User login
FDA committee approves strains for 2017-2018 flu shot
ROCKVILLE, MD. – A committee of Food and Drug Administration advisers backed the World Health Organization’s influenza vaccine recommendations for the 2017-2018 season at a meeting March 9.
In a unanimous vote, members of the Vaccines and Related Biological Products Advisory Committee recommended that trivalent vaccines for the 2017-2018 season should contain the following vaccine strains: A/Michigan/45/2015(H1N1)pdm09-like, A/Hong Kong/4801/2014(H3N2)-like, and B/Brisbane/60/2008-like.
These recommendations echo those from the 2016-2017 season, with the exception of a slight update to the H1N1 strain, which had previously been A/California/7/2009(H1N1)pdm09-like virus.
Regarding vaccine efficacy, the cell propagated A/Hong Kong strain was the strongest candidate, covering 93% of A(H3N2) viruses seen in the 2016-2017 season, according to Jacqueline Katz, PhD, director of the WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza at the Centers for Disease Control and Prevention. In comparison, the egg propagated version of the A/Hong Kong virus covered 59%.
For the influenza B virus, the Yamagata lineage and Victoria lineage strain cycled monthly as the predominant strain in the 2016-2017 season, with a split of “around 50/50,” leaning toward Yamagata in North America, Europe, and Oceana, Dr. Katz explained. The Victoria lineage, in some cases, accounted for nearly 75% of B viruses in Africa and South America.
Committee members expressed concern over the difference between strain prevalence in the United States and abroad and considered recommending a strain that did not coincide with the WHO recommendation, something that has not happened in the history of the advisory committee.
“I’m very aware of influenza vaccinations being a global enterprise, and companies manufacture vaccines for use in multiple countries,” said Committee Chair Kathryn Edwards, MD, professor of pediatrics at Vanderbilt University, Nashville, Tenn. “If we to select a B strain that differed from the WHO recommendation, would that adversely impact vaccine production for the U.S. market?”
Despite these questions, the committee continued to back the WHO recommendations.
Historically, the advisory committee has recommended flu vaccine strains earlier in the year, according to Beverly Taylor, PhD, head of influenza scientific affairs and pandemic readiness at Seqirus Vaccines. Dr. Taylor presented the vaccine manufacturers’ perspective. The delay has put added pressure on manufacturers.
“We haven’t seen impacts yet on start of vaccination dates,” said Dr. Taylor. “But the very clear message from manufacturers is if you keep squashing that manufacturing window, then there will reach a point where we are concerned we will see an impact on vaccine supply time.”
None of the committee members presented waivers of conflict of interest. While the FDA is not obligated to follow the recommendations of the advisory committee, it generally does.
ezimmerman@frontlinemedcom.com
On Twitter @EAZTweets
ROCKVILLE, MD. – A committee of Food and Drug Administration advisers backed the World Health Organization’s influenza vaccine recommendations for the 2017-2018 season at a meeting March 9.
In a unanimous vote, members of the Vaccines and Related Biological Products Advisory Committee recommended that trivalent vaccines for the 2017-2018 season should contain the following vaccine strains: A/Michigan/45/2015(H1N1)pdm09-like, A/Hong Kong/4801/2014(H3N2)-like, and B/Brisbane/60/2008-like.
These recommendations echo those from the 2016-2017 season, with the exception of a slight update to the H1N1 strain, which had previously been A/California/7/2009(H1N1)pdm09-like virus.
Regarding vaccine efficacy, the cell propagated A/Hong Kong strain was the strongest candidate, covering 93% of A(H3N2) viruses seen in the 2016-2017 season, according to Jacqueline Katz, PhD, director of the WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza at the Centers for Disease Control and Prevention. In comparison, the egg propagated version of the A/Hong Kong virus covered 59%.
For the influenza B virus, the Yamagata lineage and Victoria lineage strain cycled monthly as the predominant strain in the 2016-2017 season, with a split of “around 50/50,” leaning toward Yamagata in North America, Europe, and Oceana, Dr. Katz explained. The Victoria lineage, in some cases, accounted for nearly 75% of B viruses in Africa and South America.
Committee members expressed concern over the difference between strain prevalence in the United States and abroad and considered recommending a strain that did not coincide with the WHO recommendation, something that has not happened in the history of the advisory committee.
“I’m very aware of influenza vaccinations being a global enterprise, and companies manufacture vaccines for use in multiple countries,” said Committee Chair Kathryn Edwards, MD, professor of pediatrics at Vanderbilt University, Nashville, Tenn. “If we to select a B strain that differed from the WHO recommendation, would that adversely impact vaccine production for the U.S. market?”
Despite these questions, the committee continued to back the WHO recommendations.
Historically, the advisory committee has recommended flu vaccine strains earlier in the year, according to Beverly Taylor, PhD, head of influenza scientific affairs and pandemic readiness at Seqirus Vaccines. Dr. Taylor presented the vaccine manufacturers’ perspective. The delay has put added pressure on manufacturers.
“We haven’t seen impacts yet on start of vaccination dates,” said Dr. Taylor. “But the very clear message from manufacturers is if you keep squashing that manufacturing window, then there will reach a point where we are concerned we will see an impact on vaccine supply time.”
None of the committee members presented waivers of conflict of interest. While the FDA is not obligated to follow the recommendations of the advisory committee, it generally does.
ezimmerman@frontlinemedcom.com
On Twitter @EAZTweets
ROCKVILLE, MD. – A committee of Food and Drug Administration advisers backed the World Health Organization’s influenza vaccine recommendations for the 2017-2018 season at a meeting March 9.
In a unanimous vote, members of the Vaccines and Related Biological Products Advisory Committee recommended that trivalent vaccines for the 2017-2018 season should contain the following vaccine strains: A/Michigan/45/2015(H1N1)pdm09-like, A/Hong Kong/4801/2014(H3N2)-like, and B/Brisbane/60/2008-like.
These recommendations echo those from the 2016-2017 season, with the exception of a slight update to the H1N1 strain, which had previously been A/California/7/2009(H1N1)pdm09-like virus.
Regarding vaccine efficacy, the cell propagated A/Hong Kong strain was the strongest candidate, covering 93% of A(H3N2) viruses seen in the 2016-2017 season, according to Jacqueline Katz, PhD, director of the WHO Collaborating Center for Surveillance, Epidemiology and Control of Influenza at the Centers for Disease Control and Prevention. In comparison, the egg propagated version of the A/Hong Kong virus covered 59%.
