User login
AVAHO
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
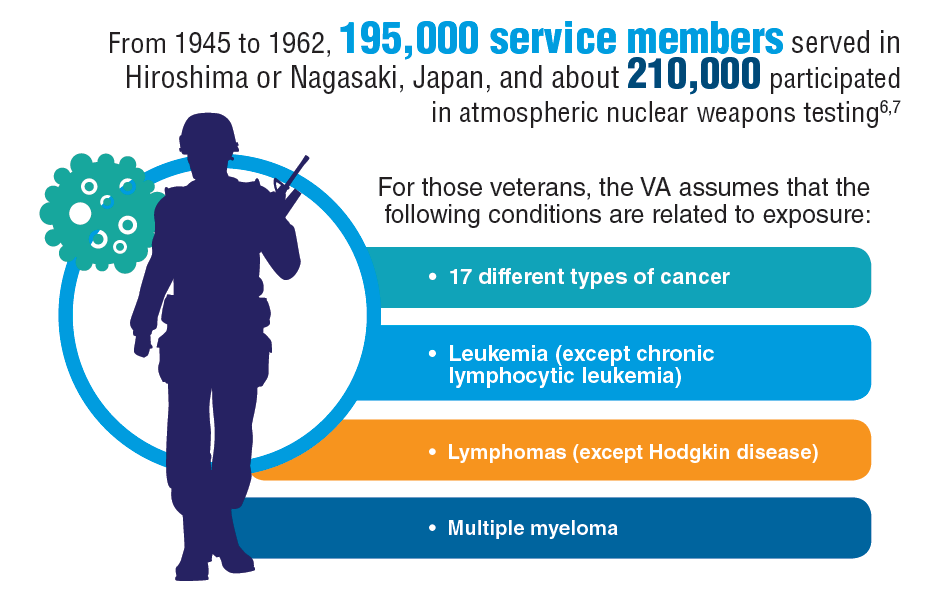
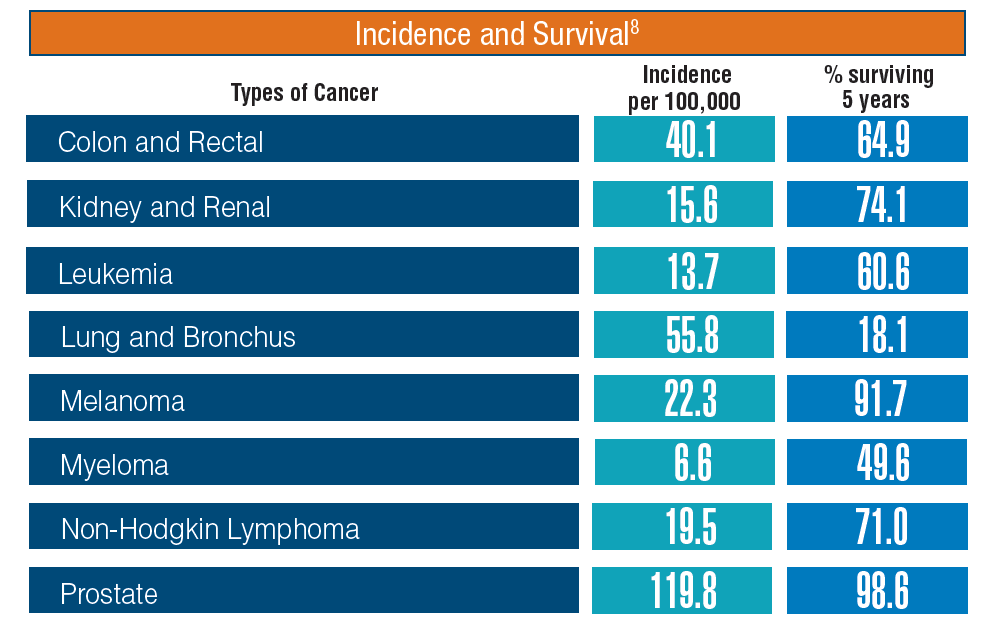
Hematology and Oncology Federal Health Data Trends (FULL)
Cancer research is a high priority for the DoD and especially for the VA. Researchers in both agencies played an important role in the early stages of the Cancer Moonshot. As part of this initiative, the VA, DoD, and National Cancer Institute joined forces in the Applied Proteogenomics Organizational Learning and Outcomes (APOLLO) project to develop a system to quickly identify unique targets and pathways of cancer for better interventions.
The VA also will provide access to the Million Veteran Program database, and > 20 years of electronic health records data for analysis using the U.S. Department of Energy’s advanced computer systems. The enhanced computational infrastructure provided by the departments will facilitate new studies of cancer genomics. The research will begin with prostate cancer, and it is hoped that the project will help researchers distinguish between those prostate cancers that require aggressive management and the more benign cancers that are less likely to progress.
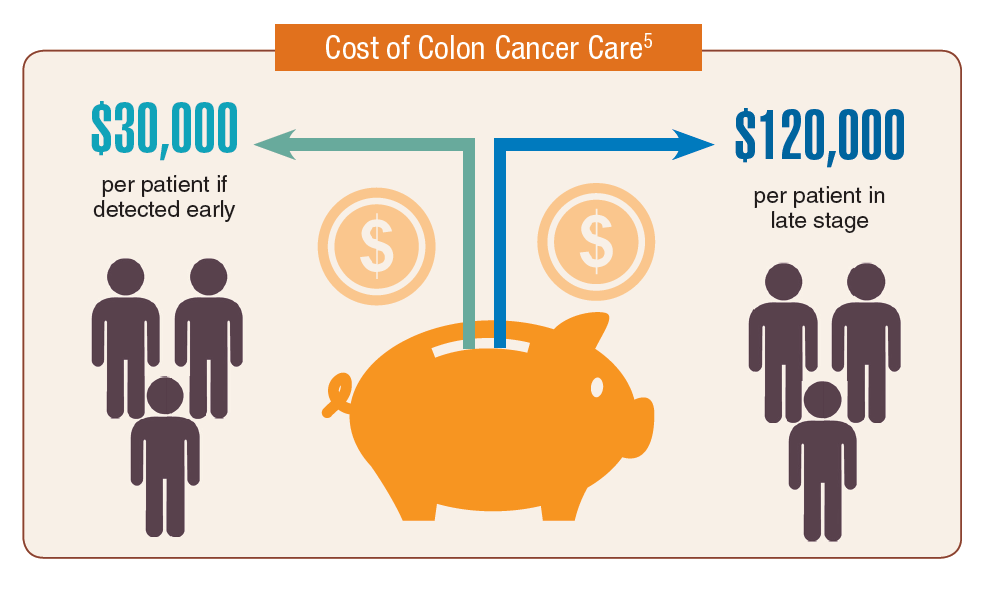
According to the latest VA budget, its researchers are conducting a broad array of research on cancers common in the veteran population, including prostate, lung, colorectal, bladder, kidney, pancreatic, skin, esophageal, and femalespecific cancers (such as breast and cervical cancer), as well as lymphomas and melanomas. For example, one study is focused on improving palliative care for patients with advanced cancer, and another will enroll 50,000 veterans to compare colorectal cancer screening strategies.
Click here to read the digital edition.
Cancer research is a high priority for the DoD and especially for the VA. Researchers in both agencies played an important role in the early stages of the Cancer Moonshot. As part of this initiative, the VA, DoD, and National Cancer Institute joined forces in the Applied Proteogenomics Organizational Learning and Outcomes (APOLLO) project to develop a system to quickly identify unique targets and pathways of cancer for better interventions.
The VA also will provide access to the Million Veteran Program database, and > 20 years of electronic health records data for analysis using the U.S. Department of Energy’s advanced computer systems. The enhanced computational infrastructure provided by the departments will facilitate new studies of cancer genomics. The research will begin with prostate cancer, and it is hoped that the project will help researchers distinguish between those prostate cancers that require aggressive management and the more benign cancers that are less likely to progress.
According to the latest VA budget, its researchers are conducting a broad array of research on cancers common in the veteran population, including prostate, lung, colorectal, bladder, kidney, pancreatic, skin, esophageal, and femalespecific cancers (such as breast and cervical cancer), as well as lymphomas and melanomas. For example, one study is focused on improving palliative care for patients with advanced cancer, and another will enroll 50,000 veterans to compare colorectal cancer screening strategies.
Click here to read the digital edition.
Cancer research is a high priority for the DoD and especially for the VA. Researchers in both agencies played an important role in the early stages of the Cancer Moonshot. As part of this initiative, the VA, DoD, and National Cancer Institute joined forces in the Applied Proteogenomics Organizational Learning and Outcomes (APOLLO) project to develop a system to quickly identify unique targets and pathways of cancer for better interventions.
The VA also will provide access to the Million Veteran Program database, and > 20 years of electronic health records data for analysis using the U.S. Department of Energy’s advanced computer systems. The enhanced computational infrastructure provided by the departments will facilitate new studies of cancer genomics. The research will begin with prostate cancer, and it is hoped that the project will help researchers distinguish between those prostate cancers that require aggressive management and the more benign cancers that are less likely to progress.
According to the latest VA budget, its researchers are conducting a broad array of research on cancers common in the veteran population, including prostate, lung, colorectal, bladder, kidney, pancreatic, skin, esophageal, and femalespecific cancers (such as breast and cervical cancer), as well as lymphomas and melanomas. For example, one study is focused on improving palliative care for patients with advanced cancer, and another will enroll 50,000 veterans to compare colorectal cancer screening strategies.
Click here to read the digital edition.
