User login
Bringing you the latest news, research and reviews, exclusive interviews, podcasts, quizzes, and more.
Powered by CHEST Physician, Clinician Reviews, MDedge Family Medicine, Internal Medicine News, and The Journal of Clinical Outcomes Management.
Poor sleep quality associated with poor cannabis cessation outcomes
Poor sleep quality was associated with higher rates of mean cannabis use and lower rates of cessation during the first 6 months following a self-guided attempt to quit, although sleep efficiency/duration was not linked to cannabis use outcomes, a study of U.S. veterans has shown.
Citing previous research associating sleep quality with cannabis quit outcomes, including a study that found 48%-77% of cannabis users reported either relapsing or turning to other substances in order to improve their sleep, Kimberly A. Babson, Ph.D., and her colleagues hypothesized that perceived sleep quality rather than actual sleep efficiency/duration would affect cannabis use following a self-guided quit attempt. The results were published recently in Addictive Behaviors (2013;38:2707-13).

Dr. Babson and her colleagues, all affiliated with the VA Palo Alto Health Care System, Menlo Park, Calif., recruited 102 veterans (95% male) with a mean age of 51 years. All participants met DSM-5 criteria for cannabis dependence, had a self-reported desire to quit of 5 or greater on a scale of 0 to 10 (0 = no interest in quitting, 10 = definite interest), and had a self-reported desire to follow a self-guided cessation program. Candidates were excluded if they already had decreased their cannabis use by 25% or more in the previous month, were pregnant or breast-feeding, or had suicidal ideation.
Anxiety disorders were present in 88% of participants, and 43% had a co-occurring mood disorder. Those with and without an anxiety disorder or mood disorder (analyzed separately) did not differ significantly in terms of perceived sleep quality (self-reported overall quality of sleep), sleep efficiency/duration (self-reported quantity of sleep), or cannabis use over the course of the study, the authors reported.
Dependence on other substances was present in 29% of the study group; 4% had a substance abuse disorder. Just more than half of the entire group reported using a sleep medication at baseline, although this group "did not differ from those who did not use a sleep medication at baseline in terms of perceived sleep quality during the study period." Participants using a sleep medication used less cannabis at baseline than did those not using a sleep medication; however, the two groups did not differ when it came to cannabis use over the course of the study.
Participants were rated on the Clinician-Administered PTSD Scale, minus evaluation for two sleep-related symptoms – nightmares and insomnia. The severity of withdrawal from cannabis symptoms also was assessed at baseline and at each follow-up assessment over the 6-month course, using the Marijuana Withdrawal Checklist-Short Form. Post-traumatic stress disorder (PTSD) symptom severity was used as a covariate; symptom withdrawal was considered a time-varying covariate.
The Pittsburgh Sleep Quality Index was used to determine perceived sleep quality in terms of subjective sleep quality, sleep latency, sleep disturbances, and daytime dysfunction. Sleep efficiency/duration was assessed in terms of the combined amount of time spent in bed, whether or not the person was asleep (efficiency), and the actual amount of time spent sleeping (duration).
To determine links between cannabis use, and pre- and post-quit period sleep disturbances, the investigators created two generalized linear mixed models, applying to each a quadratic function to account for a wider array of variables. The first model was not adjusted for covariates, while the second model was adjusted for baseline age, PTSD, and withdrawal symptom severity as measured at each follow-up over the 6-month period.
The researchers did not find a significant intercept between perceived sleep quality and cannabis use. However, they did find a significant linear and quadratic slope showing that while cannabis use declined sharply at first, it leveled off in the context of perceived sleep quality.
"This indicates that over the course of the study (i.e., aggregated across time points), lower perceived sleep quality was associated with higher mean cannabis use," the investigators wrote. "However, perceived sleep quality did not interact with the linear or quadratic slopes, meaning that the association between perceived sleep quality and mean cannabis use could not be tied to a discrete time during the follow-up period." When the investigators adjusted the results for covariates, they found the that results were consistent.
Dr. Babson and her colleagues concluded that their findings indicated one of two possibilities: Either individuals use cannabis to regulate sleep, or chronic use disrupts sleep. Further prospective studies of mechanisms such as emotional regulation would help explain the associations, they wrote. However, their findings, while potentially limited by the study’s self-report nature and the question of whether it is generalizable to a larger population, could serve to help "inform the timing of sleep interventions in cannabis treatments in order to optimize outcomes."
This study was supported by a Veterans Affairs (VA) Clinical Science Research and Development Career Development Award, and by funds from the VA Health Services Research and Development Service. The authors reported no relevant disclosures.
Poor sleep quality was associated with higher rates of mean cannabis use and lower rates of cessation during the first 6 months following a self-guided attempt to quit, although sleep efficiency/duration was not linked to cannabis use outcomes, a study of U.S. veterans has shown.
Citing previous research associating sleep quality with cannabis quit outcomes, including a study that found 48%-77% of cannabis users reported either relapsing or turning to other substances in order to improve their sleep, Kimberly A. Babson, Ph.D., and her colleagues hypothesized that perceived sleep quality rather than actual sleep efficiency/duration would affect cannabis use following a self-guided quit attempt. The results were published recently in Addictive Behaviors (2013;38:2707-13).
Dr. Babson and her colleagues, all affiliated with the VA Palo Alto Health Care System, Menlo Park, Calif., recruited 102 veterans (95% male) with a mean age of 51 years. All participants met DSM-5 criteria for cannabis dependence, had a self-reported desire to quit of 5 or greater on a scale of 0 to 10 (0 = no interest in quitting, 10 = definite interest), and had a self-reported desire to follow a self-guided cessation program. Candidates were excluded if they already had decreased their cannabis use by 25% or more in the previous month, were pregnant or breast-feeding, or had suicidal ideation.
Anxiety disorders were present in 88% of participants, and 43% had a co-occurring mood disorder. Those with and without an anxiety disorder or mood disorder (analyzed separately) did not differ significantly in terms of perceived sleep quality (self-reported overall quality of sleep), sleep efficiency/duration (self-reported quantity of sleep), or cannabis use over the course of the study, the authors reported.
Dependence on other substances was present in 29% of the study group; 4% had a substance abuse disorder. Just more than half of the entire group reported using a sleep medication at baseline, although this group "did not differ from those who did not use a sleep medication at baseline in terms of perceived sleep quality during the study period." Participants using a sleep medication used less cannabis at baseline than did those not using a sleep medication; however, the two groups did not differ when it came to cannabis use over the course of the study.
Participants were rated on the Clinician-Administered PTSD Scale, minus evaluation for two sleep-related symptoms – nightmares and insomnia. The severity of withdrawal from cannabis symptoms also was assessed at baseline and at each follow-up assessment over the 6-month course, using the Marijuana Withdrawal Checklist-Short Form. Post-traumatic stress disorder (PTSD) symptom severity was used as a covariate; symptom withdrawal was considered a time-varying covariate.
The Pittsburgh Sleep Quality Index was used to determine perceived sleep quality in terms of subjective sleep quality, sleep latency, sleep disturbances, and daytime dysfunction. Sleep efficiency/duration was assessed in terms of the combined amount of time spent in bed, whether or not the person was asleep (efficiency), and the actual amount of time spent sleeping (duration).
To determine links between cannabis use, and pre- and post-quit period sleep disturbances, the investigators created two generalized linear mixed models, applying to each a quadratic function to account for a wider array of variables. The first model was not adjusted for covariates, while the second model was adjusted for baseline age, PTSD, and withdrawal symptom severity as measured at each follow-up over the 6-month period.
The researchers did not find a significant intercept between perceived sleep quality and cannabis use. However, they did find a significant linear and quadratic slope showing that while cannabis use declined sharply at first, it leveled off in the context of perceived sleep quality.
"This indicates that over the course of the study (i.e., aggregated across time points), lower perceived sleep quality was associated with higher mean cannabis use," the investigators wrote. "However, perceived sleep quality did not interact with the linear or quadratic slopes, meaning that the association between perceived sleep quality and mean cannabis use could not be tied to a discrete time during the follow-up period." When the investigators adjusted the results for covariates, they found the that results were consistent.
Dr. Babson and her colleagues concluded that their findings indicated one of two possibilities: Either individuals use cannabis to regulate sleep, or chronic use disrupts sleep. Further prospective studies of mechanisms such as emotional regulation would help explain the associations, they wrote. However, their findings, while potentially limited by the study’s self-report nature and the question of whether it is generalizable to a larger population, could serve to help "inform the timing of sleep interventions in cannabis treatments in order to optimize outcomes."
This study was supported by a Veterans Affairs (VA) Clinical Science Research and Development Career Development Award, and by funds from the VA Health Services Research and Development Service. The authors reported no relevant disclosures.
Poor sleep quality was associated with higher rates of mean cannabis use and lower rates of cessation during the first 6 months following a self-guided attempt to quit, although sleep efficiency/duration was not linked to cannabis use outcomes, a study of U.S. veterans has shown.
Citing previous research associating sleep quality with cannabis quit outcomes, including a study that found 48%-77% of cannabis users reported either relapsing or turning to other substances in order to improve their sleep, Kimberly A. Babson, Ph.D., and her colleagues hypothesized that perceived sleep quality rather than actual sleep efficiency/duration would affect cannabis use following a self-guided quit attempt. The results were published recently in Addictive Behaviors (2013;38:2707-13).
Dr. Babson and her colleagues, all affiliated with the VA Palo Alto Health Care System, Menlo Park, Calif., recruited 102 veterans (95% male) with a mean age of 51 years. All participants met DSM-5 criteria for cannabis dependence, had a self-reported desire to quit of 5 or greater on a scale of 0 to 10 (0 = no interest in quitting, 10 = definite interest), and had a self-reported desire to follow a self-guided cessation program. Candidates were excluded if they already had decreased their cannabis use by 25% or more in the previous month, were pregnant or breast-feeding, or had suicidal ideation.
Anxiety disorders were present in 88% of participants, and 43% had a co-occurring mood disorder. Those with and without an anxiety disorder or mood disorder (analyzed separately) did not differ significantly in terms of perceived sleep quality (self-reported overall quality of sleep), sleep efficiency/duration (self-reported quantity of sleep), or cannabis use over the course of the study, the authors reported.
Dependence on other substances was present in 29% of the study group; 4% had a substance abuse disorder. Just more than half of the entire group reported using a sleep medication at baseline, although this group "did not differ from those who did not use a sleep medication at baseline in terms of perceived sleep quality during the study period." Participants using a sleep medication used less cannabis at baseline than did those not using a sleep medication; however, the two groups did not differ when it came to cannabis use over the course of the study.
Participants were rated on the Clinician-Administered PTSD Scale, minus evaluation for two sleep-related symptoms – nightmares and insomnia. The severity of withdrawal from cannabis symptoms also was assessed at baseline and at each follow-up assessment over the 6-month course, using the Marijuana Withdrawal Checklist-Short Form. Post-traumatic stress disorder (PTSD) symptom severity was used as a covariate; symptom withdrawal was considered a time-varying covariate.
The Pittsburgh Sleep Quality Index was used to determine perceived sleep quality in terms of subjective sleep quality, sleep latency, sleep disturbances, and daytime dysfunction. Sleep efficiency/duration was assessed in terms of the combined amount of time spent in bed, whether or not the person was asleep (efficiency), and the actual amount of time spent sleeping (duration).
To determine links between cannabis use, and pre- and post-quit period sleep disturbances, the investigators created two generalized linear mixed models, applying to each a quadratic function to account for a wider array of variables. The first model was not adjusted for covariates, while the second model was adjusted for baseline age, PTSD, and withdrawal symptom severity as measured at each follow-up over the 6-month period.
The researchers did not find a significant intercept between perceived sleep quality and cannabis use. However, they did find a significant linear and quadratic slope showing that while cannabis use declined sharply at first, it leveled off in the context of perceived sleep quality.
"This indicates that over the course of the study (i.e., aggregated across time points), lower perceived sleep quality was associated with higher mean cannabis use," the investigators wrote. "However, perceived sleep quality did not interact with the linear or quadratic slopes, meaning that the association between perceived sleep quality and mean cannabis use could not be tied to a discrete time during the follow-up period." When the investigators adjusted the results for covariates, they found the that results were consistent.
Dr. Babson and her colleagues concluded that their findings indicated one of two possibilities: Either individuals use cannabis to regulate sleep, or chronic use disrupts sleep. Further prospective studies of mechanisms such as emotional regulation would help explain the associations, they wrote. However, their findings, while potentially limited by the study’s self-report nature and the question of whether it is generalizable to a larger population, could serve to help "inform the timing of sleep interventions in cannabis treatments in order to optimize outcomes."
