User login
Evidence-based guideline: assessment and management of psychiatric disorders in individuals with MS. Report of the Guideline Development Subcommittee of the American Academy of Neurology
Screening and Diagnosis
What Clinical Evaluation Procedures and Screening and Diagnostic Tools Can Be Used to Accurately Identify Symptoms and Make Diagnoses of Emotional Disorders in Individuals with Multiple Sclerosis (MS)?
Conclusions and Recommendations
In individuals with MS, the Center for Neurologic Study Emotional Lability Scale (CNS-LS) is possibly effective and may be considered for screening for pseudobulbar affect (PBA) (Level C, 1 Class II study [Smith et al., 2004]). The General Health Questionnaire (GHQ) (Goldberg & Hillier, 1979) is possibly effective and may be considered for identifying individuals with broadly defined emotional disturbances (Level C, 1 Class II study [Rabins & Brooks, 1981]). The Beck Depression Inventory (BDI) (Beck et al., 1961) and a 2-question screen (Whooley et al., 1997) are possibly effective and may be considered for identifying individuals with major depressive disorder (MDD) (Level C, 1 Class II study each [Sullivan et al., 1995; Mohr et al., 2007]). There is insufficient evidence to support/refute using the Center for Epidemiologic Studies Depression Rating Scale (CES-D) (Radloff, 1977) to screen for depressive symptoms (Pandya, Metz, & Patten, 2005) or a single question to screen for MDD (Vahter et al., 2007) (Level U, 1 Class III study each); the possibility that somatic or neurovegetative symptoms negatively affect the accuracy of BDI results (Level U, 2 conflicting Class III studies) (Mohr et al., 1997; Randolph et al., 2000); and the use of specific instruments or clinical evaluation procedures to diagnose emotional disorders in individuals with MS (Level U).
Clinical Context
Because emotional disorders may be unrecognized in medical settings, validated screening tools might improve identification of individuals who could benefit from further evaluation and treatment. The true positive rate of a screening tool depends not only on its sensitivity but also on the point prevalence of the disorder in the population under study. Clinically, false-positive results are not a major concern because individuals with the conditions typically identified (e.g., adjustment and subthreshold depressive disorders) can benefit from further assessment. Administratively, however, screening tools with high false-positive rates unnecessarily increase resource use.
Treatments
What Are the Effective Treatments for Disorders of Mood in Individuals with MS?
Conclusion and Recommendations
For individuals with MS, a 16-week program of individual telephone-administered cognitive behavioral therapy (T-CBT) program is possibly effective and may be considered in treating depressive symptoms (Level C, 1 Class II study [Mohr et al., 2005], 1 Class III study [Mohr, et al., 2000]). There is insufficient evidence to support/refute the efficacy and use of 1) sertraline (Mohr et al., 2001), desipramine (Schiffer & Wineman, 1990), paroxetine (Ehde et al., 2008), individual in-person cognitive behavioral therapy (CBT) (Mohr et al., 2001), individual in-person CBT plus relaxation training (Foley et al., 1987), or CBT-based group therapy (Forman & Lincoln, 2010) for depressive symptoms; or 2) individual in-person CBT plus relaxation training (Foley et al., 1987), group relaxation and imagery (Maguire, 1996), or CBT-based group therapy (Forman & Lincoln, 2010) for anxiety (Level U, 1 Class III study each).
Clinical Context
There is evidence supporting the efficacy of pharmacologic and nonpharmacologic therapies for depressed mood and anxiety in individuals without MS. Despite the lack of evidence in individuals with MS, these therapies are frequently used to treat emotional disorders in this population.
What Are the Effective Treatments for Disorders of Affect in Individuals with MS?
Conclusion and Recommendations
Dextromethorphan and quinidine (DM/Q) is possibly effective and safe and may be considered for treating individuals with MS with PBA (Level C, 1 Class II study) (Panitch et al., 2006).
Clinical Context
DM/Q is the only drug approved by the US Food and Drug Administration for PBA treatment, although other drugs are used in clinical practice (e.g., selective serotonin reuptake inhibitors, tricyclic antidepressants). There are no randomized placebo-controlled trials of these other agents.
Definitions:
Classification of Evidence
Screening Articles
Class I: A statistical, population based sample of patients studied at a uniform point in time (usually early) during the course of the condition. All patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation that is masked to the patients' clinical presentations.
Class II: A statistical, non-referral clinic based sample of patients studied at a uniform point in time (usually early) during the course of the condition. Most patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation that is masked to the patients' clinical presentations.
Class III: A sample of patients studied during the course of the condition. Some patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation by someone other than the treating physician.
Class IV: Studies not meeting Class I, II, or III criteria, including consensus, expert opinion or a case report.
Diagnostic Articles
Class I: A cohort study with prospective data collection of a broad spectrum of persons with the suspected condition, using an acceptable reference standard for case definition. The diagnostic test is objective or performed and interpreted without knowledge of the patient's clinical status. Study results allow calculation of measures of diagnostic accuracy.
Class II: A case control study of a broad spectrum of persons with the condition established by an acceptable reference standard compared to a broad spectrum of controls or a cohort study where a broad spectrum of persons with the suspected condition where the data was collected retrospectively. The diagnostic test is objective or performed and interpreted without knowledge of disease status. Study results allow calculation of measures of diagnostic accuracy.
Class III: A case control study or cohort study where either persons with the condition or controls are of a narrow spectrum. The condition is established by an acceptable reference standard. The reference standard and diagnostic test are objective or performed and interpreted by different observers. Study results allow calculation of measures of diagnostic accuracy.
Class IV: Studies not meeting Class I, II, or III criteria including consensus, expert opinion, or a case report.
Therapeutic Articles
Class I: A randomized, controlled clinical trial of the intervention of interest with masked or objective outcome assessment, in a representative population. Relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences.
The following are also required:
- Concealed allocation
- Primary outcome(s) clearly defined
- Exclusion/inclusion criteria clearly defined
- Adequate accounting for dropouts (with at least 80% of enrolled subjects completing the study) and crossovers with numbers sufficiently low to have minimal potential for bias.
- For noninferiority or equivalence trials claiming to prove efficacy for one or both drugs, the following are also required*:
- The authors explicitly state the clinically meaningful difference to be excluded by defining the threshold for equivalence or noninferiority.
- The standard treatment used in the study is substantially similar to that used in previous studies establishing efficacy of the standard treatment (e.g., for a drug, the mode of administration, dose and dosage adjustments are similar to those previously shown to be effective).
- The inclusion and exclusion criteria for patient selection and the outcomes of patients on the standard treatment are comparable to those of previous studies establishing efficacy of the standard treatment.
- The interpretation of the results of the study is based upon a per protocol analysis that takes into account dropouts or crossovers.
Class II: A randomized controlled clinical trial of the intervention of interest in a representative population with masked or objective outcome assessment that lacks one criteria a–e above or a prospective matched cohort study with masked or objective outcome assessment in a representative population that meets b–e above. Relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences.
Class III: All other controlled trials (including well-defined natural history controls or patients serving as own controls) in a representative population, where outcome is independently assessed, or independently derived by objective outcome measurement.**
Class IV: Studies not meeting Class I, II, or III criteria including consensus or expert opinion.
*Note that numbers 1-3 in Class Ie are required for Class II in equivalence trials. If any one of the three is missing, the class is automatically downgraded to Class III.
**Objective outcome measurement: an outcome measure that is unlikely to be affected by an observer's (patient, treating physician, investigator) expectation or bias (e.g., blood tests, administrative outcome data).
Classification of Recommendations
Level A = Established as effective, ineffective or harmful (or established as useful/predictive or not useful/predictive) for the given condition in the specified population. (Level A rating requires at least two consistent Class I studies.)*
Level B = Probably effective, ineffective or harmful (or probably useful/predictive or not useful/predictive) for the given condition in the specified population. (Level B rating requires at least one Class I study or two consistent Class II studies.)
Level C = Possibly effective, ineffective or harmful (or possibly useful/predictive or not useful/predictive) for the given condition in the specified population. (Level C rating requires at least one Class II study or two consistent Class III studies.)
Level U = Data inadequate or conflicting; given current knowledge, treatment (test, predictor) is unproven.
* In exceptional cases, one convincing Class I study may suffice for an "A" recommendation if 1) all criteria are met, 2) the magnitude of effect is large (relative rate improved outcome >5 and the lower limit of the confidence interval is >2).
Screening and Diagnosis
What Clinical Evaluation Procedures and Screening and Diagnostic Tools Can Be Used to Accurately Identify Symptoms and Make Diagnoses of Emotional Disorders in Individuals with Multiple Sclerosis (MS)?
Conclusions and Recommendations
In individuals with MS, the Center for Neurologic Study Emotional Lability Scale (CNS-LS) is possibly effective and may be considered for screening for pseudobulbar affect (PBA) (Level C, 1 Class II study [Smith et al., 2004]). The General Health Questionnaire (GHQ) (Goldberg & Hillier, 1979) is possibly effective and may be considered for identifying individuals with broadly defined emotional disturbances (Level C, 1 Class II study [Rabins & Brooks, 1981]). The Beck Depression Inventory (BDI) (Beck et al., 1961) and a 2-question screen (Whooley et al., 1997) are possibly effective and may be considered for identifying individuals with major depressive disorder (MDD) (Level C, 1 Class II study each [Sullivan et al., 1995; Mohr et al., 2007]). There is insufficient evidence to support/refute using the Center for Epidemiologic Studies Depression Rating Scale (CES-D) (Radloff, 1977) to screen for depressive symptoms (Pandya, Metz, & Patten, 2005) or a single question to screen for MDD (Vahter et al., 2007) (Level U, 1 Class III study each); the possibility that somatic or neurovegetative symptoms negatively affect the accuracy of BDI results (Level U, 2 conflicting Class III studies) (Mohr et al., 1997; Randolph et al., 2000); and the use of specific instruments or clinical evaluation procedures to diagnose emotional disorders in individuals with MS (Level U).
Clinical Context
Because emotional disorders may be unrecognized in medical settings, validated screening tools might improve identification of individuals who could benefit from further evaluation and treatment. The true positive rate of a screening tool depends not only on its sensitivity but also on the point prevalence of the disorder in the population under study. Clinically, false-positive results are not a major concern because individuals with the conditions typically identified (e.g., adjustment and subthreshold depressive disorders) can benefit from further assessment. Administratively, however, screening tools with high false-positive rates unnecessarily increase resource use.
Treatments
What Are the Effective Treatments for Disorders of Mood in Individuals with MS?
Conclusion and Recommendations
For individuals with MS, a 16-week program of individual telephone-administered cognitive behavioral therapy (T-CBT) program is possibly effective and may be considered in treating depressive symptoms (Level C, 1 Class II study [Mohr et al., 2005], 1 Class III study [Mohr, et al., 2000]). There is insufficient evidence to support/refute the efficacy and use of 1) sertraline (Mohr et al., 2001), desipramine (Schiffer & Wineman, 1990), paroxetine (Ehde et al., 2008), individual in-person cognitive behavioral therapy (CBT) (Mohr et al., 2001), individual in-person CBT plus relaxation training (Foley et al., 1987), or CBT-based group therapy (Forman & Lincoln, 2010) for depressive symptoms; or 2) individual in-person CBT plus relaxation training (Foley et al., 1987), group relaxation and imagery (Maguire, 1996), or CBT-based group therapy (Forman & Lincoln, 2010) for anxiety (Level U, 1 Class III study each).
Clinical Context
There is evidence supporting the efficacy of pharmacologic and nonpharmacologic therapies for depressed mood and anxiety in individuals without MS. Despite the lack of evidence in individuals with MS, these therapies are frequently used to treat emotional disorders in this population.
What Are the Effective Treatments for Disorders of Affect in Individuals with MS?
Conclusion and Recommendations
Dextromethorphan and quinidine (DM/Q) is possibly effective and safe and may be considered for treating individuals with MS with PBA (Level C, 1 Class II study) (Panitch et al., 2006).
Clinical Context
DM/Q is the only drug approved by the US Food and Drug Administration for PBA treatment, although other drugs are used in clinical practice (e.g., selective serotonin reuptake inhibitors, tricyclic antidepressants). There are no randomized placebo-controlled trials of these other agents.
