User login
ILDS establishes guidelines for treating AK patients
The International Leagues of Dermatological Societies (ILDS) in cooperation with the European Dermatology Forum has developed consensus-based guidelines for the treatment of actinic keratosis (AK), which are published in the Journal of the European Academy of Dermatology and Venereology.
“The guidelines were elaborated along adapted recommendations by the WHO guidelines review committee and the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) working group,” say R. N. Werner and colleagues of the Medical University of Berlin. The guidelines include recommendations for treatment of different subgroups of AK patients, how to make an AK diagnosis, how to assess AK patients, and how to define AK.

The ILDS recommends or suggests the following interventions for treating patients who have single AK lesions:
• Cryotherapy
• Curettage (discrete, hyperkeratotic lesions)
• 0.5% 5-fluorouracil (5-FU)
• 5% 5-FU
• 0.5% 5-FU + 10% salicylic acid (discrete, hyperkeratotic lesions)
• 3.75% imiquimod
• 5% imiquimod
• Ingenol mebutate 0.015% (lesions on the face or scalp) and ingenol mebutate 0.05% (lesions on the trunk or extremities)
• 5-aminolevulinic acid-photodynamic therapy (ALA-PDT)
• methylaminolevulinate-photodynamic therapy (MAL-PDT)
For patients with multiple AK lesions/field cancerization, the ILDS recommends* or suggests that patients use the following therapies:
• 0.5% 5-FU*
• 3.75% imiquimod*
• Ingenol mebutate 0.015% (lesions on the face or scalp) and ingenol mebutate 0.05% (lesions on the trunk or extremities)*
• ALA-PDT*
• MAL-PDT*
• Cryotherapy (patients with multiple lesions, especially for multiple discrete lesions; not suitable for the treatment of field cancerization)
• 3% diclofenac in 2.5% hyaluronic acid gel
• 5% 5-FU
• 0.5% 5-FU + 10% salicylic acid (discrete, hyperkeratotic lesions)
• 5% imiquimod
• 2.5% imiquimod
• CO2 laser and Er:YAG laser
For immunosuppressed AK patients, the ILDS suggests the following treatments:
• Cryotherapy (especially for single lesions or multiple discrete lesions; not suitable for the treatment of field cancerization);
• Curettage (discrete, hyperkeratotic lesions)
• 5% 5-FU
• 5% imiquimod
• ALA-PDT
• MAL-PDT
The ILDS additionally recommends that immunosuppressed AK patients not use CO2 laser and Er:YAG laser.
“Deviation from the recommendations may be justified or inevitable in specific situations. The ultimate judgment regarding patient care must be individualized and must be made by the physician and patient in light of all presenting circumstances,” the authors said. “International guidelines are intended to be adapted to national or regional circumstances” (J Eur Acad Dermatol Venereol. 2015;29:2069-79).
The “long version of the guidelines” is available as an online supplement. Additionally, a methods report, results report, and declarations of interest of the guidelines development have been published at doi: 10.1111/jdv.13179 in the Journal of the European Academy of Dermatology and Venereology (2015).
The International Leagues of Dermatological Societies (ILDS) in cooperation with the European Dermatology Forum has developed consensus-based guidelines for the treatment of actinic keratosis (AK), which are published in the Journal of the European Academy of Dermatology and Venereology.
“The guidelines were elaborated along adapted recommendations by the WHO guidelines review committee and the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) working group,” say R. N. Werner and colleagues of the Medical University of Berlin. The guidelines include recommendations for treatment of different subgroups of AK patients, how to make an AK diagnosis, how to assess AK patients, and how to define AK.
The ILDS recommends or suggests the following interventions for treating patients who have single AK lesions:
• Cryotherapy
• Curettage (discrete, hyperkeratotic lesions)
• 0.5% 5-fluorouracil (5-FU)
• 5% 5-FU
• 0.5% 5-FU + 10% salicylic acid (discrete, hyperkeratotic lesions)
• 3.75% imiquimod
• 5% imiquimod
• Ingenol mebutate 0.015% (lesions on the face or scalp) and ingenol mebutate 0.05% (lesions on the trunk or extremities)
• 5-aminolevulinic acid-photodynamic therapy (ALA-PDT)
• methylaminolevulinate-photodynamic therapy (MAL-PDT)
For patients with multiple AK lesions/field cancerization, the ILDS recommends* or suggests that patients use the following therapies:
• 0.5% 5-FU*
• 3.75% imiquimod*
• Ingenol mebutate 0.015% (lesions on the face or scalp) and ingenol mebutate 0.05% (lesions on the trunk or extremities)*
• ALA-PDT*
• MAL-PDT*
• Cryotherapy (patients with multiple lesions, especially for multiple discrete lesions; not suitable for the treatment of field cancerization)
• 3% diclofenac in 2.5% hyaluronic acid gel
• 5% 5-FU
• 0.5% 5-FU + 10% salicylic acid (discrete, hyperkeratotic lesions)
• 5% imiquimod
• 2.5% imiquimod
• CO2 laser and Er:YAG laser
For immunosuppressed AK patients, the ILDS suggests the following treatments:
• Cryotherapy (especially for single lesions or multiple discrete lesions; not suitable for the treatment of field cancerization);
• Curettage (discrete, hyperkeratotic lesions)
• 5% 5-FU
• 5% imiquimod
• ALA-PDT
• MAL-PDT
The ILDS additionally recommends that immunosuppressed AK patients not use CO2 laser and Er:YAG laser.
“Deviation from the recommendations may be justified or inevitable in specific situations. The ultimate judgment regarding patient care must be individualized and must be made by the physician and patient in light of all presenting circumstances,” the authors said. “International guidelines are intended to be adapted to national or regional circumstances” (J Eur Acad Dermatol Venereol. 2015;29:2069-79).
The “long version of the guidelines” is available as an online supplement. Additionally, a methods report, results report, and declarations of interest of the guidelines development have been published at doi: 10.1111/jdv.13179 in the Journal of the European Academy of Dermatology and Venereology (2015).
The International Leagues of Dermatological Societies (ILDS) in cooperation with the European Dermatology Forum has developed consensus-based guidelines for the treatment of actinic keratosis (AK), which are published in the Journal of the European Academy of Dermatology and Venereology.
“The guidelines were elaborated along adapted recommendations by the WHO guidelines review committee and the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) working group,” say R. N. Werner and colleagues of the Medical University of Berlin. The guidelines include recommendations for treatment of different subgroups of AK patients, how to make an AK diagnosis, how to assess AK patients, and how to define AK.
The ILDS recommends or suggests the following interventions for treating patients who have single AK lesions:
• Cryotherapy
• Curettage (discrete, hyperkeratotic lesions)
• 0.5% 5-fluorouracil (5-FU)
• 5% 5-FU
• 0.5% 5-FU + 10% salicylic acid (discrete, hyperkeratotic lesions)
• 3.75% imiquimod
• 5% imiquimod
• Ingenol mebutate 0.015% (lesions on the face or scalp) and ingenol mebutate 0.05% (lesions on the trunk or extremities)
• 5-aminolevulinic acid-photodynamic therapy (ALA-PDT)
• methylaminolevulinate-photodynamic therapy (MAL-PDT)
For patients with multiple AK lesions/field cancerization, the ILDS recommends* or suggests that patients use the following therapies:
• 0.5% 5-FU*
• 3.75% imiquimod*
• Ingenol mebutate 0.015% (lesions on the face or scalp) and ingenol mebutate 0.05% (lesions on the trunk or extremities)*
• ALA-PDT*
• MAL-PDT*
• Cryotherapy (patients with multiple lesions, especially for multiple discrete lesions; not suitable for the treatment of field cancerization)
• 3% diclofenac in 2.5% hyaluronic acid gel
• 5% 5-FU
• 0.5% 5-FU + 10% salicylic acid (discrete, hyperkeratotic lesions)
• 5% imiquimod
• 2.5% imiquimod
• CO2 laser and Er:YAG laser
For immunosuppressed AK patients, the ILDS suggests the following treatments:
• Cryotherapy (especially for single lesions or multiple discrete lesions; not suitable for the treatment of field cancerization);
• Curettage (discrete, hyperkeratotic lesions)
• 5% 5-FU
• 5% imiquimod
• ALA-PDT
• MAL-PDT
The ILDS additionally recommends that immunosuppressed AK patients not use CO2 laser and Er:YAG laser.
“Deviation from the recommendations may be justified or inevitable in specific situations. The ultimate judgment regarding patient care must be individualized and must be made by the physician and patient in light of all presenting circumstances,” the authors said. “International guidelines are intended to be adapted to national or regional circumstances” (J Eur Acad Dermatol Venereol. 2015;29:2069-79).
The “long version of the guidelines” is available as an online supplement. Additionally, a methods report, results report, and declarations of interest of the guidelines development have been published at doi: 10.1111/jdv.13179 in the Journal of the European Academy of Dermatology and Venereology (2015).
FROM JOURNAL OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Increased surveillance may explain post-Fukushima pediatric thyroid cancers
LAKE BUENA VISTA, FLA. – More cases of thyroid cancer are being seen in Japanese youth after the Fukushima Daiichi nuclear power plant accident, but the increased incidence may be an artifact of heightened surveillance.
“The thyroid cancers appear to have already occurred prior to radiation exposure,” said Dr. Shinichi Suzuki of the department of thyroid and endocrinology at Fukushima (Japan) Medical University. Radiation-induced thyroid cancers take about 5 years to become detectable, so physicians should just now be seeing the earliest cases of thyroid cancer related to Fukushima radiation exposure, according to Dr. Suzuki. He presented interim results of Japan’s universal screening protocol for children potentially affected by the Fukushima incident at the International Thyroid Conference.
The protocol, designed to screen everyone residing in the Fukushima prefecture and aged 19 years or younger at the time of the 2011 incident, has been highly successful, with over 80% of those eligible receiving a baseline screening that included a thyroid ultrasound exam.
Screening consisted of an initial thyroid ultrasound exam performed with a portable ultrasound device. If no cyst or nodule was found, then the patient would be seen at the next scheduled thyroid ultrasound exam, 2 years later. Patients with cysts 20 mm or less in greatest diameter or nodules 5 mm or smaller also were deferred to the next scheduled examination. Patients with cysts larger than 20 mm or nodules larger than 5 mm received confirmatory examination by detailed ultrasound examination, blood work, and fine-needle aspiration.
Of the 300,476 patients who received the preliminary baseline survey, 2,294 (0.8%) had an abnormality that warranted confirmatory examination and 91.9% of patients went on to have the confirmatory exams. Of these, 113 were assessed as malignant or suspicious for malignancy. Ninety-nine patients had surgery, with findings of 98 cases of thyroid cancer and one benign tumor.
Patients examined after April 2014 were part of an expanded protocol. Under this protocol, 169,455 patients (44.7% participation) were examined and 1,223 patients (0.8%) had suspicious findings on thyroid ultrasound exam. Participation rate for confirmatory testing for this group was 62.7%, with 25 patients’ thyroids having malignant or potentially malignant findings. Six of these patients had surgery, and thyroid cancer was found in all six cases.
Pooling data from the 138 malignant or suspicious cases from the two groups, 105 patients in total have had surgery, 13 patients with small, noninvasive masses are being watched, and a further 20 are awaiting surgery, Dr. Suzuki said at the meeting held by the American Thyroid Association, Asia-Oceania Thyroid Association, European Thyroid Association, and Latin American Thyroid Society.
Of the 97 patients with thyroid cancer who were treated at Fukushima University, 61 were female. The mean patient age at the time of the disaster was 14.8 ± 2.7 years (range, 6-18 years), while the mean age at diagnosis was 17.4 ± 2.8 years (range, 9-22 years). All patients were asymptomatic.
Tumors were unilateral in all but two patients. Mean tumor size was 15.1 ± 0.8 mm (range, 5-53 mm). Nearly all of the tumors (94/97) were papillary thyroid carcinoma, with 86 of those being classical-type papillary thyroid carcinoma. Three patients had poorly differentiated thyroid carcinoma. Fifty-eight patients (60%) had some intraglandular spread, while 71 (73%) had calcifications.
Dr. Suzuki and his collaborators compared these 97 cases with 37 cases of pediatric thyroid cancer in an historical Japanese cohort and to the 26 cases seen in a cohort from Belarus following the Chernobyl disaster. The Fukushima patients were significantly older than either comparison group, with mean age of 11.9 years for the historical Japanese cohort and 10.6 years for the children from Belarus. Tumor size was smaller than the historical Japanese cohort’s mean of 4.1 cm but about the same as that seen in Belarus (1.4 cm). Pulmonary metastases were more common in the historical Japanese cohort (19% vs. 4% in Belarus and 2% in Fukushima).
To have reference data that use similar techniques on a similar population, Japanese researchers are conducting thyroid ultrasound examsaccording to the Fukushima protocol concurrently in three other Japanese prefectures. This is especially important, Dr. Suzuki said, because rapid technological advances in ultrasound imaging mean that screening is much more likely to detect small abnormalities in the thyroid than would have been the case even a few years ago. For this reason, and also because much more radiation was released at the site of the Chernobyl nuclear disaster, only limited comparisons can be made between pediatric thyroid cancer rates from the two nuclear accidents.
Thyroid ultrasound exam “has the ability to detect a lot of thyroid cancers,” he said, so care must be taken to avoid overdiagnosis and overtreatment in this group of young people. Information to date from the Fukushima surveillance project does not yet “give us the clear view about the influence of radiation exposure after the accident on thyroid cancer occurrence,” he said.
Dr. Suzuki reported no relevant disclosures.
On Twitter @karioakes
LAKE BUENA VISTA, FLA. – More cases of thyroid cancer are being seen in Japanese youth after the Fukushima Daiichi nuclear power plant accident, but the increased incidence may be an artifact of heightened surveillance.
“The thyroid cancers appear to have already occurred prior to radiation exposure,” said Dr. Shinichi Suzuki of the department of thyroid and endocrinology at Fukushima (Japan) Medical University. Radiation-induced thyroid cancers take about 5 years to become detectable, so physicians should just now be seeing the earliest cases of thyroid cancer related to Fukushima radiation exposure, according to Dr. Suzuki. He presented interim results of Japan’s universal screening protocol for children potentially affected by the Fukushima incident at the International Thyroid Conference.
The protocol, designed to screen everyone residing in the Fukushima prefecture and aged 19 years or younger at the time of the 2011 incident, has been highly successful, with over 80% of those eligible receiving a baseline screening that included a thyroid ultrasound exam.
Screening consisted of an initial thyroid ultrasound exam performed with a portable ultrasound device. If no cyst or nodule was found, then the patient would be seen at the next scheduled thyroid ultrasound exam, 2 years later. Patients with cysts 20 mm or less in greatest diameter or nodules 5 mm or smaller also were deferred to the next scheduled examination. Patients with cysts larger than 20 mm or nodules larger than 5 mm received confirmatory examination by detailed ultrasound examination, blood work, and fine-needle aspiration.
Of the 300,476 patients who received the preliminary baseline survey, 2,294 (0.8%) had an abnormality that warranted confirmatory examination and 91.9% of patients went on to have the confirmatory exams. Of these, 113 were assessed as malignant or suspicious for malignancy. Ninety-nine patients had surgery, with findings of 98 cases of thyroid cancer and one benign tumor.
Patients examined after April 2014 were part of an expanded protocol. Under this protocol, 169,455 patients (44.7% participation) were examined and 1,223 patients (0.8%) had suspicious findings on thyroid ultrasound exam. Participation rate for confirmatory testing for this group was 62.7%, with 25 patients’ thyroids having malignant or potentially malignant findings. Six of these patients had surgery, and thyroid cancer was found in all six cases.
Pooling data from the 138 malignant or suspicious cases from the two groups, 105 patients in total have had surgery, 13 patients with small, noninvasive masses are being watched, and a further 20 are awaiting surgery, Dr. Suzuki said at the meeting held by the American Thyroid Association, Asia-Oceania Thyroid Association, European Thyroid Association, and Latin American Thyroid Society.
Of the 97 patients with thyroid cancer who were treated at Fukushima University, 61 were female. The mean patient age at the time of the disaster was 14.8 ± 2.7 years (range, 6-18 years), while the mean age at diagnosis was 17.4 ± 2.8 years (range, 9-22 years). All patients were asymptomatic.
Tumors were unilateral in all but two patients. Mean tumor size was 15.1 ± 0.8 mm (range, 5-53 mm). Nearly all of the tumors (94/97) were papillary thyroid carcinoma, with 86 of those being classical-type papillary thyroid carcinoma. Three patients had poorly differentiated thyroid carcinoma. Fifty-eight patients (60%) had some intraglandular spread, while 71 (73%) had calcifications.
Dr. Suzuki and his collaborators compared these 97 cases with 37 cases of pediatric thyroid cancer in an historical Japanese cohort and to the 26 cases seen in a cohort from Belarus following the Chernobyl disaster. The Fukushima patients were significantly older than either comparison group, with mean age of 11.9 years for the historical Japanese cohort and 10.6 years for the children from Belarus. Tumor size was smaller than the historical Japanese cohort’s mean of 4.1 cm but about the same as that seen in Belarus (1.4 cm). Pulmonary metastases were more common in the historical Japanese cohort (19% vs. 4% in Belarus and 2% in Fukushima).
To have reference data that use similar techniques on a similar population, Japanese researchers are conducting thyroid ultrasound examsaccording to the Fukushima protocol concurrently in three other Japanese prefectures. This is especially important, Dr. Suzuki said, because rapid technological advances in ultrasound imaging mean that screening is much more likely to detect small abnormalities in the thyroid than would have been the case even a few years ago. For this reason, and also because much more radiation was released at the site of the Chernobyl nuclear disaster, only limited comparisons can be made between pediatric thyroid cancer rates from the two nuclear accidents.
Thyroid ultrasound exam “has the ability to detect a lot of thyroid cancers,” he said, so care must be taken to avoid overdiagnosis and overtreatment in this group of young people. Information to date from the Fukushima surveillance project does not yet “give us the clear view about the influence of radiation exposure after the accident on thyroid cancer occurrence,” he said.
Dr. Suzuki reported no relevant disclosures.
On Twitter @karioakes
LAKE BUENA VISTA, FLA. – More cases of thyroid cancer are being seen in Japanese youth after the Fukushima Daiichi nuclear power plant accident, but the increased incidence may be an artifact of heightened surveillance.
“The thyroid cancers appear to have already occurred prior to radiation exposure,” said Dr. Shinichi Suzuki of the department of thyroid and endocrinology at Fukushima (Japan) Medical University. Radiation-induced thyroid cancers take about 5 years to become detectable, so physicians should just now be seeing the earliest cases of thyroid cancer related to Fukushima radiation exposure, according to Dr. Suzuki. He presented interim results of Japan’s universal screening protocol for children potentially affected by the Fukushima incident at the International Thyroid Conference.
The protocol, designed to screen everyone residing in the Fukushima prefecture and aged 19 years or younger at the time of the 2011 incident, has been highly successful, with over 80% of those eligible receiving a baseline screening that included a thyroid ultrasound exam.
Screening consisted of an initial thyroid ultrasound exam performed with a portable ultrasound device. If no cyst or nodule was found, then the patient would be seen at the next scheduled thyroid ultrasound exam, 2 years later. Patients with cysts 20 mm or less in greatest diameter or nodules 5 mm or smaller also were deferred to the next scheduled examination. Patients with cysts larger than 20 mm or nodules larger than 5 mm received confirmatory examination by detailed ultrasound examination, blood work, and fine-needle aspiration.