This study was supported by a Veterans Affairs (VA) Clinical Science Research and Development Career Development Award, and by funds from the VA Health Services Research and Development Service. The authors reported no relevant disclosures.
FROM ADDICTIVE BEHAVIORS
Major finding: Lower perceived sleep quality was linked to higher mean cannabis use during cessation attempts.
Data source: A study of 102 U.S. veterans engaged in a self-guided cannabis cessation program.
Disclosures: This study was supported by a Veterans Affairs (VA) Clinical Science Research and Development Career Development Award, and by funds from the VA Health Services Research and Development Service. The authors reported no relevant disclosures.

What should physicians say about electronic cigarettes?
If you haven’t already been asked about them, you will be. So, you need to have an answer. Maybe this will help.
Electronic cigarettes (or "e-cigarettes") were reportedly invented by a Chinese pharmacist who wanted to find a safer way for smokers to inhale nicotine, after his father, a cigarette smoker, died from lung cancer. The basic e-cigarette design is a lithium battery attached to a heating element that vaporizes a solution of either propylene glycol or vegetable glycerin and liquid nicotine. Vaporization allows for inhalation, referred to as "vaping" as opposed to smoking.
There should be little debate about whether smokers should have access to these products. They do right now, and the products are unlikely to be banned in the near future, although age restriction is a moving target. Debating access seems nonproductive and a distraction from the real discussion.
The real discussion should focus on whether public health and medical health professionals should be recommending them for treatment.
Two important questions need to be answered:
• First, are they safe? Answer: We have no long-term safety data on the impact of repeated inhalation of propylene glycol or vegetable glycerin on lung tissue. Some short-term data suggest that e-cigarettes may cause airway irritation.
• Second, are they effective for increasing smoking cessation?
To help answer the second question, Dr. Christopher Bullen and his colleagues published a randomized, controlled clinical trial evaluating the comparative efficacy of 16-mg nicotine e-cigarettes, nicotine patches (21-mg patch, one daily), or placebo e-cigarettes (no nicotine) (Lancet 2013 Sept. 9 [doi:10.1016/S0140-6736(13)61842-5]). Potential participants were eligible for enrollment if they were at least 18 years of age, had smoked at least 10 cigarettes per day, and wanted to stop smoking. All participants were referred to the telephone quit line for behavioral counseling. Participants were treated for 13 weeks.
At 6 months, smoking abstinence was 7.3% with nicotine e-cigarettes, 5.8% with the nicotine patches, and 4.1% with placebo e-cigarettes. The risk difference for nicotine e-cigarettes vs. patches was 1.51; for nicotine e-cigarettes vs. placebo e-cigarettes, it was 3.16. Neither difference was statistically significant. Interestingly, e-cigarettes were associated with greater reductions in cigarette smoking, compared with nicotine patches. (None of the study’s authors reported having any relevant conflicts of interest.)
So, back to the second point. E-cigarettes are clearly not superior to nicotine patches, but this study may have been underpowered because absolute abstinence rates were low.
Currently, e-cigarette manufacturers are spending resources manufacturing and marketing rather than assisting in the creation of reliable scientific data or the creation of an international research agenda on these products.
One day, an e-cigarette device may be part of a clinical treatment program for tobacco dependence. But until that day, clinicians need to be justifiably circumspect in recommending e-cigarettes for use among cigarette smokers.
Why? Because:
• They are not clearly superior to Food and Drug Administration–approved medications for smoking cessation.
• They are not FDA approved for treatment.
• Short-term safety data suggest they may cause airway irritation.
• Long-term safety data do not exist.
• Smoking reduction is arguably not a relevant clinical outcome, because a significant increase in tobacco-related risk occurs at low levels of exposure.
For clinicians, treatment recommendations are married to the responsibility for unintended consequences. What are those unintended consequences with electronic cigarettes? We need more data.
Dr. Ebbert is professor of medicine and a primary care clinician at the Mayo Clinic in Rochester, Minn. Dr. Ebbert has received consulting fees from GlaxoSmithKline, manufacturer of nicotine replacement products, and research support from Pfizer, manufacturer of varenicline. The opinions expressed are those of the author.
If you haven’t already been asked about them, you will be. So, you need to have an answer. Maybe this will help.
Electronic cigarettes (or "e-cigarettes") were reportedly invented by a Chinese pharmacist who wanted to find a safer way for smokers to inhale nicotine, after his father, a cigarette smoker, died from lung cancer. The basic e-cigarette design is a lithium battery attached to a heating element that vaporizes a solution of either propylene glycol or vegetable glycerin and liquid nicotine. Vaporization allows for inhalation, referred to as "vaping" as opposed to smoking.
There should be little debate about whether smokers should have access to these products. They do right now, and the products are unlikely to be banned in the near future, although age restriction is a moving target. Debating access seems nonproductive and a distraction from the real discussion.
The real discussion should focus on whether public health and medical health professionals should be recommending them for treatment.
Two important questions need to be answered:
• First, are they safe? Answer: We have no long-term safety data on the impact of repeated inhalation of propylene glycol or vegetable glycerin on lung tissue. Some short-term data suggest that e-cigarettes may cause airway irritation.
• Second, are they effective for increasing smoking cessation?
To help answer the second question, Dr. Christopher Bullen and his colleagues published a randomized, controlled clinical trial evaluating the comparative efficacy of 16-mg nicotine e-cigarettes, nicotine patches (21-mg patch, one daily), or placebo e-cigarettes (no nicotine) (Lancet 2013 Sept. 9 [doi:10.1016/S0140-6736(13)61842-5]). Potential participants were eligible for enrollment if they were at least 18 years of age, had smoked at least 10 cigarettes per day, and wanted to stop smoking. All participants were referred to the telephone quit line for behavioral counseling. Participants were treated for 13 weeks.
At 6 months, smoking abstinence was 7.3% with nicotine e-cigarettes, 5.8% with the nicotine patches, and 4.1% with placebo e-cigarettes. The risk difference for nicotine e-cigarettes vs. patches was 1.51; for nicotine e-cigarettes vs. placebo e-cigarettes, it was 3.16. Neither difference was statistically significant. Interestingly, e-cigarettes were associated with greater reductions in cigarette smoking, compared with nicotine patches. (None of the study’s authors reported having any relevant conflicts of interest.)
So, back to the second point. E-cigarettes are clearly not superior to nicotine patches, but this study may have been underpowered because absolute abstinence rates were low.
Currently, e-cigarette manufacturers are spending resources manufacturing and marketing rather than assisting in the creation of reliable scientific data or the creation of an international research agenda on these products.
One day, an e-cigarette device may be part of a clinical treatment program for tobacco dependence. But until that day, clinicians need to be justifiably circumspect in recommending e-cigarettes for use among cigarette smokers.
Why? Because:
• They are not clearly superior to Food and Drug Administration–approved medications for smoking cessation.
• They are not FDA approved for treatment.
• Short-term safety data suggest they may cause airway irritation.
• Long-term safety data do not exist.
• Smoking reduction is arguably not a relevant clinical outcome, because a significant increase in tobacco-related risk occurs at low levels of exposure.
For clinicians, treatment recommendations are married to the responsibility for unintended consequences. What are those unintended consequences with electronic cigarettes? We need more data.
Dr. Ebbert is professor of medicine and a primary care clinician at the Mayo Clinic in Rochester, Minn. Dr. Ebbert has received consulting fees from GlaxoSmithKline, manufacturer of nicotine replacement products, and research support from Pfizer, manufacturer of varenicline. The opinions expressed are those of the author.
If you haven’t already been asked about them, you will be. So, you need to have an answer. Maybe this will help.
Electronic cigarettes (or "e-cigarettes") were reportedly invented by a Chinese pharmacist who wanted to find a safer way for smokers to inhale nicotine, after his father, a cigarette smoker, died from lung cancer. The basic e-cigarette design is a lithium battery attached to a heating element that vaporizes a solution of either propylene glycol or vegetable glycerin and liquid nicotine. Vaporization allows for inhalation, referred to as "vaping" as opposed to smoking.
There should be little debate about whether smokers should have access to these products. They do right now, and the products are unlikely to be banned in the near future, although age restriction is a moving target. Debating access seems nonproductive and a distraction from the real discussion.
The real discussion should focus on whether public health and medical health professionals should be recommending them for treatment.
Two important questions need to be answered:
• First, are they safe? Answer: We have no long-term safety data on the impact of repeated inhalation of propylene glycol or vegetable glycerin on lung tissue. Some short-term data suggest that e-cigarettes may cause airway irritation.
• Second, are they effective for increasing smoking cessation?
To help answer the second question, Dr. Christopher Bullen and his colleagues published a randomized, controlled clinical trial evaluating the comparative efficacy of 16-mg nicotine e-cigarettes, nicotine patches (21-mg patch, one daily), or placebo e-cigarettes (no nicotine) (Lancet 2013 Sept. 9 [doi:10.1016/S0140-6736(13)61842-5]). Potential participants were eligible for enrollment if they were at least 18 years of age, had smoked at least 10 cigarettes per day, and wanted to stop smoking. All participants were referred to the telephone quit line for behavioral counseling. Participants were treated for 13 weeks.
At 6 months, smoking abstinence was 7.3% with nicotine e-cigarettes, 5.8% with the nicotine patches, and 4.1% with placebo e-cigarettes. The risk difference for nicotine e-cigarettes vs. patches was 1.51; for nicotine e-cigarettes vs. placebo e-cigarettes, it was 3.16. Neither difference was statistically significant. Interestingly, e-cigarettes were associated with greater reductions in cigarette smoking, compared with nicotine patches. (None of the study’s authors reported having any relevant conflicts of interest.)
So, back to the second point. E-cigarettes are clearly not superior to nicotine patches, but this study may have been underpowered because absolute abstinence rates were low.
Currently, e-cigarette manufacturers are spending resources manufacturing and marketing rather than assisting in the creation of reliable scientific data or the creation of an international research agenda on these products.
One day, an e-cigarette device may be part of a clinical treatment program for tobacco dependence. But until that day, clinicians need to be justifiably circumspect in recommending e-cigarettes for use among cigarette smokers.
Why? Because:
• They are not clearly superior to Food and Drug Administration–approved medications for smoking cessation.
• They are not FDA approved for treatment.
• Short-term safety data suggest they may cause airway irritation.
• Long-term safety data do not exist.
• Smoking reduction is arguably not a relevant clinical outcome, because a significant increase in tobacco-related risk occurs at low levels of exposure.
For clinicians, treatment recommendations are married to the responsibility for unintended consequences. What are those unintended consequences with electronic cigarettes? We need more data.
Dr. Ebbert is professor of medicine and a primary care clinician at the Mayo Clinic in Rochester, Minn. Dr. Ebbert has received consulting fees from GlaxoSmithKline, manufacturer of nicotine replacement products, and research support from Pfizer, manufacturer of varenicline. The opinions expressed are those of the author.

Varenicline helps smokers with stably treated depression
Varenicline successfully promoted durable smoking cessation without exacerbating depression or anxiety in a randomized, phase IV, industry-funded study of patients with stably treated major depressive disorder.
Smoking cessation, defined as carbon monoxide–confirmed continuous abstinence, was higher at weeks 9-12 among 256 subjects in the double-blind study who were randomized to receive varenicline, compared with 269 who received placebo (35.9% vs. 15.6%; odds ratio, 3.35), Dr. Robert M. Anthenelli of the University of California, San Diego, and his colleagues reported in the Sept. 17 issue of Annals of Internal Medicine.
The differences between the groups were also significant for weeks 9-24 (20.3% vs. 10.4%; OR, 2.36) and for weeks 9-52 (25% vs. 12.3%; OR, 2.36), the investigators said (Ann. Intern. Med. 2013;159:390-400).*

No clinically relevant differences were seen between the groups in suicidal ideation or behavior as captured by the Columbia Suicide Severity Rating Scale. Furthermore, no overall worsening of depression or anxiety occurred in either group. In fact, trajectories of mood and anxiety rating trended slightly toward improvement in both groups, they said.
Participants in the multicenter study were adults aged 19-73 years who had no recent cardiovascular events, who smoked at least 10 cigarettes daily, and who had current or past stably treated unipolar major depressive disorder without psychotic features. They were recruited from 38 centers in 8 countries between March 2010 and June 2012. Randomization was stratified by antidepressant use at baseline (any vs. none) and by baseline depression score (Montgomery-Asberg Depression Rating Scale score 11 or less vs. greater than 11). Patients received either placebo or varenicline titrated to a dose of 1 mg twice daily for 12 weeks, with a 40-week nontreatment follow-up phase.
