User login
2014-2015 influenza vaccine ineffective against predominant strain
The 2014-2015 influenza vaccines offered little protection against the predominant influenza A/H3N2 virus, but were effective against influenza B, according to the vaccine effectiveness estimates provided by the U.S. Flu Vaccine Effectiveness Network.
Preferential use of the live attenuated influenza vaccine (LAIV) among young children, a recommendation previously published by the Advisory Committee on Immunization Practices, was not supported.
During the 2014-2015 influenza season, a total of 9,710 patients seeking outpatient medical treatment for acute respiratory infection with cough were enrolled into the U.S. Flu Vaccine Effectiveness study, reported Richard Zimmerman, MD, of the University of Pittsburgh, and his colleagues (Clin Infect Dis. 2016 Oct 4. doi: 10.1093/cid/ciw635).
Of these, 9,311 participants had complete data, and 7,078 (76%) tested negative for influenza. A total of 1,840 participants tested positive for influenza A – 99% of these cases were strain A/H3N2 – and 395 participants tested positive for influenza B.
Of the 4,360 vaccinated participants with known vaccine type, 39.7% received standard dose trivalent, 1.6% received high dose trivalent, 46.8% received standard dose quadrivalent, and 11.9% received quadrivalent live-attenuated vaccines. 
For influenza A and B combined, the overall adjusted vaccine effectiveness was 19% (95% Confidence Interval, 10-27%) against all medically attended influenza and was statistically significant in all age groups except 18-49 years.
Across all vaccine types, the vaccine effectiveness for the A/H3N2 strain was 6% (95% CI, -5-17%), estimates were similar across all age groups, and all vaccine types were similarly ineffective. These estimates were “consistent with a mismatch between the vaccine and circulating viruses,” the researchers noted.
Overall vaccine effectiveness for influenza B/Yamagata was 55% (95% CI, 43% to 65%) and was similarly significant in all age strata except 50-64 year olds. Trivalent vaccines were more effective at preventing influenza B and, of note, no cases of influenza B occurred among those who received a high dose trivalent flu vaccine.
The study was supported by the Centers for Disease Control and Prevention and the National Institutes of Health. Dr. Zimmerman and four other investigators reported receiving research funding from several pharmaceutical companies.
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
The 2014-2015 influenza vaccines offered little protection against the predominant influenza A/H3N2 virus, but were effective against influenza B, according to the vaccine effectiveness estimates provided by the U.S. Flu Vaccine Effectiveness Network.
Preferential use of the live attenuated influenza vaccine (LAIV) among young children, a recommendation previously published by the Advisory Committee on Immunization Practices, was not supported.
During the 2014-2015 influenza season, a total of 9,710 patients seeking outpatient medical treatment for acute respiratory infection with cough were enrolled into the U.S. Flu Vaccine Effectiveness study, reported Richard Zimmerman, MD, of the University of Pittsburgh, and his colleagues (Clin Infect Dis. 2016 Oct 4. doi: 10.1093/cid/ciw635).
Of these, 9,311 participants had complete data, and 7,078 (76%) tested negative for influenza. A total of 1,840 participants tested positive for influenza A – 99% of these cases were strain A/H3N2 – and 395 participants tested positive for influenza B.
Of the 4,360 vaccinated participants with known vaccine type, 39.7% received standard dose trivalent, 1.6% received high dose trivalent, 46.8% received standard dose quadrivalent, and 11.9% received quadrivalent live-attenuated vaccines.
For influenza A and B combined, the overall adjusted vaccine effectiveness was 19% (95% Confidence Interval, 10-27%) against all medically attended influenza and was statistically significant in all age groups except 18-49 years.
Across all vaccine types, the vaccine effectiveness for the A/H3N2 strain was 6% (95% CI, -5-17%), estimates were similar across all age groups, and all vaccine types were similarly ineffective. These estimates were “consistent with a mismatch between the vaccine and circulating viruses,” the researchers noted.
Overall vaccine effectiveness for influenza B/Yamagata was 55% (95% CI, 43% to 65%) and was similarly significant in all age strata except 50-64 year olds. Trivalent vaccines were more effective at preventing influenza B and, of note, no cases of influenza B occurred among those who received a high dose trivalent flu vaccine.
The study was supported by the Centers for Disease Control and Prevention and the National Institutes of Health. Dr. Zimmerman and four other investigators reported receiving research funding from several pharmaceutical companies.
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
The 2014-2015 influenza vaccines offered little protection against the predominant influenza A/H3N2 virus, but were effective against influenza B, according to the vaccine effectiveness estimates provided by the U.S. Flu Vaccine Effectiveness Network.
Preferential use of the live attenuated influenza vaccine (LAIV) among young children, a recommendation previously published by the Advisory Committee on Immunization Practices, was not supported.
During the 2014-2015 influenza season, a total of 9,710 patients seeking outpatient medical treatment for acute respiratory infection with cough were enrolled into the U.S. Flu Vaccine Effectiveness study, reported Richard Zimmerman, MD, of the University of Pittsburgh, and his colleagues (Clin Infect Dis. 2016 Oct 4. doi: 10.1093/cid/ciw635).
Of these, 9,311 participants had complete data, and 7,078 (76%) tested negative for influenza. A total of 1,840 participants tested positive for influenza A – 99% of these cases were strain A/H3N2 – and 395 participants tested positive for influenza B.
Of the 4,360 vaccinated participants with known vaccine type, 39.7% received standard dose trivalent, 1.6% received high dose trivalent, 46.8% received standard dose quadrivalent, and 11.9% received quadrivalent live-attenuated vaccines.
For influenza A and B combined, the overall adjusted vaccine effectiveness was 19% (95% Confidence Interval, 10-27%) against all medically attended influenza and was statistically significant in all age groups except 18-49 years.
Across all vaccine types, the vaccine effectiveness for the A/H3N2 strain was 6% (95% CI, -5-17%), estimates were similar across all age groups, and all vaccine types were similarly ineffective. These estimates were “consistent with a mismatch between the vaccine and circulating viruses,” the researchers noted.
