User login
United States nears 2,500 Zika cases in pregnant women
The week also included a report of another live-born infant with Zika-related birth defects, bringing the U.S. total to 23 for the year: 22 in the 50 states and District of Columbia and 1 in the territories, the CDC reported Oct. 6. There also have been six Zika-related pregnancy losses reported so far in the states and territories, a number that has not changed since early August.
There have been 28,019 cases of Zika reported among all Americans as of Oct. 5, with the majority (86%) coming from the territories and the majority of those cases (98%) coming from Puerto Rico, according to an update from the CDC’s Arboviral Disease Branch.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The week also included a report of another live-born infant with Zika-related birth defects, bringing the U.S. total to 23 for the year: 22 in the 50 states and District of Columbia and 1 in the territories, the CDC reported Oct. 6. There also have been six Zika-related pregnancy losses reported so far in the states and territories, a number that has not changed since early August.
There have been 28,019 cases of Zika reported among all Americans as of Oct. 5, with the majority (86%) coming from the territories and the majority of those cases (98%) coming from Puerto Rico, according to an update from the CDC’s Arboviral Disease Branch.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
The week also included a report of another live-born infant with Zika-related birth defects, bringing the U.S. total to 23 for the year: 22 in the 50 states and District of Columbia and 1 in the territories, the CDC reported Oct. 6. There also have been six Zika-related pregnancy losses reported so far in the states and territories, a number that has not changed since early August.
There have been 28,019 cases of Zika reported among all Americans as of Oct. 5, with the majority (86%) coming from the territories and the majority of those cases (98%) coming from Puerto Rico, according to an update from the CDC’s Arboviral Disease Branch.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
The pregnancy-related figures for states, territories, and D.C. reflect reporting to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System.
FDA approves ustekinumab for adult Crohn’s disease
The Food and Drug Administration has approved ustekinumab (Stelara) for the treatment of moderately to severely active Crohn’s disease in certain adults.
Specifically, the new approval for the human monoclonal antibody, which was previously approved for the treatment of psoriasis and psoriatic arthritis, is for Crohn’s disease patients aged 18 years or older who fail or cannot tolerate treatment with immunomodulators or corticosteroids but who never failed treatment with a tumor necrosis factor (TNF) blocker, or who fail or cannot tolerate treatment with one or more TNF blockers, according to a statement from the drug’s maker, Janssen Biotech.
“Crohn’s disease is a complex condition to treat, and not all therapies work for every patient. The FDA approval of Stelara represents an important advancement in treating patients with Crohn’s disease, as this therapy offers an alternate mechanism of action to induce and maintain clinical remission over time,” study investigator William J. Sandborn, MD, of the University of California, San Diego, said in the statement.
AGA Resource
AGA offers an IBD Clinical Service Line that provides tools to help you become more efficient, understand quality standards and improve the process of care for patients. Learn more at http://www.gastro.org/patient-care/conditions-diseases/ibd.
The Food and Drug Administration has approved ustekinumab (Stelara) for the treatment of moderately to severely active Crohn’s disease in certain adults.
Specifically, the new approval for the human monoclonal antibody, which was previously approved for the treatment of psoriasis and psoriatic arthritis, is for Crohn’s disease patients aged 18 years or older who fail or cannot tolerate treatment with immunomodulators or corticosteroids but who never failed treatment with a tumor necrosis factor (TNF) blocker, or who fail or cannot tolerate treatment with one or more TNF blockers, according to a statement from the drug’s maker, Janssen Biotech.
“Crohn’s disease is a complex condition to treat, and not all therapies work for every patient. The FDA approval of Stelara represents an important advancement in treating patients with Crohn’s disease, as this therapy offers an alternate mechanism of action to induce and maintain clinical remission over time,” study investigator William J. Sandborn, MD, of the University of California, San Diego, said in the statement.
AGA Resource
AGA offers an IBD Clinical Service Line that provides tools to help you become more efficient, understand quality standards and improve the process of care for patients. Learn more at http://www.gastro.org/patient-care/conditions-diseases/ibd.
The Food and Drug Administration has approved ustekinumab (Stelara) for the treatment of moderately to severely active Crohn’s disease in certain adults.
Specifically, the new approval for the human monoclonal antibody, which was previously approved for the treatment of psoriasis and psoriatic arthritis, is for Crohn’s disease patients aged 18 years or older who fail or cannot tolerate treatment with immunomodulators or corticosteroids but who never failed treatment with a tumor necrosis factor (TNF) blocker, or who fail or cannot tolerate treatment with one or more TNF blockers, according to a statement from the drug’s maker, Janssen Biotech.
“Crohn’s disease is a complex condition to treat, and not all therapies work for every patient. The FDA approval of Stelara represents an important advancement in treating patients with Crohn’s disease, as this therapy offers an alternate mechanism of action to induce and maintain clinical remission over time,” study investigator William J. Sandborn, MD, of the University of California, San Diego, said in the statement.
AGA Resource
AGA offers an IBD Clinical Service Line that provides tools to help you become more efficient, understand quality standards and improve the process of care for patients. Learn more at http://www.gastro.org/patient-care/conditions-diseases/ibd.
Four of five health care providers get flu shot
Influenza vaccine coverage among U.S. health care personnel increased very slightly during the 2015-2016 flu season, with 79% reporting that they received the shot, compared with 77% in 2014-2015.
