User login
Outpatient care 35% of mental health costs and growing
Outpatient care represents the largest share of mental health treatment expenditures, and it continues to get larger, while components such as retail drug prescriptions and inpatient care have declined, according to the National Center of Health Statistics.
In 2014, outpatient care took a $65.5-billion slice (about 35%) out of the $186-billion mental health care spending pie, compared with the $51.1 billion (27%) spent on retail drug prescriptions, which was the next-largest portion. Inpatient care was third with $30.3 billion in spending (16% of the total), followed by residential care at $23.2 billion (12%), and insurance administration at $15.9 billion (9%), the NCHS reported in “Health, United States, 2016.”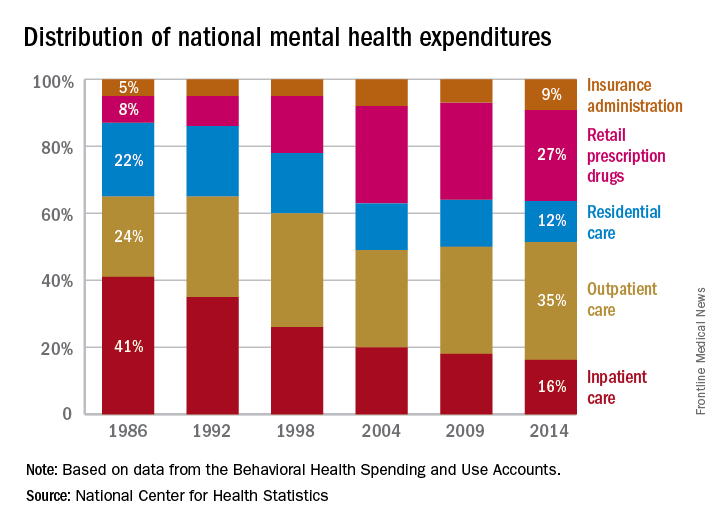
Outpatient care represents the largest share of mental health treatment expenditures, and it continues to get larger, while components such as retail drug prescriptions and inpatient care have declined, according to the National Center of Health Statistics.
In 2014, outpatient care took a $65.5-billion slice (about 35%) out of the $186-billion mental health care spending pie, compared with the $51.1 billion (27%) spent on retail drug prescriptions, which was the next-largest portion. Inpatient care was third with $30.3 billion in spending (16% of the total), followed by residential care at $23.2 billion (12%), and insurance administration at $15.9 billion (9%), the NCHS reported in “Health, United States, 2016.”
Outpatient care represents the largest share of mental health treatment expenditures, and it continues to get larger, while components such as retail drug prescriptions and inpatient care have declined, according to the National Center of Health Statistics.
In 2014, outpatient care took a $65.5-billion slice (about 35%) out of the $186-billion mental health care spending pie, compared with the $51.1 billion (27%) spent on retail drug prescriptions, which was the next-largest portion. Inpatient care was third with $30.3 billion in spending (16% of the total), followed by residential care at $23.2 billion (12%), and insurance administration at $15.9 billion (9%), the NCHS reported in “Health, United States, 2016.”
Opioid prescribing drops nationally, remains high in some counties
Opioid prescribing in the United States declined overall between 2010 and 2015, but remained stable or increased in some counties, according to a report from the Centers for Disease Control and Prevention. The findings were published online in the CDC’s Morbidity and Mortality Weekly Report.
“The bottom line remains: We have too many people getting too many prescriptions at too high a dose,” Anne Schuchat, MD, acting director of the CDC, said in a July 6 teleconference.![]()
CDC researchers calculated prescribing rates from 2006 to 2015 by dividing the number of opioid prescriptions by the population estimates from the U.S. census for each year and created quartiles using morphine milligram equivalent per capita to analyze opioid distribution. Annual opioid prescribing rates increased from 72 to 81 prescriptions per 100 persons from 2006 to 2010 and remained relatively constant from 2010 to 2012 before showing a 13% decrease to 71 prescriptions per 100 persons from 2012 to 2015 (MMWR. 2017 Jul 7;66[26]:697-704. doi: 10.15585/mmwr.mm6626a4).
But despite these overall declines, “We are now experiencing the highest overdose death rates ever recorded in the United States,” Dr. Schuchat said. Quartiles were created using MME per capita to characterize the distribution of opioids prescribed.
In the report, areas associated with higher opioid prescribing rates on a county level included small cities or towns, areas that had a higher proportion of white residents, areas with more doctors and dentists, and areas with more cases of arthritis, diabetes, or other disabilities, she said.
The findings suggest a need for more consistency among health care providers about prescription opioids, Dr. Schuchat said. “Clinical practice is all over the place, which is a sign that you need better standards; we hope the 2016 guidelines are a turning point for better prescribing,” she said.
The CDC’s guidelines on opioid prescribing were released in 2016. The guidelines recommend alternatives when possible. Clinicians should instead consider nonopioid therapy, other types of pain medication, and nondrug pain relief options, such as physical therapy and cognitive-behavioral therapy. Other concerns include the length and strength of opioid prescriptions. Even taking opioids for a few months increases the risk for addiction, Dr. Schuchat said.
“Physicians must continue to lead efforts to reverse the epidemic by using prescription drug–monitoring programs, eliminating stigma, prescribing the overdose reversal drug naloxone, and enhancing their education about safe opioid prescribing and effective pain management,” Patrice A. Harris, MD, chair of the American Medical Association Opioid Task Force, said in a statement in response to the report. “Our country must do more to provide evidence-based, comprehensive treatment for pain and for substance use disorders,” she said.
“We really encourage clinicians to look to the guidelines and the tools that are available,” Dr. Schuchat said. “We do know that internists and other primary care physicians prescribe most of the opioids, so it is important for them to be aware.” The CDC has developed a checklist and a mobile app that have been downloaded by thousands of clinicians so far, she noted. Changes in annual prescribing hold promise that practices can improve, she said.
The researchers reported no conflicts of interest.
Opioid prescribing in the United States declined overall between 2010 and 2015, but remained stable or increased in some counties, according to a report from the Centers for Disease Control and Prevention. The findings were published online in the CDC’s Morbidity and Mortality Weekly Report.
“The bottom line remains: We have too many people getting too many prescriptions at too high a dose,” Anne Schuchat, MD, acting director of the CDC, said in a July 6 teleconference.![]()
CDC researchers calculated prescribing rates from 2006 to 2015 by dividing the number of opioid prescriptions by the population estimates from the U.S. census for each year and created quartiles using morphine milligram equivalent per capita to analyze opioid distribution. Annual opioid prescribing rates increased from 72 to 81 prescriptions per 100 persons from 2006 to 2010 and remained relatively constant from 2010 to 2012 before showing a 13% decrease to 71 prescriptions per 100 persons from 2012 to 2015 (MMWR. 2017 Jul 7;66[26]:697-704. doi: 10.15585/mmwr.mm6626a4).
But despite these overall declines, “We are now experiencing the highest overdose death rates ever recorded in the United States,” Dr. Schuchat said. Quartiles were created using MME per capita to characterize the distribution of opioids prescribed.
In the report, areas associated with higher opioid prescribing rates on a county level included small cities or towns, areas that had a higher proportion of white residents, areas with more doctors and dentists, and areas with more cases of arthritis, diabetes, or other disabilities, she said.
The findings suggest a need for more consistency among health care providers about prescription opioids, Dr. Schuchat said. “Clinical practice is all over the place, which is a sign that you need better standards; we hope the 2016 guidelines are a turning point for better prescribing,” she said.
The CDC’s guidelines on opioid prescribing were released in 2016. The guidelines recommend alternatives when possible. Clinicians should instead consider nonopioid therapy, other types of pain medication, and nondrug pain relief options, such as physical therapy and cognitive-behavioral therapy. Other concerns include the length and strength of opioid prescriptions. Even taking opioids for a few months increases the risk for addiction, Dr. Schuchat said.
“Physicians must continue to lead efforts to reverse the epidemic by using prescription drug–monitoring programs, eliminating stigma, prescribing the overdose reversal drug naloxone, and enhancing their education about safe opioid prescribing and effective pain management,” Patrice A. Harris, MD, chair of the American Medical Association Opioid Task Force, said in a statement in response to the report. “Our country must do more to provide evidence-based, comprehensive treatment for pain and for substance use disorders,” she said.