Federal Health Care Data Trends: Oncology
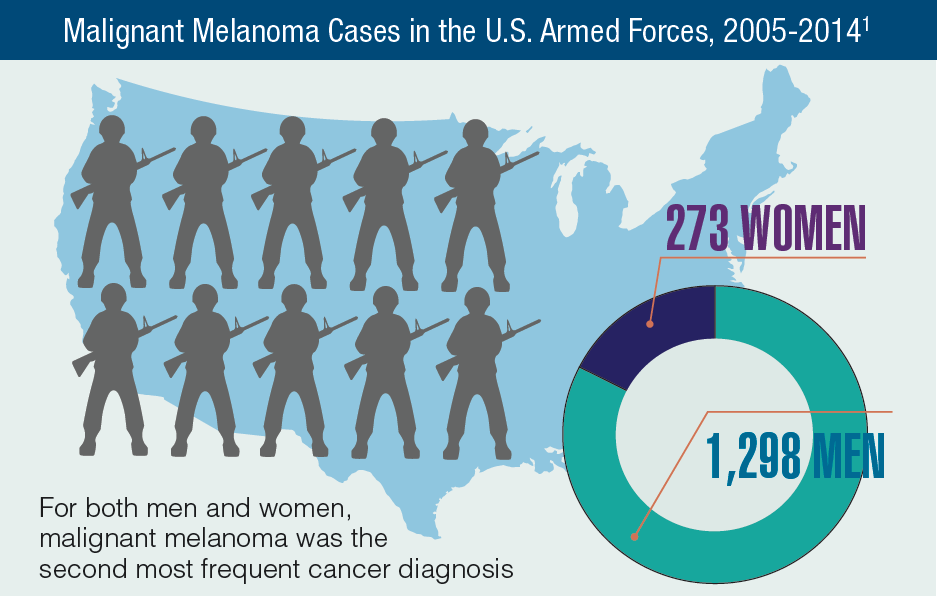
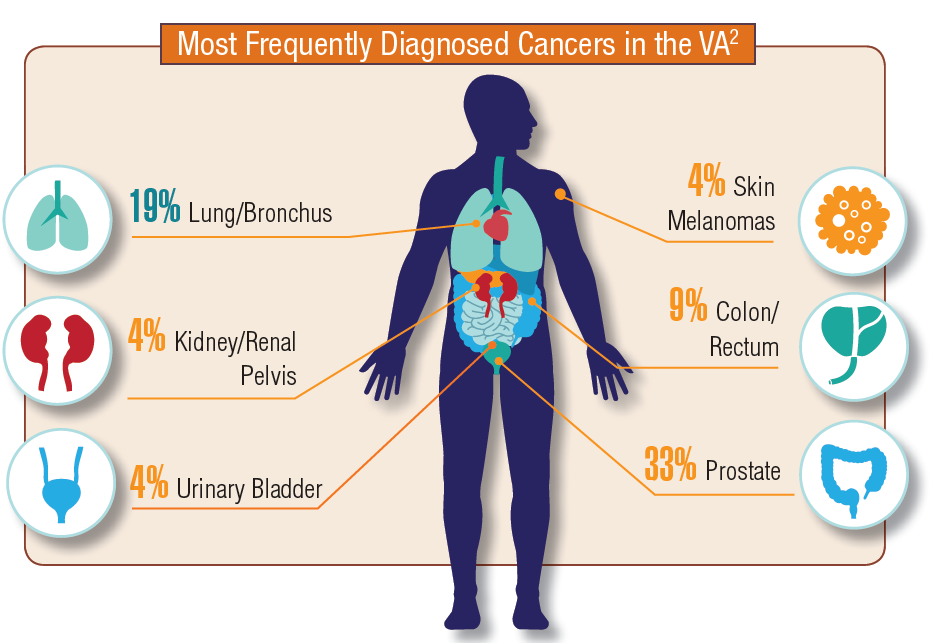
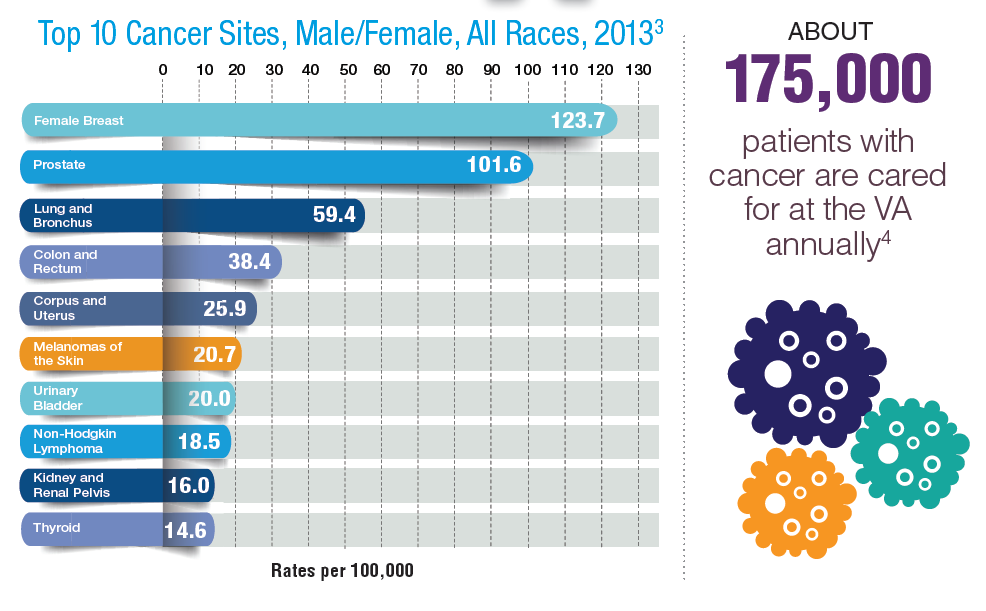
In 2012, a study conducted by Zullig and colleagues revealed that about 40,000 cancer cases are reported annually to the Veterans Affairs Central Cancer Registry.1 This represented about 3% of all cancer cases in the US. Within the VA patient population, the most commonly diagnosed cancers are prostate, lung and bronchial, colorectal, urinary and bladder cancers, and skin melanomas. This mirrors the commonly diagnosed cancers within the total US patient population.
Click here to continue reading.
In 2012, a study conducted by Zullig and colleagues revealed that about 40,000 cancer cases are reported annually to the Veterans Affairs Central Cancer Registry.1 This represented about 3% of all cancer cases in the US. Within the VA patient population, the most commonly diagnosed cancers are prostate, lung and bronchial, colorectal, urinary and bladder cancers, and skin melanomas. This mirrors the commonly diagnosed cancers within the total US patient population.
Click here to continue reading.
In 2012, a study conducted by Zullig and colleagues revealed that about 40,000 cancer cases are reported annually to the Veterans Affairs Central Cancer Registry.1 This represented about 3% of all cancer cases in the US. Within the VA patient population, the most commonly diagnosed cancers are prostate, lung and bronchial, colorectal, urinary and bladder cancers, and skin melanomas. This mirrors the commonly diagnosed cancers within the total US patient population.
Click here to continue reading.

Trio of biosimilars have good showing
CHICAGO – investigators reported at the annual meeting of the American Society of Clinical Oncology. The findings further advance the promise of new agents that have no clinically meaningful differences in efficacy and safety when compared with their reference drugs but have substantially lower cost.

Bevacizumab biosimilar
The REFLECTIONS trial (NCT02364999) was a multinational, first-line, randomized, controlled trial among 719 patients with advanced nonsquamous NSCLC. Patients were randomized to paclitaxel and carboplatin chemotherapy plus either bevacizumab (sourced from the European Union) or the candidate bevacizumab biosimilar PF-06439535 on a double-blind basis, followed by monotherapy with the same assigned agent.

The confidence interval for the risk difference fell within the equivalence margins set by European Union regulators (–13% and +13% for the 95% confidence interval). And the confidence interval for the risk ratio fell within the equivalence margins set by the Food and Drug Administration (0.73 and 1.37 for the 90% CI) and Japanese regulators (0.729 and 1.371 for the 95% CI).
Median progression-free survival was 9.0 months with the biosimilar and 7.7 months with bevacizumab (hazard ratio, 0.974; P = .814), and corresponding 1-year rates were 30.8% and 29.3%, Dr. Socinski reported. Median overall survival was 18.4 months and 17.8 months (HR, 1.001; P = .991), and corresponding 1-year rates were 66.4% and 68.8%.
Rates of grade 3 or higher hypertension, cardiac disorders, and bleeding did not differ significantly with the two agents. Patients also had similar rates of grade 3 or higher serious adverse events and of fatal (grade 5) serious adverse events (5.3% with the biosimilar and 5.9% with bevacizumab).
“Similarity between PF-06439535 and bevacizumab-EU was demonstrated for the primary efficacy endpoint of overall response rate. ... There were no clinically meaningful differences in safety profile shown in this trial, and similar pharmacokinetic and immunogenicity results were seen across treatment groups,” Dr. Socinski summarized.
“These results confirm similarity demonstrated in earlier analytical, nonclinical, and clinical studies of PF-06439535 with bevacizumab-EU,” he concluded.
Trastuzumab biosimilar
The phase 3 HERITAGE trial was a first-line, randomized, controlled trial that compared biosimilar trastuzumab-dkst (Ogivri) with trastuzumab in combination with taxane chemotherapy and then as maintenance monotherapy in 458 patients with HER2+ advanced breast cancer.
The 24-week results, previously reported (JAMA. 2017 Jan 3;317[1]:37-47), showed a similar overall response rate with each agent when combined with chemotherapy. Rates of various adverse events were essentially the same.

Presence of overall response at 24 weeks correlated with duration of progression-free survival at 48 weeks (biserial r = .752). “Additional patients achieved a response during the monotherapy portion of the treatment, which is intriguing and clearly emphasizes the importance of monotherapy, as well as the importance of having alternate agents at lower cost available,” Dr. Rugo commented.
Common adverse events through week 48 were much the same as those seen at week 24, with few additional ones occurring during monotherapy. “No new safety issues were observed, and in fact, toxicity during monotherapy was quite minor,” she noted. “One thing that’s interesting here is that there was more arthralgia during the first 24 weeks with trastuzumab-dkst than with trastuzumab, but in monotherapy, this fell down to a very low number and was identical between the two arms. Paclitaxel, which people stayed on for longer [with the biosimilar], may have been the cause of this.”
The 48-week rates of adverse events of special interest – respiratory events, cardiac disorders, and infusion-related adverse events – and of serious adverse events were similar for the two agents.
“We didn’t see any additional serious cardiac events during monotherapy,” Dr. Rugo noted. Mean and median left ventricular ejection fraction over 48 weeks were similar, as was the rate of LVEF, which dropped below 50% (4.0% with trastuzumab-dkst and 3.3% with trastuzumab). The incidences of antidrug antibody and neutralizing antibody were also comparably low in both groups.
“HERITAGE data, now at week 48, supports trastuzumab-dkst as a biosimilar to trastuzumab in all approved indications,” Dr. Rugo said. “Final overall survival will be assessed after 36 months or after 240 deaths, whichever occurs first. Based on current data, this is predicted to conclude by the end of 2018, with final overall survival data available next year.
“Trastuzumab-dkst provides an additional high-quality treatment option for patients with HER2+ breast cancers in any setting,” she added. “This study indeed shows that biosimilars offer the potential for worldwide cost savings and improved access to life-saving therapies. It’s sobering to think that the patients enrolled in this study would not otherwise have had access to continued trastuzumab therapy, and so many of them are still alive with longer follow-up.”
Filgrastim biosimilar
Investigators led by Nadia Harbeck, MD, PhD, head of the Breast Center and chair for Conservative Oncology in the department of ob&gyn at the University of Munich (Germany), compared efficacy of filgrastim-sndz (Zarxio), a biosimilar of filgrastim (recombinant granulocyte colony–stimulating factor, or G-CSF), in a trial population with that of a real-world population of women receiving chemotherapy for breast cancer.

Dr. Harbeck and her colleagues compared 217 women who had nonmetastatic breast cancer from the trial with 466 women who had any-stage breast cancer (42% metastatic) from the real-world cohort.
Results showed that the 6.2% rate of chemotherapy-induced febrile neutropenia in any cycle seen in the real-world population was much the same as the 5.1% rate seen previously in the trial population. Findings were similar for temperature exceeding 38.5˚ C in any cycle: 3.4% and 5.6%. The real-world population had a lower rate of severe neutropenia than did the trial population (19.5% vs. 74.3%) and higher rates of infection (15.5% vs. 7.9%) and hospitalization caused by febrile neutropenia (3.9% vs. 1.8%). Findings were essentially the same in cycle-level analyses.
The real-world cohort had many fewer any-severity safety events of special interest than did the trial cohort, such as musculoskeletal/connective tissue disorders (20 vs. 261 events, respectively) and skin/subcutaneous tissue disorders (5 vs. 258 events). “Seeing these data, you have to keep in mind first of all that the patients received totally different chemotherapy. TAC chemotherapy has a lot of chemotherapy-associated side effects,” Dr. Harbeck noted. “The other thing is that MONITOR was a real-world database, and one could assume that there is some underreporting of events that are not directly correlated to the events that are of particular interest.”
Additional results available only from the trial showed that no patients developed binding or neutralizing antibodies against G-CSF.
“From a clinician’s point of view, it is very reassuring that we did not see any other safety signals in the real-world data than we saw in the randomized controlled trial and the efficacy was very, very similar,” Dr. Harbeck commented. “Having seen the discrepancies in the data … I think it’s important to have randomized controlled trials to assess and monitor adverse events for registration purposes and real-world evidence to reflect the daily clinical routine,” she concluded.
Dr. Socinski disclosed that his institution receives research funding from Pfizer, among other disclosures; the REFLECTIONS trial was sponsored by Pfizer. Dr. Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan, among other disclosures; the HERITAGE trial was sponsored by Mylan. Dr. Harbeck disclosed that she has a consulting or advisory role with Sandoz, among other disclosures; the PIONEER and MONITOR-GCSF trials were both sponsored by Sandoz.
