User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Pigmenting Purpuric Dermatoses: Striking But Not a Manifestation of COVID-19 Infection
Pigmented purpuric dermatoses (PPDs) are characterized by petechiae, dusky macules representative of postinflammatory hyperpigmentation and dermal hemosiderin, and purpura generally localized to the lower extremities. They typically represent a spectrum of lymphocytic capillaritis, variable erythrocyte extravasation from papillary dermal blood vessels, and deposition of hemosiderin, yielding the classic red to orange to golden-brown findings on gross examination. Clinical overlap exists, but variants include Schamberg disease (SD), Majocchi purpura, Gougerot-Blum purpura, eczematoid purpura of Doucas and Kapetanakis (DK), and lichen aureus.1 Other forms are rarer, including linear, granulomatous, quadrantic, transitory, and familial variants. It remains controversial whether PPD may precede or have an association with cutaneous T-cell lymphoma.2 Dermoscopy usually shows copper-red pigmentation in the background, oval red dots, linear vessels, brown globules, and follicular openings. Although these findings may be useful in PPD diagnosis, they are not applicable in differentiating among the variants.
Pigmented purpuric dermatoses can easily be mistaken for stasis dermatitis or cellulitis, as these may occur concomitantly or in populations at risk for all 3 conditions, such as women older than 50 years with recent trauma or infection in the affected area. Tissue biopsy and clinical laboratory evaluation may be required to differentiate between PPD from leukocytoclastic vasculitis or the myriad causes of retiform purpura. Importantly, clinicians also should differentiate PPD from the purpuric eruptions of the lower extremities associated with COVID-19 infection.
Pigmented Purpuric Dermatoses
Schamberg Disease—In 1901, Jay Frank Schamberg, a distinguished professor of dermatology in Philadelphia, Pennsylvania, described “a peculiar progressive pigmentary disease of the skin” in a 15-year-old adolescent boy.3 Schamberg disease is the most common PPD, characterized by pruritic spots resembling cayenne pepper (Figure 1) with orange-brown pigmented macules on the legs and feet.4 Although platelet dysfunction, coagulation deficiencies, or dermal atrophy may contribute to hemorrhaging that manifests as petechiae or ecchymoses, SD typically is not associated with any laboratory abnormalities, and petechial eruption is not widespread.5 Capillary fragility can be assessed by the tourniquet test, in which pressure is applied to the forearm with a blood pressure cuff inflated between systolic and diastolic blood pressure for 5 to 10 minutes. Upon removing the cuff, a positive test is indicated by 15 or more petechiae in an area 5 cm in diameter due to poor platelet function. A positive result may be seen in SD.6

Histologically, SD is characterized by patchy parakeratosis, mild spongiosis of the stratum Malpighi, and lymphoid capillaritis (Figure 2).7 In addition to CD3+, CD4+, CD8+, CD1a+, and CD36+ lymphocytes, histology also may contain dendritic cells and cellular adhesion molecules (intercellular adhesion molecule 1, epithelial cell adhesion molecule 1) within the superficial perivascular infiltrate.8 There is no definitive therapy, but first-line interventions include emollients, topical steroids, and oral antihistamines. Nonpharmacologic management includes compression or support stockings, elevation of the lower extremities, and avoidance of offending medications (if identifiable).1

Majocchi Purpura—Domenico Majocchi was a renowned Italian dermatologist who described an entity in 1898 that he called purpura annularis telangiectodes, now also known as Majocchi purpura.9 It is more common in females, young adults, and children. Majocchi purpura has rarely been reported in families with a possible autosomal-dominant inheritance.10 Typically, bluish-red annular macules with central atrophy surrounded by hyperpigmentation may be seen on the lower extremities, potentially extending to the upper extremities.1 Treatment of Majocchi purpura remains a challenge but may respond to narrowband UVB phototherapy. Emollients and topical steroids also are used as first-line treatments. Biopsy demonstrates telangiectasia, pericapillary infiltration of mononuclear lymphocytes, and papillary dermal hemosiderin.11
Gougerot-Blum Purpura—In 1925, French dermatologists Henri Gougerot and Paul Blum described a pigmented purpuric lichenoid dermatitis known as Gougerot-Blum purpura,12 a rare PPD characterized by lichenoid papules that eventually coalesce into plaques of various colors, along with red-brown hyperpigmentation.4 As with other PPD variants, the legs are most involved, with rare extension to the trunk or thighs. The plaques may resemble and be mistaken for Kaposi sarcoma, cutaneous vasculitis, traumatic purpura, or mycosis fungoides. Dermoscopic examination reveals small, polygonal or round, red dots underlying brown scaly patches.13 Gougerot-Blum purpura is found more commonly in adult men and rarely affects children.4 Histologically, a lichenoid and superficial perivascular infiltrate composed of lymphocytes and macrophages is seen. Various therapies have been described, including topical steroids, antihistamines, psoralen plus UVA phototherapy, and cyclosporin A.14
Eczematoid Purpura of Doucas and Kapetanakis—In 1949, Greek dermatologists Christopher Doucas and John Kapetanakis observed several cases of purpuric dermatosis similar in form to the “pigmented purpuric lichenoid dermatitis” of Gougerot-Blum purpura12 and to the “progressive pigmentary dermatitis” of Schamberg disease.3 After observing a gradual disappearance of the classic yellow color from hemosiderin deposition, Doucas and Kapetanakis described a new bright red eruption with lichenification.15 Eczematoid purpura of Doucas and Kapetanakis is rare and predominantly seen in middle-aged males. Hyperpigmented or dark brown macules may develop bilaterally on the legs, progressing to the thighs and upper extremities. Unlike the other types of PPD, DK is extensive and severely pruritic.4
Although most PPD can be drug induced, DK has shown the greatest tendency for pruritic erythematous plaques following drug usage including but not limited to amlodipine, aspirin, acetaminophen, thiamine, interferon alfa, chlordiazepoxide, and isotretinoin. Additionally, DK has been associated with a contact allergy to clothing dyes and rubber.4 On histology, epidermal spongiosis may be seen, correlating with the eczematoid clinical findings. Spontaneous remission also is more common compared to the other PPDs. Treatment consists of topical corticosteroids and antihistamines.16
Lichen Aureus—Lichen aureus was first observed by the dermatologist R.H. Martin in 1958.17 It is clinically characterized by closely aggregated purpuric papules with a distinctive golden-brown color more often localized to the lower extremities and sometimes in a dermatomal distribution. Lichen aureus affects males and females equally, and similar to Majocchi purpura can be seen in children.4 Histopathologic examination reveals a prominent lichenoid plus superficial and deep perivascular lymphocytic infiltrate, extravasated erythrocytes, papillary dermal edema, hemosiderophages, and an unaffected epidermis. In rare cases, perineural infiltrates may be seen. Topical steroids usually are ineffective in lichen aureus treatment, but responses to psoralen plus UVA therapy also have been noted.17
Differential Diagnosis
COVID-19–Related Cutaneous Changes—Because COVID-19–related pathology is now a common differential diagnosis for many cutaneous eruptions, one must be mindful of the possibility for patients to have PPD, cutaneous changes from underlying COVID-19, or both.18 The microvascular changes from COVID-19 infection can be variable.19 Besides the presence of erythema along a distal digit, manifestations can include reticulated dusky erythema mimicking livedoid vasculopathy or inflammatory purpura.19
Retiform Purpura—Retiform purpura may occur in the setting of microvascular occlusion and can represent the pattern of underlying dermal vasculature. It is nonblanching and typically stellate or branching.20 The microvascular occlusion may be a result of hypercoagulability or may be secondary to cutaneous vasculitis, resulting in thrombosis and subsequent vascular occlusion.21 There are many reasons for hypercoagulability in retiform purpura, including disseminated intravascular coagulation in the setting of COVID-19 infection.22 The treatment of retiform purpura is aimed at alleviating the underlying cause and providing symptomatic relief. Conversely, the PPDs generally are benign and require minimal workup.
Leukocytoclastic Vasculitis—The hallmark of leukocytoclastic vasculitis is palpable purpura, often appearing as nonblanchable papules, typically in a dependent distribution such as the lower extremities (Figure 3). Although it primarily affects children, Henoch-Schönlein purpura is a type of leukocytoclastic vasculitis with lesions potentially similar in appearance to those of PPD.23 Palpable purpura may be painful and may ulcerate but rarely is pruritic. Leukocytoclastic vasculitis represents perivascular infiltrates composed of neutrophils, lymphocytes, and occasionally eosinophils, along with karyorrhexis, luminal fibrin, and fibrinoid degeneration of blood vessel walls, often resulting from immune complex deposition. Leukocytoclastic vasculitis may affect blood vessels of any size and requires further clinical and laboratory evaluation for infection (including COVID-19), hypercoagulability, autoimmune disease, or medication-related reactions.24

Stasis Dermatitis—Stasis dermatitis, a chronic inflammatory condition stemming from retrograde venous flow due to incompetent venous valves, mimics PPD. Stasis dermatitis initially appears as demarcated erythematous plaques, fissures, and scaling of the lower legs bilaterally, usually involving the medial malleolus.25 With time, the affected region develops overlying brawny hyperpigmentation and fibrosis (Figure 4). Pruritus or pain are common features, while fissures and superficial erosions may heal and recur, leading to lichenification.

Although both commonly appear on the lower extremities, duplex ultrasonography may be helpful to distinguish PPDs from stasis dermatitis since the latter occurs in the context of chronic venous insufficiency, varicose veins, soft tissue edema, and lymphedema.25 Additionally, pruritus, lichenification, and edema often are not seen in most PPD variants, although stasis dermatitis and PPD may occur in tandem. Conservative treatment involves elevation of the extremities, compression, and topical steroids for symptomatic relief.
Cellulitis—The key characteristics of cellulitis are redness, swelling, warmth, tenderness, fever, and leukocytosis. A history of trauma, such as a prior break in the skin, and pain in the affected area suggest cellulitis. Several skin conditions present similarly to cellulitis, including PPD, and thus approximately 30% of cases are misdiagnosed.26 Cellulitis rarely presents in a bilateral or diffusely scattered pattern as seen in PPDs. Rather, it is unilateral with smooth indistinct borders. Variables suggestive of cellulitis include immunosuppression, rapid progression, and previous occurrences. Hyperpigmented plaques or thickening of the skin are more indicative of a chronic process such as stasis dermatitis or lipodermatosclerosis rather than acute cellulitis. Purpura is not a typical finding in most cases of soft tissue cellulitis. Treatment may be case specific depending on severity, presence or absence of sepsis, findings on blood cultures, or other pathologic evaluation. Antibiotics are directed to the causative organism, typically Streptococcus and Staphylococcus species, although coverage against various gram-negative organisms may be indicated.27
Caution With Teledermatology
COVID-19 has established the value of telemedicine in providing access to health care services for at-risk or underserved individuals. The PPDs are benign, often asymptomatic, and potentially identifiable with teledermatology alone; however, they also can easily be mistaken for COVID-19–related eruptions, vasculitis, other types of purpura, stasis dermatitis, or other complications of lower extremity stasis and lymphedema, especially in an aging population. If tissue biopsy is required, as in the workup of vasculitis, the efficacy of telemedicine becomes more questionable. It is important to delineate the potentially confusing PPDs from other potentially dangerous or life-threatening inflammatory dermatoses.28
- Sardana K, Sarkar R , Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Çaytemel C, Baykut B, Ag˘ırgöl S¸, et al. Pigmented purpuric dermatosis: ten years of experience in a tertiary hospital and awareness of mycosis fungoides in differential diagnosis. J Cutan Pathol. 2021;48:611-616.
- Schamberg JF. A peculiar progressive pigmentary disease of the skin. Br J Dermatol. 1901;13:1-5.
- Martínez Pallás I, Conejero Del Mazo R, Lezcano Biosca V. Pigmented purpuric dermatosis: a review of the literature. Actas Dermosifiliogr (Engl Ed). 2020;111:196-204.
- Ozkaya DB, Emiroglu N, Su O, et al. Dermatoscopic findings of pigmented purpuric dermatosis. An Bras Dermatol. 2016;91:584-587.
- Lava SAG, Milani GP, Fossali EF, et al. Cutaneous manifestations of small-vessel leukocytoclastic vasculitides in childhood. Clin Rev Allergy Immunol. 2017;53:439-451.
- Bonnet U, Selle C, Isbruch K, et al. Recurrent purpura due to alcohol-related Schamberg’s disease and its association with serum immunoglobulins: a longitudinal observation of a heavy drinker. J Med Case Rep. 2016;10:301.
- Zaldivar Fujigaki JL, Anjum F. Schamberg Disease. StatPearls Publishing; 2021.
- Majocchi J. Purpura annularis telangiectodes. Arch Dermatol Syph. 1898;43:447.
- Sethuraman G, Sugandhan S, Bansal A, et al. Familial pigmented purpuric dermatoses. J Dermatol. 2006;33:639-641.
- Miller K, Fischer M, Kamino H, et al. Purpura annularis telangiectoides. Dermatol Online J. 2012;18:5.
- Coulombe J, Jean SE, Hatami A, et al. Pigmented purpuric dermatosis: clinicopathologic characterization in a pediatric series. Pediatr Dermatol. 2015;32:358-362.
- Park MY, Shim WH, Kim JM, et al. Dermoscopic finding in pigmented purpuric lichenoid dermatosis of Gougerot-Blum: a useful tool for clinical diagnosis. Ann Dermatol. 2018;30:245-247.
- Risikesan J, Sommerlund M, Ramsing M, et al. Successful topical treatment of pigmented purpuric lichenoid dermatitis of Gougerot-Blum in a young patient: a case report and summary of the most common pigmented purpuric dermatoses. Case Rep Dermatol. 2017;9:169-176.
- Doucas C, Kapetanakis J. Eczematid-like purpura. Dermatologica. 1953;106:86-95.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Aung PP, Burns SJ, Bhawan J. Lichen aureus: an unusual histopathological presentation: a case report and a review of literature. Am J Dermatopathol. 2014;36:E1-E4.
- Singh P, Schwartz RA. Disseminated intravascular coagulation: a devastating systemic disorder of special concern with COVID-19. Dermatol Ther. 2020;33:E14053.
- Almutairi N, Schwartz RA. COVID-19 with dermatologic manifestations and implications: an unfolding conundrum. Dermatol Ther. 2020;33:E13544.
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796.
- Torregrosa Calatayud JL, Garcías Ladaria J, De Unamuno Bustos B, et al. Retiform purpura caused by the use of cocaine, that was probably adulterated with levamisole. Ann Dermatol. 2015;27:117-119.
- Keim CK, Schwartz RA, Kapila R. Levamisole-induced and COVID-19-induced retiform purpura: two overlapping, emerging clinical syndromes. Arch Dermatol Res. 2021;22:1-9.
- González LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48:1157-1165.
- Yıldırım Bay E, Moustafa E, Semiz Y, et al. Leukocytoclastic vasculitis secondary to COVID-19 infection presenting with inclusion bodies: a histopathological correlation. J Cosmet Dermatol. 2022;21:27-29.
- Sundaresan S, Migden MR, Silapunt S. Stasis dermatitis: pathophysiology, evaluation, and management. Am J Clin Dermatol. 2017;18:383-390.
- Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part I. lower limb cellulitis. J Am Acad Dermatol. 2012;67:163.E1-E12; quiz 75-76.
- Keller EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleveland Clin J Med. 2012;79:547-552.
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816.
Pigmented purpuric dermatoses (PPDs) are characterized by petechiae, dusky macules representative of postinflammatory hyperpigmentation and dermal hemosiderin, and purpura generally localized to the lower extremities. They typically represent a spectrum of lymphocytic capillaritis, variable erythrocyte extravasation from papillary dermal blood vessels, and deposition of hemosiderin, yielding the classic red to orange to golden-brown findings on gross examination. Clinical overlap exists, but variants include Schamberg disease (SD), Majocchi purpura, Gougerot-Blum purpura, eczematoid purpura of Doucas and Kapetanakis (DK), and lichen aureus.1 Other forms are rarer, including linear, granulomatous, quadrantic, transitory, and familial variants. It remains controversial whether PPD may precede or have an association with cutaneous T-cell lymphoma.2 Dermoscopy usually shows copper-red pigmentation in the background, oval red dots, linear vessels, brown globules, and follicular openings. Although these findings may be useful in PPD diagnosis, they are not applicable in differentiating among the variants.
Pigmented purpuric dermatoses can easily be mistaken for stasis dermatitis or cellulitis, as these may occur concomitantly or in populations at risk for all 3 conditions, such as women older than 50 years with recent trauma or infection in the affected area. Tissue biopsy and clinical laboratory evaluation may be required to differentiate between PPD from leukocytoclastic vasculitis or the myriad causes of retiform purpura. Importantly, clinicians also should differentiate PPD from the purpuric eruptions of the lower extremities associated with COVID-19 infection.
Pigmented Purpuric Dermatoses
Schamberg Disease—In 1901, Jay Frank Schamberg, a distinguished professor of dermatology in Philadelphia, Pennsylvania, described “a peculiar progressive pigmentary disease of the skin” in a 15-year-old adolescent boy.3 Schamberg disease is the most common PPD, characterized by pruritic spots resembling cayenne pepper (Figure 1) with orange-brown pigmented macules on the legs and feet.4 Although platelet dysfunction, coagulation deficiencies, or dermal atrophy may contribute to hemorrhaging that manifests as petechiae or ecchymoses, SD typically is not associated with any laboratory abnormalities, and petechial eruption is not widespread.5 Capillary fragility can be assessed by the tourniquet test, in which pressure is applied to the forearm with a blood pressure cuff inflated between systolic and diastolic blood pressure for 5 to 10 minutes. Upon removing the cuff, a positive test is indicated by 15 or more petechiae in an area 5 cm in diameter due to poor platelet function. A positive result may be seen in SD.6
Histologically, SD is characterized by patchy parakeratosis, mild spongiosis of the stratum Malpighi, and lymphoid capillaritis (Figure 2).7 In addition to CD3+, CD4+, CD8+, CD1a+, and CD36+ lymphocytes, histology also may contain dendritic cells and cellular adhesion molecules (intercellular adhesion molecule 1, epithelial cell adhesion molecule 1) within the superficial perivascular infiltrate.8 There is no definitive therapy, but first-line interventions include emollients, topical steroids, and oral antihistamines. Nonpharmacologic management includes compression or support stockings, elevation of the lower extremities, and avoidance of offending medications (if identifiable).1
Majocchi Purpura—Domenico Majocchi was a renowned Italian dermatologist who described an entity in 1898 that he called purpura annularis telangiectodes, now also known as Majocchi purpura.9 It is more common in females, young adults, and children. Majocchi purpura has rarely been reported in families with a possible autosomal-dominant inheritance.10 Typically, bluish-red annular macules with central atrophy surrounded by hyperpigmentation may be seen on the lower extremities, potentially extending to the upper extremities.1 Treatment of Majocchi purpura remains a challenge but may respond to narrowband UVB phototherapy. Emollients and topical steroids also are used as first-line treatments. Biopsy demonstrates telangiectasia, pericapillary infiltration of mononuclear lymphocytes, and papillary dermal hemosiderin.11
Gougerot-Blum Purpura—In 1925, French dermatologists Henri Gougerot and Paul Blum described a pigmented purpuric lichenoid dermatitis known as Gougerot-Blum purpura,12 a rare PPD characterized by lichenoid papules that eventually coalesce into plaques of various colors, along with red-brown hyperpigmentation.4 As with other PPD variants, the legs are most involved, with rare extension to the trunk or thighs. The plaques may resemble and be mistaken for Kaposi sarcoma, cutaneous vasculitis, traumatic purpura, or mycosis fungoides. Dermoscopic examination reveals small, polygonal or round, red dots underlying brown scaly patches.13 Gougerot-Blum purpura is found more commonly in adult men and rarely affects children.4 Histologically, a lichenoid and superficial perivascular infiltrate composed of lymphocytes and macrophages is seen. Various therapies have been described, including topical steroids, antihistamines, psoralen plus UVA phototherapy, and cyclosporin A.14
Eczematoid Purpura of Doucas and Kapetanakis—In 1949, Greek dermatologists Christopher Doucas and John Kapetanakis observed several cases of purpuric dermatosis similar in form to the “pigmented purpuric lichenoid dermatitis” of Gougerot-Blum purpura12 and to the “progressive pigmentary dermatitis” of Schamberg disease.3 After observing a gradual disappearance of the classic yellow color from hemosiderin deposition, Doucas and Kapetanakis described a new bright red eruption with lichenification.15 Eczematoid purpura of Doucas and Kapetanakis is rare and predominantly seen in middle-aged males. Hyperpigmented or dark brown macules may develop bilaterally on the legs, progressing to the thighs and upper extremities. Unlike the other types of PPD, DK is extensive and severely pruritic.4
Although most PPD can be drug induced, DK has shown the greatest tendency for pruritic erythematous plaques following drug usage including but not limited to amlodipine, aspirin, acetaminophen, thiamine, interferon alfa, chlordiazepoxide, and isotretinoin. Additionally, DK has been associated with a contact allergy to clothing dyes and rubber.4 On histology, epidermal spongiosis may be seen, correlating with the eczematoid clinical findings. Spontaneous remission also is more common compared to the other PPDs. Treatment consists of topical corticosteroids and antihistamines.16
Lichen Aureus—Lichen aureus was first observed by the dermatologist R.H. Martin in 1958.17 It is clinically characterized by closely aggregated purpuric papules with a distinctive golden-brown color more often localized to the lower extremities and sometimes in a dermatomal distribution. Lichen aureus affects males and females equally, and similar to Majocchi purpura can be seen in children.4 Histopathologic examination reveals a prominent lichenoid plus superficial and deep perivascular lymphocytic infiltrate, extravasated erythrocytes, papillary dermal edema, hemosiderophages, and an unaffected epidermis. In rare cases, perineural infiltrates may be seen. Topical steroids usually are ineffective in lichen aureus treatment, but responses to psoralen plus UVA therapy also have been noted.17
Differential Diagnosis
COVID-19–Related Cutaneous Changes—Because COVID-19–related pathology is now a common differential diagnosis for many cutaneous eruptions, one must be mindful of the possibility for patients to have PPD, cutaneous changes from underlying COVID-19, or both.18 The microvascular changes from COVID-19 infection can be variable.19 Besides the presence of erythema along a distal digit, manifestations can include reticulated dusky erythema mimicking livedoid vasculopathy or inflammatory purpura.19
Retiform Purpura—Retiform purpura may occur in the setting of microvascular occlusion and can represent the pattern of underlying dermal vasculature. It is nonblanching and typically stellate or branching.20 The microvascular occlusion may be a result of hypercoagulability or may be secondary to cutaneous vasculitis, resulting in thrombosis and subsequent vascular occlusion.21 There are many reasons for hypercoagulability in retiform purpura, including disseminated intravascular coagulation in the setting of COVID-19 infection.22 The treatment of retiform purpura is aimed at alleviating the underlying cause and providing symptomatic relief. Conversely, the PPDs generally are benign and require minimal workup.
Leukocytoclastic Vasculitis—The hallmark of leukocytoclastic vasculitis is palpable purpura, often appearing as nonblanchable papules, typically in a dependent distribution such as the lower extremities (Figure 3). Although it primarily affects children, Henoch-Schönlein purpura is a type of leukocytoclastic vasculitis with lesions potentially similar in appearance to those of PPD.23 Palpable purpura may be painful and may ulcerate but rarely is pruritic. Leukocytoclastic vasculitis represents perivascular infiltrates composed of neutrophils, lymphocytes, and occasionally eosinophils, along with karyorrhexis, luminal fibrin, and fibrinoid degeneration of blood vessel walls, often resulting from immune complex deposition. Leukocytoclastic vasculitis may affect blood vessels of any size and requires further clinical and laboratory evaluation for infection (including COVID-19), hypercoagulability, autoimmune disease, or medication-related reactions.24
Stasis Dermatitis—Stasis dermatitis, a chronic inflammatory condition stemming from retrograde venous flow due to incompetent venous valves, mimics PPD. Stasis dermatitis initially appears as demarcated erythematous plaques, fissures, and scaling of the lower legs bilaterally, usually involving the medial malleolus.25 With time, the affected region develops overlying brawny hyperpigmentation and fibrosis (Figure 4). Pruritus or pain are common features, while fissures and superficial erosions may heal and recur, leading to lichenification.
Although both commonly appear on the lower extremities, duplex ultrasonography may be helpful to distinguish PPDs from stasis dermatitis since the latter occurs in the context of chronic venous insufficiency, varicose veins, soft tissue edema, and lymphedema.25 Additionally, pruritus, lichenification, and edema often are not seen in most PPD variants, although stasis dermatitis and PPD may occur in tandem. Conservative treatment involves elevation of the extremities, compression, and topical steroids for symptomatic relief.
Cellulitis—The key characteristics of cellulitis are redness, swelling, warmth, tenderness, fever, and leukocytosis. A history of trauma, such as a prior break in the skin, and pain in the affected area suggest cellulitis. Several skin conditions present similarly to cellulitis, including PPD, and thus approximately 30% of cases are misdiagnosed.26 Cellulitis rarely presents in a bilateral or diffusely scattered pattern as seen in PPDs. Rather, it is unilateral with smooth indistinct borders. Variables suggestive of cellulitis include immunosuppression, rapid progression, and previous occurrences. Hyperpigmented plaques or thickening of the skin are more indicative of a chronic process such as stasis dermatitis or lipodermatosclerosis rather than acute cellulitis. Purpura is not a typical finding in most cases of soft tissue cellulitis. Treatment may be case specific depending on severity, presence or absence of sepsis, findings on blood cultures, or other pathologic evaluation. Antibiotics are directed to the causative organism, typically Streptococcus and Staphylococcus species, although coverage against various gram-negative organisms may be indicated.27
Caution With Teledermatology
COVID-19 has established the value of telemedicine in providing access to health care services for at-risk or underserved individuals. The PPDs are benign, often asymptomatic, and potentially identifiable with teledermatology alone; however, they also can easily be mistaken for COVID-19–related eruptions, vasculitis, other types of purpura, stasis dermatitis, or other complications of lower extremity stasis and lymphedema, especially in an aging population. If tissue biopsy is required, as in the workup of vasculitis, the efficacy of telemedicine becomes more questionable. It is important to delineate the potentially confusing PPDs from other potentially dangerous or life-threatening inflammatory dermatoses.28
Pigmented purpuric dermatoses (PPDs) are characterized by petechiae, dusky macules representative of postinflammatory hyperpigmentation and dermal hemosiderin, and purpura generally localized to the lower extremities. They typically represent a spectrum of lymphocytic capillaritis, variable erythrocyte extravasation from papillary dermal blood vessels, and deposition of hemosiderin, yielding the classic red to orange to golden-brown findings on gross examination. Clinical overlap exists, but variants include Schamberg disease (SD), Majocchi purpura, Gougerot-Blum purpura, eczematoid purpura of Doucas and Kapetanakis (DK), and lichen aureus.1 Other forms are rarer, including linear, granulomatous, quadrantic, transitory, and familial variants. It remains controversial whether PPD may precede or have an association with cutaneous T-cell lymphoma.2 Dermoscopy usually shows copper-red pigmentation in the background, oval red dots, linear vessels, brown globules, and follicular openings. Although these findings may be useful in PPD diagnosis, they are not applicable in differentiating among the variants.
Pigmented purpuric dermatoses can easily be mistaken for stasis dermatitis or cellulitis, as these may occur concomitantly or in populations at risk for all 3 conditions, such as women older than 50 years with recent trauma or infection in the affected area. Tissue biopsy and clinical laboratory evaluation may be required to differentiate between PPD from leukocytoclastic vasculitis or the myriad causes of retiform purpura. Importantly, clinicians also should differentiate PPD from the purpuric eruptions of the lower extremities associated with COVID-19 infection.
Pigmented Purpuric Dermatoses
Schamberg Disease—In 1901, Jay Frank Schamberg, a distinguished professor of dermatology in Philadelphia, Pennsylvania, described “a peculiar progressive pigmentary disease of the skin” in a 15-year-old adolescent boy.3 Schamberg disease is the most common PPD, characterized by pruritic spots resembling cayenne pepper (Figure 1) with orange-brown pigmented macules on the legs and feet.4 Although platelet dysfunction, coagulation deficiencies, or dermal atrophy may contribute to hemorrhaging that manifests as petechiae or ecchymoses, SD typically is not associated with any laboratory abnormalities, and petechial eruption is not widespread.5 Capillary fragility can be assessed by the tourniquet test, in which pressure is applied to the forearm with a blood pressure cuff inflated between systolic and diastolic blood pressure for 5 to 10 minutes. Upon removing the cuff, a positive test is indicated by 15 or more petechiae in an area 5 cm in diameter due to poor platelet function. A positive result may be seen in SD.6
Histologically, SD is characterized by patchy parakeratosis, mild spongiosis of the stratum Malpighi, and lymphoid capillaritis (Figure 2).7 In addition to CD3+, CD4+, CD8+, CD1a+, and CD36+ lymphocytes, histology also may contain dendritic cells and cellular adhesion molecules (intercellular adhesion molecule 1, epithelial cell adhesion molecule 1) within the superficial perivascular infiltrate.8 There is no definitive therapy, but first-line interventions include emollients, topical steroids, and oral antihistamines. Nonpharmacologic management includes compression or support stockings, elevation of the lower extremities, and avoidance of offending medications (if identifiable).1
Majocchi Purpura—Domenico Majocchi was a renowned Italian dermatologist who described an entity in 1898 that he called purpura annularis telangiectodes, now also known as Majocchi purpura.9 It is more common in females, young adults, and children. Majocchi purpura has rarely been reported in families with a possible autosomal-dominant inheritance.10 Typically, bluish-red annular macules with central atrophy surrounded by hyperpigmentation may be seen on the lower extremities, potentially extending to the upper extremities.1 Treatment of Majocchi purpura remains a challenge but may respond to narrowband UVB phototherapy. Emollients and topical steroids also are used as first-line treatments. Biopsy demonstrates telangiectasia, pericapillary infiltration of mononuclear lymphocytes, and papillary dermal hemosiderin.11
Gougerot-Blum Purpura—In 1925, French dermatologists Henri Gougerot and Paul Blum described a pigmented purpuric lichenoid dermatitis known as Gougerot-Blum purpura,12 a rare PPD characterized by lichenoid papules that eventually coalesce into plaques of various colors, along with red-brown hyperpigmentation.4 As with other PPD variants, the legs are most involved, with rare extension to the trunk or thighs. The plaques may resemble and be mistaken for Kaposi sarcoma, cutaneous vasculitis, traumatic purpura, or mycosis fungoides. Dermoscopic examination reveals small, polygonal or round, red dots underlying brown scaly patches.13 Gougerot-Blum purpura is found more commonly in adult men and rarely affects children.4 Histologically, a lichenoid and superficial perivascular infiltrate composed of lymphocytes and macrophages is seen. Various therapies have been described, including topical steroids, antihistamines, psoralen plus UVA phototherapy, and cyclosporin A.14
Eczematoid Purpura of Doucas and Kapetanakis—In 1949, Greek dermatologists Christopher Doucas and John Kapetanakis observed several cases of purpuric dermatosis similar in form to the “pigmented purpuric lichenoid dermatitis” of Gougerot-Blum purpura12 and to the “progressive pigmentary dermatitis” of Schamberg disease.3 After observing a gradual disappearance of the classic yellow color from hemosiderin deposition, Doucas and Kapetanakis described a new bright red eruption with lichenification.15 Eczematoid purpura of Doucas and Kapetanakis is rare and predominantly seen in middle-aged males. Hyperpigmented or dark brown macules may develop bilaterally on the legs, progressing to the thighs and upper extremities. Unlike the other types of PPD, DK is extensive and severely pruritic.4
Although most PPD can be drug induced, DK has shown the greatest tendency for pruritic erythematous plaques following drug usage including but not limited to amlodipine, aspirin, acetaminophen, thiamine, interferon alfa, chlordiazepoxide, and isotretinoin. Additionally, DK has been associated with a contact allergy to clothing dyes and rubber.4 On histology, epidermal spongiosis may be seen, correlating with the eczematoid clinical findings. Spontaneous remission also is more common compared to the other PPDs. Treatment consists of topical corticosteroids and antihistamines.16
Lichen Aureus—Lichen aureus was first observed by the dermatologist R.H. Martin in 1958.17 It is clinically characterized by closely aggregated purpuric papules with a distinctive golden-brown color more often localized to the lower extremities and sometimes in a dermatomal distribution. Lichen aureus affects males and females equally, and similar to Majocchi purpura can be seen in children.4 Histopathologic examination reveals a prominent lichenoid plus superficial and deep perivascular lymphocytic infiltrate, extravasated erythrocytes, papillary dermal edema, hemosiderophages, and an unaffected epidermis. In rare cases, perineural infiltrates may be seen. Topical steroids usually are ineffective in lichen aureus treatment, but responses to psoralen plus UVA therapy also have been noted.17
Differential Diagnosis
COVID-19–Related Cutaneous Changes—Because COVID-19–related pathology is now a common differential diagnosis for many cutaneous eruptions, one must be mindful of the possibility for patients to have PPD, cutaneous changes from underlying COVID-19, or both.18 The microvascular changes from COVID-19 infection can be variable.19 Besides the presence of erythema along a distal digit, manifestations can include reticulated dusky erythema mimicking livedoid vasculopathy or inflammatory purpura.19
Retiform Purpura—Retiform purpura may occur in the setting of microvascular occlusion and can represent the pattern of underlying dermal vasculature. It is nonblanching and typically stellate or branching.20 The microvascular occlusion may be a result of hypercoagulability or may be secondary to cutaneous vasculitis, resulting in thrombosis and subsequent vascular occlusion.21 There are many reasons for hypercoagulability in retiform purpura, including disseminated intravascular coagulation in the setting of COVID-19 infection.22 The treatment of retiform purpura is aimed at alleviating the underlying cause and providing symptomatic relief. Conversely, the PPDs generally are benign and require minimal workup.
Leukocytoclastic Vasculitis—The hallmark of leukocytoclastic vasculitis is palpable purpura, often appearing as nonblanchable papules, typically in a dependent distribution such as the lower extremities (Figure 3). Although it primarily affects children, Henoch-Schönlein purpura is a type of leukocytoclastic vasculitis with lesions potentially similar in appearance to those of PPD.23 Palpable purpura may be painful and may ulcerate but rarely is pruritic. Leukocytoclastic vasculitis represents perivascular infiltrates composed of neutrophils, lymphocytes, and occasionally eosinophils, along with karyorrhexis, luminal fibrin, and fibrinoid degeneration of blood vessel walls, often resulting from immune complex deposition. Leukocytoclastic vasculitis may affect blood vessels of any size and requires further clinical and laboratory evaluation for infection (including COVID-19), hypercoagulability, autoimmune disease, or medication-related reactions.24
Stasis Dermatitis—Stasis dermatitis, a chronic inflammatory condition stemming from retrograde venous flow due to incompetent venous valves, mimics PPD. Stasis dermatitis initially appears as demarcated erythematous plaques, fissures, and scaling of the lower legs bilaterally, usually involving the medial malleolus.25 With time, the affected region develops overlying brawny hyperpigmentation and fibrosis (Figure 4). Pruritus or pain are common features, while fissures and superficial erosions may heal and recur, leading to lichenification.
Although both commonly appear on the lower extremities, duplex ultrasonography may be helpful to distinguish PPDs from stasis dermatitis since the latter occurs in the context of chronic venous insufficiency, varicose veins, soft tissue edema, and lymphedema.25 Additionally, pruritus, lichenification, and edema often are not seen in most PPD variants, although stasis dermatitis and PPD may occur in tandem. Conservative treatment involves elevation of the extremities, compression, and topical steroids for symptomatic relief.
Cellulitis—The key characteristics of cellulitis are redness, swelling, warmth, tenderness, fever, and leukocytosis. A history of trauma, such as a prior break in the skin, and pain in the affected area suggest cellulitis. Several skin conditions present similarly to cellulitis, including PPD, and thus approximately 30% of cases are misdiagnosed.26 Cellulitis rarely presents in a bilateral or diffusely scattered pattern as seen in PPDs. Rather, it is unilateral with smooth indistinct borders. Variables suggestive of cellulitis include immunosuppression, rapid progression, and previous occurrences. Hyperpigmented plaques or thickening of the skin are more indicative of a chronic process such as stasis dermatitis or lipodermatosclerosis rather than acute cellulitis. Purpura is not a typical finding in most cases of soft tissue cellulitis. Treatment may be case specific depending on severity, presence or absence of sepsis, findings on blood cultures, or other pathologic evaluation. Antibiotics are directed to the causative organism, typically Streptococcus and Staphylococcus species, although coverage against various gram-negative organisms may be indicated.27
Caution With Teledermatology
COVID-19 has established the value of telemedicine in providing access to health care services for at-risk or underserved individuals. The PPDs are benign, often asymptomatic, and potentially identifiable with teledermatology alone; however, they also can easily be mistaken for COVID-19–related eruptions, vasculitis, other types of purpura, stasis dermatitis, or other complications of lower extremity stasis and lymphedema, especially in an aging population. If tissue biopsy is required, as in the workup of vasculitis, the efficacy of telemedicine becomes more questionable. It is important to delineate the potentially confusing PPDs from other potentially dangerous or life-threatening inflammatory dermatoses.28
- Sardana K, Sarkar R , Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Çaytemel C, Baykut B, Ag˘ırgöl S¸, et al. Pigmented purpuric dermatosis: ten years of experience in a tertiary hospital and awareness of mycosis fungoides in differential diagnosis. J Cutan Pathol. 2021;48:611-616.
- Schamberg JF. A peculiar progressive pigmentary disease of the skin. Br J Dermatol. 1901;13:1-5.
- Martínez Pallás I, Conejero Del Mazo R, Lezcano Biosca V. Pigmented purpuric dermatosis: a review of the literature. Actas Dermosifiliogr (Engl Ed). 2020;111:196-204.
- Ozkaya DB, Emiroglu N, Su O, et al. Dermatoscopic findings of pigmented purpuric dermatosis. An Bras Dermatol. 2016;91:584-587.
- Lava SAG, Milani GP, Fossali EF, et al. Cutaneous manifestations of small-vessel leukocytoclastic vasculitides in childhood. Clin Rev Allergy Immunol. 2017;53:439-451.
- Bonnet U, Selle C, Isbruch K, et al. Recurrent purpura due to alcohol-related Schamberg’s disease and its association with serum immunoglobulins: a longitudinal observation of a heavy drinker. J Med Case Rep. 2016;10:301.
- Zaldivar Fujigaki JL, Anjum F. Schamberg Disease. StatPearls Publishing; 2021.
- Majocchi J. Purpura annularis telangiectodes. Arch Dermatol Syph. 1898;43:447.
- Sethuraman G, Sugandhan S, Bansal A, et al. Familial pigmented purpuric dermatoses. J Dermatol. 2006;33:639-641.
- Miller K, Fischer M, Kamino H, et al. Purpura annularis telangiectoides. Dermatol Online J. 2012;18:5.
- Coulombe J, Jean SE, Hatami A, et al. Pigmented purpuric dermatosis: clinicopathologic characterization in a pediatric series. Pediatr Dermatol. 2015;32:358-362.
- Park MY, Shim WH, Kim JM, et al. Dermoscopic finding in pigmented purpuric lichenoid dermatosis of Gougerot-Blum: a useful tool for clinical diagnosis. Ann Dermatol. 2018;30:245-247.
- Risikesan J, Sommerlund M, Ramsing M, et al. Successful topical treatment of pigmented purpuric lichenoid dermatitis of Gougerot-Blum in a young patient: a case report and summary of the most common pigmented purpuric dermatoses. Case Rep Dermatol. 2017;9:169-176.
- Doucas C, Kapetanakis J. Eczematid-like purpura. Dermatologica. 1953;106:86-95.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Aung PP, Burns SJ, Bhawan J. Lichen aureus: an unusual histopathological presentation: a case report and a review of literature. Am J Dermatopathol. 2014;36:E1-E4.
- Singh P, Schwartz RA. Disseminated intravascular coagulation: a devastating systemic disorder of special concern with COVID-19. Dermatol Ther. 2020;33:E14053.
- Almutairi N, Schwartz RA. COVID-19 with dermatologic manifestations and implications: an unfolding conundrum. Dermatol Ther. 2020;33:E13544.
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796.
- Torregrosa Calatayud JL, Garcías Ladaria J, De Unamuno Bustos B, et al. Retiform purpura caused by the use of cocaine, that was probably adulterated with levamisole. Ann Dermatol. 2015;27:117-119.
- Keim CK, Schwartz RA, Kapila R. Levamisole-induced and COVID-19-induced retiform purpura: two overlapping, emerging clinical syndromes. Arch Dermatol Res. 2021;22:1-9.
- González LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48:1157-1165.
- Yıldırım Bay E, Moustafa E, Semiz Y, et al. Leukocytoclastic vasculitis secondary to COVID-19 infection presenting with inclusion bodies: a histopathological correlation. J Cosmet Dermatol. 2022;21:27-29.
- Sundaresan S, Migden MR, Silapunt S. Stasis dermatitis: pathophysiology, evaluation, and management. Am J Clin Dermatol. 2017;18:383-390.
- Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part I. lower limb cellulitis. J Am Acad Dermatol. 2012;67:163.E1-E12; quiz 75-76.
- Keller EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleveland Clin J Med. 2012;79:547-552.
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816.
- Sardana K, Sarkar R , Sehgal VN. Pigmented purpuric dermatoses: an overview. Int J Dermatol. 2004;43:482-488.
- Çaytemel C, Baykut B, Ag˘ırgöl S¸, et al. Pigmented purpuric dermatosis: ten years of experience in a tertiary hospital and awareness of mycosis fungoides in differential diagnosis. J Cutan Pathol. 2021;48:611-616.
- Schamberg JF. A peculiar progressive pigmentary disease of the skin. Br J Dermatol. 1901;13:1-5.
- Martínez Pallás I, Conejero Del Mazo R, Lezcano Biosca V. Pigmented purpuric dermatosis: a review of the literature. Actas Dermosifiliogr (Engl Ed). 2020;111:196-204.
- Ozkaya DB, Emiroglu N, Su O, et al. Dermatoscopic findings of pigmented purpuric dermatosis. An Bras Dermatol. 2016;91:584-587.
- Lava SAG, Milani GP, Fossali EF, et al. Cutaneous manifestations of small-vessel leukocytoclastic vasculitides in childhood. Clin Rev Allergy Immunol. 2017;53:439-451.
- Bonnet U, Selle C, Isbruch K, et al. Recurrent purpura due to alcohol-related Schamberg’s disease and its association with serum immunoglobulins: a longitudinal observation of a heavy drinker. J Med Case Rep. 2016;10:301.
- Zaldivar Fujigaki JL, Anjum F. Schamberg Disease. StatPearls Publishing; 2021.
- Majocchi J. Purpura annularis telangiectodes. Arch Dermatol Syph. 1898;43:447.
- Sethuraman G, Sugandhan S, Bansal A, et al. Familial pigmented purpuric dermatoses. J Dermatol. 2006;33:639-641.
- Miller K, Fischer M, Kamino H, et al. Purpura annularis telangiectoides. Dermatol Online J. 2012;18:5.
- Coulombe J, Jean SE, Hatami A, et al. Pigmented purpuric dermatosis: clinicopathologic characterization in a pediatric series. Pediatr Dermatol. 2015;32:358-362.
- Park MY, Shim WH, Kim JM, et al. Dermoscopic finding in pigmented purpuric lichenoid dermatosis of Gougerot-Blum: a useful tool for clinical diagnosis. Ann Dermatol. 2018;30:245-247.
- Risikesan J, Sommerlund M, Ramsing M, et al. Successful topical treatment of pigmented purpuric lichenoid dermatitis of Gougerot-Blum in a young patient: a case report and summary of the most common pigmented purpuric dermatoses. Case Rep Dermatol. 2017;9:169-176.
- Doucas C, Kapetanakis J. Eczematid-like purpura. Dermatologica. 1953;106:86-95.
- Kim DH, Seo SH, Ahn HH, et al. Characteristics and clinical manifestations of pigmented purpuric dermatosis. Ann Dermatol. 2015;27:404-410.
- Aung PP, Burns SJ, Bhawan J. Lichen aureus: an unusual histopathological presentation: a case report and a review of literature. Am J Dermatopathol. 2014;36:E1-E4.
- Singh P, Schwartz RA. Disseminated intravascular coagulation: a devastating systemic disorder of special concern with COVID-19. Dermatol Ther. 2020;33:E14053.
- Almutairi N, Schwartz RA. COVID-19 with dermatologic manifestations and implications: an unfolding conundrum. Dermatol Ther. 2020;33:E13544.
- Georgesen C, Fox LP, Harp J. Retiform purpura: a diagnostic approach. J Am Acad Dermatol. 2020;82:783-796.
- Torregrosa Calatayud JL, Garcías Ladaria J, De Unamuno Bustos B, et al. Retiform purpura caused by the use of cocaine, that was probably adulterated with levamisole. Ann Dermatol. 2015;27:117-119.
- Keim CK, Schwartz RA, Kapila R. Levamisole-induced and COVID-19-induced retiform purpura: two overlapping, emerging clinical syndromes. Arch Dermatol Res. 2021;22:1-9.
- González LM, Janniger CK, Schwartz RA. Pediatric Henoch-Schönlein purpura. Int J Dermatol. 2009;48:1157-1165.
- Yıldırım Bay E, Moustafa E, Semiz Y, et al. Leukocytoclastic vasculitis secondary to COVID-19 infection presenting with inclusion bodies: a histopathological correlation. J Cosmet Dermatol. 2022;21:27-29.
- Sundaresan S, Migden MR, Silapunt S. Stasis dermatitis: pathophysiology, evaluation, and management. Am J Clin Dermatol. 2017;18:383-390.
- Hirschmann JV, Raugi GJ. Lower limb cellulitis and its mimics: part I. lower limb cellulitis. J Am Acad Dermatol. 2012;67:163.E1-E12; quiz 75-76.
- Keller EC, Tomecki KJ, Alraies MC. Distinguishing cellulitis from its mimics. Cleveland Clin J Med. 2012;79:547-552.
- Georgesen C, Fox LP, Harp J. Retiform purpura: workup and therapeutic considerations in select conditions. J Am Acad Dermatol. 2020;82:799-816.
Practice Points
- Dermatologists should be aware of the clinical presentations of pigmenting purpuric dermatoses (PPDs).
- Certain PPDs may resemble the thromboembolic events seen in COVID-19. Clinicians should especially be aware of how to differentiate these benign pigmentary disorders from other serious conditions.
- Teledermatology is widely utilized, but caution may be prudent when evaluating erythematous or purpuric dermatoses, especially those of the lower extremities.
- Pigmenting purpuric dermatoses generally are benign and do not require immediate treatment.