Hospital personnel were most likely to be covered (91%) and long-term care personnel least likely to be covered (69%), Carla L. Black, PhD, wrote in the Sept. 30 issue of Morbidity and Mortality Weekly Report (2016;65:1026-31). Coverage among health care personnel working in long-term care settings did increase, however, from 64% in the 2014-2015 seasons to 69% in the 2015-2016 season, Dr. Black and colleagues noted.
“Although low, this is the only setting with an appreciable increase in coverage, compared with last season. Influenza vaccination among health care personnel in long-term care settings is especially important because influenza vaccine effectiveness is generally lowest in the elderly,” according to Dr Black, an epidemiologist with the Centers for Disease Control and Prevention.
CDC conducted the Internet survey of 2,258 health care workers from March to April, 2016.
Physicians had the highest level of coverage (95.6%), while health care assistants and aides had the lowest (64.5%). Employers also exerted an influence on coverage. Most respondents (73%) were vaccinated at work. Coverage was highest (96.5%) at facilities where vaccination was required and lowest (45%) where vaccination was not required, promoted, or offered on site.
“Employer vaccination requirements likely contributed to the observed gradual increase in vaccination among health care personnel working in settings with the lowest coverage,” the investigators noted. “In the absence of vaccination requirements, expanding the number of health care locations offering vaccination on site, over multiple days, and at no cost might help sustain and improve influenza vaccination coverage among health care personnel, including in long-term care settings.”
As a CDC employee, Dr. Black has no financial conflicts.
Influenza vaccine coverage among U.S. health care personnel increased very slightly during the 2015-2016 flu season, with 79% reporting that they received the shot, compared with 77% in 2014-2015.
Hospital personnel were most likely to be covered (91%) and long-term care personnel least likely to be covered (69%), Carla L. Black, PhD, wrote in the Sept. 30 issue of Morbidity and Mortality Weekly Report (2016;65:1026-31). Coverage among health care personnel working in long-term care settings did increase, however, from 64% in the 2014-2015 seasons to 69% in the 2015-2016 season, Dr. Black and colleagues noted.
“Although low, this is the only setting with an appreciable increase in coverage, compared with last season. Influenza vaccination among health care personnel in long-term care settings is especially important because influenza vaccine effectiveness is generally lowest in the elderly,” according to Dr Black, an epidemiologist with the Centers for Disease Control and Prevention.
CDC conducted the Internet survey of 2,258 health care workers from March to April, 2016.
Physicians had the highest level of coverage (95.6%), while health care assistants and aides had the lowest (64.5%). Employers also exerted an influence on coverage. Most respondents (73%) were vaccinated at work. Coverage was highest (96.5%) at facilities where vaccination was required and lowest (45%) where vaccination was not required, promoted, or offered on site.
“Employer vaccination requirements likely contributed to the observed gradual increase in vaccination among health care personnel working in settings with the lowest coverage,” the investigators noted. “In the absence of vaccination requirements, expanding the number of health care locations offering vaccination on site, over multiple days, and at no cost might help sustain and improve influenza vaccination coverage among health care personnel, including in long-term care settings.”
As a CDC employee, Dr. Black has no financial conflicts.
Influenza vaccine coverage among U.S. health care personnel increased very slightly during the 2015-2016 flu season, with 79% reporting that they received the shot, compared with 77% in 2014-2015.
Hospital personnel were most likely to be covered (91%) and long-term care personnel least likely to be covered (69%), Carla L. Black, PhD, wrote in the Sept. 30 issue of Morbidity and Mortality Weekly Report (2016;65:1026-31). Coverage among health care personnel working in long-term care settings did increase, however, from 64% in the 2014-2015 seasons to 69% in the 2015-2016 season, Dr. Black and colleagues noted.
“Although low, this is the only setting with an appreciable increase in coverage, compared with last season. Influenza vaccination among health care personnel in long-term care settings is especially important because influenza vaccine effectiveness is generally lowest in the elderly,” according to Dr Black, an epidemiologist with the Centers for Disease Control and Prevention.
CDC conducted the Internet survey of 2,258 health care workers from March to April, 2016.
Physicians had the highest level of coverage (95.6%), while health care assistants and aides had the lowest (64.5%). Employers also exerted an influence on coverage. Most respondents (73%) were vaccinated at work. Coverage was highest (96.5%) at facilities where vaccination was required and lowest (45%) where vaccination was not required, promoted, or offered on site.
“Employer vaccination requirements likely contributed to the observed gradual increase in vaccination among health care personnel working in settings with the lowest coverage,” the investigators noted. “In the absence of vaccination requirements, expanding the number of health care locations offering vaccination on site, over multiple days, and at no cost might help sustain and improve influenza vaccination coverage among health care personnel, including in long-term care settings.”
As a CDC employee, Dr. Black has no financial conflicts.
Key clinical point:
Major finding: The flu vaccination rate was 79% among health care workers.
Data source: An Internet survey contained data on 2,258 people.
Disclosures: As a CDC employee, Dr. Black has no financial disclosures.
