User login
Teens’ overall tobacco use falls, but e-cigs now most popular product
Twenty percent of surveyed high school students and 7% of middle school students were using some kind of tobacco product in 2016 – and e-cigarettes were the most commonly used product among those groups, according to an analysis from federal researchers.
, although combustible tobacco product use declined in both groups, said Ahmed Jamal, MD, of the Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates. That was determined using data from the 2011-2016 National Youth Tobacco Surveys, which assessed tobacco use in the past 30 days by groups of middle school and high school students numbering from more than 17,000 to almost 25,000.
High school boys had higher use of any tobacco product, two or more tobacco products, cigars, smokeless tobacco, and pipe tobacco than did high school girls. The most commonly used tobacco product among non-Hispanic white (13.7%) and Hispanic high school students (10%) were e-cigarettes; cigars were the most commonly used tobacco product among non-Hispanic black high school students (9.5%).
Seven percent of middle school students had used any tobacco product within the past 30 days, and 3% had used two or more tobacco products. Similar to high school students, e-cigarettes were the most commonly used tobacco product among middle school students (4.3%), followed by cigarettes (2%), cigars (2%), smokeless tobacco (2%), hookahs (2%), pipe tobacco (0.7%), and bidis (0.3%).
Use of any tobacco product among middle school boys was 8.3%, and among middle school girls use was 6%. Hispanic middle school students reported higher use of any tobacco product, were more likely to use two or more tobacco products, and said they used hookahs more often than did non-Hispanic white middle school students.
Looking at the changes from 2011 to 2016, current use of any tobacco product did not change significantly among high school students (falling from 24% to 20%), but there was a reduction in current use of any combustible tobacco product (22%-14%) and in use of two or more tobacco products (12%-9.6%) over this time period.
By product type, there were increases in current use of e-cigarettes (from 1.5% to 11.3%) and hookahs (from 4.1% to 4.8%) among high school students, as well as decreases in current use of cigarettes (from 16% to 8%), cigars (from 12% to 8%), smokeless tobacco (from 8% to 6%), pipe tobacco (from 4% to 1%), and bidis (from 2% to 0.5%).
Among middle school students during 2011-2016, there was a rise in current use of e-cigarettes (from 0.6% to 4%), and in current use of hookahs (from 1% to 2%). There was a drop in current use of any combustible tobacco products (from 6% to 4%), cigarettes (from 4% to 2%), cigars (from 3.5% to 2.2%), and pipe tobacco (from 2% to 0.7%).
“Since February 2014, the Food and Drug Administration’s first national tobacco public education campaign, the Real Cost, has broadcasted tobacco education advertising designed for youths aged 12-17 years; the campaign was associated with an estimated 348,398 U.S. youths who did not initiate cigarette smoking during February 2014–March 2016,” the investigators said. “Continued implementation of these strategies can help prevent and further reduce the use of all forms of tobacco product among U.S. youths.”
Read more in MMWR (2017 Jun 16;66[23]:597-603).
Twenty percent of surveyed high school students and 7% of middle school students were using some kind of tobacco product in 2016 – and e-cigarettes were the most commonly used product among those groups, according to an analysis from federal researchers.
, although combustible tobacco product use declined in both groups, said Ahmed Jamal, MD, of the Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates. That was determined using data from the 2011-2016 National Youth Tobacco Surveys, which assessed tobacco use in the past 30 days by groups of middle school and high school students numbering from more than 17,000 to almost 25,000.
High school boys had higher use of any tobacco product, two or more tobacco products, cigars, smokeless tobacco, and pipe tobacco than did high school girls. The most commonly used tobacco product among non-Hispanic white (13.7%) and Hispanic high school students (10%) were e-cigarettes; cigars were the most commonly used tobacco product among non-Hispanic black high school students (9.5%).
Seven percent of middle school students had used any tobacco product within the past 30 days, and 3% had used two or more tobacco products. Similar to high school students, e-cigarettes were the most commonly used tobacco product among middle school students (4.3%), followed by cigarettes (2%), cigars (2%), smokeless tobacco (2%), hookahs (2%), pipe tobacco (0.7%), and bidis (0.3%).
Use of any tobacco product among middle school boys was 8.3%, and among middle school girls use was 6%. Hispanic middle school students reported higher use of any tobacco product, were more likely to use two or more tobacco products, and said they used hookahs more often than did non-Hispanic white middle school students.
Looking at the changes from 2011 to 2016, current use of any tobacco product did not change significantly among high school students (falling from 24% to 20%), but there was a reduction in current use of any combustible tobacco product (22%-14%) and in use of two or more tobacco products (12%-9.6%) over this time period.
