User login
American Heart Association (AHA): Scientific Sessions 2014
IMPROVE-IT: Ezetimibe/simvastatin further reduces cardiovascular events
CHICAGO – Adding ezetimibe to a midpotency statin in high-risk cardiovascular patients to drive their LDL cholesterol into the low 50s resulted in a further significant reduction in major cardiovascular events, compared with statin alone, in the 18,144-patient IMPROVE-IT study.
In IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), 7 years of double-blind therapy with ezetimibe 10 mg/simvastatin 40 mg (Vytorin) achieved a 6.4% lower relative risk of the primary composite endpoint, compared with patients randomized to simvastatin alone (P = 0.016).

The 7-year rate of this composite outcome – comprised of cardiovascular death, nonfatal MI, rehospitalization for unstable angina, stroke, or coronary revascularization – was 32.7% with ezetimibe/simvastatin and 34.7% with simvastatin alone, for an absolute difference of 2%, Dr. Christopher P. Cannon reported at the American Heart Association scientific sessions.
Patients on dual therapy showed relative risk reductions of 13% for MI and 21% for ischemic stroke. The number needed to treat was 50, meaning that treating 100 patients with ezetimibe/simvastatin for 7 years should prevent 2 patients from having an ischemic stroke or MI who would have had such an event on simvastatin alone. That NNT is “well within the range of our standard therapies,” noted Dr. Cannon, professor of medicine at Harvard Medical School, Boston.
Patients in IMPROVE-IT began double-blind therapy within 10 days of hospitalization for an ST-elevation MI or non-STEMI/unstable angina. Participants also had to have at least one additional high-risk feature, such as a previous MI, diabetes, or coronary artery bypass surgery.
Observers hailed IMPROVE-IT as a major study for several reasons. It’s the first-ever study to demonstrate added clinical benefit when a nonstatin having a different mechanism of action is added to a statin. Thus, it provides physicians with a new, solidly evidence-based treatment option for high-risk acute coronary syndrome patients. Also, the trial emphatically affirms the LDL hypothesis that lowering LDL prevents cardiovascular events: The mean LDL at 1 year was 53.7 mg/dL in the dual-treatment group and 69.9 mg/dL in the simvastatin arm. And, as Dr. Karol Watson noted, IMPROVE-IT, with 7-plus years of careful follow-up, confirms the safety of ezetimibe, which had been questioned earlier on the basis of smaller studies that were far smaller.
“That’s one of the most important things to me to come from this study,” said Dr. Watson, professor of medicine and codirector of the program in preventive cardiology at the University of California, Los Angeles.
Once IMPROVE-IT has been published in a peer-reviewed journal it will be reviewed by the expert panel responsible for the AHA/American College of Cardiology cholesterol management practice guidelines, which in 2013 famously abolished specific LDL target goals.
Just what impact IMPROVE-IT ought to have on the guidelines was a point of considerable contention at the Chicago AHA meeting. Late-breaking clinical trials session cochair Dr. Sidney C. Smith noted that the current guidelines (J. Am. Coll. Cardiol. 2014;63:2889-934), on the basis of very solid evidence, call for high-intensity statin therapy, such as atorvastatin at 80 mg/day, as the first-line therapy for high-risk patients such as those with acute coronary syndrome.

“There are people who can’t take atorvastatin at 80 mg or atorvastatin at all. It seems to me that these IMPROVE-IT data fit very nicely with the guidelines: In such patients, you would start with 40 mg of simvastatin along with ezetimibe because of the proven outcome you’ve demonstrated,” suggested Dr. Smith, professor of medicine at the University of North Carolina, Chapel Hill.
Dr. Cannon indicated that he sees a broader role than that for statin/ezetimibe combination therapy. Since IMPROVE-IT has now shown that the lower a patient’s LDL the better the clinical outcomes, it makes sense to consider starting high-risk patients on maximal statin therapy – atorvastatin 80 mg/day or its equivalent – along with ezetimibe, he said.
“If you’re starting out at a high baseline risk, you might go straight to both. That’s what I hope the revised guidelines will say. Whereas, if someone was at a more moderate baseline LDL level they might get a very good response with just the high-intensity statin,” the cardiologist asserted.
Dr. Neil J. Stone, chair of the cholesterol guidelines panel, wasn’t buying that argument.
“That strategy simply wasn’t tested in terms of safety and efficacy in this trial. I’m not willing to make a leap of faith. I’d like to stay as close to the evidence as we can,” said Dr. Stone, professor of medicine at Northwestern University in Chicago. He cited several fairly recent examples in which lower proved not to be better in terms of LDL reduction: Niacin, fenofibrate, estrogen/progestin for women, and torcetrapib all went down in flames in randomized trials.

“If we had made a leap of faith that lower is better, we wouldn’t have done those trials,” he observed.
Also, it’s important to recognize that the IMPROVE-IT data don’t speak to the use of ezetimibe for primary prevention in lower-risk patients. “This study is not a signal that everyone should be on ezetimibe,” Dr. Stone emphasized.
It’s estimated that, in clinical practice, up to 25% of patients are statin intolerant to varying degrees. Dr. Watson said that, after seeing the IMPROVE-IT results, “I would absolutely try to persist with statin therapy, but there are some patients who are truly statin intolerant, and for them I might add ezetimibe to a low-dose statin.”
Indeed, like many other physicians, she has been doing that all along in selected cases on the basis of clinical judgment and biologic plausibility while anxiously awaiting the IMPROVE-IT findings, which come as a great relief, she added.
IMPROVE-IT was initially planned to include 10,000 randomized patients and a minimum of 2.5 years of on-treatment follow-up. Over the years, however, it underwent four protocol amendments. As IMPROVE-IT kept growing larger and longer, some observers quipped that it appeared the organizers would just keep the trial going until it produced a positive result, although, in fact, the study leaders remained blinded to the interim safety and efficacy data.
Merck, which sponsored the study, announced that it will use the data in petitioning the Food and Drug Administration next year for a new indication for reduction of major cardiovascular events for Vytorin and ezetimibe (Zetia).
Dr. Cannon reported receiving research grants from Merck and half a dozen other pharmaceutical companies. Dr. Watson reported participating in the clinical trials adjudication committee for Merck. Dr. Smith and Dr. Stone had no disclosures.
*Correction, 11/18/14: An earlier version of this article carried an incorrect photo credit.
CHICAGO – Adding ezetimibe to a midpotency statin in high-risk cardiovascular patients to drive their LDL cholesterol into the low 50s resulted in a further significant reduction in major cardiovascular events, compared with statin alone, in the 18,144-patient IMPROVE-IT study.
In IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), 7 years of double-blind therapy with ezetimibe 10 mg/simvastatin 40 mg (Vytorin) achieved a 6.4% lower relative risk of the primary composite endpoint, compared with patients randomized to simvastatin alone (P = 0.016).
The 7-year rate of this composite outcome – comprised of cardiovascular death, nonfatal MI, rehospitalization for unstable angina, stroke, or coronary revascularization – was 32.7% with ezetimibe/simvastatin and 34.7% with simvastatin alone, for an absolute difference of 2%, Dr. Christopher P. Cannon reported at the American Heart Association scientific sessions.
Patients on dual therapy showed relative risk reductions of 13% for MI and 21% for ischemic stroke. The number needed to treat was 50, meaning that treating 100 patients with ezetimibe/simvastatin for 7 years should prevent 2 patients from having an ischemic stroke or MI who would have had such an event on simvastatin alone. That NNT is “well within the range of our standard therapies,” noted Dr. Cannon, professor of medicine at Harvard Medical School, Boston.
Patients in IMPROVE-IT began double-blind therapy within 10 days of hospitalization for an ST-elevation MI or non-STEMI/unstable angina. Participants also had to have at least one additional high-risk feature, such as a previous MI, diabetes, or coronary artery bypass surgery.
Observers hailed IMPROVE-IT as a major study for several reasons. It’s the first-ever study to demonstrate added clinical benefit when a nonstatin having a different mechanism of action is added to a statin. Thus, it provides physicians with a new, solidly evidence-based treatment option for high-risk acute coronary syndrome patients. Also, the trial emphatically affirms the LDL hypothesis that lowering LDL prevents cardiovascular events: The mean LDL at 1 year was 53.7 mg/dL in the dual-treatment group and 69.9 mg/dL in the simvastatin arm. And, as Dr. Karol Watson noted, IMPROVE-IT, with 7-plus years of careful follow-up, confirms the safety of ezetimibe, which had been questioned earlier on the basis of smaller studies that were far smaller.
“That’s one of the most important things to me to come from this study,” said Dr. Watson, professor of medicine and codirector of the program in preventive cardiology at the University of California, Los Angeles.
Once IMPROVE-IT has been published in a peer-reviewed journal it will be reviewed by the expert panel responsible for the AHA/American College of Cardiology cholesterol management practice guidelines, which in 2013 famously abolished specific LDL target goals.
Just what impact IMPROVE-IT ought to have on the guidelines was a point of considerable contention at the Chicago AHA meeting. Late-breaking clinical trials session cochair Dr. Sidney C. Smith noted that the current guidelines (J. Am. Coll. Cardiol. 2014;63:2889-934), on the basis of very solid evidence, call for high-intensity statin therapy, such as atorvastatin at 80 mg/day, as the first-line therapy for high-risk patients such as those with acute coronary syndrome.
“There are people who can’t take atorvastatin at 80 mg or atorvastatin at all. It seems to me that these IMPROVE-IT data fit very nicely with the guidelines: In such patients, you would start with 40 mg of simvastatin along with ezetimibe because of the proven outcome you’ve demonstrated,” suggested Dr. Smith, professor of medicine at the University of North Carolina, Chapel Hill.
Dr. Cannon indicated that he sees a broader role than that for statin/ezetimibe combination therapy. Since IMPROVE-IT has now shown that the lower a patient’s LDL the better the clinical outcomes, it makes sense to consider starting high-risk patients on maximal statin therapy – atorvastatin 80 mg/day or its equivalent – along with ezetimibe, he said.
“If you’re starting out at a high baseline risk, you might go straight to both. That’s what I hope the revised guidelines will say. Whereas, if someone was at a more moderate baseline LDL level they might get a very good response with just the high-intensity statin,” the cardiologist asserted.
Dr. Neil J. Stone, chair of the cholesterol guidelines panel, wasn’t buying that argument.
“That strategy simply wasn’t tested in terms of safety and efficacy in this trial. I’m not willing to make a leap of faith. I’d like to stay as close to the evidence as we can,” said Dr. Stone, professor of medicine at Northwestern University in Chicago. He cited several fairly recent examples in which lower proved not to be better in terms of LDL reduction: Niacin, fenofibrate, estrogen/progestin for women, and torcetrapib all went down in flames in randomized trials.
“If we had made a leap of faith that lower is better, we wouldn’t have done those trials,” he observed.
Also, it’s important to recognize that the IMPROVE-IT data don’t speak to the use of ezetimibe for primary prevention in lower-risk patients. “This study is not a signal that everyone should be on ezetimibe,” Dr. Stone emphasized.
It’s estimated that, in clinical practice, up to 25% of patients are statin intolerant to varying degrees. Dr. Watson said that, after seeing the IMPROVE-IT results, “I would absolutely try to persist with statin therapy, but there are some patients who are truly statin intolerant, and for them I might add ezetimibe to a low-dose statin.”
Indeed, like many other physicians, she has been doing that all along in selected cases on the basis of clinical judgment and biologic plausibility while anxiously awaiting the IMPROVE-IT findings, which come as a great relief, she added.
IMPROVE-IT was initially planned to include 10,000 randomized patients and a minimum of 2.5 years of on-treatment follow-up. Over the years, however, it underwent four protocol amendments. As IMPROVE-IT kept growing larger and longer, some observers quipped that it appeared the organizers would just keep the trial going until it produced a positive result, although, in fact, the study leaders remained blinded to the interim safety and efficacy data.
Merck, which sponsored the study, announced that it will use the data in petitioning the Food and Drug Administration next year for a new indication for reduction of major cardiovascular events for Vytorin and ezetimibe (Zetia).
Dr. Cannon reported receiving research grants from Merck and half a dozen other pharmaceutical companies. Dr. Watson reported participating in the clinical trials adjudication committee for Merck. Dr. Smith and Dr. Stone had no disclosures.
*Correction, 11/18/14: An earlier version of this article carried an incorrect photo credit.
CHICAGO – Adding ezetimibe to a midpotency statin in high-risk cardiovascular patients to drive their LDL cholesterol into the low 50s resulted in a further significant reduction in major cardiovascular events, compared with statin alone, in the 18,144-patient IMPROVE-IT study.
In IMPROVE-IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial), 7 years of double-blind therapy with ezetimibe 10 mg/simvastatin 40 mg (Vytorin) achieved a 6.4% lower relative risk of the primary composite endpoint, compared with patients randomized to simvastatin alone (P = 0.016).
The 7-year rate of this composite outcome – comprised of cardiovascular death, nonfatal MI, rehospitalization for unstable angina, stroke, or coronary revascularization – was 32.7% with ezetimibe/simvastatin and 34.7% with simvastatin alone, for an absolute difference of 2%, Dr. Christopher P. Cannon reported at the American Heart Association scientific sessions.
Patients on dual therapy showed relative risk reductions of 13% for MI and 21% for ischemic stroke. The number needed to treat was 50, meaning that treating 100 patients with ezetimibe/simvastatin for 7 years should prevent 2 patients from having an ischemic stroke or MI who would have had such an event on simvastatin alone. That NNT is “well within the range of our standard therapies,” noted Dr. Cannon, professor of medicine at Harvard Medical School, Boston.
Patients in IMPROVE-IT began double-blind therapy within 10 days of hospitalization for an ST-elevation MI or non-STEMI/unstable angina. Participants also had to have at least one additional high-risk feature, such as a previous MI, diabetes, or coronary artery bypass surgery.
Observers hailed IMPROVE-IT as a major study for several reasons. It’s the first-ever study to demonstrate added clinical benefit when a nonstatin having a different mechanism of action is added to a statin. Thus, it provides physicians with a new, solidly evidence-based treatment option for high-risk acute coronary syndrome patients. Also, the trial emphatically affirms the LDL hypothesis that lowering LDL prevents cardiovascular events: The mean LDL at 1 year was 53.7 mg/dL in the dual-treatment group and 69.9 mg/dL in the simvastatin arm. And, as Dr. Karol Watson noted, IMPROVE-IT, with 7-plus years of careful follow-up, confirms the safety of ezetimibe, which had been questioned earlier on the basis of smaller studies that were far smaller.
“That’s one of the most important things to me to come from this study,” said Dr. Watson, professor of medicine and codirector of the program in preventive cardiology at the University of California, Los Angeles.
Once IMPROVE-IT has been published in a peer-reviewed journal it will be reviewed by the expert panel responsible for the AHA/American College of Cardiology cholesterol management practice guidelines, which in 2013 famously abolished specific LDL target goals.
Just what impact IMPROVE-IT ought to have on the guidelines was a point of considerable contention at the Chicago AHA meeting. Late-breaking clinical trials session cochair Dr. Sidney C. Smith noted that the current guidelines (J. Am. Coll. Cardiol. 2014;63:2889-934), on the basis of very solid evidence, call for high-intensity statin therapy, such as atorvastatin at 80 mg/day, as the first-line therapy for high-risk patients such as those with acute coronary syndrome.
“There are people who can’t take atorvastatin at 80 mg or atorvastatin at all. It seems to me that these IMPROVE-IT data fit very nicely with the guidelines: In such patients, you would start with 40 mg of simvastatin along with ezetimibe because of the proven outcome you’ve demonstrated,” suggested Dr. Smith, professor of medicine at the University of North Carolina, Chapel Hill.
Dr. Cannon indicated that he sees a broader role than that for statin/ezetimibe combination therapy. Since IMPROVE-IT has now shown that the lower a patient’s LDL the better the clinical outcomes, it makes sense to consider starting high-risk patients on maximal statin therapy – atorvastatin 80 mg/day or its equivalent – along with ezetimibe, he said.
“If you’re starting out at a high baseline risk, you might go straight to both. That’s what I hope the revised guidelines will say. Whereas, if someone was at a more moderate baseline LDL level they might get a very good response with just the high-intensity statin,” the cardiologist asserted.
Dr. Neil J. Stone, chair of the cholesterol guidelines panel, wasn’t buying that argument.
“That strategy simply wasn’t tested in terms of safety and efficacy in this trial. I’m not willing to make a leap of faith. I’d like to stay as close to the evidence as we can,” said Dr. Stone, professor of medicine at Northwestern University in Chicago. He cited several fairly recent examples in which lower proved not to be better in terms of LDL reduction: Niacin, fenofibrate, estrogen/progestin for women, and torcetrapib all went down in flames in randomized trials.
“If we had made a leap of faith that lower is better, we wouldn’t have done those trials,” he observed.
Also, it’s important to recognize that the IMPROVE-IT data don’t speak to the use of ezetimibe for primary prevention in lower-risk patients. “This study is not a signal that everyone should be on ezetimibe,” Dr. Stone emphasized.
It’s estimated that, in clinical practice, up to 25% of patients are statin intolerant to varying degrees. Dr. Watson said that, after seeing the IMPROVE-IT results, “I would absolutely try to persist with statin therapy, but there are some patients who are truly statin intolerant, and for them I might add ezetimibe to a low-dose statin.”
Indeed, like many other physicians, she has been doing that all along in selected cases on the basis of clinical judgment and biologic plausibility while anxiously awaiting the IMPROVE-IT findings, which come as a great relief, she added.
IMPROVE-IT was initially planned to include 10,000 randomized patients and a minimum of 2.5 years of on-treatment follow-up. Over the years, however, it underwent four protocol amendments. As IMPROVE-IT kept growing larger and longer, some observers quipped that it appeared the organizers would just keep the trial going until it produced a positive result, although, in fact, the study leaders remained blinded to the interim safety and efficacy data.
Merck, which sponsored the study, announced that it will use the data in petitioning the Food and Drug Administration next year for a new indication for reduction of major cardiovascular events for Vytorin and ezetimibe (Zetia).
Dr. Cannon reported receiving research grants from Merck and half a dozen other pharmaceutical companies. Dr. Watson reported participating in the clinical trials adjudication committee for Merck. Dr. Smith and Dr. Stone had no disclosures.
*Correction, 11/18/14: An earlier version of this article carried an incorrect photo credit.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Combined therapy with ezetimibe and simvastatin reduces cardiovascular events more than simvastatin alone in high-risk patients.
Major finding: The number needed to treat with the combination therapy to prevent one MI or ischemic stroke was 50 patients for 7 years.
Data source: IMPROVE-IT was a randomized, double-blind study involving 18,144 patients with acute coronary syndrome at 1,158 centers in 39 countries.
Disclosures: The study was sponsored by Merck. The presenter reported having received research grants from Merck and several other pharmaceutical companies.

