User login
FDA approves deutetrabenazine for Huntington’s-associated chorea
The Food and Drug Administration has approved deutetrabenazine (Austedo) for the treatment of chorea associated with Huntington’s disease, according to an announcement by the drug’s manufacturer, Teva Pharmaceutical.
The random, sudden, involuntary twisting and writhing movements of chorea constitute perhaps the most striking symptom of Huntington’s disease and affect about 90% of the roughly 35,000 Americans with the fatal neurodegenerative disorder. Deutetrabenazine is only the second product approved to treat Huntington’s in any capacity, and it is the first in nearly a decade.
The most common side effects of deutetrabenazine were somnolence, diarrhea, dry mouth, and fatigue. Deutetrabenazine is contraindicated in patients with hepatic impairment; patients taking monoamine oxidase inhibitors (MAOIs), or within 14 days of discontinuing MAOI therapy; patients taking reserpine or within 20 days of discontinuing reserpine; and patients taking tetrabenazine. Deutetrabenazine can cause worsening of mood and is not recommended for suicidal patients or patients with improperly treated depression.
“Chorea associated with Huntington’s disease has a significant impact on those living with the disease and their families. The FDA’s approval of Austedo represents an important new treatment option for people with HD and highlights the need for more therapeutic resources for this underserved patient community,” Louise Vetter, CEO of the Huntington’s Disease Society of America, said in the company’s announcement.
The Food and Drug Administration has approved deutetrabenazine (Austedo) for the treatment of chorea associated with Huntington’s disease, according to an announcement by the drug’s manufacturer, Teva Pharmaceutical.
The random, sudden, involuntary twisting and writhing movements of chorea constitute perhaps the most striking symptom of Huntington’s disease and affect about 90% of the roughly 35,000 Americans with the fatal neurodegenerative disorder. Deutetrabenazine is only the second product approved to treat Huntington’s in any capacity, and it is the first in nearly a decade.
The most common side effects of deutetrabenazine were somnolence, diarrhea, dry mouth, and fatigue. Deutetrabenazine is contraindicated in patients with hepatic impairment; patients taking monoamine oxidase inhibitors (MAOIs), or within 14 days of discontinuing MAOI therapy; patients taking reserpine or within 20 days of discontinuing reserpine; and patients taking tetrabenazine. Deutetrabenazine can cause worsening of mood and is not recommended for suicidal patients or patients with improperly treated depression.
“Chorea associated with Huntington’s disease has a significant impact on those living with the disease and their families. The FDA’s approval of Austedo represents an important new treatment option for people with HD and highlights the need for more therapeutic resources for this underserved patient community,” Louise Vetter, CEO of the Huntington’s Disease Society of America, said in the company’s announcement.
The Food and Drug Administration has approved deutetrabenazine (Austedo) for the treatment of chorea associated with Huntington’s disease, according to an announcement by the drug’s manufacturer, Teva Pharmaceutical.
The random, sudden, involuntary twisting and writhing movements of chorea constitute perhaps the most striking symptom of Huntington’s disease and affect about 90% of the roughly 35,000 Americans with the fatal neurodegenerative disorder. Deutetrabenazine is only the second product approved to treat Huntington’s in any capacity, and it is the first in nearly a decade.
The most common side effects of deutetrabenazine were somnolence, diarrhea, dry mouth, and fatigue. Deutetrabenazine is contraindicated in patients with hepatic impairment; patients taking monoamine oxidase inhibitors (MAOIs), or within 14 days of discontinuing MAOI therapy; patients taking reserpine or within 20 days of discontinuing reserpine; and patients taking tetrabenazine. Deutetrabenazine can cause worsening of mood and is not recommended for suicidal patients or patients with improperly treated depression.
“Chorea associated with Huntington’s disease has a significant impact on those living with the disease and their families. The FDA’s approval of Austedo represents an important new treatment option for people with HD and highlights the need for more therapeutic resources for this underserved patient community,” Louise Vetter, CEO of the Huntington’s Disease Society of America, said in the company’s announcement.
Latest weekly flu data show no decline in visits
Outpatient visits for influenza-like illness (ILI) held steady for the week ending March 25, but the number of states at the “high” range of activity dropped from 12 from 10 the previous week, according to the Centers for Disease Prevention and Control.