Teen birth rates continue to decline in the United States
The teen birth rate of 22.3/1,000 females aged 15-19 years for 2015 was down by almost 8% from the year before and marks the seventh consecutive year of historic lows. Since 1991, when 61.8/1,000 teens aged 15-19 gave birth, the rate has fallen 64%, the NCHS reported.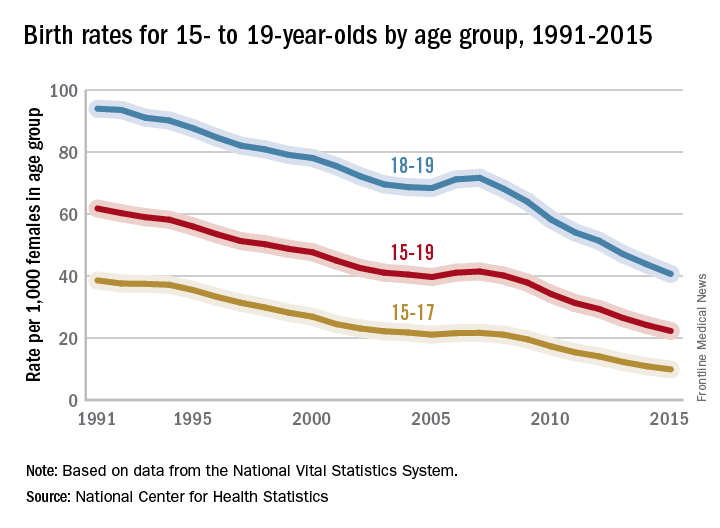
For teens aged 15-19 years, the birth rate declined for each race/ethnicity: dropping 8% for non-Hispanic whites and Hispanics, 9% for non-Hispanic blacks, 10% for Asians or Pacific Islanders, and 6% for American Indians or Alaska Natives. All rates for 2015 were historically low.
The teen birth rate of 22.3/1,000 females aged 15-19 years for 2015 was down by almost 8% from the year before and marks the seventh consecutive year of historic lows. Since 1991, when 61.8/1,000 teens aged 15-19 gave birth, the rate has fallen 64%, the NCHS reported.
For teens aged 15-19 years, the birth rate declined for each race/ethnicity: dropping 8% for non-Hispanic whites and Hispanics, 9% for non-Hispanic blacks, 10% for Asians or Pacific Islanders, and 6% for American Indians or Alaska Natives. All rates for 2015 were historically low.
The teen birth rate of 22.3/1,000 females aged 15-19 years for 2015 was down by almost 8% from the year before and marks the seventh consecutive year of historic lows. Since 1991, when 61.8/1,000 teens aged 15-19 gave birth, the rate has fallen 64%, the NCHS reported.
For teens aged 15-19 years, the birth rate declined for each race/ethnicity: dropping 8% for non-Hispanic whites and Hispanics, 9% for non-Hispanic blacks, 10% for Asians or Pacific Islanders, and 6% for American Indians or Alaska Natives. All rates for 2015 were historically low.
Fulminant HBV reactivation associated with HCV treatment
Sudden and fulminant reactivation of hepatitis B virus (HBV) infections are occurring among some patients who received direct-acting antiviral (DAA) medicines for concomitant chronic hepatitis C virus, the U.S. Food and Drug Administration has said.
HBV reactivation has been reported in 24 patients since 2013, the agency said in an Oct. 4 statement. One patient died, and one required a liver transplant, likely because of treatment delay, as HBV reactivation wasn’t a primary diagnostic candidate.
“The mechanism through which HBV reactivation occurs with DAAs is currently unknown. These medicines are not known to cause immunosuppression, but HBV reactivation may result from a complex interplay of host immunologic responses in the setting of infection with two hepatitis viruses.”
In response to the findings, the FDA will require a black box warning on all DAA medications. Before prescribing the drugs, clinicians should screen patients for evidence of current or prior HBV infection. Patients with evidence of current or prior HBV infection should be monitored for HBV surface antigen and HBV DNA, as well as serum aminotransferase bilirubin levels, and watched for signs of hepatitis flare or HBV reactivation during and after DAA treatment. Suspected cases should be reported to FDA MedWatch.
The reactivations occurred within 4-8 weeks of beginning a DAA, the FDA said. “A common sequence of events was initiation of DAA-based HCV treatment, rapid drop of HCV RNA to undetectable levels within 1-2 weeks after normalization of transaminase levels (if they were elevated), followed by a rise in HBV DNA with or without increase in transaminases between weeks 4 and 8.”
Half of the patients did eventually receive HBV antiviral treatment (tenofovir or entecavir). Treatment data were absent on six patients. The remaining six patients did not receive HBV treatment, for unclear reasons.
In eight cases, the initial transaminase increase was interpreted as a DAA drug reaction and the medicine was discontinued. These patients either failed to improve or deteriorated, prompting concerns about HBV reactivation. FDA couldn’t find any commonalities in the cases.
“The patients who developed HBV reactivation were heterogeneous in terms of HCV genotype. These patients were also heterogeneous in terms of baseline HBV disease, fitting into three general categories of patients: those with detectable HBV viral load (seven), those with positive HB surface antigen and undetectable HBV viral load (four), and those with negative HB surface antigen and undetectable HBV viral load (three).”
For the remaining 10 patients, HB surface antigen status was either not known or baseline HBV could not be interpreted.