By product type, there were increases in current use of e-cigarettes (from 1.5% to 11.3%) and hookahs (from 4.1% to 4.8%) among high school students, as well as decreases in current use of cigarettes (from 16% to 8%), cigars (from 12% to 8%), smokeless tobacco (from 8% to 6%), pipe tobacco (from 4% to 1%), and bidis (from 2% to 0.5%).
Among middle school students during 2011-2016, there was a rise in current use of e-cigarettes (from 0.6% to 4%), and in current use of hookahs (from 1% to 2%). There was a drop in current use of any combustible tobacco products (from 6% to 4%), cigarettes (from 4% to 2%), cigars (from 3.5% to 2.2%), and pipe tobacco (from 2% to 0.7%).
“Since February 2014, the Food and Drug Administration’s first national tobacco public education campaign, the Real Cost, has broadcasted tobacco education advertising designed for youths aged 12-17 years; the campaign was associated with an estimated 348,398 U.S. youths who did not initiate cigarette smoking during February 2014–March 2016,” the investigators said. “Continued implementation of these strategies can help prevent and further reduce the use of all forms of tobacco product among U.S. youths.”
Read more in MMWR (2017 Jun 16;66[23]:597-603).
Twenty percent of surveyed high school students and 7% of middle school students were using some kind of tobacco product in 2016 – and e-cigarettes were the most commonly used product among those groups, according to an analysis from federal researchers.
, although combustible tobacco product use declined in both groups, said Ahmed Jamal, MD, of the Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion, Atlanta, and his associates. That was determined using data from the 2011-2016 National Youth Tobacco Surveys, which assessed tobacco use in the past 30 days by groups of middle school and high school students numbering from more than 17,000 to almost 25,000.
High school boys had higher use of any tobacco product, two or more tobacco products, cigars, smokeless tobacco, and pipe tobacco than did high school girls. The most commonly used tobacco product among non-Hispanic white (13.7%) and Hispanic high school students (10%) were e-cigarettes; cigars were the most commonly used tobacco product among non-Hispanic black high school students (9.5%).
Seven percent of middle school students had used any tobacco product within the past 30 days, and 3% had used two or more tobacco products. Similar to high school students, e-cigarettes were the most commonly used tobacco product among middle school students (4.3%), followed by cigarettes (2%), cigars (2%), smokeless tobacco (2%), hookahs (2%), pipe tobacco (0.7%), and bidis (0.3%).
Use of any tobacco product among middle school boys was 8.3%, and among middle school girls use was 6%. Hispanic middle school students reported higher use of any tobacco product, were more likely to use two or more tobacco products, and said they used hookahs more often than did non-Hispanic white middle school students.
Looking at the changes from 2011 to 2016, current use of any tobacco product did not change significantly among high school students (falling from 24% to 20%), but there was a reduction in current use of any combustible tobacco product (22%-14%) and in use of two or more tobacco products (12%-9.6%) over this time period.
By product type, there were increases in current use of e-cigarettes (from 1.5% to 11.3%) and hookahs (from 4.1% to 4.8%) among high school students, as well as decreases in current use of cigarettes (from 16% to 8%), cigars (from 12% to 8%), smokeless tobacco (from 8% to 6%), pipe tobacco (from 4% to 1%), and bidis (from 2% to 0.5%).
Among middle school students during 2011-2016, there was a rise in current use of e-cigarettes (from 0.6% to 4%), and in current use of hookahs (from 1% to 2%). There was a drop in current use of any combustible tobacco products (from 6% to 4%), cigarettes (from 4% to 2%), cigars (from 3.5% to 2.2%), and pipe tobacco (from 2% to 0.7%).
“Since February 2014, the Food and Drug Administration’s first national tobacco public education campaign, the Real Cost, has broadcasted tobacco education advertising designed for youths aged 12-17 years; the campaign was associated with an estimated 348,398 U.S. youths who did not initiate cigarette smoking during February 2014–March 2016,” the investigators said. “Continued implementation of these strategies can help prevent and further reduce the use of all forms of tobacco product among U.S. youths.”
Read more in MMWR (2017 Jun 16;66[23]:597-603).
FROM MMWR
BMS recalls a lot of Eliquis 5 mg tablets
Bristol-Myers Squibb is recalling one lot of Eliquis (apixaban) tablets, according to a company announcement on the Food and Drug Administration website.
The recall is based on a customer complaint that a bottle labeled as containing 5 mg tablets of Eliquis actually contained 2.5 mg tablets of Eliquis. The lot in question was distributed nationwide to wholesalers and retail pharmacies in February 2017. The recall is voluntary, the company noted.