Of the 300,476 patients who received the preliminary baseline survey, 2,294 (0.8%) had an abnormality that warranted confirmatory examination and 91.9% of patients went on to have the confirmatory exams. Of these, 113 were assessed as malignant or suspicious for malignancy. Ninety-nine patients had surgery, with findings of 98 cases of thyroid cancer and one benign tumor.
Patients examined after April 2014 were part of an expanded protocol. Under this protocol, 169,455 patients (44.7% participation) were examined and 1,223 patients (0.8%) had suspicious findings on thyroid ultrasound exam. Participation rate for confirmatory testing for this group was 62.7%, with 25 patients’ thyroids having malignant or potentially malignant findings. Six of these patients had surgery, and thyroid cancer was found in all six cases.
Pooling data from the 138 malignant or suspicious cases from the two groups, 105 patients in total have had surgery, 13 patients with small, noninvasive masses are being watched, and a further 20 are awaiting surgery, Dr. Suzuki said at the meeting held by the American Thyroid Association, Asia-Oceania Thyroid Association, European Thyroid Association, and Latin American Thyroid Society.
Of the 97 patients with thyroid cancer who were treated at Fukushima University, 61 were female. The mean patient age at the time of the disaster was 14.8 ± 2.7 years (range, 6-18 years), while the mean age at diagnosis was 17.4 ± 2.8 years (range, 9-22 years). All patients were asymptomatic.
Tumors were unilateral in all but two patients. Mean tumor size was 15.1 ± 0.8 mm (range, 5-53 mm). Nearly all of the tumors (94/97) were papillary thyroid carcinoma, with 86 of those being classical-type papillary thyroid carcinoma. Three patients had poorly differentiated thyroid carcinoma. Fifty-eight patients (60%) had some intraglandular spread, while 71 (73%) had calcifications.
Dr. Suzuki and his collaborators compared these 97 cases with 37 cases of pediatric thyroid cancer in an historical Japanese cohort and to the 26 cases seen in a cohort from Belarus following the Chernobyl disaster. The Fukushima patients were significantly older than either comparison group, with mean age of 11.9 years for the historical Japanese cohort and 10.6 years for the children from Belarus. Tumor size was smaller than the historical Japanese cohort’s mean of 4.1 cm but about the same as that seen in Belarus (1.4 cm). Pulmonary metastases were more common in the historical Japanese cohort (19% vs. 4% in Belarus and 2% in Fukushima).
To have reference data that use similar techniques on a similar population, Japanese researchers are conducting thyroid ultrasound examsaccording to the Fukushima protocol concurrently in three other Japanese prefectures. This is especially important, Dr. Suzuki said, because rapid technological advances in ultrasound imaging mean that screening is much more likely to detect small abnormalities in the thyroid than would have been the case even a few years ago. For this reason, and also because much more radiation was released at the site of the Chernobyl nuclear disaster, only limited comparisons can be made between pediatric thyroid cancer rates from the two nuclear accidents.
Thyroid ultrasound exam “has the ability to detect a lot of thyroid cancers,” he said, so care must be taken to avoid overdiagnosis and overtreatment in this group of young people. Information to date from the Fukushima surveillance project does not yet “give us the clear view about the influence of radiation exposure after the accident on thyroid cancer occurrence,” he said.
Dr. Suzuki reported no relevant disclosures.
On Twitter @karioakes
AT ITC 2015
Key clinical point: The increased incidence of thyroid cancers in Japanese youth after the Fukushima nuclear accident may be an artifact of increased surveillance.
Major finding: A total of 138 thyroid cancers have been found when screening 469,931 children in Fukushima after the 2011 nuclear power plant accident.
Data source: Universal screening for thyroid cancer among individuals who were aged 18 years or younger and resident in Fukushima at the time of the accident.
Disclosures: Dr. Suzuki reported no relevant disclosures.
Heart Disease Linked to Loud Noise
NEW YORK - People with long-term exposure to loud noise at work or in leisure activities may be at increased risk of heart disease, a U.S. study finds.
Researchers found the strongest link in working-age people with high-frequency hearing loss, which is typically the result of chronic noise exposure.
"Compared with people with normal high-frequency hearing, people with bilateral high-frequency hearing loss were approximately two times more likely to have coronary heart disease," Dr. Wen Qi Gan of the University of Kentucky College of Public Health in Lexington, said by email.
Past research has already linked noise exposure, especially in workplaces, to coronary heart disease, hypertension, and other illnesses, Dr. Gan and his colleagues noted online September 15 in Occupational and Environmental Medicine. But many of these studies lacked individual information about actual noise exposure, relying instead on average decibel levels in the person's environment.
High-frequency hearing loss, the researchers wrote, is a better indicator of exposure to loud noise over time. To investigate the connection with heart disease, the researchers looked at data on 5223 individuals, ages 20 to 69, who participated in national health surveys between 1999 and 2004.
Overall, people with bilateral high-frequency hearing loss were about twice as likely to have coronary heart disease compared to those with normal high-frequency hearing. Among those age 50 and under, who were also most likely to be exposed to loud noise at work, the heart disease risk was increased four-fold.
There was no link to heart disease among people with one-sided hearing loss or loss of lower-frequency hearing, the study team noted, further supporting the idea that noise exposure is the culprit.
The study only looked at people at one time point, however, and cannot prove that noise or hearing loss are direct causes of heart disease. The researchers also acknowledged that they relied on study participants' own recollections about their work and leisure-time noise exposure.
Nonetheless, Dr. Gan said, accumulating evidence suggests that exposure to loud noise can increase the risk of coronary heart disease.
NEW YORK - People with long-term exposure to loud noise at work or in leisure activities may be at increased risk of heart disease, a U.S. study finds.
Researchers found the strongest link in working-age people with high-frequency hearing loss, which is typically the result of chronic noise exposure.
"Compared with people with normal high-frequency hearing, people with bilateral high-frequency hearing loss were approximately two times more likely to have coronary heart disease," Dr. Wen Qi Gan of the University of Kentucky College of Public Health in Lexington, said by email.
Past research has already linked noise exposure, especially in workplaces, to coronary heart disease, hypertension, and other illnesses, Dr. Gan and his colleagues noted online September 15 in Occupational and Environmental Medicine. But many of these studies lacked individual information about actual noise exposure, relying instead on average decibel levels in the person's environment.
High-frequency hearing loss, the researchers wrote, is a better indicator of exposure to loud noise over time. To investigate the connection with heart disease, the researchers looked at data on 5223 individuals, ages 20 to 69, who participated in national health surveys between 1999 and 2004.
Overall, people with bilateral high-frequency hearing loss were about twice as likely to have coronary heart disease compared to those with normal high-frequency hearing. Among those age 50 and under, who were also most likely to be exposed to loud noise at work, the heart disease risk was increased four-fold.
There was no link to heart disease among people with one-sided hearing loss or loss of lower-frequency hearing, the study team noted, further supporting the idea that noise exposure is the culprit.
The study only looked at people at one time point, however, and cannot prove that noise or hearing loss are direct causes of heart disease. The researchers also acknowledged that they relied on study participants' own recollections about their work and leisure-time noise exposure.
Nonetheless, Dr. Gan said, accumulating evidence suggests that exposure to loud noise can increase the risk of coronary heart disease.
NEW YORK - People with long-term exposure to loud noise at work or in leisure activities may be at increased risk of heart disease, a U.S. study finds.
Researchers found the strongest link in working-age people with high-frequency hearing loss, which is typically the result of chronic noise exposure.
"Compared with people with normal high-frequency hearing, people with bilateral high-frequency hearing loss were approximately two times more likely to have coronary heart disease," Dr. Wen Qi Gan of the University of Kentucky College of Public Health in Lexington, said by email.
Past research has already linked noise exposure, especially in workplaces, to coronary heart disease, hypertension, and other illnesses, Dr. Gan and his colleagues noted online September 15 in Occupational and Environmental Medicine. But many of these studies lacked individual information about actual noise exposure, relying instead on average decibel levels in the person's environment.
High-frequency hearing loss, the researchers wrote, is a better indicator of exposure to loud noise over time. To investigate the connection with heart disease, the researchers looked at data on 5223 individuals, ages 20 to 69, who participated in national health surveys between 1999 and 2004.
Overall, people with bilateral high-frequency hearing loss were about twice as likely to have coronary heart disease compared to those with normal high-frequency hearing. Among those age 50 and under, who were also most likely to be exposed to loud noise at work, the heart disease risk was increased four-fold.
There was no link to heart disease among people with one-sided hearing loss or loss of lower-frequency hearing, the study team noted, further supporting the idea that noise exposure is the culprit.
The study only looked at people at one time point, however, and cannot prove that noise or hearing loss are direct causes of heart disease. The researchers also acknowledged that they relied on study participants' own recollections about their work and leisure-time noise exposure.
Nonetheless, Dr. Gan said, accumulating evidence suggests that exposure to loud noise can increase the risk of coronary heart disease.
Could a complementary approach help your patient with insomnia?
Relaxation techniques, acupuncture, and dietary supplements are among the complementary health approaches that patients may consider when struggling with insomnia. But how effective—and safe—are they? A fact sheet from the National Center for Complementary and Alternative Medicine provides useful guidance and is available at: https://nccih.nih.gov/sites/nccam.nih.gov/files/Get_The_Facts_Sleep_Disorders_04-24-2014.pdf
Relaxation techniques, acupuncture, and dietary supplements are among the complementary health approaches that patients may consider when struggling with insomnia. But how effective—and safe—are they? A fact sheet from the National Center for Complementary and Alternative Medicine provides useful guidance and is available at: https://nccih.nih.gov/sites/nccam.nih.gov/files/Get_The_Facts_Sleep_Disorders_04-24-2014.pdf
Relaxation techniques, acupuncture, and dietary supplements are among the complementary health approaches that patients may consider when struggling with insomnia. But how effective—and safe—are they? A fact sheet from the National Center for Complementary and Alternative Medicine provides useful guidance and is available at: https://nccih.nih.gov/sites/nccam.nih.gov/files/Get_The_Facts_Sleep_Disorders_04-24-2014.pdf
Encouraging baby boomers to get tested for hepatitis C
More than 75% of adults infected with hepatitis C are baby boomers. To educate these patients on why they should get tested and how to make sense of test results, the Centers for Disease Control and Prevention has put together a fact sheet. It’s available at http://www.cdc.gov/knowmorehepatitis/media/pdfs/factsheet-boomers.pdf.
More than 75% of adults infected with hepatitis C are baby boomers. To educate these patients on why they should get tested and how to make sense of test results, the Centers for Disease Control and Prevention has put together a fact sheet. It’s available at http://www.cdc.gov/knowmorehepatitis/media/pdfs/factsheet-boomers.pdf.
More than 75% of adults infected with hepatitis C are baby boomers. To educate these patients on why they should get tested and how to make sense of test results, the Centers for Disease Control and Prevention has put together a fact sheet. It’s available at http://www.cdc.gov/knowmorehepatitis/media/pdfs/factsheet-boomers.pdf.
Surgical Management of Gorham-Stout Disease of the Pelvis Refractory to Medical and Radiation Therapy
Gorham-Stout disease (GSD) is a rare condition characterized by spontaneous idiopathic resorption of bone with lymphovascular proliferation and an absence of malignant features. It was originally described by Jackson1 in an 1838 report of a 36-year-old man whose “arm bone, between the shoulder and elbow” had completely vanished after 2 fractures. The disease was defined and its pathology characterized by Gorham and Stout2 in 1955 in a series of 24 patients. Despite about 200 reported cases in the literature,3 its etiology remains unclear. Any bone in the skeleton may be affected by GSD, although there is a predilection for the skull, humerus, clavicle, ribs, pelvis, and femur.4-6 It commonly manifests within the first 3 decades of life, but case reports range from as early as 2 months of age to the eighth decade.5,7
Gorham-Stout disease is a diagnosis of exclusion that requires careful consideration of the clinical context, radiographic findings, and histopathology. Typical histopathologic findings include benign lymphatic or vascular proliferation, involution of adipose tissue within the bone marrow, and thinning of bony trabeculae.6 Fibrous tissue may replace vascular tissue after the initial vasoproliferative, osteolytic phase.6 Some authors describe the disease as having 2 phases, the first with massive osteolysis followed by relative dormancy and the second without progression or re-ossification.8,9 Treatment remains controversial and is guided by management of the disease’s complications. Options range from careful observation and supportive management to aggressive surgical resection and reconstruction, with positive outcomes reported using many different modalities.10 Most treatment successes, however, hinge on halting bony resorption using medical and radiation therapy. Surgery is usually reserved as a salvage option for patients who have failed medical modalities and have residual symptoms or functional limitations.6
This case report describes the successful surgical management of a patient with pelvic GSD who had progressive pain and functional limitation despite exhaustive medical and radiation therapy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy 27-year-old man sought medical attention after a fall while mowing his lawn that resulted in difficulty ambulating. Radiographic studies showed discontinuous lytic lesions in the right periacetabular region and the right sacroiliac (SI) joint. Biopsy at an outside institution revealed an infiltration of thin-walled branching vascular channels involving intertrabecular marrow spaces and periosteal connective tissue. The vessels were devoid of a muscular coat and lined by flattened epithelium; these features were seen as consistent with GSD.
The patient was managed medically at the outside institution for approximately 2 years, with regimens consisting of zoledronate, denosumab, sorafenib, vincristine, sirolimus, and bevacizumab. Because there is no standard chemotherapy protocol for GSD, this broad regimen was likely an attempt by treating physicians to control disease progression before considering radiation or surgery. Zoledronate, a bisphosphonate, and denosumab, a monoclonal antibody against the receptor activator of nuclear factor κβ ligand (RANKL), both inhibit bone resorption, making them logical choices in treating an osteolytic disease. Sorafenib, vincristine, sirolimus, and bevacizumab may be of clinical benefit in GSD via inhibition of vascular proliferation, which is a key histologic feature in GSD. Sorafenib inhibits the vascular endothelial growth factor (VEGF) receptor, vincristine and sirolimus inhibit VEGF production, and bevacizumab is a monoclonal antibody targeting VEGF.
The patient’s disease continued to involve more of his right hemipelvis despite this extensive regimen of chemotherapy, and he experienced significant functional decline about 2 years after initial presentation, when he was no longer able to ambulate unassisted. Radiation therapy to the pelvis was attempted at the outside institution (6/15 MV photons, 5040 cGy, 28 fractions) without improvement. Three years after his initial injury, he presented to our clinic.
Now age 30 years, the patient ambulated only with crutches and endorsed minimal improvement in his pain over 3 years of treatment. Physical examination of the patient revealed that he was a tall, thin man in visible discomfort. Sensation was intact to light touch in the bilateral L1 to S1 nerve distributions. There was marked weakness of the right lower extremity, and his examination was limited by pain. He could not perform a straight leg raise on the right side. Right quadriceps strength was 4/5, and right hamstrings strength was 3/5. There was no weakness in the left leg. Reflexes were normal and symmetric bilaterally at the patellar and gastrocnemius soleus tendons. Distal circulatory status in both extremities was normal, and there were no deformities of the skin.
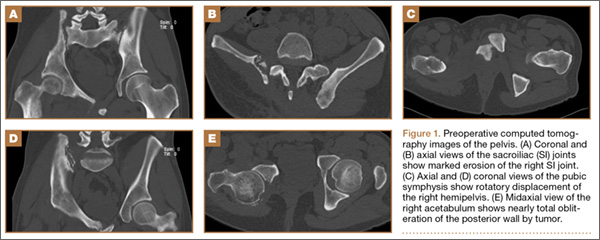
Figure 1 shows the patient’s computed tomography (CT) scan. Figures 1A and 1B reveal fragmentation of the posterior ilia and sacrum along both SI joints. Dislocation of the pubic symphysis is shown in Figures 1C and 1D, and discontinuous involvement of the ischium and posterior wall of the acetabulum is visible in Figure 1E.
Serum studies, including C-reactive protein, erythrocyte sedimentation rate, and a complete blood count, were within normal limits. A CT-guided core needle biopsy and aspiration of the right SI joint revealed no infection; pathology was nondiagnostic. Anesthetic injection of the hip joint resulted in no relief. As this man was severely functionally limited and had exhausted all medical and radiation treatment options, a collaborative decision was made to proceed with surgical management. Surgical options included spinopelvic fusion unilaterally or bilaterally, hip arthroplasty, or sacropelvic resection with or without reconstruction. The patient opted for intralesional surgery and spinopelvic fusion in place of more radical options.
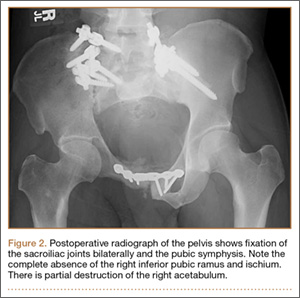
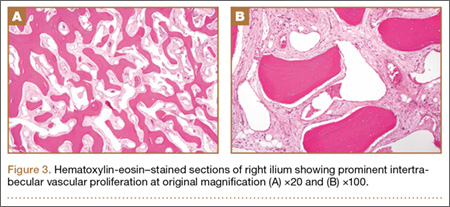
Thirty-seven months after his initial presentation, he underwent posterior spinal fusion L5 to S1, SI fusion, and anterior locking plate fixation of the pubic symphysis, as seen in Figure 2. Pathology from surgical specimens, seen at original magnification ×20 and ×100 in Figures 3A and 3B, respectively, showed prominent vascular proliferation in the right ilium, with reactive bone changes in the left ilium and right sacrum. A lytic lesion showed fibrous tissue with an embedded fragment of necrotic bone.
Six weeks after surgery, the patient had substantial improvement in his pain and was partially weight-bearing. He was able to ambulate with crutches and returned to work. The patient’s overall clinical status continued to improve throughout the postoperative course. He developed low back pain 7 months after surgery and was found to have a sacrococcygeal abscess and coccygeal fracture anterior to the sacrum. He underwent irrigation and débridement of the abscess and distal coccygectomy and was treated with 6 weeks of intravenous cefazolin and long-term suppression with levofloxacin and rifampin for methicillin-sensitive Staphylococcus aureus hardware infection and osteomyelitis. The patient’s clinical course subsequently improved. At latest follow-up 16 months after the index operation, pain was reported as manageable and mostly an annoyance. He was prescribed up to 40 mg of oxycodone daily for pain. The patient returned to work, ambulates with a cane (no other assistive devices), and reports being able to get around without any difficulty.
Discussion
Gorham-Stout disease is an exceedingly rare condition resulting in spontaneous osteolysis. Approximately 200 cases have been reported with no apparent gender, race, or familial predilection or systemic symptoms differentiating it from other etiologies of idiopathic osteolysis.6 These patients often seek medical attention after sustaining a pathologic fracture,6 when a broad differential diagnosis narrows to GSD only after biopsy excludes other possibilities and demonstrates characteristic angiomatosis without malignant features.2,4,6,8,10 Gorham-Stout disease appears more frequently at particular sites within the skeleton, and pelvic involvement is common—more than 20% of cases in 1 review.5,10 Limitations in the patient’s ability to ambulate invariably result from osteolysis of the pelvis, which is concerning considering the young age at which GSD typically presents. A variety of treatment modalities have been described for pelvic GSD, but surgery has been undertaken in relatively few cases.5
The diagnosis is one of exclusion after considering the clinical context and radiologic and pathologic findings. In this case, a pathologic fracture was discovered with osteolytic lesions throughout the hemipelvis. Biopsy excluded malignancy and demonstrated the key hemangiomatous vascular proliferation with thin-walled vessels that is classic for GSD. While our patient initially appeared to have 2 sites of disease, the surgical specimen revealed a primary site of vascular proliferation in the right ilium from which 2 apparent foci had spread, consistent with the typical monocentric presentation of GSD.11 A broad differential diagnosis must be considered at initial presentation, including osteomyelitis, metastatic disease, multiple myeloma, and primary bone sarcoma. Upon identifying a primary osteolytic process, several considerations besides GSD remain, such as Hajdu-Cheney syndrome, Winchester syndrome, multicentric osteolysis with nephropathy, familial osteolysis, Farber disease, and neurogenic osteolysis; most of these etiologies involve familial predispositions and/or systemic symptoms.