VIDEO: IMPROVE-IT exonerates ezetimibe, proves LDL hypothesis
CHICAGO – Results of the IMPROVE-IT megatrial not only show that adding ezetimibe to statin therapy lowered the risk of all cardiovascular events in high-risk patients, they also prove the LDL hypothesis, two experts commented at the American Heart Association scientific sessions.
IMPROVE-IT compared simvastatin plus placebo with simvastatin plus ezetimibe in more than 18,000 patients within 10 days of an acute coronary syndrome event. The dual-therapy patients’ LDL cholesterol went down to an average of 54 mg/dL, while the statin/placebo patients’ LDL reached 69 mg/dL.
The patients taking ezetimibe in addition to simvastatin had a 6.4% lower risk of all cardiovascular events, a 14% reduced risk of all heart attacks, and a 21% lower risk of ischemic stroke. There was no difference in mortality between groups.
Dr. Lori Mosca, professor of medicine at Columbia University, New York, and director of preventive cardiology at New York-Presbyterian Hospital, and Dr. Prakash Deedwania, professor of medicine at the University of California, San Francisco, Fresno, told us how these “exciting” results will change practice for treating very high-risk patients.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
CHICAGO – Results of the IMPROVE-IT megatrial not only show that adding ezetimibe to statin therapy lowered the risk of all cardiovascular events in high-risk patients, they also prove the LDL hypothesis, two experts commented at the American Heart Association scientific sessions.
IMPROVE-IT compared simvastatin plus placebo with simvastatin plus ezetimibe in more than 18,000 patients within 10 days of an acute coronary syndrome event. The dual-therapy patients’ LDL cholesterol went down to an average of 54 mg/dL, while the statin/placebo patients’ LDL reached 69 mg/dL.
The patients taking ezetimibe in addition to simvastatin had a 6.4% lower risk of all cardiovascular events, a 14% reduced risk of all heart attacks, and a 21% lower risk of ischemic stroke. There was no difference in mortality between groups.
Dr. Lori Mosca, professor of medicine at Columbia University, New York, and director of preventive cardiology at New York-Presbyterian Hospital, and Dr. Prakash Deedwania, professor of medicine at the University of California, San Francisco, Fresno, told us how these “exciting” results will change practice for treating very high-risk patients.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
CHICAGO – Results of the IMPROVE-IT megatrial not only show that adding ezetimibe to statin therapy lowered the risk of all cardiovascular events in high-risk patients, they also prove the LDL hypothesis, two experts commented at the American Heart Association scientific sessions.
IMPROVE-IT compared simvastatin plus placebo with simvastatin plus ezetimibe in more than 18,000 patients within 10 days of an acute coronary syndrome event. The dual-therapy patients’ LDL cholesterol went down to an average of 54 mg/dL, while the statin/placebo patients’ LDL reached 69 mg/dL.
The patients taking ezetimibe in addition to simvastatin had a 6.4% lower risk of all cardiovascular events, a 14% reduced risk of all heart attacks, and a 21% lower risk of ischemic stroke. There was no difference in mortality between groups.
Dr. Lori Mosca, professor of medicine at Columbia University, New York, and director of preventive cardiology at New York-Presbyterian Hospital, and Dr. Prakash Deedwania, professor of medicine at the University of California, San Francisco, Fresno, told us how these “exciting” results will change practice for treating very high-risk patients.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

Coronary stent patients need individualized DAPT duration
CHICAGO – Clinicians managing patients who have just received drug-eluting coronary stents need to individualize the duration of the antiplatelet therapy they prescribe and stop thinking that a single length of treatment works best for all patients, based on results from a trio of newly reported trials, as well as earlier study findings.
In general, the new findings seemed to show that following percutaneous coronary intervention (PCI) with a stent, patients with a low risk for stent thrombosis, myocardial infarction, or another type of ischemic event, as well as those with a high bleeding risk, fare best when their duration of dual antiplatelet therapy (DAPT) with aspirin plus a thienopyridine ends after 6 months, while patients with a low bleeding risk and a high risk for an ischemic event will usually do better when their DAPT continues well beyond 12 months, for 30 months and possibly longer.
“We now have results from 35,000 patients in randomized trials that addressed the question of duration of DAPT. We need to make up our minds, and it’s probably an individualized decision,” Dr. Gilles Montalescot said at the American Heart Association Scientific Sessions.

“The patients [in the three newly reported studies] were generally doing well, and so we are operating at the edge of competing risks” they face for ischemia and bleeding. “We need individualization of treatment, but what remains to be determined is, how do we make that decision? Who is more at risk for ischemic events and less at risk for bleeding, and who is more at risk for bleeding and less from ischemia?” commented Dr. Elliott M. Antman, professor of medicine and associate dean for clinical and translational research at Harvard University in Boston.
The DAPT trial
Drug-eluting stent recipients at increased risk for bleeding who are good candidates for a briefer, 6-month duration of DAPT include those with a history of a prior bleed, advanced age, pending need for surgery, patients who need anticoagulation treatment such as those with atrial fibrillation, patients with one or more comorbidities that put them at higher bleeding risk such as gastrointestinal disease or a history of stroke, and patients with a “nuisance” bleed, said Dr. Montalescot, a professor of cardiology at the University of Paris and director of the cardiac care unit at Pitié-Salpêtrière Hospital in Paris.
These conclusions emerged from a surge of new data from three studies presented at the meeting that all addressed DAPT duration following PCI.
Perhaps most noteworthy and anticipated were results from the Dual Antiplatelet Therapy study, a randomized trial that had its beginnings in 2006 when cardiologists first became alarmed by the potential risk from stent thrombosis associated with the use of first-generation DES, specifically the original sirolimus-eluting (Cypher) and paclitaxel-eluting (Taxus) models. At the urging of staffers at the Food and Drug Administration, a group of eight stent manufacturing companies and pharmaceutical companies joined together to sponsor the DAPT study, which started with nearly 23,000 patients who received a DES and, after their first year of DAPT treatment, whittled down to 9,961 patients who were eligible for randomization to either stop DAPT after 1 year or continue on DAPT for an additional 18 months (30 months total time on DAPT).
The results showed that this highly selected group of patients – who went through their first year on DAPT with no bleeding event, no ischemic event, and who were willing to be randomized – those assigned to 30 months of DAPT had a 1% absolute and 71% relative reduction in stent thrombosis, compared with patients who stopped DAPT after 12 months, and a 1.6% absolute and 29% relative reduction in their rate of major cardiovascular or cerebrovascular events, statistically significant differences for the study’s two primary endpoints. The reduction in major events was driven mainly by a 2% absolute reduction in the rate of myocardial infarctions, reported Dr. Laura Mauri at the meeting, and in a report simultaneously published online (N. Engl. J. Med. 2014 [doi:10.1056/NEJMoa1409312]).