“There are distinct visible differences between the two tablet strengths, including colors, size, and markings that distinguish the 2.5 mg and 5 mg tablets and decrease the likelihood of an incorrect dose,” Bristol-Myers Squibb noted in the press release.
Find the full Bristol-Myers Squibb press release on the FDA website.
Bristol-Myers Squibb is recalling one lot of Eliquis (apixaban) tablets, according to a company announcement on the Food and Drug Administration website.
The recall is based on a customer complaint that a bottle labeled as containing 5 mg tablets of Eliquis actually contained 2.5 mg tablets of Eliquis. The lot in question was distributed nationwide to wholesalers and retail pharmacies in February 2017. The recall is voluntary, the company noted.
“There are distinct visible differences between the two tablet strengths, including colors, size, and markings that distinguish the 2.5 mg and 5 mg tablets and decrease the likelihood of an incorrect dose,” Bristol-Myers Squibb noted in the press release.
Find the full Bristol-Myers Squibb press release on the FDA website.
Bristol-Myers Squibb is recalling one lot of Eliquis (apixaban) tablets, according to a company announcement on the Food and Drug Administration website.
The recall is based on a customer complaint that a bottle labeled as containing 5 mg tablets of Eliquis actually contained 2.5 mg tablets of Eliquis. The lot in question was distributed nationwide to wholesalers and retail pharmacies in February 2017. The recall is voluntary, the company noted.
“There are distinct visible differences between the two tablet strengths, including colors, size, and markings that distinguish the 2.5 mg and 5 mg tablets and decrease the likelihood of an incorrect dose,” Bristol-Myers Squibb noted in the press release.
Find the full Bristol-Myers Squibb press release on the FDA website.
FDA approves generic version of HIV drug Truvada
The U.S. Food and Drug Administration has approved the first generic version of emtricitabine and tenofovir disoproxil fumarate tablets, previously known by the brand name Truvada, for the treatment of HIV-1 infection.
Like Truvada, the generic version of the drug – produced by Teva Pharmaceuticals USA and approved in 200-mg/300-mg tablet form – is indicated for use in combination with other antiretrovirals for patients infected with HIV-1 and for pre-exposure prophylaxis (PrEP) to prevent sexually-acquired HIV infection in high-risk adults.
Women infected with HIV-1 should not breastfeed while taking emtricitabine and tenofovir disoproxil fumarate, the FDA said, and the drug can be used only in pediatric patients weighing more than 17 kg.
rpizzi@frontlinemedcom.com
On Twitter @richpizzi
The U.S. Food and Drug Administration has approved the first generic version of emtricitabine and tenofovir disoproxil fumarate tablets, previously known by the brand name Truvada, for the treatment of HIV-1 infection.
Like Truvada, the generic version of the drug – produced by Teva Pharmaceuticals USA and approved in 200-mg/300-mg tablet form – is indicated for use in combination with other antiretrovirals for patients infected with HIV-1 and for pre-exposure prophylaxis (PrEP) to prevent sexually-acquired HIV infection in high-risk adults.
Women infected with HIV-1 should not breastfeed while taking emtricitabine and tenofovir disoproxil fumarate, the FDA said, and the drug can be used only in pediatric patients weighing more than 17 kg.
rpizzi@frontlinemedcom.com
On Twitter @richpizzi
The U.S. Food and Drug Administration has approved the first generic version of emtricitabine and tenofovir disoproxil fumarate tablets, previously known by the brand name Truvada, for the treatment of HIV-1 infection.
Like Truvada, the generic version of the drug – produced by Teva Pharmaceuticals USA and approved in 200-mg/300-mg tablet form – is indicated for use in combination with other antiretrovirals for patients infected with HIV-1 and for pre-exposure prophylaxis (PrEP) to prevent sexually-acquired HIV infection in high-risk adults.
Women infected with HIV-1 should not breastfeed while taking emtricitabine and tenofovir disoproxil fumarate, the FDA said, and the drug can be used only in pediatric patients weighing more than 17 kg.
rpizzi@frontlinemedcom.com
On Twitter @richpizzi
Extended-release Aristada OK’d for 2-month dosing for schizophrenia
The Food and Drug Administration has expanded labeling of extended-release aripiprazole lauroxil (Aristada) to allow for administration once every 2 months at a dose of 1,064 mg, for schizophrenia.
Depending on patient needs, treatment now can be initiated at a dose of 441 mg, 662 mg, or 882 mg administered monthly; 882 mg administered every 6 weeks; or 1,064 mg administered every 2 months. For patients who have never taken aripiprazole, “establish tolerability with oral aripiprazole prior to initiating treatment with Aristada. Due to the half-life of oral aripiprazole, it may take up to 2 weeks to fully assess tolerability,” according to the updated labeling. The new 2-month option is expected to be available in mid-June.
