User login
Just the FRAX: National fracture risks estimated
Adults aged 40 years and over have a mean 10-year risk of 5.3% for major osteoporotic fracture, according to the National Center for Health Statistics.
That risk includes a 10-year probability of 0.5% for hip fracture, based on estimates using the FRAX algorithm. These nationally representative estimates, the first to use the FRAX algorithm in this population, are based on 2013-2014 data from the National Health and Nutrition Examination Survey and show that the adjusted mean risks for those aged 50 years and over are 7.4% for major osteoporotic fracture and 0.9% for hip fracture, the NCHS reported.
The risk estimates in this analysis agree in large part with guidelines from the National Osteoporosis Foundation, which “use elevated FRAX scores in combination with low bone mass to define treatment eligibility,” the investigators said. FRAX-based findings, they noted, may provide “a more global evaluation of fracture risk than can be obtained from estimates based on [bone mineral density] alone.”
Adults aged 40 years and over have a mean 10-year risk of 5.3% for major osteoporotic fracture, according to the National Center for Health Statistics.
That risk includes a 10-year probability of 0.5% for hip fracture, based on estimates using the FRAX algorithm. These nationally representative estimates, the first to use the FRAX algorithm in this population, are based on 2013-2014 data from the National Health and Nutrition Examination Survey and show that the adjusted mean risks for those aged 50 years and over are 7.4% for major osteoporotic fracture and 0.9% for hip fracture, the NCHS reported.
The risk estimates in this analysis agree in large part with guidelines from the National Osteoporosis Foundation, which “use elevated FRAX scores in combination with low bone mass to define treatment eligibility,” the investigators said. FRAX-based findings, they noted, may provide “a more global evaluation of fracture risk than can be obtained from estimates based on [bone mineral density] alone.”
Adults aged 40 years and over have a mean 10-year risk of 5.3% for major osteoporotic fracture, according to the National Center for Health Statistics.
That risk includes a 10-year probability of 0.5% for hip fracture, based on estimates using the FRAX algorithm. These nationally representative estimates, the first to use the FRAX algorithm in this population, are based on 2013-2014 data from the National Health and Nutrition Examination Survey and show that the adjusted mean risks for those aged 50 years and over are 7.4% for major osteoporotic fracture and 0.9% for hip fracture, the NCHS reported.
The risk estimates in this analysis agree in large part with guidelines from the National Osteoporosis Foundation, which “use elevated FRAX scores in combination with low bone mass to define treatment eligibility,” the investigators said. FRAX-based findings, they noted, may provide “a more global evaluation of fracture risk than can be obtained from estimates based on [bone mineral density] alone.”
CDC: Some Shigella strains show reduced ciprofloxacin susceptibility
The Centers for Disease Control and Prevention has identified an increase in Shigella isolates with reduced susceptibility to ciprofloxacin, and has released an official health advisory outlining new recommendations for clinical diagnosis, management, and reporting, as well as for laboratories and public health officials.
The Shigella isolates of concern in the United States have minimum inhibitory concentration (MIC) values of 0.12-1 mcg/mL for ciprofloxacin, which is within the range considered susceptible. These strains, however, “often have a quinolone resistance gene that may lead to clinically significant reduced susceptibility to fluoroquinolone antibiotics,” such as ciprofloxacin, according to the CDC advisory.
It is possible that strains with MIC in the 0.12-1 mcg/mL range may have worse clinical outcome or increased risk of transmission, so the CDC made the following recommendations to clinicians:
• Order a stool culture to obtain isolates for antimicrobial susceptibility testing in suspected cases.
• Order antimicrobial susceptibility testing when ordering a stool culture for Shigella.
• Avoid routine prescribing of antibiotic therapy for Shigella infection, instead reserving antibiotics for patients with a clinical indication or when advised by public health officials in an outbreak setting.
• Tailor antibiotic choice (when antibiotics are indicated) to susceptibility results as soon as possible – with special attention given to the MIC for fluoroquinolone antibiotics.
• Obtain follow-up stool cultures in shigellosis patients who have continued or worsening symptoms despite antibiotic therapy.
• Consult local or state health departments for guidance regarding when patients may return to child care, school, or work.
• Counsel patients with active diarrhea on how they can prevent spreading the infection to others, regardless of whether antibiotic treatment is prescribed.
Additionally, the CDC noted that shigellosis is a nationally notifiable condition; all cases should be reported to the local health department. If a patient with shigellosis and a ciprofloxacin MIC of 0.12-1 mcg/mL is identified, this information should be included in the report to facilitate further testing of the isolate.
The CDC reported that it is working with state and local public health departments and clinical partners to determine if outcomes are indeed worse for patients treated with ciprofloxacin for Shigella strains harboring a quinolone resistance gene, and it will continue to monitor trends in susceptibility of Shigella isolates and to perform genetic testing on select strains to confirm the presence and type of resistance genes.
The Centers for Disease Control and Prevention has identified an increase in Shigella isolates with reduced susceptibility to ciprofloxacin, and has released an official health advisory outlining new recommendations for clinical diagnosis, management, and reporting, as well as for laboratories and public health officials.
The Shigella isolates of concern in the United States have minimum inhibitory concentration (MIC) values of 0.12-1 mcg/mL for ciprofloxacin, which is within the range considered susceptible. These strains, however, “often have a quinolone resistance gene that may lead to clinically significant reduced susceptibility to fluoroquinolone antibiotics,” such as ciprofloxacin, according to the CDC advisory.
It is possible that strains with MIC in the 0.12-1 mcg/mL range may have worse clinical outcome or increased risk of transmission, so the CDC made the following recommendations to clinicians:
• Order a stool culture to obtain isolates for antimicrobial susceptibility testing in suspected cases.
• Order antimicrobial susceptibility testing when ordering a stool culture for Shigella.
• Avoid routine prescribing of antibiotic therapy for Shigella infection, instead reserving antibiotics for patients with a clinical indication or when advised by public health officials in an outbreak setting.
• Tailor antibiotic choice (when antibiotics are indicated) to susceptibility results as soon as possible – with special attention given to the MIC for fluoroquinolone antibiotics.
• Obtain follow-up stool cultures in shigellosis patients who have continued or worsening symptoms despite antibiotic therapy.
• Consult local or state health departments for guidance regarding when patients may return to child care, school, or work.
• Counsel patients with active diarrhea on how they can prevent spreading the infection to others, regardless of whether antibiotic treatment is prescribed.
Additionally, the CDC noted that shigellosis is a nationally notifiable condition; all cases should be reported to the local health department. If a patient with shigellosis and a ciprofloxacin MIC of 0.12-1 mcg/mL is identified, this information should be included in the report to facilitate further testing of the isolate.
The CDC reported that it is working with state and local public health departments and clinical partners to determine if outcomes are indeed worse for patients treated with ciprofloxacin for Shigella strains harboring a quinolone resistance gene, and it will continue to monitor trends in susceptibility of Shigella isolates and to perform genetic testing on select strains to confirm the presence and type of resistance genes.
