neutrophils to promote healing without compromising immunity. Azithromycin also attenuates airway mucus hypersecretion, improves ciliary function, and promotes pulmonary epithelial cell healing.2,3
Collectively, these effects make the drug effective in many chronic inflammatory lung conditions (Table 1).
CYSTIC FIBROSIS
Cystic fibrosis is a genetic disease affecting many organs, but its effect on the upper and lower airways has the greatest impact on quality of life and survival. Impaired mucociliary clearance and repeated respiratory infections contribute to chronic inflammation and a progressive decline in lung function.4,5
A 2012 Cochrane review of 5 studies in 549 patients found that, compared with those taking placebo, patients taking azithromycin 250–500 mg 3 times a week had improvement in forced expiratory volume in 1 second (FEV1). The mean difference at 6 months was 3.97% (95% cofidence interval [CI] 1.74– 6.19). Patients on azithromycin were free from pulmonary exacerbation approximately twice as long as patients on placebo (odds ratio 1.96, 95% CI 1.15–3.33).6,7
The Cystic Fibrosis Foundation recommends long-term azithromycin therapy to improve lung function and reduce exacerbations in patients age 6 or older who have persistent Pseudomonas aeruginosa airway cultures (level of evidence: fair).8
DIFFUSE PANBRONCHIOLITIS
Diffuse panbronchiolitis, or diffuse chronic inflammatory bronchiolitis and sinusitis, is seen mainly in patients of Asian descent.9 In the past, the mortality rate was greater than 90%, but between 1970 and 1979 the 10-year survival rate increased by more than 40% with chronic macrolide therapy, ie, with erythromycin.10,11
Later retrospective studies of azithromycin 500 mg 3 times a week showed results comparable to those with erythromycin, with improvement in symptoms, lung function, arterial partial pressure of oxygen, and radiologic findings, as well as fewer adverse effects.12 These benefits justify the current recommendation for azithromycin as the mainstay of therapy in diffuse panbronchiolitis.
BRONCHIOLITIS OBLITERANS SYNDROME
Bronchiolitis obliterans syndrome is an airflow limitation that arises without infection or imaging evidence of bronchiolitis in patients who received allogeneic hematopoietic stem cell or lung transplant. It occurs in 50% of lung transplant recipients as a form of chronic graft rejection and in 6% to 20% of allogeneic stem cell transplant recipients as a manifestation of chronic graft-vs-host disease.13,14
Azithromycin has been used in its management. A meta-analysis of lung transplant recipients found a significant improvement in the survival rate and overall lung function after an average of 7 months of treatment with azithromycin, with a mean increase in FEV1 of 8.8% (95% CI 5.1–12.47, P < .001).14 The evidence currently supports long-term azithromycin 250 mg 3 times a week after lung transplant to reduce any decline in lung function and to lower the mortality rate.14,15
In allogeneic stem cell transplant recipients, the evidence for long-term azithromycin treatment is sparse. A recent prospective multicenter study evaluated the effect of an azithromycin-based regimen (fluticasone, azithromycin, and montelukast, plus a steroid pulse) in stem cell recipients with bronchiolitis obliterans syndrome during the first 3 months after diagnosis. In the treated group, 6% had a drop in FEV1 of more than 10% at 3-month follow-up compared with 40% of historical controls (95% CI 1%–19%, P < .001). Also, treatment resulted in a 50% reduction in the dose of systemic steroids and a substantial improvement in functional status.16
Given the limited options in the management of these patients and until further studies are available, azithromycin 3 times weekly is suggested.
NON-CYSTIC FIBROSIS BRONCHIECTASIS
Non-cystic fibrosis bronchiectasis is a chronic inflammatory lung condition characterized by irreversible dilation of the bronchi and bronchioles due to a variety of causes including recurrent or old infection, immunodeficiency, autoimmune conditions, and connective tissue disease; it can also be idiopathic.17
Altenburg et al,18 in a randomized, double-blind, placebo-controlled trial, found that azithromycin 250 mg 3 times a week for 12 months reduced the number of exacerbations from a median number of 2 per patient with placebo to 0 per patient with azithromycin (P < .001). At 3 months, the FEV1 as a percent of predicted had increased by 1.03% in the azithromycin group and decreased by 0.10% in the placebo group (P = .047). The number needed to treat with azithromycin to maintain clinical stability was 3.0.
Wong et al19 randomized patients to receive azithromycin 500 mg 3 times a week or placebo for 6 months. The rate of exacerbations was 0.59 per patient in the azithromycin group and 1.57 per patient in the placebo group (P < .0001). The FEV1 remained unchanged from baseline in the azithromycin group while decreasing in the placebo group, but the difference was not significant.
EXACERBATIONS OF CHRONIC OBSTRUCTIVE PULMONARY DISEASE
Acute exacerbations of chronic obstructive pulmonary disease (COPD) are a major cause of death, poor quality of life, and healthcare expenditures.20 Prevention is therefore of the utmost importance.
Several studies have shown that azithromycin prophylaxis can reduce acute exacerbations of COPD. A recent meta-analysis showed that long-term macrolide prophylaxis significantly reduced exacerbations compared with rates in controls (risk ratio = 0.70, 95% CI 0.56–0.87, P < .01) and increased the median time to first COPD exacerbation by more than 90 days (P < .01).21 Long-term azithromycin therapy may be considered in selected patients who have frequent exacerbations despite optimal maintenance inhaler therapy.
PROPHYLAXIS IN IMMUNODEFICIENCY
Disseminated Mycobacterium avium complex (MAC) is an opportunistic infection most commonly occurring in patients with acquired immunodeficiency syndrome with CD4 counts below 50 cells/µL.22,23
In a double-blinded, randomized trial, patients who received azithromycin had a 47% reduction in the incidence of MAC infection.
Given the long half-life of azithromycin, it is effective with once-weekly dosing of 1,200 mg.23 Ideally, patients are placed on a prophylactic agent for disseminated MAC infection until the CD4 count reaches 100 cells/µL and remains at or above this level for 3 consecutive months.24
ADVERSE EFFECTS AND PRECAUTIONS
Long-term azithromycin therapy may produce bacterial resistance; the risk has been estimated at 2.7 times greater in patients who are on long-term azithromycin treatment.25 Also, patients at risk for MAC infection, such as those with cystic fibrosis, should be screened for it before starting treatment in order to prevent resistance to azithromycin.
The US Food and Drug Administration warns that azithromycin can lead to a prolonged corrected QT interval and potential fatal arrhythmias such as torsades de pointes. Major reviews have largely agreed that arrhythmias are more pronounced in patients with a coexisting cardiac risk factor such as existing QT-interval prolongation, low blood levels of potassium or magnesium, a slower than normal heart rate, or arrhythmias, or who are on class IA and III antiarrhythmic drugs.26–28
Other potential adverse effects of long-term azithromycin treatment are gastrointestinal symptoms and hearing impairment.29,30 A review of potential drug interactions is advised when patients are placed on long-term azithromycin therapy.
Although azithromycin is generally well tolerated, long-term treatment should be individualized and the benefits weighed against the risks. Patients should be monitored during treatment for any of the above adverse effects.
References
Bailly S, Pocidalo JJ, Fay M, Gougerot-Pocidalo MA. Differential modulation of cytokine production by macrolides: interleukin-6 production is increased by spiramycin and erythromycin. Antimicrob Agents Chemother 1991; 35:2016–2019.
Kanoh S, Rubin BK. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin Microbiol Rev 2010; 23:590–615.
Culić O, Eraković V, Cepelak I, et al. Azithromycin modulates neutrophil function and circulating inflammatory mediators in healthy human subjects. Eur J Pharmacol 2002; 450:277–289.
Cohen-Cymberknoh M, Kerem E, Ferkol T, Elizur A. Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax 2013; 68:1157–1162.
Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med 2012; 186:857–865.
Saiman L, Anstead M, Mayer-Hamblett N, et al; AZ0004 Azithromycin Study Group. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2010; 303:1707–1715.
Southern KW, Barker PM, Solis-Moya A, Patel L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2012;11:CD002203.
Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007; 176:957–969.
Yanagihara K, Kadoto J, Kohno S. Diffuse panbronchiolitis—pathophysiology and treatment mechanisms. Int J Antimicrob Agents 2001; 18(suppl 1):S83–S87.
Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
Schultz MJ. Macrolide activities beyond their antimicrobial effects: macrolides in diffuse panbronchiolitis and cystic fibrosis. J Antimicrob Chemother 2004; 54:21–28.
Hui D, Yan F, Chen RH. The effects of azithromycin on patients with diffuse panbronchiolitis: a retrospective study of 29 cases. J Thorac Dis 2013; 5:613–617.
Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005; 25:490–493.
Kingah PL, Muma G, Soubani A. Azithromycin improves lung function in patients with post-lung transplant bronchiolitis obliterans syndrome: a meta-analysis. Clin Transplant 2014; 28:906–910.
Corris PA, Ryan VA, Small T, et al. A randomised controlled trial of azithromycin therapy in bronchiolitis obliterans syndrome (BOS) post lung transplantation. Thorax 2015; 70:442–450.
Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016; 22:710–716.
Haworth CS, Bilton D, Elborn JS. Long-term macrolide maintenance therapy in non-CF bronchiectasis: evidence and questions. Respir Med 2014; 108:1397–1408.
Altenburg J, de Graaff CS, Stienstra Y, et al. Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: the BAT randomized controlled trial. JAMA 2013; 309:1251–1259.
Wong C, Jayaram L, Karalus N, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet 2012; 380:660–667.
Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax 2012; 67:957–963.
Ni W, Shao X, Cai X, et al. Prophylactic use of macrolide antibiotics for the prevention of chronic obstructive pulmonary disease exacerbation: a meta-analysis. PLoS One 2015; 10:e0121257.
Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416.
Havlir DV, Dubé MP, Sattler FR, et al. Prophylaxis against disseminated Mycobacterium avium complex with weekly azithromycin, daily rifabutin, or both. California Collaborative Treatment Group. N Engl J Med 1996; 335:392–398.
Uthman MM, Uthman OA, Yahaya I. Interventions for the prevention of Mycobacterium avium complex in adults and children with HIV. Cochrane Database Syst Rev 2013; 4:CD007191.
Li H, Liu DH, Chen LL, et al. Meta-analysis of the adverse effects of long-term azithromycin use in patients with chronic lung diseases. Antimicrob Agents Chemother 2014; 58:511–517.
Svanström H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013; 368:1704–1712.
Albert RK, Schuller JL; COPD Clinical Research Network. Macrolide antibiotics and the risk of cardiac arrhythmias. Am J Respir Crit Care Med 2014; 189:1173–1180.
Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366:1881–1890.
Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
Broad J, Sanger GJ. The antibiotic azithromycin is a motilin receptor agonist in human stomach: comparison with erythromycin. Br J Pharmacol 2013; 168:1859–1867.
References
Bailly S, Pocidalo JJ, Fay M, Gougerot-Pocidalo MA. Differential modulation of cytokine production by macrolides: interleukin-6 production is increased by spiramycin and erythromycin. Antimicrob Agents Chemother 1991; 35:2016–2019.
Kanoh S, Rubin BK. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin Microbiol Rev 2010; 23:590–615.
Culić O, Eraković V, Cepelak I, et al. Azithromycin modulates neutrophil function and circulating inflammatory mediators in healthy human subjects. Eur J Pharmacol 2002; 450:277–289.
Cohen-Cymberknoh M, Kerem E, Ferkol T, Elizur A. Airway inflammation in cystic fibrosis: molecular mechanisms and clinical implications. Thorax 2013; 68:1157–1162.
Sagel SD, Wagner BD, Anthony MM, Emmett P, Zemanick ET. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am J Respir Crit Care Med 2012; 186:857–865.
Saiman L, Anstead M, Mayer-Hamblett N, et al; AZ0004 Azithromycin Study Group. Effect of azithromycin on pulmonary function in patients with cystic fibrosis uninfected with Pseudomonas aeruginosa: a randomized controlled trial. JAMA 2010; 303:1707–1715.
Southern KW, Barker PM, Solis-Moya A, Patel L. Macrolide antibiotics for cystic fibrosis. Cochrane Database Syst Rev 2012;11:CD002203.
Flume PA, O’Sullivan BP, Robinson KA, et al; Cystic Fibrosis Foundation, Pulmonary Therapies Committee. Cystic fibrosis pulmonary guidelines: chronic medications for maintenance of lung health. Am J Respir Crit Care Med 2007; 176:957–969.
Yanagihara K, Kadoto J, Kohno S. Diffuse panbronchiolitis—pathophysiology and treatment mechanisms. Int J Antimicrob Agents 2001; 18(suppl 1):S83–S87.
Kudoh S, Azuma A, Yamamoto M, Izumi T, Ando M. Improvement of survival in patients with diffuse panbronchiolitis treated with low-dose erythromycin. Am J Respir Crit Care Med 1998; 157:1829–1832.
Schultz MJ. Macrolide activities beyond their antimicrobial effects: macrolides in diffuse panbronchiolitis and cystic fibrosis. J Antimicrob Chemother 2004; 54:21–28.
Hui D, Yan F, Chen RH. The effects of azithromycin on patients with diffuse panbronchiolitis: a retrospective study of 29 cases. J Thorac Dis 2013; 5:613–617.
Khalid M, Al Saghir A, Saleemi S, et al. Azithromycin in bronchiolitis obliterans complicating bone marrow transplantation: a preliminary study. Eur Respir J 2005; 25:490–493.
Kingah PL, Muma G, Soubani A. Azithromycin improves lung function in patients with post-lung transplant bronchiolitis obliterans syndrome: a meta-analysis. Clin Transplant 2014; 28:906–910.
Corris PA, Ryan VA, Small T, et al. A randomised controlled trial of azithromycin therapy in bronchiolitis obliterans syndrome (BOS) post lung transplantation. Thorax 2015; 70:442–450.
Williams KM, Cheng GS, Pusic I, et al. Fluticasone, azithromycin, and montelukast treatment for new-onset bronchiolitis obliterans syndrome after hematopoietic cell transplantation. Biol Blood Marrow Transplant 2016; 22:710–716.
Haworth CS, Bilton D, Elborn JS. Long-term macrolide maintenance therapy in non-CF bronchiectasis: evidence and questions. Respir Med 2014; 108:1397–1408.
Altenburg J, de Graaff CS, Stienstra Y, et al. Effect of azithromycin maintenance treatment on infectious exacerbations among patients with non-cystic fibrosis bronchiectasis: the BAT randomized controlled trial. JAMA 2013; 309:1251–1259.
Wong C, Jayaram L, Karalus N, et al. Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): a randomised, double-blind, placebo-controlled trial. Lancet 2012; 380:660–667.
Suissa S, Dell’Aniello S, Ernst P. Long-term natural history of chronic obstructive pulmonary disease: severe exacerbations and mortality. Thorax 2012; 67:957–963.
Ni W, Shao X, Cai X, et al. Prophylactic use of macrolide antibiotics for the prevention of chronic obstructive pulmonary disease exacerbation: a meta-analysis. PLoS One 2015; 10:e0121257.
Griffith DE, Aksamit T, Brown-Elliott BA, et al; ATS Mycobacterial Diseases Subcommittee; American Thoracic Society; Infectious Disease Society of America. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 2007; 175:367–416.
Havlir DV, Dubé MP, Sattler FR, et al. Prophylaxis against disseminated Mycobacterium avium complex with weekly azithromycin, daily rifabutin, or both. California Collaborative Treatment Group. N Engl J Med 1996; 335:392–398.
Uthman MM, Uthman OA, Yahaya I. Interventions for the prevention of Mycobacterium avium complex in adults and children with HIV. Cochrane Database Syst Rev 2013; 4:CD007191.
Li H, Liu DH, Chen LL, et al. Meta-analysis of the adverse effects of long-term azithromycin use in patients with chronic lung diseases. Antimicrob Agents Chemother 2014; 58:511–517.
Svanström H, Pasternak B, Hviid A. Use of azithromycin and death from cardiovascular causes. N Engl J Med 2013; 368:1704–1712.
Albert RK, Schuller JL; COPD Clinical Research Network. Macrolide antibiotics and the risk of cardiac arrhythmias. Am J Respir Crit Care Med 2014; 189:1173–1180.
Ray WA, Murray KT, Hall K, Arbogast PG, Stein CM. Azithromycin and the risk of cardiovascular death. N Engl J Med 2012; 366:1881–1890.
Albert RK, Connett J, Bailey WC, et al; COPD Clinical Research Network. Azithromycin for prevention of exacerbations of COPD. N Engl J Med 2011; 365:689–698.
Broad J, Sanger GJ. The antibiotic azithromycin is a motilin receptor agonist in human stomach: comparison with erythromycin. Br J Pharmacol 2013; 168:1859–1867.
African American, Hispanic, American Indian, and Alaskan Native women continue to be disproportionately affected by cervical cancer compared with white women. From 2006 to 2010, the incidence of cervical cancer in African American women was 10.3 per 100,000; in white women it was 7.2.1 The mortality rate from cervical cancer in African American women is twice that in white women.1 Although cervical cancer rates have decreased nationwide, significant racial health disparities persist.
As the first-line healthcare providers for many women, the primary care physician and the general obstetrician-gynecologist are optimally positioned to reduce these disparities.
Cervical cancer is the third most common gynecologic cancer, after uterine and ovarian cancer. Nearly 13,000 new cases are diagnosed each year in the United States, and more than 4,000 women die of it.2 Fortunately, cervical cancer can be significantly prevented with adequate screening and vaccination against human papillomavirus (HPV).
WHY ARE BLACK WOMEN MORE LIKELY TO DIE OF CERVICAL CANCER?
Later stage at diagnosis. African American women are more likely to present with advanced cervical cancer than non-Hispanic white women.3–6
Less-aggressive treatment. African American women are more likely to receive no treatment after a cancer diagnosis.6 Differences in treatment may be attributed to comorbid conditions, stage at cancer diagnosis, and patient refusal.5,7
Less access to care. A study from the Surveillance, Epidemiology, and End Results program of the National Cancer Institute looked at 7,267 women (4,431 non-Hispanic white women, 1,830 Hispanic white women, and 1,006 non-Hispanic African American women) who were diagnosed with primary invasive cervical cancer from 1992 to 1996 and followed through 2000. African American women had a 19% higher mortality rate compared with non-Hispanic white women during follow-up despite adjusting for age, stage, histology, and time of first treatment.8
However, a later study from the same program found no such difference after 1995, when the data were adjusted for marital status, disease stage, age, treatment, grade, and histology.6
Equal access to healthcare may eliminate most of the disparity.7 A study in women with cervical cancer who sought treatment within the United States military healthcare system found no difference in treatment or 5- and 10-year survival rates between African American and white women.5 Equal access to comprehensive healthcare eliminated any disparity once cervical cancer was diagnosed.
CERVICAL CANCER SCREENING
The value of cervical cancer screening and prevention is well established. In 1941, Papanicolau reported that cervical cancer could be detected from vaginal smears.9 Since the development and widespread implementation of the “Pap” smear, cervical cancer rates have decreased dramatically in the United States.
Another major advance was the discovery that persistent infection with HPV is necessary for the development of cervical cancer, precancerous lesions, and genital warts.10
With advancing research, guidelines for cervical cancer screening have changed considerably over the years. Today, combined cervical cytologic and HPV testing is the mainstay. (Isolated HPV testing is generally not available outside clinical trials.)
Who should be screened?
Previous recommendations called for women to undergo Pap testing when they first became sexually active and then every year. However, cervical lesions are likely to regress in young women.11 One study found that 28% of cervical intimal neoplasia (CIN) grade 2 and 3 lesions spontaneously regressed by 15 weeks, although lesions associated with HPV 16 infection were less likely to regress than with other HPV types.12 A study of college women found that HPV infection persisted in only 9% of women after 24 months.13
To minimize unnecessary treatment of young women with dysplasia, the American Society for Colposcopy and Cervical Pathology in 2012 recommended cytologic screening for all women 21 years or older, regardless of age at first sexual encounter.14 Screening intervals were changed from every year to every 3 years until age 30, at which time cotesting with cytology and HPV testing is performed every 5 years. Routine cotesting is not recommended for women younger than 30, who have a high likelihood of HPV infection and spontaneous regression.
In 2014, the US Food and Drug Administration approved primary HPV screening (ie, testing for HPV first, and then performing cytology in samples that test positive) for women age 25 and older.15
Patients who need further evaluation and testing should be referred for colposcopy. The current guidelines for patients who have abnormal results on cervical cancer screening16 can be reviewed at www.asccp.org/asccp-guidelines.
As screening guidelines continue to evolve, primary care physicians will need to stay current and also help educate their patients. For example, many of our patients have undergone annual Pap screening for most of their lives and may not yet know about the new testing intervals.
Are there disparities in screening and follow-up?
Disparities in screening and follow-up may exist, but the evidence is not clear-cut.
In a 2013 National Health Interview Survey report, the rates of cervical cancer screening with Pap tests did not differ between African American and white women.17 However, the information on Pap testing was based on a single question asking participants if they had had a Pap test in the last 3 years. In our experience, patients may confuse Pap tests with speculum examinations.
Once women are screened, adequate and timely follow-up of abnormal results is key.
In a study from the National Breast and Cervical Cancer Early Detection Program,18 women who had cytology findings of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesions were to undergo repeat Pap testing every 4 to 6 months for 2 years. African American women were the least likely to have a follow-up Pap smear compared with other racial groups.
On the other hand, there was no difference related to race in follow-up rates of abnormal Pap tests in women ages 47 to 64 in the South Carolina Breast and Cervical Cancer Early Detection Program.19
In a study in an urban population (predominantly African African), the overall follow-up rate was only 26% at 12 months from an initial abnormal Pap smear. This study did not find any differences in follow-up according to race or ethnicity; however, it had insufficient power to detect a difference because only 15% of the study participants were white.20
What is in a genotype?
HPV is implicated in progression to both squamous cell carcinoma and adenocarcinoma of the cervix. Worldwide, HPV genotypes 16 and 18 are associated with 73% of cases of invasive cervical cancer; most of the remainder are associated with, in order of decreasing prevalence, genotypes 58, 33, 45, 31, 52, 35, 59, 39, 51, and 56.21
High-grade cervical lesions in African American women may less often be positive for HPV 16 and 18 than in white women.22,23 On the other hand, the proportion of non-Hispanic black women infected with HPV 35 and 58 was significantly higher than in non-Hispanic white women.22 Regardless, HPV screening is recommended for women of all races and ethnicities.
The 2-valent and 4-valent HPV vaccines do not cover HPV 35 or 58. The newer 9-valent vaccine covers HPV 58 (but not 35) and so may in theory decrease any potential disparity related to infection with a specific oncogenic subtype.
THE ROLE OF PREVENTION
HPV vaccination
Currently, 3 vaccines against HPV are available in the United States, a 2-valent, a 4-valent, and since 2015, a 9-valent preparation (Table 1).
The Females United to Unilaterally Reduce Endo/Ectocervical Disease study demonstrated that the 4-valent vaccine was highly effective against cervical intraepithelial neoplasia due to HPV 16 and 18.24 In another study, the 2-valent vaccine reduced the incidence of CIN 3 or higher by 87% in women who received all 3 doses and who had no evidence of HPV infection at baseline.25
HPV vaccination is expensive. Each shot costs about $130, plus the cost of administering it. Although the Vaccines for Children program covers the HPV vaccine for uninsured and underinsured children and adolescents under age 19, Medicaid coverage varies from state to state for adults over age 21.
The Advisory Committee on Immunization Practices (ACIP)26 recommends routine vaccination for:
Males 11 or 12 years old
Females ages 9 to 26.
In October 2016, the ACIP approved a 2-dose series given 6 to 12 months apart for patients starting vaccination at ages 9 through 14 years who are not immunocompromised. Others should receive a 3-dose series, with the second dose given 1 to 2 months after the first dose and the third dose given 6 months after the first dose.27 Previously, 3 doses were recommended for everyone.
Disparities in HPV vaccination rates
HPV vaccination rates among adolescents in the United States increased from 33.6% in 2013 to 41.7% in 2014.28 However, HPV vaccination rates continue to lag behind those of other routine vaccines, such as Tdap and meningococcal conjugate.
Reagan-Steiner et al28 reported that more black than white girls age 13 through 17 received at least 1 dose of a 3-dose HPV vaccination series, but more white girls received all 3 doses (70.6% vs 61.6%). In contrast, a meta-analysis by Fisher et al29 found African American and uninsured women generally less likely to initiate the HPV vaccination series. Kessels et al30 reported similar findings.
Barriers to HPV vaccination
Barriers to HPV vaccination can be provider-dependent, parental, or institutional.
Malo et al31 surveyed Florida Medicaid providers and found that those who participated in the Vaccines for Children program were less likely to cite lack of reimbursement as a barrier to vaccination.
Meites et al32 surveyed sexually transmitted disease clinics and found that common reasons for not offering HPV vaccine were cost, staff time, and difficulty coordinating follow-up visits to complete the series.
Providers report lack of urgency or lack of perception of cervical cancer as a true public health threat, safety concerns regarding the vaccine, and the inability to coadminister vaccines as barriers.33
Studies have shown that relatively few parents (up to 18%) of parents are concerned about the effect of the vaccine on sexual activity.34 Rather, they are most likely to cite lack of information regarding the vaccine, lack of physician recommendation, and not knowing where to receive the vaccine as barriers.35,36
Guerry et al37 determined that the single most important factor in vaccine initiation was physician recommendation, a finding reiterated in other studies.35,38 A study in North Carolina identified failure of physician recommendation as one of the missed opportunities for vaccination of young women.39
Therefore, the primary care physician, as the initial contact with the child or young adult, holds a responsibility to narrow this gap. In simply discussing and recommending the vaccine, physicians could increase vaccination rates.
REPRODUCTIVE HEALTH
Although 80% of women will be infected with HPV in their lifetime, only a small proportion will develop cervical cancer, suggesting there are other cofactors in the progression to cervical cancer.40
Given the infectious etiology of cervical cancer, other contributing reproductive health factors have been described. As expected, the number of sexual partners correlates with HPV infection.41,42 Younger age at first intercourse has been linked to development of cervical neoplasia, consistent with persistent infection leading to neoplasia.41,42
Primary care physicians should provide timely and comprehensive sexual education, including information on safe sexual practices and pregnancy prevention.
Human immunodeficiency virus
In 2010, the estimated rate of new human immunodeficiency virus (HIV) infections in African American women was nearly 20 times greater than in white women.43 Previous studies have shown a clear relationship between HIV and HPV-associated cancers, including cervical neoplasia and invasive cervical cancer.44,45
Women with HIV should receive screening for cervical cancer at the time of diagnosis, 6 months after the initial diagnosis, and annually thereafter.46
Conflicting evidence exists regarding the effect of highly active antiretroviral therapy on the incidence of HPV-related disease, so aggressive screening and management of cervical neoplasia is recommended for women with HIV, regardless of CD4+ levels or viral load.47–49
Additional infectious culprits
Coinfection with other sexually transmitted infections, specifically Chlamydia, herpes, and HIV, has been associated with cervical neoplasia and invasive cervical cancer. A positive linear association exists between the number of sexually transmitted infections and cervical neoplasia.50
C trachomatis is the most common sexually transmitted infection in the United States, with a 6-times higher rate in African American women.51 Women who are seropositive for C trachomatis are at twofold higher risk of developing squamous cell cervical cancer.52,53 Women who are seropositive for Chlamydia infection, herpes virus 2, or HPV are at markedly increased risk of invasive cervical cancer.50
Tobacco use
The negative impact of smoking on numerous other cancers resulted in investigation of its role in cervical cancer.
Early case-control studies found an association between cervical cancer and smoking,54 but because these studies did not account for HPV infection status, they could not establish causality. Subsequently, several studies did control for HPV infection; the risk of squamous cervical cancer was twice as high in women who had ever smoked.55 Furthermore, the more cigarettes smoked per day, the higher the risk of cervical neoplasia.41,56
According to the US Centers for Disease Control and Prevention in 2014, the highest prevalence of smoking was among American Indian and Alaskan Native women, 32.5% of whom said they smoked every day, compared with 17.2% of white women and 13.7% of African American women.57
HOW CAN PRIMARY CARE PHYSICIANS CLOSE THE GAP?
Primary care physicians are the first point of contact for patients of all ages and so can help minimize such disparities. They can tackle 2 important cervical cancer prevention interventions first-hand: vaccination and screening (Table 2), including follow-up of abnormal screening results.
By promoting HPV vaccination to children and young adults, primary care physicians can help prevent cervical cancer. Moreover, primary care physicians will see most adolescents for a nonpreventive health visit, an optimal opportunity to discuss sexual activity practices and HPV vaccination.58 Including the HPV vaccine as routine with other vaccinations can close the gap.38
Screening and treatment of sexually transmitted infection during these visits can affect the risk that future HPV infection will progress to neoplasia or cancer. Persistent lifestyle modification counseling, especially smoking cessation through motivational interviewing, can lessen the risk of cervical cancer neoplasia progression.
Additionally, in light of recent changes in cervical cancer screening guidelines, the primary care physician’s role as educator is of utmost importance. In one study, although 99% of women had received a Pap test, 87% could not identify the purpose of the Pap test.59 The primary care physician’s role is perhaps the most influential in preventing disease and, as such, has the greatest impact on a patient’s disease process.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9–29.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5–29.
Koh WJ, Greer BE, Abu-Rustum NR, et al. Cervical cancer, version 2.2015. J Natl Compr Canc Netw 2015; 13:395-404.
Farley J, Risinger JI, Rose GS, Maxwell GL. Racial disparities in blacks with gynecologic cancers. Cancer 2007; 110:234–243.
Farley JH, Hines JF, Taylor RR, et al. Equal care ensures equal survival for African-American women with cervical carcinoma. Cancer 2001; 91:869–873.
Rauh-Hain JA, Clemmer JT, Bradford LS, et al. Racial disparities in cervical cancer survival over time. Cancer 2013; 119:3644–3652.
Collins Y, Holcomb K, Chapman-Davis E, Khabele D, Farley JH. Gynecologic cancer disparities: a report from the Health Disparities Taskforce of the Society of Gynecologic Oncology. Gynecol Oncol 2014; 133:353–361.
Patel DA, Barnholtz-Sloan JS, Patel MK, Malone JM Jr, Chuba PJ, Schwartz K. A population-based study of racial and ethnic differences in survival among women with invasive cervical cancer: analysis of surveillance, epidemiology, and end results data. Gynecol Oncol 2005; 97:550–558.
Papanicolaou GN, Traut HF. The diagnostic value of vaginal smears in carcinoma of the uterus. 1941. Arch Pathol Lab Med 1997; 121:211–224.
Walboomers JM, Jacobs M V, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
Moscicki AB, Shiboski S, Hills NK, et al. Regression of low-grade squamous intra-epithelial lesions in young women. Lancet 2004; 364:1678–1683.
Trimble CL, Piantadosi S, Gravitt P, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res 2005; 11:4717–4723.
Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med 1998; 338:423-428.
Saslow D, Solomon D, Lawson HW, et al; ACS-ASCCP-ASCP Cervical Cancer Guideline Committee. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA Cancer J Clin 2012; 62:147–172.
Huh WK, Ault KA, Chelmow D, et al. Use of primary high-risk human papillomavirus testing for cervical cancer screening: interim clinical guidance. Obstet Gynecol 2015; 125:330–337.
Massad LS, Einstein MH, Huh WK, et al; 2012 ASCCP Consensus Guidelines Conference. 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. J Low Genit Tract Dis 2013; 17(suppl 1):S1–S27.
Sabatino SA, White MC, Thompson TD, Klabunde CN. Cancer screening test use—United States, 2013. MMWR 2015; 64:464–468.
Benard VB, Lawson HW, Eheman CR, Anderson C, Helsel W. Adherence to guidelines for follow-up of low-grade cytologic abnormalities among medically underserved women. Obstet Gynecol 2005; 105:1323–1328.
Eggleston KS, Coker AL, Luchok KJ, Meyer TE. Adherence to recommendations for follow-up to abnormal Pap tests. Obstet Gynecol 2007; 109:1332–1341.
Peterson NB, Han J, Freund KM. Inadequate follow-up for abnormal Pap smears in an urban population. J Natl Med Assoc 2003; 95:825–832.
Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: variation by geographical region, histological type and year of publication. Int J Cancer 2011; 128:927–935.
Hariri S, Unger ER, Powell SE, et al; HPV-IMPACT Working Group. Human papillomavirus genotypes in high-grade cervical lesions in the United States. J Infect Dis 2012; 206:1878–1886.
Niccolai LM, Russ C, Julian PJ, et al. Individual and geographic disparities in human papillomavirus types 16/18 in high-grade cervical lesions: associations with race, ethnicity, and poverty. Cancer 2013; 119:3052–3058.
FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
Paavonen J, Naud P, Salmerón J, et al; HPV PATRICIA Study Group. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 2009; 374:301–314.
Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1705–1708.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
Reagan-Steiner S, Yankey D, Jeyarajah J, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13–17 years—United States, 2014. MMWR Morb Mortal Wkly Rep 2015; 64:784–792.
Fisher H, Trotter CL, Audrey S, MacDonald-Wallis K, Hickman M. Inequalities in the uptake of human papillomavirus vaccination: a systematic review and meta-analysis. Int J Epidemiol 2013; 42:896–908.
Kessels SJ, Marshall HS, Watson M, Braunack-Mayer AJ, Reuzel R, Tooher RL. Factors associated with HPV vaccine uptake in teenage girls: a systematic review. Vaccine 2012; 30:3546–3556.
Malo TL, Hassani D, Staras SA, Shenkman EA, Giuliano AR, Vadaparampil ST. Do Florida Medicaid providers’ barriers to HPV vaccination vary based on VFC program participation? Matern Child Health J 2013; 17:609–615.
Meites E, Llata E, Hariri S, et al. HPV vaccine implementation in STD clinics—STD Surveillance Network. Sex Transm Dis 2012; 39:32–34.
Perkins RB, Clark JA. What affects human papillomavirus vaccination rates? A qualitative analysis of providers’ perceptions. Womens Health Issues 2012; 22:e379–e386.
Holman DM, Benard V, Roland KB, Watson M, Liddon N, Stokley S. Barriers to human papillomavirus vaccination among US adolescents: a systematic review of the literature. JAMA Pediatr 2014; 168:76–82.
Dorell CG, Yankey D, Santibanez TA, Markowitz LE. Human papillomavirus vaccination series initiation and completion, 2008–2009. Pediatrics 2011; 128:830–839.
Bastani R, Glenn BA, Tsui J, et al. Understanding suboptimal human papillomavirus vaccine uptake among ethnic minority girls. Cancer Epidemiol Biomarkers Prev 2011; 20:1463–1472.
Guerry SL, De Rosa CJ, Markowitz LE, et al. Human papillomavirus vaccine initiation among adolescent girls in high-risk communities. Vaccine 2011; 29:2235–2241.
Hull PC, Williams EA, Khabele D, Dean C, Bond B, Sanderson M. HPV vaccine use among African American girls: qualitative formative research using a participatory social marketing approach. Gynecol Oncol 2014; 132(suppl 1):S13–S20.