Treatment was relatively safe; although 72.3% and 66.9% of the treatment and placebo groups, respectively, experienced treatment-emergent adverse events, most were mild or moderate. The most frequent adverse events in the treatment vs. placebo groups were nausea (27.0% vs. 10.4%, respectively), headaches (16.8% vs. 11.2%), abnormal dreams (11.3% vs. 8.2%), irritability (10.9% vs. 8.2%), and insomnia (10.9% vs. 4.8%). Two patients in the treatment group died during the nontreatment phase, but neither death was considered to be related to treatment, the investigators said.
The findings suggest that varenicline, like other FDA-approved smoking cessation aids effective in non–psychiatrically ill smokers, are similarly effective in smokers with a history of depression – without increasing depressive symptoms; in the case of varenicline, as demonstrated in this study, this also applies to patients with depression who are using antidepressant medications.
"Varenicline is a partial agonist of brain alpha4beta 2 nicotinic acetylcholine receptors and is believed to alleviate nicotine withdrawal while simultaneously blocking its rewarding effects. Because depressed smokers are prone to more severe nicotine withdrawal than nonpsychiatric smokers, mitigating withdrawal symptoms may be important in this population," the investigators said. Depressed smokers who lapse into smoking while attempting to regulate mood may find cigarettes less reinforcing while taking varenicline, thus facilitating prolonged abstinence, they noted.
Although the findings may not extrapolate to untreated or actively depressed smokers and those with other psychiatric conditions, since only stably treated patients without psychotic features and other disorders frequently associated with major depressive disorder (such as bipolar disorder and current substance use disorders) were included, the findings nevertheless suggest an important role for varenicline in smokers with a history of stably treated depression.
"With 350 million individuals having the disease worldwide and because a large proportion of smokers that seeks treatment has a lifetime history of MDD, these results have the potential to reduce morbidity and mortality in many smokers," they concluded.
The researchers noted several study limitations, including their selection of "a population of smokers who were stably treated for or remitted from depression." Thus, they wrote, "our findings may not extrapolate to untreated or actively depressed smokers, whom many consider to be poor candidates for smoking cessation until their condition stabilizes." They also excluded individuals with "with psychotic features, bipolar disorder, current substance use disorders, and other conditions frequently associated with major depressive disorder; patients receiving medication for mania or psychosis were also excluded. Missing data resulting from attrition in both treatment groups may have affected the outcomes, the authors further noted.
This study was funded by Pfizer. Individual authors reported receiving support from the Department of Veterans Affairs; the National Institute on Alcohol Abuse and Alcoholism; the University of California, San Francisco; the Smoking Cessation Leadership Center; and/or the Colorado Department of Public Health and Environment. Detailed disclosures are available at www.acponline.org/authors/icmje/ConflictOfInterestForms.do?msNum=M12-0777.
*CORRECTION, 10/10/13: An earlier version of this article misstated the citation for the journal article referenced in the story.

|
| Dr. Vera DePalo |
Dr. Vera DePalo, FCCP, comments: Smoking is a habit that contributes to several disease states and many strategies are employed to help with smoking cessation. Practitioners continue to search for the intervention that can aid the patient in success. As with all pharmacologic therapies,the prescriber considers the unintended side effects.
This industry-funded trial may be helpful to the practitioner's understanding of the possible options in non-psychiatrically ill smokers with a history of depression.
|
|
| Dr. Vera DePalo |
Dr. Vera DePalo, FCCP, comments: Smoking is a habit that contributes to several disease states and many strategies are employed to help with smoking cessation. Practitioners continue to search for the intervention that can aid the patient in success. As with all pharmacologic therapies,the prescriber considers the unintended side effects.
This industry-funded trial may be helpful to the practitioner's understanding of the possible options in non-psychiatrically ill smokers with a history of depression.
|
|
| Dr. Vera DePalo |
Dr. Vera DePalo, FCCP, comments: Smoking is a habit that contributes to several disease states and many strategies are employed to help with smoking cessation. Practitioners continue to search for the intervention that can aid the patient in success. As with all pharmacologic therapies,the prescriber considers the unintended side effects.
This industry-funded trial may be helpful to the practitioner's understanding of the possible options in non-psychiatrically ill smokers with a history of depression.
Varenicline successfully promoted durable smoking cessation without exacerbating depression or anxiety in a randomized, phase IV, industry-funded study of patients with stably treated major depressive disorder.
Smoking cessation, defined as carbon monoxide–confirmed continuous abstinence, was higher at weeks 9-12 among 256 subjects in the double-blind study who were randomized to receive varenicline, compared with 269 who received placebo (35.9% vs. 15.6%; odds ratio, 3.35), Dr. Robert M. Anthenelli of the University of California, San Diego, and his colleagues reported in the Sept. 17 issue of Annals of Internal Medicine.
The differences between the groups were also significant for weeks 9-24 (20.3% vs. 10.4%; OR, 2.36) and for weeks 9-52 (25% vs. 12.3%; OR, 2.36), the investigators said (Ann. Intern. Med. 2013;159:390-400).*
No clinically relevant differences were seen between the groups in suicidal ideation or behavior as captured by the Columbia Suicide Severity Rating Scale. Furthermore, no overall worsening of depression or anxiety occurred in either group. In fact, trajectories of mood and anxiety rating trended slightly toward improvement in both groups, they said.
Participants in the multicenter study were adults aged 19-73 years who had no recent cardiovascular events, who smoked at least 10 cigarettes daily, and who had current or past stably treated unipolar major depressive disorder without psychotic features. They were recruited from 38 centers in 8 countries between March 2010 and June 2012. Randomization was stratified by antidepressant use at baseline (any vs. none) and by baseline depression score (Montgomery-Asberg Depression Rating Scale score 11 or less vs. greater than 11). Patients received either placebo or varenicline titrated to a dose of 1 mg twice daily for 12 weeks, with a 40-week nontreatment follow-up phase.
Treatment was relatively safe; although 72.3% and 66.9% of the treatment and placebo groups, respectively, experienced treatment-emergent adverse events, most were mild or moderate. The most frequent adverse events in the treatment vs. placebo groups were nausea (27.0% vs. 10.4%, respectively), headaches (16.8% vs. 11.2%), abnormal dreams (11.3% vs. 8.2%), irritability (10.9% vs. 8.2%), and insomnia (10.9% vs. 4.8%). Two patients in the treatment group died during the nontreatment phase, but neither death was considered to be related to treatment, the investigators said.
The findings suggest that varenicline, like other FDA-approved smoking cessation aids effective in non–psychiatrically ill smokers, are similarly effective in smokers with a history of depression – without increasing depressive symptoms; in the case of varenicline, as demonstrated in this study, this also applies to patients with depression who are using antidepressant medications.
"Varenicline is a partial agonist of brain alpha4beta 2 nicotinic acetylcholine receptors and is believed to alleviate nicotine withdrawal while simultaneously blocking its rewarding effects. Because depressed smokers are prone to more severe nicotine withdrawal than nonpsychiatric smokers, mitigating withdrawal symptoms may be important in this population," the investigators said. Depressed smokers who lapse into smoking while attempting to regulate mood may find cigarettes less reinforcing while taking varenicline, thus facilitating prolonged abstinence, they noted.
Although the findings may not extrapolate to untreated or actively depressed smokers and those with other psychiatric conditions, since only stably treated patients without psychotic features and other disorders frequently associated with major depressive disorder (such as bipolar disorder and current substance use disorders) were included, the findings nevertheless suggest an important role for varenicline in smokers with a history of stably treated depression.
"With 350 million individuals having the disease worldwide and because a large proportion of smokers that seeks treatment has a lifetime history of MDD, these results have the potential to reduce morbidity and mortality in many smokers," they concluded.
The researchers noted several study limitations, including their selection of "a population of smokers who were stably treated for or remitted from depression." Thus, they wrote, "our findings may not extrapolate to untreated or actively depressed smokers, whom many consider to be poor candidates for smoking cessation until their condition stabilizes." They also excluded individuals with "with psychotic features, bipolar disorder, current substance use disorders, and other conditions frequently associated with major depressive disorder; patients receiving medication for mania or psychosis were also excluded. Missing data resulting from attrition in both treatment groups may have affected the outcomes, the authors further noted.
This study was funded by Pfizer. Individual authors reported receiving support from the Department of Veterans Affairs; the National Institute on Alcohol Abuse and Alcoholism; the University of California, San Francisco; the Smoking Cessation Leadership Center; and/or the Colorado Department of Public Health and Environment. Detailed disclosures are available at www.acponline.org/authors/icmje/ConflictOfInterestForms.do?msNum=M12-0777.
*CORRECTION, 10/10/13: An earlier version of this article misstated the citation for the journal article referenced in the story.
Varenicline successfully promoted durable smoking cessation without exacerbating depression or anxiety in a randomized, phase IV, industry-funded study of patients with stably treated major depressive disorder.
Smoking cessation, defined as carbon monoxide–confirmed continuous abstinence, was higher at weeks 9-12 among 256 subjects in the double-blind study who were randomized to receive varenicline, compared with 269 who received placebo (35.9% vs. 15.6%; odds ratio, 3.35), Dr. Robert M. Anthenelli of the University of California, San Diego, and his colleagues reported in the Sept. 17 issue of Annals of Internal Medicine.
The differences between the groups were also significant for weeks 9-24 (20.3% vs. 10.4%; OR, 2.36) and for weeks 9-52 (25% vs. 12.3%; OR, 2.36), the investigators said (Ann. Intern. Med. 2013;159:390-400).*
No clinically relevant differences were seen between the groups in suicidal ideation or behavior as captured by the Columbia Suicide Severity Rating Scale. Furthermore, no overall worsening of depression or anxiety occurred in either group. In fact, trajectories of mood and anxiety rating trended slightly toward improvement in both groups, they said.
Participants in the multicenter study were adults aged 19-73 years who had no recent cardiovascular events, who smoked at least 10 cigarettes daily, and who had current or past stably treated unipolar major depressive disorder without psychotic features. They were recruited from 38 centers in 8 countries between March 2010 and June 2012. Randomization was stratified by antidepressant use at baseline (any vs. none) and by baseline depression score (Montgomery-Asberg Depression Rating Scale score 11 or less vs. greater than 11). Patients received either placebo or varenicline titrated to a dose of 1 mg twice daily for 12 weeks, with a 40-week nontreatment follow-up phase.
Treatment was relatively safe; although 72.3% and 66.9% of the treatment and placebo groups, respectively, experienced treatment-emergent adverse events, most were mild or moderate. The most frequent adverse events in the treatment vs. placebo groups were nausea (27.0% vs. 10.4%, respectively), headaches (16.8% vs. 11.2%), abnormal dreams (11.3% vs. 8.2%), irritability (10.9% vs. 8.2%), and insomnia (10.9% vs. 4.8%). Two patients in the treatment group died during the nontreatment phase, but neither death was considered to be related to treatment, the investigators said.
The findings suggest that varenicline, like other FDA-approved smoking cessation aids effective in non–psychiatrically ill smokers, are similarly effective in smokers with a history of depression – without increasing depressive symptoms; in the case of varenicline, as demonstrated in this study, this also applies to patients with depression who are using antidepressant medications.
"Varenicline is a partial agonist of brain alpha4beta 2 nicotinic acetylcholine receptors and is believed to alleviate nicotine withdrawal while simultaneously blocking its rewarding effects. Because depressed smokers are prone to more severe nicotine withdrawal than nonpsychiatric smokers, mitigating withdrawal symptoms may be important in this population," the investigators said. Depressed smokers who lapse into smoking while attempting to regulate mood may find cigarettes less reinforcing while taking varenicline, thus facilitating prolonged abstinence, they noted.
Although the findings may not extrapolate to untreated or actively depressed smokers and those with other psychiatric conditions, since only stably treated patients without psychotic features and other disorders frequently associated with major depressive disorder (such as bipolar disorder and current substance use disorders) were included, the findings nevertheless suggest an important role for varenicline in smokers with a history of stably treated depression.
"With 350 million individuals having the disease worldwide and because a large proportion of smokers that seeks treatment has a lifetime history of MDD, these results have the potential to reduce morbidity and mortality in many smokers," they concluded.
The researchers noted several study limitations, including their selection of "a population of smokers who were stably treated for or remitted from depression." Thus, they wrote, "our findings may not extrapolate to untreated or actively depressed smokers, whom many consider to be poor candidates for smoking cessation until their condition stabilizes." They also excluded individuals with "with psychotic features, bipolar disorder, current substance use disorders, and other conditions frequently associated with major depressive disorder; patients receiving medication for mania or psychosis were also excluded. Missing data resulting from attrition in both treatment groups may have affected the outcomes, the authors further noted.
