User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
External Beam Radiotherapy of Extramammary Paget Disease
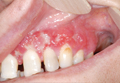
Extramammary Paget disease (EMPD) is an insidious intraepithelial neoplasm that occurs in areas with a high density of apocrine glands such as the penoscrotal area, the vulva, and occasionally the axillae. It mainly affects patients aged 50 to 80 years.1 Clinically, EMPD presents as pruritic, nonhealing, red plaques that can be mistaken for eczema. On histology, characteristic Paget cells have abundant pale cytoplasm and atypical nuclear lobuli and are adenocarcinomatous,1,2 usually infiltrating the epidermis.2 In approximately 25% of cases, EMPD is associated with neoplastic disease in adnexal structures or organs with a contiguous epithelial lining.2 Therefore, screening for an underlying malignancy when EMPD is first diagnosed is indispensable.
Because EMPD tends to be multifocal, presents in elderly patients, and affects functionally important areas such as the anal canal or genitals, treatment often is difficult.3,4 Surgery generally is considered as a first-line treatment5; however, the rate of positive margins ranges from 36% to 67%, and local recurrence is common.1
Radiotherapy has been used in EMPD patients mainly when surgery was not an option or was not effective, but several reports have indicated that it should play a more important role in the treatment of EMPD. Luk et al1 described 6 patients who were treated with different types of radiotherapy. Similar to the results of prior studies,3,5,6 they concluded that it was an effective treatment of EMPD.1
We conducted a retrospective study to analyze long-term outcomes in 7 patients who were treated with external beam radiotherapy (EBRT) for EMPD.
Methods
Seven patients (6 men and 1 woman) who had been diagnosed with EMPD and were treated with EBRT at the Department of Dermatology at the University Hospital Zurich in Switzerland (1988-2004) were evaluated. The diagnosis was confirmed by a dermatopathologist or pathologist via histology. Data regarding clinical presentation, EBRT regimen, and side effects were retrieved from the medical records. Long-term outcomes were evaluated by an attending dermatologist (1 case), a general practitioner (5 cases), or the hospital’s outpatient department (1 case). None of the patients showed an associated malignancy at the time of treatment; however, patient 5 had been diagnosed with and treated for a sigmoid colon adenocarcinoma 6 years prior to undergoing EBRT for EMPD. Three patients (patients 3, 5, and 7) received EBRT for local relapse after prior treatment of EMPD (ie, CO2 laser, multiple local treatments). One patient (patient 2) underwent surgical excision prior to EBRT. The remaining 3 patients had not undergone any prior treatment of EMPD. All patients underwent EBRT with the goal of complete remission.
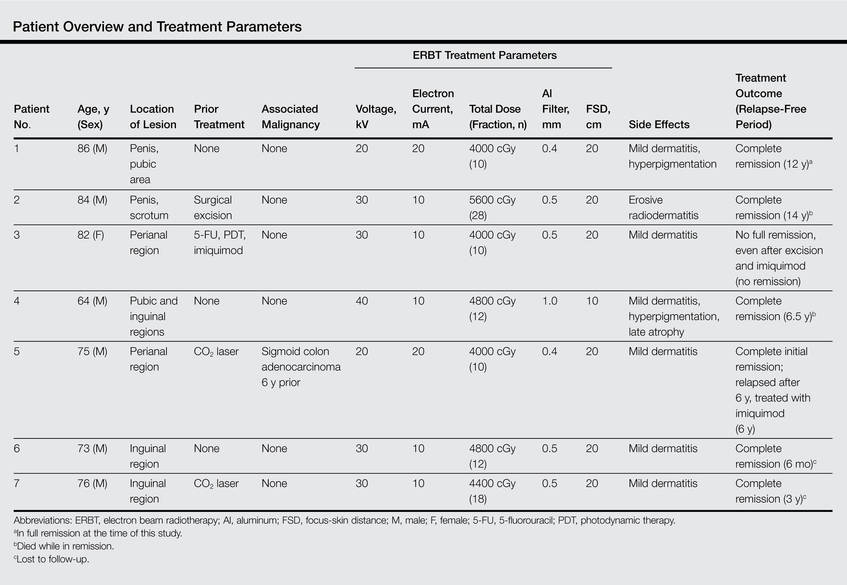
Six patients received low-energy radiotherapy of 20 to 30 kV at doses of 200 to 400 cGy per day for 2 to 5 days per week until a total dose of 4000 to 5600 cGy was completed. A 0.4- to 0.5-mm aluminum filter was used, and the focus-skin distance (FSD) was 20 cm. One patient was treated with a radiograph of 40 kV at 400 cGy per day for 2 days per week until a total dose of 4800 cGy was completed. A 1.0-mm aluminum filter was used, and the FSD was 10 cm. The field of EBRT included 2-cm margins clear of all visible disease. The treatment parameters for all patients are outlined in the Table.
Results
Complete remission was initially obtained in 6 of 7 patients. In patient 3, an erosive perianal plaque remained following treatment with EBRT that was locally treated with imiquimod cream 3%. The patient relapsed 2.5 years later with a lesion in the vaginal area that was treated with imiquimod cream 3% and later via surgical excision. Complete remission was never achieved, and the patient died 7 years after EBRT treatment due to unrelated causes. Patient 5 relapsed after 6 years of remission following treatment with EBRT and also was treated with imiquimod.
At the time of this study, 1 patient remained in full remission (patient 1: 12 years) and 2 had died while in remission (patient 2: 14 years; patient 4: 6.5 years). Two patients were lost to follow-up while in remision (patient 6: 6 months; patient 7: 3 years); however, they did not show any signs of relapse. The Figure shows patient 6 at baseline and at 4 and 8 months after starting treatment with ERBT.