Brewer NT, Gottlieb SL, Reiter PL, et al. Longitudinal predictors of human papillomavirus vaccine initiation among adolescent girls in a high-risk geographic area. Sex Transm Dis 2011; 38:197–204.
Wang SS, Zuna RE, Wentzensen N, et al. Human papillomavirus cofactors by disease progression and human papillomavirus types in the study to understand cervical cancer early endpoints and determinants. Cancer Epidemiol Biomarkers Prev 2009; 18:113–120.
Deacon JM, Evans CD, Yule R, et al. Sexual behaviour and smoking as determinants of cervical HPV infection and of CIN3 among those infected: a case-control study nested within the Manchester cohort. Br J Cancer 2000; 83:1565–1572.
International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical carcinoma and sexual behavior: collaborative reanalysis of individual data on 15,461 women with cervical carcinoma and 29,164 women without cervical carcinoma from 21 epidemiological studies. Cancer Epidemiol Biomarkers Prev 2009; 18:1060–1069.
Centers for Disease Control and Prevention (CDC). Estimated HIV incidence in the United States, 2007–2010. HIV Surveillance Supplemental Report 2012; 17(No. 4). https://www.cdc.gov/hiv/pdf/statistics_hssr_vol_17_no_4.pdf. Accessed September 12, 2017.
Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus-associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J Natl Cancer Inst 2000; 92:1500–1510.
Schäfer A, Friedmann W, Mielke M, Schwartländer B, Koch MA. The increased frequency of cervical dysplasia-neoplasia in women infected with the human immunodeficiency virus is related to the degree of immunosuppression. Am J Obstet Gynecol 1991; 164:593–599.
De Vuyst H, Lillo F, Broutet N, Smith JS. HIV, human papillomavirus, and cervical neoplasia and cancer in the era of highly active antiretroviral therapy. Eur J Cancer Prev 2008; 17:545–554.
Palefsky JM. Cervical human papillomavirus infection and cervical intraepithelial neoplasia in women positive for human immunodeficiency virus in the era of highly active antiretroviral therapy. Curr Opin Oncol 2003; 15:382–388.
Adler DH. The impact of HAART on HPV-related cervical disease. Curr HIV Res 2010; 8:493–497.
Castellsagué X, Pawlita M, Roura E, et al. Prospective seroepidemiologic study on the role of human papillomavirus and other infections in cervical carcinogenesis: evidence from the EPIC cohort. Int J Cancer 2014; 135:440–452.
Centers for Disease Control and Prevention (CDC). 2013 sexually transmitted disease surveillance. www.cdc.gov/std/stats13/exordium.htm. Accessed September 12, 2017.
Smith JS, Bosetti C, Muñoz N, et al; IARC multicentric case-control study. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case-control study. Int J Cancer 2004; 111:431–439.
Koskela P, Anttila T, Bjørge T, et al. Chlamydia trachomatis infection as a risk factor for invasive cervical cancer. Int J Cancer 2000; 85:35–39.
Office on Smoking and Health (US). Women and smoking: a report of the Surgeon General: Chapter 3. Health consequences of tobacco use among women. http://www.ncbi.nlm.nih.gov/books/NBK44312. Accessed September 12, 2017.
Plummer M, Herrero R, Franceschi S, et al; IARC Multi-centre Cervical Cancer Study Group. Smoking and cervical cancer: pooled analysis of the IARC multi-centric case—control study. Cancer Causes Control 2003; 14:805–814.
Ho GY, Kadish AS, Burk RD, et al. HPV 16 and cigarette smoking as risk factors for high-grade cervical intra-epithelial neoplasia. Int J Cancer 1998; 78:281–285.
Jamal A, Homa DM, O’Connor E, et al. Current cigarette smoking among adults - United States, 2005-2014. MMWR Morb Mortal Wkly Rep 2015; 64:1233–1240.
Nordin JD, Solberg LI, Parker ED. Adolescent primary care visit patterns. Ann Fam Med 2010; 8:511–516.
Lindau ST, Tomori C, Lyons T, Langseth L, Bennett CL, Garcia P. The association of health literacy with cervical cancer prevention knowledge and health behaviors in a multiethnic cohort of women. Am J Obstet Gynecol 2002; 186:938–943.
Cynthia Arvizo, MD Department of Obstetrics and Gynecology, Vanderbilt University Medical Center, Nashville, TN
Haider Mahdi, MD Department of Obstetrics and Gynecology, Women’s Health Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Haider Mahdi, MD, Women’s Health Institute, A81, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; mahdih@ccf.org
Cynthia Arvizo, MD Department of Obstetrics and Gynecology, Vanderbilt University Medical Center, Nashville, TN
Haider Mahdi, MD Department of Obstetrics and Gynecology, Women’s Health Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Haider Mahdi, MD, Women’s Health Institute, A81, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; mahdih@ccf.org
Author and Disclosure Information
Cynthia Arvizo, MD Department of Obstetrics and Gynecology, Vanderbilt University Medical Center, Nashville, TN
Haider Mahdi, MD Department of Obstetrics and Gynecology, Women’s Health Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Haider Mahdi, MD, Women’s Health Institute, A81, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; mahdih@ccf.org
African American, Hispanic, American Indian, and Alaskan Native women continue to be disproportionately affected by cervical cancer compared with white women. From 2006 to 2010, the incidence of cervical cancer in African American women was 10.3 per 100,000; in white women it was 7.2.1 The mortality rate from cervical cancer in African American women is twice that in white women.1 Although cervical cancer rates have decreased nationwide, significant racial health disparities persist.
As the first-line healthcare providers for many women, the primary care physician and the general obstetrician-gynecologist are optimally positioned to reduce these disparities.
Cervical cancer is the third most common gynecologic cancer, after uterine and ovarian cancer. Nearly 13,000 new cases are diagnosed each year in the United States, and more than 4,000 women die of it.2 Fortunately, cervical cancer can be significantly prevented with adequate screening and vaccination against human papillomavirus (HPV).
WHY ARE BLACK WOMEN MORE LIKELY TO DIE OF CERVICAL CANCER?
Later stage at diagnosis. African American women are more likely to present with advanced cervical cancer than non-Hispanic white women.3–6
Less-aggressive treatment. African American women are more likely to receive no treatment after a cancer diagnosis.6 Differences in treatment may be attributed to comorbid conditions, stage at cancer diagnosis, and patient refusal.5,7
Less access to care. A study from the Surveillance, Epidemiology, and End Results program of the National Cancer Institute looked at 7,267 women (4,431 non-Hispanic white women, 1,830 Hispanic white women, and 1,006 non-Hispanic African American women) who were diagnosed with primary invasive cervical cancer from 1992 to 1996 and followed through 2000. African American women had a 19% higher mortality rate compared with non-Hispanic white women during follow-up despite adjusting for age, stage, histology, and time of first treatment.8
However, a later study from the same program found no such difference after 1995, when the data were adjusted for marital status, disease stage, age, treatment, grade, and histology.6
Equal access to healthcare may eliminate most of the disparity.7 A study in women with cervical cancer who sought treatment within the United States military healthcare system found no difference in treatment or 5- and 10-year survival rates between African American and white women.5 Equal access to comprehensive healthcare eliminated any disparity once cervical cancer was diagnosed.
CERVICAL CANCER SCREENING
The value of cervical cancer screening and prevention is well established. In 1941, Papanicolau reported that cervical cancer could be detected from vaginal smears.9 Since the development and widespread implementation of the “Pap” smear, cervical cancer rates have decreased dramatically in the United States.
Another major advance was the discovery that persistent infection with HPV is necessary for the development of cervical cancer, precancerous lesions, and genital warts.10
With advancing research, guidelines for cervical cancer screening have changed considerably over the years. Today, combined cervical cytologic and HPV testing is the mainstay. (Isolated HPV testing is generally not available outside clinical trials.)
Who should be screened?
Previous recommendations called for women to undergo Pap testing when they first became sexually active and then every year. However, cervical lesions are likely to regress in young women.11 One study found that 28% of cervical intimal neoplasia (CIN) grade 2 and 3 lesions spontaneously regressed by 15 weeks, although lesions associated with HPV 16 infection were less likely to regress than with other HPV types.12 A study of college women found that HPV infection persisted in only 9% of women after 24 months.13
To minimize unnecessary treatment of young women with dysplasia, the American Society for Colposcopy and Cervical Pathology in 2012 recommended cytologic screening for all women 21 years or older, regardless of age at first sexual encounter.14 Screening intervals were changed from every year to every 3 years until age 30, at which time cotesting with cytology and HPV testing is performed every 5 years. Routine cotesting is not recommended for women younger than 30, who have a high likelihood of HPV infection and spontaneous regression.
In 2014, the US Food and Drug Administration approved primary HPV screening (ie, testing for HPV first, and then performing cytology in samples that test positive) for women age 25 and older.15
Patients who need further evaluation and testing should be referred for colposcopy. The current guidelines for patients who have abnormal results on cervical cancer screening16 can be reviewed at www.asccp.org/asccp-guidelines.
As screening guidelines continue to evolve, primary care physicians will need to stay current and also help educate their patients. For example, many of our patients have undergone annual Pap screening for most of their lives and may not yet know about the new testing intervals.
Are there disparities in screening and follow-up?
Disparities in screening and follow-up may exist, but the evidence is not clear-cut.
In a 2013 National Health Interview Survey report, the rates of cervical cancer screening with Pap tests did not differ between African American and white women.17 However, the information on Pap testing was based on a single question asking participants if they had had a Pap test in the last 3 years. In our experience, patients may confuse Pap tests with speculum examinations.
Once women are screened, adequate and timely follow-up of abnormal results is key.
In a study from the National Breast and Cervical Cancer Early Detection Program,18 women who had cytology findings of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesions were to undergo repeat Pap testing every 4 to 6 months for 2 years. African American women were the least likely to have a follow-up Pap smear compared with other racial groups.
On the other hand, there was no difference related to race in follow-up rates of abnormal Pap tests in women ages 47 to 64 in the South Carolina Breast and Cervical Cancer Early Detection Program.19
In a study in an urban population (predominantly African African), the overall follow-up rate was only 26% at 12 months from an initial abnormal Pap smear. This study did not find any differences in follow-up according to race or ethnicity; however, it had insufficient power to detect a difference because only 15% of the study participants were white.20
What is in a genotype?
HPV is implicated in progression to both squamous cell carcinoma and adenocarcinoma of the cervix. Worldwide, HPV genotypes 16 and 18 are associated with 73% of cases of invasive cervical cancer; most of the remainder are associated with, in order of decreasing prevalence, genotypes 58, 33, 45, 31, 52, 35, 59, 39, 51, and 56.21
High-grade cervical lesions in African American women may less often be positive for HPV 16 and 18 than in white women.22,23 On the other hand, the proportion of non-Hispanic black women infected with HPV 35 and 58 was significantly higher than in non-Hispanic white women.22 Regardless, HPV screening is recommended for women of all races and ethnicities.
The 2-valent and 4-valent HPV vaccines do not cover HPV 35 or 58. The newer 9-valent vaccine covers HPV 58 (but not 35) and so may in theory decrease any potential disparity related to infection with a specific oncogenic subtype.
THE ROLE OF PREVENTION
HPV vaccination
Currently, 3 vaccines against HPV are available in the United States, a 2-valent, a 4-valent, and since 2015, a 9-valent preparation (Table 1).
The Females United to Unilaterally Reduce Endo/Ectocervical Disease study demonstrated that the 4-valent vaccine was highly effective against cervical intraepithelial neoplasia due to HPV 16 and 18.24 In another study, the 2-valent vaccine reduced the incidence of CIN 3 or higher by 87% in women who received all 3 doses and who had no evidence of HPV infection at baseline.25
HPV vaccination is expensive. Each shot costs about $130, plus the cost of administering it. Although the Vaccines for Children program covers the HPV vaccine for uninsured and underinsured children and adolescents under age 19, Medicaid coverage varies from state to state for adults over age 21.
The Advisory Committee on Immunization Practices (ACIP)26 recommends routine vaccination for:
Males 11 or 12 years old
Females ages 9 to 26.
In October 2016, the ACIP approved a 2-dose series given 6 to 12 months apart for patients starting vaccination at ages 9 through 14 years who are not immunocompromised. Others should receive a 3-dose series, with the second dose given 1 to 2 months after the first dose and the third dose given 6 months after the first dose.27 Previously, 3 doses were recommended for everyone.
Disparities in HPV vaccination rates
HPV vaccination rates among adolescents in the United States increased from 33.6% in 2013 to 41.7% in 2014.28 However, HPV vaccination rates continue to lag behind those of other routine vaccines, such as Tdap and meningococcal conjugate.
Reagan-Steiner et al28 reported that more black than white girls age 13 through 17 received at least 1 dose of a 3-dose HPV vaccination series, but more white girls received all 3 doses (70.6% vs 61.6%). In contrast, a meta-analysis by Fisher et al29 found African American and uninsured women generally less likely to initiate the HPV vaccination series. Kessels et al30 reported similar findings.
Barriers to HPV vaccination
Barriers to HPV vaccination can be provider-dependent, parental, or institutional.
Malo et al31 surveyed Florida Medicaid providers and found that those who participated in the Vaccines for Children program were less likely to cite lack of reimbursement as a barrier to vaccination.
Meites et al32 surveyed sexually transmitted disease clinics and found that common reasons for not offering HPV vaccine were cost, staff time, and difficulty coordinating follow-up visits to complete the series.
Providers report lack of urgency or lack of perception of cervical cancer as a true public health threat, safety concerns regarding the vaccine, and the inability to coadminister vaccines as barriers.33
Studies have shown that relatively few parents (up to 18%) of parents are concerned about the effect of the vaccine on sexual activity.34 Rather, they are most likely to cite lack of information regarding the vaccine, lack of physician recommendation, and not knowing where to receive the vaccine as barriers.35,36
Guerry et al37 determined that the single most important factor in vaccine initiation was physician recommendation, a finding reiterated in other studies.35,38 A study in North Carolina identified failure of physician recommendation as one of the missed opportunities for vaccination of young women.39
Therefore, the primary care physician, as the initial contact with the child or young adult, holds a responsibility to narrow this gap. In simply discussing and recommending the vaccine, physicians could increase vaccination rates.
REPRODUCTIVE HEALTH
Although 80% of women will be infected with HPV in their lifetime, only a small proportion will develop cervical cancer, suggesting there are other cofactors in the progression to cervical cancer.40
Given the infectious etiology of cervical cancer, other contributing reproductive health factors have been described. As expected, the number of sexual partners correlates with HPV infection.41,42 Younger age at first intercourse has been linked to development of cervical neoplasia, consistent with persistent infection leading to neoplasia.41,42
Primary care physicians should provide timely and comprehensive sexual education, including information on safe sexual practices and pregnancy prevention.
Human immunodeficiency virus
In 2010, the estimated rate of new human immunodeficiency virus (HIV) infections in African American women was nearly 20 times greater than in white women.43 Previous studies have shown a clear relationship between HIV and HPV-associated cancers, including cervical neoplasia and invasive cervical cancer.44,45
Women with HIV should receive screening for cervical cancer at the time of diagnosis, 6 months after the initial diagnosis, and annually thereafter.46
Conflicting evidence exists regarding the effect of highly active antiretroviral therapy on the incidence of HPV-related disease, so aggressive screening and management of cervical neoplasia is recommended for women with HIV, regardless of CD4+ levels or viral load.47–49
Additional infectious culprits
Coinfection with other sexually transmitted infections, specifically Chlamydia, herpes, and HIV, has been associated with cervical neoplasia and invasive cervical cancer. A positive linear association exists between the number of sexually transmitted infections and cervical neoplasia.50
C trachomatis is the most common sexually transmitted infection in the United States, with a 6-times higher rate in African American women.51 Women who are seropositive for C trachomatis are at twofold higher risk of developing squamous cell cervical cancer.52,53 Women who are seropositive for Chlamydia infection, herpes virus 2, or HPV are at markedly increased risk of invasive cervical cancer.50
Tobacco use
The negative impact of smoking on numerous other cancers resulted in investigation of its role in cervical cancer.
Early case-control studies found an association between cervical cancer and smoking,54 but because these studies did not account for HPV infection status, they could not establish causality. Subsequently, several studies did control for HPV infection; the risk of squamous cervical cancer was twice as high in women who had ever smoked.55 Furthermore, the more cigarettes smoked per day, the higher the risk of cervical neoplasia.41,56
According to the US Centers for Disease Control and Prevention in 2014, the highest prevalence of smoking was among American Indian and Alaskan Native women, 32.5% of whom said they smoked every day, compared with 17.2% of white women and 13.7% of African American women.57
HOW CAN PRIMARY CARE PHYSICIANS CLOSE THE GAP?
Primary care physicians are the first point of contact for patients of all ages and so can help minimize such disparities. They can tackle 2 important cervical cancer prevention interventions first-hand: vaccination and screening (Table 2), including follow-up of abnormal screening results.
By promoting HPV vaccination to children and young adults, primary care physicians can help prevent cervical cancer. Moreover, primary care physicians will see most adolescents for a nonpreventive health visit, an optimal opportunity to discuss sexual activity practices and HPV vaccination.58 Including the HPV vaccine as routine with other vaccinations can close the gap.38
Screening and treatment of sexually transmitted infection during these visits can affect the risk that future HPV infection will progress to neoplasia or cancer. Persistent lifestyle modification counseling, especially smoking cessation through motivational interviewing, can lessen the risk of cervical cancer neoplasia progression.
Additionally, in light of recent changes in cervical cancer screening guidelines, the primary care physician’s role as educator is of utmost importance. In one study, although 99% of women had received a Pap test, 87% could not identify the purpose of the Pap test.59 The primary care physician’s role is perhaps the most influential in preventing disease and, as such, has the greatest impact on a patient’s disease process.
African American, Hispanic, American Indian, and Alaskan Native women continue to be disproportionately affected by cervical cancer compared with white women. From 2006 to 2010, the incidence of cervical cancer in African American women was 10.3 per 100,000; in white women it was 7.2.1 The mortality rate from cervical cancer in African American women is twice that in white women.1 Although cervical cancer rates have decreased nationwide, significant racial health disparities persist.
As the first-line healthcare providers for many women, the primary care physician and the general obstetrician-gynecologist are optimally positioned to reduce these disparities.
Cervical cancer is the third most common gynecologic cancer, after uterine and ovarian cancer. Nearly 13,000 new cases are diagnosed each year in the United States, and more than 4,000 women die of it.2 Fortunately, cervical cancer can be significantly prevented with adequate screening and vaccination against human papillomavirus (HPV).
WHY ARE BLACK WOMEN MORE LIKELY TO DIE OF CERVICAL CANCER?
Later stage at diagnosis. African American women are more likely to present with advanced cervical cancer than non-Hispanic white women.3–6
Less-aggressive treatment. African American women are more likely to receive no treatment after a cancer diagnosis.6 Differences in treatment may be attributed to comorbid conditions, stage at cancer diagnosis, and patient refusal.5,7
Less access to care. A study from the Surveillance, Epidemiology, and End Results program of the National Cancer Institute looked at 7,267 women (4,431 non-Hispanic white women, 1,830 Hispanic white women, and 1,006 non-Hispanic African American women) who were diagnosed with primary invasive cervical cancer from 1992 to 1996 and followed through 2000. African American women had a 19% higher mortality rate compared with non-Hispanic white women during follow-up despite adjusting for age, stage, histology, and time of first treatment.8
However, a later study from the same program found no such difference after 1995, when the data were adjusted for marital status, disease stage, age, treatment, grade, and histology.6
Equal access to healthcare may eliminate most of the disparity.7 A study in women with cervical cancer who sought treatment within the United States military healthcare system found no difference in treatment or 5- and 10-year survival rates between African American and white women.5 Equal access to comprehensive healthcare eliminated any disparity once cervical cancer was diagnosed.
CERVICAL CANCER SCREENING
The value of cervical cancer screening and prevention is well established. In 1941, Papanicolau reported that cervical cancer could be detected from vaginal smears.9 Since the development and widespread implementation of the “Pap” smear, cervical cancer rates have decreased dramatically in the United States.
Another major advance was the discovery that persistent infection with HPV is necessary for the development of cervical cancer, precancerous lesions, and genital warts.10
With advancing research, guidelines for cervical cancer screening have changed considerably over the years. Today, combined cervical cytologic and HPV testing is the mainstay. (Isolated HPV testing is generally not available outside clinical trials.)
Who should be screened?
Previous recommendations called for women to undergo Pap testing when they first became sexually active and then every year. However, cervical lesions are likely to regress in young women.11 One study found that 28% of cervical intimal neoplasia (CIN) grade 2 and 3 lesions spontaneously regressed by 15 weeks, although lesions associated with HPV 16 infection were less likely to regress than with other HPV types.12 A study of college women found that HPV infection persisted in only 9% of women after 24 months.13
To minimize unnecessary treatment of young women with dysplasia, the American Society for Colposcopy and Cervical Pathology in 2012 recommended cytologic screening for all women 21 years or older, regardless of age at first sexual encounter.14 Screening intervals were changed from every year to every 3 years until age 30, at which time cotesting with cytology and HPV testing is performed every 5 years. Routine cotesting is not recommended for women younger than 30, who have a high likelihood of HPV infection and spontaneous regression.
In 2014, the US Food and Drug Administration approved primary HPV screening (ie, testing for HPV first, and then performing cytology in samples that test positive) for women age 25 and older.15
Patients who need further evaluation and testing should be referred for colposcopy. The current guidelines for patients who have abnormal results on cervical cancer screening16 can be reviewed at www.asccp.org/asccp-guidelines.
As screening guidelines continue to evolve, primary care physicians will need to stay current and also help educate their patients. For example, many of our patients have undergone annual Pap screening for most of their lives and may not yet know about the new testing intervals.
Are there disparities in screening and follow-up?
Disparities in screening and follow-up may exist, but the evidence is not clear-cut.
In a 2013 National Health Interview Survey report, the rates of cervical cancer screening with Pap tests did not differ between African American and white women.17 However, the information on Pap testing was based on a single question asking participants if they had had a Pap test in the last 3 years. In our experience, patients may confuse Pap tests with speculum examinations.
Once women are screened, adequate and timely follow-up of abnormal results is key.
In a study from the National Breast and Cervical Cancer Early Detection Program,18 women who had cytology findings of atypical squamous cells of undetermined significance or low-grade squamous intraepithelial lesions were to undergo repeat Pap testing every 4 to 6 months for 2 years. African American women were the least likely to have a follow-up Pap smear compared with other racial groups.
On the other hand, there was no difference related to race in follow-up rates of abnormal Pap tests in women ages 47 to 64 in the South Carolina Breast and Cervical Cancer Early Detection Program.19
In a study in an urban population (predominantly African African), the overall follow-up rate was only 26% at 12 months from an initial abnormal Pap smear. This study did not find any differences in follow-up according to race or ethnicity; however, it had insufficient power to detect a difference because only 15% of the study participants were white.20
What is in a genotype?
HPV is implicated in progression to both squamous cell carcinoma and adenocarcinoma of the cervix. Worldwide, HPV genotypes 16 and 18 are associated with 73% of cases of invasive cervical cancer; most of the remainder are associated with, in order of decreasing prevalence, genotypes 58, 33, 45, 31, 52, 35, 59, 39, 51, and 56.21
High-grade cervical lesions in African American women may less often be positive for HPV 16 and 18 than in white women.22,23 On the other hand, the proportion of non-Hispanic black women infected with HPV 35 and 58 was significantly higher than in non-Hispanic white women.22 Regardless, HPV screening is recommended for women of all races and ethnicities.
The 2-valent and 4-valent HPV vaccines do not cover HPV 35 or 58. The newer 9-valent vaccine covers HPV 58 (but not 35) and so may in theory decrease any potential disparity related to infection with a specific oncogenic subtype.
THE ROLE OF PREVENTION
HPV vaccination
Currently, 3 vaccines against HPV are available in the United States, a 2-valent, a 4-valent, and since 2015, a 9-valent preparation (Table 1).
The Females United to Unilaterally Reduce Endo/Ectocervical Disease study demonstrated that the 4-valent vaccine was highly effective against cervical intraepithelial neoplasia due to HPV 16 and 18.24 In another study, the 2-valent vaccine reduced the incidence of CIN 3 or higher by 87% in women who received all 3 doses and who had no evidence of HPV infection at baseline.25
HPV vaccination is expensive. Each shot costs about $130, plus the cost of administering it. Although the Vaccines for Children program covers the HPV vaccine for uninsured and underinsured children and adolescents under age 19, Medicaid coverage varies from state to state for adults over age 21.
The Advisory Committee on Immunization Practices (ACIP)26 recommends routine vaccination for:
Males 11 or 12 years old
Females ages 9 to 26.
In October 2016, the ACIP approved a 2-dose series given 6 to 12 months apart for patients starting vaccination at ages 9 through 14 years who are not immunocompromised. Others should receive a 3-dose series, with the second dose given 1 to 2 months after the first dose and the third dose given 6 months after the first dose.27 Previously, 3 doses were recommended for everyone.
Disparities in HPV vaccination rates
HPV vaccination rates among adolescents in the United States increased from 33.6% in 2013 to 41.7% in 2014.28 However, HPV vaccination rates continue to lag behind those of other routine vaccines, such as Tdap and meningococcal conjugate.
Reagan-Steiner et al28 reported that more black than white girls age 13 through 17 received at least 1 dose of a 3-dose HPV vaccination series, but more white girls received all 3 doses (70.6% vs 61.6%). In contrast, a meta-analysis by Fisher et al29 found African American and uninsured women generally less likely to initiate the HPV vaccination series. Kessels et al30 reported similar findings.
Barriers to HPV vaccination
Barriers to HPV vaccination can be provider-dependent, parental, or institutional.
Malo et al31 surveyed Florida Medicaid providers and found that those who participated in the Vaccines for Children program were less likely to cite lack of reimbursement as a barrier to vaccination.
Meites et al32 surveyed sexually transmitted disease clinics and found that common reasons for not offering HPV vaccine were cost, staff time, and difficulty coordinating follow-up visits to complete the series.
Providers report lack of urgency or lack of perception of cervical cancer as a true public health threat, safety concerns regarding the vaccine, and the inability to coadminister vaccines as barriers.33
Studies have shown that relatively few parents (up to 18%) of parents are concerned about the effect of the vaccine on sexual activity.34 Rather, they are most likely to cite lack of information regarding the vaccine, lack of physician recommendation, and not knowing where to receive the vaccine as barriers.35,36
Guerry et al37 determined that the single most important factor in vaccine initiation was physician recommendation, a finding reiterated in other studies.35,38 A study in North Carolina identified failure of physician recommendation as one of the missed opportunities for vaccination of young women.39
Therefore, the primary care physician, as the initial contact with the child or young adult, holds a responsibility to narrow this gap. In simply discussing and recommending the vaccine, physicians could increase vaccination rates.
REPRODUCTIVE HEALTH
Although 80% of women will be infected with HPV in their lifetime, only a small proportion will develop cervical cancer, suggesting there are other cofactors in the progression to cervical cancer.40
Given the infectious etiology of cervical cancer, other contributing reproductive health factors have been described. As expected, the number of sexual partners correlates with HPV infection.41,42 Younger age at first intercourse has been linked to development of cervical neoplasia, consistent with persistent infection leading to neoplasia.41,42
Primary care physicians should provide timely and comprehensive sexual education, including information on safe sexual practices and pregnancy prevention.
Human immunodeficiency virus
In 2010, the estimated rate of new human immunodeficiency virus (HIV) infections in African American women was nearly 20 times greater than in white women.43 Previous studies have shown a clear relationship between HIV and HPV-associated cancers, including cervical neoplasia and invasive cervical cancer.44,45
Women with HIV should receive screening for cervical cancer at the time of diagnosis, 6 months after the initial diagnosis, and annually thereafter.46
Conflicting evidence exists regarding the effect of highly active antiretroviral therapy on the incidence of HPV-related disease, so aggressive screening and management of cervical neoplasia is recommended for women with HIV, regardless of CD4+ levels or viral load.47–49
Additional infectious culprits
Coinfection with other sexually transmitted infections, specifically Chlamydia, herpes, and HIV, has been associated with cervical neoplasia and invasive cervical cancer. A positive linear association exists between the number of sexually transmitted infections and cervical neoplasia.50
C trachomatis is the most common sexually transmitted infection in the United States, with a 6-times higher rate in African American women.51 Women who are seropositive for C trachomatis are at twofold higher risk of developing squamous cell cervical cancer.52,53 Women who are seropositive for Chlamydia infection, herpes virus 2, or HPV are at markedly increased risk of invasive cervical cancer.50
Tobacco use
The negative impact of smoking on numerous other cancers resulted in investigation of its role in cervical cancer.
Early case-control studies found an association between cervical cancer and smoking,54 but because these studies did not account for HPV infection status, they could not establish causality. Subsequently, several studies did control for HPV infection; the risk of squamous cervical cancer was twice as high in women who had ever smoked.55 Furthermore, the more cigarettes smoked per day, the higher the risk of cervical neoplasia.41,56
According to the US Centers for Disease Control and Prevention in 2014, the highest prevalence of smoking was among American Indian and Alaskan Native women, 32.5% of whom said they smoked every day, compared with 17.2% of white women and 13.7% of African American women.57
HOW CAN PRIMARY CARE PHYSICIANS CLOSE THE GAP?
Primary care physicians are the first point of contact for patients of all ages and so can help minimize such disparities. They can tackle 2 important cervical cancer prevention interventions first-hand: vaccination and screening (Table 2), including follow-up of abnormal screening results.
By promoting HPV vaccination to children and young adults, primary care physicians can help prevent cervical cancer. Moreover, primary care physicians will see most adolescents for a nonpreventive health visit, an optimal opportunity to discuss sexual activity practices and HPV vaccination.58 Including the HPV vaccine as routine with other vaccinations can close the gap.38
Screening and treatment of sexually transmitted infection during these visits can affect the risk that future HPV infection will progress to neoplasia or cancer. Persistent lifestyle modification counseling, especially smoking cessation through motivational interviewing, can lessen the risk of cervical cancer neoplasia progression.
Additionally, in light of recent changes in cervical cancer screening guidelines, the primary care physician’s role as educator is of utmost importance. In one study, although 99% of women had received a Pap test, 87% could not identify the purpose of the Pap test.59 The primary care physician’s role is perhaps the most influential in preventing disease and, as such, has the greatest impact on a patient’s disease process.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9–29.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5–29.
Koh WJ, Greer BE, Abu-Rustum NR, et al. Cervical cancer, version 2.2015. J Natl Compr Canc Netw 2015; 13:395-404.
Farley J, Risinger JI, Rose GS, Maxwell GL. Racial disparities in blacks with gynecologic cancers. Cancer 2007; 110:234–243.
Farley JH, Hines JF, Taylor RR, et al. Equal care ensures equal survival for African-American women with cervical carcinoma. Cancer 2001; 91:869–873.
Rauh-Hain JA, Clemmer JT, Bradford LS, et al. Racial disparities in cervical cancer survival over time. Cancer 2013; 119:3644–3652.
Collins Y, Holcomb K, Chapman-Davis E, Khabele D, Farley JH. Gynecologic cancer disparities: a report from the Health Disparities Taskforce of the Society of Gynecologic Oncology. Gynecol Oncol 2014; 133:353–361.
Patel DA, Barnholtz-Sloan JS, Patel MK, Malone JM Jr, Chuba PJ, Schwartz K. A population-based study of racial and ethnic differences in survival among women with invasive cervical cancer: analysis of surveillance, epidemiology, and end results data. Gynecol Oncol 2005; 97:550–558.
Papanicolaou GN, Traut HF. The diagnostic value of vaginal smears in carcinoma of the uterus. 1941. Arch Pathol Lab Med 1997; 121:211–224.
Walboomers JM, Jacobs M V, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
Moscicki AB, Shiboski S, Hills NK, et al. Regression of low-grade squamous intra-epithelial lesions in young women. Lancet 2004; 364:1678–1683.
Trimble CL, Piantadosi S, Gravitt P, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res 2005; 11:4717–4723.
Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med 1998; 338:423-428.
Saslow D, Solomon D, Lawson HW, et al; ACS-ASCCP-ASCP Cervical Cancer Guideline Committee. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA Cancer J Clin 2012; 62:147–172.
Huh WK, Ault KA, Chelmow D, et al. Use of primary high-risk human papillomavirus testing for cervical cancer screening: interim clinical guidance. Obstet Gynecol 2015; 125:330–337.
Massad LS, Einstein MH, Huh WK, et al; 2012 ASCCP Consensus Guidelines Conference. 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. J Low Genit Tract Dis 2013; 17(suppl 1):S1–S27.
Sabatino SA, White MC, Thompson TD, Klabunde CN. Cancer screening test use—United States, 2013. MMWR 2015; 64:464–468.
Benard VB, Lawson HW, Eheman CR, Anderson C, Helsel W. Adherence to guidelines for follow-up of low-grade cytologic abnormalities among medically underserved women. Obstet Gynecol 2005; 105:1323–1328.
Eggleston KS, Coker AL, Luchok KJ, Meyer TE. Adherence to recommendations for follow-up to abnormal Pap tests. Obstet Gynecol 2007; 109:1332–1341.
Peterson NB, Han J, Freund KM. Inadequate follow-up for abnormal Pap smears in an urban population. J Natl Med Assoc 2003; 95:825–832.
Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: variation by geographical region, histological type and year of publication. Int J Cancer 2011; 128:927–935.
Hariri S, Unger ER, Powell SE, et al; HPV-IMPACT Working Group. Human papillomavirus genotypes in high-grade cervical lesions in the United States. J Infect Dis 2012; 206:1878–1886.
Niccolai LM, Russ C, Julian PJ, et al. Individual and geographic disparities in human papillomavirus types 16/18 in high-grade cervical lesions: associations with race, ethnicity, and poverty. Cancer 2013; 119:3052–3058.
FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
Paavonen J, Naud P, Salmerón J, et al; HPV PATRICIA Study Group. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 2009; 374:301–314.
Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1705–1708.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
Reagan-Steiner S, Yankey D, Jeyarajah J, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13–17 years—United States, 2014. MMWR Morb Mortal Wkly Rep 2015; 64:784–792.
Fisher H, Trotter CL, Audrey S, MacDonald-Wallis K, Hickman M. Inequalities in the uptake of human papillomavirus vaccination: a systematic review and meta-analysis. Int J Epidemiol 2013; 42:896–908.
Kessels SJ, Marshall HS, Watson M, Braunack-Mayer AJ, Reuzel R, Tooher RL. Factors associated with HPV vaccine uptake in teenage girls: a systematic review. Vaccine 2012; 30:3546–3556.
Malo TL, Hassani D, Staras SA, Shenkman EA, Giuliano AR, Vadaparampil ST. Do Florida Medicaid providers’ barriers to HPV vaccination vary based on VFC program participation? Matern Child Health J 2013; 17:609–615.