Overall vaccine effectiveness for influenza B/Yamagata was 55% (95% CI, 43% to 65%) and was similarly significant in all age strata except 50-64 year olds. Trivalent vaccines were more effective at preventing influenza B and, of note, no cases of influenza B occurred among those who received a high dose trivalent flu vaccine.
The study was supported by the Centers for Disease Control and Prevention and the National Institutes of Health. Dr. Zimmerman and four other investigators reported receiving research funding from several pharmaceutical companies.
jcraig@frontlinemedcom.com
On Twitter @jessnicolecraig
Key clinical point:
Major finding: Across all vaccine types, the vaccine effectiveness for the A/H3N2 strain was 6%.
Data source: Retrospective analysis of 9,710 patients who sought outpatient medical treatment during the 2014-2015 influenza season.
Disclosures: The study was supported by the Centers for Disease Control and Prevention and the National Institutes of Health. Dr. Zimmerman and four other investigators reported receiving research funding from several pharmaceutical companies.
FDA finalizes boxed warning for Essure
The Food and Drug Administration will require the Essure permanent birth control system to carry a boxed warning about the device’s reported adverse events, including perforation of the uterus and/or fallopian tubes, identification of inserts in the abdominal or pelvic cavity, persistent pain, and suspected allergic or hypersensitivity reactions.
The boxed warning should also state that if the device needs to be removed to address an adverse event, surgery will be necessary.
FDA officials released the final guidance Oct. 28 on labeling for permanent hysteroscopically placed tubal implants intended for sterilization, following the publication of draft labeling requirements in February 2016.
The guidance includes a Patient Decision Checklist to be signed by the patient and the physician, acknowledging that the risks and benefits of the device were discussed. The checklist is divided into sections detailing other birth control options, requirements for Essure placement (including contraindications), pregnancy risks with the device, what to expect during and after the procedure based on clinical studies, and long-term risks.
The new labeling requirements follow years of controversy about the device, which is currently the only permanent contraception option for women that can be performed without surgery.
In September 2015, the FDA Obstetrics and Gynecology Devices Panel reviewed the safety of the device after receiving more than 5,000 complaints of adverse reactions. Consensus from the 19-member panel was that there was a lack of data about the risks of the device and that patients needed better counseling before choosing it as a birth control option.
The final labeling guidance document seeks to address some of those concerns, according to the FDA.
“FDA believes this will help to ensure a woman receives and understands the benefits and risks associated with her contraceptive options so that she can make an informed decision as to whether a permanent hysteroscopically placed tubal implant intended for sterilization is the right choice for her,” according to the guidance document.
In September 2016, the FDA approved a postmarket surveillance study plan from Essure’s manufacturer, Bayer. The 3-year study will compare safety and effectiveness between women who undergo hysterocopic sterilization with Essure and those who have laparoscopic tubal sterilization.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
The Food and Drug Administration will require the Essure permanent birth control system to carry a boxed warning about the device’s reported adverse events, including perforation of the uterus and/or fallopian tubes, identification of inserts in the abdominal or pelvic cavity, persistent pain, and suspected allergic or hypersensitivity reactions.
The boxed warning should also state that if the device needs to be removed to address an adverse event, surgery will be necessary.
FDA officials released the final guidance Oct. 28 on labeling for permanent hysteroscopically placed tubal implants intended for sterilization, following the publication of draft labeling requirements in February 2016.
The guidance includes a Patient Decision Checklist to be signed by the patient and the physician, acknowledging that the risks and benefits of the device were discussed. The checklist is divided into sections detailing other birth control options, requirements for Essure placement (including contraindications), pregnancy risks with the device, what to expect during and after the procedure based on clinical studies, and long-term risks.
The new labeling requirements follow years of controversy about the device, which is currently the only permanent contraception option for women that can be performed without surgery.
In September 2015, the FDA Obstetrics and Gynecology Devices Panel reviewed the safety of the device after receiving more than 5,000 complaints of adverse reactions. Consensus from the 19-member panel was that there was a lack of data about the risks of the device and that patients needed better counseling before choosing it as a birth control option.
The final labeling guidance document seeks to address some of those concerns, according to the FDA.
“FDA believes this will help to ensure a woman receives and understands the benefits and risks associated with her contraceptive options so that she can make an informed decision as to whether a permanent hysteroscopically placed tubal implant intended for sterilization is the right choice for her,” according to the guidance document.
In September 2016, the FDA approved a postmarket surveillance study plan from Essure’s manufacturer, Bayer. The 3-year study will compare safety and effectiveness between women who undergo hysterocopic sterilization with Essure and those who have laparoscopic tubal sterilization.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
The Food and Drug Administration will require the Essure permanent birth control system to carry a boxed warning about the device’s reported adverse events, including perforation of the uterus and/or fallopian tubes, identification of inserts in the abdominal or pelvic cavity, persistent pain, and suspected allergic or hypersensitivity reactions.
The boxed warning should also state that if the device needs to be removed to address an adverse event, surgery will be necessary.
FDA officials released the final guidance Oct. 28 on labeling for permanent hysteroscopically placed tubal implants intended for sterilization, following the publication of draft labeling requirements in February 2016.
The guidance includes a Patient Decision Checklist to be signed by the patient and the physician, acknowledging that the risks and benefits of the device were discussed. The checklist is divided into sections detailing other birth control options, requirements for Essure placement (including contraindications), pregnancy risks with the device, what to expect during and after the procedure based on clinical studies, and long-term risks.
The new labeling requirements follow years of controversy about the device, which is currently the only permanent contraception option for women that can be performed without surgery.
In September 2015, the FDA Obstetrics and Gynecology Devices Panel reviewed the safety of the device after receiving more than 5,000 complaints of adverse reactions. Consensus from the 19-member panel was that there was a lack of data about the risks of the device and that patients needed better counseling before choosing it as a birth control option.
The final labeling guidance document seeks to address some of those concerns, according to the FDA.