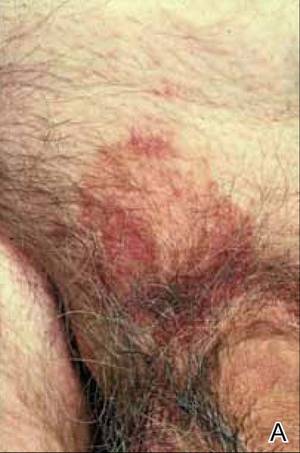 | 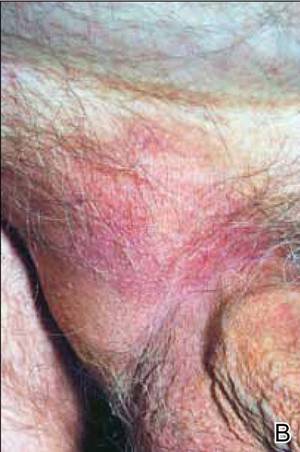 | 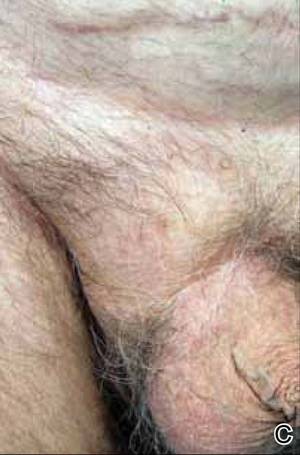 | ||
A 73-year-old man with extramammary Paget disease in the inguinal region at baseline (A) and 4 (B) and 8 (C) months after starting treatment with electron beam radiotherapy. | ||||
The most commonly reported side effect was mild dermatitis with reddening and desquamation. Patient 2 developed erosive radiodermatitis 4 days after the first treatment with EBRT. All acute reactions resolved with local treatment. Late side effects of EBRT were hyperpigmentation (patients 1 and 4) and mild skin atrophy (patient 4).
Comment
Because EMPD is such a rare disease, data regarding long-term treatment outcomes are mostly from small studies and case reports; evidence in the literature regarding treatment of EMPD with EBRT is especially limited. However, the good initial healing in most reported cases, the relatively low and late relapse rate, and the mild side effects reported in most cases make EBRT an effective treatment of EMPD. In the current study, initial complete remission was achieved in 6 of 7 patients. Patient 3 did not show complete macroscopic remission following EBRT but had a poor response to treatment in general, as she had already been unsuccessfully treated with several local treatments prior to EBRT; also, surgical and topical intervention following EBRT was not successful. Patient 5 relapsed after 6 years, but this case exceeds the follow-up period of many cases of EMPD found in the literature.
Overall, EBRT was well tolerated by the patients included in our study. All patients showed mild dermatitis following treatment as an acute reaction to EBRT. In most cases, these reactions resolved on their own or with topical treatment. Two patients developed late hyperpigmentation and one developed mild skin atrophy in the treatment area. One patient who was treated until a total dose of 5600 cGy was achieved developed erosive radiodermatitis, whereas the other patients were only treated 2 to 5 times per week. Side effects can therefore be considered as mild and/or easily controlled.
Luk et al1 also observed a low rate of long-term relapse in patients with EMPD, but consistent EBRT with similar doses and settings were applied in our study. The following parameters showed the best results in treatment response, low side effects, and relapse rate: total dose of 4000 to 4800 cGy; 20 to 30 kV; electron current of 10 to 20 mA; 0.4- to 0.5-mm aluminum filter; 20-cm FSD. This dose is at the low end of those for the standard fractionation regimen, which is a total dose of 4200 to 7000 cGy using 200-cGy fractions.1 The dose we used was slightly lower than the total dose recommended by Besa et al5 who treated 65 patients with radiotherapy for EMPD in 1992 (>50 Gy). It is equivalent to the doses used by Burrows et al6 and by Moreno-Arias et al3 (40–50 Gy). Lower radiograph doses may put treatment outcome at risk.7
Surgery is considered the first-line therapy for EMPD. Positive margin rates vary from 36% to 67% depending on the size of the lesion and the type of surgery that is used.5 Positive margin rates lead to a significant increase in recurrence rate (P<.001).8 Relapse rates for surgical intervention vary in the literature from 19% to 44%8 and 40% to 45% within 4 years of surgery.4 Wang et al8 reviewed long-term outcomes of surgical treatment in 130 Chinese patients with penoscrotal EMPD. They recommended 3-cm surgical margins and frozen section pathological examination for complicated conditions. A local recurrence rate of 9.9% was reported, which is remarkably lower than in many other studies in the literature.8 Nevertheless, the severe possible side effects of surgery cannot be easily put aside.
Electron beam radiotherapy should be considered as an alternate therapy in EMPD given its low risks and moderate side effects. In our study, the relapse rate was 28.6% (2/7), which is not remarkably higher than reports in the literature of relapse rates associated with surgical excision. Electron beam radiotherapy should be especially considered when extensive margin-controlled surgery is not an option, such as EMPD in sensitive areas or for an extensive circumference of the lesion, as surgery might then produce functional disfiguring results. Adequate limiting ray (grenz ray) or low-energy radiograph treatment has proved to preserve function, especially in the area of the vulva and glans penis.9 Furthermore, EBRT may be the treatment of choice in patients with an increased risk for morbidity from surgery, such as elderly patients5 or those with wound healing disorders (eg, diabetes mellitus).
Conclusion
Given that EMPD patients typically are elderly with multimorbidities, surgery should be carefully considered in this patient population, particularly because EMPD without underlying malignancies has an excellent survival rate.5 Highly invasive treatments should therefore be thoughtfully considered. Because of the inconsistent data on relapse rates and the small number of patients with EMPD who have been studied, further study with more cases is needed.
1. Luk NM, Yu KH, Yeung WK, et al. Extramammary Paget’s disease: outcome of radiotherapy with curative intent. Clin Exp Dermatol. 2003;28:360-363.
2. Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease. J Clin Pathol. 2000;53:742-749.
3. Moreno-Arias GA, Conill C, Castells-Mas A, et al. Radiotherapy for genital extramammary Paget’s disease in situ. Dermatol Surg. 2001;27:587-590.
4. Son SH, Lee JS, Kim YS, et al. The role of radiation therapy for the extramammary Paget’s disease of the vulva; experience of 3 cases. Cancer Res Treat. 2005;37:365-369.
5. Besa P, Rich TA, Delclos L, et al. Extramammary Paget’s disease of the perineal skin: role of radiotherapy. Int J Radiat Oncol Biol Phys. 1992;24:73-78.
6. Burrows NP, Jones DH, Hudson PM, et al. Treatment of extramammary Paget’s disease by radiotherapy. Br J Dermatol. 1995;132:970-972.
7. Jensen SL, Sjølin KE, Shokouh-Amiri MH, et al. Paget’s disease of the anal margin. Br J Surg. 1988;75:1089-1092.
8. Wang Z, Lu M, Dong GQ, et al. Penile and scrotal Paget’s disease: 130 Chinese patients with long-term follow-up. BJU Int. 2008;102:485-488.
9. Dummer R, ed. Physikalische Therapiemaßnahmen in der Dermatologie. 2nd ed. Darmstadt, Germany: Steinkopff Verlag Darmstadt; 2006.