For the influenza B virus, the Yamagata lineage and Victoria lineage strain cycled monthly as the predominant strain in the 2016-2017 season, with a split of “around 50/50,” leaning toward Yamagata in North America, Europe, and Oceana, Dr. Katz explained. The Victoria lineage, in some cases, accounted for nearly 75% of B viruses in Africa and South America.
Committee members expressed concern over the difference between strain prevalence in the United States and abroad and considered recommending a strain that did not coincide with the WHO recommendation, something that has not happened in the history of the advisory committee.
“I’m very aware of influenza vaccinations being a global enterprise, and companies manufacture vaccines for use in multiple countries,” said Committee Chair Kathryn Edwards, MD, professor of pediatrics at Vanderbilt University, Nashville, Tenn. “If we to select a B strain that differed from the WHO recommendation, would that adversely impact vaccine production for the U.S. market?”
Despite these questions, the committee continued to back the WHO recommendations.
Historically, the advisory committee has recommended flu vaccine strains earlier in the year, according to Beverly Taylor, PhD, head of influenza scientific affairs and pandemic readiness at Seqirus Vaccines. Dr. Taylor presented the vaccine manufacturers’ perspective. The delay has put added pressure on manufacturers.
“We haven’t seen impacts yet on start of vaccination dates,” said Dr. Taylor. “But the very clear message from manufacturers is if you keep squashing that manufacturing window, then there will reach a point where we are concerned we will see an impact on vaccine supply time.”
None of the committee members presented waivers of conflict of interest. While the FDA is not obligated to follow the recommendations of the advisory committee, it generally does.
ezimmerman@frontlinemedcom.com
On Twitter @EAZTweets
AT AN FDA ADVISORY COMMITTEE MEETING
FDA approves first dedicated bifurcation device to treat coronary bifurcation lesions
Tryton Medical announced on March 6 that the Food and Drug Administration has approved Tryton Side Branch Stent for the treatment of coronary bifurcation lesions involving large side branches, becoming the first dedicated bifurcation device to receive regulatory approval in the U.S.
In a post hoc analysis of a randomized clinical trial, treatment with the Tryton Side Branch Stent in the intended population of patients with large side branches (stent greater than 2.5mm) reduced the need for additional bailout stenting (0.7% vs. 5.6%, P = .02). It led to significantly lower side branch percent diameter stenosis at a 9-month follow-up (30.4% vs. 40.6%, P = .004) when compared with provisional stenting. The analysis also showed comparable major adverse cardiovascular events and myocardial infarction rates when compared with provisional stenting at 3 years.![]()
“Treatment of complex lesions at the site of a bifurcation has historically been inconsistent, with results varying depending on the procedure and the experience of the interventionist,” said Aaron Kaplan, MD, Professor of Medicine at Dartmouth Hitchcock Medical Center, Lebanon, N.H., and Chief Medical Officer of Tryton Medical, in a press release. “A predictable bifurcation solution helps alleviate some of the stress in these procedures by limiting variability and reducing the need for bailout stenting. This important FDA decision could have a profound impact on treatment protocols and guidelines for significant bifurcation lesions in the years ahead.”
There have been no randomized studies to compare the results of percutaneous coronary interventions (PCI) with coronary artery bypass grafting in a bifurcation-only patient population. But this new device should benefit results from treatment using PCI.
Coronary artery disease is the leading cause of death in the U.S. in both men and women, and often results in bifurcation. Provisional stenting of the main branch is the current standard of care, but in many cases the side branch is not stented, leaving it vulnerable to complications like occlusion requiring bailout stenting.
Read more on Tryton Side Branch Stent on Tryton’s website.
Tryton Medical announced on March 6 that the Food and Drug Administration has approved Tryton Side Branch Stent for the treatment of coronary bifurcation lesions involving large side branches, becoming the first dedicated bifurcation device to receive regulatory approval in the U.S.
In a post hoc analysis of a randomized clinical trial, treatment with the Tryton Side Branch Stent in the intended population of patients with large side branches (stent greater than 2.5mm) reduced the need for additional bailout stenting (0.7% vs. 5.6%, P = .02). It led to significantly lower side branch percent diameter stenosis at a 9-month follow-up (30.4% vs. 40.6%, P = .004) when compared with provisional stenting. The analysis also showed comparable major adverse cardiovascular events and myocardial infarction rates when compared with provisional stenting at 3 years.![]()
“Treatment of complex lesions at the site of a bifurcation has historically been inconsistent, with results varying depending on the procedure and the experience of the interventionist,” said Aaron Kaplan, MD, Professor of Medicine at Dartmouth Hitchcock Medical Center, Lebanon, N.H., and Chief Medical Officer of Tryton Medical, in a press release. “A predictable bifurcation solution helps alleviate some of the stress in these procedures by limiting variability and reducing the need for bailout stenting. This important FDA decision could have a profound impact on treatment protocols and guidelines for significant bifurcation lesions in the years ahead.”
There have been no randomized studies to compare the results of percutaneous coronary interventions (PCI) with coronary artery bypass grafting in a bifurcation-only patient population. But this new device should benefit results from treatment using PCI.
Coronary artery disease is the leading cause of death in the U.S. in both men and women, and often results in bifurcation. Provisional stenting of the main branch is the current standard of care, but in many cases the side branch is not stented, leaving it vulnerable to complications like occlusion requiring bailout stenting.
Read more on Tryton Side Branch Stent on Tryton’s website.
Tryton Medical announced on March 6 that the Food and Drug Administration has approved Tryton Side Branch Stent for the treatment of coronary bifurcation lesions involving large side branches, becoming the first dedicated bifurcation device to receive regulatory approval in the U.S.
In a post hoc analysis of a randomized clinical trial, treatment with the Tryton Side Branch Stent in the intended population of patients with large side branches (stent greater than 2.5mm) reduced the need for additional bailout stenting (0.7% vs. 5.6%, P = .02). It led to significantly lower side branch percent diameter stenosis at a 9-month follow-up (30.4% vs. 40.6%, P = .004) when compared with provisional stenting. The analysis also showed comparable major adverse cardiovascular events and myocardial infarction rates when compared with provisional stenting at 3 years.![]()
“Treatment of complex lesions at the site of a bifurcation has historically been inconsistent, with results varying depending on the procedure and the experience of the interventionist,” said Aaron Kaplan, MD, Professor of Medicine at Dartmouth Hitchcock Medical Center, Lebanon, N.H., and Chief Medical Officer of Tryton Medical, in a press release. “A predictable bifurcation solution helps alleviate some of the stress in these procedures by limiting variability and reducing the need for bailout stenting. This important FDA decision could have a profound impact on treatment protocols and guidelines for significant bifurcation lesions in the years ahead.”