Definitions:
Classification of Evidence
Screening Articles
Class I: A statistical, population based sample of patients studied at a uniform point in time (usually early) during the course of the condition. All patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation that is masked to the patients' clinical presentations.
Class II: A statistical, non-referral clinic based sample of patients studied at a uniform point in time (usually early) during the course of the condition. Most patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation that is masked to the patients' clinical presentations.
Class III: A sample of patients studied during the course of the condition. Some patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation by someone other than the treating physician.
Class IV: Studies not meeting Class I, II, or III criteria, including consensus, expert opinion or a case report.
Diagnostic Articles
Class I: A cohort study with prospective data collection of a broad spectrum of persons with the suspected condition, using an acceptable reference standard for case definition. The diagnostic test is objective or performed and interpreted without knowledge of the patient's clinical status. Study results allow calculation of measures of diagnostic accuracy.
Class II: A case control study of a broad spectrum of persons with the condition established by an acceptable reference standard compared to a broad spectrum of controls or a cohort study where a broad spectrum of persons with the suspected condition where the data was collected retrospectively. The diagnostic test is objective or performed and interpreted without knowledge of disease status. Study results allow calculation of measures of diagnostic accuracy.
Class III: A case control study or cohort study where either persons with the condition or controls are of a narrow spectrum. The condition is established by an acceptable reference standard. The reference standard and diagnostic test are objective or performed and interpreted by different observers. Study results allow calculation of measures of diagnostic accuracy.
Class IV: Studies not meeting Class I, II, or III criteria including consensus, expert opinion, or a case report.
Therapeutic Articles
Class I: A randomized, controlled clinical trial of the intervention of interest with masked or objective outcome assessment, in a representative population. Relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences.
The following are also required:
- Concealed allocation
- Primary outcome(s) clearly defined
- Exclusion/inclusion criteria clearly defined
- Adequate accounting for dropouts (with at least 80% of enrolled subjects completing the study) and crossovers with numbers sufficiently low to have minimal potential for bias.
- For noninferiority or equivalence trials claiming to prove efficacy for one or both drugs, the following are also required*:
- The authors explicitly state the clinically meaningful difference to be excluded by defining the threshold for equivalence or noninferiority.
- The standard treatment used in the study is substantially similar to that used in previous studies establishing efficacy of the standard treatment (e.g., for a drug, the mode of administration, dose and dosage adjustments are similar to those previously shown to be effective).
- The inclusion and exclusion criteria for patient selection and the outcomes of patients on the standard treatment are comparable to those of previous studies establishing efficacy of the standard treatment.
- The interpretation of the results of the study is based upon a per protocol analysis that takes into account dropouts or crossovers.
Class II: A randomized controlled clinical trial of the intervention of interest in a representative population with masked or objective outcome assessment that lacks one criteria a–e above or a prospective matched cohort study with masked or objective outcome assessment in a representative population that meets b–e above. Relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences.
Class III: All other controlled trials (including well-defined natural history controls or patients serving as own controls) in a representative population, where outcome is independently assessed, or independently derived by objective outcome measurement.**
Class IV: Studies not meeting Class I, II, or III criteria including consensus or expert opinion.
*Note that numbers 1-3 in Class Ie are required for Class II in equivalence trials. If any one of the three is missing, the class is automatically downgraded to Class III.
**Objective outcome measurement: an outcome measure that is unlikely to be affected by an observer's (patient, treating physician, investigator) expectation or bias (e.g., blood tests, administrative outcome data).
Classification of Recommendations
Level A = Established as effective, ineffective or harmful (or established as useful/predictive or not useful/predictive) for the given condition in the specified population. (Level A rating requires at least two consistent Class I studies.)*
Level B = Probably effective, ineffective or harmful (or probably useful/predictive or not useful/predictive) for the given condition in the specified population. (Level B rating requires at least one Class I study or two consistent Class II studies.)
Level C = Possibly effective, ineffective or harmful (or possibly useful/predictive or not useful/predictive) for the given condition in the specified population. (Level C rating requires at least one Class II study or two consistent Class III studies.)
Level U = Data inadequate or conflicting; given current knowledge, treatment (test, predictor) is unproven.
* In exceptional cases, one convincing Class I study may suffice for an "A" recommendation if 1) all criteria are met, 2) the magnitude of effect is large (relative rate improved outcome >5 and the lower limit of the confidence interval is >2).
Screening and Diagnosis
What Clinical Evaluation Procedures and Screening and Diagnostic Tools Can Be Used to Accurately Identify Symptoms and Make Diagnoses of Emotional Disorders in Individuals with Multiple Sclerosis (MS)?
Conclusions and Recommendations
In individuals with MS, the Center for Neurologic Study Emotional Lability Scale (CNS-LS) is possibly effective and may be considered for screening for pseudobulbar affect (PBA) (Level C, 1 Class II study [Smith et al., 2004]). The General Health Questionnaire (GHQ) (Goldberg & Hillier, 1979) is possibly effective and may be considered for identifying individuals with broadly defined emotional disturbances (Level C, 1 Class II study [Rabins & Brooks, 1981]). The Beck Depression Inventory (BDI) (Beck et al., 1961) and a 2-question screen (Whooley et al., 1997) are possibly effective and may be considered for identifying individuals with major depressive disorder (MDD) (Level C, 1 Class II study each [Sullivan et al., 1995; Mohr et al., 2007]). There is insufficient evidence to support/refute using the Center for Epidemiologic Studies Depression Rating Scale (CES-D) (Radloff, 1977) to screen for depressive symptoms (Pandya, Metz, & Patten, 2005) or a single question to screen for MDD (Vahter et al., 2007) (Level U, 1 Class III study each); the possibility that somatic or neurovegetative symptoms negatively affect the accuracy of BDI results (Level U, 2 conflicting Class III studies) (Mohr et al., 1997; Randolph et al., 2000); and the use of specific instruments or clinical evaluation procedures to diagnose emotional disorders in individuals with MS (Level U).
Clinical Context
Because emotional disorders may be unrecognized in medical settings, validated screening tools might improve identification of individuals who could benefit from further evaluation and treatment. The true positive rate of a screening tool depends not only on its sensitivity but also on the point prevalence of the disorder in the population under study. Clinically, false-positive results are not a major concern because individuals with the conditions typically identified (e.g., adjustment and subthreshold depressive disorders) can benefit from further assessment. Administratively, however, screening tools with high false-positive rates unnecessarily increase resource use.
Treatments
What Are the Effective Treatments for Disorders of Mood in Individuals with MS?
Conclusion and Recommendations
For individuals with MS, a 16-week program of individual telephone-administered cognitive behavioral therapy (T-CBT) program is possibly effective and may be considered in treating depressive symptoms (Level C, 1 Class II study [Mohr et al., 2005], 1 Class III study [Mohr, et al., 2000]). There is insufficient evidence to support/refute the efficacy and use of 1) sertraline (Mohr et al., 2001), desipramine (Schiffer & Wineman, 1990), paroxetine (Ehde et al., 2008), individual in-person cognitive behavioral therapy (CBT) (Mohr et al., 2001), individual in-person CBT plus relaxation training (Foley et al., 1987), or CBT-based group therapy (Forman & Lincoln, 2010) for depressive symptoms; or 2) individual in-person CBT plus relaxation training (Foley et al., 1987), group relaxation and imagery (Maguire, 1996), or CBT-based group therapy (Forman & Lincoln, 2010) for anxiety (Level U, 1 Class III study each).
Clinical Context
There is evidence supporting the efficacy of pharmacologic and nonpharmacologic therapies for depressed mood and anxiety in individuals without MS. Despite the lack of evidence in individuals with MS, these therapies are frequently used to treat emotional disorders in this population.
What Are the Effective Treatments for Disorders of Affect in Individuals with MS?
Conclusion and Recommendations
Dextromethorphan and quinidine (DM/Q) is possibly effective and safe and may be considered for treating individuals with MS with PBA (Level C, 1 Class II study) (Panitch et al., 2006).
Clinical Context
DM/Q is the only drug approved by the US Food and Drug Administration for PBA treatment, although other drugs are used in clinical practice (e.g., selective serotonin reuptake inhibitors, tricyclic antidepressants). There are no randomized placebo-controlled trials of these other agents.
Definitions:
Classification of Evidence
Screening Articles
Class I: A statistical, population based sample of patients studied at a uniform point in time (usually early) during the course of the condition. All patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation that is masked to the patients' clinical presentations.
Class II: A statistical, non-referral clinic based sample of patients studied at a uniform point in time (usually early) during the course of the condition. Most patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation that is masked to the patients' clinical presentations.
Class III: A sample of patients studied during the course of the condition. Some patients undergo the intervention of interest. The outcome, if not objective, is determined in an evaluation by someone other than the treating physician.
Class IV: Studies not meeting Class I, II, or III criteria, including consensus, expert opinion or a case report.
Diagnostic Articles
Class I: A cohort study with prospective data collection of a broad spectrum of persons with the suspected condition, using an acceptable reference standard for case definition. The diagnostic test is objective or performed and interpreted without knowledge of the patient's clinical status. Study results allow calculation of measures of diagnostic accuracy.
Class II: A case control study of a broad spectrum of persons with the condition established by an acceptable reference standard compared to a broad spectrum of controls or a cohort study where a broad spectrum of persons with the suspected condition where the data was collected retrospectively. The diagnostic test is objective or performed and interpreted without knowledge of disease status. Study results allow calculation of measures of diagnostic accuracy.
Class III: A case control study or cohort study where either persons with the condition or controls are of a narrow spectrum. The condition is established by an acceptable reference standard. The reference standard and diagnostic test are objective or performed and interpreted by different observers. Study results allow calculation of measures of diagnostic accuracy.
Class IV: Studies not meeting Class I, II, or III criteria including consensus, expert opinion, or a case report.
Therapeutic Articles
Class I: A randomized, controlled clinical trial of the intervention of interest with masked or objective outcome assessment, in a representative population. Relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences.
The following are also required:
- Concealed allocation
- Primary outcome(s) clearly defined
- Exclusion/inclusion criteria clearly defined
- Adequate accounting for dropouts (with at least 80% of enrolled subjects completing the study) and crossovers with numbers sufficiently low to have minimal potential for bias.
- For noninferiority or equivalence trials claiming to prove efficacy for one or both drugs, the following are also required*:
- The authors explicitly state the clinically meaningful difference to be excluded by defining the threshold for equivalence or noninferiority.
- The standard treatment used in the study is substantially similar to that used in previous studies establishing efficacy of the standard treatment (e.g., for a drug, the mode of administration, dose and dosage adjustments are similar to those previously shown to be effective).
- The inclusion and exclusion criteria for patient selection and the outcomes of patients on the standard treatment are comparable to those of previous studies establishing efficacy of the standard treatment.
- The interpretation of the results of the study is based upon a per protocol analysis that takes into account dropouts or crossovers.
Class II: A randomized controlled clinical trial of the intervention of interest in a representative population with masked or objective outcome assessment that lacks one criteria a–e above or a prospective matched cohort study with masked or objective outcome assessment in a representative population that meets b–e above. Relevant baseline characteristics are presented and substantially equivalent among treatment groups or there is appropriate statistical adjustment for differences.
Class III: All other controlled trials (including well-defined natural history controls or patients serving as own controls) in a representative population, where outcome is independently assessed, or independently derived by objective outcome measurement.**
Class IV: Studies not meeting Class I, II, or III criteria including consensus or expert opinion.
*Note that numbers 1-3 in Class Ie are required for Class II in equivalence trials. If any one of the three is missing, the class is automatically downgraded to Class III.
**Objective outcome measurement: an outcome measure that is unlikely to be affected by an observer's (patient, treating physician, investigator) expectation or bias (e.g., blood tests, administrative outcome data).
Classification of Recommendations
Level A = Established as effective, ineffective or harmful (or established as useful/predictive or not useful/predictive) for the given condition in the specified population. (Level A rating requires at least two consistent Class I studies.)*
Level B = Probably effective, ineffective or harmful (or probably useful/predictive or not useful/predictive) for the given condition in the specified population. (Level B rating requires at least one Class I study or two consistent Class II studies.)
