User login
New device that treats esophageal atresia in infants has been authorized
The FDA has authorized a medical device to treat infants up to age 1 year for esophageal atresia, called the Flourish Pediatric Esophageal Atresia Anastomosis.
The device uses magnets attached to two catheters to pull the upper and lower esophagus together, closing the gap for several days until a connection is formed. The catheters are then removed, and the infant can begin feeding via mouth.
Cook Medical provided data on 16 patients implanted with Flourish devices. All patients had successful joining of their esophagus within 3-10 days after receiving the device. A total of 13 of the 16 patients developed anastomotic stricture that required a balloon dilation procedure, a stent, or both to repair. Such strictures also occur with traditional surgery.
The Flourish device should not be used in patients older than 1 year. Other potential complications that may occur include stomach or mouth irritation near the catheter insertion sites and gastroesophageal reflux.
Learn more about the study at www.fda.gov/newsevents/newsroom/pressannouncements/ucm558241.htm.
The FDA has authorized a medical device to treat infants up to age 1 year for esophageal atresia, called the Flourish Pediatric Esophageal Atresia Anastomosis.
The device uses magnets attached to two catheters to pull the upper and lower esophagus together, closing the gap for several days until a connection is formed. The catheters are then removed, and the infant can begin feeding via mouth.
Cook Medical provided data on 16 patients implanted with Flourish devices. All patients had successful joining of their esophagus within 3-10 days after receiving the device. A total of 13 of the 16 patients developed anastomotic stricture that required a balloon dilation procedure, a stent, or both to repair. Such strictures also occur with traditional surgery.
The Flourish device should not be used in patients older than 1 year. Other potential complications that may occur include stomach or mouth irritation near the catheter insertion sites and gastroesophageal reflux.
Learn more about the study at www.fda.gov/newsevents/newsroom/pressannouncements/ucm558241.htm.
The FDA has authorized a medical device to treat infants up to age 1 year for esophageal atresia, called the Flourish Pediatric Esophageal Atresia Anastomosis.
The device uses magnets attached to two catheters to pull the upper and lower esophagus together, closing the gap for several days until a connection is formed. The catheters are then removed, and the infant can begin feeding via mouth.
Cook Medical provided data on 16 patients implanted with Flourish devices. All patients had successful joining of their esophagus within 3-10 days after receiving the device. A total of 13 of the 16 patients developed anastomotic stricture that required a balloon dilation procedure, a stent, or both to repair. Such strictures also occur with traditional surgery.
The Flourish device should not be used in patients older than 1 year. Other potential complications that may occur include stomach or mouth irritation near the catheter insertion sites and gastroesophageal reflux.
Learn more about the study at www.fda.gov/newsevents/newsroom/pressannouncements/ucm558241.htm.
Key clinical point:
Major finding: All of the 16 patients implanted with the Flourish device were successfully treated for esophageal atresia.
Data source: Data was provided by Cook Medical under a humanitarian device exemption. A total of 16 patients were implanted with the Flourish device.
Disclosures: This study was sponsored by Cook Medical.
Canagliflozin gets boxed warning for amputation
The Food and Drug Administration has added a boxed warning to the label of diabetes drug canagliflozin for the risk of lower limb amputation.
The agency cited data from two clinical trials showing nearly double the risk of leg and foot amputations in patients treated with the canagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor, compared with placebo, in a recent statement.![]()
The trials, which followed participants for an average of 5.7 and 2.1 years, respectively, showed that lower limb infections, gangrene, diabetic foot ulcers, and ischemia commonly occurred prior to the need for amputation.
The boxed warning advises physicians to consider a patient’s history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers before prescribing canagliflozin and to monitor patients for pain, tenderness, sores, ulcers, or infections on the feet or legs.
Consider discontinuing canagliflozin in these patients, as well as those with symptoms of hypotension, ketoacidosis, elevated serum potassium levels, severe urinary tract infections, hypoglycemia in combination with other prescription diabetes medicines, yeast infections, bone breaks, and increased cholesterol, according to the FDA.
The FDA first issued a safety communication on canagliflozin about a year ago but, at the time, did not advise assessing a patient’s risk for amputation.
Canagliflozin, marketed as Invokana, Invokamet, and Invokamet XR by Janssen Pharmaceuticals, was approved by the FDA in March 2013.
Adverse events involving canagliflozin – or any drug – should be reported to the FDA MedWatch program.
The Food and Drug Administration has added a boxed warning to the label of diabetes drug canagliflozin for the risk of lower limb amputation.
The agency cited data from two clinical trials showing nearly double the risk of leg and foot amputations in patients treated with the canagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor, compared with placebo, in a recent statement.![]()
The trials, which followed participants for an average of 5.7 and 2.1 years, respectively, showed that lower limb infections, gangrene, diabetic foot ulcers, and ischemia commonly occurred prior to the need for amputation.
The boxed warning advises physicians to consider a patient’s history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers before prescribing canagliflozin and to monitor patients for pain, tenderness, sores, ulcers, or infections on the feet or legs.
Consider discontinuing canagliflozin in these patients, as well as those with symptoms of hypotension, ketoacidosis, elevated serum potassium levels, severe urinary tract infections, hypoglycemia in combination with other prescription diabetes medicines, yeast infections, bone breaks, and increased cholesterol, according to the FDA.
The FDA first issued a safety communication on canagliflozin about a year ago but, at the time, did not advise assessing a patient’s risk for amputation.
Canagliflozin, marketed as Invokana, Invokamet, and Invokamet XR by Janssen Pharmaceuticals, was approved by the FDA in March 2013.
Adverse events involving canagliflozin – or any drug – should be reported to the FDA MedWatch program.
The Food and Drug Administration has added a boxed warning to the label of diabetes drug canagliflozin for the risk of lower limb amputation.