Another extended-release IM aripiprazole option, (Abilify Maintena, Otsuka) is approved only for once-a-month injection.![]()
Approval of the bimonthly dose was based on pharmacokinetic data showing long-term therapeutic aripiprazole plasma levels. Labeling did not note whether the higher dose is associated with increased adverse events. Aristada originally was approved in 2015.
As with other atypical antipsychotics, Aristada labeling notes that the drug should not be used in elderly patients with dementia because of the increased risk of fatal strokes. The labeling also warns of the possibility of tardive dyskinesia, metabolic derangements, compulsive behaviors, agranulocytosis, and other risks.
The Food and Drug Administration has expanded labeling of extended-release aripiprazole lauroxil (Aristada) to allow for administration once every 2 months at a dose of 1,064 mg, for schizophrenia.
Depending on patient needs, treatment now can be initiated at a dose of 441 mg, 662 mg, or 882 mg administered monthly; 882 mg administered every 6 weeks; or 1,064 mg administered every 2 months. For patients who have never taken aripiprazole, “establish tolerability with oral aripiprazole prior to initiating treatment with Aristada. Due to the half-life of oral aripiprazole, it may take up to 2 weeks to fully assess tolerability,” according to the updated labeling. The new 2-month option is expected to be available in mid-June.
Another extended-release IM aripiprazole option, (Abilify Maintena, Otsuka) is approved only for once-a-month injection.![]()
Approval of the bimonthly dose was based on pharmacokinetic data showing long-term therapeutic aripiprazole plasma levels. Labeling did not note whether the higher dose is associated with increased adverse events. Aristada originally was approved in 2015.
As with other atypical antipsychotics, Aristada labeling notes that the drug should not be used in elderly patients with dementia because of the increased risk of fatal strokes. The labeling also warns of the possibility of tardive dyskinesia, metabolic derangements, compulsive behaviors, agranulocytosis, and other risks.
The Food and Drug Administration has expanded labeling of extended-release aripiprazole lauroxil (Aristada) to allow for administration once every 2 months at a dose of 1,064 mg, for schizophrenia.
Depending on patient needs, treatment now can be initiated at a dose of 441 mg, 662 mg, or 882 mg administered monthly; 882 mg administered every 6 weeks; or 1,064 mg administered every 2 months. For patients who have never taken aripiprazole, “establish tolerability with oral aripiprazole prior to initiating treatment with Aristada. Due to the half-life of oral aripiprazole, it may take up to 2 weeks to fully assess tolerability,” according to the updated labeling. The new 2-month option is expected to be available in mid-June.
Another extended-release IM aripiprazole option, (Abilify Maintena, Otsuka) is approved only for once-a-month injection.![]()
Approval of the bimonthly dose was based on pharmacokinetic data showing long-term therapeutic aripiprazole plasma levels. Labeling did not note whether the higher dose is associated with increased adverse events. Aristada originally was approved in 2015.
As with other atypical antipsychotics, Aristada labeling notes that the drug should not be used in elderly patients with dementia because of the increased risk of fatal strokes. The labeling also warns of the possibility of tardive dyskinesia, metabolic derangements, compulsive behaviors, agranulocytosis, and other risks.
Infant mortality down in most states
Infant mortality in the United States was down by 15% from 2005 to 2014, with 33 states reporting significant declines, according to the National Center for Health Statistics.
The overall rate for 2014 was 5.82 infant deaths per 1,000 live births, compared with 6.84 per 1,000 in 2005. The data for individual states were grouped into 3-year periods, so between the periods of 2005-2007 and 2012-2014, there were 33 states (and the District of Columbia) with a significant decline and 17 states with no significant change. Three states – Maine, South Dakota, and Utah – had increased infant mortality, but the changes did not reach significance, the NCHS reported, using data from the National Vital Statistics System.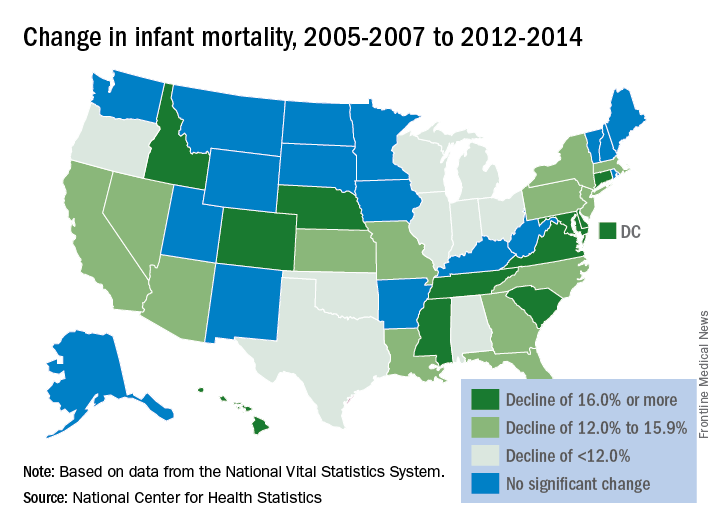
Infant mortality in the United States was down by 15% from 2005 to 2014, with 33 states reporting significant declines, according to the National Center for Health Statistics.