Level C = Possibly effective, ineffective or harmful (or possibly useful/predictive or not useful/predictive) for the given condition in the specified population. (Level C rating requires at least one Class II study or two consistent Class III studies.)
Level U = Data inadequate or conflicting; given current knowledge, treatment (test, predictor) is unproven.
* In exceptional cases, one convincing Class I study may suffice for an "A" recommendation if 1) all criteria are met, 2) the magnitude of effect is large (relative rate improved outcome >5 and the lower limit of the confidence interval is >2).
OBJECTIVE: To make evidence-based recommendations for screening, diagnosing, and treating psychiatric disorders in individuals with multiple sclerosis (MS).
METHODS: We reviewed the literature (1950 to August 2011) and evaluated the available evidence.
Guidelines are copyright © 2014 American Academy of Neurology. All rights reserved. The summary is provided by the Agency for Healthcare Research and Quality.
Varenicline combined with nicotine replacement therapy ups smoking quit rates
An estimated 42 million (18.1%) of the U.S. adult population continues to smoke cigarettes. Effective treatments exist for patients who are willing to avail themselves of such assistance. Varenicline is one of the most effective medications that we have to combat tobacco dependence. However, it doesn’t work for everybody, and questions have remained about how safe and effective it is to combine varenicline with other smoking cessation therapies such as nicotine replacement therapy.
Varenicline binds to a specific nicotine receptor, thereby partially agonizing and blocking it. The result is decreased cravings for tobacco and increased smoking quit rates. Data from early studies conducted by our group suggested that nicotine replacement therapy (NRT) combined with varenicline was safe, but questions remained about its efficacy.
One group of researchers conducted a multicenter clinical trial evaluating the efficacy of combining varenicline and the nicotine patch for increasing smoking cessation rates. Smokers were eligible if they smoked at least 10 cigarettes per day, reported F.N. Coenraad, from the Stellenbosch University, Cape Town, South Africa, and associates. Participants were randomized to active 15-mg nicotine patches or placebo patches started 2 weeks before the target quit date. All participants received varenicline for a total of 14 weeks with a 1-week ramp-up and a 1-week taper. Use of the varenicline in combination with the nicotine patch resulted in increased rates of continuous abstinence from smoking at 12 weeks (no smoking from weeks 9 to 12: 55.4% vs. 40.9%; odds ratio, 1.85; 95% confidence interval, 1.19-2.89; P = .007) and at 24 weeks (no smoking from weeks 9 to 24: 49% vs. 32.6%; OR, 1.98; 95% CI, 1.25-3.14; P = .004) (JAMA 2014;312:155-61).
This is a fantastic study answering a lingering question in tobacco control. But what is the theoretical underpinning by which this combination works? Isn’t the NRT blocked by the varenicline? It is possible that the varenicline incompletely saturates the nicotine receptors, which are additionally saturated by the supplemented nicotine. The varenicline effect is mediated through the alpha-4 beta-2 nicotinic receptor, and it is also possible that nicotine binds to nicotine receptor types that varenicline does not bind to, which decreases withdrawal symptoms.
We aren’t exactly sure how this might be working, but a near doubling of the odds of quitting is not to be disregarded. We are also not sure whether the effect holds when one uses other types of NRT such as the nicotine inhaler, nicotine lozenge, nicotine nasal spray, and nicotine gum. In practice, I tend to lean toward a combination of varenicline with the nicotine inhaler since the inhaler can help with some of the behavioral aspects of smoking while the varenicline does its heavy lifting.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author. Dr. Ebbert reports receiving research support from Pfizer, manufacturer of varenicline and the nicotine inhaler, and consulting fees from GlaxoSmithKline, manufacturer of the nicotine patch. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician.
An estimated 42 million (18.1%) of the U.S. adult population continues to smoke cigarettes. Effective treatments exist for patients who are willing to avail themselves of such assistance. Varenicline is one of the most effective medications that we have to combat tobacco dependence. However, it doesn’t work for everybody, and questions have remained about how safe and effective it is to combine varenicline with other smoking cessation therapies such as nicotine replacement therapy.
Varenicline binds to a specific nicotine receptor, thereby partially agonizing and blocking it. The result is decreased cravings for tobacco and increased smoking quit rates. Data from early studies conducted by our group suggested that nicotine replacement therapy (NRT) combined with varenicline was safe, but questions remained about its efficacy.
One group of researchers conducted a multicenter clinical trial evaluating the efficacy of combining varenicline and the nicotine patch for increasing smoking cessation rates. Smokers were eligible if they smoked at least 10 cigarettes per day, reported F.N. Coenraad, from the Stellenbosch University, Cape Town, South Africa, and associates. Participants were randomized to active 15-mg nicotine patches or placebo patches started 2 weeks before the target quit date. All participants received varenicline for a total of 14 weeks with a 1-week ramp-up and a 1-week taper. Use of the varenicline in combination with the nicotine patch resulted in increased rates of continuous abstinence from smoking at 12 weeks (no smoking from weeks 9 to 12: 55.4% vs. 40.9%; odds ratio, 1.85; 95% confidence interval, 1.19-2.89; P = .007) and at 24 weeks (no smoking from weeks 9 to 24: 49% vs. 32.6%; OR, 1.98; 95% CI, 1.25-3.14; P = .004) (JAMA 2014;312:155-61).
This is a fantastic study answering a lingering question in tobacco control. But what is the theoretical underpinning by which this combination works? Isn’t the NRT blocked by the varenicline? It is possible that the varenicline incompletely saturates the nicotine receptors, which are additionally saturated by the supplemented nicotine. The varenicline effect is mediated through the alpha-4 beta-2 nicotinic receptor, and it is also possible that nicotine binds to nicotine receptor types that varenicline does not bind to, which decreases withdrawal symptoms.
We aren’t exactly sure how this might be working, but a near doubling of the odds of quitting is not to be disregarded. We are also not sure whether the effect holds when one uses other types of NRT such as the nicotine inhaler, nicotine lozenge, nicotine nasal spray, and nicotine gum. In practice, I tend to lean toward a combination of varenicline with the nicotine inhaler since the inhaler can help with some of the behavioral aspects of smoking while the varenicline does its heavy lifting.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author. Dr. Ebbert reports receiving research support from Pfizer, manufacturer of varenicline and the nicotine inhaler, and consulting fees from GlaxoSmithKline, manufacturer of the nicotine patch. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician.
An estimated 42 million (18.1%) of the U.S. adult population continues to smoke cigarettes. Effective treatments exist for patients who are willing to avail themselves of such assistance. Varenicline is one of the most effective medications that we have to combat tobacco dependence. However, it doesn’t work for everybody, and questions have remained about how safe and effective it is to combine varenicline with other smoking cessation therapies such as nicotine replacement therapy.
Varenicline binds to a specific nicotine receptor, thereby partially agonizing and blocking it. The result is decreased cravings for tobacco and increased smoking quit rates. Data from early studies conducted by our group suggested that nicotine replacement therapy (NRT) combined with varenicline was safe, but questions remained about its efficacy.
One group of researchers conducted a multicenter clinical trial evaluating the efficacy of combining varenicline and the nicotine patch for increasing smoking cessation rates. Smokers were eligible if they smoked at least 10 cigarettes per day, reported F.N. Coenraad, from the Stellenbosch University, Cape Town, South Africa, and associates. Participants were randomized to active 15-mg nicotine patches or placebo patches started 2 weeks before the target quit date. All participants received varenicline for a total of 14 weeks with a 1-week ramp-up and a 1-week taper. Use of the varenicline in combination with the nicotine patch resulted in increased rates of continuous abstinence from smoking at 12 weeks (no smoking from weeks 9 to 12: 55.4% vs. 40.9%; odds ratio, 1.85; 95% confidence interval, 1.19-2.89; P = .007) and at 24 weeks (no smoking from weeks 9 to 24: 49% vs. 32.6%; OR, 1.98; 95% CI, 1.25-3.14; P = .004) (JAMA 2014;312:155-61).
This is a fantastic study answering a lingering question in tobacco control. But what is the theoretical underpinning by which this combination works? Isn’t the NRT blocked by the varenicline? It is possible that the varenicline incompletely saturates the nicotine receptors, which are additionally saturated by the supplemented nicotine. The varenicline effect is mediated through the alpha-4 beta-2 nicotinic receptor, and it is also possible that nicotine binds to nicotine receptor types that varenicline does not bind to, which decreases withdrawal symptoms.
We aren’t exactly sure how this might be working, but a near doubling of the odds of quitting is not to be disregarded. We are also not sure whether the effect holds when one uses other types of NRT such as the nicotine inhaler, nicotine lozenge, nicotine nasal spray, and nicotine gum. In practice, I tend to lean toward a combination of varenicline with the nicotine inhaler since the inhaler can help with some of the behavioral aspects of smoking while the varenicline does its heavy lifting.
Dr. Ebbert is professor of medicine, a general internist at the Mayo Clinic in Rochester, Minn., and a diplomate of the American Board of Addiction Medicine. The opinions expressed are those of the author. Dr. Ebbert reports receiving research support from Pfizer, manufacturer of varenicline and the nicotine inhaler, and consulting fees from GlaxoSmithKline, manufacturer of the nicotine patch. The opinions expressed in this article should not be used to diagnose or treat any medical condition, nor should they be used as a substitute for medical advice from a qualified, board-certified practicing clinician.

FDA approves ex vivo lung perfusion device that preserves donor organs
A device that preserves less-than-ideal donor lungs until they are cleared for transplantation has been approved, the Food and Drug Administration announced on Aug. 12.
The ex vivo perfusion device preserves donated lungs that initially do not meet all the criteria for a transplantable lung. The device does this by warming the donor lung to "near normal body temperature," continuously flushing the lung with a sterile solution, and ventilating the lungs, "which oxygenates the cells and makes it possible for the transplant team to examine the lungs’ airways with a bronchoscope," according to the FDA statement.

The lungs can remain in the machine for up to 4 hours, providing time for the transplant team to evaluate the lungs to determine if they meet the criteria; donor lungs that meet the criteria are then transplanted into a patient.
The device, the XVIVO Perfusion System (XPS) with STEEN Solution, is manufactured by XVIVO Perfusion.
"With this approval, there may be more lungs available for transplant, which could allow more people with end stage lung disease who have exhausted all other treatment options to be able to receive a lung transplant," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said in the statement.
About one in five donor lungs meet the standard transplantation criteria. In the United States, 1,754 lung transplants were performed in 2012 and 1,616 potential recipients were on the lung transplant waiting list at the end of 2012, according to the FDA.
In two studies, outcomes for lung-transplant recipients were similar among those who received a donor lung preserved with the device and those who received donor lungs that were considered ideal and were preserved in cold storage. "Both trials showed that recipients of the ideal and non-ideal lungs had similar survival rates up to 12 months after transplant and similar rates of organ rejection," the FDA statement said.
The manufacturer is required to conduct a long-term study of the effects of the device as a condition of approval.
This is exciting news given the shortage of available lungs which meet the current transplant criteria. Early studies showing similar 12-month survival rates and rates of organ rejection are encouraging. I would like to know if there were similar hospital lengths of stay and if there was a difference in postoperative complications. Also, how significant will the financial impact be using the device? I look forward to the results of long-term studies and hopefully this will be a viable option for our patients.
Dr. Jennifer Cox is assistant professor of pulmonary and critical care medicine critical care selective, University of South Florida, Tampa.
This is exciting news given the shortage of available lungs which meet the current transplant criteria. Early studies showing similar 12-month survival rates and rates of organ rejection are encouraging. I would like to know if there were similar hospital lengths of stay and if there was a difference in postoperative complications. Also, how significant will the financial impact be using the device? I look forward to the results of long-term studies and hopefully this will be a viable option for our patients.
Dr. Jennifer Cox is assistant professor of pulmonary and critical care medicine critical care selective, University of South Florida, Tampa.
This is exciting news given the shortage of available lungs which meet the current transplant criteria. Early studies showing similar 12-month survival rates and rates of organ rejection are encouraging. I would like to know if there were similar hospital lengths of stay and if there was a difference in postoperative complications. Also, how significant will the financial impact be using the device? I look forward to the results of long-term studies and hopefully this will be a viable option for our patients.