The agency cited data from two clinical trials showing nearly double the risk of leg and foot amputations in patients treated with the canagliflozin, a sodium-glucose cotransporter 2 (SGLT2) inhibitor, compared with placebo, in a recent statement.![]()
The trials, which followed participants for an average of 5.7 and 2.1 years, respectively, showed that lower limb infections, gangrene, diabetic foot ulcers, and ischemia commonly occurred prior to the need for amputation.
The boxed warning advises physicians to consider a patient’s history of prior amputation, peripheral vascular disease, neuropathy, and diabetic foot ulcers before prescribing canagliflozin and to monitor patients for pain, tenderness, sores, ulcers, or infections on the feet or legs.
Consider discontinuing canagliflozin in these patients, as well as those with symptoms of hypotension, ketoacidosis, elevated serum potassium levels, severe urinary tract infections, hypoglycemia in combination with other prescription diabetes medicines, yeast infections, bone breaks, and increased cholesterol, according to the FDA.
The FDA first issued a safety communication on canagliflozin about a year ago but, at the time, did not advise assessing a patient’s risk for amputation.
Canagliflozin, marketed as Invokana, Invokamet, and Invokamet XR by Janssen Pharmaceuticals, was approved by the FDA in March 2013.
Adverse events involving canagliflozin – or any drug – should be reported to the FDA MedWatch program.
Few states fully back HCV prevention, treatment
The prevalence of hepatitis C virus (HCV) varies considerably by state, and the same can be said for the state laws and policies attempting to decrease that prevalence, according to an assessment by the Centers for Disease Control and Prevention.
In 2015, incidence of acute HCV infection exceeded the national average of 0.8 per 100,000 population in 17 states, including seven with rates that at least doubled it, the report noted. New HCV infections have increased in recent years despite curative therapies “and known preventive measures to interrupt transmission.”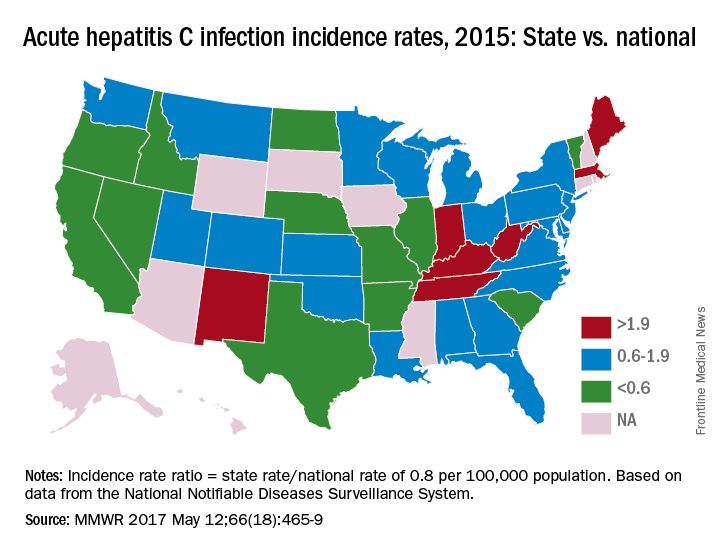
The “most comprehensive” laws on prevention through clean needle access as of 2016 were found in Maine, Nevada, and Utah, with laws in 12 other states categorized as “more comprehensive” and 18 states falling into the “least comprehensive” category. On the Medicaid side of the equation, 16 states had permissive policies that did not require sobriety or required only screening and counseling before treatment, 24 states had restrictive policies that requited sobriety, and 10 states had no policy available, the report showed (MMWR. 2017 May 12:66[18]:465-9).
Only three states – Massachusetts, New Mexico, and Washington – had a comprehensive (all three were considered “more comprehensive”) set of prevention laws and a permissive treatment policy, the investigators said, while also noting that two of the three – Massachusetts and New Mexico – were among the states with acute HCV rates that were at least twice the national average.
“Although the costs of HCV therapies have raised budgetary issues for state Medicaid programs in the past, the costs of HCV treatment have declined in recent years, increasing the cost-effectiveness of treatment, particularly among persons who inject drugs and who might serve as an ongoing source of transmission to others,” the report concluded.
The analysis examined three types of laws on access to clean needles and syringes: authorization of exchange programs, the scope of drug paraphernalia laws, and retail sale of needles and syringes. Each law was assessed for five elements, including authorization of syringe exchange statewide or in selected jurisdictions and exemption of needles or syringes from the definition of drug paraphernalia.
For the accompanying map (see “Acute hepatitis C infection incidence rates, 2015: State vs. national”), each state’s acute HCV incidence rate for 2015 was divided by the national rate to determine the incidence rate ratio, with data unavailable for 10 states.
The prevalence of hepatitis C virus (HCV) varies considerably by state, and the same can be said for the state laws and policies attempting to decrease that prevalence, according to an assessment by the Centers for Disease Control and Prevention.
In 2015, incidence of acute HCV infection exceeded the national average of 0.8 per 100,000 population in 17 states, including seven with rates that at least doubled it, the report noted. New HCV infections have increased in recent years despite curative therapies “and known preventive measures to interrupt transmission.”
The “most comprehensive” laws on prevention through clean needle access as of 2016 were found in Maine, Nevada, and Utah, with laws in 12 other states categorized as “more comprehensive” and 18 states falling into the “least comprehensive” category. On the Medicaid side of the equation, 16 states had permissive policies that did not require sobriety or required only screening and counseling before treatment, 24 states had restrictive policies that requited sobriety, and 10 states had no policy available, the report showed (MMWR. 2017 May 12:66[18]:465-9).
Only three states – Massachusetts, New Mexico, and Washington – had a comprehensive (all three were considered “more comprehensive”) set of prevention laws and a permissive treatment policy, the investigators said, while also noting that two of the three – Massachusetts and New Mexico – were among the states with acute HCV rates that were at least twice the national average.