Meites E, Llata E, Hariri S, et al. HPV vaccine implementation in STD clinics—STD Surveillance Network. Sex Transm Dis 2012; 39:32–34.
Perkins RB, Clark JA. What affects human papillomavirus vaccination rates? A qualitative analysis of providers’ perceptions. Womens Health Issues 2012; 22:e379–e386.
Holman DM, Benard V, Roland KB, Watson M, Liddon N, Stokley S. Barriers to human papillomavirus vaccination among US adolescents: a systematic review of the literature. JAMA Pediatr 2014; 168:76–82.
Dorell CG, Yankey D, Santibanez TA, Markowitz LE. Human papillomavirus vaccination series initiation and completion, 2008–2009. Pediatrics 2011; 128:830–839.
Bastani R, Glenn BA, Tsui J, et al. Understanding suboptimal human papillomavirus vaccine uptake among ethnic minority girls. Cancer Epidemiol Biomarkers Prev 2011; 20:1463–1472.
Guerry SL, De Rosa CJ, Markowitz LE, et al. Human papillomavirus vaccine initiation among adolescent girls in high-risk communities. Vaccine 2011; 29:2235–2241.
Hull PC, Williams EA, Khabele D, Dean C, Bond B, Sanderson M. HPV vaccine use among African American girls: qualitative formative research using a participatory social marketing approach. Gynecol Oncol 2014; 132(suppl 1):S13–S20.
Brewer NT, Gottlieb SL, Reiter PL, et al. Longitudinal predictors of human papillomavirus vaccine initiation among adolescent girls in a high-risk geographic area. Sex Transm Dis 2011; 38:197–204.
Wang SS, Zuna RE, Wentzensen N, et al. Human papillomavirus cofactors by disease progression and human papillomavirus types in the study to understand cervical cancer early endpoints and determinants. Cancer Epidemiol Biomarkers Prev 2009; 18:113–120.
Deacon JM, Evans CD, Yule R, et al. Sexual behaviour and smoking as determinants of cervical HPV infection and of CIN3 among those infected: a case-control study nested within the Manchester cohort. Br J Cancer 2000; 83:1565–1572.
International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical carcinoma and sexual behavior: collaborative reanalysis of individual data on 15,461 women with cervical carcinoma and 29,164 women without cervical carcinoma from 21 epidemiological studies. Cancer Epidemiol Biomarkers Prev 2009; 18:1060–1069.
Centers for Disease Control and Prevention (CDC). Estimated HIV incidence in the United States, 2007–2010. HIV Surveillance Supplemental Report 2012; 17(No. 4). https://www.cdc.gov/hiv/pdf/statistics_hssr_vol_17_no_4.pdf. Accessed September 12, 2017.
Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus-associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J Natl Cancer Inst 2000; 92:1500–1510.
Schäfer A, Friedmann W, Mielke M, Schwartländer B, Koch MA. The increased frequency of cervical dysplasia-neoplasia in women infected with the human immunodeficiency virus is related to the degree of immunosuppression. Am J Obstet Gynecol 1991; 164:593–599.
De Vuyst H, Lillo F, Broutet N, Smith JS. HIV, human papillomavirus, and cervical neoplasia and cancer in the era of highly active antiretroviral therapy. Eur J Cancer Prev 2008; 17:545–554.
Palefsky JM. Cervical human papillomavirus infection and cervical intraepithelial neoplasia in women positive for human immunodeficiency virus in the era of highly active antiretroviral therapy. Curr Opin Oncol 2003; 15:382–388.
Adler DH. The impact of HAART on HPV-related cervical disease. Curr HIV Res 2010; 8:493–497.
Castellsagué X, Pawlita M, Roura E, et al. Prospective seroepidemiologic study on the role of human papillomavirus and other infections in cervical carcinogenesis: evidence from the EPIC cohort. Int J Cancer 2014; 135:440–452.
Centers for Disease Control and Prevention (CDC). 2013 sexually transmitted disease surveillance. www.cdc.gov/std/stats13/exordium.htm. Accessed September 12, 2017.
Smith JS, Bosetti C, Muñoz N, et al; IARC multicentric case-control study. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case-control study. Int J Cancer 2004; 111:431–439.
Koskela P, Anttila T, Bjørge T, et al. Chlamydia trachomatis infection as a risk factor for invasive cervical cancer. Int J Cancer 2000; 85:35–39.
Office on Smoking and Health (US). Women and smoking: a report of the Surgeon General: Chapter 3. Health consequences of tobacco use among women. http://www.ncbi.nlm.nih.gov/books/NBK44312. Accessed September 12, 2017.
Plummer M, Herrero R, Franceschi S, et al; IARC Multi-centre Cervical Cancer Study Group. Smoking and cervical cancer: pooled analysis of the IARC multi-centric case—control study. Cancer Causes Control 2003; 14:805–814.
Ho GY, Kadish AS, Burk RD, et al. HPV 16 and cigarette smoking as risk factors for high-grade cervical intra-epithelial neoplasia. Int J Cancer 1998; 78:281–285.
Jamal A, Homa DM, O’Connor E, et al. Current cigarette smoking among adults - United States, 2005-2014. MMWR Morb Mortal Wkly Rep 2015; 64:1233–1240.
Nordin JD, Solberg LI, Parker ED. Adolescent primary care visit patterns. Ann Fam Med 2010; 8:511–516.
Lindau ST, Tomori C, Lyons T, Langseth L, Bennett CL, Garcia P. The association of health literacy with cervical cancer prevention knowledge and health behaviors in a multiethnic cohort of women. Am J Obstet Gynecol 2002; 186:938–943.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin 2014; 64:9–29.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015; 65:5–29.
Koh WJ, Greer BE, Abu-Rustum NR, et al. Cervical cancer, version 2.2015. J Natl Compr Canc Netw 2015; 13:395-404.
Farley J, Risinger JI, Rose GS, Maxwell GL. Racial disparities in blacks with gynecologic cancers. Cancer 2007; 110:234–243.
Farley JH, Hines JF, Taylor RR, et al. Equal care ensures equal survival for African-American women with cervical carcinoma. Cancer 2001; 91:869–873.
Rauh-Hain JA, Clemmer JT, Bradford LS, et al. Racial disparities in cervical cancer survival over time. Cancer 2013; 119:3644–3652.
Collins Y, Holcomb K, Chapman-Davis E, Khabele D, Farley JH. Gynecologic cancer disparities: a report from the Health Disparities Taskforce of the Society of Gynecologic Oncology. Gynecol Oncol 2014; 133:353–361.
Patel DA, Barnholtz-Sloan JS, Patel MK, Malone JM Jr, Chuba PJ, Schwartz K. A population-based study of racial and ethnic differences in survival among women with invasive cervical cancer: analysis of surveillance, epidemiology, and end results data. Gynecol Oncol 2005; 97:550–558.
Papanicolaou GN, Traut HF. The diagnostic value of vaginal smears in carcinoma of the uterus. 1941. Arch Pathol Lab Med 1997; 121:211–224.
Walboomers JM, Jacobs M V, Manos MM, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 1999; 189:12–19.
Moscicki AB, Shiboski S, Hills NK, et al. Regression of low-grade squamous intra-epithelial lesions in young women. Lancet 2004; 364:1678–1683.
Trimble CL, Piantadosi S, Gravitt P, et al. Spontaneous regression of high-grade cervical dysplasia: effects of human papillomavirus type and HLA phenotype. Clin Cancer Res 2005; 11:4717–4723.
Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med 1998; 338:423-428.
Saslow D, Solomon D, Lawson HW, et al; ACS-ASCCP-ASCP Cervical Cancer Guideline Committee. American Cancer Society, American Society for Colposcopy and Cervical Pathology, and American Society for Clinical Pathology screening guidelines for the prevention and early detection of cervical cancer. CA Cancer J Clin 2012; 62:147–172.
Huh WK, Ault KA, Chelmow D, et al. Use of primary high-risk human papillomavirus testing for cervical cancer screening: interim clinical guidance. Obstet Gynecol 2015; 125:330–337.
Massad LS, Einstein MH, Huh WK, et al; 2012 ASCCP Consensus Guidelines Conference. 2012 updated consensus guidelines for the management of abnormal cervical cancer screening tests and cancer precursors. J Low Genit Tract Dis 2013; 17(suppl 1):S1–S27.
Sabatino SA, White MC, Thompson TD, Klabunde CN. Cancer screening test use—United States, 2013. MMWR 2015; 64:464–468.
Benard VB, Lawson HW, Eheman CR, Anderson C, Helsel W. Adherence to guidelines for follow-up of low-grade cytologic abnormalities among medically underserved women. Obstet Gynecol 2005; 105:1323–1328.
Eggleston KS, Coker AL, Luchok KJ, Meyer TE. Adherence to recommendations for follow-up to abnormal Pap tests. Obstet Gynecol 2007; 109:1332–1341.
Peterson NB, Han J, Freund KM. Inadequate follow-up for abnormal Pap smears in an urban population. J Natl Med Assoc 2003; 95:825–832.
Li N, Franceschi S, Howell-Jones R, Snijders PJ, Clifford GM. Human papillomavirus type distribution in 30,848 invasive cervical cancers worldwide: variation by geographical region, histological type and year of publication. Int J Cancer 2011; 128:927–935.
Hariri S, Unger ER, Powell SE, et al; HPV-IMPACT Working Group. Human papillomavirus genotypes in high-grade cervical lesions in the United States. J Infect Dis 2012; 206:1878–1886.
Niccolai LM, Russ C, Julian PJ, et al. Individual and geographic disparities in human papillomavirus types 16/18 in high-grade cervical lesions: associations with race, ethnicity, and poverty. Cancer 2013; 119:3052–3058.
FUTURE II Study Group. Quadrivalent vaccine against human papillomavirus to prevent high-grade cervical lesions. N Engl J Med 2007; 356:1915–1927.
Paavonen J, Naud P, Salmerón J, et al; HPV PATRICIA Study Group. Efficacy of human papillomavirus (HPV)-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by oncogenic HPV types (PATRICIA): final analysis of a double-blind, randomised study in young women. Lancet 2009; 374:301–314.
Centers for Disease Control and Prevention (CDC). Recommendations on the use of quadrivalent human papillomavirus vaccine in males—Advisory Committee on Immunization Practices (ACIP), 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1705–1708.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
Reagan-Steiner S, Yankey D, Jeyarajah J, et al. National, regional, state, and selected local area vaccination coverage among adolescents aged 13–17 years—United States, 2014. MMWR Morb Mortal Wkly Rep 2015; 64:784–792.
Fisher H, Trotter CL, Audrey S, MacDonald-Wallis K, Hickman M. Inequalities in the uptake of human papillomavirus vaccination: a systematic review and meta-analysis. Int J Epidemiol 2013; 42:896–908.
Kessels SJ, Marshall HS, Watson M, Braunack-Mayer AJ, Reuzel R, Tooher RL. Factors associated with HPV vaccine uptake in teenage girls: a systematic review. Vaccine 2012; 30:3546–3556.
Malo TL, Hassani D, Staras SA, Shenkman EA, Giuliano AR, Vadaparampil ST. Do Florida Medicaid providers’ barriers to HPV vaccination vary based on VFC program participation? Matern Child Health J 2013; 17:609–615.
Meites E, Llata E, Hariri S, et al. HPV vaccine implementation in STD clinics—STD Surveillance Network. Sex Transm Dis 2012; 39:32–34.
Perkins RB, Clark JA. What affects human papillomavirus vaccination rates? A qualitative analysis of providers’ perceptions. Womens Health Issues 2012; 22:e379–e386.
Holman DM, Benard V, Roland KB, Watson M, Liddon N, Stokley S. Barriers to human papillomavirus vaccination among US adolescents: a systematic review of the literature. JAMA Pediatr 2014; 168:76–82.
Dorell CG, Yankey D, Santibanez TA, Markowitz LE. Human papillomavirus vaccination series initiation and completion, 2008–2009. Pediatrics 2011; 128:830–839.
Bastani R, Glenn BA, Tsui J, et al. Understanding suboptimal human papillomavirus vaccine uptake among ethnic minority girls. Cancer Epidemiol Biomarkers Prev 2011; 20:1463–1472.
Guerry SL, De Rosa CJ, Markowitz LE, et al. Human papillomavirus vaccine initiation among adolescent girls in high-risk communities. Vaccine 2011; 29:2235–2241.
Hull PC, Williams EA, Khabele D, Dean C, Bond B, Sanderson M. HPV vaccine use among African American girls: qualitative formative research using a participatory social marketing approach. Gynecol Oncol 2014; 132(suppl 1):S13–S20.
Brewer NT, Gottlieb SL, Reiter PL, et al. Longitudinal predictors of human papillomavirus vaccine initiation among adolescent girls in a high-risk geographic area. Sex Transm Dis 2011; 38:197–204.
Wang SS, Zuna RE, Wentzensen N, et al. Human papillomavirus cofactors by disease progression and human papillomavirus types in the study to understand cervical cancer early endpoints and determinants. Cancer Epidemiol Biomarkers Prev 2009; 18:113–120.
Deacon JM, Evans CD, Yule R, et al. Sexual behaviour and smoking as determinants of cervical HPV infection and of CIN3 among those infected: a case-control study nested within the Manchester cohort. Br J Cancer 2000; 83:1565–1572.
International Collaboration of Epidemiological Studies of Cervical Cancer. Cervical carcinoma and sexual behavior: collaborative reanalysis of individual data on 15,461 women with cervical carcinoma and 29,164 women without cervical carcinoma from 21 epidemiological studies. Cancer Epidemiol Biomarkers Prev 2009; 18:1060–1069.
Centers for Disease Control and Prevention (CDC). Estimated HIV incidence in the United States, 2007–2010. HIV Surveillance Supplemental Report 2012; 17(No. 4). https://www.cdc.gov/hiv/pdf/statistics_hssr_vol_17_no_4.pdf. Accessed September 12, 2017.
Frisch M, Biggar RJ, Goedert JJ. Human papillomavirus-associated cancers in patients with human immunodeficiency virus infection and acquired immunodeficiency syndrome. J Natl Cancer Inst 2000; 92:1500–1510.
Schäfer A, Friedmann W, Mielke M, Schwartländer B, Koch MA. The increased frequency of cervical dysplasia-neoplasia in women infected with the human immunodeficiency virus is related to the degree of immunosuppression. Am J Obstet Gynecol 1991; 164:593–599.
De Vuyst H, Lillo F, Broutet N, Smith JS. HIV, human papillomavirus, and cervical neoplasia and cancer in the era of highly active antiretroviral therapy. Eur J Cancer Prev 2008; 17:545–554.
Palefsky JM. Cervical human papillomavirus infection and cervical intraepithelial neoplasia in women positive for human immunodeficiency virus in the era of highly active antiretroviral therapy. Curr Opin Oncol 2003; 15:382–388.
Adler DH. The impact of HAART on HPV-related cervical disease. Curr HIV Res 2010; 8:493–497.
Castellsagué X, Pawlita M, Roura E, et al. Prospective seroepidemiologic study on the role of human papillomavirus and other infections in cervical carcinogenesis: evidence from the EPIC cohort. Int J Cancer 2014; 135:440–452.
Centers for Disease Control and Prevention (CDC). 2013 sexually transmitted disease surveillance. www.cdc.gov/std/stats13/exordium.htm. Accessed September 12, 2017.
Smith JS, Bosetti C, Muñoz N, et al; IARC multicentric case-control study. Chlamydia trachomatis and invasive cervical cancer: a pooled analysis of the IARC multicentric case-control study. Int J Cancer 2004; 111:431–439.
Koskela P, Anttila T, Bjørge T, et al. Chlamydia trachomatis infection as a risk factor for invasive cervical cancer. Int J Cancer 2000; 85:35–39.
Office on Smoking and Health (US). Women and smoking: a report of the Surgeon General: Chapter 3. Health consequences of tobacco use among women. http://www.ncbi.nlm.nih.gov/books/NBK44312. Accessed September 12, 2017.
Plummer M, Herrero R, Franceschi S, et al; IARC Multi-centre Cervical Cancer Study Group. Smoking and cervical cancer: pooled analysis of the IARC multi-centric case—control study. Cancer Causes Control 2003; 14:805–814.
Ho GY, Kadish AS, Burk RD, et al. HPV 16 and cigarette smoking as risk factors for high-grade cervical intra-epithelial neoplasia. Int J Cancer 1998; 78:281–285.
Jamal A, Homa DM, O’Connor E, et al. Current cigarette smoking among adults - United States, 2005-2014. MMWR Morb Mortal Wkly Rep 2015; 64:1233–1240.
Nordin JD, Solberg LI, Parker ED. Adolescent primary care visit patterns. Ann Fam Med 2010; 8:511–516.
Lindau ST, Tomori C, Lyons T, Langseth L, Bennett CL, Garcia P. The association of health literacy with cervical cancer prevention knowledge and health behaviors in a multiethnic cohort of women. Am J Obstet Gynecol 2002; 186:938–943.
Primary care providers play a crucial role in cancer control, including screening and follow-up.1,2 In particular, they are often responsible for performing the initial screening and, when necessary, discussing appropriate treatment options. However, cancer screening practices in primary care can vary significantly, leading to disparities in access to these services.3
Arvizo and Mahdi,4 in this issue of the Journal, discuss disparities in cervical cancer screening, noting that African American women have a higher risk of developing and dying of cervical cancer than white women, possibly because they are diagnosed at a later stage and have lower stage-specific survival rates. The authors state that equal access to healthcare may help mitigate these factors, and they also discuss how primary care providers can reduce these disparities.
PRIORITIZING CERVICAL CANCER SCREENING
Even in patients who have access to regular primary care, other barriers to cancer screening may exist. A 2014 study used self-reported data from the Behavioral Risk Factor Surveillance System survey to assess barriers to cervical cancer screening in older women (ages 40 to 65) who reported having health insurance and a personal healthcare provider.5 Those who were never or rarely screened for cervical cancer were more likely than those who were regularly screened to have a chronic condition, such as heart disease, chronic obstructive pulmonary disease, arthritis, depression, kidney disease, or diabetes.
This finding suggests that cancer screening may be a low priority during an adult primary care visit in which multiple chronic diseases must be addressed. To reduce disparities in cancer screening, primary care systems need to be designed to optimize delivery of preventive care and disease management using a team approach.
SYSTEMATIC FOLLOW-UP
Arvizo and Mahdi also discuss the follow-up of abnormal screening Papanicolaou (Pap) smears. While appropriate follow-up is a key factor in the management of cervical dysplasia, follow-up rates vary among African American women. System-level interventions such as the use of an electronic medical record-based tracking system in primary care settings6 with established protocols for follow-up may be effective.
But even with such systems in place, patients may face psychosocial barriers (eg, lack of health literacy, distress after receiving an abnormal cervical cytology test result7) that prevent them from seeking additional care. To improve follow-up rates, providers must be aware of these barriers and know how to address them through effective communication.
VACCINATION FOR HPV
Finally, the association between human papilloma virus (HPV) infection and cervical cancer makes HPV vaccination a crucial step in cervical cancer prevention. Continued provider education regarding HPV vaccination can improve knowledge about the HPV vaccine,8 as well as improve vaccination rates.9 The recent approval of a 2-dose vaccine schedule for younger girls10 may also help improve vaccine series completion rates.
The authors also suggest that primary care providers counsel all patients about risk factors for cervical cancer, including unsafe sex practices and tobacco use.
OPTIMIZING SCREENING AND PREVENTION
I commend the authors for their discussion of cervical cancer disparities and for raising awareness of the important role primary care providers play in reducing these disparities. Improving cervical cancer screening rates and follow-up will require providers and patients to be aware of cervical cancer risk factors. Further, system-level practice interventions will optimize primary care providers’ ability to engage patients in cancer screening conversations and ensure timely follow-up of screening tests.
References
Emery JD, Shaw K, Williams B, et al. The role of primary care in early detection and follow-up of cancer. Nat Rev Clin Oncol 2014; 11:38–48.
Rubin G, Berendsen A, Crawford SM, et al. The expanding role of primary care in cancer control. Lancet Oncol 2015; 16:1231–1272.
Martires KJ, Kurlander DE, Minwell GJ, Dahms EB, Bordeaux JS. Patterns of cancer screening in primary care from 2005 to 2010. Cancer 2014; 120:253–261.
Arvizo C, Mahdi H. Disparities in cervical cancer in African-American women: what primary care physicians can do. Cleve Clin J Med 2017; 84:788–794.
Crawford A, Benard V, King J, Thomas CC. Understanding barriers to cervical cancer screening in women with access to care, behavioral risk factor surveillance system, 2014. Prev Chronic Dis 2016; 13:E154.
Dupuis EA, White HF, Newman D, Sobieraj JE, Gokhale M, Freund KM. Tracking abnormal cervical cancer screening: evaluation of an EMR-based intervention. J Gen Intern Med 2010; 25:575–580.
Hui SK, Miller SM, Wen KY, et al. Psychosocial barriers to follow-up adherence after an abnormal cervical cytology test result among low-income, inner-city women. J Prim Care Community Health 2014; 5:234–241.
Berenson AB, Rahman M, Hirth JM, Rupp RE, Sarpong KO. A brief educational intervention increases providers’ human papillomavirus vaccine knowledge. Hum Vaccin Immunother 2015; 11:1331–1336.
Perkins RB, Zisblatt L, Legler A, Trucks E, Hanchate A, Gorin SS. Effectiveness of a provider-focused intervention to improve HPV vaccination rates in boys and girls. Vaccine 2015; 33:1223–1229.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
Anita D. Misra-Hebert MD, MPH Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, and Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Anita D. Misra-Hebert MD, MPH, Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, G10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; misraa@ccf.org
Dr. Misra-Hebert is supported by an Agency for Healthcare Research and Quality grant K08HS024128.
Anita D. Misra-Hebert MD, MPH Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, and Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Anita D. Misra-Hebert MD, MPH, Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, G10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; misraa@ccf.org
Dr. Misra-Hebert is supported by an Agency for Healthcare Research and Quality grant K08HS024128.
Author and Disclosure Information
Anita D. Misra-Hebert MD, MPH Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, and Department of Quantitative Health Sciences, Lerner Research Institute, Cleveland Clinic; Assistant Professor, Cleveland Clinic Lerner College of Medicine of Case Western Reserve University, Cleveland, OH
Address: Anita D. Misra-Hebert MD, MPH, Department of Internal Medicine, Center for Value-Based Care Research, Medicine Institute, G10, Cleveland Clinic, 9500 Euclid Avenue, Cleveland, OH 44195; misraa@ccf.org
Dr. Misra-Hebert is supported by an Agency for Healthcare Research and Quality grant K08HS024128.
Primary care providers play a crucial role in cancer control, including screening and follow-up.1,2 In particular, they are often responsible for performing the initial screening and, when necessary, discussing appropriate treatment options. However, cancer screening practices in primary care can vary significantly, leading to disparities in access to these services.3
Arvizo and Mahdi,4 in this issue of the Journal, discuss disparities in cervical cancer screening, noting that African American women have a higher risk of developing and dying of cervical cancer than white women, possibly because they are diagnosed at a later stage and have lower stage-specific survival rates. The authors state that equal access to healthcare may help mitigate these factors, and they also discuss how primary care providers can reduce these disparities.
PRIORITIZING CERVICAL CANCER SCREENING
Even in patients who have access to regular primary care, other barriers to cancer screening may exist. A 2014 study used self-reported data from the Behavioral Risk Factor Surveillance System survey to assess barriers to cervical cancer screening in older women (ages 40 to 65) who reported having health insurance and a personal healthcare provider.5 Those who were never or rarely screened for cervical cancer were more likely than those who were regularly screened to have a chronic condition, such as heart disease, chronic obstructive pulmonary disease, arthritis, depression, kidney disease, or diabetes.
This finding suggests that cancer screening may be a low priority during an adult primary care visit in which multiple chronic diseases must be addressed. To reduce disparities in cancer screening, primary care systems need to be designed to optimize delivery of preventive care and disease management using a team approach.
SYSTEMATIC FOLLOW-UP
Arvizo and Mahdi also discuss the follow-up of abnormal screening Papanicolaou (Pap) smears. While appropriate follow-up is a key factor in the management of cervical dysplasia, follow-up rates vary among African American women. System-level interventions such as the use of an electronic medical record-based tracking system in primary care settings6 with established protocols for follow-up may be effective.
But even with such systems in place, patients may face psychosocial barriers (eg, lack of health literacy, distress after receiving an abnormal cervical cytology test result7) that prevent them from seeking additional care. To improve follow-up rates, providers must be aware of these barriers and know how to address them through effective communication.
VACCINATION FOR HPV
Finally, the association between human papilloma virus (HPV) infection and cervical cancer makes HPV vaccination a crucial step in cervical cancer prevention. Continued provider education regarding HPV vaccination can improve knowledge about the HPV vaccine,8 as well as improve vaccination rates.9 The recent approval of a 2-dose vaccine schedule for younger girls10 may also help improve vaccine series completion rates.
The authors also suggest that primary care providers counsel all patients about risk factors for cervical cancer, including unsafe sex practices and tobacco use.
OPTIMIZING SCREENING AND PREVENTION
I commend the authors for their discussion of cervical cancer disparities and for raising awareness of the important role primary care providers play in reducing these disparities. Improving cervical cancer screening rates and follow-up will require providers and patients to be aware of cervical cancer risk factors. Further, system-level practice interventions will optimize primary care providers’ ability to engage patients in cancer screening conversations and ensure timely follow-up of screening tests.
Primary care providers play a crucial role in cancer control, including screening and follow-up.1,2 In particular, they are often responsible for performing the initial screening and, when necessary, discussing appropriate treatment options. However, cancer screening practices in primary care can vary significantly, leading to disparities in access to these services.3
Arvizo and Mahdi,4 in this issue of the Journal, discuss disparities in cervical cancer screening, noting that African American women have a higher risk of developing and dying of cervical cancer than white women, possibly because they are diagnosed at a later stage and have lower stage-specific survival rates. The authors state that equal access to healthcare may help mitigate these factors, and they also discuss how primary care providers can reduce these disparities.
PRIORITIZING CERVICAL CANCER SCREENING
Even in patients who have access to regular primary care, other barriers to cancer screening may exist. A 2014 study used self-reported data from the Behavioral Risk Factor Surveillance System survey to assess barriers to cervical cancer screening in older women (ages 40 to 65) who reported having health insurance and a personal healthcare provider.5 Those who were never or rarely screened for cervical cancer were more likely than those who were regularly screened to have a chronic condition, such as heart disease, chronic obstructive pulmonary disease, arthritis, depression, kidney disease, or diabetes.
This finding suggests that cancer screening may be a low priority during an adult primary care visit in which multiple chronic diseases must be addressed. To reduce disparities in cancer screening, primary care systems need to be designed to optimize delivery of preventive care and disease management using a team approach.
SYSTEMATIC FOLLOW-UP
Arvizo and Mahdi also discuss the follow-up of abnormal screening Papanicolaou (Pap) smears. While appropriate follow-up is a key factor in the management of cervical dysplasia, follow-up rates vary among African American women. System-level interventions such as the use of an electronic medical record-based tracking system in primary care settings6 with established protocols for follow-up may be effective.
But even with such systems in place, patients may face psychosocial barriers (eg, lack of health literacy, distress after receiving an abnormal cervical cytology test result7) that prevent them from seeking additional care. To improve follow-up rates, providers must be aware of these barriers and know how to address them through effective communication.
VACCINATION FOR HPV
Finally, the association between human papilloma virus (HPV) infection and cervical cancer makes HPV vaccination a crucial step in cervical cancer prevention. Continued provider education regarding HPV vaccination can improve knowledge about the HPV vaccine,8 as well as improve vaccination rates.9 The recent approval of a 2-dose vaccine schedule for younger girls10 may also help improve vaccine series completion rates.
The authors also suggest that primary care providers counsel all patients about risk factors for cervical cancer, including unsafe sex practices and tobacco use.
OPTIMIZING SCREENING AND PREVENTION
I commend the authors for their discussion of cervical cancer disparities and for raising awareness of the important role primary care providers play in reducing these disparities. Improving cervical cancer screening rates and follow-up will require providers and patients to be aware of cervical cancer risk factors. Further, system-level practice interventions will optimize primary care providers’ ability to engage patients in cancer screening conversations and ensure timely follow-up of screening tests.
References
Emery JD, Shaw K, Williams B, et al. The role of primary care in early detection and follow-up of cancer. Nat Rev Clin Oncol 2014; 11:38–48.
Rubin G, Berendsen A, Crawford SM, et al. The expanding role of primary care in cancer control. Lancet Oncol 2015; 16:1231–1272.
Martires KJ, Kurlander DE, Minwell GJ, Dahms EB, Bordeaux JS. Patterns of cancer screening in primary care from 2005 to 2010. Cancer 2014; 120:253–261.
Arvizo C, Mahdi H. Disparities in cervical cancer in African-American women: what primary care physicians can do. Cleve Clin J Med 2017; 84:788–794.
Crawford A, Benard V, King J, Thomas CC. Understanding barriers to cervical cancer screening in women with access to care, behavioral risk factor surveillance system, 2014. Prev Chronic Dis 2016; 13:E154.
Dupuis EA, White HF, Newman D, Sobieraj JE, Gokhale M, Freund KM. Tracking abnormal cervical cancer screening: evaluation of an EMR-based intervention. J Gen Intern Med 2010; 25:575–580.
Hui SK, Miller SM, Wen KY, et al. Psychosocial barriers to follow-up adherence after an abnormal cervical cytology test result among low-income, inner-city women. J Prim Care Community Health 2014; 5:234–241.
Berenson AB, Rahman M, Hirth JM, Rupp RE, Sarpong KO. A brief educational intervention increases providers’ human papillomavirus vaccine knowledge. Hum Vaccin Immunother 2015; 11:1331–1336.
Perkins RB, Zisblatt L, Legler A, Trucks E, Hanchate A, Gorin SS. Effectiveness of a provider-focused intervention to improve HPV vaccination rates in boys and girls. Vaccine 2015; 33:1223–1229.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
References
Emery JD, Shaw K, Williams B, et al. The role of primary care in early detection and follow-up of cancer. Nat Rev Clin Oncol 2014; 11:38–48.
Rubin G, Berendsen A, Crawford SM, et al. The expanding role of primary care in cancer control. Lancet Oncol 2015; 16:1231–1272.
Martires KJ, Kurlander DE, Minwell GJ, Dahms EB, Bordeaux JS. Patterns of cancer screening in primary care from 2005 to 2010. Cancer 2014; 120:253–261.
Arvizo C, Mahdi H. Disparities in cervical cancer in African-American women: what primary care physicians can do. Cleve Clin J Med 2017; 84:788–794.
Crawford A, Benard V, King J, Thomas CC. Understanding barriers to cervical cancer screening in women with access to care, behavioral risk factor surveillance system, 2014. Prev Chronic Dis 2016; 13:E154.
Dupuis EA, White HF, Newman D, Sobieraj JE, Gokhale M, Freund KM. Tracking abnormal cervical cancer screening: evaluation of an EMR-based intervention. J Gen Intern Med 2010; 25:575–580.
Hui SK, Miller SM, Wen KY, et al. Psychosocial barriers to follow-up adherence after an abnormal cervical cytology test result among low-income, inner-city women. J Prim Care Community Health 2014; 5:234–241.
Berenson AB, Rahman M, Hirth JM, Rupp RE, Sarpong KO. A brief educational intervention increases providers’ human papillomavirus vaccine knowledge. Hum Vaccin Immunother 2015; 11:1331–1336.
Perkins RB, Zisblatt L, Legler A, Trucks E, Hanchate A, Gorin SS. Effectiveness of a provider-focused intervention to improve HPV vaccination rates in boys and girls. Vaccine 2015; 33:1223–1229.
Meites E, Kempe A, Markowitz LE. Use of a 2-dose schedule for human papillomavirus vaccination—updated recommendations of the Advisory Committee on Immunization Practices. MMWR Morb Mortal Wkly Rep 2016; 65:1405–1408.
A 40-year-old man was brought to the emergency department with altered mental status. His roommate had found him lying unconscious in snow on the lawn outside his residence. When the emergency medical services team arrived, they recorded a core body temperature of 28.3°C (82.9°F) and instituted advanced cardiac life support.
During transit to the hospital, the patient’s heart rhythm changed from asystole to ventricular fibrillation, and defibrillation was performed twice.
Upon his arrival at the emergency room, advanced life support was continued, resulting in return of spontaneous circulation, with slow, wide-complex QRS rhythm noted on electrocardiography (ECG).
On examination, the patient’s pupils were fixed and dilated. The extremities were cold to palpation. The core body temperature dropped to 27.7°C (81.9°F).
Laboratory test results showed severe acidemia (arterial pH 6.8), elevated aspartate aminotransferase and alanine aminotransferase levels, and elevated creatinine and troponin. The troponin was measured 3 times and rose from 0.8 ng/mL to 0.9 ng/mL. A urine toxicology screen was positive for cannabinoids and cocaine.
Figure 1. On 12-lead electrocardiography, elevation of the J point (Osborn wave; arrows) is typically seen in precordial leads V3 to V6 and is a marker for hypothermia. The amplitude of Osborn waves is directly proportional to the degree of hypothermia.
ECG revealed J-point elevation (Osborn waves) in the precordial leads (Figure 1). A baseline electrocardiogram in the medical record from a previous admission had been normal.
An aggressive hypothermia protocol was initiated, but the patient died despite resuscitation efforts.
HYPOTHERMIA AND HEART RHYTHMS
Hypothermia—a core body temperature below 35°C (95°F)—causes generalized slowing of impulse conduction through cardiac tissues, shown on ECG as a prolongation of the PR, RR, QRS, and QT intervals.1
A characteristic feature is elevation of the J point, also called the J wave or Osborn wave, most prominent in precordial leads V2 to V5 and caused by abnormal membrane repolarization in the early phase. The degree of hypothermia correlates linearly with the amplitude of the Osborn wave.2,3
Laboratory tests can identify complications such as rhabdomyolysis, spontaneous bleeding, and lactic acidosis. Moderate to severe hypothermia may cause prolongation of all ECG intervals. Management requires resuscitation and rewarming.
Conditions to consider in the differential diagnosis are Brugada syndrome, hypercalcemia, and early repolarization syndrome.
References
Doshi HH, Giudici MC. The EKG in hypothermia and hyperthermia. J Electrocardiol 2015; 48:203–208.
Alsafwah S. Electrocardiographic changes in hypothermia. Heart Lung 2001; 30:161–163.
Graham CA, McNaughton GW, Wyatt JP. The electrocardiogram in hypothermia. Wilderness Environ Med 2001; 12:232–235.
A 40-year-old man was brought to the emergency department with altered mental status. His roommate had found him lying unconscious in snow on the lawn outside his residence. When the emergency medical services team arrived, they recorded a core body temperature of 28.3°C (82.9°F) and instituted advanced cardiac life support.
During transit to the hospital, the patient’s heart rhythm changed from asystole to ventricular fibrillation, and defibrillation was performed twice.
Upon his arrival at the emergency room, advanced life support was continued, resulting in return of spontaneous circulation, with slow, wide-complex QRS rhythm noted on electrocardiography (ECG).