The proportion of outpatient visits for ILI was 3.2% for the second consecutive week, which halted the slowdown in activity that began the week ending Feb. 18. That 3.2% represents just under 25,000 visits for ILI of the almost 747,000 total visits reported to the Outpatient Influenza-like Illness Surveillance Network (ILINet) for the week ending March 25. By age, the largest groups with ILI visits for the week were individuals aged 5-24 years (41%) and those aged 4 years and under (20%), the CDC reported.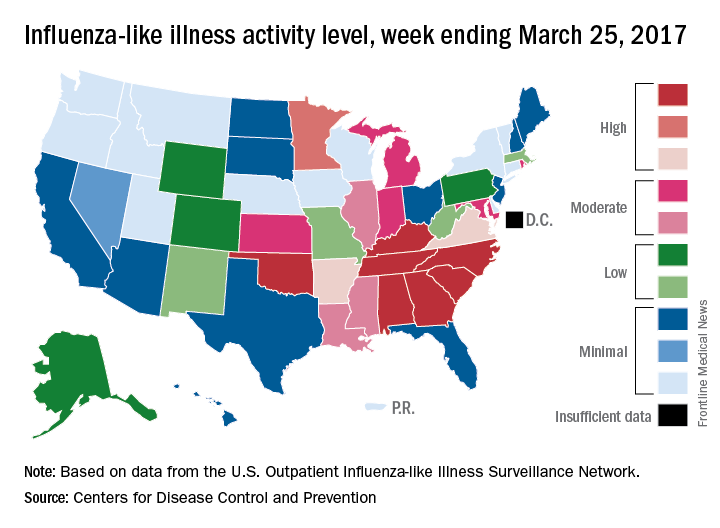
There were six flu-related pediatric deaths reported during the week ending March 25, but all occurred in earlier weeks. The total number of such deaths is now 61 for the 2016-2017 season, the CDC said.
Outpatient visits for influenza-like illness (ILI) held steady for the week ending March 25, but the number of states at the “high” range of activity dropped from 12 from 10 the previous week, according to the Centers for Disease Prevention and Control.
The proportion of outpatient visits for ILI was 3.2% for the second consecutive week, which halted the slowdown in activity that began the week ending Feb. 18. That 3.2% represents just under 25,000 visits for ILI of the almost 747,000 total visits reported to the Outpatient Influenza-like Illness Surveillance Network (ILINet) for the week ending March 25. By age, the largest groups with ILI visits for the week were individuals aged 5-24 years (41%) and those aged 4 years and under (20%), the CDC reported.
There were six flu-related pediatric deaths reported during the week ending March 25, but all occurred in earlier weeks. The total number of such deaths is now 61 for the 2016-2017 season, the CDC said.
Outpatient visits for influenza-like illness (ILI) held steady for the week ending March 25, but the number of states at the “high” range of activity dropped from 12 from 10 the previous week, according to the Centers for Disease Prevention and Control.
The proportion of outpatient visits for ILI was 3.2% for the second consecutive week, which halted the slowdown in activity that began the week ending Feb. 18. That 3.2% represents just under 25,000 visits for ILI of the almost 747,000 total visits reported to the Outpatient Influenza-like Illness Surveillance Network (ILINet) for the week ending March 25. By age, the largest groups with ILI visits for the week were individuals aged 5-24 years (41%) and those aged 4 years and under (20%), the CDC reported.
There were six flu-related pediatric deaths reported during the week ending March 25, but all occurred in earlier weeks. The total number of such deaths is now 61 for the 2016-2017 season, the CDC said.
Osimertinib receives full approval for advanced EGFR-mutated NSCLC
The Food and Drug Administration has converted accelerated approval of osimertinib to full approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation–positive non–small cell lung cancer (NSCLC), as detected by an FDA-approved test.
Also included in the indication, disease must have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy, the FDA said in a statement.
Full approval was based on an improvement in progression-free survival (PFS) in the phase III AURA3 study, which randomized 419 patients (2:1) to receive osimertinib (n = 279) 80 mg orally once daily or platinum-based doublet chemotherapy (n = 140). The hazard ratio for the investigator-assessed PFS was .30 (95% confidence interval: 0.23, 0.41; P less than .001).
The estimated median PFS was 10.1 months in the osimertinib arm and 4.4 months in the chemotherapy arm. Confirmed ORR was 65% (95% CI: 59%, 70%) and 29% (95% CI: 21%, 37%) in the osimertinib and chemotherapy arms, respectively (P less than .0001). Estimated median response durations were 11 months (95% CI: 8.6, 12.6) and 4.2 months (95% CI: 3.9, 5.9) in the osimertinib and chemotherapy arms, respectively, according to the FDA statement.
Overall survival data are immature, the FDA said.
All patients had metastatic EGFR T790M mutation–positive NSCLC, identified by the cobas EGFR mutation test performed in a central laboratory, and progressive disease following first-line EGFR TKI therapy. Patients in the chemotherapy arm received either pemetrexed, 500 mg/m2 with carboplatin AUC5, or pemetrexed, 500mg/m2 with cisplatin 75 mg/m2), on day 1 of every 21-day cycle for up to six cycles followed by pemetrexed maintenance therapy.