“Although the costs of HCV therapies have raised budgetary issues for state Medicaid programs in the past, the costs of HCV treatment have declined in recent years, increasing the cost-effectiveness of treatment, particularly among persons who inject drugs and who might serve as an ongoing source of transmission to others,” the report concluded.
The analysis examined three types of laws on access to clean needles and syringes: authorization of exchange programs, the scope of drug paraphernalia laws, and retail sale of needles and syringes. Each law was assessed for five elements, including authorization of syringe exchange statewide or in selected jurisdictions and exemption of needles or syringes from the definition of drug paraphernalia.
For the accompanying map (see “Acute hepatitis C infection incidence rates, 2015: State vs. national”), each state’s acute HCV incidence rate for 2015 was divided by the national rate to determine the incidence rate ratio, with data unavailable for 10 states.
The prevalence of hepatitis C virus (HCV) varies considerably by state, and the same can be said for the state laws and policies attempting to decrease that prevalence, according to an assessment by the Centers for Disease Control and Prevention.
In 2015, incidence of acute HCV infection exceeded the national average of 0.8 per 100,000 population in 17 states, including seven with rates that at least doubled it, the report noted. New HCV infections have increased in recent years despite curative therapies “and known preventive measures to interrupt transmission.”
The “most comprehensive” laws on prevention through clean needle access as of 2016 were found in Maine, Nevada, and Utah, with laws in 12 other states categorized as “more comprehensive” and 18 states falling into the “least comprehensive” category. On the Medicaid side of the equation, 16 states had permissive policies that did not require sobriety or required only screening and counseling before treatment, 24 states had restrictive policies that requited sobriety, and 10 states had no policy available, the report showed (MMWR. 2017 May 12:66[18]:465-9).
Only three states – Massachusetts, New Mexico, and Washington – had a comprehensive (all three were considered “more comprehensive”) set of prevention laws and a permissive treatment policy, the investigators said, while also noting that two of the three – Massachusetts and New Mexico – were among the states with acute HCV rates that were at least twice the national average.
“Although the costs of HCV therapies have raised budgetary issues for state Medicaid programs in the past, the costs of HCV treatment have declined in recent years, increasing the cost-effectiveness of treatment, particularly among persons who inject drugs and who might serve as an ongoing source of transmission to others,” the report concluded.
The analysis examined three types of laws on access to clean needles and syringes: authorization of exchange programs, the scope of drug paraphernalia laws, and retail sale of needles and syringes. Each law was assessed for five elements, including authorization of syringe exchange statewide or in selected jurisdictions and exemption of needles or syringes from the definition of drug paraphernalia.
For the accompanying map (see “Acute hepatitis C infection incidence rates, 2015: State vs. national”), each state’s acute HCV incidence rate for 2015 was divided by the national rate to determine the incidence rate ratio, with data unavailable for 10 states.
FROM MMWR
FDA approves pembrolizumab for first-line advanced NSCLC
The Food and Drug Administration has granted accelerated approval to checkpoint inhibitor pembrolizumab in combination with pemetrexed and carboplatin for the treatment of patients with previously untreated metastatic nonsquamous non–small cell lung cancer (NSCLC).
The immunotherapy pembrolizumab was approved as a second-line treatment for metastatic NSCLC in 2015.
First-line approval was based on an improved overall response rate (ORR) and progression-free survival (PFS) in a cohort of 123 patients within an open-label, multicohort study (KEYNOTE-21). Enrollees in cohort G1 had locally advanced or metastatic NSCLC and no prior systemic treatment for metastatic disease. They were randomized to receive either pembrolizumab, in combination with pemetrexed and carboplatin (PC) for four cycles followed by pembrolizumab for a maximum of 24 months (n = 60) or PC alone (n = 63). Randomization was stratified by PD-L1 tumor expression (tumor proportion score [TPS] less than 1% vs. TPS greater than or equal to 1%).![]()
The hazard ratio for PFS was 0.53 (95% CI: 0.31, 0.91, P = .0205). The median PFS was 13.0 months for the pembrolizumab plus PC arm and 8.9 months for the PC-alone arm. In the TPS less than 1% subgroup, the ORR was 57% and 13% in the pembrolizumab-plus-PC and in the PC-alone arms, respectively. In the TPS greater-than-or-equal-to-1% subgroup, the ORR was 54% in the pembrolizumab-plus-PC arm and 38% in the pembrolizumab-plus-PC arm, the FDA said.
There were serious adverse events in 41% of the patients in the pembrolizumab-plus-PC arm compared with 28% in the PC-alone arm. Pembrolizumab was discontinued for adverse reactions in 10% of patients, most commonly due to acute kidney injury. The most common grade 3-4 adverse reactions were fatigue, dyspnea, nausea, vomiting, diarrhea, and rash.
The FDA cautioned that immune-mediated adverse reactions can occur with pembrolizumab including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis. Based on the severity of the adverse reaction, pembrolizumab should be withheld or discontinued and corticosteroids administered when appropriate. The recommended dose and schedule for NSCLC is 200 mg as an intravenous infusion every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression.
Pembrolizumab is marketed as Keytruda by Merck.
The Food and Drug Administration has granted accelerated approval to checkpoint inhibitor pembrolizumab in combination with pemetrexed and carboplatin for the treatment of patients with previously untreated metastatic nonsquamous non–small cell lung cancer (NSCLC).
The immunotherapy pembrolizumab was approved as a second-line treatment for metastatic NSCLC in 2015.