On examination, the patient’s pupils were fixed and dilated. The extremities were cold to palpation. The core body temperature dropped to 27.7°C (81.9°F).
Laboratory test results showed severe acidemia (arterial pH 6.8), elevated aspartate aminotransferase and alanine aminotransferase levels, and elevated creatinine and troponin. The troponin was measured 3 times and rose from 0.8 ng/mL to 0.9 ng/mL. A urine toxicology screen was positive for cannabinoids and cocaine.
Figure 1. On 12-lead electrocardiography, elevation of the J point (Osborn wave; arrows) is typically seen in precordial leads V3 to V6 and is a marker for hypothermia. The amplitude of Osborn waves is directly proportional to the degree of hypothermia.
ECG revealed J-point elevation (Osborn waves) in the precordial leads (Figure 1). A baseline electrocardiogram in the medical record from a previous admission had been normal.
An aggressive hypothermia protocol was initiated, but the patient died despite resuscitation efforts.
HYPOTHERMIA AND HEART RHYTHMS
Hypothermia—a core body temperature below 35°C (95°F)—causes generalized slowing of impulse conduction through cardiac tissues, shown on ECG as a prolongation of the PR, RR, QRS, and QT intervals.1
A characteristic feature is elevation of the J point, also called the J wave or Osborn wave, most prominent in precordial leads V2 to V5 and caused by abnormal membrane repolarization in the early phase. The degree of hypothermia correlates linearly with the amplitude of the Osborn wave.2,3
Laboratory tests can identify complications such as rhabdomyolysis, spontaneous bleeding, and lactic acidosis. Moderate to severe hypothermia may cause prolongation of all ECG intervals. Management requires resuscitation and rewarming.
Conditions to consider in the differential diagnosis are Brugada syndrome, hypercalcemia, and early repolarization syndrome.
A 40-year-old man was brought to the emergency department with altered mental status. His roommate had found him lying unconscious in snow on the lawn outside his residence. When the emergency medical services team arrived, they recorded a core body temperature of 28.3°C (82.9°F) and instituted advanced cardiac life support.
During transit to the hospital, the patient’s heart rhythm changed from asystole to ventricular fibrillation, and defibrillation was performed twice.
Upon his arrival at the emergency room, advanced life support was continued, resulting in return of spontaneous circulation, with slow, wide-complex QRS rhythm noted on electrocardiography (ECG).
On examination, the patient’s pupils were fixed and dilated. The extremities were cold to palpation. The core body temperature dropped to 27.7°C (81.9°F).
Laboratory test results showed severe acidemia (arterial pH 6.8), elevated aspartate aminotransferase and alanine aminotransferase levels, and elevated creatinine and troponin. The troponin was measured 3 times and rose from 0.8 ng/mL to 0.9 ng/mL. A urine toxicology screen was positive for cannabinoids and cocaine.
Figure 1. On 12-lead electrocardiography, elevation of the J point (Osborn wave; arrows) is typically seen in precordial leads V3 to V6 and is a marker for hypothermia. The amplitude of Osborn waves is directly proportional to the degree of hypothermia.
ECG revealed J-point elevation (Osborn waves) in the precordial leads (Figure 1). A baseline electrocardiogram in the medical record from a previous admission had been normal.
An aggressive hypothermia protocol was initiated, but the patient died despite resuscitation efforts.
HYPOTHERMIA AND HEART RHYTHMS
Hypothermia—a core body temperature below 35°C (95°F)—causes generalized slowing of impulse conduction through cardiac tissues, shown on ECG as a prolongation of the PR, RR, QRS, and QT intervals.1
A characteristic feature is elevation of the J point, also called the J wave or Osborn wave, most prominent in precordial leads V2 to V5 and caused by abnormal membrane repolarization in the early phase. The degree of hypothermia correlates linearly with the amplitude of the Osborn wave.2,3
Laboratory tests can identify complications such as rhabdomyolysis, spontaneous bleeding, and lactic acidosis. Moderate to severe hypothermia may cause prolongation of all ECG intervals. Management requires resuscitation and rewarming.
Conditions to consider in the differential diagnosis are Brugada syndrome, hypercalcemia, and early repolarization syndrome.
References
Doshi HH, Giudici MC. The EKG in hypothermia and hyperthermia. J Electrocardiol 2015; 48:203–208.
Alsafwah S. Electrocardiographic changes in hypothermia. Heart Lung 2001; 30:161–163.
Graham CA, McNaughton GW, Wyatt JP. The electrocardiogram in hypothermia. Wilderness Environ Med 2001; 12:232–235.
References
Doshi HH, Giudici MC. The EKG in hypothermia and hyperthermia. J Electrocardiol 2015; 48:203–208.
Alsafwah S. Electrocardiographic changes in hypothermia. Heart Lung 2001; 30:161–163.
Graham CA, McNaughton GW, Wyatt JP. The electrocardiogram in hypothermia. Wilderness Environ Med 2001; 12:232–235.
A 41-year-old woman presented with coughing, wheezing, and painful subcutaneous nodules on her legs. She had presented 6 years ago with similar nodules and enlarged retroauricular, occipital, and maxillary lymph nodes. At that time, biopsy study of submaxillary lymph nodes and skin showed nonnecrotizing granulomas, with negative microbiology studies. Chest radiography and spirometry were normal. A diagnosis of sarcoidosis was made. Treatment was offered but refused.
Figure 1. Magnetic resonance imaging revealed low-intensity lesions (arrows) on T1-weighted sequences (upper panel) and high-intensity lesions on short tau inversion recovery sequences (lower panel). The lesions have defined borders, no cortical disruption, and no mass effect, features suggesting that the lesions are benign. Lack of signal homogeneity suggests chronic lesions.
Based on this history, computed tomography of the thorax and abdomen was performed and showed mediastinal and hilar lymphadenopathy, small bilateral lung nodules, and osseous cystic areas in both iliac blades. Magnetic resonance imaging (MRI) showed numerous discrete lesions in both iliac bones (Figure 1). Biopsy study of iliac bone revealed preserved architecture with no evidence of malignancy or granulomas.
Radiography of the hands showed an osseous lytic lesion in the third proximal phalanx of the right hand. No other radiographic abnormalities were noted.
The clinical and radiographic features and the patient’s clinical course were consistent with osseous sarcoidosis. She was started on methotrexate and a low-dose corticosteroid and was symptom-free at 12-month follow-up. Follow-up MRI showed reduction in the lymphadenopathies and stabilization of the bone lesions.
SARCOIDOSIS AND BONE
Sarcoidosis is a systemic granulomatous disease that involves the lung in more than 90% of cases. Skeletal involvement has been reported in 1% to 14% of patients.1,2 Typical osseous involvement is cystic osteitis of the phalangeal bones of the hands and feet, but any part of the skeleton may be involved.3
Bone sarcoidosis is usually asymptomatic and is discovered incidentally. The diagnosis of sarcoidosis has usually been established clinically before bone lesions are detected on MRI. However, sarcoidosis-related bone lesions resembling bone metastases on MRI may be the initial presentation. The presence of intralesional fat has been described as a feature that excludes malignancy.
No treatment has been shown to be of benefit.4 Sarcoidosis is a diagnosis of exclusion and radiographic lytic bone features are not specific, so a neoplastic cause (such as primary osteoblastoma, metastasis, or multiple myeloma) must always be ruled out, as well as other bone conditions such as osteomyelitis or bone cyst.
References
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet 2014; 383:1155–1167.
James DG, Neville E, Carstairs LS. Bone and joint sarcoidosis. Semin Arthritis Rheum 1976; 6:53–81.
Moore SL, Kransdorf MJ, Schweitzer ME, Murphey MD, Babb JS. Can sarcoidosis and metastatic bone lesions be reliably differentiated on routine MRI? AJR Am J Roentgenol 2012; 198:1387–1393.
Hamoud S, Srour S, Fruchter O, Vlodavsky E, Hayek T. Lytic bone lesion: presenting finding of sarcoidosis. Isr Med Assoc J 2010; 12:59–60.
Anahy M. Brandy-García Servicio de Reumatología, Hospital Universitario Central de Asturias Oviedo, Spain
Ivan Cabezas-Rodriguez Servicio de Reumatología, Hospital Universitario Central de Asturias, Oviedo, Spain
Luis Caminal-Montero Consulta-Unidad Enfermedades Autoinmunes Sistémicas, Unidad Clínica de Medicina Interna Hospital Universitario Central de Asturias, Oviedo, Spain
Carlos Suarez-Cuervo Department of General Medicine, Borders General Hospital, Melrose, Scotland, UK
Pilar Redondo-Buil Servicio de Radiología, Hospital Universitario Central de Asturias, Oviedo, Spain
Address: Anahy M. Brandy-García, Servicio de Reumatología, Hospital Universitario Central de Asturias, Av Roma s/n, 33011 Asturias, Spain; anahymbg@gmail.com
sarcoidosis, lung nodules, bone metastases, lytic osseous metastases, Anahy Brandy-Garcia, Ivan Cabezas-Rodriguez, Luis Caminal-Montero, Carlos Suarez-Cuervo, Pilar Redondo-Buil
Anahy M. Brandy-García Servicio de Reumatología, Hospital Universitario Central de Asturias Oviedo, Spain
Ivan Cabezas-Rodriguez Servicio de Reumatología, Hospital Universitario Central de Asturias, Oviedo, Spain
Luis Caminal-Montero Consulta-Unidad Enfermedades Autoinmunes Sistémicas, Unidad Clínica de Medicina Interna Hospital Universitario Central de Asturias, Oviedo, Spain
Carlos Suarez-Cuervo Department of General Medicine, Borders General Hospital, Melrose, Scotland, UK
Pilar Redondo-Buil Servicio de Radiología, Hospital Universitario Central de Asturias, Oviedo, Spain
Address: Anahy M. Brandy-García, Servicio de Reumatología, Hospital Universitario Central de Asturias, Av Roma s/n, 33011 Asturias, Spain; anahymbg@gmail.com
Author and Disclosure Information
Anahy M. Brandy-García Servicio de Reumatología, Hospital Universitario Central de Asturias Oviedo, Spain
Ivan Cabezas-Rodriguez Servicio de Reumatología, Hospital Universitario Central de Asturias, Oviedo, Spain
Luis Caminal-Montero Consulta-Unidad Enfermedades Autoinmunes Sistémicas, Unidad Clínica de Medicina Interna Hospital Universitario Central de Asturias, Oviedo, Spain
Carlos Suarez-Cuervo Department of General Medicine, Borders General Hospital, Melrose, Scotland, UK
Pilar Redondo-Buil Servicio de Radiología, Hospital Universitario Central de Asturias, Oviedo, Spain
Address: Anahy M. Brandy-García, Servicio de Reumatología, Hospital Universitario Central de Asturias, Av Roma s/n, 33011 Asturias, Spain; anahymbg@gmail.com
A 41-year-old woman presented with coughing, wheezing, and painful subcutaneous nodules on her legs. She had presented 6 years ago with similar nodules and enlarged retroauricular, occipital, and maxillary lymph nodes. At that time, biopsy study of submaxillary lymph nodes and skin showed nonnecrotizing granulomas, with negative microbiology studies. Chest radiography and spirometry were normal. A diagnosis of sarcoidosis was made. Treatment was offered but refused.
Figure 1. Magnetic resonance imaging revealed low-intensity lesions (arrows) on T1-weighted sequences (upper panel) and high-intensity lesions on short tau inversion recovery sequences (lower panel). The lesions have defined borders, no cortical disruption, and no mass effect, features suggesting that the lesions are benign. Lack of signal homogeneity suggests chronic lesions.
Based on this history, computed tomography of the thorax and abdomen was performed and showed mediastinal and hilar lymphadenopathy, small bilateral lung nodules, and osseous cystic areas in both iliac blades. Magnetic resonance imaging (MRI) showed numerous discrete lesions in both iliac bones (Figure 1). Biopsy study of iliac bone revealed preserved architecture with no evidence of malignancy or granulomas.
Radiography of the hands showed an osseous lytic lesion in the third proximal phalanx of the right hand. No other radiographic abnormalities were noted.
The clinical and radiographic features and the patient’s clinical course were consistent with osseous sarcoidosis. She was started on methotrexate and a low-dose corticosteroid and was symptom-free at 12-month follow-up. Follow-up MRI showed reduction in the lymphadenopathies and stabilization of the bone lesions.
SARCOIDOSIS AND BONE
Sarcoidosis is a systemic granulomatous disease that involves the lung in more than 90% of cases. Skeletal involvement has been reported in 1% to 14% of patients.1,2 Typical osseous involvement is cystic osteitis of the phalangeal bones of the hands and feet, but any part of the skeleton may be involved.3
Bone sarcoidosis is usually asymptomatic and is discovered incidentally. The diagnosis of sarcoidosis has usually been established clinically before bone lesions are detected on MRI. However, sarcoidosis-related bone lesions resembling bone metastases on MRI may be the initial presentation. The presence of intralesional fat has been described as a feature that excludes malignancy.
No treatment has been shown to be of benefit.4 Sarcoidosis is a diagnosis of exclusion and radiographic lytic bone features are not specific, so a neoplastic cause (such as primary osteoblastoma, metastasis, or multiple myeloma) must always be ruled out, as well as other bone conditions such as osteomyelitis or bone cyst.
A 41-year-old woman presented with coughing, wheezing, and painful subcutaneous nodules on her legs. She had presented 6 years ago with similar nodules and enlarged retroauricular, occipital, and maxillary lymph nodes. At that time, biopsy study of submaxillary lymph nodes and skin showed nonnecrotizing granulomas, with negative microbiology studies. Chest radiography and spirometry were normal. A diagnosis of sarcoidosis was made. Treatment was offered but refused.
Figure 1. Magnetic resonance imaging revealed low-intensity lesions (arrows) on T1-weighted sequences (upper panel) and high-intensity lesions on short tau inversion recovery sequences (lower panel). The lesions have defined borders, no cortical disruption, and no mass effect, features suggesting that the lesions are benign. Lack of signal homogeneity suggests chronic lesions.
Based on this history, computed tomography of the thorax and abdomen was performed and showed mediastinal and hilar lymphadenopathy, small bilateral lung nodules, and osseous cystic areas in both iliac blades. Magnetic resonance imaging (MRI) showed numerous discrete lesions in both iliac bones (Figure 1). Biopsy study of iliac bone revealed preserved architecture with no evidence of malignancy or granulomas.
Radiography of the hands showed an osseous lytic lesion in the third proximal phalanx of the right hand. No other radiographic abnormalities were noted.
The clinical and radiographic features and the patient’s clinical course were consistent with osseous sarcoidosis. She was started on methotrexate and a low-dose corticosteroid and was symptom-free at 12-month follow-up. Follow-up MRI showed reduction in the lymphadenopathies and stabilization of the bone lesions.
SARCOIDOSIS AND BONE
Sarcoidosis is a systemic granulomatous disease that involves the lung in more than 90% of cases. Skeletal involvement has been reported in 1% to 14% of patients.1,2 Typical osseous involvement is cystic osteitis of the phalangeal bones of the hands and feet, but any part of the skeleton may be involved.3
Bone sarcoidosis is usually asymptomatic and is discovered incidentally. The diagnosis of sarcoidosis has usually been established clinically before bone lesions are detected on MRI. However, sarcoidosis-related bone lesions resembling bone metastases on MRI may be the initial presentation. The presence of intralesional fat has been described as a feature that excludes malignancy.
No treatment has been shown to be of benefit.4 Sarcoidosis is a diagnosis of exclusion and radiographic lytic bone features are not specific, so a neoplastic cause (such as primary osteoblastoma, metastasis, or multiple myeloma) must always be ruled out, as well as other bone conditions such as osteomyelitis or bone cyst.
References
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet 2014; 383:1155–1167.
James DG, Neville E, Carstairs LS. Bone and joint sarcoidosis. Semin Arthritis Rheum 1976; 6:53–81.
Moore SL, Kransdorf MJ, Schweitzer ME, Murphey MD, Babb JS. Can sarcoidosis and metastatic bone lesions be reliably differentiated on routine MRI? AJR Am J Roentgenol 2012; 198:1387–1393.
Hamoud S, Srour S, Fruchter O, Vlodavsky E, Hayek T. Lytic bone lesion: presenting finding of sarcoidosis. Isr Med Assoc J 2010; 12:59–60.
References
Valeyre D, Prasse A, Nunes H, Uzunhan Y, Brillet PY, Müller-Quernheim J. Sarcoidosis. Lancet 2014; 383:1155–1167.
James DG, Neville E, Carstairs LS. Bone and joint sarcoidosis. Semin Arthritis Rheum 1976; 6:53–81.
Moore SL, Kransdorf MJ, Schweitzer ME, Murphey MD, Babb JS. Can sarcoidosis and metastatic bone lesions be reliably differentiated on routine MRI? AJR Am J Roentgenol 2012; 198:1387–1393.
Hamoud S, Srour S, Fruchter O, Vlodavsky E, Hayek T. Lytic bone lesion: presenting finding of sarcoidosis. Isr Med Assoc J 2010; 12:59–60.
sarcoidosis, lung nodules, bone metastases, lytic osseous metastases, Anahy Brandy-Garcia, Ivan Cabezas-Rodriguez, Luis Caminal-Montero, Carlos Suarez-Cuervo, Pilar Redondo-Buil
Legacy Keywords
sarcoidosis, lung nodules, bone metastases, lytic osseous metastases, Anahy Brandy-Garcia, Ivan Cabezas-Rodriguez, Luis Caminal-Montero, Carlos Suarez-Cuervo, Pilar Redondo-Buil
Raynaud phenomenon is an overactive vascular response to cold and emotional stress that results in cutaneous color changes and sensory symptoms of the digits (Figure 1). It can occur in isolation as primary Raynaud phenomenon or secondary to another disease process. It is thought to be triggered by a heightened sympathetic vasoconstrictive response of small arteriovenous anastomoses in the fingers, toes, ears, and tip of the nose. These structures play a key role in maintaining a stable core body temperature by cutaneous thermoregulation.1
Figure 1. (A) White digits with intense vasoconstriction in Raynaud phenomenon; (B) blue digits with
hypoxemic venous stasis; (C) red digits with hyperemic reperfusion.
Secondary Raynaud phenomenon can be seen with a wide array of systemic conditions as well as environmental and drug exposures. It is a frequent feature of autoimmune rheumatic conditions such as systemic sclerosis, mixed connective tissue disease, systemic lupus erythematosus, and dermatomyositis. Less commonly, cryoproteinemias, paraneoplastic syndromes, hypothyroidism, and carpal tunnel syndrome can be associated with or cause Raynaud phenomenon. Vibratory trauma (eg, from using a jackhammer) and drugs (eg, vasopressors, stimulants, ergots, chemotherapeutic agents) can also cause Raynaud phenomenon.1
A variety of disorders that cause vasospasm or vascular occlusion of the peripheral circulation can mimic typical Raynaud phenomenon, including peripheral nerve injury,2 complex regional pain syndrome,3 occlusive vascular disease, vasculitis, acrocyanosis,4 and thoracic outlet syndrome.
The prevalence of Raynaud phenomenon is not exactly known, in part due to geographic differences in climate and variation in methods of assessment. However, a 2015 systematic review and meta-analysis of primary Raynaud phenomenon determined a pooled prevalence of 4.85% (95% confidence interval [CI] 2.08%–8.71%) in the general population.5 Accordingly, accurate identification and management of this condition is a useful skill for the internist.
COLD SENSITIVITY AND COLOR CHANGES
Because there are no confirmatory diagnostic tests for this condition, there are no formal diagnostic criteria. However, many experts agree that Raynaud phenomenon can be diagnosed clinically when patients report:
Unusual sensitivity of the fingers to cold, manifesting as pain or paresthesia (eg, tingling, pricking, numbness), and
Color changes of the fingers when exposed to cold, specifically pale white or blue-black, or both.6
Provocative testing such as submerging patients’ hands in cold water is not recommended, as it is distressing to the patient and inconsistent in triggering an event.
Pain is a symptom of critical digital ischemia.
The skin color changes are due to rapid alterations in blood flow in digital skin. The pale white is due to markedly reduced or absent flow secondary to intense vasoconstriction, the blue-black is due to hypoxemic venous stasis, and the red blush is due to hyperemic reperfusion (Figure 1). However, not all patients have all 3 phases of the classic triphasic color changes, and color changes may not follow a set sequence.
Raynaud phenomenon can also occur in other areas of the body that have thermoregulatory vessels, such as the toes, ears, nipples, tongue, and nose. While some patients with Raynaud phenomenon have a finger that is more sensitive than the others, repeated isolated single-digit or asymmetric events without typical progression to all fingers suggest a secondary local structural disease requiring further investigation (see below).
Symptoms related to Raynaud often mimic sensory changes including paresthesias, numbness, aching, and clumsiness of the hand. Abnormal vascular reactivity has been implicated as a causative factor in several disorders, such as migraine headache, preeclampsia, and variant angina. While case reports, case series, and some controlled studies have linked Raynaud phenomenon and these conditions, there is no solid evidence of a systemic vasospastic disorder in patients with primary Raynaud phenomenon.
Raynaud phenomenon is triggered by more than just a cold ambient temperature. Provocation can occur during movement from warmer to relatively cooler temperatures, as well as during episodes of elevated sympathetic activity (eg, emotional distress or fear). In fact, maintaining full body warmth as well as emotional equilibrium are the most important strategies to reduce the frequency of attacks.
PRIMARY VS SECONDARY RAYNAUD PHENOMENON
To distinguish between primary and secondary Raynaud phenomenon, a careful history and physical examination are paramount.
Primary Raynaud phenomenon
In uncomplicated primary Raynaud phenomenon, the episodes typically last 15 to 20 minutes after rewarming and usually start in a single finger and spread to other digits symmetrically and bilaterally.7 The thumb is often spared, and ischemic digital ulcers do not occur. Vasoconstrictive episodes are mild.
Females under age 20 are most commonly affected. In our experience, a young woman with the above clinical picture, no signs or symptoms suggestive of connective tissue disease (see below), and normal nailfold capillaries can be diagnosed as having primary Raynaud phenomenon without any further workup.
Careful clinical follow-up is recommended, because if an occult secondary process is indeed present, most patients will begin to show additional symptoms or signs of it within 2 years of the onset of Raynaud phenomenon.
Should a clinician be unfamiliar with nailfold capillary examination, or if symptoms (eg, fatigue or arthralgia) or signs (eg, rash, arthritis) suggestive of connective tissue disease are present, referral to a rheumatologist for further evaluation is appropriate. Results of further diagnostic testing dictated by the history and physical such as a screening antinuclear antibody test can be sent before referral.
Secondary Raynaud phenomenon
Several clinical features suggest secondary Raynaud phenomenon and warrant referral to a rheumatologist:
Age 20 or older at onset
Frequent severe vasoconstrictive episodes
Male sex
Thumb involvement
Figure 2. (A) Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.
Signs of an autoimmune rheumatic disease, eg, sclerodactyly, cutaneous or mucosal matted telangiectasia, inflammatory arthritis, an abnormal lung examination, severe digital ischemia with ulceration or gangrene, or nailfold capillary dilation or dropout (Figure 2)8
Isolated single-limb or 1-finger ischemic events, seen in macrovascular occlusive disease or inflammatory disease mimicking Raynaud phenomenon (eg, atherosclerosis, vasculitis); when isolated acute ischemic events occur in the upper or lower extremity, a further workup is necessary.
Figure 3 shows our approach to evaluation.
NONPHARMACOLOGIC THERAPY
Figure 3. Our approach to diagnosis of Raynaud phenomenon and differentiating primary from secondary Raynaud phenomenon.
Cold avoidance and stress management are first-line therapies for preventing Raynaud attacks and must be part of any treatment strategy. Digital arteries and thermoregulatory vessels of the skin are predominantly under sympathetic adrenergic control, so temperature changes and emotional stressors trigger vasoconstriction. Patients should be counseled to:
Keep the whole body warm. Patients should wear multiple layers of clothing, a hat, warm gloves, and warm socks. Commercially available hand-warmers can help, especially for patients who live in cold climates.
Learn to avoid or manage stress. Good communication, attention to the patient’s needs, and regular follow-up for reassurance are paramount. For some patients, psychotropic medications to manage mood may help. Behavioral approaches have been suggested for acute stress management. One approach, autogenic training, is a form of relaxation with temperature biofeedback in which finger temperature data are provided to patients to help them learn to relax by monitoring their internal states and changes in temperature. However, there are no strong data to support the routine use of this technique or the use of one behavioral approach over another. Trials have generally been of low quality and limited by small sample size.9
Stop smoking!10
Stop a Raynaud attack should one occur, eg, place the hands under warm water or in a warm part of the body, such as under legs when sitting. This can help speed recovery.
In addition, the physician should:
Eliminate vasoconstricting agents such as nonselective beta-blockers, ergots, triptans, and amphetamines.
PHARMACOLOGIC THERAPY
For many patients, nonpharmacologic interventions are enough to decrease the severity and frequency of attacks. However, if Raynaud phenomenon continues to negatively affect quality of life, drug therapy can be added (Table 1).
Calcium channel blockers
Calcium channel blockers are first-line agents for both primary and secondary Raynaud phenomenon that does not adequately respond to nonpharmacologic interventions. These agents are effective, available, and reasonably inexpensive.
Dihydropyridine calcium channel blockers such as nifedipine and amlodipine are commonly used. Both drugs are acceptable options, though some patients may respond better to one than the other in terms of symptoms and side effects. Nondihydropyridines such as diltiazem can also be used, but they have less potent vasodilatory effects because they are less selective for vascular smooth muscle.
These medications should be started at the lowest dose and titrated up over several weeks as tolerated to achieve their maximal effect. Intermittent therapy (eg, during the winter months only) is reasonable for primary Raynaud without risk of digital ulceration, as relief of symptoms and improvement in quality of life are the main indications for therapy in this circumstance.
A 2016 Cochrane review and meta-analysis of the use of calcium channel blockers to treat primary Raynaud phenomenon included 7 randomized controlled trials with 296 patients treated with either nifedipine or nicardipine.11 There was moderate-quality evidence that these drugs minimally decreased the frequency of attacks (standardized mean difference of 0.23; 95% CI 0.08–0.38, P = .003). This translated to 1.72 fewer attacks per week with treatment than with no pharmacologic therapy (95% CI 0.60–2.84). When analyzed individually, only nifedipine was effective; nicardipine did not decrease the frequency of attacks.
Unfortunately, calcium channel blockers failed to decrease the severity of attacks (according to unvalidated severity scoring systems) or make any differences in physiologic measurement outcomes. Attacks were not completely eliminated, just less frequent than before treatment.11
Most commonly reported side effects included headache, flushing, hypotension, edema, and, rarely, gastrointestinal reflux. Use of these medications may be limited by hypotension.
The review was limited by the small sample size, short duration of treatment, and relatively low doses of calcium channel blockers used in the available studies.11
A 2005 meta-analysis also indicated a statistically significant decrease of 2.8 to 5 attacks per week with nifedipine treatment, though this study also included some patients with secondary Raynaud phenomenon.12
Phosphodiesterase type 5 inhibitors
When calcium channel blockers do not adequately control symptoms, phosphodiesterase type 5 (PDE5) inhibitors can be added or substituted. These medications work by preventing breakdown of cyclic guanosine monophosphate, which induces relaxation in vascular smooth muscle and vasodilation.
Sildenafil can be started at a low dose (20 mg daily) and up-titrated to the maximum dose (20 mg 3 times daily) as tolerated.
A 2014 meta-analysis of 6 randomized controlled trials included 244 patients with secondary Raynaud phenomenon treated with sildenafil, tadalafil, or vardenafil.13 These drugs decreased the daily frequency of attacks by about 0.5 per day vs placebo (–0.49, 95% CI –0.71 to –0.28, P < .0001). PDE5 inhibitors also decreased the severity of attacks (based on the Raynaud’s Condition Score, a popular scoring system) and the duration of attacks by a statistically significant amount.
Almost all patients in these 6 trials were on PDE5 monotherapy. Data on the cumulative benefit of calcium channel blocker and PDE5 inhibitor combination therapy are not yet available. Not all patients tolerate combination therapy, as it can cause symptomatic hypotension, but it can be a successful option in some.
There are also no data showing that either calcium channel blockers or PDE5 inhibitors are superior, though the former are less expensive. A small double-blind, randomized, crossover study of udenafil vs amlodipine in the treatment of secondary Raynaud phenomenon showed that both medications significantly decreased the frequency of attacks and had comparable efficacy.14
Cost and insurance coverage. We have generally been successful in obtaining coverage for this off-label use of PDE5 inhibitors, though additional effort may be required. No drug (not even a calcium channel blocker) is approved by the US Food and Drug Administration for use in Raynaud phenomenon. In our experience, a letter of appeal outlining the rationale for use and citing supporting publications can lead to successful coverage of a medication. If the drug is still not approved, the patient either pays for it out of pocket or another agent is selected. In certain circumstances, pharmaceutical companies may provide prescription assistance for compassionate use of these drugs in Raynaud phenomenon, although this also takes letter-writing, phone calls, or both on the part of the physician.
Topical nitrates
Patients who have an unsatisfactory response to calcium channel blockers with or without PDE5 inhibitors can try topical nitrates, available as sustained-release transdermal patches, tapes, creams, gels, and ointments.
Small trials have noted slight improvement in the Raynaud Condition Score15 and finger temperature16 with these therapies. Another trial noted decreased frequency of attacks and symptoms with the use of sustained-release glyceryl trinitrate patches, but use was limited by intolerable headache.17
In our experience, topical nitrates are most helpful for patients who have 1 or a few digits that are more severely affected than the others, and we reserve these drugs for this indication. Localized vasodilation can provide targeted rapid relief of more ischemic areas.
Topical nitroglycerin can be applied to the base of the ischemic digit for 6 to 12 hours. Preparations vary, and patients should be closely monitored for dose response and tolerance.
Combining a topical nitrate with a calcium channel blocker is safe, but the use of a nitrate with a PDE5 inhibitor is contraindicated due to the risk of hypotension. The use of topical nitrates may be limited by systemic side effects such as headache and flushing and a lack of benefit over time.
Other therapies
If the aforementioned agents are not tolerated or not effective, there is limited evidence that other therapies reduce the frequency and sometimes the severity of attacks. These are not first-line agents but may be tried when other options have been exhausted and symptoms persist. There are no data to support combining these therapies, but in our experience doing so may help some patients in whom drug-drug interactions are not prohibitive.
Prazosin, an alpha-1-adrenergic receptor antagonist, was reported to improve Raynaud phenomenon in 2 small studies in the 1980s, but we do not use it since better options are available. In addition, the vasoactive blood vessels involved do not have alpha-1 receptors, so there is no theoretical basis for using prazosin.18,19
Fluoxetine, a selective serotonin reuptake inhibitor, reduced the frequency and severity of attacks in a 6-week crossover study with nifedipine.20
Losartan, an angiotensin II receptor blocker, also reduced the severity and frequency of attacks when compared with nifedipine.21
Pentoxifylline, a nonselective phosphodiesterase inhibitor, showed some benefit in a trial in 11 patients with primary Raynaud.22
Atorvastatin, a lipid-lowering drug, reduced the number of digital ulcers in patients with secondary Raynaud already on first-line vasodilatory therapy, and might be added in this situation.23
Botulinum toxin A injections have some data to support their use, but evidence is based on uncontrolled case series.24 A controlled trial in scleroderma patients with severe Raynaud phenomenon found botulinum toxin to be no better than placebo.25
Prostacyclin preparations are available. Intermittent intravenous doses of prostacyclin analogues over several days can be used in resistant cases. Oral prostacyclin agents have not shown consistent benefit. New prostacyclin receptor agonists are under investigation.
Overall, we move to other options only in patients with persistent symptoms that impair quality of life, or in patients with recurrent digital ischemic lesions that have not responded to calcium channel blockers and PDE5 inhibitors or nitrates, either alone or in combination.
DIGITAL ULCERATION AND ACUTE DIGITAL ISCHEMIC CRISIS
Patients with secondary Raynaud phenomenon may be at risk of recurrent digital ulceration and acute digital ischemia with gangrene. These patients should be comanaged with a rheumatologist so that the underlying disease process is fully addressed. Digital ulcers should be inspected closely for signs of infection, which may require treatment with antibiotics.
Acute digital ischemia is a medical emergency and should prompt inpatient admission with warming, emotional regulation, and pain control (often with narcotics) to decrease sympathetic vasoconstriction. These patients require aggressive vasodilatory therapy to reverse the ischemic event.
A short-acting calcium channel blocker or combination therapy with a calcium channel blocker and a PDE5 inhibitor or topical nitrate should be started. If there is no benefit, then transient intravenous vasodilatory therapy with a prostacyclin (epoprostenol) or localized digital sympathectomy is used to prevent digital loss.
The endothelin receptor inhibitor bosentan has been shown to decrease recurrent digital ulcers in patients with scleroderma, and while bosentan does not decrease the frequency of Raynaud attacks, it can be used in this select group to prevent new digital ulcers.
Treatment options may be limited by insurance coverage or access to intravenous infusions.
TAKE-HOME RECOMMENDATIONS
For many patients with primary or secondary Raynaud phenomenon, nonpharmacologic interventions are all that are required to decrease the frequency of attacks and improve quality of life. The goal should not be to eliminate attacks completely, as aggressive drug treatment may cause more harm than benefit. From our perspective, the goals of treatment should be to improve quality of life and prevent ischemic complications.
Pharmacologic therapies should be added only if attacks remain poorly controlled with incapacitating symptoms, or if the patient has digital ischemic ulcers. Calcium channel blockers are first-line therapy, given proven efficacy and low cost, and should be titrated to the maximum tolerated dose before adding or substituting other agents.
References
Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med 2016; 375:556–565.
Irwin MS, Gilbert SE, Terenghi G, Smith RW, Green CJ. Cold intolerance following peripheral nerve injury. Natural history and factors predicting severity of symptoms. J Hand Surg Br 1997; 22:308–316.
Wasner G. Vasomotor disturbances in complex regional pain syndrome—a review. Pain Med 2010; 11:1267–1273.
Kurklinsky AK, Miller VM, Rooke TW. Acrocyanosis: the Flying Dutchman. Vasc Med 2011; 16:288–301.
Garner R, Kumari R, Lanyon P, Doherty M, Zhang W. Prevalence, risk factors and associations of primary Raynaud’s phenomenon: systematic review and meta-analysis of observational studies. BMJ Open 2015; 5:e006389.
Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1008.
Chikura B, Moore TL, Manning JB, Vail A, Herrick AL. Sparing of the thumb in Raynaud’s phenomenon. Rheumatology (Oxford) 2008; 47:219–221.
Kallenerg CG. Early detection of connective tissue disease in patients with Raynaud’s phenomenon. Rheum Dis Clin North Am 1990; 16:11–30.
Kwakkenbos L, Thombs BD. Non-drug approaches to treating Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA, editors. Raynaud’s Phenomenon. A Guide to Pathogenesis and Treatment. New York: Springer Science+Business Media, 2015:299–313.
