User login
FDA approves rucaparib for BRCA-positive advanced ovarian cancer
The Food and Drug Administration has granted accelerated approval to rucaparib for the treatment of women with advanced ovarian cancer who have been treated with two or more chemotherapies and whose tumors have a germline or somatic BRCA gene mutation. The FDA also approved the FoundationFocus CDxBRCA companion diagnostic for use with rucaparib to detect BRCA1 and BRCA2 gene mutations in the tumor tissue.
Approval of rucaparib (Rubraca), a poly ADP-ribose polymerase (PARP) inhibitor, was based on an objective response rate (ORR) of 54%, and a median duration of response of 9.2 months, in a pooled analysis of two, single-arm clinical trials, the FDA said in a statement.
All 106 patients in the two trials had BRCA-mutated advanced ovarian cancer and had been treated with two or more chemotherapy regimens. They received rucaparib 600 mg orally twice daily. BRCA gene mutations were confirmed in 96% of participants with available tumor tissue using the FoundationFocus CDxBRCA companion diagnostic. ORR was similar for patients with a BRCA1 gene mutation or BRCA2 gene mutation.![]()
Patients should be monitored for hematologic toxicity at baseline and monthly thereafter, and use of rucaparib should be discontinued if MDS/AML is confirmed, the FDA said on its website.
Rucaparib is marketed by Clovis Oncology. The FoundationFocus CDxBRCA companion diagnostic is marketed by Foundation Medicine.
The Food and Drug Administration has granted accelerated approval to rucaparib for the treatment of women with advanced ovarian cancer who have been treated with two or more chemotherapies and whose tumors have a germline or somatic BRCA gene mutation. The FDA also approved the FoundationFocus CDxBRCA companion diagnostic for use with rucaparib to detect BRCA1 and BRCA2 gene mutations in the tumor tissue.
Approval of rucaparib (Rubraca), a poly ADP-ribose polymerase (PARP) inhibitor, was based on an objective response rate (ORR) of 54%, and a median duration of response of 9.2 months, in a pooled analysis of two, single-arm clinical trials, the FDA said in a statement.
All 106 patients in the two trials had BRCA-mutated advanced ovarian cancer and had been treated with two or more chemotherapy regimens. They received rucaparib 600 mg orally twice daily. BRCA gene mutations were confirmed in 96% of participants with available tumor tissue using the FoundationFocus CDxBRCA companion diagnostic. ORR was similar for patients with a BRCA1 gene mutation or BRCA2 gene mutation.![]()
Patients should be monitored for hematologic toxicity at baseline and monthly thereafter, and use of rucaparib should be discontinued if MDS/AML is confirmed, the FDA said on its website.
Rucaparib is marketed by Clovis Oncology. The FoundationFocus CDxBRCA companion diagnostic is marketed by Foundation Medicine.
The Food and Drug Administration has granted accelerated approval to rucaparib for the treatment of women with advanced ovarian cancer who have been treated with two or more chemotherapies and whose tumors have a germline or somatic BRCA gene mutation. The FDA also approved the FoundationFocus CDxBRCA companion diagnostic for use with rucaparib to detect BRCA1 and BRCA2 gene mutations in the tumor tissue.
Approval of rucaparib (Rubraca), a poly ADP-ribose polymerase (PARP) inhibitor, was based on an objective response rate (ORR) of 54%, and a median duration of response of 9.2 months, in a pooled analysis of two, single-arm clinical trials, the FDA said in a statement.
All 106 patients in the two trials had BRCA-mutated advanced ovarian cancer and had been treated with two or more chemotherapy regimens. They received rucaparib 600 mg orally twice daily. BRCA gene mutations were confirmed in 96% of participants with available tumor tissue using the FoundationFocus CDxBRCA companion diagnostic. ORR was similar for patients with a BRCA1 gene mutation or BRCA2 gene mutation.![]()
Patients should be monitored for hematologic toxicity at baseline and monthly thereafter, and use of rucaparib should be discontinued if MDS/AML is confirmed, the FDA said on its website.
Rucaparib is marketed by Clovis Oncology. The FoundationFocus CDxBRCA companion diagnostic is marketed by Foundation Medicine.
Latest CDC data: Opioid deaths still rising
Opioid-related deaths continue to rise in the United States, with a 16% increase between 2014 and 2015 driven largely by overdoses of illegally manufactured fentanyl and heroin, according to a report released Dec. 16 by the Centers for Disease Control and Prevention.
CDC investigators analyzed drug-related mortality for 2010 through 2015 in a national statistics database for all 50 states and the District of Columbia, as well as drug-related deaths by subcategories of drugs in 28 states for 2014 through 2015. They found that the rapidly evolving opioid epidemic has not only continued but worsened in many ways, across all demographics and geographical regions of the country, said Rose A. Rudd, MSPH, of the CDC’s National Center for Injury Prevention and Control in Atlanta, and her associates (MMWR. 2016 Dec 16;65:1-8).![]()
• Mortality from drug overdoses rose significantly over the 5-year study period, from 12.3 per 100,000 in 2010 to 16.3 per 100,000 in 2015. It rose in 30 states and in the District of Columbia, stayed stable in 19 states, and initially decreased but then rose again in 2 states (Florida and South Carolina).
• During the last year for which data are complete (2015), deaths from drug overdoses rose by approximately 12%, “signifying a continuing trend since 1999.”
• Sixty-three percent of the 52,404 deaths from drug overdoses in 2015 involved an opioid.
• The age-adjusted opioid-related death rate rose by 16% during the last year, from 9.0 per 100,000 in 2014 to 10.4 per 100,000 in 2015.
• These significant increases were driven by a rise in deaths from synthetic opioids other than methadone – chiefly illicitly manufactured fentanyl and heroin, which rose by 72.2% and 20.6%, respectively.
