User login
Empagliflozin first antidiabetes drug to gain cardioprotective indication
Empagliflozin is the first antidiabetes medication to be approved for reducing the risk of cardiovascular death in patients with type 2 diabetes and concomitant cardiovascular disease.
The Food and Drug Administration granted the new indication based on the EMPA-REG OUTCOME study, a postmarketing analysis that found that empagliflozin (Jardiance, Boehringer-Ingelheim) reduced the risk of cardiovascular death by 38% when added to standard-of-care type 2 diabetes therapy.
When empagliflozin was approved for type 2 diabetes in 2014, the FDA required an additional postmarketing study to examine its cardiovascular safety. The 48-month, open-label EMPA-REG enrolled more than 7,000 patients who had type 2 diabetes and a high risk of cardiovascular disease.
The study’s big surprise, however, was not empagliflozin’s safety, but its striking cardioprotective qualities. It reduced by 14% the risk of the primary endpoint, a composite of cardiovascular death, nonfatal stroke, and nonfatal myocardial infarction (N Engl J Med. 2015;373:2117-28).
When examined as individual outcomes in a secondary analysis, empagliflozin significantly reduced the risk of cardiovascular death by 38%. However, risk reductions on the other endpoints were not significant. Nevertheless, experts called empagliflozin’s cardiovascular benefit a potential game-changer for the clinical challenge of managing patients with both disorders.
But the drug barely squeaked by its June FDA approval hearing for the cardioprotective indication, receiving a split 12-11 endorsement from the Endocrinologic and Metabolic Drugs Advisory Committee. The major sticking point was that EMPA-REG was a test of empagliflozin’s cardiovascular safety, not its efficacy, and that cardiovascular death was not a prespecified endpoint.
Although there were no significant cardiovascular safety issues, empagliflozin has been associated with hypotension, serious urinary tract infection, acute kidney injury, and genital infections.
“Cardiovascular disease is a leading cause of death in adults with type 2 diabetes mellitus,” Jean-Marc Guettier, MD, director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research, wrote in a press statement. “Availability of antidiabetes therapies that can help people live longer by reducing the risk of cardiovascular death is an important advance for adults with type 2 diabetes.”
msullivan@frontlinemedcom.com
On Twitter @alz_gal
Empagliflozin is the first antidiabetes medication to be approved for reducing the risk of cardiovascular death in patients with type 2 diabetes and concomitant cardiovascular disease.
The Food and Drug Administration granted the new indication based on the EMPA-REG OUTCOME study, a postmarketing analysis that found that empagliflozin (Jardiance, Boehringer-Ingelheim) reduced the risk of cardiovascular death by 38% when added to standard-of-care type 2 diabetes therapy.
When empagliflozin was approved for type 2 diabetes in 2014, the FDA required an additional postmarketing study to examine its cardiovascular safety. The 48-month, open-label EMPA-REG enrolled more than 7,000 patients who had type 2 diabetes and a high risk of cardiovascular disease.
The study’s big surprise, however, was not empagliflozin’s safety, but its striking cardioprotective qualities. It reduced by 14% the risk of the primary endpoint, a composite of cardiovascular death, nonfatal stroke, and nonfatal myocardial infarction (N Engl J Med. 2015;373:2117-28).
When examined as individual outcomes in a secondary analysis, empagliflozin significantly reduced the risk of cardiovascular death by 38%. However, risk reductions on the other endpoints were not significant. Nevertheless, experts called empagliflozin’s cardiovascular benefit a potential game-changer for the clinical challenge of managing patients with both disorders.
But the drug barely squeaked by its June FDA approval hearing for the cardioprotective indication, receiving a split 12-11 endorsement from the Endocrinologic and Metabolic Drugs Advisory Committee. The major sticking point was that EMPA-REG was a test of empagliflozin’s cardiovascular safety, not its efficacy, and that cardiovascular death was not a prespecified endpoint.
Although there were no significant cardiovascular safety issues, empagliflozin has been associated with hypotension, serious urinary tract infection, acute kidney injury, and genital infections.
“Cardiovascular disease is a leading cause of death in adults with type 2 diabetes mellitus,” Jean-Marc Guettier, MD, director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research, wrote in a press statement. “Availability of antidiabetes therapies that can help people live longer by reducing the risk of cardiovascular death is an important advance for adults with type 2 diabetes.”
msullivan@frontlinemedcom.com
On Twitter @alz_gal
Empagliflozin is the first antidiabetes medication to be approved for reducing the risk of cardiovascular death in patients with type 2 diabetes and concomitant cardiovascular disease.
The Food and Drug Administration granted the new indication based on the EMPA-REG OUTCOME study, a postmarketing analysis that found that empagliflozin (Jardiance, Boehringer-Ingelheim) reduced the risk of cardiovascular death by 38% when added to standard-of-care type 2 diabetes therapy.
When empagliflozin was approved for type 2 diabetes in 2014, the FDA required an additional postmarketing study to examine its cardiovascular safety. The 48-month, open-label EMPA-REG enrolled more than 7,000 patients who had type 2 diabetes and a high risk of cardiovascular disease.
The study’s big surprise, however, was not empagliflozin’s safety, but its striking cardioprotective qualities. It reduced by 14% the risk of the primary endpoint, a composite of cardiovascular death, nonfatal stroke, and nonfatal myocardial infarction (N Engl J Med. 2015;373:2117-28).
When examined as individual outcomes in a secondary analysis, empagliflozin significantly reduced the risk of cardiovascular death by 38%. However, risk reductions on the other endpoints were not significant. Nevertheless, experts called empagliflozin’s cardiovascular benefit a potential game-changer for the clinical challenge of managing patients with both disorders.
But the drug barely squeaked by its June FDA approval hearing for the cardioprotective indication, receiving a split 12-11 endorsement from the Endocrinologic and Metabolic Drugs Advisory Committee. The major sticking point was that EMPA-REG was a test of empagliflozin’s cardiovascular safety, not its efficacy, and that cardiovascular death was not a prespecified endpoint.