Treatment options for GSD include supportive care, medical therapy, radiation, and surgery. For pelvic GSD, numerous reports have demonstrated good outcomes with supportive management, since osteolysis often spontaneously arrests.8,9,12 Others have had success with medical treatments in attempts to halt bone resorption.6,13-15 Bisphosphonates are the cornerstone of medical therapy in GSD, as they appear to halt further osteoclastic bone breakdown. The levels of VEGF have been shown to be elevated in GSD,13 likely consistent with the vascular proliferation evident on pathology, and therapies such as bevacizumab and interferon α-2b have been used to target osteolysis via this pathway with good outcome.13,14,16 External beam-radiation therapy has been shown to prevent local progression of osteolysis in up to 80% of cases.4 However, even with arrest of bone resorption, damage to affected bone may have progressed to the point of significant functional limitation. This may be especially true in the pelvis.
We present a case of a patient who continued to deteriorate after maximal medical and radiation therapy. Many reported cases of pelvic GSD have had good outcomes with some combination of conservative management, medical therapy, and radiation. However, in our patient, the pelvis and lumbosacral spine were unstable as a result of significant bone loss and fracture, and his clinical deterioration was dramatic. We considered reasonable surgical approaches, including local intralesional débridement and massive en bloc resection with structural allograft. We chose the less radical procedure given the patient’s age, minimal surgical history, and personal preference. Although structural pelvic allograft has been successful in a few cases, there remains a high risk of complications, such as fracture, resorption, or infection.17 We considered the addition of hip arthroplasty with either scenario, but we elected not to perform this component given his young age and lack of symptomatic improvement with diagnostic anesthetic hip injection. The key to this patient’s surgical reconstruction, aside from eliminating gross disease, was the stabilization of the spinopelvic junction and pelvic ring. His functional improvement as early as 6 weeks after surgery demonstrates that surgery can have an important role for patients with pelvic GSD who fail medical and radiation therapy.
1. Jackson JBS. A boneless arm. Boston Med Surg J. 1838;18:368-369.
2. Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37(5):985-1004.
3. Lehmann G, Pfeil A, Böttcher J, et al. Benefit of a 17-year long-term bisphosphonate therapy in a patient with Gorham-Stout syndrome. Arch Orthop Trauma Surg. 2009;129(7):967-972.
4. Heyd R, Micke O, Surholt C, et al; German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179-e185.
5. Kulenkampff HA, Richter GM, Hasse WE, Adler CP. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990;14(4):361-366.
6. Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011;40(11):1391-1397.
7. Vinée P, Tanyü MO, Hauenstein KH, Sigmund G, Stöver B, Adler CP. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994;18(6):985-989.
8. Boyer P, Bourgeois P, Boyer O, Catonné Y, Saillant G. Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol. 2005;24(5):551-555.
9. Malde R, Agrawal HM, Ghosh SL, Dinshaw KA. Vanishing bone disease involving the pelvis. J Cancer Res Ther. 2005;1(4):227-228.
10. Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-α: a case report and literature review. J Pediatr Hematol Oncol. 2010;32(8):579-584.
11. Tie ML, Poland GA, Rosenow EC III. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. 1994;105(1):208-213.
12. Möller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501-506.
13. Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis—Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010;46(3):873-876.
14. Wang JD, Chang TK, Cheng YY, et al. A child with dyspnea and unstable gait. Pediatr Hemat Oncol. 2007;24(4):321-324.
15. Zheng MW, Yang M, Qiu JX, et al. Gorham-Stout syndrome presenting in a 5-year-old girl with a successful bisphosphonate therapeutic effect. Exp Ther Med. 2012;4(3):449-451.
16. Timke C, Krause MF, Oppermann HC, Leuschner I, Claviez A. Interferon alpha 2b treatment in an eleven-year-old boy with disseminated lymphangiomatosis. Pediatr Blood Cancer. 2007;48(1):108-111.
17. Stöve J, Reichelt A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch Orthop Trauma Surg. 1995;114(4):207-210.
Gorham-Stout disease (GSD) is a rare condition characterized by spontaneous idiopathic resorption of bone with lymphovascular proliferation and an absence of malignant features. It was originally described by Jackson1 in an 1838 report of a 36-year-old man whose “arm bone, between the shoulder and elbow” had completely vanished after 2 fractures. The disease was defined and its pathology characterized by Gorham and Stout2 in 1955 in a series of 24 patients. Despite about 200 reported cases in the literature,3 its etiology remains unclear. Any bone in the skeleton may be affected by GSD, although there is a predilection for the skull, humerus, clavicle, ribs, pelvis, and femur.4-6 It commonly manifests within the first 3 decades of life, but case reports range from as early as 2 months of age to the eighth decade.5,7
Gorham-Stout disease is a diagnosis of exclusion that requires careful consideration of the clinical context, radiographic findings, and histopathology. Typical histopathologic findings include benign lymphatic or vascular proliferation, involution of adipose tissue within the bone marrow, and thinning of bony trabeculae.6 Fibrous tissue may replace vascular tissue after the initial vasoproliferative, osteolytic phase.6 Some authors describe the disease as having 2 phases, the first with massive osteolysis followed by relative dormancy and the second without progression or re-ossification.8,9 Treatment remains controversial and is guided by management of the disease’s complications. Options range from careful observation and supportive management to aggressive surgical resection and reconstruction, with positive outcomes reported using many different modalities.10 Most treatment successes, however, hinge on halting bony resorption using medical and radiation therapy. Surgery is usually reserved as a salvage option for patients who have failed medical modalities and have residual symptoms or functional limitations.6
This case report describes the successful surgical management of a patient with pelvic GSD who had progressive pain and functional limitation despite exhaustive medical and radiation therapy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy 27-year-old man sought medical attention after a fall while mowing his lawn that resulted in difficulty ambulating. Radiographic studies showed discontinuous lytic lesions in the right periacetabular region and the right sacroiliac (SI) joint. Biopsy at an outside institution revealed an infiltration of thin-walled branching vascular channels involving intertrabecular marrow spaces and periosteal connective tissue. The vessels were devoid of a muscular coat and lined by flattened epithelium; these features were seen as consistent with GSD.
The patient was managed medically at the outside institution for approximately 2 years, with regimens consisting of zoledronate, denosumab, sorafenib, vincristine, sirolimus, and bevacizumab. Because there is no standard chemotherapy protocol for GSD, this broad regimen was likely an attempt by treating physicians to control disease progression before considering radiation or surgery. Zoledronate, a bisphosphonate, and denosumab, a monoclonal antibody against the receptor activator of nuclear factor κβ ligand (RANKL), both inhibit bone resorption, making them logical choices in treating an osteolytic disease. Sorafenib, vincristine, sirolimus, and bevacizumab may be of clinical benefit in GSD via inhibition of vascular proliferation, which is a key histologic feature in GSD. Sorafenib inhibits the vascular endothelial growth factor (VEGF) receptor, vincristine and sirolimus inhibit VEGF production, and bevacizumab is a monoclonal antibody targeting VEGF.
The patient’s disease continued to involve more of his right hemipelvis despite this extensive regimen of chemotherapy, and he experienced significant functional decline about 2 years after initial presentation, when he was no longer able to ambulate unassisted. Radiation therapy to the pelvis was attempted at the outside institution (6/15 MV photons, 5040 cGy, 28 fractions) without improvement. Three years after his initial injury, he presented to our clinic.
Now age 30 years, the patient ambulated only with crutches and endorsed minimal improvement in his pain over 3 years of treatment. Physical examination of the patient revealed that he was a tall, thin man in visible discomfort. Sensation was intact to light touch in the bilateral L1 to S1 nerve distributions. There was marked weakness of the right lower extremity, and his examination was limited by pain. He could not perform a straight leg raise on the right side. Right quadriceps strength was 4/5, and right hamstrings strength was 3/5. There was no weakness in the left leg. Reflexes were normal and symmetric bilaterally at the patellar and gastrocnemius soleus tendons. Distal circulatory status in both extremities was normal, and there were no deformities of the skin.
Figure 1 shows the patient’s computed tomography (CT) scan. Figures 1A and 1B reveal fragmentation of the posterior ilia and sacrum along both SI joints. Dislocation of the pubic symphysis is shown in Figures 1C and 1D, and discontinuous involvement of the ischium and posterior wall of the acetabulum is visible in Figure 1E.
Serum studies, including C-reactive protein, erythrocyte sedimentation rate, and a complete blood count, were within normal limits. A CT-guided core needle biopsy and aspiration of the right SI joint revealed no infection; pathology was nondiagnostic. Anesthetic injection of the hip joint resulted in no relief. As this man was severely functionally limited and had exhausted all medical and radiation treatment options, a collaborative decision was made to proceed with surgical management. Surgical options included spinopelvic fusion unilaterally or bilaterally, hip arthroplasty, or sacropelvic resection with or without reconstruction. The patient opted for intralesional surgery and spinopelvic fusion in place of more radical options.
Thirty-seven months after his initial presentation, he underwent posterior spinal fusion L5 to S1, SI fusion, and anterior locking plate fixation of the pubic symphysis, as seen in Figure 2. Pathology from surgical specimens, seen at original magnification ×20 and ×100 in Figures 3A and 3B, respectively, showed prominent vascular proliferation in the right ilium, with reactive bone changes in the left ilium and right sacrum. A lytic lesion showed fibrous tissue with an embedded fragment of necrotic bone.
Six weeks after surgery, the patient had substantial improvement in his pain and was partially weight-bearing. He was able to ambulate with crutches and returned to work. The patient’s overall clinical status continued to improve throughout the postoperative course. He developed low back pain 7 months after surgery and was found to have a sacrococcygeal abscess and coccygeal fracture anterior to the sacrum. He underwent irrigation and débridement of the abscess and distal coccygectomy and was treated with 6 weeks of intravenous cefazolin and long-term suppression with levofloxacin and rifampin for methicillin-sensitive Staphylococcus aureus hardware infection and osteomyelitis. The patient’s clinical course subsequently improved. At latest follow-up 16 months after the index operation, pain was reported as manageable and mostly an annoyance. He was prescribed up to 40 mg of oxycodone daily for pain. The patient returned to work, ambulates with a cane (no other assistive devices), and reports being able to get around without any difficulty.
Discussion
Gorham-Stout disease is an exceedingly rare condition resulting in spontaneous osteolysis. Approximately 200 cases have been reported with no apparent gender, race, or familial predilection or systemic symptoms differentiating it from other etiologies of idiopathic osteolysis.6 These patients often seek medical attention after sustaining a pathologic fracture,6 when a broad differential diagnosis narrows to GSD only after biopsy excludes other possibilities and demonstrates characteristic angiomatosis without malignant features.2,4,6,8,10 Gorham-Stout disease appears more frequently at particular sites within the skeleton, and pelvic involvement is common—more than 20% of cases in 1 review.5,10 Limitations in the patient’s ability to ambulate invariably result from osteolysis of the pelvis, which is concerning considering the young age at which GSD typically presents. A variety of treatment modalities have been described for pelvic GSD, but surgery has been undertaken in relatively few cases.5
The diagnosis is one of exclusion after considering the clinical context and radiologic and pathologic findings. In this case, a pathologic fracture was discovered with osteolytic lesions throughout the hemipelvis. Biopsy excluded malignancy and demonstrated the key hemangiomatous vascular proliferation with thin-walled vessels that is classic for GSD. While our patient initially appeared to have 2 sites of disease, the surgical specimen revealed a primary site of vascular proliferation in the right ilium from which 2 apparent foci had spread, consistent with the typical monocentric presentation of GSD.11 A broad differential diagnosis must be considered at initial presentation, including osteomyelitis, metastatic disease, multiple myeloma, and primary bone sarcoma. Upon identifying a primary osteolytic process, several considerations besides GSD remain, such as Hajdu-Cheney syndrome, Winchester syndrome, multicentric osteolysis with nephropathy, familial osteolysis, Farber disease, and neurogenic osteolysis; most of these etiologies involve familial predispositions and/or systemic symptoms.
Treatment options for GSD include supportive care, medical therapy, radiation, and surgery. For pelvic GSD, numerous reports have demonstrated good outcomes with supportive management, since osteolysis often spontaneously arrests.8,9,12 Others have had success with medical treatments in attempts to halt bone resorption.6,13-15 Bisphosphonates are the cornerstone of medical therapy in GSD, as they appear to halt further osteoclastic bone breakdown. The levels of VEGF have been shown to be elevated in GSD,13 likely consistent with the vascular proliferation evident on pathology, and therapies such as bevacizumab and interferon α-2b have been used to target osteolysis via this pathway with good outcome.13,14,16 External beam-radiation therapy has been shown to prevent local progression of osteolysis in up to 80% of cases.4 However, even with arrest of bone resorption, damage to affected bone may have progressed to the point of significant functional limitation. This may be especially true in the pelvis.
We present a case of a patient who continued to deteriorate after maximal medical and radiation therapy. Many reported cases of pelvic GSD have had good outcomes with some combination of conservative management, medical therapy, and radiation. However, in our patient, the pelvis and lumbosacral spine were unstable as a result of significant bone loss and fracture, and his clinical deterioration was dramatic. We considered reasonable surgical approaches, including local intralesional débridement and massive en bloc resection with structural allograft. We chose the less radical procedure given the patient’s age, minimal surgical history, and personal preference. Although structural pelvic allograft has been successful in a few cases, there remains a high risk of complications, such as fracture, resorption, or infection.17 We considered the addition of hip arthroplasty with either scenario, but we elected not to perform this component given his young age and lack of symptomatic improvement with diagnostic anesthetic hip injection. The key to this patient’s surgical reconstruction, aside from eliminating gross disease, was the stabilization of the spinopelvic junction and pelvic ring. His functional improvement as early as 6 weeks after surgery demonstrates that surgery can have an important role for patients with pelvic GSD who fail medical and radiation therapy.
Gorham-Stout disease (GSD) is a rare condition characterized by spontaneous idiopathic resorption of bone with lymphovascular proliferation and an absence of malignant features. It was originally described by Jackson1 in an 1838 report of a 36-year-old man whose “arm bone, between the shoulder and elbow” had completely vanished after 2 fractures. The disease was defined and its pathology characterized by Gorham and Stout2 in 1955 in a series of 24 patients. Despite about 200 reported cases in the literature,3 its etiology remains unclear. Any bone in the skeleton may be affected by GSD, although there is a predilection for the skull, humerus, clavicle, ribs, pelvis, and femur.4-6 It commonly manifests within the first 3 decades of life, but case reports range from as early as 2 months of age to the eighth decade.5,7
Gorham-Stout disease is a diagnosis of exclusion that requires careful consideration of the clinical context, radiographic findings, and histopathology. Typical histopathologic findings include benign lymphatic or vascular proliferation, involution of adipose tissue within the bone marrow, and thinning of bony trabeculae.6 Fibrous tissue may replace vascular tissue after the initial vasoproliferative, osteolytic phase.6 Some authors describe the disease as having 2 phases, the first with massive osteolysis followed by relative dormancy and the second without progression or re-ossification.8,9 Treatment remains controversial and is guided by management of the disease’s complications. Options range from careful observation and supportive management to aggressive surgical resection and reconstruction, with positive outcomes reported using many different modalities.10 Most treatment successes, however, hinge on halting bony resorption using medical and radiation therapy. Surgery is usually reserved as a salvage option for patients who have failed medical modalities and have residual symptoms or functional limitations.6
This case report describes the successful surgical management of a patient with pelvic GSD who had progressive pain and functional limitation despite exhaustive medical and radiation therapy. The patient provided written informed consent for print and electronic publication of this case report.
Case Report
A healthy 27-year-old man sought medical attention after a fall while mowing his lawn that resulted in difficulty ambulating. Radiographic studies showed discontinuous lytic lesions in the right periacetabular region and the right sacroiliac (SI) joint. Biopsy at an outside institution revealed an infiltration of thin-walled branching vascular channels involving intertrabecular marrow spaces and periosteal connective tissue. The vessels were devoid of a muscular coat and lined by flattened epithelium; these features were seen as consistent with GSD.
The patient was managed medically at the outside institution for approximately 2 years, with regimens consisting of zoledronate, denosumab, sorafenib, vincristine, sirolimus, and bevacizumab. Because there is no standard chemotherapy protocol for GSD, this broad regimen was likely an attempt by treating physicians to control disease progression before considering radiation or surgery. Zoledronate, a bisphosphonate, and denosumab, a monoclonal antibody against the receptor activator of nuclear factor κβ ligand (RANKL), both inhibit bone resorption, making them logical choices in treating an osteolytic disease. Sorafenib, vincristine, sirolimus, and bevacizumab may be of clinical benefit in GSD via inhibition of vascular proliferation, which is a key histologic feature in GSD. Sorafenib inhibits the vascular endothelial growth factor (VEGF) receptor, vincristine and sirolimus inhibit VEGF production, and bevacizumab is a monoclonal antibody targeting VEGF.
The patient’s disease continued to involve more of his right hemipelvis despite this extensive regimen of chemotherapy, and he experienced significant functional decline about 2 years after initial presentation, when he was no longer able to ambulate unassisted. Radiation therapy to the pelvis was attempted at the outside institution (6/15 MV photons, 5040 cGy, 28 fractions) without improvement. Three years after his initial injury, he presented to our clinic.
Now age 30 years, the patient ambulated only with crutches and endorsed minimal improvement in his pain over 3 years of treatment. Physical examination of the patient revealed that he was a tall, thin man in visible discomfort. Sensation was intact to light touch in the bilateral L1 to S1 nerve distributions. There was marked weakness of the right lower extremity, and his examination was limited by pain. He could not perform a straight leg raise on the right side. Right quadriceps strength was 4/5, and right hamstrings strength was 3/5. There was no weakness in the left leg. Reflexes were normal and symmetric bilaterally at the patellar and gastrocnemius soleus tendons. Distal circulatory status in both extremities was normal, and there were no deformities of the skin.
Figure 1 shows the patient’s computed tomography (CT) scan. Figures 1A and 1B reveal fragmentation of the posterior ilia and sacrum along both SI joints. Dislocation of the pubic symphysis is shown in Figures 1C and 1D, and discontinuous involvement of the ischium and posterior wall of the acetabulum is visible in Figure 1E.
Serum studies, including C-reactive protein, erythrocyte sedimentation rate, and a complete blood count, were within normal limits. A CT-guided core needle biopsy and aspiration of the right SI joint revealed no infection; pathology was nondiagnostic. Anesthetic injection of the hip joint resulted in no relief. As this man was severely functionally limited and had exhausted all medical and radiation treatment options, a collaborative decision was made to proceed with surgical management. Surgical options included spinopelvic fusion unilaterally or bilaterally, hip arthroplasty, or sacropelvic resection with or without reconstruction. The patient opted for intralesional surgery and spinopelvic fusion in place of more radical options.
Thirty-seven months after his initial presentation, he underwent posterior spinal fusion L5 to S1, SI fusion, and anterior locking plate fixation of the pubic symphysis, as seen in Figure 2. Pathology from surgical specimens, seen at original magnification ×20 and ×100 in Figures 3A and 3B, respectively, showed prominent vascular proliferation in the right ilium, with reactive bone changes in the left ilium and right sacrum. A lytic lesion showed fibrous tissue with an embedded fragment of necrotic bone.