Counterbalancing this benefit was a statistically significant excess of moderately severe bleeding events among patients on longer-term DAPT, who had a 0.7% absolute increased rate, and a trend toward increased severe bleeds, with a 0.2% absolute increased rate. Patients on longer-term DAPT also showed an unexpected, 0.5% increased rate of both all cause death and noncardiovascular death. Detailed analysis indicated that this seemed to result from a “chance imbalance” in randomization that put an excess of patients with cancer into the 30-month DAPT subgroup, said Dr. Mauri, professor of medicine at Harvard Medical School and an interventional cardiologist at Brigham and Women’s Hospital, both in Boston.
DAPT treatment extending longer than 12 months switches the focus from preventing stent thrombosis to secondary prevention of all ischemic events, including those that occur elsewhere in a patient’s coronary arteries, noted Dr. Montalescot, who served as the meeting’s designated discussant for the DAPT trial and the other reported DAPT studies. This option, which can now be offered to appropriate patients, also raised the possibility of continuing DAPT indefinitely beyond 30 months in patients who continue to show no bleeding risk and can pay for ongoing treatment with a thienopyridine. But he advised waiting on the use of very prolonged DAPT until results arrive next year from the PEGASUS study.
ITALIC and ISAR-SAFE
Results from two other studies reported at the meeting looked at the other end of the spectrum, the safety of stopping DAPT after just 6 months. One of these, the ITALIC study, randomized 1,850 patients to 6 months or 24 months of treatment with aspirin and clopidogrel, and exclusively enrolled patients who received the Xience V everolimus-eluting, third-generation coronary stent. The results showed low and nearly identical 1.5% and 1.6% rates of the combined endpoint of major adverse events, including bleeds, in the two comparator arms after 24 months of follow-up. The results also appeared in an article published simultaneously online (J. Am. Coll. Card. 2014 [doi:10.1016/j.jacc.2014.11.008]).
A similarly designed study, ISAR-SAFE, randomized 4,005 PCI patients who received a DES to DAPT with aspirin and clopidogrel for either 6 or 12 months, and also showed a similar and low, roughly 1.5% rate of combined major adverse cardiovascular events and bleeds in both treatment arms, reported Dr. Stefanie Schulz-Schüpke, an interventional cardiologist at the German Heart Center in Munich.

These two studies, along with five prior reports that collectively randomized nearly 16,000 patients to 6 months of DAPT or a longer duration show a statistically significant reduction in major bleeds with good protection against ischemic events when DAPT stopped after 6 months, said Dr. Montalescot. “We can accept that 6 months is safer than 12 months of DAPT,” and it makes sense to stop after 6 months in a patient with a bleeding risk, but not in a patient with a high ischemia risk such as those with left main coronary disease, a history of stent thrombosis or myocardial infarction, a patient who received a first-generation DES, patients with extensive coronary disease or coronary stenting.*
While Dr. Montalescot, Dr. Antman, and others who heard these results agreed that they all pointed to the need for individualized decisions on DAPT duration, they also said the new findings were unlikely to change current guidelines. Existing PCI guidelines from the American Heart Association and American College of Cardiology call for a standard 12 months of DAPT in DES recipients, but offer clinicians to reduce or extend the duration when judged appropriate (J. Am. Coll. Cardiol. 2011;58:e44-e122). Guidelines from the European College of Cardiology call for 6 months of DAPT as standard, but also suggest that clinicians can reduce or lengthen the duration when appropriate (Eur. Heart J. 2014 [doi.org/10.1093/eurheartj/ehu278]).
According to three staffers from the FDA who also spoke at the session, the agency is also unlikely to change its guidance on DAPT in the immediate future, even though it triggered launch of the DAPT trial.
The DAPT trial “is large enough to allow us to answer some really critical public-health questions, and to potentially expand the indications for drug-eluting coronary stents,” said Dr. Bram D. Zuckerman, director of the division of cardiovascular devices of the FDA. But he and his colleagues who spoke during the session said that the agency will not update its recommendations or labels until the FDA staff has had a chance to study in detail the full data set collected in the DAPT trial. Concurrent with Dr. Mauri’s report, the agency issued a statement calling on physicians to continue to prescribe DAPT as they have in the past and for patients to continue to take all their prescribed medications.
Many physicians have already concluded that 6 months of DAPT is a practical way to enhance patient compliance with their regimen and minimize the money they spend on these medications, commented Dr. Richard C. Becker, professor of medicine and director of the Cardiovascular Institute at the University of Cincinnati. The three trials reported at the meeting “do not answer the question [of whether 6 months is adequate] because in each trial patients had already tolerated a period of DAPT before entering the study” Dr. Becker said in an interview.
The question of DAPT duration “is a question that clinicians will need to have with each of their patients, especially those who are tolerating DAPT,” commented Dr. Richard A. Chazal, an interventionalist and medical director of the Heart and Vascular Institute at the Lee Memorial Health System in Fort Myers, Fla.
Dr. Montalescot has been a consultant to more than a dozen pharmaceutical and device companies, including companies that market antiplatelet drugs. Dr. Antman has received grant support from Daiichi Sankyo. Dr. Mauri received personal fees from Medtronic, Recur, St. Jude, and Biotronik, and grant support from nine device manufacturer and drug companies. Dr. Schulz-Schüpke, Dr. Zuckerman, Dr. Becker, and Dr. Chazal had no relevant disclosures.
On Twitter@mitchelzoler
*CORRECTION, 11/18/2014: An earlier version of this article misstated the treatment length in patients with a high ischemia risk.
CHICAGO – Clinicians managing patients who have just received drug-eluting coronary stents need to individualize the duration of the antiplatelet therapy they prescribe and stop thinking that a single length of treatment works best for all patients, based on results from a trio of newly reported trials, as well as earlier study findings.
In general, the new findings seemed to show that following percutaneous coronary intervention (PCI) with a stent, patients with a low risk for stent thrombosis, myocardial infarction, or another type of ischemic event, as well as those with a high bleeding risk, fare best when their duration of dual antiplatelet therapy (DAPT) with aspirin plus a thienopyridine ends after 6 months, while patients with a low bleeding risk and a high risk for an ischemic event will usually do better when their DAPT continues well beyond 12 months, for 30 months and possibly longer.
“We now have results from 35,000 patients in randomized trials that addressed the question of duration of DAPT. We need to make up our minds, and it’s probably an individualized decision,” Dr. Gilles Montalescot said at the American Heart Association Scientific Sessions.
“The patients [in the three newly reported studies] were generally doing well, and so we are operating at the edge of competing risks” they face for ischemia and bleeding. “We need individualization of treatment, but what remains to be determined is, how do we make that decision? Who is more at risk for ischemic events and less at risk for bleeding, and who is more at risk for bleeding and less from ischemia?” commented Dr. Elliott M. Antman, professor of medicine and associate dean for clinical and translational research at Harvard University in Boston.
The DAPT trial
Drug-eluting stent recipients at increased risk for bleeding who are good candidates for a briefer, 6-month duration of DAPT include those with a history of a prior bleed, advanced age, pending need for surgery, patients who need anticoagulation treatment such as those with atrial fibrillation, patients with one or more comorbidities that put them at higher bleeding risk such as gastrointestinal disease or a history of stroke, and patients with a “nuisance” bleed, said Dr. Montalescot, a professor of cardiology at the University of Paris and director of the cardiac care unit at Pitié-Salpêtrière Hospital in Paris.
These conclusions emerged from a surge of new data from three studies presented at the meeting that all addressed DAPT duration following PCI.
Perhaps most noteworthy and anticipated were results from the Dual Antiplatelet Therapy study, a randomized trial that had its beginnings in 2006 when cardiologists first became alarmed by the potential risk from stent thrombosis associated with the use of first-generation DES, specifically the original sirolimus-eluting (Cypher) and paclitaxel-eluting (Taxus) models. At the urging of staffers at the Food and Drug Administration, a group of eight stent manufacturing companies and pharmaceutical companies joined together to sponsor the DAPT study, which started with nearly 23,000 patients who received a DES and, after their first year of DAPT treatment, whittled down to 9,961 patients who were eligible for randomization to either stop DAPT after 1 year or continue on DAPT for an additional 18 months (30 months total time on DAPT).
The results showed that this highly selected group of patients – who went through their first year on DAPT with no bleeding event, no ischemic event, and who were willing to be randomized – those assigned to 30 months of DAPT had a 1% absolute and 71% relative reduction in stent thrombosis, compared with patients who stopped DAPT after 12 months, and a 1.6% absolute and 29% relative reduction in their rate of major cardiovascular or cerebrovascular events, statistically significant differences for the study’s two primary endpoints. The reduction in major events was driven mainly by a 2% absolute reduction in the rate of myocardial infarctions, reported Dr. Laura Mauri at the meeting, and in a report simultaneously published online (N. Engl. J. Med. 2014 [doi:10.1056/NEJMoa1409312]).
Counterbalancing this benefit was a statistically significant excess of moderately severe bleeding events among patients on longer-term DAPT, who had a 0.7% absolute increased rate, and a trend toward increased severe bleeds, with a 0.2% absolute increased rate. Patients on longer-term DAPT also showed an unexpected, 0.5% increased rate of both all cause death and noncardiovascular death. Detailed analysis indicated that this seemed to result from a “chance imbalance” in randomization that put an excess of patients with cancer into the 30-month DAPT subgroup, said Dr. Mauri, professor of medicine at Harvard Medical School and an interventional cardiologist at Brigham and Women’s Hospital, both in Boston.
DAPT treatment extending longer than 12 months switches the focus from preventing stent thrombosis to secondary prevention of all ischemic events, including those that occur elsewhere in a patient’s coronary arteries, noted Dr. Montalescot, who served as the meeting’s designated discussant for the DAPT trial and the other reported DAPT studies. This option, which can now be offered to appropriate patients, also raised the possibility of continuing DAPT indefinitely beyond 30 months in patients who continue to show no bleeding risk and can pay for ongoing treatment with a thienopyridine. But he advised waiting on the use of very prolonged DAPT until results arrive next year from the PEGASUS study.
ITALIC and ISAR-SAFE
Results from two other studies reported at the meeting looked at the other end of the spectrum, the safety of stopping DAPT after just 6 months. One of these, the ITALIC study, randomized 1,850 patients to 6 months or 24 months of treatment with aspirin and clopidogrel, and exclusively enrolled patients who received the Xience V everolimus-eluting, third-generation coronary stent. The results showed low and nearly identical 1.5% and 1.6% rates of the combined endpoint of major adverse events, including bleeds, in the two comparator arms after 24 months of follow-up. The results also appeared in an article published simultaneously online (J. Am. Coll. Card. 2014 [doi:10.1016/j.jacc.2014.11.008]).
A similarly designed study, ISAR-SAFE, randomized 4,005 PCI patients who received a DES to DAPT with aspirin and clopidogrel for either 6 or 12 months, and also showed a similar and low, roughly 1.5% rate of combined major adverse cardiovascular events and bleeds in both treatment arms, reported Dr. Stefanie Schulz-Schüpke, an interventional cardiologist at the German Heart Center in Munich.
These two studies, along with five prior reports that collectively randomized nearly 16,000 patients to 6 months of DAPT or a longer duration show a statistically significant reduction in major bleeds with good protection against ischemic events when DAPT stopped after 6 months, said Dr. Montalescot. “We can accept that 6 months is safer than 12 months of DAPT,” and it makes sense to stop after 6 months in a patient with a bleeding risk, but not in a patient with a high ischemia risk such as those with left main coronary disease, a history of stent thrombosis or myocardial infarction, a patient who received a first-generation DES, patients with extensive coronary disease or coronary stenting.*