Goodfield MJ, Hume A, Rowell NR. The acute effects of cigarette smoking on cutaneous blood flow in smoking and non-smoking subjects with and without Raynaud’s phenomenon. Br J Rheumatol 1990; 29:89–91.
Ennis H, Hughes M, Anderson ME, Wilkinson J, Herrick AL. Calcium channel blockers for primary Raynaud’s phenomenon. Cochrane Database Sys Review 2016; 2:CD002069.
Thompson AE, Pope JE. Calcium channel blockers for primary Raynaud’s phenomenon: a meta-analysis. Rheumatology (Oxford) 2005; 44:145–150.
Roustit M, Blaise S, Allanore Y, Carpentier P, Caglayan E, Cracowski J. Phosphodiesterase-5 inhibitors for the treatment of secondary Raynaud’s phenomenon: systematic review and meta-analysis of randomized trials. Ann Rheum Dis 2013; 72:1696–1699.
Lee EY, Park JK, Lee W, et al. Head-to-head comparison of udenafil vs amlodipine in the treatment of secondary Raynaud's phenomenon: a double-blind, randomized, cross-over study. Rheumatology (Oxford) 2014; 53:658–664.
Chung L, Shapiro L, Fiorentino D, et al. MQX-503, a novel formulation of nitroglycerin, improves the severity of Raynaud’s phenomenon: a randomized, controlled trial. Arthritis Rheum 2009; 60:870–877.
Kan C, Akimoto S, Abe M, Okada K, Ishikawa O. Preliminary thermographic evaluation of a new nitroglycerine tape on the peripheral circulatory disturbance in systemic sclerosis. Ann Rheum Dis 2002; 61:177–179.
Teh LS, Manning J, Moore T, Tully MP, O’Reilly D, Jayson MI. Sustained-release transdermal glyceryl trinitrate patches as a treatment for primary and secondary Raynaud’s phenomenon. Br J Rheumatol 1995; 34:636–641.
Russell IJ, Lessard JA. Prazosin treatment of Raynaud’s phenomenon: a double blind single crossover study. J Rheumatol 1985; 12:94–98.
Wollersheim H, Thien T, Fennis J, van Elteren P, van ‘t Laar A. Double-blind, placebo-controlled study of prazosin in Raynaud’s phenomenon. Clin Pharmacol Ther 1986; 40:219–225.
Coleiro B, Marshall SE, Denton CP, et al. Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford) 2001; 40:1038–1043.
Didazio M, Denton CP, Smith R, et al. Losartan therapy for Raynaud’s phenomenon and scleroderma: clinical and biochemical findings in a fifteen-week randomized, parallel-group, controlled trial. Arthritis Rheum 1999; 42:2646–2655.
Neirotti M, Longo F, Molaschi M, Macchione C, Pernigotti L. Functional vascular disorders: treatment with pentoxifylline. Angiology 1987; 38:575–580.
Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35:1801–1808.
Iorio ML, Masden DL, Higgins JP. Botulinum toxin A treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum 2012; 41: 599–603.
Bello RJ, Cooney CM, Melamed E, et al. The therapeutic efficacy of botulinum toxin in treating scleroderma-associated Raynaud’s phenomenon: a randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol 2017. Epub ahead of print.
Samantha C. Shapiro, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Postdoctoral Fellow, Johns Hopkins Division of Rheumatology, Baltimore, MD
Fredrick M. Wigley, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Martha McCrory Professor of Medicine, Johns Hopkins Division of Rheumatology, Baltimore, MD
Address: Samantha C. Shapiro, MD, Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; 5200 Eastern Avenue, Suite 4100, Mason F. Lord Building, Center Tower, Baltimore, MD 21224; sshapi28@jhmi.edu
Samantha C. Shapiro, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Postdoctoral Fellow, Johns Hopkins Division of Rheumatology, Baltimore, MD
Fredrick M. Wigley, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Martha McCrory Professor of Medicine, Johns Hopkins Division of Rheumatology, Baltimore, MD
Address: Samantha C. Shapiro, MD, Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; 5200 Eastern Avenue, Suite 4100, Mason F. Lord Building, Center Tower, Baltimore, MD 21224; sshapi28@jhmi.edu
Author and Disclosure Information
Samantha C. Shapiro, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Postdoctoral Fellow, Johns Hopkins Division of Rheumatology, Baltimore, MD
Fredrick M. Wigley, MD Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; Martha McCrory Professor of Medicine, Johns Hopkins Division of Rheumatology, Baltimore, MD
Address: Samantha C. Shapiro, MD, Department of Medicine, Division of Rheumatology, Johns Hopkins University School of Medicine; 5200 Eastern Avenue, Suite 4100, Mason F. Lord Building, Center Tower, Baltimore, MD 21224; sshapi28@jhmi.edu
Raynaud phenomenon is an overactive vascular response to cold and emotional stress that results in cutaneous color changes and sensory symptoms of the digits (Figure 1). It can occur in isolation as primary Raynaud phenomenon or secondary to another disease process. It is thought to be triggered by a heightened sympathetic vasoconstrictive response of small arteriovenous anastomoses in the fingers, toes, ears, and tip of the nose. These structures play a key role in maintaining a stable core body temperature by cutaneous thermoregulation.1
Figure 1. (A) White digits with intense vasoconstriction in Raynaud phenomenon; (B) blue digits with
hypoxemic venous stasis; (C) red digits with hyperemic reperfusion.
Secondary Raynaud phenomenon can be seen with a wide array of systemic conditions as well as environmental and drug exposures. It is a frequent feature of autoimmune rheumatic conditions such as systemic sclerosis, mixed connective tissue disease, systemic lupus erythematosus, and dermatomyositis. Less commonly, cryoproteinemias, paraneoplastic syndromes, hypothyroidism, and carpal tunnel syndrome can be associated with or cause Raynaud phenomenon. Vibratory trauma (eg, from using a jackhammer) and drugs (eg, vasopressors, stimulants, ergots, chemotherapeutic agents) can also cause Raynaud phenomenon.1
A variety of disorders that cause vasospasm or vascular occlusion of the peripheral circulation can mimic typical Raynaud phenomenon, including peripheral nerve injury,2 complex regional pain syndrome,3 occlusive vascular disease, vasculitis, acrocyanosis,4 and thoracic outlet syndrome.
The prevalence of Raynaud phenomenon is not exactly known, in part due to geographic differences in climate and variation in methods of assessment. However, a 2015 systematic review and meta-analysis of primary Raynaud phenomenon determined a pooled prevalence of 4.85% (95% confidence interval [CI] 2.08%–8.71%) in the general population.5 Accordingly, accurate identification and management of this condition is a useful skill for the internist.
COLD SENSITIVITY AND COLOR CHANGES
Because there are no confirmatory diagnostic tests for this condition, there are no formal diagnostic criteria. However, many experts agree that Raynaud phenomenon can be diagnosed clinically when patients report:
Unusual sensitivity of the fingers to cold, manifesting as pain or paresthesia (eg, tingling, pricking, numbness), and
Color changes of the fingers when exposed to cold, specifically pale white or blue-black, or both.6
Provocative testing such as submerging patients’ hands in cold water is not recommended, as it is distressing to the patient and inconsistent in triggering an event.
Pain is a symptom of critical digital ischemia.
The skin color changes are due to rapid alterations in blood flow in digital skin. The pale white is due to markedly reduced or absent flow secondary to intense vasoconstriction, the blue-black is due to hypoxemic venous stasis, and the red blush is due to hyperemic reperfusion (Figure 1). However, not all patients have all 3 phases of the classic triphasic color changes, and color changes may not follow a set sequence.
Raynaud phenomenon can also occur in other areas of the body that have thermoregulatory vessels, such as the toes, ears, nipples, tongue, and nose. While some patients with Raynaud phenomenon have a finger that is more sensitive than the others, repeated isolated single-digit or asymmetric events without typical progression to all fingers suggest a secondary local structural disease requiring further investigation (see below).
Symptoms related to Raynaud often mimic sensory changes including paresthesias, numbness, aching, and clumsiness of the hand. Abnormal vascular reactivity has been implicated as a causative factor in several disorders, such as migraine headache, preeclampsia, and variant angina. While case reports, case series, and some controlled studies have linked Raynaud phenomenon and these conditions, there is no solid evidence of a systemic vasospastic disorder in patients with primary Raynaud phenomenon.
Raynaud phenomenon is triggered by more than just a cold ambient temperature. Provocation can occur during movement from warmer to relatively cooler temperatures, as well as during episodes of elevated sympathetic activity (eg, emotional distress or fear). In fact, maintaining full body warmth as well as emotional equilibrium are the most important strategies to reduce the frequency of attacks.
PRIMARY VS SECONDARY RAYNAUD PHENOMENON
To distinguish between primary and secondary Raynaud phenomenon, a careful history and physical examination are paramount.
Primary Raynaud phenomenon
In uncomplicated primary Raynaud phenomenon, the episodes typically last 15 to 20 minutes after rewarming and usually start in a single finger and spread to other digits symmetrically and bilaterally.7 The thumb is often spared, and ischemic digital ulcers do not occur. Vasoconstrictive episodes are mild.
Females under age 20 are most commonly affected. In our experience, a young woman with the above clinical picture, no signs or symptoms suggestive of connective tissue disease (see below), and normal nailfold capillaries can be diagnosed as having primary Raynaud phenomenon without any further workup.
Careful clinical follow-up is recommended, because if an occult secondary process is indeed present, most patients will begin to show additional symptoms or signs of it within 2 years of the onset of Raynaud phenomenon.
Should a clinician be unfamiliar with nailfold capillary examination, or if symptoms (eg, fatigue or arthralgia) or signs (eg, rash, arthritis) suggestive of connective tissue disease are present, referral to a rheumatologist for further evaluation is appropriate. Results of further diagnostic testing dictated by the history and physical such as a screening antinuclear antibody test can be sent before referral.
Secondary Raynaud phenomenon
Several clinical features suggest secondary Raynaud phenomenon and warrant referral to a rheumatologist:
Age 20 or older at onset
Frequent severe vasoconstrictive episodes
Male sex
Thumb involvement
Figure 2. (A) Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.
Signs of an autoimmune rheumatic disease, eg, sclerodactyly, cutaneous or mucosal matted telangiectasia, inflammatory arthritis, an abnormal lung examination, severe digital ischemia with ulceration or gangrene, or nailfold capillary dilation or dropout (Figure 2)8
Isolated single-limb or 1-finger ischemic events, seen in macrovascular occlusive disease or inflammatory disease mimicking Raynaud phenomenon (eg, atherosclerosis, vasculitis); when isolated acute ischemic events occur in the upper or lower extremity, a further workup is necessary.
Figure 3 shows our approach to evaluation.
NONPHARMACOLOGIC THERAPY
Figure 3. Our approach to diagnosis of Raynaud phenomenon and differentiating primary from secondary Raynaud phenomenon.
Cold avoidance and stress management are first-line therapies for preventing Raynaud attacks and must be part of any treatment strategy. Digital arteries and thermoregulatory vessels of the skin are predominantly under sympathetic adrenergic control, so temperature changes and emotional stressors trigger vasoconstriction. Patients should be counseled to:
Keep the whole body warm. Patients should wear multiple layers of clothing, a hat, warm gloves, and warm socks. Commercially available hand-warmers can help, especially for patients who live in cold climates.
Learn to avoid or manage stress. Good communication, attention to the patient’s needs, and regular follow-up for reassurance are paramount. For some patients, psychotropic medications to manage mood may help. Behavioral approaches have been suggested for acute stress management. One approach, autogenic training, is a form of relaxation with temperature biofeedback in which finger temperature data are provided to patients to help them learn to relax by monitoring their internal states and changes in temperature. However, there are no strong data to support the routine use of this technique or the use of one behavioral approach over another. Trials have generally been of low quality and limited by small sample size.9
Stop smoking!10
Stop a Raynaud attack should one occur, eg, place the hands under warm water or in a warm part of the body, such as under legs when sitting. This can help speed recovery.
In addition, the physician should:
Eliminate vasoconstricting agents such as nonselective beta-blockers, ergots, triptans, and amphetamines.
PHARMACOLOGIC THERAPY
For many patients, nonpharmacologic interventions are enough to decrease the severity and frequency of attacks. However, if Raynaud phenomenon continues to negatively affect quality of life, drug therapy can be added (Table 1).
Calcium channel blockers
Calcium channel blockers are first-line agents for both primary and secondary Raynaud phenomenon that does not adequately respond to nonpharmacologic interventions. These agents are effective, available, and reasonably inexpensive.
Dihydropyridine calcium channel blockers such as nifedipine and amlodipine are commonly used. Both drugs are acceptable options, though some patients may respond better to one than the other in terms of symptoms and side effects. Nondihydropyridines such as diltiazem can also be used, but they have less potent vasodilatory effects because they are less selective for vascular smooth muscle.
These medications should be started at the lowest dose and titrated up over several weeks as tolerated to achieve their maximal effect. Intermittent therapy (eg, during the winter months only) is reasonable for primary Raynaud without risk of digital ulceration, as relief of symptoms and improvement in quality of life are the main indications for therapy in this circumstance.
A 2016 Cochrane review and meta-analysis of the use of calcium channel blockers to treat primary Raynaud phenomenon included 7 randomized controlled trials with 296 patients treated with either nifedipine or nicardipine.11 There was moderate-quality evidence that these drugs minimally decreased the frequency of attacks (standardized mean difference of 0.23; 95% CI 0.08–0.38, P = .003). This translated to 1.72 fewer attacks per week with treatment than with no pharmacologic therapy (95% CI 0.60–2.84). When analyzed individually, only nifedipine was effective; nicardipine did not decrease the frequency of attacks.
Unfortunately, calcium channel blockers failed to decrease the severity of attacks (according to unvalidated severity scoring systems) or make any differences in physiologic measurement outcomes. Attacks were not completely eliminated, just less frequent than before treatment.11
Most commonly reported side effects included headache, flushing, hypotension, edema, and, rarely, gastrointestinal reflux. Use of these medications may be limited by hypotension.
The review was limited by the small sample size, short duration of treatment, and relatively low doses of calcium channel blockers used in the available studies.11
A 2005 meta-analysis also indicated a statistically significant decrease of 2.8 to 5 attacks per week with nifedipine treatment, though this study also included some patients with secondary Raynaud phenomenon.12
Phosphodiesterase type 5 inhibitors
When calcium channel blockers do not adequately control symptoms, phosphodiesterase type 5 (PDE5) inhibitors can be added or substituted. These medications work by preventing breakdown of cyclic guanosine monophosphate, which induces relaxation in vascular smooth muscle and vasodilation.
Sildenafil can be started at a low dose (20 mg daily) and up-titrated to the maximum dose (20 mg 3 times daily) as tolerated.
A 2014 meta-analysis of 6 randomized controlled trials included 244 patients with secondary Raynaud phenomenon treated with sildenafil, tadalafil, or vardenafil.13 These drugs decreased the daily frequency of attacks by about 0.5 per day vs placebo (–0.49, 95% CI –0.71 to –0.28, P < .0001). PDE5 inhibitors also decreased the severity of attacks (based on the Raynaud’s Condition Score, a popular scoring system) and the duration of attacks by a statistically significant amount.
Almost all patients in these 6 trials were on PDE5 monotherapy. Data on the cumulative benefit of calcium channel blocker and PDE5 inhibitor combination therapy are not yet available. Not all patients tolerate combination therapy, as it can cause symptomatic hypotension, but it can be a successful option in some.
There are also no data showing that either calcium channel blockers or PDE5 inhibitors are superior, though the former are less expensive. A small double-blind, randomized, crossover study of udenafil vs amlodipine in the treatment of secondary Raynaud phenomenon showed that both medications significantly decreased the frequency of attacks and had comparable efficacy.14
Cost and insurance coverage. We have generally been successful in obtaining coverage for this off-label use of PDE5 inhibitors, though additional effort may be required. No drug (not even a calcium channel blocker) is approved by the US Food and Drug Administration for use in Raynaud phenomenon. In our experience, a letter of appeal outlining the rationale for use and citing supporting publications can lead to successful coverage of a medication. If the drug is still not approved, the patient either pays for it out of pocket or another agent is selected. In certain circumstances, pharmaceutical companies may provide prescription assistance for compassionate use of these drugs in Raynaud phenomenon, although this also takes letter-writing, phone calls, or both on the part of the physician.
Topical nitrates
Patients who have an unsatisfactory response to calcium channel blockers with or without PDE5 inhibitors can try topical nitrates, available as sustained-release transdermal patches, tapes, creams, gels, and ointments.
Small trials have noted slight improvement in the Raynaud Condition Score15 and finger temperature16 with these therapies. Another trial noted decreased frequency of attacks and symptoms with the use of sustained-release glyceryl trinitrate patches, but use was limited by intolerable headache.17
In our experience, topical nitrates are most helpful for patients who have 1 or a few digits that are more severely affected than the others, and we reserve these drugs for this indication. Localized vasodilation can provide targeted rapid relief of more ischemic areas.
Topical nitroglycerin can be applied to the base of the ischemic digit for 6 to 12 hours. Preparations vary, and patients should be closely monitored for dose response and tolerance.
Combining a topical nitrate with a calcium channel blocker is safe, but the use of a nitrate with a PDE5 inhibitor is contraindicated due to the risk of hypotension. The use of topical nitrates may be limited by systemic side effects such as headache and flushing and a lack of benefit over time.
Other therapies
If the aforementioned agents are not tolerated or not effective, there is limited evidence that other therapies reduce the frequency and sometimes the severity of attacks. These are not first-line agents but may be tried when other options have been exhausted and symptoms persist. There are no data to support combining these therapies, but in our experience doing so may help some patients in whom drug-drug interactions are not prohibitive.
Prazosin, an alpha-1-adrenergic receptor antagonist, was reported to improve Raynaud phenomenon in 2 small studies in the 1980s, but we do not use it since better options are available. In addition, the vasoactive blood vessels involved do not have alpha-1 receptors, so there is no theoretical basis for using prazosin.18,19
Fluoxetine, a selective serotonin reuptake inhibitor, reduced the frequency and severity of attacks in a 6-week crossover study with nifedipine.20
Losartan, an angiotensin II receptor blocker, also reduced the severity and frequency of attacks when compared with nifedipine.21
Pentoxifylline, a nonselective phosphodiesterase inhibitor, showed some benefit in a trial in 11 patients with primary Raynaud.22
Atorvastatin, a lipid-lowering drug, reduced the number of digital ulcers in patients with secondary Raynaud already on first-line vasodilatory therapy, and might be added in this situation.23
Botulinum toxin A injections have some data to support their use, but evidence is based on uncontrolled case series.24 A controlled trial in scleroderma patients with severe Raynaud phenomenon found botulinum toxin to be no better than placebo.25
Prostacyclin preparations are available. Intermittent intravenous doses of prostacyclin analogues over several days can be used in resistant cases. Oral prostacyclin agents have not shown consistent benefit. New prostacyclin receptor agonists are under investigation.
Overall, we move to other options only in patients with persistent symptoms that impair quality of life, or in patients with recurrent digital ischemic lesions that have not responded to calcium channel blockers and PDE5 inhibitors or nitrates, either alone or in combination.
DIGITAL ULCERATION AND ACUTE DIGITAL ISCHEMIC CRISIS
Patients with secondary Raynaud phenomenon may be at risk of recurrent digital ulceration and acute digital ischemia with gangrene. These patients should be comanaged with a rheumatologist so that the underlying disease process is fully addressed. Digital ulcers should be inspected closely for signs of infection, which may require treatment with antibiotics.
Acute digital ischemia is a medical emergency and should prompt inpatient admission with warming, emotional regulation, and pain control (often with narcotics) to decrease sympathetic vasoconstriction. These patients require aggressive vasodilatory therapy to reverse the ischemic event.
A short-acting calcium channel blocker or combination therapy with a calcium channel blocker and a PDE5 inhibitor or topical nitrate should be started. If there is no benefit, then transient intravenous vasodilatory therapy with a prostacyclin (epoprostenol) or localized digital sympathectomy is used to prevent digital loss.
The endothelin receptor inhibitor bosentan has been shown to decrease recurrent digital ulcers in patients with scleroderma, and while bosentan does not decrease the frequency of Raynaud attacks, it can be used in this select group to prevent new digital ulcers.
Treatment options may be limited by insurance coverage or access to intravenous infusions.
TAKE-HOME RECOMMENDATIONS
For many patients with primary or secondary Raynaud phenomenon, nonpharmacologic interventions are all that are required to decrease the frequency of attacks and improve quality of life. The goal should not be to eliminate attacks completely, as aggressive drug treatment may cause more harm than benefit. From our perspective, the goals of treatment should be to improve quality of life and prevent ischemic complications.
Pharmacologic therapies should be added only if attacks remain poorly controlled with incapacitating symptoms, or if the patient has digital ischemic ulcers. Calcium channel blockers are first-line therapy, given proven efficacy and low cost, and should be titrated to the maximum tolerated dose before adding or substituting other agents.
Raynaud phenomenon is an overactive vascular response to cold and emotional stress that results in cutaneous color changes and sensory symptoms of the digits (Figure 1). It can occur in isolation as primary Raynaud phenomenon or secondary to another disease process. It is thought to be triggered by a heightened sympathetic vasoconstrictive response of small arteriovenous anastomoses in the fingers, toes, ears, and tip of the nose. These structures play a key role in maintaining a stable core body temperature by cutaneous thermoregulation.1
Figure 1. (A) White digits with intense vasoconstriction in Raynaud phenomenon; (B) blue digits with
hypoxemic venous stasis; (C) red digits with hyperemic reperfusion.
Secondary Raynaud phenomenon can be seen with a wide array of systemic conditions as well as environmental and drug exposures. It is a frequent feature of autoimmune rheumatic conditions such as systemic sclerosis, mixed connective tissue disease, systemic lupus erythematosus, and dermatomyositis. Less commonly, cryoproteinemias, paraneoplastic syndromes, hypothyroidism, and carpal tunnel syndrome can be associated with or cause Raynaud phenomenon. Vibratory trauma (eg, from using a jackhammer) and drugs (eg, vasopressors, stimulants, ergots, chemotherapeutic agents) can also cause Raynaud phenomenon.1
A variety of disorders that cause vasospasm or vascular occlusion of the peripheral circulation can mimic typical Raynaud phenomenon, including peripheral nerve injury,2 complex regional pain syndrome,3 occlusive vascular disease, vasculitis, acrocyanosis,4 and thoracic outlet syndrome.
The prevalence of Raynaud phenomenon is not exactly known, in part due to geographic differences in climate and variation in methods of assessment. However, a 2015 systematic review and meta-analysis of primary Raynaud phenomenon determined a pooled prevalence of 4.85% (95% confidence interval [CI] 2.08%–8.71%) in the general population.5 Accordingly, accurate identification and management of this condition is a useful skill for the internist.
COLD SENSITIVITY AND COLOR CHANGES
Because there are no confirmatory diagnostic tests for this condition, there are no formal diagnostic criteria. However, many experts agree that Raynaud phenomenon can be diagnosed clinically when patients report:
Unusual sensitivity of the fingers to cold, manifesting as pain or paresthesia (eg, tingling, pricking, numbness), and
Color changes of the fingers when exposed to cold, specifically pale white or blue-black, or both.6
Provocative testing such as submerging patients’ hands in cold water is not recommended, as it is distressing to the patient and inconsistent in triggering an event.
Pain is a symptom of critical digital ischemia.
The skin color changes are due to rapid alterations in blood flow in digital skin. The pale white is due to markedly reduced or absent flow secondary to intense vasoconstriction, the blue-black is due to hypoxemic venous stasis, and the red blush is due to hyperemic reperfusion (Figure 1). However, not all patients have all 3 phases of the classic triphasic color changes, and color changes may not follow a set sequence.
Raynaud phenomenon can also occur in other areas of the body that have thermoregulatory vessels, such as the toes, ears, nipples, tongue, and nose. While some patients with Raynaud phenomenon have a finger that is more sensitive than the others, repeated isolated single-digit or asymmetric events without typical progression to all fingers suggest a secondary local structural disease requiring further investigation (see below).
Symptoms related to Raynaud often mimic sensory changes including paresthesias, numbness, aching, and clumsiness of the hand. Abnormal vascular reactivity has been implicated as a causative factor in several disorders, such as migraine headache, preeclampsia, and variant angina. While case reports, case series, and some controlled studies have linked Raynaud phenomenon and these conditions, there is no solid evidence of a systemic vasospastic disorder in patients with primary Raynaud phenomenon.
Raynaud phenomenon is triggered by more than just a cold ambient temperature. Provocation can occur during movement from warmer to relatively cooler temperatures, as well as during episodes of elevated sympathetic activity (eg, emotional distress or fear). In fact, maintaining full body warmth as well as emotional equilibrium are the most important strategies to reduce the frequency of attacks.
PRIMARY VS SECONDARY RAYNAUD PHENOMENON
To distinguish between primary and secondary Raynaud phenomenon, a careful history and physical examination are paramount.
Primary Raynaud phenomenon
In uncomplicated primary Raynaud phenomenon, the episodes typically last 15 to 20 minutes after rewarming and usually start in a single finger and spread to other digits symmetrically and bilaterally.7 The thumb is often spared, and ischemic digital ulcers do not occur. Vasoconstrictive episodes are mild.
Females under age 20 are most commonly affected. In our experience, a young woman with the above clinical picture, no signs or symptoms suggestive of connective tissue disease (see below), and normal nailfold capillaries can be diagnosed as having primary Raynaud phenomenon without any further workup.
Careful clinical follow-up is recommended, because if an occult secondary process is indeed present, most patients will begin to show additional symptoms or signs of it within 2 years of the onset of Raynaud phenomenon.
Should a clinician be unfamiliar with nailfold capillary examination, or if symptoms (eg, fatigue or arthralgia) or signs (eg, rash, arthritis) suggestive of connective tissue disease are present, referral to a rheumatologist for further evaluation is appropriate. Results of further diagnostic testing dictated by the history and physical such as a screening antinuclear antibody test can be sent before referral.
Secondary Raynaud phenomenon
Several clinical features suggest secondary Raynaud phenomenon and warrant referral to a rheumatologist:
Age 20 or older at onset
Frequent severe vasoconstrictive episodes
Male sex
Thumb involvement
Figure 2. (A) Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.
Signs of an autoimmune rheumatic disease, eg, sclerodactyly, cutaneous or mucosal matted telangiectasia, inflammatory arthritis, an abnormal lung examination, severe digital ischemia with ulceration or gangrene, or nailfold capillary dilation or dropout (Figure 2)8
Isolated single-limb or 1-finger ischemic events, seen in macrovascular occlusive disease or inflammatory disease mimicking Raynaud phenomenon (eg, atherosclerosis, vasculitis); when isolated acute ischemic events occur in the upper or lower extremity, a further workup is necessary.
Figure 3 shows our approach to evaluation.
NONPHARMACOLOGIC THERAPY
Figure 3. Our approach to diagnosis of Raynaud phenomenon and differentiating primary from secondary Raynaud phenomenon.
Cold avoidance and stress management are first-line therapies for preventing Raynaud attacks and must be part of any treatment strategy. Digital arteries and thermoregulatory vessels of the skin are predominantly under sympathetic adrenergic control, so temperature changes and emotional stressors trigger vasoconstriction. Patients should be counseled to:
Keep the whole body warm. Patients should wear multiple layers of clothing, a hat, warm gloves, and warm socks. Commercially available hand-warmers can help, especially for patients who live in cold climates.
Learn to avoid or manage stress. Good communication, attention to the patient’s needs, and regular follow-up for reassurance are paramount. For some patients, psychotropic medications to manage mood may help. Behavioral approaches have been suggested for acute stress management. One approach, autogenic training, is a form of relaxation with temperature biofeedback in which finger temperature data are provided to patients to help them learn to relax by monitoring their internal states and changes in temperature. However, there are no strong data to support the routine use of this technique or the use of one behavioral approach over another. Trials have generally been of low quality and limited by small sample size.9
Stop smoking!10
Stop a Raynaud attack should one occur, eg, place the hands under warm water or in a warm part of the body, such as under legs when sitting. This can help speed recovery.
In addition, the physician should:
Eliminate vasoconstricting agents such as nonselective beta-blockers, ergots, triptans, and amphetamines.
PHARMACOLOGIC THERAPY
For many patients, nonpharmacologic interventions are enough to decrease the severity and frequency of attacks. However, if Raynaud phenomenon continues to negatively affect quality of life, drug therapy can be added (Table 1).
Calcium channel blockers
Calcium channel blockers are first-line agents for both primary and secondary Raynaud phenomenon that does not adequately respond to nonpharmacologic interventions. These agents are effective, available, and reasonably inexpensive.
Dihydropyridine calcium channel blockers such as nifedipine and amlodipine are commonly used. Both drugs are acceptable options, though some patients may respond better to one than the other in terms of symptoms and side effects. Nondihydropyridines such as diltiazem can also be used, but they have less potent vasodilatory effects because they are less selective for vascular smooth muscle.
These medications should be started at the lowest dose and titrated up over several weeks as tolerated to achieve their maximal effect. Intermittent therapy (eg, during the winter months only) is reasonable for primary Raynaud without risk of digital ulceration, as relief of symptoms and improvement in quality of life are the main indications for therapy in this circumstance.
A 2016 Cochrane review and meta-analysis of the use of calcium channel blockers to treat primary Raynaud phenomenon included 7 randomized controlled trials with 296 patients treated with either nifedipine or nicardipine.11 There was moderate-quality evidence that these drugs minimally decreased the frequency of attacks (standardized mean difference of 0.23; 95% CI 0.08–0.38, P = .003). This translated to 1.72 fewer attacks per week with treatment than with no pharmacologic therapy (95% CI 0.60–2.84). When analyzed individually, only nifedipine was effective; nicardipine did not decrease the frequency of attacks.
Unfortunately, calcium channel blockers failed to decrease the severity of attacks (according to unvalidated severity scoring systems) or make any differences in physiologic measurement outcomes. Attacks were not completely eliminated, just less frequent than before treatment.11
Most commonly reported side effects included headache, flushing, hypotension, edema, and, rarely, gastrointestinal reflux. Use of these medications may be limited by hypotension.
The review was limited by the small sample size, short duration of treatment, and relatively low doses of calcium channel blockers used in the available studies.11
A 2005 meta-analysis also indicated a statistically significant decrease of 2.8 to 5 attacks per week with nifedipine treatment, though this study also included some patients with secondary Raynaud phenomenon.12
Phosphodiesterase type 5 inhibitors
When calcium channel blockers do not adequately control symptoms, phosphodiesterase type 5 (PDE5) inhibitors can be added or substituted. These medications work by preventing breakdown of cyclic guanosine monophosphate, which induces relaxation in vascular smooth muscle and vasodilation.
Sildenafil can be started at a low dose (20 mg daily) and up-titrated to the maximum dose (20 mg 3 times daily) as tolerated.
A 2014 meta-analysis of 6 randomized controlled trials included 244 patients with secondary Raynaud phenomenon treated with sildenafil, tadalafil, or vardenafil.13 These drugs decreased the daily frequency of attacks by about 0.5 per day vs placebo (–0.49, 95% CI –0.71 to –0.28, P < .0001). PDE5 inhibitors also decreased the severity of attacks (based on the Raynaud’s Condition Score, a popular scoring system) and the duration of attacks by a statistically significant amount.
Almost all patients in these 6 trials were on PDE5 monotherapy. Data on the cumulative benefit of calcium channel blocker and PDE5 inhibitor combination therapy are not yet available. Not all patients tolerate combination therapy, as it can cause symptomatic hypotension, but it can be a successful option in some.
There are also no data showing that either calcium channel blockers or PDE5 inhibitors are superior, though the former are less expensive. A small double-blind, randomized, crossover study of udenafil vs amlodipine in the treatment of secondary Raynaud phenomenon showed that both medications significantly decreased the frequency of attacks and had comparable efficacy.14
Cost and insurance coverage. We have generally been successful in obtaining coverage for this off-label use of PDE5 inhibitors, though additional effort may be required. No drug (not even a calcium channel blocker) is approved by the US Food and Drug Administration for use in Raynaud phenomenon. In our experience, a letter of appeal outlining the rationale for use and citing supporting publications can lead to successful coverage of a medication. If the drug is still not approved, the patient either pays for it out of pocket or another agent is selected. In certain circumstances, pharmaceutical companies may provide prescription assistance for compassionate use of these drugs in Raynaud phenomenon, although this also takes letter-writing, phone calls, or both on the part of the physician.
Topical nitrates
Patients who have an unsatisfactory response to calcium channel blockers with or without PDE5 inhibitors can try topical nitrates, available as sustained-release transdermal patches, tapes, creams, gels, and ointments.
Small trials have noted slight improvement in the Raynaud Condition Score15 and finger temperature16 with these therapies. Another trial noted decreased frequency of attacks and symptoms with the use of sustained-release glyceryl trinitrate patches, but use was limited by intolerable headache.17
In our experience, topical nitrates are most helpful for patients who have 1 or a few digits that are more severely affected than the others, and we reserve these drugs for this indication. Localized vasodilation can provide targeted rapid relief of more ischemic areas.
Topical nitroglycerin can be applied to the base of the ischemic digit for 6 to 12 hours. Preparations vary, and patients should be closely monitored for dose response and tolerance.
Combining a topical nitrate with a calcium channel blocker is safe, but the use of a nitrate with a PDE5 inhibitor is contraindicated due to the risk of hypotension. The use of topical nitrates may be limited by systemic side effects such as headache and flushing and a lack of benefit over time.
Other therapies
If the aforementioned agents are not tolerated or not effective, there is limited evidence that other therapies reduce the frequency and sometimes the severity of attacks. These are not first-line agents but may be tried when other options have been exhausted and symptoms persist. There are no data to support combining these therapies, but in our experience doing so may help some patients in whom drug-drug interactions are not prohibitive.
Prazosin, an alpha-1-adrenergic receptor antagonist, was reported to improve Raynaud phenomenon in 2 small studies in the 1980s, but we do not use it since better options are available. In addition, the vasoactive blood vessels involved do not have alpha-1 receptors, so there is no theoretical basis for using prazosin.18,19
Fluoxetine, a selective serotonin reuptake inhibitor, reduced the frequency and severity of attacks in a 6-week crossover study with nifedipine.20
Losartan, an angiotensin II receptor blocker, also reduced the severity and frequency of attacks when compared with nifedipine.21
Pentoxifylline, a nonselective phosphodiesterase inhibitor, showed some benefit in a trial in 11 patients with primary Raynaud.22
Atorvastatin, a lipid-lowering drug, reduced the number of digital ulcers in patients with secondary Raynaud already on first-line vasodilatory therapy, and might be added in this situation.23
Botulinum toxin A injections have some data to support their use, but evidence is based on uncontrolled case series.24 A controlled trial in scleroderma patients with severe Raynaud phenomenon found botulinum toxin to be no better than placebo.25
Prostacyclin preparations are available. Intermittent intravenous doses of prostacyclin analogues over several days can be used in resistant cases. Oral prostacyclin agents have not shown consistent benefit. New prostacyclin receptor agonists are under investigation.