Penile Herpes Vegetans in a Patient With Well-controlled HIV
To the Editor:
Herpes vegetans (HV) is an uncommon infection caused by human herpesvirus (HHV) in patients who are immunocompromised, such as those who are HIV positive.1 Unlike typical HHV infection, HV can present with exophytic exudative ulcers and papillomatous vegetations. The presentation of ulcerated genital nodules, especially in an immunocompromised patient, yields an array of disorders in the differential diagnosis, including condyloma latum, condyloma acuminatum, pyogenic granuloma (PG), and verrucous carcinoma.2,3 Histopathology of HV reveals pseudoepitheliomatous hyperplasia, plasma cell infiltration, and positivity for HHV type 1 (HHV-1) and/or HHV type 2 (HHV-2). Herpes vegetans lesions typically require a multimodal treatment approach because many cases are resistant to acyclovir. Treatment options include the nucleoside analogues foscarnet and cidofovir; immunomodulators such as topical imiquimod; and the topical antiviral trifluridine.1,4-6 We describe a case of HV in a patient with a history of well-controlled HIV infection who presented with a painful fungating penile lesion.

A 55-year-old man presented to the hospital with a painful expanding mass on the distal aspect of the penis of 3 months’ duration. He had a history of HIV infection that was well-controlled by antiretroviral therapy, prior hepatitis B virus infection and acyclovir-resistant genital HHV-2 infection. Physical examination revealed a large, firm, circumferential, exophytic, verrucous plaque with various areas of ulceration and purulent drainage on the distal shaft and glans of the penis (Figure 1). The patient’s most recent absolute CD4 count was 425 cells/mm3 (reference range, 500–1500 cells/mm3). His HIV viral load was undetectable at less than 30 copies/mL. Histopathology with hematoxylin and eosin staining of biopsy material from the penile lesion demonstrated pseudoepitheliomatous epidermal hyperplasia with focal ulceration and a mixed inflammatory infiltrate (Figure 2A). At higher magnification, clear viral cytopathic changes of HHV were noted, including multinucleation, nuclear molding, and homogenous gray nuclei (Figure 2B). Additional staining for fungi, mycobacteria, and spirochetes was negative. In-situ hybridization was negative for human papillomavirus subtypes. A bacterial culture of swabs of the purulent drainage was positive for Staphylococcus aureus and Proteus mirabilis.
.")
Given the patient’s known history of acyclovir-resistant HHV-2 infection, he received a 28-day course of intravenous foscarnet 40 mg/kg every 12 hours. He also was given a 14-day course of intravenous ampicillin-sulbactam 3 g every 6 hours. The patient gradually improved during a 35-day hospital stay. He was discharged with cidofovir cream 1% and oral valacyclovir; the latter was subsequently discontinued by dermatology because of his known history of acyclovir resistance. Four months after discharge, the patient underwent a circumcision performed by urology to decrease the risk for recurrence and achieve the best cosmetic outcome. At the 6-month follow-up visit, dramatic clinical improvement was evident, with complete resolution of the plaque and only isolated areas of scarring (Figure 3). The patient reported that penile function was preserved.