While Dr. Montalescot, Dr. Antman, and others who heard these results agreed that they all pointed to the need for individualized decisions on DAPT duration, they also said the new findings were unlikely to change current guidelines. Existing PCI guidelines from the American Heart Association and American College of Cardiology call for a standard 12 months of DAPT in DES recipients, but offer clinicians to reduce or extend the duration when judged appropriate (J. Am. Coll. Cardiol. 2011;58:e44-e122). Guidelines from the European College of Cardiology call for 6 months of DAPT as standard, but also suggest that clinicians can reduce or lengthen the duration when appropriate (Eur. Heart J. 2014 [doi.org/10.1093/eurheartj/ehu278]).
According to three staffers from the FDA who also spoke at the session, the agency is also unlikely to change its guidance on DAPT in the immediate future, even though it triggered launch of the DAPT trial.
The DAPT trial “is large enough to allow us to answer some really critical public-health questions, and to potentially expand the indications for drug-eluting coronary stents,” said Dr. Bram D. Zuckerman, director of the division of cardiovascular devices of the FDA. But he and his colleagues who spoke during the session said that the agency will not update its recommendations or labels until the FDA staff has had a chance to study in detail the full data set collected in the DAPT trial. Concurrent with Dr. Mauri’s report, the agency issued a statement calling on physicians to continue to prescribe DAPT as they have in the past and for patients to continue to take all their prescribed medications.
Many physicians have already concluded that 6 months of DAPT is a practical way to enhance patient compliance with their regimen and minimize the money they spend on these medications, commented Dr. Richard C. Becker, professor of medicine and director of the Cardiovascular Institute at the University of Cincinnati. The three trials reported at the meeting “do not answer the question [of whether 6 months is adequate] because in each trial patients had already tolerated a period of DAPT before entering the study” Dr. Becker said in an interview.
The question of DAPT duration “is a question that clinicians will need to have with each of their patients, especially those who are tolerating DAPT,” commented Dr. Richard A. Chazal, an interventionalist and medical director of the Heart and Vascular Institute at the Lee Memorial Health System in Fort Myers, Fla.
Dr. Montalescot has been a consultant to more than a dozen pharmaceutical and device companies, including companies that market antiplatelet drugs. Dr. Antman has received grant support from Daiichi Sankyo. Dr. Mauri received personal fees from Medtronic, Recur, St. Jude, and Biotronik, and grant support from nine device manufacturer and drug companies. Dr. Schulz-Schüpke, Dr. Zuckerman, Dr. Becker, and Dr. Chazal had no relevant disclosures.
On Twitter@mitchelzoler
*CORRECTION, 11/18/2014: An earlier version of this article misstated the treatment length in patients with a high ischemia risk.
CHICAGO – Clinicians managing patients who have just received drug-eluting coronary stents need to individualize the duration of the antiplatelet therapy they prescribe and stop thinking that a single length of treatment works best for all patients, based on results from a trio of newly reported trials, as well as earlier study findings.
In general, the new findings seemed to show that following percutaneous coronary intervention (PCI) with a stent, patients with a low risk for stent thrombosis, myocardial infarction, or another type of ischemic event, as well as those with a high bleeding risk, fare best when their duration of dual antiplatelet therapy (DAPT) with aspirin plus a thienopyridine ends after 6 months, while patients with a low bleeding risk and a high risk for an ischemic event will usually do better when their DAPT continues well beyond 12 months, for 30 months and possibly longer.
“We now have results from 35,000 patients in randomized trials that addressed the question of duration of DAPT. We need to make up our minds, and it’s probably an individualized decision,” Dr. Gilles Montalescot said at the American Heart Association Scientific Sessions.
“The patients [in the three newly reported studies] were generally doing well, and so we are operating at the edge of competing risks” they face for ischemia and bleeding. “We need individualization of treatment, but what remains to be determined is, how do we make that decision? Who is more at risk for ischemic events and less at risk for bleeding, and who is more at risk for bleeding and less from ischemia?” commented Dr. Elliott M. Antman, professor of medicine and associate dean for clinical and translational research at Harvard University in Boston.
The DAPT trial
Drug-eluting stent recipients at increased risk for bleeding who are good candidates for a briefer, 6-month duration of DAPT include those with a history of a prior bleed, advanced age, pending need for surgery, patients who need anticoagulation treatment such as those with atrial fibrillation, patients with one or more comorbidities that put them at higher bleeding risk such as gastrointestinal disease or a history of stroke, and patients with a “nuisance” bleed, said Dr. Montalescot, a professor of cardiology at the University of Paris and director of the cardiac care unit at Pitié-Salpêtrière Hospital in Paris.
These conclusions emerged from a surge of new data from three studies presented at the meeting that all addressed DAPT duration following PCI.
Perhaps most noteworthy and anticipated were results from the Dual Antiplatelet Therapy study, a randomized trial that had its beginnings in 2006 when cardiologists first became alarmed by the potential risk from stent thrombosis associated with the use of first-generation DES, specifically the original sirolimus-eluting (Cypher) and paclitaxel-eluting (Taxus) models. At the urging of staffers at the Food and Drug Administration, a group of eight stent manufacturing companies and pharmaceutical companies joined together to sponsor the DAPT study, which started with nearly 23,000 patients who received a DES and, after their first year of DAPT treatment, whittled down to 9,961 patients who were eligible for randomization to either stop DAPT after 1 year or continue on DAPT for an additional 18 months (30 months total time on DAPT).
The results showed that this highly selected group of patients – who went through their first year on DAPT with no bleeding event, no ischemic event, and who were willing to be randomized – those assigned to 30 months of DAPT had a 1% absolute and 71% relative reduction in stent thrombosis, compared with patients who stopped DAPT after 12 months, and a 1.6% absolute and 29% relative reduction in their rate of major cardiovascular or cerebrovascular events, statistically significant differences for the study’s two primary endpoints. The reduction in major events was driven mainly by a 2% absolute reduction in the rate of myocardial infarctions, reported Dr. Laura Mauri at the meeting, and in a report simultaneously published online (N. Engl. J. Med. 2014 [doi:10.1056/NEJMoa1409312]).
Counterbalancing this benefit was a statistically significant excess of moderately severe bleeding events among patients on longer-term DAPT, who had a 0.7% absolute increased rate, and a trend toward increased severe bleeds, with a 0.2% absolute increased rate. Patients on longer-term DAPT also showed an unexpected, 0.5% increased rate of both all cause death and noncardiovascular death. Detailed analysis indicated that this seemed to result from a “chance imbalance” in randomization that put an excess of patients with cancer into the 30-month DAPT subgroup, said Dr. Mauri, professor of medicine at Harvard Medical School and an interventional cardiologist at Brigham and Women’s Hospital, both in Boston.
DAPT treatment extending longer than 12 months switches the focus from preventing stent thrombosis to secondary prevention of all ischemic events, including those that occur elsewhere in a patient’s coronary arteries, noted Dr. Montalescot, who served as the meeting’s designated discussant for the DAPT trial and the other reported DAPT studies. This option, which can now be offered to appropriate patients, also raised the possibility of continuing DAPT indefinitely beyond 30 months in patients who continue to show no bleeding risk and can pay for ongoing treatment with a thienopyridine. But he advised waiting on the use of very prolonged DAPT until results arrive next year from the PEGASUS study.
ITALIC and ISAR-SAFE
Results from two other studies reported at the meeting looked at the other end of the spectrum, the safety of stopping DAPT after just 6 months. One of these, the ITALIC study, randomized 1,850 patients to 6 months or 24 months of treatment with aspirin and clopidogrel, and exclusively enrolled patients who received the Xience V everolimus-eluting, third-generation coronary stent. The results showed low and nearly identical 1.5% and 1.6% rates of the combined endpoint of major adverse events, including bleeds, in the two comparator arms after 24 months of follow-up. The results also appeared in an article published simultaneously online (J. Am. Coll. Card. 2014 [doi:10.1016/j.jacc.2014.11.008]).
A similarly designed study, ISAR-SAFE, randomized 4,005 PCI patients who received a DES to DAPT with aspirin and clopidogrel for either 6 or 12 months, and also showed a similar and low, roughly 1.5% rate of combined major adverse cardiovascular events and bleeds in both treatment arms, reported Dr. Stefanie Schulz-Schüpke, an interventional cardiologist at the German Heart Center in Munich.
These two studies, along with five prior reports that collectively randomized nearly 16,000 patients to 6 months of DAPT or a longer duration show a statistically significant reduction in major bleeds with good protection against ischemic events when DAPT stopped after 6 months, said Dr. Montalescot. “We can accept that 6 months is safer than 12 months of DAPT,” and it makes sense to stop after 6 months in a patient with a bleeding risk, but not in a patient with a high ischemia risk such as those with left main coronary disease, a history of stent thrombosis or myocardial infarction, a patient who received a first-generation DES, patients with extensive coronary disease or coronary stenting.*
While Dr. Montalescot, Dr. Antman, and others who heard these results agreed that they all pointed to the need for individualized decisions on DAPT duration, they also said the new findings were unlikely to change current guidelines. Existing PCI guidelines from the American Heart Association and American College of Cardiology call for a standard 12 months of DAPT in DES recipients, but offer clinicians to reduce or extend the duration when judged appropriate (J. Am. Coll. Cardiol. 2011;58:e44-e122). Guidelines from the European College of Cardiology call for 6 months of DAPT as standard, but also suggest that clinicians can reduce or lengthen the duration when appropriate (Eur. Heart J. 2014 [doi.org/10.1093/eurheartj/ehu278]).
According to three staffers from the FDA who also spoke at the session, the agency is also unlikely to change its guidance on DAPT in the immediate future, even though it triggered launch of the DAPT trial.
The DAPT trial “is large enough to allow us to answer some really critical public-health questions, and to potentially expand the indications for drug-eluting coronary stents,” said Dr. Bram D. Zuckerman, director of the division of cardiovascular devices of the FDA. But he and his colleagues who spoke during the session said that the agency will not update its recommendations or labels until the FDA staff has had a chance to study in detail the full data set collected in the DAPT trial. Concurrent with Dr. Mauri’s report, the agency issued a statement calling on physicians to continue to prescribe DAPT as they have in the past and for patients to continue to take all their prescribed medications.
Many physicians have already concluded that 6 months of DAPT is a practical way to enhance patient compliance with their regimen and minimize the money they spend on these medications, commented Dr. Richard C. Becker, professor of medicine and director of the Cardiovascular Institute at the University of Cincinnati. The three trials reported at the meeting “do not answer the question [of whether 6 months is adequate] because in each trial patients had already tolerated a period of DAPT before entering the study” Dr. Becker said in an interview.
The question of DAPT duration “is a question that clinicians will need to have with each of their patients, especially those who are tolerating DAPT,” commented Dr. Richard A. Chazal, an interventionalist and medical director of the Heart and Vascular Institute at the Lee Memorial Health System in Fort Myers, Fla.
Dr. Montalescot has been a consultant to more than a dozen pharmaceutical and device companies, including companies that market antiplatelet drugs. Dr. Antman has received grant support from Daiichi Sankyo. Dr. Mauri received personal fees from Medtronic, Recur, St. Jude, and Biotronik, and grant support from nine device manufacturer and drug companies. Dr. Schulz-Schüpke, Dr. Zuckerman, Dr. Becker, and Dr. Chazal had no relevant disclosures.
On Twitter@mitchelzoler
*CORRECTION, 11/18/2014: An earlier version of this article misstated the treatment length in patients with a high ischemia risk.
EXPERT OPINION FROM THE AHA SCIENTIFIC SESSIONS