SOURCE: Socinski MA et al. ASCO 2018, Abstract 109. Manikhas A et al. ASCO 2018, Abstract 110. Harbeck N et al. ASCO 2018, Abstract 111.
A variety of issues are influencing whether and how clinicians incorporate biosimilars into cancer care, according to Michael A. Thompson, MD, PhD, medical director of the Early Phase Cancer Research Program and the Oncology Precision Medicine Program at Aurora Health Care in Milwaukee, Wis., who spoke at the annual meeting of the American Society of Clinical Oncology.
“The issue of competition is highly relevant to biosimilars,” he said. Among important questions here: Is the oncology drug market a free market? Who owns the biosimilar companies? Does competition lower drug prices? And if biosimilars don’t decrease drug cost, why bother pursuing them? “We are seeing examples where the biosimilars have been developed, they appear to work, they appear safe, and really the proof will be how much is this pushing the market to decrease cost,” he noted.
Real-world data provide some insight into how biosimilars are being incorporated into oncology care. For example, in patients with non-Hodgkin lymphoma, hematologists tend to use rituximab (Rituxan) biosimilars in later lines of therapy, in patients with a better performance status and fewer comorbidities, and in cases of indolent or incurable disease (J Clin Oncol. 2018;36[suppl; abstr 112]). “So it appears that prescribers are acting tentatively to cautiously test the waters,” Dr. Thompson said.
Use will be influenced by clinical decision support and pathways, whether those are developed by institutions or insurers. These tools generally look at efficacy first, safety second, and cost third.
The relevance of patient choice (especially when physicians decreasingly have a choice) and perception of biosimilars may, or may not, be important, according to Dr. Thompson. In some areas of medicine, there is evidence of a nocebo effect: Patients perceive worsening of symptoms when they believe they are getting a nonbranded medication. But “I am not sure if this is valid in oncology, where we are already using many older chemotherapy drugs, the generics,” he said.
The American Society of Clinical Oncology recently published a statement on the use of biosimilars and related issues, such as safety and efficacy; naming and labeling; interchangeability, switching, and substitution; and the value proposition of these agents (J Clin Oncol. 2018 Apr 20;36[12]:1260-5). “The ASCO statement and guidelines are a great resource for really digging deeply into this area,” Dr. Thompson commented.
One concern surrounding uptake of biosimilars is the possibility of an actual increase in patient cost related to single sources and potentially differing reimbursement rates, which could diminish the financial benefit of these drugs. Technically, if biosimilars have similar efficacy and safety, and lower cost, they provide greater value than the reference drugs.
But there may still be reasons for not using a higher-value drug, according to Dr. Thompson. Clinicians may have lingering questions about efficacy and safety despite trial data, a situation that is being addressed in Europe by postmarketing pharmacovigilance. Other issues include delays in pathway implementation, the contracting of pharmacies with companies, and creation of new chemotherapy builds in electronic medical records. “These are all minor but potential barriers to as fast an implementation as possible,” he said.
A variety of issues are influencing whether and how clinicians incorporate biosimilars into cancer care, according to Michael A. Thompson, MD, PhD, medical director of the Early Phase Cancer Research Program and the Oncology Precision Medicine Program at Aurora Health Care in Milwaukee, Wis., who spoke at the annual meeting of the American Society of Clinical Oncology.
“The issue of competition is highly relevant to biosimilars,” he said. Among important questions here: Is the oncology drug market a free market? Who owns the biosimilar companies? Does competition lower drug prices? And if biosimilars don’t decrease drug cost, why bother pursuing them? “We are seeing examples where the biosimilars have been developed, they appear to work, they appear safe, and really the proof will be how much is this pushing the market to decrease cost,” he noted.
Real-world data provide some insight into how biosimilars are being incorporated into oncology care. For example, in patients with non-Hodgkin lymphoma, hematologists tend to use rituximab (Rituxan) biosimilars in later lines of therapy, in patients with a better performance status and fewer comorbidities, and in cases of indolent or incurable disease (J Clin Oncol. 2018;36[suppl; abstr 112]). “So it appears that prescribers are acting tentatively to cautiously test the waters,” Dr. Thompson said.
Use will be influenced by clinical decision support and pathways, whether those are developed by institutions or insurers. These tools generally look at efficacy first, safety second, and cost third.
The relevance of patient choice (especially when physicians decreasingly have a choice) and perception of biosimilars may, or may not, be important, according to Dr. Thompson. In some areas of medicine, there is evidence of a nocebo effect: Patients perceive worsening of symptoms when they believe they are getting a nonbranded medication. But “I am not sure if this is valid in oncology, where we are already using many older chemotherapy drugs, the generics,” he said.
The American Society of Clinical Oncology recently published a statement on the use of biosimilars and related issues, such as safety and efficacy; naming and labeling; interchangeability, switching, and substitution; and the value proposition of these agents (J Clin Oncol. 2018 Apr 20;36[12]:1260-5). “The ASCO statement and guidelines are a great resource for really digging deeply into this area,” Dr. Thompson commented.
One concern surrounding uptake of biosimilars is the possibility of an actual increase in patient cost related to single sources and potentially differing reimbursement rates, which could diminish the financial benefit of these drugs. Technically, if biosimilars have similar efficacy and safety, and lower cost, they provide greater value than the reference drugs.
But there may still be reasons for not using a higher-value drug, according to Dr. Thompson. Clinicians may have lingering questions about efficacy and safety despite trial data, a situation that is being addressed in Europe by postmarketing pharmacovigilance. Other issues include delays in pathway implementation, the contracting of pharmacies with companies, and creation of new chemotherapy builds in electronic medical records. “These are all minor but potential barriers to as fast an implementation as possible,” he said.
A variety of issues are influencing whether and how clinicians incorporate biosimilars into cancer care, according to Michael A. Thompson, MD, PhD, medical director of the Early Phase Cancer Research Program and the Oncology Precision Medicine Program at Aurora Health Care in Milwaukee, Wis., who spoke at the annual meeting of the American Society of Clinical Oncology.
“The issue of competition is highly relevant to biosimilars,” he said. Among important questions here: Is the oncology drug market a free market? Who owns the biosimilar companies? Does competition lower drug prices? And if biosimilars don’t decrease drug cost, why bother pursuing them? “We are seeing examples where the biosimilars have been developed, they appear to work, they appear safe, and really the proof will be how much is this pushing the market to decrease cost,” he noted.
Real-world data provide some insight into how biosimilars are being incorporated into oncology care. For example, in patients with non-Hodgkin lymphoma, hematologists tend to use rituximab (Rituxan) biosimilars in later lines of therapy, in patients with a better performance status and fewer comorbidities, and in cases of indolent or incurable disease (J Clin Oncol. 2018;36[suppl; abstr 112]). “So it appears that prescribers are acting tentatively to cautiously test the waters,” Dr. Thompson said.
Use will be influenced by clinical decision support and pathways, whether those are developed by institutions or insurers. These tools generally look at efficacy first, safety second, and cost third.
The relevance of patient choice (especially when physicians decreasingly have a choice) and perception of biosimilars may, or may not, be important, according to Dr. Thompson. In some areas of medicine, there is evidence of a nocebo effect: Patients perceive worsening of symptoms when they believe they are getting a nonbranded medication. But “I am not sure if this is valid in oncology, where we are already using many older chemotherapy drugs, the generics,” he said.
The American Society of Clinical Oncology recently published a statement on the use of biosimilars and related issues, such as safety and efficacy; naming and labeling; interchangeability, switching, and substitution; and the value proposition of these agents (J Clin Oncol. 2018 Apr 20;36[12]:1260-5). “The ASCO statement and guidelines are a great resource for really digging deeply into this area,” Dr. Thompson commented.
One concern surrounding uptake of biosimilars is the possibility of an actual increase in patient cost related to single sources and potentially differing reimbursement rates, which could diminish the financial benefit of these drugs. Technically, if biosimilars have similar efficacy and safety, and lower cost, they provide greater value than the reference drugs.
But there may still be reasons for not using a higher-value drug, according to Dr. Thompson. Clinicians may have lingering questions about efficacy and safety despite trial data, a situation that is being addressed in Europe by postmarketing pharmacovigilance. Other issues include delays in pathway implementation, the contracting of pharmacies with companies, and creation of new chemotherapy builds in electronic medical records. “These are all minor but potential barriers to as fast an implementation as possible,” he said.
CHICAGO – investigators reported at the annual meeting of the American Society of Clinical Oncology. The findings further advance the promise of new agents that have no clinically meaningful differences in efficacy and safety when compared with their reference drugs but have substantially lower cost.
Bevacizumab biosimilar
The REFLECTIONS trial (NCT02364999) was a multinational, first-line, randomized, controlled trial among 719 patients with advanced nonsquamous NSCLC. Patients were randomized to paclitaxel and carboplatin chemotherapy plus either bevacizumab (sourced from the European Union) or the candidate bevacizumab biosimilar PF-06439535 on a double-blind basis, followed by monotherapy with the same assigned agent.
The confidence interval for the risk difference fell within the equivalence margins set by European Union regulators (–13% and +13% for the 95% confidence interval). And the confidence interval for the risk ratio fell within the equivalence margins set by the Food and Drug Administration (0.73 and 1.37 for the 90% CI) and Japanese regulators (0.729 and 1.371 for the 95% CI).
Median progression-free survival was 9.0 months with the biosimilar and 7.7 months with bevacizumab (hazard ratio, 0.974; P = .814), and corresponding 1-year rates were 30.8% and 29.3%, Dr. Socinski reported. Median overall survival was 18.4 months and 17.8 months (HR, 1.001; P = .991), and corresponding 1-year rates were 66.4% and 68.8%.
Rates of grade 3 or higher hypertension, cardiac disorders, and bleeding did not differ significantly with the two agents. Patients also had similar rates of grade 3 or higher serious adverse events and of fatal (grade 5) serious adverse events (5.3% with the biosimilar and 5.9% with bevacizumab).
“Similarity between PF-06439535 and bevacizumab-EU was demonstrated for the primary efficacy endpoint of overall response rate. ... There were no clinically meaningful differences in safety profile shown in this trial, and similar pharmacokinetic and immunogenicity results were seen across treatment groups,” Dr. Socinski summarized.
“These results confirm similarity demonstrated in earlier analytical, nonclinical, and clinical studies of PF-06439535 with bevacizumab-EU,” he concluded.
Trastuzumab biosimilar
The phase 3 HERITAGE trial was a first-line, randomized, controlled trial that compared biosimilar trastuzumab-dkst (Ogivri) with trastuzumab in combination with taxane chemotherapy and then as maintenance monotherapy in 458 patients with HER2+ advanced breast cancer.
The 24-week results, previously reported (JAMA. 2017 Jan 3;317[1]:37-47), showed a similar overall response rate with each agent when combined with chemotherapy. Rates of various adverse events were essentially the same.
Presence of overall response at 24 weeks correlated with duration of progression-free survival at 48 weeks (biserial r = .752). “Additional patients achieved a response during the monotherapy portion of the treatment, which is intriguing and clearly emphasizes the importance of monotherapy, as well as the importance of having alternate agents at lower cost available,” Dr. Rugo commented.
Common adverse events through week 48 were much the same as those seen at week 24, with few additional ones occurring during monotherapy. “No new safety issues were observed, and in fact, toxicity during monotherapy was quite minor,” she noted. “One thing that’s interesting here is that there was more arthralgia during the first 24 weeks with trastuzumab-dkst than with trastuzumab, but in monotherapy, this fell down to a very low number and was identical between the two arms. Paclitaxel, which people stayed on for longer [with the biosimilar], may have been the cause of this.”