Herpes vegetans represents a rare infection with HHV-1 or HHV-2, typically in patients who are considerably immunosuppressed, such as those with cancer, those undergoing transplantation, and those with uncontrolled HIV infection.1 Few cases of HV have been described in an immunocompetent patient.2 Our case is unique because the patient’s HIV infection was well controlled at the time HV was diagnosed, demonstrated by his modestly low CD4 count and undetectable HIV viral load.
Patients with HV can present diagnostic and therapeutic challenges. Typically, a diagnosis of cutaneous HHV infection does not require a biopsy; most cases appear as clustered vesicular lesions, making the disease easy to diagnose clinically. However, biopsies and cultures are necessary to identify the underlying cause of atypical verrucous exophytic lesions. Other conditions with clinical features similar to HV include squamous cell carcinoma, condyloma acuminatum, and deep fungal and mycobacterial infections.2,3 A tissue biopsy, histologic staining, and tissue culture should be performed to identify the causative pathogen and potential targets for treatment. Definitive diagnosis is vital to deliver proper treatment modalities, which often involve a multimodal multidisciplinary approach.
Several pathogenic mechanisms of HV have been proposed. One theory suggests that in an immunocompetent patient, HHV typically triggers a lymphocytic response, which leads to activation of interferon alpha. However, in an immunocompromised patient, such as an individual with AIDS, this interferon response is diminished, which explains why these patients typically have a chronic and resistant HHV infection. HIV has an affinity for infecting dermal dendritic cells, which signals activation of tumor necrosis factor and interleukin.6 Both cytokines contribute to an antiapoptotic environment that promotes continued proliferation of these viral cells in the epidermis. Over time, propagation of disinhibited cells can lead to the verrucous and hyperkeratotic-appearing skin that is common in patients with HV.7
Another theorized mechanism underlying hypertrophic herpetic lesions was described in the context of HHV-1 infection and subsequent PG. El Hayderi et al8 reported that histologic and immunohistochemical examination of a patient’s lesion revealed sparse epithelial cell aggregates within PG as well as HHV-1 antigens in the nuclei and cytoplasm of normal-appearing and cytopathic epithelial cells. Immunohistochemical examination also revealed vascular endothelial growth factor within HHV-1–infected epithelial cells and PG endothelial cells, suggesting that PG formation may be indirectly driven by vascular endothelial growth factor and its proangiogenic properties. The pathogenesis of PG in the setting of HHV-1 infection displays many similarities to hyperkeratotic lesions observed in atypical cutaneous manifestations of HHV-2.8
The management of patients with HV continues to be complex, often requiring a multimodal regimen. Although acyclovir has been shown to be highly effective for treating and preventing most HHV infections, acyclovir resistance frequently has been reported in immunocompromised populations.5 Acyclovir resistance can be correlated with the severity of immunodeficiency as well as the duration of acyclovir exposure. Resistance to acyclovir often results from deficient intracellular phosphorylation, which is required for activation of the drug. If patients show resistance to acyclovir and its derivatives, alternate drug classes that do not depend on thymidine kinase phosphorylation should be considered.
Our patient received a combination of intravenous foscarnet and a course of ampicillin-sulbactam while an inpatient due to his documented history of acyclovir-resistant HHV-2 infection, and he was discharged on cidofovir cream 1%. Cidofovir is US Food and Drug Administration approved for treating cytomegalovirus retinitis in patients with AIDS. Although data are limited, topical and intralesional cidofovir have been used to treat acyclovir-resistant cases of HV with documented success.1,9 In refractory HV or when the disease is slow to resolve, intralesional cidofovir has been documented to be an additional treatment option. Intralesional and topical cidofovir carry a much lower risk for adverse effects such as kidney dysfunction compared to intravenous cidofovir1 and can be considered in patients with minimal clinical improvement and those at increased risk for side effects.
Our case demonstrated how a patient with HV may require a complex and prolonged hospital course for appropriate treatment. Our patient required an array of both medical and surgical modalities to reach the desired outcome. Here, a multitude of specialties including infectious disease, dermatology, and urology worked together to reach a positive clinical and cosmetic outcome for this patient.
- Castelo-Soccio L, Bernardin R, Stern J, et al. Successful treatment of acyclovir-resistant herpes simplex virus with intralesional cidofovir. Arch Dermatol. 2010;146:124-126. doi:10.1001/archdermatol.2009.363
- Bae-Harboe Y-SC, Khachemoune A. Verrucous herpetic infection of the scrotum and the groin in an immuno-competent patient: case report and review of the literature. Dermatol Online J. 2012;18. https://doi.org/10.5070/D30sv058j6
- Elosiebo RI, Koubek VA, Patel TS, et al. Vegetative sacral plaque in a patient with human immunodeficiency virus. Cutis. 2015;96:E7-E9.
- Saling C, Slim J, Szabela ME. A case of an atypical resistant granulomatous HHV-1 and HHV-2 ulceration in an AIDS patient treated with intralesional cidofovir. SAGE Open Med Case Rep. 2019;7:2050313X19847029. doi:10.1177/2050313X19847029
- Martinez V, Molina J-M, Scieux C, et al. Topical imiquimod for recurrent acyclovir-resistant HHV infection. Am J Med. 2006 May;119:E9-E11. doi:10.1016/j.amjmed.2005.06.037
- Ronkainen SD, Rothenberger M. Herpes vegetans: an unusual and acyclovir-resistant form of HHV. J Gen Intern Med. 2018;33:393. doi:10.1007/s11606-017-4256-y
- Quesada AE, Galfione S, Colome M, et al. Verrucous herpes of the scrotum presenting clinically as verrucous squamous cell carcinoma: case report and review of the literature. Ann Clin Lab Sci. 2014;44:208-212.
- El Hayderi L, Paurobally D, Fassotte MF, et al. Herpes simplex virus type-I and pyogenic granuloma: a vascular endothelial growth factor-mediated association? Case Rep Dermatol. 2013;5:236-243. doi:10.1159/000354570
- Toro JR, Sanchez S, Turiansky G, et al. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol Clin. 2003;21:301-319. doi:10.1016/s0733-8635(02)00116-x
To the Editor:
Herpes vegetans (HV) is an uncommon infection caused by human herpesvirus (HHV) in patients who are immunocompromised, such as those who are HIV positive.1 Unlike typical HHV infection, HV can present with exophytic exudative ulcers and papillomatous vegetations. The presentation of ulcerated genital nodules, especially in an immunocompromised patient, yields an array of disorders in the differential diagnosis, including condyloma latum, condyloma acuminatum, pyogenic granuloma (PG), and verrucous carcinoma.2,3 Histopathology of HV reveals pseudoepitheliomatous hyperplasia, plasma cell infiltration, and positivity for HHV type 1 (HHV-1) and/or HHV type 2 (HHV-2). Herpes vegetans lesions typically require a multimodal treatment approach because many cases are resistant to acyclovir. Treatment options include the nucleoside analogues foscarnet and cidofovir; immunomodulators such as topical imiquimod; and the topical antiviral trifluridine.1,4-6 We describe a case of HV in a patient with a history of well-controlled HIV infection who presented with a painful fungating penile lesion.
A 55-year-old man presented to the hospital with a painful expanding mass on the distal aspect of the penis of 3 months’ duration. He had a history of HIV infection that was well-controlled by antiretroviral therapy, prior hepatitis B virus infection and acyclovir-resistant genital HHV-2 infection. Physical examination revealed a large, firm, circumferential, exophytic, verrucous plaque with various areas of ulceration and purulent drainage on the distal shaft and glans of the penis (Figure 1). The patient’s most recent absolute CD4 count was 425 cells/mm3 (reference range, 500–1500 cells/mm3). His HIV viral load was undetectable at less than 30 copies/mL. Histopathology with hematoxylin and eosin staining of biopsy material from the penile lesion demonstrated pseudoepitheliomatous epidermal hyperplasia with focal ulceration and a mixed inflammatory infiltrate (Figure 2A). At higher magnification, clear viral cytopathic changes of HHV were noted, including multinucleation, nuclear molding, and homogenous gray nuclei (Figure 2B). Additional staining for fungi, mycobacteria, and spirochetes was negative. In-situ hybridization was negative for human papillomavirus subtypes. A bacterial culture of swabs of the purulent drainage was positive for Staphylococcus aureus and Proteus mirabilis.
Given the patient’s known history of acyclovir-resistant HHV-2 infection, he received a 28-day course of intravenous foscarnet 40 mg/kg every 12 hours. He also was given a 14-day course of intravenous ampicillin-sulbactam 3 g every 6 hours. The patient gradually improved during a 35-day hospital stay. He was discharged with cidofovir cream 1% and oral valacyclovir; the latter was subsequently discontinued by dermatology because of his known history of acyclovir resistance. Four months after discharge, the patient underwent a circumcision performed by urology to decrease the risk for recurrence and achieve the best cosmetic outcome. At the 6-month follow-up visit, dramatic clinical improvement was evident, with complete resolution of the plaque and only isolated areas of scarring (Figure 3). The patient reported that penile function was preserved.
Herpes vegetans represents a rare infection with HHV-1 or HHV-2, typically in patients who are considerably immunosuppressed, such as those with cancer, those undergoing transplantation, and those with uncontrolled HIV infection.1 Few cases of HV have been described in an immunocompetent patient.2 Our case is unique because the patient’s HIV infection was well controlled at the time HV was diagnosed, demonstrated by his modestly low CD4 count and undetectable HIV viral load.
Patients with HV can present diagnostic and therapeutic challenges. Typically, a diagnosis of cutaneous HHV infection does not require a biopsy; most cases appear as clustered vesicular lesions, making the disease easy to diagnose clinically. However, biopsies and cultures are necessary to identify the underlying cause of atypical verrucous exophytic lesions. Other conditions with clinical features similar to HV include squamous cell carcinoma, condyloma acuminatum, and deep fungal and mycobacterial infections.2,3 A tissue biopsy, histologic staining, and tissue culture should be performed to identify the causative pathogen and potential targets for treatment. Definitive diagnosis is vital to deliver proper treatment modalities, which often involve a multimodal multidisciplinary approach.
Several pathogenic mechanisms of HV have been proposed. One theory suggests that in an immunocompetent patient, HHV typically triggers a lymphocytic response, which leads to activation of interferon alpha. However, in an immunocompromised patient, such as an individual with AIDS, this interferon response is diminished, which explains why these patients typically have a chronic and resistant HHV infection. HIV has an affinity for infecting dermal dendritic cells, which signals activation of tumor necrosis factor and interleukin.6 Both cytokines contribute to an antiapoptotic environment that promotes continued proliferation of these viral cells in the epidermis. Over time, propagation of disinhibited cells can lead to the verrucous and hyperkeratotic-appearing skin that is common in patients with HV.7
Another theorized mechanism underlying hypertrophic herpetic lesions was described in the context of HHV-1 infection and subsequent PG. El Hayderi et al8 reported that histologic and immunohistochemical examination of a patient’s lesion revealed sparse epithelial cell aggregates within PG as well as HHV-1 antigens in the nuclei and cytoplasm of normal-appearing and cytopathic epithelial cells. Immunohistochemical examination also revealed vascular endothelial growth factor within HHV-1–infected epithelial cells and PG endothelial cells, suggesting that PG formation may be indirectly driven by vascular endothelial growth factor and its proangiogenic properties. The pathogenesis of PG in the setting of HHV-1 infection displays many similarities to hyperkeratotic lesions observed in atypical cutaneous manifestations of HHV-2.8
The management of patients with HV continues to be complex, often requiring a multimodal regimen. Although acyclovir has been shown to be highly effective for treating and preventing most HHV infections, acyclovir resistance frequently has been reported in immunocompromised populations.5 Acyclovir resistance can be correlated with the severity of immunodeficiency as well as the duration of acyclovir exposure. Resistance to acyclovir often results from deficient intracellular phosphorylation, which is required for activation of the drug. If patients show resistance to acyclovir and its derivatives, alternate drug classes that do not depend on thymidine kinase phosphorylation should be considered.
Our patient received a combination of intravenous foscarnet and a course of ampicillin-sulbactam while an inpatient due to his documented history of acyclovir-resistant HHV-2 infection, and he was discharged on cidofovir cream 1%. Cidofovir is US Food and Drug Administration approved for treating cytomegalovirus retinitis in patients with AIDS. Although data are limited, topical and intralesional cidofovir have been used to treat acyclovir-resistant cases of HV with documented success.1,9 In refractory HV or when the disease is slow to resolve, intralesional cidofovir has been documented to be an additional treatment option. Intralesional and topical cidofovir carry a much lower risk for adverse effects such as kidney dysfunction compared to intravenous cidofovir1 and can be considered in patients with minimal clinical improvement and those at increased risk for side effects.
Our case demonstrated how a patient with HV may require a complex and prolonged hospital course for appropriate treatment. Our patient required an array of both medical and surgical modalities to reach the desired outcome. Here, a multitude of specialties including infectious disease, dermatology, and urology worked together to reach a positive clinical and cosmetic outcome for this patient.
To the Editor:
Herpes vegetans (HV) is an uncommon infection caused by human herpesvirus (HHV) in patients who are immunocompromised, such as those who are HIV positive.1 Unlike typical HHV infection, HV can present with exophytic exudative ulcers and papillomatous vegetations. The presentation of ulcerated genital nodules, especially in an immunocompromised patient, yields an array of disorders in the differential diagnosis, including condyloma latum, condyloma acuminatum, pyogenic granuloma (PG), and verrucous carcinoma.2,3 Histopathology of HV reveals pseudoepitheliomatous hyperplasia, plasma cell infiltration, and positivity for HHV type 1 (HHV-1) and/or HHV type 2 (HHV-2). Herpes vegetans lesions typically require a multimodal treatment approach because many cases are resistant to acyclovir. Treatment options include the nucleoside analogues foscarnet and cidofovir; immunomodulators such as topical imiquimod; and the topical antiviral trifluridine.1,4-6 We describe a case of HV in a patient with a history of well-controlled HIV infection who presented with a painful fungating penile lesion.
A 55-year-old man presented to the hospital with a painful expanding mass on the distal aspect of the penis of 3 months’ duration. He had a history of HIV infection that was well-controlled by antiretroviral therapy, prior hepatitis B virus infection and acyclovir-resistant genital HHV-2 infection. Physical examination revealed a large, firm, circumferential, exophytic, verrucous plaque with various areas of ulceration and purulent drainage on the distal shaft and glans of the penis (Figure 1). The patient’s most recent absolute CD4 count was 425 cells/mm3 (reference range, 500–1500 cells/mm3). His HIV viral load was undetectable at less than 30 copies/mL. Histopathology with hematoxylin and eosin staining of biopsy material from the penile lesion demonstrated pseudoepitheliomatous epidermal hyperplasia with focal ulceration and a mixed inflammatory infiltrate (Figure 2A). At higher magnification, clear viral cytopathic changes of HHV were noted, including multinucleation, nuclear molding, and homogenous gray nuclei (Figure 2B). Additional staining for fungi, mycobacteria, and spirochetes was negative. In-situ hybridization was negative for human papillomavirus subtypes. A bacterial culture of swabs of the purulent drainage was positive for Staphylococcus aureus and Proteus mirabilis.
Given the patient’s known history of acyclovir-resistant HHV-2 infection, he received a 28-day course of intravenous foscarnet 40 mg/kg every 12 hours. He also was given a 14-day course of intravenous ampicillin-sulbactam 3 g every 6 hours. The patient gradually improved during a 35-day hospital stay. He was discharged with cidofovir cream 1% and oral valacyclovir; the latter was subsequently discontinued by dermatology because of his known history of acyclovir resistance. Four months after discharge, the patient underwent a circumcision performed by urology to decrease the risk for recurrence and achieve the best cosmetic outcome. At the 6-month follow-up visit, dramatic clinical improvement was evident, with complete resolution of the plaque and only isolated areas of scarring (Figure 3). The patient reported that penile function was preserved.
Herpes vegetans represents a rare infection with HHV-1 or HHV-2, typically in patients who are considerably immunosuppressed, such as those with cancer, those undergoing transplantation, and those with uncontrolled HIV infection.1 Few cases of HV have been described in an immunocompetent patient.2 Our case is unique because the patient’s HIV infection was well controlled at the time HV was diagnosed, demonstrated by his modestly low CD4 count and undetectable HIV viral load.
Patients with HV can present diagnostic and therapeutic challenges. Typically, a diagnosis of cutaneous HHV infection does not require a biopsy; most cases appear as clustered vesicular lesions, making the disease easy to diagnose clinically. However, biopsies and cultures are necessary to identify the underlying cause of atypical verrucous exophytic lesions. Other conditions with clinical features similar to HV include squamous cell carcinoma, condyloma acuminatum, and deep fungal and mycobacterial infections.2,3 A tissue biopsy, histologic staining, and tissue culture should be performed to identify the causative pathogen and potential targets for treatment. Definitive diagnosis is vital to deliver proper treatment modalities, which often involve a multimodal multidisciplinary approach.
Several pathogenic mechanisms of HV have been proposed. One theory suggests that in an immunocompetent patient, HHV typically triggers a lymphocytic response, which leads to activation of interferon alpha. However, in an immunocompromised patient, such as an individual with AIDS, this interferon response is diminished, which explains why these patients typically have a chronic and resistant HHV infection. HIV has an affinity for infecting dermal dendritic cells, which signals activation of tumor necrosis factor and interleukin.6 Both cytokines contribute to an antiapoptotic environment that promotes continued proliferation of these viral cells in the epidermis. Over time, propagation of disinhibited cells can lead to the verrucous and hyperkeratotic-appearing skin that is common in patients with HV.7
Another theorized mechanism underlying hypertrophic herpetic lesions was described in the context of HHV-1 infection and subsequent PG. El Hayderi et al8 reported that histologic and immunohistochemical examination of a patient’s lesion revealed sparse epithelial cell aggregates within PG as well as HHV-1 antigens in the nuclei and cytoplasm of normal-appearing and cytopathic epithelial cells. Immunohistochemical examination also revealed vascular endothelial growth factor within HHV-1–infected epithelial cells and PG endothelial cells, suggesting that PG formation may be indirectly driven by vascular endothelial growth factor and its proangiogenic properties. The pathogenesis of PG in the setting of HHV-1 infection displays many similarities to hyperkeratotic lesions observed in atypical cutaneous manifestations of HHV-2.8
The management of patients with HV continues to be complex, often requiring a multimodal regimen. Although acyclovir has been shown to be highly effective for treating and preventing most HHV infections, acyclovir resistance frequently has been reported in immunocompromised populations.5 Acyclovir resistance can be correlated with the severity of immunodeficiency as well as the duration of acyclovir exposure. Resistance to acyclovir often results from deficient intracellular phosphorylation, which is required for activation of the drug. If patients show resistance to acyclovir and its derivatives, alternate drug classes that do not depend on thymidine kinase phosphorylation should be considered.
Our patient received a combination of intravenous foscarnet and a course of ampicillin-sulbactam while an inpatient due to his documented history of acyclovir-resistant HHV-2 infection, and he was discharged on cidofovir cream 1%. Cidofovir is US Food and Drug Administration approved for treating cytomegalovirus retinitis in patients with AIDS. Although data are limited, topical and intralesional cidofovir have been used to treat acyclovir-resistant cases of HV with documented success.1,9 In refractory HV or when the disease is slow to resolve, intralesional cidofovir has been documented to be an additional treatment option. Intralesional and topical cidofovir carry a much lower risk for adverse effects such as kidney dysfunction compared to intravenous cidofovir1 and can be considered in patients with minimal clinical improvement and those at increased risk for side effects.
Our case demonstrated how a patient with HV may require a complex and prolonged hospital course for appropriate treatment. Our patient required an array of both medical and surgical modalities to reach the desired outcome. Here, a multitude of specialties including infectious disease, dermatology, and urology worked together to reach a positive clinical and cosmetic outcome for this patient.
- Castelo-Soccio L, Bernardin R, Stern J, et al. Successful treatment of acyclovir-resistant herpes simplex virus with intralesional cidofovir. Arch Dermatol. 2010;146:124-126. doi:10.1001/archdermatol.2009.363
- Bae-Harboe Y-SC, Khachemoune A. Verrucous herpetic infection of the scrotum and the groin in an immuno-competent patient: case report and review of the literature. Dermatol Online J. 2012;18. https://doi.org/10.5070/D30sv058j6
- Elosiebo RI, Koubek VA, Patel TS, et al. Vegetative sacral plaque in a patient with human immunodeficiency virus. Cutis. 2015;96:E7-E9.
- Saling C, Slim J, Szabela ME. A case of an atypical resistant granulomatous HHV-1 and HHV-2 ulceration in an AIDS patient treated with intralesional cidofovir. SAGE Open Med Case Rep. 2019;7:2050313X19847029. doi:10.1177/2050313X19847029
- Martinez V, Molina J-M, Scieux C, et al. Topical imiquimod for recurrent acyclovir-resistant HHV infection. Am J Med. 2006 May;119:E9-E11. doi:10.1016/j.amjmed.2005.06.037
- Ronkainen SD, Rothenberger M. Herpes vegetans: an unusual and acyclovir-resistant form of HHV. J Gen Intern Med. 2018;33:393. doi:10.1007/s11606-017-4256-y
- Quesada AE, Galfione S, Colome M, et al. Verrucous herpes of the scrotum presenting clinically as verrucous squamous cell carcinoma: case report and review of the literature. Ann Clin Lab Sci. 2014;44:208-212.
- El Hayderi L, Paurobally D, Fassotte MF, et al. Herpes simplex virus type-I and pyogenic granuloma: a vascular endothelial growth factor-mediated association? Case Rep Dermatol. 2013;5:236-243. doi:10.1159/000354570
- Toro JR, Sanchez S, Turiansky G, et al. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol Clin. 2003;21:301-319. doi:10.1016/s0733-8635(02)00116-x
- Castelo-Soccio L, Bernardin R, Stern J, et al. Successful treatment of acyclovir-resistant herpes simplex virus with intralesional cidofovir. Arch Dermatol. 2010;146:124-126. doi:10.1001/archdermatol.2009.363
- Bae-Harboe Y-SC, Khachemoune A. Verrucous herpetic infection of the scrotum and the groin in an immuno-competent patient: case report and review of the literature. Dermatol Online J. 2012;18. https://doi.org/10.5070/D30sv058j6
- Elosiebo RI, Koubek VA, Patel TS, et al. Vegetative sacral plaque in a patient with human immunodeficiency virus. Cutis. 2015;96:E7-E9.
- Saling C, Slim J, Szabela ME. A case of an atypical resistant granulomatous HHV-1 and HHV-2 ulceration in an AIDS patient treated with intralesional cidofovir. SAGE Open Med Case Rep. 2019;7:2050313X19847029. doi:10.1177/2050313X19847029
- Martinez V, Molina J-M, Scieux C, et al. Topical imiquimod for recurrent acyclovir-resistant HHV infection. Am J Med. 2006 May;119:E9-E11. doi:10.1016/j.amjmed.2005.06.037
- Ronkainen SD, Rothenberger M. Herpes vegetans: an unusual and acyclovir-resistant form of HHV. J Gen Intern Med. 2018;33:393. doi:10.1007/s11606-017-4256-y
- Quesada AE, Galfione S, Colome M, et al. Verrucous herpes of the scrotum presenting clinically as verrucous squamous cell carcinoma: case report and review of the literature. Ann Clin Lab Sci. 2014;44:208-212.
- El Hayderi L, Paurobally D, Fassotte MF, et al. Herpes simplex virus type-I and pyogenic granuloma: a vascular endothelial growth factor-mediated association? Case Rep Dermatol. 2013;5:236-243. doi:10.1159/000354570
- Toro JR, Sanchez S, Turiansky G, et al. Topical cidofovir for the treatment of dermatologic conditions: verruca, condyloma, intraepithelial neoplasia, herpes simplex and its potential use in smallpox. Dermatol Clin. 2003;21:301-319. doi:10.1016/s0733-8635(02)00116-x
Practice Points
- Maintain a high clinical suspicion for herpes vegetans (HV) in a patient who has a history of immunosuppression and presents with exophytic genital lesions.
- A history of resistance to acyclovir requires a multimodal approach to treatment of HV lesions, including medical and surgical therapies.

Treatment of an Unresectable Cutaneous Squamous Cell Carcinoma With ED&C and 5-FU
To the Editor:
Most cutaneous squamous cell carcinomas (cSCCs) are successfully treated with standard modalities such as surgical excision; however, a subset of tumors is not amenable to surgical resection.1,2 Patients who are not able to undergo surgical treatment may instead receive radiation therapy, topical 5-fluorouracil (5-FU), imiquimod, cryosurgery, photodynamic therapy, or systemic treatment (eg, immunotherapy) in addition to intralesional approaches for localized disease.1-4 However, the adverse effects associated with these treatments and their modest effect in preventing the recurrence of cutaneous lesions limit their efficacy against unresectable cSCC.4-6 We present a case that demonstrates the efficacy of electrodesiccation and curettage (ED&C) followed by topical 5-FU for an invasive cSCC not amenable to surgical therapy.
A 58-year-old woman presented for evaluation of a 3.5×3.4-cm, incisional biopsy–proven, invasive stage T2a cSCC (Brigham and Women’s Hospital tumor staging system [Boston, Massachusetts]) on the dorsal aspect of the left foot, which had developed over several months (Figure 1A). She had a history of treatment with psoralen plus UV light therapy for erythroderma of unknown cause and peripheral neuropathy. She was not a surgical candidate because of suspected underlying cutaneous sclerosis and a history of poor wound healing on the lower legs.
 on the dorsal aspect of the left foot at presentation. Margins are inked and highlighted with a dashed circle")
Prior to presentation to dermatology, the patient had been treated with intralesional methotrexate, intralesional 5-FU, and the antiangiogenic and antiproliferative combination agent OLCAT-0053—consisting of equal parts [by volume] of diclofenac gel 3%, imiquimod cream 5%, hydrocortisone valerate cream 0.2%, calcipotriene cream 0.005%, and tretinoin cream 0.05—which failed, and the patient reported that OLCAT-005 made the pain from the cSCC worse.
Upon growth of the lesion over several months, the patient was referred to the High-Risk Skin Cancer Clinic at Massachusetts General Hospital (Boston, Massachusetts). A repeat biopsy demonstrated an invasive well-differentiated cSCC (Figure 2). The size and invasive features of the lesion on clinical examination prompted a referral to surgical oncology for a wide local excision. However, surgical oncology concluded she was not a surgical candidate.
(H&E, original magnification ×40).")
Magnetic resonance imaging showed no deep invasion of the cSCC to the tendons or bones. Electrodesiccation and curettage was performed to debulk the tumor, followed by twice-daily application of topical 5-FU for 4 weeks to improve the odds of tumor clearance (Figure 1B). Fourteen weeks after completion of 5-FU treatment, the cSCC showed complete clinical regression (Figure 1C). No recurrence has been detected clinically more than 3 years following treatment.
Prior to the advent of Mohs micrographic surgery, ED&C commonly was used to treat skin cancer, with a lower cost and a cure rate close to 95%.7,8 We postulate that the mechanism of tumor regression in our patient was ED&C-mediated removal and necrosis of neoplastic tissue combined with 5-FU–induced cancer-cell DNA damage and apoptosis. An antitumor immune response also may have contributed to the complete regression of the cSCC.
Although antiangiogenic and antiproliferative agents are suitable for primary cSCC treatment, it is possible that this patient’s prior therapies alone—in the absence of debulking by ED&C to sufficiently reduce disease burden—did not allow for tumor clearance and were ineffective. Many clinicians are reluctant to apply 5-FU to a wound bed because it can impede wound healing.9 In this case, re-epithelialization likely occurred primarily after completion of 5-FU treatment.
We recommend consideration of ED&C with 5-FU for similar malignant lesions that are not amenable to surgical excision. Nevertheless, Mohs micrographic surgery and wide local excision remain the gold standards for definitive treatment of invasive skin cancer in a patient who is a candidate for surgical treatment.
- Nehal KS, Bichakjian CK. Update on keratinocyte carcinomas. N Engl J Med. 2018;379:363-374. doi:10.1056/NEJMra1708701
- de Jong E, Lammerts MUPA, Genders RE, et al. Update of advanced cutaneous squamous cell carcinoma. J Eur Acad Dermatol Venereol. 2022;36(suppl 1):6-10. doi:10.1111/jdv.17728
- Li VW, Ball RA, Vasan N, et al. Antiangiogenic therapy for squamous cell carcinoma using combinatorial agents [abstract]. J Clin Oncol. 2005;23(16 suppl):3032. doi:10.1200/jco.2005.23.16_suppl.3032
- Lansbury L, Bath-Hextall F, Perkins W, et al. Interventions for non-metastatic squamous cell carcinoma of the skin: systematic review and pooled analysis of observational studies. BMJ. 2013;347:f6153. doi:10.1136/bmj.f6153
- Behshad R, Garcia‐Zuazaga J, Bordeaux J. Systemic treatment of locally advanced nonmetastatic cutaneous squamous cell carcinoma: a review of the literature. Br J Dermatol. 2011;165:1169-1177. doi:10.1111/j.1365-2133.2011.10524.x
- Rowe DE, Carroll RJ, Day CL Jr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. implications for treatment modality selection. J Am Acad Dermatol. 1992;26:976-990. doi:10.1016/0190-9622(92)70144-5
- Knox JM, Lyles TW, Shapiro EM, et al. Curettage and electrodesiccation in the treatment of skin cancer. Arch Dermatol. 1960;82:197-204.
- Chren M-M, Linos E, Torres JS, et al. Tumor recurrence 5 years after treatment of cutaneous basal cell carcinoma and squamous cell carcinoma. J Invest Dermatol. 2013;133:1188-1196. doi:10.1038/jid.2012.403
- Berman B, Maderal A, Raphael B. Keloids and hypertrophic scars: pathophysiology, classification, and treatment. Dermatologic Surgery. 2017;43:S3-S18.
To the Editor:
Most cutaneous squamous cell carcinomas (cSCCs) are successfully treated with standard modalities such as surgical excision; however, a subset of tumors is not amenable to surgical resection.1,2 Patients who are not able to undergo surgical treatment may instead receive radiation therapy, topical 5-fluorouracil (5-FU), imiquimod, cryosurgery, photodynamic therapy, or systemic treatment (eg, immunotherapy) in addition to intralesional approaches for localized disease.1-4 However, the adverse effects associated with these treatments and their modest effect in preventing the recurrence of cutaneous lesions limit their efficacy against unresectable cSCC.4-6 We present a case that demonstrates the efficacy of electrodesiccation and curettage (ED&C) followed by topical 5-FU for an invasive cSCC not amenable to surgical therapy.
A 58-year-old woman presented for evaluation of a 3.5×3.4-cm, incisional biopsy–proven, invasive stage T2a cSCC (Brigham and Women’s Hospital tumor staging system [Boston, Massachusetts]) on the dorsal aspect of the left foot, which had developed over several months (Figure 1A). She had a history of treatment with psoralen plus UV light therapy for erythroderma of unknown cause and peripheral neuropathy. She was not a surgical candidate because of suspected underlying cutaneous sclerosis and a history of poor wound healing on the lower legs.
Prior to presentation to dermatology, the patient had been treated with intralesional methotrexate, intralesional 5-FU, and the antiangiogenic and antiproliferative combination agent OLCAT-0053—consisting of equal parts [by volume] of diclofenac gel 3%, imiquimod cream 5%, hydrocortisone valerate cream 0.2%, calcipotriene cream 0.005%, and tretinoin cream 0.05—which failed, and the patient reported that OLCAT-005 made the pain from the cSCC worse.
Upon growth of the lesion over several months, the patient was referred to the High-Risk Skin Cancer Clinic at Massachusetts General Hospital (Boston, Massachusetts). A repeat biopsy demonstrated an invasive well-differentiated cSCC (Figure 2). The size and invasive features of the lesion on clinical examination prompted a referral to surgical oncology for a wide local excision. However, surgical oncology concluded she was not a surgical candidate.
Magnetic resonance imaging showed no deep invasion of the cSCC to the tendons or bones. Electrodesiccation and curettage was performed to debulk the tumor, followed by twice-daily application of topical 5-FU for 4 weeks to improve the odds of tumor clearance (Figure 1B). Fourteen weeks after completion of 5-FU treatment, the cSCC showed complete clinical regression (Figure 1C). No recurrence has been detected clinically more than 3 years following treatment.
Prior to the advent of Mohs micrographic surgery, ED&C commonly was used to treat skin cancer, with a lower cost and a cure rate close to 95%.7,8 We postulate that the mechanism of tumor regression in our patient was ED&C-mediated removal and necrosis of neoplastic tissue combined with 5-FU–induced cancer-cell DNA damage and apoptosis. An antitumor immune response also may have contributed to the complete regression of the cSCC.
Although antiangiogenic and antiproliferative agents are suitable for primary cSCC treatment, it is possible that this patient’s prior therapies alone—in the absence of debulking by ED&C to sufficiently reduce disease burden—did not allow for tumor clearance and were ineffective. Many clinicians are reluctant to apply 5-FU to a wound bed because it can impede wound healing.9 In this case, re-epithelialization likely occurred primarily after completion of 5-FU treatment.
We recommend consideration of ED&C with 5-FU for similar malignant lesions that are not amenable to surgical excision. Nevertheless, Mohs micrographic surgery and wide local excision remain the gold standards for definitive treatment of invasive skin cancer in a patient who is a candidate for surgical treatment.
To the Editor:
Most cutaneous squamous cell carcinomas (cSCCs) are successfully treated with standard modalities such as surgical excision; however, a subset of tumors is not amenable to surgical resection.1,2 Patients who are not able to undergo surgical treatment may instead receive radiation therapy, topical 5-fluorouracil (5-FU), imiquimod, cryosurgery, photodynamic therapy, or systemic treatment (eg, immunotherapy) in addition to intralesional approaches for localized disease.1-4 However, the adverse effects associated with these treatments and their modest effect in preventing the recurrence of cutaneous lesions limit their efficacy against unresectable cSCC.4-6 We present a case that demonstrates the efficacy of electrodesiccation and curettage (ED&C) followed by topical 5-FU for an invasive cSCC not amenable to surgical therapy.
A 58-year-old woman presented for evaluation of a 3.5×3.4-cm, incisional biopsy–proven, invasive stage T2a cSCC (Brigham and Women’s Hospital tumor staging system [Boston, Massachusetts]) on the dorsal aspect of the left foot, which had developed over several months (Figure 1A). She had a history of treatment with psoralen plus UV light therapy for erythroderma of unknown cause and peripheral neuropathy. She was not a surgical candidate because of suspected underlying cutaneous sclerosis and a history of poor wound healing on the lower legs.
Prior to presentation to dermatology, the patient had been treated with intralesional methotrexate, intralesional 5-FU, and the antiangiogenic and antiproliferative combination agent OLCAT-0053—consisting of equal parts [by volume] of diclofenac gel 3%, imiquimod cream 5%, hydrocortisone valerate cream 0.2%, calcipotriene cream 0.005%, and tretinoin cream 0.05—which failed, and the patient reported that OLCAT-005 made the pain from the cSCC worse.
Upon growth of the lesion over several months, the patient was referred to the High-Risk Skin Cancer Clinic at Massachusetts General Hospital (Boston, Massachusetts). A repeat biopsy demonstrated an invasive well-differentiated cSCC (Figure 2). The size and invasive features of the lesion on clinical examination prompted a referral to surgical oncology for a wide local excision. However, surgical oncology concluded she was not a surgical candidate.
Magnetic resonance imaging showed no deep invasion of the cSCC to the tendons or bones. Electrodesiccation and curettage was performed to debulk the tumor, followed by twice-daily application of topical 5-FU for 4 weeks to improve the odds of tumor clearance (Figure 1B). Fourteen weeks after completion of 5-FU treatment, the cSCC showed complete clinical regression (Figure 1C). No recurrence has been detected clinically more than 3 years following treatment.
Prior to the advent of Mohs micrographic surgery, ED&C commonly was used to treat skin cancer, with a lower cost and a cure rate close to 95%.7,8 We postulate that the mechanism of tumor regression in our patient was ED&C-mediated removal and necrosis of neoplastic tissue combined with 5-FU–induced cancer-cell DNA damage and apoptosis. An antitumor immune response also may have contributed to the complete regression of the cSCC.
Although antiangiogenic and antiproliferative agents are suitable for primary cSCC treatment, it is possible that this patient’s prior therapies alone—in the absence of debulking by ED&C to sufficiently reduce disease burden—did not allow for tumor clearance and were ineffective. Many clinicians are reluctant to apply 5-FU to a wound bed because it can impede wound healing.9 In this case, re-epithelialization likely occurred primarily after completion of 5-FU treatment.
We recommend consideration of ED&C with 5-FU for similar malignant lesions that are not amenable to surgical excision. Nevertheless, Mohs micrographic surgery and wide local excision remain the gold standards for definitive treatment of invasive skin cancer in a patient who is a candidate for surgical treatment.
- Nehal KS, Bichakjian CK. Update on keratinocyte carcinomas. N Engl J Med. 2018;379:363-374. doi:10.1056/NEJMra1708701
- de Jong E, Lammerts MUPA, Genders RE, et al. Update of advanced cutaneous squamous cell carcinoma. J Eur Acad Dermatol Venereol. 2022;36(suppl 1):6-10. doi:10.1111/jdv.17728
- Li VW, Ball RA, Vasan N, et al. Antiangiogenic therapy for squamous cell carcinoma using combinatorial agents [abstract]. J Clin Oncol. 2005;23(16 suppl):3032. doi:10.1200/jco.2005.23.16_suppl.3032
- Lansbury L, Bath-Hextall F, Perkins W, et al. Interventions for non-metastatic squamous cell carcinoma of the skin: systematic review and pooled analysis of observational studies. BMJ. 2013;347:f6153. doi:10.1136/bmj.f6153
- Behshad R, Garcia‐Zuazaga J, Bordeaux J. Systemic treatment of locally advanced nonmetastatic cutaneous squamous cell carcinoma: a review of the literature. Br J Dermatol. 2011;165:1169-1177. doi:10.1111/j.1365-2133.2011.10524.x
- Rowe DE, Carroll RJ, Day CL Jr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. implications for treatment modality selection. J Am Acad Dermatol. 1992;26:976-990. doi:10.1016/0190-9622(92)70144-5
- Knox JM, Lyles TW, Shapiro EM, et al. Curettage and electrodesiccation in the treatment of skin cancer. Arch Dermatol. 1960;82:197-204.
- Chren M-M, Linos E, Torres JS, et al. Tumor recurrence 5 years after treatment of cutaneous basal cell carcinoma and squamous cell carcinoma. J Invest Dermatol. 2013;133:1188-1196. doi:10.1038/jid.2012.403
- Berman B, Maderal A, Raphael B. Keloids and hypertrophic scars: pathophysiology, classification, and treatment. Dermatologic Surgery. 2017;43:S3-S18.
- Nehal KS, Bichakjian CK. Update on keratinocyte carcinomas. N Engl J Med. 2018;379:363-374. doi:10.1056/NEJMra1708701
- de Jong E, Lammerts MUPA, Genders RE, et al. Update of advanced cutaneous squamous cell carcinoma. J Eur Acad Dermatol Venereol. 2022;36(suppl 1):6-10. doi:10.1111/jdv.17728
- Li VW, Ball RA, Vasan N, et al. Antiangiogenic therapy for squamous cell carcinoma using combinatorial agents [abstract]. J Clin Oncol. 2005;23(16 suppl):3032. doi:10.1200/jco.2005.23.16_suppl.3032
- Lansbury L, Bath-Hextall F, Perkins W, et al. Interventions for non-metastatic squamous cell carcinoma of the skin: systematic review and pooled analysis of observational studies. BMJ. 2013;347:f6153. doi:10.1136/bmj.f6153
- Behshad R, Garcia‐Zuazaga J, Bordeaux J. Systemic treatment of locally advanced nonmetastatic cutaneous squamous cell carcinoma: a review of the literature. Br J Dermatol. 2011;165:1169-1177. doi:10.1111/j.1365-2133.2011.10524.x
- Rowe DE, Carroll RJ, Day CL Jr. Prognostic factors for local recurrence, metastasis, and survival rates in squamous cell carcinoma of the skin, ear, and lip. implications for treatment modality selection. J Am Acad Dermatol. 1992;26:976-990. doi:10.1016/0190-9622(92)70144-5
- Knox JM, Lyles TW, Shapiro EM, et al. Curettage and electrodesiccation in the treatment of skin cancer. Arch Dermatol. 1960;82:197-204.
- Chren M-M, Linos E, Torres JS, et al. Tumor recurrence 5 years after treatment of cutaneous basal cell carcinoma and squamous cell carcinoma. J Invest Dermatol. 2013;133:1188-1196. doi:10.1038/jid.2012.403
- Berman B, Maderal A, Raphael B. Keloids and hypertrophic scars: pathophysiology, classification, and treatment. Dermatologic Surgery. 2017;43:S3-S18.
Practice Points
- In a subset of cases of cutaneous squamous cell carcinoma (cSCC), the tumor is not amenable to surgical resection or other standard treatment modalities.
- Electrodesiccation and curettage followed by topical 5-fluorouracil may be an effective option in eliminating unresectable primary cSCCs that do not respond to intralesional treatment.
 on the dorsal aspect of the left foot at presentation. Margins are inked and highlighted with a dashed circle.")
Porcelain White, Crinkled, Violaceous Patches on the Inner Thighs
The Diagnosis: Extragenital Lichen Sclerosus et Atrophicus
A punch biopsy of the lesion revealed epidermal hyperkeratosis, atrophy, follicular plugs with basal vacuolar degeneration, and homogenous dense fibrosis in the papillary dermis with a dense lymphocytic infiltrate beneath the fibrosis (Figure 1). Dermoscopic examination was remarkable for a distinctive rainbow pattern. Clinical, histopathologic, and dermoscopic findings led to the diagnosis of extragenital lichen sclerosus et atrophicus (LSEA). A potent corticosteroid cream was prescribed twice daily for 2 months, after which the lesions completely resolved. At 2-year follow-up, a relapse was not observed (Figure 2).