“We really encourage clinicians to look to the guidelines and the tools that are available,” Dr. Schuchat said. “We do know that internists and other primary care physicians prescribe most of the opioids, so it is important for them to be aware.” The CDC has developed a checklist and a mobile app that have been downloaded by thousands of clinicians so far, she noted. Changes in annual prescribing hold promise that practices can improve, she said.
The researchers reported no conflicts of interest.
Opioid prescribing in the United States declined overall between 2010 and 2015, but remained stable or increased in some counties, according to a report from the Centers for Disease Control and Prevention. The findings were published online in the CDC’s Morbidity and Mortality Weekly Report.
“The bottom line remains: We have too many people getting too many prescriptions at too high a dose,” Anne Schuchat, MD, acting director of the CDC, said in a July 6 teleconference.![]()
CDC researchers calculated prescribing rates from 2006 to 2015 by dividing the number of opioid prescriptions by the population estimates from the U.S. census for each year and created quartiles using morphine milligram equivalent per capita to analyze opioid distribution. Annual opioid prescribing rates increased from 72 to 81 prescriptions per 100 persons from 2006 to 2010 and remained relatively constant from 2010 to 2012 before showing a 13% decrease to 71 prescriptions per 100 persons from 2012 to 2015 (MMWR. 2017 Jul 7;66[26]:697-704. doi: 10.15585/mmwr.mm6626a4).
But despite these overall declines, “We are now experiencing the highest overdose death rates ever recorded in the United States,” Dr. Schuchat said. Quartiles were created using MME per capita to characterize the distribution of opioids prescribed.
In the report, areas associated with higher opioid prescribing rates on a county level included small cities or towns, areas that had a higher proportion of white residents, areas with more doctors and dentists, and areas with more cases of arthritis, diabetes, or other disabilities, she said.
The findings suggest a need for more consistency among health care providers about prescription opioids, Dr. Schuchat said. “Clinical practice is all over the place, which is a sign that you need better standards; we hope the 2016 guidelines are a turning point for better prescribing,” she said.
The CDC’s guidelines on opioid prescribing were released in 2016. The guidelines recommend alternatives when possible. Clinicians should instead consider nonopioid therapy, other types of pain medication, and nondrug pain relief options, such as physical therapy and cognitive-behavioral therapy. Other concerns include the length and strength of opioid prescriptions. Even taking opioids for a few months increases the risk for addiction, Dr. Schuchat said.
“Physicians must continue to lead efforts to reverse the epidemic by using prescription drug–monitoring programs, eliminating stigma, prescribing the overdose reversal drug naloxone, and enhancing their education about safe opioid prescribing and effective pain management,” Patrice A. Harris, MD, chair of the American Medical Association Opioid Task Force, said in a statement in response to the report. “Our country must do more to provide evidence-based, comprehensive treatment for pain and for substance use disorders,” she said.
“We really encourage clinicians to look to the guidelines and the tools that are available,” Dr. Schuchat said. “We do know that internists and other primary care physicians prescribe most of the opioids, so it is important for them to be aware.” The CDC has developed a checklist and a mobile app that have been downloaded by thousands of clinicians so far, she noted. Changes in annual prescribing hold promise that practices can improve, she said.
The researchers reported no conflicts of interest.
FROM MMWR
Endo removes Opana ER from market
Even as it defended the product’s safety when appropriately used, Endo International withdrew from the market its long-acting opioid agonist Opana ER, in compliance with a June 8 Food and Drug Administration request. The company “continues to believe in the safety, efficacy, and favorable benefit-risk profile of Opana ER (oxymorphone hydrochloride extended release) when used as intended, and notes that the company has taken significant steps over the years to combat misuse and abuse,” according to a news release posted on Endo’s website. “Nevertheless, after careful consideration and consultation with the FDA following [its] June 2017 withdrawal request, the company has decided to voluntarily remove Opana ER from the market.”

In fact, the data showed a significant shift in the route of abuse of Opana ER from nasal to injection following the product’s reformulation. Injection abuse of reformulated Opana ER has been associated with a serious outbreak of HIV and hepatitis C, as well as cases of a thrombotic microangiopathy.
Endo said it will work with FDA to coordinate a smooth removal of the product, and insisted that the drug is safe and effective.
“Endo reiterates that neither the FDA’s withdrawal request nor Endo’s decision to voluntarily remove Opana ER from the market reflect a finding that the product is not safe or effective when taken as prescribed. To the contrary, Endo remains confident in the clinical research and other data demonstrating Opana ER’s safety and efficacy, as well as its favorable risk-benefit profile when used as intended in appropriate patients.”
Opana ER was first approved in 2006 for the management of moderate to severe pain when a continuous, around-the-clock opioid analgesic is needed for an extended period of time. It was reformulated in 2012, with the intent of making it “resistant to physical and chemical manipulation for abuse by snorting or injecting,” according to the FDA release.
On Twitter @Alz_gal
Even as it defended the product’s safety when appropriately used, Endo International withdrew from the market its long-acting opioid agonist Opana ER, in compliance with a June 8 Food and Drug Administration request. The company “continues to believe in the safety, efficacy, and favorable benefit-risk profile of Opana ER (oxymorphone hydrochloride extended release) when used as intended, and notes that the company has taken significant steps over the years to combat misuse and abuse,” according to a news release posted on Endo’s website. “Nevertheless, after careful consideration and consultation with the FDA following [its] June 2017 withdrawal request, the company has decided to voluntarily remove Opana ER from the market.”
In fact, the data showed a significant shift in the route of abuse of Opana ER from nasal to injection following the product’s reformulation. Injection abuse of reformulated Opana ER has been associated with a serious outbreak of HIV and hepatitis C, as well as cases of a thrombotic microangiopathy.
Endo said it will work with FDA to coordinate a smooth removal of the product, and insisted that the drug is safe and effective.
“Endo reiterates that neither the FDA’s withdrawal request nor Endo’s decision to voluntarily remove Opana ER from the market reflect a finding that the product is not safe or effective when taken as prescribed. To the contrary, Endo remains confident in the clinical research and other data demonstrating Opana ER’s safety and efficacy, as well as its favorable risk-benefit profile when used as intended in appropriate patients.”
Opana ER was first approved in 2006 for the management of moderate to severe pain when a continuous, around-the-clock opioid analgesic is needed for an extended period of time. It was reformulated in 2012, with the intent of making it “resistant to physical and chemical manipulation for abuse by snorting or injecting,” according to the FDA release.
On Twitter @Alz_gal
Even as it defended the product’s safety when appropriately used, Endo International withdrew from the market its long-acting opioid agonist Opana ER, in compliance with a June 8 Food and Drug Administration request. The company “continues to believe in the safety, efficacy, and favorable benefit-risk profile of Opana ER (oxymorphone hydrochloride extended release) when used as intended, and notes that the company has taken significant steps over the years to combat misuse and abuse,” according to a news release posted on Endo’s website. “Nevertheless, after careful consideration and consultation with the FDA following [its] June 2017 withdrawal request, the company has decided to voluntarily remove Opana ER from the market.”
In fact, the data showed a significant shift in the route of abuse of Opana ER from nasal to injection following the product’s reformulation. Injection abuse of reformulated Opana ER has been associated with a serious outbreak of HIV and hepatitis C, as well as cases of a thrombotic microangiopathy.
Endo said it will work with FDA to coordinate a smooth removal of the product, and insisted that the drug is safe and effective.
“Endo reiterates that neither the FDA’s withdrawal request nor Endo’s decision to voluntarily remove Opana ER from the market reflect a finding that the product is not safe or effective when taken as prescribed. To the contrary, Endo remains confident in the clinical research and other data demonstrating Opana ER’s safety and efficacy, as well as its favorable risk-benefit profile when used as intended in appropriate patients.”
Opana ER was first approved in 2006 for the management of moderate to severe pain when a continuous, around-the-clock opioid analgesic is needed for an extended period of time. It was reformulated in 2012, with the intent of making it “resistant to physical and chemical manipulation for abuse by snorting or injecting,” according to the FDA release.
On Twitter @Alz_gal
Adult diabetes up 35% over 25 years
The overall prevalence of diabetes increased 35% from 1988-1994 to 2011-2014 among adults aged 20 years and over, according the National Center for Health Statistics.