Sudden and fulminant reactivation of hepatitis B virus (HBV) infections are occurring among some patients who received direct-acting antiviral (DAA) medicines for concomitant chronic hepatitis C virus, the U.S. Food and Drug Administration has said.
HBV reactivation has been reported in 24 patients since 2013, the agency said in an Oct. 4 statement. One patient died, and one required a liver transplant, likely because of treatment delay, as HBV reactivation wasn’t a primary diagnostic candidate.
“The mechanism through which HBV reactivation occurs with DAAs is currently unknown. These medicines are not known to cause immunosuppression, but HBV reactivation may result from a complex interplay of host immunologic responses in the setting of infection with two hepatitis viruses.”
In response to the findings, the FDA will require a black box warning on all DAA medications. Before prescribing the drugs, clinicians should screen patients for evidence of current or prior HBV infection. Patients with evidence of current or prior HBV infection should be monitored for HBV surface antigen and HBV DNA, as well as serum aminotransferase bilirubin levels, and watched for signs of hepatitis flare or HBV reactivation during and after DAA treatment. Suspected cases should be reported to FDA MedWatch.
The reactivations occurred within 4-8 weeks of beginning a DAA, the FDA said. “A common sequence of events was initiation of DAA-based HCV treatment, rapid drop of HCV RNA to undetectable levels within 1-2 weeks after normalization of transaminase levels (if they were elevated), followed by a rise in HBV DNA with or without increase in transaminases between weeks 4 and 8.”
Half of the patients did eventually receive HBV antiviral treatment (tenofovir or entecavir). Treatment data were absent on six patients. The remaining six patients did not receive HBV treatment, for unclear reasons.
In eight cases, the initial transaminase increase was interpreted as a DAA drug reaction and the medicine was discontinued. These patients either failed to improve or deteriorated, prompting concerns about HBV reactivation. FDA couldn’t find any commonalities in the cases.
“The patients who developed HBV reactivation were heterogeneous in terms of HCV genotype. These patients were also heterogeneous in terms of baseline HBV disease, fitting into three general categories of patients: those with detectable HBV viral load (seven), those with positive HB surface antigen and undetectable HBV viral load (four), and those with negative HB surface antigen and undetectable HBV viral load (three).”
For the remaining 10 patients, HB surface antigen status was either not known or baseline HBV could not be interpreted.
Sudden and fulminant reactivation of hepatitis B virus (HBV) infections are occurring among some patients who received direct-acting antiviral (DAA) medicines for concomitant chronic hepatitis C virus, the U.S. Food and Drug Administration has said.
HBV reactivation has been reported in 24 patients since 2013, the agency said in an Oct. 4 statement. One patient died, and one required a liver transplant, likely because of treatment delay, as HBV reactivation wasn’t a primary diagnostic candidate.
“The mechanism through which HBV reactivation occurs with DAAs is currently unknown. These medicines are not known to cause immunosuppression, but HBV reactivation may result from a complex interplay of host immunologic responses in the setting of infection with two hepatitis viruses.”
In response to the findings, the FDA will require a black box warning on all DAA medications. Before prescribing the drugs, clinicians should screen patients for evidence of current or prior HBV infection. Patients with evidence of current or prior HBV infection should be monitored for HBV surface antigen and HBV DNA, as well as serum aminotransferase bilirubin levels, and watched for signs of hepatitis flare or HBV reactivation during and after DAA treatment. Suspected cases should be reported to FDA MedWatch.
The reactivations occurred within 4-8 weeks of beginning a DAA, the FDA said. “A common sequence of events was initiation of DAA-based HCV treatment, rapid drop of HCV RNA to undetectable levels within 1-2 weeks after normalization of transaminase levels (if they were elevated), followed by a rise in HBV DNA with or without increase in transaminases between weeks 4 and 8.”
Half of the patients did eventually receive HBV antiviral treatment (tenofovir or entecavir). Treatment data were absent on six patients. The remaining six patients did not receive HBV treatment, for unclear reasons.
In eight cases, the initial transaminase increase was interpreted as a DAA drug reaction and the medicine was discontinued. These patients either failed to improve or deteriorated, prompting concerns about HBV reactivation. FDA couldn’t find any commonalities in the cases.
“The patients who developed HBV reactivation were heterogeneous in terms of HCV genotype. These patients were also heterogeneous in terms of baseline HBV disease, fitting into three general categories of patients: those with detectable HBV viral load (seven), those with positive HB surface antigen and undetectable HBV viral load (four), and those with negative HB surface antigen and undetectable HBV viral load (three).”
For the remaining 10 patients, HB surface antigen status was either not known or baseline HBV could not be interpreted.
Key clinical point:
Major finding: HBV reactivation has been reported in 24 patients since 2013.
Data source: Reports were made to the FDA MedWatch system.
Disclosures: No conflicts of interest were reported.
FDA provides clearance for new colonoscope system
The U.S Food and Drug Administration provided 510(k) clearance for the new Aer-O-Scope colonoscope system, according to GI View of Ramat Gan, Israel.
The Aer-O-Scope is a self-propelled, disposable colonoscope with a 360-degree view of the colon that is controlled with a joystick.