Lichen sclerosus et atrophicus is an inflammatory dermatosis that clinically presents as atrophic or hypertrophic plaques that may show pigmentation changes with anogenital and extragenital involvement. It is common among females and predominantly occurs in prepubescent girls and postmenopausal women. The exact etiology is unclear; however, it is hypothesized to occur secondary to autoimmunity with an underlying genetic predisposition. Local trauma, hormonal influences, and infections are other suspected etiologic factors. Genital lesions often lead to itching, pain, and dyspareunia, whereas extragenital lesions predominantly are asymptomatic. When symptomatic, itching usually is the main concern. Unlike genital LSEA, extragenital lesions are not associated with squamous cell carcinoma development. Reported dermoscopic features of LSEA are white structureless areas with scaling, comedolike openings, follicular plugs, white shiny streaks, blue-gray peppering, pigment network, and red-purple globules.1 In our case, the dermoscopic finding of a rainbow pattern in LSEA is rare.2 Although the mechanism behind this appearance unclear, it can be the result of the birefringence effect—local variations in refractive index—influenced by the direction of structures within the dermis such as collagen. In this case, there was diffuse and dense homogenous fibrosis in the superficial dermis that corresponded to dermoscopic white polygonal clods.

Extragenital LSEA commonly is located on the neck, shoulders, wrists, and upper trunk and manifests clinically as whitish papules coalescing into scarlike plaques. Of all patients who have LSEA, 20% have extragenital lesions, and most of these lesions are seen in patients who also have genital LSEA. Approximately 6% of all LSEA patients have extragenital LSEA without genital involvement.3
For experienced dermatologists, clinical symptoms and lesion characteristics usually are sufficient for diagnosis; however, a differential diagnosis of atypical lesions and isolated extragenital presentations such as morphea, lichen simplex chronicus, lichen planus, and vitiligo requires the correlation of clinical findings with histopathology and dermoscopy. Morphea, known as localized scleroderma, is an idiopathic inflammatory skin disease with sclerotic changes. It manifests as inflammatory plaques that vary in color from red to purple. If there is moderate sclerosis in the center of this plaque, the color progressively fades to white, leaving a purplish ring around the edges. Dermoscopic features of morphea are reported as areas of erythema; red-focused vessels of linear, irregular, or dotted morphology; white fibrotic beams; and pigmentary structures.2 Lichen simplex chronicus is characterized by single or multiple dry and patchy skin lesions that are intensely pruritic. It commonly occurs on the neck, scalp, extremities, genital areas, and buttocks. Scratching the lesions leads to scarring, thickening of the skin, and increased frequency of itching. Histopathology of lichen simplex chronicus most frequently demonstrates a thickening of the epidermis and papillary dermis, irregularly elongated rete ridges, and fibroplasia with stellate or multinucleated fibroblasts completed by perivascular lymphocytic inflammation.4 Lichen planus presents with pruritic, polygonal, purple papules and/or plaques that can present in a variety of clinical forms, including atrophic and hypertrophic lichen planus.5 Lichen planus was an unlikely diagnosis for our patient due to the presence of patchy scarlike lesions and dermoscopic features that are well described in patients with LSEA. Lichen sclerosus et atrophicus presents with hypopigmented and/or hyperpigmented patches and plaques, distinguishing itself from vitiligo, which has flat lesions.
Topical steroids are the first-line therapeutic agents in the treatment of LSEA.6 Despite frequent use in this setting, common side effects such as localized scarring and atrophic degenerations have led to debate about their use. In our patient, the lesions resolved almost completely in 2 months, and no relapse was observed in the following 2 years. In the setting of topical steroid resistance, topical calcineurin inhibitors, UVA/UVB phototherapy, and topical tacrolimus can be used for treatment.6
The diagnosis of isolated extragenital LSEA may be a clinical challenge and generally requires further workup. When evaluating extragenital lesions, dermatologists should keep in mind extragenital LSEA as a differential diagnosis in the presence of a dermoscopic rainbow pattern arranged over white polygonal clods.
- Wang Y-K, Hao J-C, Liu J, et al. Dermoscopic features of morphea and extragenital lichen sclerosus in Chinese patients. Chin Med J (Engl). 2020;133:2109-2111.
- Errichetti E, Lallas A, Apalla Z, et al. Dermoscopy of morphea and cutaneous lichen sclerosus: clinicopathological correlation study and comparative analysis. Dermatology. 2017;233:462-470.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Kirtschig G, Becker K, Günthert A, et al. Evidence-based (S3) guideline on (anogenital) lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29:E1-E43.
The Diagnosis: Extragenital Lichen Sclerosus et Atrophicus
A punch biopsy of the lesion revealed epidermal hyperkeratosis, atrophy, follicular plugs with basal vacuolar degeneration, and homogenous dense fibrosis in the papillary dermis with a dense lymphocytic infiltrate beneath the fibrosis (Figure 1). Dermoscopic examination was remarkable for a distinctive rainbow pattern. Clinical, histopathologic, and dermoscopic findings led to the diagnosis of extragenital lichen sclerosus et atrophicus (LSEA). A potent corticosteroid cream was prescribed twice daily for 2 months, after which the lesions completely resolved. At 2-year follow-up, a relapse was not observed (Figure 2).
Lichen sclerosus et atrophicus is an inflammatory dermatosis that clinically presents as atrophic or hypertrophic plaques that may show pigmentation changes with anogenital and extragenital involvement. It is common among females and predominantly occurs in prepubescent girls and postmenopausal women. The exact etiology is unclear; however, it is hypothesized to occur secondary to autoimmunity with an underlying genetic predisposition. Local trauma, hormonal influences, and infections are other suspected etiologic factors. Genital lesions often lead to itching, pain, and dyspareunia, whereas extragenital lesions predominantly are asymptomatic. When symptomatic, itching usually is the main concern. Unlike genital LSEA, extragenital lesions are not associated with squamous cell carcinoma development. Reported dermoscopic features of LSEA are white structureless areas with scaling, comedolike openings, follicular plugs, white shiny streaks, blue-gray peppering, pigment network, and red-purple globules.1 In our case, the dermoscopic finding of a rainbow pattern in LSEA is rare.2 Although the mechanism behind this appearance unclear, it can be the result of the birefringence effect—local variations in refractive index—influenced by the direction of structures within the dermis such as collagen. In this case, there was diffuse and dense homogenous fibrosis in the superficial dermis that corresponded to dermoscopic white polygonal clods.
Extragenital LSEA commonly is located on the neck, shoulders, wrists, and upper trunk and manifests clinically as whitish papules coalescing into scarlike plaques. Of all patients who have LSEA, 20% have extragenital lesions, and most of these lesions are seen in patients who also have genital LSEA. Approximately 6% of all LSEA patients have extragenital LSEA without genital involvement.3
For experienced dermatologists, clinical symptoms and lesion characteristics usually are sufficient for diagnosis; however, a differential diagnosis of atypical lesions and isolated extragenital presentations such as morphea, lichen simplex chronicus, lichen planus, and vitiligo requires the correlation of clinical findings with histopathology and dermoscopy. Morphea, known as localized scleroderma, is an idiopathic inflammatory skin disease with sclerotic changes. It manifests as inflammatory plaques that vary in color from red to purple. If there is moderate sclerosis in the center of this plaque, the color progressively fades to white, leaving a purplish ring around the edges. Dermoscopic features of morphea are reported as areas of erythema; red-focused vessels of linear, irregular, or dotted morphology; white fibrotic beams; and pigmentary structures.2 Lichen simplex chronicus is characterized by single or multiple dry and patchy skin lesions that are intensely pruritic. It commonly occurs on the neck, scalp, extremities, genital areas, and buttocks. Scratching the lesions leads to scarring, thickening of the skin, and increased frequency of itching. Histopathology of lichen simplex chronicus most frequently demonstrates a thickening of the epidermis and papillary dermis, irregularly elongated rete ridges, and fibroplasia with stellate or multinucleated fibroblasts completed by perivascular lymphocytic inflammation.4 Lichen planus presents with pruritic, polygonal, purple papules and/or plaques that can present in a variety of clinical forms, including atrophic and hypertrophic lichen planus.5 Lichen planus was an unlikely diagnosis for our patient due to the presence of patchy scarlike lesions and dermoscopic features that are well described in patients with LSEA. Lichen sclerosus et atrophicus presents with hypopigmented and/or hyperpigmented patches and plaques, distinguishing itself from vitiligo, which has flat lesions.
Topical steroids are the first-line therapeutic agents in the treatment of LSEA.6 Despite frequent use in this setting, common side effects such as localized scarring and atrophic degenerations have led to debate about their use. In our patient, the lesions resolved almost completely in 2 months, and no relapse was observed in the following 2 years. In the setting of topical steroid resistance, topical calcineurin inhibitors, UVA/UVB phototherapy, and topical tacrolimus can be used for treatment.6
The diagnosis of isolated extragenital LSEA may be a clinical challenge and generally requires further workup. When evaluating extragenital lesions, dermatologists should keep in mind extragenital LSEA as a differential diagnosis in the presence of a dermoscopic rainbow pattern arranged over white polygonal clods.
The Diagnosis: Extragenital Lichen Sclerosus et Atrophicus
A punch biopsy of the lesion revealed epidermal hyperkeratosis, atrophy, follicular plugs with basal vacuolar degeneration, and homogenous dense fibrosis in the papillary dermis with a dense lymphocytic infiltrate beneath the fibrosis (Figure 1). Dermoscopic examination was remarkable for a distinctive rainbow pattern. Clinical, histopathologic, and dermoscopic findings led to the diagnosis of extragenital lichen sclerosus et atrophicus (LSEA). A potent corticosteroid cream was prescribed twice daily for 2 months, after which the lesions completely resolved. At 2-year follow-up, a relapse was not observed (Figure 2).
Lichen sclerosus et atrophicus is an inflammatory dermatosis that clinically presents as atrophic or hypertrophic plaques that may show pigmentation changes with anogenital and extragenital involvement. It is common among females and predominantly occurs in prepubescent girls and postmenopausal women. The exact etiology is unclear; however, it is hypothesized to occur secondary to autoimmunity with an underlying genetic predisposition. Local trauma, hormonal influences, and infections are other suspected etiologic factors. Genital lesions often lead to itching, pain, and dyspareunia, whereas extragenital lesions predominantly are asymptomatic. When symptomatic, itching usually is the main concern. Unlike genital LSEA, extragenital lesions are not associated with squamous cell carcinoma development. Reported dermoscopic features of LSEA are white structureless areas with scaling, comedolike openings, follicular plugs, white shiny streaks, blue-gray peppering, pigment network, and red-purple globules.1 In our case, the dermoscopic finding of a rainbow pattern in LSEA is rare.2 Although the mechanism behind this appearance unclear, it can be the result of the birefringence effect—local variations in refractive index—influenced by the direction of structures within the dermis such as collagen. In this case, there was diffuse and dense homogenous fibrosis in the superficial dermis that corresponded to dermoscopic white polygonal clods.
Extragenital LSEA commonly is located on the neck, shoulders, wrists, and upper trunk and manifests clinically as whitish papules coalescing into scarlike plaques. Of all patients who have LSEA, 20% have extragenital lesions, and most of these lesions are seen in patients who also have genital LSEA. Approximately 6% of all LSEA patients have extragenital LSEA without genital involvement.3
For experienced dermatologists, clinical symptoms and lesion characteristics usually are sufficient for diagnosis; however, a differential diagnosis of atypical lesions and isolated extragenital presentations such as morphea, lichen simplex chronicus, lichen planus, and vitiligo requires the correlation of clinical findings with histopathology and dermoscopy. Morphea, known as localized scleroderma, is an idiopathic inflammatory skin disease with sclerotic changes. It manifests as inflammatory plaques that vary in color from red to purple. If there is moderate sclerosis in the center of this plaque, the color progressively fades to white, leaving a purplish ring around the edges. Dermoscopic features of morphea are reported as areas of erythema; red-focused vessels of linear, irregular, or dotted morphology; white fibrotic beams; and pigmentary structures.2 Lichen simplex chronicus is characterized by single or multiple dry and patchy skin lesions that are intensely pruritic. It commonly occurs on the neck, scalp, extremities, genital areas, and buttocks. Scratching the lesions leads to scarring, thickening of the skin, and increased frequency of itching. Histopathology of lichen simplex chronicus most frequently demonstrates a thickening of the epidermis and papillary dermis, irregularly elongated rete ridges, and fibroplasia with stellate or multinucleated fibroblasts completed by perivascular lymphocytic inflammation.4 Lichen planus presents with pruritic, polygonal, purple papules and/or plaques that can present in a variety of clinical forms, including atrophic and hypertrophic lichen planus.5 Lichen planus was an unlikely diagnosis for our patient due to the presence of patchy scarlike lesions and dermoscopic features that are well described in patients with LSEA. Lichen sclerosus et atrophicus presents with hypopigmented and/or hyperpigmented patches and plaques, distinguishing itself from vitiligo, which has flat lesions.
Topical steroids are the first-line therapeutic agents in the treatment of LSEA.6 Despite frequent use in this setting, common side effects such as localized scarring and atrophic degenerations have led to debate about their use. In our patient, the lesions resolved almost completely in 2 months, and no relapse was observed in the following 2 years. In the setting of topical steroid resistance, topical calcineurin inhibitors, UVA/UVB phototherapy, and topical tacrolimus can be used for treatment.6
The diagnosis of isolated extragenital LSEA may be a clinical challenge and generally requires further workup. When evaluating extragenital lesions, dermatologists should keep in mind extragenital LSEA as a differential diagnosis in the presence of a dermoscopic rainbow pattern arranged over white polygonal clods.
- Wang Y-K, Hao J-C, Liu J, et al. Dermoscopic features of morphea and extragenital lichen sclerosus in Chinese patients. Chin Med J (Engl). 2020;133:2109-2111.
- Errichetti E, Lallas A, Apalla Z, et al. Dermoscopy of morphea and cutaneous lichen sclerosus: clinicopathological correlation study and comparative analysis. Dermatology. 2017;233:462-470.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Kirtschig G, Becker K, Günthert A, et al. Evidence-based (S3) guideline on (anogenital) lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29:E1-E43.
- Wang Y-K, Hao J-C, Liu J, et al. Dermoscopic features of morphea and extragenital lichen sclerosus in Chinese patients. Chin Med J (Engl). 2020;133:2109-2111.
- Errichetti E, Lallas A, Apalla Z, et al. Dermoscopy of morphea and cutaneous lichen sclerosus: clinicopathological correlation study and comparative analysis. Dermatology. 2017;233:462-470.
- Wallace HJ. Lichen sclerosus et atrophicus. Trans St Johns Hosp Dermatol Soc. 1971;57:9-30.
- Balan R, Grigoras¸ A, Popovici D, et al. The histopathological landscape of the major psoriasiform dermatoses. Arch Clin Cases. 2021;6:59-68.
- Weston G, Payette M. Update on lichen planus and its clinical variants. Int J Womens Dermatol. 2015;1:140-149.
- Kirtschig G, Becker K, Günthert A, et al. Evidence-based (S3) guideline on (anogenital) lichen sclerosus. J Eur Acad Dermatol Venereol. 2015;29:E1-E43.
A 50-year-old woman presented with multiple pruritic lesions on the right inner thigh of 2 years’ duration. Physical examination revealed porcelain white, crinkled, violaceous patches extending from the right inner thigh to the inguinal fold (top). Dermoscopic examination revealed follicular plugs, white structureless areas, white lines, and a rainbow pattern arranged over white polygonal clods on polarized mode (bottom).


Porocarcinoma Development in a Prior Trauma Site
To the Editor:
Porocarcinoma, or malignant poroma, is a rare adnexal malignancy of a predominantly glandular origin that comprises less than 0.01% of all cutaneous neoplasms.1,2 Although exposure to UV radiation and immunosuppression have been implicated in the malignant degeneration of benign poromas into porocarcinomas, at least half of all malignant variants will arise de novo.3,4 Patients present with an evolving nodule or plaque and often are in their seventh or eighth decade of life at the time of diagnosis.2 Localized trauma from burns or radiation exposure has been causatively linked to de novo porocarcinoma formation.2,5 These suppressive and traumatic stimuli drive increased genetic heterogeneity along with characteristic gene mutations in known tumor suppressor genes.6
A 62-year-old man presented with a nonhealing wound on the right hand of 5 years’ duration that had previously been attributed to a penetrating injury with a piece of copper from a refrigerant coolant system. The wound initially blistered and then eventually callused and developed areas of ulceration. The patient consulted multiple physicians for treatment of the intensely pruritic and ulcerated lesion. He received prescriptions for cephalexin, trimethoprim-sulfamethoxazole, doxycycline, clindamycin, and clobetasol cream, all of which offered minimal improvement. Home therapies including vitamin E and tea tree oil yielded no benefit. The lesion roughly quadrupled in size over the last 5 years.