There have been no randomized studies to compare the results of percutaneous coronary interventions (PCI) with coronary artery bypass grafting in a bifurcation-only patient population. But this new device should benefit results from treatment using PCI.
Coronary artery disease is the leading cause of death in the U.S. in both men and women, and often results in bifurcation. Provisional stenting of the main branch is the current standard of care, but in many cases the side branch is not stented, leaving it vulnerable to complications like occlusion requiring bailout stenting.
Read more on Tryton Side Branch Stent on Tryton’s website.
Zika-infected pregnancies continue to rise in U.S.
Reports of new cases of Zika infection in pregnant women held steady during the 2 weeks ending Feb. 21 as the number of new cases dropped in the territories and rose in the 50 states and D.C., according to the Centers for Disease Control and Prevention.
Compared with the previous 2-week period (Jan. 25-Feb.7), reports of new cases of pregnant women with laboratory evidence of Zika virus infection were up from 146 to 148, an increase from 61 to 79 in the states/D.C. and a decrease from 85 to 69 in the territories. The total number of Zika cases among pregnant women in the United States for 2016-2017 is 4,759, with 1,534 occurring in the states/D.C. and 3,225 in the territories, the CDC reported March 2.
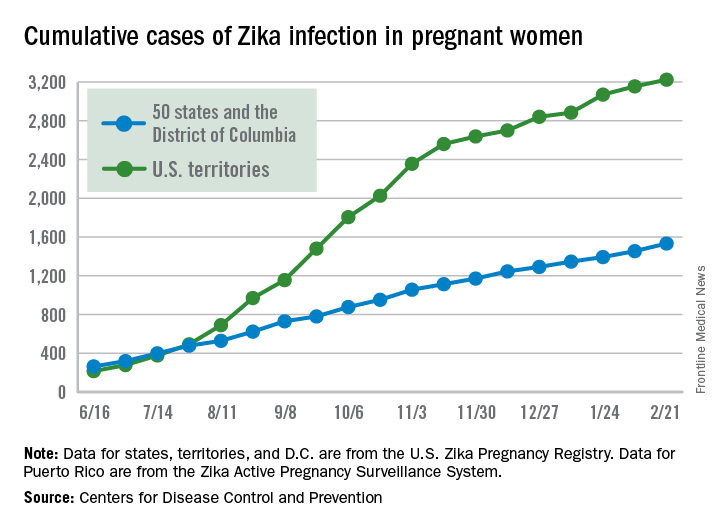
Among all Americans, the number of Zika cases reported is now up to 43,380 since Jan. 1, 2015, with 38,306 occurring in the territories and 5,074 in the states and D.C. The state with the most cases is Florida at 1,107, followed by New York at 1,007 and California with 431. Puerto Rico has reported 37,515 cases so far, and the U.S. Virgin Islands have reported 989, the CDC said.
The figures for states, territories, and the District of Columbia are reported to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System. These are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes.
Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
Reports of new cases of Zika infection in pregnant women held steady during the 2 weeks ending Feb. 21 as the number of new cases dropped in the territories and rose in the 50 states and D.C., according to the Centers for Disease Control and Prevention.
Compared with the previous 2-week period (Jan. 25-Feb.7), reports of new cases of pregnant women with laboratory evidence of Zika virus infection were up from 146 to 148, an increase from 61 to 79 in the states/D.C. and a decrease from 85 to 69 in the territories. The total number of Zika cases among pregnant women in the United States for 2016-2017 is 4,759, with 1,534 occurring in the states/D.C. and 3,225 in the territories, the CDC reported March 2.
Among all Americans, the number of Zika cases reported is now up to 43,380 since Jan. 1, 2015, with 38,306 occurring in the territories and 5,074 in the states and D.C. The state with the most cases is Florida at 1,107, followed by New York at 1,007 and California with 431. Puerto Rico has reported 37,515 cases so far, and the U.S. Virgin Islands have reported 989, the CDC said.
The figures for states, territories, and the District of Columbia are reported to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System. These are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes.
Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
Reports of new cases of Zika infection in pregnant women held steady during the 2 weeks ending Feb. 21 as the number of new cases dropped in the territories and rose in the 50 states and D.C., according to the Centers for Disease Control and Prevention.
Compared with the previous 2-week period (Jan. 25-Feb.7), reports of new cases of pregnant women with laboratory evidence of Zika virus infection were up from 146 to 148, an increase from 61 to 79 in the states/D.C. and a decrease from 85 to 69 in the territories. The total number of Zika cases among pregnant women in the United States for 2016-2017 is 4,759, with 1,534 occurring in the states/D.C. and 3,225 in the territories, the CDC reported March 2.
Among all Americans, the number of Zika cases reported is now up to 43,380 since Jan. 1, 2015, with 38,306 occurring in the territories and 5,074 in the states and D.C. The state with the most cases is Florida at 1,107, followed by New York at 1,007 and California with 431. Puerto Rico has reported 37,515 cases so far, and the U.S. Virgin Islands have reported 989, the CDC said.
The figures for states, territories, and the District of Columbia are reported to the U.S. Zika Pregnancy Registry; data for Puerto Rico are reported to the U.S. Zika Active Pregnancy Surveillance System. These are not real-time data and reflect only pregnancy outcomes for women with any laboratory evidence of possible Zika virus infection, although it is not known if Zika virus was the cause of the poor outcomes.
Zika-related birth defects recorded by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, or termination with evidence of birth defects.
Birth defects in United States up 20-fold since Zika outbreak began
Birth defects potentially linked to cases of Zika virus in the United States have increased by a factor of nearly 20 since the virus first made its way into the country, according to new findings by the Centers for Disease Control and Prevention.