• In contrast, death rates tied to natural or semisynthetic opioids increased by only 2.6%, while those tied to methadone decreased by 9.1%.
Dr. Rudd and her associates cited several limitations. One is that some drug overdose death certificates did not identify specific drugs. Another is that heroin and morphine are metabolized similarly, which means that some heroin deaths might have been misclassified. Also, it could be problematic to generalize the findings, because the “state-specific analyses of opioid deaths are restricted to 28 states.”
“The ongoing epidemic of opioid deaths requires intense attention and action,” Dr. Rudd and her associates wrote. “Intensifying efforts to distribute naloxone (an antidote to reverse an opioid overdose), enhancing access to treatment ... and implementing harm reduction services are urgently needed.”
The study was conducted by the CDC.
Opioid-related deaths continue to rise in the United States, with a 16% increase between 2014 and 2015 driven largely by overdoses of illegally manufactured fentanyl and heroin, according to a report released Dec. 16 by the Centers for Disease Control and Prevention.
CDC investigators analyzed drug-related mortality for 2010 through 2015 in a national statistics database for all 50 states and the District of Columbia, as well as drug-related deaths by subcategories of drugs in 28 states for 2014 through 2015. They found that the rapidly evolving opioid epidemic has not only continued but worsened in many ways, across all demographics and geographical regions of the country, said Rose A. Rudd, MSPH, of the CDC’s National Center for Injury Prevention and Control in Atlanta, and her associates (MMWR. 2016 Dec 16;65:1-8).![]()
• Mortality from drug overdoses rose significantly over the 5-year study period, from 12.3 per 100,000 in 2010 to 16.3 per 100,000 in 2015. It rose in 30 states and in the District of Columbia, stayed stable in 19 states, and initially decreased but then rose again in 2 states (Florida and South Carolina).
• During the last year for which data are complete (2015), deaths from drug overdoses rose by approximately 12%, “signifying a continuing trend since 1999.”
• Sixty-three percent of the 52,404 deaths from drug overdoses in 2015 involved an opioid.
• The age-adjusted opioid-related death rate rose by 16% during the last year, from 9.0 per 100,000 in 2014 to 10.4 per 100,000 in 2015.
• These significant increases were driven by a rise in deaths from synthetic opioids other than methadone – chiefly illicitly manufactured fentanyl and heroin, which rose by 72.2% and 20.6%, respectively.
• In contrast, death rates tied to natural or semisynthetic opioids increased by only 2.6%, while those tied to methadone decreased by 9.1%.
Dr. Rudd and her associates cited several limitations. One is that some drug overdose death certificates did not identify specific drugs. Another is that heroin and morphine are metabolized similarly, which means that some heroin deaths might have been misclassified. Also, it could be problematic to generalize the findings, because the “state-specific analyses of opioid deaths are restricted to 28 states.”
“The ongoing epidemic of opioid deaths requires intense attention and action,” Dr. Rudd and her associates wrote. “Intensifying efforts to distribute naloxone (an antidote to reverse an opioid overdose), enhancing access to treatment ... and implementing harm reduction services are urgently needed.”
The study was conducted by the CDC.
Opioid-related deaths continue to rise in the United States, with a 16% increase between 2014 and 2015 driven largely by overdoses of illegally manufactured fentanyl and heroin, according to a report released Dec. 16 by the Centers for Disease Control and Prevention.
CDC investigators analyzed drug-related mortality for 2010 through 2015 in a national statistics database for all 50 states and the District of Columbia, as well as drug-related deaths by subcategories of drugs in 28 states for 2014 through 2015. They found that the rapidly evolving opioid epidemic has not only continued but worsened in many ways, across all demographics and geographical regions of the country, said Rose A. Rudd, MSPH, of the CDC’s National Center for Injury Prevention and Control in Atlanta, and her associates (MMWR. 2016 Dec 16;65:1-8).![]()
• Mortality from drug overdoses rose significantly over the 5-year study period, from 12.3 per 100,000 in 2010 to 16.3 per 100,000 in 2015. It rose in 30 states and in the District of Columbia, stayed stable in 19 states, and initially decreased but then rose again in 2 states (Florida and South Carolina).
• During the last year for which data are complete (2015), deaths from drug overdoses rose by approximately 12%, “signifying a continuing trend since 1999.”
• Sixty-three percent of the 52,404 deaths from drug overdoses in 2015 involved an opioid.
• The age-adjusted opioid-related death rate rose by 16% during the last year, from 9.0 per 100,000 in 2014 to 10.4 per 100,000 in 2015.
• These significant increases were driven by a rise in deaths from synthetic opioids other than methadone – chiefly illicitly manufactured fentanyl and heroin, which rose by 72.2% and 20.6%, respectively.
• In contrast, death rates tied to natural or semisynthetic opioids increased by only 2.6%, while those tied to methadone decreased by 9.1%.
Dr. Rudd and her associates cited several limitations. One is that some drug overdose death certificates did not identify specific drugs. Another is that heroin and morphine are metabolized similarly, which means that some heroin deaths might have been misclassified. Also, it could be problematic to generalize the findings, because the “state-specific analyses of opioid deaths are restricted to 28 states.”
“The ongoing epidemic of opioid deaths requires intense attention and action,” Dr. Rudd and her associates wrote. “Intensifying efforts to distribute naloxone (an antidote to reverse an opioid overdose), enhancing access to treatment ... and implementing harm reduction services are urgently needed.”
The study was conducted by the CDC.
FROM MMWR
Key clinical point: The most recent Centers for Disease Control and Prevention data show that opioid-related deaths continue to rise, with a 16% increase from 2014 to 2015, which was driven largely by overdoses of illegally manufactured fentanyl and heroin.
Major finding: Sixty-three percent of the 52,404 deaths from drug overdoses in 2015 involved opioid overdoses.