Although there were no significant cardiovascular safety issues, empagliflozin has been associated with hypotension, serious urinary tract infection, acute kidney injury, and genital infections.
“Cardiovascular disease is a leading cause of death in adults with type 2 diabetes mellitus,” Jean-Marc Guettier, MD, director of the Division of Metabolism and Endocrinology Products in the FDA’s Center for Drug Evaluation and Research, wrote in a press statement. “Availability of antidiabetes therapies that can help people live longer by reducing the risk of cardiovascular death is an important advance for adults with type 2 diabetes.”
msullivan@frontlinemedcom.com
On Twitter @alz_gal
FDA approves daratumumab in combination with standard therapy for multiple myeloma
The Food and Drug Administration has approved daratumumab in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy.
The drug was approved last year as monotherapy for patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double refractory to a proteasome inhibitor and an immunomodulatory agent.
In the POLLUX trial, median PFS had not been reached in the daratumumab plus lenalidomide and dexamethasone arm and was 18.4 months among patients getting lenalidomide and dexamethasone alone (HR=0.37; 95% CI: 0.27, 0.52; P less than.0001).
In the CASTOR trial, which compared the combination of daratumumab, bortezomib, and dexamethasone with bortezomib and dexamethasone, the estimated median PFS had not been reached in the daratumumab arm and was 7.2 months in the control arm (hazard ratio, 0.39; 95% confidence interval, 0.28-0.53; P less than .0001).
Updated results for both trials will be presented at the upcoming annual meeting of the American Society of Hematology (abstract #1150, abstract #1151).
The most frequently reported adverse reactions in POLLUX were infusion reactions, diarrhea, nausea, fatigue, pyrexia, upper respiratory tract infection, muscle spasm, cough, and dyspnea. The most frequently reported adverse reactions in CASTOR were infusion reactions, diarrhea, peripheral edema, upper respiratory tract infection, peripheral sensory neuropathy, cough, and dyspnea.
The recommended dose of daratumumab is 16 mg/kg IV (calculated on actual body weight), the FDA said.
Full prescribing information is available here.
The Food and Drug Administration has approved daratumumab in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy.
The drug was approved last year as monotherapy for patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double refractory to a proteasome inhibitor and an immunomodulatory agent.
In the POLLUX trial, median PFS had not been reached in the daratumumab plus lenalidomide and dexamethasone arm and was 18.4 months among patients getting lenalidomide and dexamethasone alone (HR=0.37; 95% CI: 0.27, 0.52; P less than.0001).
In the CASTOR trial, which compared the combination of daratumumab, bortezomib, and dexamethasone with bortezomib and dexamethasone, the estimated median PFS had not been reached in the daratumumab arm and was 7.2 months in the control arm (hazard ratio, 0.39; 95% confidence interval, 0.28-0.53; P less than .0001).
Updated results for both trials will be presented at the upcoming annual meeting of the American Society of Hematology (abstract #1150, abstract #1151).
The most frequently reported adverse reactions in POLLUX were infusion reactions, diarrhea, nausea, fatigue, pyrexia, upper respiratory tract infection, muscle spasm, cough, and dyspnea. The most frequently reported adverse reactions in CASTOR were infusion reactions, diarrhea, peripheral edema, upper respiratory tract infection, peripheral sensory neuropathy, cough, and dyspnea.
The recommended dose of daratumumab is 16 mg/kg IV (calculated on actual body weight), the FDA said.
Full prescribing information is available here.
The Food and Drug Administration has approved daratumumab in combination with lenalidomide and dexamethasone, or bortezomib and dexamethasone, for the treatment of patients with multiple myeloma who have received at least one prior therapy.
The drug was approved last year as monotherapy for patients with multiple myeloma who have received at least three prior lines of therapy, including a proteasome inhibitor and an immunomodulatory agent, or who are double refractory to a proteasome inhibitor and an immunomodulatory agent.
In the POLLUX trial, median PFS had not been reached in the daratumumab plus lenalidomide and dexamethasone arm and was 18.4 months among patients getting lenalidomide and dexamethasone alone (HR=0.37; 95% CI: 0.27, 0.52; P less than.0001).
In the CASTOR trial, which compared the combination of daratumumab, bortezomib, and dexamethasone with bortezomib and dexamethasone, the estimated median PFS had not been reached in the daratumumab arm and was 7.2 months in the control arm (hazard ratio, 0.39; 95% confidence interval, 0.28-0.53; P less than .0001).
Updated results for both trials will be presented at the upcoming annual meeting of the American Society of Hematology (abstract #1150, abstract #1151).
The most frequently reported adverse reactions in POLLUX were infusion reactions, diarrhea, nausea, fatigue, pyrexia, upper respiratory tract infection, muscle spasm, cough, and dyspnea. The most frequently reported adverse reactions in CASTOR were infusion reactions, diarrhea, peripheral edema, upper respiratory tract infection, peripheral sensory neuropathy, cough, and dyspnea.
The recommended dose of daratumumab is 16 mg/kg IV (calculated on actual body weight), the FDA said.
Full prescribing information is available here.
FDA approves vaginal insert to treat dyspareunia in menopause
Prasterone (Intrarosa), a vaginal insert containing dehydroepiandrosterone (DHEA) to treat dyspareunia in menopause caused by vulvar and vaginal atrophy, has won Food and Drug Administration approval.
It’s the first FDA-approved product containing the active ingredient prasterone, known also as DHEA. It will be marketed by Endoceutics Inc., a Quebec-based pharmaceutical company focused on women’s health.![]()
The approval is based on the results of two 12-week placebo-controlled trials of 406 healthy, postmenopausal women, ranging in age from 40 to 80 years, who identified dyspareunia as their most bothersome symptom of VVA. During the trials, prasterone reduced the severity of pain experienced during sexual intercourse, when compared with placebo. Safety of the treatment was established in four 12-week placebo-controlled trials and one 52-week open-label trial. The most common adverse events were vaginal discharge and abnormal Pap smear, according to the FDA.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
Prasterone (Intrarosa), a vaginal insert containing dehydroepiandrosterone (DHEA) to treat dyspareunia in menopause caused by vulvar and vaginal atrophy, has won Food and Drug Administration approval.