“FDA believes this will help to ensure a woman receives and understands the benefits and risks associated with her contraceptive options so that she can make an informed decision as to whether a permanent hysteroscopically placed tubal implant intended for sterilization is the right choice for her,” according to the guidance document.
In September 2016, the FDA approved a postmarket surveillance study plan from Essure’s manufacturer, Bayer. The 3-year study will compare safety and effectiveness between women who undergo hysterocopic sterilization with Essure and those who have laparoscopic tubal sterilization.
mschneider@frontlinemedcom.com
On Twitter @maryellenny
United States nears 3,000 Zika-infected pregnancies
There were 54 new cases of pregnant women with laboratory evidence of Zika virus in the 50 states and the District of Columbia reported during the week ending Oct. 20 – the largest weekly increase in a month, according to the Centers for Disease Control and Prevention.
The number of cases reported in the U.S. territories, 100, was lower for the second week in a row, however, so the U.S. total for the week was a fairly average 154. The total number of pregnant women with laboratory evidence of Zika virus infection is now 2,980 for the year: 953 in the states/D.C. and 2,027 in the territories, the CDC reported.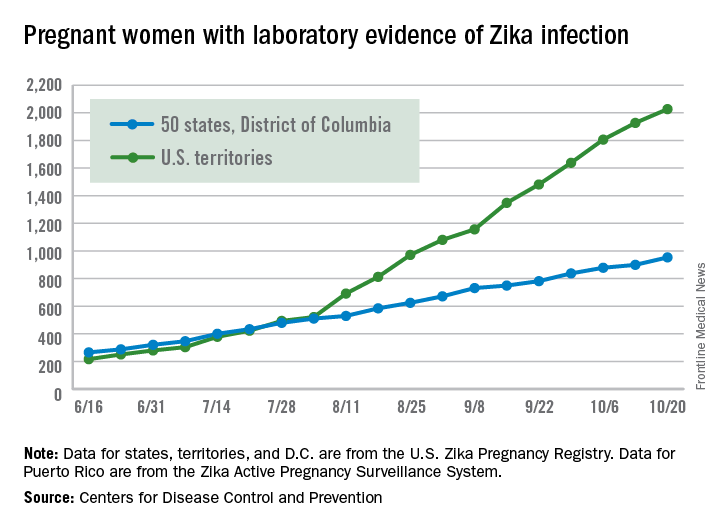
Among all Americans, the number of cases for 2015-2016 is now up to 32,814, with 1,396 new cases reported for the week ending Oct. 26: 75 in the states/D.C. and 1,321 in the territories. Almost all (98%) of the territorial cases have occurred in Puerto Rico, which continues to retroactively report cases, the CDC Arboviral Disease Branch noted.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
There were 54 new cases of pregnant women with laboratory evidence of Zika virus in the 50 states and the District of Columbia reported during the week ending Oct. 20 – the largest weekly increase in a month, according to the Centers for Disease Control and Prevention.
The number of cases reported in the U.S. territories, 100, was lower for the second week in a row, however, so the U.S. total for the week was a fairly average 154. The total number of pregnant women with laboratory evidence of Zika virus infection is now 2,980 for the year: 953 in the states/D.C. and 2,027 in the territories, the CDC reported.
Among all Americans, the number of cases for 2015-2016 is now up to 32,814, with 1,396 new cases reported for the week ending Oct. 26: 75 in the states/D.C. and 1,321 in the territories. Almost all (98%) of the territorial cases have occurred in Puerto Rico, which continues to retroactively report cases, the CDC Arboviral Disease Branch noted.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
There were 54 new cases of pregnant women with laboratory evidence of Zika virus in the 50 states and the District of Columbia reported during the week ending Oct. 20 – the largest weekly increase in a month, according to the Centers for Disease Control and Prevention.
The number of cases reported in the U.S. territories, 100, was lower for the second week in a row, however, so the U.S. total for the week was a fairly average 154. The total number of pregnant women with laboratory evidence of Zika virus infection is now 2,980 for the year: 953 in the states/D.C. and 2,027 in the territories, the CDC reported.
Among all Americans, the number of cases for 2015-2016 is now up to 32,814, with 1,396 new cases reported for the week ending Oct. 26: 75 in the states/D.C. and 1,321 in the territories. Almost all (98%) of the territorial cases have occurred in Puerto Rico, which continues to retroactively report cases, the CDC Arboviral Disease Branch noted.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
FDA expands indication for pembrolizumab in NSCLC
The Food and Drug Administration has approved pembrolizumab for the treatment of patients with metastatic non–small cell lung cancer (NSCLC) whose tumors express PD-L1 as determined by an FDA-approved test. This is the first approval of a checkpoint inhibitor for first-line treatment of the disease.
Pembrolizumab (Keytruda) is now approved to treat patients with metastatic NSCLC whose tumors have high PD-L1 expression (Tumor Proportion Score [TPS] greater than or equal to 50%), with no EGFR or ALK genomic tumor aberrations, and no prior systemic chemotherapy treatment for metastatic NSCLC, the FDA said in a written statement.![]()
The FDA based its approval on improvement in overall survival in two trials comparing treatment with pembrolizumab to treatment from chemotherapy. In one trial of 305 patients who had no prior treatment for metastatic NSCLC and TPS greater than or equal to 50%, those who received pembrolizumab (200 mg every 3 weeks) had a statistically significant improvement in overall survival, compared with patients randomized to receive chemotherapy (hazard ratio, 0.60; 95% confidence interval, 0.41-0.89; P less than .005). There was also significant improvement in progression-free survival for those receiving the checkpoint inhibitor (HR, 0.50; 95% CI, 0.37-0.68; P less than .001).
In the second trial, a three-arm trial of 1,033 patients who were previously treated for metastatic NSCLC with a TPS greater than or equal to 1%, those randomized to pembrolizumab 2 mg/kg every 3 weeks (HR, 0.71; 95% CI, 0.58-0.88; P less than .001) or pembrolizumab 10 mg/kg every 3 weeks (HR, 0.61; 95% CI, 0.49-0.75; P less than .001) had an improved overall survival, compared with patients receiving docetaxel. The median survival was 10.4 months in the pembrolizumab 2 mg/kg arm, 12.7 months in the pembrolizumab 10 mg/kg arm, and 8.5 months in the docetaxel arm.