The Centers for Disease Control and Prevention has identified an increase in Shigella isolates with reduced susceptibility to ciprofloxacin, and has released an official health advisory outlining new recommendations for clinical diagnosis, management, and reporting, as well as for laboratories and public health officials.
The Shigella isolates of concern in the United States have minimum inhibitory concentration (MIC) values of 0.12-1 mcg/mL for ciprofloxacin, which is within the range considered susceptible. These strains, however, “often have a quinolone resistance gene that may lead to clinically significant reduced susceptibility to fluoroquinolone antibiotics,” such as ciprofloxacin, according to the CDC advisory.
It is possible that strains with MIC in the 0.12-1 mcg/mL range may have worse clinical outcome or increased risk of transmission, so the CDC made the following recommendations to clinicians:
• Order a stool culture to obtain isolates for antimicrobial susceptibility testing in suspected cases.
• Order antimicrobial susceptibility testing when ordering a stool culture for Shigella.
• Avoid routine prescribing of antibiotic therapy for Shigella infection, instead reserving antibiotics for patients with a clinical indication or when advised by public health officials in an outbreak setting.
• Tailor antibiotic choice (when antibiotics are indicated) to susceptibility results as soon as possible – with special attention given to the MIC for fluoroquinolone antibiotics.
• Obtain follow-up stool cultures in shigellosis patients who have continued or worsening symptoms despite antibiotic therapy.
• Consult local or state health departments for guidance regarding when patients may return to child care, school, or work.
• Counsel patients with active diarrhea on how they can prevent spreading the infection to others, regardless of whether antibiotic treatment is prescribed.
Additionally, the CDC noted that shigellosis is a nationally notifiable condition; all cases should be reported to the local health department. If a patient with shigellosis and a ciprofloxacin MIC of 0.12-1 mcg/mL is identified, this information should be included in the report to facilitate further testing of the isolate.
The CDC reported that it is working with state and local public health departments and clinical partners to determine if outcomes are indeed worse for patients treated with ciprofloxacin for Shigella strains harboring a quinolone resistance gene, and it will continue to monitor trends in susceptibility of Shigella isolates and to perform genetic testing on select strains to confirm the presence and type of resistance genes.
FDA warns against use of codeine, tramadol in children
The Food and Drug Administration is restricting the use of two opiates – codeine and tramadol – in children, and also warns they are potentially dangerous to infants of breastfeeding women.
Codeine is approved to treat pain and cough; tramadol is approved to treat pain.
“We understand that there are limited options when it comes to treating pain or cough in children, and that these changes may raise some questions for health care providers and parents. However, please know that our decision today was made based on the latest evidence and with this goal in mind: keeping our kids safe,” Douglas Throckmorton, MD, the deputy center director for regulatory programs at the FDA Center for Drug Evaluation and Research, said in a statement.
In 2013, the FDA updated prescription codeine labeling to contain a boxed warning and contraindication for children up to age 18 years regarding the risk of slowed breathing from codeine prescribed after tonsillectomy and/or adenoidectomy. And in 2015, the agency issued warnings about the risk of serious breathing problems in children who had ultrarapid metabolism of codeine and tramadol.
In the current safety statement, the FDA said it will require additional labeling changes, including contraindications for use of codeine or tramadol in all children younger than 12 years of age and a new contraindication for tramadol in children younger than 18 years being treated for pain after tonsillectomy and/or adenoidectomy, warnings about their use in children 12-18 years of age with certain medical conditions, and a stronger warning recommending against their use in nursing mothers.
Single-ingredient codeine and all tramadol-containing products are FDA-approved only for use in adults, the agency noted.
The agency cited particular concerns over those who are “ultra-rapid metabolizers” of substrates of the cytochrome P450 isoenzyme 2D6 (CYP2D6) genotype. These people more quickly convert codeine into potentially dangerously high levels of morphine, which can lead to fatal respiratory depression.
Supporting the restrictions and warning were data on 64 cases of respiratory depression that occurred worldwide between 1969 and 2015, when a codeine-based medicine was used in children younger than 18 years. Twenty-four of these children died.
The most frequently cited medicines in the cases were acetaminophen with codeine and promethazine with codeine with or without phenylephrine, either given to soothe postoperative pain, general pain, sore or strep throat pain, or cough and cold.
Ten of the 64 cases implicated the CYP2D6 genotype. Seven of the patients were “ultrarapid metabolizers,” five of whom died.
Data cited on tramadol included nine worldwide cases of tramadol-related respiratory depression occurring between 1969 and 2016, resulting in the deaths of three children, all under the age of 6 years, and all of whom were taking the drug for postoperative pain or fever.
All but one case of respiratory depression occurred within the first 24 hours of taking the drug. One of the patients was genotyped for CYP2D6, and found to have three functional alleles consistent with ultrarapid metabolism.
Mothers who are ultrarapid metabolizers of codeine can have high levels of morphine in their breast milk that are dangerous to their breastfed infants, whereas there is less of a threat posed by women with normal codeine metabolism because the amount of codeine secreted into the breast milk is low and dose dependent.
The FDA stated that data reveal numerous reports of respiratory depression and at least one death in infants of breastfeeding mothers taking these medicines, particularly mothers with the CYP2D6 genotype. Although there are FDA-cleared tests for ultrarapid metabolism, they are not commonly used.
The agency stated that breastfeeding women taking any opioid pain medicine, including codeine or tramadol should contact their health care provider if they notice the infant is sleeping more than 4 hours at a time, or if the infant is having difficulty breastfeeding or seems limp.
Clinicians are urged to report side effects that occur while using codeine or tramadol to the FDA’s MedWatch Adverse Event Reporting program.
The Food and Drug Administration is restricting the use of two opiates – codeine and tramadol – in children, and also warns they are potentially dangerous to infants of breastfeeding women.
Codeine is approved to treat pain and cough; tramadol is approved to treat pain.
“We understand that there are limited options when it comes to treating pain or cough in children, and that these changes may raise some questions for health care providers and parents. However, please know that our decision today was made based on the latest evidence and with this goal in mind: keeping our kids safe,” Douglas Throckmorton, MD, the deputy center director for regulatory programs at the FDA Center for Drug Evaluation and Research, said in a statement.
In 2013, the FDA updated prescription codeine labeling to contain a boxed warning and contraindication for children up to age 18 years regarding the risk of slowed breathing from codeine prescribed after tonsillectomy and/or adenoidectomy. And in 2015, the agency issued warnings about the risk of serious breathing problems in children who had ultrarapid metabolism of codeine and tramadol.
In the current safety statement, the FDA said it will require additional labeling changes, including contraindications for use of codeine or tramadol in all children younger than 12 years of age and a new contraindication for tramadol in children younger than 18 years being treated for pain after tonsillectomy and/or adenoidectomy, warnings about their use in children 12-18 years of age with certain medical conditions, and a stronger warning recommending against their use in nursing mothers.