It’s the first FDA-approved product containing the active ingredient prasterone, known also as DHEA. It will be marketed by Endoceutics Inc., a Quebec-based pharmaceutical company focused on women’s health.![]()
The approval is based on the results of two 12-week placebo-controlled trials of 406 healthy, postmenopausal women, ranging in age from 40 to 80 years, who identified dyspareunia as their most bothersome symptom of VVA. During the trials, prasterone reduced the severity of pain experienced during sexual intercourse, when compared with placebo. Safety of the treatment was established in four 12-week placebo-controlled trials and one 52-week open-label trial. The most common adverse events were vaginal discharge and abnormal Pap smear, according to the FDA.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
Prasterone (Intrarosa), a vaginal insert containing dehydroepiandrosterone (DHEA) to treat dyspareunia in menopause caused by vulvar and vaginal atrophy, has won Food and Drug Administration approval.
It’s the first FDA-approved product containing the active ingredient prasterone, known also as DHEA. It will be marketed by Endoceutics Inc., a Quebec-based pharmaceutical company focused on women’s health.![]()
The approval is based on the results of two 12-week placebo-controlled trials of 406 healthy, postmenopausal women, ranging in age from 40 to 80 years, who identified dyspareunia as their most bothersome symptom of VVA. During the trials, prasterone reduced the severity of pain experienced during sexual intercourse, when compared with placebo. Safety of the treatment was established in four 12-week placebo-controlled trials and one 52-week open-label trial. The most common adverse events were vaginal discharge and abnormal Pap smear, according to the FDA.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
Fewer Zika-infected pregnancies reported for second week in a row
The number of new Zika cases reported among pregnant women in the United States dropped for the second week in a row, according to data from the Centers for Disease Control and Prevention.
For the week ending Nov. 10, there were 124 new cases of pregnant women with laboratory evidence of Zika virus infection reported to the CDC: 30 in the states and the District of Columbia and 94 in the U.S. territories. The previous week (Nov. 3), the number of new cases was 146, which came on the heels of a new weekly high of 288 for the week of Oct. 27.
The total number of pregnant women with Zika now stands at 3,538 for the year: 1,087 for the states and 2,451 for the territories, the CDC said.
Among all Americans, there have been 36,323 cases of Zika reported to the CDC: 4,255 have occurred in the states/D.C. and 32,068 in the territories. About 98% of territorial cases have occurred in Puerto Rico, the CDC said.
No new cases of infants with Zika-related birth defects were reported for the week ending Nov. 10, so the totals hold at 26 infants born with Zika-related birth defects and five Zika-related pregnancy losses, according to the CDC.
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The number of new Zika cases reported among pregnant women in the United States dropped for the second week in a row, according to data from the Centers for Disease Control and Prevention.
For the week ending Nov. 10, there were 124 new cases of pregnant women with laboratory evidence of Zika virus infection reported to the CDC: 30 in the states and the District of Columbia and 94 in the U.S. territories. The previous week (Nov. 3), the number of new cases was 146, which came on the heels of a new weekly high of 288 for the week of Oct. 27.
The total number of pregnant women with Zika now stands at 3,538 for the year: 1,087 for the states and 2,451 for the territories, the CDC said.
Among all Americans, there have been 36,323 cases of Zika reported to the CDC: 4,255 have occurred in the states/D.C. and 32,068 in the territories. About 98% of territorial cases have occurred in Puerto Rico, the CDC said.
No new cases of infants with Zika-related birth defects were reported for the week ending Nov. 10, so the totals hold at 26 infants born with Zika-related birth defects and five Zika-related pregnancy losses, according to the CDC.
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The number of new Zika cases reported among pregnant women in the United States dropped for the second week in a row, according to data from the Centers for Disease Control and Prevention.
For the week ending Nov. 10, there were 124 new cases of pregnant women with laboratory evidence of Zika virus infection reported to the CDC: 30 in the states and the District of Columbia and 94 in the U.S. territories. The previous week (Nov. 3), the number of new cases was 146, which came on the heels of a new weekly high of 288 for the week of Oct. 27.
The total number of pregnant women with Zika now stands at 3,538 for the year: 1,087 for the states and 2,451 for the territories, the CDC said.
Among all Americans, there have been 36,323 cases of Zika reported to the CDC: 4,255 have occurred in the states/D.C. and 32,068 in the territories. About 98% of territorial cases have occurred in Puerto Rico, the CDC said.
No new cases of infants with Zika-related birth defects were reported for the week ending Nov. 10, so the totals hold at 26 infants born with Zika-related birth defects and five Zika-related pregnancy losses, according to the CDC.
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
FDA approves nivolumab for advanced squamous cell carcinoma of the head and neck
The Food and Drug Administration has approved the immune checkpoint inhibitor nivolumab for the treatment of patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after a platinum-based therapy.
The FDA based its approval on an improvement in overall survival demonstrated in CheckMate-141, a randomized trial comparing nivolumab with the investigator’s choice of standard therapy, the FDA said in a written statement.
Earlier this year, the FDA granted accelerated approval to another checkpoint inhibitor targeting the PD-1/PD-L1 pathway, pembrolizumab, for the same indication, based on an objective response rate of 16% in the nonrandomized KEYNOTE-012 trial. Merck Sharp & Dohme, maker of pembrolizumab, is looking to demonstrate an improvement in overall survival with the ongoing KEYNOTE-040 study.
Checkmate-141 enrolled 361 patients with recurrent or metastatic SCCHN with disease progression on or within 6 months of receiving platinum-based chemotherapy and randomized (2:1) to nivolumab 3 mg/kg every 2 weeks intravenously or the investigator’s choice of cetuximab 400 mg/m2 IV once, then 250 mg/m2 IV weekly; methotrexate 40 mg/m2 IV weekly; or docetaxel 30 mg/m2 IV weekly until disease progression or unacceptable toxicity.