Data source: Analysis of drug-related mortality data from 2010 to 2015 in the National Vital Statistics System for all 50 states and the District of Columbia, plus an analysis of drug-related deaths by subcategories of drugs in 28 states from 2014 to 2015.
Disclosures: The study was conducted by the CDC.
FDA bans powdered gloves
The Food and Drug Administration has banned powdered gloves for use in health care settings, citing “numerous risks to patients and health care workers.” The ban extends to gloves currently in commercial distribution and in the hands of the ultimate user, meaning powdered gloves will have to be pulled from examination rooms and operating theaters.
“A thorough review of all currently available information supports FDA’s conclusion that powdered surgeon’s gloves, powdered patient examination gloves, and absorbable powder for lubricating a surgeon’s glove should be banned,” according to a FDA final rule available now online and scheduled for publication in the Federal Register on Dec. 19, 2016. The ban will become effective 30 days after the document’s publication in the Federal Register.
Notes the final document, “The benefits of powdered gloves appear to only include greater ease of donning and doffing, decreased tackiness, and a degree of added comfort, which FDA believes are nominal when compared to the risks posed by these devices.”
Since viable nonpowdered alternatives exist, the FDA believes that the ban would not have significant economic impact and that shortages should not affect care delivery.
Many nonpowdered gloves, said the FDA, now “have the same level of protection, dexterity, and performance” as powdered gloves.
Powder may still be used in the glove manufacturing process, but the FDA continues to recommend that no more than 2 mg of residual powder per glove remains after the manufacturing process.
Though the final document banning powdered gloves notes that the FDA received many comments asking for a ban of all natural rubber latex (NRL) gloves, the ban applied only to powdered gloves. The FDA noted that NRL gloves already must carry a statement alerting users to the risks of allergic reaction, and also noted that eliminating powder from NRL gloves reduces the risk of latex sensitization.
In explaining its analysis of the costs and benefits of the powdered glove ban, the FDA estimated that the annual net benefits would range between $26.8 million and $31.8 million.
When this ban comes into force, it will be only the second such ban; the first was the 1983 ban of prosthetic hair fibers, which were found to provide no public health benefit.
koakes@frontlinemedcom.com
On Twitter @karioakes
The Food and Drug Administration has banned powdered gloves for use in health care settings, citing “numerous risks to patients and health care workers.” The ban extends to gloves currently in commercial distribution and in the hands of the ultimate user, meaning powdered gloves will have to be pulled from examination rooms and operating theaters.
“A thorough review of all currently available information supports FDA’s conclusion that powdered surgeon’s gloves, powdered patient examination gloves, and absorbable powder for lubricating a surgeon’s glove should be banned,” according to a FDA final rule available now online and scheduled for publication in the Federal Register on Dec. 19, 2016. The ban will become effective 30 days after the document’s publication in the Federal Register.
Notes the final document, “The benefits of powdered gloves appear to only include greater ease of donning and doffing, decreased tackiness, and a degree of added comfort, which FDA believes are nominal when compared to the risks posed by these devices.”
Since viable nonpowdered alternatives exist, the FDA believes that the ban would not have significant economic impact and that shortages should not affect care delivery.
Many nonpowdered gloves, said the FDA, now “have the same level of protection, dexterity, and performance” as powdered gloves.
Powder may still be used in the glove manufacturing process, but the FDA continues to recommend that no more than 2 mg of residual powder per glove remains after the manufacturing process.
Though the final document banning powdered gloves notes that the FDA received many comments asking for a ban of all natural rubber latex (NRL) gloves, the ban applied only to powdered gloves. The FDA noted that NRL gloves already must carry a statement alerting users to the risks of allergic reaction, and also noted that eliminating powder from NRL gloves reduces the risk of latex sensitization.
In explaining its analysis of the costs and benefits of the powdered glove ban, the FDA estimated that the annual net benefits would range between $26.8 million and $31.8 million.
When this ban comes into force, it will be only the second such ban; the first was the 1983 ban of prosthetic hair fibers, which were found to provide no public health benefit.
koakes@frontlinemedcom.com
On Twitter @karioakes
The Food and Drug Administration has banned powdered gloves for use in health care settings, citing “numerous risks to patients and health care workers.” The ban extends to gloves currently in commercial distribution and in the hands of the ultimate user, meaning powdered gloves will have to be pulled from examination rooms and operating theaters.
“A thorough review of all currently available information supports FDA’s conclusion that powdered surgeon’s gloves, powdered patient examination gloves, and absorbable powder for lubricating a surgeon’s glove should be banned,” according to a FDA final rule available now online and scheduled for publication in the Federal Register on Dec. 19, 2016. The ban will become effective 30 days after the document’s publication in the Federal Register.
Notes the final document, “The benefits of powdered gloves appear to only include greater ease of donning and doffing, decreased tackiness, and a degree of added comfort, which FDA believes are nominal when compared to the risks posed by these devices.”
Since viable nonpowdered alternatives exist, the FDA believes that the ban would not have significant economic impact and that shortages should not affect care delivery.
Many nonpowdered gloves, said the FDA, now “have the same level of protection, dexterity, and performance” as powdered gloves.
Powder may still be used in the glove manufacturing process, but the FDA continues to recommend that no more than 2 mg of residual powder per glove remains after the manufacturing process.
Though the final document banning powdered gloves notes that the FDA received many comments asking for a ban of all natural rubber latex (NRL) gloves, the ban applied only to powdered gloves. The FDA noted that NRL gloves already must carry a statement alerting users to the risks of allergic reaction, and also noted that eliminating powder from NRL gloves reduces the risk of latex sensitization.
In explaining its analysis of the costs and benefits of the powdered glove ban, the FDA estimated that the annual net benefits would range between $26.8 million and $31.8 million.