“The higher proportion of these defects among pregnancies with laboratory evidence of Zika infection in USZPR [U.S. Zika Pregnancy Registry] supports the relationship between congenital Zika virus infection and these birth defects,” wrote the authors of a new report led by Janet D. Cragan, MD, of the National Center on Birth Defects and Developmental Disabilities at the CDC (MMWR Morb Mortal Wkly Rep. 2017;66:219-22).![]()
[[{"attributes":{},"fields":{}}]]
Dr. Cragan and her coauthors retrospectively examined data on birth defects in three regions of the country: Massachusetts during 2013, North Carolina during 2013, and Atlanta during 2013-2014. The investigators focused on birth defects associated with prenatal Zika virus infections, mainly brain abnormalities and microcephaly.
The rate of total birth defects across the three regions was 2.86 per 1,000 live births, with 747 infants and fetuses identified as having one or more defects. Microcephaly and brain abnormalities alone occurred at a rate of 1.50 per 1,000 live births, with eye abnormalities and central nervous system dysfunction also occurring.
These numbers are relatively low when compared with data from Jan. 15 through Sept. 22, 2016. The birth defect rate jumped up to 58.8 per 1,000 live births, according to data from the USZPR, which found evidence of 26 infants and fetuses with brain or cranial defects in 442 completed pregnancies. These infants were all born to mothers with laboratory-confirmed Zika virus infections.
“Among 410 (55%) infants or fetuses with information on the earliest age a birth defect was recorded, 371 (90%) had evidence of a birth defect meeting the Zika definition before age 3 months,” the authors explained. “More than half of those with brain abnormalities or microcephaly or with neural tube defects and other early brain malformations had evidence of these defects noted prenatally (55% and 89%, respectively).”
Dr. Cragan and her colleagues hope that this evidence will further solidify the link between Zika virus and birth defects and pave the way for more population-based studies.
“These data demonstrate the critical contribution of population-based birth defects surveillance to understanding the impact of Zika virus infection during pregnancy,” the authors concluded. “In 2016, CDC provided funding for 45 local, state, and territorial health departments to conduct rapid population-based surveillance for defects potentially related to Zika virus infection, which will provide essential data to monitor the impact of Zika virus infection in the United States.”
Birth defects potentially linked to cases of Zika virus in the United States have increased by a factor of nearly 20 since the virus first made its way into the country, according to new findings by the Centers for Disease Control and Prevention.
“The higher proportion of these defects among pregnancies with laboratory evidence of Zika infection in USZPR [U.S. Zika Pregnancy Registry] supports the relationship between congenital Zika virus infection and these birth defects,” wrote the authors of a new report led by Janet D. Cragan, MD, of the National Center on Birth Defects and Developmental Disabilities at the CDC (MMWR Morb Mortal Wkly Rep. 2017;66:219-22).![]()
[[{"attributes":{},"fields":{}}]]
Dr. Cragan and her coauthors retrospectively examined data on birth defects in three regions of the country: Massachusetts during 2013, North Carolina during 2013, and Atlanta during 2013-2014. The investigators focused on birth defects associated with prenatal Zika virus infections, mainly brain abnormalities and microcephaly.
The rate of total birth defects across the three regions was 2.86 per 1,000 live births, with 747 infants and fetuses identified as having one or more defects. Microcephaly and brain abnormalities alone occurred at a rate of 1.50 per 1,000 live births, with eye abnormalities and central nervous system dysfunction also occurring.
These numbers are relatively low when compared with data from Jan. 15 through Sept. 22, 2016. The birth defect rate jumped up to 58.8 per 1,000 live births, according to data from the USZPR, which found evidence of 26 infants and fetuses with brain or cranial defects in 442 completed pregnancies. These infants were all born to mothers with laboratory-confirmed Zika virus infections.
“Among 410 (55%) infants or fetuses with information on the earliest age a birth defect was recorded, 371 (90%) had evidence of a birth defect meeting the Zika definition before age 3 months,” the authors explained. “More than half of those with brain abnormalities or microcephaly or with neural tube defects and other early brain malformations had evidence of these defects noted prenatally (55% and 89%, respectively).”
Dr. Cragan and her colleagues hope that this evidence will further solidify the link between Zika virus and birth defects and pave the way for more population-based studies.
“These data demonstrate the critical contribution of population-based birth defects surveillance to understanding the impact of Zika virus infection during pregnancy,” the authors concluded. “In 2016, CDC provided funding for 45 local, state, and territorial health departments to conduct rapid population-based surveillance for defects potentially related to Zika virus infection, which will provide essential data to monitor the impact of Zika virus infection in the United States.”
Birth defects potentially linked to cases of Zika virus in the United States have increased by a factor of nearly 20 since the virus first made its way into the country, according to new findings by the Centers for Disease Control and Prevention.
“The higher proportion of these defects among pregnancies with laboratory evidence of Zika infection in USZPR [U.S. Zika Pregnancy Registry] supports the relationship between congenital Zika virus infection and these birth defects,” wrote the authors of a new report led by Janet D. Cragan, MD, of the National Center on Birth Defects and Developmental Disabilities at the CDC (MMWR Morb Mortal Wkly Rep. 2017;66:219-22).![]()
[[{"attributes":{},"fields":{}}]]
Dr. Cragan and her coauthors retrospectively examined data on birth defects in three regions of the country: Massachusetts during 2013, North Carolina during 2013, and Atlanta during 2013-2014. The investigators focused on birth defects associated with prenatal Zika virus infections, mainly brain abnormalities and microcephaly.
The rate of total birth defects across the three regions was 2.86 per 1,000 live births, with 747 infants and fetuses identified as having one or more defects. Microcephaly and brain abnormalities alone occurred at a rate of 1.50 per 1,000 live births, with eye abnormalities and central nervous system dysfunction also occurring.
These numbers are relatively low when compared with data from Jan. 15 through Sept. 22, 2016. The birth defect rate jumped up to 58.8 per 1,000 live births, according to data from the USZPR, which found evidence of 26 infants and fetuses with brain or cranial defects in 442 completed pregnancies. These infants were all born to mothers with laboratory-confirmed Zika virus infections.
“Among 410 (55%) infants or fetuses with information on the earliest age a birth defect was recorded, 371 (90%) had evidence of a birth defect meeting the Zika definition before age 3 months,” the authors explained. “More than half of those with brain abnormalities or microcephaly or with neural tube defects and other early brain malformations had evidence of these defects noted prenatally (55% and 89%, respectively).”