First-line approval was based on an improved overall response rate (ORR) and progression-free survival (PFS) in a cohort of 123 patients within an open-label, multicohort study (KEYNOTE-21). Enrollees in cohort G1 had locally advanced or metastatic NSCLC and no prior systemic treatment for metastatic disease. They were randomized to receive either pembrolizumab, in combination with pemetrexed and carboplatin (PC) for four cycles followed by pembrolizumab for a maximum of 24 months (n = 60) or PC alone (n = 63). Randomization was stratified by PD-L1 tumor expression (tumor proportion score [TPS] less than 1% vs. TPS greater than or equal to 1%).![]()
The hazard ratio for PFS was 0.53 (95% CI: 0.31, 0.91, P = .0205). The median PFS was 13.0 months for the pembrolizumab plus PC arm and 8.9 months for the PC-alone arm. In the TPS less than 1% subgroup, the ORR was 57% and 13% in the pembrolizumab-plus-PC and in the PC-alone arms, respectively. In the TPS greater-than-or-equal-to-1% subgroup, the ORR was 54% in the pembrolizumab-plus-PC arm and 38% in the pembrolizumab-plus-PC arm, the FDA said.
There were serious adverse events in 41% of the patients in the pembrolizumab-plus-PC arm compared with 28% in the PC-alone arm. Pembrolizumab was discontinued for adverse reactions in 10% of patients, most commonly due to acute kidney injury. The most common grade 3-4 adverse reactions were fatigue, dyspnea, nausea, vomiting, diarrhea, and rash.
The FDA cautioned that immune-mediated adverse reactions can occur with pembrolizumab including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis. Based on the severity of the adverse reaction, pembrolizumab should be withheld or discontinued and corticosteroids administered when appropriate. The recommended dose and schedule for NSCLC is 200 mg as an intravenous infusion every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression.
Pembrolizumab is marketed as Keytruda by Merck.
The Food and Drug Administration has granted accelerated approval to checkpoint inhibitor pembrolizumab in combination with pemetrexed and carboplatin for the treatment of patients with previously untreated metastatic nonsquamous non–small cell lung cancer (NSCLC).
The immunotherapy pembrolizumab was approved as a second-line treatment for metastatic NSCLC in 2015.
First-line approval was based on an improved overall response rate (ORR) and progression-free survival (PFS) in a cohort of 123 patients within an open-label, multicohort study (KEYNOTE-21). Enrollees in cohort G1 had locally advanced or metastatic NSCLC and no prior systemic treatment for metastatic disease. They were randomized to receive either pembrolizumab, in combination with pemetrexed and carboplatin (PC) for four cycles followed by pembrolizumab for a maximum of 24 months (n = 60) or PC alone (n = 63). Randomization was stratified by PD-L1 tumor expression (tumor proportion score [TPS] less than 1% vs. TPS greater than or equal to 1%).![]()
The hazard ratio for PFS was 0.53 (95% CI: 0.31, 0.91, P = .0205). The median PFS was 13.0 months for the pembrolizumab plus PC arm and 8.9 months for the PC-alone arm. In the TPS less than 1% subgroup, the ORR was 57% and 13% in the pembrolizumab-plus-PC and in the PC-alone arms, respectively. In the TPS greater-than-or-equal-to-1% subgroup, the ORR was 54% in the pembrolizumab-plus-PC arm and 38% in the pembrolizumab-plus-PC arm, the FDA said.
There were serious adverse events in 41% of the patients in the pembrolizumab-plus-PC arm compared with 28% in the PC-alone arm. Pembrolizumab was discontinued for adverse reactions in 10% of patients, most commonly due to acute kidney injury. The most common grade 3-4 adverse reactions were fatigue, dyspnea, nausea, vomiting, diarrhea, and rash.
The FDA cautioned that immune-mediated adverse reactions can occur with pembrolizumab including pneumonitis, colitis, hepatitis, endocrinopathies, and nephritis. Based on the severity of the adverse reaction, pembrolizumab should be withheld or discontinued and corticosteroids administered when appropriate. The recommended dose and schedule for NSCLC is 200 mg as an intravenous infusion every 3 weeks until disease progression, unacceptable toxicity, or up to 24 months in patients without disease progression.
Pembrolizumab is marketed as Keytruda by Merck.
FDA: Fluoroquinolone use not linked to retina detachment, aortic problems
The Food and Drug Administration has found no evidence of a link between fluoroquinolone usage and retinal detachment or aortic aneurysm and dissection, according to a new Drug Safety Communication update on potential serious, disabling side effects of oral and injectable fluoroquinolone antibiotics.
Fluoroquinolones are used to treat acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections.
However, after reviewing patient cases and study findings, the FDA said the evidence didn’t support an association between fluoroquinolone usage and potential retinal or aortic dangers, according to its May 10, 2017, Drug Safety Communication update.
Serious side effects associated with fluoroquinolone use include hallucination, depression, suicidal thoughts, tendinitis and tendon rupture, a “pins and needles” feeling in the arms and legs, joint pain and swelling, skin rash, and severe diarrhea.
“We will continue to assess safety issues with fluoroquinolones, and will update the public if additional actions are needed,” the FDA said in a statement.
The Food and Drug Administration has found no evidence of a link between fluoroquinolone usage and retinal detachment or aortic aneurysm and dissection, according to a new Drug Safety Communication update on potential serious, disabling side effects of oral and injectable fluoroquinolone antibiotics.
Fluoroquinolones are used to treat acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections.
However, after reviewing patient cases and study findings, the FDA said the evidence didn’t support an association between fluoroquinolone usage and potential retinal or aortic dangers, according to its May 10, 2017, Drug Safety Communication update.
Serious side effects associated with fluoroquinolone use include hallucination, depression, suicidal thoughts, tendinitis and tendon rupture, a “pins and needles” feeling in the arms and legs, joint pain and swelling, skin rash, and severe diarrhea.
“We will continue to assess safety issues with fluoroquinolones, and will update the public if additional actions are needed,” the FDA said in a statement.