The 48-week rates of adverse events of special interest – respiratory events, cardiac disorders, and infusion-related adverse events – and of serious adverse events were similar for the two agents.
“We didn’t see any additional serious cardiac events during monotherapy,” Dr. Rugo noted. Mean and median left ventricular ejection fraction over 48 weeks were similar, as was the rate of LVEF, which dropped below 50% (4.0% with trastuzumab-dkst and 3.3% with trastuzumab). The incidences of antidrug antibody and neutralizing antibody were also comparably low in both groups.
“HERITAGE data, now at week 48, supports trastuzumab-dkst as a biosimilar to trastuzumab in all approved indications,” Dr. Rugo said. “Final overall survival will be assessed after 36 months or after 240 deaths, whichever occurs first. Based on current data, this is predicted to conclude by the end of 2018, with final overall survival data available next year.
“Trastuzumab-dkst provides an additional high-quality treatment option for patients with HER2+ breast cancers in any setting,” she added. “This study indeed shows that biosimilars offer the potential for worldwide cost savings and improved access to life-saving therapies. It’s sobering to think that the patients enrolled in this study would not otherwise have had access to continued trastuzumab therapy, and so many of them are still alive with longer follow-up.”
Filgrastim biosimilar
Investigators led by Nadia Harbeck, MD, PhD, head of the Breast Center and chair for Conservative Oncology in the department of ob&gyn at the University of Munich (Germany), compared efficacy of filgrastim-sndz (Zarxio), a biosimilar of filgrastim (recombinant granulocyte colony–stimulating factor, or G-CSF), in a trial population with that of a real-world population of women receiving chemotherapy for breast cancer.
Dr. Harbeck and her colleagues compared 217 women who had nonmetastatic breast cancer from the trial with 466 women who had any-stage breast cancer (42% metastatic) from the real-world cohort.
Results showed that the 6.2% rate of chemotherapy-induced febrile neutropenia in any cycle seen in the real-world population was much the same as the 5.1% rate seen previously in the trial population. Findings were similar for temperature exceeding 38.5˚ C in any cycle: 3.4% and 5.6%. The real-world population had a lower rate of severe neutropenia than did the trial population (19.5% vs. 74.3%) and higher rates of infection (15.5% vs. 7.9%) and hospitalization caused by febrile neutropenia (3.9% vs. 1.8%). Findings were essentially the same in cycle-level analyses.
The real-world cohort had many fewer any-severity safety events of special interest than did the trial cohort, such as musculoskeletal/connective tissue disorders (20 vs. 261 events, respectively) and skin/subcutaneous tissue disorders (5 vs. 258 events). “Seeing these data, you have to keep in mind first of all that the patients received totally different chemotherapy. TAC chemotherapy has a lot of chemotherapy-associated side effects,” Dr. Harbeck noted. “The other thing is that MONITOR was a real-world database, and one could assume that there is some underreporting of events that are not directly correlated to the events that are of particular interest.”
Additional results available only from the trial showed that no patients developed binding or neutralizing antibodies against G-CSF.
“From a clinician’s point of view, it is very reassuring that we did not see any other safety signals in the real-world data than we saw in the randomized controlled trial and the efficacy was very, very similar,” Dr. Harbeck commented. “Having seen the discrepancies in the data … I think it’s important to have randomized controlled trials to assess and monitor adverse events for registration purposes and real-world evidence to reflect the daily clinical routine,” she concluded.
Dr. Socinski disclosed that his institution receives research funding from Pfizer, among other disclosures; the REFLECTIONS trial was sponsored by Pfizer. Dr. Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan, among other disclosures; the HERITAGE trial was sponsored by Mylan. Dr. Harbeck disclosed that she has a consulting or advisory role with Sandoz, among other disclosures; the PIONEER and MONITOR-GCSF trials were both sponsored by Sandoz.
SOURCE: Socinski MA et al. ASCO 2018, Abstract 109. Manikhas A et al. ASCO 2018, Abstract 110. Harbeck N et al. ASCO 2018, Abstract 111.
CHICAGO – investigators reported at the annual meeting of the American Society of Clinical Oncology. The findings further advance the promise of new agents that have no clinically meaningful differences in efficacy and safety when compared with their reference drugs but have substantially lower cost.
Bevacizumab biosimilar
The REFLECTIONS trial (NCT02364999) was a multinational, first-line, randomized, controlled trial among 719 patients with advanced nonsquamous NSCLC. Patients were randomized to paclitaxel and carboplatin chemotherapy plus either bevacizumab (sourced from the European Union) or the candidate bevacizumab biosimilar PF-06439535 on a double-blind basis, followed by monotherapy with the same assigned agent.
The confidence interval for the risk difference fell within the equivalence margins set by European Union regulators (–13% and +13% for the 95% confidence interval). And the confidence interval for the risk ratio fell within the equivalence margins set by the Food and Drug Administration (0.73 and 1.37 for the 90% CI) and Japanese regulators (0.729 and 1.371 for the 95% CI).
Median progression-free survival was 9.0 months with the biosimilar and 7.7 months with bevacizumab (hazard ratio, 0.974; P = .814), and corresponding 1-year rates were 30.8% and 29.3%, Dr. Socinski reported. Median overall survival was 18.4 months and 17.8 months (HR, 1.001; P = .991), and corresponding 1-year rates were 66.4% and 68.8%.
Rates of grade 3 or higher hypertension, cardiac disorders, and bleeding did not differ significantly with the two agents. Patients also had similar rates of grade 3 or higher serious adverse events and of fatal (grade 5) serious adverse events (5.3% with the biosimilar and 5.9% with bevacizumab).
“Similarity between PF-06439535 and bevacizumab-EU was demonstrated for the primary efficacy endpoint of overall response rate. ... There were no clinically meaningful differences in safety profile shown in this trial, and similar pharmacokinetic and immunogenicity results were seen across treatment groups,” Dr. Socinski summarized.
“These results confirm similarity demonstrated in earlier analytical, nonclinical, and clinical studies of PF-06439535 with bevacizumab-EU,” he concluded.
Trastuzumab biosimilar
The phase 3 HERITAGE trial was a first-line, randomized, controlled trial that compared biosimilar trastuzumab-dkst (Ogivri) with trastuzumab in combination with taxane chemotherapy and then as maintenance monotherapy in 458 patients with HER2+ advanced breast cancer.
The 24-week results, previously reported (JAMA. 2017 Jan 3;317[1]:37-47), showed a similar overall response rate with each agent when combined with chemotherapy. Rates of various adverse events were essentially the same.
Presence of overall response at 24 weeks correlated with duration of progression-free survival at 48 weeks (biserial r = .752). “Additional patients achieved a response during the monotherapy portion of the treatment, which is intriguing and clearly emphasizes the importance of monotherapy, as well as the importance of having alternate agents at lower cost available,” Dr. Rugo commented.
Common adverse events through week 48 were much the same as those seen at week 24, with few additional ones occurring during monotherapy. “No new safety issues were observed, and in fact, toxicity during monotherapy was quite minor,” she noted. “One thing that’s interesting here is that there was more arthralgia during the first 24 weeks with trastuzumab-dkst than with trastuzumab, but in monotherapy, this fell down to a very low number and was identical between the two arms. Paclitaxel, which people stayed on for longer [with the biosimilar], may have been the cause of this.”
The 48-week rates of adverse events of special interest – respiratory events, cardiac disorders, and infusion-related adverse events – and of serious adverse events were similar for the two agents.
“We didn’t see any additional serious cardiac events during monotherapy,” Dr. Rugo noted. Mean and median left ventricular ejection fraction over 48 weeks were similar, as was the rate of LVEF, which dropped below 50% (4.0% with trastuzumab-dkst and 3.3% with trastuzumab). The incidences of antidrug antibody and neutralizing antibody were also comparably low in both groups.
“HERITAGE data, now at week 48, supports trastuzumab-dkst as a biosimilar to trastuzumab in all approved indications,” Dr. Rugo said. “Final overall survival will be assessed after 36 months or after 240 deaths, whichever occurs first. Based on current data, this is predicted to conclude by the end of 2018, with final overall survival data available next year.
“Trastuzumab-dkst provides an additional high-quality treatment option for patients with HER2+ breast cancers in any setting,” she added. “This study indeed shows that biosimilars offer the potential for worldwide cost savings and improved access to life-saving therapies. It’s sobering to think that the patients enrolled in this study would not otherwise have had access to continued trastuzumab therapy, and so many of them are still alive with longer follow-up.”
Filgrastim biosimilar
Investigators led by Nadia Harbeck, MD, PhD, head of the Breast Center and chair for Conservative Oncology in the department of ob&gyn at the University of Munich (Germany), compared efficacy of filgrastim-sndz (Zarxio), a biosimilar of filgrastim (recombinant granulocyte colony–stimulating factor, or G-CSF), in a trial population with that of a real-world population of women receiving chemotherapy for breast cancer.
Dr. Harbeck and her colleagues compared 217 women who had nonmetastatic breast cancer from the trial with 466 women who had any-stage breast cancer (42% metastatic) from the real-world cohort.
Results showed that the 6.2% rate of chemotherapy-induced febrile neutropenia in any cycle seen in the real-world population was much the same as the 5.1% rate seen previously in the trial population. Findings were similar for temperature exceeding 38.5˚ C in any cycle: 3.4% and 5.6%. The real-world population had a lower rate of severe neutropenia than did the trial population (19.5% vs. 74.3%) and higher rates of infection (15.5% vs. 7.9%) and hospitalization caused by febrile neutropenia (3.9% vs. 1.8%). Findings were essentially the same in cycle-level analyses.
The real-world cohort had many fewer any-severity safety events of special interest than did the trial cohort, such as musculoskeletal/connective tissue disorders (20 vs. 261 events, respectively) and skin/subcutaneous tissue disorders (5 vs. 258 events). “Seeing these data, you have to keep in mind first of all that the patients received totally different chemotherapy. TAC chemotherapy has a lot of chemotherapy-associated side effects,” Dr. Harbeck noted. “The other thing is that MONITOR was a real-world database, and one could assume that there is some underreporting of events that are not directly correlated to the events that are of particular interest.”
Additional results available only from the trial showed that no patients developed binding or neutralizing antibodies against G-CSF.
“From a clinician’s point of view, it is very reassuring that we did not see any other safety signals in the real-world data than we saw in the randomized controlled trial and the efficacy was very, very similar,” Dr. Harbeck commented. “Having seen the discrepancies in the data … I think it’s important to have randomized controlled trials to assess and monitor adverse events for registration purposes and real-world evidence to reflect the daily clinical routine,” she concluded.
Dr. Socinski disclosed that his institution receives research funding from Pfizer, among other disclosures; the REFLECTIONS trial was sponsored by Pfizer. Dr. Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan, among other disclosures; the HERITAGE trial was sponsored by Mylan. Dr. Harbeck disclosed that she has a consulting or advisory role with Sandoz, among other disclosures; the PIONEER and MONITOR-GCSF trials were both sponsored by Sandoz.
SOURCE: Socinski MA et al. ASCO 2018, Abstract 109. Manikhas A et al. ASCO 2018, Abstract 110. Harbeck N et al. ASCO 2018, Abstract 111.
REPORTING FROM ASCO 2018
Key clinical point: Biosimilars for bevacizumab, trastuzumab, and filgrastim showed similar efficacy and safety.
Major finding: In patients with advanced nonsquamous NSCLC, the overall response rate was 45.3% with a candidate bevacizumab biosimilar and 44.6% with bevacizumab. In patients with HER2+ advanced breast cancer, 48-week median progression-free survival was 11.1 months for both trastuzumab-dkst and trastuzumab. The rate of chemotherapy-induced febrile neutropenia among breast cancer patients given a biosimilar for filgrastim was 5.1% in a trial population and 6.2% in a real-world population.