The new system, which is an improvement on the earlier Aer-O-Scope colonoscope, also FDA approved, will have two working channels in order to provide therapeutic access for standard tools to take biopsies or perform polypectomies. It uses a soft multilumen tube designed to significantly reduce pressure on the colon wall, which may increase patient safety. It is also hydrophilic and reduces the friction between bowel and scope by more than 90%. The system has a self-propelled intubation process, created with balloons and low pressure CO2 gas. And, like all colonoscopes, the Aer-O-Scope provides insufflation, irrigation, and suction.
“The new Aer-O-Scope system with therapeutic access has many significant clinical benefits including enabling physicians to more easily, effectively, and efficiently identify and remove polyps and prevent colon cancer,” said GI View CEO Tal Simchony, PhD, in a press release. “We are now working on U.S. market introduction of the Aer-O-Scope. Postmarket studies are also in the plans.”
Colorectal cancer is one of the biggest causes of cancer death worldwide. It can be prevented by early detection and removal of polyps. Read more about Aer-O-Scope on GI View’s website.
The U.S Food and Drug Administration provided 510(k) clearance for the new Aer-O-Scope colonoscope system, according to GI View of Ramat Gan, Israel.
The Aer-O-Scope is a self-propelled, disposable colonoscope with a 360-degree view of the colon that is controlled with a joystick.
The new system, which is an improvement on the earlier Aer-O-Scope colonoscope, also FDA approved, will have two working channels in order to provide therapeutic access for standard tools to take biopsies or perform polypectomies. It uses a soft multilumen tube designed to significantly reduce pressure on the colon wall, which may increase patient safety. It is also hydrophilic and reduces the friction between bowel and scope by more than 90%. The system has a self-propelled intubation process, created with balloons and low pressure CO2 gas. And, like all colonoscopes, the Aer-O-Scope provides insufflation, irrigation, and suction.
“The new Aer-O-Scope system with therapeutic access has many significant clinical benefits including enabling physicians to more easily, effectively, and efficiently identify and remove polyps and prevent colon cancer,” said GI View CEO Tal Simchony, PhD, in a press release. “We are now working on U.S. market introduction of the Aer-O-Scope. Postmarket studies are also in the plans.”
Colorectal cancer is one of the biggest causes of cancer death worldwide. It can be prevented by early detection and removal of polyps. Read more about Aer-O-Scope on GI View’s website.
The U.S Food and Drug Administration provided 510(k) clearance for the new Aer-O-Scope colonoscope system, according to GI View of Ramat Gan, Israel.
The Aer-O-Scope is a self-propelled, disposable colonoscope with a 360-degree view of the colon that is controlled with a joystick.
The new system, which is an improvement on the earlier Aer-O-Scope colonoscope, also FDA approved, will have two working channels in order to provide therapeutic access for standard tools to take biopsies or perform polypectomies. It uses a soft multilumen tube designed to significantly reduce pressure on the colon wall, which may increase patient safety. It is also hydrophilic and reduces the friction between bowel and scope by more than 90%. The system has a self-propelled intubation process, created with balloons and low pressure CO2 gas. And, like all colonoscopes, the Aer-O-Scope provides insufflation, irrigation, and suction.
“The new Aer-O-Scope system with therapeutic access has many significant clinical benefits including enabling physicians to more easily, effectively, and efficiently identify and remove polyps and prevent colon cancer,” said GI View CEO Tal Simchony, PhD, in a press release. “We are now working on U.S. market introduction of the Aer-O-Scope. Postmarket studies are also in the plans.”
Colorectal cancer is one of the biggest causes of cancer death worldwide. It can be prevented by early detection and removal of polyps. Read more about Aer-O-Scope on GI View’s website.

FDA approves biosimilar adalimumab
There are “no clinically meaningful differences” between Amgen’s biosimilar adalimumab (Amjevita) and AbbVie’s branded product Humira, the Food and Drug Administration noted it its Sept. 23 announcement of Amjevita’s approval.
Although Amjevita (adalimumab-atto) is expected to cost less than Humira, Amgen has not released price information or a launch date pending ongoing litigation with AbbVie over intellectual property rights, an Amgen spokeswoman said.
The products carry an identical black box warning of tuberculosis and other serious infections, as well as lymphoma and other malignancies “reported in children and adolescent patients treated with [tumor necrosis factor] blockers including adalimumab.” As with Humira, “the most common expected adverse reactions with Amjevita are infections and injection site reactions,” the FDA said. Both products are approved in 20 mg/0.4 mL and 40 mg/0.8 mL prefilled injections, but Humira also has a 10 mg/0.2 mL option.
Amjevita was unanimously recommended for approval by an FDA review panel in July. Although “the biosimilar pathway is still a new frontier,” it’s likely to “enhance access to treatment for patients with serious medical conditions,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in the agency statement.
The approval follows the FDA’s approval of biosimilar infliximab (Inflectra) in April 2016 and biosimilar etanercept (Erelzi) in August 2016. Inflectra has not hit the U.S. market yet, but the European experience with biosimilar infliximab – generally positive – may give an indication of how Amjevita will fare in the United States. It’s perhaps a third or more less expensive than the original product (Remicade) and often used for new starts. There is uncertainty, however, about switching patients already established on Remicade, especially when it’s forced by cost issues.