The most serious adverse reactions, evaluated in 833 patients receiving osimertinib, were interstitial lung disease/pneumonitis (3.5%), QTc interval prolongation (0.7%), cardiomyopathy (1.9%), and keratitis (0.7%). The most common adverse reactions were diarrhea, rash, dry skin, nail toxicity, and fatigue.
The recommended dose of osimertinib, to be marketed as Tagrisso by AstraZeneca, is 80 mg orally once daily, with or without food, until disease progression or unacceptable toxicity. The presence of an EGFR T790M mutation in a tumor specimen, or plasma specimen (if tumor tissue is unavailable), should be confirmed by an FDA-approved test prior to initiation of treatment.
Full prescribing information is available here.
The Food and Drug Administration has converted accelerated approval of osimertinib to full approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation–positive non–small cell lung cancer (NSCLC), as detected by an FDA-approved test.
Also included in the indication, disease must have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy, the FDA said in a statement.
Full approval was based on an improvement in progression-free survival (PFS) in the phase III AURA3 study, which randomized 419 patients (2:1) to receive osimertinib (n = 279) 80 mg orally once daily or platinum-based doublet chemotherapy (n = 140). The hazard ratio for the investigator-assessed PFS was .30 (95% confidence interval: 0.23, 0.41; P less than .001).
The estimated median PFS was 10.1 months in the osimertinib arm and 4.4 months in the chemotherapy arm. Confirmed ORR was 65% (95% CI: 59%, 70%) and 29% (95% CI: 21%, 37%) in the osimertinib and chemotherapy arms, respectively (P less than .0001). Estimated median response durations were 11 months (95% CI: 8.6, 12.6) and 4.2 months (95% CI: 3.9, 5.9) in the osimertinib and chemotherapy arms, respectively, according to the FDA statement.
Overall survival data are immature, the FDA said.
All patients had metastatic EGFR T790M mutation–positive NSCLC, identified by the cobas EGFR mutation test performed in a central laboratory, and progressive disease following first-line EGFR TKI therapy. Patients in the chemotherapy arm received either pemetrexed, 500 mg/m2 with carboplatin AUC5, or pemetrexed, 500mg/m2 with cisplatin 75 mg/m2), on day 1 of every 21-day cycle for up to six cycles followed by pemetrexed maintenance therapy.
The most serious adverse reactions, evaluated in 833 patients receiving osimertinib, were interstitial lung disease/pneumonitis (3.5%), QTc interval prolongation (0.7%), cardiomyopathy (1.9%), and keratitis (0.7%). The most common adverse reactions were diarrhea, rash, dry skin, nail toxicity, and fatigue.
The recommended dose of osimertinib, to be marketed as Tagrisso by AstraZeneca, is 80 mg orally once daily, with or without food, until disease progression or unacceptable toxicity. The presence of an EGFR T790M mutation in a tumor specimen, or plasma specimen (if tumor tissue is unavailable), should be confirmed by an FDA-approved test prior to initiation of treatment.
Full prescribing information is available here.
The Food and Drug Administration has converted accelerated approval of osimertinib to full approval for the treatment of patients with metastatic epidermal growth factor receptor (EGFR) T790M mutation–positive non–small cell lung cancer (NSCLC), as detected by an FDA-approved test.
Also included in the indication, disease must have progressed on or after EGFR tyrosine kinase inhibitor (TKI) therapy, the FDA said in a statement.
Full approval was based on an improvement in progression-free survival (PFS) in the phase III AURA3 study, which randomized 419 patients (2:1) to receive osimertinib (n = 279) 80 mg orally once daily or platinum-based doublet chemotherapy (n = 140). The hazard ratio for the investigator-assessed PFS was .30 (95% confidence interval: 0.23, 0.41; P less than .001).
The estimated median PFS was 10.1 months in the osimertinib arm and 4.4 months in the chemotherapy arm. Confirmed ORR was 65% (95% CI: 59%, 70%) and 29% (95% CI: 21%, 37%) in the osimertinib and chemotherapy arms, respectively (P less than .0001). Estimated median response durations were 11 months (95% CI: 8.6, 12.6) and 4.2 months (95% CI: 3.9, 5.9) in the osimertinib and chemotherapy arms, respectively, according to the FDA statement.
Overall survival data are immature, the FDA said.
All patients had metastatic EGFR T790M mutation–positive NSCLC, identified by the cobas EGFR mutation test performed in a central laboratory, and progressive disease following first-line EGFR TKI therapy. Patients in the chemotherapy arm received either pemetrexed, 500 mg/m2 with carboplatin AUC5, or pemetrexed, 500mg/m2 with cisplatin 75 mg/m2), on day 1 of every 21-day cycle for up to six cycles followed by pemetrexed maintenance therapy.
The most serious adverse reactions, evaluated in 833 patients receiving osimertinib, were interstitial lung disease/pneumonitis (3.5%), QTc interval prolongation (0.7%), cardiomyopathy (1.9%), and keratitis (0.7%). The most common adverse reactions were diarrhea, rash, dry skin, nail toxicity, and fatigue.