The Food and Drug Administration has found no evidence of a link between fluoroquinolone usage and retinal detachment or aortic aneurysm and dissection, according to a new Drug Safety Communication update on potential serious, disabling side effects of oral and injectable fluoroquinolone antibiotics.
Fluoroquinolones are used to treat acute bacterial sinusitis, acute bacterial exacerbation of chronic bronchitis, and uncomplicated urinary tract infections.
However, after reviewing patient cases and study findings, the FDA said the evidence didn’t support an association between fluoroquinolone usage and potential retinal or aortic dangers, according to its May 10, 2017, Drug Safety Communication update.
Serious side effects associated with fluoroquinolone use include hallucination, depression, suicidal thoughts, tendinitis and tendon rupture, a “pins and needles” feeling in the arms and legs, joint pain and swelling, skin rash, and severe diarrhea.
“We will continue to assess safety issues with fluoroquinolones, and will update the public if additional actions are needed,” the FDA said in a statement.
HCV pregnancy increase suggests need for broader screening
Hepatitis C virus (HCV) infection among women giving birth increased 89% from 1.8 cases per 1,000 live births in 2009 to 3.4 cases in 2014, translating to about 35 infants exposed to the virus per day in the United States in 2014, according to the May 11 Morbidity and Mortality Weekly Report.
The highest rate in 2014 was in West Virginia, with 22.6 cases per 1,000 live births. The lowest was in Hawaii, with 0.7 cases.![]()
“The prevalence of maternal HCV infection appears to have increased sharply in the United States. ... Ensuring that women of childbearing age have access to HCV testing and treatment and consideration of universal screening among women of reproductive age residing in areas with high HCV prevalence might mitigate risk and prevent transmission,” said investigators led by Stephen Patrick, MD, of the division of neonatology at Vanderbilt University, Nashville (MMWR. 2017;66[18]:470-3).
Both the Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists recommend selective screening of pregnant women at high risk for HCV infection, including intravenous drug users and long-term hemodialysis patients.
“These data might inform expansion of the definition of women at risk.” Generally, about 6% of infants will pick up HCV from their mother during delivery. Screening and treating women of childbearing could reduce the risk, the investigators said.
The analysis was based on U.S. birth certificate data. Risk factors were identified using Tennessee birth certificates.
Hepatitis C virus (HCV) infection among women giving birth increased 89% from 1.8 cases per 1,000 live births in 2009 to 3.4 cases in 2014, translating to about 35 infants exposed to the virus per day in the United States in 2014, according to the May 11 Morbidity and Mortality Weekly Report.
The highest rate in 2014 was in West Virginia, with 22.6 cases per 1,000 live births. The lowest was in Hawaii, with 0.7 cases.![]()
“The prevalence of maternal HCV infection appears to have increased sharply in the United States. ... Ensuring that women of childbearing age have access to HCV testing and treatment and consideration of universal screening among women of reproductive age residing in areas with high HCV prevalence might mitigate risk and prevent transmission,” said investigators led by Stephen Patrick, MD, of the division of neonatology at Vanderbilt University, Nashville (MMWR. 2017;66[18]:470-3).
Both the Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists recommend selective screening of pregnant women at high risk for HCV infection, including intravenous drug users and long-term hemodialysis patients.
“These data might inform expansion of the definition of women at risk.” Generally, about 6% of infants will pick up HCV from their mother during delivery. Screening and treating women of childbearing could reduce the risk, the investigators said.
The analysis was based on U.S. birth certificate data. Risk factors were identified using Tennessee birth certificates.
Hepatitis C virus (HCV) infection among women giving birth increased 89% from 1.8 cases per 1,000 live births in 2009 to 3.4 cases in 2014, translating to about 35 infants exposed to the virus per day in the United States in 2014, according to the May 11 Morbidity and Mortality Weekly Report.
The highest rate in 2014 was in West Virginia, with 22.6 cases per 1,000 live births. The lowest was in Hawaii, with 0.7 cases.![]()
“The prevalence of maternal HCV infection appears to have increased sharply in the United States. ... Ensuring that women of childbearing age have access to HCV testing and treatment and consideration of universal screening among women of reproductive age residing in areas with high HCV prevalence might mitigate risk and prevent transmission,” said investigators led by Stephen Patrick, MD, of the division of neonatology at Vanderbilt University, Nashville (MMWR. 2017;66[18]:470-3).
Both the Centers for Disease Control and Prevention and the American College of Obstetricians and Gynecologists recommend selective screening of pregnant women at high risk for HCV infection, including intravenous drug users and long-term hemodialysis patients.
“These data might inform expansion of the definition of women at risk.” Generally, about 6% of infants will pick up HCV from their mother during delivery. Screening and treating women of childbearing could reduce the risk, the investigators said.
The analysis was based on U.S. birth certificate data. Risk factors were identified using Tennessee birth certificates.
FROM MMWR
Fewer adults unaware of their hypertension
Hypertension awareness has improved significantly in adults since 2002, according to the National Center for Health Statistics.