Single-ingredient codeine and all tramadol-containing products are FDA-approved only for use in adults, the agency noted.
The agency cited particular concerns over those who are “ultra-rapid metabolizers” of substrates of the cytochrome P450 isoenzyme 2D6 (CYP2D6) genotype. These people more quickly convert codeine into potentially dangerously high levels of morphine, which can lead to fatal respiratory depression.
Supporting the restrictions and warning were data on 64 cases of respiratory depression that occurred worldwide between 1969 and 2015, when a codeine-based medicine was used in children younger than 18 years. Twenty-four of these children died.
The most frequently cited medicines in the cases were acetaminophen with codeine and promethazine with codeine with or without phenylephrine, either given to soothe postoperative pain, general pain, sore or strep throat pain, or cough and cold.
Ten of the 64 cases implicated the CYP2D6 genotype. Seven of the patients were “ultrarapid metabolizers,” five of whom died.
Data cited on tramadol included nine worldwide cases of tramadol-related respiratory depression occurring between 1969 and 2016, resulting in the deaths of three children, all under the age of 6 years, and all of whom were taking the drug for postoperative pain or fever.
All but one case of respiratory depression occurred within the first 24 hours of taking the drug. One of the patients was genotyped for CYP2D6, and found to have three functional alleles consistent with ultrarapid metabolism.
Mothers who are ultrarapid metabolizers of codeine can have high levels of morphine in their breast milk that are dangerous to their breastfed infants, whereas there is less of a threat posed by women with normal codeine metabolism because the amount of codeine secreted into the breast milk is low and dose dependent.
The FDA stated that data reveal numerous reports of respiratory depression and at least one death in infants of breastfeeding mothers taking these medicines, particularly mothers with the CYP2D6 genotype. Although there are FDA-cleared tests for ultrarapid metabolism, they are not commonly used.
The agency stated that breastfeeding women taking any opioid pain medicine, including codeine or tramadol should contact their health care provider if they notice the infant is sleeping more than 4 hours at a time, or if the infant is having difficulty breastfeeding or seems limp.
Clinicians are urged to report side effects that occur while using codeine or tramadol to the FDA’s MedWatch Adverse Event Reporting program.
The Food and Drug Administration is restricting the use of two opiates – codeine and tramadol – in children, and also warns they are potentially dangerous to infants of breastfeeding women.
Codeine is approved to treat pain and cough; tramadol is approved to treat pain.
“We understand that there are limited options when it comes to treating pain or cough in children, and that these changes may raise some questions for health care providers and parents. However, please know that our decision today was made based on the latest evidence and with this goal in mind: keeping our kids safe,” Douglas Throckmorton, MD, the deputy center director for regulatory programs at the FDA Center for Drug Evaluation and Research, said in a statement.
In 2013, the FDA updated prescription codeine labeling to contain a boxed warning and contraindication for children up to age 18 years regarding the risk of slowed breathing from codeine prescribed after tonsillectomy and/or adenoidectomy. And in 2015, the agency issued warnings about the risk of serious breathing problems in children who had ultrarapid metabolism of codeine and tramadol.
In the current safety statement, the FDA said it will require additional labeling changes, including contraindications for use of codeine or tramadol in all children younger than 12 years of age and a new contraindication for tramadol in children younger than 18 years being treated for pain after tonsillectomy and/or adenoidectomy, warnings about their use in children 12-18 years of age with certain medical conditions, and a stronger warning recommending against their use in nursing mothers.
Single-ingredient codeine and all tramadol-containing products are FDA-approved only for use in adults, the agency noted.
The agency cited particular concerns over those who are “ultra-rapid metabolizers” of substrates of the cytochrome P450 isoenzyme 2D6 (CYP2D6) genotype. These people more quickly convert codeine into potentially dangerously high levels of morphine, which can lead to fatal respiratory depression.
Supporting the restrictions and warning were data on 64 cases of respiratory depression that occurred worldwide between 1969 and 2015, when a codeine-based medicine was used in children younger than 18 years. Twenty-four of these children died.
The most frequently cited medicines in the cases were acetaminophen with codeine and promethazine with codeine with or without phenylephrine, either given to soothe postoperative pain, general pain, sore or strep throat pain, or cough and cold.
Ten of the 64 cases implicated the CYP2D6 genotype. Seven of the patients were “ultrarapid metabolizers,” five of whom died.
Data cited on tramadol included nine worldwide cases of tramadol-related respiratory depression occurring between 1969 and 2016, resulting in the deaths of three children, all under the age of 6 years, and all of whom were taking the drug for postoperative pain or fever.
All but one case of respiratory depression occurred within the first 24 hours of taking the drug. One of the patients was genotyped for CYP2D6, and found to have three functional alleles consistent with ultrarapid metabolism.
Mothers who are ultrarapid metabolizers of codeine can have high levels of morphine in their breast milk that are dangerous to their breastfed infants, whereas there is less of a threat posed by women with normal codeine metabolism because the amount of codeine secreted into the breast milk is low and dose dependent.
The FDA stated that data reveal numerous reports of respiratory depression and at least one death in infants of breastfeeding mothers taking these medicines, particularly mothers with the CYP2D6 genotype. Although there are FDA-cleared tests for ultrarapid metabolism, they are not commonly used.
The agency stated that breastfeeding women taking any opioid pain medicine, including codeine or tramadol should contact their health care provider if they notice the infant is sleeping more than 4 hours at a time, or if the infant is having difficulty breastfeeding or seems limp.
Clinicians are urged to report side effects that occur while using codeine or tramadol to the FDA’s MedWatch Adverse Event Reporting program.
Genital HPV prevalence tops 42% in adults
The overall prevalence of genital human papillomavirus (HPV) among adults aged 18-59 years was 42.5% in 2013-2014, with more than half of that representing infection with high-risk types, according to the National Center for Health Statistics.