Extramammary Paget disease (EMPD) is an insidious intraepithelial neoplasm that occurs in areas with a high density of apocrine glands such as the penoscrotal area, the vulva, and occasionally the axillae. It mainly affects patients aged 50 to 80 years.1 Clinically, EMPD presents as pruritic, nonhealing, red plaques that can be mistaken for eczema. On histology, characteristic Paget cells have abundant pale cytoplasm and atypical nuclear lobuli and are adenocarcinomatous,1,2 usually infiltrating the epidermis.2 In approximately 25% of cases, EMPD is associated with neoplastic disease in adnexal structures or organs with a contiguous epithelial lining.2 Therefore, screening for an underlying malignancy when EMPD is first diagnosed is indispensable.
Because EMPD tends to be multifocal, presents in elderly patients, and affects functionally important areas such as the anal canal or genitals, treatment often is difficult.3,4 Surgery generally is considered as a first-line treatment5; however, the rate of positive margins ranges from 36% to 67%, and local recurrence is common.1
Radiotherapy has been used in EMPD patients mainly when surgery was not an option or was not effective, but several reports have indicated that it should play a more important role in the treatment of EMPD. Luk et al1 described 6 patients who were treated with different types of radiotherapy. Similar to the results of prior studies,3,5,6 they concluded that it was an effective treatment of EMPD.1
We conducted a retrospective study to analyze long-term outcomes in 7 patients who were treated with external beam radiotherapy (EBRT) for EMPD.
Methods
Seven patients (6 men and 1 woman) who had been diagnosed with EMPD and were treated with EBRT at the Department of Dermatology at the University Hospital Zurich in Switzerland (1988-2004) were evaluated. The diagnosis was confirmed by a dermatopathologist or pathologist via histology. Data regarding clinical presentation, EBRT regimen, and side effects were retrieved from the medical records. Long-term outcomes were evaluated by an attending dermatologist (1 case), a general practitioner (5 cases), or the hospital’s outpatient department (1 case). None of the patients showed an associated malignancy at the time of treatment; however, patient 5 had been diagnosed with and treated for a sigmoid colon adenocarcinoma 6 years prior to undergoing EBRT for EMPD. Three patients (patients 3, 5, and 7) received EBRT for local relapse after prior treatment of EMPD (ie, CO2 laser, multiple local treatments). One patient (patient 2) underwent surgical excision prior to EBRT. The remaining 3 patients had not undergone any prior treatment of EMPD. All patients underwent EBRT with the goal of complete remission.
Six patients received low-energy radiotherapy of 20 to 30 kV at doses of 200 to 400 cGy per day for 2 to 5 days per week until a total dose of 4000 to 5600 cGy was completed. A 0.4- to 0.5-mm aluminum filter was used, and the focus-skin distance (FSD) was 20 cm. One patient was treated with a radiograph of 40 kV at 400 cGy per day for 2 days per week until a total dose of 4800 cGy was completed. A 1.0-mm aluminum filter was used, and the FSD was 10 cm. The field of EBRT included 2-cm margins clear of all visible disease. The treatment parameters for all patients are outlined in the Table.
Results
Complete remission was initially obtained in 6 of 7 patients. In patient 3, an erosive perianal plaque remained following treatment with EBRT that was locally treated with imiquimod cream 3%. The patient relapsed 2.5 years later with a lesion in the vaginal area that was treated with imiquimod cream 3% and later via surgical excision. Complete remission was never achieved, and the patient died 7 years after EBRT treatment due to unrelated causes. Patient 5 relapsed after 6 years of remission following treatment with EBRT and also was treated with imiquimod.
At the time of this study, 1 patient remained in full remission (patient 1: 12 years) and 2 had died while in remission (patient 2: 14 years; patient 4: 6.5 years). Two patients were lost to follow-up while in remision (patient 6: 6 months; patient 7: 3 years); however, they did not show any signs of relapse. The Figure shows patient 6 at baseline and at 4 and 8 months after starting treatment with ERBT.
| | | ||
A 73-year-old man with extramammary Paget disease in the inguinal region at baseline (A) and 4 (B) and 8 (C) months after starting treatment with electron beam radiotherapy. | ||||
The most commonly reported side effect was mild dermatitis with reddening and desquamation. Patient 2 developed erosive radiodermatitis 4 days after the first treatment with EBRT. All acute reactions resolved with local treatment. Late side effects of EBRT were hyperpigmentation (patients 1 and 4) and mild skin atrophy (patient 4).
Comment
Because EMPD is such a rare disease, data regarding long-term treatment outcomes are mostly from small studies and case reports; evidence in the literature regarding treatment of EMPD with EBRT is especially limited. However, the good initial healing in most reported cases, the relatively low and late relapse rate, and the mild side effects reported in most cases make EBRT an effective treatment of EMPD. In the current study, initial complete remission was achieved in 6 of 7 patients. Patient 3 did not show complete macroscopic remission following EBRT but had a poor response to treatment in general, as she had already been unsuccessfully treated with several local treatments prior to EBRT; also, surgical and topical intervention following EBRT was not successful. Patient 5 relapsed after 6 years, but this case exceeds the follow-up period of many cases of EMPD found in the literature.
Overall, EBRT was well tolerated by the patients included in our study. All patients showed mild dermatitis following treatment as an acute reaction to EBRT. In most cases, these reactions resolved on their own or with topical treatment. Two patients developed late hyperpigmentation and one developed mild skin atrophy in the treatment area. One patient who was treated until a total dose of 5600 cGy was achieved developed erosive radiodermatitis, whereas the other patients were only treated 2 to 5 times per week. Side effects can therefore be considered as mild and/or easily controlled.
Luk et al1 also observed a low rate of long-term relapse in patients with EMPD, but consistent EBRT with similar doses and settings were applied in our study. The following parameters showed the best results in treatment response, low side effects, and relapse rate: total dose of 4000 to 4800 cGy; 20 to 30 kV; electron current of 10 to 20 mA; 0.4- to 0.5-mm aluminum filter; 20-cm FSD. This dose is at the low end of those for the standard fractionation regimen, which is a total dose of 4200 to 7000 cGy using 200-cGy fractions.1 The dose we used was slightly lower than the total dose recommended by Besa et al5 who treated 65 patients with radiotherapy for EMPD in 1992 (>50 Gy). It is equivalent to the doses used by Burrows et al6 and by Moreno-Arias et al3 (40–50 Gy). Lower radiograph doses may put treatment outcome at risk.7
Surgery is considered the first-line therapy for EMPD. Positive margin rates vary from 36% to 67% depending on the size of the lesion and the type of surgery that is used.5 Positive margin rates lead to a significant increase in recurrence rate (P<.001).8 Relapse rates for surgical intervention vary in the literature from 19% to 44%8 and 40% to 45% within 4 years of surgery.4 Wang et al8 reviewed long-term outcomes of surgical treatment in 130 Chinese patients with penoscrotal EMPD. They recommended 3-cm surgical margins and frozen section pathological examination for complicated conditions. A local recurrence rate of 9.9% was reported, which is remarkably lower than in many other studies in the literature.8 Nevertheless, the severe possible side effects of surgery cannot be easily put aside.