The most common side effects of treatment with pembrolizumab included decreased appetite, fatigue, nausea, dyspnea, cough, and constipation. Rare but serious adverse events included immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis, the FDA said.
The recommended dose and schedule of pembrolizumab for NSCLC is 200 mg intravenously every 3 weeks. Full prescribing information is available here.
The Food and Drug Administration has approved pembrolizumab for the treatment of patients with metastatic non–small cell lung cancer (NSCLC) whose tumors express PD-L1 as determined by an FDA-approved test. This is the first approval of a checkpoint inhibitor for first-line treatment of the disease.
Pembrolizumab (Keytruda) is now approved to treat patients with metastatic NSCLC whose tumors have high PD-L1 expression (Tumor Proportion Score [TPS] greater than or equal to 50%), with no EGFR or ALK genomic tumor aberrations, and no prior systemic chemotherapy treatment for metastatic NSCLC, the FDA said in a written statement.![]()
The FDA based its approval on improvement in overall survival in two trials comparing treatment with pembrolizumab to treatment from chemotherapy. In one trial of 305 patients who had no prior treatment for metastatic NSCLC and TPS greater than or equal to 50%, those who received pembrolizumab (200 mg every 3 weeks) had a statistically significant improvement in overall survival, compared with patients randomized to receive chemotherapy (hazard ratio, 0.60; 95% confidence interval, 0.41-0.89; P less than .005). There was also significant improvement in progression-free survival for those receiving the checkpoint inhibitor (HR, 0.50; 95% CI, 0.37-0.68; P less than .001).
In the second trial, a three-arm trial of 1,033 patients who were previously treated for metastatic NSCLC with a TPS greater than or equal to 1%, those randomized to pembrolizumab 2 mg/kg every 3 weeks (HR, 0.71; 95% CI, 0.58-0.88; P less than .001) or pembrolizumab 10 mg/kg every 3 weeks (HR, 0.61; 95% CI, 0.49-0.75; P less than .001) had an improved overall survival, compared with patients receiving docetaxel. The median survival was 10.4 months in the pembrolizumab 2 mg/kg arm, 12.7 months in the pembrolizumab 10 mg/kg arm, and 8.5 months in the docetaxel arm.
The most common side effects of treatment with pembrolizumab included decreased appetite, fatigue, nausea, dyspnea, cough, and constipation. Rare but serious adverse events included immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis, the FDA said.
The recommended dose and schedule of pembrolizumab for NSCLC is 200 mg intravenously every 3 weeks. Full prescribing information is available here.
The Food and Drug Administration has approved pembrolizumab for the treatment of patients with metastatic non–small cell lung cancer (NSCLC) whose tumors express PD-L1 as determined by an FDA-approved test. This is the first approval of a checkpoint inhibitor for first-line treatment of the disease.
Pembrolizumab (Keytruda) is now approved to treat patients with metastatic NSCLC whose tumors have high PD-L1 expression (Tumor Proportion Score [TPS] greater than or equal to 50%), with no EGFR or ALK genomic tumor aberrations, and no prior systemic chemotherapy treatment for metastatic NSCLC, the FDA said in a written statement.![]()
The FDA based its approval on improvement in overall survival in two trials comparing treatment with pembrolizumab to treatment from chemotherapy. In one trial of 305 patients who had no prior treatment for metastatic NSCLC and TPS greater than or equal to 50%, those who received pembrolizumab (200 mg every 3 weeks) had a statistically significant improvement in overall survival, compared with patients randomized to receive chemotherapy (hazard ratio, 0.60; 95% confidence interval, 0.41-0.89; P less than .005). There was also significant improvement in progression-free survival for those receiving the checkpoint inhibitor (HR, 0.50; 95% CI, 0.37-0.68; P less than .001).
In the second trial, a three-arm trial of 1,033 patients who were previously treated for metastatic NSCLC with a TPS greater than or equal to 1%, those randomized to pembrolizumab 2 mg/kg every 3 weeks (HR, 0.71; 95% CI, 0.58-0.88; P less than .001) or pembrolizumab 10 mg/kg every 3 weeks (HR, 0.61; 95% CI, 0.49-0.75; P less than .001) had an improved overall survival, compared with patients receiving docetaxel. The median survival was 10.4 months in the pembrolizumab 2 mg/kg arm, 12.7 months in the pembrolizumab 10 mg/kg arm, and 8.5 months in the docetaxel arm.
The most common side effects of treatment with pembrolizumab included decreased appetite, fatigue, nausea, dyspnea, cough, and constipation. Rare but serious adverse events included immune-mediated pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis, the FDA said.
The recommended dose and schedule of pembrolizumab for NSCLC is 200 mg intravenously every 3 weeks. Full prescribing information is available here.
Zika increase slows slightly in pregnant women
The total number of pregnant women in the United States and its territories with laboratory evidence of Zika infection rose by 142 for the week ending Oct. 13 – the smallest increase since early September, according to the Centers for Disease Control and Prevention.