During 2011-2014, the age-adjusted prevalence of diabetes was 11.9% in adults aged 20 years and over, compared with 8.8% in 1988-1994. That 35% increase came despite a decrease in undiagnosed diabetes from 3.6% to 2.9% over that time period, which was not enough to offset a jump in physician-diagnosed disease from 5.2% to 9%, the NCHS reported in “Health, United States, 2016.”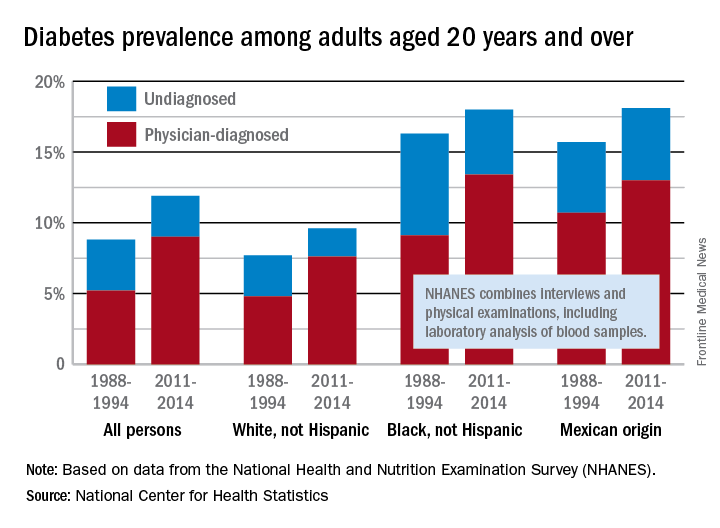
Undiagnosed diabetes dropped from 2.9% to 2% in whites and from 7.2% to 4.6% in blacks, but adults of Mexican origin saw a slight increase from 5% in 1988-1994 to 5.1% in 2011-2014. The physician-diagnosed side of the equation rose from 4.8% to 7.6% in whites, 9.1% to 13.4% in blacks, and 10.7% to 13% in those of Mexican origin, according to data from the National Health and Nutrition Examination Survey, which combines interviews and physical examinations, including laboratory analysis of blood samples.
The overall prevalence of diabetes increased 35% from 1988-1994 to 2011-2014 among adults aged 20 years and over, according the National Center for Health Statistics.
During 2011-2014, the age-adjusted prevalence of diabetes was 11.9% in adults aged 20 years and over, compared with 8.8% in 1988-1994. That 35% increase came despite a decrease in undiagnosed diabetes from 3.6% to 2.9% over that time period, which was not enough to offset a jump in physician-diagnosed disease from 5.2% to 9%, the NCHS reported in “Health, United States, 2016.”
Undiagnosed diabetes dropped from 2.9% to 2% in whites and from 7.2% to 4.6% in blacks, but adults of Mexican origin saw a slight increase from 5% in 1988-1994 to 5.1% in 2011-2014. The physician-diagnosed side of the equation rose from 4.8% to 7.6% in whites, 9.1% to 13.4% in blacks, and 10.7% to 13% in those of Mexican origin, according to data from the National Health and Nutrition Examination Survey, which combines interviews and physical examinations, including laboratory analysis of blood samples.
The overall prevalence of diabetes increased 35% from 1988-1994 to 2011-2014 among adults aged 20 years and over, according the National Center for Health Statistics.
During 2011-2014, the age-adjusted prevalence of diabetes was 11.9% in adults aged 20 years and over, compared with 8.8% in 1988-1994. That 35% increase came despite a decrease in undiagnosed diabetes from 3.6% to 2.9% over that time period, which was not enough to offset a jump in physician-diagnosed disease from 5.2% to 9%, the NCHS reported in “Health, United States, 2016.”
Undiagnosed diabetes dropped from 2.9% to 2% in whites and from 7.2% to 4.6% in blacks, but adults of Mexican origin saw a slight increase from 5% in 1988-1994 to 5.1% in 2011-2014. The physician-diagnosed side of the equation rose from 4.8% to 7.6% in whites, 9.1% to 13.4% in blacks, and 10.7% to 13% in those of Mexican origin, according to data from the National Health and Nutrition Examination Survey, which combines interviews and physical examinations, including laboratory analysis of blood samples.
U.S. goals for earlier HIV diagnosis and treatment may be out of reach
In 2015, only 66% of U.S. youth who were diagnosed with HIV in a Centers for Disease Control and Prevention program were introduced to proper care within 90 days of diagnosis, falling far short of the 2020 national goal to introduce 85% of HIV-affected youth to proper care within 30 days.
In an analysis of data from a CDC-funded program covering 61 state and local health departments and 123 community-based organizations in the United States, Puerto Rico, and the U.S. Virgin Islands, the CDC looked at HIV tests, new positive diagnoses, and linkage between patient and care within 90 days of diagnosis. Of 2,973 youths who were newly diagnosed with HIV, 1,955 (66%) were connected to care within 90 days, and 1,871 were interviewed for partner services, according to the CDC. Of 1,911 youths who had been previously diagnosed, 1,749 (92%) were not in medical care at the time of CDC testing.![]()
“A health care provider’s testing recommendation is the most important predictor of testing among adolescents at risk for HIV infection,” the researchers said. “Increasing the number of HIV tests among youths at risk for HIV and increasing regular retesting among these youths is essential for reducing HIV infection in this vulnerable population.”
No conflicts of interest were reported by the authors.
Read more in MMWR (2017 Jun 23. doi: 10.15585/mmwr.mm6624a2).
In 2015, only 66% of U.S. youth who were diagnosed with HIV in a Centers for Disease Control and Prevention program were introduced to proper care within 90 days of diagnosis, falling far short of the 2020 national goal to introduce 85% of HIV-affected youth to proper care within 30 days.
In an analysis of data from a CDC-funded program covering 61 state and local health departments and 123 community-based organizations in the United States, Puerto Rico, and the U.S. Virgin Islands, the CDC looked at HIV tests, new positive diagnoses, and linkage between patient and care within 90 days of diagnosis. Of 2,973 youths who were newly diagnosed with HIV, 1,955 (66%) were connected to care within 90 days, and 1,871 were interviewed for partner services, according to the CDC. Of 1,911 youths who had been previously diagnosed, 1,749 (92%) were not in medical care at the time of CDC testing.![]()
“A health care provider’s testing recommendation is the most important predictor of testing among adolescents at risk for HIV infection,” the researchers said. “Increasing the number of HIV tests among youths at risk for HIV and increasing regular retesting among these youths is essential for reducing HIV infection in this vulnerable population.”
No conflicts of interest were reported by the authors.
Read more in MMWR (2017 Jun 23. doi: 10.15585/mmwr.mm6624a2).
In 2015, only 66% of U.S. youth who were diagnosed with HIV in a Centers for Disease Control and Prevention program were introduced to proper care within 90 days of diagnosis, falling far short of the 2020 national goal to introduce 85% of HIV-affected youth to proper care within 30 days.
In an analysis of data from a CDC-funded program covering 61 state and local health departments and 123 community-based organizations in the United States, Puerto Rico, and the U.S. Virgin Islands, the CDC looked at HIV tests, new positive diagnoses, and linkage between patient and care within 90 days of diagnosis. Of 2,973 youths who were newly diagnosed with HIV, 1,955 (66%) were connected to care within 90 days, and 1,871 were interviewed for partner services, according to the CDC. Of 1,911 youths who had been previously diagnosed, 1,749 (92%) were not in medical care at the time of CDC testing.![]()
“A health care provider’s testing recommendation is the most important predictor of testing among adolescents at risk for HIV infection,” the researchers said. “Increasing the number of HIV tests among youths at risk for HIV and increasing regular retesting among these youths is essential for reducing HIV infection in this vulnerable population.”
No conflicts of interest were reported by the authors.
Read more in MMWR (2017 Jun 23. doi: 10.15585/mmwr.mm6624a2).
FROM MMWR
‘Chronic Lyme’: Serious bacterial infections reported with unproven treatments
Unproven treatments for so-called chronic Lyme disease can cause serious, even fatal, complications, according to five case reports published in the Morbidity and Mortality Weekly Report.
“Patients, clinicians, and public health practitioners should be aware that treatments for chronic Lyme disease can carry serious risks,” the authors wrote.