Electron beam radiotherapy should be considered as an alternate therapy in EMPD given its low risks and moderate side effects. In our study, the relapse rate was 28.6% (2/7), which is not remarkably higher than reports in the literature of relapse rates associated with surgical excision. Electron beam radiotherapy should be especially considered when extensive margin-controlled surgery is not an option, such as EMPD in sensitive areas or for an extensive circumference of the lesion, as surgery might then produce functional disfiguring results. Adequate limiting ray (grenz ray) or low-energy radiograph treatment has proved to preserve function, especially in the area of the vulva and glans penis.9 Furthermore, EBRT may be the treatment of choice in patients with an increased risk for morbidity from surgery, such as elderly patients5 or those with wound healing disorders (eg, diabetes mellitus).
Conclusion
Given that EMPD patients typically are elderly with multimorbidities, surgery should be carefully considered in this patient population, particularly because EMPD without underlying malignancies has an excellent survival rate.5 Highly invasive treatments should therefore be thoughtfully considered. Because of the inconsistent data on relapse rates and the small number of patients with EMPD who have been studied, further study with more cases is needed.
Extramammary Paget disease (EMPD) is an insidious intraepithelial neoplasm that occurs in areas with a high density of apocrine glands such as the penoscrotal area, the vulva, and occasionally the axillae. It mainly affects patients aged 50 to 80 years.1 Clinically, EMPD presents as pruritic, nonhealing, red plaques that can be mistaken for eczema. On histology, characteristic Paget cells have abundant pale cytoplasm and atypical nuclear lobuli and are adenocarcinomatous,1,2 usually infiltrating the epidermis.2 In approximately 25% of cases, EMPD is associated with neoplastic disease in adnexal structures or organs with a contiguous epithelial lining.2 Therefore, screening for an underlying malignancy when EMPD is first diagnosed is indispensable.
Because EMPD tends to be multifocal, presents in elderly patients, and affects functionally important areas such as the anal canal or genitals, treatment often is difficult.3,4 Surgery generally is considered as a first-line treatment5; however, the rate of positive margins ranges from 36% to 67%, and local recurrence is common.1
Radiotherapy has been used in EMPD patients mainly when surgery was not an option or was not effective, but several reports have indicated that it should play a more important role in the treatment of EMPD. Luk et al1 described 6 patients who were treated with different types of radiotherapy. Similar to the results of prior studies,3,5,6 they concluded that it was an effective treatment of EMPD.1
We conducted a retrospective study to analyze long-term outcomes in 7 patients who were treated with external beam radiotherapy (EBRT) for EMPD.
Methods
Seven patients (6 men and 1 woman) who had been diagnosed with EMPD and were treated with EBRT at the Department of Dermatology at the University Hospital Zurich in Switzerland (1988-2004) were evaluated. The diagnosis was confirmed by a dermatopathologist or pathologist via histology. Data regarding clinical presentation, EBRT regimen, and side effects were retrieved from the medical records. Long-term outcomes were evaluated by an attending dermatologist (1 case), a general practitioner (5 cases), or the hospital’s outpatient department (1 case). None of the patients showed an associated malignancy at the time of treatment; however, patient 5 had been diagnosed with and treated for a sigmoid colon adenocarcinoma 6 years prior to undergoing EBRT for EMPD. Three patients (patients 3, 5, and 7) received EBRT for local relapse after prior treatment of EMPD (ie, CO2 laser, multiple local treatments). One patient (patient 2) underwent surgical excision prior to EBRT. The remaining 3 patients had not undergone any prior treatment of EMPD. All patients underwent EBRT with the goal of complete remission.
Six patients received low-energy radiotherapy of 20 to 30 kV at doses of 200 to 400 cGy per day for 2 to 5 days per week until a total dose of 4000 to 5600 cGy was completed. A 0.4- to 0.5-mm aluminum filter was used, and the focus-skin distance (FSD) was 20 cm. One patient was treated with a radiograph of 40 kV at 400 cGy per day for 2 days per week until a total dose of 4800 cGy was completed. A 1.0-mm aluminum filter was used, and the FSD was 10 cm. The field of EBRT included 2-cm margins clear of all visible disease. The treatment parameters for all patients are outlined in the Table.
Results
Complete remission was initially obtained in 6 of 7 patients. In patient 3, an erosive perianal plaque remained following treatment with EBRT that was locally treated with imiquimod cream 3%. The patient relapsed 2.5 years later with a lesion in the vaginal area that was treated with imiquimod cream 3% and later via surgical excision. Complete remission was never achieved, and the patient died 7 years after EBRT treatment due to unrelated causes. Patient 5 relapsed after 6 years of remission following treatment with EBRT and also was treated with imiquimod.
At the time of this study, 1 patient remained in full remission (patient 1: 12 years) and 2 had died while in remission (patient 2: 14 years; patient 4: 6.5 years). Two patients were lost to follow-up while in remision (patient 6: 6 months; patient 7: 3 years); however, they did not show any signs of relapse. The Figure shows patient 6 at baseline and at 4 and 8 months after starting treatment with ERBT.
| | | ||
A 73-year-old man with extramammary Paget disease in the inguinal region at baseline (A) and 4 (B) and 8 (C) months after starting treatment with electron beam radiotherapy. | ||||
The most commonly reported side effect was mild dermatitis with reddening and desquamation. Patient 2 developed erosive radiodermatitis 4 days after the first treatment with EBRT. All acute reactions resolved with local treatment. Late side effects of EBRT were hyperpigmentation (patients 1 and 4) and mild skin atrophy (patient 4).
Comment
Because EMPD is such a rare disease, data regarding long-term treatment outcomes are mostly from small studies and case reports; evidence in the literature regarding treatment of EMPD with EBRT is especially limited. However, the good initial healing in most reported cases, the relatively low and late relapse rate, and the mild side effects reported in most cases make EBRT an effective treatment of EMPD. In the current study, initial complete remission was achieved in 6 of 7 patients. Patient 3 did not show complete macroscopic remission following EBRT but had a poor response to treatment in general, as she had already been unsuccessfully treated with several local treatments prior to EBRT; also, surgical and topical intervention following EBRT was not successful. Patient 5 relapsed after 6 years, but this case exceeds the follow-up period of many cases of EMPD found in the literature.