The overall rate for 2014 was 5.82 infant deaths per 1,000 live births, compared with 6.84 per 1,000 in 2005. The data for individual states were grouped into 3-year periods, so between the periods of 2005-2007 and 2012-2014, there were 33 states (and the District of Columbia) with a significant decline and 17 states with no significant change. Three states – Maine, South Dakota, and Utah – had increased infant mortality, but the changes did not reach significance, the NCHS reported, using data from the National Vital Statistics System.
Infant mortality in the United States was down by 15% from 2005 to 2014, with 33 states reporting significant declines, according to the National Center for Health Statistics.
The overall rate for 2014 was 5.82 infant deaths per 1,000 live births, compared with 6.84 per 1,000 in 2005. The data for individual states were grouped into 3-year periods, so between the periods of 2005-2007 and 2012-2014, there were 33 states (and the District of Columbia) with a significant decline and 17 states with no significant change. Three states – Maine, South Dakota, and Utah – had increased infant mortality, but the changes did not reach significance, the NCHS reported, using data from the National Vital Statistics System.
FDA asks drug maker to shelve Opana ER
The Food and Drug Administration has asked Endo Pharmaceuticals to voluntarily remove its opioid pain medication, reformulated Opana ER (oxymorphone hydrochloride), from the market in the United States, citing the potential for its abuse as a concern.
“We are facing an opioid epidemic – a public health crisis, and we must take all necessary steps to reduce the scope of opioid misuse and abuse,” FDA Commissioner Scott Gottlieb, MD, said in a June 8 press release . “We will continue to take regulatory steps when we see situations where an opioid product’s risks outweigh its benefits, not only for its intended patient population but also in regard to its potential for misuse and abuse.”
Opana ER was first approved in 2006 for the management of moderate to severe pain when a continuous, around-the-clock opioid analgesic is needed for an extended period of time. It was reformulated in 2012, with the intent of making it “resistant to physical and chemical manipulation for abuse by snorting or injecting,” according to the FDA release.
The Food and Drug Administration has asked Endo Pharmaceuticals to voluntarily remove its opioid pain medication, reformulated Opana ER (oxymorphone hydrochloride), from the market in the United States, citing the potential for its abuse as a concern.
“We are facing an opioid epidemic – a public health crisis, and we must take all necessary steps to reduce the scope of opioid misuse and abuse,” FDA Commissioner Scott Gottlieb, MD, said in a June 8 press release . “We will continue to take regulatory steps when we see situations where an opioid product’s risks outweigh its benefits, not only for its intended patient population but also in regard to its potential for misuse and abuse.”
Opana ER was first approved in 2006 for the management of moderate to severe pain when a continuous, around-the-clock opioid analgesic is needed for an extended period of time. It was reformulated in 2012, with the intent of making it “resistant to physical and chemical manipulation for abuse by snorting or injecting,” according to the FDA release.
The Food and Drug Administration has asked Endo Pharmaceuticals to voluntarily remove its opioid pain medication, reformulated Opana ER (oxymorphone hydrochloride), from the market in the United States, citing the potential for its abuse as a concern.
“We are facing an opioid epidemic – a public health crisis, and we must take all necessary steps to reduce the scope of opioid misuse and abuse,” FDA Commissioner Scott Gottlieb, MD, said in a June 8 press release . “We will continue to take regulatory steps when we see situations where an opioid product’s risks outweigh its benefits, not only for its intended patient population but also in regard to its potential for misuse and abuse.”
Opana ER was first approved in 2006 for the management of moderate to severe pain when a continuous, around-the-clock opioid analgesic is needed for an extended period of time. It was reformulated in 2012, with the intent of making it “resistant to physical and chemical manipulation for abuse by snorting or injecting,” according to the FDA release.
Zika-related birth defects up in recent weeks
Zika virus infection has been occurring in pregnant women at a slow but steady clip over the last couple of months, but cases of liveborn infants with Zika-related birth defects have jumped in recent weeks, according to the Centers for Disease Control and Prevention.