As reported at the European Society of Medical Oncology Congress and in the New England Journal of Medicine (2016;375:1856-67), the median overall survival was 7.5 months for patients on nivolumab, compared with 5.1 months for those on standard chemotherapy. The hazard ratio for death with nivolumab was 0.70 (P = .01). Estimates of 1-year survival were 36% vs. 16.6%, respectively.
Treatment-related adverse events of grade 3 or 4 occurred in 13.1% of patients on nivolumab, compared with 35.1% of those on standard therapy. The most frequent serious adverse reactions reported in at least 2% of patients receiving nivolumab were pneumonia, dyspnea, respiratory failure, respiratory tract infection, and sepsis.
The most common adverse reactions occurring in more than 10% of nivolumab-treated patients and at a higher incidence than with standard therapy were cough and dyspnea. The most common laboratory abnormalities occurring in 10% or more nivolumab-treated patients and at a higher incidence than with standard therapy were increased alkaline phosphatase level, increased amylase level, hypercalcemia, hyperkalemia, and increased thyroid-stimulating hormone level, the FDA said.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and previously has been approved to treat classical Hodgkin’s lymphoma, advanced renal cell carcinoma, lung cancer, and melanoma.
The Food and Drug Administration has approved the immune checkpoint inhibitor nivolumab for the treatment of patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after a platinum-based therapy.
The FDA based its approval on an improvement in overall survival demonstrated in CheckMate-141, a randomized trial comparing nivolumab with the investigator’s choice of standard therapy, the FDA said in a written statement.
Earlier this year, the FDA granted accelerated approval to another checkpoint inhibitor targeting the PD-1/PD-L1 pathway, pembrolizumab, for the same indication, based on an objective response rate of 16% in the nonrandomized KEYNOTE-012 trial. Merck Sharp & Dohme, maker of pembrolizumab, is looking to demonstrate an improvement in overall survival with the ongoing KEYNOTE-040 study.
Checkmate-141 enrolled 361 patients with recurrent or metastatic SCCHN with disease progression on or within 6 months of receiving platinum-based chemotherapy and randomized (2:1) to nivolumab 3 mg/kg every 2 weeks intravenously or the investigator’s choice of cetuximab 400 mg/m2 IV once, then 250 mg/m2 IV weekly; methotrexate 40 mg/m2 IV weekly; or docetaxel 30 mg/m2 IV weekly until disease progression or unacceptable toxicity.
As reported at the European Society of Medical Oncology Congress and in the New England Journal of Medicine (2016;375:1856-67), the median overall survival was 7.5 months for patients on nivolumab, compared with 5.1 months for those on standard chemotherapy. The hazard ratio for death with nivolumab was 0.70 (P = .01). Estimates of 1-year survival were 36% vs. 16.6%, respectively.
Treatment-related adverse events of grade 3 or 4 occurred in 13.1% of patients on nivolumab, compared with 35.1% of those on standard therapy. The most frequent serious adverse reactions reported in at least 2% of patients receiving nivolumab were pneumonia, dyspnea, respiratory failure, respiratory tract infection, and sepsis.
The most common adverse reactions occurring in more than 10% of nivolumab-treated patients and at a higher incidence than with standard therapy were cough and dyspnea. The most common laboratory abnormalities occurring in 10% or more nivolumab-treated patients and at a higher incidence than with standard therapy were increased alkaline phosphatase level, increased amylase level, hypercalcemia, hyperkalemia, and increased thyroid-stimulating hormone level, the FDA said.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and previously has been approved to treat classical Hodgkin’s lymphoma, advanced renal cell carcinoma, lung cancer, and melanoma.
The Food and Drug Administration has approved the immune checkpoint inhibitor nivolumab for the treatment of patients with recurrent or metastatic squamous cell carcinoma of the head and neck (SCCHN) with disease progression on or after a platinum-based therapy.
The FDA based its approval on an improvement in overall survival demonstrated in CheckMate-141, a randomized trial comparing nivolumab with the investigator’s choice of standard therapy, the FDA said in a written statement.
Earlier this year, the FDA granted accelerated approval to another checkpoint inhibitor targeting the PD-1/PD-L1 pathway, pembrolizumab, for the same indication, based on an objective response rate of 16% in the nonrandomized KEYNOTE-012 trial. Merck Sharp & Dohme, maker of pembrolizumab, is looking to demonstrate an improvement in overall survival with the ongoing KEYNOTE-040 study.
Checkmate-141 enrolled 361 patients with recurrent or metastatic SCCHN with disease progression on or within 6 months of receiving platinum-based chemotherapy and randomized (2:1) to nivolumab 3 mg/kg every 2 weeks intravenously or the investigator’s choice of cetuximab 400 mg/m2 IV once, then 250 mg/m2 IV weekly; methotrexate 40 mg/m2 IV weekly; or docetaxel 30 mg/m2 IV weekly until disease progression or unacceptable toxicity.
As reported at the European Society of Medical Oncology Congress and in the New England Journal of Medicine (2016;375:1856-67), the median overall survival was 7.5 months for patients on nivolumab, compared with 5.1 months for those on standard chemotherapy. The hazard ratio for death with nivolumab was 0.70 (P = .01). Estimates of 1-year survival were 36% vs. 16.6%, respectively.
Treatment-related adverse events of grade 3 or 4 occurred in 13.1% of patients on nivolumab, compared with 35.1% of those on standard therapy. The most frequent serious adverse reactions reported in at least 2% of patients receiving nivolumab were pneumonia, dyspnea, respiratory failure, respiratory tract infection, and sepsis.
The most common adverse reactions occurring in more than 10% of nivolumab-treated patients and at a higher incidence than with standard therapy were cough and dyspnea. The most common laboratory abnormalities occurring in 10% or more nivolumab-treated patients and at a higher incidence than with standard therapy were increased alkaline phosphatase level, increased amylase level, hypercalcemia, hyperkalemia, and increased thyroid-stimulating hormone level, the FDA said.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and previously has been approved to treat classical Hodgkin’s lymphoma, advanced renal cell carcinoma, lung cancer, and melanoma.
