User login
Zika slowing, but not going away
New cases of pregnant women with laboratory evidence of Zika virus infection were down for the 2 weeks ending Feb. 7, with a big drop in the U.S. territories offsetting an increase among the 50 states and the District of Columbia, according to the Centers for Disease Control and Prevention.
There were 146 new cases of Zika infection reported in pregnant women over that period, compared with 233 over the previous 2 weeks. Of the new cases, 85 were reported in the U.S. territories (down from 186) and 61 were reported in the 50 states and D.C. (up from 47), the CDC reported. These are not real-time estimates, the CDC noted. They reflect only the number of reports received, so the cases may have occurred during earlier reporting periods.
Of the cases in the states/D.C., 1,047 pregnancies have been completed with or without birth defects. There have been 43 liveborn infants with Zika-related birth defects and five pregnancy losses with Zika-related defects, the CDC said. The number of liveborn infants was up from 38 for the previous 2-week span, which was the largest increase since mid-November.
The Zika caseload for all Americans is 42,063 (from Jan. 1, 2015 to Feb. 15, 2017), of which 5,040 cases were reported in the 50 states/D.C. and 37,023 were reported in the territories (98% in Puerto Rico), the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
New cases of pregnant women with laboratory evidence of Zika virus infection were down for the 2 weeks ending Feb. 7, with a big drop in the U.S. territories offsetting an increase among the 50 states and the District of Columbia, according to the Centers for Disease Control and Prevention.
There were 146 new cases of Zika infection reported in pregnant women over that period, compared with 233 over the previous 2 weeks. Of the new cases, 85 were reported in the U.S. territories (down from 186) and 61 were reported in the 50 states and D.C. (up from 47), the CDC reported. These are not real-time estimates, the CDC noted. They reflect only the number of reports received, so the cases may have occurred during earlier reporting periods.
Of the cases in the states/D.C., 1,047 pregnancies have been completed with or without birth defects. There have been 43 liveborn infants with Zika-related birth defects and five pregnancy losses with Zika-related defects, the CDC said. The number of liveborn infants was up from 38 for the previous 2-week span, which was the largest increase since mid-November.
The Zika caseload for all Americans is 42,063 (from Jan. 1, 2015 to Feb. 15, 2017), of which 5,040 cases were reported in the 50 states/D.C. and 37,023 were reported in the territories (98% in Puerto Rico), the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
New cases of pregnant women with laboratory evidence of Zika virus infection were down for the 2 weeks ending Feb. 7, with a big drop in the U.S. territories offsetting an increase among the 50 states and the District of Columbia, according to the Centers for Disease Control and Prevention.
There were 146 new cases of Zika infection reported in pregnant women over that period, compared with 233 over the previous 2 weeks. Of the new cases, 85 were reported in the U.S. territories (down from 186) and 61 were reported in the 50 states and D.C. (up from 47), the CDC reported. These are not real-time estimates, the CDC noted. They reflect only the number of reports received, so the cases may have occurred during earlier reporting periods.
Of the cases in the states/D.C., 1,047 pregnancies have been completed with or without birth defects. There have been 43 liveborn infants with Zika-related birth defects and five pregnancy losses with Zika-related defects, the CDC said. The number of liveborn infants was up from 38 for the previous 2-week span, which was the largest increase since mid-November.
The Zika caseload for all Americans is 42,063 (from Jan. 1, 2015 to Feb. 15, 2017), of which 5,040 cases were reported in the 50 states/D.C. and 37,023 were reported in the territories (98% in Puerto Rico), the CDC reported.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
CDC: More preterm births after fertility treatments
Infants conceived through assisted reproductive technology (ART) represented less than 2% of all children born in the United States in 2014, but they experienced disproportionately large shares of adverse perinatal outcomes, according to the Centers for Disease Control and Prevention.
Of the 4.02 million children born in the United States in 2014, a total of 65,296 (1.6%) were conceived with ART procedures. ART-conceived children, however, represented 5.5% of all low-birth-weight infants (less than 2,500 g) and 5.6% of all very-low-birth-weight infants (less than 1,500 g). ART-conceived children also represented 4.7% of all preterm births (less than 37 weeks) in 2014 and 5.0% of all very preterm (less than 32 weeks) infants nationally, the CDC reported (MMWR Surveill Summ. 2017;66[SS-6]:1-24).
These poor outcomes are related, at least in part, to the effect of infertility treatment on multiple births. The percentage of triplets or higher-order infants among ART-conceived infants has decreased since 2000, but “the percentage of twin infants has remained persistently high,” the CDC noted. Multiple-birth infants represent 39.4% of all ART infants but only 3.5% of all infants, and ART-conceived multiple-birth infants represent 18.3% of all multiple-birth infants, the CDC said.
The multiple-birth effect varied considerably by state, however. In Hawaii, ART-conceived multiple-birth infants accounted for 37.3% of all multiple births in 2014, while Alaska and West Virginia both had rates of 5.5%. Poor outcomes also varied by state: In Massachusetts, ART infants represented 14.2% of all low-birth-weight infants, compared with 1.2% in West Virginia. Massachusetts had the highest rate of ART preterm infants to all infants, 13.4%, and Puerto Rico had the lowest, 1.2%, according to the CDC report, which was based on data from the National ART Surveillance System.
Puerto Rico’s rate of 364 ART procedures per 1 million women aged 15-44 years was the lowest in the country, and the highest rate belonged to Massachusetts at 6,726 per 1 million women aged 15-44 years.
Infants conceived through assisted reproductive technology (ART) represented less than 2% of all children born in the United States in 2014, but they experienced disproportionately large shares of adverse perinatal outcomes, according to the Centers for Disease Control and Prevention.