This study was funded by Pfizer. Individual authors reported receiving support from the Department of Veterans Affairs; the National Institute on Alcohol Abuse and Alcoholism; the University of California, San Francisco; the Smoking Cessation Leadership Center; and/or the Colorado Department of Public Health and Environment. Detailed disclosures are available at www.acponline.org/authors/icmje/ConflictOfInterestForms.do?msNum=M12-0777.
*CORRECTION, 10/10/13: An earlier version of this article misstated the citation for the journal article referenced in the story.
FROM ANNALS OF INTERNAL MEDICINE
Major finding: Smoking cessation rates among patients with stably treated major depressive disorder were higher at 9-12 weeks in those treated with varenicline than in those treated with placebo (35.9% vs. 15.6%).
Data source: A randomized, double-blind, placebo-controlled phase IV study.
Disclosures: This study was funded by Pfizer. Individual authors reported receiving support from the Department of Veterans Affairs; the National Institute on Alcohol Abuse and Alcoholism; the University of California, San Francisco; the Smoking Cessation Leadership Center; and/or the Colorado Department of Public Health and Environment.
Dexamethasone improves outcomes for infants with bronchiolitis, atopy history
A 5-day course of dexamethasone significantly shortened hospital stays for infants with bronchiolitis who had eczema or close relatives with asthma.
The randomized, placebo-controlled study suggests that a family history of atopy could identify a subset of babies who would benefit from the addition of a corticosteroid to the usual salbutamol therapy for acute bronchiolitis, according to Dr. Khalid Alansari and colleagues. The report was published in the Sept. 16 issue of Pediatrics.
The researchers examined 7-day outcomes in 200 infants with acute bronchiolitis who were at a high risk of asthma, as determined by having at least one first-degree relative with either asthma or eczema. All of the children (mean age 3.5 months) were admitted to a pediatric hospital for treatment, wrote Dr. Alansari of Weill Cornell Medical College, Doha, Qatar, and coauthors. Infants who received dexamethasone were discharged 8 hours earlier than were those receiving standard treatment. The mean duration of symptoms was 4.5 days (Pediatrics 2013 Sept. 13 [doi: 10.1542/peds.2012-3746]).
The study’s primary outcome was time until discharge. Secondary outcomes included the number of patients who needed epinephrine treatment, readmission for a shorter stay in an infirmary site, and revisiting the emergency department or another clinic for the same illness. A study nurse made daily calls to assess the patients after discharge.
Infants in the dexamethasone group were discharged at a mean of 18.6 hours – significantly sooner than those in the control group (27 hours). Epinephrine was necessary for 19 infants in the dexamethasone group and 31 in the placebo group – again a significant difference.
Similar numbers in each group needed readmission and additional outpatient visits in the week after discharge. During the follow-up week, 22% of the dexamethasone group needed infirmary care and the mean stay was 17 hours, compared with 21% of the placebo group with a mean stay of 18 hours.
Nineteen in the dexamethasone group and 11 in the placebo group made a clinic visit (18.6% vs. 11%); this difference was not significant.
The chest radiograph was normal in about 37% of infants studied. About half showed lesser infiltrates; 15% had a lobar collapse or consolidation.
More than 70% had a full sibling with asthma. About 20% had a parent with the disease; in 5%, both parents had it. About 20% of patients had both eczema and first-degree relative with asthma.
All of the infants received 2.5 mg salbutamol nebulization at baseline and at 30, 60, and 120 minutes, and then every 2 hours until discharge. Nebulized epinephrine (0.5 mL/kg with a maximum dose of 5 mL) was available if needed. In addition, they were randomized to either placebo or to a 5-day course of dexamethasone 1 mg/mL, at a rate of 1 mL/kg on day 1, reduced to 0.6 mL/kg for days 2-5.
The study was sponsored by Hamad Medical Corporation. The authors reported no financial conflicts.
A 5-day course of dexamethasone significantly shortened hospital stays for infants with bronchiolitis who had eczema or close relatives with asthma.
The randomized, placebo-controlled study suggests that a family history of atopy could identify a subset of babies who would benefit from the addition of a corticosteroid to the usual salbutamol therapy for acute bronchiolitis, according to Dr. Khalid Alansari and colleagues. The report was published in the Sept. 16 issue of Pediatrics.
The researchers examined 7-day outcomes in 200 infants with acute bronchiolitis who were at a high risk of asthma, as determined by having at least one first-degree relative with either asthma or eczema. All of the children (mean age 3.5 months) were admitted to a pediatric hospital for treatment, wrote Dr. Alansari of Weill Cornell Medical College, Doha, Qatar, and coauthors. Infants who received dexamethasone were discharged 8 hours earlier than were those receiving standard treatment. The mean duration of symptoms was 4.5 days (Pediatrics 2013 Sept. 13 [doi: 10.1542/peds.2012-3746]).
The study’s primary outcome was time until discharge. Secondary outcomes included the number of patients who needed epinephrine treatment, readmission for a shorter stay in an infirmary site, and revisiting the emergency department or another clinic for the same illness. A study nurse made daily calls to assess the patients after discharge.
Infants in the dexamethasone group were discharged at a mean of 18.6 hours – significantly sooner than those in the control group (27 hours). Epinephrine was necessary for 19 infants in the dexamethasone group and 31 in the placebo group – again a significant difference.
Similar numbers in each group needed readmission and additional outpatient visits in the week after discharge. During the follow-up week, 22% of the dexamethasone group needed infirmary care and the mean stay was 17 hours, compared with 21% of the placebo group with a mean stay of 18 hours.
Nineteen in the dexamethasone group and 11 in the placebo group made a clinic visit (18.6% vs. 11%); this difference was not significant.
The chest radiograph was normal in about 37% of infants studied. About half showed lesser infiltrates; 15% had a lobar collapse or consolidation.
More than 70% had a full sibling with asthma. About 20% had a parent with the disease; in 5%, both parents had it. About 20% of patients had both eczema and first-degree relative with asthma.
All of the infants received 2.5 mg salbutamol nebulization at baseline and at 30, 60, and 120 minutes, and then every 2 hours until discharge. Nebulized epinephrine (0.5 mL/kg with a maximum dose of 5 mL) was available if needed. In addition, they were randomized to either placebo or to a 5-day course of dexamethasone 1 mg/mL, at a rate of 1 mL/kg on day 1, reduced to 0.6 mL/kg for days 2-5.
The study was sponsored by Hamad Medical Corporation. The authors reported no financial conflicts.
A 5-day course of dexamethasone significantly shortened hospital stays for infants with bronchiolitis who had eczema or close relatives with asthma.
The randomized, placebo-controlled study suggests that a family history of atopy could identify a subset of babies who would benefit from the addition of a corticosteroid to the usual salbutamol therapy for acute bronchiolitis, according to Dr. Khalid Alansari and colleagues. The report was published in the Sept. 16 issue of Pediatrics.
The researchers examined 7-day outcomes in 200 infants with acute bronchiolitis who were at a high risk of asthma, as determined by having at least one first-degree relative with either asthma or eczema. All of the children (mean age 3.5 months) were admitted to a pediatric hospital for treatment, wrote Dr. Alansari of Weill Cornell Medical College, Doha, Qatar, and coauthors. Infants who received dexamethasone were discharged 8 hours earlier than were those receiving standard treatment. The mean duration of symptoms was 4.5 days (Pediatrics 2013 Sept. 13 [doi: 10.1542/peds.2012-3746]).
The study’s primary outcome was time until discharge. Secondary outcomes included the number of patients who needed epinephrine treatment, readmission for a shorter stay in an infirmary site, and revisiting the emergency department or another clinic for the same illness. A study nurse made daily calls to assess the patients after discharge.
Infants in the dexamethasone group were discharged at a mean of 18.6 hours – significantly sooner than those in the control group (27 hours). Epinephrine was necessary for 19 infants in the dexamethasone group and 31 in the placebo group – again a significant difference.
Similar numbers in each group needed readmission and additional outpatient visits in the week after discharge. During the follow-up week, 22% of the dexamethasone group needed infirmary care and the mean stay was 17 hours, compared with 21% of the placebo group with a mean stay of 18 hours.
Nineteen in the dexamethasone group and 11 in the placebo group made a clinic visit (18.6% vs. 11%); this difference was not significant.
The chest radiograph was normal in about 37% of infants studied. About half showed lesser infiltrates; 15% had a lobar collapse or consolidation.
More than 70% had a full sibling with asthma. About 20% had a parent with the disease; in 5%, both parents had it. About 20% of patients had both eczema and first-degree relative with asthma.
All of the infants received 2.5 mg salbutamol nebulization at baseline and at 30, 60, and 120 minutes, and then every 2 hours until discharge. Nebulized epinephrine (0.5 mL/kg with a maximum dose of 5 mL) was available if needed. In addition, they were randomized to either placebo or to a 5-day course of dexamethasone 1 mg/mL, at a rate of 1 mL/kg on day 1, reduced to 0.6 mL/kg for days 2-5.
The study was sponsored by Hamad Medical Corporation. The authors reported no financial conflicts.
Dexamethasone improves outcomes for infants with bronchiolitis, atopy history
A 5-day course of dexamethasone significantly shortened hospital stays for infants with bronchiolitis who had eczema or close relatives with asthma.
The randomized, placebo-controlled study suggests that a family history of atopy could identify a subset of babies who would benefit from the addition of a corticosteroid to the usual salbutamol therapy for acute bronchiolitis, according to Dr. Khalid Alansari and colleagues. The report was published in the Sept. 16 issue of Pediatrics.
The researchers examined 7-day outcomes in 200 infants with acute bronchiolitis who were at a high risk of asthma, as determined by having at least one first-degree relative with either asthma or eczema. All of the children (mean age 3.5 months) were admitted to a pediatric hospital for treatment, wrote Dr. Alansari of Weill Cornell Medical College, Doha, Qatar, and coauthors. Infants who received dexamethasone were discharged 8 hours earlier than were those receiving standard treatment. The mean duration of symptoms was 4.5 days (Pediatrics 2013 Sept. 13 [doi: 10.1542/peds.2012-3746]).
The study’s primary outcome was time until discharge. Secondary outcomes included the number of patients who needed epinephrine treatment, readmission for a shorter stay in an infirmary site, and revisiting the emergency department or another clinic for the same illness. A study nurse made daily calls to assess the patients after discharge.
Infants in the dexamethasone group were discharged at a mean of 18.6 hours – significantly sooner than those in the control group (27 hours). Epinephrine was necessary for 19 infants in the dexamethasone group and 31 in the placebo group – again a significant difference.
Similar numbers in each group needed readmission and additional outpatient visits in the week after discharge. During the follow-up week, 22% of the dexamethasone group needed infirmary care and the mean stay was 17 hours, compared with 21% of the placebo group with a mean stay of 18 hours.
Nineteen in the dexamethasone group and 11 in the placebo group made a clinic visit (18.6% vs. 11%); this difference was not significant.
The chest radiograph was normal in about 37% of infants studied. About half showed lesser infiltrates; 15% had a lobar collapse or consolidation.
More than 70% had a full sibling with asthma. About 20% had a parent with the disease; in 5%, both parents had it. About 20% of patients had both eczema and first-degree relative with asthma.
All of the infants received 2.5 mg salbutamol nebulization at baseline and at 30, 60, and 120 minutes, and then every 2 hours until discharge. Nebulized epinephrine (0.5 mL/kg with a maximum dose of 5 mL) was available if needed. In addition, they were randomized to either placebo or to a 5-day course of dexamethasone 1 mg/mL, at a rate of 1 mL/kg on day 1, reduced to 0.6 mL/kg for days 2-5.
The study was sponsored by Hamad Medical Corporation. The authors reported no financial conflicts.
A 5-day course of dexamethasone significantly shortened hospital stays for infants with bronchiolitis who had eczema or close relatives with asthma.
The randomized, placebo-controlled study suggests that a family history of atopy could identify a subset of babies who would benefit from the addition of a corticosteroid to the usual salbutamol therapy for acute bronchiolitis, according to Dr. Khalid Alansari and colleagues. The report was published in the Sept. 16 issue of Pediatrics.
The researchers examined 7-day outcomes in 200 infants with acute bronchiolitis who were at a high risk of asthma, as determined by having at least one first-degree relative with either asthma or eczema. All of the children (mean age 3.5 months) were admitted to a pediatric hospital for treatment, wrote Dr. Alansari of Weill Cornell Medical College, Doha, Qatar, and coauthors. Infants who received dexamethasone were discharged 8 hours earlier than were those receiving standard treatment. The mean duration of symptoms was 4.5 days (Pediatrics 2013 Sept. 13 [doi: 10.1542/peds.2012-3746]).