Dr. Cragan and her colleagues hope that this evidence will further solidify the link between Zika virus and birth defects and pave the way for more population-based studies.
“These data demonstrate the critical contribution of population-based birth defects surveillance to understanding the impact of Zika virus infection during pregnancy,” the authors concluded. “In 2016, CDC provided funding for 45 local, state, and territorial health departments to conduct rapid population-based surveillance for defects potentially related to Zika virus infection, which will provide essential data to monitor the impact of Zika virus infection in the United States.”
FDA approves sublingual immunotherapy for dust mite allergies
Odactra (Merck, Sharp & Dohme) had been approved in adults aged 18-65 years, with allergic rhinitis with or without conjunctivitis. The tablets offer an alternative to subcutaneous injections, the FDA said in a statement issued March 1.
The sublingual tablets are intended to be taken daily, year-round, and the first dose must be taken under physician supervision to monitor for adverse reactions, according to the FDA. As with other sublingual immunotherapies, patients using the tablets should be simultaneously prescribed autoinjectable epinephrine.
The approval was based on results from randomized trials enrolling about 2,500 patients in Europe and the United States, according to the FDA. Patients taking the tablets saw a 16%-18% reduction in symptoms across studies, compared with placebo. Clinical benefit may be delayed by 8-14 weeks after starting the therapy, the agency said. Common adverse reactions reported in the studies included nausea, itching of the ears and mouth, and swelling of the lips and tongue.
Odactra is the fourth sublingual immunotherapy to be approved in the United States since 2014. Other approved therapies target grass and ragweed allergies.
Odactra (Merck, Sharp & Dohme) had been approved in adults aged 18-65 years, with allergic rhinitis with or without conjunctivitis. The tablets offer an alternative to subcutaneous injections, the FDA said in a statement issued March 1.
The sublingual tablets are intended to be taken daily, year-round, and the first dose must be taken under physician supervision to monitor for adverse reactions, according to the FDA. As with other sublingual immunotherapies, patients using the tablets should be simultaneously prescribed autoinjectable epinephrine.
The approval was based on results from randomized trials enrolling about 2,500 patients in Europe and the United States, according to the FDA. Patients taking the tablets saw a 16%-18% reduction in symptoms across studies, compared with placebo. Clinical benefit may be delayed by 8-14 weeks after starting the therapy, the agency said. Common adverse reactions reported in the studies included nausea, itching of the ears and mouth, and swelling of the lips and tongue.
Odactra is the fourth sublingual immunotherapy to be approved in the United States since 2014. Other approved therapies target grass and ragweed allergies.
Odactra (Merck, Sharp & Dohme) had been approved in adults aged 18-65 years, with allergic rhinitis with or without conjunctivitis. The tablets offer an alternative to subcutaneous injections, the FDA said in a statement issued March 1.
The sublingual tablets are intended to be taken daily, year-round, and the first dose must be taken under physician supervision to monitor for adverse reactions, according to the FDA. As with other sublingual immunotherapies, patients using the tablets should be simultaneously prescribed autoinjectable epinephrine.
The approval was based on results from randomized trials enrolling about 2,500 patients in Europe and the United States, according to the FDA. Patients taking the tablets saw a 16%-18% reduction in symptoms across studies, compared with placebo. Clinical benefit may be delayed by 8-14 weeks after starting the therapy, the agency said. Common adverse reactions reported in the studies included nausea, itching of the ears and mouth, and swelling of the lips and tongue.
Odactra is the fourth sublingual immunotherapy to be approved in the United States since 2014. Other approved therapies target grass and ragweed allergies.
Outpatient flu visits down slightly
The overall national measure of outpatient flu activity was down for the week ending Feb. 18, and the number of states at the highest level of activity dropped from 25 to 24, according to the Centers for Disease Control and Prevention.
The national proportion of outpatient visits for influenza-like illness (ILI) decreased from 5.2% the previous week to 4.8% for the week ending Feb. 18, the CDC reported.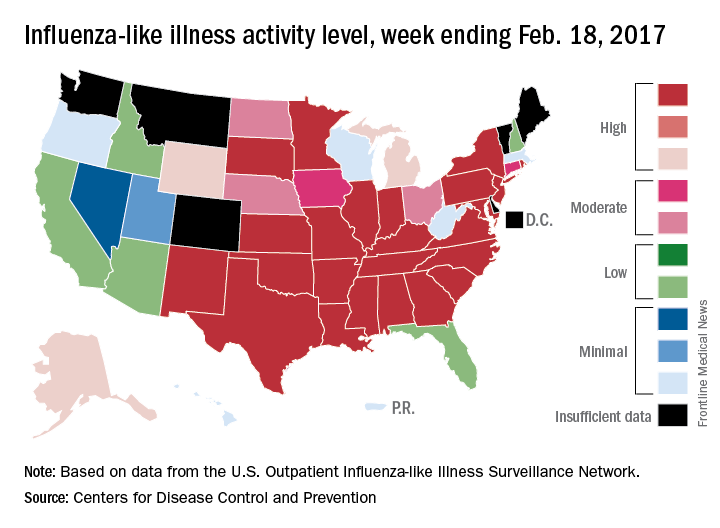
There were 5 ILI-related pediatric deaths reported during the week, bringing the total to 34 for the season so far, but none of the 5 occurred in the current week, the CDC said. There were 89 pediatric deaths reported during the 2015-2016 season, with the peak week occurring in late March/early April (11 deaths). During the 2014-2015 season, there were 148 deaths reported, and 111 were reported in 2013-2014.
The overall national measure of outpatient flu activity was down for the week ending Feb. 18, and the number of states at the highest level of activity dropped from 25 to 24, according to the Centers for Disease Control and Prevention.
The national proportion of outpatient visits for influenza-like illness (ILI) decreased from 5.2% the previous week to 4.8% for the week ending Feb. 18, the CDC reported.
There were 5 ILI-related pediatric deaths reported during the week, bringing the total to 34 for the season so far, but none of the 5 occurred in the current week, the CDC said. There were 89 pediatric deaths reported during the 2015-2016 season, with the peak week occurring in late March/early April (11 deaths). During the 2014-2015 season, there were 148 deaths reported, and 111 were reported in 2013-2014.
The overall national measure of outpatient flu activity was down for the week ending Feb. 18, and the number of states at the highest level of activity dropped from 25 to 24, according to the Centers for Disease Control and Prevention.