Chronic Lyme disease is a nonspecific diagnosis that has no consistent definition. Some clinicians use this label for patients who have a variety of debilitating conditions such as fatigue, generalized pain, and neurologic symptoms, even in the absence of laboratory evidence of Borrelia burgdorferi infection, objective signs of infection, or a history of tick exposure. People who support this diagnosis mistakenly believe that B. burgdorferi can cause longstanding disabling symptoms even when standard testing for the organism is negative, when the truth is that tests for the organism become more sensitive the longer the infection persists, according to Natalie S. Marzec, MD, a resident in preventive medicine at the University of Colorado, Aurora, and her associates.

Patients who cannot obtain symptom relief with conventional clinicians may consult “practitioners who might identify themselves as Lyme disease specialists (‘Lyme literate’ doctors) or from complementary and alternative medicine clinics,” where they are diagnosed with chronic Lyme disease. Such patients have been offered unproven treatments, including extended courses of intravenous antibiotics, infusions of hydrogen peroxide, immunoglobulin therapy, hyperbaric oxygen treatment, electromagnetic frequency therapy, garlic supplements, colloidal silver, and stem-cell transplantation.
Dr. Marzec and her associates presented case reports of five such patients diagnosed with chronic Lyme disease who sustained serious harm from such treatments (MMWR 2017;66[23]:607-9).
A woman in her late 30s with fatigue and joint pain was given a peripherally inserted central catheter (PICC) for IV delivery of ceftriaxone and cefotaxime. After 3 weeks, she developed fever, rash, hypotension, and tachycardia. In the intensive care unit (ICU), she was given broad-spectrum IV antibiotics and vasopressors, and was mechanically ventilated, but died of septic shock related to catheter-associated bacteremia.
An adolescent was told at an alternative medicine clinic that her years of muscle and joint pain, backaches, headaches, and lethargy were due to chronic Lyme disease. A PICC was placed to deliver IV antibiotics for 5 months. She developed pallor, chills, fever, hypotension, and tachycardia consistent with septic shock. Cultures demonstrated Acinetobacter species in her blood and on the PICC, and she required several weeks of ICU care.
A woman in her late 40s was diagnosed as having chronic Lyme disease based on unvalidated tests and was treated for months with intramuscular penicillin, IV ceftriazone, and IV azithromycin administered through a tunneled IV catheter; as well as doxycycline, and the antiparasitic drug tinidazole. She was hospitalized for back pain, shortness of breath, and malaise, and cultures of the catheter and her blood yielded Pseudomonas aeruginosa. She was found to have osteodiscitis caused by the same organism, with destruction of the 9th and 10th vertebrae, and was treated, and her back pain eventually improved.
A woman in her 50s was diagnosed as having amyotrophic lateral sclerosis, but sought a second opinion and was told that she had chronic Lyme disease (along with babesiosis, and Rocky Mountain spotted fever). She was treated with herbs and homeopathic remedies, followed by intensive antimicrobial and antiviral therapies. She developed intractable Clostridium difficile colitis that lasted for 2 years until she died from complications related to amyotrophic lateral sclerosis.
A woman in her 60s who had autoimmune neutropenia, mixed connective tissue disease, and degenerative arthritis was told her neuropathy was due to chronic Lyme disease. She was treated with immunoglobulin administered through an implanted subcutaneous port. After years of treatments, she was hospitalized for fever and back pain, had blood cultures positive for methicillin-sensitive Staphylococcus aureus, was found to have inflammation of the lumbar facet joints, epidural space, and paraspinal muscles – and eventually required surgical drainage of a paraspinal abscess.
“These cases highlight the severity and scope of adverse effects that can be caused by the use of unproven treatments for chronic Lyme disease,” Dr. Marzec and her associates said.
This work was supported by the CDC. Dr. Marzec and her associates reported having no relevant financial disclosures.
Unproven treatments for so-called chronic Lyme disease can cause serious, even fatal, complications, according to five case reports published in the Morbidity and Mortality Weekly Report.
“Patients, clinicians, and public health practitioners should be aware that treatments for chronic Lyme disease can carry serious risks,” the authors wrote.
Chronic Lyme disease is a nonspecific diagnosis that has no consistent definition. Some clinicians use this label for patients who have a variety of debilitating conditions such as fatigue, generalized pain, and neurologic symptoms, even in the absence of laboratory evidence of Borrelia burgdorferi infection, objective signs of infection, or a history of tick exposure. People who support this diagnosis mistakenly believe that B. burgdorferi can cause longstanding disabling symptoms even when standard testing for the organism is negative, when the truth is that tests for the organism become more sensitive the longer the infection persists, according to Natalie S. Marzec, MD, a resident in preventive medicine at the University of Colorado, Aurora, and her associates.
Patients who cannot obtain symptom relief with conventional clinicians may consult “practitioners who might identify themselves as Lyme disease specialists (‘Lyme literate’ doctors) or from complementary and alternative medicine clinics,” where they are diagnosed with chronic Lyme disease. Such patients have been offered unproven treatments, including extended courses of intravenous antibiotics, infusions of hydrogen peroxide, immunoglobulin therapy, hyperbaric oxygen treatment, electromagnetic frequency therapy, garlic supplements, colloidal silver, and stem-cell transplantation.
Dr. Marzec and her associates presented case reports of five such patients diagnosed with chronic Lyme disease who sustained serious harm from such treatments (MMWR 2017;66[23]:607-9).
A woman in her late 30s with fatigue and joint pain was given a peripherally inserted central catheter (PICC) for IV delivery of ceftriaxone and cefotaxime. After 3 weeks, she developed fever, rash, hypotension, and tachycardia. In the intensive care unit (ICU), she was given broad-spectrum IV antibiotics and vasopressors, and was mechanically ventilated, but died of septic shock related to catheter-associated bacteremia.
An adolescent was told at an alternative medicine clinic that her years of muscle and joint pain, backaches, headaches, and lethargy were due to chronic Lyme disease. A PICC was placed to deliver IV antibiotics for 5 months. She developed pallor, chills, fever, hypotension, and tachycardia consistent with septic shock. Cultures demonstrated Acinetobacter species in her blood and on the PICC, and she required several weeks of ICU care.
A woman in her late 40s was diagnosed as having chronic Lyme disease based on unvalidated tests and was treated for months with intramuscular penicillin, IV ceftriazone, and IV azithromycin administered through a tunneled IV catheter; as well as doxycycline, and the antiparasitic drug tinidazole. She was hospitalized for back pain, shortness of breath, and malaise, and cultures of the catheter and her blood yielded Pseudomonas aeruginosa. She was found to have osteodiscitis caused by the same organism, with destruction of the 9th and 10th vertebrae, and was treated, and her back pain eventually improved.
A woman in her 50s was diagnosed as having amyotrophic lateral sclerosis, but sought a second opinion and was told that she had chronic Lyme disease (along with babesiosis, and Rocky Mountain spotted fever). She was treated with herbs and homeopathic remedies, followed by intensive antimicrobial and antiviral therapies. She developed intractable Clostridium difficile colitis that lasted for 2 years until she died from complications related to amyotrophic lateral sclerosis.
A woman in her 60s who had autoimmune neutropenia, mixed connective tissue disease, and degenerative arthritis was told her neuropathy was due to chronic Lyme disease. She was treated with immunoglobulin administered through an implanted subcutaneous port. After years of treatments, she was hospitalized for fever and back pain, had blood cultures positive for methicillin-sensitive Staphylococcus aureus, was found to have inflammation of the lumbar facet joints, epidural space, and paraspinal muscles – and eventually required surgical drainage of a paraspinal abscess.
“These cases highlight the severity and scope of adverse effects that can be caused by the use of unproven treatments for chronic Lyme disease,” Dr. Marzec and her associates said.
This work was supported by the CDC. Dr. Marzec and her associates reported having no relevant financial disclosures.
Unproven treatments for so-called chronic Lyme disease can cause serious, even fatal, complications, according to five case reports published in the Morbidity and Mortality Weekly Report.
“Patients, clinicians, and public health practitioners should be aware that treatments for chronic Lyme disease can carry serious risks,” the authors wrote.