The study’s primary outcome was time until discharge. Secondary outcomes included the number of patients who needed epinephrine treatment, readmission for a shorter stay in an infirmary site, and revisiting the emergency department or another clinic for the same illness. A study nurse made daily calls to assess the patients after discharge.
Infants in the dexamethasone group were discharged at a mean of 18.6 hours – significantly sooner than those in the control group (27 hours). Epinephrine was necessary for 19 infants in the dexamethasone group and 31 in the placebo group – again a significant difference.
Similar numbers in each group needed readmission and additional outpatient visits in the week after discharge. During the follow-up week, 22% of the dexamethasone group needed infirmary care and the mean stay was 17 hours, compared with 21% of the placebo group with a mean stay of 18 hours.
Nineteen in the dexamethasone group and 11 in the placebo group made a clinic visit (18.6% vs. 11%); this difference was not significant.
The chest radiograph was normal in about 37% of infants studied. About half showed lesser infiltrates; 15% had a lobar collapse or consolidation.
More than 70% had a full sibling with asthma. About 20% had a parent with the disease; in 5%, both parents had it. About 20% of patients had both eczema and first-degree relative with asthma.
All of the infants received 2.5 mg salbutamol nebulization at baseline and at 30, 60, and 120 minutes, and then every 2 hours until discharge. Nebulized epinephrine (0.5 mL/kg with a maximum dose of 5 mL) was available if needed. In addition, they were randomized to either placebo or to a 5-day course of dexamethasone 1 mg/mL, at a rate of 1 mL/kg on day 1, reduced to 0.6 mL/kg for days 2-5.
The study was sponsored by Hamad Medical Corporation. The authors reported no financial conflicts.
A 5-day course of dexamethasone significantly shortened hospital stays for infants with bronchiolitis who had eczema or close relatives with asthma.
The randomized, placebo-controlled study suggests that a family history of atopy could identify a subset of babies who would benefit from the addition of a corticosteroid to the usual salbutamol therapy for acute bronchiolitis, according to Dr. Khalid Alansari and colleagues. The report was published in the Sept. 16 issue of Pediatrics.
The researchers examined 7-day outcomes in 200 infants with acute bronchiolitis who were at a high risk of asthma, as determined by having at least one first-degree relative with either asthma or eczema. All of the children (mean age 3.5 months) were admitted to a pediatric hospital for treatment, wrote Dr. Alansari of Weill Cornell Medical College, Doha, Qatar, and coauthors. Infants who received dexamethasone were discharged 8 hours earlier than were those receiving standard treatment. The mean duration of symptoms was 4.5 days (Pediatrics 2013 Sept. 13 [doi: 10.1542/peds.2012-3746]).
The study’s primary outcome was time until discharge. Secondary outcomes included the number of patients who needed epinephrine treatment, readmission for a shorter stay in an infirmary site, and revisiting the emergency department or another clinic for the same illness. A study nurse made daily calls to assess the patients after discharge.
Infants in the dexamethasone group were discharged at a mean of 18.6 hours – significantly sooner than those in the control group (27 hours). Epinephrine was necessary for 19 infants in the dexamethasone group and 31 in the placebo group – again a significant difference.
Similar numbers in each group needed readmission and additional outpatient visits in the week after discharge. During the follow-up week, 22% of the dexamethasone group needed infirmary care and the mean stay was 17 hours, compared with 21% of the placebo group with a mean stay of 18 hours.
Nineteen in the dexamethasone group and 11 in the placebo group made a clinic visit (18.6% vs. 11%); this difference was not significant.
The chest radiograph was normal in about 37% of infants studied. About half showed lesser infiltrates; 15% had a lobar collapse or consolidation.
More than 70% had a full sibling with asthma. About 20% had a parent with the disease; in 5%, both parents had it. About 20% of patients had both eczema and first-degree relative with asthma.
All of the infants received 2.5 mg salbutamol nebulization at baseline and at 30, 60, and 120 minutes, and then every 2 hours until discharge. Nebulized epinephrine (0.5 mL/kg with a maximum dose of 5 mL) was available if needed. In addition, they were randomized to either placebo or to a 5-day course of dexamethasone 1 mg/mL, at a rate of 1 mL/kg on day 1, reduced to 0.6 mL/kg for days 2-5.
The study was sponsored by Hamad Medical Corporation. The authors reported no financial conflicts.
FROM PEDIATRICS
Major finding: Infants with bronchiolitis who had a family history of atopy who received dexamethasone added to salbutamol were discharged significantly sooner than were those receiving usual therapy (18.6 hours vs. 27 hours).
Data source: The randomized, placebo controlled study comprised 200 infants with acute bronchiolitis.
Disclosures: The study was sponsored by Hamad Medical Corporation. The authors reported no financial conflicts.
Hypertonic saline a washout in bronchiolitis
NEW ORLEANS – Hypertonic saline alone does not affect length of stay among infants hospitalized for bronchiolitis, according to preliminary results of an ongoing randomized, prospective U.S. trial.
Among 127 evaluable patients, the median length of stay was 2.2 days with hypertonic saline and 2.0 days with normal saline (P = .65).
Infants in one still-blinded group were significantly younger (3.3 vs. 4.4 months), but hospital stay did not differ between groups, even when adjusted for age.
"Preliminary results suggest little utility for the routine use of hypertonic saline in treating infants hospitalized with bronchiolitis," Dr. Alyssa Silver said at Pediatric Hospital Medicine 2013.
However, she added, the results do show that 3% hypertonic saline without adjunctive bronchodilators is safe for infants, including those with a history of prior wheeze, even in a population with a high endemic prevalence of asthma.

A theoretical risk of bronchospasm has been identified in patients with cystic fibrosis at much higher concentrations and quantities.
This is the first prospective, randomized, double-blind, controlled clinical trial of nebulized hypertonic saline conducted in the United States.
Several studies have been conducted outside the United States, but adjuvant bronchodilators were used in all but one, patients with a history of wheeze were excluded, and they had long lengths of stay that make the data difficult to generalize to the United States, said Dr. Silver, a pediatric hospitalist with the Children’s Hospital at Montefiore in New York.
Dr. Shawn Ralston, cochair of the American Academy of Pediatrics subcommittee on diagnosis and management of bronchiolitis, which is revising the 2006 AAP clinical practice guidelines for bronchiolitis,said in an interview that a clearer picture of the utility of hypertonic saline is beginning to emerge. All of the studies where hypertonic saline is used in the ED or short-stay units show no utility, and the only inpatient studies that show benefit seem to be in settings where there is longer length of stay. Further, Montefiore has a very short length of stay, which is typical of U.S. hospitals.
"I think what we’re learning about hypertonic saline as the studies come in is that any treatment effect, if there does prove to be one, will be seen over several days," said Dr. Ralston, chief of the pediatrics section, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
"From a guidelines standpoint, it is very important for us to keep up with the emerging data, and we are unlikely to be able to recommend the use of hypertonic saline given what has been published or presented within the last year. That said, I would actually welcome studies of outpatient use of hypertonic saline, as I think that is where the real utility may lie, based on my review of the literature in preparation for the guideline."
The current study included 156 infants, less than 12 months of age, admitted with a diagnosis of bronchiolitis from November 2011 through January 2013 and randomized by the pharmacy to 4 mL of hypertonic saline or 4 mL of normal saline every 4 hours until discharge.
The readmission rate was identical at 6.3% in both study groups (P = 1.0), Dr. Silver reported. Also similar were clinically important adverse events, including readmission (four events in each group) and worsening respiratory status (six events in each group; both P = 1.0).
A subgroup analysis showed no difference in length of stay among patients treated with hypertonic saline vs. normal saline who were positive for respiratory syncytial virus (2.5 vs. 2.0 days; P = .35), had a history of previous wheeze (1.8 vs. 2.0 days; P = .58), or had a history of prematurity (3.5 vs. 2.5 days; P = .11).
The study was limited by the single-center design and the inability to randomize all patients within 12 hours of admission. Despite the lack of funding, however, the study captured 80% of bronchiolitis admissions among infants during the study period, Dr. Silver noted.
"These findings cast significant doubt on the utility of nebulized 3% hypertonic saline for the routine treatment of infants hospitalized with bronchiolitis," she said in an interview. "Unfortunately, the mainstay of routine inpatient treatment remains supportive care."
Future directions include enrolling patients directly from the emergency department with a minimum severity score for study entry, use of higher concentrations of sodium chloride, and possibly an outpatient study.
Dr. Silver reported having no relevant financial disclosures.
NEW ORLEANS – Hypertonic saline alone does not affect length of stay among infants hospitalized for bronchiolitis, according to preliminary results of an ongoing randomized, prospective U.S. trial.
Among 127 evaluable patients, the median length of stay was 2.2 days with hypertonic saline and 2.0 days with normal saline (P = .65).
Infants in one still-blinded group were significantly younger (3.3 vs. 4.4 months), but hospital stay did not differ between groups, even when adjusted for age.
"Preliminary results suggest little utility for the routine use of hypertonic saline in treating infants hospitalized with bronchiolitis," Dr. Alyssa Silver said at Pediatric Hospital Medicine 2013.
However, she added, the results do show that 3% hypertonic saline without adjunctive bronchodilators is safe for infants, including those with a history of prior wheeze, even in a population with a high endemic prevalence of asthma.
A theoretical risk of bronchospasm has been identified in patients with cystic fibrosis at much higher concentrations and quantities.
This is the first prospective, randomized, double-blind, controlled clinical trial of nebulized hypertonic saline conducted in the United States.
Several studies have been conducted outside the United States, but adjuvant bronchodilators were used in all but one, patients with a history of wheeze were excluded, and they had long lengths of stay that make the data difficult to generalize to the United States, said Dr. Silver, a pediatric hospitalist with the Children’s Hospital at Montefiore in New York.
Dr. Shawn Ralston, cochair of the American Academy of Pediatrics subcommittee on diagnosis and management of bronchiolitis, which is revising the 2006 AAP clinical practice guidelines for bronchiolitis,said in an interview that a clearer picture of the utility of hypertonic saline is beginning to emerge. All of the studies where hypertonic saline is used in the ED or short-stay units show no utility, and the only inpatient studies that show benefit seem to be in settings where there is longer length of stay. Further, Montefiore has a very short length of stay, which is typical of U.S. hospitals.
"I think what we’re learning about hypertonic saline as the studies come in is that any treatment effect, if there does prove to be one, will be seen over several days," said Dr. Ralston, chief of the pediatrics section, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
"From a guidelines standpoint, it is very important for us to keep up with the emerging data, and we are unlikely to be able to recommend the use of hypertonic saline given what has been published or presented within the last year. That said, I would actually welcome studies of outpatient use of hypertonic saline, as I think that is where the real utility may lie, based on my review of the literature in preparation for the guideline."
The current study included 156 infants, less than 12 months of age, admitted with a diagnosis of bronchiolitis from November 2011 through January 2013 and randomized by the pharmacy to 4 mL of hypertonic saline or 4 mL of normal saline every 4 hours until discharge.
The readmission rate was identical at 6.3% in both study groups (P = 1.0), Dr. Silver reported. Also similar were clinically important adverse events, including readmission (four events in each group) and worsening respiratory status (six events in each group; both P = 1.0).
A subgroup analysis showed no difference in length of stay among patients treated with hypertonic saline vs. normal saline who were positive for respiratory syncytial virus (2.5 vs. 2.0 days; P = .35), had a history of previous wheeze (1.8 vs. 2.0 days; P = .58), or had a history of prematurity (3.5 vs. 2.5 days; P = .11).
The study was limited by the single-center design and the inability to randomize all patients within 12 hours of admission. Despite the lack of funding, however, the study captured 80% of bronchiolitis admissions among infants during the study period, Dr. Silver noted.
"These findings cast significant doubt on the utility of nebulized 3% hypertonic saline for the routine treatment of infants hospitalized with bronchiolitis," she said in an interview. "Unfortunately, the mainstay of routine inpatient treatment remains supportive care."
Future directions include enrolling patients directly from the emergency department with a minimum severity score for study entry, use of higher concentrations of sodium chloride, and possibly an outpatient study.
Dr. Silver reported having no relevant financial disclosures.
NEW ORLEANS – Hypertonic saline alone does not affect length of stay among infants hospitalized for bronchiolitis, according to preliminary results of an ongoing randomized, prospective U.S. trial.