Prevalence of the 14 HPV types (out of 37 total) considered to be high risk was almost 23% in 2013-2014. Men were significantly more likely than women to have any genital HPV (45% vs. 40%) and high-risk genital HPV (25% vs. 20%), the NCHS reported.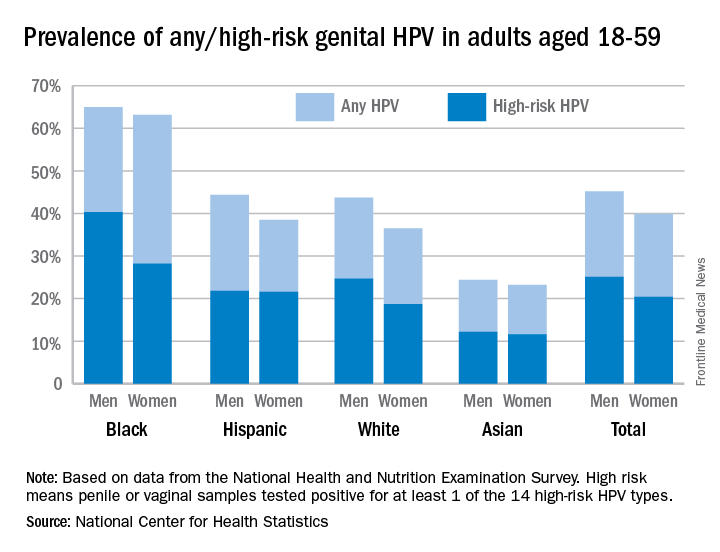
Oral rinse samples were collected as well for adults aged 18-69 years, and the overall prevalence was 7.3% for any oral HPV and 4% for high-risk HPV in 2011-2014. Prevalence among men was significantly higher than among women for all oral HPV (11.5% vs. 3.3%) and for high-risk HPV (6.8% vs. 1.2%), the NCHS said.
The overall prevalence of genital human papillomavirus (HPV) among adults aged 18-59 years was 42.5% in 2013-2014, with more than half of that representing infection with high-risk types, according to the National Center for Health Statistics.
Prevalence of the 14 HPV types (out of 37 total) considered to be high risk was almost 23% in 2013-2014. Men were significantly more likely than women to have any genital HPV (45% vs. 40%) and high-risk genital HPV (25% vs. 20%), the NCHS reported.
Oral rinse samples were collected as well for adults aged 18-69 years, and the overall prevalence was 7.3% for any oral HPV and 4% for high-risk HPV in 2011-2014. Prevalence among men was significantly higher than among women for all oral HPV (11.5% vs. 3.3%) and for high-risk HPV (6.8% vs. 1.2%), the NCHS said.
The overall prevalence of genital human papillomavirus (HPV) among adults aged 18-59 years was 42.5% in 2013-2014, with more than half of that representing infection with high-risk types, according to the National Center for Health Statistics.
Prevalence of the 14 HPV types (out of 37 total) considered to be high risk was almost 23% in 2013-2014. Men were significantly more likely than women to have any genital HPV (45% vs. 40%) and high-risk genital HPV (25% vs. 20%), the NCHS reported.
Oral rinse samples were collected as well for adults aged 18-69 years, and the overall prevalence was 7.3% for any oral HPV and 4% for high-risk HPV in 2011-2014. Prevalence among men was significantly higher than among women for all oral HPV (11.5% vs. 3.3%) and for high-risk HPV (6.8% vs. 1.2%), the NCHS said.
FDA approves new treatment for episodic cluster headaches in adults
The Food and Drug Administration announced April 18 the approval of the gammaCore device to treat pain associated with episodic cluster headache in adult patients.
The agency based its approval of the noninvasive vagus nerve stimulator device on subgroup analyses of episodic cluster headache patients in the ACT1 and ACT2 trials, which were double-blind, placebo-controlled, randomized studies.
In the ACT1 trial of 85 patients, 34.2% experienced a reduction in pain (defined as the percentage of patients who reported mild or no pain 15 minutes after treatment initiation with gammaCore), compared with 10.6% of patients treated with placebo (P = .008). Of 27 patients in the ACT2 trial, 47.5% of patients using the device were pain free at 15 minutes after the onset of pain from cluster headache, with no use of rescue medication through the 30-minute treatment period, which was significantly greater than for placebo (6.2%; P = .003).
“Cluster headache is a rare, debilitating, and difficult to treat disorder with few effective acute therapies,” Stephen Silberstein, MD, director of the Headache Center at Jefferson University, Philadelphia, said in a statement from the manufacturer, electroCore. “The FDA release of gammaCore is an important advance in the treatment of the pain associated with cluster headache. It is a way for patients to treat their symptoms as often as they need to use the device. It does not have the side effects or dose limitations of commonly prescribed treatments or the need for invasive implantation procedures, which can be inconvenient, costly, and high-risk.”
The gammaCore device works by transmitting a mild electrical stimulation to the vagus nerve through the skin, resulting in a reduction of pain. Currently, gammaCore is in use only outside of the United States, including in the European Union. Commercial availability of gammaCore in the United States is expected to begin early in the third quarter of 2017, the company said.
The Food and Drug Administration announced April 18 the approval of the gammaCore device to treat pain associated with episodic cluster headache in adult patients.
The agency based its approval of the noninvasive vagus nerve stimulator device on subgroup analyses of episodic cluster headache patients in the ACT1 and ACT2 trials, which were double-blind, placebo-controlled, randomized studies.
In the ACT1 trial of 85 patients, 34.2% experienced a reduction in pain (defined as the percentage of patients who reported mild or no pain 15 minutes after treatment initiation with gammaCore), compared with 10.6% of patients treated with placebo (P = .008). Of 27 patients in the ACT2 trial, 47.5% of patients using the device were pain free at 15 minutes after the onset of pain from cluster headache, with no use of rescue medication through the 30-minute treatment period, which was significantly greater than for placebo (6.2%; P = .003).
“Cluster headache is a rare, debilitating, and difficult to treat disorder with few effective acute therapies,” Stephen Silberstein, MD, director of the Headache Center at Jefferson University, Philadelphia, said in a statement from the manufacturer, electroCore. “The FDA release of gammaCore is an important advance in the treatment of the pain associated with cluster headache. It is a way for patients to treat their symptoms as often as they need to use the device. It does not have the side effects or dose limitations of commonly prescribed treatments or the need for invasive implantation procedures, which can be inconvenient, costly, and high-risk.”
The gammaCore device works by transmitting a mild electrical stimulation to the vagus nerve through the skin, resulting in a reduction of pain. Currently, gammaCore is in use only outside of the United States, including in the European Union. Commercial availability of gammaCore in the United States is expected to begin early in the third quarter of 2017, the company said.
The Food and Drug Administration announced April 18 the approval of the gammaCore device to treat pain associated with episodic cluster headache in adult patients.
The agency based its approval of the noninvasive vagus nerve stimulator device on subgroup analyses of episodic cluster headache patients in the ACT1 and ACT2 trials, which were double-blind, placebo-controlled, randomized studies.
In the ACT1 trial of 85 patients, 34.2% experienced a reduction in pain (defined as the percentage of patients who reported mild or no pain 15 minutes after treatment initiation with gammaCore), compared with 10.6% of patients treated with placebo (P = .008). Of 27 patients in the ACT2 trial, 47.5% of patients using the device were pain free at 15 minutes after the onset of pain from cluster headache, with no use of rescue medication through the 30-minute treatment period, which was significantly greater than for placebo (6.2%; P = .003).