Overall, we move to other options only in patients with persistent symptoms that impair quality of life, or in patients with recurrent digital ischemic lesions that have not responded to calcium channel blockers and PDE5 inhibitors or nitrates, either alone or in combination.
DIGITAL ULCERATION AND ACUTE DIGITAL ISCHEMIC CRISIS
Patients with secondary Raynaud phenomenon may be at risk of recurrent digital ulceration and acute digital ischemia with gangrene. These patients should be comanaged with a rheumatologist so that the underlying disease process is fully addressed. Digital ulcers should be inspected closely for signs of infection, which may require treatment with antibiotics.
Acute digital ischemia is a medical emergency and should prompt inpatient admission with warming, emotional regulation, and pain control (often with narcotics) to decrease sympathetic vasoconstriction. These patients require aggressive vasodilatory therapy to reverse the ischemic event.
A short-acting calcium channel blocker or combination therapy with a calcium channel blocker and a PDE5 inhibitor or topical nitrate should be started. If there is no benefit, then transient intravenous vasodilatory therapy with a prostacyclin (epoprostenol) or localized digital sympathectomy is used to prevent digital loss.
The endothelin receptor inhibitor bosentan has been shown to decrease recurrent digital ulcers in patients with scleroderma, and while bosentan does not decrease the frequency of Raynaud attacks, it can be used in this select group to prevent new digital ulcers.
Treatment options may be limited by insurance coverage or access to intravenous infusions.
TAKE-HOME RECOMMENDATIONS
For many patients with primary or secondary Raynaud phenomenon, nonpharmacologic interventions are all that are required to decrease the frequency of attacks and improve quality of life. The goal should not be to eliminate attacks completely, as aggressive drug treatment may cause more harm than benefit. From our perspective, the goals of treatment should be to improve quality of life and prevent ischemic complications.
Pharmacologic therapies should be added only if attacks remain poorly controlled with incapacitating symptoms, or if the patient has digital ischemic ulcers. Calcium channel blockers are first-line therapy, given proven efficacy and low cost, and should be titrated to the maximum tolerated dose before adding or substituting other agents.
References
Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med 2016; 375:556–565.
Irwin MS, Gilbert SE, Terenghi G, Smith RW, Green CJ. Cold intolerance following peripheral nerve injury. Natural history and factors predicting severity of symptoms. J Hand Surg Br 1997; 22:308–316.
Wasner G. Vasomotor disturbances in complex regional pain syndrome—a review. Pain Med 2010; 11:1267–1273.
Kurklinsky AK, Miller VM, Rooke TW. Acrocyanosis: the Flying Dutchman. Vasc Med 2011; 16:288–301.
Garner R, Kumari R, Lanyon P, Doherty M, Zhang W. Prevalence, risk factors and associations of primary Raynaud’s phenomenon: systematic review and meta-analysis of observational studies. BMJ Open 2015; 5:e006389.
Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1008.
Chikura B, Moore TL, Manning JB, Vail A, Herrick AL. Sparing of the thumb in Raynaud’s phenomenon. Rheumatology (Oxford) 2008; 47:219–221.
Kallenerg CG. Early detection of connective tissue disease in patients with Raynaud’s phenomenon. Rheum Dis Clin North Am 1990; 16:11–30.
Kwakkenbos L, Thombs BD. Non-drug approaches to treating Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA, editors. Raynaud’s Phenomenon. A Guide to Pathogenesis and Treatment. New York: Springer Science+Business Media, 2015:299–313.
Goodfield MJ, Hume A, Rowell NR. The acute effects of cigarette smoking on cutaneous blood flow in smoking and non-smoking subjects with and without Raynaud’s phenomenon. Br J Rheumatol 1990; 29:89–91.
Ennis H, Hughes M, Anderson ME, Wilkinson J, Herrick AL. Calcium channel blockers for primary Raynaud’s phenomenon. Cochrane Database Sys Review 2016; 2:CD002069.
Thompson AE, Pope JE. Calcium channel blockers for primary Raynaud’s phenomenon: a meta-analysis. Rheumatology (Oxford) 2005; 44:145–150.
Roustit M, Blaise S, Allanore Y, Carpentier P, Caglayan E, Cracowski J. Phosphodiesterase-5 inhibitors for the treatment of secondary Raynaud’s phenomenon: systematic review and meta-analysis of randomized trials. Ann Rheum Dis 2013; 72:1696–1699.
Lee EY, Park JK, Lee W, et al. Head-to-head comparison of udenafil vs amlodipine in the treatment of secondary Raynaud's phenomenon: a double-blind, randomized, cross-over study. Rheumatology (Oxford) 2014; 53:658–664.
Chung L, Shapiro L, Fiorentino D, et al. MQX-503, a novel formulation of nitroglycerin, improves the severity of Raynaud’s phenomenon: a randomized, controlled trial. Arthritis Rheum 2009; 60:870–877.
Kan C, Akimoto S, Abe M, Okada K, Ishikawa O. Preliminary thermographic evaluation of a new nitroglycerine tape on the peripheral circulatory disturbance in systemic sclerosis. Ann Rheum Dis 2002; 61:177–179.
Teh LS, Manning J, Moore T, Tully MP, O’Reilly D, Jayson MI. Sustained-release transdermal glyceryl trinitrate patches as a treatment for primary and secondary Raynaud’s phenomenon. Br J Rheumatol 1995; 34:636–641.
Russell IJ, Lessard JA. Prazosin treatment of Raynaud’s phenomenon: a double blind single crossover study. J Rheumatol 1985; 12:94–98.
Wollersheim H, Thien T, Fennis J, van Elteren P, van ‘t Laar A. Double-blind, placebo-controlled study of prazosin in Raynaud’s phenomenon. Clin Pharmacol Ther 1986; 40:219–225.
Coleiro B, Marshall SE, Denton CP, et al. Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford) 2001; 40:1038–1043.
Didazio M, Denton CP, Smith R, et al. Losartan therapy for Raynaud’s phenomenon and scleroderma: clinical and biochemical findings in a fifteen-week randomized, parallel-group, controlled trial. Arthritis Rheum 1999; 42:2646–2655.
Neirotti M, Longo F, Molaschi M, Macchione C, Pernigotti L. Functional vascular disorders: treatment with pentoxifylline. Angiology 1987; 38:575–580.
Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35:1801–1808.
Iorio ML, Masden DL, Higgins JP. Botulinum toxin A treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum 2012; 41: 599–603.
Bello RJ, Cooney CM, Melamed E, et al. The therapeutic efficacy of botulinum toxin in treating scleroderma-associated Raynaud’s phenomenon: a randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol 2017. Epub ahead of print.
References
Wigley FM, Flavahan NA. Raynaud’s phenomenon. N Engl J Med 2016; 375:556–565.
Irwin MS, Gilbert SE, Terenghi G, Smith RW, Green CJ. Cold intolerance following peripheral nerve injury. Natural history and factors predicting severity of symptoms. J Hand Surg Br 1997; 22:308–316.
Wasner G. Vasomotor disturbances in complex regional pain syndrome—a review. Pain Med 2010; 11:1267–1273.
Kurklinsky AK, Miller VM, Rooke TW. Acrocyanosis: the Flying Dutchman. Vasc Med 2011; 16:288–301.
Garner R, Kumari R, Lanyon P, Doherty M, Zhang W. Prevalence, risk factors and associations of primary Raynaud’s phenomenon: systematic review and meta-analysis of observational studies. BMJ Open 2015; 5:e006389.
Wigley FM. Clinical practice. Raynaud’s phenomenon. N Engl J Med 2002; 347:1001–1008.
Chikura B, Moore TL, Manning JB, Vail A, Herrick AL. Sparing of the thumb in Raynaud’s phenomenon. Rheumatology (Oxford) 2008; 47:219–221.
Kallenerg CG. Early detection of connective tissue disease in patients with Raynaud’s phenomenon. Rheum Dis Clin North Am 1990; 16:11–30.
Kwakkenbos L, Thombs BD. Non-drug approaches to treating Raynaud’s phenomenon. In: Wigley FM, Herrick AL, Flavahan NA, editors. Raynaud’s Phenomenon. A Guide to Pathogenesis and Treatment. New York: Springer Science+Business Media, 2015:299–313.
Goodfield MJ, Hume A, Rowell NR. The acute effects of cigarette smoking on cutaneous blood flow in smoking and non-smoking subjects with and without Raynaud’s phenomenon. Br J Rheumatol 1990; 29:89–91.
Ennis H, Hughes M, Anderson ME, Wilkinson J, Herrick AL. Calcium channel blockers for primary Raynaud’s phenomenon. Cochrane Database Sys Review 2016; 2:CD002069.
Thompson AE, Pope JE. Calcium channel blockers for primary Raynaud’s phenomenon: a meta-analysis. Rheumatology (Oxford) 2005; 44:145–150.
Roustit M, Blaise S, Allanore Y, Carpentier P, Caglayan E, Cracowski J. Phosphodiesterase-5 inhibitors for the treatment of secondary Raynaud’s phenomenon: systematic review and meta-analysis of randomized trials. Ann Rheum Dis 2013; 72:1696–1699.
Lee EY, Park JK, Lee W, et al. Head-to-head comparison of udenafil vs amlodipine in the treatment of secondary Raynaud's phenomenon: a double-blind, randomized, cross-over study. Rheumatology (Oxford) 2014; 53:658–664.
Chung L, Shapiro L, Fiorentino D, et al. MQX-503, a novel formulation of nitroglycerin, improves the severity of Raynaud’s phenomenon: a randomized, controlled trial. Arthritis Rheum 2009; 60:870–877.
Kan C, Akimoto S, Abe M, Okada K, Ishikawa O. Preliminary thermographic evaluation of a new nitroglycerine tape on the peripheral circulatory disturbance in systemic sclerosis. Ann Rheum Dis 2002; 61:177–179.
Teh LS, Manning J, Moore T, Tully MP, O’Reilly D, Jayson MI. Sustained-release transdermal glyceryl trinitrate patches as a treatment for primary and secondary Raynaud’s phenomenon. Br J Rheumatol 1995; 34:636–641.
Russell IJ, Lessard JA. Prazosin treatment of Raynaud’s phenomenon: a double blind single crossover study. J Rheumatol 1985; 12:94–98.
Wollersheim H, Thien T, Fennis J, van Elteren P, van ‘t Laar A. Double-blind, placebo-controlled study of prazosin in Raynaud’s phenomenon. Clin Pharmacol Ther 1986; 40:219–225.
Coleiro B, Marshall SE, Denton CP, et al. Treatment of Raynaud’s phenomenon with the selective serotonin reuptake inhibitor fluoxetine. Rheumatology (Oxford) 2001; 40:1038–1043.
Didazio M, Denton CP, Smith R, et al. Losartan therapy for Raynaud’s phenomenon and scleroderma: clinical and biochemical findings in a fifteen-week randomized, parallel-group, controlled trial. Arthritis Rheum 1999; 42:2646–2655.
Neirotti M, Longo F, Molaschi M, Macchione C, Pernigotti L. Functional vascular disorders: treatment with pentoxifylline. Angiology 1987; 38:575–580.
Abou-Raya A, Abou-Raya S, Helmii M. Statins: potentially useful in therapy of systemic sclerosis-related Raynaud’s phenomenon and digital ulcers. J Rheumatol 2008; 35:1801–1808.
Iorio ML, Masden DL, Higgins JP. Botulinum toxin A treatment of Raynaud’s phenomenon: a review. Semin Arthritis Rheum 2012; 41: 599–603.
Bello RJ, Cooney CM, Melamed E, et al. The therapeutic efficacy of botulinum toxin in treating scleroderma-associated Raynaud’s phenomenon: a randomized, double-blind, placebo-controlled clinical trial. Arthritis Rheumatol 2017. Epub ahead of print.
Primary Raynaud phenomenon occurs in the absence of any underlying disease process. Secondary Raynaud phenomenon occurs in concert with another disease, frequently rheumatic.
Young patients with mild Raynaud phenomenon, normal nailfold capillaries, and no additional symptoms or signs to suggest a rheumatic or other underlying disease can be followed carefully by the primary care doctor and do not require further serologic workup or referral to a specialist.
Nonpharmacologic interventions, ie, cold avoidance and stress management, are first-line for all patients.
Calcium channel blockers are first-line drugs and should be titrated to the maximum tolerated dose before adding or switching to other agents.
The goal of treatment should not be to eliminate Raynaud attacks completely but to improve quality of life and prevent ischemic complications.
Disallow All Ads
Content Gating
No Gating (article Unlocked/Free)
Alternative CME
Disqus Comments
Default
Consolidated Pubs: Do Not Show Source Publication Logo
For practitioners who see a lot of patients, particularly a lot of young women, patients describing cold-induced color changes of the fingers sometimes accompanied by tingling or burning are common. For most patients it is mild, but for some the discoloration or dysesthesia may be striking and disconcerting. For a minority, this reversible vasoconstrictive phenomenon (Raynaud “disease” if it occurs in isolation, without any associated underlying condition) may be the presenting sign of a systemic disorder.
For many patients, Raynaud symptoms are mild enough to not even mention to their primary care provider, and conversely, there is little reason for most clinicians to routinely inquire about such symptoms. So it may surprise some readers to read about the nuances of diagnosis and treatment discussed by Shapiro and Wigley in this issue of the Journal.
To a rheumatologist, Raynaud phenomenon, particularly of recent onset in an adult, raises the specter of an underlying systemic inflammatory disease. The phenomenon is not linked to a specific diagnosis; it is associated with lupus, rheumatoid arthritis, cryoglobulinemia, inflammatory myopathy, Sjögren syndrome, and, in its severe form, with the scleroderma syndromes. We focus on differentiating between these rheumatic disorders once we have discarded nonrheumatic causes such as atherosclerotic arterial disease, carcinoma, embolism, Buerger disease, medications, smoking, or thrombosis.
But rheumatologists are toward the bottom of the diagnostic funnel—we see these patients when an underlying disease is already suspected. The real challenge is for the primary care providers who first recognize the digital vasospasm on examination or are told of the symptoms by their patient. These clinicians need to know which initial reflexive actions are warranted and which can wait, for, as noted by Shapiro and Wigley, there are several options.
The first action is to try to determine the timeline, although Raynaud disease often has an insidious onset or the patient doesn’t recall the onset. New and sudden onset likely has a stronger association with an underlying disease. A focused physical examination should look for digital stigmata of ischemic damage; the presence of digital ulcers or healed digital pits indicates a possible vascular occlusive component in addition to the vascular spasm. This strongly suggests scleroderma or Buerger disease, as tissue damage doesn’t occur in (primary) Raynaud disease or generally even with Raynaud phenomenon associated with lupus or other rheumatic disorders. Sclerodactyly should be looked for: diffuse finger puffiness, skin-tightening, or early signs such as loss of the usual finger skin creases. Telangiectasia (not vascular spiders or cherry angiomata) should be searched for, particularly on the palms, face, and inner lips, as these vascular lesions are common in patients with limited scleroderma. Careful auscultation for basilar lung crackles should be done. Distal pulses should all be assessed, and bruits in the neck, abdomen and inguinal areas should be carefully sought.
Patients should be questioned about any symptom-associated reduction in exercise tolerance and particularly about trouble swallowing, “heartburn,” and symptoms of reflux. Although patients with Raynaud disease may have demonstrable esophageal dysmotility, the presence of significant, new, or worsened symptoms raises the concern of scleroderma. Patients should be asked about symptoms of malabsorption. Specific questioning should be directed at eliciting a history of joint stiffness and especially muscle weakness. The latter can be approached by inquiring about new or progressive difficulty in specific tasks such as walking up steps, brushing hair, and arising from low chairs or the toilet. Distinguishing muscle weakness from general fatigue is not always easy, but it is important.
Shapiro and Wigley discuss the extremely useful evaluation of nailfold capillaries, which can be done with a standard magnifier or ophthalmoscope. This is very valuable to help predict the development or current presence of a systemic rheumatic disease. But this is not a technique that most clinicians are familiar with. A potentially useful surrogate or adjunctive test, especially in the setting of new-onset Raynaud, is the antinuclear antibody (ANA) test; I prefer the immunofluorescent assay. While a positive test alone (with Raynaud) does not define the presence of any rheumatic disease, several older studies suggest that patients with a new onset of Raynaud phenomenon and a positive ANA test are more likely to develop a systemic autoimmune disorder than if the test is negative. Those who do so (and this is far from all) are most likely to have the disease manifest within a few years. Hence, if the ANA test is positive but the history, physical examination, and limited laboratory testing (complete blood cell count with differential, complete metabolic panel, creatine kinase, and urinalysis) are normal, it is reasonable to reexamine the patient in 3 months and then every 6 months for 2 to 3 years, repeating the focused history and physical examination. It is also reasonable at some point to refer these patients to a rheumatologist.
Since Raynaud phenomenon is common, and the associated severe rheumatic disorders associated with it are rare, it is easy to not recognize Raynaud phenomenon as a clue to the onset of a potentially severe systemic disease. Yet with a few simple questions, a focused examination, and minimal laboratory testing, patients who are more likely to harbor a systemic disease can usually be treated symptomatically if necessary, and appropriately triaged to observation or for subspecialty referral.
For practitioners who see a lot of patients, particularly a lot of young women, patients describing cold-induced color changes of the fingers sometimes accompanied by tingling or burning are common. For most patients it is mild, but for some the discoloration or dysesthesia may be striking and disconcerting. For a minority, this reversible vasoconstrictive phenomenon (Raynaud “disease” if it occurs in isolation, without any associated underlying condition) may be the presenting sign of a systemic disorder.
For many patients, Raynaud symptoms are mild enough to not even mention to their primary care provider, and conversely, there is little reason for most clinicians to routinely inquire about such symptoms. So it may surprise some readers to read about the nuances of diagnosis and treatment discussed by Shapiro and Wigley in this issue of the Journal.
To a rheumatologist, Raynaud phenomenon, particularly of recent onset in an adult, raises the specter of an underlying systemic inflammatory disease. The phenomenon is not linked to a specific diagnosis; it is associated with lupus, rheumatoid arthritis, cryoglobulinemia, inflammatory myopathy, Sjögren syndrome, and, in its severe form, with the scleroderma syndromes. We focus on differentiating between these rheumatic disorders once we have discarded nonrheumatic causes such as atherosclerotic arterial disease, carcinoma, embolism, Buerger disease, medications, smoking, or thrombosis.
But rheumatologists are toward the bottom of the diagnostic funnel—we see these patients when an underlying disease is already suspected. The real challenge is for the primary care providers who first recognize the digital vasospasm on examination or are told of the symptoms by their patient. These clinicians need to know which initial reflexive actions are warranted and which can wait, for, as noted by Shapiro and Wigley, there are several options.
The first action is to try to determine the timeline, although Raynaud disease often has an insidious onset or the patient doesn’t recall the onset. New and sudden onset likely has a stronger association with an underlying disease. A focused physical examination should look for digital stigmata of ischemic damage; the presence of digital ulcers or healed digital pits indicates a possible vascular occlusive component in addition to the vascular spasm. This strongly suggests scleroderma or Buerger disease, as tissue damage doesn’t occur in (primary) Raynaud disease or generally even with Raynaud phenomenon associated with lupus or other rheumatic disorders. Sclerodactyly should be looked for: diffuse finger puffiness, skin-tightening, or early signs such as loss of the usual finger skin creases. Telangiectasia (not vascular spiders or cherry angiomata) should be searched for, particularly on the palms, face, and inner lips, as these vascular lesions are common in patients with limited scleroderma. Careful auscultation for basilar lung crackles should be done. Distal pulses should all be assessed, and bruits in the neck, abdomen and inguinal areas should be carefully sought.
Patients should be questioned about any symptom-associated reduction in exercise tolerance and particularly about trouble swallowing, “heartburn,” and symptoms of reflux. Although patients with Raynaud disease may have demonstrable esophageal dysmotility, the presence of significant, new, or worsened symptoms raises the concern of scleroderma. Patients should be asked about symptoms of malabsorption. Specific questioning should be directed at eliciting a history of joint stiffness and especially muscle weakness. The latter can be approached by inquiring about new or progressive difficulty in specific tasks such as walking up steps, brushing hair, and arising from low chairs or the toilet. Distinguishing muscle weakness from general fatigue is not always easy, but it is important.
Shapiro and Wigley discuss the extremely useful evaluation of nailfold capillaries, which can be done with a standard magnifier or ophthalmoscope. This is very valuable to help predict the development or current presence of a systemic rheumatic disease. But this is not a technique that most clinicians are familiar with. A potentially useful surrogate or adjunctive test, especially in the setting of new-onset Raynaud, is the antinuclear antibody (ANA) test; I prefer the immunofluorescent assay. While a positive test alone (with Raynaud) does not define the presence of any rheumatic disease, several older studies suggest that patients with a new onset of Raynaud phenomenon and a positive ANA test are more likely to develop a systemic autoimmune disorder than if the test is negative. Those who do so (and this is far from all) are most likely to have the disease manifest within a few years. Hence, if the ANA test is positive but the history, physical examination, and limited laboratory testing (complete blood cell count with differential, complete metabolic panel, creatine kinase, and urinalysis) are normal, it is reasonable to reexamine the patient in 3 months and then every 6 months for 2 to 3 years, repeating the focused history and physical examination. It is also reasonable at some point to refer these patients to a rheumatologist.
Since Raynaud phenomenon is common, and the associated severe rheumatic disorders associated with it are rare, it is easy to not recognize Raynaud phenomenon as a clue to the onset of a potentially severe systemic disease. Yet with a few simple questions, a focused examination, and minimal laboratory testing, patients who are more likely to harbor a systemic disease can usually be treated symptomatically if necessary, and appropriately triaged to observation or for subspecialty referral.
For practitioners who see a lot of patients, particularly a lot of young women, patients describing cold-induced color changes of the fingers sometimes accompanied by tingling or burning are common. For most patients it is mild, but for some the discoloration or dysesthesia may be striking and disconcerting. For a minority, this reversible vasoconstrictive phenomenon (Raynaud “disease” if it occurs in isolation, without any associated underlying condition) may be the presenting sign of a systemic disorder.
For many patients, Raynaud symptoms are mild enough to not even mention to their primary care provider, and conversely, there is little reason for most clinicians to routinely inquire about such symptoms. So it may surprise some readers to read about the nuances of diagnosis and treatment discussed by Shapiro and Wigley in this issue of the Journal.
To a rheumatologist, Raynaud phenomenon, particularly of recent onset in an adult, raises the specter of an underlying systemic inflammatory disease. The phenomenon is not linked to a specific diagnosis; it is associated with lupus, rheumatoid arthritis, cryoglobulinemia, inflammatory myopathy, Sjögren syndrome, and, in its severe form, with the scleroderma syndromes. We focus on differentiating between these rheumatic disorders once we have discarded nonrheumatic causes such as atherosclerotic arterial disease, carcinoma, embolism, Buerger disease, medications, smoking, or thrombosis.
But rheumatologists are toward the bottom of the diagnostic funnel—we see these patients when an underlying disease is already suspected. The real challenge is for the primary care providers who first recognize the digital vasospasm on examination or are told of the symptoms by their patient. These clinicians need to know which initial reflexive actions are warranted and which can wait, for, as noted by Shapiro and Wigley, there are several options.
The first action is to try to determine the timeline, although Raynaud disease often has an insidious onset or the patient doesn’t recall the onset. New and sudden onset likely has a stronger association with an underlying disease. A focused physical examination should look for digital stigmata of ischemic damage; the presence of digital ulcers or healed digital pits indicates a possible vascular occlusive component in addition to the vascular spasm. This strongly suggests scleroderma or Buerger disease, as tissue damage doesn’t occur in (primary) Raynaud disease or generally even with Raynaud phenomenon associated with lupus or other rheumatic disorders. Sclerodactyly should be looked for: diffuse finger puffiness, skin-tightening, or early signs such as loss of the usual finger skin creases. Telangiectasia (not vascular spiders or cherry angiomata) should be searched for, particularly on the palms, face, and inner lips, as these vascular lesions are common in patients with limited scleroderma. Careful auscultation for basilar lung crackles should be done. Distal pulses should all be assessed, and bruits in the neck, abdomen and inguinal areas should be carefully sought.
Patients should be questioned about any symptom-associated reduction in exercise tolerance and particularly about trouble swallowing, “heartburn,” and symptoms of reflux. Although patients with Raynaud disease may have demonstrable esophageal dysmotility, the presence of significant, new, or worsened symptoms raises the concern of scleroderma. Patients should be asked about symptoms of malabsorption. Specific questioning should be directed at eliciting a history of joint stiffness and especially muscle weakness. The latter can be approached by inquiring about new or progressive difficulty in specific tasks such as walking up steps, brushing hair, and arising from low chairs or the toilet. Distinguishing muscle weakness from general fatigue is not always easy, but it is important.
Shapiro and Wigley discuss the extremely useful evaluation of nailfold capillaries, which can be done with a standard magnifier or ophthalmoscope. This is very valuable to help predict the development or current presence of a systemic rheumatic disease. But this is not a technique that most clinicians are familiar with. A potentially useful surrogate or adjunctive test, especially in the setting of new-onset Raynaud, is the antinuclear antibody (ANA) test; I prefer the immunofluorescent assay. While a positive test alone (with Raynaud) does not define the presence of any rheumatic disease, several older studies suggest that patients with a new onset of Raynaud phenomenon and a positive ANA test are more likely to develop a systemic autoimmune disorder than if the test is negative. Those who do so (and this is far from all) are most likely to have the disease manifest within a few years. Hence, if the ANA test is positive but the history, physical examination, and limited laboratory testing (complete blood cell count with differential, complete metabolic panel, creatine kinase, and urinalysis) are normal, it is reasonable to reexamine the patient in 3 months and then every 6 months for 2 to 3 years, repeating the focused history and physical examination. It is also reasonable at some point to refer these patients to a rheumatologist.
Since Raynaud phenomenon is common, and the associated severe rheumatic disorders associated with it are rare, it is easy to not recognize Raynaud phenomenon as a clue to the onset of a potentially severe systemic disease. Yet with a few simple questions, a focused examination, and minimal laboratory testing, patients who are more likely to harbor a systemic disease can usually be treated symptomatically if necessary, and appropriately triaged to observation or for subspecialty referral.
The overall response rate to immune checkpoint inhibitors was 45% among cancer patients who had more than three variants of unknown significance in their circulating tumor DNA; among those with three or fewer, the response rate was 15%, according to a University of California, San Diego, investigation with 69 subjects.
Higher mutation burdens in circulating tumor DNA (ctDNA) also correlated with improved progression-free and overall survival across 20 cancer types, the investigators reported (Clin Cancer Res. 2017 Oct. 1. doi: 10.1158/1078-0432.CCR-17-1439).
Tumor mutation burdens can predict response to checkpoint inhibitors, but they are usually assessed by tissue biopsy, which is costly and invasive. The findings suggest that blood tests could replace tissue biopsies to green-light immune checkpoint inhibitor treatment.
“Our current results may be clinically exploitable. ... Liquid biopsies that assess blood-derived ctDNA are noninvasive, easily acquired, and inexpensive. The ctDNA derived from blood may also represent shed DNA from multiple metastatic sites, whereas tissue genomics reflects only the piece of tissue removed,” said investigators led by Yulian Khagi, MD, a hematology-oncology fellow at the university.
In a press statement, Dr. Khagi said “If verified by further studies, clinicians will be able to utilize the ... results of this simple blood test to make determinations about whether to use checkpoint inhibitor–based immune therapy in a variety of tumor types.”
The 69 patients were a median of 56 years old, and 43 (62.3%) were men. Melanoma, lung cancer, and head and neck cancer were the most common malignancies. The majority of patients had anti–PD-1 or PD-L1 monotherapy.
For most patients, blood samples were drawn a month or 2 before treatment. Next-generation sequencing (Guardant360) was done on ctDNA to detect alterations in cancer genes. Of the 69 patients, 20 (29%) had more than three variants of unknown significance (VUS); the rest had three or fewer.
The median overall survival was 15.3 months from the start of immunotherapy. For patients with three or fewer VUS, median overall survival was 10.72 months; for patients with more, median overall survival could not be calculated because more than half were alive at the study’s conclusion.
Median progression-fee survival was 2.07 months with three or fewer VUS, versus 3.84 months with more. The findings were statistically significant.
Similar results were found when all genomic alterations, not just VUS, were examined and dichotomized as six or more versus fewer than six.
“The number of genes assayed in our ctDNA analysis was only between 54 and 70. Unlike targeted NGS [next-generation sequencing] of tumor tissue, which often tests for hundreds of genes and allows a relatively accurate estimate of total mutational burden, targeted NGS of plasma ctDNA provides only a limited snapshot of the cancer genome. More extensive ctDNA gene panels merit investigation to determine if they increase the correlative value of our findings,” the investigators said.
The work was funded by the Joan and Irwin Jacobs Fund and the National Cancer Institute. Dr. Khagi had no industry disclosures. Three authors reported financial ties to a number of companies, including Boehringer, Merck, Guardant, and Pfizer. The senior author has ownership interests in CureMatch.
The overall response rate to immune checkpoint inhibitors was 45% among cancer patients who had more than three variants of unknown significance in their circulating tumor DNA; among those with three or fewer, the response rate was 15%, according to a University of California, San Diego, investigation with 69 subjects.
Higher mutation burdens in circulating tumor DNA (ctDNA) also correlated with improved progression-free and overall survival across 20 cancer types, the investigators reported (Clin Cancer Res. 2017 Oct. 1. doi: 10.1158/1078-0432.CCR-17-1439).
Tumor mutation burdens can predict response to checkpoint inhibitors, but they are usually assessed by tissue biopsy, which is costly and invasive. The findings suggest that blood tests could replace tissue biopsies to green-light immune checkpoint inhibitor treatment.
“Our current results may be clinically exploitable. ... Liquid biopsies that assess blood-derived ctDNA are noninvasive, easily acquired, and inexpensive. The ctDNA derived from blood may also represent shed DNA from multiple metastatic sites, whereas tissue genomics reflects only the piece of tissue removed,” said investigators led by Yulian Khagi, MD, a hematology-oncology fellow at the university.
In a press statement, Dr. Khagi said “If verified by further studies, clinicians will be able to utilize the ... results of this simple blood test to make determinations about whether to use checkpoint inhibitor–based immune therapy in a variety of tumor types.”
The 69 patients were a median of 56 years old, and 43 (62.3%) were men. Melanoma, lung cancer, and head and neck cancer were the most common malignancies. The majority of patients had anti–PD-1 or PD-L1 monotherapy.
For most patients, blood samples were drawn a month or 2 before treatment. Next-generation sequencing (Guardant360) was done on ctDNA to detect alterations in cancer genes. Of the 69 patients, 20 (29%) had more than three variants of unknown significance (VUS); the rest had three or fewer.
The median overall survival was 15.3 months from the start of immunotherapy. For patients with three or fewer VUS, median overall survival was 10.72 months; for patients with more, median overall survival could not be calculated because more than half were alive at the study’s conclusion.
Median progression-fee survival was 2.07 months with three or fewer VUS, versus 3.84 months with more. The findings were statistically significant.
Similar results were found when all genomic alterations, not just VUS, were examined and dichotomized as six or more versus fewer than six.
“The number of genes assayed in our ctDNA analysis was only between 54 and 70. Unlike targeted NGS [next-generation sequencing] of tumor tissue, which often tests for hundreds of genes and allows a relatively accurate estimate of total mutational burden, targeted NGS of plasma ctDNA provides only a limited snapshot of the cancer genome. More extensive ctDNA gene panels merit investigation to determine if they increase the correlative value of our findings,” the investigators said.
The work was funded by the Joan and Irwin Jacobs Fund and the National Cancer Institute. Dr. Khagi had no industry disclosures. Three authors reported financial ties to a number of companies, including Boehringer, Merck, Guardant, and Pfizer. The senior author has ownership interests in CureMatch.
The overall response rate to immune checkpoint inhibitors was 45% among cancer patients who had more than three variants of unknown significance in their circulating tumor DNA; among those with three or fewer, the response rate was 15%, according to a University of California, San Diego, investigation with 69 subjects.
Higher mutation burdens in circulating tumor DNA (ctDNA) also correlated with improved progression-free and overall survival across 20 cancer types, the investigators reported (Clin Cancer Res. 2017 Oct. 1. doi: 10.1158/1078-0432.CCR-17-1439).
Tumor mutation burdens can predict response to checkpoint inhibitors, but they are usually assessed by tissue biopsy, which is costly and invasive. The findings suggest that blood tests could replace tissue biopsies to green-light immune checkpoint inhibitor treatment.
“Our current results may be clinically exploitable. ... Liquid biopsies that assess blood-derived ctDNA are noninvasive, easily acquired, and inexpensive. The ctDNA derived from blood may also represent shed DNA from multiple metastatic sites, whereas tissue genomics reflects only the piece of tissue removed,” said investigators led by Yulian Khagi, MD, a hematology-oncology fellow at the university.
In a press statement, Dr. Khagi said “If verified by further studies, clinicians will be able to utilize the ... results of this simple blood test to make determinations about whether to use checkpoint inhibitor–based immune therapy in a variety of tumor types.”
The 69 patients were a median of 56 years old, and 43 (62.3%) were men. Melanoma, lung cancer, and head and neck cancer were the most common malignancies. The majority of patients had anti–PD-1 or PD-L1 monotherapy.
For most patients, blood samples were drawn a month or 2 before treatment. Next-generation sequencing (Guardant360) was done on ctDNA to detect alterations in cancer genes. Of the 69 patients, 20 (29%) had more than three variants of unknown significance (VUS); the rest had three or fewer.
The median overall survival was 15.3 months from the start of immunotherapy. For patients with three or fewer VUS, median overall survival was 10.72 months; for patients with more, median overall survival could not be calculated because more than half were alive at the study’s conclusion.
Median progression-fee survival was 2.07 months with three or fewer VUS, versus 3.84 months with more. The findings were statistically significant.
Similar results were found when all genomic alterations, not just VUS, were examined and dichotomized as six or more versus fewer than six.
“The number of genes assayed in our ctDNA analysis was only between 54 and 70. Unlike targeted NGS [next-generation sequencing] of tumor tissue, which often tests for hundreds of genes and allows a relatively accurate estimate of total mutational burden, targeted NGS of plasma ctDNA provides only a limited snapshot of the cancer genome. More extensive ctDNA gene panels merit investigation to determine if they increase the correlative value of our findings,” the investigators said.
The work was funded by the Joan and Irwin Jacobs Fund and the National Cancer Institute. Dr. Khagi had no industry disclosures. Three authors reported financial ties to a number of companies, including Boehringer, Merck, Guardant, and Pfizer. The senior author has ownership interests in CureMatch.
Key clinical point: A simple blood test might soon replace tissue biopsy to green-light immune checkpoint inhibitor treatment.