Eight liveborn infants with Zika-related birth defects were reported to the U.S. Zika Pregnancy Registry during the 2 weeks ending May 23, more than any other 2-week period this year, and that was after six such infants were reported for the 2 weeks ending May 9. The total for the 50 states and the District of Columbia is now 72 for 2016-2017. No new pregnancy losses with birth defects were reported over the same 4-week span, so the 50 state/D.C. total remained at eight for 2016-2017, CDC data show.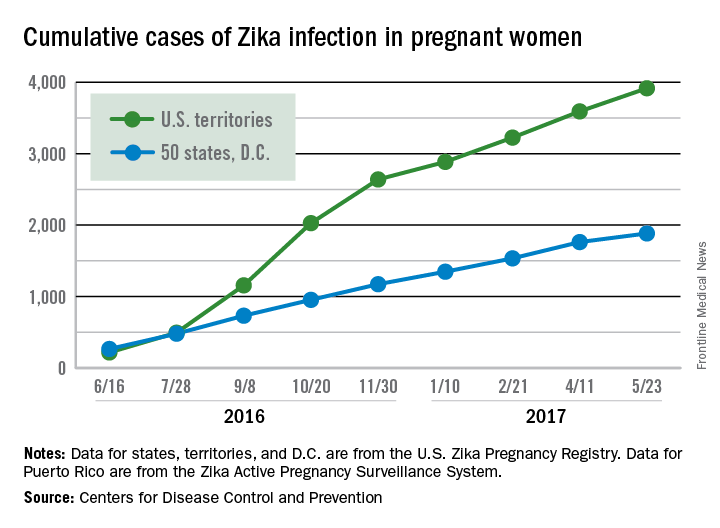
The CDC notes that these are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes. Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
Zika virus infection has been occurring in pregnant women at a slow but steady clip over the last couple of months, but cases of liveborn infants with Zika-related birth defects have jumped in recent weeks, according to the Centers for Disease Control and Prevention.
Eight liveborn infants with Zika-related birth defects were reported to the U.S. Zika Pregnancy Registry during the 2 weeks ending May 23, more than any other 2-week period this year, and that was after six such infants were reported for the 2 weeks ending May 9. The total for the 50 states and the District of Columbia is now 72 for 2016-2017. No new pregnancy losses with birth defects were reported over the same 4-week span, so the 50 state/D.C. total remained at eight for 2016-2017, CDC data show.
The CDC notes that these are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes. Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
Zika virus infection has been occurring in pregnant women at a slow but steady clip over the last couple of months, but cases of liveborn infants with Zika-related birth defects have jumped in recent weeks, according to the Centers for Disease Control and Prevention.
Eight liveborn infants with Zika-related birth defects were reported to the U.S. Zika Pregnancy Registry during the 2 weeks ending May 23, more than any other 2-week period this year, and that was after six such infants were reported for the 2 weeks ending May 9. The total for the 50 states and the District of Columbia is now 72 for 2016-2017. No new pregnancy losses with birth defects were reported over the same 4-week span, so the 50 state/D.C. total remained at eight for 2016-2017, CDC data show.
The CDC notes that these are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes. Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
FDA approves generic Strattera for pediatric, adult ADHD patients
The Food and Drug Administration has approved the first generic versions of Strattera (atomoxetine) for the treatment of attention-deficit/hyperactivity disorder, the agency announced May 30.
Apotex, Teva Pharmaceuticals USA, Aurobindo Pharma, and Glenmark Pharmaceuticals all gained approval to market generic atomoxetine at various strengths. All versions must be sold with a patient medication guide describing the uses and risks of atomoxetine and must also include a boxed warning detailing the potential for increased risk of suicidal ideation in children and adolescents.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards. Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA,” Kathleen Uhl, MD, director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generic versions of Strattera (atomoxetine) for the treatment of attention-deficit/hyperactivity disorder, the agency announced May 30.
Apotex, Teva Pharmaceuticals USA, Aurobindo Pharma, and Glenmark Pharmaceuticals all gained approval to market generic atomoxetine at various strengths. All versions must be sold with a patient medication guide describing the uses and risks of atomoxetine and must also include a boxed warning detailing the potential for increased risk of suicidal ideation in children and adolescents.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards. Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA,” Kathleen Uhl, MD, director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved the first generic versions of Strattera (atomoxetine) for the treatment of attention-deficit/hyperactivity disorder, the agency announced May 30.
Apotex, Teva Pharmaceuticals USA, Aurobindo Pharma, and Glenmark Pharmaceuticals all gained approval to market generic atomoxetine at various strengths. All versions must be sold with a patient medication guide describing the uses and risks of atomoxetine and must also include a boxed warning detailing the potential for increased risk of suicidal ideation in children and adolescents.