Chronic Lyme disease is a nonspecific diagnosis that has no consistent definition. Some clinicians use this label for patients who have a variety of debilitating conditions such as fatigue, generalized pain, and neurologic symptoms, even in the absence of laboratory evidence of Borrelia burgdorferi infection, objective signs of infection, or a history of tick exposure. People who support this diagnosis mistakenly believe that B. burgdorferi can cause longstanding disabling symptoms even when standard testing for the organism is negative, when the truth is that tests for the organism become more sensitive the longer the infection persists, according to Natalie S. Marzec, MD, a resident in preventive medicine at the University of Colorado, Aurora, and her associates.
Patients who cannot obtain symptom relief with conventional clinicians may consult “practitioners who might identify themselves as Lyme disease specialists (‘Lyme literate’ doctors) or from complementary and alternative medicine clinics,” where they are diagnosed with chronic Lyme disease. Such patients have been offered unproven treatments, including extended courses of intravenous antibiotics, infusions of hydrogen peroxide, immunoglobulin therapy, hyperbaric oxygen treatment, electromagnetic frequency therapy, garlic supplements, colloidal silver, and stem-cell transplantation.
Dr. Marzec and her associates presented case reports of five such patients diagnosed with chronic Lyme disease who sustained serious harm from such treatments (MMWR 2017;66[23]:607-9).
A woman in her late 30s with fatigue and joint pain was given a peripherally inserted central catheter (PICC) for IV delivery of ceftriaxone and cefotaxime. After 3 weeks, she developed fever, rash, hypotension, and tachycardia. In the intensive care unit (ICU), she was given broad-spectrum IV antibiotics and vasopressors, and was mechanically ventilated, but died of septic shock related to catheter-associated bacteremia.
An adolescent was told at an alternative medicine clinic that her years of muscle and joint pain, backaches, headaches, and lethargy were due to chronic Lyme disease. A PICC was placed to deliver IV antibiotics for 5 months. She developed pallor, chills, fever, hypotension, and tachycardia consistent with septic shock. Cultures demonstrated Acinetobacter species in her blood and on the PICC, and she required several weeks of ICU care.
A woman in her late 40s was diagnosed as having chronic Lyme disease based on unvalidated tests and was treated for months with intramuscular penicillin, IV ceftriazone, and IV azithromycin administered through a tunneled IV catheter; as well as doxycycline, and the antiparasitic drug tinidazole. She was hospitalized for back pain, shortness of breath, and malaise, and cultures of the catheter and her blood yielded Pseudomonas aeruginosa. She was found to have osteodiscitis caused by the same organism, with destruction of the 9th and 10th vertebrae, and was treated, and her back pain eventually improved.
A woman in her 50s was diagnosed as having amyotrophic lateral sclerosis, but sought a second opinion and was told that she had chronic Lyme disease (along with babesiosis, and Rocky Mountain spotted fever). She was treated with herbs and homeopathic remedies, followed by intensive antimicrobial and antiviral therapies. She developed intractable Clostridium difficile colitis that lasted for 2 years until she died from complications related to amyotrophic lateral sclerosis.
A woman in her 60s who had autoimmune neutropenia, mixed connective tissue disease, and degenerative arthritis was told her neuropathy was due to chronic Lyme disease. She was treated with immunoglobulin administered through an implanted subcutaneous port. After years of treatments, she was hospitalized for fever and back pain, had blood cultures positive for methicillin-sensitive Staphylococcus aureus, was found to have inflammation of the lumbar facet joints, epidural space, and paraspinal muscles – and eventually required surgical drainage of a paraspinal abscess.
“These cases highlight the severity and scope of adverse effects that can be caused by the use of unproven treatments for chronic Lyme disease,” Dr. Marzec and her associates said.
This work was supported by the CDC. Dr. Marzec and her associates reported having no relevant financial disclosures.
FROM MMWR
Key clinical point: Unproven treatments for so-called chronic Lyme disease cause serious, even fatal, complications.
Major finding:
Data source: Five case reports submitted to the CDC by clinicians, health departments, and individual patients.
Disclosures: This work was supported by the CDC. Dr. Marzec and her associates reported having no relevant financial disclosures.
FDA approves dabrafenib and trametinib for BRAF V600E+ metastatic NSCLC
The FDA also approved a diagnostic, the Oncomine Dx Target Test, a next-generation sequencing test to detect gene mutations or alterations, including BRAF, from a single tissue specimen, the FDA reported in a statement.
The approvals are based on overall response rate (ORR) for the combination in a phase II, nonrandomized, noncomparative, open-label trial of patients with locally confirmed BRAF V600E mutation-positive metastatic NSCLC. The ORR for the combination treatment was 61% (95% confidence interval, 44%-77%) among 36 patients who had received no prior systemic therapy for metastatic NSCLC, and 63% (95% CI, 49%-76%) among 57 patients who had received at least one platinum-based chemotherapy regimen with demonstrated disease progression before enrollment. Those 93 patients were all treated with the combination of dabrafenib (150 mg orally twice daily) and trametinib (2 mg orally once daily).
The ORR was 27% (95% CI, 18%-38%) among a third cohort of 78 patients with previously treated BRAF V600E mutation-positive NSCLC who received single-agent dabrafenib.
The most common adverse reactions were similar to those reported in prior approvals for patients with melanoma, including pyrexia, fatigue, nausea, vomiting, diarrhea, dry skin, decreased appetite, edema, rash, chills, hemorrhage, cough, and dyspnea. The most common grade 3-4 adverse reactions were pyrexia, fatigue, dyspnea, vomiting, rash, hemorrhage, and diarrhea. The most common grade 3-4 laboratory abnormalities were hyponatremia, lymphopenia, anemia, hyperglycemia, neutropenia, leukopenia, hypophosphatemia, and increased alanine aminotransferase. Dabrafenib and trametinib were discontinued for adverse reactions in 18% and 19% of patients, respectively, the FDA said.
Novartis is marketing Dabrafenib as Tafinlar and trametinib as Mekinist.
The recommended doses are dabrafenib 150 mg orally twice daily, approximately 12 hours apart, with trametinib 2 mg orally once daily, following confirmation of BRAF V600E mutation in a tumor specimen by an FDA-approved test.
The FDA also approved a diagnostic, the Oncomine Dx Target Test, a next-generation sequencing test to detect gene mutations or alterations, including BRAF, from a single tissue specimen, the FDA reported in a statement.
The approvals are based on overall response rate (ORR) for the combination in a phase II, nonrandomized, noncomparative, open-label trial of patients with locally confirmed BRAF V600E mutation-positive metastatic NSCLC. The ORR for the combination treatment was 61% (95% confidence interval, 44%-77%) among 36 patients who had received no prior systemic therapy for metastatic NSCLC, and 63% (95% CI, 49%-76%) among 57 patients who had received at least one platinum-based chemotherapy regimen with demonstrated disease progression before enrollment. Those 93 patients were all treated with the combination of dabrafenib (150 mg orally twice daily) and trametinib (2 mg orally once daily).
The ORR was 27% (95% CI, 18%-38%) among a third cohort of 78 patients with previously treated BRAF V600E mutation-positive NSCLC who received single-agent dabrafenib.
The most common adverse reactions were similar to those reported in prior approvals for patients with melanoma, including pyrexia, fatigue, nausea, vomiting, diarrhea, dry skin, decreased appetite, edema, rash, chills, hemorrhage, cough, and dyspnea. The most common grade 3-4 adverse reactions were pyrexia, fatigue, dyspnea, vomiting, rash, hemorrhage, and diarrhea. The most common grade 3-4 laboratory abnormalities were hyponatremia, lymphopenia, anemia, hyperglycemia, neutropenia, leukopenia, hypophosphatemia, and increased alanine aminotransferase. Dabrafenib and trametinib were discontinued for adverse reactions in 18% and 19% of patients, respectively, the FDA said.
Novartis is marketing Dabrafenib as Tafinlar and trametinib as Mekinist.
The recommended doses are dabrafenib 150 mg orally twice daily, approximately 12 hours apart, with trametinib 2 mg orally once daily, following confirmation of BRAF V600E mutation in a tumor specimen by an FDA-approved test.
The FDA also approved a diagnostic, the Oncomine Dx Target Test, a next-generation sequencing test to detect gene mutations or alterations, including BRAF, from a single tissue specimen, the FDA reported in a statement.