Physical examination revealed a 7.5×4.2-cm ulcerated plaque with ragged borders and abundant central neoepithelialization on the right palmar surface (Figure 1). No gross motor or sensory defects were identified. There was no epitrochlear, axillary, cervical, or supraclavicular lymphadenopathy. A shave biopsy of the plaque’s edge was performed, which demonstrated a hyperplastic epidermis comprising atypical poroid cells with frequent mitoses, scant necrosis, and regular ductal structures confined to the epidermis (Figure 2). Immunohistochemical profiling results were positive for anticytokeratin (CAM 5.2) and Ber-EP4 (Figure 3). When evaluated in aggregate, these findings were consistent with porocarcinoma in situ.
.")
The patient was referred to a surgical oncologist for evaluation. At that time, an exophytic mass had developed in the central lesion. Although no lymphadenopathy was identified upon examination, the patient had developed tremoring and a contracture deformity of the right hand. Extensive imaging and urgent surgical resection were recommended, but the patient did not wish to pursue these options, opting instead to continue home remedies. At a 15-month follow-up via telephone, the patient reported that the home therapy had failed and he had moved back to Vietnam. Partial limb amputation had been recommended by a local provider. Unfortunately, the patient was subsequently lost to follow-up, and his current status is unknown.
.")
Porocarcinomas are rare tumors, comprising just 0.005% to 0.01% of all cutaneous epithelial tumors.1,2,5 They affect men and women equally, with an average age at diagnosis of 60 to 70 years.1,2 At least half of all porocarcinomas develop de novo, while 18% to 50% arise from the degeneration of an existing poroma.2,3 Exposure to UV light and immunosuppression, particularly following organ transplantation, represent 2 commonly suspected catalysts for this malignant transformation.4 De novo porocarcinomas are most causatively linked to localized trauma from burns or radiation exposure.5 Gene mutations in classic tumor suppressor genes—tumor protein p53 (TP53), phosphatase and tensin homolog (PTEN), rearranged during transfection (RET), adenomatous polyposis coli (APC)—and increased genetic heterogeneity follow these stimuli.6
The morphologic presentation of porocarcinoma is highly variable and may manifest as papules, nodules, or plaques in various states of erosion, ulceration, or excoriation. Diagnoses of basal and squamous cell carcinoma, primary adnexal tumors, seborrheic keratosis, pyogenic granuloma, and melanoma must all be considered and methodically ruled out.7 Porocarcinomas may arise nearly anywhere on the body, with a particular predilection for the lower extremities (35%), head/neck (24%), and upper extremities (14%).3,4 Primary lesions arising from the extremities, genitalia, or buttocks herald a higher risk for lymphatic invasion and distant metastasis, while head and neck tumors more commonly remain localized.8 Bleeding, ulceration, or rapid expansion of a preexisting poroma is suggestive of malignant transformation and may portend a more aggressive disease pattern.2,9
Unequivocal diagnosis relies on histological and immunohistochemical studies due to the marked clinical variance of this neoplasm.7 An irregular histologic pattern of poromatous basaloid cells with ductal differentiation and cytologic atypia commonly are seen with porocarcinomas.2,8 Nuclear pleomorphism with cellular necrosis, increased mitotic figures, and abortive ductal formation with a distinct lack of retraction around cellular aggregates often are found. Immunohistochemical staining is needed to confirm the primary tumor diagnosis. Histochemical stains commonly employed include carcinoembryonic antigen (CEA), cytokeratin AE1/AE3, epithelial membrane antigen, p53, p63, Ki67, and periodic acid-Schiff.10 The use of BerEP4 has been reported as efficacious in highlighting sweat structures, which can be particularly useful in cases when basal cell carcinoma is not in the histologic differential.11 These staining profiles afford confirmation of ductal differentiation with CEA, epithelial membrane antigen, and BerEP4, while p63 and Ki67 are used as surrogates for primary cutaneous neoplasia and cell proliferation, respectively.5,11 Porocarcinoma lesions may be most sensitive to CEA and most specific to CK19 (a component of cytokeratin AE1/AE3), though these findings have not been widely reproduced.7
The treatment and prognosis of porocarcinoma vary widely. Surgically excised lesions recur in roughly 20% of cases, though these rates likely include tumors that were incompletely resected in the primary attempt. Although wide local excision with an average 1-cm margin remains the most employed removal technique, Mohs micrographic surgery may more effectively limit recurrence and metastasis of localized disease.7,8,12 Metastatic disease foretells a mortality rate of at least 65%, which is problematic in that 10% to 20% of patients have metastatic disease at the time of diagnosis and another 20% will show metastasis following primary tumor excision.8,10 Neoplasms with high mitotic rates and depths greater than 7 mm should prompt thorough diagnostic imaging, such as positron emission tomography or magnetic resonance imaging. A sentinel lymph node biopsy should be strongly considered and discussed with the patient.10 Treatment options for nodal and distant metastases include a combination of localized surgery, lymphadenectomy, radiotherapy, and chemotherapeutic agents.2,4,5 The response to systemic treatment and radiotherapy often is quite poor, though the use of combinations of docetaxel, paclitaxel, cetuximab, and immunotherapy have been efficacious in smaller studies.8,10 The highest rates of morbidity and mortality are seen in patients with metastases on presentation or with localized tumors in the groin and buttocks.8
The diagnosis of porocarcinoma may be elusive due to its relatively rare occurrence. Therefore, it is critical to consider this neoplasm in high-risk sites in older patients who present with an evolving nodule or tumor on an extremity. Routine histology and astute histochemical profiling are necessary to exclude diseases that mimic porocarcinoma. Once diagnosis is confirmed, management with prompt excision and diagnostic imaging is recommended, including a lymph node biopsy if appropriate. Due to its high metastatic potential and associated morbidity and mortality, patients with porocarcinoma should be followed closely by a multidisciplinary care team.
- Belin E, Ezzedine K, Stanislas S, et al. Factors in the surgical management of primary eccrine porocarcinoma: prognostic histological factors can guide the surgical procedure. Br J Dermatol. 2011;165:985-989.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Spencer DM, Bigler LR, Hearne DW, et al. Pedal papule. eccrine porocarcinoma (EPC) in association with poroma. Arch Dermatol. 1995;131:211, 214.
- Salih AM, Kakamad FH, Essa RA, et al. Porocarcinoma: a systematic review of literature with a single case report. Int J Surg Case Rep. 2017;30:13-16.
- Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Mosby Elsevier; 2018.
- Bosic M, Kirchner M, Brasanac D, et al. Targeted molecular profiling reveals genetic heterogeneity of poromas and porocarcinomas. Pathology. 2018;50:327-332.
- Mahalingam M, Richards JE, Selim MA, et al. An immunohistochemical comparison of cytokeratin 7, cytokeratin 15, cytokeratin 19, CAM 5.2, carcinoembryonic antigen, and nestin in differentiating porocarcinoma from squamous cell carcinoma. Hum Pathol. 2012;43:1265-1272.
- Nazemi A, Higgins S, Swift R, et al. Eccrine porocarcinoma: new insights and a systematic review of the literature. Dermatol Surg. 2018;44:1247-1261.
- Wen SY. Case report of eccrine porocarcinoma in situ associated with eccrine poroma on the forehead. J Dermatol. 2012;39:649-651.
- Gerber PA, Schulte KW, Ruzicka T, et al. Eccrine porocarcinoma of the head: an important differential diagnosis in the elderly patient. Dermatology. 2008;216:229-233.
- Afshar M, Deroide F, Robson A. BerEP4 is widely expressed in tumors of the sweat apparatus: a source of potential diagnostic error. J Cutan Pathol. 2013;40:259-264.
- Tolkachjov SN, Hocker TL, Camilleri MJ, et al. Treatment of porocarcinoma with Mohs micrographic surgery: the Mayo clinic experience. Dermatol Surg. 2016;42:745-750.
To the Editor:
Porocarcinoma, or malignant poroma, is a rare adnexal malignancy of a predominantly glandular origin that comprises less than 0.01% of all cutaneous neoplasms.1,2 Although exposure to UV radiation and immunosuppression have been implicated in the malignant degeneration of benign poromas into porocarcinomas, at least half of all malignant variants will arise de novo.3,4 Patients present with an evolving nodule or plaque and often are in their seventh or eighth decade of life at the time of diagnosis.2 Localized trauma from burns or radiation exposure has been causatively linked to de novo porocarcinoma formation.2,5 These suppressive and traumatic stimuli drive increased genetic heterogeneity along with characteristic gene mutations in known tumor suppressor genes.6
A 62-year-old man presented with a nonhealing wound on the right hand of 5 years’ duration that had previously been attributed to a penetrating injury with a piece of copper from a refrigerant coolant system. The wound initially blistered and then eventually callused and developed areas of ulceration. The patient consulted multiple physicians for treatment of the intensely pruritic and ulcerated lesion. He received prescriptions for cephalexin, trimethoprim-sulfamethoxazole, doxycycline, clindamycin, and clobetasol cream, all of which offered minimal improvement. Home therapies including vitamin E and tea tree oil yielded no benefit. The lesion roughly quadrupled in size over the last 5 years.
Physical examination revealed a 7.5×4.2-cm ulcerated plaque with ragged borders and abundant central neoepithelialization on the right palmar surface (Figure 1). No gross motor or sensory defects were identified. There was no epitrochlear, axillary, cervical, or supraclavicular lymphadenopathy. A shave biopsy of the plaque’s edge was performed, which demonstrated a hyperplastic epidermis comprising atypical poroid cells with frequent mitoses, scant necrosis, and regular ductal structures confined to the epidermis (Figure 2). Immunohistochemical profiling results were positive for anticytokeratin (CAM 5.2) and Ber-EP4 (Figure 3). When evaluated in aggregate, these findings were consistent with porocarcinoma in situ.
The patient was referred to a surgical oncologist for evaluation. At that time, an exophytic mass had developed in the central lesion. Although no lymphadenopathy was identified upon examination, the patient had developed tremoring and a contracture deformity of the right hand. Extensive imaging and urgent surgical resection were recommended, but the patient did not wish to pursue these options, opting instead to continue home remedies. At a 15-month follow-up via telephone, the patient reported that the home therapy had failed and he had moved back to Vietnam. Partial limb amputation had been recommended by a local provider. Unfortunately, the patient was subsequently lost to follow-up, and his current status is unknown.
Porocarcinomas are rare tumors, comprising just 0.005% to 0.01% of all cutaneous epithelial tumors.1,2,5 They affect men and women equally, with an average age at diagnosis of 60 to 70 years.1,2 At least half of all porocarcinomas develop de novo, while 18% to 50% arise from the degeneration of an existing poroma.2,3 Exposure to UV light and immunosuppression, particularly following organ transplantation, represent 2 commonly suspected catalysts for this malignant transformation.4 De novo porocarcinomas are most causatively linked to localized trauma from burns or radiation exposure.5 Gene mutations in classic tumor suppressor genes—tumor protein p53 (TP53), phosphatase and tensin homolog (PTEN), rearranged during transfection (RET), adenomatous polyposis coli (APC)—and increased genetic heterogeneity follow these stimuli.6
The morphologic presentation of porocarcinoma is highly variable and may manifest as papules, nodules, or plaques in various states of erosion, ulceration, or excoriation. Diagnoses of basal and squamous cell carcinoma, primary adnexal tumors, seborrheic keratosis, pyogenic granuloma, and melanoma must all be considered and methodically ruled out.7 Porocarcinomas may arise nearly anywhere on the body, with a particular predilection for the lower extremities (35%), head/neck (24%), and upper extremities (14%).3,4 Primary lesions arising from the extremities, genitalia, or buttocks herald a higher risk for lymphatic invasion and distant metastasis, while head and neck tumors more commonly remain localized.8 Bleeding, ulceration, or rapid expansion of a preexisting poroma is suggestive of malignant transformation and may portend a more aggressive disease pattern.2,9
Unequivocal diagnosis relies on histological and immunohistochemical studies due to the marked clinical variance of this neoplasm.7 An irregular histologic pattern of poromatous basaloid cells with ductal differentiation and cytologic atypia commonly are seen with porocarcinomas.2,8 Nuclear pleomorphism with cellular necrosis, increased mitotic figures, and abortive ductal formation with a distinct lack of retraction around cellular aggregates often are found. Immunohistochemical staining is needed to confirm the primary tumor diagnosis. Histochemical stains commonly employed include carcinoembryonic antigen (CEA), cytokeratin AE1/AE3, epithelial membrane antigen, p53, p63, Ki67, and periodic acid-Schiff.10 The use of BerEP4 has been reported as efficacious in highlighting sweat structures, which can be particularly useful in cases when basal cell carcinoma is not in the histologic differential.11 These staining profiles afford confirmation of ductal differentiation with CEA, epithelial membrane antigen, and BerEP4, while p63 and Ki67 are used as surrogates for primary cutaneous neoplasia and cell proliferation, respectively.5,11 Porocarcinoma lesions may be most sensitive to CEA and most specific to CK19 (a component of cytokeratin AE1/AE3), though these findings have not been widely reproduced.7
The treatment and prognosis of porocarcinoma vary widely. Surgically excised lesions recur in roughly 20% of cases, though these rates likely include tumors that were incompletely resected in the primary attempt. Although wide local excision with an average 1-cm margin remains the most employed removal technique, Mohs micrographic surgery may more effectively limit recurrence and metastasis of localized disease.7,8,12 Metastatic disease foretells a mortality rate of at least 65%, which is problematic in that 10% to 20% of patients have metastatic disease at the time of diagnosis and another 20% will show metastasis following primary tumor excision.8,10 Neoplasms with high mitotic rates and depths greater than 7 mm should prompt thorough diagnostic imaging, such as positron emission tomography or magnetic resonance imaging. A sentinel lymph node biopsy should be strongly considered and discussed with the patient.10 Treatment options for nodal and distant metastases include a combination of localized surgery, lymphadenectomy, radiotherapy, and chemotherapeutic agents.2,4,5 The response to systemic treatment and radiotherapy often is quite poor, though the use of combinations of docetaxel, paclitaxel, cetuximab, and immunotherapy have been efficacious in smaller studies.8,10 The highest rates of morbidity and mortality are seen in patients with metastases on presentation or with localized tumors in the groin and buttocks.8
The diagnosis of porocarcinoma may be elusive due to its relatively rare occurrence. Therefore, it is critical to consider this neoplasm in high-risk sites in older patients who present with an evolving nodule or tumor on an extremity. Routine histology and astute histochemical profiling are necessary to exclude diseases that mimic porocarcinoma. Once diagnosis is confirmed, management with prompt excision and diagnostic imaging is recommended, including a lymph node biopsy if appropriate. Due to its high metastatic potential and associated morbidity and mortality, patients with porocarcinoma should be followed closely by a multidisciplinary care team.
To the Editor:
Porocarcinoma, or malignant poroma, is a rare adnexal malignancy of a predominantly glandular origin that comprises less than 0.01% of all cutaneous neoplasms.1,2 Although exposure to UV radiation and immunosuppression have been implicated in the malignant degeneration of benign poromas into porocarcinomas, at least half of all malignant variants will arise de novo.3,4 Patients present with an evolving nodule or plaque and often are in their seventh or eighth decade of life at the time of diagnosis.2 Localized trauma from burns or radiation exposure has been causatively linked to de novo porocarcinoma formation.2,5 These suppressive and traumatic stimuli drive increased genetic heterogeneity along with characteristic gene mutations in known tumor suppressor genes.6
A 62-year-old man presented with a nonhealing wound on the right hand of 5 years’ duration that had previously been attributed to a penetrating injury with a piece of copper from a refrigerant coolant system. The wound initially blistered and then eventually callused and developed areas of ulceration. The patient consulted multiple physicians for treatment of the intensely pruritic and ulcerated lesion. He received prescriptions for cephalexin, trimethoprim-sulfamethoxazole, doxycycline, clindamycin, and clobetasol cream, all of which offered minimal improvement. Home therapies including vitamin E and tea tree oil yielded no benefit. The lesion roughly quadrupled in size over the last 5 years.
Physical examination revealed a 7.5×4.2-cm ulcerated plaque with ragged borders and abundant central neoepithelialization on the right palmar surface (Figure 1). No gross motor or sensory defects were identified. There was no epitrochlear, axillary, cervical, or supraclavicular lymphadenopathy. A shave biopsy of the plaque’s edge was performed, which demonstrated a hyperplastic epidermis comprising atypical poroid cells with frequent mitoses, scant necrosis, and regular ductal structures confined to the epidermis (Figure 2). Immunohistochemical profiling results were positive for anticytokeratin (CAM 5.2) and Ber-EP4 (Figure 3). When evaluated in aggregate, these findings were consistent with porocarcinoma in situ.
The patient was referred to a surgical oncologist for evaluation. At that time, an exophytic mass had developed in the central lesion. Although no lymphadenopathy was identified upon examination, the patient had developed tremoring and a contracture deformity of the right hand. Extensive imaging and urgent surgical resection were recommended, but the patient did not wish to pursue these options, opting instead to continue home remedies. At a 15-month follow-up via telephone, the patient reported that the home therapy had failed and he had moved back to Vietnam. Partial limb amputation had been recommended by a local provider. Unfortunately, the patient was subsequently lost to follow-up, and his current status is unknown.
Porocarcinomas are rare tumors, comprising just 0.005% to 0.01% of all cutaneous epithelial tumors.1,2,5 They affect men and women equally, with an average age at diagnosis of 60 to 70 years.1,2 At least half of all porocarcinomas develop de novo, while 18% to 50% arise from the degeneration of an existing poroma.2,3 Exposure to UV light and immunosuppression, particularly following organ transplantation, represent 2 commonly suspected catalysts for this malignant transformation.4 De novo porocarcinomas are most causatively linked to localized trauma from burns or radiation exposure.5 Gene mutations in classic tumor suppressor genes—tumor protein p53 (TP53), phosphatase and tensin homolog (PTEN), rearranged during transfection (RET), adenomatous polyposis coli (APC)—and increased genetic heterogeneity follow these stimuli.6
The morphologic presentation of porocarcinoma is highly variable and may manifest as papules, nodules, or plaques in various states of erosion, ulceration, or excoriation. Diagnoses of basal and squamous cell carcinoma, primary adnexal tumors, seborrheic keratosis, pyogenic granuloma, and melanoma must all be considered and methodically ruled out.7 Porocarcinomas may arise nearly anywhere on the body, with a particular predilection for the lower extremities (35%), head/neck (24%), and upper extremities (14%).3,4 Primary lesions arising from the extremities, genitalia, or buttocks herald a higher risk for lymphatic invasion and distant metastasis, while head and neck tumors more commonly remain localized.8 Bleeding, ulceration, or rapid expansion of a preexisting poroma is suggestive of malignant transformation and may portend a more aggressive disease pattern.2,9
Unequivocal diagnosis relies on histological and immunohistochemical studies due to the marked clinical variance of this neoplasm.7 An irregular histologic pattern of poromatous basaloid cells with ductal differentiation and cytologic atypia commonly are seen with porocarcinomas.2,8 Nuclear pleomorphism with cellular necrosis, increased mitotic figures, and abortive ductal formation with a distinct lack of retraction around cellular aggregates often are found. Immunohistochemical staining is needed to confirm the primary tumor diagnosis. Histochemical stains commonly employed include carcinoembryonic antigen (CEA), cytokeratin AE1/AE3, epithelial membrane antigen, p53, p63, Ki67, and periodic acid-Schiff.10 The use of BerEP4 has been reported as efficacious in highlighting sweat structures, which can be particularly useful in cases when basal cell carcinoma is not in the histologic differential.11 These staining profiles afford confirmation of ductal differentiation with CEA, epithelial membrane antigen, and BerEP4, while p63 and Ki67 are used as surrogates for primary cutaneous neoplasia and cell proliferation, respectively.5,11 Porocarcinoma lesions may be most sensitive to CEA and most specific to CK19 (a component of cytokeratin AE1/AE3), though these findings have not been widely reproduced.7
The treatment and prognosis of porocarcinoma vary widely. Surgically excised lesions recur in roughly 20% of cases, though these rates likely include tumors that were incompletely resected in the primary attempt. Although wide local excision with an average 1-cm margin remains the most employed removal technique, Mohs micrographic surgery may more effectively limit recurrence and metastasis of localized disease.7,8,12 Metastatic disease foretells a mortality rate of at least 65%, which is problematic in that 10% to 20% of patients have metastatic disease at the time of diagnosis and another 20% will show metastasis following primary tumor excision.8,10 Neoplasms with high mitotic rates and depths greater than 7 mm should prompt thorough diagnostic imaging, such as positron emission tomography or magnetic resonance imaging. A sentinel lymph node biopsy should be strongly considered and discussed with the patient.10 Treatment options for nodal and distant metastases include a combination of localized surgery, lymphadenectomy, radiotherapy, and chemotherapeutic agents.2,4,5 The response to systemic treatment and radiotherapy often is quite poor, though the use of combinations of docetaxel, paclitaxel, cetuximab, and immunotherapy have been efficacious in smaller studies.8,10 The highest rates of morbidity and mortality are seen in patients with metastases on presentation or with localized tumors in the groin and buttocks.8
The diagnosis of porocarcinoma may be elusive due to its relatively rare occurrence. Therefore, it is critical to consider this neoplasm in high-risk sites in older patients who present with an evolving nodule or tumor on an extremity. Routine histology and astute histochemical profiling are necessary to exclude diseases that mimic porocarcinoma. Once diagnosis is confirmed, management with prompt excision and diagnostic imaging is recommended, including a lymph node biopsy if appropriate. Due to its high metastatic potential and associated morbidity and mortality, patients with porocarcinoma should be followed closely by a multidisciplinary care team.
- Belin E, Ezzedine K, Stanislas S, et al. Factors in the surgical management of primary eccrine porocarcinoma: prognostic histological factors can guide the surgical procedure. Br J Dermatol. 2011;165:985-989.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Spencer DM, Bigler LR, Hearne DW, et al. Pedal papule. eccrine porocarcinoma (EPC) in association with poroma. Arch Dermatol. 1995;131:211, 214.
- Salih AM, Kakamad FH, Essa RA, et al. Porocarcinoma: a systematic review of literature with a single case report. Int J Surg Case Rep. 2017;30:13-16.
- Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Mosby Elsevier; 2018.
- Bosic M, Kirchner M, Brasanac D, et al. Targeted molecular profiling reveals genetic heterogeneity of poromas and porocarcinomas. Pathology. 2018;50:327-332.
- Mahalingam M, Richards JE, Selim MA, et al. An immunohistochemical comparison of cytokeratin 7, cytokeratin 15, cytokeratin 19, CAM 5.2, carcinoembryonic antigen, and nestin in differentiating porocarcinoma from squamous cell carcinoma. Hum Pathol. 2012;43:1265-1272.
- Nazemi A, Higgins S, Swift R, et al. Eccrine porocarcinoma: new insights and a systematic review of the literature. Dermatol Surg. 2018;44:1247-1261.
- Wen SY. Case report of eccrine porocarcinoma in situ associated with eccrine poroma on the forehead. J Dermatol. 2012;39:649-651.
- Gerber PA, Schulte KW, Ruzicka T, et al. Eccrine porocarcinoma of the head: an important differential diagnosis in the elderly patient. Dermatology. 2008;216:229-233.
- Afshar M, Deroide F, Robson A. BerEP4 is widely expressed in tumors of the sweat apparatus: a source of potential diagnostic error. J Cutan Pathol. 2013;40:259-264.
- Tolkachjov SN, Hocker TL, Camilleri MJ, et al. Treatment of porocarcinoma with Mohs micrographic surgery: the Mayo clinic experience. Dermatol Surg. 2016;42:745-750.
- Belin E, Ezzedine K, Stanislas S, et al. Factors in the surgical management of primary eccrine porocarcinoma: prognostic histological factors can guide the surgical procedure. Br J Dermatol. 2011;165:985-989.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Spencer DM, Bigler LR, Hearne DW, et al. Pedal papule. eccrine porocarcinoma (EPC) in association with poroma. Arch Dermatol. 1995;131:211, 214.
- Salih AM, Kakamad FH, Essa RA, et al. Porocarcinoma: a systematic review of literature with a single case report. Int J Surg Case Rep. 2017;30:13-16.
- Bolognia JL, Schaffer JV, Cerroni L, eds. Dermatology. Mosby Elsevier; 2018.
- Bosic M, Kirchner M, Brasanac D, et al. Targeted molecular profiling reveals genetic heterogeneity of poromas and porocarcinomas. Pathology. 2018;50:327-332.
- Mahalingam M, Richards JE, Selim MA, et al. An immunohistochemical comparison of cytokeratin 7, cytokeratin 15, cytokeratin 19, CAM 5.2, carcinoembryonic antigen, and nestin in differentiating porocarcinoma from squamous cell carcinoma. Hum Pathol. 2012;43:1265-1272.
- Nazemi A, Higgins S, Swift R, et al. Eccrine porocarcinoma: new insights and a systematic review of the literature. Dermatol Surg. 2018;44:1247-1261.
- Wen SY. Case report of eccrine porocarcinoma in situ associated with eccrine poroma on the forehead. J Dermatol. 2012;39:649-651.
- Gerber PA, Schulte KW, Ruzicka T, et al. Eccrine porocarcinoma of the head: an important differential diagnosis in the elderly patient. Dermatology. 2008;216:229-233.
- Afshar M, Deroide F, Robson A. BerEP4 is widely expressed in tumors of the sweat apparatus: a source of potential diagnostic error. J Cutan Pathol. 2013;40:259-264.
- Tolkachjov SN, Hocker TL, Camilleri MJ, et al. Treatment of porocarcinoma with Mohs micrographic surgery: the Mayo clinic experience. Dermatol Surg. 2016;42:745-750.
Practice Points
- Porocarcinoma is a rare, potentially aggressive, glandular malignancy that should be a clinical consideration in patients presenting with a cutaneous neoplasm.
- Although wide local excision historically has been the treatment of choice for porocarcinoma, Mohs micrographic surgery has demonstrated excellent cure rates.
- Patients with unresectable or metastatic porocarcinomas have a poor prognosis but may respond to combination chemotherapy regimens.

Genital Ulcerations With Swelling
The Diagnosis: Mpox (Monkeypox)
Tests for active herpes simplex virus (HHV), gonorrhea, chlamydia, HIV, and syphilis were negative. Swabs from the penile lesion demonstrated positivity for the West African clade of mpox (monkeypox) virus (MPXV) by polymerase chain reaction. The patient was treated supportively without the addition of antiviral therapy, and he experienced a complete recovery.
Mpox virus was first isolated in 1958 in a research facility and was named after the laboratory animals that were housed there. The first human documentation of the disease occurred in 1970, and it was first documented in the United States in 2003 in an infection that was traced to a shipment of small mammals from Ghana to Texas.1 The disease has always been endemic to Africa; however, the incidence has been increasing.2 A new MPXV outbreak was reported in many countries in early 2022, including the United States.1
The MPXV is a double-stranded DNA virus of the genus Orthopoxvirus, and 2 genetic clades have been identified: clade I (formerly the Central African clade) and clade II (formerly the West African clade). The virus has the capability to infect many mammals; however, its host remains unidentified.1 The exact mechanism of transmission from infected animals to humans largely is unknown; however, direct or indirect contact with infected animals likely is responsible. Human-to-human transmission can occur by many mechanisms including contact with large respiratory droplets, bodily fluids, and contaminated surfaces. The incubation period is 5 to 21 days, and the symptoms last 2 to 5 weeks.1
.")
The clinical manifestations of MPXV during the most recent outbreak differ from prior outbreaks. Patients are more likely to experience minimal to no systemic symptoms, and cutaneous lesions can be few and localized to a focal area, especially on the face and in the anogenital region,3 similar to the presentation in our patient (Figure 1). Cutaneous lesions of the most recent MPXV outbreak also include painless ulcerations similar to syphilitic chancres and lesions that are in various stages of healing.3 Lesions often begin as pseudopustules, which are firm white papules with or without a necrotic center that resemble pustules; unlike true pustules, there is no identifiable purulent material within it. Bacterial superinfection of the lesions is not uncommon.4 Over time, a secondary pustular eruption resembling folliculitis also may occur,4 as noted in our patient (Figure 2).
 on the arms.")
Although we did not have a biopsy to support the diagnosis of associated erythema multiforme (EM) in our patient, features supportive of this diagnosis included the classic clinical appearance of typical, well-defined, targetoid plaques with 3 distinct zones (Figure 3); the association with a known infection; the distribution on the arms with truncal sparing; and self-limited lesions. More than 90% of EM cases are associated with infection, with HHV representing the most common culprit5; therefore, the relationship with a different virus is not an unreasonable suggestion. Additionally, there have been rare reports of EM in association with MPXV.4
.")
Histopathology of MPXV may have distinctive features. Lesions often demonstrate keratinocytic necrosis and basal layer vacuolization with an associated superficial and deep perivascular lymphohistiocytic infiltrate. When the morphology of the lesion is vesicular, histopathology reveals spongiosis and ballooning degeneration with epidermal necrosis. Viral inclusion bodies within keratinocytes may be identified.1 Death rates from MPXV has been reported from 1% to 11%, with increased mortality among high-risk populations including children and immunocompromised individuals. Treatment of the disease largely consists of supportive care and management of any associated complications including bacterial infection, pneumonia, and encephalitis.1
The differential diagnosis of MPXV includes other ulcerative lesions that can occur on the genital skin. Fixed drug eruptions often present on the penis,6 but there was no identifiable inciting drug in our patient. Herpes simplex virus infection was very high on the differential given our patient’s history of recurrent infections and association with a targetoid rash, but HHV type 1 and HHV type 2 testing of the lesion was negative. A syphilitic chancre also may present with the nontender genital ulceration7 that was seen in our patient, but serology did not support this diagnosis. Cutaneous Crohn disease also may manifest with genital ulceration even before a diagnosis of Crohn disease is made, but these lesions often present as linear knife-cut ulcerations of the anogenital region.8
Our case further supports a clinical presentation that diverges from the more traditional cases of MPXV. Additionally, associated EM may be a clue to infection, especially in cases of negative HHV and other sexually transmitted infection testing.
- Bunge EM, Hoet B, Chen L, et al. The changing epidemiology of human monkeypox—a potential threat? a systematic review. PLoS Negl Trop Dis. 2022;16:E0010141.
- Kumar N, Acharya A, Gendelman HE, et al. The 2022 outbreak and the pathobiology of the monkeypox virus. J Autoimmun. 2022;131:102855.
- Eisenstadt R, Liszewski WJ, Nguyen CV. Recognizing minimal cutaneous involvement or systemic symptoms in monkeypox. JAMA Dermatol. 2022;158:1457-1458.
- Català A, Clavo-Escribano P, Riera-Monroig J, et al. Monkeypox outbreak in Spain: clinical and epidemiological findings in a prospective cross-sectional study of 185 cases [published online August 2, 2022]. Br J Dermatol. 2022;187:765-772.
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902.
- Waleryie-Allanore L, Obeid G, Revuz J. Drug reactions. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:348-375.
- Stary G, Stary A. Sexually transmitted infections. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1447-1469.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1644-1663.
The Diagnosis: Mpox (Monkeypox)
Tests for active herpes simplex virus (HHV), gonorrhea, chlamydia, HIV, and syphilis were negative. Swabs from the penile lesion demonstrated positivity for the West African clade of mpox (monkeypox) virus (MPXV) by polymerase chain reaction. The patient was treated supportively without the addition of antiviral therapy, and he experienced a complete recovery.
Mpox virus was first isolated in 1958 in a research facility and was named after the laboratory animals that were housed there. The first human documentation of the disease occurred in 1970, and it was first documented in the United States in 2003 in an infection that was traced to a shipment of small mammals from Ghana to Texas.1 The disease has always been endemic to Africa; however, the incidence has been increasing.2 A new MPXV outbreak was reported in many countries in early 2022, including the United States.1
The MPXV is a double-stranded DNA virus of the genus Orthopoxvirus, and 2 genetic clades have been identified: clade I (formerly the Central African clade) and clade II (formerly the West African clade). The virus has the capability to infect many mammals; however, its host remains unidentified.1 The exact mechanism of transmission from infected animals to humans largely is unknown; however, direct or indirect contact with infected animals likely is responsible. Human-to-human transmission can occur by many mechanisms including contact with large respiratory droplets, bodily fluids, and contaminated surfaces. The incubation period is 5 to 21 days, and the symptoms last 2 to 5 weeks.1
The clinical manifestations of MPXV during the most recent outbreak differ from prior outbreaks. Patients are more likely to experience minimal to no systemic symptoms, and cutaneous lesions can be few and localized to a focal area, especially on the face and in the anogenital region,3 similar to the presentation in our patient (Figure 1). Cutaneous lesions of the most recent MPXV outbreak also include painless ulcerations similar to syphilitic chancres and lesions that are in various stages of healing.3 Lesions often begin as pseudopustules, which are firm white papules with or without a necrotic center that resemble pustules; unlike true pustules, there is no identifiable purulent material within it. Bacterial superinfection of the lesions is not uncommon.4 Over time, a secondary pustular eruption resembling folliculitis also may occur,4 as noted in our patient (Figure 2).
Although we did not have a biopsy to support the diagnosis of associated erythema multiforme (EM) in our patient, features supportive of this diagnosis included the classic clinical appearance of typical, well-defined, targetoid plaques with 3 distinct zones (Figure 3); the association with a known infection; the distribution on the arms with truncal sparing; and self-limited lesions. More than 90% of EM cases are associated with infection, with HHV representing the most common culprit5; therefore, the relationship with a different virus is not an unreasonable suggestion. Additionally, there have been rare reports of EM in association with MPXV.4
Histopathology of MPXV may have distinctive features. Lesions often demonstrate keratinocytic necrosis and basal layer vacuolization with an associated superficial and deep perivascular lymphohistiocytic infiltrate. When the morphology of the lesion is vesicular, histopathology reveals spongiosis and ballooning degeneration with epidermal necrosis. Viral inclusion bodies within keratinocytes may be identified.1 Death rates from MPXV has been reported from 1% to 11%, with increased mortality among high-risk populations including children and immunocompromised individuals. Treatment of the disease largely consists of supportive care and management of any associated complications including bacterial infection, pneumonia, and encephalitis.1
The differential diagnosis of MPXV includes other ulcerative lesions that can occur on the genital skin. Fixed drug eruptions often present on the penis,6 but there was no identifiable inciting drug in our patient. Herpes simplex virus infection was very high on the differential given our patient’s history of recurrent infections and association with a targetoid rash, but HHV type 1 and HHV type 2 testing of the lesion was negative. A syphilitic chancre also may present with the nontender genital ulceration7 that was seen in our patient, but serology did not support this diagnosis. Cutaneous Crohn disease also may manifest with genital ulceration even before a diagnosis of Crohn disease is made, but these lesions often present as linear knife-cut ulcerations of the anogenital region.8
Our case further supports a clinical presentation that diverges from the more traditional cases of MPXV. Additionally, associated EM may be a clue to infection, especially in cases of negative HHV and other sexually transmitted infection testing.
The Diagnosis: Mpox (Monkeypox)
Tests for active herpes simplex virus (HHV), gonorrhea, chlamydia, HIV, and syphilis were negative. Swabs from the penile lesion demonstrated positivity for the West African clade of mpox (monkeypox) virus (MPXV) by polymerase chain reaction. The patient was treated supportively without the addition of antiviral therapy, and he experienced a complete recovery.
Mpox virus was first isolated in 1958 in a research facility and was named after the laboratory animals that were housed there. The first human documentation of the disease occurred in 1970, and it was first documented in the United States in 2003 in an infection that was traced to a shipment of small mammals from Ghana to Texas.1 The disease has always been endemic to Africa; however, the incidence has been increasing.2 A new MPXV outbreak was reported in many countries in early 2022, including the United States.1
The MPXV is a double-stranded DNA virus of the genus Orthopoxvirus, and 2 genetic clades have been identified: clade I (formerly the Central African clade) and clade II (formerly the West African clade). The virus has the capability to infect many mammals; however, its host remains unidentified.1 The exact mechanism of transmission from infected animals to humans largely is unknown; however, direct or indirect contact with infected animals likely is responsible. Human-to-human transmission can occur by many mechanisms including contact with large respiratory droplets, bodily fluids, and contaminated surfaces. The incubation period is 5 to 21 days, and the symptoms last 2 to 5 weeks.1
The clinical manifestations of MPXV during the most recent outbreak differ from prior outbreaks. Patients are more likely to experience minimal to no systemic symptoms, and cutaneous lesions can be few and localized to a focal area, especially on the face and in the anogenital region,3 similar to the presentation in our patient (Figure 1). Cutaneous lesions of the most recent MPXV outbreak also include painless ulcerations similar to syphilitic chancres and lesions that are in various stages of healing.3 Lesions often begin as pseudopustules, which are firm white papules with or without a necrotic center that resemble pustules; unlike true pustules, there is no identifiable purulent material within it. Bacterial superinfection of the lesions is not uncommon.4 Over time, a secondary pustular eruption resembling folliculitis also may occur,4 as noted in our patient (Figure 2).
Although we did not have a biopsy to support the diagnosis of associated erythema multiforme (EM) in our patient, features supportive of this diagnosis included the classic clinical appearance of typical, well-defined, targetoid plaques with 3 distinct zones (Figure 3); the association with a known infection; the distribution on the arms with truncal sparing; and self-limited lesions. More than 90% of EM cases are associated with infection, with HHV representing the most common culprit5; therefore, the relationship with a different virus is not an unreasonable suggestion. Additionally, there have been rare reports of EM in association with MPXV.4
Histopathology of MPXV may have distinctive features. Lesions often demonstrate keratinocytic necrosis and basal layer vacuolization with an associated superficial and deep perivascular lymphohistiocytic infiltrate. When the morphology of the lesion is vesicular, histopathology reveals spongiosis and ballooning degeneration with epidermal necrosis. Viral inclusion bodies within keratinocytes may be identified.1 Death rates from MPXV has been reported from 1% to 11%, with increased mortality among high-risk populations including children and immunocompromised individuals. Treatment of the disease largely consists of supportive care and management of any associated complications including bacterial infection, pneumonia, and encephalitis.1
The differential diagnosis of MPXV includes other ulcerative lesions that can occur on the genital skin. Fixed drug eruptions often present on the penis,6 but there was no identifiable inciting drug in our patient. Herpes simplex virus infection was very high on the differential given our patient’s history of recurrent infections and association with a targetoid rash, but HHV type 1 and HHV type 2 testing of the lesion was negative. A syphilitic chancre also may present with the nontender genital ulceration7 that was seen in our patient, but serology did not support this diagnosis. Cutaneous Crohn disease also may manifest with genital ulceration even before a diagnosis of Crohn disease is made, but these lesions often present as linear knife-cut ulcerations of the anogenital region.8
Our case further supports a clinical presentation that diverges from the more traditional cases of MPXV. Additionally, associated EM may be a clue to infection, especially in cases of negative HHV and other sexually transmitted infection testing.
- Bunge EM, Hoet B, Chen L, et al. The changing epidemiology of human monkeypox—a potential threat? a systematic review. PLoS Negl Trop Dis. 2022;16:E0010141.
- Kumar N, Acharya A, Gendelman HE, et al. The 2022 outbreak and the pathobiology of the monkeypox virus. J Autoimmun. 2022;131:102855.
- Eisenstadt R, Liszewski WJ, Nguyen CV. Recognizing minimal cutaneous involvement or systemic symptoms in monkeypox. JAMA Dermatol. 2022;158:1457-1458.
- Català A, Clavo-Escribano P, Riera-Monroig J, et al. Monkeypox outbreak in Spain: clinical and epidemiological findings in a prospective cross-sectional study of 185 cases [published online August 2, 2022]. Br J Dermatol. 2022;187:765-772.
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902.
- Waleryie-Allanore L, Obeid G, Revuz J. Drug reactions. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:348-375.
- Stary G, Stary A. Sexually transmitted infections. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1447-1469.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1644-1663.
- Bunge EM, Hoet B, Chen L, et al. The changing epidemiology of human monkeypox—a potential threat? a systematic review. PLoS Negl Trop Dis. 2022;16:E0010141.
- Kumar N, Acharya A, Gendelman HE, et al. The 2022 outbreak and the pathobiology of the monkeypox virus. J Autoimmun. 2022;131:102855.
- Eisenstadt R, Liszewski WJ, Nguyen CV. Recognizing minimal cutaneous involvement or systemic symptoms in monkeypox. JAMA Dermatol. 2022;158:1457-1458.
- Català A, Clavo-Escribano P, Riera-Monroig J, et al. Monkeypox outbreak in Spain: clinical and epidemiological findings in a prospective cross-sectional study of 185 cases [published online August 2, 2022]. Br J Dermatol. 2022;187:765-772.
- Sokumbi O, Wetter DA. Clinical features, diagnosis, and treatment of erythema multiforme: a review for the practicing dermatologist. Int J Dermatol. 2012;51:889-902.
- Waleryie-Allanore L, Obeid G, Revuz J. Drug reactions. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:348-375.
- Stary G, Stary A. Sexually transmitted infections. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1447-1469.
- Rosenbach MA, Wanat KA, Reisenauer A, et al. Non-infectious granulomas. In: Bolognia J, Schaffer J, Cerroni L, eds. Dermatology. Elsevier; 2018:1644-1663.
A 50-year-old man with a history of recurrent genital herpes simplex virus infections presented to the hospital with genital lesions and swelling of 5 days’ duration. Prior to admission, the patient was treated with a course of valacyclovir by an urgent care physician without improvement. Physical examination revealed a 3-cm, nontender, shallow, ulcerative plaque with irregular borders and a purulent yellow base distributed on the distal shaft of the penis with extension into the coronal sulcus. A few other scattered erosions were noted on the distal penile shaft. He had associated diffuse nonpitting edema of the penis and scrotum as well as tender bilateral inguinal lymphadenopathy. Three days after the genital ulcerations began, the patient developed a nontender erythematous papule with a necrotic center on the right jaw followed by an eruption of erythematous papulopustules on the arms and trunk. The patient denied dysuria, purulent penile discharge, fevers, chills, headaches, myalgia, arthralgia, nausea, vomiting, or diarrhea. The patient was sexually active exclusively with females and had more than 10 partners in the prior year. Shortly after hospital admission, the patient developed red targetoid plaques on the groin, trunk, and arms. No oral mucosal lesions were identified.