Study details: Randomized, controlled trials of first-line therapy among 719 patients with advanced nonsquamous NSCLC (REFLECTIONS trial) and among 458 patients with HER2+ advanced breast cancer (HERITAGE trial). Comparison of outcomes in a randomized, controlled trial among 217 patients with nonmetastatic breast cancer (PIONEER trial) and a real-world cohort study of 466 patients with any-stage breast cancer (MONITOR-GCSF).
Disclosures: Dr. Socinski disclosed that his institution receives research funding from Pfizer, among other disclosures; the REFLECTIONS trial was sponsored by Pfizer. Dr. Rugo disclosed that she receives travel, accommodations, and/or expenses from Mylan, among other disclosures; the HERITAGE trial was sponsored by Mylan. Dr. Harbeck disclosed that she has a consulting or advisory role with Sandoz, among other disclosures; the PIONEER and MONITOR-GCSF trials were sponsored by Sandoz.
Source: Socinski MA et al. ASCO 2018, Abstract 109. Manikhas A et al. ASCO 2018, Abstract 110. Harbeck N et al. ASCO 2018, Abstract 111.
Could High BMI Reduce Premenopausal Breast Cancer Risk?
Young women may not want to hear it, but fat could be their friend. Researchers from the Premenopausal Breast Cancer Collaborative Group have found that women aged 18 – 24 years with high body fat have a lower risk of developing breast cancer before menopause.
The researchers pooled data from 19 different studies, involving about 800,000 women from around the world. Overall, 1.7% of the women developed breast cancer. The researchers found that the relative risk of premenopausal breast cancer dropped 12% to 23% for each 5-unit increase in body mass index, depending on age. They saw the strongest effect at ages 18 – 24 years: Very obese women in this age group were 4.2 times less likely to develop premenopausal breast cancer than women with low body mass index (BMI) at the same age.
The researchers do not know why high BMI might protect against breast cancer in some women. Breast cancer is relatively rare before menopause, although previous studies have suggested that the risk factors might be different for younger vs older women, says Dale Sandler, PhD, co-author of the group and head of the Epidemiology Branch at the National Institute of Environmental Health Sciences. For instance, it is well known that women who gain weight, particularly after menopause, have a higher risk. The fact that this study found that the risk not only is not increased, but actually decreased, in younger women points to the possibility that different biologic mechanisms are at work, Sandler says.
Nonetheless, the researchers caution that young women should not intentionally gain weight to offset the risk.
Source:
National Institutes of Health. https://www.nih.gov/news-events/news-releases/nih-study-associates-obesity-lower-breast-cancer-risk-young-women. Published June 27, 2018. Accessed July 18, 2018.
Young women may not want to hear it, but fat could be their friend. Researchers from the Premenopausal Breast Cancer Collaborative Group have found that women aged 18 – 24 years with high body fat have a lower risk of developing breast cancer before menopause.
The researchers pooled data from 19 different studies, involving about 800,000 women from around the world. Overall, 1.7% of the women developed breast cancer. The researchers found that the relative risk of premenopausal breast cancer dropped 12% to 23% for each 5-unit increase in body mass index, depending on age. They saw the strongest effect at ages 18 – 24 years: Very obese women in this age group were 4.2 times less likely to develop premenopausal breast cancer than women with low body mass index (BMI) at the same age.
The researchers do not know why high BMI might protect against breast cancer in some women. Breast cancer is relatively rare before menopause, although previous studies have suggested that the risk factors might be different for younger vs older women, says Dale Sandler, PhD, co-author of the group and head of the Epidemiology Branch at the National Institute of Environmental Health Sciences. For instance, it is well known that women who gain weight, particularly after menopause, have a higher risk. The fact that this study found that the risk not only is not increased, but actually decreased, in younger women points to the possibility that different biologic mechanisms are at work, Sandler says.
Nonetheless, the researchers caution that young women should not intentionally gain weight to offset the risk.
Source:
National Institutes of Health. https://www.nih.gov/news-events/news-releases/nih-study-associates-obesity-lower-breast-cancer-risk-young-women. Published June 27, 2018. Accessed July 18, 2018.
Young women may not want to hear it, but fat could be their friend. Researchers from the Premenopausal Breast Cancer Collaborative Group have found that women aged 18 – 24 years with high body fat have a lower risk of developing breast cancer before menopause.
The researchers pooled data from 19 different studies, involving about 800,000 women from around the world. Overall, 1.7% of the women developed breast cancer. The researchers found that the relative risk of premenopausal breast cancer dropped 12% to 23% for each 5-unit increase in body mass index, depending on age. They saw the strongest effect at ages 18 – 24 years: Very obese women in this age group were 4.2 times less likely to develop premenopausal breast cancer than women with low body mass index (BMI) at the same age.
The researchers do not know why high BMI might protect against breast cancer in some women. Breast cancer is relatively rare before menopause, although previous studies have suggested that the risk factors might be different for younger vs older women, says Dale Sandler, PhD, co-author of the group and head of the Epidemiology Branch at the National Institute of Environmental Health Sciences. For instance, it is well known that women who gain weight, particularly after menopause, have a higher risk. The fact that this study found that the risk not only is not increased, but actually decreased, in younger women points to the possibility that different biologic mechanisms are at work, Sandler says.
Nonetheless, the researchers caution that young women should not intentionally gain weight to offset the risk.
Source:
National Institutes of Health. https://www.nih.gov/news-events/news-releases/nih-study-associates-obesity-lower-breast-cancer-risk-young-women. Published June 27, 2018. Accessed July 18, 2018.
Stress balls, hand-holding no help during dermatology procedures
according to a randomized trial of 135 patients at Northwestern University, Chicago.

In all three groups, anxiety levels were a little over 3 points on a 10-point Visual Analog Scale (VAS) before surgery and around 2 points during it. The 6-item State Trait Anxiety Inventory score was just under 9 in all three groups right after the procedure, meaning patients weren’t very anxious. Physiological measures did not change from before to after the procedure or between groups. Postoperative pain scores were all under 1 on a 10-point scale, and patients in all three groups were highly satisfied with their encounter, the researchers said in JAMA Dermatology.
“Many patients commented anecdotally on the calming effect of hand-holding or stress ball use,” so “it was surprising that the total data did not show these interventions to preferentially decrease anxiety or alleviate pain,” Arianna F. Yanes, a medical student at Northwestern University, and her coinvestigators said.
It could be that standard measures – giving patients an opportunity to ask questions, making sure they feel comfortable, and the like – are enough. However, “hand-holding and stress balls may still provide stress relief in patients who are particularly anxious before the procedure.” Perhaps patients would have preferred having their hand held by a loved one instead of a stranger, the investigators said.
Meanwhile, patients who researched their operation online beforehand had higher preoperative anxiety scores (3.84 vs. 2.62 points on the VAS; P = .04), but they could have been more anxious from the start.
The mean subject age was 66 years, and 62% were men.
The work was funded by Northwestern University and a grant from Merz. The investigators had no relevant disclosures.
SOURCE: Yanes AF et al. JAMA Dermatol. 2018 Jul 18. doi:10.1001/jamadermatol.2018.1783.
according to a randomized trial of 135 patients at Northwestern University, Chicago.
In all three groups, anxiety levels were a little over 3 points on a 10-point Visual Analog Scale (VAS) before surgery and around 2 points during it. The 6-item State Trait Anxiety Inventory score was just under 9 in all three groups right after the procedure, meaning patients weren’t very anxious. Physiological measures did not change from before to after the procedure or between groups. Postoperative pain scores were all under 1 on a 10-point scale, and patients in all three groups were highly satisfied with their encounter, the researchers said in JAMA Dermatology.
“Many patients commented anecdotally on the calming effect of hand-holding or stress ball use,” so “it was surprising that the total data did not show these interventions to preferentially decrease anxiety or alleviate pain,” Arianna F. Yanes, a medical student at Northwestern University, and her coinvestigators said.
It could be that standard measures – giving patients an opportunity to ask questions, making sure they feel comfortable, and the like – are enough. However, “hand-holding and stress balls may still provide stress relief in patients who are particularly anxious before the procedure.” Perhaps patients would have preferred having their hand held by a loved one instead of a stranger, the investigators said.
Meanwhile, patients who researched their operation online beforehand had higher preoperative anxiety scores (3.84 vs. 2.62 points on the VAS; P = .04), but they could have been more anxious from the start.
The mean subject age was 66 years, and 62% were men.
The work was funded by Northwestern University and a grant from Merz. The investigators had no relevant disclosures.
SOURCE: Yanes AF et al. JAMA Dermatol. 2018 Jul 18. doi:10.1001/jamadermatol.2018.1783.
according to a randomized trial of 135 patients at Northwestern University, Chicago.
In all three groups, anxiety levels were a little over 3 points on a 10-point Visual Analog Scale (VAS) before surgery and around 2 points during it. The 6-item State Trait Anxiety Inventory score was just under 9 in all three groups right after the procedure, meaning patients weren’t very anxious. Physiological measures did not change from before to after the procedure or between groups. Postoperative pain scores were all under 1 on a 10-point scale, and patients in all three groups were highly satisfied with their encounter, the researchers said in JAMA Dermatology.
“Many patients commented anecdotally on the calming effect of hand-holding or stress ball use,” so “it was surprising that the total data did not show these interventions to preferentially decrease anxiety or alleviate pain,” Arianna F. Yanes, a medical student at Northwestern University, and her coinvestigators said.
It could be that standard measures – giving patients an opportunity to ask questions, making sure they feel comfortable, and the like – are enough. However, “hand-holding and stress balls may still provide stress relief in patients who are particularly anxious before the procedure.” Perhaps patients would have preferred having their hand held by a loved one instead of a stranger, the investigators said.
Meanwhile, patients who researched their operation online beforehand had higher preoperative anxiety scores (3.84 vs. 2.62 points on the VAS; P = .04), but they could have been more anxious from the start.
The mean subject age was 66 years, and 62% were men.
The work was funded by Northwestern University and a grant from Merz. The investigators had no relevant disclosures.
SOURCE: Yanes AF et al. JAMA Dermatol. 2018 Jul 18. doi:10.1001/jamadermatol.2018.1783.
FROM JAMA DERMATOLOGY
About half of FDA expedited approvals lack double-blind trials
About half of the drugs approved under the Food and Drug Administrations’s Breakthrough Therapy designation have lacked the gold-standard evidence of a double-blind, randomized, placebo-controlled trial, according to a new JAMA report.
“This study of all FDA approvals granted Breakthrough Therapy designation from 2012 through 2017 suggests that pivotal trials supporting these approvals commonly lacked randomization, double-blinding, and control groups, used surrogate markers as primary end points, and enrolled small numbers of patients,” wrote Jeremy Puthumana and his coauthors. “Furthermore, more than half were based on a single, pivotal trial.”
The average premarket development time was about 5 years, but regulatory review of these agents took less than 7 months on average, the report found.
Mr. Puthumana, of Yale University, New Haven, Conn., and his coauthors, reviewed all 46 of the drugs and biologics approved by the FDA from 2012 to 2017 under the designation. The Breakthrough Therapy designation allows for the rapid review of drugs and biologics for serious or life-threatening conditions where there is preliminary evidence demonstrating a substantial improvement over existing therapies. The researchers identified all pivotal trials supporting approval, looking at randomization, blinding, comparator group, primary endpoint, and patient numbers.
Of these drugs, most (25) were oncologic agents. Other indications were infectious disease (8), genetic or metabolic disorders (5), and other unspecified purposes (8). The median number of patients enrolled among all pivotal trials supporting an indication approval was 222.