There were 121 new cases of Zika virus infection in pregnant women reported in the U.S. territories and 21 new cases in the states and the District of Columbia, bringing the corresponding totals for the year to 1,927 cases in the territories and 899 in the states/D.C. – a total of 2,826 pregnant women with Zika in the United States, the CDC reported Oct. 20.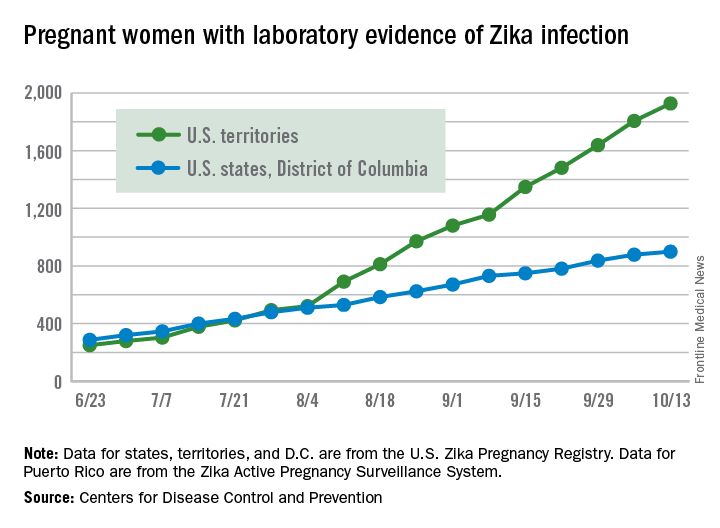
The Zika caseload among all Americans was 31,418 as of Oct. 19. An additional 80 cases reported from Oct. 14 through Oct. 19 brought the state/D.C. total to 4,016, and 1,447 new cases for the week brings the territorial total to 27,402 for 2015-2016, the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The total number of pregnant women in the United States and its territories with laboratory evidence of Zika infection rose by 142 for the week ending Oct. 13 – the smallest increase since early September, according to the Centers for Disease Control and Prevention.
There were 121 new cases of Zika virus infection in pregnant women reported in the U.S. territories and 21 new cases in the states and the District of Columbia, bringing the corresponding totals for the year to 1,927 cases in the territories and 899 in the states/D.C. – a total of 2,826 pregnant women with Zika in the United States, the CDC reported Oct. 20.
The Zika caseload among all Americans was 31,418 as of Oct. 19. An additional 80 cases reported from Oct. 14 through Oct. 19 brought the state/D.C. total to 4,016, and 1,447 new cases for the week brings the territorial total to 27,402 for 2015-2016, the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The total number of pregnant women in the United States and its territories with laboratory evidence of Zika infection rose by 142 for the week ending Oct. 13 – the smallest increase since early September, according to the Centers for Disease Control and Prevention.
There were 121 new cases of Zika virus infection in pregnant women reported in the U.S. territories and 21 new cases in the states and the District of Columbia, bringing the corresponding totals for the year to 1,927 cases in the territories and 899 in the states/D.C. – a total of 2,826 pregnant women with Zika in the United States, the CDC reported Oct. 20.
The Zika caseload among all Americans was 31,418 as of Oct. 19. An additional 80 cases reported from Oct. 14 through Oct. 19 brought the state/D.C. total to 4,016, and 1,447 new cases for the week brings the territorial total to 27,402 for 2015-2016, the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
Benefits of dental sealant programs for low-income children exceed costs
Cavities and fillings were approximately three times more likely among low-income children aged 7-11 years whose teeth were not treated with dental sealants, compared to those treated with sealants, based on data from the National Health and Nutrition Examination Survey (NHANES) from 2011-2014.
Overall, 43% of children and 39% of low-income children in the United States were treated at least once with dental sealant.
Although sealant use in children increased overall from 31% to 44% in a comparison of 1999-2004 and 2011-2014 NHANES data, sealant use was less common among low-income children compared with high-income children (39% vs. 48%) in the 2011-2014 period after increases of 16% and 9%, respectively, during 2011-2014.
School-based programs to provide sealants could increase their use among low-income children, wrote Susan O. Griffin, PhD, of the Centers for Disease Control and Prevention in Atlanta, and her associates. The researchers reviewed data from 1,371 low-income children aged 6-11 years, and found that 60% (approximately 6.5 million children), had not been treated with dental sealants. “The systematic review of economic evaluations of school-based sealant programs (SBSP) conducted for the Task Force found that SBSP became cost-saving within 2 years of placing sealants,” the researchers noted.
The data were published in the CDC’s Morbidity and Mortality Weekly Report (MMWR 2016 Oct 21;65[41];1141-5).
Cavities and fillings were approximately three times more likely among low-income children aged 7-11 years whose teeth were not treated with dental sealants, compared to those treated with sealants, based on data from the National Health and Nutrition Examination Survey (NHANES) from 2011-2014.
Overall, 43% of children and 39% of low-income children in the United States were treated at least once with dental sealant.
Although sealant use in children increased overall from 31% to 44% in a comparison of 1999-2004 and 2011-2014 NHANES data, sealant use was less common among low-income children compared with high-income children (39% vs. 48%) in the 2011-2014 period after increases of 16% and 9%, respectively, during 2011-2014.
School-based programs to provide sealants could increase their use among low-income children, wrote Susan O. Griffin, PhD, of the Centers for Disease Control and Prevention in Atlanta, and her associates. The researchers reviewed data from 1,371 low-income children aged 6-11 years, and found that 60% (approximately 6.5 million children), had not been treated with dental sealants. “The systematic review of economic evaluations of school-based sealant programs (SBSP) conducted for the Task Force found that SBSP became cost-saving within 2 years of placing sealants,” the researchers noted.
The data were published in the CDC’s Morbidity and Mortality Weekly Report (MMWR 2016 Oct 21;65[41];1141-5).
Cavities and fillings were approximately three times more likely among low-income children aged 7-11 years whose teeth were not treated with dental sealants, compared to those treated with sealants, based on data from the National Health and Nutrition Examination Survey (NHANES) from 2011-2014.
Overall, 43% of children and 39% of low-income children in the United States were treated at least once with dental sealant.
Although sealant use in children increased overall from 31% to 44% in a comparison of 1999-2004 and 2011-2014 NHANES data, sealant use was less common among low-income children compared with high-income children (39% vs. 48%) in the 2011-2014 period after increases of 16% and 9%, respectively, during 2011-2014.
School-based programs to provide sealants could increase their use among low-income children, wrote Susan O. Griffin, PhD, of the Centers for Disease Control and Prevention in Atlanta, and her associates. The researchers reviewed data from 1,371 low-income children aged 6-11 years, and found that 60% (approximately 6.5 million children), had not been treated with dental sealants. “The systematic review of economic evaluations of school-based sealant programs (SBSP) conducted for the Task Force found that SBSP became cost-saving within 2 years of placing sealants,” the researchers noted.