Six weeks after surgery, the patient had substantial improvement in his pain and was partially weight-bearing. He was able to ambulate with crutches and returned to work. The patient’s overall clinical status continued to improve throughout the postoperative course. He developed low back pain 7 months after surgery and was found to have a sacrococcygeal abscess and coccygeal fracture anterior to the sacrum. He underwent irrigation and débridement of the abscess and distal coccygectomy and was treated with 6 weeks of intravenous cefazolin and long-term suppression with levofloxacin and rifampin for methicillin-sensitive Staphylococcus aureus hardware infection and osteomyelitis. The patient’s clinical course subsequently improved. At latest follow-up 16 months after the index operation, pain was reported as manageable and mostly an annoyance. He was prescribed up to 40 mg of oxycodone daily for pain. The patient returned to work, ambulates with a cane (no other assistive devices), and reports being able to get around without any difficulty.
Discussion
Gorham-Stout disease is an exceedingly rare condition resulting in spontaneous osteolysis. Approximately 200 cases have been reported with no apparent gender, race, or familial predilection or systemic symptoms differentiating it from other etiologies of idiopathic osteolysis.6 These patients often seek medical attention after sustaining a pathologic fracture,6 when a broad differential diagnosis narrows to GSD only after biopsy excludes other possibilities and demonstrates characteristic angiomatosis without malignant features.2,4,6,8,10 Gorham-Stout disease appears more frequently at particular sites within the skeleton, and pelvic involvement is common—more than 20% of cases in 1 review.5,10 Limitations in the patient’s ability to ambulate invariably result from osteolysis of the pelvis, which is concerning considering the young age at which GSD typically presents. A variety of treatment modalities have been described for pelvic GSD, but surgery has been undertaken in relatively few cases.5
The diagnosis is one of exclusion after considering the clinical context and radiologic and pathologic findings. In this case, a pathologic fracture was discovered with osteolytic lesions throughout the hemipelvis. Biopsy excluded malignancy and demonstrated the key hemangiomatous vascular proliferation with thin-walled vessels that is classic for GSD. While our patient initially appeared to have 2 sites of disease, the surgical specimen revealed a primary site of vascular proliferation in the right ilium from which 2 apparent foci had spread, consistent with the typical monocentric presentation of GSD.11 A broad differential diagnosis must be considered at initial presentation, including osteomyelitis, metastatic disease, multiple myeloma, and primary bone sarcoma. Upon identifying a primary osteolytic process, several considerations besides GSD remain, such as Hajdu-Cheney syndrome, Winchester syndrome, multicentric osteolysis with nephropathy, familial osteolysis, Farber disease, and neurogenic osteolysis; most of these etiologies involve familial predispositions and/or systemic symptoms.
Treatment options for GSD include supportive care, medical therapy, radiation, and surgery. For pelvic GSD, numerous reports have demonstrated good outcomes with supportive management, since osteolysis often spontaneously arrests.8,9,12 Others have had success with medical treatments in attempts to halt bone resorption.6,13-15 Bisphosphonates are the cornerstone of medical therapy in GSD, as they appear to halt further osteoclastic bone breakdown. The levels of VEGF have been shown to be elevated in GSD,13 likely consistent with the vascular proliferation evident on pathology, and therapies such as bevacizumab and interferon α-2b have been used to target osteolysis via this pathway with good outcome.13,14,16 External beam-radiation therapy has been shown to prevent local progression of osteolysis in up to 80% of cases.4 However, even with arrest of bone resorption, damage to affected bone may have progressed to the point of significant functional limitation. This may be especially true in the pelvis.
We present a case of a patient who continued to deteriorate after maximal medical and radiation therapy. Many reported cases of pelvic GSD have had good outcomes with some combination of conservative management, medical therapy, and radiation. However, in our patient, the pelvis and lumbosacral spine were unstable as a result of significant bone loss and fracture, and his clinical deterioration was dramatic. We considered reasonable surgical approaches, including local intralesional débridement and massive en bloc resection with structural allograft. We chose the less radical procedure given the patient’s age, minimal surgical history, and personal preference. Although structural pelvic allograft has been successful in a few cases, there remains a high risk of complications, such as fracture, resorption, or infection.17 We considered the addition of hip arthroplasty with either scenario, but we elected not to perform this component given his young age and lack of symptomatic improvement with diagnostic anesthetic hip injection. The key to this patient’s surgical reconstruction, aside from eliminating gross disease, was the stabilization of the spinopelvic junction and pelvic ring. His functional improvement as early as 6 weeks after surgery demonstrates that surgery can have an important role for patients with pelvic GSD who fail medical and radiation therapy.
1. Jackson JBS. A boneless arm. Boston Med Surg J. 1838;18:368-369.
2. Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37(5):985-1004.
3. Lehmann G, Pfeil A, Böttcher J, et al. Benefit of a 17-year long-term bisphosphonate therapy in a patient with Gorham-Stout syndrome. Arch Orthop Trauma Surg. 2009;129(7):967-972.
4. Heyd R, Micke O, Surholt C, et al; German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179-e185.
5. Kulenkampff HA, Richter GM, Hasse WE, Adler CP. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990;14(4):361-366.
6. Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011;40(11):1391-1397.
7. Vinée P, Tanyü MO, Hauenstein KH, Sigmund G, Stöver B, Adler CP. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994;18(6):985-989.
8. Boyer P, Bourgeois P, Boyer O, Catonné Y, Saillant G. Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol. 2005;24(5):551-555.
9. Malde R, Agrawal HM, Ghosh SL, Dinshaw KA. Vanishing bone disease involving the pelvis. J Cancer Res Ther. 2005;1(4):227-228.
10. Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-α: a case report and literature review. J Pediatr Hematol Oncol. 2010;32(8):579-584.
11. Tie ML, Poland GA, Rosenow EC III. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. 1994;105(1):208-213.
12. Möller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501-506.
13. Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis—Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010;46(3):873-876.
14. Wang JD, Chang TK, Cheng YY, et al. A child with dyspnea and unstable gait. Pediatr Hemat Oncol. 2007;24(4):321-324.
15. Zheng MW, Yang M, Qiu JX, et al. Gorham-Stout syndrome presenting in a 5-year-old girl with a successful bisphosphonate therapeutic effect. Exp Ther Med. 2012;4(3):449-451.
16. Timke C, Krause MF, Oppermann HC, Leuschner I, Claviez A. Interferon alpha 2b treatment in an eleven-year-old boy with disseminated lymphangiomatosis. Pediatr Blood Cancer. 2007;48(1):108-111.
17. Stöve J, Reichelt A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch Orthop Trauma Surg. 1995;114(4):207-210.
1. Jackson JBS. A boneless arm. Boston Med Surg J. 1838;18:368-369.
2. Gorham LW, Stout AP. Massive osteolysis (acute spontaneous absorption of bone, phantom bone, disappearing bone): its relation to hemangiomatosis. J Bone Joint Surg Am. 1955;37(5):985-1004.
3. Lehmann G, Pfeil A, Böttcher J, et al. Benefit of a 17-year long-term bisphosphonate therapy in a patient with Gorham-Stout syndrome. Arch Orthop Trauma Surg. 2009;129(7):967-972.
4. Heyd R, Micke O, Surholt C, et al; German Cooperative Group on Radiotherapy for Benign Diseases (GCG-BD). Radiation therapy for Gorham-Stout syndrome: results of a national patterns-of-care study and literature review. Int J Radiat Oncol Biol Phys. 2011;81(3):e179-e185.
5. Kulenkampff HA, Richter GM, Hasse WE, Adler CP. Massive pelvic osteolysis in the Gorham-Stout syndrome. Int Orthop. 1990;14(4):361-366.
6. Ruggieri P, Montalti M, Angelini A, Alberghini M, Mercuri M. Gorham-Stout disease: the experience of the Rizzoli Institute and review of the literature. Skeletal Radiol. 2011;40(11):1391-1397.
7. Vinée P, Tanyü MO, Hauenstein KH, Sigmund G, Stöver B, Adler CP. CT and MRI of Gorham syndrome. J Comput Assist Tomogr. 1994;18(6):985-989.
8. Boyer P, Bourgeois P, Boyer O, Catonné Y, Saillant G. Massive Gorham-Stout syndrome of the pelvis. Clin Rheumatol. 2005;24(5):551-555.
9. Malde R, Agrawal HM, Ghosh SL, Dinshaw KA. Vanishing bone disease involving the pelvis. J Cancer Res Ther. 2005;1(4):227-228.
10. Kuriyama DK, McElligott SC, Glaser DW, Thompson KS. Treatment of Gorham-Stout disease with zoledronic acid and interferon-α: a case report and literature review. J Pediatr Hematol Oncol. 2010;32(8):579-584.
11. Tie ML, Poland GA, Rosenow EC III. Chylothorax in Gorham’s syndrome. A common complication of a rare disease. Chest. 1994;105(1):208-213.
12. Möller G, Priemel M, Amling M, Werner M, Kuhlmey AS, Delling G. The Gorham-Stout syndrome (Gorham’s massive osteolysis). A report of six cases with histopathological findings. J Bone Joint Surg Br. 1999;81(3):501-506.
13. Dupond JL, Bermont L, Runge M, de Billy M. Plasma VEGF determination in disseminated lymphangiomatosis—Gorham-Stout syndrome: a marker of activity? A case report with a 5-year follow-up. Bone. 2010;46(3):873-876.
14. Wang JD, Chang TK, Cheng YY, et al. A child with dyspnea and unstable gait. Pediatr Hemat Oncol. 2007;24(4):321-324.
15. Zheng MW, Yang M, Qiu JX, et al. Gorham-Stout syndrome presenting in a 5-year-old girl with a successful bisphosphonate therapeutic effect. Exp Ther Med. 2012;4(3):449-451.
16. Timke C, Krause MF, Oppermann HC, Leuschner I, Claviez A. Interferon alpha 2b treatment in an eleven-year-old boy with disseminated lymphangiomatosis. Pediatr Blood Cancer. 2007;48(1):108-111.
17. Stöve J, Reichelt A. Massive osteolysis of the pelvis, femur and sacral bone with a Gorham-Stout syndrome. Arch Orthop Trauma Surg. 1995;114(4):207-210.
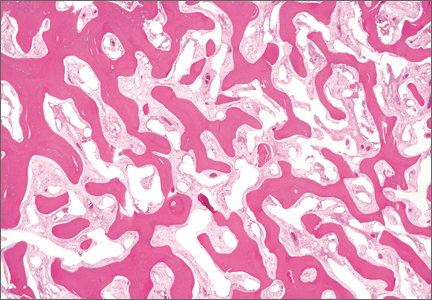
Simultaneous equal to sequential treatment for actinic keratoses
Patients with multiple actinic keratoses (AKs) may be treated either sequentially or simultaneously with ingenol mebutate gel, according to the authors of a study that found no difference in outcomes or adverse effects from either treatment approach.
The phase IIIb study conducted in Italy and Spain enrolled 199 patients with two separate areas of clinically visible, nonhyperkeratotic AKs. Subjects were randomized to have the two areas (face/scalp and trunk/extremities) treated simultaneously (101 patients) or sequentially (98 patients) with 0.015% and 0.05% ingenol mebutate gel.

There were no significant differences in localized skin responses between the simultaneous and sequential treatment groups, based on the mean composite local skin response scores 3 days after treatment started, which were similar between the groups for the face/scalp and trunk/extremities applications. About 22% of patients in each group experienced adverse events.
At 8 weeks, the complete clearance rates also were not statistically different between the simultaneous and sequential groups (52.7% and 46.9%, respectively; P = .34), and patient satisfaction with treatment was similar for both treatment approaches. At that time, the number of AKs had dropped by a mean of 83.4% among those in the simultaneous group and 79.1% in the sequential group (P = .20).
“The favorable rate of complete clearance in the simultaneous treatment group means that patients can receive their treatment for both areas in one visit, rather than having to return to the clinic for a second cycle of treatment,” wrote Dr. Giovanni Pellacani of the department of dermatology at the University of Modena (Italy) and Reggio Emilia, and his coauthors (J Eur Acad Dermatol Venereol. 2015;29[11]:2192-8).
“Ultimately, the treatment schedule is based on agreement between the physician and the patient; this study helps to support the selection of the most appropriate regimen to treat AK in individual patients,” they commented.
The study was funded by LEO Pharma, the manufacturer of ingenol mebutate gel (Picato). Dr. Pellacani has received consultant fees from the company; three authors are employees of the company; and the other authors declared consultancies, honoraria and, grants from LEO Pharma and other pharmaceutical companies.
Patients with multiple actinic keratoses (AKs) may be treated either sequentially or simultaneously with ingenol mebutate gel, according to the authors of a study that found no difference in outcomes or adverse effects from either treatment approach.
The phase IIIb study conducted in Italy and Spain enrolled 199 patients with two separate areas of clinically visible, nonhyperkeratotic AKs. Subjects were randomized to have the two areas (face/scalp and trunk/extremities) treated simultaneously (101 patients) or sequentially (98 patients) with 0.015% and 0.05% ingenol mebutate gel.
There were no significant differences in localized skin responses between the simultaneous and sequential treatment groups, based on the mean composite local skin response scores 3 days after treatment started, which were similar between the groups for the face/scalp and trunk/extremities applications. About 22% of patients in each group experienced adverse events.
At 8 weeks, the complete clearance rates also were not statistically different between the simultaneous and sequential groups (52.7% and 46.9%, respectively; P = .34), and patient satisfaction with treatment was similar for both treatment approaches. At that time, the number of AKs had dropped by a mean of 83.4% among those in the simultaneous group and 79.1% in the sequential group (P = .20).
“The favorable rate of complete clearance in the simultaneous treatment group means that patients can receive their treatment for both areas in one visit, rather than having to return to the clinic for a second cycle of treatment,” wrote Dr. Giovanni Pellacani of the department of dermatology at the University of Modena (Italy) and Reggio Emilia, and his coauthors (J Eur Acad Dermatol Venereol. 2015;29[11]:2192-8).
“Ultimately, the treatment schedule is based on agreement between the physician and the patient; this study helps to support the selection of the most appropriate regimen to treat AK in individual patients,” they commented.
The study was funded by LEO Pharma, the manufacturer of ingenol mebutate gel (Picato). Dr. Pellacani has received consultant fees from the company; three authors are employees of the company; and the other authors declared consultancies, honoraria and, grants from LEO Pharma and other pharmaceutical companies.
Patients with multiple actinic keratoses (AKs) may be treated either sequentially or simultaneously with ingenol mebutate gel, according to the authors of a study that found no difference in outcomes or adverse effects from either treatment approach.
The phase IIIb study conducted in Italy and Spain enrolled 199 patients with two separate areas of clinically visible, nonhyperkeratotic AKs. Subjects were randomized to have the two areas (face/scalp and trunk/extremities) treated simultaneously (101 patients) or sequentially (98 patients) with 0.015% and 0.05% ingenol mebutate gel.
There were no significant differences in localized skin responses between the simultaneous and sequential treatment groups, based on the mean composite local skin response scores 3 days after treatment started, which were similar between the groups for the face/scalp and trunk/extremities applications. About 22% of patients in each group experienced adverse events.
At 8 weeks, the complete clearance rates also were not statistically different between the simultaneous and sequential groups (52.7% and 46.9%, respectively; P = .34), and patient satisfaction with treatment was similar for both treatment approaches. At that time, the number of AKs had dropped by a mean of 83.4% among those in the simultaneous group and 79.1% in the sequential group (P = .20).
“The favorable rate of complete clearance in the simultaneous treatment group means that patients can receive their treatment for both areas in one visit, rather than having to return to the clinic for a second cycle of treatment,” wrote Dr. Giovanni Pellacani of the department of dermatology at the University of Modena (Italy) and Reggio Emilia, and his coauthors (J Eur Acad Dermatol Venereol. 2015;29[11]:2192-8).
“Ultimately, the treatment schedule is based on agreement between the physician and the patient; this study helps to support the selection of the most appropriate regimen to treat AK in individual patients,” they commented.
The study was funded by LEO Pharma, the manufacturer of ingenol mebutate gel (Picato). Dr. Pellacani has received consultant fees from the company; three authors are employees of the company; and the other authors declared consultancies, honoraria and, grants from LEO Pharma and other pharmaceutical companies.
FROM THE JOURNAL OF THE EUROPEAN ACADEMY OF DERMATOLOGY AND VENEREOLOGY
Key clinical point: Patients with multiple AKs may be treated either sequentially or simultaneously with ingenol mebutate gel, with similar efficacy and safety outcomes.
Major finding: The incidence of localized skin responses, complete clearance rates, and patient treatment satisfaction were similar for simultaneous and sequential treatment approaches.
Data source: A phase IIIb randomized, multicenter, open-label, parallel-group study evaluated 199 patients with two separate areas of clinically visible, nonhyperkeratotic AKs.
Disclosures: The study was funded by ingenol mebutate gel manufacturer LEO Pharma. Three authors are employees of the company; the other authors declared consultancies, honoraria, and/or grants from LEO Pharma and other pharmaceutical companies.
Posterior Reversible Encephalopathy Syndrome: Temporary Visual Loss After Spinal Deformity Surgery
First described in 1996, posterior reversible encephalopathy syndrome (PRES) exhibits a wide clinical spectrum and is definitively diagnosed through computed tomography (CT) and/or magnetic resonance imaging (MRI) studies of the brain.1 Clinical presentation may include a spectrum of symptoms, including nausea, emesis, seizures, visual loss, paralysis, and headaches.2,3 The most common imaging finding of PRES is bilateral foci of vasogenic edema located in the parieto-occipital white matter.2-6 Other areas of the brain are frequently affected as well, with the frontal and temporal lobes and the basal or cortical ganglia showing signs of distinctly noncytotoxic edema in 12.5% to 54.2% of all cases.3 With the symptom of visual loss being present in 20% to 62.5% of patients with PRES, the syndrome constitutes a rare potential cause for postoperative visual loss (POVL) after spinal surgery, which has a generally good prognosis because most patients will completely regain their eyesight.2,3
We present a unique account of 2 patients who underwent extensive spinal surgery and received a timely diagnosis and treatment of PRES at a single institution. We aim to elucidate the difference in clinical and radiographic presentation of PRES in relation to other known causes of POVL after spinal surgery. The patients provided written informed consent for print and electronic publication of these case reports.
Case Reports
Case 1
Clinical Presentation. A 78-year-old woman presented to the outpatient clinic with disability due to severe lower back pain. Her surgical history was significant for breast lumpectomy and cataract excision. Her medical history was significant for hypertension, obesity (body mass index, 31.5), hypercholesterolemia, emphysema, and anemia. She had undergone spinal surgery, specifically laminectomies from L2 to S1. The radiographic examination showed degenerative thoracolumbar scoliosis with severe spondylosis, disc space collapse, and ankylosis of L4-L5 (Figure 1).
Operative Procedure. The patient underwent transpsoas lumbar interbody fusion (XLIF, NuVasive) from L1 to L4 and posterior spinal fusion from T10 to pelvis (Expedium, Depuy Synthes) (Figure 2). Operative time was 553 minutes; estimated blood loss was 2000 mL due to intraoperative coagulopathy (platelets, 40,000/µL) near the end of the posterior portion of the procedure. Intraoperative hypotension was treated by volume resuscitation and transient use of vasopressor agents. She was transfused with 1700 mL of blood, 150 mL of saline solution, and 420 mL of Lactated Ringer’s solution. No intraoperative complications occurred. The patient was extubated uneventfully on postoperative day 1 and was at baseline neurologically with no visual disturbances.