“Today’s approvals mark an important step forward in bringing consumers additional treatments that have met the FDA’s rigorous standards. Quickly bringing generics to market so patients have more options to treat their conditions is a top priority for the FDA,” Kathleen Uhl, MD, director of the Office of Generic Drugs in the FDA’s Center for Drug Evaluation and Research, said in a press release.
Find the full press release on the FDA website.
Alzheimer’s mortality in U.S. grew from 1999 to 2014
The rate of death attributable to Alzheimer’s disease increased by more than 50% from 1999 to 2014 in the United States, according to a report from the Centers for Disease Control and Prevention.
According to data collected from the National Vital Statistics System, the total number of Alzheimer’s deaths was 44,536 in 1999 and 93,541 in 2014. The mortality in 1999 was 16.5 per 100,000 people, and in 2014 it was 25.4 per 100,000 people, a rate increase of 54.5%. Alzheimer’s was the sixth most common cause of death in 2014, accounting for 3.6% of all U.S. deaths.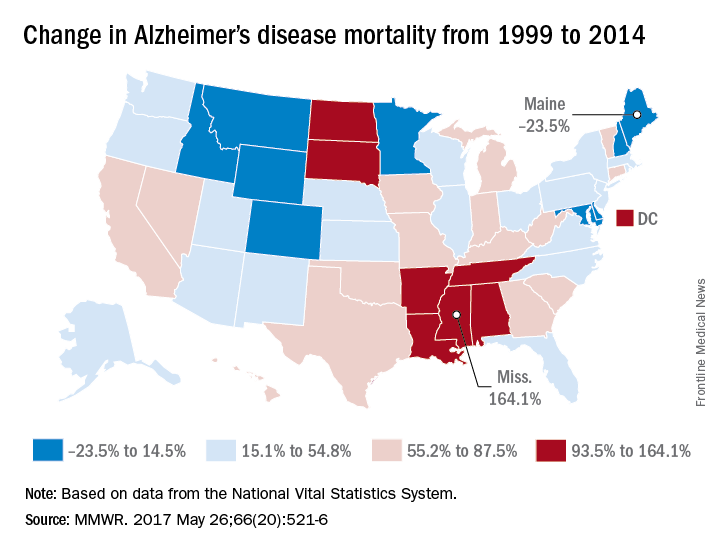
Mississippi, Louisiana, Arkansas, South Dakota, and Tennessee had mortality increases of more than 100%; Mississippi’s 164.1% increase from 13.3 to 35.2 per 100,000 people was the greatest during the study period. Maine, Montana, and Maryland saw decreases in mortality, with the percentages falling 23.5%, 9.9%, and 6.1%, respectively.
“An increasing number of Alzheimer’s deaths coupled with an increasing number of patients dying at home suggests that there is an increasing number of caregivers of persons with Alzheimer’s. It is likely that these caregivers might benefit from interventions such as education, respite care, and case management that can lessen the potential burden of caregiving,” the CDC investigators concluded.
Find the full study in MMWR (2017;66[20]:521-6).
The rate of death attributable to Alzheimer’s disease increased by more than 50% from 1999 to 2014 in the United States, according to a report from the Centers for Disease Control and Prevention.
According to data collected from the National Vital Statistics System, the total number of Alzheimer’s deaths was 44,536 in 1999 and 93,541 in 2014. The mortality in 1999 was 16.5 per 100,000 people, and in 2014 it was 25.4 per 100,000 people, a rate increase of 54.5%. Alzheimer’s was the sixth most common cause of death in 2014, accounting for 3.6% of all U.S. deaths.
Mississippi, Louisiana, Arkansas, South Dakota, and Tennessee had mortality increases of more than 100%; Mississippi’s 164.1% increase from 13.3 to 35.2 per 100,000 people was the greatest during the study period. Maine, Montana, and Maryland saw decreases in mortality, with the percentages falling 23.5%, 9.9%, and 6.1%, respectively.
“An increasing number of Alzheimer’s deaths coupled with an increasing number of patients dying at home suggests that there is an increasing number of caregivers of persons with Alzheimer’s. It is likely that these caregivers might benefit from interventions such as education, respite care, and case management that can lessen the potential burden of caregiving,” the CDC investigators concluded.
Find the full study in MMWR (2017;66[20]:521-6).
The rate of death attributable to Alzheimer’s disease increased by more than 50% from 1999 to 2014 in the United States, according to a report from the Centers for Disease Control and Prevention.