Overall, EBRT was well tolerated by the patients included in our study. All patients showed mild dermatitis following treatment as an acute reaction to EBRT. In most cases, these reactions resolved on their own or with topical treatment. Two patients developed late hyperpigmentation and one developed mild skin atrophy in the treatment area. One patient who was treated until a total dose of 5600 cGy was achieved developed erosive radiodermatitis, whereas the other patients were only treated 2 to 5 times per week. Side effects can therefore be considered as mild and/or easily controlled.
Luk et al1 also observed a low rate of long-term relapse in patients with EMPD, but consistent EBRT with similar doses and settings were applied in our study. The following parameters showed the best results in treatment response, low side effects, and relapse rate: total dose of 4000 to 4800 cGy; 20 to 30 kV; electron current of 10 to 20 mA; 0.4- to 0.5-mm aluminum filter; 20-cm FSD. This dose is at the low end of those for the standard fractionation regimen, which is a total dose of 4200 to 7000 cGy using 200-cGy fractions.1 The dose we used was slightly lower than the total dose recommended by Besa et al5 who treated 65 patients with radiotherapy for EMPD in 1992 (>50 Gy). It is equivalent to the doses used by Burrows et al6 and by Moreno-Arias et al3 (40–50 Gy). Lower radiograph doses may put treatment outcome at risk.7
Surgery is considered the first-line therapy for EMPD. Positive margin rates vary from 36% to 67% depending on the size of the lesion and the type of surgery that is used.5 Positive margin rates lead to a significant increase in recurrence rate (P<.001).8 Relapse rates for surgical intervention vary in the literature from 19% to 44%8 and 40% to 45% within 4 years of surgery.4 Wang et al8 reviewed long-term outcomes of surgical treatment in 130 Chinese patients with penoscrotal EMPD. They recommended 3-cm surgical margins and frozen section pathological examination for complicated conditions. A local recurrence rate of 9.9% was reported, which is remarkably lower than in many other studies in the literature.8 Nevertheless, the severe possible side effects of surgery cannot be easily put aside.
Electron beam radiotherapy should be considered as an alternate therapy in EMPD given its low risks and moderate side effects. In our study, the relapse rate was 28.6% (2/7), which is not remarkably higher than reports in the literature of relapse rates associated with surgical excision. Electron beam radiotherapy should be especially considered when extensive margin-controlled surgery is not an option, such as EMPD in sensitive areas or for an extensive circumference of the lesion, as surgery might then produce functional disfiguring results. Adequate limiting ray (grenz ray) or low-energy radiograph treatment has proved to preserve function, especially in the area of the vulva and glans penis.9 Furthermore, EBRT may be the treatment of choice in patients with an increased risk for morbidity from surgery, such as elderly patients5 or those with wound healing disorders (eg, diabetes mellitus).
Conclusion
Given that EMPD patients typically are elderly with multimorbidities, surgery should be carefully considered in this patient population, particularly because EMPD without underlying malignancies has an excellent survival rate.5 Highly invasive treatments should therefore be thoughtfully considered. Because of the inconsistent data on relapse rates and the small number of patients with EMPD who have been studied, further study with more cases is needed.
1. Luk NM, Yu KH, Yeung WK, et al. Extramammary Paget’s disease: outcome of radiotherapy with curative intent. Clin Exp Dermatol. 2003;28:360-363.
2. Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease. J Clin Pathol. 2000;53:742-749.
3. Moreno-Arias GA, Conill C, Castells-Mas A, et al. Radiotherapy for genital extramammary Paget’s disease in situ. Dermatol Surg. 2001;27:587-590.
4. Son SH, Lee JS, Kim YS, et al. The role of radiation therapy for the extramammary Paget’s disease of the vulva; experience of 3 cases. Cancer Res Treat. 2005;37:365-369.
5. Besa P, Rich TA, Delclos L, et al. Extramammary Paget’s disease of the perineal skin: role of radiotherapy. Int J Radiat Oncol Biol Phys. 1992;24:73-78.
6. Burrows NP, Jones DH, Hudson PM, et al. Treatment of extramammary Paget’s disease by radiotherapy. Br J Dermatol. 1995;132:970-972.
7. Jensen SL, Sjølin KE, Shokouh-Amiri MH, et al. Paget’s disease of the anal margin. Br J Surg. 1988;75:1089-1092.
8. Wang Z, Lu M, Dong GQ, et al. Penile and scrotal Paget’s disease: 130 Chinese patients with long-term follow-up. BJU Int. 2008;102:485-488.
9. Dummer R, ed. Physikalische Therapiemaßnahmen in der Dermatologie. 2nd ed. Darmstadt, Germany: Steinkopff Verlag Darmstadt; 2006.
1. Luk NM, Yu KH, Yeung WK, et al. Extramammary Paget’s disease: outcome of radiotherapy with curative intent. Clin Exp Dermatol. 2003;28:360-363.
2. Lloyd J, Flanagan AM. Mammary and extramammary Paget’s disease. J Clin Pathol. 2000;53:742-749.
3. Moreno-Arias GA, Conill C, Castells-Mas A, et al. Radiotherapy for genital extramammary Paget’s disease in situ. Dermatol Surg. 2001;27:587-590.
4. Son SH, Lee JS, Kim YS, et al. The role of radiation therapy for the extramammary Paget’s disease of the vulva; experience of 3 cases. Cancer Res Treat. 2005;37:365-369.
5. Besa P, Rich TA, Delclos L, et al. Extramammary Paget’s disease of the perineal skin: role of radiotherapy. Int J Radiat Oncol Biol Phys. 1992;24:73-78.
6. Burrows NP, Jones DH, Hudson PM, et al. Treatment of extramammary Paget’s disease by radiotherapy. Br J Dermatol. 1995;132:970-972.
7. Jensen SL, Sjølin KE, Shokouh-Amiri MH, et al. Paget’s disease of the anal margin. Br J Surg. 1988;75:1089-1092.
8. Wang Z, Lu M, Dong GQ, et al. Penile and scrotal Paget’s disease: 130 Chinese patients with long-term follow-up. BJU Int. 2008;102:485-488.
9. Dummer R, ed. Physikalische Therapiemaßnahmen in der Dermatologie. 2nd ed. Darmstadt, Germany: Steinkopff Verlag Darmstadt; 2006.