Of the 4.02 million children born in the United States in 2014, a total of 65,296 (1.6%) were conceived with ART procedures. ART-conceived children, however, represented 5.5% of all low-birth-weight infants (less than 2,500 g) and 5.6% of all very-low-birth-weight infants (less than 1,500 g). ART-conceived children also represented 4.7% of all preterm births (less than 37 weeks) in 2014 and 5.0% of all very preterm (less than 32 weeks) infants nationally, the CDC reported (MMWR Surveill Summ. 2017;66[SS-6]:1-24).
These poor outcomes are related, at least in part, to the effect of infertility treatment on multiple births. The percentage of triplets or higher-order infants among ART-conceived infants has decreased since 2000, but “the percentage of twin infants has remained persistently high,” the CDC noted. Multiple-birth infants represent 39.4% of all ART infants but only 3.5% of all infants, and ART-conceived multiple-birth infants represent 18.3% of all multiple-birth infants, the CDC said.
The multiple-birth effect varied considerably by state, however. In Hawaii, ART-conceived multiple-birth infants accounted for 37.3% of all multiple births in 2014, while Alaska and West Virginia both had rates of 5.5%. Poor outcomes also varied by state: In Massachusetts, ART infants represented 14.2% of all low-birth-weight infants, compared with 1.2% in West Virginia. Massachusetts had the highest rate of ART preterm infants to all infants, 13.4%, and Puerto Rico had the lowest, 1.2%, according to the CDC report, which was based on data from the National ART Surveillance System.
Puerto Rico’s rate of 364 ART procedures per 1 million women aged 15-44 years was the lowest in the country, and the highest rate belonged to Massachusetts at 6,726 per 1 million women aged 15-44 years.
Infants conceived through assisted reproductive technology (ART) represented less than 2% of all children born in the United States in 2014, but they experienced disproportionately large shares of adverse perinatal outcomes, according to the Centers for Disease Control and Prevention.
Of the 4.02 million children born in the United States in 2014, a total of 65,296 (1.6%) were conceived with ART procedures. ART-conceived children, however, represented 5.5% of all low-birth-weight infants (less than 2,500 g) and 5.6% of all very-low-birth-weight infants (less than 1,500 g). ART-conceived children also represented 4.7% of all preterm births (less than 37 weeks) in 2014 and 5.0% of all very preterm (less than 32 weeks) infants nationally, the CDC reported (MMWR Surveill Summ. 2017;66[SS-6]:1-24).
These poor outcomes are related, at least in part, to the effect of infertility treatment on multiple births. The percentage of triplets or higher-order infants among ART-conceived infants has decreased since 2000, but “the percentage of twin infants has remained persistently high,” the CDC noted. Multiple-birth infants represent 39.4% of all ART infants but only 3.5% of all infants, and ART-conceived multiple-birth infants represent 18.3% of all multiple-birth infants, the CDC said.
The multiple-birth effect varied considerably by state, however. In Hawaii, ART-conceived multiple-birth infants accounted for 37.3% of all multiple births in 2014, while Alaska and West Virginia both had rates of 5.5%. Poor outcomes also varied by state: In Massachusetts, ART infants represented 14.2% of all low-birth-weight infants, compared with 1.2% in West Virginia. Massachusetts had the highest rate of ART preterm infants to all infants, 13.4%, and Puerto Rico had the lowest, 1.2%, according to the CDC report, which was based on data from the National ART Surveillance System.
Puerto Rico’s rate of 364 ART procedures per 1 million women aged 15-44 years was the lowest in the country, and the highest rate belonged to Massachusetts at 6,726 per 1 million women aged 15-44 years.
FROM MMWR SURVEILLANCE SUMMARIES
Brodalumab approved for psoriasis with REMS required
The Food and Drug Administration has approved brodalumab, an interleukin-17 receptor A-antagonist, for treating adults with moderate to severe plaque psoriasis, with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the “observed risk of suicidal ideation and behavior” in clinical trials, the agency announced on Feb. 16.
In the three pivotal phase III studies of 4,373 adults with moderate to severe plaque psoriasis who were candidates for systemic therapy or phototherapy, 83%-86% of those treated with brodalumab achieved Psoriasis Area and Severity Index (PASI 75) scores at 12 weeks of treatment, compared with 3%-8% of those on placebo. In addition, 37%-44% of those on brodalumab achieved PASI 100 scores, compared with 1% or fewer of those on placebo. In the psoriasis clinical trials, suicidal ideation and behavior, which included four completed suicides, occurred in patients treated with brodalumab, according to the prescribing information
In the statement, the FDA points out that “a causal association between treatment with Siliq and increased risk of suicidal ideation and behavior has not been established,” but that “suicidal ideation and behavior, including completed suicides, have occurred in patients treated with Siliq during clinical trials. Siliq users with a history of suicidality or depression had an increased incidence of suicidal ideation and behavior, compared to users without this history.”
At a meeting in July 2016, the FDA’s Dermatologic and Ophthalmic Drugs Advisory Committee voted 18-0 in favor of approving brodalumab, with the majority (14) recommending risk management options beyond labeling to address these concerns.
Elements of the REMS include requirements for prescribers, pharmacy certification, and a medication guide for patients with information about the risks of suicidal ideation and behavior. Prescribers are required to counsel patients about this risk, and patients are required to sign a “Patient-Prescriber Agreement Form and be made aware of the need to seek medical attention should they experience new or worsening suicidal thoughts or behavior, feelings of depression, anxiety or other mood changes,” the FDA said. The prescribing information also includes a boxed warning about suicidal ideation and behavior.