Development and Diagnosis of PRES. The patient made significant progress with physical therapy and developed episodes of hypertension at night on postoperative days 4 to 6. Her mean peak systolic blood pressure was 180 mm Hg. This improved after oral beta-blocker therapy. On postoperative day 6, the patient was ambulating with physical therapy and the aid of a walker. She was found to be neurologically intact, was resting comfortable in a chair reading a book, and was cleared for transfer to a rehabilitation facility the next day. During the morning on postoperative day 7, she developed confusion and visual loss. The patient reported blurry vision followed by complete bilateral painless loss of vision aside from mild light perception. She was unable to identify any objects. She had extinction to double simultaneous stimuli and evidence of agraphesthesia in the left hand. Her neurologic examination was otherwise at baseline. Upon emergent imaging, head CT showed bilateral symmetric areas of hypodensity involving the cortical and subcortical white matter of both occipital lobes (Figure 3). MRI showed extensive bilateral cortical and subcortical signal hyperintensity involving the parietal and occipital lobes (Figure 4). No evidence of petechial or lobar hemorrhage was found.
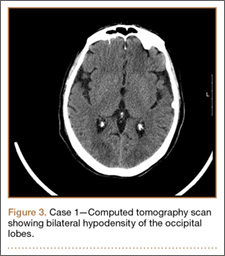
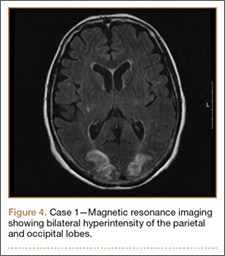
Treatment and Clinical Course. The patient was transferred to the neurology intensive care unit for neurologic monitoring. She was treated aggressively for recurrent hypertensive episodes. Twenty-four hours after initial blood pressure optimization therapy, she partially recovered her eyesight. She exhibited complete recovery after 48 hours. The patient was discharged to a rehabilitation facility in stable condition on postoperative day 11.
Case 2
Clinical Presentation. A 51-year-old woman presented to the outpatient clinic with progressive low back pain and decompensation due to degenerative adult scoliosis. Her surgical history was significant for an uneventful Caesarean section. Her medical history was significant for borderline hypertension and obesity (body mass index, 34.4). The radiographic examination showed an S-shaped thoracolumbar curve from T4 to L4 (Figure 5).
Operative Procedure. After discussions about the risks and benefits of the procedure, the patient underwent posterior spinal fusion from T3 to pelvis (Mesa, K2M) and interbody fusion from L4 to S1 via a presacral approach using the AxiaLIF system (TranS1) (Figure 6). The operation spanned 507 minutes. The patient lost approximately 2200 mL of blood. She was transfused with 1690 mL of blood, 1250 mL of Lactated Ringer’s solution, and 1 unit (50 mL) of albumin. No intraoperative complications occurred.
Development and Diagnosis of PRES. The patient was ambulatory with physical therapy and a walker on postoperative day 1. Her albumin levels were noted to be decreased postoperatively (28 mg/mL; normal, >35 mg/mL). She developed intermittent hypertensive episodes and experienced transient peripheral vision loss. After her ophthalmologic symptoms cleared, she was discharged and transferred to a rehabilitation facility on postoperative day 9. Eleven days later, the patient was emergently readmitted for a deep spine wound infection after an onset of wound swelling and fever. She underwent irrigation and débridement of the spine wound with an estimated blood loss of 400 mL. The patient continued to have fevers and was placed on ciprofloxacin and vancomycin, which was changed to levofloxacin on postoperative day 5. Elevated creatinine was noted, and the patient was diagnosed with acute renal failure. On postoperative day 7, oxacillin therapy was commenced. After her cultures grew methicillin-resistant Staphylococcus aureus, a peripherally inserted central catheter line was placed on postoperative day 9. As a result of nausea and constipation, the patient received feeding tubes on postoperative day 11. Additionally, she was diagnosed with a pleural effusion on postoperative day 14. Although her creatinine levels were decreasing, she continued to experience intermittent hypertensive episodes with a mean peak systolic blood pressure of 148 mm Hg. On postoperative day 15, she had a seizure and again developed visual loss. The patient was lethargic and followed only simple commands. She moved all extremities and withdrew symmetrically to noxious stimuli. Upon emergent imaging, head CT showed posterior subcortical white matter hypodensity within the occipital and parietal lobes bilaterally (Figure 7). MRI showed focal regions of symmetric hemispheric edema involving the parietal and occipital lobes in a predominantly subcortical white-matter distribution. Additionally, extensive involvement of the splenium and of the corpus callosum, left greater than right, was observed (Figure 8).
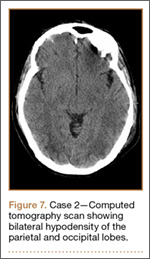
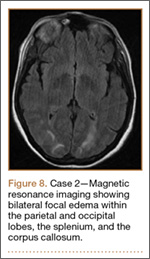
Treatment and Clinical Course. The patient was transferred to the intensive care unit for neuromonitoring. Her hypokalemia and hypertension were treated aggressively to normalize her potassium levels and blood pressure. Her oxacillin therapy was changed to daptomycin. On postoperative day 17, the patient was transferred to another institution for further medical management after achieving full recovery of her eyesight after electrolyte and blood pressure corrections.
Discussion
Posterior reversible encephalopathy syndrome is a rare but frequently devastating complication of spinal surgery, with an estimated incidence of 0.094% to 0.2%.7,8 Pediatric patients, as well as patients undergoing deformity correction surgery and posterior lumbar fusion, which necessitate prone positioning, have a significantly increased risk of POVL after spinal surgery.9 There are several causes of POVL after spinal surgery, each with a unique pathophysiology, clinical presentation, and prognosis.
The most common cause of POVL, accounting for 89% of all cases, is ischemic neuropathy.10 Ischemic neuropathy refers to a hypoperfusion or infarction of the anterior or posterior portion of the optic nerve and presents as painless bilateral vision loss or complete blindness on waking from the surgical procedure.11 Risk factors associated with anterior ischemic neuropathy are primarily diabetes mellitus, prone positioning, nocturnal hypotension, and blood loss.11 Posterior ischemic neuropathy has been most strongly correlated with anemia and hypotension.12 The exact etiology of this complication has not been established, although the prognosis is generally unfavorable, with most vision loss being permanent.10-12
Another potential cause of POVL after spinal surgery is retinal artery occlusion. It is most commonly observed in patients who were improperly positioned, resulting in compression of an orbit on the surface of the headrest or the operating table.13 Retinal artery occlusion characteristically presents as an irreversible unilateral complete loss of vision with a red spot on the macula and an afferent pupillary defect.14
Cortical blindness, another possible common cause of POVL, results from the hypoperfusion of the occipital cortex and has a slightly better prognosis. Cortical blindness generally results from an embolic event that can be visualized through neuroimaging and may be unilateral or bilateral, ranging from mild peripheral vision loss to complete blindness.15
Posterior reversible encephalopathy syndrome, the cause of POVL diagnosed in the 2 patients in this case report, is a neurologic syndrome that differs significantly in its clinical presentation and pathophysiology from the more well-known etiologies. The precise pathophysiologic mechanism of the syndrome is yet to be elucidated. One theory revolves around the failure of cerebral vascular autoregulation. It postulates that intracerebellar hypertension leads to the extravasation of proteins and fluid, resulting in the characteristic vasogenic edema.16,17 The other equally discussed theory postulates that cerebellar vasospasm and subsequent hypoperfusion leading to cellular hypoxemia and ischemia may be responsible.18-20 Posterior reversible encephalopathy syndrome has been reported with increasing frequency, particularly in connection with hypertension, acute renal failure associated with malignancy, cytotoxicity, and corticosteroids, as well as preeclampsia, eclampsia, and autoimmune disorders.1-3,21-23 Traditionally, patients display a combination of different symptoms, including vision changes ranging from slightly decreased perception to complete blindness. Unlike retinal artery occlusion and ischemic optic neuropathy, the onset of vision loss often does not happen immediately after surgery and may occur several hours to days after surgery. Visual disturbance may progressively worsen if the medical cause for the syndrome is not determined and corrected.2,3 In contrast to other known etiologies of POVL, PRES has a relatively favorable prognosis if managed appropriately. Reported case series determined a resolution of the characteristic parieto-occipital vasogenic edema in 83% to 88% of all patients in follow-up neuroimaging after aggressive control of seizures and arterial hypertension.2-3
Both patients undergoing spinal deformity surgery in this report suffered from intermittent hypertensive episodes in the postoperative period. One patient also developed acute renal failure during her hospital stay, and demonstrated low albumin levels postoperatively, which has also been associated with PRES.24 Through the immediate diagnosis and primary control of hypertension, both patients achieved complete neurologic recovery after a mean of 1.5 days (range, 1-2 days); this compares to a recovery period of an average 6.2 days (range, 1-14 days) reported by Ni and colleagues.3 The catastrophic effects of a misdiagnosis and incorrect or untimely treatment were well described in this case report. Several patients who were incorrectly diagnosed with demyelinating disorders or lupus encephalopathy received high doses of immunosuppressants and corticosteroids, known risk factors for the development of PRES.3 The patients subsequently rapidly deteriorated; no patients had a full recovery of their preoperative eyesight, and 1 patient developed complete permanent blindness.3 Optimized multidisciplinary collaboration allowing for a rapid neuro-ophthalmic examination and appropriate neuroimaging will permit an accurate and rapid diagnosis, leading to timely intervention and restoration of vision.
Conclusion
Temporary POVL is a potentially devastating complication of spinal surgery and general anesthesia. The more frequent causes such as ischemic optic neuropathy, retinal artery occlusion, and cortical blindness have very limited effective options for treatment and an overall poor prognosis. The inclusion of PRES in the differential diagnosis of POVL may allow early detection, management, and restoration of vision.
1. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334(8):494-500.
2. Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85(5):427-432.
3. Ni J, Zhou LX, Hao HL, et al. The clinical and radiological spectrum of posterior reversible encephalopathy syndrome: a retrospective series of 24 patients. J Neuroimaging. 2011;21(3):219-224.
4. Stevens CJ, Heran MK. The many faces of posterior reversible encephalopathy syndrome. Br J Radiol. 2012;85(1020):1566-1575.
5. Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29(6):1036-1042.
6. Yoon SD, Cho BM, Oh SM, et al. Clinical and radiological spectrum of posterior reversible encephalopathy syndrome. J Cerebrovasc Endovasc Neurosurg. 2013;15(3):206-213.
7. Patil CG, Lad EM, Lad SP, Ho C, Boakye M. Visual loss after spine surgery: a population-based study. Spine (Phila Pa 1976). 2008;33(13):1491-1496.
8. Stevens WR, Glazer PA, Kelley SD, Lietman TM, Bradford DS. Ophthalmic complications after spinal surgery. Spine (Phila Pa 1976). 1997;22(12):1319-1324.
9. Shen Y, Drum M, Roth S. The prevalence of perioperative visual loss in the United States: a 10-year study from 1996 to 2005 of spinal, orthopedic, cardiac, and general surgery. Anesth Analg. 2009;109(5):1534-1545.
10. Lee LA, Roth S, Posner KL, et al. The American Society of Anesthesiologists Postoperative Visual Loss Registry: analysis of 93 spine surgery cases with postoperative visual loss. Anesthesiology. 2006;105(4):652-659; quiz 867-868.
11. Hayreh SS. Ischemic optic neuropathies - where are we now? Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1873-1884.
12. Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol. 2005;50(1):15-26.
13. Katz DA, Karlin LI. Visual field defect after posterior spine fusion. Spine (Phila Pa 1976). 2005;30(3):E83-E85.
14. Hayreh SS, Kolder HE, Weingeist TA. Central retinal artery occlusion and retinal tolerance time. Ophthalmology. 1980;87(1):75-78.
15. Berg KT, Harrison AR, Lee MS. Perioperative visual loss in ocular and nonocular surgery. Clin Ophthalmol. 2010;4:531-546.
16. Primavera A, Audenino D, Mavilio N, Cocito L. Reversible posterior leucoencephalopathy syndrome in systemic lupus and vasculitis. Ann Rheum Dis. 2001;60(5):534-537.
17. Bartynski WS, Boardman JF. Catheter angiography, MR angiography, and MR perfusion in posterior reversible encephalopathy syndrome. AJNR Am J Neuroradiol. 2008;29(3):447-455.
18. Ito T, Sakai T, Inagawa S, Utsu M, Bun T. MR angiography of cerebral vasospasm in preeclampsia. AJNR Am J Neuroradiol. 1995;16(6):1344-1346.
19. Agarwal R, Davis C, Altinok D, Serajee FJ. Posterior reversible encephalopathy and cerebral vasoconstriction in a patient with hemolytic uremic syndrome. Pediatr Neurol. 2014;50(5):518-521.
20. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29(6):1043-1049.
21. Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;65(2):205-210.
22. Ekawa Y, Shiota M, Tobiume T, et al. Reversible posterior leukoencephalopathy syndrome accompanying eclampsia: correct diagnosis using preoperative MRI. Tohoku J Exp Med. 2012;226(1):55-58.
23. Kur JK, Esdaile JM. Posterior reversible encephalopathy syndrome--an underrecognized manifestation of systemic lupus erythematosus. J Rheumatol. 2006;33(11):2178-2183.
24. Pirker A, Kramer L, Voller B, et al. Type of edema in posterior reversible encephalopathy syndrome depends on serum albumin levels: an MR imaging study in 28 patients. AJNR Am J Neuroradiol. 2011;32(3):527-531.
First described in 1996, posterior reversible encephalopathy syndrome (PRES) exhibits a wide clinical spectrum and is definitively diagnosed through computed tomography (CT) and/or magnetic resonance imaging (MRI) studies of the brain.1 Clinical presentation may include a spectrum of symptoms, including nausea, emesis, seizures, visual loss, paralysis, and headaches.2,3 The most common imaging finding of PRES is bilateral foci of vasogenic edema located in the parieto-occipital white matter.2-6 Other areas of the brain are frequently affected as well, with the frontal and temporal lobes and the basal or cortical ganglia showing signs of distinctly noncytotoxic edema in 12.5% to 54.2% of all cases.3 With the symptom of visual loss being present in 20% to 62.5% of patients with PRES, the syndrome constitutes a rare potential cause for postoperative visual loss (POVL) after spinal surgery, which has a generally good prognosis because most patients will completely regain their eyesight.2,3
We present a unique account of 2 patients who underwent extensive spinal surgery and received a timely diagnosis and treatment of PRES at a single institution. We aim to elucidate the difference in clinical and radiographic presentation of PRES in relation to other known causes of POVL after spinal surgery. The patients provided written informed consent for print and electronic publication of these case reports.
Case Reports
Case 1
Clinical Presentation. A 78-year-old woman presented to the outpatient clinic with disability due to severe lower back pain. Her surgical history was significant for breast lumpectomy and cataract excision. Her medical history was significant for hypertension, obesity (body mass index, 31.5), hypercholesterolemia, emphysema, and anemia. She had undergone spinal surgery, specifically laminectomies from L2 to S1. The radiographic examination showed degenerative thoracolumbar scoliosis with severe spondylosis, disc space collapse, and ankylosis of L4-L5 (Figure 1).
Operative Procedure. The patient underwent transpsoas lumbar interbody fusion (XLIF, NuVasive) from L1 to L4 and posterior spinal fusion from T10 to pelvis (Expedium, Depuy Synthes) (Figure 2). Operative time was 553 minutes; estimated blood loss was 2000 mL due to intraoperative coagulopathy (platelets, 40,000/µL) near the end of the posterior portion of the procedure. Intraoperative hypotension was treated by volume resuscitation and transient use of vasopressor agents. She was transfused with 1700 mL of blood, 150 mL of saline solution, and 420 mL of Lactated Ringer’s solution. No intraoperative complications occurred. The patient was extubated uneventfully on postoperative day 1 and was at baseline neurologically with no visual disturbances.
Development and Diagnosis of PRES. The patient made significant progress with physical therapy and developed episodes of hypertension at night on postoperative days 4 to 6. Her mean peak systolic blood pressure was 180 mm Hg. This improved after oral beta-blocker therapy. On postoperative day 6, the patient was ambulating with physical therapy and the aid of a walker. She was found to be neurologically intact, was resting comfortable in a chair reading a book, and was cleared for transfer to a rehabilitation facility the next day. During the morning on postoperative day 7, she developed confusion and visual loss. The patient reported blurry vision followed by complete bilateral painless loss of vision aside from mild light perception. She was unable to identify any objects. She had extinction to double simultaneous stimuli and evidence of agraphesthesia in the left hand. Her neurologic examination was otherwise at baseline. Upon emergent imaging, head CT showed bilateral symmetric areas of hypodensity involving the cortical and subcortical white matter of both occipital lobes (Figure 3). MRI showed extensive bilateral cortical and subcortical signal hyperintensity involving the parietal and occipital lobes (Figure 4). No evidence of petechial or lobar hemorrhage was found.
Treatment and Clinical Course. The patient was transferred to the neurology intensive care unit for neurologic monitoring. She was treated aggressively for recurrent hypertensive episodes. Twenty-four hours after initial blood pressure optimization therapy, she partially recovered her eyesight. She exhibited complete recovery after 48 hours. The patient was discharged to a rehabilitation facility in stable condition on postoperative day 11.
Case 2
Clinical Presentation. A 51-year-old woman presented to the outpatient clinic with progressive low back pain and decompensation due to degenerative adult scoliosis. Her surgical history was significant for an uneventful Caesarean section. Her medical history was significant for borderline hypertension and obesity (body mass index, 34.4). The radiographic examination showed an S-shaped thoracolumbar curve from T4 to L4 (Figure 5).
Operative Procedure. After discussions about the risks and benefits of the procedure, the patient underwent posterior spinal fusion from T3 to pelvis (Mesa, K2M) and interbody fusion from L4 to S1 via a presacral approach using the AxiaLIF system (TranS1) (Figure 6). The operation spanned 507 minutes. The patient lost approximately 2200 mL of blood. She was transfused with 1690 mL of blood, 1250 mL of Lactated Ringer’s solution, and 1 unit (50 mL) of albumin. No intraoperative complications occurred.
Development and Diagnosis of PRES. The patient was ambulatory with physical therapy and a walker on postoperative day 1. Her albumin levels were noted to be decreased postoperatively (28 mg/mL; normal, >35 mg/mL). She developed intermittent hypertensive episodes and experienced transient peripheral vision loss. After her ophthalmologic symptoms cleared, she was discharged and transferred to a rehabilitation facility on postoperative day 9. Eleven days later, the patient was emergently readmitted for a deep spine wound infection after an onset of wound swelling and fever. She underwent irrigation and débridement of the spine wound with an estimated blood loss of 400 mL. The patient continued to have fevers and was placed on ciprofloxacin and vancomycin, which was changed to levofloxacin on postoperative day 5. Elevated creatinine was noted, and the patient was diagnosed with acute renal failure. On postoperative day 7, oxacillin therapy was commenced. After her cultures grew methicillin-resistant Staphylococcus aureus, a peripherally inserted central catheter line was placed on postoperative day 9. As a result of nausea and constipation, the patient received feeding tubes on postoperative day 11. Additionally, she was diagnosed with a pleural effusion on postoperative day 14. Although her creatinine levels were decreasing, she continued to experience intermittent hypertensive episodes with a mean peak systolic blood pressure of 148 mm Hg. On postoperative day 15, she had a seizure and again developed visual loss. The patient was lethargic and followed only simple commands. She moved all extremities and withdrew symmetrically to noxious stimuli. Upon emergent imaging, head CT showed posterior subcortical white matter hypodensity within the occipital and parietal lobes bilaterally (Figure 7). MRI showed focal regions of symmetric hemispheric edema involving the parietal and occipital lobes in a predominantly subcortical white-matter distribution. Additionally, extensive involvement of the splenium and of the corpus callosum, left greater than right, was observed (Figure 8).