Dr. Jennifer Cox is assistant professor of pulmonary and critical care medicine critical care selective, University of South Florida, Tampa.
A device that preserves less-than-ideal donor lungs until they are cleared for transplantation has been approved, the Food and Drug Administration announced on Aug. 12.
The ex vivo perfusion device preserves donated lungs that initially do not meet all the criteria for a transplantable lung. The device does this by warming the donor lung to "near normal body temperature," continuously flushing the lung with a sterile solution, and ventilating the lungs, "which oxygenates the cells and makes it possible for the transplant team to examine the lungs’ airways with a bronchoscope," according to the FDA statement.
The lungs can remain in the machine for up to 4 hours, providing time for the transplant team to evaluate the lungs to determine if they meet the criteria; donor lungs that meet the criteria are then transplanted into a patient.
The device, the XVIVO Perfusion System (XPS) with STEEN Solution, is manufactured by XVIVO Perfusion.
"With this approval, there may be more lungs available for transplant, which could allow more people with end stage lung disease who have exhausted all other treatment options to be able to receive a lung transplant," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said in the statement.
About one in five donor lungs meet the standard transplantation criteria. In the United States, 1,754 lung transplants were performed in 2012 and 1,616 potential recipients were on the lung transplant waiting list at the end of 2012, according to the FDA.
In two studies, outcomes for lung-transplant recipients were similar among those who received a donor lung preserved with the device and those who received donor lungs that were considered ideal and were preserved in cold storage. "Both trials showed that recipients of the ideal and non-ideal lungs had similar survival rates up to 12 months after transplant and similar rates of organ rejection," the FDA statement said.
The manufacturer is required to conduct a long-term study of the effects of the device as a condition of approval.
A device that preserves less-than-ideal donor lungs until they are cleared for transplantation has been approved, the Food and Drug Administration announced on Aug. 12.
The ex vivo perfusion device preserves donated lungs that initially do not meet all the criteria for a transplantable lung. The device does this by warming the donor lung to "near normal body temperature," continuously flushing the lung with a sterile solution, and ventilating the lungs, "which oxygenates the cells and makes it possible for the transplant team to examine the lungs’ airways with a bronchoscope," according to the FDA statement.
The lungs can remain in the machine for up to 4 hours, providing time for the transplant team to evaluate the lungs to determine if they meet the criteria; donor lungs that meet the criteria are then transplanted into a patient.
The device, the XVIVO Perfusion System (XPS) with STEEN Solution, is manufactured by XVIVO Perfusion.
"With this approval, there may be more lungs available for transplant, which could allow more people with end stage lung disease who have exhausted all other treatment options to be able to receive a lung transplant," Christy Foreman, director of the Office of Device Evaluation in the FDA’s Center for Devices and Radiological Health, Silver Spring, Md., said in the statement.
About one in five donor lungs meet the standard transplantation criteria. In the United States, 1,754 lung transplants were performed in 2012 and 1,616 potential recipients were on the lung transplant waiting list at the end of 2012, according to the FDA.
In two studies, outcomes for lung-transplant recipients were similar among those who received a donor lung preserved with the device and those who received donor lungs that were considered ideal and were preserved in cold storage. "Both trials showed that recipients of the ideal and non-ideal lungs had similar survival rates up to 12 months after transplant and similar rates of organ rejection," the FDA statement said.
The manufacturer is required to conduct a long-term study of the effects of the device as a condition of approval.

Drug decreases need for blood transfusions
![]()
Credit: UAB Hospital
Results of a retrospective study suggest an antifibrinolytic agent can significantly reduce the need for blood transfusions after surgery, without increasing the risk of complications.
The agent, tranexamic acid, has been shown to reduce blood loss during or shortly after major joint surgery, but safety concerns remain because large-scale effectiveness studies are lacking.
So researchers set out to evaluate tranexamic acid in a large sample of surgical patients.
The team recounted their efforts in BMJ.
Stavros Memtsoudis, MD, PhD, of the Hospital for Special Surgery in New York, New York, and his colleagues analyzed data from 872,416 patients who underwent total hip or knee replacement procedures at 510 US hospitals between 2006 and 2012.
The researchers compared patients who received tranexamic acid (at 1000 mg,
2000 mg, or 3000 mg) on the day of surgery to patients who did not. The team adjusted their analysis for factors such as patient age and sex, hospital size and location, the type of procedure, and the anesthesia used.
Results showed that use of tranexamic acid was associated with an up to 69% reduction in the need for blood transfusions. Overall, the rate of allogeneic or autologous transfusion was 7.7% among patients who received tranexamic acid and 20.1% among those who did not (P<0.01).
Tranexamic acid use was also linked to a decreased risk of all complications (1.9% vs 2.6%, P<0.001), thromboembolic events (0.6% vs 0.8%, P=0.0057), the need for mechanical ventilation (0.1% vs 0.2%, P=0.0003), and admission to an intensive care unit (3.1% vs 7.5%, P<0.001).
The median length of hospital stay was the same for treated and untreated patients—3 days. But the median cost of hospital stay was lower among tranexamic acid-treated patients—$14,890 vs $15,110 (P<0.001).
A tranexamic acid dose of 2000 mg appeared to have the best effectiveness and safety profile. But the researchers said additional studies are needed to establish optimal dosing schemes and assess subgroup-specific effectiveness and safety. ![]()
![]()
Credit: UAB Hospital
Results of a retrospective study suggest an antifibrinolytic agent can significantly reduce the need for blood transfusions after surgery, without increasing the risk of complications.
The agent, tranexamic acid, has been shown to reduce blood loss during or shortly after major joint surgery, but safety concerns remain because large-scale effectiveness studies are lacking.
So researchers set out to evaluate tranexamic acid in a large sample of surgical patients.
The team recounted their efforts in BMJ.
Stavros Memtsoudis, MD, PhD, of the Hospital for Special Surgery in New York, New York, and his colleagues analyzed data from 872,416 patients who underwent total hip or knee replacement procedures at 510 US hospitals between 2006 and 2012.
The researchers compared patients who received tranexamic acid (at 1000 mg,
2000 mg, or 3000 mg) on the day of surgery to patients who did not. The team adjusted their analysis for factors such as patient age and sex, hospital size and location, the type of procedure, and the anesthesia used.
Results showed that use of tranexamic acid was associated with an up to 69% reduction in the need for blood transfusions. Overall, the rate of allogeneic or autologous transfusion was 7.7% among patients who received tranexamic acid and 20.1% among those who did not (P<0.01).
Tranexamic acid use was also linked to a decreased risk of all complications (1.9% vs 2.6%, P<0.001), thromboembolic events (0.6% vs 0.8%, P=0.0057), the need for mechanical ventilation (0.1% vs 0.2%, P=0.0003), and admission to an intensive care unit (3.1% vs 7.5%, P<0.001).
The median length of hospital stay was the same for treated and untreated patients—3 days. But the median cost of hospital stay was lower among tranexamic acid-treated patients—$14,890 vs $15,110 (P<0.001).
A tranexamic acid dose of 2000 mg appeared to have the best effectiveness and safety profile. But the researchers said additional studies are needed to establish optimal dosing schemes and assess subgroup-specific effectiveness and safety. ![]()
![]()
Credit: UAB Hospital
Results of a retrospective study suggest an antifibrinolytic agent can significantly reduce the need for blood transfusions after surgery, without increasing the risk of complications.
The agent, tranexamic acid, has been shown to reduce blood loss during or shortly after major joint surgery, but safety concerns remain because large-scale effectiveness studies are lacking.
So researchers set out to evaluate tranexamic acid in a large sample of surgical patients.
The team recounted their efforts in BMJ.
Stavros Memtsoudis, MD, PhD, of the Hospital for Special Surgery in New York, New York, and his colleagues analyzed data from 872,416 patients who underwent total hip or knee replacement procedures at 510 US hospitals between 2006 and 2012.
The researchers compared patients who received tranexamic acid (at 1000 mg,
2000 mg, or 3000 mg) on the day of surgery to patients who did not. The team adjusted their analysis for factors such as patient age and sex, hospital size and location, the type of procedure, and the anesthesia used.
Results showed that use of tranexamic acid was associated with an up to 69% reduction in the need for blood transfusions. Overall, the rate of allogeneic or autologous transfusion was 7.7% among patients who received tranexamic acid and 20.1% among those who did not (P<0.01).
Tranexamic acid use was also linked to a decreased risk of all complications (1.9% vs 2.6%, P<0.001), thromboembolic events (0.6% vs 0.8%, P=0.0057), the need for mechanical ventilation (0.1% vs 0.2%, P=0.0003), and admission to an intensive care unit (3.1% vs 7.5%, P<0.001).
The median length of hospital stay was the same for treated and untreated patients—3 days. But the median cost of hospital stay was lower among tranexamic acid-treated patients—$14,890 vs $15,110 (P<0.001).
A tranexamic acid dose of 2000 mg appeared to have the best effectiveness and safety profile. But the researchers said additional studies are needed to establish optimal dosing schemes and assess subgroup-specific effectiveness and safety. ![]()
Molecule is active against MYC-driven malignancies

Credit: Ed Uthman
A small molecule can disrupt the interactions between MYC and its binding partner MAX in MYC-driven cancers, according to research published in PNAS.
The molecule, KJ-Pyr-9, inhibited MYC-induced oncogenic transformation in cell culture but had little to no effect on the oncogenic activity of several unrelated oncoproteins.
KJ-Pyr-9 preferentially interfered with proliferation in a range of cells that overexpressed MYC, including leukemia and lymphoma cells.
In vivo, the molecule inhibited the growth of MYC-amplified human cancer cells.
“We finally hit a home run with this—maybe a grand slam,” said study author Kim Janda, PhD, of The Scripps Research Institute in La Jolla, California.
For years, MYC has challenged researchers seeking to disrupt its activity in cancer cells.
“At room temperature or body temperature, MYC without any binding partners is random and constantly shifting,” said study author Jonathan Ross Hart, PhD, also of The Scripps Research Institute. “It’s like a piece of spaghetti.”
So instead of designing a compound to target the structure of MYC, the researchers tested a range of compounds from a library to see if any could disrupt the interactions between MYC and other proteins important in cell proliferation. One did—the small molecule KJ-Pyr-9.
To further investigate, the researchers ran tests in a variety of cell lines, including chronic myeloid leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, Burkitt lymphoma, and solid tumors. And they tested KJ-Pyr-9 in mouse models of breast cancer.
The experiments showed that MYC-dependent cells die if treated with KJ-Pyr-9. In fact, a dose of KJ-Pyr-9 made it seem as if MYC was not present at all.
When mice with MYC-dependent tumors received KJ-Pyr-9, the tumors showed no growth after 31 days, compared with significant tumor growth in untreated mice.
Dr Janda said he hopes further research will reveal exactly how KJ-Pyr-9 interacts with MYC and how the compound can more effectively reach tumor cells. ![]()
Credit: Ed Uthman
A small molecule can disrupt the interactions between MYC and its binding partner MAX in MYC-driven cancers, according to research published in PNAS.
The molecule, KJ-Pyr-9, inhibited MYC-induced oncogenic transformation in cell culture but had little to no effect on the oncogenic activity of several unrelated oncoproteins.
KJ-Pyr-9 preferentially interfered with proliferation in a range of cells that overexpressed MYC, including leukemia and lymphoma cells.
In vivo, the molecule inhibited the growth of MYC-amplified human cancer cells.
“We finally hit a home run with this—maybe a grand slam,” said study author Kim Janda, PhD, of The Scripps Research Institute in La Jolla, California.
For years, MYC has challenged researchers seeking to disrupt its activity in cancer cells.
“At room temperature or body temperature, MYC without any binding partners is random and constantly shifting,” said study author Jonathan Ross Hart, PhD, also of The Scripps Research Institute. “It’s like a piece of spaghetti.”
So instead of designing a compound to target the structure of MYC, the researchers tested a range of compounds from a library to see if any could disrupt the interactions between MYC and other proteins important in cell proliferation. One did—the small molecule KJ-Pyr-9.
To further investigate, the researchers ran tests in a variety of cell lines, including chronic myeloid leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, Burkitt lymphoma, and solid tumors. And they tested KJ-Pyr-9 in mouse models of breast cancer.