The recommended dose of osimertinib, to be marketed as Tagrisso by AstraZeneca, is 80 mg orally once daily, with or without food, until disease progression or unacceptable toxicity. The presence of an EGFR T790M mutation in a tumor specimen, or plasma specimen (if tumor tissue is unavailable), should be confirmed by an FDA-approved test prior to initiation of treatment.
Full prescribing information is available here.
FDA grants breakthrough therapy status to rituximab for pemphigus vulgaris
The Food and Drug Administration has granted breakthrough therapy status to rituximab (Rituxan) for treating pemphigus vulgaris, according to the manufacturer.
Rituximab, a CD20-directed cytolytic antibody approved in 1997, is currently in a phase III study evaluating its efficacy for the pemphigus indication. It is approved in the United States for treating non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis (with methotrexate), granulomatosis with polyangiitis (Wegener’s granulomatosis), and microscopic polyangiitis (with glucocorticoids).![]()
The patients, who were experiencing their first episode of pemphigus vulgaris, were randomized to daily oral prednisone, tapered over a 12- to 18-month period, or rituximab administered intravenously (at days 0 and 14, and months 12 and 18), plus daily oral prednisone, tapered over 3 or 6 months. At 2 years, when they were no longer on therapy, 89% of those treated with rituximab and prednisone were in complete remission, compared with 34% of those treated with prednisone alone (P less than .0001).
The breakthrough therapy process is “designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s),” according to the FDA.
The study was supported by the French Ministry of Health, the French Society of Dermatology, and Roche, which owns Genentech. Genentech markets rituximab in the United States with Biogen and is conducting the phase III study.
The Food and Drug Administration has granted breakthrough therapy status to rituximab (Rituxan) for treating pemphigus vulgaris, according to the manufacturer.
Rituximab, a CD20-directed cytolytic antibody approved in 1997, is currently in a phase III study evaluating its efficacy for the pemphigus indication. It is approved in the United States for treating non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis (with methotrexate), granulomatosis with polyangiitis (Wegener’s granulomatosis), and microscopic polyangiitis (with glucocorticoids).![]()
The patients, who were experiencing their first episode of pemphigus vulgaris, were randomized to daily oral prednisone, tapered over a 12- to 18-month period, or rituximab administered intravenously (at days 0 and 14, and months 12 and 18), plus daily oral prednisone, tapered over 3 or 6 months. At 2 years, when they were no longer on therapy, 89% of those treated with rituximab and prednisone were in complete remission, compared with 34% of those treated with prednisone alone (P less than .0001).
The breakthrough therapy process is “designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s),” according to the FDA.
The study was supported by the French Ministry of Health, the French Society of Dermatology, and Roche, which owns Genentech. Genentech markets rituximab in the United States with Biogen and is conducting the phase III study.
The Food and Drug Administration has granted breakthrough therapy status to rituximab (Rituxan) for treating pemphigus vulgaris, according to the manufacturer.
Rituximab, a CD20-directed cytolytic antibody approved in 1997, is currently in a phase III study evaluating its efficacy for the pemphigus indication. It is approved in the United States for treating non-Hodgkin lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis (with methotrexate), granulomatosis with polyangiitis (Wegener’s granulomatosis), and microscopic polyangiitis (with glucocorticoids).![]()
The patients, who were experiencing their first episode of pemphigus vulgaris, were randomized to daily oral prednisone, tapered over a 12- to 18-month period, or rituximab administered intravenously (at days 0 and 14, and months 12 and 18), plus daily oral prednisone, tapered over 3 or 6 months. At 2 years, when they were no longer on therapy, 89% of those treated with rituximab and prednisone were in complete remission, compared with 34% of those treated with prednisone alone (P less than .0001).
The breakthrough therapy process is “designed to expedite the development and review of drugs that are intended to treat a serious condition and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over available therapy on a clinically significant endpoint(s),” according to the FDA.
The study was supported by the French Ministry of Health, the French Society of Dermatology, and Roche, which owns Genentech. Genentech markets rituximab in the United States with Biogen and is conducting the phase III study.