The most common adverse events associated with brodalumab, the FDA statement noted, include arthralgia, headache, fatigue, diarrhea, oropharyngeal pain, nausea, myalgia, injection site reactions, neutropenia, and fungal infections. The recommended dose of brodalumab is 210 mg, administered subcutaneously, at weeks 0, 1, and 2, followed by 210 mg every 2 weeks.
The Food and Drug Administration has approved brodalumab, an interleukin-17 receptor A-antagonist, for treating adults with moderate to severe plaque psoriasis, with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the “observed risk of suicidal ideation and behavior” in clinical trials, the agency announced on Feb. 16.
In the three pivotal phase III studies of 4,373 adults with moderate to severe plaque psoriasis who were candidates for systemic therapy or phototherapy, 83%-86% of those treated with brodalumab achieved Psoriasis Area and Severity Index (PASI 75) scores at 12 weeks of treatment, compared with 3%-8% of those on placebo. In addition, 37%-44% of those on brodalumab achieved PASI 100 scores, compared with 1% or fewer of those on placebo. In the psoriasis clinical trials, suicidal ideation and behavior, which included four completed suicides, occurred in patients treated with brodalumab, according to the prescribing information
In the statement, the FDA points out that “a causal association between treatment with Siliq and increased risk of suicidal ideation and behavior has not been established,” but that “suicidal ideation and behavior, including completed suicides, have occurred in patients treated with Siliq during clinical trials. Siliq users with a history of suicidality or depression had an increased incidence of suicidal ideation and behavior, compared to users without this history.”
At a meeting in July 2016, the FDA’s Dermatologic and Ophthalmic Drugs Advisory Committee voted 18-0 in favor of approving brodalumab, with the majority (14) recommending risk management options beyond labeling to address these concerns.
Elements of the REMS include requirements for prescribers, pharmacy certification, and a medication guide for patients with information about the risks of suicidal ideation and behavior. Prescribers are required to counsel patients about this risk, and patients are required to sign a “Patient-Prescriber Agreement Form and be made aware of the need to seek medical attention should they experience new or worsening suicidal thoughts or behavior, feelings of depression, anxiety or other mood changes,” the FDA said. The prescribing information also includes a boxed warning about suicidal ideation and behavior.
The most common adverse events associated with brodalumab, the FDA statement noted, include arthralgia, headache, fatigue, diarrhea, oropharyngeal pain, nausea, myalgia, injection site reactions, neutropenia, and fungal infections. The recommended dose of brodalumab is 210 mg, administered subcutaneously, at weeks 0, 1, and 2, followed by 210 mg every 2 weeks.
The Food and Drug Administration has approved brodalumab, an interleukin-17 receptor A-antagonist, for treating adults with moderate to severe plaque psoriasis, with a Risk Evaluation and Mitigation Strategy (REMS) that addresses the “observed risk of suicidal ideation and behavior” in clinical trials, the agency announced on Feb. 16.
In the three pivotal phase III studies of 4,373 adults with moderate to severe plaque psoriasis who were candidates for systemic therapy or phototherapy, 83%-86% of those treated with brodalumab achieved Psoriasis Area and Severity Index (PASI 75) scores at 12 weeks of treatment, compared with 3%-8% of those on placebo. In addition, 37%-44% of those on brodalumab achieved PASI 100 scores, compared with 1% or fewer of those on placebo. In the psoriasis clinical trials, suicidal ideation and behavior, which included four completed suicides, occurred in patients treated with brodalumab, according to the prescribing information
In the statement, the FDA points out that “a causal association between treatment with Siliq and increased risk of suicidal ideation and behavior has not been established,” but that “suicidal ideation and behavior, including completed suicides, have occurred in patients treated with Siliq during clinical trials. Siliq users with a history of suicidality or depression had an increased incidence of suicidal ideation and behavior, compared to users without this history.”
At a meeting in July 2016, the FDA’s Dermatologic and Ophthalmic Drugs Advisory Committee voted 18-0 in favor of approving brodalumab, with the majority (14) recommending risk management options beyond labeling to address these concerns.
Elements of the REMS include requirements for prescribers, pharmacy certification, and a medication guide for patients with information about the risks of suicidal ideation and behavior. Prescribers are required to counsel patients about this risk, and patients are required to sign a “Patient-Prescriber Agreement Form and be made aware of the need to seek medical attention should they experience new or worsening suicidal thoughts or behavior, feelings of depression, anxiety or other mood changes,” the FDA said. The prescribing information also includes a boxed warning about suicidal ideation and behavior.
The most common adverse events associated with brodalumab, the FDA statement noted, include arthralgia, headache, fatigue, diarrhea, oropharyngeal pain, nausea, myalgia, injection site reactions, neutropenia, and fungal infections. The recommended dose of brodalumab is 210 mg, administered subcutaneously, at weeks 0, 1, and 2, followed by 210 mg every 2 weeks.
Flu activity levels high in 23 states
The number of states at the highest level of flu activity jumped from 7 to 19 during the week ending Feb. 4, 2017, according to the Centers for Disease Control and Prevention.