The approvals are based on overall response rate (ORR) for the combination in a phase II, nonrandomized, noncomparative, open-label trial of patients with locally confirmed BRAF V600E mutation-positive metastatic NSCLC. The ORR for the combination treatment was 61% (95% confidence interval, 44%-77%) among 36 patients who had received no prior systemic therapy for metastatic NSCLC, and 63% (95% CI, 49%-76%) among 57 patients who had received at least one platinum-based chemotherapy regimen with demonstrated disease progression before enrollment. Those 93 patients were all treated with the combination of dabrafenib (150 mg orally twice daily) and trametinib (2 mg orally once daily).
The ORR was 27% (95% CI, 18%-38%) among a third cohort of 78 patients with previously treated BRAF V600E mutation-positive NSCLC who received single-agent dabrafenib.
The most common adverse reactions were similar to those reported in prior approvals for patients with melanoma, including pyrexia, fatigue, nausea, vomiting, diarrhea, dry skin, decreased appetite, edema, rash, chills, hemorrhage, cough, and dyspnea. The most common grade 3-4 adverse reactions were pyrexia, fatigue, dyspnea, vomiting, rash, hemorrhage, and diarrhea. The most common grade 3-4 laboratory abnormalities were hyponatremia, lymphopenia, anemia, hyperglycemia, neutropenia, leukopenia, hypophosphatemia, and increased alanine aminotransferase. Dabrafenib and trametinib were discontinued for adverse reactions in 18% and 19% of patients, respectively, the FDA said.
Novartis is marketing Dabrafenib as Tafinlar and trametinib as Mekinist.
The recommended doses are dabrafenib 150 mg orally twice daily, approximately 12 hours apart, with trametinib 2 mg orally once daily, following confirmation of BRAF V600E mutation in a tumor specimen by an FDA-approved test.
Cotempla XR-ODT approved for children, adolescents with ADHD
The Food and Drug Administration has approved the first methylphenidate extended-release orally disintegrating tablet for treating ADHD in patients aged 6-17 years old, Neos Therapeutics announced June 19.
The company said the approval came after a phase III trial showed that treatment in a laboratory classroom with the drug, called Cotempla XR-ODT, showed a significant improvement in attention-deficit/hyperactivity disorder symptom control when compared with a placebo across the classroom day (placebo-subtracted difference of –11). The onset of effect was shown at 1 hour post-dose and lasted through 12 hours. No serious adverse events were reported during the trial, and the adverse event profile was consistent with the established safety profile for other extended-release methylphenidate products.
Cotempla XR-ODT will be available commercially in a portable, child-resistant blister pack in the fall of 2017.
Find the full press release on Neos Therapeutics website.
The Food and Drug Administration has approved the first methylphenidate extended-release orally disintegrating tablet for treating ADHD in patients aged 6-17 years old, Neos Therapeutics announced June 19.
The company said the approval came after a phase III trial showed that treatment in a laboratory classroom with the drug, called Cotempla XR-ODT, showed a significant improvement in attention-deficit/hyperactivity disorder symptom control when compared with a placebo across the classroom day (placebo-subtracted difference of –11). The onset of effect was shown at 1 hour post-dose and lasted through 12 hours. No serious adverse events were reported during the trial, and the adverse event profile was consistent with the established safety profile for other extended-release methylphenidate products.
Cotempla XR-ODT will be available commercially in a portable, child-resistant blister pack in the fall of 2017.
Find the full press release on Neos Therapeutics website.
The Food and Drug Administration has approved the first methylphenidate extended-release orally disintegrating tablet for treating ADHD in patients aged 6-17 years old, Neos Therapeutics announced June 19.
The company said the approval came after a phase III trial showed that treatment in a laboratory classroom with the drug, called Cotempla XR-ODT, showed a significant improvement in attention-deficit/hyperactivity disorder symptom control when compared with a placebo across the classroom day (placebo-subtracted difference of –11). The onset of effect was shown at 1 hour post-dose and lasted through 12 hours. No serious adverse events were reported during the trial, and the adverse event profile was consistent with the established safety profile for other extended-release methylphenidate products.
Cotempla XR-ODT will be available commercially in a portable, child-resistant blister pack in the fall of 2017.
Find the full press release on Neos Therapeutics website.
FDA advisory committee supports new CV liraglutide indication
A Food and Drug Administration advisory committee voted 17-2 in support of a supplemental new drug application for liraglutide (Victoza) injections to reduce the risk of major adverse cardiovascular events in adults with type 2 diabetes and established cardiovascular disease.
Novo Nordisk, the maker of the glucagon-like peptide-1 (GLP-1) analogue, proposed the additional indication for liraglutide as an adjunct to standard treatment of cardiovascular risk factors in such patients based solely on the results of the randomized, placebo-controlled postmarketing LEADER trial.
If this additional indication for liraglutide is approved by the FDA, the drug would join the antidiabetic drug empagliflozin (Jardiance) in having a second indication for the reduction of the risk of cardiovascular death. The supplemental new drug application for Jardiance was approved by the FDA in December 2016 – also based on the results of a single trial (the EMPA-REG outcomes trial). Of note, the American Diabetes Association in its 2017 Standards of Medical Care has already called for consideration of both liraglutide and empagliflozin to reduce the risk of cardiovascular death in patients with type 2 diabetes and documented cardiovascular disease.
Liraglutide is currently approved for blood glucose lowering in adults with type 2 diabetes and is marketed as Saxenda for the treatment of overweight and obese adults with at least one weight-related comorbidity. It was shown in the LEADER trial to be associated with a significant 13% lower risk vs. placebo for a composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke in patients with type 2 diabetes.
All 19 voting members of the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) agreed that the LEADER results confirm there is no excess cardiovascular risk associated with liraglutide in patients with type 2 diabetes, but, on the question of whether the results provide “the substantial evidence required to establish that liraglutide 1.8 mg reduces cardiovascular risk in patients with type 2 diabetes mellitus and established cardiovascular disease,” almost all voting members expressed concerns about subgroup analyses showing reduced benefit among U.S. patients, compared with those from other countries.
“I think probably the most influential finding for me was the overall cardiovascular mortality finding, followed by the consistency of the results,” said biostatistics expert James D. Neaton, PhD, of the University of Minnesota, Minneapolis. He added that the indication should focus on patients at high cardiovascular event risk, as the LEADER population was a very high risk group.
Similarly, temporary voting member Marvin A. Konstam, MD, of Tufts University, Boston, said “the primary trial results are very robust and substantiated.
“And I think the cardiovascular mortality is the biggest contributor to that, which is obviously a very important finding,” he said, also stressing that the indication should focus on patients with established cardiovascular disease.
“I am concerned about the U.S. population, but at the end of the day, it’s a subgroup, and I just can’t overrate that to diminish the overall finding,” he added.
Peter W.F. Wilson, MD, EMDAC chairperson, said he “wrestles with exactly who benefits the most because of overlapping of some of the groupings.
“But people who really have atherosclerotic cardiovascular disease ... are probably the people who will benefit the most, and I hope those are the people who will get it,” said Dr. Wilson, professor of medicine at Emory University, professor of public health at Rollins School of Public Health, and director of epidemiology and genomic medicine at the Atlanta VA Medical Center.
In explaining his “no” vote, Daniel Budnitz, MD, of the Centers for Disease Control and Prevention, Atlanta, said his was a tough decision, but that ultimately, since the U.S. population is the one the FDA is addressing with its labeling, the subgroup concerns weighed heavily.
“And I do worry about a slippery slope of using single-trial data for new indications, when there are questions and when you do have an interaction term for the U.S. vs. the rest of the world,” he said, adding that he would like to see either another international trial where the United States population does not differ from the rest of the world, or a U.S. trial.
Carmen J. Allegra, MD, of the University of Florida, Gainesville, also voted no, and said he, too, was concerned by the subgroup analysis.
“I was very much concerned and swayed by the subgroup analysis. The U.S. target population is a pretty darn important population for us to consider, and we saw a significant interaction with outcomes vs. the region by the FDA’s analysis,” he said. “I was really swayed by the fact that we really didn’t see evidence of superiority in the U.S. population.”