When this ban comes into force, it will be only the second such ban; the first was the 1983 ban of prosthetic hair fibers, which were found to provide no public health benefit.
koakes@frontlinemedcom.com
On Twitter @karioakes
FDA warning: General anesthetics may damage young brains
The Food and Drug Administration has issued a warning that repeated or lengthy use of general anesthetic and sedation drugs during surgeries or procedures in children younger than 3 years or in pregnant women during their third trimester may affect the development of children’s brains.
“Recent human studies suggest that a single, relatively short exposure to general anesthetic and sedation drugs in infants or toddlers is unlikely to have negative effects on behavior or learning.” The studies suggesting a problem with longer or repeat exposures “had limitations, and it is unclear whether any negative effects seen in children’s learning or behavior were due to the drugs or to other factors, such as the underlying medical condition that led to the need for the surgery or procedure.” Further research is needed, the agency said.
FDA is adding its warning to the labels of 11 general anesthetics and sedatives, including desflurane, halothane, ketamine, lorazepam injection, methohexital, pentobarbital, and propofol. The drugs block N-methyl-D-aspartate (NMDA) receptors and/or potentiate gamma-aminobutyric acid (GABA) activity. No specific medications have been shown to be safer than any other, the agency said.
FDA will continue to monitor the situation, and update its warning as additional information comes in. “We urge health care professionals, patients, parents, and caregivers to report side effects involving anesthetic and sedation drugs or other medicines to the FDA MedWatch program,” the FDA said.
The Food and Drug Administration has issued a warning that repeated or lengthy use of general anesthetic and sedation drugs during surgeries or procedures in children younger than 3 years or in pregnant women during their third trimester may affect the development of children’s brains.
“Recent human studies suggest that a single, relatively short exposure to general anesthetic and sedation drugs in infants or toddlers is unlikely to have negative effects on behavior or learning.” The studies suggesting a problem with longer or repeat exposures “had limitations, and it is unclear whether any negative effects seen in children’s learning or behavior were due to the drugs or to other factors, such as the underlying medical condition that led to the need for the surgery or procedure.” Further research is needed, the agency said.
FDA is adding its warning to the labels of 11 general anesthetics and sedatives, including desflurane, halothane, ketamine, lorazepam injection, methohexital, pentobarbital, and propofol. The drugs block N-methyl-D-aspartate (NMDA) receptors and/or potentiate gamma-aminobutyric acid (GABA) activity. No specific medications have been shown to be safer than any other, the agency said.
FDA will continue to monitor the situation, and update its warning as additional information comes in. “We urge health care professionals, patients, parents, and caregivers to report side effects involving anesthetic and sedation drugs or other medicines to the FDA MedWatch program,” the FDA said.
The Food and Drug Administration has issued a warning that repeated or lengthy use of general anesthetic and sedation drugs during surgeries or procedures in children younger than 3 years or in pregnant women during their third trimester may affect the development of children’s brains.
“Recent human studies suggest that a single, relatively short exposure to general anesthetic and sedation drugs in infants or toddlers is unlikely to have negative effects on behavior or learning.” The studies suggesting a problem with longer or repeat exposures “had limitations, and it is unclear whether any negative effects seen in children’s learning or behavior were due to the drugs or to other factors, such as the underlying medical condition that led to the need for the surgery or procedure.” Further research is needed, the agency said.
FDA is adding its warning to the labels of 11 general anesthetics and sedatives, including desflurane, halothane, ketamine, lorazepam injection, methohexital, pentobarbital, and propofol. The drugs block N-methyl-D-aspartate (NMDA) receptors and/or potentiate gamma-aminobutyric acid (GABA) activity. No specific medications have been shown to be safer than any other, the agency said.
FDA will continue to monitor the situation, and update its warning as additional information comes in. “We urge health care professionals, patients, parents, and caregivers to report side effects involving anesthetic and sedation drugs or other medicines to the FDA MedWatch program,” the FDA said.
FDA gives nod to crisaborole for atopic dermatitis
The Food and Drug Administration has approved crisaborole topical ointment, 2%, to treat mild to moderate atopic dermatitis in patients 2 years of age and older. The boron-based phosphodiesterase 4 inhibitor, which will be marketed as Eucrisa, was developed by Anacor Pharmaceuticals, which Pfizer acquired in May of 2016.
“Today’s approval provides another treatment option for patients dealing with mild to moderate atopic dermatitis,” Amy Egan, deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research, said in a prepared statement. Safety and efficacy of the drug were established in two placebo-controlled trials with a total of 1,522 participants aged 2-79 years with mild to moderate atopic dermatitis.

At the annual meeting of the Pacific Dermatologic Association in August 2016, Dr. Kelly Cordoro, a pediatric dermatologist at the University of California, San Francisco, described crisaborole as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways. “Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said.
The results of the two phase III studies were recently published in the Journal of the American Academy of Dermatology (2016 Sept;75[3]:494-503).
The Food and Drug Administration has approved crisaborole topical ointment, 2%, to treat mild to moderate atopic dermatitis in patients 2 years of age and older. The boron-based phosphodiesterase 4 inhibitor, which will be marketed as Eucrisa, was developed by Anacor Pharmaceuticals, which Pfizer acquired in May of 2016.
“Today’s approval provides another treatment option for patients dealing with mild to moderate atopic dermatitis,” Amy Egan, deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research, said in a prepared statement. Safety and efficacy of the drug were established in two placebo-controlled trials with a total of 1,522 participants aged 2-79 years with mild to moderate atopic dermatitis.
At the annual meeting of the Pacific Dermatologic Association in August 2016, Dr. Kelly Cordoro, a pediatric dermatologist at the University of California, San Francisco, described crisaborole as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways. “Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said.
The results of the two phase III studies were recently published in the Journal of the American Academy of Dermatology (2016 Sept;75[3]:494-503).
The Food and Drug Administration has approved crisaborole topical ointment, 2%, to treat mild to moderate atopic dermatitis in patients 2 years of age and older. The boron-based phosphodiesterase 4 inhibitor, which will be marketed as Eucrisa, was developed by Anacor Pharmaceuticals, which Pfizer acquired in May of 2016.