The data were published in the CDC’s Morbidity and Mortality Weekly Report (MMWR 2016 Oct 21;65[41];1141-5).
FROM MMWR
FDA approves atezolizumab for advanced NSCLC
The Food and Drug Administration has approved the programmed death-ligand 1 (PD-L1) blocking antibody atezolizumab for the treatment of patients with metastatic non–small cell lung cancer (NSCLC) whose disease has progressed during or following platinum-containing chemotherapy.
The FDA previously approved atezolizumab (Tecentriq) for the treatment of locally advanced or metastatic urothelial carcinoma that has progressed after platinum-containing chemotherapy.
Approval for treatment of NSCLC was based on results from the phase III OAK and phase II POPLAR trials that enrolled a total of 1,137 patients with NSCLC whose disease had progressed on platinum-containing chemotherapy. In OAK, median overall survival for patients assigned to atezolizumab was 13.8 months, compared with 9.6 months for patients assigned to docetaxel, as recently reported at the European Society for Medical Oncology Congress.
In POPLAR, overall survival was 12.6 months for patients receiving atezolizumab versus 9.7 months for those assigned to docetaxel, as reported at the European Cancer Congress in 2015.
The most common (greater than or equal to 20%) adverse reactions in patients treated with atezolizumab were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation, according to the FDA website. The most common (greater than or equal to 2%) grade 3-4 adverse events in patients treated with atezolizumab were dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, anemia, musculoskeletal pain, AST increase, ALT increase, dysphagia, and arthralgia. Clinically significant immune-related adverse events for patients receiving atezolizumab have included pneumonitis, hepatitis, colitis, and thyroid disease.
The recommended dose is 1,200 mg administered as an intravenous infusion over 60 minutes every 3 weeks until disease progression or unacceptable toxicity.
Patients with EGFR or ALK genomic tumor aberrations should not receive atezolizumab before having disease progression on FDA-approved therapy for these aberrations, the FDA said.
Full prescribing information is available on the FDA website.
The Food and Drug Administration has approved the programmed death-ligand 1 (PD-L1) blocking antibody atezolizumab for the treatment of patients with metastatic non–small cell lung cancer (NSCLC) whose disease has progressed during or following platinum-containing chemotherapy.
The FDA previously approved atezolizumab (Tecentriq) for the treatment of locally advanced or metastatic urothelial carcinoma that has progressed after platinum-containing chemotherapy.
Approval for treatment of NSCLC was based on results from the phase III OAK and phase II POPLAR trials that enrolled a total of 1,137 patients with NSCLC whose disease had progressed on platinum-containing chemotherapy. In OAK, median overall survival for patients assigned to atezolizumab was 13.8 months, compared with 9.6 months for patients assigned to docetaxel, as recently reported at the European Society for Medical Oncology Congress.
In POPLAR, overall survival was 12.6 months for patients receiving atezolizumab versus 9.7 months for those assigned to docetaxel, as reported at the European Cancer Congress in 2015.
The most common (greater than or equal to 20%) adverse reactions in patients treated with atezolizumab were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation, according to the FDA website. The most common (greater than or equal to 2%) grade 3-4 adverse events in patients treated with atezolizumab were dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, anemia, musculoskeletal pain, AST increase, ALT increase, dysphagia, and arthralgia. Clinically significant immune-related adverse events for patients receiving atezolizumab have included pneumonitis, hepatitis, colitis, and thyroid disease.
The recommended dose is 1,200 mg administered as an intravenous infusion over 60 minutes every 3 weeks until disease progression or unacceptable toxicity.
Patients with EGFR or ALK genomic tumor aberrations should not receive atezolizumab before having disease progression on FDA-approved therapy for these aberrations, the FDA said.
Full prescribing information is available on the FDA website.
The Food and Drug Administration has approved the programmed death-ligand 1 (PD-L1) blocking antibody atezolizumab for the treatment of patients with metastatic non–small cell lung cancer (NSCLC) whose disease has progressed during or following platinum-containing chemotherapy.
The FDA previously approved atezolizumab (Tecentriq) for the treatment of locally advanced or metastatic urothelial carcinoma that has progressed after platinum-containing chemotherapy.
Approval for treatment of NSCLC was based on results from the phase III OAK and phase II POPLAR trials that enrolled a total of 1,137 patients with NSCLC whose disease had progressed on platinum-containing chemotherapy. In OAK, median overall survival for patients assigned to atezolizumab was 13.8 months, compared with 9.6 months for patients assigned to docetaxel, as recently reported at the European Society for Medical Oncology Congress.
In POPLAR, overall survival was 12.6 months for patients receiving atezolizumab versus 9.7 months for those assigned to docetaxel, as reported at the European Cancer Congress in 2015.
The most common (greater than or equal to 20%) adverse reactions in patients treated with atezolizumab were fatigue, decreased appetite, dyspnea, cough, nausea, musculoskeletal pain, and constipation, according to the FDA website. The most common (greater than or equal to 2%) grade 3-4 adverse events in patients treated with atezolizumab were dyspnea, pneumonia, hypoxia, hyponatremia, fatigue, anemia, musculoskeletal pain, AST increase, ALT increase, dysphagia, and arthralgia. Clinically significant immune-related adverse events for patients receiving atezolizumab have included pneumonitis, hepatitis, colitis, and thyroid disease.
The recommended dose is 1,200 mg administered as an intravenous infusion over 60 minutes every 3 weeks until disease progression or unacceptable toxicity.
Patients with EGFR or ALK genomic tumor aberrations should not receive atezolizumab before having disease progression on FDA-approved therapy for these aberrations, the FDA said.
Full prescribing information is available on the FDA website.
FDA grants accelerated approval to olaratumab for soft tissue sarcoma
The Food and Drug Administration has granted accelerated approval to olaratumab with doxorubicin for the treatment of adult patients with certain types of soft tissue sarcoma.