The 19 states at level 10 on the CDC’s 1-10 scale of influenza-like illness (ILI) activity were largely concentrated in the South and lower Midwest, with another grouping mainly in the Mid-Atlantic. They were joined in the “high” range of ILI activity by four other states: Indiana and Minnesota at level 9 and New Mexico and Virginia at level 8, the CDC reported.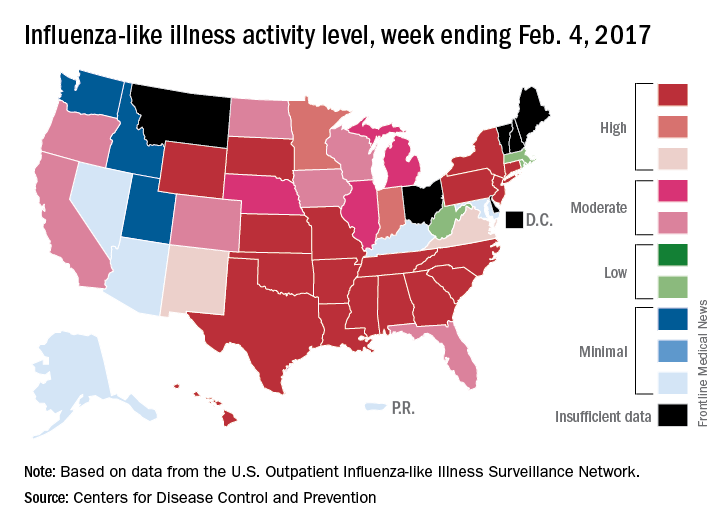
For the 2016-2017 season, 20 flu-related pediatric deaths have been reported: 5 were reported during the week ending Feb. 4, but 4 occurred during the week ending Jan. 28 and 1 occurred during the week ending Jan. 14, according to the CDC.
Since Oct. 1, 2016, there have been 6,804 laboratory-confirmed flu-related hospitalizations reported in the 13 states – including California, Georgia, New York, and Ohio – of the CDC’s Influenza Hospitalization Surveillance Network, for an overall hospitalization rate of 24.3 per 100,000 population. That rate, however, is “likely to be an underestimate as influenza-related hospitalizations can be missed, either because testing is not performed, or because cases may be attributed to other causes of pneumonia or other common influenza-related complications,” the CDC said.
The number of states at the highest level of flu activity jumped from 7 to 19 during the week ending Feb. 4, 2017, according to the Centers for Disease Control and Prevention.
The 19 states at level 10 on the CDC’s 1-10 scale of influenza-like illness (ILI) activity were largely concentrated in the South and lower Midwest, with another grouping mainly in the Mid-Atlantic. They were joined in the “high” range of ILI activity by four other states: Indiana and Minnesota at level 9 and New Mexico and Virginia at level 8, the CDC reported.
For the 2016-2017 season, 20 flu-related pediatric deaths have been reported: 5 were reported during the week ending Feb. 4, but 4 occurred during the week ending Jan. 28 and 1 occurred during the week ending Jan. 14, according to the CDC.
Since Oct. 1, 2016, there have been 6,804 laboratory-confirmed flu-related hospitalizations reported in the 13 states – including California, Georgia, New York, and Ohio – of the CDC’s Influenza Hospitalization Surveillance Network, for an overall hospitalization rate of 24.3 per 100,000 population. That rate, however, is “likely to be an underestimate as influenza-related hospitalizations can be missed, either because testing is not performed, or because cases may be attributed to other causes of pneumonia or other common influenza-related complications,” the CDC said.
The number of states at the highest level of flu activity jumped from 7 to 19 during the week ending Feb. 4, 2017, according to the Centers for Disease Control and Prevention.
The 19 states at level 10 on the CDC’s 1-10 scale of influenza-like illness (ILI) activity were largely concentrated in the South and lower Midwest, with another grouping mainly in the Mid-Atlantic. They were joined in the “high” range of ILI activity by four other states: Indiana and Minnesota at level 9 and New Mexico and Virginia at level 8, the CDC reported.
For the 2016-2017 season, 20 flu-related pediatric deaths have been reported: 5 were reported during the week ending Feb. 4, but 4 occurred during the week ending Jan. 28 and 1 occurred during the week ending Jan. 14, according to the CDC.
Since Oct. 1, 2016, there have been 6,804 laboratory-confirmed flu-related hospitalizations reported in the 13 states – including California, Georgia, New York, and Ohio – of the CDC’s Influenza Hospitalization Surveillance Network, for an overall hospitalization rate of 24.3 per 100,000 population. That rate, however, is “likely to be an underestimate as influenza-related hospitalizations can be missed, either because testing is not performed, or because cases may be attributed to other causes of pneumonia or other common influenza-related complications,” the CDC said.
FDA approves Emflaza for Duchenne muscular dystrophy
Emflaza, a tablet and oral suspension corticosteroid, has been approved by the Food and Drug Administration for the treatment of Duchenne muscular dystrophy in patients aged 5 years and older.
The agency’s Feb. 9 announcement notes that similar corticosteroids have been used around the world to treat Duchenne muscular dystrophy (DMD), but this is the first to gain approval in the United States. Emflaza (deflazacort) works by decreasing inflammation and immune system activity.
DMD is the most common form of muscular dystrophy but is still rare, occurring in about 1 in 3,600 male infants worldwide. One study found that patients taking deflazacort had some improvements in muscle strength at 12 weeks, compared with those taking placebo, and maintained muscle strength stability through 52 weeks. A longer-term study showed that patients who took deflazacort had better average muscle strength than did those taking placebo and suggested that deflazacort helped prolong patients’ ability to walk.
Side effects experienced by patients taking Emflaza are similar to those associated with other corticosteroids, such as facial puffiness (cushingoid appearance), weight gain, increased appetite, upper respiratory tract infection, cough, extraordinary daytime urinary frequency (pollakiuria), unwanted hair growth (hirsutism), and excessive fat around the stomach (central obesity).
In the FDA’s announcement, Billy Dunn, MD, director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, said, “We hope that this treatment option will benefit many patients with DMD.”
Emflaza, a tablet and oral suspension corticosteroid, has been approved by the Food and Drug Administration for the treatment of Duchenne muscular dystrophy in patients aged 5 years and older.