Novel oral hyperkalemia drug shows promise in phase III trial
The oral drug sodium zirconium cyclosilicate may offer an effective new treatment option for outpatients with mild hyperkalemia, according to results of the phase III, open-label, randomized HARMONIZE trial presented at the American Heart Association scientific sessions.
The sodium-potassium cation exchanger zirconium cyclosilicate reduced potassium levels in 258 patients with mild hyperkalemia to normal levels within 48 hours, compared with placebo, and effectively maintained potassium levels for up to 4 weeks. The findings were published simultaneously in JAMA (JAMA 2014 Nov. 17 [doi: 10.1001/jama.2014.15688]). However, more long-term studies are needed to assess the efficacy and safety of the drug beyond 4 weeks and in patients with more severe hyperkalemia, reported Dr. Mikhail Kosiborod of Saint Luke’s Mid America Heart Institute in Kansas City, Mo., and his colleagues.

In the open-label phase of the study, 258 outpatients with hyperkalemia (serum potassium ≥5.1 mEq/L) were given 10 g of zirconium cyclosilicate three times a day for 48 hours. Mean serum potassium levels significantly declined by 0.2 mEq/L 1 hour after the initial dose and decreased an average 1.1 mEq/L at 48 hours, with 98% (n = 237) of patients achieving normokalemia (3.5-5.0 mEq/L). These patients were then randomized to receive 5 g (n = 45), 10 g (n = 51), or 15 g (n = 56) of zirconium cyclosilicate or placebo (n = 85) daily, and were followed for 28 days.
Zirconium cyclosilicate lowered serum potassium to 4.8, 4.5, and 4.4 mEq/L at 5, 10, and 15 g, respectively, compared with 5.1 mEq/L for placebo. Adverse events were comparable between the treatment and placebo groups, but edema was more common in the 15-g group. Hyperkalemia developed in 10% of the 10-g group and 11% of the 15-g group, compared with no patients in the 5-g or placebo groups.
In an accompanying editorial, Dr. Bradley S. Dixon of the University of Iowa, Iowa City, said the findings suggest that zirconium cyclosilicate may offer an important new treatment for the acute and short-term treatment of outpatients with mild hyperkalemia. “Longer-term studies are needed to assess the clinical benefits and risks that may be related to more extended use of this product, especially among hospitalized patients, as well as those with more severe hyperkalemia, other medical conditions, and other medications that affect potassium homeostasis,” he wrote.
The study was sponsored and funded by ZS Pharma, maker of sodium zirconium cyclosilicate. Dr. Kosiborod reported serving as a consultant for ZS Pharma and as an investigator on the HARMONIZE study. Several coauthors also declared financial interests with ZS Pharma. Dr. Dixon reported financial relationships with multiple companies, but not with ZS Pharma.
The oral drug sodium zirconium cyclosilicate may offer an effective new treatment option for outpatients with mild hyperkalemia, according to results of the phase III, open-label, randomized HARMONIZE trial presented at the American Heart Association scientific sessions.
The sodium-potassium cation exchanger zirconium cyclosilicate reduced potassium levels in 258 patients with mild hyperkalemia to normal levels within 48 hours, compared with placebo, and effectively maintained potassium levels for up to 4 weeks. The findings were published simultaneously in JAMA (JAMA 2014 Nov. 17 [doi: 10.1001/jama.2014.15688]). However, more long-term studies are needed to assess the efficacy and safety of the drug beyond 4 weeks and in patients with more severe hyperkalemia, reported Dr. Mikhail Kosiborod of Saint Luke’s Mid America Heart Institute in Kansas City, Mo., and his colleagues.
In the open-label phase of the study, 258 outpatients with hyperkalemia (serum potassium ≥5.1 mEq/L) were given 10 g of zirconium cyclosilicate three times a day for 48 hours. Mean serum potassium levels significantly declined by 0.2 mEq/L 1 hour after the initial dose and decreased an average 1.1 mEq/L at 48 hours, with 98% (n = 237) of patients achieving normokalemia (3.5-5.0 mEq/L). These patients were then randomized to receive 5 g (n = 45), 10 g (n = 51), or 15 g (n = 56) of zirconium cyclosilicate or placebo (n = 85) daily, and were followed for 28 days.
Zirconium cyclosilicate lowered serum potassium to 4.8, 4.5, and 4.4 mEq/L at 5, 10, and 15 g, respectively, compared with 5.1 mEq/L for placebo. Adverse events were comparable between the treatment and placebo groups, but edema was more common in the 15-g group. Hyperkalemia developed in 10% of the 10-g group and 11% of the 15-g group, compared with no patients in the 5-g or placebo groups.
In an accompanying editorial, Dr. Bradley S. Dixon of the University of Iowa, Iowa City, said the findings suggest that zirconium cyclosilicate may offer an important new treatment for the acute and short-term treatment of outpatients with mild hyperkalemia. “Longer-term studies are needed to assess the clinical benefits and risks that may be related to more extended use of this product, especially among hospitalized patients, as well as those with more severe hyperkalemia, other medical conditions, and other medications that affect potassium homeostasis,” he wrote.
The study was sponsored and funded by ZS Pharma, maker of sodium zirconium cyclosilicate. Dr. Kosiborod reported serving as a consultant for ZS Pharma and as an investigator on the HARMONIZE study. Several coauthors also declared financial interests with ZS Pharma. Dr. Dixon reported financial relationships with multiple companies, but not with ZS Pharma.
The oral drug sodium zirconium cyclosilicate may offer an effective new treatment option for outpatients with mild hyperkalemia, according to results of the phase III, open-label, randomized HARMONIZE trial presented at the American Heart Association scientific sessions.
The sodium-potassium cation exchanger zirconium cyclosilicate reduced potassium levels in 258 patients with mild hyperkalemia to normal levels within 48 hours, compared with placebo, and effectively maintained potassium levels for up to 4 weeks. The findings were published simultaneously in JAMA (JAMA 2014 Nov. 17 [doi: 10.1001/jama.2014.15688]). However, more long-term studies are needed to assess the efficacy and safety of the drug beyond 4 weeks and in patients with more severe hyperkalemia, reported Dr. Mikhail Kosiborod of Saint Luke’s Mid America Heart Institute in Kansas City, Mo., and his colleagues.
In the open-label phase of the study, 258 outpatients with hyperkalemia (serum potassium ≥5.1 mEq/L) were given 10 g of zirconium cyclosilicate three times a day for 48 hours. Mean serum potassium levels significantly declined by 0.2 mEq/L 1 hour after the initial dose and decreased an average 1.1 mEq/L at 48 hours, with 98% (n = 237) of patients achieving normokalemia (3.5-5.0 mEq/L). These patients were then randomized to receive 5 g (n = 45), 10 g (n = 51), or 15 g (n = 56) of zirconium cyclosilicate or placebo (n = 85) daily, and were followed for 28 days.
Zirconium cyclosilicate lowered serum potassium to 4.8, 4.5, and 4.4 mEq/L at 5, 10, and 15 g, respectively, compared with 5.1 mEq/L for placebo. Adverse events were comparable between the treatment and placebo groups, but edema was more common in the 15-g group. Hyperkalemia developed in 10% of the 10-g group and 11% of the 15-g group, compared with no patients in the 5-g or placebo groups.
In an accompanying editorial, Dr. Bradley S. Dixon of the University of Iowa, Iowa City, said the findings suggest that zirconium cyclosilicate may offer an important new treatment for the acute and short-term treatment of outpatients with mild hyperkalemia. “Longer-term studies are needed to assess the clinical benefits and risks that may be related to more extended use of this product, especially among hospitalized patients, as well as those with more severe hyperkalemia, other medical conditions, and other medications that affect potassium homeostasis,” he wrote.
The study was sponsored and funded by ZS Pharma, maker of sodium zirconium cyclosilicate. Dr. Kosiborod reported serving as a consultant for ZS Pharma and as an investigator on the HARMONIZE study. Several coauthors also declared financial interests with ZS Pharma. Dr. Dixon reported financial relationships with multiple companies, but not with ZS Pharma.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: A new oral agent shows promise in outpatients with mild hyperkalemia in the short term, but more studies are needed to determine its use beyond 4 weeks and in patients with severe hyperkalemia.
Major finding: Zirconium cyclosilicate reduced potassium levels in outpatients with mild hyperkalemia to normal levels within 48 hours compared with placebo, effectively maintaining potassium levels for up to 4 weeks.
Data source: Phase III, multicenter, randomized, double-blind, placebo-controlled trial of 258 outpatients with mild hyperkalemia.
Disclosures: The study was sponsored and funded by ZS Pharma, the makers of sodium zirconium cyclosilicate. Dr. Kosiborod reported serving as a consultant for ZS Pharma and as an investigator on the HARMONIZE study. Several coauthors also declared financial interests with ZS Pharma.

VIDEO: DAPT trials send mixed messages on treatment duration
CHICAGO – The four studies reported during the American Heart Association Scientific Sessions that looked at the safety and efficacy of different durations of dual antiplatelet therapy following coronary stenting addressed two different issues.
Two of the studies, ISAR-SAFE and ITALIC, both based in Europe, addressed the question of whether 6 months of dual antiplatelet therapy (DAPT) was as safe and effective as either 12 or 24 months of treatment, and the results from both studies indicated that a 6-month duration is as effective as longer term treatment. This means that especially for patients with an increased bleeding risk or another good reason to stop DAPT in the short term, such as patients who need surgery or those with atrial fibrillation who need treatment with an oral anticoagulant, stopping DAPT after 6 months is a reasonable and safe approach, Dr. Gilles Montalescot said during an interview at the meeting. More than half the patients who undergo coronary artery stenting likely fall into this subgroup, and perhaps as many as 60% or 70% of patients, estimated Dr. Montalescot, a professor of cardiology at the University of Paris and director of the cardiac care unit at Pitié-Salpêtrière Hospital in Paris.
But results from the other two DAPT trials reported at the meeting, TAXUS Liberté and DAPT, showed that for the subgroup of patients who can reasonably continue on DAPT beyond 6 months continuing the treatment out to 30 months led to substantially fewer ischemic events compared with patients who came off DAPT after 12 months, with a modest increase in bleeding events. Hence, for patients with a reduced risk for bleeding, more prolonged DAPT treatment, for as long as 30 months, produced a net benefit.
Many of the ischemic events prevented by more prolonged DAPT occurred in coronary sites outside of the placed stents, suggesting that this benefit is due more to secondary coronary disease prevention rather than prevention of late stent thrombosis, noted Dr. Montalescot. This raises the question of whether DAPT should continue indefinitely in coronary disease patients with a low bleeding risk. The current trial results do not fully resolve this, but if results from another study, PEGASUS, expected next March show similar findings, the accumulated data may support very long-term DAPT treatment in selected patients with coronary artery disease.
Dr. Montalescot said that he has been a consultant to more than a dozen pharmaceutical and device companies, including companies that market antiplatelet drugs.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
CHICAGO – The four studies reported during the American Heart Association Scientific Sessions that looked at the safety and efficacy of different durations of dual antiplatelet therapy following coronary stenting addressed two different issues.
Two of the studies, ISAR-SAFE and ITALIC, both based in Europe, addressed the question of whether 6 months of dual antiplatelet therapy (DAPT) was as safe and effective as either 12 or 24 months of treatment, and the results from both studies indicated that a 6-month duration is as effective as longer term treatment. This means that especially for patients with an increased bleeding risk or another good reason to stop DAPT in the short term, such as patients who need surgery or those with atrial fibrillation who need treatment with an oral anticoagulant, stopping DAPT after 6 months is a reasonable and safe approach, Dr. Gilles Montalescot said during an interview at the meeting. More than half the patients who undergo coronary artery stenting likely fall into this subgroup, and perhaps as many as 60% or 70% of patients, estimated Dr. Montalescot, a professor of cardiology at the University of Paris and director of the cardiac care unit at Pitié-Salpêtrière Hospital in Paris.
But results from the other two DAPT trials reported at the meeting, TAXUS Liberté and DAPT, showed that for the subgroup of patients who can reasonably continue on DAPT beyond 6 months continuing the treatment out to 30 months led to substantially fewer ischemic events compared with patients who came off DAPT after 12 months, with a modest increase in bleeding events. Hence, for patients with a reduced risk for bleeding, more prolonged DAPT treatment, for as long as 30 months, produced a net benefit.
Many of the ischemic events prevented by more prolonged DAPT occurred in coronary sites outside of the placed stents, suggesting that this benefit is due more to secondary coronary disease prevention rather than prevention of late stent thrombosis, noted Dr. Montalescot. This raises the question of whether DAPT should continue indefinitely in coronary disease patients with a low bleeding risk. The current trial results do not fully resolve this, but if results from another study, PEGASUS, expected next March show similar findings, the accumulated data may support very long-term DAPT treatment in selected patients with coronary artery disease.
Dr. Montalescot said that he has been a consultant to more than a dozen pharmaceutical and device companies, including companies that market antiplatelet drugs.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
CHICAGO – The four studies reported during the American Heart Association Scientific Sessions that looked at the safety and efficacy of different durations of dual antiplatelet therapy following coronary stenting addressed two different issues.
Two of the studies, ISAR-SAFE and ITALIC, both based in Europe, addressed the question of whether 6 months of dual antiplatelet therapy (DAPT) was as safe and effective as either 12 or 24 months of treatment, and the results from both studies indicated that a 6-month duration is as effective as longer term treatment. This means that especially for patients with an increased bleeding risk or another good reason to stop DAPT in the short term, such as patients who need surgery or those with atrial fibrillation who need treatment with an oral anticoagulant, stopping DAPT after 6 months is a reasonable and safe approach, Dr. Gilles Montalescot said during an interview at the meeting. More than half the patients who undergo coronary artery stenting likely fall into this subgroup, and perhaps as many as 60% or 70% of patients, estimated Dr. Montalescot, a professor of cardiology at the University of Paris and director of the cardiac care unit at Pitié-Salpêtrière Hospital in Paris.
But results from the other two DAPT trials reported at the meeting, TAXUS Liberté and DAPT, showed that for the subgroup of patients who can reasonably continue on DAPT beyond 6 months continuing the treatment out to 30 months led to substantially fewer ischemic events compared with patients who came off DAPT after 12 months, with a modest increase in bleeding events. Hence, for patients with a reduced risk for bleeding, more prolonged DAPT treatment, for as long as 30 months, produced a net benefit.
Many of the ischemic events prevented by more prolonged DAPT occurred in coronary sites outside of the placed stents, suggesting that this benefit is due more to secondary coronary disease prevention rather than prevention of late stent thrombosis, noted Dr. Montalescot. This raises the question of whether DAPT should continue indefinitely in coronary disease patients with a low bleeding risk. The current trial results do not fully resolve this, but if results from another study, PEGASUS, expected next March show similar findings, the accumulated data may support very long-term DAPT treatment in selected patients with coronary artery disease.
Dr. Montalescot said that he has been a consultant to more than a dozen pharmaceutical and device companies, including companies that market antiplatelet drugs.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
On Twitter @mitchelzoler
EXPERT ANALYSIS FROM AHA 2014

Telephone CPR training boosts cardiac arrest survival
CHICAGO – Systematic implementation of a comprehensive telephone CPR bundle of care targeting EMS dispatch services resulted in substantial improvements in rates of survival to hospital discharge with good neurologic outcomes in patients with out-of-hospital cardiac arrest in a major Arizona statewide public health initiative.
How big was the intervention’s impact? The rate of survival to hospital discharge showed a 33% relative increase compared to preintervention, and survival with a favorable Cerebral Performance Category score of 0 or 1 increased by 42%, Dr. Bentley J. Bobrow reported at the American Heart Association Scientific Sessions.
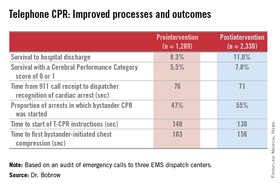
“These results suggest that when deliberately implemented and measured, telephone CPR is a targeted, effective method to increase bystander CPR and survival on a vast scale with minimal capital expense. This is why we believe telephone CPR along with public training may be the most efficient way to move the needle on cardiac arrest survival,” declared Dr. Bobrow, professor of emergency medicine at the University of Arizona College of Medicine-Phoenix Campus and chair of the AHA Basic Life Support Subcommittee.
Telephone CPR (T-CPR) entails the provision of CPR instruction to bystanders who have called 911 regarding an out of hospital cardiac arrest (OHCA). It’s well established that bystander CPR commenced before EMS personnel arrive on the scene doubles or even triples OHCA survival, but it is provided in only about one-third of OHCA events. And while T-CPR is independently associated with increased rates of bystander CPR as well as patient survival, its utilization varies widely throughout the country and few EMS services measure performance.
Dr. Bobrow reported on an ambitious undertaking that involved systematic training in T-CPR for dispatchers, 911 managers, and medical directors at all nine of the regional emergency dispatch centers in Arizona, which together with 190 EMS agencies and 40 cardiac care hospitals participate in a statewide resuscitation program.
The training was designed to implement the latest AHA guidelines on T-CPR (Circulation 2012;125:648-55). The program entailed a half-day in-person training session plus completion of a 1-hour web-based interactive video. The protocol emphasizes asking two key questions of the 911 caller: “Is the patient conscious?” and “Is the patient breathing normally?” If the response is no to both, the dispatcher is to start issuing bystander CPR instructions without delay – no further questions – and continue the coaching until EMS personnel arrive on the scene to take over.
A core aspect of the T-CPR bundle is performance measurement for quality improvement, with auditing of 911 calls to learn the time from the start of the call to the bystander’s first chest compression and five other key performance metrics. Feedback is provided to the 911 call center regarding system- and case-level performance reports in a continuing education, quality improvement process. Individual dispatchers are singled out for exemplary performance, Dr. Bobrow explained.
He presented a prospective before-and-after study conducted at the three EMS dispatch centers serving Arizona’s Maricopa County, home to two-thirds of the state’s population. The study entailed auditing nearly 6,000 911 calls, each averaging 6.5 minutes in length. After excluding calls where CPR wasn’t indicated or the OHCA involved a patient less than 8 years old, investigators were left with two groups for comparison comprised of 1,289 pre- and 2,330 post-intervention events.