“Today’s approval provides another treatment option for patients dealing with mild to moderate atopic dermatitis,” Amy Egan, deputy director of the Office of Drug Evaluation III in the FDA’s Center for Drug Evaluation and Research, said in a prepared statement. Safety and efficacy of the drug were established in two placebo-controlled trials with a total of 1,522 participants aged 2-79 years with mild to moderate atopic dermatitis.
At the annual meeting of the Pacific Dermatologic Association in August 2016, Dr. Kelly Cordoro, a pediatric dermatologist at the University of California, San Francisco, described crisaborole as an anti-inflammatory agent that modifies inflammation by inhibiting the degradation of cAMP by PDE4, resulting in downstream modification of nuclear factor-kB and T-cell signaling pathways. “Crisaborole has shown promising results from four clinical studies in patients 2 years of age and older, with notable improvements in all atopic dermatitis parameters,” she said.
The results of the two phase III studies were recently published in the Journal of the American Academy of Dermatology (2016 Sept;75[3]:494-503).
XR version of Synjardy gets FDA’s nod
for use in the management of blood sugar in adults with type 2 diabetes. The drug, to be marketed as Synjardy XR by Boehringer Ingelheim and Eli Lilly, is intended for use with a diet and exercise program.
Its standard release formulation was approved in August 2015.
FDA approved the extended-release version on Dec. 12. The updated label includes a boxed warning regarding the risk of metformin-associated lactic acidosis
Empagliflozin recently received an FDA indication as a medication for use in reducing the risk of cardiovascular death in patients with type 2 diabetes and concomitant cardiovascular disease, the first drug for type 2 diabetes to do so.
The label includes information of the drug’s four doses, all of which contain 1,000 mg of metformin plus empagliflozin in strengths of 5 mg, 10 mg, 12.5 mg, and 25 mg.
Among patients who are already taking metformin and plan to begin a regimen of Synjardy XR, their best choice is to switch to the formulation containing a similar daily dose of metformin and a total daily dose of 10 mg empagliflozin. Those already on empagliflozin should switch to Synjardy XR containing the same daily dose of empagliflozin and a total daily dose of 1,000 mg metformin. Dosing may be adjusted upward on the basis of effectiveness and tolerability up to a maximum of 25 mg empagliflozin/2,000 mg metformin, taken once daily with a meal in the morning.
Synjardy XR is contraindicated in patients with an estimated glomerular filtration rate of less than 45 mL/min per 1.73 m2. Renal function should be assessed in patients with impaired renal function before taking Synjardy XR.
Several clinical trials tested coadministration of metformin and empagliflozin. In the largest, 637 patients with type 2 diabetes who were inadequately controlled (hemoglobin A1c of 7%-10%) with 1,500 mg metformin were randomized to receive placebo, 10 mg empagliflozin, or 25 mg empagliflozin for 24 weeks. Both the 10-mg and 25-mg dosages of empagliflozin resulted in significant reductions compared with placebo in HbA1c (–0.7% and –0.8%, respectively) fasting glucose (–26 mg/dL and –29 mg/dL), and body weight (–2 kg and –2.5 kg).
The single-pill formulation of Synjardy XR was not tested in clinical trials for efficacy; however, several trials performed in healthy subjects showed bioequivalence of the fixed dose combination, compared with separate pills.
skubetin@frontlinemedcom.com
for use in the management of blood sugar in adults with type 2 diabetes. The drug, to be marketed as Synjardy XR by Boehringer Ingelheim and Eli Lilly, is intended for use with a diet and exercise program.
Its standard release formulation was approved in August 2015.
FDA approved the extended-release version on Dec. 12. The updated label includes a boxed warning regarding the risk of metformin-associated lactic acidosis
Empagliflozin recently received an FDA indication as a medication for use in reducing the risk of cardiovascular death in patients with type 2 diabetes and concomitant cardiovascular disease, the first drug for type 2 diabetes to do so.
The label includes information of the drug’s four doses, all of which contain 1,000 mg of metformin plus empagliflozin in strengths of 5 mg, 10 mg, 12.5 mg, and 25 mg.
Among patients who are already taking metformin and plan to begin a regimen of Synjardy XR, their best choice is to switch to the formulation containing a similar daily dose of metformin and a total daily dose of 10 mg empagliflozin. Those already on empagliflozin should switch to Synjardy XR containing the same daily dose of empagliflozin and a total daily dose of 1,000 mg metformin. Dosing may be adjusted upward on the basis of effectiveness and tolerability up to a maximum of 25 mg empagliflozin/2,000 mg metformin, taken once daily with a meal in the morning.
Synjardy XR is contraindicated in patients with an estimated glomerular filtration rate of less than 45 mL/min per 1.73 m2. Renal function should be assessed in patients with impaired renal function before taking Synjardy XR.
Several clinical trials tested coadministration of metformin and empagliflozin. In the largest, 637 patients with type 2 diabetes who were inadequately controlled (hemoglobin A1c of 7%-10%) with 1,500 mg metformin were randomized to receive placebo, 10 mg empagliflozin, or 25 mg empagliflozin for 24 weeks. Both the 10-mg and 25-mg dosages of empagliflozin resulted in significant reductions compared with placebo in HbA1c (–0.7% and –0.8%, respectively) fasting glucose (–26 mg/dL and –29 mg/dL), and body weight (–2 kg and –2.5 kg).
The single-pill formulation of Synjardy XR was not tested in clinical trials for efficacy; however, several trials performed in healthy subjects showed bioequivalence of the fixed dose combination, compared with separate pills.
skubetin@frontlinemedcom.com
for use in the management of blood sugar in adults with type 2 diabetes. The drug, to be marketed as Synjardy XR by Boehringer Ingelheim and Eli Lilly, is intended for use with a diet and exercise program.