The percentage of adults aged 18-39 years who had hypertension but reported not being told about it by a health care provider dropped 36% from 1999-2002, when it was 48.3%, to 2011-2014, when it was down to 30.8%. For adults aged 40-59 years, 26.6% were unaware of their hypertension in 1999-2002, compared with 17.4% in 2011-2014, a relative decrease of almost 35%. The decline was an even larger 55% for those aged 60 years and over – from 27.6% in 1999-2002 to 12.5% in 2011-2014, the NCHS reported.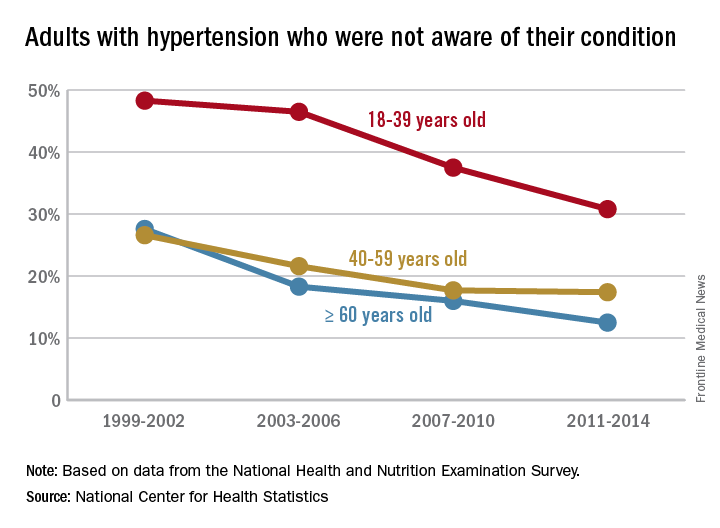
Hypertension awareness has improved significantly in adults since 2002, according to the National Center for Health Statistics.
The percentage of adults aged 18-39 years who had hypertension but reported not being told about it by a health care provider dropped 36% from 1999-2002, when it was 48.3%, to 2011-2014, when it was down to 30.8%. For adults aged 40-59 years, 26.6% were unaware of their hypertension in 1999-2002, compared with 17.4% in 2011-2014, a relative decrease of almost 35%. The decline was an even larger 55% for those aged 60 years and over – from 27.6% in 1999-2002 to 12.5% in 2011-2014, the NCHS reported.
Hypertension awareness has improved significantly in adults since 2002, according to the National Center for Health Statistics.
The percentage of adults aged 18-39 years who had hypertension but reported not being told about it by a health care provider dropped 36% from 1999-2002, when it was 48.3%, to 2011-2014, when it was down to 30.8%. For adults aged 40-59 years, 26.6% were unaware of their hypertension in 1999-2002, compared with 17.4% in 2011-2014, a relative decrease of almost 35%. The decline was an even larger 55% for those aged 60 years and over – from 27.6% in 1999-2002 to 12.5% in 2011-2014, the NCHS reported.
Preeclampsia/eclampsia rate highest in black women
The rate of preeclampsia and eclampsia for black women is 61% higher than it is for white women and 50% higher than for women overall, according to the Agency for Healthcare Research and Quality.
In 2014, black women experienced preeclampsia/eclampsia in 69.8 of every 1,000 deliveries, compared with 43.3 per 1,000 deliveries in white women and 46.6 per 1,000 for all women. Hispanic women were just above the overall rate at 46.8 per 1,000 deliveries, and Asian/Pacific Islander women were 38% lower than the overall rate at 28.8 per 1,000 deliveries, AHRQ said in its report. The overall rate was up 21% over the 38.4 per 1,000 reported in 2005.
Looking at degree of severity, 1.7% of all preeclampsia/eclampsia births in black women were eclampsia, compared with 1.4% for white and Hispanic women and 0.9% for Asian/Pacific Islanders. Severe preeclampsia was most common in Asian/Pacific Islanders – 40.4% of those diagnosed – with Hispanics at 40.3%, blacks at 38.5%, and whites at 34.3%. Mild or unspecified preeclampsia was diagnosed in 52% of preeclamptic/eclamptic white women, 46% of Hispanic women, 45% of Asians/Pacific Islanders, and 37% of black women, the AHRQ said in its analysis of data from the National Inpatient Sample.
Altogether, there were almost 177,000 delivery hospitalizations with preeclampsia/eclampsia in 2014, representing 4.7% of all deliveries and making it the most common hypertension-related diagnosis, followed by gestational hypertension (3.8%) and preexisting hypertension (1.7%). Compared with hospital stays for all other deliveries, those complicated by preeclampsia/eclampsia were 70% longer (mean, 4.4 vs. 2.6 days) and 70% more expensive (mean, $7,500 vs. $4,400), according to the report. 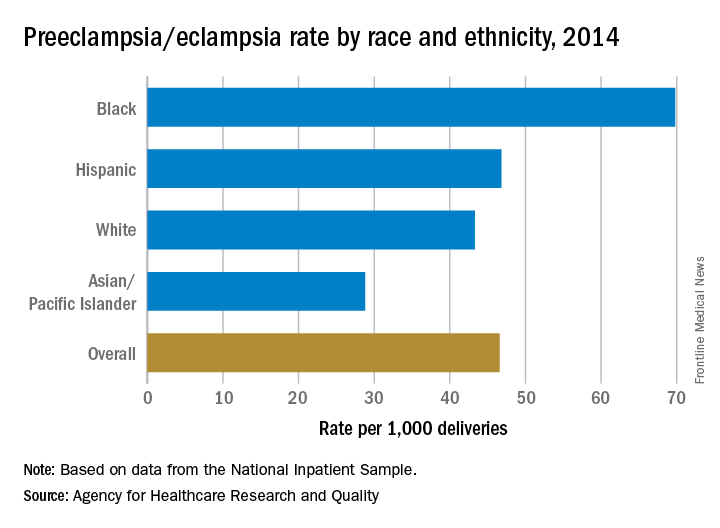
The rate of preeclampsia and eclampsia for black women is 61% higher than it is for white women and 50% higher than for women overall, according to the Agency for Healthcare Research and Quality.
In 2014, black women experienced preeclampsia/eclampsia in 69.8 of every 1,000 deliveries, compared with 43.3 per 1,000 deliveries in white women and 46.6 per 1,000 for all women. Hispanic women were just above the overall rate at 46.8 per 1,000 deliveries, and Asian/Pacific Islander women were 38% lower than the overall rate at 28.8 per 1,000 deliveries, AHRQ said in its report. The overall rate was up 21% over the 38.4 per 1,000 reported in 2005.