Treatment and Clinical Course. The patient was transferred to the intensive care unit for neuromonitoring. Her hypokalemia and hypertension were treated aggressively to normalize her potassium levels and blood pressure. Her oxacillin therapy was changed to daptomycin. On postoperative day 17, the patient was transferred to another institution for further medical management after achieving full recovery of her eyesight after electrolyte and blood pressure corrections.
Discussion
Posterior reversible encephalopathy syndrome is a rare but frequently devastating complication of spinal surgery, with an estimated incidence of 0.094% to 0.2%.7,8 Pediatric patients, as well as patients undergoing deformity correction surgery and posterior lumbar fusion, which necessitate prone positioning, have a significantly increased risk of POVL after spinal surgery.9 There are several causes of POVL after spinal surgery, each with a unique pathophysiology, clinical presentation, and prognosis.
The most common cause of POVL, accounting for 89% of all cases, is ischemic neuropathy.10 Ischemic neuropathy refers to a hypoperfusion or infarction of the anterior or posterior portion of the optic nerve and presents as painless bilateral vision loss or complete blindness on waking from the surgical procedure.11 Risk factors associated with anterior ischemic neuropathy are primarily diabetes mellitus, prone positioning, nocturnal hypotension, and blood loss.11 Posterior ischemic neuropathy has been most strongly correlated with anemia and hypotension.12 The exact etiology of this complication has not been established, although the prognosis is generally unfavorable, with most vision loss being permanent.10-12
Another potential cause of POVL after spinal surgery is retinal artery occlusion. It is most commonly observed in patients who were improperly positioned, resulting in compression of an orbit on the surface of the headrest or the operating table.13 Retinal artery occlusion characteristically presents as an irreversible unilateral complete loss of vision with a red spot on the macula and an afferent pupillary defect.14
Cortical blindness, another possible common cause of POVL, results from the hypoperfusion of the occipital cortex and has a slightly better prognosis. Cortical blindness generally results from an embolic event that can be visualized through neuroimaging and may be unilateral or bilateral, ranging from mild peripheral vision loss to complete blindness.15
Posterior reversible encephalopathy syndrome, the cause of POVL diagnosed in the 2 patients in this case report, is a neurologic syndrome that differs significantly in its clinical presentation and pathophysiology from the more well-known etiologies. The precise pathophysiologic mechanism of the syndrome is yet to be elucidated. One theory revolves around the failure of cerebral vascular autoregulation. It postulates that intracerebellar hypertension leads to the extravasation of proteins and fluid, resulting in the characteristic vasogenic edema.16,17 The other equally discussed theory postulates that cerebellar vasospasm and subsequent hypoperfusion leading to cellular hypoxemia and ischemia may be responsible.18-20 Posterior reversible encephalopathy syndrome has been reported with increasing frequency, particularly in connection with hypertension, acute renal failure associated with malignancy, cytotoxicity, and corticosteroids, as well as preeclampsia, eclampsia, and autoimmune disorders.1-3,21-23 Traditionally, patients display a combination of different symptoms, including vision changes ranging from slightly decreased perception to complete blindness. Unlike retinal artery occlusion and ischemic optic neuropathy, the onset of vision loss often does not happen immediately after surgery and may occur several hours to days after surgery. Visual disturbance may progressively worsen if the medical cause for the syndrome is not determined and corrected.2,3 In contrast to other known etiologies of POVL, PRES has a relatively favorable prognosis if managed appropriately. Reported case series determined a resolution of the characteristic parieto-occipital vasogenic edema in 83% to 88% of all patients in follow-up neuroimaging after aggressive control of seizures and arterial hypertension.2-3
Both patients undergoing spinal deformity surgery in this report suffered from intermittent hypertensive episodes in the postoperative period. One patient also developed acute renal failure during her hospital stay, and demonstrated low albumin levels postoperatively, which has also been associated with PRES.24 Through the immediate diagnosis and primary control of hypertension, both patients achieved complete neurologic recovery after a mean of 1.5 days (range, 1-2 days); this compares to a recovery period of an average 6.2 days (range, 1-14 days) reported by Ni and colleagues.3 The catastrophic effects of a misdiagnosis and incorrect or untimely treatment were well described in this case report. Several patients who were incorrectly diagnosed with demyelinating disorders or lupus encephalopathy received high doses of immunosuppressants and corticosteroids, known risk factors for the development of PRES.3 The patients subsequently rapidly deteriorated; no patients had a full recovery of their preoperative eyesight, and 1 patient developed complete permanent blindness.3 Optimized multidisciplinary collaboration allowing for a rapid neuro-ophthalmic examination and appropriate neuroimaging will permit an accurate and rapid diagnosis, leading to timely intervention and restoration of vision.
Conclusion
Temporary POVL is a potentially devastating complication of spinal surgery and general anesthesia. The more frequent causes such as ischemic optic neuropathy, retinal artery occlusion, and cortical blindness have very limited effective options for treatment and an overall poor prognosis. The inclusion of PRES in the differential diagnosis of POVL may allow early detection, management, and restoration of vision.
First described in 1996, posterior reversible encephalopathy syndrome (PRES) exhibits a wide clinical spectrum and is definitively diagnosed through computed tomography (CT) and/or magnetic resonance imaging (MRI) studies of the brain.1 Clinical presentation may include a spectrum of symptoms, including nausea, emesis, seizures, visual loss, paralysis, and headaches.2,3 The most common imaging finding of PRES is bilateral foci of vasogenic edema located in the parieto-occipital white matter.2-6 Other areas of the brain are frequently affected as well, with the frontal and temporal lobes and the basal or cortical ganglia showing signs of distinctly noncytotoxic edema in 12.5% to 54.2% of all cases.3 With the symptom of visual loss being present in 20% to 62.5% of patients with PRES, the syndrome constitutes a rare potential cause for postoperative visual loss (POVL) after spinal surgery, which has a generally good prognosis because most patients will completely regain their eyesight.2,3
We present a unique account of 2 patients who underwent extensive spinal surgery and received a timely diagnosis and treatment of PRES at a single institution. We aim to elucidate the difference in clinical and radiographic presentation of PRES in relation to other known causes of POVL after spinal surgery. The patients provided written informed consent for print and electronic publication of these case reports.
Case Reports
Case 1
Clinical Presentation. A 78-year-old woman presented to the outpatient clinic with disability due to severe lower back pain. Her surgical history was significant for breast lumpectomy and cataract excision. Her medical history was significant for hypertension, obesity (body mass index, 31.5), hypercholesterolemia, emphysema, and anemia. She had undergone spinal surgery, specifically laminectomies from L2 to S1. The radiographic examination showed degenerative thoracolumbar scoliosis with severe spondylosis, disc space collapse, and ankylosis of L4-L5 (Figure 1).
Operative Procedure. The patient underwent transpsoas lumbar interbody fusion (XLIF, NuVasive) from L1 to L4 and posterior spinal fusion from T10 to pelvis (Expedium, Depuy Synthes) (Figure 2). Operative time was 553 minutes; estimated blood loss was 2000 mL due to intraoperative coagulopathy (platelets, 40,000/µL) near the end of the posterior portion of the procedure. Intraoperative hypotension was treated by volume resuscitation and transient use of vasopressor agents. She was transfused with 1700 mL of blood, 150 mL of saline solution, and 420 mL of Lactated Ringer’s solution. No intraoperative complications occurred. The patient was extubated uneventfully on postoperative day 1 and was at baseline neurologically with no visual disturbances.
Development and Diagnosis of PRES. The patient made significant progress with physical therapy and developed episodes of hypertension at night on postoperative days 4 to 6. Her mean peak systolic blood pressure was 180 mm Hg. This improved after oral beta-blocker therapy. On postoperative day 6, the patient was ambulating with physical therapy and the aid of a walker. She was found to be neurologically intact, was resting comfortable in a chair reading a book, and was cleared for transfer to a rehabilitation facility the next day. During the morning on postoperative day 7, she developed confusion and visual loss. The patient reported blurry vision followed by complete bilateral painless loss of vision aside from mild light perception. She was unable to identify any objects. She had extinction to double simultaneous stimuli and evidence of agraphesthesia in the left hand. Her neurologic examination was otherwise at baseline. Upon emergent imaging, head CT showed bilateral symmetric areas of hypodensity involving the cortical and subcortical white matter of both occipital lobes (Figure 3). MRI showed extensive bilateral cortical and subcortical signal hyperintensity involving the parietal and occipital lobes (Figure 4). No evidence of petechial or lobar hemorrhage was found.
Treatment and Clinical Course. The patient was transferred to the neurology intensive care unit for neurologic monitoring. She was treated aggressively for recurrent hypertensive episodes. Twenty-four hours after initial blood pressure optimization therapy, she partially recovered her eyesight. She exhibited complete recovery after 48 hours. The patient was discharged to a rehabilitation facility in stable condition on postoperative day 11.
Case 2
Clinical Presentation. A 51-year-old woman presented to the outpatient clinic with progressive low back pain and decompensation due to degenerative adult scoliosis. Her surgical history was significant for an uneventful Caesarean section. Her medical history was significant for borderline hypertension and obesity (body mass index, 34.4). The radiographic examination showed an S-shaped thoracolumbar curve from T4 to L4 (Figure 5).
Operative Procedure. After discussions about the risks and benefits of the procedure, the patient underwent posterior spinal fusion from T3 to pelvis (Mesa, K2M) and interbody fusion from L4 to S1 via a presacral approach using the AxiaLIF system (TranS1) (Figure 6). The operation spanned 507 minutes. The patient lost approximately 2200 mL of blood. She was transfused with 1690 mL of blood, 1250 mL of Lactated Ringer’s solution, and 1 unit (50 mL) of albumin. No intraoperative complications occurred.
Development and Diagnosis of PRES. The patient was ambulatory with physical therapy and a walker on postoperative day 1. Her albumin levels were noted to be decreased postoperatively (28 mg/mL; normal, >35 mg/mL). She developed intermittent hypertensive episodes and experienced transient peripheral vision loss. After her ophthalmologic symptoms cleared, she was discharged and transferred to a rehabilitation facility on postoperative day 9. Eleven days later, the patient was emergently readmitted for a deep spine wound infection after an onset of wound swelling and fever. She underwent irrigation and débridement of the spine wound with an estimated blood loss of 400 mL. The patient continued to have fevers and was placed on ciprofloxacin and vancomycin, which was changed to levofloxacin on postoperative day 5. Elevated creatinine was noted, and the patient was diagnosed with acute renal failure. On postoperative day 7, oxacillin therapy was commenced. After her cultures grew methicillin-resistant Staphylococcus aureus, a peripherally inserted central catheter line was placed on postoperative day 9. As a result of nausea and constipation, the patient received feeding tubes on postoperative day 11. Additionally, she was diagnosed with a pleural effusion on postoperative day 14. Although her creatinine levels were decreasing, she continued to experience intermittent hypertensive episodes with a mean peak systolic blood pressure of 148 mm Hg. On postoperative day 15, she had a seizure and again developed visual loss. The patient was lethargic and followed only simple commands. She moved all extremities and withdrew symmetrically to noxious stimuli. Upon emergent imaging, head CT showed posterior subcortical white matter hypodensity within the occipital and parietal lobes bilaterally (Figure 7). MRI showed focal regions of symmetric hemispheric edema involving the parietal and occipital lobes in a predominantly subcortical white-matter distribution. Additionally, extensive involvement of the splenium and of the corpus callosum, left greater than right, was observed (Figure 8).
Treatment and Clinical Course. The patient was transferred to the intensive care unit for neuromonitoring. Her hypokalemia and hypertension were treated aggressively to normalize her potassium levels and blood pressure. Her oxacillin therapy was changed to daptomycin. On postoperative day 17, the patient was transferred to another institution for further medical management after achieving full recovery of her eyesight after electrolyte and blood pressure corrections.
Discussion
Posterior reversible encephalopathy syndrome is a rare but frequently devastating complication of spinal surgery, with an estimated incidence of 0.094% to 0.2%.7,8 Pediatric patients, as well as patients undergoing deformity correction surgery and posterior lumbar fusion, which necessitate prone positioning, have a significantly increased risk of POVL after spinal surgery.9 There are several causes of POVL after spinal surgery, each with a unique pathophysiology, clinical presentation, and prognosis.
The most common cause of POVL, accounting for 89% of all cases, is ischemic neuropathy.10 Ischemic neuropathy refers to a hypoperfusion or infarction of the anterior or posterior portion of the optic nerve and presents as painless bilateral vision loss or complete blindness on waking from the surgical procedure.11 Risk factors associated with anterior ischemic neuropathy are primarily diabetes mellitus, prone positioning, nocturnal hypotension, and blood loss.11 Posterior ischemic neuropathy has been most strongly correlated with anemia and hypotension.12 The exact etiology of this complication has not been established, although the prognosis is generally unfavorable, with most vision loss being permanent.10-12
Another potential cause of POVL after spinal surgery is retinal artery occlusion. It is most commonly observed in patients who were improperly positioned, resulting in compression of an orbit on the surface of the headrest or the operating table.13 Retinal artery occlusion characteristically presents as an irreversible unilateral complete loss of vision with a red spot on the macula and an afferent pupillary defect.14
Cortical blindness, another possible common cause of POVL, results from the hypoperfusion of the occipital cortex and has a slightly better prognosis. Cortical blindness generally results from an embolic event that can be visualized through neuroimaging and may be unilateral or bilateral, ranging from mild peripheral vision loss to complete blindness.15
Posterior reversible encephalopathy syndrome, the cause of POVL diagnosed in the 2 patients in this case report, is a neurologic syndrome that differs significantly in its clinical presentation and pathophysiology from the more well-known etiologies. The precise pathophysiologic mechanism of the syndrome is yet to be elucidated. One theory revolves around the failure of cerebral vascular autoregulation. It postulates that intracerebellar hypertension leads to the extravasation of proteins and fluid, resulting in the characteristic vasogenic edema.16,17 The other equally discussed theory postulates that cerebellar vasospasm and subsequent hypoperfusion leading to cellular hypoxemia and ischemia may be responsible.18-20 Posterior reversible encephalopathy syndrome has been reported with increasing frequency, particularly in connection with hypertension, acute renal failure associated with malignancy, cytotoxicity, and corticosteroids, as well as preeclampsia, eclampsia, and autoimmune disorders.1-3,21-23 Traditionally, patients display a combination of different symptoms, including vision changes ranging from slightly decreased perception to complete blindness. Unlike retinal artery occlusion and ischemic optic neuropathy, the onset of vision loss often does not happen immediately after surgery and may occur several hours to days after surgery. Visual disturbance may progressively worsen if the medical cause for the syndrome is not determined and corrected.2,3 In contrast to other known etiologies of POVL, PRES has a relatively favorable prognosis if managed appropriately. Reported case series determined a resolution of the characteristic parieto-occipital vasogenic edema in 83% to 88% of all patients in follow-up neuroimaging after aggressive control of seizures and arterial hypertension.2-3
Both patients undergoing spinal deformity surgery in this report suffered from intermittent hypertensive episodes in the postoperative period. One patient also developed acute renal failure during her hospital stay, and demonstrated low albumin levels postoperatively, which has also been associated with PRES.24 Through the immediate diagnosis and primary control of hypertension, both patients achieved complete neurologic recovery after a mean of 1.5 days (range, 1-2 days); this compares to a recovery period of an average 6.2 days (range, 1-14 days) reported by Ni and colleagues.3 The catastrophic effects of a misdiagnosis and incorrect or untimely treatment were well described in this case report. Several patients who were incorrectly diagnosed with demyelinating disorders or lupus encephalopathy received high doses of immunosuppressants and corticosteroids, known risk factors for the development of PRES.3 The patients subsequently rapidly deteriorated; no patients had a full recovery of their preoperative eyesight, and 1 patient developed complete permanent blindness.3 Optimized multidisciplinary collaboration allowing for a rapid neuro-ophthalmic examination and appropriate neuroimaging will permit an accurate and rapid diagnosis, leading to timely intervention and restoration of vision.
Conclusion
Temporary POVL is a potentially devastating complication of spinal surgery and general anesthesia. The more frequent causes such as ischemic optic neuropathy, retinal artery occlusion, and cortical blindness have very limited effective options for treatment and an overall poor prognosis. The inclusion of PRES in the differential diagnosis of POVL may allow early detection, management, and restoration of vision.
1. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334(8):494-500.
2. Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85(5):427-432.
3. Ni J, Zhou LX, Hao HL, et al. The clinical and radiological spectrum of posterior reversible encephalopathy syndrome: a retrospective series of 24 patients. J Neuroimaging. 2011;21(3):219-224.
4. Stevens CJ, Heran MK. The many faces of posterior reversible encephalopathy syndrome. Br J Radiol. 2012;85(1020):1566-1575.
5. Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29(6):1036-1042.
6. Yoon SD, Cho BM, Oh SM, et al. Clinical and radiological spectrum of posterior reversible encephalopathy syndrome. J Cerebrovasc Endovasc Neurosurg. 2013;15(3):206-213.
7. Patil CG, Lad EM, Lad SP, Ho C, Boakye M. Visual loss after spine surgery: a population-based study. Spine (Phila Pa 1976). 2008;33(13):1491-1496.
8. Stevens WR, Glazer PA, Kelley SD, Lietman TM, Bradford DS. Ophthalmic complications after spinal surgery. Spine (Phila Pa 1976). 1997;22(12):1319-1324.
9. Shen Y, Drum M, Roth S. The prevalence of perioperative visual loss in the United States: a 10-year study from 1996 to 2005 of spinal, orthopedic, cardiac, and general surgery. Anesth Analg. 2009;109(5):1534-1545.
10. Lee LA, Roth S, Posner KL, et al. The American Society of Anesthesiologists Postoperative Visual Loss Registry: analysis of 93 spine surgery cases with postoperative visual loss. Anesthesiology. 2006;105(4):652-659; quiz 867-868.
11. Hayreh SS. Ischemic optic neuropathies - where are we now? Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1873-1884.
12. Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol. 2005;50(1):15-26.
13. Katz DA, Karlin LI. Visual field defect after posterior spine fusion. Spine (Phila Pa 1976). 2005;30(3):E83-E85.
14. Hayreh SS, Kolder HE, Weingeist TA. Central retinal artery occlusion and retinal tolerance time. Ophthalmology. 1980;87(1):75-78.
15. Berg KT, Harrison AR, Lee MS. Perioperative visual loss in ocular and nonocular surgery. Clin Ophthalmol. 2010;4:531-546.
16. Primavera A, Audenino D, Mavilio N, Cocito L. Reversible posterior leucoencephalopathy syndrome in systemic lupus and vasculitis. Ann Rheum Dis. 2001;60(5):534-537.
17. Bartynski WS, Boardman JF. Catheter angiography, MR angiography, and MR perfusion in posterior reversible encephalopathy syndrome. AJNR Am J Neuroradiol. 2008;29(3):447-455.
18. Ito T, Sakai T, Inagawa S, Utsu M, Bun T. MR angiography of cerebral vasospasm in preeclampsia. AJNR Am J Neuroradiol. 1995;16(6):1344-1346.
19. Agarwal R, Davis C, Altinok D, Serajee FJ. Posterior reversible encephalopathy and cerebral vasoconstriction in a patient with hemolytic uremic syndrome. Pediatr Neurol. 2014;50(5):518-521.
20. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29(6):1043-1049.
21. Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;65(2):205-210.