Among 127 evaluable patients, the median length of stay was 2.2 days with hypertonic saline and 2.0 days with normal saline (P = .65).
Infants in one still-blinded group were significantly younger (3.3 vs. 4.4 months), but hospital stay did not differ between groups, even when adjusted for age.
"Preliminary results suggest little utility for the routine use of hypertonic saline in treating infants hospitalized with bronchiolitis," Dr. Alyssa Silver said at Pediatric Hospital Medicine 2013.
However, she added, the results do show that 3% hypertonic saline without adjunctive bronchodilators is safe for infants, including those with a history of prior wheeze, even in a population with a high endemic prevalence of asthma.
A theoretical risk of bronchospasm has been identified in patients with cystic fibrosis at much higher concentrations and quantities.
This is the first prospective, randomized, double-blind, controlled clinical trial of nebulized hypertonic saline conducted in the United States.
Several studies have been conducted outside the United States, but adjuvant bronchodilators were used in all but one, patients with a history of wheeze were excluded, and they had long lengths of stay that make the data difficult to generalize to the United States, said Dr. Silver, a pediatric hospitalist with the Children’s Hospital at Montefiore in New York.
Dr. Shawn Ralston, cochair of the American Academy of Pediatrics subcommittee on diagnosis and management of bronchiolitis, which is revising the 2006 AAP clinical practice guidelines for bronchiolitis,said in an interview that a clearer picture of the utility of hypertonic saline is beginning to emerge. All of the studies where hypertonic saline is used in the ED or short-stay units show no utility, and the only inpatient studies that show benefit seem to be in settings where there is longer length of stay. Further, Montefiore has a very short length of stay, which is typical of U.S. hospitals.
"I think what we’re learning about hypertonic saline as the studies come in is that any treatment effect, if there does prove to be one, will be seen over several days," said Dr. Ralston, chief of the pediatrics section, Dartmouth-Hitchcock Medical Center, Lebanon, N.H.
"From a guidelines standpoint, it is very important for us to keep up with the emerging data, and we are unlikely to be able to recommend the use of hypertonic saline given what has been published or presented within the last year. That said, I would actually welcome studies of outpatient use of hypertonic saline, as I think that is where the real utility may lie, based on my review of the literature in preparation for the guideline."
The current study included 156 infants, less than 12 months of age, admitted with a diagnosis of bronchiolitis from November 2011 through January 2013 and randomized by the pharmacy to 4 mL of hypertonic saline or 4 mL of normal saline every 4 hours until discharge.
The readmission rate was identical at 6.3% in both study groups (P = 1.0), Dr. Silver reported. Also similar were clinically important adverse events, including readmission (four events in each group) and worsening respiratory status (six events in each group; both P = 1.0).
A subgroup analysis showed no difference in length of stay among patients treated with hypertonic saline vs. normal saline who were positive for respiratory syncytial virus (2.5 vs. 2.0 days; P = .35), had a history of previous wheeze (1.8 vs. 2.0 days; P = .58), or had a history of prematurity (3.5 vs. 2.5 days; P = .11).
The study was limited by the single-center design and the inability to randomize all patients within 12 hours of admission. Despite the lack of funding, however, the study captured 80% of bronchiolitis admissions among infants during the study period, Dr. Silver noted.
"These findings cast significant doubt on the utility of nebulized 3% hypertonic saline for the routine treatment of infants hospitalized with bronchiolitis," she said in an interview. "Unfortunately, the mainstay of routine inpatient treatment remains supportive care."
Future directions include enrolling patients directly from the emergency department with a minimum severity score for study entry, use of higher concentrations of sodium chloride, and possibly an outpatient study.
Dr. Silver reported having no relevant financial disclosures.
AT PEDIATRIC HOSPITAL MEDICINE 2013
Major finding: Median length of stay was 2.2 days with hypertonic saline and 2.0 days with normal saline (P = .65).
Data source: A prospective, double-blind randomized controlled trial in 127 infants with bronchiolitis.
Disclosures: Dr. Silver reported having no relevant financial disclosures.

Sleep aid use reported by 16% of those with sleep disorders
Use of prescription sleep aids was more than five times higher in those who reported a physician’s diagnosis of a sleep disorder, compared with those who did not have such a diagnosis, the National Center for Health Statistics reported.
In 2005-2010, sleep-aid use in the past 30 days was reported by 16.3% of adults who had a diagnosed sleep disorder, compared with 3.1% of those who did not have a sleep disorder, according to data from the National Health and Nutrition Examination Survey.
Past 30-day use of sleep aids was 12.7% among those who had told a physician that they had trouble sleeping and 1.1% for those who did not report any trouble sleeping, according to the report.
The sleep aids covered in the report included all hypnotic drugs and four antidepressant or sedative medications commonly prescribed for insomnia or depression: amitriptyline, doxepin, mirtazapine, and trazodone.
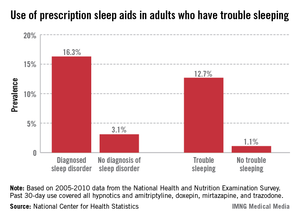
Use of prescription sleep aids was more than five times higher in those who reported a physician’s diagnosis of a sleep disorder, compared with those who did not have such a diagnosis, the National Center for Health Statistics reported.
In 2005-2010, sleep-aid use in the past 30 days was reported by 16.3% of adults who had a diagnosed sleep disorder, compared with 3.1% of those who did not have a sleep disorder, according to data from the National Health and Nutrition Examination Survey.
Past 30-day use of sleep aids was 12.7% among those who had told a physician that they had trouble sleeping and 1.1% for those who did not report any trouble sleeping, according to the report.
The sleep aids covered in the report included all hypnotic drugs and four antidepressant or sedative medications commonly prescribed for insomnia or depression: amitriptyline, doxepin, mirtazapine, and trazodone.
Use of prescription sleep aids was more than five times higher in those who reported a physician’s diagnosis of a sleep disorder, compared with those who did not have such a diagnosis, the National Center for Health Statistics reported.
In 2005-2010, sleep-aid use in the past 30 days was reported by 16.3% of adults who had a diagnosed sleep disorder, compared with 3.1% of those who did not have a sleep disorder, according to data from the National Health and Nutrition Examination Survey.
Past 30-day use of sleep aids was 12.7% among those who had told a physician that they had trouble sleeping and 1.1% for those who did not report any trouble sleeping, according to the report.
The sleep aids covered in the report included all hypnotic drugs and four antidepressant or sedative medications commonly prescribed for insomnia or depression: amitriptyline, doxepin, mirtazapine, and trazodone.
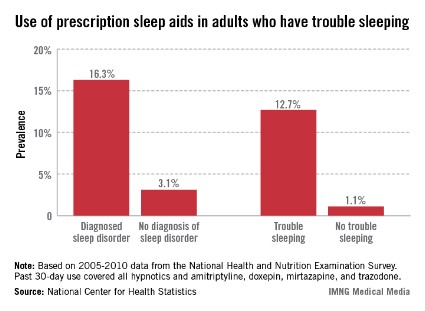
FDA panel backs bronchodilator combo for COPD, with precautions
SILVER SPRING, MD. – A fixed-dose combination of umeclidinium bromide, a new long-acting antimuscarinic agent (LAMA), and vilanterol, a relatively new long-acting beta agonist (LABA), in an inhaled powder formulation, should be approved for treatment of chronic obstructive pulmonary disease, with a recommendation for a postmarketing safety study, according to the majority of a Food and Drug Administration advisory panel.
At a meeting on Sept. 10, the FDA’s Pulmonary-Allergy Drugs Advisory Committee voted 11-2 that the safety and efficacy data on the combined treatment, administered once a day, supported approval for the proposed indication: the long-term once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD, including chronic bronchitis and/or emphysema. The daily dose is 62.5 mcg of umeclidinium and 25 mcg of vilanterol (UMEC/VI 62.5/25), provided in one inhalation.
In another vote, the panel unanimously agreed that treatment with UMEC/VI 62.5/25 provided clinically meaningful benefits for this indication, based on the clinical trial results provided by GlaxoSmithKline. However, all the panelists were concerned about potential safety issues in patients with severe heart disease. There was also concern over the generalizability of the use of the combination because of a cardiovascular safety signal in the studies and the types of patients excluded in the research.
Those voting in favor of approval recommended a postmarketing study to evaluate the safety signal further, and a statement in the precautions and warnings section of the label about possible risks. The two panelists voting against approval said that the study should be done before approval.
If approved, this would be the first LAMA/LABA combination product to become available in the United States. Umeclidinium is not yet available in any product, although the FDA is currently reviewing the 62.5-mcg dose as monotherapy. Vilanterol is available only as the LABA component in Breo Ellipta, the combination of vilanterol with the inhaled corticosteroid fluticasone, which was approved in May 2013 for COPD. The same device is used to deliver UMEC/VI 62.5/25, which, if approved, will be marketed as Anoro Ellipta, by GSK and Theravance.
Research details
The four main safety and efficacy studies compared two doses of umeclidinium (125 mcg and 62.5 mcg) combined with 25 mcg of vilanterol to the separate components alone and to placebo (in two studies) or to the approved long-acting antimuscarinic agent tiotropium (two studies) in about 4,700 patients – most of whom were male and white – with moderate to severe COPD. Their mean age was 63 years, almost half were still smoking, and almost 30% had had at least one exacerbation during the previous year that required treatment with corticosteroids and/or antibiotics; about 10% said they had been hospitalized for an exacerbation during the previous year (25% were enrolled in the United States).
In the two placebo-controlled studies, the combination and the two separate components (UMEC and vilanterol) were associated with statistically significant improvements in mean trough forced expiratory volume in 1 second (FEV1) from baseline at 24 weeks (the primary endpoint), compared with placebo. Improvements were also statistically significant for the two combinations over the individual components. In the active controlled study, the two combinations showed improvements in trough FEV1 that were superior to vilanterol alone and to tiotropium alone. The results with the higher dose of umeclidinium and vilanterol were similar to the 62.5-mcg dose, the proposed dose for approval.
The mortality rate and nonfatal serious adverse events were even in the treatment arms, in the four primary efficacy studies, and in long-term safety studies, and adverse events were consistent with those associated with the LAMAs and LABAs, according to GSK. The total number of cardiovascular-related events was fairly low in the safety database that included the four main studies and a long-term safety study. But there were numerical imbalances in ischemia-related events in the primary efficacy studies that were not seen in the long-term safety study, an issue raised by the FDA, although the overall number of events was low, according to the agency.
Panelists raised several safety issues, including the generalizability of the cardiac safety data to patients with more severe cardiac disease, because of exclusion criteria that may have eliminated these patients.
The panel chair, Dr. David Jacoby, professor of medicine in the division of pulmonary and critical care medicine at Oregon Health and Science University, Portland, supported approval but recommended that safety should be studied further in patients with COPD who have more severe cardiac disease.
Also voting for approval, Dr. Nizar Jarjour, professor and head of the section of allergy, pulmonary, and critical care at the University of Wisconsin, Madison, said the data indicated that the product was reasonably safe. But if it were to be used outside the parameters studied in the trials, such as for patients with recent exacerbations of COPD, or severe heart disease, he said he have would "no real confidence" that it would have the same safety profile as seen in the trials.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but that was unnecessary at this meeting.
SILVER SPRING, MD. – A fixed-dose combination of umeclidinium bromide, a new long-acting antimuscarinic agent (LAMA), and vilanterol, a relatively new long-acting beta agonist (LABA), in an inhaled powder formulation, should be approved for treatment of chronic obstructive pulmonary disease, with a recommendation for a postmarketing safety study, according to the majority of a Food and Drug Administration advisory panel.
At a meeting on Sept. 10, the FDA’s Pulmonary-Allergy Drugs Advisory Committee voted 11-2 that the safety and efficacy data on the combined treatment, administered once a day, supported approval for the proposed indication: the long-term once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD, including chronic bronchitis and/or emphysema. The daily dose is 62.5 mcg of umeclidinium and 25 mcg of vilanterol (UMEC/VI 62.5/25), provided in one inhalation.
In another vote, the panel unanimously agreed that treatment with UMEC/VI 62.5/25 provided clinically meaningful benefits for this indication, based on the clinical trial results provided by GlaxoSmithKline. However, all the panelists were concerned about potential safety issues in patients with severe heart disease. There was also concern over the generalizability of the use of the combination because of a cardiovascular safety signal in the studies and the types of patients excluded in the research.
Those voting in favor of approval recommended a postmarketing study to evaluate the safety signal further, and a statement in the precautions and warnings section of the label about possible risks. The two panelists voting against approval said that the study should be done before approval.