“Cluster headache is a rare, debilitating, and difficult to treat disorder with few effective acute therapies,” Stephen Silberstein, MD, director of the Headache Center at Jefferson University, Philadelphia, said in a statement from the manufacturer, electroCore. “The FDA release of gammaCore is an important advance in the treatment of the pain associated with cluster headache. It is a way for patients to treat their symptoms as often as they need to use the device. It does not have the side effects or dose limitations of commonly prescribed treatments or the need for invasive implantation procedures, which can be inconvenient, costly, and high-risk.”
The gammaCore device works by transmitting a mild electrical stimulation to the vagus nerve through the skin, resulting in a reduction of pain. Currently, gammaCore is in use only outside of the United States, including in the European Union. Commercial availability of gammaCore in the United States is expected to begin early in the third quarter of 2017, the company said.
Dupilumab approval marks the first targeted treatment for AD
The approval of dupilumab, the first targeted biologic therapy approved in the United States for treatment of atopic dermatitis, provides a welcome, long-awaited alternative to currently available therapies for moderate to severe disease, according to two of the pivotal trial investigators.
In late March, the Food and Drug Administration approved dupilumab, a monoclonal antibody that inhibits signaling of both interleukin-4 and interleukin-13, for the treatment of moderate to severe AD in adults “whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.” It is administered subcutaneously, with an initial 600-mg dose, followed by 300 mg every other week, according to the prescribing information.
Dupilumab (Dupixent) was the second novel treatment approved for AD in less than 4 months – after years of no approvals of new therapies for this indication. In December, crisaborole ointment (Eucrisa) was approved for treatment of mild to moderate AD in patients aged 2 years and older. Crisaborole is a topical phosphodiesterase-4 inhibitor.
Dupilumab’s approval was based on three phase III studies of adults with moderate to severe AD: SOLO-1 and SOLO-2, which evaluated dupilumab as monotherapy, and the CHRONOS study, which compared dupilumab with topical corticosteroids to treatment with topical corticosteroids alone.
At 16 weeks in the SOLO trials, those treated with dupilumab had improvements in signs and symptoms of AD, including pruritus, anxiety and depression, and quality of life, compared with placebo. Almost 40% of those treated with dupilumab met the primary outcome – a score of clear or almost clear on the Investigator’s Global Assessment and a reduction of 2 points or more in that score from baseline – compared with 8%-10% of those on placebo (P less than .001). Injection-site reactions and conjunctivitis were more common among those treated with dupilumab (N Engl J Med. 2016;375:2335-48).

Emma Guttman-Yassky, MD, PhD, also an investigator in the SOLO 1 and SOLO 2 trials, said that prior to the approval, “we had nothing to treat our patients safely long term.”

“With dupilumab, because it’s specific, it provides the safety that we need,” while providing efficacy that is similar to or better than that of cyclosporine, with the caveat that dupilumab and cyclosporine have not been evaluated in a head-to-head study, Dr. Guttman-Yassky, professor and vice-chair of dermatology at the Icahn School of Medicine at Mount Sinai, New York, said in an interview.
About 5%-10% of those treated at her site had allergic conjunctivitis; most cases resolved with eye drops. So far, “it looks very safe but we need to have long-term data,” she added.
Dr. Simpson said that he was pleased to see the maintenance of therapeutic effects with no emergence of new side effects with the report of 52-week CHRONOS data in March at the American Academy of Dermatology annual meeting.
“We’re entering the era and the decade of atopic dermatitis,” he said. “Dermatologists should look out for many new topical and systemic therapies now that we’re uncovering and figuring out the pathophysiology of atopic dermatitis.”
Dupilumab, which costs $37,000 per year, is marketed by Regeneron and Sanofi. Studies in children and adolescents with AD are underway.
Dr. Guttman-Yassky has received funding from Regeneron for mechanistic studies and is working with most companies developing AD treatments. Dr. Simpson has received research grants from and serves as a consultant to Regeneron, as well as other pharmaceutical companies.
The approval of dupilumab, the first targeted biologic therapy approved in the United States for treatment of atopic dermatitis, provides a welcome, long-awaited alternative to currently available therapies for moderate to severe disease, according to two of the pivotal trial investigators.
In late March, the Food and Drug Administration approved dupilumab, a monoclonal antibody that inhibits signaling of both interleukin-4 and interleukin-13, for the treatment of moderate to severe AD in adults “whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.” It is administered subcutaneously, with an initial 600-mg dose, followed by 300 mg every other week, according to the prescribing information.
Dupilumab (Dupixent) was the second novel treatment approved for AD in less than 4 months – after years of no approvals of new therapies for this indication. In December, crisaborole ointment (Eucrisa) was approved for treatment of mild to moderate AD in patients aged 2 years and older. Crisaborole is a topical phosphodiesterase-4 inhibitor.
Dupilumab’s approval was based on three phase III studies of adults with moderate to severe AD: SOLO-1 and SOLO-2, which evaluated dupilumab as monotherapy, and the CHRONOS study, which compared dupilumab with topical corticosteroids to treatment with topical corticosteroids alone.
At 16 weeks in the SOLO trials, those treated with dupilumab had improvements in signs and symptoms of AD, including pruritus, anxiety and depression, and quality of life, compared with placebo. Almost 40% of those treated with dupilumab met the primary outcome – a score of clear or almost clear on the Investigator’s Global Assessment and a reduction of 2 points or more in that score from baseline – compared with 8%-10% of those on placebo (P less than .001). Injection-site reactions and conjunctivitis were more common among those treated with dupilumab (N Engl J Med. 2016;375:2335-48).
Emma Guttman-Yassky, MD, PhD, also an investigator in the SOLO 1 and SOLO 2 trials, said that prior to the approval, “we had nothing to treat our patients safely long term.”
“With dupilumab, because it’s specific, it provides the safety that we need,” while providing efficacy that is similar to or better than that of cyclosporine, with the caveat that dupilumab and cyclosporine have not been evaluated in a head-to-head study, Dr. Guttman-Yassky, professor and vice-chair of dermatology at the Icahn School of Medicine at Mount Sinai, New York, said in an interview.
About 5%-10% of those treated at her site had allergic conjunctivitis; most cases resolved with eye drops. So far, “it looks very safe but we need to have long-term data,” she added.
Dr. Simpson said that he was pleased to see the maintenance of therapeutic effects with no emergence of new side effects with the report of 52-week CHRONOS data in March at the American Academy of Dermatology annual meeting.
“We’re entering the era and the decade of atopic dermatitis,” he said. “Dermatologists should look out for many new topical and systemic therapies now that we’re uncovering and figuring out the pathophysiology of atopic dermatitis.”
Dupilumab, which costs $37,000 per year, is marketed by Regeneron and Sanofi. Studies in children and adolescents with AD are underway.
Dr. Guttman-Yassky has received funding from Regeneron for mechanistic studies and is working with most companies developing AD treatments. Dr. Simpson has received research grants from and serves as a consultant to Regeneron, as well as other pharmaceutical companies.
The approval of dupilumab, the first targeted biologic therapy approved in the United States for treatment of atopic dermatitis, provides a welcome, long-awaited alternative to currently available therapies for moderate to severe disease, according to two of the pivotal trial investigators.