Most of the approvals (27) were based on randomized trials, 21 (45.7%) were based on double-blind randomization, 25 (54.3%) employed an active or placebo comparator group, and 10 (21.7%) used a clinical primary endpoint.
Compared with drugs without accelerated approval, drugs with accelerated approval status were less likely to be examined in randomized or double-blinded trials(24 vs. 3 and 20 vs. 1, respectively), and were less likely to include a control group (32 vs. 3).
However, all drugs with Accelerated Approval status underwent at least one clinical safety or efficacy-focused postmarketing requirement, as did 64.3% of those without that status.
“Patients and physicians may have misconceptions about the strength of evidence supporting breakthrough approvals,” the authors wrote. “FDA-required postmarketing studies will be critical to confirm the clinical benefit and safety of these promising, newly approved therapies.”
Mr. Puthumana reported having no financial disclosures.
SOURCE: Puthumana J et al. JAMA. 2018;320(3):301-3.
Expedited drug approvals raise concerns that important questions about safety and effectiveness might be insufficiently answered before an agent makes it to pharmacy shelves, Austin B. Frakt, PhD, wrote in an accompanying editorial.
Several key facts suggest that the Food and Drug Administration’s expedited review programs may invite greater risks than benefits, he wrote.
Most new drugs are approved with relatively little data about long-term outcomes.
More than two-thirds of approvals are based on studies lasting less than 6 months.
The FDA approves novel therapeutic agents more quickly than do similar regulatory bodies in Europe and Canada, with a median time of 6 months for cancer drug approval.
Expedited reviews have increased in the last 2 decades. The increase is driven by drugs that are not first in their class, implying that they aren’t addressing unmet needs.
“The idea that doing something more quickly means it is not done as well has considerable face validity,” Dr. Frakt wrote. Nevertheless, at least one study suggests that expedited FDA approvals do confer substantial gains in quality of life. “[The study suggests] that the FDA’s expedited drug review programs include drugs that provide greater benefits than those undergoing conventional review. Indeed, to the extent the expedited programs handle drugs for conditions for which there is unmet medical need, relatively larger QALY [quality-adjusted life-year] gains are to be expected.”
However, drugs subject to less FDA scrutiny are more likely to exhibit safety problems, be withdrawn from the market, or carry black box warnings. But in some cases, at least, the trade-off seems worth it.
“Because expedited review programs are intended for drugs that treat serious conditions and address unmet medical needs, accepting greater risk may be reasonable and more consistent with patients’ preferences,” he said. “However, because many of these drugs also come with high price tags, financed with public funds through Medicare, Medicaid, and other programs, the patients’ point of view is not the only one of relevance. A consideration of cost is also reasonable from the point of view of taxpayers.”
Dr. Frakt is director of the Partnered Evidence-based Policy Resource Center at the Boston Veterans Affairs Healthcare System. His remarks are adapted from an accompanying editorial (JAMA. 2018;320[3]:225-6).
Expedited drug approvals raise concerns that important questions about safety and effectiveness might be insufficiently answered before an agent makes it to pharmacy shelves, Austin B. Frakt, PhD, wrote in an accompanying editorial.
Several key facts suggest that the Food and Drug Administration’s expedited review programs may invite greater risks than benefits, he wrote.
Most new drugs are approved with relatively little data about long-term outcomes.
More than two-thirds of approvals are based on studies lasting less than 6 months.
The FDA approves novel therapeutic agents more quickly than do similar regulatory bodies in Europe and Canada, with a median time of 6 months for cancer drug approval.
Expedited reviews have increased in the last 2 decades. The increase is driven by drugs that are not first in their class, implying that they aren’t addressing unmet needs.
“The idea that doing something more quickly means it is not done as well has considerable face validity,” Dr. Frakt wrote. Nevertheless, at least one study suggests that expedited FDA approvals do confer substantial gains in quality of life. “[The study suggests] that the FDA’s expedited drug review programs include drugs that provide greater benefits than those undergoing conventional review. Indeed, to the extent the expedited programs handle drugs for conditions for which there is unmet medical need, relatively larger QALY [quality-adjusted life-year] gains are to be expected.”
However, drugs subject to less FDA scrutiny are more likely to exhibit safety problems, be withdrawn from the market, or carry black box warnings. But in some cases, at least, the trade-off seems worth it.
“Because expedited review programs are intended for drugs that treat serious conditions and address unmet medical needs, accepting greater risk may be reasonable and more consistent with patients’ preferences,” he said. “However, because many of these drugs also come with high price tags, financed with public funds through Medicare, Medicaid, and other programs, the patients’ point of view is not the only one of relevance. A consideration of cost is also reasonable from the point of view of taxpayers.”
Dr. Frakt is director of the Partnered Evidence-based Policy Resource Center at the Boston Veterans Affairs Healthcare System. His remarks are adapted from an accompanying editorial (JAMA. 2018;320[3]:225-6).
Expedited drug approvals raise concerns that important questions about safety and effectiveness might be insufficiently answered before an agent makes it to pharmacy shelves, Austin B. Frakt, PhD, wrote in an accompanying editorial.
Several key facts suggest that the Food and Drug Administration’s expedited review programs may invite greater risks than benefits, he wrote.
Most new drugs are approved with relatively little data about long-term outcomes.
More than two-thirds of approvals are based on studies lasting less than 6 months.
The FDA approves novel therapeutic agents more quickly than do similar regulatory bodies in Europe and Canada, with a median time of 6 months for cancer drug approval.
Expedited reviews have increased in the last 2 decades. The increase is driven by drugs that are not first in their class, implying that they aren’t addressing unmet needs.
“The idea that doing something more quickly means it is not done as well has considerable face validity,” Dr. Frakt wrote. Nevertheless, at least one study suggests that expedited FDA approvals do confer substantial gains in quality of life. “[The study suggests] that the FDA’s expedited drug review programs include drugs that provide greater benefits than those undergoing conventional review. Indeed, to the extent the expedited programs handle drugs for conditions for which there is unmet medical need, relatively larger QALY [quality-adjusted life-year] gains are to be expected.”
However, drugs subject to less FDA scrutiny are more likely to exhibit safety problems, be withdrawn from the market, or carry black box warnings. But in some cases, at least, the trade-off seems worth it.
“Because expedited review programs are intended for drugs that treat serious conditions and address unmet medical needs, accepting greater risk may be reasonable and more consistent with patients’ preferences,” he said. “However, because many of these drugs also come with high price tags, financed with public funds through Medicare, Medicaid, and other programs, the patients’ point of view is not the only one of relevance. A consideration of cost is also reasonable from the point of view of taxpayers.”
Dr. Frakt is director of the Partnered Evidence-based Policy Resource Center at the Boston Veterans Affairs Healthcare System. His remarks are adapted from an accompanying editorial (JAMA. 2018;320[3]:225-6).
About half of the drugs approved under the Food and Drug Administrations’s Breakthrough Therapy designation have lacked the gold-standard evidence of a double-blind, randomized, placebo-controlled trial, according to a new JAMA report.
“This study of all FDA approvals granted Breakthrough Therapy designation from 2012 through 2017 suggests that pivotal trials supporting these approvals commonly lacked randomization, double-blinding, and control groups, used surrogate markers as primary end points, and enrolled small numbers of patients,” wrote Jeremy Puthumana and his coauthors. “Furthermore, more than half were based on a single, pivotal trial.”
The average premarket development time was about 5 years, but regulatory review of these agents took less than 7 months on average, the report found.
Mr. Puthumana, of Yale University, New Haven, Conn., and his coauthors, reviewed all 46 of the drugs and biologics approved by the FDA from 2012 to 2017 under the designation. The Breakthrough Therapy designation allows for the rapid review of drugs and biologics for serious or life-threatening conditions where there is preliminary evidence demonstrating a substantial improvement over existing therapies. The researchers identified all pivotal trials supporting approval, looking at randomization, blinding, comparator group, primary endpoint, and patient numbers.
Of these drugs, most (25) were oncologic agents. Other indications were infectious disease (8), genetic or metabolic disorders (5), and other unspecified purposes (8). The median number of patients enrolled among all pivotal trials supporting an indication approval was 222.
Most of the approvals (27) were based on randomized trials, 21 (45.7%) were based on double-blind randomization, 25 (54.3%) employed an active or placebo comparator group, and 10 (21.7%) used a clinical primary endpoint.
Compared with drugs without accelerated approval, drugs with accelerated approval status were less likely to be examined in randomized or double-blinded trials(24 vs. 3 and 20 vs. 1, respectively), and were less likely to include a control group (32 vs. 3).
However, all drugs with Accelerated Approval status underwent at least one clinical safety or efficacy-focused postmarketing requirement, as did 64.3% of those without that status.
“Patients and physicians may have misconceptions about the strength of evidence supporting breakthrough approvals,” the authors wrote. “FDA-required postmarketing studies will be critical to confirm the clinical benefit and safety of these promising, newly approved therapies.”
Mr. Puthumana reported having no financial disclosures.
SOURCE: Puthumana J et al. JAMA. 2018;320(3):301-3.
About half of the drugs approved under the Food and Drug Administrations’s Breakthrough Therapy designation have lacked the gold-standard evidence of a double-blind, randomized, placebo-controlled trial, according to a new JAMA report.
“This study of all FDA approvals granted Breakthrough Therapy designation from 2012 through 2017 suggests that pivotal trials supporting these approvals commonly lacked randomization, double-blinding, and control groups, used surrogate markers as primary end points, and enrolled small numbers of patients,” wrote Jeremy Puthumana and his coauthors. “Furthermore, more than half were based on a single, pivotal trial.”
The average premarket development time was about 5 years, but regulatory review of these agents took less than 7 months on average, the report found.
Mr. Puthumana, of Yale University, New Haven, Conn., and his coauthors, reviewed all 46 of the drugs and biologics approved by the FDA from 2012 to 2017 under the designation. The Breakthrough Therapy designation allows for the rapid review of drugs and biologics for serious or life-threatening conditions where there is preliminary evidence demonstrating a substantial improvement over existing therapies. The researchers identified all pivotal trials supporting approval, looking at randomization, blinding, comparator group, primary endpoint, and patient numbers.
Of these drugs, most (25) were oncologic agents. Other indications were infectious disease (8), genetic or metabolic disorders (5), and other unspecified purposes (8). The median number of patients enrolled among all pivotal trials supporting an indication approval was 222.
Most of the approvals (27) were based on randomized trials, 21 (45.7%) were based on double-blind randomization, 25 (54.3%) employed an active or placebo comparator group, and 10 (21.7%) used a clinical primary endpoint.
Compared with drugs without accelerated approval, drugs with accelerated approval status were less likely to be examined in randomized or double-blinded trials(24 vs. 3 and 20 vs. 1, respectively), and were less likely to include a control group (32 vs. 3).
However, all drugs with Accelerated Approval status underwent at least one clinical safety or efficacy-focused postmarketing requirement, as did 64.3% of those without that status.
“Patients and physicians may have misconceptions about the strength of evidence supporting breakthrough approvals,” the authors wrote. “FDA-required postmarketing studies will be critical to confirm the clinical benefit and safety of these promising, newly approved therapies.”
Mr. Puthumana reported having no financial disclosures.
SOURCE: Puthumana J et al. JAMA. 2018;320(3):301-3.
FROM JAMA
Key clinical point:
Major finding: Just 45.7% of drugs granted Breakthrough Therapy approval by the Food and Drug Administration went through a double-blind, randomized study.
Study details: The review comprised 46 drugs granted Breakthrough status from 2012 to 2017.
Disclosures: Mr. Puthumana reported having no financial disclosures.
Source: Puthumana J et al. JAMA. 2018;320[3]:301-3.
Lab results may help predict complications in ALL treatment
(ALL) who were treated with four-drug induction therapy.