The improvements in process and clinical outcomes were dramatic. In 2012, after introduction of the T-CPR training program, the bystander CPR rate crossed the 50% threshold for the first time ever in Maricopa County. The rate of survival of OHCA to hospital discharge improved from 8.3% to 11%, a highly statistically significant 33% relative increase. Survival with a Cerebral Performance Category score of 0 or 1 climbed from 5.5% to 7.8%, a 42% relative increase. In a multivariate analysis adjusted for potential confounders, the adjusted odds ratio for survival of OHCA was 2.25-fold greater for all cases after implementation of the T-CPR program and similarly increased for arrests of cardiac origin.
Dr. Bobrow observed that this was not a randomized trial, which he considered would be both unethical and impractical.
“We controlled for known risk factors and confounders, and while we cannot prove that better outcomes resulted directly from the process improvements, the two are independently associated in this controlled study,” said the emergency physician, who is medical director of the Bureau of Emergency Medical Services and Trauma Systems at the Arizona Dept. of Health Services.
Audience members rose to praise the “fantastic” achievements in Arizona and ask why they’re not having similar success rates in their own districts, given that the AHA guidelines are readily available.
“A lot of places say they’re doing this,” Dr. Bobrow replied, “but when they realize what ‘this’ is, they understand that they really weren’t doing it in this type of depth. When we showed them the data on how marginal their performance was, I think that really made the difference.”
“I think that once most dispatch centers really understand the issues and their importance and the power that they have, and you can engage them, I’m confident that people would see the same changes,” he added.
His study was honored as the best oral abstract presentation at the AHA resuscitation science symposium.
Dr. Bobrow reported serving as co-principal investigator of the HeartRescue Project, funded by Medtronic Philanthropy.
CHICAGO – Systematic implementation of a comprehensive telephone CPR bundle of care targeting EMS dispatch services resulted in substantial improvements in rates of survival to hospital discharge with good neurologic outcomes in patients with out-of-hospital cardiac arrest in a major Arizona statewide public health initiative.
How big was the intervention’s impact? The rate of survival to hospital discharge showed a 33% relative increase compared to preintervention, and survival with a favorable Cerebral Performance Category score of 0 or 1 increased by 42%, Dr. Bentley J. Bobrow reported at the American Heart Association Scientific Sessions.
“These results suggest that when deliberately implemented and measured, telephone CPR is a targeted, effective method to increase bystander CPR and survival on a vast scale with minimal capital expense. This is why we believe telephone CPR along with public training may be the most efficient way to move the needle on cardiac arrest survival,” declared Dr. Bobrow, professor of emergency medicine at the University of Arizona College of Medicine-Phoenix Campus and chair of the AHA Basic Life Support Subcommittee.
Telephone CPR (T-CPR) entails the provision of CPR instruction to bystanders who have called 911 regarding an out of hospital cardiac arrest (OHCA). It’s well established that bystander CPR commenced before EMS personnel arrive on the scene doubles or even triples OHCA survival, but it is provided in only about one-third of OHCA events. And while T-CPR is independently associated with increased rates of bystander CPR as well as patient survival, its utilization varies widely throughout the country and few EMS services measure performance.
Dr. Bobrow reported on an ambitious undertaking that involved systematic training in T-CPR for dispatchers, 911 managers, and medical directors at all nine of the regional emergency dispatch centers in Arizona, which together with 190 EMS agencies and 40 cardiac care hospitals participate in a statewide resuscitation program.
The training was designed to implement the latest AHA guidelines on T-CPR (Circulation 2012;125:648-55). The program entailed a half-day in-person training session plus completion of a 1-hour web-based interactive video. The protocol emphasizes asking two key questions of the 911 caller: “Is the patient conscious?” and “Is the patient breathing normally?” If the response is no to both, the dispatcher is to start issuing bystander CPR instructions without delay – no further questions – and continue the coaching until EMS personnel arrive on the scene to take over.
A core aspect of the T-CPR bundle is performance measurement for quality improvement, with auditing of 911 calls to learn the time from the start of the call to the bystander’s first chest compression and five other key performance metrics. Feedback is provided to the 911 call center regarding system- and case-level performance reports in a continuing education, quality improvement process. Individual dispatchers are singled out for exemplary performance, Dr. Bobrow explained.
He presented a prospective before-and-after study conducted at the three EMS dispatch centers serving Arizona’s Maricopa County, home to two-thirds of the state’s population. The study entailed auditing nearly 6,000 911 calls, each averaging 6.5 minutes in length. After excluding calls where CPR wasn’t indicated or the OHCA involved a patient less than 8 years old, investigators were left with two groups for comparison comprised of 1,289 pre- and 2,330 post-intervention events.
The improvements in process and clinical outcomes were dramatic. In 2012, after introduction of the T-CPR training program, the bystander CPR rate crossed the 50% threshold for the first time ever in Maricopa County. The rate of survival of OHCA to hospital discharge improved from 8.3% to 11%, a highly statistically significant 33% relative increase. Survival with a Cerebral Performance Category score of 0 or 1 climbed from 5.5% to 7.8%, a 42% relative increase. In a multivariate analysis adjusted for potential confounders, the adjusted odds ratio for survival of OHCA was 2.25-fold greater for all cases after implementation of the T-CPR program and similarly increased for arrests of cardiac origin.
Dr. Bobrow observed that this was not a randomized trial, which he considered would be both unethical and impractical.
“We controlled for known risk factors and confounders, and while we cannot prove that better outcomes resulted directly from the process improvements, the two are independently associated in this controlled study,” said the emergency physician, who is medical director of the Bureau of Emergency Medical Services and Trauma Systems at the Arizona Dept. of Health Services.
Audience members rose to praise the “fantastic” achievements in Arizona and ask why they’re not having similar success rates in their own districts, given that the AHA guidelines are readily available.
“A lot of places say they’re doing this,” Dr. Bobrow replied, “but when they realize what ‘this’ is, they understand that they really weren’t doing it in this type of depth. When we showed them the data on how marginal their performance was, I think that really made the difference.”
“I think that once most dispatch centers really understand the issues and their importance and the power that they have, and you can engage them, I’m confident that people would see the same changes,” he added.
His study was honored as the best oral abstract presentation at the AHA resuscitation science symposium.
Dr. Bobrow reported serving as co-principal investigator of the HeartRescue Project, funded by Medtronic Philanthropy.
CHICAGO – Systematic implementation of a comprehensive telephone CPR bundle of care targeting EMS dispatch services resulted in substantial improvements in rates of survival to hospital discharge with good neurologic outcomes in patients with out-of-hospital cardiac arrest in a major Arizona statewide public health initiative.
How big was the intervention’s impact? The rate of survival to hospital discharge showed a 33% relative increase compared to preintervention, and survival with a favorable Cerebral Performance Category score of 0 or 1 increased by 42%, Dr. Bentley J. Bobrow reported at the American Heart Association Scientific Sessions.
“These results suggest that when deliberately implemented and measured, telephone CPR is a targeted, effective method to increase bystander CPR and survival on a vast scale with minimal capital expense. This is why we believe telephone CPR along with public training may be the most efficient way to move the needle on cardiac arrest survival,” declared Dr. Bobrow, professor of emergency medicine at the University of Arizona College of Medicine-Phoenix Campus and chair of the AHA Basic Life Support Subcommittee.
Telephone CPR (T-CPR) entails the provision of CPR instruction to bystanders who have called 911 regarding an out of hospital cardiac arrest (OHCA). It’s well established that bystander CPR commenced before EMS personnel arrive on the scene doubles or even triples OHCA survival, but it is provided in only about one-third of OHCA events. And while T-CPR is independently associated with increased rates of bystander CPR as well as patient survival, its utilization varies widely throughout the country and few EMS services measure performance.
Dr. Bobrow reported on an ambitious undertaking that involved systematic training in T-CPR for dispatchers, 911 managers, and medical directors at all nine of the regional emergency dispatch centers in Arizona, which together with 190 EMS agencies and 40 cardiac care hospitals participate in a statewide resuscitation program.
The training was designed to implement the latest AHA guidelines on T-CPR (Circulation 2012;125:648-55). The program entailed a half-day in-person training session plus completion of a 1-hour web-based interactive video. The protocol emphasizes asking two key questions of the 911 caller: “Is the patient conscious?” and “Is the patient breathing normally?” If the response is no to both, the dispatcher is to start issuing bystander CPR instructions without delay – no further questions – and continue the coaching until EMS personnel arrive on the scene to take over.
A core aspect of the T-CPR bundle is performance measurement for quality improvement, with auditing of 911 calls to learn the time from the start of the call to the bystander’s first chest compression and five other key performance metrics. Feedback is provided to the 911 call center regarding system- and case-level performance reports in a continuing education, quality improvement process. Individual dispatchers are singled out for exemplary performance, Dr. Bobrow explained.
He presented a prospective before-and-after study conducted at the three EMS dispatch centers serving Arizona’s Maricopa County, home to two-thirds of the state’s population. The study entailed auditing nearly 6,000 911 calls, each averaging 6.5 minutes in length. After excluding calls where CPR wasn’t indicated or the OHCA involved a patient less than 8 years old, investigators were left with two groups for comparison comprised of 1,289 pre- and 2,330 post-intervention events.
The improvements in process and clinical outcomes were dramatic. In 2012, after introduction of the T-CPR training program, the bystander CPR rate crossed the 50% threshold for the first time ever in Maricopa County. The rate of survival of OHCA to hospital discharge improved from 8.3% to 11%, a highly statistically significant 33% relative increase. Survival with a Cerebral Performance Category score of 0 or 1 climbed from 5.5% to 7.8%, a 42% relative increase. In a multivariate analysis adjusted for potential confounders, the adjusted odds ratio for survival of OHCA was 2.25-fold greater for all cases after implementation of the T-CPR program and similarly increased for arrests of cardiac origin.
Dr. Bobrow observed that this was not a randomized trial, which he considered would be both unethical and impractical.
“We controlled for known risk factors and confounders, and while we cannot prove that better outcomes resulted directly from the process improvements, the two are independently associated in this controlled study,” said the emergency physician, who is medical director of the Bureau of Emergency Medical Services and Trauma Systems at the Arizona Dept. of Health Services.
Audience members rose to praise the “fantastic” achievements in Arizona and ask why they’re not having similar success rates in their own districts, given that the AHA guidelines are readily available.
“A lot of places say they’re doing this,” Dr. Bobrow replied, “but when they realize what ‘this’ is, they understand that they really weren’t doing it in this type of depth. When we showed them the data on how marginal their performance was, I think that really made the difference.”
“I think that once most dispatch centers really understand the issues and their importance and the power that they have, and you can engage them, I’m confident that people would see the same changes,” he added.
His study was honored as the best oral abstract presentation at the AHA resuscitation science symposium.
Dr. Bobrow reported serving as co-principal investigator of the HeartRescue Project, funded by Medtronic Philanthropy.
AT THE AHA SCIENTIFIC SESSIONS
Key clinical point: Adoption of the most recent AHA guidelines on telephone CPR by EMS dispatchers will lead to vast improvement in survival for patients with out-of-hospital cardiac arrest.
Major finding: Following implementation of an Arizona statewide program to improve telephone CPR by 911 dispatchers to bystanders at the scene of out-of-hospital cardiac arrest, survival to hospital discharge climbed from 8.3% to 11.0%.
Data source: This was a prospective study comparing the outcomes of 1,289 calls to Arizona 911 centers regarding out-of-hospital cardiac arrests before introduction of a comprehensive statewide telephone CPR program to 2,330 calls received post-intervention.
Disclosures: The study was financially supported by Medtronic Philanthropy as part of the HeartRescue Project. The presenter is co-principal investigator of the project.