In late March, the Food and Drug Administration approved dupilumab, a monoclonal antibody that inhibits signaling of both interleukin-4 and interleukin-13, for the treatment of moderate to severe AD in adults “whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable.” It is administered subcutaneously, with an initial 600-mg dose, followed by 300 mg every other week, according to the prescribing information.
Dupilumab (Dupixent) was the second novel treatment approved for AD in less than 4 months – after years of no approvals of new therapies for this indication. In December, crisaborole ointment (Eucrisa) was approved for treatment of mild to moderate AD in patients aged 2 years and older. Crisaborole is a topical phosphodiesterase-4 inhibitor.
Dupilumab’s approval was based on three phase III studies of adults with moderate to severe AD: SOLO-1 and SOLO-2, which evaluated dupilumab as monotherapy, and the CHRONOS study, which compared dupilumab with topical corticosteroids to treatment with topical corticosteroids alone.
At 16 weeks in the SOLO trials, those treated with dupilumab had improvements in signs and symptoms of AD, including pruritus, anxiety and depression, and quality of life, compared with placebo. Almost 40% of those treated with dupilumab met the primary outcome – a score of clear or almost clear on the Investigator’s Global Assessment and a reduction of 2 points or more in that score from baseline – compared with 8%-10% of those on placebo (P less than .001). Injection-site reactions and conjunctivitis were more common among those treated with dupilumab (N Engl J Med. 2016;375:2335-48).
Emma Guttman-Yassky, MD, PhD, also an investigator in the SOLO 1 and SOLO 2 trials, said that prior to the approval, “we had nothing to treat our patients safely long term.”
“With dupilumab, because it’s specific, it provides the safety that we need,” while providing efficacy that is similar to or better than that of cyclosporine, with the caveat that dupilumab and cyclosporine have not been evaluated in a head-to-head study, Dr. Guttman-Yassky, professor and vice-chair of dermatology at the Icahn School of Medicine at Mount Sinai, New York, said in an interview.
About 5%-10% of those treated at her site had allergic conjunctivitis; most cases resolved with eye drops. So far, “it looks very safe but we need to have long-term data,” she added.
Dr. Simpson said that he was pleased to see the maintenance of therapeutic effects with no emergence of new side effects with the report of 52-week CHRONOS data in March at the American Academy of Dermatology annual meeting.
“We’re entering the era and the decade of atopic dermatitis,” he said. “Dermatologists should look out for many new topical and systemic therapies now that we’re uncovering and figuring out the pathophysiology of atopic dermatitis.”
Dupilumab, which costs $37,000 per year, is marketed by Regeneron and Sanofi. Studies in children and adolescents with AD are underway.
Dr. Guttman-Yassky has received funding from Regeneron for mechanistic studies and is working with most companies developing AD treatments. Dr. Simpson has received research grants from and serves as a consultant to Regeneron, as well as other pharmaceutical companies.
Crohn’s hospitalizations up significantly … or not
Hospitalizations for Crohn’s disease were up by a statistically significant 35% from 2003 to 2013 … or they were up just 5%, according to the Centers for Disease Control and Prevention.
It depends on how you look at it. In 2013, age-adjusted hospitalization was 59.7 stays per 100,000 population for Crohn’s as any-listed diagnosis – indicating that patients had Crohn’s disease but that it was not necessarily the main reason they were being hospitalized – compared with 44.2 per 100,000 in 2003. That’s an increase of 35%, the CDC said (MMWR. 2017 Apr 14;66[14]:377-81).
Hospitalizations with Crohn’s as the first-listed diagnosis – making it the main reason for the admission – were up 5% over the same period: 18.2 stays per 100,000 pop. in 2003 and 19.1 per 100,000 in 2013. That increase is not statistically significant, the CDC noted.
The increase in any-listed diagnosis “might represent greater physician awareness and diagnosis of Crohn’s disease or more complete coding of secondary diagnoses by physicians,” CDC investigators suggested.
Increases in hospitalizations for any-listed Crohn’s were significant for both men (40.5%) and women (31.6%). For first-listed Crohn’s, women saw a slight but not significant decrease of 1% while men had a significant 14.5% increase, according to the report, which was based on data from the National Inpatient Sample.
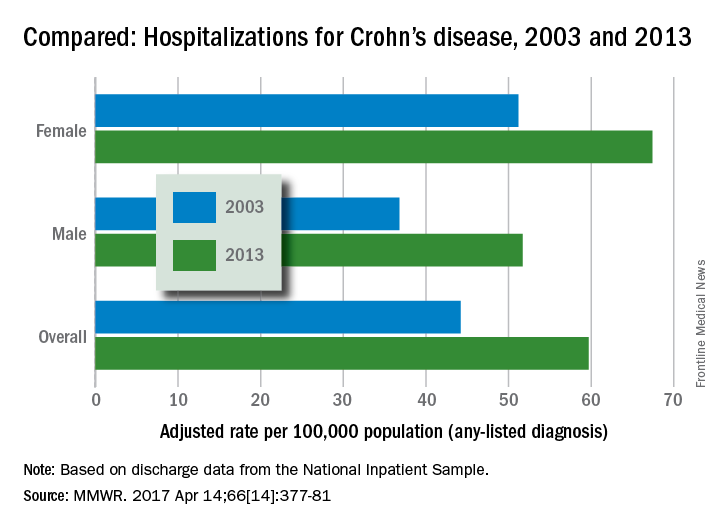
Hospitalizations for Crohn’s disease were up by a statistically significant 35% from 2003 to 2013 … or they were up just 5%, according to the Centers for Disease Control and Prevention.
It depends on how you look at it. In 2013, age-adjusted hospitalization was 59.7 stays per 100,000 population for Crohn’s as any-listed diagnosis – indicating that patients had Crohn’s disease but that it was not necessarily the main reason they were being hospitalized – compared with 44.2 per 100,000 in 2003. That’s an increase of 35%, the CDC said (MMWR. 2017 Apr 14;66[14]:377-81).
Hospitalizations with Crohn’s as the first-listed diagnosis – making it the main reason for the admission – were up 5% over the same period: 18.2 stays per 100,000 pop. in 2003 and 19.1 per 100,000 in 2013. That increase is not statistically significant, the CDC noted.
The increase in any-listed diagnosis “might represent greater physician awareness and diagnosis of Crohn’s disease or more complete coding of secondary diagnoses by physicians,” CDC investigators suggested.
Increases in hospitalizations for any-listed Crohn’s were significant for both men (40.5%) and women (31.6%). For first-listed Crohn’s, women saw a slight but not significant decrease of 1% while men had a significant 14.5% increase, according to the report, which was based on data from the National Inpatient Sample.
Hospitalizations for Crohn’s disease were up by a statistically significant 35% from 2003 to 2013 … or they were up just 5%, according to the Centers for Disease Control and Prevention.