Practice Points
- Elderly patients with extramammary Paget disease (EMPD) usually are multimorbid and frail.
- Nonsurgical options for treatment of EMPD can be advantageous. External beam radiotherapy is a good option for EMPD.
Factors Affecting Academic Leadership in Dermatology
Leadership is widely recognized as a key component in the role of a physician,1 which is especially true in dermatology, a specialty that faces severe challenges in the recruitment and retention of academic faculty.2 A study of the dermatology workforce found that academic institutions are more likely to be seeking to hire new faculty2 and that many dermatology residency programs often are looking to replace chairpersons (chairs) and/or chiefs.3 Although fewer dermatology residents are pursuing academic careers than careers in private practice, full-time faculty members also are leaving their academic posts. This shift is demonstrated by the younger mean age of academic dermatologists2 and the increased rate of departure from academia prior to pursuing more formalized leadership roles.4
It has been suggested that the number of full-time faculty and number of faculty publications positively influence graduates of dermatology residency programs to pursue academic careers; however, variables affecting the likelihood of graduates of dermatology residency programs becoming academic leaders later in their career have not been well studied.3 The purpose of this study is to determine the factors that influence the development of program chairs/chiefs and program directors (PDs) of dermatology residency programs.
Methods
Data were collected from all accredited dermatology residency programs in the United States as of December 31, 2008. Residency programs that were started after 2004 were excluded from the study, as it was thought that these programs may not have graduated a sufficient number of residents for assessment. Military residency programs also were excluded, as graduates from these programs often do not freely choose their careers after residency.
Primary end points were the number of chairs/chiefs and PDs who had graduated from each dermatology residency program. Variables included the number of years the program had been in existence, status of the program as a department or division, number of full-time faculty members, number of residents, National Institutes of Health funding received in 2008 (http://www.report.nih.gov/award/index.cfm), Dermatology Foundation (DF) funding received (http://www.dermatologyfoundation.org/rap/), number of publications from full-time faculty members in 2008 (http://www.ncbi.nlm.nih.gov/pubmed/), number of faculty lectures given at annual meetings of 5 societies in 2008 (American Academy of Dermatology, the Society for Investigative Dermatology, the American Society of Dermatopathology, the Society for Pediatric Dermatology, and American Society for Dermatologic Surgery), and the number of faculty members on the editorial boards of 6 major dermatology journals (Journal of the American Academy of Dermatology, Journal of Investigative Dermatology, Archives of Dermatology [currently known as JAMA Dermatology], Dermatologic Surgery, Pediatric Dermatology, and Journal of Cutaneous Pathology). Data regarding faculty and residents were obtained from program Web sites and inquiries from individual programs. The year 1974 was used as a cutoff for the number of years a program had been in existence. Years of existence of a program was controlled for in the analysis. The ratio of faculty to residents was calculated per year and categorized as 4 or more or less than 4 to minimize the effect of changing program size over the years. For faculty members who split time between 2 residency programs, each program was given credit for the duration of time spent at that program. Faculty members who hold a PhD only and those who completed their residencies in non-US dermatology residency programs were excluded from the outcome variables. To avoid duplicate faculty publications, collections for each residency program were created within PubMed (ie, if 2 authors from the same program coauthored an article, it was only counted once toward the total number of faculty publications from that program).
Because the data were skewed (ie, there were a large number of programs with 0 graduating chairs/chiefs and PDs), nonparametric analyses were utilized. Logistic regression was used to calculate the odds of producing chairs/chiefs or PDs (yes vs no). Multiple logistic regression helped to determine those variables that were most closely associated with odds of graduating a chair/chief or PD. Variables with a significance level of P<.10 were considered in the multiple logistic regression, and backward selection was used to determine a model. Multiple linear regression was used to determine correlation coefficients for each of the variables and the number of chairs/chiefs or PDs graduated, controlling for the estimated number of graduates from the program and number of years the program had been in existence. Analyses for graduating chairs/chiefs and PDs were conducted separately. The final significance level used was P<.05. Data were analyzed using SAS version 9.3. This study was approved by the institutional review board at Kaiser Permanente Southern California.
Results
Data from 103 dermatology residency programs were included in the analysis. Of these programs, 47 had graduated at least 1 chair/chief and 55 had graduated at least 1 PD. Among the programs graduating any chairs/chiefs, they produced an average of 2.04 chairs/chiefs and 1.86 PDs. The 5 dermatology residency programs that graduated the highest total number of chairs/chiefs and PDs were Harvard University (Cambridge, Massachusetts), the University of Michigan (Ann Arbor, Michigan), New York University (New York, New York), Yale-New Haven Hospital (New Haven, Connecticut), and the University of Minnesota (Minneapolis, Minnesota).
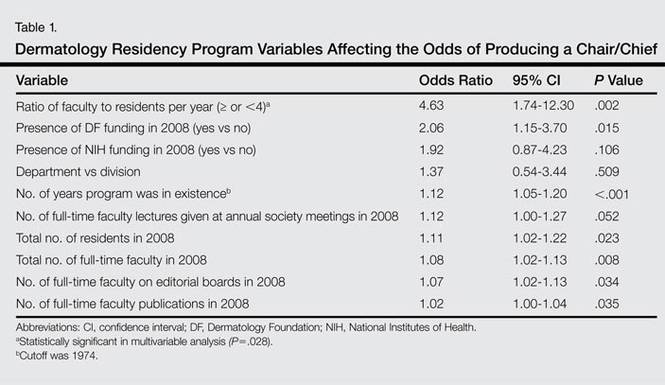
Factors that had the highest effect on the odds of a program graduating a chair/chief included the ratio of faculty to residents per year, presence of DF funding in 2008, number of years program was in existence, number of residents, number of full-time faculty, and number of full-time faculty on editorial boards of 6 major dermatology journals (Table 1). When controlling for each of these variables in the final multivariable analysis, programs with 4 or more faculty per resident had 3.31 times the odds of producing a chair/chief (95% confidence interval [CI], 1.14-9.66; P=.028).