22. Ekawa Y, Shiota M, Tobiume T, et al. Reversible posterior leukoencephalopathy syndrome accompanying eclampsia: correct diagnosis using preoperative MRI. Tohoku J Exp Med. 2012;226(1):55-58.
23. Kur JK, Esdaile JM. Posterior reversible encephalopathy syndrome--an underrecognized manifestation of systemic lupus erythematosus. J Rheumatol. 2006;33(11):2178-2183.
24. Pirker A, Kramer L, Voller B, et al. Type of edema in posterior reversible encephalopathy syndrome depends on serum albumin levels: an MR imaging study in 28 patients. AJNR Am J Neuroradiol. 2011;32(3):527-531.
1. Hinchey J, Chaves C, Appignani B, et al. A reversible posterior leukoencephalopathy syndrome. N Engl J Med. 1996;334(8):494-500.
2. Fugate JE, Claassen DO, Cloft HJ, et al. Posterior reversible encephalopathy syndrome: associated clinical and radiologic findings. Mayo Clin Proc. 2010;85(5):427-432.
3. Ni J, Zhou LX, Hao HL, et al. The clinical and radiological spectrum of posterior reversible encephalopathy syndrome: a retrospective series of 24 patients. J Neuroimaging. 2011;21(3):219-224.
4. Stevens CJ, Heran MK. The many faces of posterior reversible encephalopathy syndrome. Br J Radiol. 2012;85(1020):1566-1575.
5. Bartynski WS. Posterior reversible encephalopathy syndrome, part 1: fundamental imaging and clinical features. AJNR Am J Neuroradiol. 2008;29(6):1036-1042.
6. Yoon SD, Cho BM, Oh SM, et al. Clinical and radiological spectrum of posterior reversible encephalopathy syndrome. J Cerebrovasc Endovasc Neurosurg. 2013;15(3):206-213.
7. Patil CG, Lad EM, Lad SP, Ho C, Boakye M. Visual loss after spine surgery: a population-based study. Spine (Phila Pa 1976). 2008;33(13):1491-1496.
8. Stevens WR, Glazer PA, Kelley SD, Lietman TM, Bradford DS. Ophthalmic complications after spinal surgery. Spine (Phila Pa 1976). 1997;22(12):1319-1324.
9. Shen Y, Drum M, Roth S. The prevalence of perioperative visual loss in the United States: a 10-year study from 1996 to 2005 of spinal, orthopedic, cardiac, and general surgery. Anesth Analg. 2009;109(5):1534-1545.
10. Lee LA, Roth S, Posner KL, et al. The American Society of Anesthesiologists Postoperative Visual Loss Registry: analysis of 93 spine surgery cases with postoperative visual loss. Anesthesiology. 2006;105(4):652-659; quiz 867-868.
11. Hayreh SS. Ischemic optic neuropathies - where are we now? Graefes Arch Clin Exp Ophthalmol. 2013;251(8):1873-1884.
12. Buono LM, Foroozan R. Perioperative posterior ischemic optic neuropathy: review of the literature. Surv Ophthalmol. 2005;50(1):15-26.
13. Katz DA, Karlin LI. Visual field defect after posterior spine fusion. Spine (Phila Pa 1976). 2005;30(3):E83-E85.
14. Hayreh SS, Kolder HE, Weingeist TA. Central retinal artery occlusion and retinal tolerance time. Ophthalmology. 1980;87(1):75-78.
15. Berg KT, Harrison AR, Lee MS. Perioperative visual loss in ocular and nonocular surgery. Clin Ophthalmol. 2010;4:531-546.
16. Primavera A, Audenino D, Mavilio N, Cocito L. Reversible posterior leucoencephalopathy syndrome in systemic lupus and vasculitis. Ann Rheum Dis. 2001;60(5):534-537.
17. Bartynski WS, Boardman JF. Catheter angiography, MR angiography, and MR perfusion in posterior reversible encephalopathy syndrome. AJNR Am J Neuroradiol. 2008;29(3):447-455.
18. Ito T, Sakai T, Inagawa S, Utsu M, Bun T. MR angiography of cerebral vasospasm in preeclampsia. AJNR Am J Neuroradiol. 1995;16(6):1344-1346.
19. Agarwal R, Davis C, Altinok D, Serajee FJ. Posterior reversible encephalopathy and cerebral vasoconstriction in a patient with hemolytic uremic syndrome. Pediatr Neurol. 2014;50(5):518-521.
20. Bartynski WS. Posterior reversible encephalopathy syndrome, part 2: controversies surrounding pathophysiology of vasogenic edema. AJNR Am J Neuroradiol. 2008;29(6):1043-1049.
21. Lee VH, Wijdicks EF, Manno EM, Rabinstein AA. Clinical spectrum of reversible posterior leukoencephalopathy syndrome. Arch Neurol. 2008;65(2):205-210.
22. Ekawa Y, Shiota M, Tobiume T, et al. Reversible posterior leukoencephalopathy syndrome accompanying eclampsia: correct diagnosis using preoperative MRI. Tohoku J Exp Med. 2012;226(1):55-58.
23. Kur JK, Esdaile JM. Posterior reversible encephalopathy syndrome--an underrecognized manifestation of systemic lupus erythematosus. J Rheumatol. 2006;33(11):2178-2183.
24. Pirker A, Kramer L, Voller B, et al. Type of edema in posterior reversible encephalopathy syndrome depends on serum albumin levels: an MR imaging study in 28 patients. AJNR Am J Neuroradiol. 2011;32(3):527-531.
Canada may shorten deferral for MSM blood donors

Photo by Charles Haymond
ANAHEIM, CA—Lifting the lifetime ban on blood donations from men who have sex with men (MSM) has not altered the safety of the blood supply in Canada, according to a new study.
The study showed no increase in the rate of HIV-positive blood donations since Canada changed its policy regarding MSM blood donors, allowing MSM to donate if they have not had sexual contact with another man in the last 5 years.
Because of this finding, Canada may shorten the deferral period for MSM blood donors to 1 year, according to Sheila F. O’Brien, PhD, of Canadian Blood Services in Ottawa, Ontario, Canada.
Dr O’Brien mentioned this possibility and presented data from the study at the 2015 AABB Annual Meeting (abstract S35-030E*).
Prior to 2013, MSM in Canada were not allowed to donate blood if they had any sexual contact with another male since 1977. Females were barred from donating if, in the last year, they had sexual contact with a man who had sex with another man after 1977.
On July 22, 2013, Canada changed this policy so that MSM can donate blood if they have abstained from sexual contact with another man for the past 5 years. The deferral period for females is still 12 months if they have had sex with a man who has had sex with another man in the last 5 years, but there is no deferral if the man had sex with another man more than 5 years before.
To evaluate the impact of this policy change, Dr O’Brien and her colleagues assessed compliance with the MSM criteria before and after the change, as well as the number of HIV-positive blood donations before and after the change.
The researchers also assessed the number of donors who would have been deferred according to the old MSM criteria but donated blood under the new criteria.
MSM history
The researchers selected random male donors of whole blood each month from October 2012 to February 2013 (pre-change) and from October 2014 to February 2015 (post-change). These donors were invited to complete an anonymous online survey about their MSM history.
The survey was completed by 9669 donors before the policy change and 6881 donors after the change. There were 77 donors with MSM history before the change (20% first-time donors, 80% repeat) and 75 donors with MSM history after the change (22% first-time, 78% repeat).
Compliance with policy
After the change in policy for MSM blood donors, there was no significant change in the proportion of donors who had recent MSM history but donated anyway (non-compliant). Before the change, 0.37% of blood donors had an MSM partner in the last 5 years, compared to 0.43% after the change (P=0.54).
However, there was a significant change in the proportion of blood donors with MSM history further in the past. Before the MSM policy change, 0.42% of donors had an MSM partner but not in the last 5 years, compared to 0.66% of donors after the change (P=0.04).
“So we have an improvement in compliance, but it’s mainly because the donors are no longer deferrable,” Dr O’Brien explained.
“Donating while ineligible because of MSM history is actually quite rare, and the percentage of donors with MSM history in the last 5 years did not change when we changed the criteria. But we did see a modest increase in newly eligible MSM, so those that had more than 5 years since their last male-to-male sex.”
In all, there were 112 donors who were newly eligible due to the policy change and did, in fact, donate blood between July 22, 2013 and July 21, 2015. Five of these donors were females who had sexual contact with MSMs.
There were 70 “reinstated” donors in the first year after the policy change and 42 in the second year.
HIV-positive donations
The researchers monitored HIV rates in all blood donations from January 2010 to March 2015.
The rates of HIV-positive donations were as follows: 0.20 for 2010 (2/989,916), 0.50 for 2011 (5/995,122), 0.51 for 2012 (5/987,527), 0 (0/525,337) from January 1, 2013 to July 21, 2013 (before the policy change), 0.54 from July 22, 2013 to July 21, 2014 (5/929,656), and 0.22 from July 22, 2014 to July 21, 2015 (2/893,513).
“So absolutely no change in HIV rate following implementation of our 5-year deferral,” Dr O’Brien said.
In all, there were 7 HIV-positive donations after the policy change. Four were from male donors, and 3 were from females.
Three of the male donors (2 first-time donors, 1 repeat) denied having MSM risk factors, and 1 first-time male donor was aware he was HIV-positive at the time of donation. This man said he donated to determine if his HIV medication was working.
Two of the females were repeat donors, and 1 was a first-timer. The first-time donor did not acknowledge any MSM risk factors. One of the repeat donors had a sexual relationship with a bisexual male who was HIV-positive. The other repeat donor had multiple sexual partners, 1 of whom was known to be hepatitis C-positive.
Future policy change
Dr O’Brien noted that the LGBTQ community in Canada has advocated abolishing the deferral period for MSM blood donors or changing to a risk-based policy that would allow more individuals with MSM history to donate blood.
She said the combined blood services in Canada—Canadian Blood Services and Héma-Québec—are now considering a 12-month deferral period for individuals with MSM history.
“We’re pretty sure we’re going to go ahead,” she noted.
However, the groups must submit a policy request to Health Canada, which will ultimately make the decision. ![]()
*Data in the abstract differ from data presented at the meeting.
Photo by Charles Haymond
ANAHEIM, CA—Lifting the lifetime ban on blood donations from men who have sex with men (MSM) has not altered the safety of the blood supply in Canada, according to a new study.
The study showed no increase in the rate of HIV-positive blood donations since Canada changed its policy regarding MSM blood donors, allowing MSM to donate if they have not had sexual contact with another man in the last 5 years.
Because of this finding, Canada may shorten the deferral period for MSM blood donors to 1 year, according to Sheila F. O’Brien, PhD, of Canadian Blood Services in Ottawa, Ontario, Canada.
Dr O’Brien mentioned this possibility and presented data from the study at the 2015 AABB Annual Meeting (abstract S35-030E*).
Prior to 2013, MSM in Canada were not allowed to donate blood if they had any sexual contact with another male since 1977. Females were barred from donating if, in the last year, they had sexual contact with a man who had sex with another man after 1977.
On July 22, 2013, Canada changed this policy so that MSM can donate blood if they have abstained from sexual contact with another man for the past 5 years. The deferral period for females is still 12 months if they have had sex with a man who has had sex with another man in the last 5 years, but there is no deferral if the man had sex with another man more than 5 years before.
To evaluate the impact of this policy change, Dr O’Brien and her colleagues assessed compliance with the MSM criteria before and after the change, as well as the number of HIV-positive blood donations before and after the change.
The researchers also assessed the number of donors who would have been deferred according to the old MSM criteria but donated blood under the new criteria.
MSM history
The researchers selected random male donors of whole blood each month from October 2012 to February 2013 (pre-change) and from October 2014 to February 2015 (post-change). These donors were invited to complete an anonymous online survey about their MSM history.
The survey was completed by 9669 donors before the policy change and 6881 donors after the change. There were 77 donors with MSM history before the change (20% first-time donors, 80% repeat) and 75 donors with MSM history after the change (22% first-time, 78% repeat).
Compliance with policy
After the change in policy for MSM blood donors, there was no significant change in the proportion of donors who had recent MSM history but donated anyway (non-compliant). Before the change, 0.37% of blood donors had an MSM partner in the last 5 years, compared to 0.43% after the change (P=0.54).
However, there was a significant change in the proportion of blood donors with MSM history further in the past. Before the MSM policy change, 0.42% of donors had an MSM partner but not in the last 5 years, compared to 0.66% of donors after the change (P=0.04).
“So we have an improvement in compliance, but it’s mainly because the donors are no longer deferrable,” Dr O’Brien explained.
“Donating while ineligible because of MSM history is actually quite rare, and the percentage of donors with MSM history in the last 5 years did not change when we changed the criteria. But we did see a modest increase in newly eligible MSM, so those that had more than 5 years since their last male-to-male sex.”
In all, there were 112 donors who were newly eligible due to the policy change and did, in fact, donate blood between July 22, 2013 and July 21, 2015. Five of these donors were females who had sexual contact with MSMs.
There were 70 “reinstated” donors in the first year after the policy change and 42 in the second year.
HIV-positive donations
The researchers monitored HIV rates in all blood donations from January 2010 to March 2015.
The rates of HIV-positive donations were as follows: 0.20 for 2010 (2/989,916), 0.50 for 2011 (5/995,122), 0.51 for 2012 (5/987,527), 0 (0/525,337) from January 1, 2013 to July 21, 2013 (before the policy change), 0.54 from July 22, 2013 to July 21, 2014 (5/929,656), and 0.22 from July 22, 2014 to July 21, 2015 (2/893,513).
“So absolutely no change in HIV rate following implementation of our 5-year deferral,” Dr O’Brien said.
In all, there were 7 HIV-positive donations after the policy change. Four were from male donors, and 3 were from females.
Three of the male donors (2 first-time donors, 1 repeat) denied having MSM risk factors, and 1 first-time male donor was aware he was HIV-positive at the time of donation. This man said he donated to determine if his HIV medication was working.
Two of the females were repeat donors, and 1 was a first-timer. The first-time donor did not acknowledge any MSM risk factors. One of the repeat donors had a sexual relationship with a bisexual male who was HIV-positive. The other repeat donor had multiple sexual partners, 1 of whom was known to be hepatitis C-positive.
Future policy change
Dr O’Brien noted that the LGBTQ community in Canada has advocated abolishing the deferral period for MSM blood donors or changing to a risk-based policy that would allow more individuals with MSM history to donate blood.
She said the combined blood services in Canada—Canadian Blood Services and Héma-Québec—are now considering a 12-month deferral period for individuals with MSM history.
“We’re pretty sure we’re going to go ahead,” she noted.
However, the groups must submit a policy request to Health Canada, which will ultimately make the decision. ![]()
*Data in the abstract differ from data presented at the meeting.
Photo by Charles Haymond
ANAHEIM, CA—Lifting the lifetime ban on blood donations from men who have sex with men (MSM) has not altered the safety of the blood supply in Canada, according to a new study.
The study showed no increase in the rate of HIV-positive blood donations since Canada changed its policy regarding MSM blood donors, allowing MSM to donate if they have not had sexual contact with another man in the last 5 years.
Because of this finding, Canada may shorten the deferral period for MSM blood donors to 1 year, according to Sheila F. O’Brien, PhD, of Canadian Blood Services in Ottawa, Ontario, Canada.
Dr O’Brien mentioned this possibility and presented data from the study at the 2015 AABB Annual Meeting (abstract S35-030E*).
Prior to 2013, MSM in Canada were not allowed to donate blood if they had any sexual contact with another male since 1977. Females were barred from donating if, in the last year, they had sexual contact with a man who had sex with another man after 1977.
On July 22, 2013, Canada changed this policy so that MSM can donate blood if they have abstained from sexual contact with another man for the past 5 years. The deferral period for females is still 12 months if they have had sex with a man who has had sex with another man in the last 5 years, but there is no deferral if the man had sex with another man more than 5 years before.
To evaluate the impact of this policy change, Dr O’Brien and her colleagues assessed compliance with the MSM criteria before and after the change, as well as the number of HIV-positive blood donations before and after the change.
The researchers also assessed the number of donors who would have been deferred according to the old MSM criteria but donated blood under the new criteria.
MSM history
The researchers selected random male donors of whole blood each month from October 2012 to February 2013 (pre-change) and from October 2014 to February 2015 (post-change). These donors were invited to complete an anonymous online survey about their MSM history.
The survey was completed by 9669 donors before the policy change and 6881 donors after the change. There were 77 donors with MSM history before the change (20% first-time donors, 80% repeat) and 75 donors with MSM history after the change (22% first-time, 78% repeat).
Compliance with policy
After the change in policy for MSM blood donors, there was no significant change in the proportion of donors who had recent MSM history but donated anyway (non-compliant). Before the change, 0.37% of blood donors had an MSM partner in the last 5 years, compared to 0.43% after the change (P=0.54).
However, there was a significant change in the proportion of blood donors with MSM history further in the past. Before the MSM policy change, 0.42% of donors had an MSM partner but not in the last 5 years, compared to 0.66% of donors after the change (P=0.04).
“So we have an improvement in compliance, but it’s mainly because the donors are no longer deferrable,” Dr O’Brien explained.
“Donating while ineligible because of MSM history is actually quite rare, and the percentage of donors with MSM history in the last 5 years did not change when we changed the criteria. But we did see a modest increase in newly eligible MSM, so those that had more than 5 years since their last male-to-male sex.”
In all, there were 112 donors who were newly eligible due to the policy change and did, in fact, donate blood between July 22, 2013 and July 21, 2015. Five of these donors were females who had sexual contact with MSMs.
There were 70 “reinstated” donors in the first year after the policy change and 42 in the second year.
HIV-positive donations
The researchers monitored HIV rates in all blood donations from January 2010 to March 2015.
The rates of HIV-positive donations were as follows: 0.20 for 2010 (2/989,916), 0.50 for 2011 (5/995,122), 0.51 for 2012 (5/987,527), 0 (0/525,337) from January 1, 2013 to July 21, 2013 (before the policy change), 0.54 from July 22, 2013 to July 21, 2014 (5/929,656), and 0.22 from July 22, 2014 to July 21, 2015 (2/893,513).
“So absolutely no change in HIV rate following implementation of our 5-year deferral,” Dr O’Brien said.
In all, there were 7 HIV-positive donations after the policy change. Four were from male donors, and 3 were from females.
Three of the male donors (2 first-time donors, 1 repeat) denied having MSM risk factors, and 1 first-time male donor was aware he was HIV-positive at the time of donation. This man said he donated to determine if his HIV medication was working.
Two of the females were repeat donors, and 1 was a first-timer. The first-time donor did not acknowledge any MSM risk factors. One of the repeat donors had a sexual relationship with a bisexual male who was HIV-positive. The other repeat donor had multiple sexual partners, 1 of whom was known to be hepatitis C-positive.
Future policy change
Dr O’Brien noted that the LGBTQ community in Canada has advocated abolishing the deferral period for MSM blood donors or changing to a risk-based policy that would allow more individuals with MSM history to donate blood.
She said the combined blood services in Canada—Canadian Blood Services and Héma-Québec—are now considering a 12-month deferral period for individuals with MSM history.
“We’re pretty sure we’re going to go ahead,” she noted.
However, the groups must submit a policy request to Health Canada, which will ultimately make the decision. ![]()
*Data in the abstract differ from data presented at the meeting.
Median DOR, PFS not yet reached for ibrutinib in CLL

Photo courtesy of
Janssen Biotech, Inc.
NEW YORK—Long-term follow-up of single-agent ibrutinib at the approved dose of 420 mg daily confirms that the Bruton’s tyrosine kinase inhibitor produces rapid and durable responses in patients with chronic lymphocytic leukemia (CLL), according to an update presented at Lymphoma & Myeloma 2015.