“This is the first new therapy approved by the FDA for the initial treatment of soft tissue sarcoma since doxorubicin’s approval more than 40 years ago,” Richard Pazdur, MD, director of the office of hematology and oncology products in the FDA Center for Drug Evaluation and Research and acting director of the FDA Oncology Center of Excellence, said in a statement.
Approval was based on a statistically significant improvement in survival in a randomized trial involving 133 patients with more than 25 different subtypes of metastatic soft tissue sarcoma. Patients received olaratumab (Lartruvo) with doxorubicin or doxorubicin alone. Median survival was 26.5 months for patients who received both drugs, compared with 14.7 months for patients who received doxorubicin alone. Median progression-free survival was 8.2 months for patients who received both drugs and 4.4 months for patients who received doxorubicin alone.
The most common adverse reactions from olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. There are serious risks of infusion-related reactions and embryo-fetal harm, the FDA warned.
Olaratumab was approved under the FDA’s accelerated approval program after receiving fast track designation, breakthrough therapy designation, and a priority review status. The drug also received an orphan drug designation. Drug maker Eli Lilly is currently conducting a larger study of olaratumab across multiple subtypes of soft tissue sarcoma.
The Food and Drug Administration has granted accelerated approval to olaratumab with doxorubicin for the treatment of adult patients with certain types of soft tissue sarcoma.
“This is the first new therapy approved by the FDA for the initial treatment of soft tissue sarcoma since doxorubicin’s approval more than 40 years ago,” Richard Pazdur, MD, director of the office of hematology and oncology products in the FDA Center for Drug Evaluation and Research and acting director of the FDA Oncology Center of Excellence, said in a statement.
Approval was based on a statistically significant improvement in survival in a randomized trial involving 133 patients with more than 25 different subtypes of metastatic soft tissue sarcoma. Patients received olaratumab (Lartruvo) with doxorubicin or doxorubicin alone. Median survival was 26.5 months for patients who received both drugs, compared with 14.7 months for patients who received doxorubicin alone. Median progression-free survival was 8.2 months for patients who received both drugs and 4.4 months for patients who received doxorubicin alone.
The most common adverse reactions from olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. There are serious risks of infusion-related reactions and embryo-fetal harm, the FDA warned.
Olaratumab was approved under the FDA’s accelerated approval program after receiving fast track designation, breakthrough therapy designation, and a priority review status. The drug also received an orphan drug designation. Drug maker Eli Lilly is currently conducting a larger study of olaratumab across multiple subtypes of soft tissue sarcoma.
The Food and Drug Administration has granted accelerated approval to olaratumab with doxorubicin for the treatment of adult patients with certain types of soft tissue sarcoma.
“This is the first new therapy approved by the FDA for the initial treatment of soft tissue sarcoma since doxorubicin’s approval more than 40 years ago,” Richard Pazdur, MD, director of the office of hematology and oncology products in the FDA Center for Drug Evaluation and Research and acting director of the FDA Oncology Center of Excellence, said in a statement.
Approval was based on a statistically significant improvement in survival in a randomized trial involving 133 patients with more than 25 different subtypes of metastatic soft tissue sarcoma. Patients received olaratumab (Lartruvo) with doxorubicin or doxorubicin alone. Median survival was 26.5 months for patients who received both drugs, compared with 14.7 months for patients who received doxorubicin alone. Median progression-free survival was 8.2 months for patients who received both drugs and 4.4 months for patients who received doxorubicin alone.
The most common adverse reactions from olaratumab were nausea, fatigue, neutropenia, musculoskeletal pain, mucositis, alopecia, vomiting, diarrhea, decreased appetite, abdominal pain, neuropathy, and headache. There are serious risks of infusion-related reactions and embryo-fetal harm, the FDA warned.
Olaratumab was approved under the FDA’s accelerated approval program after receiving fast track designation, breakthrough therapy designation, and a priority review status. The drug also received an orphan drug designation. Drug maker Eli Lilly is currently conducting a larger study of olaratumab across multiple subtypes of soft tissue sarcoma.
Number of Zika-infected pregnant women continues to climb
The number of pregnant women with laboratory evidence of Zika infection increased by 209 for the week ending Oct. 6, the second-largest weekly increase so far in the United States in 2016, according to the Centers for Disease Control and Prevention.
Another liveborn infant with Zika-related birth defects also was reported for the week, bringing the total for the year to 23 for the 50 states and the District of Columbia. There were no new cases of Zika-related pregnancy losses reported, so the 50-state/DC total remains at five.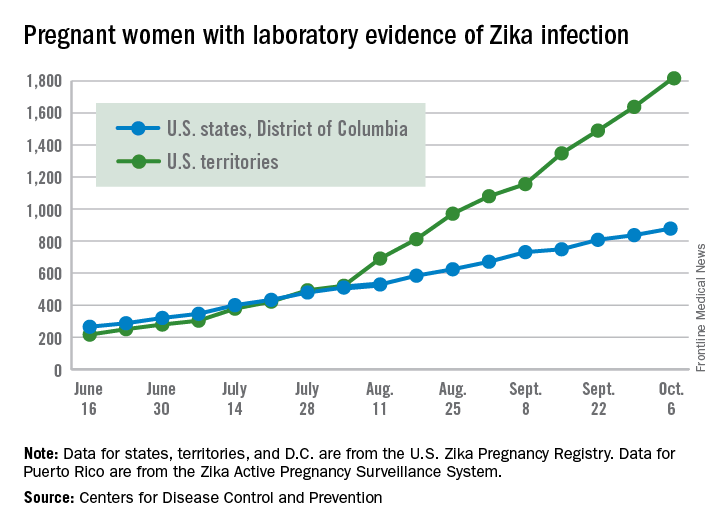
Of the 209 new cases reported for the week ending Oct. 6, 41 were in the states/D.C. and 168 were in the territories. The total number of U.S. Zika cases in pregnant women for the year is 2,684: 878 in the states/D.C. and 1,806 in the territories, the CDC said.