The agency’s Feb. 9 announcement notes that similar corticosteroids have been used around the world to treat Duchenne muscular dystrophy (DMD), but this is the first to gain approval in the United States. Emflaza (deflazacort) works by decreasing inflammation and immune system activity.
DMD is the most common form of muscular dystrophy but is still rare, occurring in about 1 in 3,600 male infants worldwide. One study found that patients taking deflazacort had some improvements in muscle strength at 12 weeks, compared with those taking placebo, and maintained muscle strength stability through 52 weeks. A longer-term study showed that patients who took deflazacort had better average muscle strength than did those taking placebo and suggested that deflazacort helped prolong patients’ ability to walk.
Side effects experienced by patients taking Emflaza are similar to those associated with other corticosteroids, such as facial puffiness (cushingoid appearance), weight gain, increased appetite, upper respiratory tract infection, cough, extraordinary daytime urinary frequency (pollakiuria), unwanted hair growth (hirsutism), and excessive fat around the stomach (central obesity).
In the FDA’s announcement, Billy Dunn, MD, director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, said, “We hope that this treatment option will benefit many patients with DMD.”
Emflaza, a tablet and oral suspension corticosteroid, has been approved by the Food and Drug Administration for the treatment of Duchenne muscular dystrophy in patients aged 5 years and older.
The agency’s Feb. 9 announcement notes that similar corticosteroids have been used around the world to treat Duchenne muscular dystrophy (DMD), but this is the first to gain approval in the United States. Emflaza (deflazacort) works by decreasing inflammation and immune system activity.
DMD is the most common form of muscular dystrophy but is still rare, occurring in about 1 in 3,600 male infants worldwide. One study found that patients taking deflazacort had some improvements in muscle strength at 12 weeks, compared with those taking placebo, and maintained muscle strength stability through 52 weeks. A longer-term study showed that patients who took deflazacort had better average muscle strength than did those taking placebo and suggested that deflazacort helped prolong patients’ ability to walk.
Side effects experienced by patients taking Emflaza are similar to those associated with other corticosteroids, such as facial puffiness (cushingoid appearance), weight gain, increased appetite, upper respiratory tract infection, cough, extraordinary daytime urinary frequency (pollakiuria), unwanted hair growth (hirsutism), and excessive fat around the stomach (central obesity).
In the FDA’s announcement, Billy Dunn, MD, director of the Division of Neurology Products in the FDA’s Center for Drug Evaluation and Research, said, “We hope that this treatment option will benefit many patients with DMD.”
ACIP releases updated guidance for adult vaccinations
, according to the newly issued 2017 adult Recommended Immunization Schedule released by the CDC’s Advisory Committee on Immunization Practices (ACIP).
“Changes are related to concerns regarding low effectiveness of [LAIV] (FluMist, MedImmune) against influenza A(H1N1)pdm09 in the United States during the 2013-2014 and 2015-2016 influenza seasons,” wrote the authors of the report, published in Annals of Internal Medicine and led by David K. Kim, MD, of the CDC’s Immunization Services Division in Atlanta. ![]()
Another major change involves vaccination of adults with mild or severe egg allergy. The new guidance states that those with a mild egg allergy should receive either inactivated influenza vaccine or recombinant influenza vaccine, while those with severe allergies should be given one of the same vaccinations but only in a health care setting, so a clinician can monitor any signs of reaction and treat the patient accordingly.
Vaccine doses should be administered based on the patient’s age; a patient with “severe” egg allergy is one who exhibits angioedema, respiratory distress, lightheadedness, or recurrent emesis; requires epinephrine; or requires emergency medical care of any kind after consuming egg products.
Human papillomavirus (HPV) schedules have also been noticeably altered, with the CDC now considering all men and women through the ages of 21 and 26 years, respectively, who received a two-dose series of HPV vaccinations before the age of 15 to be adequately protected. Those who took only one of those two doses still need to take another dose, while men and women who have not been vaccinated should received a three-dose series at 0, 1-2, and 6 months.
Hepatitis B recommendations were also updated, with the CDC now advising: “Adults with chronic liver disease, including, but not limited to, hepatitis C virus infection, cirrhosis, fatty liver disease, alcoholic liver disease, autoimmune hepatitis, and an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) level greater than twice the upper limit of normal, should receive a HepB series.”
Meningococcal vaccination guidelines also underwent a number of small changes pertaining to adults with anatomical or functional asplenia and human immunodeficiency virus, among other risk factors.
A number of small changes to the schedule chart were implemented to help make the immunization schedule more “clean and streamlined,” according to the CDC.
“Physicians should pay careful attention to the details found in the footnotes,” the CDC said in a statement. “The footnotes clarify who needs what vaccine, when, and at what dose.”
, according to the newly issued 2017 adult Recommended Immunization Schedule released by the CDC’s Advisory Committee on Immunization Practices (ACIP).
“Changes are related to concerns regarding low effectiveness of [LAIV] (FluMist, MedImmune) against influenza A(H1N1)pdm09 in the United States during the 2013-2014 and 2015-2016 influenza seasons,” wrote the authors of the report, published in Annals of Internal Medicine and led by David K. Kim, MD, of the CDC’s Immunization Services Division in Atlanta. ![]()
Another major change involves vaccination of adults with mild or severe egg allergy. The new guidance states that those with a mild egg allergy should receive either inactivated influenza vaccine or recombinant influenza vaccine, while those with severe allergies should be given one of the same vaccinations but only in a health care setting, so a clinician can monitor any signs of reaction and treat the patient accordingly.
Vaccine doses should be administered based on the patient’s age; a patient with “severe” egg allergy is one who exhibits angioedema, respiratory distress, lightheadedness, or recurrent emesis; requires epinephrine; or requires emergency medical care of any kind after consuming egg products.