At up to 44 months of follow-up, the median duration of response (DOR) and progression-free survival (PFS) have not yet been reached.
At 30 months, the PFS rate was 96% for treatment-naïve patients and 76% for relapsed or refractory patients. Patients with del 17p had a median PFS of 32.4 months.
“Virtually all the patients do respond to treatment,” said Steven Coutre, MD, of Stanford University School of Medicine in California.
“Only a handful of patients achieve less than CR [complete response] or PR [partial response],” he said during his presentation at the meeting.
Phase 1/2b and extension studies
Ninety-four patients enrolled in the phase 1/2b (PCYC-1102) and extension (PCYC-1103) studies received 420 mg of ibrutinib once daily.
“We initially enrolled patients with relapsed/refractory CLL,” Dr Coutre clarified. “Then, because of the significant efficacy and safety that was observed, we added a second cohort of treatment-naïve patients age 65 and older.”
The treatment-naïve (TN) cohort consisted of 27 CLL patients. The relapsed or refractory (R/R) cohort consisted of 67 patients with CLL or small lymphocytic lymphoma, including patients with high-risk disease, which was defined as disease progression less than 24 months after the start of a chemoimmunotherapy regimen or refractory to the most recent regimen.
The median time on study was 32 months (range, 0–44).
In the TN cohort, the median age was 71, 78% were ECOG performance status 0, and most had advanced disease as indicated by Rai stage.
In the R/R cohort, the median age was 66, 40% were ECOG performance status 0, 57% were ECOG performance status 1, and 52% had bulky nodes greater than 5 cm.
“We had a significant representation of high-risk cytogenetic abnormalities,” Dr Coutre noted.
In the R/R group, 34% of patients had del 17p, and 33% had del 11q. In the TN cohort, 7% of patients had del 17p, and none had del 11q.
“There were also a significant number of cytopenias,” Dr Coutre said, “as one might expect in a heavily pretreated patient population.”
The number of prior therapies was also “quite significant,” he said, with 55% having a median of 4 or more therapies (range, 1–12).
“It really stretches the imagination to figure out what those 12 different regimens were,” he commented.
All R/R patients had prior chemotherapy, 94% a nucleoside analog, 90% an alkylator (including bendamustine), 99% anti-CD20-based therapy, 97% anti-CD20-based chemoimmunotherapy, 24% alemtuzumab, and 6% idelalisib.
The median time on treatment was 30.4 months (range, 1.3–44.2) for TN patients and 21.9 months (range, 0.3–44.6) for R/R patients. The majority of patients in both groups remain on ibrutinib—81% of the TN patients and 60% of R/R patients.
Safety
“Only 1 patient in the treatment-naïve cohort has progressed,” Dr Coutre noted. “That was a patient with deletion 17p [who progressed in about 8 months].”
The primary reasons for discontinuing therapy were progressive disease (1 TN, 11 R/R), adverse events (AEs; 3 TN, 9 R/R), consent withdrawal (1 TN, 2 R/R), investigators’ decision (0 TN, 4 R/R), and other reasons (0 TN, 1 R/R).
“Discontinuations due to AEs occurred predominantly early,” Dr Coutre observed. “So of the 12 patients [who discontinued due to AEs], 7 discontinued in the first year, 3 in the second year, and only 2 beyond year 3.”
Grade 3 or higher AEs occurred in 55 R/R patients (82%) and 17 TN patients (63%). Infection occurred in 48% of R/R patients and 11% of TN patients. Dr Coutre pointed out that most of these AEs were not related to ibrutinib.
Grade 3 or higher ibrutinib-related AEs occurred in 6 TN patients (22%) and 25 R/R patients (37%). One TN patient and 8 R/R patients experienced grade 3 or higher serious ibrutinib-related AEs.
One TN patient and 7 R/R patients required a dose reduction due to an AE. However, the dose reductions occurred predominantly during the first year, Dr Coutre noted.
Regarding time to onset of grade 3 or higher AEs, Dr Coutre said most of the events occurred early and decreased with time. Pneumonia and atrial fibrillation followed this pattern, as did neutropenia and thrombocytopenia. Hypertension was the exception, occurring during all years.
Nonhematologic AEs of grade 3 or higher that occurred in at least 5% of patients were pneumonia, hypertension, diarrhea, hyponatremia, and atrial fibrillation in TN patients, and sepsis, cellulitis, dehydration, and fatigue in R/R patients.
Hematologic AEs of grade 3 or higher in each cohort included neutropenia, thrombocytopenia, and anemia.
“The drug doesn’t seem to be myelosuppressive,” Dr Coutre noted. “We don’t have prolonged cytopenias as patients stay on treatment.”
One TN patient and 7 R/R patients died during the study.
Response and survival
The response rate (as assessed by the investigators) was 85% for TN patients. Twenty-six percent of patients achieved a complete response, 52% a partial response (PR), and 7% a PR with lymphocytosis.
The response rate for R/R patients was 94%. Nine percent achieved a complete response, 82% a PR, and 3% a PR with lymphocytosis.
The median time to the best response was 7.4 months for both cohorts.
The median DOR has not been reached in either cohort, but the 30-month DOR was 95.2% for TN patients and 79.1% for R/R patients.
The 30-month PFS was 95.8% for TN patients and 75.9% for R/R patients.
At 30 months, the PFS rate was 59.6% for patients with del 17p and 82.4% for patients with del 11q. The median PFS for patients with del 17p was 32.4 months, and it was not reached for patients with del 11q. For patients with neither of these abnormalities, the median PFS has not been reached.
“Overall survival was equally impressive,” Dr Coutre said.
The median overall survival has not been reached for any group, and 30-month overall survival is 81.3% for del 17p patients, 88.2% for patients with del 11q, and 90.3% for patients with neither abnormality.
“[I]brutinib induces rapid and durable responses that continue to improve over time . . . ,” Dr Coutre said.
He added that the drug is well-tolerated, “allowing us to continue patients on treatment, which, I think, is particularly important for these types of drugs because we clearly see that patients have significant clinical benefit, despite the fact that they still often have easily detectable disease, particularly in the bone marrow.”
“So one of the challenges is going to be [to determine] how to use these drugs on a long-term basis and [see if we can] use them in a more time-limited fashion.”
Ibrutinib is approved by the US Food and Drug Administration for 4 indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
Photo courtesy of
Janssen Biotech, Inc.
NEW YORK—Long-term follow-up of single-agent ibrutinib at the approved dose of 420 mg daily confirms that the Bruton’s tyrosine kinase inhibitor produces rapid and durable responses in patients with chronic lymphocytic leukemia (CLL), according to an update presented at Lymphoma & Myeloma 2015.
At up to 44 months of follow-up, the median duration of response (DOR) and progression-free survival (PFS) have not yet been reached.
At 30 months, the PFS rate was 96% for treatment-naïve patients and 76% for relapsed or refractory patients. Patients with del 17p had a median PFS of 32.4 months.
“Virtually all the patients do respond to treatment,” said Steven Coutre, MD, of Stanford University School of Medicine in California.
“Only a handful of patients achieve less than CR [complete response] or PR [partial response],” he said during his presentation at the meeting.
Phase 1/2b and extension studies
Ninety-four patients enrolled in the phase 1/2b (PCYC-1102) and extension (PCYC-1103) studies received 420 mg of ibrutinib once daily.
“We initially enrolled patients with relapsed/refractory CLL,” Dr Coutre clarified. “Then, because of the significant efficacy and safety that was observed, we added a second cohort of treatment-naïve patients age 65 and older.”
The treatment-naïve (TN) cohort consisted of 27 CLL patients. The relapsed or refractory (R/R) cohort consisted of 67 patients with CLL or small lymphocytic lymphoma, including patients with high-risk disease, which was defined as disease progression less than 24 months after the start of a chemoimmunotherapy regimen or refractory to the most recent regimen.
The median time on study was 32 months (range, 0–44).
In the TN cohort, the median age was 71, 78% were ECOG performance status 0, and most had advanced disease as indicated by Rai stage.
In the R/R cohort, the median age was 66, 40% were ECOG performance status 0, 57% were ECOG performance status 1, and 52% had bulky nodes greater than 5 cm.
“We had a significant representation of high-risk cytogenetic abnormalities,” Dr Coutre noted.
In the R/R group, 34% of patients had del 17p, and 33% had del 11q. In the TN cohort, 7% of patients had del 17p, and none had del 11q.
“There were also a significant number of cytopenias,” Dr Coutre said, “as one might expect in a heavily pretreated patient population.”
The number of prior therapies was also “quite significant,” he said, with 55% having a median of 4 or more therapies (range, 1–12).
“It really stretches the imagination to figure out what those 12 different regimens were,” he commented.
All R/R patients had prior chemotherapy, 94% a nucleoside analog, 90% an alkylator (including bendamustine), 99% anti-CD20-based therapy, 97% anti-CD20-based chemoimmunotherapy, 24% alemtuzumab, and 6% idelalisib.
The median time on treatment was 30.4 months (range, 1.3–44.2) for TN patients and 21.9 months (range, 0.3–44.6) for R/R patients. The majority of patients in both groups remain on ibrutinib—81% of the TN patients and 60% of R/R patients.
Safety
“Only 1 patient in the treatment-naïve cohort has progressed,” Dr Coutre noted. “That was a patient with deletion 17p [who progressed in about 8 months].”
The primary reasons for discontinuing therapy were progressive disease (1 TN, 11 R/R), adverse events (AEs; 3 TN, 9 R/R), consent withdrawal (1 TN, 2 R/R), investigators’ decision (0 TN, 4 R/R), and other reasons (0 TN, 1 R/R).
“Discontinuations due to AEs occurred predominantly early,” Dr Coutre observed. “So of the 12 patients [who discontinued due to AEs], 7 discontinued in the first year, 3 in the second year, and only 2 beyond year 3.”
Grade 3 or higher AEs occurred in 55 R/R patients (82%) and 17 TN patients (63%). Infection occurred in 48% of R/R patients and 11% of TN patients. Dr Coutre pointed out that most of these AEs were not related to ibrutinib.
Grade 3 or higher ibrutinib-related AEs occurred in 6 TN patients (22%) and 25 R/R patients (37%). One TN patient and 8 R/R patients experienced grade 3 or higher serious ibrutinib-related AEs.
One TN patient and 7 R/R patients required a dose reduction due to an AE. However, the dose reductions occurred predominantly during the first year, Dr Coutre noted.
Regarding time to onset of grade 3 or higher AEs, Dr Coutre said most of the events occurred early and decreased with time. Pneumonia and atrial fibrillation followed this pattern, as did neutropenia and thrombocytopenia. Hypertension was the exception, occurring during all years.
Nonhematologic AEs of grade 3 or higher that occurred in at least 5% of patients were pneumonia, hypertension, diarrhea, hyponatremia, and atrial fibrillation in TN patients, and sepsis, cellulitis, dehydration, and fatigue in R/R patients.
Hematologic AEs of grade 3 or higher in each cohort included neutropenia, thrombocytopenia, and anemia.
“The drug doesn’t seem to be myelosuppressive,” Dr Coutre noted. “We don’t have prolonged cytopenias as patients stay on treatment.”
One TN patient and 7 R/R patients died during the study.
Response and survival
The response rate (as assessed by the investigators) was 85% for TN patients. Twenty-six percent of patients achieved a complete response, 52% a partial response (PR), and 7% a PR with lymphocytosis.
The response rate for R/R patients was 94%. Nine percent achieved a complete response, 82% a PR, and 3% a PR with lymphocytosis.
The median time to the best response was 7.4 months for both cohorts.
The median DOR has not been reached in either cohort, but the 30-month DOR was 95.2% for TN patients and 79.1% for R/R patients.
The 30-month PFS was 95.8% for TN patients and 75.9% for R/R patients.
At 30 months, the PFS rate was 59.6% for patients with del 17p and 82.4% for patients with del 11q. The median PFS for patients with del 17p was 32.4 months, and it was not reached for patients with del 11q. For patients with neither of these abnormalities, the median PFS has not been reached.
“Overall survival was equally impressive,” Dr Coutre said.
The median overall survival has not been reached for any group, and 30-month overall survival is 81.3% for del 17p patients, 88.2% for patients with del 11q, and 90.3% for patients with neither abnormality.
“[I]brutinib induces rapid and durable responses that continue to improve over time . . . ,” Dr Coutre said.
He added that the drug is well-tolerated, “allowing us to continue patients on treatment, which, I think, is particularly important for these types of drugs because we clearly see that patients have significant clinical benefit, despite the fact that they still often have easily detectable disease, particularly in the bone marrow.”
“So one of the challenges is going to be [to determine] how to use these drugs on a long-term basis and [see if we can] use them in a more time-limited fashion.”
Ibrutinib is approved by the US Food and Drug Administration for 4 indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()
Photo courtesy of
Janssen Biotech, Inc.
NEW YORK—Long-term follow-up of single-agent ibrutinib at the approved dose of 420 mg daily confirms that the Bruton’s tyrosine kinase inhibitor produces rapid and durable responses in patients with chronic lymphocytic leukemia (CLL), according to an update presented at Lymphoma & Myeloma 2015.
At up to 44 months of follow-up, the median duration of response (DOR) and progression-free survival (PFS) have not yet been reached.
At 30 months, the PFS rate was 96% for treatment-naïve patients and 76% for relapsed or refractory patients. Patients with del 17p had a median PFS of 32.4 months.
“Virtually all the patients do respond to treatment,” said Steven Coutre, MD, of Stanford University School of Medicine in California.
“Only a handful of patients achieve less than CR [complete response] or PR [partial response],” he said during his presentation at the meeting.
Phase 1/2b and extension studies
Ninety-four patients enrolled in the phase 1/2b (PCYC-1102) and extension (PCYC-1103) studies received 420 mg of ibrutinib once daily.
“We initially enrolled patients with relapsed/refractory CLL,” Dr Coutre clarified. “Then, because of the significant efficacy and safety that was observed, we added a second cohort of treatment-naïve patients age 65 and older.”
The treatment-naïve (TN) cohort consisted of 27 CLL patients. The relapsed or refractory (R/R) cohort consisted of 67 patients with CLL or small lymphocytic lymphoma, including patients with high-risk disease, which was defined as disease progression less than 24 months after the start of a chemoimmunotherapy regimen or refractory to the most recent regimen.
The median time on study was 32 months (range, 0–44).
In the TN cohort, the median age was 71, 78% were ECOG performance status 0, and most had advanced disease as indicated by Rai stage.
In the R/R cohort, the median age was 66, 40% were ECOG performance status 0, 57% were ECOG performance status 1, and 52% had bulky nodes greater than 5 cm.
“We had a significant representation of high-risk cytogenetic abnormalities,” Dr Coutre noted.
In the R/R group, 34% of patients had del 17p, and 33% had del 11q. In the TN cohort, 7% of patients had del 17p, and none had del 11q.
“There were also a significant number of cytopenias,” Dr Coutre said, “as one might expect in a heavily pretreated patient population.”
The number of prior therapies was also “quite significant,” he said, with 55% having a median of 4 or more therapies (range, 1–12).
“It really stretches the imagination to figure out what those 12 different regimens were,” he commented.
All R/R patients had prior chemotherapy, 94% a nucleoside analog, 90% an alkylator (including bendamustine), 99% anti-CD20-based therapy, 97% anti-CD20-based chemoimmunotherapy, 24% alemtuzumab, and 6% idelalisib.
The median time on treatment was 30.4 months (range, 1.3–44.2) for TN patients and 21.9 months (range, 0.3–44.6) for R/R patients. The majority of patients in both groups remain on ibrutinib—81% of the TN patients and 60% of R/R patients.
Safety
“Only 1 patient in the treatment-naïve cohort has progressed,” Dr Coutre noted. “That was a patient with deletion 17p [who progressed in about 8 months].”
The primary reasons for discontinuing therapy were progressive disease (1 TN, 11 R/R), adverse events (AEs; 3 TN, 9 R/R), consent withdrawal (1 TN, 2 R/R), investigators’ decision (0 TN, 4 R/R), and other reasons (0 TN, 1 R/R).
“Discontinuations due to AEs occurred predominantly early,” Dr Coutre observed. “So of the 12 patients [who discontinued due to AEs], 7 discontinued in the first year, 3 in the second year, and only 2 beyond year 3.”
Grade 3 or higher AEs occurred in 55 R/R patients (82%) and 17 TN patients (63%). Infection occurred in 48% of R/R patients and 11% of TN patients. Dr Coutre pointed out that most of these AEs were not related to ibrutinib.
Grade 3 or higher ibrutinib-related AEs occurred in 6 TN patients (22%) and 25 R/R patients (37%). One TN patient and 8 R/R patients experienced grade 3 or higher serious ibrutinib-related AEs.
One TN patient and 7 R/R patients required a dose reduction due to an AE. However, the dose reductions occurred predominantly during the first year, Dr Coutre noted.
Regarding time to onset of grade 3 or higher AEs, Dr Coutre said most of the events occurred early and decreased with time. Pneumonia and atrial fibrillation followed this pattern, as did neutropenia and thrombocytopenia. Hypertension was the exception, occurring during all years.
Nonhematologic AEs of grade 3 or higher that occurred in at least 5% of patients were pneumonia, hypertension, diarrhea, hyponatremia, and atrial fibrillation in TN patients, and sepsis, cellulitis, dehydration, and fatigue in R/R patients.
Hematologic AEs of grade 3 or higher in each cohort included neutropenia, thrombocytopenia, and anemia.
“The drug doesn’t seem to be myelosuppressive,” Dr Coutre noted. “We don’t have prolonged cytopenias as patients stay on treatment.”
One TN patient and 7 R/R patients died during the study.
Response and survival
The response rate (as assessed by the investigators) was 85% for TN patients. Twenty-six percent of patients achieved a complete response, 52% a partial response (PR), and 7% a PR with lymphocytosis.
The response rate for R/R patients was 94%. Nine percent achieved a complete response, 82% a PR, and 3% a PR with lymphocytosis.
The median time to the best response was 7.4 months for both cohorts.
The median DOR has not been reached in either cohort, but the 30-month DOR was 95.2% for TN patients and 79.1% for R/R patients.
The 30-month PFS was 95.8% for TN patients and 75.9% for R/R patients.
At 30 months, the PFS rate was 59.6% for patients with del 17p and 82.4% for patients with del 11q. The median PFS for patients with del 17p was 32.4 months, and it was not reached for patients with del 11q. For patients with neither of these abnormalities, the median PFS has not been reached.
“Overall survival was equally impressive,” Dr Coutre said.
The median overall survival has not been reached for any group, and 30-month overall survival is 81.3% for del 17p patients, 88.2% for patients with del 11q, and 90.3% for patients with neither abnormality.
“[I]brutinib induces rapid and durable responses that continue to improve over time . . . ,” Dr Coutre said.
He added that the drug is well-tolerated, “allowing us to continue patients on treatment, which, I think, is particularly important for these types of drugs because we clearly see that patients have significant clinical benefit, despite the fact that they still often have easily detectable disease, particularly in the bone marrow.”
“So one of the challenges is going to be [to determine] how to use these drugs on a long-term basis and [see if we can] use them in a more time-limited fashion.”
Ibrutinib is approved by the US Food and Drug Administration for 4 indications: patients with CLL who have received at least 1 prior therapy, CLL patients with del 17p, patients with mantle cell lymphoma, and patients with Waldenström’s macroglobulinemia.
Ibrutinib is distributed and marketed as Imbruvica by Pharmacyclics and also marketed by Janssen Biotech, Inc. ![]()