2016-2017 flu season continues to wind down
Influenza activity took another healthy step down as outpatient visits continued to drop, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was down to 3.2% for the week ending March 18, 2017, the CDC reported, compared with 3.6% the week before. (The figure of 3.7% previously reported for last week has been adjusted this week, so the halt in the decline in outpatient visits was actually more of a slowdown.) The national baseline for outpatient ILI visits is 2.2%.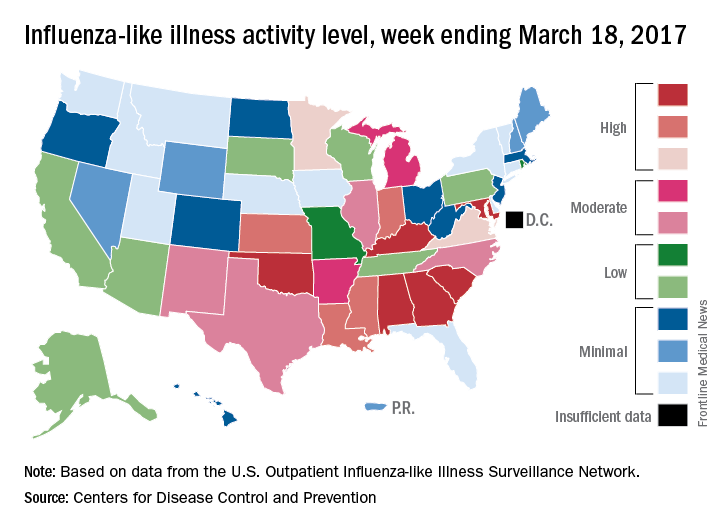
Two flu-related pediatric deaths were reported during the week of March 18, but both occurred earlier: one during the week ending Feb. 18 and the other in the week ending Feb. 25, the CDC reported. The total number of pediatric flu deaths reported is now 55 for the 2016-2017 season.
Influenza activity took another healthy step down as outpatient visits continued to drop, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was down to 3.2% for the week ending March 18, 2017, the CDC reported, compared with 3.6% the week before. (The figure of 3.7% previously reported for last week has been adjusted this week, so the halt in the decline in outpatient visits was actually more of a slowdown.) The national baseline for outpatient ILI visits is 2.2%.
Two flu-related pediatric deaths were reported during the week of March 18, but both occurred earlier: one during the week ending Feb. 18 and the other in the week ending Feb. 25, the CDC reported. The total number of pediatric flu deaths reported is now 55 for the 2016-2017 season.
Influenza activity took another healthy step down as outpatient visits continued to drop, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was down to 3.2% for the week ending March 18, 2017, the CDC reported, compared with 3.6% the week before. (The figure of 3.7% previously reported for last week has been adjusted this week, so the halt in the decline in outpatient visits was actually more of a slowdown.) The national baseline for outpatient ILI visits is 2.2%.
Two flu-related pediatric deaths were reported during the week of March 18, but both occurred earlier: one during the week ending Feb. 18 and the other in the week ending Feb. 25, the CDC reported. The total number of pediatric flu deaths reported is now 55 for the 2016-2017 season.
CDC reports two new Zika-related pregnancy losses
The first pregnancy losses with Zika-related birth defects since last summer were reported March 14 by the Centers for Disease Control and Prevention.
Two new cases were reported to the U.S. Zika Pregnancy Registry between Feb. 21 and March 14 – the time period covered by the most recent release of data from the CDC. That brings the total number of Zika virus–related pregnancy losses to seven since the beginning of 2016 in the 50 states and the District of Columbia, along with 54 liveborn infants with Zika-related birth defects. The CDC stopped reporting adverse pregnancy outcomes in Puerto Rico and the other U.S. territories in early October 2016 because Puerto Rico’s Zika Active Pregnancy Surveillance System is not using the same inclusion criteria as its mainland counterpart.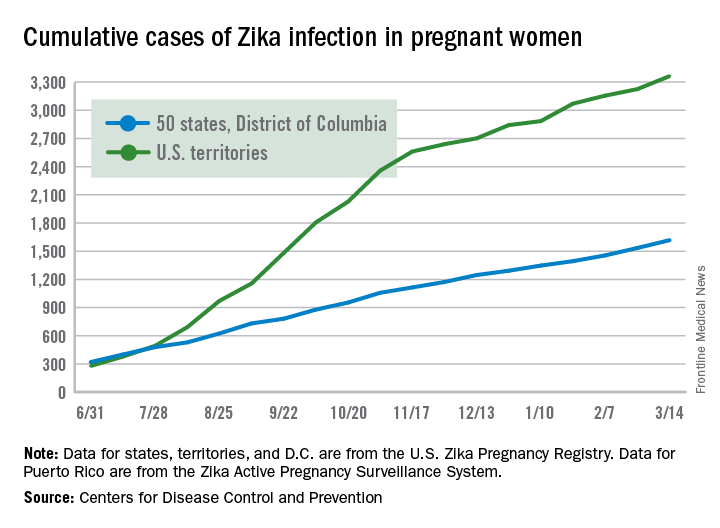
These are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes. Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
The first pregnancy losses with Zika-related birth defects since last summer were reported March 14 by the Centers for Disease Control and Prevention.