The LEADER trial, which was designed in accordance with FDA Guidance issued in 2008 to demonstrate that new antidiabetes drugs do not result in unacceptably increased cardiovascular risk, included 9,340 patients who were randomized to receive liraglutide or placebo as add-on to standard of care treatment and who were followed for a median of 3.8 years. Those randomized to receive liraglutide experienced significantly lower risk of the composite primary outcome (hazard ratio, 0.87), Notably, the effect was diminished among U.S. patients, compared with the overall benefit.
However, after hearing LEADER analyses from Novo Nordisk representatives and FDA representatives, and testimony from numerous individuals, including patients, physicians, and patient advocates who spoke overwhelmingly in favor of approval of the supplemental drug application, the committee recommended that approval.
“This was not a slam dunk. I think the subgroup analysis was interesting discussion, but in the end you have to take the data and the primary outcome measure as what you move on,” said temporary voting member David C. Robbins, MD, of the University of Kansas, Kansas City, adding that “the good is outweighing the bad on this.
“I’m glad to see diabetes management moving toward more than lowering blood sugar. It’s a good step in the right direction,” he said.
The FDA, which usually follows the recommendations of its advisory committees, will now consider the supplemental new drug application for liraglutide.
In a statement released after the vote, Todd Hobbs, MD, vice president and U.S. chief medical officer of Novo Nordisk, noted that cardiovascular disease remains the leading cause of death for people with type 2 diabetes. The discussion during the EMDAC meeting is “an important reminder that there is an unmet need to provide benefits beyond HbA1c control” in patients with type 2 diabetes.
EMDAC committee members were screened and found to be in compliance with federal ethics and conflict of interest laws; one (Dr. Konstam) was granted a waiver in accordance with rules allowing such waivers when the need for an individual’s service outweighs any potential financial conflicts of interest. Dr. Konstam reported financial relationships with competing firms.
A Food and Drug Administration advisory committee voted 17-2 in support of a supplemental new drug application for liraglutide (Victoza) injections to reduce the risk of major adverse cardiovascular events in adults with type 2 diabetes and established cardiovascular disease.
Novo Nordisk, the maker of the glucagon-like peptide-1 (GLP-1) analogue, proposed the additional indication for liraglutide as an adjunct to standard treatment of cardiovascular risk factors in such patients based solely on the results of the randomized, placebo-controlled postmarketing LEADER trial.
If this additional indication for liraglutide is approved by the FDA, the drug would join the antidiabetic drug empagliflozin (Jardiance) in having a second indication for the reduction of the risk of cardiovascular death. The supplemental new drug application for Jardiance was approved by the FDA in December 2016 – also based on the results of a single trial (the EMPA-REG outcomes trial). Of note, the American Diabetes Association in its 2017 Standards of Medical Care has already called for consideration of both liraglutide and empagliflozin to reduce the risk of cardiovascular death in patients with type 2 diabetes and documented cardiovascular disease.
Liraglutide is currently approved for blood glucose lowering in adults with type 2 diabetes and is marketed as Saxenda for the treatment of overweight and obese adults with at least one weight-related comorbidity. It was shown in the LEADER trial to be associated with a significant 13% lower risk vs. placebo for a composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke in patients with type 2 diabetes.
All 19 voting members of the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) agreed that the LEADER results confirm there is no excess cardiovascular risk associated with liraglutide in patients with type 2 diabetes, but, on the question of whether the results provide “the substantial evidence required to establish that liraglutide 1.8 mg reduces cardiovascular risk in patients with type 2 diabetes mellitus and established cardiovascular disease,” almost all voting members expressed concerns about subgroup analyses showing reduced benefit among U.S. patients, compared with those from other countries.
“I think probably the most influential finding for me was the overall cardiovascular mortality finding, followed by the consistency of the results,” said biostatistics expert James D. Neaton, PhD, of the University of Minnesota, Minneapolis. He added that the indication should focus on patients at high cardiovascular event risk, as the LEADER population was a very high risk group.
Similarly, temporary voting member Marvin A. Konstam, MD, of Tufts University, Boston, said “the primary trial results are very robust and substantiated.
“And I think the cardiovascular mortality is the biggest contributor to that, which is obviously a very important finding,” he said, also stressing that the indication should focus on patients with established cardiovascular disease.
“I am concerned about the U.S. population, but at the end of the day, it’s a subgroup, and I just can’t overrate that to diminish the overall finding,” he added.
Peter W.F. Wilson, MD, EMDAC chairperson, said he “wrestles with exactly who benefits the most because of overlapping of some of the groupings.
“But people who really have atherosclerotic cardiovascular disease ... are probably the people who will benefit the most, and I hope those are the people who will get it,” said Dr. Wilson, professor of medicine at Emory University, professor of public health at Rollins School of Public Health, and director of epidemiology and genomic medicine at the Atlanta VA Medical Center.
In explaining his “no” vote, Daniel Budnitz, MD, of the Centers for Disease Control and Prevention, Atlanta, said his was a tough decision, but that ultimately, since the U.S. population is the one the FDA is addressing with its labeling, the subgroup concerns weighed heavily.
“And I do worry about a slippery slope of using single-trial data for new indications, when there are questions and when you do have an interaction term for the U.S. vs. the rest of the world,” he said, adding that he would like to see either another international trial where the United States population does not differ from the rest of the world, or a U.S. trial.
Carmen J. Allegra, MD, of the University of Florida, Gainesville, also voted no, and said he, too, was concerned by the subgroup analysis.
“I was very much concerned and swayed by the subgroup analysis. The U.S. target population is a pretty darn important population for us to consider, and we saw a significant interaction with outcomes vs. the region by the FDA’s analysis,” he said. “I was really swayed by the fact that we really didn’t see evidence of superiority in the U.S. population.”
The LEADER trial, which was designed in accordance with FDA Guidance issued in 2008 to demonstrate that new antidiabetes drugs do not result in unacceptably increased cardiovascular risk, included 9,340 patients who were randomized to receive liraglutide or placebo as add-on to standard of care treatment and who were followed for a median of 3.8 years. Those randomized to receive liraglutide experienced significantly lower risk of the composite primary outcome (hazard ratio, 0.87), Notably, the effect was diminished among U.S. patients, compared with the overall benefit.
However, after hearing LEADER analyses from Novo Nordisk representatives and FDA representatives, and testimony from numerous individuals, including patients, physicians, and patient advocates who spoke overwhelmingly in favor of approval of the supplemental drug application, the committee recommended that approval.
“This was not a slam dunk. I think the subgroup analysis was interesting discussion, but in the end you have to take the data and the primary outcome measure as what you move on,” said temporary voting member David C. Robbins, MD, of the University of Kansas, Kansas City, adding that “the good is outweighing the bad on this.
“I’m glad to see diabetes management moving toward more than lowering blood sugar. It’s a good step in the right direction,” he said.
The FDA, which usually follows the recommendations of its advisory committees, will now consider the supplemental new drug application for liraglutide.
In a statement released after the vote, Todd Hobbs, MD, vice president and U.S. chief medical officer of Novo Nordisk, noted that cardiovascular disease remains the leading cause of death for people with type 2 diabetes. The discussion during the EMDAC meeting is “an important reminder that there is an unmet need to provide benefits beyond HbA1c control” in patients with type 2 diabetes.
EMDAC committee members were screened and found to be in compliance with federal ethics and conflict of interest laws; one (Dr. Konstam) was granted a waiver in accordance with rules allowing such waivers when the need for an individual’s service outweighs any potential financial conflicts of interest. Dr. Konstam reported financial relationships with competing firms.
A Food and Drug Administration advisory committee voted 17-2 in support of a supplemental new drug application for liraglutide (Victoza) injections to reduce the risk of major adverse cardiovascular events in adults with type 2 diabetes and established cardiovascular disease.
Novo Nordisk, the maker of the glucagon-like peptide-1 (GLP-1) analogue, proposed the additional indication for liraglutide as an adjunct to standard treatment of cardiovascular risk factors in such patients based solely on the results of the randomized, placebo-controlled postmarketing LEADER trial.
If this additional indication for liraglutide is approved by the FDA, the drug would join the antidiabetic drug empagliflozin (Jardiance) in having a second indication for the reduction of the risk of cardiovascular death. The supplemental new drug application for Jardiance was approved by the FDA in December 2016 – also based on the results of a single trial (the EMPA-REG outcomes trial). Of note, the American Diabetes Association in its 2017 Standards of Medical Care has already called for consideration of both liraglutide and empagliflozin to reduce the risk of cardiovascular death in patients with type 2 diabetes and documented cardiovascular disease.