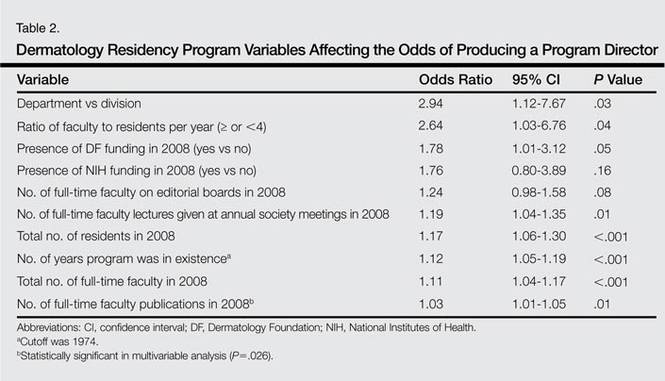
Factors that had the highest effect on the odds of a program graduating a PD included status as department versus division, ratio of faculty to residents per year, presence of DF funding in 2008, number of lectures given by full-time faculty members at annual society meetings, number of residents, number of years program was in existence, number of full-time faculty, and number of publications from full-time faculty members (Table 2). The most significant factor associated with graduating PDs after controlling for other variables was the number of publications from full-time faculty members. The odds increased by 3.2% for every 1 additional publication and 32% for every 10 additional publications (95% CI, 1.01-1.06; P=.026).
Multiple linear regression demonstrated a positive relationship between the number of graduating chairs/chiefs and total full-time faculty members (R2=0.26; P=.034) and ratio of full-time faculty to residents (R2=0.29; P<.001). Marginally significant correlations were seen between the number of PDs and ratio of full-time faculty to residents (R2=0.32; P=.05) as well as the number of publications from full-time faculty members (R2=0.32; P=.05).
Comment
The ratio of full-time faculty to residents increased a program’s odds of graduating a chair/chief. More faculty members may lead to more opportunities for mentorship of residents and young faculty. Mentors are widely perceived to be integral to the learning and development of residents, not only in dermatology5 but across all specialties.6 Mentors also have been noted to play a key role in bolstering and maintaining interest in academics,7 which is true not only with regard to recruiting new residents but for retaining young faculty members. In a study (N=109) that examined factors associated with residents’ loss of interest in academic careers, half of the participants reported a lack of effective mentors, role models, and professional guidance.8 Mentors provide teaching, supervision, and advice, especially with regard to research and career paths.9 A large number of faculty members provides more opportunities for direct mentorship and offers residents more exposure to research, specialty clinics, and academic philosophies, which may positively influence and even inspire academic pursuits and leadership.3
Although the solution to producing future chairs/chiefs and PDs may lie in faculty guidance, finding and retaining faculty members as mentors amidst a shortage of academic dermatologists presents an underlying issue.3 In addition to a lack of mentorship, residents cite bureaucracy, salary differentials, and location to explain a loss of interest in academic careers.8 Several programs have been developed to address the recruitment of dermatology residents for academic careers, including combined medical-dermatology programs, 2+2 programs (2 years of clinical residency plus 2 additional research years), clinical research fellowships,10 and the Society for Investigative Dermatology’s Dermatology Resident Retreat for Future Academicians (http://www.sidnet.org/fortraineesandresidents).11 Perhaps recruitment should even start at the medical student level. In light of the academic strength of the current pool of dermatology residency applicants,12 training programs should continue to screen for applicants with sincere interests in academia.13 Students with more research and publications may be more likely to pursue academic careers, in accordance with prior studies of dermatology trainees.3,14 Studies also have shown that graduates of foreign dermatology residencies15 and individuals who hold both MD and PhD degrees may be more likely to enter into academic careers.16
For creating future chairs/chiefs and PDs, retention of young faculty in academics is as important as recruiting residents.17 At the mid-career level, the decline of funds for research has generated pressure for academic physicians to see increasing numbers of patients, leaving insufficient time for the many duties that accompany academic posts,2 including teaching and publishing. Other reasons that faculty members leave their posts before 40 years of age include financial and family concerns18 as well as the desire for more autonomy.4 Formalized training is seen with the American Academy of Dermatology’s Academic Dermatology Leadership Program (https://www.aad.org/members/leadership-institute/mentoring/aad-mentoring-opportunities/academic-dermatology-leadership-program-mentee), which promotes advanced leadership training to recent graduates.5 Other methods include support of young faculty with mentorship; grant applications; and administration at the department, hospital, and government levels.17 Recruitment of faculty from private practice may represent another potential source of faculty who wish to pursue more scholarly endeavors.4 Teaching has been cited as a primary reason for faculty members to remain in academia,18 and thus time for teaching must be protected. Such a strategy is in accordance with our findings that amount of annual DF funding received, number of full-time faculty publications, number of faculty members on editorial boards of major dermatology journals, and number of lectures given by full-time faculty factors at annual society meetings are positively associated with the odds of producing chairs/chiefs or PDs. In particular, the number of full-time faculty publications is directly related to increased odds of graduates becoming PDs. Residents and young faculty members who take part in research and attend national conferences may find inspiration or develop a passion for academic leadership.
A limitation to this study is that the ratio of faculty to graduated residents for some programs likely has fluctuated over the last 35 years. This study assumed that certain programs remained generally small or large during the course of their existence, which was controlled by using the ratio between faculty and residents. Additionally, the number of years that a program has been in existence influences the likelihood of that program to graduate higher numbers of residents who become chairs/chiefs or PDs. As a result, we used multiple linear regression to control for the number of residents and number of years that a program had been in existence. Finally, while the relationship between academic leaders and research may be explained by the increased likelihood of faculty with more funding, publications, or lectures to be selected for leadership roles, this finding supports the notion that research can contribute to leadership. This analysis identifies modifiable factors among residency training programs to improve the odds of graduating future academic leaders.
Conclusion
As the present study shows, the ratio of faculty to residents and the number of full-time faculty publications are key to graduating academic leaders in dermatology. Retention of faculty as leaders in academic dermatology is as crucial to the field as recruitment of residents into academic dermatology. Mentorship should be highly encouraged through the creation of formal programs and should not end at the resident level. Emphasizing the intellectually stimulating aspects of academia and providing administrative resources may help decrease the burden of academic duties, allowing the pursuit of teaching and research and ultimately the resources to become candidates for leadership positions in academia.
1. Wood LD, Miller JJ, Marks JG Jr. The need for leadership: how can we better train the next generation of dermatologists? J Am Acad Dermatol. 2008;58:715-717.
2. Resneck JS Jr, Tierney EP, Kimball AB. Challenges facing academic dermatology: survey data on the faculty workforce. J Am Acad Dermatol. 2006;54:211-216.
3. Wu JJ, Ramirez CC, Alonso CA, et al. Dermatology residency program characteristics that correlate with graduates selecting an academic dermatology career. Arch Dermatol. 2006;142:845-850.
4. Loo DS, Liu CL, Geller AC, et al. Academic dermatology manpower: issues of recruitment and retention. Arch Dermatol. 2007;143:341-347.