Among all Americans, there were 29,891 cases reported as of Oct. 12: 3,936 in the states/D.C. and 25,955 in the territories. Of the territorial cases, 98% have been in Puerto Rico, the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The number of pregnant women with laboratory evidence of Zika infection increased by 209 for the week ending Oct. 6, the second-largest weekly increase so far in the United States in 2016, according to the Centers for Disease Control and Prevention.
Another liveborn infant with Zika-related birth defects also was reported for the week, bringing the total for the year to 23 for the 50 states and the District of Columbia. There were no new cases of Zika-related pregnancy losses reported, so the 50-state/DC total remains at five.
Of the 209 new cases reported for the week ending Oct. 6, 41 were in the states/D.C. and 168 were in the territories. The total number of U.S. Zika cases in pregnant women for the year is 2,684: 878 in the states/D.C. and 1,806 in the territories, the CDC said.
Among all Americans, there were 29,891 cases reported as of Oct. 12: 3,936 in the states/D.C. and 25,955 in the territories. Of the territorial cases, 98% have been in Puerto Rico, the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The number of pregnant women with laboratory evidence of Zika infection increased by 209 for the week ending Oct. 6, the second-largest weekly increase so far in the United States in 2016, according to the Centers for Disease Control and Prevention.
Another liveborn infant with Zika-related birth defects also was reported for the week, bringing the total for the year to 23 for the 50 states and the District of Columbia. There were no new cases of Zika-related pregnancy losses reported, so the 50-state/DC total remains at five.
Of the 209 new cases reported for the week ending Oct. 6, 41 were in the states/D.C. and 168 were in the territories. The total number of U.S. Zika cases in pregnant women for the year is 2,684: 878 in the states/D.C. and 1,806 in the territories, the CDC said.
Among all Americans, there were 29,891 cases reported as of Oct. 12: 3,936 in the states/D.C. and 25,955 in the territories. Of the territorial cases, 98% have been in Puerto Rico, the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
FDA reaffirms rivaroxaban’s atrial fib efficacy in ROCKET AF
The Food and Drug Administration reaffirmed its confidence in the data supporting the claim that rivaroxaban (Xarelto) is a safe and effective alternative to warfarin for preventing strokes and blood clots in patients with nonvalvular atrial fibrillation.
“The FDA concludes that Xarelto is a safe and effective alternative to warfarin in patients with atrial fibrillation,” the agency said in a statement released on Oct. 11.
In response to these events the FDA “completed a variety of analyses to assess the impact that this faulty monitoring device had on the ROCKET AF study results. The agency has determined that effects on strokes or bleeding, including bleeding in the head, were minimal,” the agency said in its statement.
Researchers associated with ROCKET AF published their own analysis of the impact of the faulty device on bleeding rates among patients treated with warfarin in the trial and concluded that device malfunction did not appear to influence the results (N Engl J Med. 2016 Feb 25;374[8]:785-8).
Rivaroxaban is one of four new oral anticoagulants (NOACs) on the U.S. market that are alternatives to warfarin for stroke and clot prevention in patients with nonvalvular atrial fibrillation. An analysis of 2014 data on U.S. office-based prescriptions for NOACs in atrial fibrillation patients showed that rivaroxaban was by far the most commonly prescribed drug in the class, prescribed for patients during 48% of physician office visits that led to a NOAC prescription (Am J Med. 2015 Dec;128[12]:1300-5).
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
The Food and Drug Administration reaffirmed its confidence in the data supporting the claim that rivaroxaban (Xarelto) is a safe and effective alternative to warfarin for preventing strokes and blood clots in patients with nonvalvular atrial fibrillation.
“The FDA concludes that Xarelto is a safe and effective alternative to warfarin in patients with atrial fibrillation,” the agency said in a statement released on Oct. 11.
In response to these events the FDA “completed a variety of analyses to assess the impact that this faulty monitoring device had on the ROCKET AF study results. The agency has determined that effects on strokes or bleeding, including bleeding in the head, were minimal,” the agency said in its statement.
Researchers associated with ROCKET AF published their own analysis of the impact of the faulty device on bleeding rates among patients treated with warfarin in the trial and concluded that device malfunction did not appear to influence the results (N Engl J Med. 2016 Feb 25;374[8]:785-8).
Rivaroxaban is one of four new oral anticoagulants (NOACs) on the U.S. market that are alternatives to warfarin for stroke and clot prevention in patients with nonvalvular atrial fibrillation. An analysis of 2014 data on U.S. office-based prescriptions for NOACs in atrial fibrillation patients showed that rivaroxaban was by far the most commonly prescribed drug in the class, prescribed for patients during 48% of physician office visits that led to a NOAC prescription (Am J Med. 2015 Dec;128[12]:1300-5).
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler
The Food and Drug Administration reaffirmed its confidence in the data supporting the claim that rivaroxaban (Xarelto) is a safe and effective alternative to warfarin for preventing strokes and blood clots in patients with nonvalvular atrial fibrillation.
“The FDA concludes that Xarelto is a safe and effective alternative to warfarin in patients with atrial fibrillation,” the agency said in a statement released on Oct. 11.
In response to these events the FDA “completed a variety of analyses to assess the impact that this faulty monitoring device had on the ROCKET AF study results. The agency has determined that effects on strokes or bleeding, including bleeding in the head, were minimal,” the agency said in its statement.
Researchers associated with ROCKET AF published their own analysis of the impact of the faulty device on bleeding rates among patients treated with warfarin in the trial and concluded that device malfunction did not appear to influence the results (N Engl J Med. 2016 Feb 25;374[8]:785-8).
Rivaroxaban is one of four new oral anticoagulants (NOACs) on the U.S. market that are alternatives to warfarin for stroke and clot prevention in patients with nonvalvular atrial fibrillation. An analysis of 2014 data on U.S. office-based prescriptions for NOACs in atrial fibrillation patients showed that rivaroxaban was by far the most commonly prescribed drug in the class, prescribed for patients during 48% of physician office visits that led to a NOAC prescription (Am J Med. 2015 Dec;128[12]:1300-5).
mzoler@frontlinemedcom.com
On Twitter @mitchelzoler