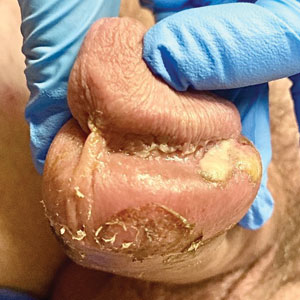
Racial Disparities in Hidradenitis Suppurativa–Related Pain: A Cross-sectional Analysis
Hidradenitis suppurativa (HS), a chronic inflammatory disease that is characterized by tender inflamed nodules of the skin and subcutaneous tissue, disproportionately affects postpubertal females as well as Black/African American individuals. The nodules can rupture, form sinus tracts, and scar. 1 Hidradenitis suppurativa has been associated with cardiovascular disease, type 2 diabetes mellitus, polycystic ovary syndrome, depression, suicide, and substance use disorders. Because of the symptom burden and associated conditions, HS can be a painful and distressing disease that substantially impairs the quality of life for individuals with this condition. 2
Pain is a commonly reported symptom in HS that often goes untreated. Furthermore, HS-related pain is complex due to the involvement of different pain types that require various treatment modalities.3 According to Savage et al,4 recognizing whether HS-related pain is acute, chronic, neuropathic, or nociceptive is vital in establishing a framework for an effective pain management scheme. Currently, such established multimodal pain management strategies in dermatology do not exist. In 2021, dermatology-specific pain management strategies proposed the use of a multimodal regimen to address the multifaceted nature of HS-related pain.4 However, these strategies failed to recognize the systemic racial and ethnic biases in the US health care system that undermine pain management care for minority groups.5,6 One approach to combatting racial disparities in pain management is determining average pain levels across racial groups.7 This study sought to compare HS-related pain scores by racial groups. Furthermore, we assessed differences in perception of patients’ respective pain management regimens by race. We hypothesized that the average HS-related pain intensities and pain management would differ between self-reported racial groups.
Methods
This cross-sectional study took place over 5 months (August through December 2021). A survey was emailed to 2198 adult patients with HS in the University of Alabama Health System. The survey consisted of demographic and general questions about a patient’s HS. Pain scores were captured using the numeric rating scale (NRS), a measurement tool for pain intensity on a scale from 0 to 10. 8 Age at diagnosis, gender, education level, household income, total body areas affected by HS, disease severity (categorized as mild, moderate, and severe), comorbidities including mood disorders, tobacco use, and HS and HS-related pain medication regimens also were collected. Additionally, participants were asked about their level of agreement with the following statements: “I am satisfied with how my pain related to HS is being managed by my doctors” and “My pain related to HS is under control.” The level of agreement was measured using a 5-point Likert scale, with responses ranging from strongly disagree to strongly agree. All data included in the analysis were self-reported. The study received institutional review board approval for the University of Alabama at Birmingham.
Statistical Analysis—Descriptive statistics were used to assess statistical differences in patient characteristics of Black/African American participants compared to other participants, including White, Asian, and Hispanic/Latino participants. Thirteen participants were excluded from the final analysis: 2 participants were missing data, and 11 biracial participants were excluded due to overlapping White and Black/African American races that may have confounded the analysis. Categorical variables were reported as frequencies and percentages, and χ2 and Fisher exact tests, when necessary, were used to test for statistically significant differences. Continuous variables were summarized with means and standard deviations, and a t test was used for statistically significant differences.
Logistic regression was performed to assess the relationship between race and pain after adjusting for confounding variables such as obesity, current tobacco use, self-reported HS severity, and the presence of comorbidities. A total of 204 patient records were included in the analysis, of which 70 (34.3%) had a pain score of 8 or higher, which indicates very severe pain intensity levels on the NRS,8 and were selected as a cut point based on the distribution of responses. For this cross-sectional cohort, our approach was to compare characteristics of those classified with a top score of 8 or higher (n=70) vs a top score of 0 to 7 (n=134)(cases vs noncases). Statistical analyses were performed using JMP Pro 16 (JMP Statistical Discovery LLC) at an α=.05 significance level; logistic regression was performed using SPSS Statistics (IBM). For the logistic regression, we grouped patient race into 2 categories: Black/African American and Other, which included White, Asian, and Hispanic/Latino participants.
Crude and adjusted multivariable logistic regression analyses were used to calculate prevalence odds ratios with 95% confidence intervals. Covariate inclusion in the multivariable logistic regression was based on a priori hypothesis/knowledge and was meant to estimate the independent effect of race after adjustment for income, HS severity, and history of prescription pain medication use. Other variables, including tobacco use, obesity, mood disorders, and current HS treatments, were all individually tested in the multivariate analysis and did not significantly impact the odds ratio for high pain. Statistical adjustment slightly decreased (19%) the magnitude between crude and adjusted prevalence odds ratios for the association between Black/African American race and high pain score.
Results
Survey Demographics —The final analysis included 204 survey respondents. Most respondents were Black/African American (58.82%), and nearly all were female (89.71%)(Table 1). The mean age (SD) of respondents was 37.37 (11.29) years (range, 19-70 years). Many respondents reported having completed some college (36.27%) or receiving a bachelor’s degree (19.12%). Of patients who were not Black/African American, 10.71% had higher than a master’s degree, whereas no Black/African American patients held a degree higher than a master’s ( P = .0052). Additionally, more Black/African American respondents (35.83%) reported an annual household income level of less than $25,000 compared with respondents who were not Black/African American (19.05%, P = .0001). Most respondents rated the severity of their HS as moderate or severe (46.57% and 41.67%, respectively), and there was no significant difference in reported severity of HS between racial groups ( P = .5395).


Pain Scores—As documented in the Methods, respondents were asked to rate their HS-related pain intensity from 0 to 10 using the NRS. The average pain score (SD)—the level of pain intensity over the prior month—was 6.39 (2.56)(range, 0–10). The mean pain score (SD) at the time of the survey was 3.61 (2.98)(range, 0–10)(Table 1). These data revealed that Black/African American patients had a significantly higher average pain score (SD) than patients with HS who were not Black/African American (7.08 [2.49] and 5.40 [2.35], respectively; P<.0001). After adjustment with multivariable logistical regression, Black/African American patients had 4-fold increased odds for very severe levels of pain (score of ≥8) compared with patients who were not Black/African American.
Pain Management—Although pain scores were higher for Black/African American patients with HS, there was no significant difference in the perception of pain control between racial groups (P=.0761). Additionally, we found low income (adjusted prevalence odds ratio [POR], 0.22; 95% CI, 0.05-0.91), a history of prescription pain medication use (adjusted POR, 2.25; 95% CI, 1.13-4.51), and HS severity (adjusted POR, 4.40; 95% CI, 1.11-17.36) all to be independent risk factors contributing to higher pain scores in patients with HS (Table 2). Lastly, we noted current or reported history of pain medication use was significantly correlated with higher pain scores (P=.0280 and P=.0213, respectively).
")
Satisfaction With Pain Management—The level of satisfaction with physician management of HS-related pain was significantly different between Black/African American patients and those who were not Black/African American (P=.0129). Of those who identified as Black/African American, 26.7% (n=32) strongly disagreed with the statement, “I am satisfied with how my pain related to HS is being managed by my doctors,” whereas only 15.5% (n=13) of patients who were not Black/African American strongly disagreed.
Comment
There is no cure for HS, and a large focus of treatment is pain management. Because racial disparities in the treatment of chronic pain will affect those with HS, we conducted a cross-sectional analysis of pain and pain management among HS patients. We found that Black/African American patients with HS have higher average pain scores than those who are not Black/African American and were 4 times more likely to experience very severe pain. Prior studies have established that patients with HS often report higher pain levels than patients with other chronic inflammatory skin conditions, 7,8 and our study identified racial disparities in HS-related pain management.
Measuring pain is challenging because of its multidimensional and subjective nature, making it essential to consider underlying causes and patients’ emotional responses to pain.9 By adjusting for confounding factors that may influence pain, such as mood disorders, disease severity, comorbidities, and medication use, we were able to gain better insight into fundamental differences in average pain intensity levels among racial groups and assess what factors may be contributing to a patient’s pain perception. Our study determined that lower income levels, higher HS disease severity, and a history of prescription pain medication use were all independent risk factors for high pain. Of note, obesity, tobacco use, and mood disorders such as anxiety and depression did not significantly differ between racial groups or increase the odds of high pain between racial groups identified.
With low income being an independent risk factor for high pain, we must consider the social determinants of health and how they may influence the pain experience in HS. We speculate that low income may be associated with other social determinants of health for the patients assessed in this study, such as lack of social and community support or limited health care access that contribute to worse health outcomes.10,11 In addition, low income contributes to limited access to medical care or treatments12; without access to effective HS management, lower-income patients may be at risk for higher disease severity and thus higher pain levels. However, economic stability is only a part of the whole picture; therefore, assessing the other social determinants of health in patients with HS may lead to better health outcomes and quality of life.
Another identified risk factor for high pain was a reported history of prescription pain medication use. This finding suggests that patients with moderate to severe pain likely have required stronger analgesic medications in the past. We further speculate that high pain levels in patients who have received prescription pain medications indicate either undertreatment, mistreatment, or recalcitrant pain. More research is needed to assess the relationship between HS-related pain intensity, analgesic medications, and providers who manage HS-related pain.
We also found that Black/African American patients with HS had a significantly higher dissatisfaction with their physician’s management of their pain, which could be attributable to several factors, including biological differences in medication metabolism (in which the patient has medication-resistant HS), undertreatment of pain, and/or poor doctor-patient relations. These reasons coincide with other diseases where health disparities are found.13-15 Recognizing these factors will be key to dismantling the disparities in HS that are noted within this study. The limitations of this work include the cross-sectional study design and its inability to evaluate causal factors of high pain levels across racial groups, the NRS lack of insight on pain chronicity or pain experience,7 the lack of provider or institution perspectives, and self-reported data. Additionally, only patients with email access were included, which may have excluded vulnerable populations with more pain associated with their HS.
Our findings highlight an area for further investigation to assess why these racial differences exist in HS-related pain. The results also emphasize the need for research evaluating whether systemic or health care provider biases contribute to racial differences in HS-related pain management.
Acknowledgment — Dr. Weir was supported by the Predoctoral Clinical/Translational Research Program (TL1), a National Institutes of Health Ruth L. Kirschstein National Research Service Award (NRSA), through the University of Alabama at Birmingham (UAB) Center for Clinical and Translational Science (CCTS).
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764. doi:10.1001/jamadermatol.2017.0201
- Nguyen TV, Damiani G, Orenstein LAV, et al. Hidradenitis suppurativa: an update on epidemiology, phenotypes, diagnosis, pathogenesis, comorbidities and quality of life. J Eur Acad Dermatol Venereol. 2021;35:50-61. doi:10.1111/jdv.16677
- Krajewski PK, Matusiak Ł, von Stebut E, et al. Pain in hidradenitis suppurativa: a cross-sectional study of 1,795 patients. Acta Derm Venereol. 2021;101:adv00364. doi:10.2340/00015555-3724
- Savage KT, Singh V, Patel ZS, et al. Pain management in hidradenitis suppurativa and a proposed treatment algorithm. J Am Acad Dermatol. 2021;85:187-199. doi:10.1016/j.jaad.2020.09.039
- Morales ME, Yong RJ. Racial and ethnic disparities in the treatment of chronic pain. Pain Med. 2021;22:75-90. doi:10.1093/pm/pnaa427
- US Department of Health and Human Services. 2019 National Healthcare Quality and Disparities Report. December 2020. Accessed June 21, 2023. https://www.ahrq.gov/sites/default/files/wysiwyg/research/findings/nhqrdr/2019qdr.pdf
- Hoffman KM, Trawalter S, Axt JR, et al. Racial bias in pain assessment and treatment recommendations, and false beliefs about biological differences between blacks and whites. Proc Natl Acad Sci U S A. 2016;113:4296-4301. doi:10.1073/pnas.1516047113
- Patel ZS, Hoffman LK, Buse DC, et al. Pain, psychological comorbidities, disability, and impaired quality of life in hidradenitis suppurativa. Curr Pain Headache Rep. 2017;21:49. doi:10.1007/s11916-017-0647-3. Published correction appears in Curr Pain Headache Rep. 2017;21:52.
- McDowell I. Pain measurements. In: Measuring Health: A Guide to Rating Scales and Questionnaires. Oxford University Press; 2006:477-478.
- Singh GK, Daus GP, Allender M, et al. Social determinants of health in the United States: addressing major health inequality trends for the nation, 1935-2016. Int J MCH AIDS. 2017;6:139-164. doi:10.21106/ijma.236
- Sulley S, Bayssie M. Social determinants of health: an evaluation of risk factors associated with inpatient presentations in the United States. Cureus. 2021;13:E13287. doi:10.7759/cureus.13287
- Lazar M, Davenport L. Barriers to health care access for low income families: a review of literature. J Community Health Nurs. 2018;35:28-37. doi:10.1080/07370016.2018.1404832
- Ghoshal M, Shapiro H, Todd K, et al. Chronic noncancer pain management and systemic racism: time to move toward equal care standards.J Pain Res. 2020;13:2825-2836. doi:10.214/JPR.S287314
- Cintron A, Morrison RS. Pain and ethnicity in the United States: a systematic review. J Palliat Med. 2006;9:1454-1473. doi:10.1089/jpm.2006.9.1454
- Green CR, Anderson KO, Baker TA, et al. The unequal burden of pain: confronting racial and ethnic disparities in pain. Pain Med. 2003;4:277-294. doi:10.1046/j.1526-4637.2003.03034.x. Published correction appears in Pain Med. 2005;6:99.
Hidradenitis suppurativa (HS), a chronic inflammatory disease that is characterized by tender inflamed nodules of the skin and subcutaneous tissue, disproportionately affects postpubertal females as well as Black/African American individuals. The nodules can rupture, form sinus tracts, and scar. 1 Hidradenitis suppurativa has been associated with cardiovascular disease, type 2 diabetes mellitus, polycystic ovary syndrome, depression, suicide, and substance use disorders. Because of the symptom burden and associated conditions, HS can be a painful and distressing disease that substantially impairs the quality of life for individuals with this condition. 2
Pain is a commonly reported symptom in HS that often goes untreated. Furthermore, HS-related pain is complex due to the involvement of different pain types that require various treatment modalities.3 According to Savage et al,4 recognizing whether HS-related pain is acute, chronic, neuropathic, or nociceptive is vital in establishing a framework for an effective pain management scheme. Currently, such established multimodal pain management strategies in dermatology do not exist. In 2021, dermatology-specific pain management strategies proposed the use of a multimodal regimen to address the multifaceted nature of HS-related pain.4 However, these strategies failed to recognize the systemic racial and ethnic biases in the US health care system that undermine pain management care for minority groups.5,6 One approach to combatting racial disparities in pain management is determining average pain levels across racial groups.7 This study sought to compare HS-related pain scores by racial groups. Furthermore, we assessed differences in perception of patients’ respective pain management regimens by race. We hypothesized that the average HS-related pain intensities and pain management would differ between self-reported racial groups.
Methods
This cross-sectional study took place over 5 months (August through December 2021). A survey was emailed to 2198 adult patients with HS in the University of Alabama Health System. The survey consisted of demographic and general questions about a patient’s HS. Pain scores were captured using the numeric rating scale (NRS), a measurement tool for pain intensity on a scale from 0 to 10. 8 Age at diagnosis, gender, education level, household income, total body areas affected by HS, disease severity (categorized as mild, moderate, and severe), comorbidities including mood disorders, tobacco use, and HS and HS-related pain medication regimens also were collected. Additionally, participants were asked about their level of agreement with the following statements: “I am satisfied with how my pain related to HS is being managed by my doctors” and “My pain related to HS is under control.” The level of agreement was measured using a 5-point Likert scale, with responses ranging from strongly disagree to strongly agree. All data included in the analysis were self-reported. The study received institutional review board approval for the University of Alabama at Birmingham.
Statistical Analysis—Descriptive statistics were used to assess statistical differences in patient characteristics of Black/African American participants compared to other participants, including White, Asian, and Hispanic/Latino participants. Thirteen participants were excluded from the final analysis: 2 participants were missing data, and 11 biracial participants were excluded due to overlapping White and Black/African American races that may have confounded the analysis. Categorical variables were reported as frequencies and percentages, and χ2 and Fisher exact tests, when necessary, were used to test for statistically significant differences. Continuous variables were summarized with means and standard deviations, and a t test was used for statistically significant differences.
Logistic regression was performed to assess the relationship between race and pain after adjusting for confounding variables such as obesity, current tobacco use, self-reported HS severity, and the presence of comorbidities. A total of 204 patient records were included in the analysis, of which 70 (34.3%) had a pain score of 8 or higher, which indicates very severe pain intensity levels on the NRS,8 and were selected as a cut point based on the distribution of responses. For this cross-sectional cohort, our approach was to compare characteristics of those classified with a top score of 8 or higher (n=70) vs a top score of 0 to 7 (n=134)(cases vs noncases). Statistical analyses were performed using JMP Pro 16 (JMP Statistical Discovery LLC) at an α=.05 significance level; logistic regression was performed using SPSS Statistics (IBM). For the logistic regression, we grouped patient race into 2 categories: Black/African American and Other, which included White, Asian, and Hispanic/Latino participants.
Crude and adjusted multivariable logistic regression analyses were used to calculate prevalence odds ratios with 95% confidence intervals. Covariate inclusion in the multivariable logistic regression was based on a priori hypothesis/knowledge and was meant to estimate the independent effect of race after adjustment for income, HS severity, and history of prescription pain medication use. Other variables, including tobacco use, obesity, mood disorders, and current HS treatments, were all individually tested in the multivariate analysis and did not significantly impact the odds ratio for high pain. Statistical adjustment slightly decreased (19%) the magnitude between crude and adjusted prevalence odds ratios for the association between Black/African American race and high pain score.
Results
Survey Demographics —The final analysis included 204 survey respondents. Most respondents were Black/African American (58.82%), and nearly all were female (89.71%)(Table 1). The mean age (SD) of respondents was 37.37 (11.29) years (range, 19-70 years). Many respondents reported having completed some college (36.27%) or receiving a bachelor’s degree (19.12%). Of patients who were not Black/African American, 10.71% had higher than a master’s degree, whereas no Black/African American patients held a degree higher than a master’s ( P = .0052). Additionally, more Black/African American respondents (35.83%) reported an annual household income level of less than $25,000 compared with respondents who were not Black/African American (19.05%, P = .0001). Most respondents rated the severity of their HS as moderate or severe (46.57% and 41.67%, respectively), and there was no significant difference in reported severity of HS between racial groups ( P = .5395).
Pain Scores—As documented in the Methods, respondents were asked to rate their HS-related pain intensity from 0 to 10 using the NRS. The average pain score (SD)—the level of pain intensity over the prior month—was 6.39 (2.56)(range, 0–10). The mean pain score (SD) at the time of the survey was 3.61 (2.98)(range, 0–10)(Table 1). These data revealed that Black/African American patients had a significantly higher average pain score (SD) than patients with HS who were not Black/African American (7.08 [2.49] and 5.40 [2.35], respectively; P<.0001). After adjustment with multivariable logistical regression, Black/African American patients had 4-fold increased odds for very severe levels of pain (score of ≥8) compared with patients who were not Black/African American.
Pain Management—Although pain scores were higher for Black/African American patients with HS, there was no significant difference in the perception of pain control between racial groups (P=.0761). Additionally, we found low income (adjusted prevalence odds ratio [POR], 0.22; 95% CI, 0.05-0.91), a history of prescription pain medication use (adjusted POR, 2.25; 95% CI, 1.13-4.51), and HS severity (adjusted POR, 4.40; 95% CI, 1.11-17.36) all to be independent risk factors contributing to higher pain scores in patients with HS (Table 2). Lastly, we noted current or reported history of pain medication use was significantly correlated with higher pain scores (P=.0280 and P=.0213, respectively).
Satisfaction With Pain Management—The level of satisfaction with physician management of HS-related pain was significantly different between Black/African American patients and those who were not Black/African American (P=.0129). Of those who identified as Black/African American, 26.7% (n=32) strongly disagreed with the statement, “I am satisfied with how my pain related to HS is being managed by my doctors,” whereas only 15.5% (n=13) of patients who were not Black/African American strongly disagreed.
Comment
There is no cure for HS, and a large focus of treatment is pain management. Because racial disparities in the treatment of chronic pain will affect those with HS, we conducted a cross-sectional analysis of pain and pain management among HS patients. We found that Black/African American patients with HS have higher average pain scores than those who are not Black/African American and were 4 times more likely to experience very severe pain. Prior studies have established that patients with HS often report higher pain levels than patients with other chronic inflammatory skin conditions, 7,8 and our study identified racial disparities in HS-related pain management.
Measuring pain is challenging because of its multidimensional and subjective nature, making it essential to consider underlying causes and patients’ emotional responses to pain.9 By adjusting for confounding factors that may influence pain, such as mood disorders, disease severity, comorbidities, and medication use, we were able to gain better insight into fundamental differences in average pain intensity levels among racial groups and assess what factors may be contributing to a patient’s pain perception. Our study determined that lower income levels, higher HS disease severity, and a history of prescription pain medication use were all independent risk factors for high pain. Of note, obesity, tobacco use, and mood disorders such as anxiety and depression did not significantly differ between racial groups or increase the odds of high pain between racial groups identified.
With low income being an independent risk factor for high pain, we must consider the social determinants of health and how they may influence the pain experience in HS. We speculate that low income may be associated with other social determinants of health for the patients assessed in this study, such as lack of social and community support or limited health care access that contribute to worse health outcomes.10,11 In addition, low income contributes to limited access to medical care or treatments12; without access to effective HS management, lower-income patients may be at risk for higher disease severity and thus higher pain levels. However, economic stability is only a part of the whole picture; therefore, assessing the other social determinants of health in patients with HS may lead to better health outcomes and quality of life.
Another identified risk factor for high pain was a reported history of prescription pain medication use. This finding suggests that patients with moderate to severe pain likely have required stronger analgesic medications in the past. We further speculate that high pain levels in patients who have received prescription pain medications indicate either undertreatment, mistreatment, or recalcitrant pain. More research is needed to assess the relationship between HS-related pain intensity, analgesic medications, and providers who manage HS-related pain.
We also found that Black/African American patients with HS had a significantly higher dissatisfaction with their physician’s management of their pain, which could be attributable to several factors, including biological differences in medication metabolism (in which the patient has medication-resistant HS), undertreatment of pain, and/or poor doctor-patient relations. These reasons coincide with other diseases where health disparities are found.13-15 Recognizing these factors will be key to dismantling the disparities in HS that are noted within this study. The limitations of this work include the cross-sectional study design and its inability to evaluate causal factors of high pain levels across racial groups, the NRS lack of insight on pain chronicity or pain experience,7 the lack of provider or institution perspectives, and self-reported data. Additionally, only patients with email access were included, which may have excluded vulnerable populations with more pain associated with their HS.
Our findings highlight an area for further investigation to assess why these racial differences exist in HS-related pain. The results also emphasize the need for research evaluating whether systemic or health care provider biases contribute to racial differences in HS-related pain management.
Acknowledgment — Dr. Weir was supported by the Predoctoral Clinical/Translational Research Program (TL1), a National Institutes of Health Ruth L. Kirschstein National Research Service Award (NRSA), through the University of Alabama at Birmingham (UAB) Center for Clinical and Translational Science (CCTS).
Hidradenitis suppurativa (HS), a chronic inflammatory disease that is characterized by tender inflamed nodules of the skin and subcutaneous tissue, disproportionately affects postpubertal females as well as Black/African American individuals. The nodules can rupture, form sinus tracts, and scar. 1 Hidradenitis suppurativa has been associated with cardiovascular disease, type 2 diabetes mellitus, polycystic ovary syndrome, depression, suicide, and substance use disorders. Because of the symptom burden and associated conditions, HS can be a painful and distressing disease that substantially impairs the quality of life for individuals with this condition. 2
Pain is a commonly reported symptom in HS that often goes untreated. Furthermore, HS-related pain is complex due to the involvement of different pain types that require various treatment modalities.3 According to Savage et al,4 recognizing whether HS-related pain is acute, chronic, neuropathic, or nociceptive is vital in establishing a framework for an effective pain management scheme. Currently, such established multimodal pain management strategies in dermatology do not exist. In 2021, dermatology-specific pain management strategies proposed the use of a multimodal regimen to address the multifaceted nature of HS-related pain.4 However, these strategies failed to recognize the systemic racial and ethnic biases in the US health care system that undermine pain management care for minority groups.5,6 One approach to combatting racial disparities in pain management is determining average pain levels across racial groups.7 This study sought to compare HS-related pain scores by racial groups. Furthermore, we assessed differences in perception of patients’ respective pain management regimens by race. We hypothesized that the average HS-related pain intensities and pain management would differ between self-reported racial groups.
Methods
This cross-sectional study took place over 5 months (August through December 2021). A survey was emailed to 2198 adult patients with HS in the University of Alabama Health System. The survey consisted of demographic and general questions about a patient’s HS. Pain scores were captured using the numeric rating scale (NRS), a measurement tool for pain intensity on a scale from 0 to 10. 8 Age at diagnosis, gender, education level, household income, total body areas affected by HS, disease severity (categorized as mild, moderate, and severe), comorbidities including mood disorders, tobacco use, and HS and HS-related pain medication regimens also were collected. Additionally, participants were asked about their level of agreement with the following statements: “I am satisfied with how my pain related to HS is being managed by my doctors” and “My pain related to HS is under control.” The level of agreement was measured using a 5-point Likert scale, with responses ranging from strongly disagree to strongly agree. All data included in the analysis were self-reported. The study received institutional review board approval for the University of Alabama at Birmingham.
Statistical Analysis—Descriptive statistics were used to assess statistical differences in patient characteristics of Black/African American participants compared to other participants, including White, Asian, and Hispanic/Latino participants. Thirteen participants were excluded from the final analysis: 2 participants were missing data, and 11 biracial participants were excluded due to overlapping White and Black/African American races that may have confounded the analysis. Categorical variables were reported as frequencies and percentages, and χ2 and Fisher exact tests, when necessary, were used to test for statistically significant differences. Continuous variables were summarized with means and standard deviations, and a t test was used for statistically significant differences.
Logistic regression was performed to assess the relationship between race and pain after adjusting for confounding variables such as obesity, current tobacco use, self-reported HS severity, and the presence of comorbidities. A total of 204 patient records were included in the analysis, of which 70 (34.3%) had a pain score of 8 or higher, which indicates very severe pain intensity levels on the NRS,8 and were selected as a cut point based on the distribution of responses. For this cross-sectional cohort, our approach was to compare characteristics of those classified with a top score of 8 or higher (n=70) vs a top score of 0 to 7 (n=134)(cases vs noncases). Statistical analyses were performed using JMP Pro 16 (JMP Statistical Discovery LLC) at an α=.05 significance level; logistic regression was performed using SPSS Statistics (IBM). For the logistic regression, we grouped patient race into 2 categories: Black/African American and Other, which included White, Asian, and Hispanic/Latino participants.
Crude and adjusted multivariable logistic regression analyses were used to calculate prevalence odds ratios with 95% confidence intervals. Covariate inclusion in the multivariable logistic regression was based on a priori hypothesis/knowledge and was meant to estimate the independent effect of race after adjustment for income, HS severity, and history of prescription pain medication use. Other variables, including tobacco use, obesity, mood disorders, and current HS treatments, were all individually tested in the multivariate analysis and did not significantly impact the odds ratio for high pain. Statistical adjustment slightly decreased (19%) the magnitude between crude and adjusted prevalence odds ratios for the association between Black/African American race and high pain score.
Results
Survey Demographics —The final analysis included 204 survey respondents. Most respondents were Black/African American (58.82%), and nearly all were female (89.71%)(Table 1). The mean age (SD) of respondents was 37.37 (11.29) years (range, 19-70 years). Many respondents reported having completed some college (36.27%) or receiving a bachelor’s degree (19.12%). Of patients who were not Black/African American, 10.71% had higher than a master’s degree, whereas no Black/African American patients held a degree higher than a master’s ( P = .0052). Additionally, more Black/African American respondents (35.83%) reported an annual household income level of less than $25,000 compared with respondents who were not Black/African American (19.05%, P = .0001). Most respondents rated the severity of their HS as moderate or severe (46.57% and 41.67%, respectively), and there was no significant difference in reported severity of HS between racial groups ( P = .5395).
Pain Scores—As documented in the Methods, respondents were asked to rate their HS-related pain intensity from 0 to 10 using the NRS. The average pain score (SD)—the level of pain intensity over the prior month—was 6.39 (2.56)(range, 0–10). The mean pain score (SD) at the time of the survey was 3.61 (2.98)(range, 0–10)(Table 1). These data revealed that Black/African American patients had a significantly higher average pain score (SD) than patients with HS who were not Black/African American (7.08 [2.49] and 5.40 [2.35], respectively; P<.0001). After adjustment with multivariable logistical regression, Black/African American patients had 4-fold increased odds for very severe levels of pain (score of ≥8) compared with patients who were not Black/African American.
Pain Management—Although pain scores were higher for Black/African American patients with HS, there was no significant difference in the perception of pain control between racial groups (P=.0761). Additionally, we found low income (adjusted prevalence odds ratio [POR], 0.22; 95% CI, 0.05-0.91), a history of prescription pain medication use (adjusted POR, 2.25; 95% CI, 1.13-4.51), and HS severity (adjusted POR, 4.40; 95% CI, 1.11-17.36) all to be independent risk factors contributing to higher pain scores in patients with HS (Table 2). Lastly, we noted current or reported history of pain medication use was significantly correlated with higher pain scores (P=.0280 and P=.0213, respectively).
Satisfaction With Pain Management—The level of satisfaction with physician management of HS-related pain was significantly different between Black/African American patients and those who were not Black/African American (P=.0129). Of those who identified as Black/African American, 26.7% (n=32) strongly disagreed with the statement, “I am satisfied with how my pain related to HS is being managed by my doctors,” whereas only 15.5% (n=13) of patients who were not Black/African American strongly disagreed.
Comment
There is no cure for HS, and a large focus of treatment is pain management. Because racial disparities in the treatment of chronic pain will affect those with HS, we conducted a cross-sectional analysis of pain and pain management among HS patients. We found that Black/African American patients with HS have higher average pain scores than those who are not Black/African American and were 4 times more likely to experience very severe pain. Prior studies have established that patients with HS often report higher pain levels than patients with other chronic inflammatory skin conditions, 7,8 and our study identified racial disparities in HS-related pain management.
Measuring pain is challenging because of its multidimensional and subjective nature, making it essential to consider underlying causes and patients’ emotional responses to pain.9 By adjusting for confounding factors that may influence pain, such as mood disorders, disease severity, comorbidities, and medication use, we were able to gain better insight into fundamental differences in average pain intensity levels among racial groups and assess what factors may be contributing to a patient’s pain perception. Our study determined that lower income levels, higher HS disease severity, and a history of prescription pain medication use were all independent risk factors for high pain. Of note, obesity, tobacco use, and mood disorders such as anxiety and depression did not significantly differ between racial groups or increase the odds of high pain between racial groups identified.
With low income being an independent risk factor for high pain, we must consider the social determinants of health and how they may influence the pain experience in HS. We speculate that low income may be associated with other social determinants of health for the patients assessed in this study, such as lack of social and community support or limited health care access that contribute to worse health outcomes.10,11 In addition, low income contributes to limited access to medical care or treatments12; without access to effective HS management, lower-income patients may be at risk for higher disease severity and thus higher pain levels. However, economic stability is only a part of the whole picture; therefore, assessing the other social determinants of health in patients with HS may lead to better health outcomes and quality of life.
Another identified risk factor for high pain was a reported history of prescription pain medication use. This finding suggests that patients with moderate to severe pain likely have required stronger analgesic medications in the past. We further speculate that high pain levels in patients who have received prescription pain medications indicate either undertreatment, mistreatment, or recalcitrant pain. More research is needed to assess the relationship between HS-related pain intensity, analgesic medications, and providers who manage HS-related pain.
We also found that Black/African American patients with HS had a significantly higher dissatisfaction with their physician’s management of their pain, which could be attributable to several factors, including biological differences in medication metabolism (in which the patient has medication-resistant HS), undertreatment of pain, and/or poor doctor-patient relations. These reasons coincide with other diseases where health disparities are found.13-15 Recognizing these factors will be key to dismantling the disparities in HS that are noted within this study. The limitations of this work include the cross-sectional study design and its inability to evaluate causal factors of high pain levels across racial groups, the NRS lack of insight on pain chronicity or pain experience,7 the lack of provider or institution perspectives, and self-reported data. Additionally, only patients with email access were included, which may have excluded vulnerable populations with more pain associated with their HS.
Our findings highlight an area for further investigation to assess why these racial differences exist in HS-related pain. The results also emphasize the need for research evaluating whether systemic or health care provider biases contribute to racial differences in HS-related pain management.
Acknowledgment — Dr. Weir was supported by the Predoctoral Clinical/Translational Research Program (TL1), a National Institutes of Health Ruth L. Kirschstein National Research Service Award (NRSA), through the University of Alabama at Birmingham (UAB) Center for Clinical and Translational Science (CCTS).
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764. doi:10.1001/jamadermatol.2017.0201
- Nguyen TV, Damiani G, Orenstein LAV, et al. Hidradenitis suppurativa: an update on epidemiology, phenotypes, diagnosis, pathogenesis, comorbidities and quality of life. J Eur Acad Dermatol Venereol. 2021;35:50-61. doi:10.1111/jdv.16677
- Krajewski PK, Matusiak Ł, von Stebut E, et al. Pain in hidradenitis suppurativa: a cross-sectional study of 1,795 patients. Acta Derm Venereol. 2021;101:adv00364. doi:10.2340/00015555-3724
- Savage KT, Singh V, Patel ZS, et al. Pain management in hidradenitis suppurativa and a proposed treatment algorithm. J Am Acad Dermatol. 2021;85:187-199. doi:10.1016/j.jaad.2020.09.039
- Morales ME, Yong RJ. Racial and ethnic disparities in the treatment of chronic pain. Pain Med. 2021;22:75-90. doi:10.1093/pm/pnaa427
- US Department of Health and Human Services. 2019 National Healthcare Quality and Disparities Report. December 2020. Accessed June 21, 2023. https://www.ahrq.gov/sites/default/files/wysiwyg/research/findings/nhqrdr/2019qdr.pdf
- Hoffman KM, Trawalter S, Axt JR, et al. Racial bias in pain assessment and treatment recommendations, and false beliefs about biological differences between blacks and whites. Proc Natl Acad Sci U S A. 2016;113:4296-4301. doi:10.1073/pnas.1516047113
- Patel ZS, Hoffman LK, Buse DC, et al. Pain, psychological comorbidities, disability, and impaired quality of life in hidradenitis suppurativa. Curr Pain Headache Rep. 2017;21:49. doi:10.1007/s11916-017-0647-3. Published correction appears in Curr Pain Headache Rep. 2017;21:52.
- McDowell I. Pain measurements. In: Measuring Health: A Guide to Rating Scales and Questionnaires. Oxford University Press; 2006:477-478.
- Singh GK, Daus GP, Allender M, et al. Social determinants of health in the United States: addressing major health inequality trends for the nation, 1935-2016. Int J MCH AIDS. 2017;6:139-164. doi:10.21106/ijma.236
- Sulley S, Bayssie M. Social determinants of health: an evaluation of risk factors associated with inpatient presentations in the United States. Cureus. 2021;13:E13287. doi:10.7759/cureus.13287
- Lazar M, Davenport L. Barriers to health care access for low income families: a review of literature. J Community Health Nurs. 2018;35:28-37. doi:10.1080/07370016.2018.1404832
- Ghoshal M, Shapiro H, Todd K, et al. Chronic noncancer pain management and systemic racism: time to move toward equal care standards.J Pain Res. 2020;13:2825-2836. doi:10.214/JPR.S287314
- Cintron A, Morrison RS. Pain and ethnicity in the United States: a systematic review. J Palliat Med. 2006;9:1454-1473. doi:10.1089/jpm.2006.9.1454
- Green CR, Anderson KO, Baker TA, et al. The unequal burden of pain: confronting racial and ethnic disparities in pain. Pain Med. 2003;4:277-294. doi:10.1046/j.1526-4637.2003.03034.x. Published correction appears in Pain Med. 2005;6:99.
- Garg A, Kirby JS, Lavian J, et al. Sex- and age-adjusted population analysis of prevalence estimates for hidradenitis suppurativa in the United States. JAMA Dermatol. 2017;153:760-764. doi:10.1001/jamadermatol.2017.0201
- Nguyen TV, Damiani G, Orenstein LAV, et al. Hidradenitis suppurativa: an update on epidemiology, phenotypes, diagnosis, pathogenesis, comorbidities and quality of life. J Eur Acad Dermatol Venereol. 2021;35:50-61. doi:10.1111/jdv.16677
- Krajewski PK, Matusiak Ł, von Stebut E, et al. Pain in hidradenitis suppurativa: a cross-sectional study of 1,795 patients. Acta Derm Venereol. 2021;101:adv00364. doi:10.2340/00015555-3724
- Savage KT, Singh V, Patel ZS, et al. Pain management in hidradenitis suppurativa and a proposed treatment algorithm. J Am Acad Dermatol. 2021;85:187-199. doi:10.1016/j.jaad.2020.09.039
- Morales ME, Yong RJ. Racial and ethnic disparities in the treatment of chronic pain. Pain Med. 2021;22:75-90. doi:10.1093/pm/pnaa427
- US Department of Health and Human Services. 2019 National Healthcare Quality and Disparities Report. December 2020. Accessed June 21, 2023. https://www.ahrq.gov/sites/default/files/wysiwyg/research/findings/nhqrdr/2019qdr.pdf
- Hoffman KM, Trawalter S, Axt JR, et al. Racial bias in pain assessment and treatment recommendations, and false beliefs about biological differences between blacks and whites. Proc Natl Acad Sci U S A. 2016;113:4296-4301. doi:10.1073/pnas.1516047113
- Patel ZS, Hoffman LK, Buse DC, et al. Pain, psychological comorbidities, disability, and impaired quality of life in hidradenitis suppurativa. Curr Pain Headache Rep. 2017;21:49. doi:10.1007/s11916-017-0647-3. Published correction appears in Curr Pain Headache Rep. 2017;21:52.
- McDowell I. Pain measurements. In: Measuring Health: A Guide to Rating Scales and Questionnaires. Oxford University Press; 2006:477-478.
- Singh GK, Daus GP, Allender M, et al. Social determinants of health in the United States: addressing major health inequality trends for the nation, 1935-2016. Int J MCH AIDS. 2017;6:139-164. doi:10.21106/ijma.236
- Sulley S, Bayssie M. Social determinants of health: an evaluation of risk factors associated with inpatient presentations in the United States. Cureus. 2021;13:E13287. doi:10.7759/cureus.13287
- Lazar M, Davenport L. Barriers to health care access for low income families: a review of literature. J Community Health Nurs. 2018;35:28-37. doi:10.1080/07370016.2018.1404832
- Ghoshal M, Shapiro H, Todd K, et al. Chronic noncancer pain management and systemic racism: time to move toward equal care standards.J Pain Res. 2020;13:2825-2836. doi:10.214/JPR.S287314
- Cintron A, Morrison RS. Pain and ethnicity in the United States: a systematic review. J Palliat Med. 2006;9:1454-1473. doi:10.1089/jpm.2006.9.1454
- Green CR, Anderson KO, Baker TA, et al. The unequal burden of pain: confronting racial and ethnic disparities in pain. Pain Med. 2003;4:277-294. doi:10.1046/j.1526-4637.2003.03034.x. Published correction appears in Pain Med. 2005;6:99.
Practice Points
- Racial disparities exist in the management of hidradenitis suppurativa (HS)–related pain.
- Black/African American patients with HS are 4 times more likely to experience very severe pain than patients of other races or ethnicities.
- Lower income levels, higher HS disease severity, and a history of prescription pain medication use are all independent risk factors for very severe pain in patients with HS.
Association Between Psoriasis and Obesity Among US Adults in the 2009-2014 National Health and Nutrition Examination Survey
To the Editor:
Psoriasis is an immune-mediated dermatologic condition that is associated with various comorbidities, including obesity.1 The underlying pathophysiology of psoriasis has been extensively studied, and recent research has discussed the role of obesity in IL-17 secretion.2 The relationship between being overweight/obese and having psoriasis has been documented in the literature.1,2 However, this association in a recent population is lacking. We sought to investigate the association between psoriasis and obesity utilizing a representative US population of adults—the 2009-2014 National Health and Nutrition Examination Survey (NHANES) data,3 which contains the most recent psoriasis data.
We conducted a population-based, cross-sectional study focused on patients 20 years and older with psoriasis from the 2009-2014 NHANES database. Three 2-year cycles of NHANES data were combined to create our 2009 to 2014 dataset. In the Table, numerous variables including age, sex, household income, race/ethnicity, education, diabetes status, tobacco use, body mass index (BMI), waist circumference, and being called overweight by a health care provider were analyzed using χ2 or t test analyses to evaluate for differences among those with and without psoriasis. Diabetes status was assessed by the question “Other than during pregnancy, have you ever been told by a doctor or health professional that you have diabetes or sugar diabetes?” Tobacco use was assessed by the question “Have you smoked at least 100 cigarettes in your entire life?” Psoriasis status was assessed by a self-reported response to the question “Have you ever been told by a doctor or other health care professional that you had psoriasis?” Three different outcome variables were used to determine if patients were overweight or obese: BMI, waist circumference, and response to the question “Has a doctor or other health professional ever told you that you were overweight?” Obesity was defined as having a BMI of 30 or higher or waist circumference of 102 cm or more in males and 88 cm or more in females.4 Being overweight was defined as having a BMI of 25 to 29.99 or response of Yes to “Has a doctor or other health professional ever told you that you were overweight?”
")
Initially, there were 17,547 participants 20 years and older from 2009 to 2014, but 1654 participants were excluded because of missing data for obesity or psoriasis; therefore, 15,893 patients were included in our analysis. Multivariable logistic regressions were utilized to examine the association between psoriasis and being overweight/obese (eTable). Additionally, the models were adjusted based on age, sex, household income, race/ethnicity, diabetes status, and tobacco use. All data processing and analysis were performed in Stata/MP 17 (StataCorp LLC). P<.05 was considered statistically significant.