The experiments showed that MYC-dependent cells die if treated with KJ-Pyr-9. In fact, a dose of KJ-Pyr-9 made it seem as if MYC was not present at all.
When mice with MYC-dependent tumors received KJ-Pyr-9, the tumors showed no growth after 31 days, compared with significant tumor growth in untreated mice.
Dr Janda said he hopes further research will reveal exactly how KJ-Pyr-9 interacts with MYC and how the compound can more effectively reach tumor cells. ![]()
Credit: Ed Uthman
A small molecule can disrupt the interactions between MYC and its binding partner MAX in MYC-driven cancers, according to research published in PNAS.
The molecule, KJ-Pyr-9, inhibited MYC-induced oncogenic transformation in cell culture but had little to no effect on the oncogenic activity of several unrelated oncoproteins.
KJ-Pyr-9 preferentially interfered with proliferation in a range of cells that overexpressed MYC, including leukemia and lymphoma cells.
In vivo, the molecule inhibited the growth of MYC-amplified human cancer cells.
“We finally hit a home run with this—maybe a grand slam,” said study author Kim Janda, PhD, of The Scripps Research Institute in La Jolla, California.
For years, MYC has challenged researchers seeking to disrupt its activity in cancer cells.
“At room temperature or body temperature, MYC without any binding partners is random and constantly shifting,” said study author Jonathan Ross Hart, PhD, also of The Scripps Research Institute. “It’s like a piece of spaghetti.”
So instead of designing a compound to target the structure of MYC, the researchers tested a range of compounds from a library to see if any could disrupt the interactions between MYC and other proteins important in cell proliferation. One did—the small molecule KJ-Pyr-9.
To further investigate, the researchers ran tests in a variety of cell lines, including chronic myeloid leukemia, acute lymphoblastic leukemia, acute myeloid leukemia, Burkitt lymphoma, and solid tumors. And they tested KJ-Pyr-9 in mouse models of breast cancer.
The experiments showed that MYC-dependent cells die if treated with KJ-Pyr-9. In fact, a dose of KJ-Pyr-9 made it seem as if MYC was not present at all.
When mice with MYC-dependent tumors received KJ-Pyr-9, the tumors showed no growth after 31 days, compared with significant tumor growth in untreated mice.
Dr Janda said he hopes further research will reveal exactly how KJ-Pyr-9 interacts with MYC and how the compound can more effectively reach tumor cells. ![]()
Viruses can protect mice from malaria
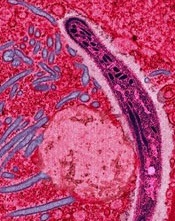
Credit: Ute Frevert
and Margaret Shear
In a new study, genetically altered viruses produced long-lasting antimalaria antibodies in mice and protected many of them from the disease.
The approach, known as vector immunoprophylaxis (VIP), produced antibodies against the Plasmodium falciparum circumsporozoite protein (CSP) and prevented malaria infection in 10% to 100% of mice, depending on the dose and type of viral vector used.
Researchers recounted these results in PNAS.
“We need better ways to fight malaria,” said study author Gary Ketner, PhD, of the Johns Hopkins Bloomberg School of Public Health in Baltimore, Maryland. “And our research suggests [VIP] could be a promising approach.”
To test the approach, Dr Ketner and his colleagues constructed adeno-associated virus (AAV) vectors encoding human immunoglobulin G (hIgG) specific for the P falciparum CSP central repeat by inserting the variable regions of mouse monoclonal antibodies (mAbs) 2A10 and 2C11 into the hIgG framework of the VIP expression plasmid.
The team then injected mice with 1 x 1011 genome copies (GC) of 2A10-AAV, 2C11-AAV, b12-AAV (which protects against HIV), or with buffer.
Within a week, the AAV-transduced mice expressed hIgG at 50 μg/mL to 500 μg/mL in serum. The expression increased until about the 4-week mark, when it reached 1000 μg/mL in some mice.
The mice that received 2A10-AAV or 2C11-AAV expressed antibodies that bound recombinant CSP and recognized whole P falciparum sporozoites. The b12-AAV-transduced mice and buffer-transduced mice did not.
In all AAV-transduced mice, serum antibody concentrations plateaued at 4 to 8 weeks and remained at that level through the end of the study, which was 52 weeks after transduction.
At the 8-week mark, the researchers tested the efficacy of VIP. They introduced—either intravenously or through a mosquito-bite challenge—transgenic Plasmodium berghei rodent sporozoites that incorporate the P falciparum target of the antibody in their CSP.
In the intravenously challenged group, 70% of 2A10-AAV-transduced mice were protected from malaria. In the mosquito-bite-challenged group, 60% of 2A10-AAV-transduced mice and 30% of 2C11-AAV-transduced mice were protected from malaria.
Role of dose and antibody level
To examine the effects of vector dose on mAb production and malaria protection, the researchers compared varying doses of 2A10-AAV to b12-AAV. They tested mice transduced with 3 x 1011 GC of b12-AAV or doses of 2A10-AAV ranging from 3 x 109 GC to 3 x 1011 GC.
The team conducted a mosquito-bite challenge at 11 weeks after transduction. And they found that 70% of the mice that received the highest AAV dose (1 x 1011 GC) were protected, as were 40% of mice that received 3 x 1010 GC and 10% of mice that received 1 x 1010 GC.
All mice transduced with 3 x 109 GC were parasitemic by day 7, and all b12-AAV mice were parasitemic by day 6. There was a signficant correlation between 2A10 antibody concentration and day to parasitemia.
In a subset of mice that produced higher levels of antibodies, the antimalaria protection was 100%. These mice expressed CSP-specific mAb 2A10 at 1 mg/mL or more.
So it seems the protection from malaria is dose-dependent, said study author Cailin Deal, PhD, of the Ragon Institute of MGH, MIT and Harvard in Cambridge, Massachusetts.
“Of course, we don’t know what the human dosage would be,” she added, “but it’s conceivable that the right dosage could completely protect against malaria.” ![]()
Credit: Ute Frevert
and Margaret Shear
In a new study, genetically altered viruses produced long-lasting antimalaria antibodies in mice and protected many of them from the disease.
The approach, known as vector immunoprophylaxis (VIP), produced antibodies against the Plasmodium falciparum circumsporozoite protein (CSP) and prevented malaria infection in 10% to 100% of mice, depending on the dose and type of viral vector used.
Researchers recounted these results in PNAS.
“We need better ways to fight malaria,” said study author Gary Ketner, PhD, of the Johns Hopkins Bloomberg School of Public Health in Baltimore, Maryland. “And our research suggests [VIP] could be a promising approach.”
To test the approach, Dr Ketner and his colleagues constructed adeno-associated virus (AAV) vectors encoding human immunoglobulin G (hIgG) specific for the P falciparum CSP central repeat by inserting the variable regions of mouse monoclonal antibodies (mAbs) 2A10 and 2C11 into the hIgG framework of the VIP expression plasmid.
The team then injected mice with 1 x 1011 genome copies (GC) of 2A10-AAV, 2C11-AAV, b12-AAV (which protects against HIV), or with buffer.
Within a week, the AAV-transduced mice expressed hIgG at 50 μg/mL to 500 μg/mL in serum. The expression increased until about the 4-week mark, when it reached 1000 μg/mL in some mice.
The mice that received 2A10-AAV or 2C11-AAV expressed antibodies that bound recombinant CSP and recognized whole P falciparum sporozoites. The b12-AAV-transduced mice and buffer-transduced mice did not.
In all AAV-transduced mice, serum antibody concentrations plateaued at 4 to 8 weeks and remained at that level through the end of the study, which was 52 weeks after transduction.
At the 8-week mark, the researchers tested the efficacy of VIP. They introduced—either intravenously or through a mosquito-bite challenge—transgenic Plasmodium berghei rodent sporozoites that incorporate the P falciparum target of the antibody in their CSP.
In the intravenously challenged group, 70% of 2A10-AAV-transduced mice were protected from malaria. In the mosquito-bite-challenged group, 60% of 2A10-AAV-transduced mice and 30% of 2C11-AAV-transduced mice were protected from malaria.
Role of dose and antibody level
To examine the effects of vector dose on mAb production and malaria protection, the researchers compared varying doses of 2A10-AAV to b12-AAV. They tested mice transduced with 3 x 1011 GC of b12-AAV or doses of 2A10-AAV ranging from 3 x 109 GC to 3 x 1011 GC.
The team conducted a mosquito-bite challenge at 11 weeks after transduction. And they found that 70% of the mice that received the highest AAV dose (1 x 1011 GC) were protected, as were 40% of mice that received 3 x 1010 GC and 10% of mice that received 1 x 1010 GC.
All mice transduced with 3 x 109 GC were parasitemic by day 7, and all b12-AAV mice were parasitemic by day 6. There was a signficant correlation between 2A10 antibody concentration and day to parasitemia.
In a subset of mice that produced higher levels of antibodies, the antimalaria protection was 100%. These mice expressed CSP-specific mAb 2A10 at 1 mg/mL or more.
So it seems the protection from malaria is dose-dependent, said study author Cailin Deal, PhD, of the Ragon Institute of MGH, MIT and Harvard in Cambridge, Massachusetts.
“Of course, we don’t know what the human dosage would be,” she added, “but it’s conceivable that the right dosage could completely protect against malaria.” ![]()
Credit: Ute Frevert
and Margaret Shear
In a new study, genetically altered viruses produced long-lasting antimalaria antibodies in mice and protected many of them from the disease.
The approach, known as vector immunoprophylaxis (VIP), produced antibodies against the Plasmodium falciparum circumsporozoite protein (CSP) and prevented malaria infection in 10% to 100% of mice, depending on the dose and type of viral vector used.
Researchers recounted these results in PNAS.
“We need better ways to fight malaria,” said study author Gary Ketner, PhD, of the Johns Hopkins Bloomberg School of Public Health in Baltimore, Maryland. “And our research suggests [VIP] could be a promising approach.”
To test the approach, Dr Ketner and his colleagues constructed adeno-associated virus (AAV) vectors encoding human immunoglobulin G (hIgG) specific for the P falciparum CSP central repeat by inserting the variable regions of mouse monoclonal antibodies (mAbs) 2A10 and 2C11 into the hIgG framework of the VIP expression plasmid.
The team then injected mice with 1 x 1011 genome copies (GC) of 2A10-AAV, 2C11-AAV, b12-AAV (which protects against HIV), or with buffer.
Within a week, the AAV-transduced mice expressed hIgG at 50 μg/mL to 500 μg/mL in serum. The expression increased until about the 4-week mark, when it reached 1000 μg/mL in some mice.
The mice that received 2A10-AAV or 2C11-AAV expressed antibodies that bound recombinant CSP and recognized whole P falciparum sporozoites. The b12-AAV-transduced mice and buffer-transduced mice did not.
In all AAV-transduced mice, serum antibody concentrations plateaued at 4 to 8 weeks and remained at that level through the end of the study, which was 52 weeks after transduction.
At the 8-week mark, the researchers tested the efficacy of VIP. They introduced—either intravenously or through a mosquito-bite challenge—transgenic Plasmodium berghei rodent sporozoites that incorporate the P falciparum target of the antibody in their CSP.
In the intravenously challenged group, 70% of 2A10-AAV-transduced mice were protected from malaria. In the mosquito-bite-challenged group, 60% of 2A10-AAV-transduced mice and 30% of 2C11-AAV-transduced mice were protected from malaria.
Role of dose and antibody level
To examine the effects of vector dose on mAb production and malaria protection, the researchers compared varying doses of 2A10-AAV to b12-AAV. They tested mice transduced with 3 x 1011 GC of b12-AAV or doses of 2A10-AAV ranging from 3 x 109 GC to 3 x 1011 GC.
The team conducted a mosquito-bite challenge at 11 weeks after transduction. And they found that 70% of the mice that received the highest AAV dose (1 x 1011 GC) were protected, as were 40% of mice that received 3 x 1010 GC and 10% of mice that received 1 x 1010 GC.
All mice transduced with 3 x 109 GC were parasitemic by day 7, and all b12-AAV mice were parasitemic by day 6. There was a signficant correlation between 2A10 antibody concentration and day to parasitemia.