Human papillomavirus (HPV) schedules have also been noticeably altered, with the CDC now considering all men and women through the ages of 21 and 26 years, respectively, who received a two-dose series of HPV vaccinations before the age of 15 to be adequately protected. Those who took only one of those two doses still need to take another dose, while men and women who have not been vaccinated should received a three-dose series at 0, 1-2, and 6 months.
Hepatitis B recommendations were also updated, with the CDC now advising: “Adults with chronic liver disease, including, but not limited to, hepatitis C virus infection, cirrhosis, fatty liver disease, alcoholic liver disease, autoimmune hepatitis, and an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) level greater than twice the upper limit of normal, should receive a HepB series.”
Meningococcal vaccination guidelines also underwent a number of small changes pertaining to adults with anatomical or functional asplenia and human immunodeficiency virus, among other risk factors.
A number of small changes to the schedule chart were implemented to help make the immunization schedule more “clean and streamlined,” according to the CDC.
“Physicians should pay careful attention to the details found in the footnotes,” the CDC said in a statement. “The footnotes clarify who needs what vaccine, when, and at what dose.”
, according to the newly issued 2017 adult Recommended Immunization Schedule released by the CDC’s Advisory Committee on Immunization Practices (ACIP).
“Changes are related to concerns regarding low effectiveness of [LAIV] (FluMist, MedImmune) against influenza A(H1N1)pdm09 in the United States during the 2013-2014 and 2015-2016 influenza seasons,” wrote the authors of the report, published in Annals of Internal Medicine and led by David K. Kim, MD, of the CDC’s Immunization Services Division in Atlanta. ![]()
Another major change involves vaccination of adults with mild or severe egg allergy. The new guidance states that those with a mild egg allergy should receive either inactivated influenza vaccine or recombinant influenza vaccine, while those with severe allergies should be given one of the same vaccinations but only in a health care setting, so a clinician can monitor any signs of reaction and treat the patient accordingly.
Vaccine doses should be administered based on the patient’s age; a patient with “severe” egg allergy is one who exhibits angioedema, respiratory distress, lightheadedness, or recurrent emesis; requires epinephrine; or requires emergency medical care of any kind after consuming egg products.
Human papillomavirus (HPV) schedules have also been noticeably altered, with the CDC now considering all men and women through the ages of 21 and 26 years, respectively, who received a two-dose series of HPV vaccinations before the age of 15 to be adequately protected. Those who took only one of those two doses still need to take another dose, while men and women who have not been vaccinated should received a three-dose series at 0, 1-2, and 6 months.
Hepatitis B recommendations were also updated, with the CDC now advising: “Adults with chronic liver disease, including, but not limited to, hepatitis C virus infection, cirrhosis, fatty liver disease, alcoholic liver disease, autoimmune hepatitis, and an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) level greater than twice the upper limit of normal, should receive a HepB series.”
Meningococcal vaccination guidelines also underwent a number of small changes pertaining to adults with anatomical or functional asplenia and human immunodeficiency virus, among other risk factors.
A number of small changes to the schedule chart were implemented to help make the immunization schedule more “clean and streamlined,” according to the CDC.
“Physicians should pay careful attention to the details found in the footnotes,” the CDC said in a statement. “The footnotes clarify who needs what vaccine, when, and at what dose.”
FROM ANNALS OF INTERNAL MEDICINE
High levels of flu activity reported in 15 states
Fifteen states experienced high levels of flu activity for the week ending Jan. 28 as the 2016-2017 season moved past last season’s peak, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 3.9% for the week ending Jan. 28, 2017, the CDC reported. That level is higher than the national baseline level of 2.2% and higher than the peak of 3.6% for the 2015-2016 season.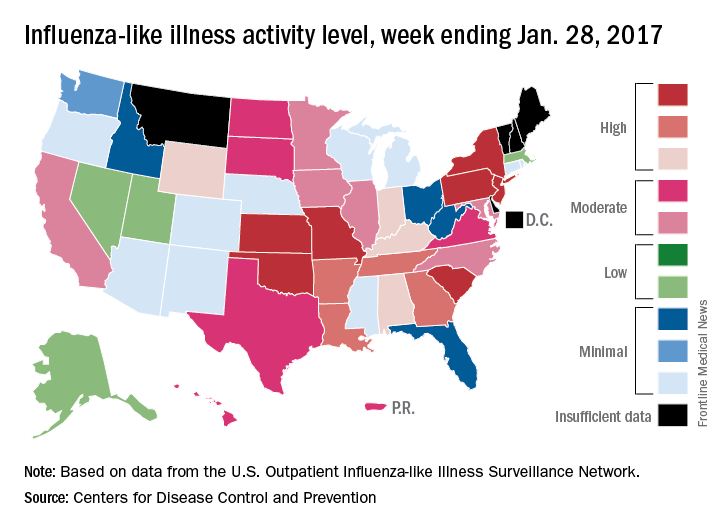
The week also brought reports of seven flu-related pediatric deaths, although five actually occurred during previous weeks. There have now been 15 flu-related pediatric deaths during the 2016-2017 season, the CDC reported.
Fifteen states experienced high levels of flu activity for the week ending Jan. 28 as the 2016-2017 season moved past last season’s peak, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 3.9% for the week ending Jan. 28, 2017, the CDC reported. That level is higher than the national baseline level of 2.2% and higher than the peak of 3.6% for the 2015-2016 season.
The week also brought reports of seven flu-related pediatric deaths, although five actually occurred during previous weeks. There have now been 15 flu-related pediatric deaths during the 2016-2017 season, the CDC reported.