If approved, this would be the first LAMA/LABA combination product to become available in the United States. Umeclidinium is not yet available in any product, although the FDA is currently reviewing the 62.5-mcg dose as monotherapy. Vilanterol is available only as the LABA component in Breo Ellipta, the combination of vilanterol with the inhaled corticosteroid fluticasone, which was approved in May 2013 for COPD. The same device is used to deliver UMEC/VI 62.5/25, which, if approved, will be marketed as Anoro Ellipta, by GSK and Theravance.
Research details
The four main safety and efficacy studies compared two doses of umeclidinium (125 mcg and 62.5 mcg) combined with 25 mcg of vilanterol to the separate components alone and to placebo (in two studies) or to the approved long-acting antimuscarinic agent tiotropium (two studies) in about 4,700 patients – most of whom were male and white – with moderate to severe COPD. Their mean age was 63 years, almost half were still smoking, and almost 30% had had at least one exacerbation during the previous year that required treatment with corticosteroids and/or antibiotics; about 10% said they had been hospitalized for an exacerbation during the previous year (25% were enrolled in the United States).
In the two placebo-controlled studies, the combination and the two separate components (UMEC and vilanterol) were associated with statistically significant improvements in mean trough forced expiratory volume in 1 second (FEV1) from baseline at 24 weeks (the primary endpoint), compared with placebo. Improvements were also statistically significant for the two combinations over the individual components. In the active controlled study, the two combinations showed improvements in trough FEV1 that were superior to vilanterol alone and to tiotropium alone. The results with the higher dose of umeclidinium and vilanterol were similar to the 62.5-mcg dose, the proposed dose for approval.
The mortality rate and nonfatal serious adverse events were even in the treatment arms, in the four primary efficacy studies, and in long-term safety studies, and adverse events were consistent with those associated with the LAMAs and LABAs, according to GSK. The total number of cardiovascular-related events was fairly low in the safety database that included the four main studies and a long-term safety study. But there were numerical imbalances in ischemia-related events in the primary efficacy studies that were not seen in the long-term safety study, an issue raised by the FDA, although the overall number of events was low, according to the agency.
Panelists raised several safety issues, including the generalizability of the cardiac safety data to patients with more severe cardiac disease, because of exclusion criteria that may have eliminated these patients.
The panel chair, Dr. David Jacoby, professor of medicine in the division of pulmonary and critical care medicine at Oregon Health and Science University, Portland, supported approval but recommended that safety should be studied further in patients with COPD who have more severe cardiac disease.
Also voting for approval, Dr. Nizar Jarjour, professor and head of the section of allergy, pulmonary, and critical care at the University of Wisconsin, Madison, said the data indicated that the product was reasonably safe. But if it were to be used outside the parameters studied in the trials, such as for patients with recent exacerbations of COPD, or severe heart disease, he said he have would "no real confidence" that it would have the same safety profile as seen in the trials.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but that was unnecessary at this meeting.
SILVER SPRING, MD. – A fixed-dose combination of umeclidinium bromide, a new long-acting antimuscarinic agent (LAMA), and vilanterol, a relatively new long-acting beta agonist (LABA), in an inhaled powder formulation, should be approved for treatment of chronic obstructive pulmonary disease, with a recommendation for a postmarketing safety study, according to the majority of a Food and Drug Administration advisory panel.
At a meeting on Sept. 10, the FDA’s Pulmonary-Allergy Drugs Advisory Committee voted 11-2 that the safety and efficacy data on the combined treatment, administered once a day, supported approval for the proposed indication: the long-term once-daily maintenance bronchodilator treatment of airflow obstruction in patients with COPD, including chronic bronchitis and/or emphysema. The daily dose is 62.5 mcg of umeclidinium and 25 mcg of vilanterol (UMEC/VI 62.5/25), provided in one inhalation.
In another vote, the panel unanimously agreed that treatment with UMEC/VI 62.5/25 provided clinically meaningful benefits for this indication, based on the clinical trial results provided by GlaxoSmithKline. However, all the panelists were concerned about potential safety issues in patients with severe heart disease. There was also concern over the generalizability of the use of the combination because of a cardiovascular safety signal in the studies and the types of patients excluded in the research.
Those voting in favor of approval recommended a postmarketing study to evaluate the safety signal further, and a statement in the precautions and warnings section of the label about possible risks. The two panelists voting against approval said that the study should be done before approval.
If approved, this would be the first LAMA/LABA combination product to become available in the United States. Umeclidinium is not yet available in any product, although the FDA is currently reviewing the 62.5-mcg dose as monotherapy. Vilanterol is available only as the LABA component in Breo Ellipta, the combination of vilanterol with the inhaled corticosteroid fluticasone, which was approved in May 2013 for COPD. The same device is used to deliver UMEC/VI 62.5/25, which, if approved, will be marketed as Anoro Ellipta, by GSK and Theravance.
Research details
The four main safety and efficacy studies compared two doses of umeclidinium (125 mcg and 62.5 mcg) combined with 25 mcg of vilanterol to the separate components alone and to placebo (in two studies) or to the approved long-acting antimuscarinic agent tiotropium (two studies) in about 4,700 patients – most of whom were male and white – with moderate to severe COPD. Their mean age was 63 years, almost half were still smoking, and almost 30% had had at least one exacerbation during the previous year that required treatment with corticosteroids and/or antibiotics; about 10% said they had been hospitalized for an exacerbation during the previous year (25% were enrolled in the United States).
In the two placebo-controlled studies, the combination and the two separate components (UMEC and vilanterol) were associated with statistically significant improvements in mean trough forced expiratory volume in 1 second (FEV1) from baseline at 24 weeks (the primary endpoint), compared with placebo. Improvements were also statistically significant for the two combinations over the individual components. In the active controlled study, the two combinations showed improvements in trough FEV1 that were superior to vilanterol alone and to tiotropium alone. The results with the higher dose of umeclidinium and vilanterol were similar to the 62.5-mcg dose, the proposed dose for approval.
The mortality rate and nonfatal serious adverse events were even in the treatment arms, in the four primary efficacy studies, and in long-term safety studies, and adverse events were consistent with those associated with the LAMAs and LABAs, according to GSK. The total number of cardiovascular-related events was fairly low in the safety database that included the four main studies and a long-term safety study. But there were numerical imbalances in ischemia-related events in the primary efficacy studies that were not seen in the long-term safety study, an issue raised by the FDA, although the overall number of events was low, according to the agency.
Panelists raised several safety issues, including the generalizability of the cardiac safety data to patients with more severe cardiac disease, because of exclusion criteria that may have eliminated these patients.
The panel chair, Dr. David Jacoby, professor of medicine in the division of pulmonary and critical care medicine at Oregon Health and Science University, Portland, supported approval but recommended that safety should be studied further in patients with COPD who have more severe cardiac disease.
Also voting for approval, Dr. Nizar Jarjour, professor and head of the section of allergy, pulmonary, and critical care at the University of Wisconsin, Madison, said the data indicated that the product was reasonably safe. But if it were to be used outside the parameters studied in the trials, such as for patients with recent exacerbations of COPD, or severe heart disease, he said he have would "no real confidence" that it would have the same safety profile as seen in the trials.
The FDA usually follows the recommendations of its advisory panels. Panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver, but that was unnecessary at this meeting.
AT A FOOD AND DRUG ADMINISTRATION ADVISORY PANEL MEETING
Novel breath test helped identify invasive aspergillosis
DENVER – A novel breathalyzerlike test detected fungal gas metabolites in the breath of immunocompromised patients with suspected invasive aspergillosis, with excellent sensitivity and specificity, a single-center study demonstrated.
"We envision this work can be adapted to a rapid, noninvasive point-of-care detection system for real-time surveillance of patient breath for the emergence of aspergillosis and potentially for the diagnosis of lung infections caused by other fungal and bacterial pathogens," Dr. Sophia Koo said in an interview prior to a poster session at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy, where the study was presented.

"An urgent need" exists for better diagnostic tests for invasive aspergillosis, a life-threatening fungal pneumonia in immunocompromised patients, explained Dr. Koo of Harvard Medical School, Boston. Current diagnostic tests – respiratory tract cultures, serum and bronchoalveolar lavage for fungal antigen testing, and nucleic acid detection assays – "have significant limitations in their sensitivity and specificity, and the turnaround time of these assays in clinical practice is often days," she said.
Dr. Koo and her colleagues developed a new method to detect volatile fungal metabolites directly in patient breath. First, they defined the fungal volatile metabolite profile of Aspergillus fumigatus, the most common cause of invasive aspergillosis, in in vitro culture. Next, they used gas chromatography–mass spectrometry to detect those metabolites in the breath of 54 immunocompromised patients with suspected invasive aspergillosis pneumonia. Of those 54 patients, 29 ultimately had invasive aspergillosis, and 25 had other causes of pneumonia.
The breathalyzerlike test correctly identified 27 of 29 patients with invasive aspergillosis and 24 of 25 patients without invasive aspergillosis, for an overall diagnostic sensitivity of 93% and a diagnostic specificity of 96%.
"We were a bit surprised by how clearly we were able to discriminate patients with invasive aspergillosis from patients with other pneumonias using this approach," Dr. Koo noted. "When we made our initial plot of breath fungal volatile metabolites, clustered by patients with and without invasive aspergillosis, there was a clear line demarcating the two groups. We expected some differences in the fungal volatile metabolite profile in vitro and in vivo, with the overlay of the host-pathogen interaction, and we did indeed see some fungal metabolites in vivo that we did not see in any of our in vitro experiments."
While Dr. Koo described gas chromatography–mass spectrometry as "labor intensive," she characterized the findings of the current study as "easily translatable to a point of care platform for the detection of these fungal breath metabolites and potentially the diagnosis of other bacterial and fungal pneumonias at the bedside."
Dr. Koo acknowledged certain limitations of the study, including its single-center design and the fact that it will require validation in a larger cohort of patients.
The National Institutes of Health and the Harvard Catalyst/the Harvard Clinical and Translational Science Center Pilot Grant Program supported the study. The authors stated that they have no relevant financial conflicts.

|
| Dr. Marcos Restrepo |
Dr. Marcos I. Restrepo, FCCP, comments: This promising and novel breath test that identifies invasive aspergillosis may have important implications in the care of immuncompromised patients with invasive fungal infections. Further developments are necessary before this technology can be considered as a point-of-care testing in the real world.
|
|
| Dr. Marcos Restrepo |
Dr. Marcos I. Restrepo, FCCP, comments: This promising and novel breath test that identifies invasive aspergillosis may have important implications in the care of immuncompromised patients with invasive fungal infections. Further developments are necessary before this technology can be considered as a point-of-care testing in the real world.
|
|
| Dr. Marcos Restrepo |
Dr. Marcos I. Restrepo, FCCP, comments: This promising and novel breath test that identifies invasive aspergillosis may have important implications in the care of immuncompromised patients with invasive fungal infections. Further developments are necessary before this technology can be considered as a point-of-care testing in the real world.
DENVER – A novel breathalyzerlike test detected fungal gas metabolites in the breath of immunocompromised patients with suspected invasive aspergillosis, with excellent sensitivity and specificity, a single-center study demonstrated.
"We envision this work can be adapted to a rapid, noninvasive point-of-care detection system for real-time surveillance of patient breath for the emergence of aspergillosis and potentially for the diagnosis of lung infections caused by other fungal and bacterial pathogens," Dr. Sophia Koo said in an interview prior to a poster session at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy, where the study was presented.
"An urgent need" exists for better diagnostic tests for invasive aspergillosis, a life-threatening fungal pneumonia in immunocompromised patients, explained Dr. Koo of Harvard Medical School, Boston. Current diagnostic tests – respiratory tract cultures, serum and bronchoalveolar lavage for fungal antigen testing, and nucleic acid detection assays – "have significant limitations in their sensitivity and specificity, and the turnaround time of these assays in clinical practice is often days," she said.
Dr. Koo and her colleagues developed a new method to detect volatile fungal metabolites directly in patient breath. First, they defined the fungal volatile metabolite profile of Aspergillus fumigatus, the most common cause of invasive aspergillosis, in in vitro culture. Next, they used gas chromatography–mass spectrometry to detect those metabolites in the breath of 54 immunocompromised patients with suspected invasive aspergillosis pneumonia. Of those 54 patients, 29 ultimately had invasive aspergillosis, and 25 had other causes of pneumonia.
The breathalyzerlike test correctly identified 27 of 29 patients with invasive aspergillosis and 24 of 25 patients without invasive aspergillosis, for an overall diagnostic sensitivity of 93% and a diagnostic specificity of 96%.