In a subset of mice that produced higher levels of antibodies, the antimalaria protection was 100%. These mice expressed CSP-specific mAb 2A10 at 1 mg/mL or more.
So it seems the protection from malaria is dose-dependent, said study author Cailin Deal, PhD, of the Ragon Institute of MGH, MIT and Harvard in Cambridge, Massachusetts.
“Of course, we don’t know what the human dosage would be,” she added, “but it’s conceivable that the right dosage could completely protect against malaria.” ![]()
HDAC inhibitor gets orphan status for DLBCL

The US Food and Drug Administration (FDA) has granted orphan designation for the histone deacetylase (HDAC) inhibitor mocetinostat to treat diffuse large B-cell lymphoma (DLBCL). The drug already had orphan designation as a treatment for myelodysplastic syndrome (MDS).
The FDA grants orphan status to support the development of drugs for underserved patient populations or rare disorders affecting fewer than 200,000 people in the US.
Orphan designation provides the drug’s developer, Mirati Therapeutics, Inc., with certain benefits, including market exclusivity upon regulatory approval, exemption of FDA application fees, and tax credits for qualified clinical trials.
Mocetinostat works by reversing aberrant acetylation resulting from mutations in histone acetyltransferases (HATs).
The drug is being developed as a single-agent treatment for patients with DLBCL or bladder cancer characterized by HAT mutations that Mirati believes are critical in the pathogenesis and progression of these cancers.
“We have identified genetic alterations in histone acetylation pathways (CREBBP and EP300) in approximately one-third of DLBCL and bladder tumors,” said Charles Baum, MD, PhD, president and CEO of Mirati.
He added that nonclinical tumor models with these mutations have proven responsive to mocetinostat, so Mirati predicts the HDAC inhibitor will halt tumor progression and reduce tumor burden in patients.
Mocetinostat is also under investigation in phase 2 studies in combination with azacitidine (Vidaza) as a treatment for intermediate- and high-risk MDS.
Mocetinostat previously demonstrated activity, as well as toxicity, in patients with Hodgkin lymphoma. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation for the histone deacetylase (HDAC) inhibitor mocetinostat to treat diffuse large B-cell lymphoma (DLBCL). The drug already had orphan designation as a treatment for myelodysplastic syndrome (MDS).
The FDA grants orphan status to support the development of drugs for underserved patient populations or rare disorders affecting fewer than 200,000 people in the US.
Orphan designation provides the drug’s developer, Mirati Therapeutics, Inc., with certain benefits, including market exclusivity upon regulatory approval, exemption of FDA application fees, and tax credits for qualified clinical trials.
Mocetinostat works by reversing aberrant acetylation resulting from mutations in histone acetyltransferases (HATs).
The drug is being developed as a single-agent treatment for patients with DLBCL or bladder cancer characterized by HAT mutations that Mirati believes are critical in the pathogenesis and progression of these cancers.
“We have identified genetic alterations in histone acetylation pathways (CREBBP and EP300) in approximately one-third of DLBCL and bladder tumors,” said Charles Baum, MD, PhD, president and CEO of Mirati.
He added that nonclinical tumor models with these mutations have proven responsive to mocetinostat, so Mirati predicts the HDAC inhibitor will halt tumor progression and reduce tumor burden in patients.
Mocetinostat is also under investigation in phase 2 studies in combination with azacitidine (Vidaza) as a treatment for intermediate- and high-risk MDS.
Mocetinostat previously demonstrated activity, as well as toxicity, in patients with Hodgkin lymphoma. ![]()
The US Food and Drug Administration (FDA) has granted orphan designation for the histone deacetylase (HDAC) inhibitor mocetinostat to treat diffuse large B-cell lymphoma (DLBCL). The drug already had orphan designation as a treatment for myelodysplastic syndrome (MDS).
The FDA grants orphan status to support the development of drugs for underserved patient populations or rare disorders affecting fewer than 200,000 people in the US.
Orphan designation provides the drug’s developer, Mirati Therapeutics, Inc., with certain benefits, including market exclusivity upon regulatory approval, exemption of FDA application fees, and tax credits for qualified clinical trials.
Mocetinostat works by reversing aberrant acetylation resulting from mutations in histone acetyltransferases (HATs).
The drug is being developed as a single-agent treatment for patients with DLBCL or bladder cancer characterized by HAT mutations that Mirati believes are critical in the pathogenesis and progression of these cancers.
“We have identified genetic alterations in histone acetylation pathways (CREBBP and EP300) in approximately one-third of DLBCL and bladder tumors,” said Charles Baum, MD, PhD, president and CEO of Mirati.
He added that nonclinical tumor models with these mutations have proven responsive to mocetinostat, so Mirati predicts the HDAC inhibitor will halt tumor progression and reduce tumor burden in patients.
Mocetinostat is also under investigation in phase 2 studies in combination with azacitidine (Vidaza) as a treatment for intermediate- and high-risk MDS.
Mocetinostat previously demonstrated activity, as well as toxicity, in patients with Hodgkin lymphoma. ![]()
What do the guidelines say?
Atopic dermatitis remains a challenging condition.
The 2014 guidelines of care for the management of atopic dermatitis (AD) are being published by the American Academy of Dermatology in a series of four parts. Each part begins with a disclaimer stating that, "the ultimate judgment regarding the propriety of any specific therapy must be made by the physician and the patient in light of all the circumstances presented by the individual patient and the known variability and biologic behavior of the disease." The disclaimer continues, "This guideline reflects the best available data at the time the guideline was prepared. The results of future studies may require revisions to the recommendations in this guideline to reflect new data."
• Section 1: Diagnosis and assessment of atopic dermatitis. This section includes risk factors for the development of AD, diagnostic and monitoring techniques, assessment and outcomes, and clinical associations in AD patients (J. Am. Acad. Dermatol. 2014;70:338-51).
• Section 2: Management and treatment of atopic dermatitis with topical therapies. This section focuses on recommendations for the use of nonpharmacologic and topical therapies in the management of AD (J. Am. Acad. Dermatol. 2014;71:116-32).
• Section 3: Management and treatment with phototherapy and systemic agents. This section reviews indications for the use of phototherapy and systemic immunomodulators for treating AD, including side-effect profiles and clinical considerations for treating children (J. Am. Acad. Dermatol. 2014;71:327-49).
• Section 4: The fourth and final section of the guidelines is expected to be published in the September 2014 issue of the Journal of the American Academy of Dermatology.
No outside funding sources were involved in the creation of the guidelines. Disclosures of members of the guidelines committee are available following full text of each guidelines section in print and online.
Atopic dermatitis remains a challenging condition.
The 2014 guidelines of care for the management of atopic dermatitis (AD) are being published by the American Academy of Dermatology in a series of four parts. Each part begins with a disclaimer stating that, "the ultimate judgment regarding the propriety of any specific therapy must be made by the physician and the patient in light of all the circumstances presented by the individual patient and the known variability and biologic behavior of the disease." The disclaimer continues, "This guideline reflects the best available data at the time the guideline was prepared. The results of future studies may require revisions to the recommendations in this guideline to reflect new data."
• Section 1: Diagnosis and assessment of atopic dermatitis. This section includes risk factors for the development of AD, diagnostic and monitoring techniques, assessment and outcomes, and clinical associations in AD patients (J. Am. Acad. Dermatol. 2014;70:338-51).
• Section 2: Management and treatment of atopic dermatitis with topical therapies. This section focuses on recommendations for the use of nonpharmacologic and topical therapies in the management of AD (J. Am. Acad. Dermatol. 2014;71:116-32).
• Section 3: Management and treatment with phototherapy and systemic agents. This section reviews indications for the use of phototherapy and systemic immunomodulators for treating AD, including side-effect profiles and clinical considerations for treating children (J. Am. Acad. Dermatol. 2014;71:327-49).
• Section 4: The fourth and final section of the guidelines is expected to be published in the September 2014 issue of the Journal of the American Academy of Dermatology.
No outside funding sources were involved in the creation of the guidelines. Disclosures of members of the guidelines committee are available following full text of each guidelines section in print and online.
Atopic dermatitis remains a challenging condition.
The 2014 guidelines of care for the management of atopic dermatitis (AD) are being published by the American Academy of Dermatology in a series of four parts. Each part begins with a disclaimer stating that, "the ultimate judgment regarding the propriety of any specific therapy must be made by the physician and the patient in light of all the circumstances presented by the individual patient and the known variability and biologic behavior of the disease." The disclaimer continues, "This guideline reflects the best available data at the time the guideline was prepared. The results of future studies may require revisions to the recommendations in this guideline to reflect new data."
• Section 1: Diagnosis and assessment of atopic dermatitis. This section includes risk factors for the development of AD, diagnostic and monitoring techniques, assessment and outcomes, and clinical associations in AD patients (J. Am. Acad. Dermatol. 2014;70:338-51).
• Section 2: Management and treatment of atopic dermatitis with topical therapies. This section focuses on recommendations for the use of nonpharmacologic and topical therapies in the management of AD (J. Am. Acad. Dermatol. 2014;71:116-32).
• Section 3: Management and treatment with phototherapy and systemic agents. This section reviews indications for the use of phototherapy and systemic immunomodulators for treating AD, including side-effect profiles and clinical considerations for treating children (J. Am. Acad. Dermatol. 2014;71:327-49).
• Section 4: The fourth and final section of the guidelines is expected to be published in the September 2014 issue of the Journal of the American Academy of Dermatology.
No outside funding sources were involved in the creation of the guidelines. Disclosures of members of the guidelines committee are available following full text of each guidelines section in print and online.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Caring for an expectant mother
"Mrs. Jones in ED room 12? Septic with severe multilobar pneumonia and hypoxemic respiratory failure? Got it. I’m on my way down to the ED now." The heart races in anticipation of caring for this critically ill patient near death’s door. But it’s okay. You know exactly what you have to do. You have done it many times before. No problem. Just as you are about to hang up the phone you learn more ...
"What’s that? She is 22 weeks pregnant? Uh, all right."
Suddenly, what initially seemed to be a routine case takes a complicated twist. You are no longer caring for one life, you are caring for two (or more), and one of those is extremely frail and vulnerable. The bugs, the drugs, maintaining adequate perfusion of vital organs – both hers and her baby’s – the questions of "What if ... ?" and "Should I ... ?" race through your mind. Should you get infectious disease and pulmonary consultations for an added layer of protection or should you treat her as you have treated so many others before her, the caveat, of course, being the need to check every single drug for its teratogenicity.
If you have ever felt at least a little queasiness in the pit of your stomach when called upon to care for an expectant mother, you are notalone. I think it is natural to feel a bit uneasy when we care for pregnant patients because most of us do it rather infrequently and there may not be room for a do-over even if you make the tiniest mistake. Each drug we order has the potential to do harm and any missed or delayed diagnosis may be tolerated well by mom, but maybe not much by the tiny baby growing inside her. Get it right and the family may live a storybook fantasy. Yet, a single miscalculation, an honest mistake, and that innocent child’s future may be compromised or destroyed.
Fortunately, these days, horror stories seem to be less frequent than in the past. But we must remain vigilant to not only optimally treat our patient (mommy), but also to protect our patients (mommy and baby) from future complications of treatment. If there is any doubt, go ahead and call an infectious disease and pulmonary consultation. Sometimes, everyone sleeps better when we do.
Dr. A. Maria Hester is a hospitalist with Baltimore-Washington Medical Center, Glen Burnie, Md., who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS.
"Mrs. Jones in ED room 12? Septic with severe multilobar pneumonia and hypoxemic respiratory failure? Got it. I’m on my way down to the ED now." The heart races in anticipation of caring for this critically ill patient near death’s door. But it’s okay. You know exactly what you have to do. You have done it many times before. No problem. Just as you are about to hang up the phone you learn more ...
"What’s that? She is 22 weeks pregnant? Uh, all right."
Suddenly, what initially seemed to be a routine case takes a complicated twist. You are no longer caring for one life, you are caring for two (or more), and one of those is extremely frail and vulnerable. The bugs, the drugs, maintaining adequate perfusion of vital organs – both hers and her baby’s – the questions of "What if ... ?" and "Should I ... ?" race through your mind. Should you get infectious disease and pulmonary consultations for an added layer of protection or should you treat her as you have treated so many others before her, the caveat, of course, being the need to check every single drug for its teratogenicity.