Interchangeability is a concern in the United States as well. The FDA is working on the issue but has not yet released guidance, and the agency was careful to note in its statement that Amjevita was “approved as a biosimilar, not as an interchangeable product.” Biosimilar adalimumab, meanwhile, is under review in Europe, according to an Amgen statement.
The FDA approved Amjevita after reviewing structural and functional characteristics, pharmacokinetics and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrated similarity to Humira, including two phase III trials for plaque psoriasis and rheumatoid arthritis.
An AbbVie spokesperson said the company “anticipated Amgen’s product would be approved,” but noted the ongoing litigation.
There are “no clinically meaningful differences” between Amgen’s biosimilar adalimumab (Amjevita) and AbbVie’s branded product Humira, the Food and Drug Administration noted it its Sept. 23 announcement of Amjevita’s approval.
Although Amjevita (adalimumab-atto) is expected to cost less than Humira, Amgen has not released price information or a launch date pending ongoing litigation with AbbVie over intellectual property rights, an Amgen spokeswoman said.
The products carry an identical black box warning of tuberculosis and other serious infections, as well as lymphoma and other malignancies “reported in children and adolescent patients treated with [tumor necrosis factor] blockers including adalimumab.” As with Humira, “the most common expected adverse reactions with Amjevita are infections and injection site reactions,” the FDA said. Both products are approved in 20 mg/0.4 mL and 40 mg/0.8 mL prefilled injections, but Humira also has a 10 mg/0.2 mL option.
Amjevita was unanimously recommended for approval by an FDA review panel in July. Although “the biosimilar pathway is still a new frontier,” it’s likely to “enhance access to treatment for patients with serious medical conditions,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in the agency statement.
The approval follows the FDA’s approval of biosimilar infliximab (Inflectra) in April 2016 and biosimilar etanercept (Erelzi) in August 2016. Inflectra has not hit the U.S. market yet, but the European experience with biosimilar infliximab – generally positive – may give an indication of how Amjevita will fare in the United States. It’s perhaps a third or more less expensive than the original product (Remicade) and often used for new starts. There is uncertainty, however, about switching patients already established on Remicade, especially when it’s forced by cost issues.
Interchangeability is a concern in the United States as well. The FDA is working on the issue but has not yet released guidance, and the agency was careful to note in its statement that Amjevita was “approved as a biosimilar, not as an interchangeable product.” Biosimilar adalimumab, meanwhile, is under review in Europe, according to an Amgen statement.
The FDA approved Amjevita after reviewing structural and functional characteristics, pharmacokinetics and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrated similarity to Humira, including two phase III trials for plaque psoriasis and rheumatoid arthritis.
An AbbVie spokesperson said the company “anticipated Amgen’s product would be approved,” but noted the ongoing litigation.
There are “no clinically meaningful differences” between Amgen’s biosimilar adalimumab (Amjevita) and AbbVie’s branded product Humira, the Food and Drug Administration noted it its Sept. 23 announcement of Amjevita’s approval.
Although Amjevita (adalimumab-atto) is expected to cost less than Humira, Amgen has not released price information or a launch date pending ongoing litigation with AbbVie over intellectual property rights, an Amgen spokeswoman said.
The products carry an identical black box warning of tuberculosis and other serious infections, as well as lymphoma and other malignancies “reported in children and adolescent patients treated with [tumor necrosis factor] blockers including adalimumab.” As with Humira, “the most common expected adverse reactions with Amjevita are infections and injection site reactions,” the FDA said. Both products are approved in 20 mg/0.4 mL and 40 mg/0.8 mL prefilled injections, but Humira also has a 10 mg/0.2 mL option.
Amjevita was unanimously recommended for approval by an FDA review panel in July. Although “the biosimilar pathway is still a new frontier,” it’s likely to “enhance access to treatment for patients with serious medical conditions,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in the agency statement.
The approval follows the FDA’s approval of biosimilar infliximab (Inflectra) in April 2016 and biosimilar etanercept (Erelzi) in August 2016. Inflectra has not hit the U.S. market yet, but the European experience with biosimilar infliximab – generally positive – may give an indication of how Amjevita will fare in the United States. It’s perhaps a third or more less expensive than the original product (Remicade) and often used for new starts. There is uncertainty, however, about switching patients already established on Remicade, especially when it’s forced by cost issues.
Interchangeability is a concern in the United States as well. The FDA is working on the issue but has not yet released guidance, and the agency was careful to note in its statement that Amjevita was “approved as a biosimilar, not as an interchangeable product.” Biosimilar adalimumab, meanwhile, is under review in Europe, according to an Amgen statement.
The FDA approved Amjevita after reviewing structural and functional characteristics, pharmacokinetics and pharmacodynamics data, clinical immunogenicity data, and other clinical safety and effectiveness data that demonstrated similarity to Humira, including two phase III trials for plaque psoriasis and rheumatoid arthritis.
An AbbVie spokesperson said the company “anticipated Amgen’s product would be approved,” but noted the ongoing litigation.
FDA approves ustekinumab (Stelara) for adult Crohn’s disease
The Food and Drug Administration has approved ustekinumab (Stelara) for the treatment of moderately to severely active Crohn’s disease in certain adults.