Looking at degree of severity, 1.7% of all preeclampsia/eclampsia births in black women were eclampsia, compared with 1.4% for white and Hispanic women and 0.9% for Asian/Pacific Islanders. Severe preeclampsia was most common in Asian/Pacific Islanders – 40.4% of those diagnosed – with Hispanics at 40.3%, blacks at 38.5%, and whites at 34.3%. Mild or unspecified preeclampsia was diagnosed in 52% of preeclamptic/eclamptic white women, 46% of Hispanic women, 45% of Asians/Pacific Islanders, and 37% of black women, the AHRQ said in its analysis of data from the National Inpatient Sample.
Altogether, there were almost 177,000 delivery hospitalizations with preeclampsia/eclampsia in 2014, representing 4.7% of all deliveries and making it the most common hypertension-related diagnosis, followed by gestational hypertension (3.8%) and preexisting hypertension (1.7%). Compared with hospital stays for all other deliveries, those complicated by preeclampsia/eclampsia were 70% longer (mean, 4.4 vs. 2.6 days) and 70% more expensive (mean, $7,500 vs. $4,400), according to the report.
The rate of preeclampsia and eclampsia for black women is 61% higher than it is for white women and 50% higher than for women overall, according to the Agency for Healthcare Research and Quality.
In 2014, black women experienced preeclampsia/eclampsia in 69.8 of every 1,000 deliveries, compared with 43.3 per 1,000 deliveries in white women and 46.6 per 1,000 for all women. Hispanic women were just above the overall rate at 46.8 per 1,000 deliveries, and Asian/Pacific Islander women were 38% lower than the overall rate at 28.8 per 1,000 deliveries, AHRQ said in its report. The overall rate was up 21% over the 38.4 per 1,000 reported in 2005.
Looking at degree of severity, 1.7% of all preeclampsia/eclampsia births in black women were eclampsia, compared with 1.4% for white and Hispanic women and 0.9% for Asian/Pacific Islanders. Severe preeclampsia was most common in Asian/Pacific Islanders – 40.4% of those diagnosed – with Hispanics at 40.3%, blacks at 38.5%, and whites at 34.3%. Mild or unspecified preeclampsia was diagnosed in 52% of preeclamptic/eclamptic white women, 46% of Hispanic women, 45% of Asians/Pacific Islanders, and 37% of black women, the AHRQ said in its analysis of data from the National Inpatient Sample.
Altogether, there were almost 177,000 delivery hospitalizations with preeclampsia/eclampsia in 2014, representing 4.7% of all deliveries and making it the most common hypertension-related diagnosis, followed by gestational hypertension (3.8%) and preexisting hypertension (1.7%). Compared with hospital stays for all other deliveries, those complicated by preeclampsia/eclampsia were 70% longer (mean, 4.4 vs. 2.6 days) and 70% more expensive (mean, $7,500 vs. $4,400), according to the report.
FDA approves brigatinib for second-line advanced ALK-positive NSCLC
The Food and Drug Administration has granted accelerated approval to brigatinib for the treatment of patients with metastatic anaplastic lymphoma kinase (ALK)–positive non–small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib.
Approval was based on a meaningful and durable overall response rate in a two-arm, open-label, multicenter trial of 222 patients with locally advanced or metastatic ALK-positive NSCLC who had progressed on crizotinib. Patients were randomized to brigatinib orally either 90 mg once daily (112 patients) or 180 mg once daily following a 7-day lead-in at 90 mg once daily (110 patients).
The median duration of response was 13.8 months in both arms.
The most common adverse reactions in 219 patients who received at least one dose were nausea, diarrhea, fatigue, cough, and headache. The most common serious adverse reactions were pneumonia and interstitial lung disease/pneumonitis. The rate of fatal adverse reactions was 3.7%; they were pneumonia in two patients and sudden death, dyspnea, respiratory failure, pulmonary embolism, bacterial meningitis, and urosepsis in one patient each. Visual disturbances also occurred in patients receiving brigatinib. The FDA cautions that patients receiving brigatinib should be monitored for new or worsening respiratory symptoms; hypertension; bradycardia; visual symptoms; and elevations in amylase, lipase, blood glucose, and creatine phosphokinase.
The recommended dosing of brigatinib, marketed as Alunbrig by Takeda Pharmaceutical, is 90 mg orally once daily for the first 7 days then, if tolerated, increase to 180 mg orally once daily.
Full prescribing information is available here.
The Food and Drug Administration has granted accelerated approval to brigatinib for the treatment of patients with metastatic anaplastic lymphoma kinase (ALK)–positive non–small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib.
Approval was based on a meaningful and durable overall response rate in a two-arm, open-label, multicenter trial of 222 patients with locally advanced or metastatic ALK-positive NSCLC who had progressed on crizotinib. Patients were randomized to brigatinib orally either 90 mg once daily (112 patients) or 180 mg once daily following a 7-day lead-in at 90 mg once daily (110 patients).
The median duration of response was 13.8 months in both arms.
The most common adverse reactions in 219 patients who received at least one dose were nausea, diarrhea, fatigue, cough, and headache. The most common serious adverse reactions were pneumonia and interstitial lung disease/pneumonitis. The rate of fatal adverse reactions was 3.7%; they were pneumonia in two patients and sudden death, dyspnea, respiratory failure, pulmonary embolism, bacterial meningitis, and urosepsis in one patient each. Visual disturbances also occurred in patients receiving brigatinib. The FDA cautions that patients receiving brigatinib should be monitored for new or worsening respiratory symptoms; hypertension; bradycardia; visual symptoms; and elevations in amylase, lipase, blood glucose, and creatine phosphokinase.
The recommended dosing of brigatinib, marketed as Alunbrig by Takeda Pharmaceutical, is 90 mg orally once daily for the first 7 days then, if tolerated, increase to 180 mg orally once daily.
Full prescribing information is available here.