If you have ever felt at least a little queasiness in the pit of your stomach when called upon to care for an expectant mother, you are notalone. I think it is natural to feel a bit uneasy when we care for pregnant patients because most of us do it rather infrequently and there may not be room for a do-over even if you make the tiniest mistake. Each drug we order has the potential to do harm and any missed or delayed diagnosis may be tolerated well by mom, but maybe not much by the tiny baby growing inside her. Get it right and the family may live a storybook fantasy. Yet, a single miscalculation, an honest mistake, and that innocent child’s future may be compromised or destroyed.
Fortunately, these days, horror stories seem to be less frequent than in the past. But we must remain vigilant to not only optimally treat our patient (mommy), but also to protect our patients (mommy and baby) from future complications of treatment. If there is any doubt, go ahead and call an infectious disease and pulmonary consultation. Sometimes, everyone sleeps better when we do.
Dr. A. Maria Hester is a hospitalist with Baltimore-Washington Medical Center, Glen Burnie, Md., who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS.
"Mrs. Jones in ED room 12? Septic with severe multilobar pneumonia and hypoxemic respiratory failure? Got it. I’m on my way down to the ED now." The heart races in anticipation of caring for this critically ill patient near death’s door. But it’s okay. You know exactly what you have to do. You have done it many times before. No problem. Just as you are about to hang up the phone you learn more ...
"What’s that? She is 22 weeks pregnant? Uh, all right."
Suddenly, what initially seemed to be a routine case takes a complicated twist. You are no longer caring for one life, you are caring for two (or more), and one of those is extremely frail and vulnerable. The bugs, the drugs, maintaining adequate perfusion of vital organs – both hers and her baby’s – the questions of "What if ... ?" and "Should I ... ?" race through your mind. Should you get infectious disease and pulmonary consultations for an added layer of protection or should you treat her as you have treated so many others before her, the caveat, of course, being the need to check every single drug for its teratogenicity.
If you have ever felt at least a little queasiness in the pit of your stomach when called upon to care for an expectant mother, you are notalone. I think it is natural to feel a bit uneasy when we care for pregnant patients because most of us do it rather infrequently and there may not be room for a do-over even if you make the tiniest mistake. Each drug we order has the potential to do harm and any missed or delayed diagnosis may be tolerated well by mom, but maybe not much by the tiny baby growing inside her. Get it right and the family may live a storybook fantasy. Yet, a single miscalculation, an honest mistake, and that innocent child’s future may be compromised or destroyed.
Fortunately, these days, horror stories seem to be less frequent than in the past. But we must remain vigilant to not only optimally treat our patient (mommy), but also to protect our patients (mommy and baby) from future complications of treatment. If there is any doubt, go ahead and call an infectious disease and pulmonary consultation. Sometimes, everyone sleeps better when we do.
Dr. A. Maria Hester is a hospitalist with Baltimore-Washington Medical Center, Glen Burnie, Md., who has a passion for empowering patients to partner in their health care. She is the creator of the Patient Whiz, a patient-engagement app for iOS.
Almonds
A tree species belonging to the Rosaceae family and native to the Middle East and South Asia, Prunus dulcis (also called Prunus amygdalus), popularly known as almond, is cultivated widely in warm, arid locations and used primarily for its edible seeds (J. Agric. Food Chem. 2007;55:8498-507; J. Agric. Food Chem. 2006;54:312-8). Almonds and almond skins are rich in polyphenols and are an important source of these phytonutrients, particularly flavan-3-ols and flavonols (J. Proteome Res. 2010;9:5859-67). Almonds also are known to contain an abundance of fiber (Anaerobe 2014;26:1-6). Almond extracts are used in cosmetic formulations because of their high concentration of polyphenols, particularly flavonoids and phenolic acids (Int. J. Curr. Pharm. Res. 2011;3:57-9).
Key components
In 2006, Milbury et al. determined that the main flavonoids and phenolic acids in Prunus dulcis skins and kernels are isorhamnetin-3-O-rutinoside and isorhamnetin-3-O-glucoside (in combination), catechin, kaempferol-3-O-rutinoside, epicatechin, quercetin-3-O-galactoside, and isorhamnetin-3-O-galactoside (J. Agric. Food Chem. 2006;54:5027-33).

In a separate study that year, Wijeratne et al. identified quercetin, isorhamnetin, quercitrin, kaempferol 3-O-rutinoside, isorhamnetin 3-O-glucoside, and morin as the primary flavonoids in various whole-seed, brown, skin, and green-shell-cover almond extracts (J. Agric. Food Chem. 2006;54:312-8). Almond seed skin also has been shown to contain highly polymerized polyphenols that exhibit potent alpha-amylase inhibitory activity, thus slowing the absorption of carbohydrate (J. Agric. Food Chem. 2013;61:4570-6).
In 2010, Bartolomé et al. identified A- and B-type procyanidin, propelargonidin, and prodelphinidin polymers in almond skins. In human plasma and urine samples taken after consumption of almond skin polyphenols, the investigators detected O-methyl glucuronide sulfate and O-methyl sulfate derivatives of (epi)catechin; the glucuronide conjugates of naringenin and isorhamnetin; and sulfate conjugates of isorhamnetin, together with conjugates of hydroxyphenylvalerolactones. They also identified various microbial-derived metabolites, including hydroxyphenylpropionic, hydroxyphenylacetic, hydroxycinnamic, hydroxybenzoic, and hydroxyhippuric acids (Arch. Biochem. Biophys. 2010;501:124-33).
A functional food: Antioxidant constituents and activity
In a study of two human subjects conducted in 2009, Urpi-Sarda et al. profiled microbial-derived phenolic metabolites in plasma and urine samples before and after the consumption of almond skins. They detected glucuronide, O-methyl glucuronide sulfate, and O-methyl sulfate derivatives of epicatechin, the glucuronide conjugates of naringenin and glucuronide, and sulfate conjugates of isorhamnetin. The researchers also detected, in their glucuronide and sulfate forms, microbial-derived metabolites of flavanols, including 5-(dihydroxyphenyl)-gamma-valerolactone and 5-(hydroxymethoxyphenyl)-gamma-valerolactone (J. Agric. Food Chem. 2009;57:10134-42).
A 2010 study with 24 volunteers conducted by Llorach et al. investigated urinary metabolome changes during the 24-hour period following ingestion of a single dose of almond skin extract. The participants, who were following a polyphenol-free diet for 48 hours, were divided into two groups: the almond skin group or a placebo group. Thirty-four metabolites were linked to the consumption of the almond extract (J. Proteome Res. 2010;9:5859-67).

Monagas et al. previously led a comprehensive investigation of the phenolic composition of almond skins to elucidate its potential as a functional food. They identified 33 compounds, including flavanols, flavonols, dihydroflavonols, and flavanones, as well as nonflavonoid substances. The most abundant phenols were flavanols (38%-57%) and flavonol glycosides (14%-35%). Further, the researchers measured the oxygen radical absorbance capacity of almond skins at 0.398-0.500 mmol Trolox/g, a range that suggests its antioxidant potency (J. Agric. Food Chem. 2007;55:8498-507).
In 2008, Garrido et al. investigated the phenolic composition and antioxidant activity of almond skins (Prunus dulcis) obtained from three almond varieties and through various industrial processes, including blanching, blanching and drying, as well as roasting. Identified were 31 phenolic compounds, including flavan-3-ols, flavonol glycosides, hydroxybenzoic acids and aldehydes, flavonol aglycones, flavanone glycosides, flavanone aglycones, hydroxycinnamic acids, and dihydroflavonol aglycones. Phenolic content as well as antioxidant activity were significantly higher in the roasted samples (J. Food Sci. 2008;73:C106-15).
In 2014, Liu et al. conducted a study in healthy humans of the potential prebiotic effects of consuming almonds and almond skins. For 6 weeks, 48 adult volunteers ingested a daily dose of roasted almonds, almond skins, or a positive control (commercial fructo-oligosaccharides). The investigators collected fecal samples and analyzed them for microbiota composition. They observed significant increases in populations of Bifidobacterium spp. and Lactobacillus spp. in fecal samples after almond or almond skin supplementation; substantial inhibition of the growth of the pathogen Clostridium perfringens; and favorable changes in bacterial enzyme activities. They concluded that almonds and almond skins appear to exhibit potential prebiotic qualities (Anaerobe 2014;26:1-6).
Topical antiaging potential
In 2011, Sachdeva and Katyal assessed the antioxidant and antiwrinkle effects of almond skin extracts in UV-induced photoaging in mice. Twenty-five mice were used as an unirradiated control, receiving neither UV exposure nor almond skin treatment. A second group of 24 mice received only UV exposure of 5 minutes twice a day, and served as an irradiated control. Further groups of 24 mice received both UV radiation and treatment with prepared formulation. The treatment groups received various topical almond skin extract doses 2 hours prior to the same level of UV exposure. Significant decreases in malondialdehyde and increases in glutathione levels, respectively, suggested to the investigators that the almond skin extracts effectively scavenged free radicals while also enhancing moisturization. They concluded that almond skin extracts display potential as antiaging ingredients in topical cosmetic formulations (Int. J. Curr. Pharm. Res. 2011;3:57-9).
Conclusion
Almonds are believed to be a healthy addition to the human diet, with their regular consumption thought to confer cardiovascular benefits. The healthful effects of dietary intake of almonds are often attributed to the presence of several polyphenolic constituents. While it is speculated that such ingredients also play a role in imparting cutaneous benefits, the body of evidence supporting such claims remains sparse to date. Nevertheless, P. dulcis is incorporated in various cosmetic formulations. More research is necessary to ascertain whether such inclusion is warranted.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in Miami Beach. She founded the cosmetic dermatology center at the University of Miami in 1997. Dr. Baumann wrote the textbook "Cosmetic Dermatology: Principles and Practice" (McGraw-Hill, April 2002), and a book for consumers, "The Skin Type Solution" (Bantam, 2006). She has contributed to the Cosmeceutical Critique column in Skin & Allergy News since January 2001. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Galderma, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Stiefel, Topix Pharmaceuticals, and Unilever.
A tree species belonging to the Rosaceae family and native to the Middle East and South Asia, Prunus dulcis (also called Prunus amygdalus), popularly known as almond, is cultivated widely in warm, arid locations and used primarily for its edible seeds (J. Agric. Food Chem. 2007;55:8498-507; J. Agric. Food Chem. 2006;54:312-8). Almonds and almond skins are rich in polyphenols and are an important source of these phytonutrients, particularly flavan-3-ols and flavonols (J. Proteome Res. 2010;9:5859-67). Almonds also are known to contain an abundance of fiber (Anaerobe 2014;26:1-6). Almond extracts are used in cosmetic formulations because of their high concentration of polyphenols, particularly flavonoids and phenolic acids (Int. J. Curr. Pharm. Res. 2011;3:57-9).
Key components
In 2006, Milbury et al. determined that the main flavonoids and phenolic acids in Prunus dulcis skins and kernels are isorhamnetin-3-O-rutinoside and isorhamnetin-3-O-glucoside (in combination), catechin, kaempferol-3-O-rutinoside, epicatechin, quercetin-3-O-galactoside, and isorhamnetin-3-O-galactoside (J. Agric. Food Chem. 2006;54:5027-33).
In a separate study that year, Wijeratne et al. identified quercetin, isorhamnetin, quercitrin, kaempferol 3-O-rutinoside, isorhamnetin 3-O-glucoside, and morin as the primary flavonoids in various whole-seed, brown, skin, and green-shell-cover almond extracts (J. Agric. Food Chem. 2006;54:312-8). Almond seed skin also has been shown to contain highly polymerized polyphenols that exhibit potent alpha-amylase inhibitory activity, thus slowing the absorption of carbohydrate (J. Agric. Food Chem. 2013;61:4570-6).