Major finding: The overall response rate to immune checkpoint inhibitors was 45% among cancer patients who had more than three variants of unknown significance in their circulating tumor DNA; among those with three or fewer, the response rate was 15%.
Data source: Review of 69 cancer patients.
Disclosures: The work was funded by the Joan and Irwin Jacobs Fund and the National Cancer Institute. Three investigators reported financial ties to a number of companies, including Boehringer, Merck, Guardant, and Pfizer. The senior author has ownership interests in CureMatch.
Modest weight loss of 5% to 10% among patients who are overweight or obese can result in a clinically relevant reduction in cardiovascular (CV) disease risk.1 This amount of weight loss can increase insulin sensitivity in adipose tissue, liver, and muscle, and have a positive impact on blood sugar, blood pressure, triglycerides, and high-density lipoprotein cholesterol.1,2
All patients who are obese or overweight with increased CV risk should be counseled on diet, exercise, and other behavioral interventions.3 Weight loss secondary to lifestyle modification alone, however, leads to adaptive physiologic responses, which increase appetite and reduce energy expenditure.4-6
Pharmacotherapy can counteract this metabolic adaptation and lead to sustained weight loss. Antiobesity medication can be considered if a patient has a body mass index (BMI) ≥30 kg/m2 or ≥27 kg/m2 with obesity-related comorbidities such as hypertension, type 2 diabetes, dyslipidemia, or obstructive sleep apnea.3,7
Until recently, there were few pharmacologic options approved by the US Food and Drug Administration (FDA) for the management of obesity. The mainstays of treatment were phentermine (Adipex-P, Ionamin, Suprenza) and orlistat (Alli, Xenical). Since 2012, however, 4 agents have been approved as adjuncts to a reduced-calorie diet and increased physical activity for long-term weight management.8,9 Phentermine/topiramate extended-release (ER) (Qsymia) and lorcaserin (Belviq) were approved in 2012,10,11 and naltrexone sustained release (SR)/bupropion SR (Contrave) and liraglutide 3 mg (Saxenda) were approved in 201412,13 (TABLE9,14-39). These medications have the potential to not only limit weight gain, but also promote weight loss and, thus, improve blood pressure, cholesterol, glucose, and insulin.40
Despite the growing obesity epidemic and the availability of several additional medications for chronic weight management, use of antiobesity pharmacotherapy has been limited. Barriers to use include inadequate training of health care professionals, poor insurance coverage for new agents, and low reimbursement for office visits to address weight.41
Weight loss secondary to lifestyle changes can lead to adaptive physiologic responses, which increase appetite and reduce energy expenditure. Pharmacotherapy can counteract this.
In addition, the number of obesity medicine specialists, while increasing, is still not sufficient. Therefore, it is imperative for other health care professionals—namely family practitioners—to be aware of the treatment options available to patients who are overweight or obese and to be adept at using them.
In this review, we present 4 cases that depict patients who could benefit from the addition of antiobesity pharmacotherapy to a comprehensive treatment plan that includes diet, physical activity, and behavioral modification.
[polldaddy:9840472]
CASE 1 Melissa C, a 27-year-old woman with obesity (BMI 33 kg/m2), hyperlipidemia, and migraine headaches, presents for weight management. Despite a calorie-reduced diet and 200 minutes per week of exercise for the past 6 months, she has been unable to lose weight. The only medications she’s taking are oral contraceptive pills and sumatriptan, as needed. She suffers from migraines 3 times a month and has no anxiety. Laboratory test results are normal with the exception of an elevated low-density lipoprotein (LDL) level.
Which medication is an appropriate next step for Ms. C?
Discussion
When considering an antiobesity agent for any patient, there are 2 important questions to ask:
Are there contraindications, drug-drug interactions, or undesirable adverse effects associated with this medication that could be problematic for the patient?
Can this medication improve other symptoms or conditions the patient has?
In addition, see “Before prescribing antiobesity medication . . .”
SIDEBAR Before prescribing antiobesity medication . . .Have a frank discussion with the patient and be sure to cover the following points:
The rationale for pharmacologic treatment is to counteract adaptive physiologic responses, which increase appetite and reduce energy expenditure, in response to diet-induced weight loss.
Antiobesity medication is only one component of a comprehensive treatment plan, which also includes diet, physical activity, and behavior modification.
Antiobesity agents are intended for long-term use, as obesity is a chronic disease. If/when you stop the medication, there may be some weight regain, similar to an increase in blood pressure after discontinuing an antihypertensive agent.
Because antiobesity medications improve many parameters including glucose/hemoglobin A1c, lipids, blood pressure, and waist circumference, it is possible that the addition of one antiobesity medication can reduce, or even eliminate, the need for several other medications.
Remember that many patients who present for obesity management have experienced weight bias. It is important to not be judgmental, but rather explain why obesity is a chronic disease. If patients understand the physiology of their condition, they will understand that their limited success with weight loss in the past is not just a matter of willpower. Lifestyle change and weight loss are extremely difficult, so it is important to provide encouragement and support for ongoing behavioral modification.
Phentermine/topiramate ER is a good first choice for this young patient with class I (BMI 30-34.9 kg/m2) obesity and migraines, as she can likely tolerate a stimulant and her migraines might improve with topiramate. Before starting the medication, ask about insomnia and nephrolithiasis in addition to anxiety and other contraindications (ie, glaucoma, hyperthyroidism, recent monoamine oxidase inhibitor use, or a known hypersensitivity or idiosyncrasy to sympathomimetic amines).23The most common adverse events reported in phase III trials were dry mouth, paresthesia, and constipation.24-26
Not for pregnant women.Women of childbearing age must have a negative pregnancy test before starting phentermine/topiramate ER and every month while taking the medication. The FDA requires a Risk Evaluation and Mitigation Strategy (REMS) to inform prescribers and patients about the increased risk of congenital malformation, specifically orofacial clefts, in infants exposed to topiramate during the first trimester of pregnancy.42 REMS focuses on the importance of pregnancy prevention, the consistent use of birth control, and the need to discontinue phentermine/topiramate ER immediately if pregnancy occurs.
Flexible dosing. Phentermine/topiramate ER is available in 4 dosages: phentermine 3.75 mg/topiramate 23 mg ER; phentermine 7.5 mg/topiramate 46 mg ER; phentermine 11.25 mg/topiramate 69 mg ER; and phentermine 15 mg/topiramate 92 mg ER. Gradual dose escalation minimizes risks and adverse events.23
Monitor patients frequently to evaluate for adverse effects and ensure adherence to diet, exercise, and lifestyle modifications. If weight loss is slower or less robust than expected, check for dietary indiscretion, as medications have limited efficacy without appropriate behavioral changes.
Discontinue phentermine/topiramate ER if the patient does not achieve 5% weight loss after 12 weeks on the maximum dose, as it is unlikely that she will achieve and sustain clinically meaningful weight loss with continued treatment.23 In this case, consider another agent with a different mechanism of action. Any of the other antiobesity medications could be appropriate for this patient.
CASE 2 Norman S, a 52-year-old overweight man (BMI 29 kg/m2) with type 2 diabetes, hyperlipidemia, osteoarthritis, and glaucoma, has recently hit a plateau with his weight loss. He lost 45 pounds secondary to diet and exercise, but hasn’t been able to lose any more. He also struggles with constant hunger. His medications include metformin 1000 mg bid, atorvastatin 10 mg/d, and occasional acetaminophen/oxycodone for knee pain until he undergoes a left knee replacement. Laboratory values are normal except for a hemoglobin A1c of 7.2%.
Mr. S is afraid of needles and cannot tolerate stimulants due to anxiety. Which medication is an appropriate next step for this patient?
Discussion
Lorcaserin is a good choice for this patient who is overweight and has several weight-related comorbidities. He has worked hard to lose a significant number of pounds, and is now at high risk of regaining them. That’s because his appetite has increased with his new exercise regimen, but his energy expenditure has decreased secondary to metabolic adaptation.
Narrowing the field. Naltrexone SR/bupropion SR cannot be used because of his opioid use. Phentermine/topiramate ER is contraindicated for patients with glaucoma, and liraglutide 3 mg is not appropriate given the patient’s fear of needles.
He could try orlistat, especially if he struggles with constipation, but the gastrointestinal adverse effects are difficult for many patients to tolerate. While not an antiobesity medication, a sodium-glucose co-transporter 2 (SGLT2) inhibitor could be prescribed for his diabetes and may also promote weight loss.43
An appealing choice.The glucose-lowering effect of lorcaserin could provide an added benefit for the patient. The BLOOM-DM (Behavioral modification and lorcaserin for overweight and obesity management in diabetes mellitus) study reported a mean reduction in hemoglobin A1c of 0.9% in the treatment group compared with a 0.4% reduction in the placebo group,30 and the effect of lorcaserin on A1c appeared to be independent of weight loss.
Mechanism of action: Cause for concern?Although lorcaserin selectively binds to serotonin 5-HT2C receptors, the theoretical risk of cardiac valvulopathy was evaluated in phase III studies, as fenfluramine, a 5-HT2B-receptor agonist, was withdrawn from the US market in 1997 for this reason.44 Both the BLOOM (Behavioral modification and lorcaserin for overweight and obesity management) and BLOSSOM (Behavioral modification and lorcaserin second study for obesity management) studies found that lorcaserin did not increase the incidence of FDA-defined cardiac valvulopathy.28,29
Formulations/adverse effects. Lorcaserin is available in 2 formulations: 10-mg tablets, which are taken twice daily, or 20-mg XR tablets, which are taken once daily. Both are generally well tolerated.27,45 The most common adverse event reported in phase III trials was headache.28,30,43 Discontinue lorcaserin if the patient does not lose 5% of his initial weight after 12 weeks, as weight loss at this stage is a good predictor of longer-term success.46
Some patients don’t respond.Interestingly, a subset of patients do not respond to lorcaserin. The most likely explanation for different responses to the medication is that there are many causes of obesity, only some of which respond to 5-HT2C agonism. Currently, we do not perform pharmacogenomic testing before prescribing lorcaserin, but perhaps an inexpensive test to identify responders will be available in the future.
CASE 3 Kathryn M, a 38-year-old woman with obesity (BMI 42 kg/m2), obstructive sleep apnea, gastroesophageal reflux disease, and depression, is eager to get better control over her weight. Her medications include lansoprazole 30 mg/d and a multivitamin. She reports constantly thinking about food and not being able to control her impulses to buy large quantities of unhealthy snacks. She is so preoccupied by thoughts of food that she has difficulty concentrating at work.
Naltrexone SR/bupropion SR is a good choice for patients who describe debilitating cravings and addictive behavior surrounding food.
Ms. M smokes a quarter of a pack of cigarettes daily, but she is ready to quit. She views bariatric surgery as a “last resort” and has no anxiety, pain, or history of seizures. Which medication is appropriate for this patient?
Discussion
This patient with class III obesity (BMI ≥40 kg/m2) is eligible for bariatric surgery; however, she is not interested in pursuing it at this time. It is important to discuss all of her options before deciding on a treatment plan. For patients like Ms. M, who would benefit from more than modest weight loss, consider a multidisciplinary approach including lifestyle modifications, pharmacotherapy, devices (eg, an intragastric balloon), and/or surgery. You would need to make clear to Ms. M that she may still be eligible for insurance coverage for surgery if she changes her mind after pursuing other treatments as long as her BMI remains ≥35 kg/m2 with obesity-related comorbidities.
Naltrexone SR/bupropion SR is a good choice for Ms. M because she describes debilitating cravings and addictive behavior surrounding food. Patients taking naltrexone SR/bupropion SR in the Contrave Obesity Research (COR)-I and COR-II phase III trials experienced a reduced frequency of food cravings, reduced difficulty in resisting food cravings, and an increased ability to control eating compared with those assigned to placebo.32,33
Added benefits.Bupropion could also help Ms. M quit smoking and improve her mood, as it is FDA-approved for smoking cessation and depression. She denies anxiety and seizures, so bupropion is not contraindicated. Even if a patient denies a history of seizure, ask about any conditions that predispose to seizures, such as anorexia nervosa or bulimia or the abrupt discontinuation of alcohol, benzodiazepines, barbiturates, or antiepileptic drugs.
Opioid use.Although the patient denies pain, ask about potential opioid use, as naltrexone is an opioid receptor antagonist. Patients should be informed that opioids may be ineffective if they are required unexpectedly (eg, for trauma) and that naltrexone SR/bupropion SR should be withheld for any planned surgical procedure potentially requiring opioid use.
Other options.While naltrexone SR/bupropion SR is the most appropriate choice for this patient because it addresses Ms. M’s problematic eating behaviors while potentially improving mood and assisting with smoking cessation, phentermine/topiramate ER, lorcaserin, and liraglutide 3 mg could also be used and should certainly be tried if naltrexone SR/bupropion SR does not produce the desired weight loss.
Adverse effects. Titrate naltrexone SR/bupropion SR slowly to the treatment dose to minimize risks and adverse events.31 The most common adverse effects reported in phase III trials were nausea, constipation, and headache.34,35,45,46 Discontinue naltrexone SR/bupropion SR if the patient does not achieve 5% weight loss at 16 weeks (after 12 weeks at the maintenance dose).31
CASE 4 William P, a 65-year-old man with obesity (BMI 39 kg/m2) who underwent Roux-en-Y gastric bypass surgery and who has type 2 diabetes, congestive heart failure, coronary artery disease, hypertension, and hyperlipidemia, remains concerned about his weight. He lost 100 lbs following surgery and maintained his weight for 3 years, but then regained 30 lbs. He comes in for an office visit because he’s concerned about his increasing blood sugar and wants to prevent further weight gain. His medications include metformin 1000 mg bid, lisinopril 5 mg/d, carvedilol 12.5 mg bid, simvastatin 20 mg/d, and aspirin 81 mg/d. Laboratory test results are normal except for a hemoglobin A1c of 8%. He denies pancreatitis and a personal or family history of thyroid cancer.
Which medication is an appropriate next step for Mr. P?
Discussion
Pharmacotherapy is a great option for this patient, who is regaining weight following bariatric surgery. Phentermine/topiramate ER is the only medication that would be contraindicated because of his heart disease. Lorcaserin and naltrexone SR/bupropion SR could be considered, but liraglutide 3 mg is the most appropriate option, given his need for further glucose control.
Medication is a great option for patients who are regaining weight after bariatric surgery.
Furthermore, the recent LEADER (Liraglutide effect and action in diabetes: evaluation of CV outcome results) trial reported a significant mortality benefit with liraglutide 1.8 mg/d among patients with type 2 diabetes and high CV risk.47 The study found that liraglutide was superior to placebo in reducing CV events.
Contraindications. Ask patients about a history of pancreatitis before starting liraglutide 3 mg given the possible increased risk. In addition, liraglutide is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with multiple endocrine neoplasia syndrome type 2. Thyroid C-cell tumors have been found in rodents given supratherapeutic doses of liraglutide;48however, there is no evidence of liraglutide causing C-cell tumors in humans.
For patients taking a medication that can cause hypoglycemia, such as insulin or a sulfonylurea, monitor blood sugar and consider reducing the dose of that medication when starting liraglutide.
Administration and titration.Liraglutide is injected subcutaneously once daily. The dose is titrated up weekly to reduce gastrointestinal symptoms.36 The most common adverse effects reported in phase III trials were nausea, diarrhea, and constipation.37-39 Discontinue liraglutide 3 mg if the patient does not lose at least 4% of baseline body weight after 16 weeks.49
CORRESPONDENCE Katherine H. Saunders, MD, DABOM, Comprehensive Weight Control Center, Division of Endocrinology, Diabetes and Metabolism, Weill Cornell Medicine, 1165 York Avenue, New York, NY 10065; kph2001@med.cornell.edu.
References
1. Wing RR, Lang W, Wadden TA, et al. Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care. 2011;34:1481-1486.
2. Magkos F, Fraterrigo G, Yoshino J. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. 2016;23:591-601.
3. Jensen MD, Ryan DH, Apovian CM, et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol. 2014;63(25 Pt B):2985-3023.
4. Sumithran P, Predergast LA, Delbridge E, et al. Long-term persistence of hormonal adaptations to weight loss. N Engl J Med. 2011;365:1597-1604.
5. Greenway FL. Physiological adaptations to weight loss and factors favouring weight regain.Int J Obes (Lond). 2015;39:1188-1196.
6. Fothergill E, Guo J, Howard L, et al. Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity (Silver Spring). 2016;24:1612-1619.
7. Apovian CM, Aronne LJ, Bessesen DH, et al. Pharmacological management of obesity: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015;100:342-362.
8. Saunders KH, Shukla AP, Igel LI, et al. Pharmacotherapy for obesity. Endocrinol Metab Clin North Am. 2016;45:521-538.
9. Saunders KH, Kumar RB, Igel LI, et al. Pharmacologic approaches to weight management: recent gains and shortfalls in combating obesity. Curr Atheroscler Rep. 2016;18:36.
11. Arena Pharmaceuticals. Arena Pharmaceuticals and Eisai announce FDA approval of BELVIQ® (lorcaserin HCl) for chronic weight management in adults who are overweight with a comorbidity or obese. Available at: http://invest.arenapharm.com/releasedetail.cfm?ReleaseID=687182. Accessed August 28, 2017.
19. Aronne LJ, Wadden TA, Peterson C, et al. Evaluation of phentermine and topiramate versus phentermine/topiramate extended-release in obese adults. Obesity (Silver Spring). 2013;21:2163-2171.
22. Torgerson JS, Hauptman J, Boldrin MN, et al. XENical in the prevention of Diabetes in Obese Subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27:155-161.
24. Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20:330-342.
25. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomized, placebo-controlled, phase 3 trial. Lancet. 2011;377:1341-1352.
26. Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95:297-308.
28. Smith SR, Weissman NJ, Anderson CM, et al. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363:245-256.
29. Fidler MC, Sanchez M, Raether B, et al. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96:3067-3077.
30. O’Neil PM, Smith SR, Weissman NJ, et al. Randomized placebo controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring). 2012;20:1426-1436.
32. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376:595-605.
33. Apovian CM, Aronne L, Rubino D, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring). 2013;21:935-943.
34. Wadden TA, Foreyt JP, Foster GD, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring). 2011;19:110-120.
35. Hollander P, Gupta AK, Plodkowski R, et al. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36:4022-4029.
37. Pi-Sunyer X, Astrup A, Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373:11-22.
38. Davies MJ, Bergenstal R, Bode B, et al. Efficacy of liraglutide for weight loss among patients with type 2 diabetes: the SCALE Diabetes randomized clinical trial. JAMA. 2015;314:687-699.
39. Wadden TA, Hollander P, Klein S, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37:1443-1451.
40. Saunders KH, Igel LI, Aronne LJ. An update on naltrexone/bupropion extended-release in the treatment of obesity. Expert Opin Pharmacother. 2016. [Epub ahead of print]
41. Thomas CE, Mauer EA, Shukla AP, et al. Low adoption of weight loss medications: a comparison of prescribing patterns of antiobesity pharmacotherapies and SGLT2s. Obesity (Silver Spring). 2016;24:1955-1961.
42. Qsymia Risk Evaluation and Mitigation Strategy (REMS). VIVUS, Inc. Available at: http://www.qsymiarems.com. Accessed January 16, 2017.
43. Zinman B, Wanner C, Lachin JM, et al. Empaglifozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
46. Smith SR, O’Neil PM, Astrup A. Early weight loss while on lorcaserin, diet and exercise as a predictor of week 52 weight-loss outcomes. Obesity (Silver Spring). 2014;22:2137-2146.
47. Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
48. Madsen LW, Knauf JA, Gotfredsen C, et al. GLP-1 receptor agonists and the thyroid: C-cell effects in mice are mediated via the GLP-1 receptor and not associated with RET activation. Endocrinology. 2012;153:1538-1547.
49. Fujioka K, O’Neil PM, Davies M, et al. Early weight loss with liraglutide 3.0 mg predicts 1-year weight loss and is associated with improvements in clinical markers. Obesity (Silver Spring). 2016;24:2278-2288.
Comprehensive Weight Control Center, Division of Endocrinology, Diabetes and Metabolism, Cornell Medicine, New York, NY kph2001@med.cornell.edu
Drs. Saunders, Shukla, and Igel reported no potential conflicts of interest relevant to this article. Dr. Aronne reported various financial relationships with Aspire Bariatrics, AstraZeneca, BMIQ, Eisai, Gelesis, GI Dynamics, Jamieson Laboratories, Janssen Pharmaceuticals, MYOS RENS Technology Inc., Novo Nordisk, Pfizer, Real Appeal, UnitedHealth Group Ventures, and Zafgen.
Comprehensive Weight Control Center, Division of Endocrinology, Diabetes and Metabolism, Cornell Medicine, New York, NY kph2001@med.cornell.edu
Drs. Saunders, Shukla, and Igel reported no potential conflicts of interest relevant to this article. Dr. Aronne reported various financial relationships with Aspire Bariatrics, AstraZeneca, BMIQ, Eisai, Gelesis, GI Dynamics, Jamieson Laboratories, Janssen Pharmaceuticals, MYOS RENS Technology Inc., Novo Nordisk, Pfizer, Real Appeal, UnitedHealth Group Ventures, and Zafgen.
Author and Disclosure Information
Comprehensive Weight Control Center, Division of Endocrinology, Diabetes and Metabolism, Cornell Medicine, New York, NY kph2001@med.cornell.edu
Drs. Saunders, Shukla, and Igel reported no potential conflicts of interest relevant to this article. Dr. Aronne reported various financial relationships with Aspire Bariatrics, AstraZeneca, BMIQ, Eisai, Gelesis, GI Dynamics, Jamieson Laboratories, Janssen Pharmaceuticals, MYOS RENS Technology Inc., Novo Nordisk, Pfizer, Real Appeal, UnitedHealth Group Ventures, and Zafgen.
Modest weight loss of 5% to 10% among patients who are overweight or obese can result in a clinically relevant reduction in cardiovascular (CV) disease risk.1 This amount of weight loss can increase insulin sensitivity in adipose tissue, liver, and muscle, and have a positive impact on blood sugar, blood pressure, triglycerides, and high-density lipoprotein cholesterol.1,2
All patients who are obese or overweight with increased CV risk should be counseled on diet, exercise, and other behavioral interventions.3 Weight loss secondary to lifestyle modification alone, however, leads to adaptive physiologic responses, which increase appetite and reduce energy expenditure.4-6
Pharmacotherapy can counteract this metabolic adaptation and lead to sustained weight loss. Antiobesity medication can be considered if a patient has a body mass index (BMI) ≥30 kg/m2 or ≥27 kg/m2 with obesity-related comorbidities such as hypertension, type 2 diabetes, dyslipidemia, or obstructive sleep apnea.3,7
Until recently, there were few pharmacologic options approved by the US Food and Drug Administration (FDA) for the management of obesity. The mainstays of treatment were phentermine (Adipex-P, Ionamin, Suprenza) and orlistat (Alli, Xenical). Since 2012, however, 4 agents have been approved as adjuncts to a reduced-calorie diet and increased physical activity for long-term weight management.8,9 Phentermine/topiramate extended-release (ER) (Qsymia) and lorcaserin (Belviq) were approved in 2012,10,11 and naltrexone sustained release (SR)/bupropion SR (Contrave) and liraglutide 3 mg (Saxenda) were approved in 201412,13 (TABLE9,14-39). These medications have the potential to not only limit weight gain, but also promote weight loss and, thus, improve blood pressure, cholesterol, glucose, and insulin.40
Despite the growing obesity epidemic and the availability of several additional medications for chronic weight management, use of antiobesity pharmacotherapy has been limited. Barriers to use include inadequate training of health care professionals, poor insurance coverage for new agents, and low reimbursement for office visits to address weight.41
Weight loss secondary to lifestyle changes can lead to adaptive physiologic responses, which increase appetite and reduce energy expenditure. Pharmacotherapy can counteract this.
In addition, the number of obesity medicine specialists, while increasing, is still not sufficient. Therefore, it is imperative for other health care professionals—namely family practitioners—to be aware of the treatment options available to patients who are overweight or obese and to be adept at using them.
In this review, we present 4 cases that depict patients who could benefit from the addition of antiobesity pharmacotherapy to a comprehensive treatment plan that includes diet, physical activity, and behavioral modification.
[polldaddy:9840472]
CASE 1 Melissa C, a 27-year-old woman with obesity (BMI 33 kg/m2), hyperlipidemia, and migraine headaches, presents for weight management. Despite a calorie-reduced diet and 200 minutes per week of exercise for the past 6 months, she has been unable to lose weight. The only medications she’s taking are oral contraceptive pills and sumatriptan, as needed. She suffers from migraines 3 times a month and has no anxiety. Laboratory test results are normal with the exception of an elevated low-density lipoprotein (LDL) level.
Which medication is an appropriate next step for Ms. C?
Discussion
When considering an antiobesity agent for any patient, there are 2 important questions to ask:
Are there contraindications, drug-drug interactions, or undesirable adverse effects associated with this medication that could be problematic for the patient?
Can this medication improve other symptoms or conditions the patient has?
In addition, see “Before prescribing antiobesity medication . . .”
SIDEBAR Before prescribing antiobesity medication . . .Have a frank discussion with the patient and be sure to cover the following points:
The rationale for pharmacologic treatment is to counteract adaptive physiologic responses, which increase appetite and reduce energy expenditure, in response to diet-induced weight loss.
Antiobesity medication is only one component of a comprehensive treatment plan, which also includes diet, physical activity, and behavior modification.
Antiobesity agents are intended for long-term use, as obesity is a chronic disease. If/when you stop the medication, there may be some weight regain, similar to an increase in blood pressure after discontinuing an antihypertensive agent.
Because antiobesity medications improve many parameters including glucose/hemoglobin A1c, lipids, blood pressure, and waist circumference, it is possible that the addition of one antiobesity medication can reduce, or even eliminate, the need for several other medications.
Remember that many patients who present for obesity management have experienced weight bias. It is important to not be judgmental, but rather explain why obesity is a chronic disease. If patients understand the physiology of their condition, they will understand that their limited success with weight loss in the past is not just a matter of willpower. Lifestyle change and weight loss are extremely difficult, so it is important to provide encouragement and support for ongoing behavioral modification.
Phentermine/topiramate ER is a good first choice for this young patient with class I (BMI 30-34.9 kg/m2) obesity and migraines, as she can likely tolerate a stimulant and her migraines might improve with topiramate. Before starting the medication, ask about insomnia and nephrolithiasis in addition to anxiety and other contraindications (ie, glaucoma, hyperthyroidism, recent monoamine oxidase inhibitor use, or a known hypersensitivity or idiosyncrasy to sympathomimetic amines).23The most common adverse events reported in phase III trials were dry mouth, paresthesia, and constipation.24-26
Not for pregnant women.Women of childbearing age must have a negative pregnancy test before starting phentermine/topiramate ER and every month while taking the medication. The FDA requires a Risk Evaluation and Mitigation Strategy (REMS) to inform prescribers and patients about the increased risk of congenital malformation, specifically orofacial clefts, in infants exposed to topiramate during the first trimester of pregnancy.42 REMS focuses on the importance of pregnancy prevention, the consistent use of birth control, and the need to discontinue phentermine/topiramate ER immediately if pregnancy occurs.
Flexible dosing. Phentermine/topiramate ER is available in 4 dosages: phentermine 3.75 mg/topiramate 23 mg ER; phentermine 7.5 mg/topiramate 46 mg ER; phentermine 11.25 mg/topiramate 69 mg ER; and phentermine 15 mg/topiramate 92 mg ER. Gradual dose escalation minimizes risks and adverse events.23
Monitor patients frequently to evaluate for adverse effects and ensure adherence to diet, exercise, and lifestyle modifications. If weight loss is slower or less robust than expected, check for dietary indiscretion, as medications have limited efficacy without appropriate behavioral changes.
Discontinue phentermine/topiramate ER if the patient does not achieve 5% weight loss after 12 weeks on the maximum dose, as it is unlikely that she will achieve and sustain clinically meaningful weight loss with continued treatment.23 In this case, consider another agent with a different mechanism of action. Any of the other antiobesity medications could be appropriate for this patient.
CASE 2 Norman S, a 52-year-old overweight man (BMI 29 kg/m2) with type 2 diabetes, hyperlipidemia, osteoarthritis, and glaucoma, has recently hit a plateau with his weight loss. He lost 45 pounds secondary to diet and exercise, but hasn’t been able to lose any more. He also struggles with constant hunger. His medications include metformin 1000 mg bid, atorvastatin 10 mg/d, and occasional acetaminophen/oxycodone for knee pain until he undergoes a left knee replacement. Laboratory values are normal except for a hemoglobin A1c of 7.2%.
Mr. S is afraid of needles and cannot tolerate stimulants due to anxiety. Which medication is an appropriate next step for this patient?
Discussion
Lorcaserin is a good choice for this patient who is overweight and has several weight-related comorbidities. He has worked hard to lose a significant number of pounds, and is now at high risk of regaining them. That’s because his appetite has increased with his new exercise regimen, but his energy expenditure has decreased secondary to metabolic adaptation.
Narrowing the field. Naltrexone SR/bupropion SR cannot be used because of his opioid use. Phentermine/topiramate ER is contraindicated for patients with glaucoma, and liraglutide 3 mg is not appropriate given the patient’s fear of needles.
He could try orlistat, especially if he struggles with constipation, but the gastrointestinal adverse effects are difficult for many patients to tolerate. While not an antiobesity medication, a sodium-glucose co-transporter 2 (SGLT2) inhibitor could be prescribed for his diabetes and may also promote weight loss.43
An appealing choice.The glucose-lowering effect of lorcaserin could provide an added benefit for the patient. The BLOOM-DM (Behavioral modification and lorcaserin for overweight and obesity management in diabetes mellitus) study reported a mean reduction in hemoglobin A1c of 0.9% in the treatment group compared with a 0.4% reduction in the placebo group,30 and the effect of lorcaserin on A1c appeared to be independent of weight loss.
Mechanism of action: Cause for concern?Although lorcaserin selectively binds to serotonin 5-HT2C receptors, the theoretical risk of cardiac valvulopathy was evaluated in phase III studies, as fenfluramine, a 5-HT2B-receptor agonist, was withdrawn from the US market in 1997 for this reason.44 Both the BLOOM (Behavioral modification and lorcaserin for overweight and obesity management) and BLOSSOM (Behavioral modification and lorcaserin second study for obesity management) studies found that lorcaserin did not increase the incidence of FDA-defined cardiac valvulopathy.28,29
Formulations/adverse effects. Lorcaserin is available in 2 formulations: 10-mg tablets, which are taken twice daily, or 20-mg XR tablets, which are taken once daily. Both are generally well tolerated.27,45 The most common adverse event reported in phase III trials was headache.28,30,43 Discontinue lorcaserin if the patient does not lose 5% of his initial weight after 12 weeks, as weight loss at this stage is a good predictor of longer-term success.46
Some patients don’t respond.Interestingly, a subset of patients do not respond to lorcaserin. The most likely explanation for different responses to the medication is that there are many causes of obesity, only some of which respond to 5-HT2C agonism. Currently, we do not perform pharmacogenomic testing before prescribing lorcaserin, but perhaps an inexpensive test to identify responders will be available in the future.
CASE 3 Kathryn M, a 38-year-old woman with obesity (BMI 42 kg/m2), obstructive sleep apnea, gastroesophageal reflux disease, and depression, is eager to get better control over her weight. Her medications include lansoprazole 30 mg/d and a multivitamin. She reports constantly thinking about food and not being able to control her impulses to buy large quantities of unhealthy snacks. She is so preoccupied by thoughts of food that she has difficulty concentrating at work.
Naltrexone SR/bupropion SR is a good choice for patients who describe debilitating cravings and addictive behavior surrounding food.
Ms. M smokes a quarter of a pack of cigarettes daily, but she is ready to quit. She views bariatric surgery as a “last resort” and has no anxiety, pain, or history of seizures. Which medication is appropriate for this patient?
Discussion
This patient with class III obesity (BMI ≥40 kg/m2) is eligible for bariatric surgery; however, she is not interested in pursuing it at this time. It is important to discuss all of her options before deciding on a treatment plan. For patients like Ms. M, who would benefit from more than modest weight loss, consider a multidisciplinary approach including lifestyle modifications, pharmacotherapy, devices (eg, an intragastric balloon), and/or surgery. You would need to make clear to Ms. M that she may still be eligible for insurance coverage for surgery if she changes her mind after pursuing other treatments as long as her BMI remains ≥35 kg/m2 with obesity-related comorbidities.
Naltrexone SR/bupropion SR is a good choice for Ms. M because she describes debilitating cravings and addictive behavior surrounding food. Patients taking naltrexone SR/bupropion SR in the Contrave Obesity Research (COR)-I and COR-II phase III trials experienced a reduced frequency of food cravings, reduced difficulty in resisting food cravings, and an increased ability to control eating compared with those assigned to placebo.32,33
Added benefits.Bupropion could also help Ms. M quit smoking and improve her mood, as it is FDA-approved for smoking cessation and depression. She denies anxiety and seizures, so bupropion is not contraindicated. Even if a patient denies a history of seizure, ask about any conditions that predispose to seizures, such as anorexia nervosa or bulimia or the abrupt discontinuation of alcohol, benzodiazepines, barbiturates, or antiepileptic drugs.
Opioid use.Although the patient denies pain, ask about potential opioid use, as naltrexone is an opioid receptor antagonist. Patients should be informed that opioids may be ineffective if they are required unexpectedly (eg, for trauma) and that naltrexone SR/bupropion SR should be withheld for any planned surgical procedure potentially requiring opioid use.
Other options.While naltrexone SR/bupropion SR is the most appropriate choice for this patient because it addresses Ms. M’s problematic eating behaviors while potentially improving mood and assisting with smoking cessation, phentermine/topiramate ER, lorcaserin, and liraglutide 3 mg could also be used and should certainly be tried if naltrexone SR/bupropion SR does not produce the desired weight loss.
Adverse effects. Titrate naltrexone SR/bupropion SR slowly to the treatment dose to minimize risks and adverse events.31 The most common adverse effects reported in phase III trials were nausea, constipation, and headache.34,35,45,46 Discontinue naltrexone SR/bupropion SR if the patient does not achieve 5% weight loss at 16 weeks (after 12 weeks at the maintenance dose).31
CASE 4 William P, a 65-year-old man with obesity (BMI 39 kg/m2) who underwent Roux-en-Y gastric bypass surgery and who has type 2 diabetes, congestive heart failure, coronary artery disease, hypertension, and hyperlipidemia, remains concerned about his weight. He lost 100 lbs following surgery and maintained his weight for 3 years, but then regained 30 lbs. He comes in for an office visit because he’s concerned about his increasing blood sugar and wants to prevent further weight gain. His medications include metformin 1000 mg bid, lisinopril 5 mg/d, carvedilol 12.5 mg bid, simvastatin 20 mg/d, and aspirin 81 mg/d. Laboratory test results are normal except for a hemoglobin A1c of 8%. He denies pancreatitis and a personal or family history of thyroid cancer.
Which medication is an appropriate next step for Mr. P?