The Table shows characteristics of US adults with and without psoriasis in NHANES 2009-2014. We found that the variables of interest evaluating body weight that were significantly different on analysis between patients with and without psoriasis included waist circumference—patients with psoriasis had a significantly higher waist circumference (P=.009)—and being told by a health care provider that they are overweight (P<.0001), which supports the findings by Love et al,5 who reported abdominal obesity was the most common feature of metabolic syndrome exhibited among patients with psoriasis.
Multivariable logistic regression analysis (eTable) revealed that there was a significant association between psoriasis and BMI of 25 to 29.99 (adjusted odds ratio [AOR], 1.34; 95% CI, 1.02-1.76; P=.04) and being told by a health care provider that they are overweight (AOR, 1.91; 95% CI, 1.44-2.52; P<.001). After adjusting for confounding variables, there was no significant association between psoriasis and a BMI of 30 or higher (AOR, 1.00; 95% CI, 0.73-1.38; P=.99) or a waist circumference of 102 cm or more in males and 88 cm or more in females (AOR, 1.15; 95% CI, 0.86-1.53; P=.3).
Our findings suggest that a few variables indicative of being overweight or obese are associated with psoriasis. This relationship most likely is due to increased adipokine, including resistin, levels in overweight individuals, resulting in a proinflammatory state.6 It has been suggested that BMI alone is not a definitive marker for measuring fat storage levels in individuals. People can have a normal or slightly elevated BMI but possess excessive adiposity, resulting in chronic inflammation.6 Therefore, our findings of a significant association between psoriasis and being told by a health care provider that they are overweight might be a stronger measurement for possessing excessive fat, as this is likely due to clinical judgment rather than BMI measurement.
Moreover, it should be noted that the potential reason for the lack of association between BMI of 30 or higher and psoriasis in our analysis may be a result of BMI serving as a poor measurement for adiposity. Additionally, Armstrong and colleagues7 discussed that the association between BMI and psoriasis was stronger for patients with moderate to severe psoriasis. Our study consisted of NHANES data for self-reported psoriasis diagnoses without a psoriasis severity index, making it difficult to extrapolate which individuals had mild or moderate to severe psoriasis, which may have contributed to our finding of no association between BMI of 30 or higher and psoriasis.
The self-reported nature of the survey questions and lack of questions regarding psoriasis severity serve as limitations to the study. Both obesity and psoriasis can have various systemic consequences, such as cardiovascular disease, due to the development of an inflammatory state.8 Future studies may explore other body measurements that indicate being overweight or obese and the potential synergistic relationship of obesity and psoriasis severity, optimizing the development of effective treatment plans.
- Jensen P, Skov L. Psoriasis and obesity. Dermatology. 2016;232:633-639.
- Xu C, Ji J, Su T, et al. The association of psoriasis and obesity: focusing on IL-17A-related immunological mechanisms. Int J Dermatol Venereol. 2021;4:116-121.
- National Center for Health Statistics. NHANES questionnaires, datasets, and related documentation. Centers for Disease Control and Prevention website. Accessed June 22, 2023. https://wwwn.cdc.govnchs/nhanes/Default.aspx
- Ross R, Neeland IJ, Yamashita S, et al. Waist circumference as a vital sign in clinical practice: a Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat Rev Endocrinol. 2020;16:177-189.
- Love TJ, Qureshi AA, Karlson EW, et al. Prevalence of the metabolic syndrome in psoriasis: results from the National Health and Nutrition Examination Survey, 2003-2006. Arch Dermatol. 2011;147:419-424.
- Paroutoglou K, Papadavid E, Christodoulatos GS, et al. Deciphering the association between psoriasis and obesity: current evidence and treatment considerations. Curr Obes Rep. 2020;9:165-178.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:E54.
- Hamminga EA, van der Lely AJ, Neumann HAM, et al. Chronic inflammation in psoriasis and obesity: implications for therapy. Med Hypotheses. 2006;67:768-773.
To the Editor:
Psoriasis is an immune-mediated dermatologic condition that is associated with various comorbidities, including obesity.1 The underlying pathophysiology of psoriasis has been extensively studied, and recent research has discussed the role of obesity in IL-17 secretion.2 The relationship between being overweight/obese and having psoriasis has been documented in the literature.1,2 However, this association in a recent population is lacking. We sought to investigate the association between psoriasis and obesity utilizing a representative US population of adults—the 2009-2014 National Health and Nutrition Examination Survey (NHANES) data,3 which contains the most recent psoriasis data.
We conducted a population-based, cross-sectional study focused on patients 20 years and older with psoriasis from the 2009-2014 NHANES database. Three 2-year cycles of NHANES data were combined to create our 2009 to 2014 dataset. In the Table, numerous variables including age, sex, household income, race/ethnicity, education, diabetes status, tobacco use, body mass index (BMI), waist circumference, and being called overweight by a health care provider were analyzed using χ2 or t test analyses to evaluate for differences among those with and without psoriasis. Diabetes status was assessed by the question “Other than during pregnancy, have you ever been told by a doctor or health professional that you have diabetes or sugar diabetes?” Tobacco use was assessed by the question “Have you smoked at least 100 cigarettes in your entire life?” Psoriasis status was assessed by a self-reported response to the question “Have you ever been told by a doctor or other health care professional that you had psoriasis?” Three different outcome variables were used to determine if patients were overweight or obese: BMI, waist circumference, and response to the question “Has a doctor or other health professional ever told you that you were overweight?” Obesity was defined as having a BMI of 30 or higher or waist circumference of 102 cm or more in males and 88 cm or more in females.4 Being overweight was defined as having a BMI of 25 to 29.99 or response of Yes to “Has a doctor or other health professional ever told you that you were overweight?”
Initially, there were 17,547 participants 20 years and older from 2009 to 2014, but 1654 participants were excluded because of missing data for obesity or psoriasis; therefore, 15,893 patients were included in our analysis. Multivariable logistic regressions were utilized to examine the association between psoriasis and being overweight/obese (eTable). Additionally, the models were adjusted based on age, sex, household income, race/ethnicity, diabetes status, and tobacco use. All data processing and analysis were performed in Stata/MP 17 (StataCorp LLC). P<.05 was considered statistically significant.
The Table shows characteristics of US adults with and without psoriasis in NHANES 2009-2014. We found that the variables of interest evaluating body weight that were significantly different on analysis between patients with and without psoriasis included waist circumference—patients with psoriasis had a significantly higher waist circumference (P=.009)—and being told by a health care provider that they are overweight (P<.0001), which supports the findings by Love et al,5 who reported abdominal obesity was the most common feature of metabolic syndrome exhibited among patients with psoriasis.
Multivariable logistic regression analysis (eTable) revealed that there was a significant association between psoriasis and BMI of 25 to 29.99 (adjusted odds ratio [AOR], 1.34; 95% CI, 1.02-1.76; P=.04) and being told by a health care provider that they are overweight (AOR, 1.91; 95% CI, 1.44-2.52; P<.001). After adjusting for confounding variables, there was no significant association between psoriasis and a BMI of 30 or higher (AOR, 1.00; 95% CI, 0.73-1.38; P=.99) or a waist circumference of 102 cm or more in males and 88 cm or more in females (AOR, 1.15; 95% CI, 0.86-1.53; P=.3).
Our findings suggest that a few variables indicative of being overweight or obese are associated with psoriasis. This relationship most likely is due to increased adipokine, including resistin, levels in overweight individuals, resulting in a proinflammatory state.6 It has been suggested that BMI alone is not a definitive marker for measuring fat storage levels in individuals. People can have a normal or slightly elevated BMI but possess excessive adiposity, resulting in chronic inflammation.6 Therefore, our findings of a significant association between psoriasis and being told by a health care provider that they are overweight might be a stronger measurement for possessing excessive fat, as this is likely due to clinical judgment rather than BMI measurement.
Moreover, it should be noted that the potential reason for the lack of association between BMI of 30 or higher and psoriasis in our analysis may be a result of BMI serving as a poor measurement for adiposity. Additionally, Armstrong and colleagues7 discussed that the association between BMI and psoriasis was stronger for patients with moderate to severe psoriasis. Our study consisted of NHANES data for self-reported psoriasis diagnoses without a psoriasis severity index, making it difficult to extrapolate which individuals had mild or moderate to severe psoriasis, which may have contributed to our finding of no association between BMI of 30 or higher and psoriasis.
The self-reported nature of the survey questions and lack of questions regarding psoriasis severity serve as limitations to the study. Both obesity and psoriasis can have various systemic consequences, such as cardiovascular disease, due to the development of an inflammatory state.8 Future studies may explore other body measurements that indicate being overweight or obese and the potential synergistic relationship of obesity and psoriasis severity, optimizing the development of effective treatment plans.
To the Editor:
Psoriasis is an immune-mediated dermatologic condition that is associated with various comorbidities, including obesity.1 The underlying pathophysiology of psoriasis has been extensively studied, and recent research has discussed the role of obesity in IL-17 secretion.2 The relationship between being overweight/obese and having psoriasis has been documented in the literature.1,2 However, this association in a recent population is lacking. We sought to investigate the association between psoriasis and obesity utilizing a representative US population of adults—the 2009-2014 National Health and Nutrition Examination Survey (NHANES) data,3 which contains the most recent psoriasis data.
We conducted a population-based, cross-sectional study focused on patients 20 years and older with psoriasis from the 2009-2014 NHANES database. Three 2-year cycles of NHANES data were combined to create our 2009 to 2014 dataset. In the Table, numerous variables including age, sex, household income, race/ethnicity, education, diabetes status, tobacco use, body mass index (BMI), waist circumference, and being called overweight by a health care provider were analyzed using χ2 or t test analyses to evaluate for differences among those with and without psoriasis. Diabetes status was assessed by the question “Other than during pregnancy, have you ever been told by a doctor or health professional that you have diabetes or sugar diabetes?” Tobacco use was assessed by the question “Have you smoked at least 100 cigarettes in your entire life?” Psoriasis status was assessed by a self-reported response to the question “Have you ever been told by a doctor or other health care professional that you had psoriasis?” Three different outcome variables were used to determine if patients were overweight or obese: BMI, waist circumference, and response to the question “Has a doctor or other health professional ever told you that you were overweight?” Obesity was defined as having a BMI of 30 or higher or waist circumference of 102 cm or more in males and 88 cm or more in females.4 Being overweight was defined as having a BMI of 25 to 29.99 or response of Yes to “Has a doctor or other health professional ever told you that you were overweight?”
Initially, there were 17,547 participants 20 years and older from 2009 to 2014, but 1654 participants were excluded because of missing data for obesity or psoriasis; therefore, 15,893 patients were included in our analysis. Multivariable logistic regressions were utilized to examine the association between psoriasis and being overweight/obese (eTable). Additionally, the models were adjusted based on age, sex, household income, race/ethnicity, diabetes status, and tobacco use. All data processing and analysis were performed in Stata/MP 17 (StataCorp LLC). P<.05 was considered statistically significant.
The Table shows characteristics of US adults with and without psoriasis in NHANES 2009-2014. We found that the variables of interest evaluating body weight that were significantly different on analysis between patients with and without psoriasis included waist circumference—patients with psoriasis had a significantly higher waist circumference (P=.009)—and being told by a health care provider that they are overweight (P<.0001), which supports the findings by Love et al,5 who reported abdominal obesity was the most common feature of metabolic syndrome exhibited among patients with psoriasis.
Multivariable logistic regression analysis (eTable) revealed that there was a significant association between psoriasis and BMI of 25 to 29.99 (adjusted odds ratio [AOR], 1.34; 95% CI, 1.02-1.76; P=.04) and being told by a health care provider that they are overweight (AOR, 1.91; 95% CI, 1.44-2.52; P<.001). After adjusting for confounding variables, there was no significant association between psoriasis and a BMI of 30 or higher (AOR, 1.00; 95% CI, 0.73-1.38; P=.99) or a waist circumference of 102 cm or more in males and 88 cm or more in females (AOR, 1.15; 95% CI, 0.86-1.53; P=.3).
Our findings suggest that a few variables indicative of being overweight or obese are associated with psoriasis. This relationship most likely is due to increased adipokine, including resistin, levels in overweight individuals, resulting in a proinflammatory state.6 It has been suggested that BMI alone is not a definitive marker for measuring fat storage levels in individuals. People can have a normal or slightly elevated BMI but possess excessive adiposity, resulting in chronic inflammation.6 Therefore, our findings of a significant association between psoriasis and being told by a health care provider that they are overweight might be a stronger measurement for possessing excessive fat, as this is likely due to clinical judgment rather than BMI measurement.
Moreover, it should be noted that the potential reason for the lack of association between BMI of 30 or higher and psoriasis in our analysis may be a result of BMI serving as a poor measurement for adiposity. Additionally, Armstrong and colleagues7 discussed that the association between BMI and psoriasis was stronger for patients with moderate to severe psoriasis. Our study consisted of NHANES data for self-reported psoriasis diagnoses without a psoriasis severity index, making it difficult to extrapolate which individuals had mild or moderate to severe psoriasis, which may have contributed to our finding of no association between BMI of 30 or higher and psoriasis.
The self-reported nature of the survey questions and lack of questions regarding psoriasis severity serve as limitations to the study. Both obesity and psoriasis can have various systemic consequences, such as cardiovascular disease, due to the development of an inflammatory state.8 Future studies may explore other body measurements that indicate being overweight or obese and the potential synergistic relationship of obesity and psoriasis severity, optimizing the development of effective treatment plans.
- Jensen P, Skov L. Psoriasis and obesity. Dermatology. 2016;232:633-639.
- Xu C, Ji J, Su T, et al. The association of psoriasis and obesity: focusing on IL-17A-related immunological mechanisms. Int J Dermatol Venereol. 2021;4:116-121.
- National Center for Health Statistics. NHANES questionnaires, datasets, and related documentation. Centers for Disease Control and Prevention website. Accessed June 22, 2023. https://wwwn.cdc.govnchs/nhanes/Default.aspx
- Ross R, Neeland IJ, Yamashita S, et al. Waist circumference as a vital sign in clinical practice: a Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat Rev Endocrinol. 2020;16:177-189.
- Love TJ, Qureshi AA, Karlson EW, et al. Prevalence of the metabolic syndrome in psoriasis: results from the National Health and Nutrition Examination Survey, 2003-2006. Arch Dermatol. 2011;147:419-424.
- Paroutoglou K, Papadavid E, Christodoulatos GS, et al. Deciphering the association between psoriasis and obesity: current evidence and treatment considerations. Curr Obes Rep. 2020;9:165-178.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:E54.
- Hamminga EA, van der Lely AJ, Neumann HAM, et al. Chronic inflammation in psoriasis and obesity: implications for therapy. Med Hypotheses. 2006;67:768-773.
- Jensen P, Skov L. Psoriasis and obesity. Dermatology. 2016;232:633-639.
- Xu C, Ji J, Su T, et al. The association of psoriasis and obesity: focusing on IL-17A-related immunological mechanisms. Int J Dermatol Venereol. 2021;4:116-121.
- National Center for Health Statistics. NHANES questionnaires, datasets, and related documentation. Centers for Disease Control and Prevention website. Accessed June 22, 2023. https://wwwn.cdc.govnchs/nhanes/Default.aspx
- Ross R, Neeland IJ, Yamashita S, et al. Waist circumference as a vital sign in clinical practice: a Consensus Statement from the IAS and ICCR Working Group on Visceral Obesity. Nat Rev Endocrinol. 2020;16:177-189.
- Love TJ, Qureshi AA, Karlson EW, et al. Prevalence of the metabolic syndrome in psoriasis: results from the National Health and Nutrition Examination Survey, 2003-2006. Arch Dermatol. 2011;147:419-424.
- Paroutoglou K, Papadavid E, Christodoulatos GS, et al. Deciphering the association between psoriasis and obesity: current evidence and treatment considerations. Curr Obes Rep. 2020;9:165-178.
- Armstrong AW, Harskamp CT, Armstrong EJ. The association between psoriasis and obesity: a systematic review and meta-analysis of observational studies. Nutr Diabetes. 2012;2:E54.
- Hamminga EA, van der Lely AJ, Neumann HAM, et al. Chronic inflammation in psoriasis and obesity: implications for therapy. Med Hypotheses. 2006;67:768-773.
Practice Points
- There are many comorbidities that are associated with psoriasis, making it crucial to evaluate for these diseases in patients with psoriasis.
- Obesity may be a contributing factor to psoriasis development due to the role of IL-17 secretion.