The Food and Drug Administration has granted accelerated approval to brigatinib for the treatment of patients with metastatic anaplastic lymphoma kinase (ALK)–positive non–small cell lung cancer (NSCLC) who have progressed on or are intolerant to crizotinib.
Approval was based on a meaningful and durable overall response rate in a two-arm, open-label, multicenter trial of 222 patients with locally advanced or metastatic ALK-positive NSCLC who had progressed on crizotinib. Patients were randomized to brigatinib orally either 90 mg once daily (112 patients) or 180 mg once daily following a 7-day lead-in at 90 mg once daily (110 patients).
The median duration of response was 13.8 months in both arms.
The most common adverse reactions in 219 patients who received at least one dose were nausea, diarrhea, fatigue, cough, and headache. The most common serious adverse reactions were pneumonia and interstitial lung disease/pneumonitis. The rate of fatal adverse reactions was 3.7%; they were pneumonia in two patients and sudden death, dyspnea, respiratory failure, pulmonary embolism, bacterial meningitis, and urosepsis in one patient each. Visual disturbances also occurred in patients receiving brigatinib. The FDA cautions that patients receiving brigatinib should be monitored for new or worsening respiratory symptoms; hypertension; bradycardia; visual symptoms; and elevations in amylase, lipase, blood glucose, and creatine phosphokinase.
The recommended dosing of brigatinib, marketed as Alunbrig by Takeda Pharmaceutical, is 90 mg orally once daily for the first 7 days then, if tolerated, increase to 180 mg orally once daily.
Full prescribing information is available here.
FDA approves midostaurin for adult patients with FLT3+ AML
The Food and Drug Administration has approved midostaurin for the treatment of FLT3 mutation–positive acute myeloid leukemia (FLT3+ AML) in adult patients in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation.
Approval was based on results from a randomized, double-blind, placebo-controlled trial of 717 patients with previously untreated FLT3+ AML. The hazard ratio for overall survival in patients receiving midostaurin, compared with a placebo, was 0.77 (P = .016). A companion diagnostic tool, the LeukoStrat CDx FLT3 Mutation Assay manufactured by Invivoscribe Technologies, was also approved.
Midostaurin was also approved for the treatment of aggressive systemic mastocytosis, SM with associated hematological neoplasm, or mast cell leukemia. This indication approval was based on a single-arm, open-label study of midostaurin 100 mg, taken orally twice daily. Complete plus incomplete remission rates were 38% for ASM and 16% for SM with associated hematological neoplasm. Common adverse events included nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, fever, headache, and dyspnea.
The recommended dose of midostaurin in AML is 50 mg twice daily with food on days 8 to 21 of each cycle of induction and consolidation chemotherapy followed by 50 mg with food as a single agent for up to 12 months. The recommended dose for the treatment of adults with aggressive SM, SM with associated hematological neoplasm, or mast cell leukemia is 100 mg twice daily with food, the FDA said.
Midostaurin will be marketed as Rydapt by Novartis Pharmaceuticals.
The Food and Drug Administration has approved midostaurin for the treatment of FLT3 mutation–positive acute myeloid leukemia (FLT3+ AML) in adult patients in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation.
Approval was based on results from a randomized, double-blind, placebo-controlled trial of 717 patients with previously untreated FLT3+ AML. The hazard ratio for overall survival in patients receiving midostaurin, compared with a placebo, was 0.77 (P = .016). A companion diagnostic tool, the LeukoStrat CDx FLT3 Mutation Assay manufactured by Invivoscribe Technologies, was also approved.
Midostaurin was also approved for the treatment of aggressive systemic mastocytosis, SM with associated hematological neoplasm, or mast cell leukemia. This indication approval was based on a single-arm, open-label study of midostaurin 100 mg, taken orally twice daily. Complete plus incomplete remission rates were 38% for ASM and 16% for SM with associated hematological neoplasm. Common adverse events included nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, fever, headache, and dyspnea.
The recommended dose of midostaurin in AML is 50 mg twice daily with food on days 8 to 21 of each cycle of induction and consolidation chemotherapy followed by 50 mg with food as a single agent for up to 12 months. The recommended dose for the treatment of adults with aggressive SM, SM with associated hematological neoplasm, or mast cell leukemia is 100 mg twice daily with food, the FDA said.
Midostaurin will be marketed as Rydapt by Novartis Pharmaceuticals.
The Food and Drug Administration has approved midostaurin for the treatment of FLT3 mutation–positive acute myeloid leukemia (FLT3+ AML) in adult patients in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation.
Approval was based on results from a randomized, double-blind, placebo-controlled trial of 717 patients with previously untreated FLT3+ AML. The hazard ratio for overall survival in patients receiving midostaurin, compared with a placebo, was 0.77 (P = .016). A companion diagnostic tool, the LeukoStrat CDx FLT3 Mutation Assay manufactured by Invivoscribe Technologies, was also approved.
Midostaurin was also approved for the treatment of aggressive systemic mastocytosis, SM with associated hematological neoplasm, or mast cell leukemia. This indication approval was based on a single-arm, open-label study of midostaurin 100 mg, taken orally twice daily. Complete plus incomplete remission rates were 38% for ASM and 16% for SM with associated hematological neoplasm. Common adverse events included nausea, vomiting, diarrhea, edema, musculoskeletal pain, abdominal pain, fatigue, upper respiratory tract infection, fever, headache, and dyspnea.
The recommended dose of midostaurin in AML is 50 mg twice daily with food on days 8 to 21 of each cycle of induction and consolidation chemotherapy followed by 50 mg with food as a single agent for up to 12 months. The recommended dose for the treatment of adults with aggressive SM, SM with associated hematological neoplasm, or mast cell leukemia is 100 mg twice daily with food, the FDA said.
Midostaurin will be marketed as Rydapt by Novartis Pharmaceuticals.