Stress More Dangerous for Women With Heart Disease Than for Men
Mental stress triggered significantly more myocardial ischemia in younger women with stable coronary heart disease than in younger men with CHD, according to a study presented at the American Heart Association’s Scientific Sessions.
However, physical stress generated little or no difference in blood flow to the heart between women and men with stable coronary heart disease (CHD).
“Women who develop heart disease at a younger age make up a special high-risk group, because they are disproportionally vulnerable to emotional stress,” said Dr. Viola Vaccarino, chair of cardiovascular research and epidemiology at the Rollins School of Public Health at Emory University, Atlanta, and her colleagues in a statement.
In a population-based study, Dr. Vaccarino and her associates analyzed 534 patients (including 151 women) aged 38-79 years. All patients had CHD, which was verified by their medical records, and were divided into three age-based groups: 55 years or younger, 56-64 years, and 65 years or older.
Men and women in each age group underwent two stress tests, one mental and one physical, along with a third test while at rest. The mental test consisted of giving a public address about a real-life stressful scenario, while the physical test was a treadmill exercise conducted in accordance with the Bruce protocol. Patients who were unable to achieve the target heart rate in the physical test underwent pharmacological stress with regadenoson.
During each test, patients’ heart rates and blood pressures were monitored, from which total severity scores were derived by quantifying myocardial perfusion deficit (size and severity) across 17 myocardial segments. Total ischemic perfusion deficits (IPD) were calculated by taking the difference between a patient’s severity score at rest and severity score during mental/physical stress.
Overall, women had greater IPD with mental stress than men did, and younger age exacerbated the difference.
Women aged 55 years or younger experiencing 3.5 times the mean IPD as did men in the same age group (139 vs. 40; P < .0001). Mean IPD was 1.8 times greater in women aged 56-64 years than in men (101 vs. 56; P = .03), while mean IPD in women 65 years or older was almost identical to that of their male counterparts (69 vs. 61; P = 0.5).
Conversely, physical stress yielded no significant difference between men and women across all three age groups. Those aged 55 years or younger showed the largest, albeit nonsignificant, differences – with women having 1.5 times more IPD under physical stress than similarly aged men (123 vs. 84; P = .22). In the other two age groups, however, differences between women and men were minimal and nonsignificant.
“Health care providers should be aware of young and middle-age women’s special vulnerability to stress, and ask the questions about psychological stress that often don’t get asked,” the authors advised. “If they note that their patient is under psychological stress or is depressed, they should advise the woman to get relevant help or support from mental health providers, stress-reduction programs, or other means.”
The National Heart, Lung, and Blood Institute funded the study. Dr. Vaccarino had no relevant disclosures.
Mental stress triggered significantly more myocardial ischemia in younger women with stable coronary heart disease than in younger men with CHD, according to a study presented at the American Heart Association’s Scientific Sessions.
However, physical stress generated little or no difference in blood flow to the heart between women and men with stable coronary heart disease (CHD).
“Women who develop heart disease at a younger age make up a special high-risk group, because they are disproportionally vulnerable to emotional stress,” said Dr. Viola Vaccarino, chair of cardiovascular research and epidemiology at the Rollins School of Public Health at Emory University, Atlanta, and her colleagues in a statement.
In a population-based study, Dr. Vaccarino and her associates analyzed 534 patients (including 151 women) aged 38-79 years. All patients had CHD, which was verified by their medical records, and were divided into three age-based groups: 55 years or younger, 56-64 years, and 65 years or older.
Men and women in each age group underwent two stress tests, one mental and one physical, along with a third test while at rest. The mental test consisted of giving a public address about a real-life stressful scenario, while the physical test was a treadmill exercise conducted in accordance with the Bruce protocol. Patients who were unable to achieve the target heart rate in the physical test underwent pharmacological stress with regadenoson.
During each test, patients’ heart rates and blood pressures were monitored, from which total severity scores were derived by quantifying myocardial perfusion deficit (size and severity) across 17 myocardial segments. Total ischemic perfusion deficits (IPD) were calculated by taking the difference between a patient’s severity score at rest and severity score during mental/physical stress.
Overall, women had greater IPD with mental stress than men did, and younger age exacerbated the difference.
Women aged 55 years or younger experiencing 3.5 times the mean IPD as did men in the same age group (139 vs. 40; P < .0001). Mean IPD was 1.8 times greater in women aged 56-64 years than in men (101 vs. 56; P = .03), while mean IPD in women 65 years or older was almost identical to that of their male counterparts (69 vs. 61; P = 0.5).
Conversely, physical stress yielded no significant difference between men and women across all three age groups. Those aged 55 years or younger showed the largest, albeit nonsignificant, differences – with women having 1.5 times more IPD under physical stress than similarly aged men (123 vs. 84; P = .22). In the other two age groups, however, differences between women and men were minimal and nonsignificant.
“Health care providers should be aware of young and middle-age women’s special vulnerability to stress, and ask the questions about psychological stress that often don’t get asked,” the authors advised. “If they note that their patient is under psychological stress or is depressed, they should advise the woman to get relevant help or support from mental health providers, stress-reduction programs, or other means.”
The National Heart, Lung, and Blood Institute funded the study. Dr. Vaccarino had no relevant disclosures.
Mental stress triggered significantly more myocardial ischemia in younger women with stable coronary heart disease than in younger men with CHD, according to a study presented at the American Heart Association’s Scientific Sessions.
However, physical stress generated little or no difference in blood flow to the heart between women and men with stable coronary heart disease (CHD).
“Women who develop heart disease at a younger age make up a special high-risk group, because they are disproportionally vulnerable to emotional stress,” said Dr. Viola Vaccarino, chair of cardiovascular research and epidemiology at the Rollins School of Public Health at Emory University, Atlanta, and her colleagues in a statement.
In a population-based study, Dr. Vaccarino and her associates analyzed 534 patients (including 151 women) aged 38-79 years. All patients had CHD, which was verified by their medical records, and were divided into three age-based groups: 55 years or younger, 56-64 years, and 65 years or older.
Men and women in each age group underwent two stress tests, one mental and one physical, along with a third test while at rest. The mental test consisted of giving a public address about a real-life stressful scenario, while the physical test was a treadmill exercise conducted in accordance with the Bruce protocol. Patients who were unable to achieve the target heart rate in the physical test underwent pharmacological stress with regadenoson.
During each test, patients’ heart rates and blood pressures were monitored, from which total severity scores were derived by quantifying myocardial perfusion deficit (size and severity) across 17 myocardial segments. Total ischemic perfusion deficits (IPD) were calculated by taking the difference between a patient’s severity score at rest and severity score during mental/physical stress.
Overall, women had greater IPD with mental stress than men did, and younger age exacerbated the difference.
Women aged 55 years or younger experiencing 3.5 times the mean IPD as did men in the same age group (139 vs. 40; P < .0001). Mean IPD was 1.8 times greater in women aged 56-64 years than in men (101 vs. 56; P = .03), while mean IPD in women 65 years or older was almost identical to that of their male counterparts (69 vs. 61; P = 0.5).
Conversely, physical stress yielded no significant difference between men and women across all three age groups. Those aged 55 years or younger showed the largest, albeit nonsignificant, differences – with women having 1.5 times more IPD under physical stress than similarly aged men (123 vs. 84; P = .22). In the other two age groups, however, differences between women and men were minimal and nonsignificant.
“Health care providers should be aware of young and middle-age women’s special vulnerability to stress, and ask the questions about psychological stress that often don’t get asked,” the authors advised. “If they note that their patient is under psychological stress or is depressed, they should advise the woman to get relevant help or support from mental health providers, stress-reduction programs, or other means.”
The National Heart, Lung, and Blood Institute funded the study. Dr. Vaccarino had no relevant disclosures.

Stress more dangerous for women with heart disease than for men
Mental stress triggered significantly more myocardial ischemia in younger women with stable coronary heart disease than in younger men with CHD, according to a study presented at the American Heart Association’s Scientific Sessions.
However, physical stress generated little or no difference in blood flow to the heart between women and men with stable coronary heart disease (CHD).

“Women who develop heart disease at a younger age make up a special high-risk group, because they are disproportionally vulnerable to emotional stress,” said Dr. Viola Vaccarino, chair of cardiovascular research and epidemiology at the Rollins School of Public Health at Emory University, Atlanta, and her colleagues in a statement.
In a population-based study, Dr. Vaccarino and her associates analyzed 534 patients (including 151 women) aged 38-79 years. All patients had CHD, which was verified by their medical records, and were divided into three age-based groups: 55 years or younger, 56-64 years, and 65 years or older.
Men and women in each age group underwent two stress tests, one mental and one physical, along with a third test while at rest. The mental test consisted of giving a public address about a real-life stressful scenario, while the physical test was a treadmill exercise conducted in accordance with the Bruce protocol. Patients who were unable to achieve the target heart rate in the physical test underwent pharmacological stress with regadenoson.
During each test, patients’ heart rates and blood pressures were monitored, from which total severity scores were derived by quantifying myocardial perfusion deficit (size and severity) across 17 myocardial segments. Total ischemic perfusion deficits (IPD) were calculated by taking the difference between a patient’s severity score at rest and severity score during mental/physical stress.
Overall, women had greater IPD with mental stress than men did, and younger age exacerbated the difference.
Women aged 55 years or younger experiencing 3.5 times the mean IPD as did men in the same age group (139 vs. 40; P < .0001). Mean IPD was 1.8 times greater in women aged 56-64 years than in men (101 vs. 56; P = .03), while mean IPD in women 65 years or older was almost identical to that of their male counterparts (69 vs. 61; P = 0.5).
Conversely, physical stress yielded no significant difference between men and women across all three age groups. Those aged 55 years or younger showed the largest, albeit nonsignificant, differences – with women having 1.5 times more IPD under physical stress than similarly aged men (123 vs. 84; P = .22). In the other two age groups, however, differences between women and men were minimal and nonsignificant.
“Health care providers should be aware of young and middle-age women’s special vulnerability to stress, and ask the questions about psychological stress that often don’t get asked,” the authors advised. “If they note that their patient is under psychological stress or is depressed, they should advise the woman to get relevant help or support from mental health providers, stress-reduction programs, or other means.”
The National Heart, Lung, and Blood Institute funded the study. Dr. Vaccarino had no relevant disclosures.
Mental stress triggered significantly more myocardial ischemia in younger women with stable coronary heart disease than in younger men with CHD, according to a study presented at the American Heart Association’s Scientific Sessions.
However, physical stress generated little or no difference in blood flow to the heart between women and men with stable coronary heart disease (CHD).
“Women who develop heart disease at a younger age make up a special high-risk group, because they are disproportionally vulnerable to emotional stress,” said Dr. Viola Vaccarino, chair of cardiovascular research and epidemiology at the Rollins School of Public Health at Emory University, Atlanta, and her colleagues in a statement.
In a population-based study, Dr. Vaccarino and her associates analyzed 534 patients (including 151 women) aged 38-79 years. All patients had CHD, which was verified by their medical records, and were divided into three age-based groups: 55 years or younger, 56-64 years, and 65 years or older.
Men and women in each age group underwent two stress tests, one mental and one physical, along with a third test while at rest. The mental test consisted of giving a public address about a real-life stressful scenario, while the physical test was a treadmill exercise conducted in accordance with the Bruce protocol. Patients who were unable to achieve the target heart rate in the physical test underwent pharmacological stress with regadenoson.
During each test, patients’ heart rates and blood pressures were monitored, from which total severity scores were derived by quantifying myocardial perfusion deficit (size and severity) across 17 myocardial segments. Total ischemic perfusion deficits (IPD) were calculated by taking the difference between a patient’s severity score at rest and severity score during mental/physical stress.
Overall, women had greater IPD with mental stress than men did, and younger age exacerbated the difference.
Women aged 55 years or younger experiencing 3.5 times the mean IPD as did men in the same age group (139 vs. 40; P < .0001). Mean IPD was 1.8 times greater in women aged 56-64 years than in men (101 vs. 56; P = .03), while mean IPD in women 65 years or older was almost identical to that of their male counterparts (69 vs. 61; P = 0.5).
Conversely, physical stress yielded no significant difference between men and women across all three age groups. Those aged 55 years or younger showed the largest, albeit nonsignificant, differences – with women having 1.5 times more IPD under physical stress than similarly aged men (123 vs. 84; P = .22). In the other two age groups, however, differences between women and men were minimal and nonsignificant.
“Health care providers should be aware of young and middle-age women’s special vulnerability to stress, and ask the questions about psychological stress that often don’t get asked,” the authors advised. “If they note that their patient is under psychological stress or is depressed, they should advise the woman to get relevant help or support from mental health providers, stress-reduction programs, or other means.”
The National Heart, Lung, and Blood Institute funded the study. Dr. Vaccarino had no relevant disclosures.
Mental stress triggered significantly more myocardial ischemia in younger women with stable coronary heart disease than in younger men with CHD, according to a study presented at the American Heart Association’s Scientific Sessions.
However, physical stress generated little or no difference in blood flow to the heart between women and men with stable coronary heart disease (CHD).
“Women who develop heart disease at a younger age make up a special high-risk group, because they are disproportionally vulnerable to emotional stress,” said Dr. Viola Vaccarino, chair of cardiovascular research and epidemiology at the Rollins School of Public Health at Emory University, Atlanta, and her colleagues in a statement.
In a population-based study, Dr. Vaccarino and her associates analyzed 534 patients (including 151 women) aged 38-79 years. All patients had CHD, which was verified by their medical records, and were divided into three age-based groups: 55 years or younger, 56-64 years, and 65 years or older.
Men and women in each age group underwent two stress tests, one mental and one physical, along with a third test while at rest. The mental test consisted of giving a public address about a real-life stressful scenario, while the physical test was a treadmill exercise conducted in accordance with the Bruce protocol. Patients who were unable to achieve the target heart rate in the physical test underwent pharmacological stress with regadenoson.
During each test, patients’ heart rates and blood pressures were monitored, from which total severity scores were derived by quantifying myocardial perfusion deficit (size and severity) across 17 myocardial segments. Total ischemic perfusion deficits (IPD) were calculated by taking the difference between a patient’s severity score at rest and severity score during mental/physical stress.
Overall, women had greater IPD with mental stress than men did, and younger age exacerbated the difference.
Women aged 55 years or younger experiencing 3.5 times the mean IPD as did men in the same age group (139 vs. 40; P < .0001). Mean IPD was 1.8 times greater in women aged 56-64 years than in men (101 vs. 56; P = .03), while mean IPD in women 65 years or older was almost identical to that of their male counterparts (69 vs. 61; P = 0.5).
Conversely, physical stress yielded no significant difference between men and women across all three age groups. Those aged 55 years or younger showed the largest, albeit nonsignificant, differences – with women having 1.5 times more IPD under physical stress than similarly aged men (123 vs. 84; P = .22). In the other two age groups, however, differences between women and men were minimal and nonsignificant.
“Health care providers should be aware of young and middle-age women’s special vulnerability to stress, and ask the questions about psychological stress that often don’t get asked,” the authors advised. “If they note that their patient is under psychological stress or is depressed, they should advise the woman to get relevant help or support from mental health providers, stress-reduction programs, or other means.”
The National Heart, Lung, and Blood Institute funded the study. Dr. Vaccarino had no relevant disclosures.
Key clinical point: Younger women with stable coronary heart disease suffer greater stress-induced myocardial ischemia than men do.
Major finding: After mental stress, ischemic perfusion deficits were 3.5 times greater in women aged 55 years or younger with CHD than in men of similar age with CHD.
Data source: A population-based study.
Disclosures: The National Heart, Lung, and Blood Institute funded the study. Dr. Vaccarino had no relevant disclosures.