According to data collected from the National Vital Statistics System, the total number of Alzheimer’s deaths was 44,536 in 1999 and 93,541 in 2014. The mortality in 1999 was 16.5 per 100,000 people, and in 2014 it was 25.4 per 100,000 people, a rate increase of 54.5%. Alzheimer’s was the sixth most common cause of death in 2014, accounting for 3.6% of all U.S. deaths.
Mississippi, Louisiana, Arkansas, South Dakota, and Tennessee had mortality increases of more than 100%; Mississippi’s 164.1% increase from 13.3 to 35.2 per 100,000 people was the greatest during the study period. Maine, Montana, and Maryland saw decreases in mortality, with the percentages falling 23.5%, 9.9%, and 6.1%, respectively.
“An increasing number of Alzheimer’s deaths coupled with an increasing number of patients dying at home suggests that there is an increasing number of caregivers of persons with Alzheimer’s. It is likely that these caregivers might benefit from interventions such as education, respite care, and case management that can lessen the potential burden of caregiving,” the CDC investigators concluded.
Find the full study in MMWR (2017;66[20]:521-6).
FROM MMWR
FDA approves first specific treatment for giant cell arteritis
The Food and Drug Administration has approved subcutaneous tocilizumab (Actemra) for the treatment of giant cell arteritis, according to a May 22 announcement from the agency.
Giant cell arteritis is a type of vasculitis that inflames blood vessels in the head, causing arteries to narrow or become irregular. The temporal arteries are the most commonly affected blood vessels, but giant cell arteritis can also affect other large blood vessels such as the aorta. Tocilizumab is the first drug specifically intended to treat giant cell arteritis. The standard treatment has typically been high doses of corticosteroids, tapered over time.![]()
The FDA’s approval of tocilizumab was based on results from a double-blind, placebo-controlled study of 251 patients with giant cell arteritis. After 1 year, patients who received tocilizumab and tapered prednisone achieved remission at a higher rate than did patients who received placebo and tapered prednisone. Safety was consistent with tocilizumab’s known safety profile.
Subcutaneous tocilizumab is also approved for moderate to severely active rheumatoid arthritis. The intravenous formulation is approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis, and polyarticular juvenile idiopathic arthritis.
The FDA granted both Breakthrough Therapy and Priority Review designations to this supplemental new drug application of tocilizumab.
The Food and Drug Administration has approved subcutaneous tocilizumab (Actemra) for the treatment of giant cell arteritis, according to a May 22 announcement from the agency.
Giant cell arteritis is a type of vasculitis that inflames blood vessels in the head, causing arteries to narrow or become irregular. The temporal arteries are the most commonly affected blood vessels, but giant cell arteritis can also affect other large blood vessels such as the aorta. Tocilizumab is the first drug specifically intended to treat giant cell arteritis. The standard treatment has typically been high doses of corticosteroids, tapered over time.![]()
The FDA’s approval of tocilizumab was based on results from a double-blind, placebo-controlled study of 251 patients with giant cell arteritis. After 1 year, patients who received tocilizumab and tapered prednisone achieved remission at a higher rate than did patients who received placebo and tapered prednisone. Safety was consistent with tocilizumab’s known safety profile.
Subcutaneous tocilizumab is also approved for moderate to severely active rheumatoid arthritis. The intravenous formulation is approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis, and polyarticular juvenile idiopathic arthritis.
The FDA granted both Breakthrough Therapy and Priority Review designations to this supplemental new drug application of tocilizumab.
The Food and Drug Administration has approved subcutaneous tocilizumab (Actemra) for the treatment of giant cell arteritis, according to a May 22 announcement from the agency.
Giant cell arteritis is a type of vasculitis that inflames blood vessels in the head, causing arteries to narrow or become irregular. The temporal arteries are the most commonly affected blood vessels, but giant cell arteritis can also affect other large blood vessels such as the aorta. Tocilizumab is the first drug specifically intended to treat giant cell arteritis. The standard treatment has typically been high doses of corticosteroids, tapered over time.![]()
The FDA’s approval of tocilizumab was based on results from a double-blind, placebo-controlled study of 251 patients with giant cell arteritis. After 1 year, patients who received tocilizumab and tapered prednisone achieved remission at a higher rate than did patients who received placebo and tapered prednisone. Safety was consistent with tocilizumab’s known safety profile.
Subcutaneous tocilizumab is also approved for moderate to severely active rheumatoid arthritis. The intravenous formulation is approved for the treatment of moderate to severely active rheumatoid arthritis, systemic juvenile idiopathic arthritis, and polyarticular juvenile idiopathic arthritis.
The FDA granted both Breakthrough Therapy and Priority Review designations to this supplemental new drug application of tocilizumab.