Two new cases were reported to the U.S. Zika Pregnancy Registry between Feb. 21 and March 14 – the time period covered by the most recent release of data from the CDC. That brings the total number of Zika virus–related pregnancy losses to seven since the beginning of 2016 in the 50 states and the District of Columbia, along with 54 liveborn infants with Zika-related birth defects. The CDC stopped reporting adverse pregnancy outcomes in Puerto Rico and the other U.S. territories in early October 2016 because Puerto Rico’s Zika Active Pregnancy Surveillance System is not using the same inclusion criteria as its mainland counterpart.
These are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes. Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
The first pregnancy losses with Zika-related birth defects since last summer were reported March 14 by the Centers for Disease Control and Prevention.
Two new cases were reported to the U.S. Zika Pregnancy Registry between Feb. 21 and March 14 – the time period covered by the most recent release of data from the CDC. That brings the total number of Zika virus–related pregnancy losses to seven since the beginning of 2016 in the 50 states and the District of Columbia, along with 54 liveborn infants with Zika-related birth defects. The CDC stopped reporting adverse pregnancy outcomes in Puerto Rico and the other U.S. territories in early October 2016 because Puerto Rico’s Zika Active Pregnancy Surveillance System is not using the same inclusion criteria as its mainland counterpart.
These are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes. Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
FDA approves safinamide to treat Parkinson’s disease
The U.S. Food and Drug Administration approved safinamide tablets on March 21 as an add-on treatment for patients with Parkinson’s disease who are currently taking levodopa/carbidopa and experiencing “off” episodes.
Newron Pharmaceuticals will market safinamide under the brand name Xadago.
The most common adverse reactions observed in patients taking safinamide were uncontrolled involuntary movement, falls, nausea, and insomnia.
In its announcement of the approval, the FDA noted that patients should not take safinamide if they have severe liver problems, take dextromethorphan, or take a monoamine oxidase inhibitor, because the two together may cause a sudden severe increase in blood pressure. Safinamide also should not be taken by patients who use a opioid drug, St. John’s wort, certain antidepressants (such as serotonin-norepinephrine reuptake inhibitors, tricyclics, tetracyclics, and triazolopyridines), or cyclobenzaprine, because it may cause a life-threatening reaction called serotonin syndrome.
The U.S. Food and Drug Administration approved safinamide tablets on March 21 as an add-on treatment for patients with Parkinson’s disease who are currently taking levodopa/carbidopa and experiencing “off” episodes.
Newron Pharmaceuticals will market safinamide under the brand name Xadago.
The most common adverse reactions observed in patients taking safinamide were uncontrolled involuntary movement, falls, nausea, and insomnia.
In its announcement of the approval, the FDA noted that patients should not take safinamide if they have severe liver problems, take dextromethorphan, or take a monoamine oxidase inhibitor, because the two together may cause a sudden severe increase in blood pressure. Safinamide also should not be taken by patients who use a opioid drug, St. John’s wort, certain antidepressants (such as serotonin-norepinephrine reuptake inhibitors, tricyclics, tetracyclics, and triazolopyridines), or cyclobenzaprine, because it may cause a life-threatening reaction called serotonin syndrome.
The U.S. Food and Drug Administration approved safinamide tablets on March 21 as an add-on treatment for patients with Parkinson’s disease who are currently taking levodopa/carbidopa and experiencing “off” episodes.
Newron Pharmaceuticals will market safinamide under the brand name Xadago.
The most common adverse reactions observed in patients taking safinamide were uncontrolled involuntary movement, falls, nausea, and insomnia.
In its announcement of the approval, the FDA noted that patients should not take safinamide if they have severe liver problems, take dextromethorphan, or take a monoamine oxidase inhibitor, because the two together may cause a sudden severe increase in blood pressure. Safinamide also should not be taken by patients who use a opioid drug, St. John’s wort, certain antidepressants (such as serotonin-norepinephrine reuptake inhibitors, tricyclics, tetracyclics, and triazolopyridines), or cyclobenzaprine, because it may cause a life-threatening reaction called serotonin syndrome.
U.S. influenza activity remains steady
The decline in U.S. influenza activity that started in February paused during the week ending March 11, according to the U.S. Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) stayed at 3.7% for a second consecutive week after declining for 3 weeks in a row. The peak for the season, 5.2%, came during the week ending Feb. 11, CDC data show. The national baseline is 2.2%.