Specifically, the new approval for the human monoclonal antibody, which was previously approved for the treatment of psoriasis and psoriatic arthritis, is for Crohn’s disease patients aged 18 years or older who fail or cannot tolerate treatment with immunomodulators or corticosteroids but who never failed treatment with a tumor necrosis factor (TNF) blocker, or who fail or cannot tolerate treatment with one or more TNF blockers, according to a statement from the drug’s maker, Janssen Biotech.

The drug is the first biologic agent to target interleukin (IL)-12 and IL-23 in Crohn’s disease, and its approval was based on findings in more than 1,300 patients across three Janssen Biotech–sponsored phase III studies (UNITI-1, UNITI-2, IM-UNITI) demonstrating its efficacy with respect to clinical response and remission rates.
“Crohn’s disease is a complex condition to treat, and not all therapies work for every patient. The FDA approval of Stelara represents an important advancement in treating patients with Crohn’s disease, as this therapy offers an alternate mechanism of action to induce and maintain clinical remission over time,” study investigator William J. Sandborn, MD, of the University of California, San Diego, said in the statement.
The Food and Drug Administration has approved ustekinumab (Stelara) for the treatment of moderately to severely active Crohn’s disease in certain adults.
Specifically, the new approval for the human monoclonal antibody, which was previously approved for the treatment of psoriasis and psoriatic arthritis, is for Crohn’s disease patients aged 18 years or older who fail or cannot tolerate treatment with immunomodulators or corticosteroids but who never failed treatment with a tumor necrosis factor (TNF) blocker, or who fail or cannot tolerate treatment with one or more TNF blockers, according to a statement from the drug’s maker, Janssen Biotech.
The drug is the first biologic agent to target interleukin (IL)-12 and IL-23 in Crohn’s disease, and its approval was based on findings in more than 1,300 patients across three Janssen Biotech–sponsored phase III studies (UNITI-1, UNITI-2, IM-UNITI) demonstrating its efficacy with respect to clinical response and remission rates.
“Crohn’s disease is a complex condition to treat, and not all therapies work for every patient. The FDA approval of Stelara represents an important advancement in treating patients with Crohn’s disease, as this therapy offers an alternate mechanism of action to induce and maintain clinical remission over time,” study investigator William J. Sandborn, MD, of the University of California, San Diego, said in the statement.
The Food and Drug Administration has approved ustekinumab (Stelara) for the treatment of moderately to severely active Crohn’s disease in certain adults.
Specifically, the new approval for the human monoclonal antibody, which was previously approved for the treatment of psoriasis and psoriatic arthritis, is for Crohn’s disease patients aged 18 years or older who fail or cannot tolerate treatment with immunomodulators or corticosteroids but who never failed treatment with a tumor necrosis factor (TNF) blocker, or who fail or cannot tolerate treatment with one or more TNF blockers, according to a statement from the drug’s maker, Janssen Biotech.
The drug is the first biologic agent to target interleukin (IL)-12 and IL-23 in Crohn’s disease, and its approval was based on findings in more than 1,300 patients across three Janssen Biotech–sponsored phase III studies (UNITI-1, UNITI-2, IM-UNITI) demonstrating its efficacy with respect to clinical response and remission rates.
“Crohn’s disease is a complex condition to treat, and not all therapies work for every patient. The FDA approval of Stelara represents an important advancement in treating patients with Crohn’s disease, as this therapy offers an alternate mechanism of action to induce and maintain clinical remission over time,” study investigator William J. Sandborn, MD, of the University of California, San Diego, said in the statement.

FDA gives orphan drug designation to BIV201 for ascites treatment
The Food and Drug Administration has given an orphan drug designation to the compound BIV201 for the treatment of ascites, according to a press release from the drug’s manufacturer, BioVie.
The FDA designation for BIV201 is for the treatment of ascites due to all etiologies except cancer. Clinical trials could commence by 2017, if accepted by the FDA. If trials are successful, other applications for BIV201 could be tested. BIV201 is a vasoconstricter, and could also be used to treat diseases like type 1 hepatorenal syndrome, esophageal variceal bleeds, and sepsis.
![]()
Ascites, a common complication of liver cirrhosis, has no specific approved treatment, and 40% of patients diagnosed with ascites die within 2 years. Other treatments may work initially, but become ineffective as the patient worsens. Treatment costs in the United States for liver cirrhosis and related complications, including ascites, totals over $4 billion.
“Orphan drug designation represents a major milestone supporting the clinical development and eventual commercialization of BIV201 therapy. It recognizes the importance of pioneering a new therapeutic approach for this relatively small group of desperately ill patients,” Jonathan Adams, BioVie CEO, said in the press release.
Find the full press release on the BioVie website.
The Food and Drug Administration has given an orphan drug designation to the compound BIV201 for the treatment of ascites, according to a press release from the drug’s manufacturer, BioVie.
The FDA designation for BIV201 is for the treatment of ascites due to all etiologies except cancer. Clinical trials could commence by 2017, if accepted by the FDA. If trials are successful, other applications for BIV201 could be tested. BIV201 is a vasoconstricter, and could also be used to treat diseases like type 1 hepatorenal syndrome, esophageal variceal bleeds, and sepsis.
![]()
Ascites, a common complication of liver cirrhosis, has no specific approved treatment, and 40% of patients diagnosed with ascites die within 2 years. Other treatments may work initially, but become ineffective as the patient worsens. Treatment costs in the United States for liver cirrhosis and related complications, including ascites, totals over $4 billion.