The national proportion of outpatient visits for influenza-like illness (ILI) decreased from 5.2% the previous week to 4.8% for the week ending Feb. 18, the CDC reported.
There were 5 ILI-related pediatric deaths reported during the week, bringing the total to 34 for the season so far, but none of the 5 occurred in the current week, the CDC said. There were 89 pediatric deaths reported during the 2015-2016 season, with the peak week occurring in late March/early April (11 deaths). During the 2014-2015 season, there were 148 deaths reported, and 111 were reported in 2013-2014.
FDA clears procalcitonin test to hone antibiotic use in LRTI, sepsis
The Food and Drug Administration has cleared the expanded use of a procalcitonin test to help determine antibiotic use in patients with lower respiratory tract infections (LRTI) and sepsis.
The Vidas Brahms PCT Assay (bioMérieux) uses procalcitonin levels to determine whether a patient with a lower respiratory tract infection (LRTI) should begin or remain on antibiotics and when antibiotics should be withdrawn in a patient with sepsis.
The test will be used primarily in hospital settings and emergency departments, according to the FDA. Test levels that are high levels suggest bacterial infection and the need for antibiotics while low levels indicate viral or noninfectious processes. However, concerns exist regarding false-positive or false-negative test results, which can prompt clinicians to prematurely stop or unnecessarily continue an antibiotic regimen in certain patients.
“Health care providers should not rely solely on PCT test results when making treatment decisions but should interpret test results in the context of a patient’s clinical status and other laboratory results,” according to the FDA statement.
The expanded use of the test was approved based on promising data from clinical trials that was presented at an FDA advisory committee meeting in November 2016. The Vidas Brahms test was already approved by the FDA for use in determining a patient’s risk of dying from sepsis. The test was cleared via the FDA 510(k) regulatory pathway, which is meant for tests or devices for which there is already something similar on the market.
Support for the test’s expanded usage comes from published prospective, randomized clinical trials that compared PCT-guided therapy with standard therapy. In those studies, patients who had received PCT-guided therapy experienced significant decreases in antibiotic use without significant affects to their safety.
The Food and Drug Administration has cleared the expanded use of a procalcitonin test to help determine antibiotic use in patients with lower respiratory tract infections (LRTI) and sepsis.
The Vidas Brahms PCT Assay (bioMérieux) uses procalcitonin levels to determine whether a patient with a lower respiratory tract infection (LRTI) should begin or remain on antibiotics and when antibiotics should be withdrawn in a patient with sepsis.
The test will be used primarily in hospital settings and emergency departments, according to the FDA. Test levels that are high levels suggest bacterial infection and the need for antibiotics while low levels indicate viral or noninfectious processes. However, concerns exist regarding false-positive or false-negative test results, which can prompt clinicians to prematurely stop or unnecessarily continue an antibiotic regimen in certain patients.
“Health care providers should not rely solely on PCT test results when making treatment decisions but should interpret test results in the context of a patient’s clinical status and other laboratory results,” according to the FDA statement.
The expanded use of the test was approved based on promising data from clinical trials that was presented at an FDA advisory committee meeting in November 2016. The Vidas Brahms test was already approved by the FDA for use in determining a patient’s risk of dying from sepsis. The test was cleared via the FDA 510(k) regulatory pathway, which is meant for tests or devices for which there is already something similar on the market.
Support for the test’s expanded usage comes from published prospective, randomized clinical trials that compared PCT-guided therapy with standard therapy. In those studies, patients who had received PCT-guided therapy experienced significant decreases in antibiotic use without significant affects to their safety.
The Food and Drug Administration has cleared the expanded use of a procalcitonin test to help determine antibiotic use in patients with lower respiratory tract infections (LRTI) and sepsis.
The Vidas Brahms PCT Assay (bioMérieux) uses procalcitonin levels to determine whether a patient with a lower respiratory tract infection (LRTI) should begin or remain on antibiotics and when antibiotics should be withdrawn in a patient with sepsis.
The test will be used primarily in hospital settings and emergency departments, according to the FDA. Test levels that are high levels suggest bacterial infection and the need for antibiotics while low levels indicate viral or noninfectious processes. However, concerns exist regarding false-positive or false-negative test results, which can prompt clinicians to prematurely stop or unnecessarily continue an antibiotic regimen in certain patients.
“Health care providers should not rely solely on PCT test results when making treatment decisions but should interpret test results in the context of a patient’s clinical status and other laboratory results,” according to the FDA statement.
The expanded use of the test was approved based on promising data from clinical trials that was presented at an FDA advisory committee meeting in November 2016. The Vidas Brahms test was already approved by the FDA for use in determining a patient’s risk of dying from sepsis. The test was cleared via the FDA 510(k) regulatory pathway, which is meant for tests or devices for which there is already something similar on the market.
Support for the test’s expanded usage comes from published prospective, randomized clinical trials that compared PCT-guided therapy with standard therapy. In those studies, patients who had received PCT-guided therapy experienced significant decreases in antibiotic use without significant affects to their safety.
Multiple myeloma: Lenalidomide approved as maintenance therapy after auto-HSCT
The Food and Drug Administration has approved the use of lenalidomide (Revlimid) for maintenance therapy following autologous hematopoietic stem cell transplant in patients with multiple myeloma.
The expanded indication, announced Feb. 22, makes the immunomodulatory agent the first and only approved treatment for post autologous hematopoietic stem cell transplant (auto-HSCT) maintenance. It was initially approved in 2006 for use in combination with dexamethasone in patients with multiple myeloma who have received at least one prior therapy, and that indication was expanded in 2015 to include those with newly diagnosed multiple myeloma.
According to Celgene, the maker of Revlimid, the latest approval was based on data showing that lenalidomide maintenance therapy delays disease progression following auto-HSCT. Updated phase III randomized controlled trial data from two studies including more than 1,000 patients demonstrated median progression-free survival (PFS) advantages with lenalidomide maintenance vs. no maintenance. In one study – the U.S.-based CALGB 1001014 – median PFS was 5.7 vs. 1.9 years for a difference of 3.8 years (hazard ratio, 0.38). In the second study – the European IFM 2005-02 – median PFS was 3.9 vs. 2 years, for a difference of 1.9 years (HR, 0.53).![]()
In both studies lenalidomide was given as a 10-mg daily oral dose (increased to 15 mg daily after 3 months if tolerated) until disease progression or unacceptable toxicity after auto-HSCT.