Energy drink poison control calls involved cardiac, neurologic effects
Children under 6 years old are among those at the highest risk for poison control calls related to energy drinks, yet those most likely to experience moderate or major clinical effects from energy drinks are adults and often involve mixing with alcohol, according to a recent study.
Approximately 40% of all calls involving single-product energy drinks (not mixed with alcohol) were unintentional exposures by children under 6 years old, according to researchers led by Dr. Steven E. Lipshultz, chair of the department of pediatrics at Wayne State University in Detroit.

“We recommend improved labeling of caffeine content and continued efforts to decrease pediatric exposures to these products,” the researchers concluded in a presentation at the American Heart Association scientific sessions Nov. 16.
Emergency department visits involving caffeinated energy drinks in children under 12 years of age increased “exponentially” between 2005 and 2011, from 840 cases of adverse reactions in 2005 to 14,042 cases in 2011, according to Drug Abuse Warning Network background information in the presentation.
For their study, the researchers analyzed all cases of energy drink exposure reported to the U.S. National Poison Data System during October 2010 and September 2013. These include calls to 55 poison control centers in the United States.
Among the total 10,610 cases reported, 5,156 (49%) were single-product exposures with ingredients that could be identified, thereby excluding exposures related to alcoholic mixes or other combinations.
Mixtures of energy drinks and alcohol produced the most serious problems, with moderate to major effects occurring in 42% of cases involving alcohol and 19% of cases without alcohol (P < .001). Exposures involving alcohol have fallen sharply, though, since a 2010 Food and Drug Administration ban on prepackaged energy drinks containing alcohol, the researchers reported.
Both pharmaceutical-grade caffeine and caffeine from other sources may be in energy drinks, according to a statement from the American Heart Association. In this study, 23% of cases related to drinks with both pharmaceutical and additional caffeine resulted in moderate or major outcomes, compared with 15% of cases involving drinks with only pharmaceutical-grade caffeine (P < .001).
Children were least likely to experience major problems, with only 4% of cases with major or moderate effects involving children under age 12 years. Meanwhile, 64% of cases resulting in moderate or major clinical effects involved adults aged 20 years and older.
The most commonly affected organ system for non–alcohol-related energy drink exposures was neurologic, composing 20% of all clinical effects reported. These symptoms included agitation/irritability, dizziness, lethargy, headache, seizures, and tremors.
In addition, 15% of the cases involved gastrointestinal effects, such as abdominal pain, diarrhea, nausea, and vomiting, and 12% involved cardiovascular effects, which included chest pain, hypertension, arrhythmia, and tachycardia, among others.
Among the cases involving major clinical effects, 57% were cardiovascular, 54% were neurologic, and 14% were gastrointestinal. Some patients had multiple effects in more than one organ system.
Children younger than age 6 years represented the highest proportion of unintentional exposures, 75% of the total, while 10% of unintentional exposures occurred to children aged 6-12 years. Among intentional exposures, on the other hand, 45% included teenagers aged 13-19 years, and 41% included people aged 20 years and older.
These findings, based on poison control center reports, underestimate the problem, the researchers noted, because “many people who become ill from energy drinks don’t call the hotlines and emergency room visits are not included.” Those that are reported may, however, be biased in favor of greater toxicity, they said.
Information on funding and disclosures was unavailable.
Children under 6 years old are among those at the highest risk for poison control calls related to energy drinks, yet those most likely to experience moderate or major clinical effects from energy drinks are adults and often involve mixing with alcohol, according to a recent study.
Approximately 40% of all calls involving single-product energy drinks (not mixed with alcohol) were unintentional exposures by children under 6 years old, according to researchers led by Dr. Steven E. Lipshultz, chair of the department of pediatrics at Wayne State University in Detroit.
“We recommend improved labeling of caffeine content and continued efforts to decrease pediatric exposures to these products,” the researchers concluded in a presentation at the American Heart Association scientific sessions Nov. 16.
Emergency department visits involving caffeinated energy drinks in children under 12 years of age increased “exponentially” between 2005 and 2011, from 840 cases of adverse reactions in 2005 to 14,042 cases in 2011, according to Drug Abuse Warning Network background information in the presentation.
For their study, the researchers analyzed all cases of energy drink exposure reported to the U.S. National Poison Data System during October 2010 and September 2013. These include calls to 55 poison control centers in the United States.
Among the total 10,610 cases reported, 5,156 (49%) were single-product exposures with ingredients that could be identified, thereby excluding exposures related to alcoholic mixes or other combinations.
Mixtures of energy drinks and alcohol produced the most serious problems, with moderate to major effects occurring in 42% of cases involving alcohol and 19% of cases without alcohol (P < .001). Exposures involving alcohol have fallen sharply, though, since a 2010 Food and Drug Administration ban on prepackaged energy drinks containing alcohol, the researchers reported.
Both pharmaceutical-grade caffeine and caffeine from other sources may be in energy drinks, according to a statement from the American Heart Association. In this study, 23% of cases related to drinks with both pharmaceutical and additional caffeine resulted in moderate or major outcomes, compared with 15% of cases involving drinks with only pharmaceutical-grade caffeine (P < .001).
Children were least likely to experience major problems, with only 4% of cases with major or moderate effects involving children under age 12 years. Meanwhile, 64% of cases resulting in moderate or major clinical effects involved adults aged 20 years and older.
The most commonly affected organ system for non–alcohol-related energy drink exposures was neurologic, composing 20% of all clinical effects reported. These symptoms included agitation/irritability, dizziness, lethargy, headache, seizures, and tremors.
In addition, 15% of the cases involved gastrointestinal effects, such as abdominal pain, diarrhea, nausea, and vomiting, and 12% involved cardiovascular effects, which included chest pain, hypertension, arrhythmia, and tachycardia, among others.
Among the cases involving major clinical effects, 57% were cardiovascular, 54% were neurologic, and 14% were gastrointestinal. Some patients had multiple effects in more than one organ system.
Children younger than age 6 years represented the highest proportion of unintentional exposures, 75% of the total, while 10% of unintentional exposures occurred to children aged 6-12 years. Among intentional exposures, on the other hand, 45% included teenagers aged 13-19 years, and 41% included people aged 20 years and older.
These findings, based on poison control center reports, underestimate the problem, the researchers noted, because “many people who become ill from energy drinks don’t call the hotlines and emergency room visits are not included.” Those that are reported may, however, be biased in favor of greater toxicity, they said.
Information on funding and disclosures was unavailable.
Children under 6 years old are among those at the highest risk for poison control calls related to energy drinks, yet those most likely to experience moderate or major clinical effects from energy drinks are adults and often involve mixing with alcohol, according to a recent study.
Approximately 40% of all calls involving single-product energy drinks (not mixed with alcohol) were unintentional exposures by children under 6 years old, according to researchers led by Dr. Steven E. Lipshultz, chair of the department of pediatrics at Wayne State University in Detroit.
“We recommend improved labeling of caffeine content and continued efforts to decrease pediatric exposures to these products,” the researchers concluded in a presentation at the American Heart Association scientific sessions Nov. 16.
Emergency department visits involving caffeinated energy drinks in children under 12 years of age increased “exponentially” between 2005 and 2011, from 840 cases of adverse reactions in 2005 to 14,042 cases in 2011, according to Drug Abuse Warning Network background information in the presentation.
For their study, the researchers analyzed all cases of energy drink exposure reported to the U.S. National Poison Data System during October 2010 and September 2013. These include calls to 55 poison control centers in the United States.
Among the total 10,610 cases reported, 5,156 (49%) were single-product exposures with ingredients that could be identified, thereby excluding exposures related to alcoholic mixes or other combinations.
Mixtures of energy drinks and alcohol produced the most serious problems, with moderate to major effects occurring in 42% of cases involving alcohol and 19% of cases without alcohol (P < .001). Exposures involving alcohol have fallen sharply, though, since a 2010 Food and Drug Administration ban on prepackaged energy drinks containing alcohol, the researchers reported.
Both pharmaceutical-grade caffeine and caffeine from other sources may be in energy drinks, according to a statement from the American Heart Association. In this study, 23% of cases related to drinks with both pharmaceutical and additional caffeine resulted in moderate or major outcomes, compared with 15% of cases involving drinks with only pharmaceutical-grade caffeine (P < .001).
Children were least likely to experience major problems, with only 4% of cases with major or moderate effects involving children under age 12 years. Meanwhile, 64% of cases resulting in moderate or major clinical effects involved adults aged 20 years and older.
The most commonly affected organ system for non–alcohol-related energy drink exposures was neurologic, composing 20% of all clinical effects reported. These symptoms included agitation/irritability, dizziness, lethargy, headache, seizures, and tremors.
In addition, 15% of the cases involved gastrointestinal effects, such as abdominal pain, diarrhea, nausea, and vomiting, and 12% involved cardiovascular effects, which included chest pain, hypertension, arrhythmia, and tachycardia, among others.
Among the cases involving major clinical effects, 57% were cardiovascular, 54% were neurologic, and 14% were gastrointestinal. Some patients had multiple effects in more than one organ system.
Children younger than age 6 years represented the highest proportion of unintentional exposures, 75% of the total, while 10% of unintentional exposures occurred to children aged 6-12 years. Among intentional exposures, on the other hand, 45% included teenagers aged 13-19 years, and 41% included people aged 20 years and older.
These findings, based on poison control center reports, underestimate the problem, the researchers noted, because “many people who become ill from energy drinks don’t call the hotlines and emergency room visits are not included.” Those that are reported may, however, be biased in favor of greater toxicity, they said.
Information on funding and disclosures was unavailable.
FROM THE AHA SCIENTIFIC SESSIONS
Key clinical point: Energy drink exposures commonly involve children and may lead to serious cardiac and neurologic effects in adults.
Major finding: A total of 40% of unintentional energy drink exposures involved children under 6 years; 42% of energy drink exposures involving alcohol and 19% without alcohol resulted in moderate to major clinical effects.
Data source: Retrospective analysis of 10,610 exposures reported to the U.S. National Poison Data System between October 2010 and September 2013.
Disclosures: Information on funding and disclosures was unavailable.