Palliative Care: Utilization Patterns in Inpatient Dermatology
Palliative care (PC) is a field of medicine that focuses on improving quality of life by managing physical symptoms as well as mental and spiritual well-being in patients with severe illnesses.1,2 Despite cases of severe dermatologic disease, the use of PC in the field of dermatology is limited, often leaving patients with a range of unmet needs.2,3 In one study that explored PC in patients with melanoma, only one-third of patients with advanced melanoma had a PC consultation.4 Reasons behind the lack of utilization of PC in dermatology include time constraints and limited training in addressing the complex psychosocial needs of patients with severe dermatologic illnesses.1 We conducted a retrospective, cross-sectional, single-institution study of specific inpatient dermatology consultations over a 5-year period to describe PC utilization among patients who were hospitalized with select severe dermatologic diseases.
Methods
A retrospective, cross-sectional study of inpatient dermatology consultations over a 5-year period (October 2016 to October 2021) was performed at Atrium Health Wake Forest Baptist Medical Center (Winston-Salem, North Carolina). Patients’ medical records were reviewed if they had one of the following diseases: bullous pemphigoid, calciphylaxis, cutaneous T-cell lymphoma (CTCL), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, erythrodermic psoriasis, graft-vs-host disease, pemphigus vulgaris (PV), purpura fulminans, pyoderma gangrenosum, and Stevens-Johnson syndrome/toxic epidermal necrolysis. These diseases were selected for inclusion because they have been associated with a documented increase in inpatient mortality and have been described in the published literature on PC in dermatology.2 This study was reviewed and approved by the Wake Forest University institutional review board.
Use of PC consultative services along with other associated consultative care (ie, recreation therapy [RT], acute pain management, pastoral care) was assessed for each patient. Recreation therapy included specific interventions such as music therapy, arts/craft therapy, pet therapy, and other services with the goal of improving patient cognitive, emotional, and social function. For patients with a completed PC consultation, goals for PC intervention were recorded.
Results
The total study sample included 193 inpatient dermatology consultations. The mean age of the patients was 58.9 years (range, 2–100 years); 66.8% (129/193) were White and 28.5% (55/193) were Black (Table). Palliative care was consulted in 5.7% of cases, with consultations being requested by the primary care team. Reasons for PC consultation included assessment of the patient’s goals of care (4.1% [8/193]), pain management (3.6% [7/193]), non–pain symptom management (2.6% [5/193]), psychosocial support (1.6% [3/193]), and transitions of care (1.0% [2/193]). The average length of patients’ hospital stay prior to PC consultation was 11.5 days(range, 1–32 days). Acute pain management was the reason for consultation in 15.0% of cases (29/193), RT in 21.8% (42/193), and pastoral care in 13.5% (26/193) of cases. Patients with calciphylaxis received the most PC and pain consultations, but fewer than half received these services. Patients with calciphylaxis, PV, purpura fulminans, and CTCL received a higher percentage of PC consultations than the overall cohort, while patients with calciphylaxis, DRESS syndrome, PV, and pyoderma gangrenosum received relatively more pain consultations than the overall cohort (Figure).

Comment
Clinical practice guidelines for quality PC stress the importance of specialists being familiar with these services and the ability to involve PC as part of the treatment plan to achieve better care for patients with serious illnesses.5 Our results demonstrated low rates of PC consultation services for dermatology patients, which supports the existing literature and suggests that PC may be highly underutilized in inpatient settings for patients with serious skin diseases. Use of PC was infrequent and was initiated relatively late in the course of hospital admission, which can negatively impact a patient’s well-being and care experience and can increase the care burden on their caregivers and families.2
, acute pain management, recreation therapy (RT), or pastoral care consultations during hospitalization.")
Our results suggest a discrepancy in the frequency of formal PC and other palliative consultative services used for dermatologic diseases, with non-PC services including RT, acute pain management, and pastoral care more likely to be utilized. Impacting this finding may be that RT, pastoral care, and acute pain management are provided by nonphysician providers at our institution, not attending faculty staffing PC services. Patients with calciphylaxis were more likely to have PC consultations, potentially due to medicine providers’ familiarity with its morbidity and mortality, as it is commonly associated with end-stage renal disease. Similarly, internal medicine providers may be more familiar with pain classically associated with PG and PV and may be more likely to engage pain experts. Some diseases with notable morbidity and potential mortality were underrepresented including SJS/TEN, erythrodermic psoriasis, CTCL, and GVHD.
Limitations of our study included examination of data from a single institution, as well as the small sample sizes in specific subgroups, which prevented us from making comparisons between diseases. The cross-sectional design also limited our ability to control for confounding variables.
Conclusion
We urge dermatology consultation services to advocate for patients with serious skin diseases andinclude PC consultation as part of their recommendations to primary care teams. Further research should characterize the specific needs of patients that may be addressed by PC services and explore ways dermatologists and others can identify and provide specialty care to hospitalized patients.
- Kelley AS, Morrison RS. Palliative care for the seriously ill. N Engl J Med. 2015;373:747-755.
- Thompson LL, Chen ST, Lawton A, et al. Palliative care in dermatology: a clinical primer, review of the literature, and needs assessment. J Am Acad Dermatol. 2021;85:708-717. doi:10.1016/j.jaad.2020.08.029
- Yang CS, Quan VL, Charrow A. The power of a palliative perspective in dermatology. JAMA Dermatol. 2022;158:609-610. doi:10.1001/jamadermatol.2022.1298
- Osagiede O, Colibaseanu DT, Spaulding AC, et al. Palliative care use among patients with solid cancer tumors. J Palliat Care. 2018;33:149-158.
- Clinical Practice Guidelines for Quality Palliative Care. 4th ed. National Coalition for Hospice and Palliative Care; 2018. Accessed June 21, 2023. https://www.nationalcoalitionhpc.org/wp-content/uploads/2018/10/NCHPC-NCPGuidelines_4thED_web_FINAL.pdf
Palliative care (PC) is a field of medicine that focuses on improving quality of life by managing physical symptoms as well as mental and spiritual well-being in patients with severe illnesses.1,2 Despite cases of severe dermatologic disease, the use of PC in the field of dermatology is limited, often leaving patients with a range of unmet needs.2,3 In one study that explored PC in patients with melanoma, only one-third of patients with advanced melanoma had a PC consultation.4 Reasons behind the lack of utilization of PC in dermatology include time constraints and limited training in addressing the complex psychosocial needs of patients with severe dermatologic illnesses.1 We conducted a retrospective, cross-sectional, single-institution study of specific inpatient dermatology consultations over a 5-year period to describe PC utilization among patients who were hospitalized with select severe dermatologic diseases.
Methods
A retrospective, cross-sectional study of inpatient dermatology consultations over a 5-year period (October 2016 to October 2021) was performed at Atrium Health Wake Forest Baptist Medical Center (Winston-Salem, North Carolina). Patients’ medical records were reviewed if they had one of the following diseases: bullous pemphigoid, calciphylaxis, cutaneous T-cell lymphoma (CTCL), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, erythrodermic psoriasis, graft-vs-host disease, pemphigus vulgaris (PV), purpura fulminans, pyoderma gangrenosum, and Stevens-Johnson syndrome/toxic epidermal necrolysis. These diseases were selected for inclusion because they have been associated with a documented increase in inpatient mortality and have been described in the published literature on PC in dermatology.2 This study was reviewed and approved by the Wake Forest University institutional review board.
Use of PC consultative services along with other associated consultative care (ie, recreation therapy [RT], acute pain management, pastoral care) was assessed for each patient. Recreation therapy included specific interventions such as music therapy, arts/craft therapy, pet therapy, and other services with the goal of improving patient cognitive, emotional, and social function. For patients with a completed PC consultation, goals for PC intervention were recorded.
Results
The total study sample included 193 inpatient dermatology consultations. The mean age of the patients was 58.9 years (range, 2–100 years); 66.8% (129/193) were White and 28.5% (55/193) were Black (Table). Palliative care was consulted in 5.7% of cases, with consultations being requested by the primary care team. Reasons for PC consultation included assessment of the patient’s goals of care (4.1% [8/193]), pain management (3.6% [7/193]), non–pain symptom management (2.6% [5/193]), psychosocial support (1.6% [3/193]), and transitions of care (1.0% [2/193]). The average length of patients’ hospital stay prior to PC consultation was 11.5 days(range, 1–32 days). Acute pain management was the reason for consultation in 15.0% of cases (29/193), RT in 21.8% (42/193), and pastoral care in 13.5% (26/193) of cases. Patients with calciphylaxis received the most PC and pain consultations, but fewer than half received these services. Patients with calciphylaxis, PV, purpura fulminans, and CTCL received a higher percentage of PC consultations than the overall cohort, while patients with calciphylaxis, DRESS syndrome, PV, and pyoderma gangrenosum received relatively more pain consultations than the overall cohort (Figure).
Comment
Clinical practice guidelines for quality PC stress the importance of specialists being familiar with these services and the ability to involve PC as part of the treatment plan to achieve better care for patients with serious illnesses.5 Our results demonstrated low rates of PC consultation services for dermatology patients, which supports the existing literature and suggests that PC may be highly underutilized in inpatient settings for patients with serious skin diseases. Use of PC was infrequent and was initiated relatively late in the course of hospital admission, which can negatively impact a patient’s well-being and care experience and can increase the care burden on their caregivers and families.2
Our results suggest a discrepancy in the frequency of formal PC and other palliative consultative services used for dermatologic diseases, with non-PC services including RT, acute pain management, and pastoral care more likely to be utilized. Impacting this finding may be that RT, pastoral care, and acute pain management are provided by nonphysician providers at our institution, not attending faculty staffing PC services. Patients with calciphylaxis were more likely to have PC consultations, potentially due to medicine providers’ familiarity with its morbidity and mortality, as it is commonly associated with end-stage renal disease. Similarly, internal medicine providers may be more familiar with pain classically associated with PG and PV and may be more likely to engage pain experts. Some diseases with notable morbidity and potential mortality were underrepresented including SJS/TEN, erythrodermic psoriasis, CTCL, and GVHD.
Limitations of our study included examination of data from a single institution, as well as the small sample sizes in specific subgroups, which prevented us from making comparisons between diseases. The cross-sectional design also limited our ability to control for confounding variables.
Conclusion
We urge dermatology consultation services to advocate for patients with serious skin diseases andinclude PC consultation as part of their recommendations to primary care teams. Further research should characterize the specific needs of patients that may be addressed by PC services and explore ways dermatologists and others can identify and provide specialty care to hospitalized patients.
Palliative care (PC) is a field of medicine that focuses on improving quality of life by managing physical symptoms as well as mental and spiritual well-being in patients with severe illnesses.1,2 Despite cases of severe dermatologic disease, the use of PC in the field of dermatology is limited, often leaving patients with a range of unmet needs.2,3 In one study that explored PC in patients with melanoma, only one-third of patients with advanced melanoma had a PC consultation.4 Reasons behind the lack of utilization of PC in dermatology include time constraints and limited training in addressing the complex psychosocial needs of patients with severe dermatologic illnesses.1 We conducted a retrospective, cross-sectional, single-institution study of specific inpatient dermatology consultations over a 5-year period to describe PC utilization among patients who were hospitalized with select severe dermatologic diseases.
Methods
A retrospective, cross-sectional study of inpatient dermatology consultations over a 5-year period (October 2016 to October 2021) was performed at Atrium Health Wake Forest Baptist Medical Center (Winston-Salem, North Carolina). Patients’ medical records were reviewed if they had one of the following diseases: bullous pemphigoid, calciphylaxis, cutaneous T-cell lymphoma (CTCL), drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, erythrodermic psoriasis, graft-vs-host disease, pemphigus vulgaris (PV), purpura fulminans, pyoderma gangrenosum, and Stevens-Johnson syndrome/toxic epidermal necrolysis. These diseases were selected for inclusion because they have been associated with a documented increase in inpatient mortality and have been described in the published literature on PC in dermatology.2 This study was reviewed and approved by the Wake Forest University institutional review board.
Use of PC consultative services along with other associated consultative care (ie, recreation therapy [RT], acute pain management, pastoral care) was assessed for each patient. Recreation therapy included specific interventions such as music therapy, arts/craft therapy, pet therapy, and other services with the goal of improving patient cognitive, emotional, and social function. For patients with a completed PC consultation, goals for PC intervention were recorded.
Results
The total study sample included 193 inpatient dermatology consultations. The mean age of the patients was 58.9 years (range, 2–100 years); 66.8% (129/193) were White and 28.5% (55/193) were Black (Table). Palliative care was consulted in 5.7% of cases, with consultations being requested by the primary care team. Reasons for PC consultation included assessment of the patient’s goals of care (4.1% [8/193]), pain management (3.6% [7/193]), non–pain symptom management (2.6% [5/193]), psychosocial support (1.6% [3/193]), and transitions of care (1.0% [2/193]). The average length of patients’ hospital stay prior to PC consultation was 11.5 days(range, 1–32 days). Acute pain management was the reason for consultation in 15.0% of cases (29/193), RT in 21.8% (42/193), and pastoral care in 13.5% (26/193) of cases. Patients with calciphylaxis received the most PC and pain consultations, but fewer than half received these services. Patients with calciphylaxis, PV, purpura fulminans, and CTCL received a higher percentage of PC consultations than the overall cohort, while patients with calciphylaxis, DRESS syndrome, PV, and pyoderma gangrenosum received relatively more pain consultations than the overall cohort (Figure).
Comment
Clinical practice guidelines for quality PC stress the importance of specialists being familiar with these services and the ability to involve PC as part of the treatment plan to achieve better care for patients with serious illnesses.5 Our results demonstrated low rates of PC consultation services for dermatology patients, which supports the existing literature and suggests that PC may be highly underutilized in inpatient settings for patients with serious skin diseases. Use of PC was infrequent and was initiated relatively late in the course of hospital admission, which can negatively impact a patient’s well-being and care experience and can increase the care burden on their caregivers and families.2
Our results suggest a discrepancy in the frequency of formal PC and other palliative consultative services used for dermatologic diseases, with non-PC services including RT, acute pain management, and pastoral care more likely to be utilized. Impacting this finding may be that RT, pastoral care, and acute pain management are provided by nonphysician providers at our institution, not attending faculty staffing PC services. Patients with calciphylaxis were more likely to have PC consultations, potentially due to medicine providers’ familiarity with its morbidity and mortality, as it is commonly associated with end-stage renal disease. Similarly, internal medicine providers may be more familiar with pain classically associated with PG and PV and may be more likely to engage pain experts. Some diseases with notable morbidity and potential mortality were underrepresented including SJS/TEN, erythrodermic psoriasis, CTCL, and GVHD.
Limitations of our study included examination of data from a single institution, as well as the small sample sizes in specific subgroups, which prevented us from making comparisons between diseases. The cross-sectional design also limited our ability to control for confounding variables.
Conclusion
We urge dermatology consultation services to advocate for patients with serious skin diseases andinclude PC consultation as part of their recommendations to primary care teams. Further research should characterize the specific needs of patients that may be addressed by PC services and explore ways dermatologists and others can identify and provide specialty care to hospitalized patients.
- Kelley AS, Morrison RS. Palliative care for the seriously ill. N Engl J Med. 2015;373:747-755.
- Thompson LL, Chen ST, Lawton A, et al. Palliative care in dermatology: a clinical primer, review of the literature, and needs assessment. J Am Acad Dermatol. 2021;85:708-717. doi:10.1016/j.jaad.2020.08.029
- Yang CS, Quan VL, Charrow A. The power of a palliative perspective in dermatology. JAMA Dermatol. 2022;158:609-610. doi:10.1001/jamadermatol.2022.1298
- Osagiede O, Colibaseanu DT, Spaulding AC, et al. Palliative care use among patients with solid cancer tumors. J Palliat Care. 2018;33:149-158.
- Clinical Practice Guidelines for Quality Palliative Care. 4th ed. National Coalition for Hospice and Palliative Care; 2018. Accessed June 21, 2023. https://www.nationalcoalitionhpc.org/wp-content/uploads/2018/10/NCHPC-NCPGuidelines_4thED_web_FINAL.pdf
- Kelley AS, Morrison RS. Palliative care for the seriously ill. N Engl J Med. 2015;373:747-755.
- Thompson LL, Chen ST, Lawton A, et al. Palliative care in dermatology: a clinical primer, review of the literature, and needs assessment. J Am Acad Dermatol. 2021;85:708-717. doi:10.1016/j.jaad.2020.08.029
- Yang CS, Quan VL, Charrow A. The power of a palliative perspective in dermatology. JAMA Dermatol. 2022;158:609-610. doi:10.1001/jamadermatol.2022.1298
- Osagiede O, Colibaseanu DT, Spaulding AC, et al. Palliative care use among patients with solid cancer tumors. J Palliat Care. 2018;33:149-158.
- Clinical Practice Guidelines for Quality Palliative Care. 4th ed. National Coalition for Hospice and Palliative Care; 2018. Accessed June 21, 2023. https://www.nationalcoalitionhpc.org/wp-content/uploads/2018/10/NCHPC-NCPGuidelines_4thED_web_FINAL.pdf
Practice Points
- Although severe dermatologic disease negatively impacts patients’ quality of life, palliative care may be underutilized in this population.
- Palliative care should be an integral part of caring for patients who are admitted to the hospital with serious dermatologic illnesses.
Painful, Nonhealing, Violaceus Plaque on the Right Breast
The Diagnosis: Diffuse Dermal Angiomatosis
Diffuse dermal angiomatosis (DDA) is an acquired reactive vascular proliferation in the spectrum of cutaneous reactive angioendotheliomatoses. Clinically, DDA presents as violaceous reticulated plaques, often with secondary ulceration and sometimes necrosis.1-3 Diffuse dermal angiomatosis more commonly presents in patients with a history of severe peripheral vascular disease, coagulopathies, or infection, and it frequently arises on the extremities. Diffuse dermal angiomatosis also has been shown to develop on the breasts, particularly in patients with pendulous breast tissue. Vascular proliferation in DDA is hypothesized to be from ischemia and hypoxia, leading to angiogenesis.1-3 Diffuse dermal angiomatosis is characterized histologically by the presence of a diffuse proliferation of spindled endothelial cells distributed between the collagen bundles throughout the dermis (quiz image and Figure 1). Spindle-shaped endothelial cells exhibit a vacuolated cytoplasm. On immunohistochemistry, these dermal spindle cells classically stain positive for CD31, CD34, and erythroblast transformation specific–related gene (Erg) and stain negative for both human herpesvirus 8 (HHV-8) and factor XIIIa.

Cutaneous fibrous histiocytoma, more commonly referred to as dermatofibroma, is a common benign lesion that presents clinically as a solitary firm nodule most commonly on the extremities in areas of repetitive trauma or pressure. It classically exhibits dimpling of the overlaying skin with lateral pressure on the lesion, known as the dimple sign.4 Histologically, dermatofibromas share similar features to DDA and demonstrate the presence of bland-appearing spindle cells within the dermis between the collagen bundles, resulting in collagen trapping. However, a distinguishing histologic feature of a dermatofibroma in comparison to DDA is the presence of epidermal hyperplasia overlying the dermatofibroma, leading to tabled rete ridges (Figure 2). Spindle cells in dermatofibromas are fibroblasts and have a distinct immunophenotype that includes factor XIIIa positivity and negative staining for CD31, CD34, and Erg.4,5

Dermatofibrosarcoma protuberans (DFSP) is a rare malignant soft-tissue sarcoma that clinically presents as a firm, flesh-colored, dermal plaque on the trunk, proximal extremities, head, or neck.5 Histologically, DFSP can be distinguished from DDA by the high density of spindle cells that are arranged in a storiform pattern, extending and infiltrating the underlying subcutaneous fat in a honeycomblike pattern (Figure 3). Spindle cells in DFSP typically show expression of CD34 but are negative for CD31, Erg, and factor XIIIa.5

Kaposi sarcoma (KS) is an endothelial cell–driven angioproliferative neoplasm that is associated with HHV-8 infection.6 The clinical presentation of KS can range from isolated pink or purple papules and patches to more extensive ulcerated plaques or nodules. Histopathology exhibits proliferation of monomorphic spindled endothelial cells within the dermis staining positive for HHV-8, Erg, CD31, and CD34, in conjunction with extravasated erythrocytes arranged within slitlike vascular spaces (Figure 4). Additionally, KS classically exhibits aberrant endothelial cell proliferation and vessel formation around preexisting vessels, which is referred to as the promontory sign (Figure 4).

Angiosarcoma is a rare and highly aggressive vascular tumor arising from endothelial cells lining the blood vessels and lymphatics.7,8 Clinically, angiosarcoma presents as ulcerated violaceous nodules or plaques on the head, neck, or trunk. Histologic evaluation of angiosarcoma reveals a complex and poorly demarcated vascular network dissecting between collagen bundles in the dermis (Figure 5). Multilayering of endothelial cells, papillary projections extending into the vessel lumina, and mitoses frequently are seen. On immunohistochemistry, endothelial cells demonstrate prominent cellular atypia and stain positive with CD31, CD34, and Erg.

- Touloei K, Tongdee E, Smirnov B, et al. Diffuse dermal angiomatosis. Cutis. 2019;103:181-184.
- Nguyen N, Silfvast-Kaiser AS, Frieder J, et al. Diffuse dermal angiomatosis of the breast. Baylor Univ Med Cent Proc. 2020;33:273-275.
- Frikha F, Boudaya S, Abid N, et al. Diffuse dermal angiomatosis of the breast with adjacent fat necrosis: a case report and review of the literature. Dermatol Online J. 2018;24:13030/qt1vq114n7.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours—an update. Histopathology. 2010;56:148-165.
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752.
- Etemad SA, Dewan AK. Kaposi sarcoma updates. Dermatol Clin. 2019;37:505-517.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
The Diagnosis: Diffuse Dermal Angiomatosis
Diffuse dermal angiomatosis (DDA) is an acquired reactive vascular proliferation in the spectrum of cutaneous reactive angioendotheliomatoses. Clinically, DDA presents as violaceous reticulated plaques, often with secondary ulceration and sometimes necrosis.1-3 Diffuse dermal angiomatosis more commonly presents in patients with a history of severe peripheral vascular disease, coagulopathies, or infection, and it frequently arises on the extremities. Diffuse dermal angiomatosis also has been shown to develop on the breasts, particularly in patients with pendulous breast tissue. Vascular proliferation in DDA is hypothesized to be from ischemia and hypoxia, leading to angiogenesis.1-3 Diffuse dermal angiomatosis is characterized histologically by the presence of a diffuse proliferation of spindled endothelial cells distributed between the collagen bundles throughout the dermis (quiz image and Figure 1). Spindle-shaped endothelial cells exhibit a vacuolated cytoplasm. On immunohistochemistry, these dermal spindle cells classically stain positive for CD31, CD34, and erythroblast transformation specific–related gene (Erg) and stain negative for both human herpesvirus 8 (HHV-8) and factor XIIIa.
Cutaneous fibrous histiocytoma, more commonly referred to as dermatofibroma, is a common benign lesion that presents clinically as a solitary firm nodule most commonly on the extremities in areas of repetitive trauma or pressure. It classically exhibits dimpling of the overlaying skin with lateral pressure on the lesion, known as the dimple sign.4 Histologically, dermatofibromas share similar features to DDA and demonstrate the presence of bland-appearing spindle cells within the dermis between the collagen bundles, resulting in collagen trapping. However, a distinguishing histologic feature of a dermatofibroma in comparison to DDA is the presence of epidermal hyperplasia overlying the dermatofibroma, leading to tabled rete ridges (Figure 2). Spindle cells in dermatofibromas are fibroblasts and have a distinct immunophenotype that includes factor XIIIa positivity and negative staining for CD31, CD34, and Erg.4,5
Dermatofibrosarcoma protuberans (DFSP) is a rare malignant soft-tissue sarcoma that clinically presents as a firm, flesh-colored, dermal plaque on the trunk, proximal extremities, head, or neck.5 Histologically, DFSP can be distinguished from DDA by the high density of spindle cells that are arranged in a storiform pattern, extending and infiltrating the underlying subcutaneous fat in a honeycomblike pattern (Figure 3). Spindle cells in DFSP typically show expression of CD34 but are negative for CD31, Erg, and factor XIIIa.5
Kaposi sarcoma (KS) is an endothelial cell–driven angioproliferative neoplasm that is associated with HHV-8 infection.6 The clinical presentation of KS can range from isolated pink or purple papules and patches to more extensive ulcerated plaques or nodules. Histopathology exhibits proliferation of monomorphic spindled endothelial cells within the dermis staining positive for HHV-8, Erg, CD31, and CD34, in conjunction with extravasated erythrocytes arranged within slitlike vascular spaces (Figure 4). Additionally, KS classically exhibits aberrant endothelial cell proliferation and vessel formation around preexisting vessels, which is referred to as the promontory sign (Figure 4).
Angiosarcoma is a rare and highly aggressive vascular tumor arising from endothelial cells lining the blood vessels and lymphatics.7,8 Clinically, angiosarcoma presents as ulcerated violaceous nodules or plaques on the head, neck, or trunk. Histologic evaluation of angiosarcoma reveals a complex and poorly demarcated vascular network dissecting between collagen bundles in the dermis (Figure 5). Multilayering of endothelial cells, papillary projections extending into the vessel lumina, and mitoses frequently are seen. On immunohistochemistry, endothelial cells demonstrate prominent cellular atypia and stain positive with CD31, CD34, and Erg.
The Diagnosis: Diffuse Dermal Angiomatosis
Diffuse dermal angiomatosis (DDA) is an acquired reactive vascular proliferation in the spectrum of cutaneous reactive angioendotheliomatoses. Clinically, DDA presents as violaceous reticulated plaques, often with secondary ulceration and sometimes necrosis.1-3 Diffuse dermal angiomatosis more commonly presents in patients with a history of severe peripheral vascular disease, coagulopathies, or infection, and it frequently arises on the extremities. Diffuse dermal angiomatosis also has been shown to develop on the breasts, particularly in patients with pendulous breast tissue. Vascular proliferation in DDA is hypothesized to be from ischemia and hypoxia, leading to angiogenesis.1-3 Diffuse dermal angiomatosis is characterized histologically by the presence of a diffuse proliferation of spindled endothelial cells distributed between the collagen bundles throughout the dermis (quiz image and Figure 1). Spindle-shaped endothelial cells exhibit a vacuolated cytoplasm. On immunohistochemistry, these dermal spindle cells classically stain positive for CD31, CD34, and erythroblast transformation specific–related gene (Erg) and stain negative for both human herpesvirus 8 (HHV-8) and factor XIIIa.
Cutaneous fibrous histiocytoma, more commonly referred to as dermatofibroma, is a common benign lesion that presents clinically as a solitary firm nodule most commonly on the extremities in areas of repetitive trauma or pressure. It classically exhibits dimpling of the overlaying skin with lateral pressure on the lesion, known as the dimple sign.4 Histologically, dermatofibromas share similar features to DDA and demonstrate the presence of bland-appearing spindle cells within the dermis between the collagen bundles, resulting in collagen trapping. However, a distinguishing histologic feature of a dermatofibroma in comparison to DDA is the presence of epidermal hyperplasia overlying the dermatofibroma, leading to tabled rete ridges (Figure 2). Spindle cells in dermatofibromas are fibroblasts and have a distinct immunophenotype that includes factor XIIIa positivity and negative staining for CD31, CD34, and Erg.4,5
Dermatofibrosarcoma protuberans (DFSP) is a rare malignant soft-tissue sarcoma that clinically presents as a firm, flesh-colored, dermal plaque on the trunk, proximal extremities, head, or neck.5 Histologically, DFSP can be distinguished from DDA by the high density of spindle cells that are arranged in a storiform pattern, extending and infiltrating the underlying subcutaneous fat in a honeycomblike pattern (Figure 3). Spindle cells in DFSP typically show expression of CD34 but are negative for CD31, Erg, and factor XIIIa.5
Kaposi sarcoma (KS) is an endothelial cell–driven angioproliferative neoplasm that is associated with HHV-8 infection.6 The clinical presentation of KS can range from isolated pink or purple papules and patches to more extensive ulcerated plaques or nodules. Histopathology exhibits proliferation of monomorphic spindled endothelial cells within the dermis staining positive for HHV-8, Erg, CD31, and CD34, in conjunction with extravasated erythrocytes arranged within slitlike vascular spaces (Figure 4). Additionally, KS classically exhibits aberrant endothelial cell proliferation and vessel formation around preexisting vessels, which is referred to as the promontory sign (Figure 4).
Angiosarcoma is a rare and highly aggressive vascular tumor arising from endothelial cells lining the blood vessels and lymphatics.7,8 Clinically, angiosarcoma presents as ulcerated violaceous nodules or plaques on the head, neck, or trunk. Histologic evaluation of angiosarcoma reveals a complex and poorly demarcated vascular network dissecting between collagen bundles in the dermis (Figure 5). Multilayering of endothelial cells, papillary projections extending into the vessel lumina, and mitoses frequently are seen. On immunohistochemistry, endothelial cells demonstrate prominent cellular atypia and stain positive with CD31, CD34, and Erg.
- Touloei K, Tongdee E, Smirnov B, et al. Diffuse dermal angiomatosis. Cutis. 2019;103:181-184.
- Nguyen N, Silfvast-Kaiser AS, Frieder J, et al. Diffuse dermal angiomatosis of the breast. Baylor Univ Med Cent Proc. 2020;33:273-275.
- Frikha F, Boudaya S, Abid N, et al. Diffuse dermal angiomatosis of the breast with adjacent fat necrosis: a case report and review of the literature. Dermatol Online J. 2018;24:13030/qt1vq114n7.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours—an update. Histopathology. 2010;56:148-165.
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752.
- Etemad SA, Dewan AK. Kaposi sarcoma updates. Dermatol Clin. 2019;37:505-517.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
- Touloei K, Tongdee E, Smirnov B, et al. Diffuse dermal angiomatosis. Cutis. 2019;103:181-184.
- Nguyen N, Silfvast-Kaiser AS, Frieder J, et al. Diffuse dermal angiomatosis of the breast. Baylor Univ Med Cent Proc. 2020;33:273-275.
- Frikha F, Boudaya S, Abid N, et al. Diffuse dermal angiomatosis of the breast with adjacent fat necrosis: a case report and review of the literature. Dermatol Online J. 2018;24:13030/qt1vq114n7.
- Luzar B, Calonje E. Cutaneous fibrohistiocytic tumours—an update. Histopathology. 2010;56:148-165.
- Hao X, Billings SD, Wu F, et al. Dermatofibrosarcoma protuberans: update on the diagnosis and treatment. J Clin Med. 2020;9:1752.
- Etemad SA, Dewan AK. Kaposi sarcoma updates. Dermatol Clin. 2019;37:505-517.
- Cao J, Wang J, He C, et al. Angiosarcoma: a review of diagnosis and current treatment. Am J Cancer Res. 2019;9:2303-2313.
- Shon W, Billings SD. Cutaneous malignant vascular neoplasms. Clin Lab Med. 2017;37:633-646.
A 42-year-old woman with a medical history of hypertension and smoking tobacco (5 pack years) presented with a painful, nonhealing, violaceous, reticulated plaque with ulceration on the right breast of 3 months’ duration. Histopathology revealed diffuse, interstitial, bland-appearing spindle cells throughout the papillary and reticular dermis that were distributed between the collagen bundles. Dermal interstitial spindle cells were positive for CD31, CD34, and erythroblast transformation specific–related gene immunostains. Factor XIIIa and human herpesvirus 8 immunostaining was negative.