FDA approves tenofovir alafenamide for patients with chronic hepatitis B and liver disease
The Food and Drug Administration has approved tenofovir alafenamide (marketed as Vemlidy by Gilead Sciences) for the treatment of adults with chronic hepatitis B virus infection with compensated liver disease.
Tenofovir alafenamide is a novel, targeted prodrug of tenofovir that has demonstrated antiviral efficacy similar to tenofovir disoproxil fumarate (Viread) at significantly lower doses.
Compared with tenofovir disoproxil fumarate, tenofovir alafenamide has “greater plasma stability and more efficiently delivers tenofovir to hepatocytes” which allows tenofovir alafenamide to be administered in daily doses of 25mg while tenofovir disoproxil fumarate requires a dose of 300 mg to be as effective.
In addition, patients treated with tenofovir alafenamide demonstrated “improvements in certain bone and renal laboratory parameters.”
Overall, tenofovir alafenamide was well tolerated. Only 1% of patients discontinued treatment because of adverse events, and the most common adverse events were headache, abdominal pain, fatigue, cough, nausea, and back pain. Vemlidy has a boxed warning in its product label regarding the risks of lactic acidosis/severe hepatomegaly with steatosis and severe acute exacerbation of hepatitis B with discontinuation.
“Vemlidy is the first medication approved to treat this disease in nearly a decade,” said President and Chief Executive Officer of Gilead Sciences John Milligan. “We are excited to offer a new, effective option to help advance long-term care for patients.”
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
The Food and Drug Administration has approved tenofovir alafenamide (marketed as Vemlidy by Gilead Sciences) for the treatment of adults with chronic hepatitis B virus infection with compensated liver disease.
Tenofovir alafenamide is a novel, targeted prodrug of tenofovir that has demonstrated antiviral efficacy similar to tenofovir disoproxil fumarate (Viread) at significantly lower doses.
Compared with tenofovir disoproxil fumarate, tenofovir alafenamide has “greater plasma stability and more efficiently delivers tenofovir to hepatocytes” which allows tenofovir alafenamide to be administered in daily doses of 25mg while tenofovir disoproxil fumarate requires a dose of 300 mg to be as effective.
In addition, patients treated with tenofovir alafenamide demonstrated “improvements in certain bone and renal laboratory parameters.”
Overall, tenofovir alafenamide was well tolerated. Only 1% of patients discontinued treatment because of adverse events, and the most common adverse events were headache, abdominal pain, fatigue, cough, nausea, and back pain. Vemlidy has a boxed warning in its product label regarding the risks of lactic acidosis/severe hepatomegaly with steatosis and severe acute exacerbation of hepatitis B with discontinuation.
“Vemlidy is the first medication approved to treat this disease in nearly a decade,” said President and Chief Executive Officer of Gilead Sciences John Milligan. “We are excited to offer a new, effective option to help advance long-term care for patients.”
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
The Food and Drug Administration has approved tenofovir alafenamide (marketed as Vemlidy by Gilead Sciences) for the treatment of adults with chronic hepatitis B virus infection with compensated liver disease.
Tenofovir alafenamide is a novel, targeted prodrug of tenofovir that has demonstrated antiviral efficacy similar to tenofovir disoproxil fumarate (Viread) at significantly lower doses.
Compared with tenofovir disoproxil fumarate, tenofovir alafenamide has “greater plasma stability and more efficiently delivers tenofovir to hepatocytes” which allows tenofovir alafenamide to be administered in daily doses of 25mg while tenofovir disoproxil fumarate requires a dose of 300 mg to be as effective.
In addition, patients treated with tenofovir alafenamide demonstrated “improvements in certain bone and renal laboratory parameters.”
Overall, tenofovir alafenamide was well tolerated. Only 1% of patients discontinued treatment because of adverse events, and the most common adverse events were headache, abdominal pain, fatigue, cough, nausea, and back pain. Vemlidy has a boxed warning in its product label regarding the risks of lactic acidosis/severe hepatomegaly with steatosis and severe acute exacerbation of hepatitis B with discontinuation.
“Vemlidy is the first medication approved to treat this disease in nearly a decade,” said President and Chief Executive Officer of Gilead Sciences John Milligan. “We are excited to offer a new, effective option to help advance long-term care for patients.”
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
Weekly number of Zika-infected pregnancies drops by half
There were 146 new cases of pregnant women with laboratory evidence of Zika infection reported in the United States for the week ending Nov. 3 – just about half of the 288-case increase reported the week before, according to Centers for Disease Control and Prevention.
For the year so far, there have been 1,057 cases of pregnant women with Zika in the states and the District of Columbia – 52 for the week ending Nov. 3 – and 2,357 cases in the territories, the CDC announced, with 94 reported in the most recent week. The total number of U.S. cases – 3,414 – is up by 4.4% over the previous week.
The CDC also reported one new infant born with Zika-related birth defects for the week ending Nov. 3, bringing the total for the year to 26 in the states/D.C. There were no new Zika-related pregnancy losses reported, so the total remains at five. State-level data are not being reported to protect the privacy of affected women and children.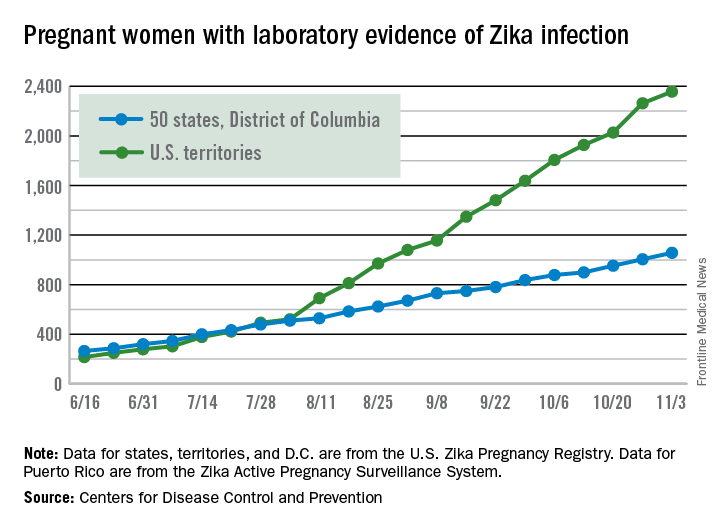
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
There were 146 new cases of pregnant women with laboratory evidence of Zika infection reported in the United States for the week ending Nov. 3 – just about half of the 288-case increase reported the week before, according to Centers for Disease Control and Prevention.