Its standard release formulation was approved in August 2015.
FDA approved the extended-release version on Dec. 12. The updated label includes a boxed warning regarding the risk of metformin-associated lactic acidosis
Empagliflozin recently received an FDA indication as a medication for use in reducing the risk of cardiovascular death in patients with type 2 diabetes and concomitant cardiovascular disease, the first drug for type 2 diabetes to do so.
The label includes information of the drug’s four doses, all of which contain 1,000 mg of metformin plus empagliflozin in strengths of 5 mg, 10 mg, 12.5 mg, and 25 mg.
Among patients who are already taking metformin and plan to begin a regimen of Synjardy XR, their best choice is to switch to the formulation containing a similar daily dose of metformin and a total daily dose of 10 mg empagliflozin. Those already on empagliflozin should switch to Synjardy XR containing the same daily dose of empagliflozin and a total daily dose of 1,000 mg metformin. Dosing may be adjusted upward on the basis of effectiveness and tolerability up to a maximum of 25 mg empagliflozin/2,000 mg metformin, taken once daily with a meal in the morning.
Synjardy XR is contraindicated in patients with an estimated glomerular filtration rate of less than 45 mL/min per 1.73 m2. Renal function should be assessed in patients with impaired renal function before taking Synjardy XR.
Several clinical trials tested coadministration of metformin and empagliflozin. In the largest, 637 patients with type 2 diabetes who were inadequately controlled (hemoglobin A1c of 7%-10%) with 1,500 mg metformin were randomized to receive placebo, 10 mg empagliflozin, or 25 mg empagliflozin for 24 weeks. Both the 10-mg and 25-mg dosages of empagliflozin resulted in significant reductions compared with placebo in HbA1c (–0.7% and –0.8%, respectively) fasting glucose (–26 mg/dL and –29 mg/dL), and body weight (–2 kg and –2.5 kg).
The single-pill formulation of Synjardy XR was not tested in clinical trials for efficacy; however, several trials performed in healthy subjects showed bioequivalence of the fixed dose combination, compared with separate pills.
skubetin@frontlinemedcom.com
FDA affirms bladder cancer warning with diabetes drug
Though a 10-year epidemiologic study did not find an increased risk of bladder cancer with pioglitazone use, the Food and Drug Administration has chosen to affirm the warning on the label of the type 2 diabetes drug following an updated review of several studies.
The FDA issued a warning about the possible risk of bladder cancer based on interim results from the 10-year epidemiologic study in 2010, and it changed the labels of pioglitazone-containing medicines in 2011 to include warnings about this risk.
There was a modest trend toward higher risk with increasing duration of use, but the trend was not statistically significant. Compared with the interim 5-year results, these final 10-year results found weaker associations that were not statistically significant.
The directions of the associations, however, remained unchanged. Based on these findings and other reviewed studies with conflicting results, the FDA has concluded that use of pioglitazone may be linked to an increased risk of bladder cancer.
The labels of pioglitazone-containing medicines already contain warnings about this risk, but the FDA has now approved label updates to describe the additional studies reviewed.
Read the FDA update and data summary here.
Though a 10-year epidemiologic study did not find an increased risk of bladder cancer with pioglitazone use, the Food and Drug Administration has chosen to affirm the warning on the label of the type 2 diabetes drug following an updated review of several studies.
The FDA issued a warning about the possible risk of bladder cancer based on interim results from the 10-year epidemiologic study in 2010, and it changed the labels of pioglitazone-containing medicines in 2011 to include warnings about this risk.
There was a modest trend toward higher risk with increasing duration of use, but the trend was not statistically significant. Compared with the interim 5-year results, these final 10-year results found weaker associations that were not statistically significant.
The directions of the associations, however, remained unchanged. Based on these findings and other reviewed studies with conflicting results, the FDA has concluded that use of pioglitazone may be linked to an increased risk of bladder cancer.
The labels of pioglitazone-containing medicines already contain warnings about this risk, but the FDA has now approved label updates to describe the additional studies reviewed.
Read the FDA update and data summary here.
Though a 10-year epidemiologic study did not find an increased risk of bladder cancer with pioglitazone use, the Food and Drug Administration has chosen to affirm the warning on the label of the type 2 diabetes drug following an updated review of several studies.
The FDA issued a warning about the possible risk of bladder cancer based on interim results from the 10-year epidemiologic study in 2010, and it changed the labels of pioglitazone-containing medicines in 2011 to include warnings about this risk.
There was a modest trend toward higher risk with increasing duration of use, but the trend was not statistically significant. Compared with the interim 5-year results, these final 10-year results found weaker associations that were not statistically significant.
The directions of the associations, however, remained unchanged. Based on these findings and other reviewed studies with conflicting results, the FDA has concluded that use of pioglitazone may be linked to an increased risk of bladder cancer.
The labels of pioglitazone-containing medicines already contain warnings about this risk, but the FDA has now approved label updates to describe the additional studies reviewed.
Read the FDA update and data summary here.
Spike in Colombian microcephaly cases linked to Zika infection early in pregnancy
Colombia experienced a fourfold increase in cases of microcephaly following the Zika virus outbreak in 2016, with temporal evidence suggesting that infection in the first months of pregnancy poses the greatest risk to the fetus of microcephaly.
From January 31 through mid-November 2016, there were 476 cases of microcephaly reported in Colombia, four times the rate of cases reported during the same period in 2015. In July 2016, there was a ninefold increase in Colombian microcephaly cases reported, compared with July 2015, according to data published in the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report.![]()
Based on an average full-term gestation – because the 24-week period from the peak of Colombia’s Zika virus outbreak occurred simultaneously with the peak in reported microcephaly cases – there is a temporal suggestion that the greatest risk for microcephaly is associated with Zika virus infection during the first half of pregnancy, according to the authors of the report. Of the reported cases of microcephaly in 2016, 432 were live-born infants and 44 were pregnancy losses (MMWR. 2016 Dec 9. doi: 10.15585/mmwr.mm6549e1).