“Orphan drug designation represents a major milestone supporting the clinical development and eventual commercialization of BIV201 therapy. It recognizes the importance of pioneering a new therapeutic approach for this relatively small group of desperately ill patients,” Jonathan Adams, BioVie CEO, said in the press release.
Find the full press release on the BioVie website.
The Food and Drug Administration has given an orphan drug designation to the compound BIV201 for the treatment of ascites, according to a press release from the drug’s manufacturer, BioVie.
The FDA designation for BIV201 is for the treatment of ascites due to all etiologies except cancer. Clinical trials could commence by 2017, if accepted by the FDA. If trials are successful, other applications for BIV201 could be tested. BIV201 is a vasoconstricter, and could also be used to treat diseases like type 1 hepatorenal syndrome, esophageal variceal bleeds, and sepsis.
![]()
Ascites, a common complication of liver cirrhosis, has no specific approved treatment, and 40% of patients diagnosed with ascites die within 2 years. Other treatments may work initially, but become ineffective as the patient worsens. Treatment costs in the United States for liver cirrhosis and related complications, including ascites, totals over $4 billion.
“Orphan drug designation represents a major milestone supporting the clinical development and eventual commercialization of BIV201 therapy. It recognizes the importance of pioneering a new therapeutic approach for this relatively small group of desperately ill patients,” Jonathan Adams, BioVie CEO, said in the press release.
Find the full press release on the BioVie website.
FDA approves first-in-class oral antiplatelet drug vorapaxar
The novel antiplatelet drug vorapaxar has been approved for reducing the risk of myocardial infarction, stroke, cardiovascular death, and need for coronary revascularization procedures in patients with a previous MI or peripheral arterial disease, the Food and Drug Administration announced on May 8.
Vorapaxar is an antagonist of protease-activated receptor-1 (PAR-1), which inhibits the action of thrombin on the platelet, and is the first drug in this class to be approved. Merck Sharp & Dohme Corp. will market the drug as Zontivity. It comes in a tablet formulation.
![]()
At a meeting in January, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 10-1 to recommend approval of vorapaxar.
"In patients who have had a heart attack or who have peripheral arterial disease, this drug will lower the risk of heart attack, stroke, and cardiovascular death," Dr. Ellis F. Unger, director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. "In the study that supported the drug’s approval, Zontivity lowered this risk from 9.5% to 7.9% over a 3-year period – about 0.5% per year," he noted.
Because of the increased risk of bleeding, including life-threatening and fatal bleeding, the drug’s prescribing information includes a boxed warning about this risk and a medication guide informing patients about these risks and how to use the drug will be provided with each dispensed prescription. A history of stroke, or transient ischemic attack, and a history of intracranial bleeding are contraindications.
The novel antiplatelet drug vorapaxar has been approved for reducing the risk of myocardial infarction, stroke, cardiovascular death, and need for coronary revascularization procedures in patients with a previous MI or peripheral arterial disease, the Food and Drug Administration announced on May 8.
Vorapaxar is an antagonist of protease-activated receptor-1 (PAR-1), which inhibits the action of thrombin on the platelet, and is the first drug in this class to be approved. Merck Sharp & Dohme Corp. will market the drug as Zontivity. It comes in a tablet formulation.
![]()
At a meeting in January, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 10-1 to recommend approval of vorapaxar.
"In patients who have had a heart attack or who have peripheral arterial disease, this drug will lower the risk of heart attack, stroke, and cardiovascular death," Dr. Ellis F. Unger, director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. "In the study that supported the drug’s approval, Zontivity lowered this risk from 9.5% to 7.9% over a 3-year period – about 0.5% per year," he noted.
Because of the increased risk of bleeding, including life-threatening and fatal bleeding, the drug’s prescribing information includes a boxed warning about this risk and a medication guide informing patients about these risks and how to use the drug will be provided with each dispensed prescription. A history of stroke, or transient ischemic attack, and a history of intracranial bleeding are contraindications.
The novel antiplatelet drug vorapaxar has been approved for reducing the risk of myocardial infarction, stroke, cardiovascular death, and need for coronary revascularization procedures in patients with a previous MI or peripheral arterial disease, the Food and Drug Administration announced on May 8.
Vorapaxar is an antagonist of protease-activated receptor-1 (PAR-1), which inhibits the action of thrombin on the platelet, and is the first drug in this class to be approved. Merck Sharp & Dohme Corp. will market the drug as Zontivity. It comes in a tablet formulation.
![]()
At a meeting in January, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 10-1 to recommend approval of vorapaxar.
"In patients who have had a heart attack or who have peripheral arterial disease, this drug will lower the risk of heart attack, stroke, and cardiovascular death," Dr. Ellis F. Unger, director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research, said in the FDA statement. "In the study that supported the drug’s approval, Zontivity lowered this risk from 9.5% to 7.9% over a 3-year period – about 0.5% per year," he noted.
Because of the increased risk of bleeding, including life-threatening and fatal bleeding, the drug’s prescribing information includes a boxed warning about this risk and a medication guide informing patients about these risks and how to use the drug will be provided with each dispensed prescription. A history of stroke, or transient ischemic attack, and a history of intracranial bleeding are contraindications.