Lenalidomide, a derivative of thalidomide, can cause fetal harm and is contraindicated in women who are pregnant. It is available only through a restricted distribution program.
The most frequently reported adverse reactions in the two studies were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, muscle spasm, and pyrexia. The most frequently reported grade 3 or 4 reactions occurring in more than 20% of patients in the lenalidomide arms included neutropenia, thrombocytopenia, and leukopenia.
“Autologous stem cell transplant after induction therapy is part of the continuum of care for transplant-eligible multiple myeloma patients. However, most patients will still see their disease recur or progress after this treatment,” Philip McCarthy, MD, of the Roswell Park Cancer Institute in Buffalo, N.Y., said in a Celgene press statement. “Lenalidomide maintenance therapy ... can be considered a standard of care for these patients.”
The Food and Drug Administration has approved the use of lenalidomide (Revlimid) for maintenance therapy following autologous hematopoietic stem cell transplant in patients with multiple myeloma.
The expanded indication, announced Feb. 22, makes the immunomodulatory agent the first and only approved treatment for post autologous hematopoietic stem cell transplant (auto-HSCT) maintenance. It was initially approved in 2006 for use in combination with dexamethasone in patients with multiple myeloma who have received at least one prior therapy, and that indication was expanded in 2015 to include those with newly diagnosed multiple myeloma.
According to Celgene, the maker of Revlimid, the latest approval was based on data showing that lenalidomide maintenance therapy delays disease progression following auto-HSCT. Updated phase III randomized controlled trial data from two studies including more than 1,000 patients demonstrated median progression-free survival (PFS) advantages with lenalidomide maintenance vs. no maintenance. In one study – the U.S.-based CALGB 1001014 – median PFS was 5.7 vs. 1.9 years for a difference of 3.8 years (hazard ratio, 0.38). In the second study – the European IFM 2005-02 – median PFS was 3.9 vs. 2 years, for a difference of 1.9 years (HR, 0.53).![]()
In both studies lenalidomide was given as a 10-mg daily oral dose (increased to 15 mg daily after 3 months if tolerated) until disease progression or unacceptable toxicity after auto-HSCT.
Lenalidomide, a derivative of thalidomide, can cause fetal harm and is contraindicated in women who are pregnant. It is available only through a restricted distribution program.
The most frequently reported adverse reactions in the two studies were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, muscle spasm, and pyrexia. The most frequently reported grade 3 or 4 reactions occurring in more than 20% of patients in the lenalidomide arms included neutropenia, thrombocytopenia, and leukopenia.
“Autologous stem cell transplant after induction therapy is part of the continuum of care for transplant-eligible multiple myeloma patients. However, most patients will still see their disease recur or progress after this treatment,” Philip McCarthy, MD, of the Roswell Park Cancer Institute in Buffalo, N.Y., said in a Celgene press statement. “Lenalidomide maintenance therapy ... can be considered a standard of care for these patients.”
The Food and Drug Administration has approved the use of lenalidomide (Revlimid) for maintenance therapy following autologous hematopoietic stem cell transplant in patients with multiple myeloma.
The expanded indication, announced Feb. 22, makes the immunomodulatory agent the first and only approved treatment for post autologous hematopoietic stem cell transplant (auto-HSCT) maintenance. It was initially approved in 2006 for use in combination with dexamethasone in patients with multiple myeloma who have received at least one prior therapy, and that indication was expanded in 2015 to include those with newly diagnosed multiple myeloma.
According to Celgene, the maker of Revlimid, the latest approval was based on data showing that lenalidomide maintenance therapy delays disease progression following auto-HSCT. Updated phase III randomized controlled trial data from two studies including more than 1,000 patients demonstrated median progression-free survival (PFS) advantages with lenalidomide maintenance vs. no maintenance. In one study – the U.S.-based CALGB 1001014 – median PFS was 5.7 vs. 1.9 years for a difference of 3.8 years (hazard ratio, 0.38). In the second study – the European IFM 2005-02 – median PFS was 3.9 vs. 2 years, for a difference of 1.9 years (HR, 0.53).![]()
In both studies lenalidomide was given as a 10-mg daily oral dose (increased to 15 mg daily after 3 months if tolerated) until disease progression or unacceptable toxicity after auto-HSCT.
Lenalidomide, a derivative of thalidomide, can cause fetal harm and is contraindicated in women who are pregnant. It is available only through a restricted distribution program.
The most frequently reported adverse reactions in the two studies were neutropenia, thrombocytopenia, leukopenia, anemia, upper respiratory tract infection, bronchitis, nasopharyngitis, cough, gastroenteritis, diarrhea, rash, fatigue, muscle spasm, and pyrexia. The most frequently reported grade 3 or 4 reactions occurring in more than 20% of patients in the lenalidomide arms included neutropenia, thrombocytopenia, and leukopenia.
“Autologous stem cell transplant after induction therapy is part of the continuum of care for transplant-eligible multiple myeloma patients. However, most patients will still see their disease recur or progress after this treatment,” Philip McCarthy, MD, of the Roswell Park Cancer Institute in Buffalo, N.Y., said in a Celgene press statement. “Lenalidomide maintenance therapy ... can be considered a standard of care for these patients.”
FDA confirms complications from intragastric balloons
Complications from overinflation and acute pancreatitis can create problems for obesity patients treated with intragastric balloons, according to a statement from the Food and Drug Administration. In a letter to health care providers published on February 9, 2017, the FDA warned of the two specific issues that have been the subject of multiple adverse event reports.
“We recommend that you closely monitor patients with these devices for these adverse events, and to submit reports to help us better understand any complications from the use of these obesity treatment devices,” the letter said.
Most of the overinflation reports involved the Orbera Intragastric Balloon System (Apollo Endosurgery) that uses a single balloon, although some reports involved the ReShape Integrated Dual Balloon System (ReShape Medical) that uses two balloons. Neither product mentions overinflation risk in its labeling. “At this moment there is not enough information to determine what is causing the balloon to overinflate,” according to the FDA letter.
A separate set of adverse event reports noted the development of acute pancreatitis caused when the balloons compressed other gastrointestinal structures. Both the Orbera and ReShape products were associated with pancreatitis, although neither lists pancreatitis as a potential complication on their labels. Pancreatitis was reported as early as 3 days after implantation, and symptoms included severe back and abdominal pain.