It depends on how you look at it. In 2013, age-adjusted hospitalization was 59.7 stays per 100,000 population for Crohn’s as any-listed diagnosis – indicating that patients had Crohn’s disease but that it was not necessarily the main reason they were being hospitalized – compared with 44.2 per 100,000 in 2003. That’s an increase of 35%, the CDC said (MMWR. 2017 Apr 14;66[14]:377-81).
Hospitalizations with Crohn’s as the first-listed diagnosis – making it the main reason for the admission – were up 5% over the same period: 18.2 stays per 100,000 pop. in 2003 and 19.1 per 100,000 in 2013. That increase is not statistically significant, the CDC noted.
The increase in any-listed diagnosis “might represent greater physician awareness and diagnosis of Crohn’s disease or more complete coding of secondary diagnoses by physicians,” CDC investigators suggested.
Increases in hospitalizations for any-listed Crohn’s were significant for both men (40.5%) and women (31.6%). For first-listed Crohn’s, women saw a slight but not significant decrease of 1% while men had a significant 14.5% increase, according to the report, which was based on data from the National Inpatient Sample.
FROM MORBIDITY AND MORTALITY WEEKLY REPORT
FDA approves Ingrezza for tardive dyskinesia in adults
The Food and Drug Administration has approved valbenazine capsules for the treatment of tardive dyskinesia in adults.
The approval, announced April 11, came after a 6-week, placebo-controlled trial of 234 participants that compared valbenazine to placebo. At the end of the trial, those who took valbenazine experienced improvement in the severity of involuntary movements, compared with those who received placebo. As a vesicular monoamine transporter type 2 inhibitor, valbenazine works by regulating the amount of dopamine that is released into nerve cells by blocking a protein in the brain. The drug, the first to receive approval for tardive dyskinesia in adults, will be marketed as Ingrezza by Neurocrine Biosciences. Valbenazine is reportedly being studied as a treatment for Tourette syndrome in children and adolescents.![]()
In a statement, Mitchell Mathis, MD, director of the FDA’s division of psychiatry products in the Center for Drug Evaluation and Research, praised the approval as “an important advance for patients suffering with tardive dyskinesia.”
Characterized by uncontrolled movement of the face and body, tardive dyskinesia is a side effect in up to 8% of patients taking typical and atypical antipsychotics. The movement disorder also can cause other debilitating problems, including difficulty with chewing, speaking, and swallowing. Some data show that in about 87% of cases, the condition is irreversible even 3 years after the inciting agent has been discontinued (Parkinsonism Relat Disord. 2016. 32;124-6).
“Tardive dyskinesia can be disabling and can further stigmatize patients with mental illness,” Dr. Mathis said in the statement.
The side effects of valbenazine include sleepiness and QT prolongation. The agency said the patients taking the drug should not drive, operate heavy machinery, or engage in other potentially dangerous activities. Likewise, the drug should be avoided in patients with congenital long QT syndrome or in those with abnormal heartbeats associated with a prolonged QT interval, the agency said.
The Food and Drug Administration has approved valbenazine capsules for the treatment of tardive dyskinesia in adults.
The approval, announced April 11, came after a 6-week, placebo-controlled trial of 234 participants that compared valbenazine to placebo. At the end of the trial, those who took valbenazine experienced improvement in the severity of involuntary movements, compared with those who received placebo. As a vesicular monoamine transporter type 2 inhibitor, valbenazine works by regulating the amount of dopamine that is released into nerve cells by blocking a protein in the brain. The drug, the first to receive approval for tardive dyskinesia in adults, will be marketed as Ingrezza by Neurocrine Biosciences. Valbenazine is reportedly being studied as a treatment for Tourette syndrome in children and adolescents.![]()
In a statement, Mitchell Mathis, MD, director of the FDA’s division of psychiatry products in the Center for Drug Evaluation and Research, praised the approval as “an important advance for patients suffering with tardive dyskinesia.”
Characterized by uncontrolled movement of the face and body, tardive dyskinesia is a side effect in up to 8% of patients taking typical and atypical antipsychotics. The movement disorder also can cause other debilitating problems, including difficulty with chewing, speaking, and swallowing. Some data show that in about 87% of cases, the condition is irreversible even 3 years after the inciting agent has been discontinued (Parkinsonism Relat Disord. 2016. 32;124-6).
“Tardive dyskinesia can be disabling and can further stigmatize patients with mental illness,” Dr. Mathis said in the statement.
The side effects of valbenazine include sleepiness and QT prolongation. The agency said the patients taking the drug should not drive, operate heavy machinery, or engage in other potentially dangerous activities. Likewise, the drug should be avoided in patients with congenital long QT syndrome or in those with abnormal heartbeats associated with a prolonged QT interval, the agency said.
The Food and Drug Administration has approved valbenazine capsules for the treatment of tardive dyskinesia in adults.
The approval, announced April 11, came after a 6-week, placebo-controlled trial of 234 participants that compared valbenazine to placebo. At the end of the trial, those who took valbenazine experienced improvement in the severity of involuntary movements, compared with those who received placebo. As a vesicular monoamine transporter type 2 inhibitor, valbenazine works by regulating the amount of dopamine that is released into nerve cells by blocking a protein in the brain. The drug, the first to receive approval for tardive dyskinesia in adults, will be marketed as Ingrezza by Neurocrine Biosciences. Valbenazine is reportedly being studied as a treatment for Tourette syndrome in children and adolescents.![]()
In a statement, Mitchell Mathis, MD, director of the FDA’s division of psychiatry products in the Center for Drug Evaluation and Research, praised the approval as “an important advance for patients suffering with tardive dyskinesia.”
Characterized by uncontrolled movement of the face and body, tardive dyskinesia is a side effect in up to 8% of patients taking typical and atypical antipsychotics. The movement disorder also can cause other debilitating problems, including difficulty with chewing, speaking, and swallowing. Some data show that in about 87% of cases, the condition is irreversible even 3 years after the inciting agent has been discontinued (Parkinsonism Relat Disord. 2016. 32;124-6).
“Tardive dyskinesia can be disabling and can further stigmatize patients with mental illness,” Dr. Mathis said in the statement.
The side effects of valbenazine include sleepiness and QT prolongation. The agency said the patients taking the drug should not drive, operate heavy machinery, or engage in other potentially dangerous activities. Likewise, the drug should be avoided in patients with congenital long QT syndrome or in those with abnormal heartbeats associated with a prolonged QT interval, the agency said.
Decline in U.S. flu activity puts end of season within sight
Outpatient visits for influenza were down again in the United States during the week ending April 1, and the number of states at the highest level of flu activity dropped from seven to four, according to the Centers for Disease Control and Prevention.