For the year so far, there have been 1,057 cases of pregnant women with Zika in the states and the District of Columbia – 52 for the week ending Nov. 3 – and 2,357 cases in the territories, the CDC announced, with 94 reported in the most recent week. The total number of U.S. cases – 3,414 – is up by 4.4% over the previous week.
The CDC also reported one new infant born with Zika-related birth defects for the week ending Nov. 3, bringing the total for the year to 26 in the states/D.C. There were no new Zika-related pregnancy losses reported, so the total remains at five. State-level data are not being reported to protect the privacy of affected women and children.
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
There were 146 new cases of pregnant women with laboratory evidence of Zika infection reported in the United States for the week ending Nov. 3 – just about half of the 288-case increase reported the week before, according to Centers for Disease Control and Prevention.
For the year so far, there have been 1,057 cases of pregnant women with Zika in the states and the District of Columbia – 52 for the week ending Nov. 3 – and 2,357 cases in the territories, the CDC announced, with 94 reported in the most recent week. The total number of U.S. cases – 3,414 – is up by 4.4% over the previous week.
The CDC also reported one new infant born with Zika-related birth defects for the week ending Nov. 3, bringing the total for the year to 26 in the states/D.C. There were no new Zika-related pregnancy losses reported, so the total remains at five. State-level data are not being reported to protect the privacy of affected women and children.
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
CDC: Seven cases of multidrug resistant C. auris have occurred in United States
The Centers for Disease Control and Prevention have reported the first cases of the multidrug-resistant fungal infection Candida auris in the United States, with evidence suggesting transmission may have occurred within U.S. health care facilities.
The report, published in the Nov. 4 edition of Morbidity and Mortality Weekly Report, described seven cases of patients infected with C. auris, which was isolated from blood in five cases, urine in one, and the ear in one. All the patients with bloodstream infections had central venous catheters at the time of diagnosis, and four of these patients died in the weeks and months after diagnosis of the infection.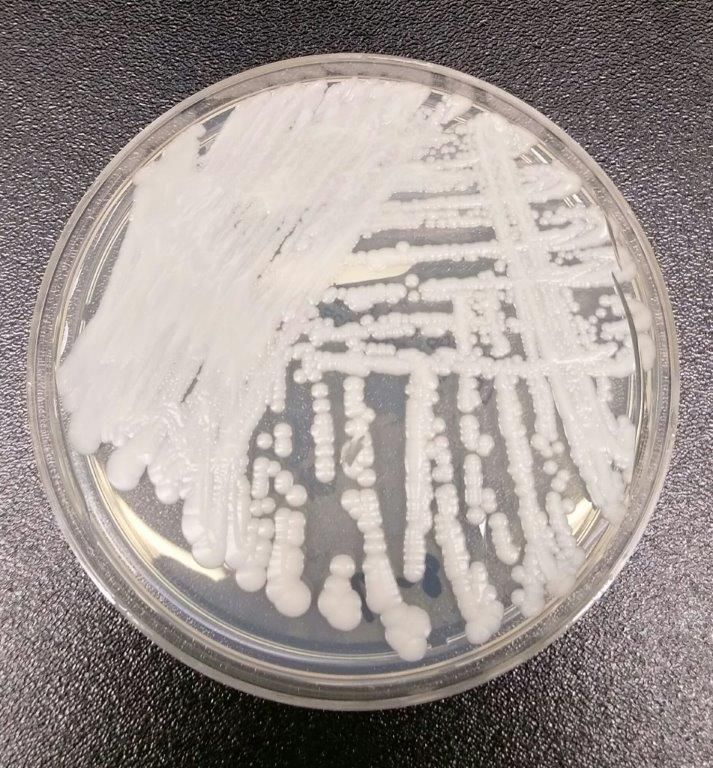
Patients’ underlying conditions usually involved immune system suppression resulting from corticisteroid therapy, malignancty, short gut syndrome, or parapleglia with a long-term, indwelling Foley catheter.
C. auris was first isolated in 2009 in Japan, but has since been reported in countries including Colombia, India, South Africa, Israel, and the United Kingdom. Snigdha Vallabhaneni, MD, of the mycotic diseases branch of CDC’s division of food water and environmental diseases, and her coauthors, said its appearance in the United States is a cause for serious concern (MMWR. 2016 Nov 4. doi: 0.15585/mmwr.mm6544e1).
“First, many isolates are multidrug resistant, with some strains having elevated minimum inhibitory concentrations to drugs in all three major classes of antifungal medications, a feature not found in other clinically relevant Candida species,” the authors wrote. All the patients with bloodstream infections were treated with antifungal echinocandins, and one also received liposomal amphotericin B.
“Second, C. auris is challenging to identify, requiring specialized methods such as matrix-assisted laser desorption/ionization time-of-flight or molecular identification based on sequencing the D1-D2 region of the 28s ribosomal DNA.”
They also highlighted that C. auris is known to cause outbreaks in health care settings. Samples taken from the mattress, bedside table, bed rail, chair, and windowsill in the room of one patient all tested positive for C. auris.
The authors also sequenced the genome of the isolates and found that isolates taken from patients admitted to the same hospital in New Jersey or the same Illinois hospital were nearly identical.
“Facilities should ensure thorough daily and terminal cleaning of rooms of patients with C. auris infections, including use of an [Environmental Protection Agency]–registered disinfectant with a fungal claim,” the authors wrote, stressing that facilities and laboratories should continue to report cases and forward suspicious unidentified Candida isolates to state or local health authorities and the CDC.