Fifteen states experienced high levels of flu activity for the week ending Jan. 28 as the 2016-2017 season moved past last season’s peak, according to the Centers for Disease Control and Prevention.
The proportion of outpatient visits for influenza-like illness (ILI) was 3.9% for the week ending Jan. 28, 2017, the CDC reported. That level is higher than the national baseline level of 2.2% and higher than the peak of 3.6% for the 2015-2016 season.
The week also brought reports of seven flu-related pediatric deaths, although five actually occurred during previous weeks. There have now been 15 flu-related pediatric deaths during the 2016-2017 season, the CDC reported.
FDA opens abbreviated approval pathway for interchangeable biosimilars
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique treatments.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference product is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such biosimilars have come on the market, at an average price of about 30% less than the reference product. Prices have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive treatments. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer treatments (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those products to make up for the money they were losing on the Russian market.

“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive treatments, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).

“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the product was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American Gastroenterological Association has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing treatments, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer products are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the treatment maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older product, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique treatments.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference product is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such biosimilars have come on the market, at an average price of about 30% less than the reference product. Prices have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive treatments. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer treatments (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those products to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive treatments, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the product was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American Gastroenterological Association has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing treatments, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer products are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the treatment maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older product, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique treatments.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference product is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such biosimilars have come on the market, at an average price of about 30% less than the reference product. Prices have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive treatments. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer treatments (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those products to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive treatments, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the product was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American Gastroenterological Association has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing treatments, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer products are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the treatment maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older product, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
New Zika-infected pregnancies down for most of United States
New cases of Zika infection in pregnant women were down again for the 50 states over the 2-week reporting period ending Jan. 24, but U.S. territories saw a big increase, according to the Centers for Disease Control and Prevention.
It is important to note, however, that Puerto Rico, where most U.S. Zika cases are occurring, has been retroactively reporting cases for months, which can result in larger-than-normal increases.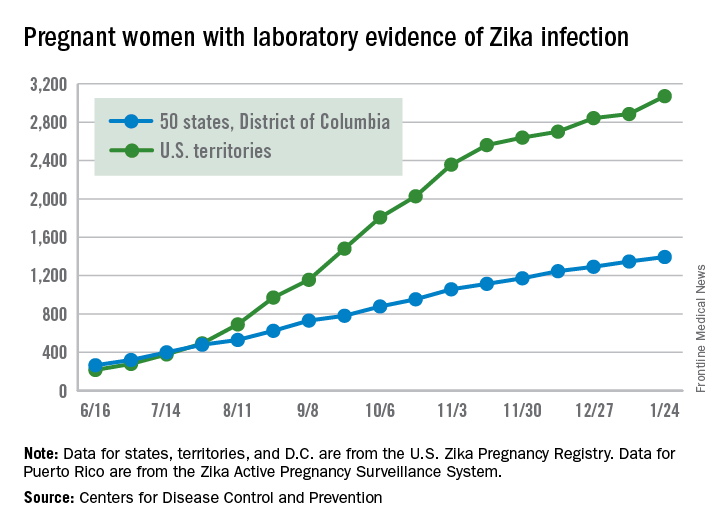
The territories had 186 new cases of pregnant women with laboratory evidence of Zika virus infection for the 2 weeks ending Jan. 24, compared with 43 for the previous 2-week period. The corresponding numbers for the 50 states and the District of Columbia were 47 (Jan. 24) and 55 (Jan. 10), the CDC data show.
So far for 2016-2017, there have been 4,465 pregnant women reported to have Zika virus infection in the United States: 3,071 in the territories and 1,394 in the 50 states and D.C., according to the U.S. Zika Pregnancy Registry (states, territories, and the District of Columbia) and the U.S. Zika Active Pregnancy Surveillance System (Puerto Rico).
Of the nearly 1,400 state/D.C. Zika-infected pregnancies, 999 have been completed, with 38 infants born with birth defects and 5 defect-related pregnancy losses, the CDC said. There have been no Zika-related pregnancy losses reported in the states/D.C. since late June. The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same inclusion criteria.
Among all Americans in 2015-2017, there have been 41,387 cases of Zika infection as of Feb. 1. Of that total, 35,334 cases, which is more than 85%, have occurred in Puerto Rico, according to data from the CDC’s Arboviral Disease Branch.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
New cases of Zika infection in pregnant women were down again for the 50 states over the 2-week reporting period ending Jan. 24, but U.S. territories saw a big increase, according to the Centers for Disease Control and Prevention.
It is important to note, however, that Puerto Rico, where most U.S. Zika cases are occurring, has been retroactively reporting cases for months, which can result in larger-than-normal increases.
The territories had 186 new cases of pregnant women with laboratory evidence of Zika virus infection for the 2 weeks ending Jan. 24, compared with 43 for the previous 2-week period. The corresponding numbers for the 50 states and the District of Columbia were 47 (Jan. 24) and 55 (Jan. 10), the CDC data show.
So far for 2016-2017, there have been 4,465 pregnant women reported to have Zika virus infection in the United States: 3,071 in the territories and 1,394 in the 50 states and D.C., according to the U.S. Zika Pregnancy Registry (states, territories, and the District of Columbia) and the U.S. Zika Active Pregnancy Surveillance System (Puerto Rico).
Of the nearly 1,400 state/D.C. Zika-infected pregnancies, 999 have been completed, with 38 infants born with birth defects and 5 defect-related pregnancy losses, the CDC said. There have been no Zika-related pregnancy losses reported in the states/D.C. since late June. The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same inclusion criteria.