Discussion
Pharmacotherapy is a great option for this patient, who is regaining weight following bariatric surgery. Phentermine/topiramate ER is the only medication that would be contraindicated because of his heart disease. Lorcaserin and naltrexone SR/bupropion SR could be considered, but liraglutide 3 mg is the most appropriate option, given his need for further glucose control.
Medication is a great option for patients who are regaining weight after bariatric surgery.
Furthermore, the recent LEADER (Liraglutide effect and action in diabetes: evaluation of CV outcome results) trial reported a significant mortality benefit with liraglutide 1.8 mg/d among patients with type 2 diabetes and high CV risk.47 The study found that liraglutide was superior to placebo in reducing CV events.
Contraindications. Ask patients about a history of pancreatitis before starting liraglutide 3 mg given the possible increased risk. In addition, liraglutide is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with multiple endocrine neoplasia syndrome type 2. Thyroid C-cell tumors have been found in rodents given supratherapeutic doses of liraglutide;48however, there is no evidence of liraglutide causing C-cell tumors in humans.
For patients taking a medication that can cause hypoglycemia, such as insulin or a sulfonylurea, monitor blood sugar and consider reducing the dose of that medication when starting liraglutide.
Administration and titration.Liraglutide is injected subcutaneously once daily. The dose is titrated up weekly to reduce gastrointestinal symptoms.36 The most common adverse effects reported in phase III trials were nausea, diarrhea, and constipation.37-39 Discontinue liraglutide 3 mg if the patient does not lose at least 4% of baseline body weight after 16 weeks.49
CORRESPONDENCE Katherine H. Saunders, MD, DABOM, Comprehensive Weight Control Center, Division of Endocrinology, Diabetes and Metabolism, Weill Cornell Medicine, 1165 York Avenue, New York, NY 10065; kph2001@med.cornell.edu.
Modest weight loss of 5% to 10% among patients who are overweight or obese can result in a clinically relevant reduction in cardiovascular (CV) disease risk.1 This amount of weight loss can increase insulin sensitivity in adipose tissue, liver, and muscle, and have a positive impact on blood sugar, blood pressure, triglycerides, and high-density lipoprotein cholesterol.1,2
All patients who are obese or overweight with increased CV risk should be counseled on diet, exercise, and other behavioral interventions.3 Weight loss secondary to lifestyle modification alone, however, leads to adaptive physiologic responses, which increase appetite and reduce energy expenditure.4-6
Pharmacotherapy can counteract this metabolic adaptation and lead to sustained weight loss. Antiobesity medication can be considered if a patient has a body mass index (BMI) ≥30 kg/m2 or ≥27 kg/m2 with obesity-related comorbidities such as hypertension, type 2 diabetes, dyslipidemia, or obstructive sleep apnea.3,7
Until recently, there were few pharmacologic options approved by the US Food and Drug Administration (FDA) for the management of obesity. The mainstays of treatment were phentermine (Adipex-P, Ionamin, Suprenza) and orlistat (Alli, Xenical). Since 2012, however, 4 agents have been approved as adjuncts to a reduced-calorie diet and increased physical activity for long-term weight management.8,9 Phentermine/topiramate extended-release (ER) (Qsymia) and lorcaserin (Belviq) were approved in 2012,10,11 and naltrexone sustained release (SR)/bupropion SR (Contrave) and liraglutide 3 mg (Saxenda) were approved in 201412,13 (TABLE9,14-39). These medications have the potential to not only limit weight gain, but also promote weight loss and, thus, improve blood pressure, cholesterol, glucose, and insulin.40
Despite the growing obesity epidemic and the availability of several additional medications for chronic weight management, use of antiobesity pharmacotherapy has been limited. Barriers to use include inadequate training of health care professionals, poor insurance coverage for new agents, and low reimbursement for office visits to address weight.41
Weight loss secondary to lifestyle changes can lead to adaptive physiologic responses, which increase appetite and reduce energy expenditure. Pharmacotherapy can counteract this.
In addition, the number of obesity medicine specialists, while increasing, is still not sufficient. Therefore, it is imperative for other health care professionals—namely family practitioners—to be aware of the treatment options available to patients who are overweight or obese and to be adept at using them.
In this review, we present 4 cases that depict patients who could benefit from the addition of antiobesity pharmacotherapy to a comprehensive treatment plan that includes diet, physical activity, and behavioral modification.
[polldaddy:9840472]
CASE 1 Melissa C, a 27-year-old woman with obesity (BMI 33 kg/m2), hyperlipidemia, and migraine headaches, presents for weight management. Despite a calorie-reduced diet and 200 minutes per week of exercise for the past 6 months, she has been unable to lose weight. The only medications she’s taking are oral contraceptive pills and sumatriptan, as needed. She suffers from migraines 3 times a month and has no anxiety. Laboratory test results are normal with the exception of an elevated low-density lipoprotein (LDL) level.
Which medication is an appropriate next step for Ms. C?
Discussion
When considering an antiobesity agent for any patient, there are 2 important questions to ask:
Are there contraindications, drug-drug interactions, or undesirable adverse effects associated with this medication that could be problematic for the patient?
Can this medication improve other symptoms or conditions the patient has?
In addition, see “Before prescribing antiobesity medication . . .”
SIDEBAR Before prescribing antiobesity medication . . .Have a frank discussion with the patient and be sure to cover the following points:
The rationale for pharmacologic treatment is to counteract adaptive physiologic responses, which increase appetite and reduce energy expenditure, in response to diet-induced weight loss.
Antiobesity medication is only one component of a comprehensive treatment plan, which also includes diet, physical activity, and behavior modification.
Antiobesity agents are intended for long-term use, as obesity is a chronic disease. If/when you stop the medication, there may be some weight regain, similar to an increase in blood pressure after discontinuing an antihypertensive agent.
Because antiobesity medications improve many parameters including glucose/hemoglobin A1c, lipids, blood pressure, and waist circumference, it is possible that the addition of one antiobesity medication can reduce, or even eliminate, the need for several other medications.
Remember that many patients who present for obesity management have experienced weight bias. It is important to not be judgmental, but rather explain why obesity is a chronic disease. If patients understand the physiology of their condition, they will understand that their limited success with weight loss in the past is not just a matter of willpower. Lifestyle change and weight loss are extremely difficult, so it is important to provide encouragement and support for ongoing behavioral modification.
Phentermine/topiramate ER is a good first choice for this young patient with class I (BMI 30-34.9 kg/m2) obesity and migraines, as she can likely tolerate a stimulant and her migraines might improve with topiramate. Before starting the medication, ask about insomnia and nephrolithiasis in addition to anxiety and other contraindications (ie, glaucoma, hyperthyroidism, recent monoamine oxidase inhibitor use, or a known hypersensitivity or idiosyncrasy to sympathomimetic amines).23The most common adverse events reported in phase III trials were dry mouth, paresthesia, and constipation.24-26
Not for pregnant women.Women of childbearing age must have a negative pregnancy test before starting phentermine/topiramate ER and every month while taking the medication. The FDA requires a Risk Evaluation and Mitigation Strategy (REMS) to inform prescribers and patients about the increased risk of congenital malformation, specifically orofacial clefts, in infants exposed to topiramate during the first trimester of pregnancy.42 REMS focuses on the importance of pregnancy prevention, the consistent use of birth control, and the need to discontinue phentermine/topiramate ER immediately if pregnancy occurs.
Flexible dosing. Phentermine/topiramate ER is available in 4 dosages: phentermine 3.75 mg/topiramate 23 mg ER; phentermine 7.5 mg/topiramate 46 mg ER; phentermine 11.25 mg/topiramate 69 mg ER; and phentermine 15 mg/topiramate 92 mg ER. Gradual dose escalation minimizes risks and adverse events.23
Monitor patients frequently to evaluate for adverse effects and ensure adherence to diet, exercise, and lifestyle modifications. If weight loss is slower or less robust than expected, check for dietary indiscretion, as medications have limited efficacy without appropriate behavioral changes.
Discontinue phentermine/topiramate ER if the patient does not achieve 5% weight loss after 12 weeks on the maximum dose, as it is unlikely that she will achieve and sustain clinically meaningful weight loss with continued treatment.23 In this case, consider another agent with a different mechanism of action. Any of the other antiobesity medications could be appropriate for this patient.
CASE 2 Norman S, a 52-year-old overweight man (BMI 29 kg/m2) with type 2 diabetes, hyperlipidemia, osteoarthritis, and glaucoma, has recently hit a plateau with his weight loss. He lost 45 pounds secondary to diet and exercise, but hasn’t been able to lose any more. He also struggles with constant hunger. His medications include metformin 1000 mg bid, atorvastatin 10 mg/d, and occasional acetaminophen/oxycodone for knee pain until he undergoes a left knee replacement. Laboratory values are normal except for a hemoglobin A1c of 7.2%.
Mr. S is afraid of needles and cannot tolerate stimulants due to anxiety. Which medication is an appropriate next step for this patient?
Discussion
Lorcaserin is a good choice for this patient who is overweight and has several weight-related comorbidities. He has worked hard to lose a significant number of pounds, and is now at high risk of regaining them. That’s because his appetite has increased with his new exercise regimen, but his energy expenditure has decreased secondary to metabolic adaptation.
Narrowing the field. Naltrexone SR/bupropion SR cannot be used because of his opioid use. Phentermine/topiramate ER is contraindicated for patients with glaucoma, and liraglutide 3 mg is not appropriate given the patient’s fear of needles.
He could try orlistat, especially if he struggles with constipation, but the gastrointestinal adverse effects are difficult for many patients to tolerate. While not an antiobesity medication, a sodium-glucose co-transporter 2 (SGLT2) inhibitor could be prescribed for his diabetes and may also promote weight loss.43
An appealing choice.The glucose-lowering effect of lorcaserin could provide an added benefit for the patient. The BLOOM-DM (Behavioral modification and lorcaserin for overweight and obesity management in diabetes mellitus) study reported a mean reduction in hemoglobin A1c of 0.9% in the treatment group compared with a 0.4% reduction in the placebo group,30 and the effect of lorcaserin on A1c appeared to be independent of weight loss.
Mechanism of action: Cause for concern?Although lorcaserin selectively binds to serotonin 5-HT2C receptors, the theoretical risk of cardiac valvulopathy was evaluated in phase III studies, as fenfluramine, a 5-HT2B-receptor agonist, was withdrawn from the US market in 1997 for this reason.44 Both the BLOOM (Behavioral modification and lorcaserin for overweight and obesity management) and BLOSSOM (Behavioral modification and lorcaserin second study for obesity management) studies found that lorcaserin did not increase the incidence of FDA-defined cardiac valvulopathy.28,29
Formulations/adverse effects. Lorcaserin is available in 2 formulations: 10-mg tablets, which are taken twice daily, or 20-mg XR tablets, which are taken once daily. Both are generally well tolerated.27,45 The most common adverse event reported in phase III trials was headache.28,30,43 Discontinue lorcaserin if the patient does not lose 5% of his initial weight after 12 weeks, as weight loss at this stage is a good predictor of longer-term success.46
Some patients don’t respond.Interestingly, a subset of patients do not respond to lorcaserin. The most likely explanation for different responses to the medication is that there are many causes of obesity, only some of which respond to 5-HT2C agonism. Currently, we do not perform pharmacogenomic testing before prescribing lorcaserin, but perhaps an inexpensive test to identify responders will be available in the future.
CASE 3 Kathryn M, a 38-year-old woman with obesity (BMI 42 kg/m2), obstructive sleep apnea, gastroesophageal reflux disease, and depression, is eager to get better control over her weight. Her medications include lansoprazole 30 mg/d and a multivitamin. She reports constantly thinking about food and not being able to control her impulses to buy large quantities of unhealthy snacks. She is so preoccupied by thoughts of food that she has difficulty concentrating at work.
Naltrexone SR/bupropion SR is a good choice for patients who describe debilitating cravings and addictive behavior surrounding food.
Ms. M smokes a quarter of a pack of cigarettes daily, but she is ready to quit. She views bariatric surgery as a “last resort” and has no anxiety, pain, or history of seizures. Which medication is appropriate for this patient?
Discussion
This patient with class III obesity (BMI ≥40 kg/m2) is eligible for bariatric surgery; however, she is not interested in pursuing it at this time. It is important to discuss all of her options before deciding on a treatment plan. For patients like Ms. M, who would benefit from more than modest weight loss, consider a multidisciplinary approach including lifestyle modifications, pharmacotherapy, devices (eg, an intragastric balloon), and/or surgery. You would need to make clear to Ms. M that she may still be eligible for insurance coverage for surgery if she changes her mind after pursuing other treatments as long as her BMI remains ≥35 kg/m2 with obesity-related comorbidities.
Naltrexone SR/bupropion SR is a good choice for Ms. M because she describes debilitating cravings and addictive behavior surrounding food. Patients taking naltrexone SR/bupropion SR in the Contrave Obesity Research (COR)-I and COR-II phase III trials experienced a reduced frequency of food cravings, reduced difficulty in resisting food cravings, and an increased ability to control eating compared with those assigned to placebo.32,33
Added benefits.Bupropion could also help Ms. M quit smoking and improve her mood, as it is FDA-approved for smoking cessation and depression. She denies anxiety and seizures, so bupropion is not contraindicated. Even if a patient denies a history of seizure, ask about any conditions that predispose to seizures, such as anorexia nervosa or bulimia or the abrupt discontinuation of alcohol, benzodiazepines, barbiturates, or antiepileptic drugs.
Opioid use.Although the patient denies pain, ask about potential opioid use, as naltrexone is an opioid receptor antagonist. Patients should be informed that opioids may be ineffective if they are required unexpectedly (eg, for trauma) and that naltrexone SR/bupropion SR should be withheld for any planned surgical procedure potentially requiring opioid use.
Other options.While naltrexone SR/bupropion SR is the most appropriate choice for this patient because it addresses Ms. M’s problematic eating behaviors while potentially improving mood and assisting with smoking cessation, phentermine/topiramate ER, lorcaserin, and liraglutide 3 mg could also be used and should certainly be tried if naltrexone SR/bupropion SR does not produce the desired weight loss.
Adverse effects. Titrate naltrexone SR/bupropion SR slowly to the treatment dose to minimize risks and adverse events.31 The most common adverse effects reported in phase III trials were nausea, constipation, and headache.34,35,45,46 Discontinue naltrexone SR/bupropion SR if the patient does not achieve 5% weight loss at 16 weeks (after 12 weeks at the maintenance dose).31
CASE 4 William P, a 65-year-old man with obesity (BMI 39 kg/m2) who underwent Roux-en-Y gastric bypass surgery and who has type 2 diabetes, congestive heart failure, coronary artery disease, hypertension, and hyperlipidemia, remains concerned about his weight. He lost 100 lbs following surgery and maintained his weight for 3 years, but then regained 30 lbs. He comes in for an office visit because he’s concerned about his increasing blood sugar and wants to prevent further weight gain. His medications include metformin 1000 mg bid, lisinopril 5 mg/d, carvedilol 12.5 mg bid, simvastatin 20 mg/d, and aspirin 81 mg/d. Laboratory test results are normal except for a hemoglobin A1c of 8%. He denies pancreatitis and a personal or family history of thyroid cancer.
Which medication is an appropriate next step for Mr. P?
Discussion
Pharmacotherapy is a great option for this patient, who is regaining weight following bariatric surgery. Phentermine/topiramate ER is the only medication that would be contraindicated because of his heart disease. Lorcaserin and naltrexone SR/bupropion SR could be considered, but liraglutide 3 mg is the most appropriate option, given his need for further glucose control.
Medication is a great option for patients who are regaining weight after bariatric surgery.
Furthermore, the recent LEADER (Liraglutide effect and action in diabetes: evaluation of CV outcome results) trial reported a significant mortality benefit with liraglutide 1.8 mg/d among patients with type 2 diabetes and high CV risk.47 The study found that liraglutide was superior to placebo in reducing CV events.
Contraindications. Ask patients about a history of pancreatitis before starting liraglutide 3 mg given the possible increased risk. In addition, liraglutide is contraindicated in patients with a personal or family history of medullary thyroid carcinoma or in patients with multiple endocrine neoplasia syndrome type 2. Thyroid C-cell tumors have been found in rodents given supratherapeutic doses of liraglutide;48however, there is no evidence of liraglutide causing C-cell tumors in humans.
For patients taking a medication that can cause hypoglycemia, such as insulin or a sulfonylurea, monitor blood sugar and consider reducing the dose of that medication when starting liraglutide.
Administration and titration.Liraglutide is injected subcutaneously once daily. The dose is titrated up weekly to reduce gastrointestinal symptoms.36 The most common adverse effects reported in phase III trials were nausea, diarrhea, and constipation.37-39 Discontinue liraglutide 3 mg if the patient does not lose at least 4% of baseline body weight after 16 weeks.49
CORRESPONDENCE Katherine H. Saunders, MD, DABOM, Comprehensive Weight Control Center, Division of Endocrinology, Diabetes and Metabolism, Weill Cornell Medicine, 1165 York Avenue, New York, NY 10065; kph2001@med.cornell.edu.
References
1. Wing RR, Lang W, Wadden TA, et al. Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care. 2011;34:1481-1486.
2. Magkos F, Fraterrigo G, Yoshino J. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. 2016;23:591-601.
3. Jensen MD, Ryan DH, Apovian CM, et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol. 2014;63(25 Pt B):2985-3023.
4. Sumithran P, Predergast LA, Delbridge E, et al. Long-term persistence of hormonal adaptations to weight loss. N Engl J Med. 2011;365:1597-1604.
5. Greenway FL. Physiological adaptations to weight loss and factors favouring weight regain.Int J Obes (Lond). 2015;39:1188-1196.
6. Fothergill E, Guo J, Howard L, et al. Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity (Silver Spring). 2016;24:1612-1619.
7. Apovian CM, Aronne LJ, Bessesen DH, et al. Pharmacological management of obesity: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015;100:342-362.
8. Saunders KH, Shukla AP, Igel LI, et al. Pharmacotherapy for obesity. Endocrinol Metab Clin North Am. 2016;45:521-538.
9. Saunders KH, Kumar RB, Igel LI, et al. Pharmacologic approaches to weight management: recent gains and shortfalls in combating obesity. Curr Atheroscler Rep. 2016;18:36.
11. Arena Pharmaceuticals. Arena Pharmaceuticals and Eisai announce FDA approval of BELVIQ® (lorcaserin HCl) for chronic weight management in adults who are overweight with a comorbidity or obese. Available at: http://invest.arenapharm.com/releasedetail.cfm?ReleaseID=687182. Accessed August 28, 2017.
19. Aronne LJ, Wadden TA, Peterson C, et al. Evaluation of phentermine and topiramate versus phentermine/topiramate extended-release in obese adults. Obesity (Silver Spring). 2013;21:2163-2171.
22. Torgerson JS, Hauptman J, Boldrin MN, et al. XENical in the prevention of Diabetes in Obese Subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27:155-161.
24. Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20:330-342.
25. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomized, placebo-controlled, phase 3 trial. Lancet. 2011;377:1341-1352.
26. Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95:297-308.
28. Smith SR, Weissman NJ, Anderson CM, et al. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363:245-256.
29. Fidler MC, Sanchez M, Raether B, et al. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96:3067-3077.
30. O’Neil PM, Smith SR, Weissman NJ, et al. Randomized placebo controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring). 2012;20:1426-1436.
32. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376:595-605.
33. Apovian CM, Aronne L, Rubino D, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring). 2013;21:935-943.
34. Wadden TA, Foreyt JP, Foster GD, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring). 2011;19:110-120.
35. Hollander P, Gupta AK, Plodkowski R, et al. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36:4022-4029.
37. Pi-Sunyer X, Astrup A, Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373:11-22.
38. Davies MJ, Bergenstal R, Bode B, et al. Efficacy of liraglutide for weight loss among patients with type 2 diabetes: the SCALE Diabetes randomized clinical trial. JAMA. 2015;314:687-699.
39. Wadden TA, Hollander P, Klein S, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37:1443-1451.
40. Saunders KH, Igel LI, Aronne LJ. An update on naltrexone/bupropion extended-release in the treatment of obesity. Expert Opin Pharmacother. 2016. [Epub ahead of print]
41. Thomas CE, Mauer EA, Shukla AP, et al. Low adoption of weight loss medications: a comparison of prescribing patterns of antiobesity pharmacotherapies and SGLT2s. Obesity (Silver Spring). 2016;24:1955-1961.
42. Qsymia Risk Evaluation and Mitigation Strategy (REMS). VIVUS, Inc. Available at: http://www.qsymiarems.com. Accessed January 16, 2017.
43. Zinman B, Wanner C, Lachin JM, et al. Empaglifozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
46. Smith SR, O’Neil PM, Astrup A. Early weight loss while on lorcaserin, diet and exercise as a predictor of week 52 weight-loss outcomes. Obesity (Silver Spring). 2014;22:2137-2146.
47. Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
48. Madsen LW, Knauf JA, Gotfredsen C, et al. GLP-1 receptor agonists and the thyroid: C-cell effects in mice are mediated via the GLP-1 receptor and not associated with RET activation. Endocrinology. 2012;153:1538-1547.
49. Fujioka K, O’Neil PM, Davies M, et al. Early weight loss with liraglutide 3.0 mg predicts 1-year weight loss and is associated with improvements in clinical markers. Obesity (Silver Spring). 2016;24:2278-2288.
References
1. Wing RR, Lang W, Wadden TA, et al. Benefits of modest weight loss in improving cardiovascular risk factors in overweight and obese individuals with type 2 diabetes. Diabetes Care. 2011;34:1481-1486.
2. Magkos F, Fraterrigo G, Yoshino J. Effects of moderate and subsequent progressive weight loss on metabolic function and adipose tissue biology in humans with obesity. Cell Metab. 2016;23:591-601.
3. Jensen MD, Ryan DH, Apovian CM, et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. J Am Coll Cardiol. 2014;63(25 Pt B):2985-3023.
4. Sumithran P, Predergast LA, Delbridge E, et al. Long-term persistence of hormonal adaptations to weight loss. N Engl J Med. 2011;365:1597-1604.
5. Greenway FL. Physiological adaptations to weight loss and factors favouring weight regain.Int J Obes (Lond). 2015;39:1188-1196.
6. Fothergill E, Guo J, Howard L, et al. Persistent metabolic adaptation 6 years after “The Biggest Loser” competition. Obesity (Silver Spring). 2016;24:1612-1619.
7. Apovian CM, Aronne LJ, Bessesen DH, et al. Pharmacological management of obesity: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2015;100:342-362.
8. Saunders KH, Shukla AP, Igel LI, et al. Pharmacotherapy for obesity. Endocrinol Metab Clin North Am. 2016;45:521-538.
9. Saunders KH, Kumar RB, Igel LI, et al. Pharmacologic approaches to weight management: recent gains and shortfalls in combating obesity. Curr Atheroscler Rep. 2016;18:36.
11. Arena Pharmaceuticals. Arena Pharmaceuticals and Eisai announce FDA approval of BELVIQ® (lorcaserin HCl) for chronic weight management in adults who are overweight with a comorbidity or obese. Available at: http://invest.arenapharm.com/releasedetail.cfm?ReleaseID=687182. Accessed August 28, 2017.
19. Aronne LJ, Wadden TA, Peterson C, et al. Evaluation of phentermine and topiramate versus phentermine/topiramate extended-release in obese adults. Obesity (Silver Spring). 2013;21:2163-2171.
22. Torgerson JS, Hauptman J, Boldrin MN, et al. XENical in the prevention of Diabetes in Obese Subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care. 2004;27:155-161.
24. Allison DB, Gadde KM, Garvey WT, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity (Silver Spring). 2012;20:330-342.
25. Gadde KM, Allison DB, Ryan DH, et al. Effects of low-dose, controlled-release, phentermine plus topiramate combination on weight and associated comorbidities in overweight and obese adults (CONQUER): a randomized, placebo-controlled, phase 3 trial. Lancet. 2011;377:1341-1352.
26. Garvey WT, Ryan DH, Look M, et al. Two-year sustained weight loss and metabolic benefits with controlled-release phentermine/topiramate in obese and overweight adults (SEQUEL): a randomized, placebo-controlled, phase 3 extension study. Am J Clin Nutr. 2012;95:297-308.
28. Smith SR, Weissman NJ, Anderson CM, et al. Multicenter, placebo-controlled trial of lorcaserin for weight management. N Engl J Med. 2010;363:245-256.
29. Fidler MC, Sanchez M, Raether B, et al. A one-year randomized trial of lorcaserin for weight loss in obese and overweight adults: the BLOSSOM trial. J Clin Endocrinol Metab. 2011;96:3067-3077.
30. O’Neil PM, Smith SR, Weissman NJ, et al. Randomized placebo controlled clinical trial of lorcaserin for weight loss in type 2 diabetes mellitus: the BLOOM-DM study. Obesity (Silver Spring). 2012;20:1426-1436.
32. Greenway FL, Fujioka K, Plodkowski RA, et al. Effect of naltrexone plus bupropion on weight loss in overweight and obese adults (COR-I): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2010;376:595-605.
33. Apovian CM, Aronne L, Rubino D, et al. A randomized, phase 3 trial of naltrexone SR/bupropion SR on weight and obesity-related risk factors (COR-II). Obesity (Silver Spring). 2013;21:935-943.
34. Wadden TA, Foreyt JP, Foster GD, et al. Weight loss with naltrexone SR/bupropion SR combination therapy as an adjunct to behavior modification: the COR-BMOD trial. Obesity (Silver Spring). 2011;19:110-120.
35. Hollander P, Gupta AK, Plodkowski R, et al. Effects of naltrexone sustained-release/bupropion sustained-release combination therapy on body weight and glycemic parameters in overweight and obese patients with type 2 diabetes. Diabetes Care. 2013;36:4022-4029.
37. Pi-Sunyer X, Astrup A, Fujioka K, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. 2015;373:11-22.
38. Davies MJ, Bergenstal R, Bode B, et al. Efficacy of liraglutide for weight loss among patients with type 2 diabetes: the SCALE Diabetes randomized clinical trial. JAMA. 2015;314:687-699.
39. Wadden TA, Hollander P, Klein S, et al. Weight maintenance and additional weight loss with liraglutide after low-calorie-diet induced weight loss: the SCALE Maintenance randomized study. Int J Obes (Lond). 2013;37:1443-1451.
40. Saunders KH, Igel LI, Aronne LJ. An update on naltrexone/bupropion extended-release in the treatment of obesity. Expert Opin Pharmacother. 2016. [Epub ahead of print]
41. Thomas CE, Mauer EA, Shukla AP, et al. Low adoption of weight loss medications: a comparison of prescribing patterns of antiobesity pharmacotherapies and SGLT2s. Obesity (Silver Spring). 2016;24:1955-1961.
42. Qsymia Risk Evaluation and Mitigation Strategy (REMS). VIVUS, Inc. Available at: http://www.qsymiarems.com. Accessed January 16, 2017.
43. Zinman B, Wanner C, Lachin JM, et al. Empaglifozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117-2128.
46. Smith SR, O’Neil PM, Astrup A. Early weight loss while on lorcaserin, diet and exercise as a predictor of week 52 weight-loss outcomes. Obesity (Silver Spring). 2014;22:2137-2146.
47. Marso SP, Daniels GH, Brown-Frandsen K, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311-322.
48. Madsen LW, Knauf JA, Gotfredsen C, et al. GLP-1 receptor agonists and the thyroid: C-cell effects in mice are mediated via the GLP-1 receptor and not associated with RET activation. Endocrinology. 2012;153:1538-1547.
49. Fujioka K, O’Neil PM, Davies M, et al. Early weight loss with liraglutide 3.0 mg predicts 1-year weight loss and is associated with improvements in clinical markers. Obesity (Silver Spring). 2016;24:2278-2288.
From The Journal of Family Practice | 2017;66(10):608-616.
Inside the Article
PRACTICE RECOMMENDATIONS
For patients with a body mass index (BMI) ≥30 kg/m2 or BMI ≥27 kg/m2 with weight-related comorbidities:
› Consider antiobesity pharmacotherapy when diet, exercise, and behavior modification do not produce sufficient weight loss. A
› Continue an antiobesity medication if it is deemed effective and well tolerated. A
Strength of recommendation (SOR)
A Good-quality patient-oriented evidence B Inconsistent or limited-quality patient-oriented evidence C Consensus, usual practice, opinion, disease-oriented evidence, case series
Recently, the US has seen a decline in the use of cord blood in transplants, according to a study by the RAND Corporation.
The research revealed a significant increase in the national cord blood inventory but also suggested many cord blood units may go unused because they have a total nucleated cell (TNC) count of less than 1.25 billion cells per unit.
RAND researchers conducted this study by examining past research about cord blood banks, analyzing data on public cord blood bank collection and use, and interviewing stakeholders such as blood bank leaders and transplant center physicians.
The data showed the number of hematopoietic stem cell transplants (HSCTs) performed in the US has increased in recent years.
However, the percentage of HSCTs in which cord blood is used has declined, from about 12% (n=822) of all HSCTs in 2010 to about 8% (n=718) of all HSCTs in 2015.
At the same time, public cord blood banks in the US have greatly increased their inventory. This is, at least in part, because the federal government began offering subsidies to public contractor banks with the goal of helping them obtain 150,000 new units of high-quality cord blood.
The numeric goal has been met, as the inventory currently exceeds 200,000 units. However, many of the units cannot be considered high-quality because they have a TNC count of less than 1.25 billion cells.
The researchers noted that the government subsidies are not structured to discourage the processing and banking of cord blood units with lower TNC counts.
In fact, the team found that about half of the national inventory is made up of cord blood units with TNC counts of less than 1.25 billion cells. They said the probability that such a unit will be used in a given year is about 0.1%, and there’s an 11% chance it will ever be used.
On the other hand, there is a 1% to 3% chance a cord blood unit with a TNC count of more than 1.5 billion will be used in a given year, and there’s a 61% chance it will ever be used.
Therefore, the researchers recommend the federal government find ways to encourage public cord blood banks to collect samples with higher TNC counts, as they will help expand the utility of the national inventory.
Support for this research was provided by the US Department of Health and Human Services, Office of the Assistant Secretary of Health.
Recently, the US has seen a decline in the use of cord blood in transplants, according to a study by the RAND Corporation.
The research revealed a significant increase in the national cord blood inventory but also suggested many cord blood units may go unused because they have a total nucleated cell (TNC) count of less than 1.25 billion cells per unit.
RAND researchers conducted this study by examining past research about cord blood banks, analyzing data on public cord blood bank collection and use, and interviewing stakeholders such as blood bank leaders and transplant center physicians.
The data showed the number of hematopoietic stem cell transplants (HSCTs) performed in the US has increased in recent years.
However, the percentage of HSCTs in which cord blood is used has declined, from about 12% (n=822) of all HSCTs in 2010 to about 8% (n=718) of all HSCTs in 2015.
At the same time, public cord blood banks in the US have greatly increased their inventory. This is, at least in part, because the federal government began offering subsidies to public contractor banks with the goal of helping them obtain 150,000 new units of high-quality cord blood.
The numeric goal has been met, as the inventory currently exceeds 200,000 units. However, many of the units cannot be considered high-quality because they have a TNC count of less than 1.25 billion cells.
The researchers noted that the government subsidies are not structured to discourage the processing and banking of cord blood units with lower TNC counts.
In fact, the team found that about half of the national inventory is made up of cord blood units with TNC counts of less than 1.25 billion cells. They said the probability that such a unit will be used in a given year is about 0.1%, and there’s an 11% chance it will ever be used.
On the other hand, there is a 1% to 3% chance a cord blood unit with a TNC count of more than 1.5 billion will be used in a given year, and there’s a 61% chance it will ever be used.
Therefore, the researchers recommend the federal government find ways to encourage public cord blood banks to collect samples with higher TNC counts, as they will help expand the utility of the national inventory.
Support for this research was provided by the US Department of Health and Human Services, Office of the Assistant Secretary of Health.
Photo courtesy of NHS
Cord blood donation
Recently, the US has seen a decline in the use of cord blood in transplants, according to a study by the RAND Corporation.
The research revealed a significant increase in the national cord blood inventory but also suggested many cord blood units may go unused because they have a total nucleated cell (TNC) count of less than 1.25 billion cells per unit.
RAND researchers conducted this study by examining past research about cord blood banks, analyzing data on public cord blood bank collection and use, and interviewing stakeholders such as blood bank leaders and transplant center physicians.
The data showed the number of hematopoietic stem cell transplants (HSCTs) performed in the US has increased in recent years.
However, the percentage of HSCTs in which cord blood is used has declined, from about 12% (n=822) of all HSCTs in 2010 to about 8% (n=718) of all HSCTs in 2015.
At the same time, public cord blood banks in the US have greatly increased their inventory. This is, at least in part, because the federal government began offering subsidies to public contractor banks with the goal of helping them obtain 150,000 new units of high-quality cord blood.
The numeric goal has been met, as the inventory currently exceeds 200,000 units. However, many of the units cannot be considered high-quality because they have a TNC count of less than 1.25 billion cells.
The researchers noted that the government subsidies are not structured to discourage the processing and banking of cord blood units with lower TNC counts.
In fact, the team found that about half of the national inventory is made up of cord blood units with TNC counts of less than 1.25 billion cells. They said the probability that such a unit will be used in a given year is about 0.1%, and there’s an 11% chance it will ever be used.
On the other hand, there is a 1% to 3% chance a cord blood unit with a TNC count of more than 1.5 billion will be used in a given year, and there’s a 61% chance it will ever be used.
Therefore, the researchers recommend the federal government find ways to encourage public cord blood banks to collect samples with higher TNC counts, as they will help expand the utility of the national inventory.
Support for this research was provided by the US Department of Health and Human Services, Office of the Assistant Secretary of Health.

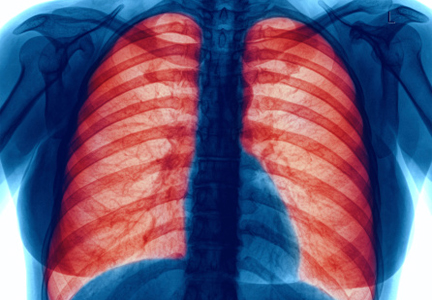




 is typically seen in precordial leads V3 to V6 and is a marker for hypothermia.")
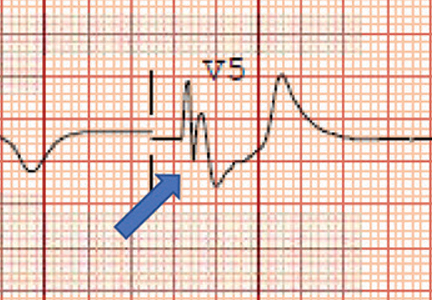
 White digits with intense vasoconstriction in Raynaud phenomenon; (B) blue digits with hypoxemic venous stasis; (C) red digits with hyperemic reperfusion.")
 Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.")
 Dilated nailfold capillaries in a patient with scleroderma (blue arrow); (B) dilation and dropout of nailfold capillaries (white arrow) viewed with a magnifier.")