Kasper Warrick, MD, and his colleagues at Indiana University in Indianapolis reported findings from a retrospective study of 73 ALL patients at their hospital. They performed chart reviews comparing a cohort of 42 patients who were discharged on day 4 of their induction treatment with 31 similar patients who had a longer hospital stay or admission to the intensive care unit. The report was published in Leukemia Research.
Univariate analysis found that patients with a longer length of stay were more likely to have a fever, pretransfusion hemoglobin of less than 8 g/dL, lower serum bicarbonate values, abnormal serum calcium, and abnormal serum phosphate. Multivariate stepwise logistic regression found that low serum bicarbonate and a lower platelet count on day 4 of admission was predictive of a prolonged hospital stay. About a third of patients from each group had an unplanned readmission within 30 days.
The researchers concluded that early discharge is safe in only a subgroup of high-risk ALL patients undergoing induction chemotherapy. “Treating physicians could opt for a discharge only after normalization of electrolyte abnormalities and renal functions, and when no transfusion support is needed (stable hematocrit and platelet count),” they wrote. Even in those cases, they recommended “aggressive and close outpatient follow” since patients are vulnerable to complications and readmissions.
SOURCE: Warrick K et al. Leuk Res. 2018 Jun 30:71:36-42.
(ALL) who were treated with four-drug induction therapy.
Kasper Warrick, MD, and his colleagues at Indiana University in Indianapolis reported findings from a retrospective study of 73 ALL patients at their hospital. They performed chart reviews comparing a cohort of 42 patients who were discharged on day 4 of their induction treatment with 31 similar patients who had a longer hospital stay or admission to the intensive care unit. The report was published in Leukemia Research.
Univariate analysis found that patients with a longer length of stay were more likely to have a fever, pretransfusion hemoglobin of less than 8 g/dL, lower serum bicarbonate values, abnormal serum calcium, and abnormal serum phosphate. Multivariate stepwise logistic regression found that low serum bicarbonate and a lower platelet count on day 4 of admission was predictive of a prolonged hospital stay. About a third of patients from each group had an unplanned readmission within 30 days.
The researchers concluded that early discharge is safe in only a subgroup of high-risk ALL patients undergoing induction chemotherapy. “Treating physicians could opt for a discharge only after normalization of electrolyte abnormalities and renal functions, and when no transfusion support is needed (stable hematocrit and platelet count),” they wrote. Even in those cases, they recommended “aggressive and close outpatient follow” since patients are vulnerable to complications and readmissions.
SOURCE: Warrick K et al. Leuk Res. 2018 Jun 30:71:36-42.
(ALL) who were treated with four-drug induction therapy.
Kasper Warrick, MD, and his colleagues at Indiana University in Indianapolis reported findings from a retrospective study of 73 ALL patients at their hospital. They performed chart reviews comparing a cohort of 42 patients who were discharged on day 4 of their induction treatment with 31 similar patients who had a longer hospital stay or admission to the intensive care unit. The report was published in Leukemia Research.
Univariate analysis found that patients with a longer length of stay were more likely to have a fever, pretransfusion hemoglobin of less than 8 g/dL, lower serum bicarbonate values, abnormal serum calcium, and abnormal serum phosphate. Multivariate stepwise logistic regression found that low serum bicarbonate and a lower platelet count on day 4 of admission was predictive of a prolonged hospital stay. About a third of patients from each group had an unplanned readmission within 30 days.
The researchers concluded that early discharge is safe in only a subgroup of high-risk ALL patients undergoing induction chemotherapy. “Treating physicians could opt for a discharge only after normalization of electrolyte abnormalities and renal functions, and when no transfusion support is needed (stable hematocrit and platelet count),” they wrote. Even in those cases, they recommended “aggressive and close outpatient follow” since patients are vulnerable to complications and readmissions.
SOURCE: Warrick K et al. Leuk Res. 2018 Jun 30:71:36-42.
FROM LEUKEMIA RESEARCH
Are We Beating Cancer—Finally?
Cancer death rates continue to decline in the US in all major racial and ethnic groups, according to the National Cancer Institute’s (NCI) latest Annual Report to the Nation on the Status of Cancer. The data are an “encouraging indicator of progress” in cancer research, says NCI Director Ned Sharpless, MD. “It’s clear that interventions are having an impact.”
Overall incidence, or rates of new cancers, dropped by 1.8% in men and 1.4% in women from 1999 to 2015. Between 2011 and 2015, death rates dropped for 11 of the 18 most common cancer types in men and 14 of the 20 most common types in women. The researchers say the “significant declines” also hold “significant differences” in rate by sex, race, and ethnicity. For example, black men and white women had the highest incidence rates, and black men and black women had the highest death rates.
However, over the same period, death rates for cancers of the liver, pancreas, and brain and nervous system rose in both men and women. Death rates for cancer of the uterus rose (the researchers say obesity is thought to be a contributing factor) and death rates for cancers of the oral cavity and pharynx and soft tissue increased in men, perhaps associated with human papillomavirus infection.
In a companion study, when researchers explored prostate cancer trends in more detail they found overall prostate cancer incidence rates declined an average of 6.5% each year between 2007 and 2014, from 163 new cases per 100,000 men to 104 new cases. Still, after a 2-decade steady decline, rates leveled off. Incidence of distant disease rose from 7.8 new cases per 100,000 to 9.2, but there was no increase in the rates of cases with aggressive histologic grade.
Interestingly, the researchers also report a decline in recent prostate-specific antigen screening between 2010 and 2013 national surveys. “The increase in late-stage disease and the flattening of the mortality trended occurred contemporaneously with the observed decrease in PSA screening,” said Serban Negoita, MD, DrPH, of NCI’s Surveillance Research Program. However, while “suggestive,” Negoita adds, their observation does not demonstrate causality: many factors contribute to incidence and mortality, such as improvements in staging and treating cancer.
Cancer death rates continue to decline in the US in all major racial and ethnic groups, according to the National Cancer Institute’s (NCI) latest Annual Report to the Nation on the Status of Cancer. The data are an “encouraging indicator of progress” in cancer research, says NCI Director Ned Sharpless, MD. “It’s clear that interventions are having an impact.”
Overall incidence, or rates of new cancers, dropped by 1.8% in men and 1.4% in women from 1999 to 2015. Between 2011 and 2015, death rates dropped for 11 of the 18 most common cancer types in men and 14 of the 20 most common types in women. The researchers say the “significant declines” also hold “significant differences” in rate by sex, race, and ethnicity. For example, black men and white women had the highest incidence rates, and black men and black women had the highest death rates.
However, over the same period, death rates for cancers of the liver, pancreas, and brain and nervous system rose in both men and women. Death rates for cancer of the uterus rose (the researchers say obesity is thought to be a contributing factor) and death rates for cancers of the oral cavity and pharynx and soft tissue increased in men, perhaps associated with human papillomavirus infection.
In a companion study, when researchers explored prostate cancer trends in more detail they found overall prostate cancer incidence rates declined an average of 6.5% each year between 2007 and 2014, from 163 new cases per 100,000 men to 104 new cases. Still, after a 2-decade steady decline, rates leveled off. Incidence of distant disease rose from 7.8 new cases per 100,000 to 9.2, but there was no increase in the rates of cases with aggressive histologic grade.
Interestingly, the researchers also report a decline in recent prostate-specific antigen screening between 2010 and 2013 national surveys. “The increase in late-stage disease and the flattening of the mortality trended occurred contemporaneously with the observed decrease in PSA screening,” said Serban Negoita, MD, DrPH, of NCI’s Surveillance Research Program. However, while “suggestive,” Negoita adds, their observation does not demonstrate causality: many factors contribute to incidence and mortality, such as improvements in staging and treating cancer.
Cancer death rates continue to decline in the US in all major racial and ethnic groups, according to the National Cancer Institute’s (NCI) latest Annual Report to the Nation on the Status of Cancer. The data are an “encouraging indicator of progress” in cancer research, says NCI Director Ned Sharpless, MD. “It’s clear that interventions are having an impact.”
Overall incidence, or rates of new cancers, dropped by 1.8% in men and 1.4% in women from 1999 to 2015. Between 2011 and 2015, death rates dropped for 11 of the 18 most common cancer types in men and 14 of the 20 most common types in women. The researchers say the “significant declines” also hold “significant differences” in rate by sex, race, and ethnicity. For example, black men and white women had the highest incidence rates, and black men and black women had the highest death rates.
However, over the same period, death rates for cancers of the liver, pancreas, and brain and nervous system rose in both men and women. Death rates for cancer of the uterus rose (the researchers say obesity is thought to be a contributing factor) and death rates for cancers of the oral cavity and pharynx and soft tissue increased in men, perhaps associated with human papillomavirus infection.
In a companion study, when researchers explored prostate cancer trends in more detail they found overall prostate cancer incidence rates declined an average of 6.5% each year between 2007 and 2014, from 163 new cases per 100,000 men to 104 new cases. Still, after a 2-decade steady decline, rates leveled off. Incidence of distant disease rose from 7.8 new cases per 100,000 to 9.2, but there was no increase in the rates of cases with aggressive histologic grade.
Interestingly, the researchers also report a decline in recent prostate-specific antigen screening between 2010 and 2013 national surveys. “The increase in late-stage disease and the flattening of the mortality trended occurred contemporaneously with the observed decrease in PSA screening,” said Serban Negoita, MD, DrPH, of NCI’s Surveillance Research Program. However, while “suggestive,” Negoita adds, their observation does not demonstrate causality: many factors contribute to incidence and mortality, such as improvements in staging and treating cancer.
Promising phase 3 results for ixazomib in multiple myeloma
who had responded to high-dose therapy and autologous stem cell transplant.
The drug’s sponsor, Takeda, announced that the oral proteasome inhibitor had met the primary endpoint – progression-free survival versus placebo – in the randomized, phase 3 TOURMALINE-MM3 study. They also reported that adverse events were consistent with previously reported results for single-agent use of ixazomib and that there were no new safety signals.
Full study results will be presented at the annual meeting of the American Society of Hematology. Company officials plan to submit the trial data to the Food and Drug Administration and regulatory agencies around the world to gain approval of ixazomib as a single-agent maintenance therapy, according to a Takeda announcement.
The TOURMALINE-MM3 study is a double-blind study of 656 patients with multiple myeloma who have had complete response, very good partial response, or partial response to induction therapy followed by high-dose therapy and autologous stem cell transplant. In addition to progression-free survival, the trial assessed overall survival.
who had responded to high-dose therapy and autologous stem cell transplant.
The drug’s sponsor, Takeda, announced that the oral proteasome inhibitor had met the primary endpoint – progression-free survival versus placebo – in the randomized, phase 3 TOURMALINE-MM3 study. They also reported that adverse events were consistent with previously reported results for single-agent use of ixazomib and that there were no new safety signals.
Full study results will be presented at the annual meeting of the American Society of Hematology. Company officials plan to submit the trial data to the Food and Drug Administration and regulatory agencies around the world to gain approval of ixazomib as a single-agent maintenance therapy, according to a Takeda announcement.
The TOURMALINE-MM3 study is a double-blind study of 656 patients with multiple myeloma who have had complete response, very good partial response, or partial response to induction therapy followed by high-dose therapy and autologous stem cell transplant. In addition to progression-free survival, the trial assessed overall survival.
who had responded to high-dose therapy and autologous stem cell transplant.
The drug’s sponsor, Takeda, announced that the oral proteasome inhibitor had met the primary endpoint – progression-free survival versus placebo – in the randomized, phase 3 TOURMALINE-MM3 study. They also reported that adverse events were consistent with previously reported results for single-agent use of ixazomib and that there were no new safety signals.
Full study results will be presented at the annual meeting of the American Society of Hematology. Company officials plan to submit the trial data to the Food and Drug Administration and regulatory agencies around the world to gain approval of ixazomib as a single-agent maintenance therapy, according to a Takeda announcement.