The FDA letter recommends that health care providers consider overinflation and pancreatitis in their differential diagnoses of obesity patients with intragastric balloons who present with the symptoms described, and to report any type of serious adverse events associated with intragastric balloons to the FDA through the MedWatch program. For more information about reporting adverse events to the FDA, visit the MedWatch site.
Complications from overinflation and acute pancreatitis can create problems for obesity patients treated with intragastric balloons, according to a statement from the Food and Drug Administration. In a letter to health care providers published on February 9, 2017, the FDA warned of the two specific issues that have been the subject of multiple adverse event reports.
“We recommend that you closely monitor patients with these devices for these adverse events, and to submit reports to help us better understand any complications from the use of these obesity treatment devices,” the letter said.
Most of the overinflation reports involved the Orbera Intragastric Balloon System (Apollo Endosurgery) that uses a single balloon, although some reports involved the ReShape Integrated Dual Balloon System (ReShape Medical) that uses two balloons. Neither product mentions overinflation risk in its labeling. “At this moment there is not enough information to determine what is causing the balloon to overinflate,” according to the FDA letter.
A separate set of adverse event reports noted the development of acute pancreatitis caused when the balloons compressed other gastrointestinal structures. Both the Orbera and ReShape products were associated with pancreatitis, although neither lists pancreatitis as a potential complication on their labels. Pancreatitis was reported as early as 3 days after implantation, and symptoms included severe back and abdominal pain.
The FDA letter recommends that health care providers consider overinflation and pancreatitis in their differential diagnoses of obesity patients with intragastric balloons who present with the symptoms described, and to report any type of serious adverse events associated with intragastric balloons to the FDA through the MedWatch program. For more information about reporting adverse events to the FDA, visit the MedWatch site.
Complications from overinflation and acute pancreatitis can create problems for obesity patients treated with intragastric balloons, according to a statement from the Food and Drug Administration. In a letter to health care providers published on February 9, 2017, the FDA warned of the two specific issues that have been the subject of multiple adverse event reports.
“We recommend that you closely monitor patients with these devices for these adverse events, and to submit reports to help us better understand any complications from the use of these obesity treatment devices,” the letter said.
Most of the overinflation reports involved the Orbera Intragastric Balloon System (Apollo Endosurgery) that uses a single balloon, although some reports involved the ReShape Integrated Dual Balloon System (ReShape Medical) that uses two balloons. Neither product mentions overinflation risk in its labeling. “At this moment there is not enough information to determine what is causing the balloon to overinflate,” according to the FDA letter.
A separate set of adverse event reports noted the development of acute pancreatitis caused when the balloons compressed other gastrointestinal structures. Both the Orbera and ReShape products were associated with pancreatitis, although neither lists pancreatitis as a potential complication on their labels. Pancreatitis was reported as early as 3 days after implantation, and symptoms included severe back and abdominal pain.
The FDA letter recommends that health care providers consider overinflation and pancreatitis in their differential diagnoses of obesity patients with intragastric balloons who present with the symptoms described, and to report any type of serious adverse events associated with intragastric balloons to the FDA through the MedWatch program. For more information about reporting adverse events to the FDA, visit the MedWatch site.
Twenty-five states at highest flu activity level
Flu activity in the United States continued to increase as half of the states reached the highest level of influenza-like illness (ILI) activity in the week ending Feb. 11, according to the Centers for Disease Control and Prevention.
For the week, the 25 states at level 10 on the CDC’s 1-10 scale of ILI activity were joined in the high range by Illinois and Kentucky at level 9 and Iowa at level 8, the CDC reported. The previous week, there were 23 states in the high range.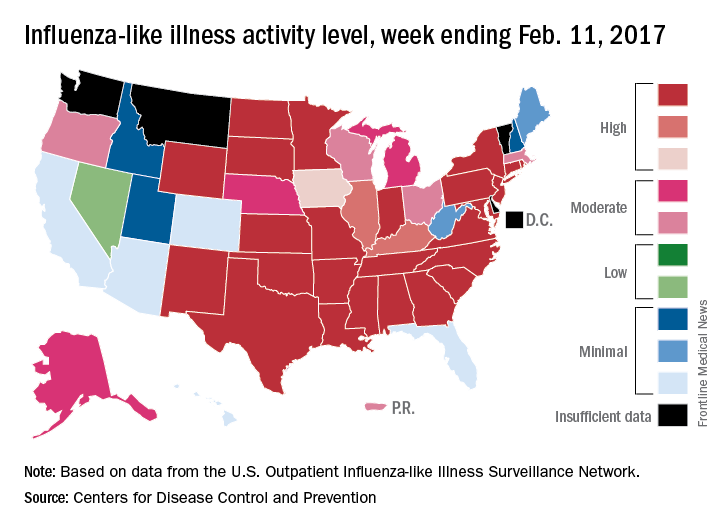
Of the nine flu-related pediatric deaths reported to the CDC during the latest week, eight occurred in earlier weeks. For the 2016-2017 season so far, 29 flu-related pediatric deaths have been reported, the CDC said.
Flu activity in the United States continued to increase as half of the states reached the highest level of influenza-like illness (ILI) activity in the week ending Feb. 11, according to the Centers for Disease Control and Prevention.
For the week, the 25 states at level 10 on the CDC’s 1-10 scale of ILI activity were joined in the high range by Illinois and Kentucky at level 9 and Iowa at level 8, the CDC reported. The previous week, there were 23 states in the high range.
Of the nine flu-related pediatric deaths reported to the CDC during the latest week, eight occurred in earlier weeks. For the 2016-2017 season so far, 29 flu-related pediatric deaths have been reported, the CDC said.
Flu activity in the United States continued to increase as half of the states reached the highest level of influenza-like illness (ILI) activity in the week ending Feb. 11, according to the Centers for Disease Control and Prevention.
For the week, the 25 states at level 10 on the CDC’s 1-10 scale of ILI activity were joined in the high range by Illinois and Kentucky at level 9 and Iowa at level 8, the CDC reported. The previous week, there were 23 states in the high range.
Of the nine flu-related pediatric deaths reported to the CDC during the latest week, eight occurred in earlier weeks. For the 2016-2017 season so far, 29 flu-related pediatric deaths have been reported, the CDC said.