The national proportion of outpatient visits for influenza-like illness (ILI) was 2.9% for the week ending April 1, compared with 3.2% the week before, the CDC’s Outpatient Influenza-like Illness Surveillance Network reported. The national baseline level is 2.2%.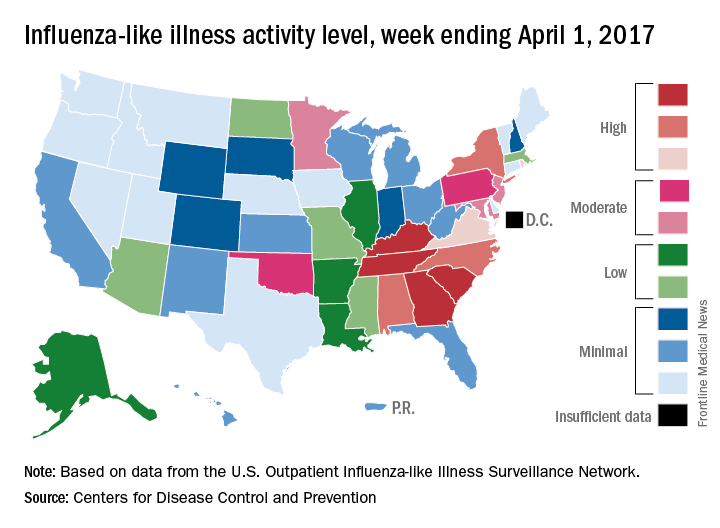
There were 7 flu-related pediatric deaths reported for the week ending April 1 – six of the deaths occurred in previous weeks – which brings the total for the 2016-2017 season to 68, the CDC said. The largest share of those deaths by age group has been among 5- to 11-year-olds (36.8%), followed by those aged 12-17 years (26.5%), 6-23 months (16.2%), 2-4 years (14.7%), and 0-5 months (5.9%).
Outpatient visits for influenza were down again in the United States during the week ending April 1, and the number of states at the highest level of flu activity dropped from seven to four, according to the Centers for Disease Control and Prevention.
The national proportion of outpatient visits for influenza-like illness (ILI) was 2.9% for the week ending April 1, compared with 3.2% the week before, the CDC’s Outpatient Influenza-like Illness Surveillance Network reported. The national baseline level is 2.2%.
There were 7 flu-related pediatric deaths reported for the week ending April 1 – six of the deaths occurred in previous weeks – which brings the total for the 2016-2017 season to 68, the CDC said. The largest share of those deaths by age group has been among 5- to 11-year-olds (36.8%), followed by those aged 12-17 years (26.5%), 6-23 months (16.2%), 2-4 years (14.7%), and 0-5 months (5.9%).
Outpatient visits for influenza were down again in the United States during the week ending April 1, and the number of states at the highest level of flu activity dropped from seven to four, according to the Centers for Disease Control and Prevention.
The national proportion of outpatient visits for influenza-like illness (ILI) was 2.9% for the week ending April 1, compared with 3.2% the week before, the CDC’s Outpatient Influenza-like Illness Surveillance Network reported. The national baseline level is 2.2%.
There were 7 flu-related pediatric deaths reported for the week ending April 1 – six of the deaths occurred in previous weeks – which brings the total for the 2016-2017 season to 68, the CDC said. The largest share of those deaths by age group has been among 5- to 11-year-olds (36.8%), followed by those aged 12-17 years (26.5%), 6-23 months (16.2%), 2-4 years (14.7%), and 0-5 months (5.9%).
FDA approves Sovaldi, Harvoni for HCV in ages 12-plus
The Food and Drug Administration has approved the use of Sovaldi (sofosbuvir) and Harvoni (ledipasvir and sofosbuvir) for the treatment of hepatitis C virus (HCV) in children aged 12 years and older.
The drugs – the first direct-acting, potentially curative antiviral treatments approved for children and adolescents with HCV – previously were approved for adults. The supplemental applications submitted by Gilead Sciences, which markets the drugs, were approved by the FDA on April 7 and expand the use of these drugs to pediatric patients aged 12 and up who weigh at least 77 pounds, and who have either mild or no cirrhosis; Sovaldi is indicated for those with HCV genotypes 2 or 3, and Harvoni is indicated for those with HCV genotypes 1, 4, 5, or 6.
“These approvals will help change the landscape for HCV treatment by addressing an unmet need in children and adolescents,” Edward Cox, MD, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, said in a press statement.![]()
Of note, hepatitis B virus (HBV) reactivation has been reported in adults with HCV/HBV coinfection who were treated with these drugs, but who were not receiving HBV antiviral therapy; therefore, all patients should be screened for evidence of current or prior HBV infection before starting treatment with Harvoni or Sovaldi, according to the FDA statement.
The Food and Drug Administration has approved the use of Sovaldi (sofosbuvir) and Harvoni (ledipasvir and sofosbuvir) for the treatment of hepatitis C virus (HCV) in children aged 12 years and older.
The drugs – the first direct-acting, potentially curative antiviral treatments approved for children and adolescents with HCV – previously were approved for adults. The supplemental applications submitted by Gilead Sciences, which markets the drugs, were approved by the FDA on April 7 and expand the use of these drugs to pediatric patients aged 12 and up who weigh at least 77 pounds, and who have either mild or no cirrhosis; Sovaldi is indicated for those with HCV genotypes 2 or 3, and Harvoni is indicated for those with HCV genotypes 1, 4, 5, or 6.
“These approvals will help change the landscape for HCV treatment by addressing an unmet need in children and adolescents,” Edward Cox, MD, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, said in a press statement.![]()
Of note, hepatitis B virus (HBV) reactivation has been reported in adults with HCV/HBV coinfection who were treated with these drugs, but who were not receiving HBV antiviral therapy; therefore, all patients should be screened for evidence of current or prior HBV infection before starting treatment with Harvoni or Sovaldi, according to the FDA statement.
The Food and Drug Administration has approved the use of Sovaldi (sofosbuvir) and Harvoni (ledipasvir and sofosbuvir) for the treatment of hepatitis C virus (HCV) in children aged 12 years and older.
The drugs – the first direct-acting, potentially curative antiviral treatments approved for children and adolescents with HCV – previously were approved for adults. The supplemental applications submitted by Gilead Sciences, which markets the drugs, were approved by the FDA on April 7 and expand the use of these drugs to pediatric patients aged 12 and up who weigh at least 77 pounds, and who have either mild or no cirrhosis; Sovaldi is indicated for those with HCV genotypes 2 or 3, and Harvoni is indicated for those with HCV genotypes 1, 4, 5, or 6.
“These approvals will help change the landscape for HCV treatment by addressing an unmet need in children and adolescents,” Edward Cox, MD, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, said in a press statement.![]()
Of note, hepatitis B virus (HBV) reactivation has been reported in adults with HCV/HBV coinfection who were treated with these drugs, but who were not receiving HBV antiviral therapy; therefore, all patients should be screened for evidence of current or prior HBV infection before starting treatment with Harvoni or Sovaldi, according to the FDA statement.