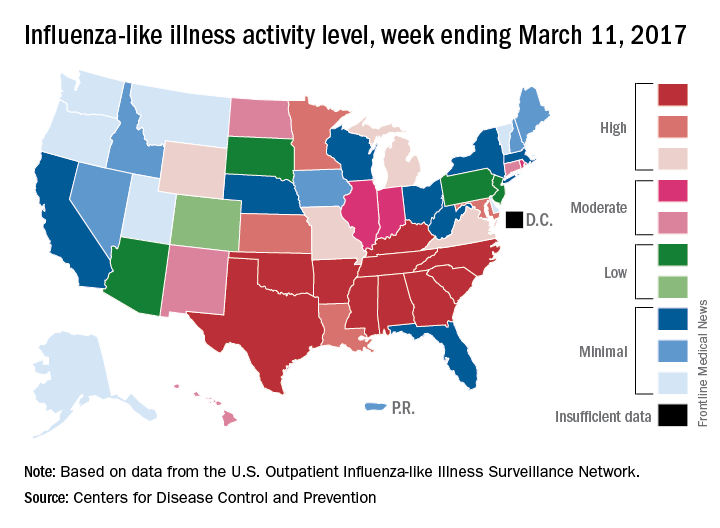
Five ILI-related pediatric deaths were reported to the CDC for the week – all of which occurred during previous weeks – bringing the total to 53 for the 2016-2017 season, the CDC said.
The decline in U.S. influenza activity that started in February paused during the week ending March 11, according to the U.S. Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) stayed at 3.7% for a second consecutive week after declining for 3 weeks in a row. The peak for the season, 5.2%, came during the week ending Feb. 11, CDC data show. The national baseline is 2.2%.
Five ILI-related pediatric deaths were reported to the CDC for the week – all of which occurred during previous weeks – bringing the total to 53 for the 2016-2017 season, the CDC said.
The decline in U.S. influenza activity that started in February paused during the week ending March 11, according to the U.S. Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) stayed at 3.7% for a second consecutive week after declining for 3 weeks in a row. The peak for the season, 5.2%, came during the week ending Feb. 11, CDC data show. The national baseline is 2.2%.
Five ILI-related pediatric deaths were reported to the CDC for the week – all of which occurred during previous weeks – bringing the total to 53 for the 2016-2017 season, the CDC said.
FDA grants Hodgkin lymphoma indication for pembrolizumab
Pembrolizumab is now approved for the treatment of adults and children who have refractory classical Hodgkin lymphoma, or who have relapsed after three or more prior lines of therapy, the Food and Drug Administration announced on March 14.
Pembrolizumab (Keytruda) is a humanized monoclonal antibody administered intravenously. According to a press release from Merck, the manufacturer of Keytruda, the approval is based on data from 210 patients aged 18 years and older in the KEYNOTE-087 trial, which found an overall response rate of 69% among patients who received 200 mg of the drug every 3 weeks. Among responders, the median duration of response was 11.1 months.
“For the patients with classical Hodgkin lymphoma who are not cured with existing treatments, there are limited options, and treating their disease becomes more challenging,” Craig H. Moskowitz, MD, clinical director of the division of hematologic oncology at Memorial Sloan Kettering Cancer Center, New York, said in the press release. “This approval is an important step forward in treating these patients, who are generally young and have a particularly poor prognosis.”
According to Merck, continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The revised drug label information can be found here.
dbrunk@frontlinemedcom.com
Pembrolizumab is now approved for the treatment of adults and children who have refractory classical Hodgkin lymphoma, or who have relapsed after three or more prior lines of therapy, the Food and Drug Administration announced on March 14.
Pembrolizumab (Keytruda) is a humanized monoclonal antibody administered intravenously. According to a press release from Merck, the manufacturer of Keytruda, the approval is based on data from 210 patients aged 18 years and older in the KEYNOTE-087 trial, which found an overall response rate of 69% among patients who received 200 mg of the drug every 3 weeks. Among responders, the median duration of response was 11.1 months.
“For the patients with classical Hodgkin lymphoma who are not cured with existing treatments, there are limited options, and treating their disease becomes more challenging,” Craig H. Moskowitz, MD, clinical director of the division of hematologic oncology at Memorial Sloan Kettering Cancer Center, New York, said in the press release. “This approval is an important step forward in treating these patients, who are generally young and have a particularly poor prognosis.”
According to Merck, continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The revised drug label information can be found here.
dbrunk@frontlinemedcom.com
Pembrolizumab is now approved for the treatment of adults and children who have refractory classical Hodgkin lymphoma, or who have relapsed after three or more prior lines of therapy, the Food and Drug Administration announced on March 14.
Pembrolizumab (Keytruda) is a humanized monoclonal antibody administered intravenously. According to a press release from Merck, the manufacturer of Keytruda, the approval is based on data from 210 patients aged 18 years and older in the KEYNOTE-087 trial, which found an overall response rate of 69% among patients who received 200 mg of the drug every 3 weeks. Among responders, the median duration of response was 11.1 months.
“For the patients with classical Hodgkin lymphoma who are not cured with existing treatments, there are limited options, and treating their disease becomes more challenging,” Craig H. Moskowitz, MD, clinical director of the division of hematologic oncology at Memorial Sloan Kettering Cancer Center, New York, said in the press release. “This approval is an important step forward in treating these patients, who are generally young and have a particularly poor prognosis.”