Among all Americans in 2015-2017, there have been 41,387 cases of Zika infection as of Feb. 1. Of that total, 35,334 cases, which is more than 85%, have occurred in Puerto Rico, according to data from the CDC’s Arboviral Disease Branch.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
New cases of Zika infection in pregnant women were down again for the 50 states over the 2-week reporting period ending Jan. 24, but U.S. territories saw a big increase, according to the Centers for Disease Control and Prevention.
It is important to note, however, that Puerto Rico, where most U.S. Zika cases are occurring, has been retroactively reporting cases for months, which can result in larger-than-normal increases.
The territories had 186 new cases of pregnant women with laboratory evidence of Zika virus infection for the 2 weeks ending Jan. 24, compared with 43 for the previous 2-week period. The corresponding numbers for the 50 states and the District of Columbia were 47 (Jan. 24) and 55 (Jan. 10), the CDC data show.
So far for 2016-2017, there have been 4,465 pregnant women reported to have Zika virus infection in the United States: 3,071 in the territories and 1,394 in the 50 states and D.C., according to the U.S. Zika Pregnancy Registry (states, territories, and the District of Columbia) and the U.S. Zika Active Pregnancy Surveillance System (Puerto Rico).
Of the nearly 1,400 state/D.C. Zika-infected pregnancies, 999 have been completed, with 38 infants born with birth defects and 5 defect-related pregnancy losses, the CDC said. There have been no Zika-related pregnancy losses reported in the states/D.C. since late June. The CDC is no longer reporting adverse pregnancy outcomes for the territories because Puerto Rico is not using the same inclusion criteria.
Among all Americans in 2015-2017, there have been 41,387 cases of Zika infection as of Feb. 1. Of that total, 35,334 cases, which is more than 85%, have occurred in Puerto Rico, according to data from the CDC’s Arboviral Disease Branch.
Zika-related birth defects reported by the CDC could include microcephaly, calcium deposits in the brain indicating possible brain damage, excess fluid in the brain cavities and surrounding the brain, absent or poorly formed brain structures, abnormal eye development, or other problems resulting from brain damage that affect nerves, muscles, and bones. The pregnancy losses encompass any miscarriage, stillbirth, and termination with evidence of birth defects.
FDA approves nivolumab for advanced urothelial carcinoma
The Food and Drug Administration has granted accelerated approval to nivolumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with a platinum-containing chemotherapy.
Approval was based on objective response rate (ORR) in a single-arm study of 270 patients with locally advanced or metastatic urothelial carcinoma who progressed during or following platinum-containing chemotherapy, or progressed within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. Patients received nivolumab, 3 mg/kg every 2 weeks, until disease progression or unacceptable toxicity. The ORR was 19.6% (53/270; 95% confidence interval, 15.1-24.9), according to a written statement from the FDA.
Fourteen patients died from causes other than disease progression, including four who died from pneumonitis or cardiovascular failure attributed to nivolumab, the FDA said.
The most common adverse reactions were fatigue, musculoskeletal pain, nausea, and decreased appetite. Adverse reactions led to dose discontinuation in 17% of patients.
The recommended dose and schedule for nivolumab for the above indication is 240 mg intravenously every 2 weeks.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and previously has been approved to treat classical Hodgkin’s lymphoma, advanced renal cell carcinoma, lung cancer, melanoma, and squamous cell carcinoma of the head and neck.
The Food and Drug Administration has granted accelerated approval to nivolumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with a platinum-containing chemotherapy.
Approval was based on objective response rate (ORR) in a single-arm study of 270 patients with locally advanced or metastatic urothelial carcinoma who progressed during or following platinum-containing chemotherapy, or progressed within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. Patients received nivolumab, 3 mg/kg every 2 weeks, until disease progression or unacceptable toxicity. The ORR was 19.6% (53/270; 95% confidence interval, 15.1-24.9), according to a written statement from the FDA.
Fourteen patients died from causes other than disease progression, including four who died from pneumonitis or cardiovascular failure attributed to nivolumab, the FDA said.
The most common adverse reactions were fatigue, musculoskeletal pain, nausea, and decreased appetite. Adverse reactions led to dose discontinuation in 17% of patients.
The recommended dose and schedule for nivolumab for the above indication is 240 mg intravenously every 2 weeks.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and previously has been approved to treat classical Hodgkin’s lymphoma, advanced renal cell carcinoma, lung cancer, melanoma, and squamous cell carcinoma of the head and neck.
The Food and Drug Administration has granted accelerated approval to nivolumab for treatment of patients with locally advanced or metastatic urothelial carcinoma who have disease progression during or following platinum-containing chemotherapy or have disease progression within 12 months of neoadjuvant or adjuvant treatment with a platinum-containing chemotherapy.
Approval was based on objective response rate (ORR) in a single-arm study of 270 patients with locally advanced or metastatic urothelial carcinoma who progressed during or following platinum-containing chemotherapy, or progressed within 12 months of neoadjuvant or adjuvant treatment with platinum-containing chemotherapy. Patients received nivolumab, 3 mg/kg every 2 weeks, until disease progression or unacceptable toxicity. The ORR was 19.6% (53/270; 95% confidence interval, 15.1-24.9), according to a written statement from the FDA.
Fourteen patients died from causes other than disease progression, including four who died from pneumonitis or cardiovascular failure attributed to nivolumab, the FDA said.
The most common adverse reactions were fatigue, musculoskeletal pain, nausea, and decreased appetite. Adverse reactions led to dose discontinuation in 17% of patients.
The recommended dose and schedule for nivolumab for the above indication is 240 mg intravenously every 2 weeks.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and previously has been approved to treat classical Hodgkin’s lymphoma, advanced renal cell carcinoma, lung cancer, melanoma, and squamous cell carcinoma of the head and neck.