Liraglutide is currently approved for blood glucose lowering in adults with type 2 diabetes and is marketed as Saxenda for the treatment of overweight and obese adults with at least one weight-related comorbidity. It was shown in the LEADER trial to be associated with a significant 13% lower risk vs. placebo for a composite outcome of death from cardiovascular causes, nonfatal myocardial infarction, and nonfatal stroke in patients with type 2 diabetes.
All 19 voting members of the Endocrinologic and Metabolic Drugs Advisory Committee (EMDAC) agreed that the LEADER results confirm there is no excess cardiovascular risk associated with liraglutide in patients with type 2 diabetes, but, on the question of whether the results provide “the substantial evidence required to establish that liraglutide 1.8 mg reduces cardiovascular risk in patients with type 2 diabetes mellitus and established cardiovascular disease,” almost all voting members expressed concerns about subgroup analyses showing reduced benefit among U.S. patients, compared with those from other countries.
“I think probably the most influential finding for me was the overall cardiovascular mortality finding, followed by the consistency of the results,” said biostatistics expert James D. Neaton, PhD, of the University of Minnesota, Minneapolis. He added that the indication should focus on patients at high cardiovascular event risk, as the LEADER population was a very high risk group.
Similarly, temporary voting member Marvin A. Konstam, MD, of Tufts University, Boston, said “the primary trial results are very robust and substantiated.
“And I think the cardiovascular mortality is the biggest contributor to that, which is obviously a very important finding,” he said, also stressing that the indication should focus on patients with established cardiovascular disease.
“I am concerned about the U.S. population, but at the end of the day, it’s a subgroup, and I just can’t overrate that to diminish the overall finding,” he added.
Peter W.F. Wilson, MD, EMDAC chairperson, said he “wrestles with exactly who benefits the most because of overlapping of some of the groupings.
“But people who really have atherosclerotic cardiovascular disease ... are probably the people who will benefit the most, and I hope those are the people who will get it,” said Dr. Wilson, professor of medicine at Emory University, professor of public health at Rollins School of Public Health, and director of epidemiology and genomic medicine at the Atlanta VA Medical Center.
In explaining his “no” vote, Daniel Budnitz, MD, of the Centers for Disease Control and Prevention, Atlanta, said his was a tough decision, but that ultimately, since the U.S. population is the one the FDA is addressing with its labeling, the subgroup concerns weighed heavily.
“And I do worry about a slippery slope of using single-trial data for new indications, when there are questions and when you do have an interaction term for the U.S. vs. the rest of the world,” he said, adding that he would like to see either another international trial where the United States population does not differ from the rest of the world, or a U.S. trial.
Carmen J. Allegra, MD, of the University of Florida, Gainesville, also voted no, and said he, too, was concerned by the subgroup analysis.
“I was very much concerned and swayed by the subgroup analysis. The U.S. target population is a pretty darn important population for us to consider, and we saw a significant interaction with outcomes vs. the region by the FDA’s analysis,” he said. “I was really swayed by the fact that we really didn’t see evidence of superiority in the U.S. population.”
The LEADER trial, which was designed in accordance with FDA Guidance issued in 2008 to demonstrate that new antidiabetes drugs do not result in unacceptably increased cardiovascular risk, included 9,340 patients who were randomized to receive liraglutide or placebo as add-on to standard of care treatment and who were followed for a median of 3.8 years. Those randomized to receive liraglutide experienced significantly lower risk of the composite primary outcome (hazard ratio, 0.87), Notably, the effect was diminished among U.S. patients, compared with the overall benefit.
However, after hearing LEADER analyses from Novo Nordisk representatives and FDA representatives, and testimony from numerous individuals, including patients, physicians, and patient advocates who spoke overwhelmingly in favor of approval of the supplemental drug application, the committee recommended that approval.
“This was not a slam dunk. I think the subgroup analysis was interesting discussion, but in the end you have to take the data and the primary outcome measure as what you move on,” said temporary voting member David C. Robbins, MD, of the University of Kansas, Kansas City, adding that “the good is outweighing the bad on this.
“I’m glad to see diabetes management moving toward more than lowering blood sugar. It’s a good step in the right direction,” he said.
The FDA, which usually follows the recommendations of its advisory committees, will now consider the supplemental new drug application for liraglutide.
In a statement released after the vote, Todd Hobbs, MD, vice president and U.S. chief medical officer of Novo Nordisk, noted that cardiovascular disease remains the leading cause of death for people with type 2 diabetes. The discussion during the EMDAC meeting is “an important reminder that there is an unmet need to provide benefits beyond HbA1c control” in patients with type 2 diabetes.
EMDAC committee members were screened and found to be in compliance with federal ethics and conflict of interest laws; one (Dr. Konstam) was granted a waiver in accordance with rules allowing such waivers when the need for an individual’s service outweighs any potential financial conflicts of interest. Dr. Konstam reported financial relationships with competing firms.
Mydayis approved for teens, adults with ADHD
The Food and Drug Administration has approved a once-a-day treatment for patients aged 13 years and older with ADHD, Shire announced June 20 in a press release.
The approval of Mydayis was based on results from 16 clinical studies evaluating the medication in more than 1,600 adolescents (aged 13-17 years) and adults with attention-deficit/hyperactivity disorder. In the placebo-controlled clinical studies, Mydayis significantly improved symptoms of ADHD, as measured by the ADHD-RS-IV and the Permanent Product Measure of Performance (PERMP), in adults and adolescents. Improvement on the PERMP reached statistical significance beginning at 2 or 4 hours post dose and lasting up to 16 hours post dose.
The medication, an amphetamine product, consists of three different types of drug-releasing beads.
“With this approval, we hope to help patients who need a once-daily treatment option,” Flemming Ornskov, MD, MPH, said in a press release. Dr. Ornskov is CEO of Shire.
It is estimated that 4.4% of adults have ADHD in the United States, and 50%-66% of children with ADHD may continue to have symptoms of the disorder as adults.
Mydayis will be commercially available in the United States in the third quarter of 2017.
Read the full press release here.
The Food and Drug Administration has approved a once-a-day treatment for patients aged 13 years and older with ADHD, Shire announced June 20 in a press release.
The approval of Mydayis was based on results from 16 clinical studies evaluating the medication in more than 1,600 adolescents (aged 13-17 years) and adults with attention-deficit/hyperactivity disorder. In the placebo-controlled clinical studies, Mydayis significantly improved symptoms of ADHD, as measured by the ADHD-RS-IV and the Permanent Product Measure of Performance (PERMP), in adults and adolescents. Improvement on the PERMP reached statistical significance beginning at 2 or 4 hours post dose and lasting up to 16 hours post dose.
The medication, an amphetamine product, consists of three different types of drug-releasing beads.
“With this approval, we hope to help patients who need a once-daily treatment option,” Flemming Ornskov, MD, MPH, said in a press release. Dr. Ornskov is CEO of Shire.
It is estimated that 4.4% of adults have ADHD in the United States, and 50%-66% of children with ADHD may continue to have symptoms of the disorder as adults.
Mydayis will be commercially available in the United States in the third quarter of 2017.
Read the full press release here.
The Food and Drug Administration has approved a once-a-day treatment for patients aged 13 years and older with ADHD, Shire announced June 20 in a press release.
The approval of Mydayis was based on results from 16 clinical studies evaluating the medication in more than 1,600 adolescents (aged 13-17 years) and adults with attention-deficit/hyperactivity disorder. In the placebo-controlled clinical studies, Mydayis significantly improved symptoms of ADHD, as measured by the ADHD-RS-IV and the Permanent Product Measure of Performance (PERMP), in adults and adolescents. Improvement on the PERMP reached statistical significance beginning at 2 or 4 hours post dose and lasting up to 16 hours post dose.
The medication, an amphetamine product, consists of three different types of drug-releasing beads.
“With this approval, we hope to help patients who need a once-daily treatment option,” Flemming Ornskov, MD, MPH, said in a press release. Dr. Ornskov is CEO of Shire.
It is estimated that 4.4% of adults have ADHD in the United States, and 50%-66% of children with ADHD may continue to have symptoms of the disorder as adults.
Mydayis will be commercially available in the United States in the third quarter of 2017.
Read the full press release here.