In 2010, Bartolomé et al. identified A- and B-type procyanidin, propelargonidin, and prodelphinidin polymers in almond skins. In human plasma and urine samples taken after consumption of almond skin polyphenols, the investigators detected O-methyl glucuronide sulfate and O-methyl sulfate derivatives of (epi)catechin; the glucuronide conjugates of naringenin and isorhamnetin; and sulfate conjugates of isorhamnetin, together with conjugates of hydroxyphenylvalerolactones. They also identified various microbial-derived metabolites, including hydroxyphenylpropionic, hydroxyphenylacetic, hydroxycinnamic, hydroxybenzoic, and hydroxyhippuric acids (Arch. Biochem. Biophys. 2010;501:124-33).
A functional food: Antioxidant constituents and activity
In a study of two human subjects conducted in 2009, Urpi-Sarda et al. profiled microbial-derived phenolic metabolites in plasma and urine samples before and after the consumption of almond skins. They detected glucuronide, O-methyl glucuronide sulfate, and O-methyl sulfate derivatives of epicatechin, the glucuronide conjugates of naringenin and glucuronide, and sulfate conjugates of isorhamnetin. The researchers also detected, in their glucuronide and sulfate forms, microbial-derived metabolites of flavanols, including 5-(dihydroxyphenyl)-gamma-valerolactone and 5-(hydroxymethoxyphenyl)-gamma-valerolactone (J. Agric. Food Chem. 2009;57:10134-42).
A 2010 study with 24 volunteers conducted by Llorach et al. investigated urinary metabolome changes during the 24-hour period following ingestion of a single dose of almond skin extract. The participants, who were following a polyphenol-free diet for 48 hours, were divided into two groups: the almond skin group or a placebo group. Thirty-four metabolites were linked to the consumption of the almond extract (J. Proteome Res. 2010;9:5859-67).
Monagas et al. previously led a comprehensive investigation of the phenolic composition of almond skins to elucidate its potential as a functional food. They identified 33 compounds, including flavanols, flavonols, dihydroflavonols, and flavanones, as well as nonflavonoid substances. The most abundant phenols were flavanols (38%-57%) and flavonol glycosides (14%-35%). Further, the researchers measured the oxygen radical absorbance capacity of almond skins at 0.398-0.500 mmol Trolox/g, a range that suggests its antioxidant potency (J. Agric. Food Chem. 2007;55:8498-507).
In 2008, Garrido et al. investigated the phenolic composition and antioxidant activity of almond skins (Prunus dulcis) obtained from three almond varieties and through various industrial processes, including blanching, blanching and drying, as well as roasting. Identified were 31 phenolic compounds, including flavan-3-ols, flavonol glycosides, hydroxybenzoic acids and aldehydes, flavonol aglycones, flavanone glycosides, flavanone aglycones, hydroxycinnamic acids, and dihydroflavonol aglycones. Phenolic content as well as antioxidant activity were significantly higher in the roasted samples (J. Food Sci. 2008;73:C106-15).
In 2014, Liu et al. conducted a study in healthy humans of the potential prebiotic effects of consuming almonds and almond skins. For 6 weeks, 48 adult volunteers ingested a daily dose of roasted almonds, almond skins, or a positive control (commercial fructo-oligosaccharides). The investigators collected fecal samples and analyzed them for microbiota composition. They observed significant increases in populations of Bifidobacterium spp. and Lactobacillus spp. in fecal samples after almond or almond skin supplementation; substantial inhibition of the growth of the pathogen Clostridium perfringens; and favorable changes in bacterial enzyme activities. They concluded that almonds and almond skins appear to exhibit potential prebiotic qualities (Anaerobe 2014;26:1-6).
Topical antiaging potential
In 2011, Sachdeva and Katyal assessed the antioxidant and antiwrinkle effects of almond skin extracts in UV-induced photoaging in mice. Twenty-five mice were used as an unirradiated control, receiving neither UV exposure nor almond skin treatment. A second group of 24 mice received only UV exposure of 5 minutes twice a day, and served as an irradiated control. Further groups of 24 mice received both UV radiation and treatment with prepared formulation. The treatment groups received various topical almond skin extract doses 2 hours prior to the same level of UV exposure. Significant decreases in malondialdehyde and increases in glutathione levels, respectively, suggested to the investigators that the almond skin extracts effectively scavenged free radicals while also enhancing moisturization. They concluded that almond skin extracts display potential as antiaging ingredients in topical cosmetic formulations (Int. J. Curr. Pharm. Res. 2011;3:57-9).
Conclusion
Almonds are believed to be a healthy addition to the human diet, with their regular consumption thought to confer cardiovascular benefits. The healthful effects of dietary intake of almonds are often attributed to the presence of several polyphenolic constituents. While it is speculated that such ingredients also play a role in imparting cutaneous benefits, the body of evidence supporting such claims remains sparse to date. Nevertheless, P. dulcis is incorporated in various cosmetic formulations. More research is necessary to ascertain whether such inclusion is warranted.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in Miami Beach. She founded the cosmetic dermatology center at the University of Miami in 1997. Dr. Baumann wrote the textbook "Cosmetic Dermatology: Principles and Practice" (McGraw-Hill, April 2002), and a book for consumers, "The Skin Type Solution" (Bantam, 2006). She has contributed to the Cosmeceutical Critique column in Skin & Allergy News since January 2001. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Galderma, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Stiefel, Topix Pharmaceuticals, and Unilever.
A tree species belonging to the Rosaceae family and native to the Middle East and South Asia, Prunus dulcis (also called Prunus amygdalus), popularly known as almond, is cultivated widely in warm, arid locations and used primarily for its edible seeds (J. Agric. Food Chem. 2007;55:8498-507; J. Agric. Food Chem. 2006;54:312-8). Almonds and almond skins are rich in polyphenols and are an important source of these phytonutrients, particularly flavan-3-ols and flavonols (J. Proteome Res. 2010;9:5859-67). Almonds also are known to contain an abundance of fiber (Anaerobe 2014;26:1-6). Almond extracts are used in cosmetic formulations because of their high concentration of polyphenols, particularly flavonoids and phenolic acids (Int. J. Curr. Pharm. Res. 2011;3:57-9).
Key components
In 2006, Milbury et al. determined that the main flavonoids and phenolic acids in Prunus dulcis skins and kernels are isorhamnetin-3-O-rutinoside and isorhamnetin-3-O-glucoside (in combination), catechin, kaempferol-3-O-rutinoside, epicatechin, quercetin-3-O-galactoside, and isorhamnetin-3-O-galactoside (J. Agric. Food Chem. 2006;54:5027-33).
In a separate study that year, Wijeratne et al. identified quercetin, isorhamnetin, quercitrin, kaempferol 3-O-rutinoside, isorhamnetin 3-O-glucoside, and morin as the primary flavonoids in various whole-seed, brown, skin, and green-shell-cover almond extracts (J. Agric. Food Chem. 2006;54:312-8). Almond seed skin also has been shown to contain highly polymerized polyphenols that exhibit potent alpha-amylase inhibitory activity, thus slowing the absorption of carbohydrate (J. Agric. Food Chem. 2013;61:4570-6).
In 2010, Bartolomé et al. identified A- and B-type procyanidin, propelargonidin, and prodelphinidin polymers in almond skins. In human plasma and urine samples taken after consumption of almond skin polyphenols, the investigators detected O-methyl glucuronide sulfate and O-methyl sulfate derivatives of (epi)catechin; the glucuronide conjugates of naringenin and isorhamnetin; and sulfate conjugates of isorhamnetin, together with conjugates of hydroxyphenylvalerolactones. They also identified various microbial-derived metabolites, including hydroxyphenylpropionic, hydroxyphenylacetic, hydroxycinnamic, hydroxybenzoic, and hydroxyhippuric acids (Arch. Biochem. Biophys. 2010;501:124-33).
A functional food: Antioxidant constituents and activity
In a study of two human subjects conducted in 2009, Urpi-Sarda et al. profiled microbial-derived phenolic metabolites in plasma and urine samples before and after the consumption of almond skins. They detected glucuronide, O-methyl glucuronide sulfate, and O-methyl sulfate derivatives of epicatechin, the glucuronide conjugates of naringenin and glucuronide, and sulfate conjugates of isorhamnetin. The researchers also detected, in their glucuronide and sulfate forms, microbial-derived metabolites of flavanols, including 5-(dihydroxyphenyl)-gamma-valerolactone and 5-(hydroxymethoxyphenyl)-gamma-valerolactone (J. Agric. Food Chem. 2009;57:10134-42).
A 2010 study with 24 volunteers conducted by Llorach et al. investigated urinary metabolome changes during the 24-hour period following ingestion of a single dose of almond skin extract. The participants, who were following a polyphenol-free diet for 48 hours, were divided into two groups: the almond skin group or a placebo group. Thirty-four metabolites were linked to the consumption of the almond extract (J. Proteome Res. 2010;9:5859-67).
Monagas et al. previously led a comprehensive investigation of the phenolic composition of almond skins to elucidate its potential as a functional food. They identified 33 compounds, including flavanols, flavonols, dihydroflavonols, and flavanones, as well as nonflavonoid substances. The most abundant phenols were flavanols (38%-57%) and flavonol glycosides (14%-35%). Further, the researchers measured the oxygen radical absorbance capacity of almond skins at 0.398-0.500 mmol Trolox/g, a range that suggests its antioxidant potency (J. Agric. Food Chem. 2007;55:8498-507).
In 2008, Garrido et al. investigated the phenolic composition and antioxidant activity of almond skins (Prunus dulcis) obtained from three almond varieties and through various industrial processes, including blanching, blanching and drying, as well as roasting. Identified were 31 phenolic compounds, including flavan-3-ols, flavonol glycosides, hydroxybenzoic acids and aldehydes, flavonol aglycones, flavanone glycosides, flavanone aglycones, hydroxycinnamic acids, and dihydroflavonol aglycones. Phenolic content as well as antioxidant activity were significantly higher in the roasted samples (J. Food Sci. 2008;73:C106-15).
In 2014, Liu et al. conducted a study in healthy humans of the potential prebiotic effects of consuming almonds and almond skins. For 6 weeks, 48 adult volunteers ingested a daily dose of roasted almonds, almond skins, or a positive control (commercial fructo-oligosaccharides). The investigators collected fecal samples and analyzed them for microbiota composition. They observed significant increases in populations of Bifidobacterium spp. and Lactobacillus spp. in fecal samples after almond or almond skin supplementation; substantial inhibition of the growth of the pathogen Clostridium perfringens; and favorable changes in bacterial enzyme activities. They concluded that almonds and almond skins appear to exhibit potential prebiotic qualities (Anaerobe 2014;26:1-6).
Topical antiaging potential
In 2011, Sachdeva and Katyal assessed the antioxidant and antiwrinkle effects of almond skin extracts in UV-induced photoaging in mice. Twenty-five mice were used as an unirradiated control, receiving neither UV exposure nor almond skin treatment. A second group of 24 mice received only UV exposure of 5 minutes twice a day, and served as an irradiated control. Further groups of 24 mice received both UV radiation and treatment with prepared formulation. The treatment groups received various topical almond skin extract doses 2 hours prior to the same level of UV exposure. Significant decreases in malondialdehyde and increases in glutathione levels, respectively, suggested to the investigators that the almond skin extracts effectively scavenged free radicals while also enhancing moisturization. They concluded that almond skin extracts display potential as antiaging ingredients in topical cosmetic formulations (Int. J. Curr. Pharm. Res. 2011;3:57-9).
Conclusion
Almonds are believed to be a healthy addition to the human diet, with their regular consumption thought to confer cardiovascular benefits. The healthful effects of dietary intake of almonds are often attributed to the presence of several polyphenolic constituents. While it is speculated that such ingredients also play a role in imparting cutaneous benefits, the body of evidence supporting such claims remains sparse to date. Nevertheless, P. dulcis is incorporated in various cosmetic formulations. More research is necessary to ascertain whether such inclusion is warranted.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in Miami Beach. She founded the cosmetic dermatology center at the University of Miami in 1997. Dr. Baumann wrote the textbook "Cosmetic Dermatology: Principles and Practice" (McGraw-Hill, April 2002), and a book for consumers, "The Skin Type Solution" (Bantam, 2006). She has contributed to the Cosmeceutical Critique column in Skin & Allergy News since January 2001. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Galderma, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Stiefel, Topix Pharmaceuticals, and Unilever.