No conflicts of interest were declared.
The Centers for Disease Control and Prevention have reported the first cases of the multidrug-resistant fungal infection Candida auris in the United States, with evidence suggesting transmission may have occurred within U.S. health care facilities.
The report, published in the Nov. 4 edition of Morbidity and Mortality Weekly Report, described seven cases of patients infected with C. auris, which was isolated from blood in five cases, urine in one, and the ear in one. All the patients with bloodstream infections had central venous catheters at the time of diagnosis, and four of these patients died in the weeks and months after diagnosis of the infection.
Patients’ underlying conditions usually involved immune system suppression resulting from corticisteroid therapy, malignancty, short gut syndrome, or parapleglia with a long-term, indwelling Foley catheter.
C. auris was first isolated in 2009 in Japan, but has since been reported in countries including Colombia, India, South Africa, Israel, and the United Kingdom. Snigdha Vallabhaneni, MD, of the mycotic diseases branch of CDC’s division of food water and environmental diseases, and her coauthors, said its appearance in the United States is a cause for serious concern (MMWR. 2016 Nov 4. doi: 0.15585/mmwr.mm6544e1).
“First, many isolates are multidrug resistant, with some strains having elevated minimum inhibitory concentrations to drugs in all three major classes of antifungal medications, a feature not found in other clinically relevant Candida species,” the authors wrote. All the patients with bloodstream infections were treated with antifungal echinocandins, and one also received liposomal amphotericin B.
“Second, C. auris is challenging to identify, requiring specialized methods such as matrix-assisted laser desorption/ionization time-of-flight or molecular identification based on sequencing the D1-D2 region of the 28s ribosomal DNA.”
They also highlighted that C. auris is known to cause outbreaks in health care settings. Samples taken from the mattress, bedside table, bed rail, chair, and windowsill in the room of one patient all tested positive for C. auris.
The authors also sequenced the genome of the isolates and found that isolates taken from patients admitted to the same hospital in New Jersey or the same Illinois hospital were nearly identical.
“Facilities should ensure thorough daily and terminal cleaning of rooms of patients with C. auris infections, including use of an [Environmental Protection Agency]–registered disinfectant with a fungal claim,” the authors wrote, stressing that facilities and laboratories should continue to report cases and forward suspicious unidentified Candida isolates to state or local health authorities and the CDC.
No conflicts of interest were declared.
The Centers for Disease Control and Prevention have reported the first cases of the multidrug-resistant fungal infection Candida auris in the United States, with evidence suggesting transmission may have occurred within U.S. health care facilities.
The report, published in the Nov. 4 edition of Morbidity and Mortality Weekly Report, described seven cases of patients infected with C. auris, which was isolated from blood in five cases, urine in one, and the ear in one. All the patients with bloodstream infections had central venous catheters at the time of diagnosis, and four of these patients died in the weeks and months after diagnosis of the infection.
Patients’ underlying conditions usually involved immune system suppression resulting from corticisteroid therapy, malignancty, short gut syndrome, or parapleglia with a long-term, indwelling Foley catheter.
C. auris was first isolated in 2009 in Japan, but has since been reported in countries including Colombia, India, South Africa, Israel, and the United Kingdom. Snigdha Vallabhaneni, MD, of the mycotic diseases branch of CDC’s division of food water and environmental diseases, and her coauthors, said its appearance in the United States is a cause for serious concern (MMWR. 2016 Nov 4. doi: 0.15585/mmwr.mm6544e1).
“First, many isolates are multidrug resistant, with some strains having elevated minimum inhibitory concentrations to drugs in all three major classes of antifungal medications, a feature not found in other clinically relevant Candida species,” the authors wrote. All the patients with bloodstream infections were treated with antifungal echinocandins, and one also received liposomal amphotericin B.
“Second, C. auris is challenging to identify, requiring specialized methods such as matrix-assisted laser desorption/ionization time-of-flight or molecular identification based on sequencing the D1-D2 region of the 28s ribosomal DNA.”
They also highlighted that C. auris is known to cause outbreaks in health care settings. Samples taken from the mattress, bedside table, bed rail, chair, and windowsill in the room of one patient all tested positive for C. auris.
The authors also sequenced the genome of the isolates and found that isolates taken from patients admitted to the same hospital in New Jersey or the same Illinois hospital were nearly identical.
“Facilities should ensure thorough daily and terminal cleaning of rooms of patients with C. auris infections, including use of an [Environmental Protection Agency]–registered disinfectant with a fungal claim,” the authors wrote, stressing that facilities and laboratories should continue to report cases and forward suspicious unidentified Candida isolates to state or local health authorities and the CDC.
No conflicts of interest were declared.
Key clinical point: The first cases of the multidrug-resistant fungal infection C. auris have been reported in the United States.
Major finding: Seven cases of infection with the multidrug-resistant emerging fungal infection C. auris have been reported in the United States, five of which were bloodstream infections.
Data source: Case series.
Disclosures: No conflicts of interest were declared.
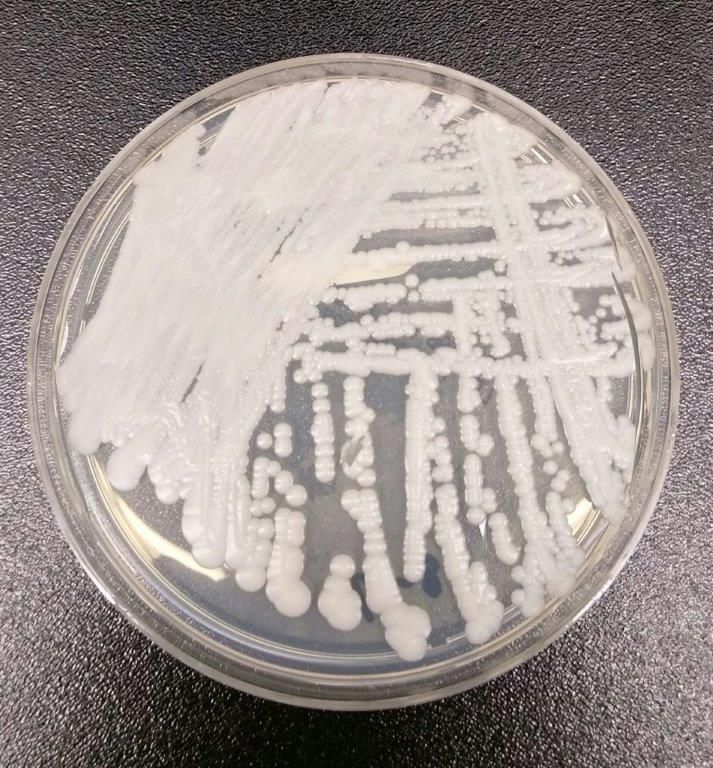
FDA: Etanercept first biologic approved for pediatric psoriasis
Etanercept has been received Food and Drug Administration approval for treating chronic moderate to severe plaque psoriasis in children and adolescents, aged 4-17 years, making this the first biologic and first systemic treatment approved in the United States for pediatric psoriasis.