According to Merck, continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials. The revised drug label information can be found here.
dbrunk@frontlinemedcom.com
U.S. flu activity continues to decline
The 2016-2017 U.S. influenza season appears to have peaked, as activity measures dropped for the third consecutive week, the Centers for Disease Control and Prevention reported.
For the week ending March 4, there were 11 states at level 10 on the CDC’s 1-10 scale of influenza-like illness (ILI) activity, with another three in the “high” range at levels 8 and 9. The previous week (Feb. 25), there were 22 states at level 10, with a total of 27 in the high range of ILI activity. At the peak of activity during the week of Feb. 11, there were 25 states at level 10, data from the CDC’s Outpatient ILI Surveillance Network show.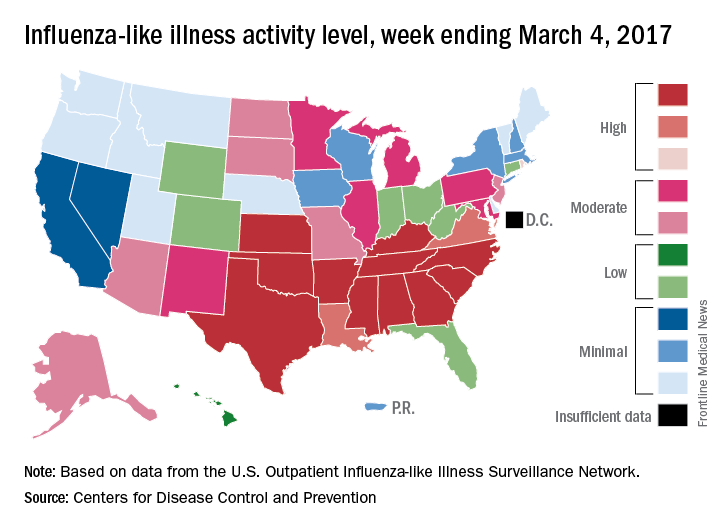
There were eight ILI-related pediatric deaths reported during the week ending March 4, although all occurred in earlier weeks. For the 2016-2017 season so far, 48 ILI-related pediatric deaths have been reported, the CDC said.
For the 70 counties in 13 states that report to the Influenza Hospitalization Surveillance Network, the flu-related hospitalization rate for the season is 43.5 per 100,000 population. The highest rate by age group is for those 65 years and over at 198.8 per 100,000, followed by 50- to 64-year-olds at 42.2 per 100,000 and children aged 0-4 years at 28.8 per 100,000, according to the CDC.
The 2016-2017 U.S. influenza season appears to have peaked, as activity measures dropped for the third consecutive week, the Centers for Disease Control and Prevention reported.
For the week ending March 4, there were 11 states at level 10 on the CDC’s 1-10 scale of influenza-like illness (ILI) activity, with another three in the “high” range at levels 8 and 9. The previous week (Feb. 25), there were 22 states at level 10, with a total of 27 in the high range of ILI activity. At the peak of activity during the week of Feb. 11, there were 25 states at level 10, data from the CDC’s Outpatient ILI Surveillance Network show.
There were eight ILI-related pediatric deaths reported during the week ending March 4, although all occurred in earlier weeks. For the 2016-2017 season so far, 48 ILI-related pediatric deaths have been reported, the CDC said.
For the 70 counties in 13 states that report to the Influenza Hospitalization Surveillance Network, the flu-related hospitalization rate for the season is 43.5 per 100,000 population. The highest rate by age group is for those 65 years and over at 198.8 per 100,000, followed by 50- to 64-year-olds at 42.2 per 100,000 and children aged 0-4 years at 28.8 per 100,000, according to the CDC.
The 2016-2017 U.S. influenza season appears to have peaked, as activity measures dropped for the third consecutive week, the Centers for Disease Control and Prevention reported.
For the week ending March 4, there were 11 states at level 10 on the CDC’s 1-10 scale of influenza-like illness (ILI) activity, with another three in the “high” range at levels 8 and 9. The previous week (Feb. 25), there were 22 states at level 10, with a total of 27 in the high range of ILI activity. At the peak of activity during the week of Feb. 11, there were 25 states at level 10, data from the CDC’s Outpatient ILI Surveillance Network show.
There were eight ILI-related pediatric deaths reported during the week ending March 4, although all occurred in earlier weeks. For the 2016-2017 season so far, 48 ILI-related pediatric deaths have been reported, the CDC said.
For the 70 counties in 13 states that report to the Influenza Hospitalization Surveillance Network, the flu-related hospitalization rate for the season is 43.5 per 100,000 population. The highest rate by age group is for those 65 years and over at 198.8 per 100,000, followed by 50- to 64-year-olds at 42.2 per 100,000 and children aged 0-4 years at 28.8 per 100,000, according to the CDC.