The TOURMALINE-MM3 study is a double-blind study of 656 patients with multiple myeloma who have had complete response, very good partial response, or partial response to induction therapy followed by high-dose therapy and autologous stem cell transplant. In addition to progression-free survival, the trial assessed overall survival.
IVF linked to slight increase in risk of some cancers
A large observational study from Great Britain paints a mixed picture of cancer risks associated with assisted reproduction technologies such as in vitro fertilization (IVF).
Among nearly 256,000 women who had assisted reproduction over a 2-decade period and were followed for more than 2.25 million person-years, there was no significant increase in risk of uterine cancers or invasive breast cancers.
However, these women were at slight but significantly increased risk for in situ breast cancers and invasive or borderline ovarian cancers, although the ovarian tumors may be related more to patient factors than to the assisted reproduction technology itself, reported Alastair Sutcliffe, MD, PhD, from University College London, and his colleagues.
“We were not able to distinguish between a genuine increase in risk of borderline ovarian tumors and other explanations including surveillance bias. Further investigation of this and longer follow-up is warranted to continue monitoring these important outcomes in this ever-growing population,” they wrote in BMJ.
Women who undergo IVF and related procedures are typically exposed to high levels of estradiol, luteinizing hormone, follicle-stimulating hormone, and multiple ovarian punctures, all of which are potentially carcinogenic, the authors noted.
Previous studies of cancer risk in women who undergo assisted reproduction treatment have had conflicting or inconsistent results and lacked information on potential confounders, they pointed out.
To get a better sense of the potential risks, the investigators looked at linked data from the United Kingdom’s Human Fertilization and Embryology Authority and national cancer registries. They identified a total of 255,786 women treated from 1991 through 2010. Total follow-up was for 2,257,789 person-years.
They found that, during an average 8.8 years of follow-up, women had no significant increase in risk of tumors of the uterine corpus, with 164 cancers observed vs. 146.9 expected, for a standardized incidence ratio (SIR) of 1.12 (95% confidence interval, 0.95-1.30).
Similarly, there was no significant increase in risk of either breast cancer overall (2,578 observed vs. 2,641.2 expected; SIR, 0.98; 95% CI, 0.94-1.01) or invasive breast cancer (2,272 observed vs. 2,371.4 expected; SIR, 0.96; 95% CI, 0.92-1.00).
As noted, however, there was an increased risk of in situ breast cancer with 291 vs. 253.5 cases, respectively, translating into a SIR of 1.15 (95% CI, 1.02-1.29), for an absolute excess risk (AER) of 1.7 cases per 100,000 person-years, associated with an increasing number of treatment cycles (P = .03).
Additionally, the women in the study had an increased risk of ovarian cancer with an observed incidence of 405 vs. 291.82 expected, for an SIR of 1.39 (95% CI, 1.26-1.53) and an AER of 5.0 cases per 100,000 person-years.
There was a significantly increased risk for both invasive tumors, with 264 cases vs. 188.1 expected, for an SIR of 1.40 (95% CI, 1.24-1.58) and an AER of 3.4 cases per 100,000 person-years, and borderline ovarian cancers, with 141 vs. 103.7, for an SIR of 1.36 (95% CI, 1.15-1.60) and an AER of 1.7 cases per 100,000 person-years.
Increased risks of ovarian tumors were limited to women with endometriosis, low parity, or both. This study found no increased risk of any ovarian tumor in women treated because of only male-factor or unexplained infertility.
Women who had previously given birth and did not have a diagnosis of endometriosis had no increase in risk for ovarian cancer. Instead, increased risk appeared to be limited to women with endometriosis, few or no births (low parity), or both.
The authors emphasized that ongoing monitoring of outcomes for women who have undergone assisted reproduction is essential.
The study was funded by Cancer Research UK and the National Institute for Health Research, and supported by the NIHR Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. Three authors reported personal fees or royalties from entities not involved in the study.
SOURCE: Williams CL et al. BMJ. 2018;362:k2644.
A large observational study from Great Britain paints a mixed picture of cancer risks associated with assisted reproduction technologies such as in vitro fertilization (IVF).
Among nearly 256,000 women who had assisted reproduction over a 2-decade period and were followed for more than 2.25 million person-years, there was no significant increase in risk of uterine cancers or invasive breast cancers.
However, these women were at slight but significantly increased risk for in situ breast cancers and invasive or borderline ovarian cancers, although the ovarian tumors may be related more to patient factors than to the assisted reproduction technology itself, reported Alastair Sutcliffe, MD, PhD, from University College London, and his colleagues.
“We were not able to distinguish between a genuine increase in risk of borderline ovarian tumors and other explanations including surveillance bias. Further investigation of this and longer follow-up is warranted to continue monitoring these important outcomes in this ever-growing population,” they wrote in BMJ.
Women who undergo IVF and related procedures are typically exposed to high levels of estradiol, luteinizing hormone, follicle-stimulating hormone, and multiple ovarian punctures, all of which are potentially carcinogenic, the authors noted.
Previous studies of cancer risk in women who undergo assisted reproduction treatment have had conflicting or inconsistent results and lacked information on potential confounders, they pointed out.
To get a better sense of the potential risks, the investigators looked at linked data from the United Kingdom’s Human Fertilization and Embryology Authority and national cancer registries. They identified a total of 255,786 women treated from 1991 through 2010. Total follow-up was for 2,257,789 person-years.
They found that, during an average 8.8 years of follow-up, women had no significant increase in risk of tumors of the uterine corpus, with 164 cancers observed vs. 146.9 expected, for a standardized incidence ratio (SIR) of 1.12 (95% confidence interval, 0.95-1.30).
Similarly, there was no significant increase in risk of either breast cancer overall (2,578 observed vs. 2,641.2 expected; SIR, 0.98; 95% CI, 0.94-1.01) or invasive breast cancer (2,272 observed vs. 2,371.4 expected; SIR, 0.96; 95% CI, 0.92-1.00).
As noted, however, there was an increased risk of in situ breast cancer with 291 vs. 253.5 cases, respectively, translating into a SIR of 1.15 (95% CI, 1.02-1.29), for an absolute excess risk (AER) of 1.7 cases per 100,000 person-years, associated with an increasing number of treatment cycles (P = .03).
Additionally, the women in the study had an increased risk of ovarian cancer with an observed incidence of 405 vs. 291.82 expected, for an SIR of 1.39 (95% CI, 1.26-1.53) and an AER of 5.0 cases per 100,000 person-years.
There was a significantly increased risk for both invasive tumors, with 264 cases vs. 188.1 expected, for an SIR of 1.40 (95% CI, 1.24-1.58) and an AER of 3.4 cases per 100,000 person-years, and borderline ovarian cancers, with 141 vs. 103.7, for an SIR of 1.36 (95% CI, 1.15-1.60) and an AER of 1.7 cases per 100,000 person-years.
Increased risks of ovarian tumors were limited to women with endometriosis, low parity, or both. This study found no increased risk of any ovarian tumor in women treated because of only male-factor or unexplained infertility.
Women who had previously given birth and did not have a diagnosis of endometriosis had no increase in risk for ovarian cancer. Instead, increased risk appeared to be limited to women with endometriosis, few or no births (low parity), or both.
The authors emphasized that ongoing monitoring of outcomes for women who have undergone assisted reproduction is essential.
The study was funded by Cancer Research UK and the National Institute for Health Research, and supported by the NIHR Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. Three authors reported personal fees or royalties from entities not involved in the study.
SOURCE: Williams CL et al. BMJ. 2018;362:k2644.
A large observational study from Great Britain paints a mixed picture of cancer risks associated with assisted reproduction technologies such as in vitro fertilization (IVF).
Among nearly 256,000 women who had assisted reproduction over a 2-decade period and were followed for more than 2.25 million person-years, there was no significant increase in risk of uterine cancers or invasive breast cancers.
However, these women were at slight but significantly increased risk for in situ breast cancers and invasive or borderline ovarian cancers, although the ovarian tumors may be related more to patient factors than to the assisted reproduction technology itself, reported Alastair Sutcliffe, MD, PhD, from University College London, and his colleagues.
“We were not able to distinguish between a genuine increase in risk of borderline ovarian tumors and other explanations including surveillance bias. Further investigation of this and longer follow-up is warranted to continue monitoring these important outcomes in this ever-growing population,” they wrote in BMJ.
Women who undergo IVF and related procedures are typically exposed to high levels of estradiol, luteinizing hormone, follicle-stimulating hormone, and multiple ovarian punctures, all of which are potentially carcinogenic, the authors noted.
Previous studies of cancer risk in women who undergo assisted reproduction treatment have had conflicting or inconsistent results and lacked information on potential confounders, they pointed out.
To get a better sense of the potential risks, the investigators looked at linked data from the United Kingdom’s Human Fertilization and Embryology Authority and national cancer registries. They identified a total of 255,786 women treated from 1991 through 2010. Total follow-up was for 2,257,789 person-years.
They found that, during an average 8.8 years of follow-up, women had no significant increase in risk of tumors of the uterine corpus, with 164 cancers observed vs. 146.9 expected, for a standardized incidence ratio (SIR) of 1.12 (95% confidence interval, 0.95-1.30).
Similarly, there was no significant increase in risk of either breast cancer overall (2,578 observed vs. 2,641.2 expected; SIR, 0.98; 95% CI, 0.94-1.01) or invasive breast cancer (2,272 observed vs. 2,371.4 expected; SIR, 0.96; 95% CI, 0.92-1.00).
As noted, however, there was an increased risk of in situ breast cancer with 291 vs. 253.5 cases, respectively, translating into a SIR of 1.15 (95% CI, 1.02-1.29), for an absolute excess risk (AER) of 1.7 cases per 100,000 person-years, associated with an increasing number of treatment cycles (P = .03).
Additionally, the women in the study had an increased risk of ovarian cancer with an observed incidence of 405 vs. 291.82 expected, for an SIR of 1.39 (95% CI, 1.26-1.53) and an AER of 5.0 cases per 100,000 person-years.
There was a significantly increased risk for both invasive tumors, with 264 cases vs. 188.1 expected, for an SIR of 1.40 (95% CI, 1.24-1.58) and an AER of 3.4 cases per 100,000 person-years, and borderline ovarian cancers, with 141 vs. 103.7, for an SIR of 1.36 (95% CI, 1.15-1.60) and an AER of 1.7 cases per 100,000 person-years.
Increased risks of ovarian tumors were limited to women with endometriosis, low parity, or both. This study found no increased risk of any ovarian tumor in women treated because of only male-factor or unexplained infertility.
Women who had previously given birth and did not have a diagnosis of endometriosis had no increase in risk for ovarian cancer. Instead, increased risk appeared to be limited to women with endometriosis, few or no births (low parity), or both.
The authors emphasized that ongoing monitoring of outcomes for women who have undergone assisted reproduction is essential.
The study was funded by Cancer Research UK and the National Institute for Health Research, and supported by the NIHR Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. Three authors reported personal fees or royalties from entities not involved in the study.
SOURCE: Williams CL et al. BMJ. 2018;362:k2644.
FROM BMJ
Key clinical point: Assisted reproductive technologies may be linked to increased risk for in situ breast and ovarian cancers.
Major finding: The standardized incidence ratios (observed vs. expected cases) for invasive and borderline ovarian tumors were 1.40 and 1.41, respectively, both statistically significant.
Study details: Observational study with linked fertility clinic and cancer registry data on 255,786 women with total follow-up of 2,257,789 person-years.
Disclosures: The study was funded by Cancer Research UK and the National Institute for Health Research, and supported by the NIHR Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. Three authors reported personal fees or royalties from entities not involved in the study.
Source: Williams CL et al. BMJ. 2018;362:k2644.