![]()
Etanercept, a tumor necrosis factor blocker marketed as Enbrel, was approved in 1998 for treating moderately to severely active rheumatoid arthritis and has been approved for several other indications since then, including psoriatic arthritis and moderate to severe psoriasis in adults, and polyarticular juvenile idiopathic arthritis in patients aged 2 years and older.
Etanercept has been received Food and Drug Administration approval for treating chronic moderate to severe plaque psoriasis in children and adolescents, aged 4-17 years, making this the first biologic and first systemic treatment approved in the United States for pediatric psoriasis.
![]()
Etanercept, a tumor necrosis factor blocker marketed as Enbrel, was approved in 1998 for treating moderately to severely active rheumatoid arthritis and has been approved for several other indications since then, including psoriatic arthritis and moderate to severe psoriasis in adults, and polyarticular juvenile idiopathic arthritis in patients aged 2 years and older.
Etanercept has been received Food and Drug Administration approval for treating chronic moderate to severe plaque psoriasis in children and adolescents, aged 4-17 years, making this the first biologic and first systemic treatment approved in the United States for pediatric psoriasis.
![]()
Etanercept, a tumor necrosis factor blocker marketed as Enbrel, was approved in 1998 for treating moderately to severely active rheumatoid arthritis and has been approved for several other indications since then, including psoriatic arthritis and moderate to severe psoriasis in adults, and polyarticular juvenile idiopathic arthritis in patients aged 2 years and older.
Large increase seen in number of Zika-infected pregnancies
The 288 new cases of pregnant women in the United States with laboratory evidence of Zika infection reported for the week ending Oct. 27, 2016, represent the largest weekly increase so far this year, according to new data from the Centers for Disease Control and Prevention.
The 52 new cases in the 50 states and the District of Columbia and the 236 cases in the U.S. territories bring the totals for the year to 1,005 and 2,263, respectively, and to 3,268 for the United States as a whole, the CDC reported.
The week ending Oct. 27 also brought reports of two more infants born with Zika-related birth defects in the 50 states/D.C., but there were no reports of Zika-related pregnancy losses. So far in 2016, there have been 25 liveborn infants with Zika-related birth defects and five pregnancy losses in the states.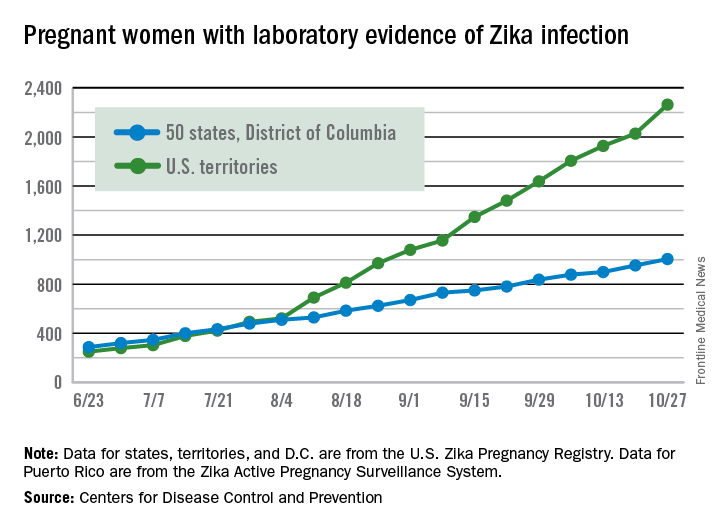
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The 288 new cases of pregnant women in the United States with laboratory evidence of Zika infection reported for the week ending Oct. 27, 2016, represent the largest weekly increase so far this year, according to new data from the Centers for Disease Control and Prevention.
The 52 new cases in the 50 states and the District of Columbia and the 236 cases in the U.S. territories bring the totals for the year to 1,005 and 2,263, respectively, and to 3,268 for the United States as a whole, the CDC reported.
The week ending Oct. 27 also brought reports of two more infants born with Zika-related birth defects in the 50 states/D.C., but there were no reports of Zika-related pregnancy losses. So far in 2016, there have been 25 liveborn infants with Zika-related birth defects and five pregnancy losses in the states.
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The 288 new cases of pregnant women in the United States with laboratory evidence of Zika infection reported for the week ending Oct. 27, 2016, represent the largest weekly increase so far this year, according to new data from the Centers for Disease Control and Prevention.
The 52 new cases in the 50 states and the District of Columbia and the 236 cases in the U.S. territories bring the totals for the year to 1,005 and 2,263, respectively, and to 3,268 for the United States as a whole, the CDC reported.
The week ending Oct. 27 also brought reports of two more infants born with Zika-related birth defects in the 50 states/D.C., but there were no reports of Zika-related pregnancy losses. So far in 2016, there have been 25 liveborn infants with Zika-related birth defects and five pregnancy losses in the states.
The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same “inclusion criteria to monitor brain abnormalities and other adverse pregnancy outcomes.” As of Sept. 29 – the date of the last territorial report – there had been one liveborn infant and one pregnancy loss related to Zika.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.