"We were a bit surprised by how clearly we were able to discriminate patients with invasive aspergillosis from patients with other pneumonias using this approach," Dr. Koo noted. "When we made our initial plot of breath fungal volatile metabolites, clustered by patients with and without invasive aspergillosis, there was a clear line demarcating the two groups. We expected some differences in the fungal volatile metabolite profile in vitro and in vivo, with the overlay of the host-pathogen interaction, and we did indeed see some fungal metabolites in vivo that we did not see in any of our in vitro experiments."
While Dr. Koo described gas chromatography–mass spectrometry as "labor intensive," she characterized the findings of the current study as "easily translatable to a point of care platform for the detection of these fungal breath metabolites and potentially the diagnosis of other bacterial and fungal pneumonias at the bedside."
Dr. Koo acknowledged certain limitations of the study, including its single-center design and the fact that it will require validation in a larger cohort of patients.
The National Institutes of Health and the Harvard Catalyst/the Harvard Clinical and Translational Science Center Pilot Grant Program supported the study. The authors stated that they have no relevant financial conflicts.
DENVER – A novel breathalyzerlike test detected fungal gas metabolites in the breath of immunocompromised patients with suspected invasive aspergillosis, with excellent sensitivity and specificity, a single-center study demonstrated.
"We envision this work can be adapted to a rapid, noninvasive point-of-care detection system for real-time surveillance of patient breath for the emergence of aspergillosis and potentially for the diagnosis of lung infections caused by other fungal and bacterial pathogens," Dr. Sophia Koo said in an interview prior to a poster session at the annual Interscience Conference on Antimicrobial Agents and Chemotherapy, where the study was presented.
"An urgent need" exists for better diagnostic tests for invasive aspergillosis, a life-threatening fungal pneumonia in immunocompromised patients, explained Dr. Koo of Harvard Medical School, Boston. Current diagnostic tests – respiratory tract cultures, serum and bronchoalveolar lavage for fungal antigen testing, and nucleic acid detection assays – "have significant limitations in their sensitivity and specificity, and the turnaround time of these assays in clinical practice is often days," she said.
Dr. Koo and her colleagues developed a new method to detect volatile fungal metabolites directly in patient breath. First, they defined the fungal volatile metabolite profile of Aspergillus fumigatus, the most common cause of invasive aspergillosis, in in vitro culture. Next, they used gas chromatography–mass spectrometry to detect those metabolites in the breath of 54 immunocompromised patients with suspected invasive aspergillosis pneumonia. Of those 54 patients, 29 ultimately had invasive aspergillosis, and 25 had other causes of pneumonia.
The breathalyzerlike test correctly identified 27 of 29 patients with invasive aspergillosis and 24 of 25 patients without invasive aspergillosis, for an overall diagnostic sensitivity of 93% and a diagnostic specificity of 96%.
"We were a bit surprised by how clearly we were able to discriminate patients with invasive aspergillosis from patients with other pneumonias using this approach," Dr. Koo noted. "When we made our initial plot of breath fungal volatile metabolites, clustered by patients with and without invasive aspergillosis, there was a clear line demarcating the two groups. We expected some differences in the fungal volatile metabolite profile in vitro and in vivo, with the overlay of the host-pathogen interaction, and we did indeed see some fungal metabolites in vivo that we did not see in any of our in vitro experiments."
While Dr. Koo described gas chromatography–mass spectrometry as "labor intensive," she characterized the findings of the current study as "easily translatable to a point of care platform for the detection of these fungal breath metabolites and potentially the diagnosis of other bacterial and fungal pneumonias at the bedside."
Dr. Koo acknowledged certain limitations of the study, including its single-center design and the fact that it will require validation in a larger cohort of patients.
The National Institutes of Health and the Harvard Catalyst/the Harvard Clinical and Translational Science Center Pilot Grant Program supported the study. The authors stated that they have no relevant financial conflicts.
AT ICAAC 2013
Major finding: A new breathalyzerlike test correctly identified 27 of 29 patients with invasive aspergillosis and 24 of 25 patients without invasive aspergillosis, for an overall diagnostic sensitivity of 93% and a diagnostic specificity of 96%.
Data source: A study that used gas chromatography–mass spectrometry to detect fungal gas metabolites in the breath of 54 immunocompromised patients with suspected invasive aspergillosis pneumonia.
Disclosures: The National Institutes of Health and the Harvard Catalyst/the Harvard Clinical and Translational Science Center Pilot Grant Program supported the study. The authors stated that they have no relevant financial conflicts.

Vascular risk factors increased likelihood of restless legs syndrome in women
Vascular risk factors such as diabetes, hypercholesterolemia, body mass index, smoking status, and exercise were associated with the prevalence of restless legs syndrome in 12% of women aged older than 45 years, although a link was not found with cardiovascular events such as stroke or myocardial infarction, according to a study.
Previous studies have indicated that an unhealthy vascular profile might be the mechanism linking restless legs syndrome with cardiovascular disease (CVD) in women, but the data are inconsistent.

Dr. Tobias Kurth of Inserm Research Center for Epidemiology and Biostatistics and the University of Bordeaux, France, and his colleagues, sought a more definitive connection in a cross-sectional study of 30,262 women who participated in the Women’s Health Study (WHS). In the 9th year of the study, validated criteria from the International Restless Legs Study Group were added to the follow-up survey.
Dr. Kurth and his investigators applied age-adjusted and multivariable-adjusted logistic regression models to the complete data provided by 30,262 respondents (mean age 63.6 years) to evaluate the association between restless legs syndrome and independent vascular risk factors such as diabetes, hypertension, hypercholesterolemia, BMI, alcohol, smoking, exercise, and family history of MI. They adjusted for age, aspirin use, postmenopausal status, postmenopausal hormone use, and history of oral contraceptive use (Am. J. Med. 2013;126:220-7 [doi:10.1016/j.amjmed.2012.06.040]).
Patient-reported data on cardiovascular events were confirmed by medical record review.
A total of 3,634 (12%) of the women had restless legs syndrome. Women with several vascular risk factors were at greater odds of having restless legs syndrome, BMI greater than 35 (OR 1.35), diabetes (OR 1.19), hypercholesterolemia (OR 1.17), smoking 15 or more cigarettes per day (OR 1.41), and exercise less than four times per week (OR 0.84). Those who underwent coronary revascularization had a multivariable-adjusted OR of 1.39 for restless legs syndrome.
Noting that genetics, endocrine disturbances, and lifestyle-related comorbidities such as smoking have been theorized as mechanisms of restless legs syndrome, the authors wrote that "the cross-sectional design of our study and previous reports does not allow drawing conclusions regarding direction and causality of the association."
Furthermore, while reports cited by the investigators found similar prevalence rates between restless legs syndrome and vascular risk, the authors concluded they "could not confirm results from previous studies indicating an association between prevalent cardiovascular disease and restless legs syndrome."
The study was funded by the National Heart, Lung, and Blood Institute. The Women’s Health Study was underwritten by NHBLI and the National Cancer Institute. The investigators listed multiple financial conflicts of interest regarding restless legs syndrome research and treatments.
Vascular risk factors such as diabetes, hypercholesterolemia, body mass index, smoking status, and exercise were associated with the prevalence of restless legs syndrome in 12% of women aged older than 45 years, although a link was not found with cardiovascular events such as stroke or myocardial infarction, according to a study.
Previous studies have indicated that an unhealthy vascular profile might be the mechanism linking restless legs syndrome with cardiovascular disease (CVD) in women, but the data are inconsistent.
Dr. Tobias Kurth of Inserm Research Center for Epidemiology and Biostatistics and the University of Bordeaux, France, and his colleagues, sought a more definitive connection in a cross-sectional study of 30,262 women who participated in the Women’s Health Study (WHS). In the 9th year of the study, validated criteria from the International Restless Legs Study Group were added to the follow-up survey.
Dr. Kurth and his investigators applied age-adjusted and multivariable-adjusted logistic regression models to the complete data provided by 30,262 respondents (mean age 63.6 years) to evaluate the association between restless legs syndrome and independent vascular risk factors such as diabetes, hypertension, hypercholesterolemia, BMI, alcohol, smoking, exercise, and family history of MI. They adjusted for age, aspirin use, postmenopausal status, postmenopausal hormone use, and history of oral contraceptive use (Am. J. Med. 2013;126:220-7 [doi:10.1016/j.amjmed.2012.06.040]).
Patient-reported data on cardiovascular events were confirmed by medical record review.
A total of 3,634 (12%) of the women had restless legs syndrome. Women with several vascular risk factors were at greater odds of having restless legs syndrome, BMI greater than 35 (OR 1.35), diabetes (OR 1.19), hypercholesterolemia (OR 1.17), smoking 15 or more cigarettes per day (OR 1.41), and exercise less than four times per week (OR 0.84). Those who underwent coronary revascularization had a multivariable-adjusted OR of 1.39 for restless legs syndrome.
Noting that genetics, endocrine disturbances, and lifestyle-related comorbidities such as smoking have been theorized as mechanisms of restless legs syndrome, the authors wrote that "the cross-sectional design of our study and previous reports does not allow drawing conclusions regarding direction and causality of the association."
Furthermore, while reports cited by the investigators found similar prevalence rates between restless legs syndrome and vascular risk, the authors concluded they "could not confirm results from previous studies indicating an association between prevalent cardiovascular disease and restless legs syndrome."
The study was funded by the National Heart, Lung, and Blood Institute. The Women’s Health Study was underwritten by NHBLI and the National Cancer Institute. The investigators listed multiple financial conflicts of interest regarding restless legs syndrome research and treatments.
Vascular risk factors such as diabetes, hypercholesterolemia, body mass index, smoking status, and exercise were associated with the prevalence of restless legs syndrome in 12% of women aged older than 45 years, although a link was not found with cardiovascular events such as stroke or myocardial infarction, according to a study.
Previous studies have indicated that an unhealthy vascular profile might be the mechanism linking restless legs syndrome with cardiovascular disease (CVD) in women, but the data are inconsistent.
Dr. Tobias Kurth of Inserm Research Center for Epidemiology and Biostatistics and the University of Bordeaux, France, and his colleagues, sought a more definitive connection in a cross-sectional study of 30,262 women who participated in the Women’s Health Study (WHS). In the 9th year of the study, validated criteria from the International Restless Legs Study Group were added to the follow-up survey.
Dr. Kurth and his investigators applied age-adjusted and multivariable-adjusted logistic regression models to the complete data provided by 30,262 respondents (mean age 63.6 years) to evaluate the association between restless legs syndrome and independent vascular risk factors such as diabetes, hypertension, hypercholesterolemia, BMI, alcohol, smoking, exercise, and family history of MI. They adjusted for age, aspirin use, postmenopausal status, postmenopausal hormone use, and history of oral contraceptive use (Am. J. Med. 2013;126:220-7 [doi:10.1016/j.amjmed.2012.06.040]).
Patient-reported data on cardiovascular events were confirmed by medical record review.
A total of 3,634 (12%) of the women had restless legs syndrome. Women with several vascular risk factors were at greater odds of having restless legs syndrome, BMI greater than 35 (OR 1.35), diabetes (OR 1.19), hypercholesterolemia (OR 1.17), smoking 15 or more cigarettes per day (OR 1.41), and exercise less than four times per week (OR 0.84). Those who underwent coronary revascularization had a multivariable-adjusted OR of 1.39 for restless legs syndrome.
Noting that genetics, endocrine disturbances, and lifestyle-related comorbidities such as smoking have been theorized as mechanisms of restless legs syndrome, the authors wrote that "the cross-sectional design of our study and previous reports does not allow drawing conclusions regarding direction and causality of the association."
Furthermore, while reports cited by the investigators found similar prevalence rates between restless legs syndrome and vascular risk, the authors concluded they "could not confirm results from previous studies indicating an association between prevalent cardiovascular disease and restless legs syndrome."
The study was funded by the National Heart, Lung, and Blood Institute. The Women’s Health Study was underwritten by NHBLI and the National Cancer Institute. The investigators listed multiple financial conflicts of interest regarding restless legs syndrome research and treatments.
FROM THE JOURNAL OF AMERICAN MEDICINE
Major finding: Diabetes, hypercholesterolemia, BMI, smoking, and lack of exercise were linked to restless legs syndrome in 12% of women aged 45 years and over.
Data source: Logistic regression analysis of cross-sectional, cohort study of 30,262 participants in the Women’s Health Study.
Disclosures: The study was underwritten by a grant from the National Heart, Lung, and Blood Institute. The Women’s Health Study was underwritten by NHBLI and the National Cancer Institute. The investigators listed multiple financial conflicts of interest regarding restless legs syndrome research and treatments.