Of Colombia’s reported 105,000 cases of Zika virus occurring between August 9, 2015, and November 12, 2016, nearly 20,000 cases were in pregnant women, according to the Instituto Nacional de Salud.
The findings reinforce previous data indicating the correlation between early Zika virus-infection and microcephaly (N Engl J Med. 2016;374:1981-7. doi: 10.1056/NEJMsr1604338), although the report’s authors cautioned there are several confounders to these data, including that the surveillance was passive, not all reported cases of Zika virus infection were confirmed by a laboratory, and not all cases might have been reported.
The authors reported no relevant disclosures.
Colombia experienced a fourfold increase in cases of microcephaly following the Zika virus outbreak in 2016, with temporal evidence suggesting that infection in the first months of pregnancy poses the greatest risk to the fetus of microcephaly.
From January 31 through mid-November 2016, there were 476 cases of microcephaly reported in Colombia, four times the rate of cases reported during the same period in 2015. In July 2016, there was a ninefold increase in Colombian microcephaly cases reported, compared with July 2015, according to data published in the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report.![]()
Based on an average full-term gestation – because the 24-week period from the peak of Colombia’s Zika virus outbreak occurred simultaneously with the peak in reported microcephaly cases – there is a temporal suggestion that the greatest risk for microcephaly is associated with Zika virus infection during the first half of pregnancy, according to the authors of the report. Of the reported cases of microcephaly in 2016, 432 were live-born infants and 44 were pregnancy losses (MMWR. 2016 Dec 9. doi: 10.15585/mmwr.mm6549e1).
Of Colombia’s reported 105,000 cases of Zika virus occurring between August 9, 2015, and November 12, 2016, nearly 20,000 cases were in pregnant women, according to the Instituto Nacional de Salud.
The findings reinforce previous data indicating the correlation between early Zika virus-infection and microcephaly (N Engl J Med. 2016;374:1981-7. doi: 10.1056/NEJMsr1604338), although the report’s authors cautioned there are several confounders to these data, including that the surveillance was passive, not all reported cases of Zika virus infection were confirmed by a laboratory, and not all cases might have been reported.
The authors reported no relevant disclosures.
Colombia experienced a fourfold increase in cases of microcephaly following the Zika virus outbreak in 2016, with temporal evidence suggesting that infection in the first months of pregnancy poses the greatest risk to the fetus of microcephaly.
From January 31 through mid-November 2016, there were 476 cases of microcephaly reported in Colombia, four times the rate of cases reported during the same period in 2015. In July 2016, there was a ninefold increase in Colombian microcephaly cases reported, compared with July 2015, according to data published in the Centers for Disease Control and Prevention’s Morbidity and Mortality Weekly Report.![]()
Based on an average full-term gestation – because the 24-week period from the peak of Colombia’s Zika virus outbreak occurred simultaneously with the peak in reported microcephaly cases – there is a temporal suggestion that the greatest risk for microcephaly is associated with Zika virus infection during the first half of pregnancy, according to the authors of the report. Of the reported cases of microcephaly in 2016, 432 were live-born infants and 44 were pregnancy losses (MMWR. 2016 Dec 9. doi: 10.15585/mmwr.mm6549e1).
Of Colombia’s reported 105,000 cases of Zika virus occurring between August 9, 2015, and November 12, 2016, nearly 20,000 cases were in pregnant women, according to the Instituto Nacional de Salud.
The findings reinforce previous data indicating the correlation between early Zika virus-infection and microcephaly (N Engl J Med. 2016;374:1981-7. doi: 10.1056/NEJMsr1604338), although the report’s authors cautioned there are several confounders to these data, including that the surveillance was passive, not all reported cases of Zika virus infection were confirmed by a laboratory, and not all cases might have been reported.
The authors reported no relevant disclosures.
FROM MMWR
Key clinical point:
Major finding: In 2016, a fourfold increase in microcephaly cases occurred in Colombia compared with 2015. Most cases coincided with women who were in their first trimester at the Zika outbreak’s inception.
Data source: National passive surveillance data of reported birth defects from Colombia’s Instituto Nacional de Salud.
Disclosures: The authors reported no relevant disclosures.
New Zika cases in pregnant women continue to drop
There were 136 new cases of pregnant women with laboratory evidence of Zika infection reported during the 2-week period ending Nov. 30, along with four liveborn infants with Zika-related birth defects, according to the Centers for Disease Control and Prevention.
The CDC did not report new totals for pregnant women and pregnancy outcomes for the week ending Nov. 23, so the most recent data release covers the 2-week period from Nov. 17-30. That 2-week total was barely more than the 124 reported for the week ending Nov. 10.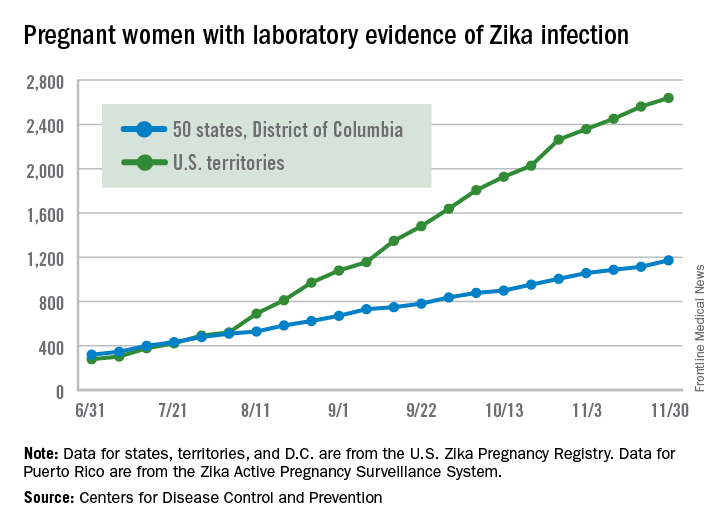
The four infants born with Zika-related birth defects were all born in the 50 states and D.C., as the CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika. There were no new pregnancy losses with Zika-related birth defects in the states/D.C., so that number remains at five, while the total number of liveborn infants with Zika-related birth defects is now 32, the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
There were 136 new cases of pregnant women with laboratory evidence of Zika infection reported during the 2-week period ending Nov. 30, along with four liveborn infants with Zika-related birth defects, according to the Centers for Disease Control and Prevention.
The CDC did not report new totals for pregnant women and pregnancy outcomes for the week ending Nov. 23, so the most recent data release covers the 2-week period from Nov. 17-30. That 2-week total was barely more than the 124 reported for the week ending Nov. 10.
The four infants born with Zika-related birth defects were all born in the 50 states and D.C., as the CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika. There were no new pregnancy losses with Zika-related birth defects in the states/D.C., so that number remains at five, while the total number of liveborn infants with Zika-related birth defects is now 32, the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
There were 136 new cases of pregnant women with laboratory evidence of Zika infection reported during the 2-week period ending Nov. 30, along with four liveborn infants with Zika-related birth defects, according to the Centers for Disease Control and Prevention.
The CDC did not report new totals for pregnant women and pregnancy outcomes for the week ending Nov. 23, so the most recent data release covers the 2-week period from Nov. 17-30. That 2-week total was barely more than the 124 reported for the week ending Nov. 10.
The four infants born with Zika-related birth defects were all born in the 50 states and D.C., as the CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika. There were no new pregnancy losses with Zika-related birth defects in the states/D.C., so that number remains at five, while the total number of liveborn infants with Zika-related birth defects is now 32, the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
FDA approves treatment for platinum-sensitive ovarian cancer
The Food and Drug Administration approved bevacizumab (Avastin), used in combination with certain types of chemotherapy, for the treatment of patients with platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary perioneal cancer.
The FDA approved its use in combination with either carboplatin and paclitaxel or carboplatin and gemcitabine chemotherapy, followed by bevacizumab alone. In November 2014, the FDA approved bevacizumab for use in combination with paclitaxel, pegylated liposomal doxorubicin, or topotecan chemotherapy for women with platinum-resistant recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer.
The GOG-0213 study showed an overall survival improvement of 5 months with bevacizumab plus chemotherapy, compared with chemotherapy alone (hazard ratio, 0.84). The study also showed improved progression-free survival of 3.4 months (HR, 0.61). In the OCEANS study, progression-free survival was 12.4 months for bevacizumab plus chemotherapy, compared with 8.4 months for placebo plus chemotherapy (HR, 0.46). Overall survival was not significantly improved by the addition of bevacizumab in the OCEANS study.
Some adverse events observed in the trials included fatigue, hypertension, febrile neutropenia, proteinuria, abdominal pain, hyponatremia, headache, nausea, and pain in the extremity.
Bevacizumab is manufactured by Genentech, and full prescribing information and a boxed warning are available on the drug’s website, www.avastin.com.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
The Food and Drug Administration approved bevacizumab (Avastin), used in combination with certain types of chemotherapy, for the treatment of patients with platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary perioneal cancer.
The FDA approved its use in combination with either carboplatin and paclitaxel or carboplatin and gemcitabine chemotherapy, followed by bevacizumab alone. In November 2014, the FDA approved bevacizumab for use in combination with paclitaxel, pegylated liposomal doxorubicin, or topotecan chemotherapy for women with platinum-resistant recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer.
The GOG-0213 study showed an overall survival improvement of 5 months with bevacizumab plus chemotherapy, compared with chemotherapy alone (hazard ratio, 0.84). The study also showed improved progression-free survival of 3.4 months (HR, 0.61). In the OCEANS study, progression-free survival was 12.4 months for bevacizumab plus chemotherapy, compared with 8.4 months for placebo plus chemotherapy (HR, 0.46). Overall survival was not significantly improved by the addition of bevacizumab in the OCEANS study.
Some adverse events observed in the trials included fatigue, hypertension, febrile neutropenia, proteinuria, abdominal pain, hyponatremia, headache, nausea, and pain in the extremity.
Bevacizumab is manufactured by Genentech, and full prescribing information and a boxed warning are available on the drug’s website, www.avastin.com.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
The Food and Drug Administration approved bevacizumab (Avastin), used in combination with certain types of chemotherapy, for the treatment of patients with platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary perioneal cancer.
The FDA approved its use in combination with either carboplatin and paclitaxel or carboplatin and gemcitabine chemotherapy, followed by bevacizumab alone. In November 2014, the FDA approved bevacizumab for use in combination with paclitaxel, pegylated liposomal doxorubicin, or topotecan chemotherapy for women with platinum-resistant recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer.
The GOG-0213 study showed an overall survival improvement of 5 months with bevacizumab plus chemotherapy, compared with chemotherapy alone (hazard ratio, 0.84). The study also showed improved progression-free survival of 3.4 months (HR, 0.61). In the OCEANS study, progression-free survival was 12.4 months for bevacizumab plus chemotherapy, compared with 8.4 months for placebo plus chemotherapy (HR, 0.46). Overall survival was not significantly improved by the addition of bevacizumab in the OCEANS study.
Some adverse events observed in the trials included fatigue, hypertension, febrile neutropenia, proteinuria, abdominal pain, hyponatremia, headache, nausea, and pain in the extremity.
Bevacizumab is manufactured by Genentech, and full prescribing information and a boxed warning are available on the drug’s website, www.avastin.com.
mschneider@frontlinemedcom.com
On Twitter @maryellenny