5. Donovan JC. A survey of dermatology residency program directors’ views on mentorship. Dermatol Online J. 2009;15:1.
6. Sambunjak D, Straus SE, Marusi´c A. Mentoring in academic medicine: a systematic review. JAMA. 2006;296:1103-1115.
7. Rubenstein DS, Blauvelt A, Chen SC, et al. The future of academic dermatology in the United States: report on the resident retreat for future physician-scientists, June 15-17, 2001. J Am Acad Dermatol. 2002;47:300-303.
8. Reck SJ, Stratman EJ, Vogel C, et al. Assessment of residents’ loss of interest in academic careers and identification of correctable factors. Arch Dermatol. 2006;142:855-858.
9. Clark RA. Teacher, supervisor, adviser, or mentor? J Invest Dermatol. 2011;131:1779-1780.
10. Wu JJ. Current strategies to address the ongoing shortage of academic dermatologists. J Am Acad Dermatol. 2007;56:1065-1066.
11. Culton DA, Rubenstein DS, Diaz LA. The resident retreat for future academicians. J Invest Dermatol. 2010;130:1775-1777.
12. Wu JJ, Tyring SK. The academic strength of current dermatology residency applicants. Dermatol Online J. 2003;9:22.
13. Kia KF, Gielczyk RA, Ellis CN. Academia is the life for me, I’m sure. Arch Dermatol. 2006;142:911-913.
14. Miller CJ, Wood GC, Miller JJ, et al. Academics or private practice? the future of dermatologic surgery education. Dermatol Surg. 2006;32:70-75, discussion 76-78.
15. Wu JJ, Davis KF, Ramirez CC, et al. Graduates-of-foreign-dermatology residencies and military dermatology residencies and women in academic dermatology. Dermatol Online J. 2009;15:2.
16. Wu JJ, Davis KF, Ramirez CC, et al. MD/PhDs are more likely than MDs to choose a career in academic dermatology. Dermatol Online J. 2008;14:27.
17. Olerud JE. Academic workforce in dermatology. Arch Dermatol. 2007;143:409-410.
18. Turner E, Yoo J, Salter S, et al. Leadership workforce in academic dermatology. Arch Dermatol. 2007;143:948-949.
Leadership is widely recognized as a key component in the role of a physician,1 which is especially true in dermatology, a specialty that faces severe challenges in the recruitment and retention of academic faculty.2 A study of the dermatology workforce found that academic institutions are more likely to be seeking to hire new faculty2 and that many dermatology residency programs often are looking to replace chairpersons (chairs) and/or chiefs.3 Although fewer dermatology residents are pursuing academic careers than careers in private practice, full-time faculty members also are leaving their academic posts. This shift is demonstrated by the younger mean age of academic dermatologists2 and the increased rate of departure from academia prior to pursuing more formalized leadership roles.4
It has been suggested that the number of full-time faculty and number of faculty publications positively influence graduates of dermatology residency programs to pursue academic careers; however, variables affecting the likelihood of graduates of dermatology residency programs becoming academic leaders later in their career have not been well studied.3 The purpose of this study is to determine the factors that influence the development of program chairs/chiefs and program directors (PDs) of dermatology residency programs.
Methods
Data were collected from all accredited dermatology residency programs in the United States as of December 31, 2008. Residency programs that were started after 2004 were excluded from the study, as it was thought that these programs may not have graduated a sufficient number of residents for assessment. Military residency programs also were excluded, as graduates from these programs often do not freely choose their careers after residency.
Primary end points were the number of chairs/chiefs and PDs who had graduated from each dermatology residency program. Variables included the number of years the program had been in existence, status of the program as a department or division, number of full-time faculty members, number of residents, National Institutes of Health funding received in 2008 (http://www.report.nih.gov/award/index.cfm), Dermatology Foundation (DF) funding received (http://www.dermatologyfoundation.org/rap/), number of publications from full-time faculty members in 2008 (http://www.ncbi.nlm.nih.gov/pubmed/), number of faculty lectures given at annual meetings of 5 societies in 2008 (American Academy of Dermatology, the Society for Investigative Dermatology, the American Society of Dermatopathology, the Society for Pediatric Dermatology, and American Society for Dermatologic Surgery), and the number of faculty members on the editorial boards of 6 major dermatology journals (Journal of the American Academy of Dermatology, Journal of Investigative Dermatology, Archives of Dermatology [currently known as JAMA Dermatology], Dermatologic Surgery, Pediatric Dermatology, and Journal of Cutaneous Pathology). Data regarding faculty and residents were obtained from program Web sites and inquiries from individual programs. The year 1974 was used as a cutoff for the number of years a program had been in existence. Years of existence of a program was controlled for in the analysis. The ratio of faculty to residents was calculated per year and categorized as 4 or more or less than 4 to minimize the effect of changing program size over the years. For faculty members who split time between 2 residency programs, each program was given credit for the duration of time spent at that program. Faculty members who hold a PhD only and those who completed their residencies in non-US dermatology residency programs were excluded from the outcome variables. To avoid duplicate faculty publications, collections for each residency program were created within PubMed (ie, if 2 authors from the same program coauthored an article, it was only counted once toward the total number of faculty publications from that program).
Because the data were skewed (ie, there were a large number of programs with 0 graduating chairs/chiefs and PDs), nonparametric analyses were utilized. Logistic regression was used to calculate the odds of producing chairs/chiefs or PDs (yes vs no). Multiple logistic regression helped to determine those variables that were most closely associated with odds of graduating a chair/chief or PD. Variables with a significance level of P<.10 were considered in the multiple logistic regression, and backward selection was used to determine a model. Multiple linear regression was used to determine correlation coefficients for each of the variables and the number of chairs/chiefs or PDs graduated, controlling for the estimated number of graduates from the program and number of years the program had been in existence. Analyses for graduating chairs/chiefs and PDs were conducted separately. The final significance level used was P<.05. Data were analyzed using SAS version 9.3. This study was approved by the institutional review board at Kaiser Permanente Southern California.
Results
Data from 103 dermatology residency programs were included in the analysis. Of these programs, 47 had graduated at least 1 chair/chief and 55 had graduated at least 1 PD. Among the programs graduating any chairs/chiefs, they produced an average of 2.04 chairs/chiefs and 1.86 PDs. The 5 dermatology residency programs that graduated the highest total number of chairs/chiefs and PDs were Harvard University (Cambridge, Massachusetts), the University of Michigan (Ann Arbor, Michigan), New York University (New York, New York), Yale-New Haven Hospital (New Haven, Connecticut), and the University of Minnesota (Minneapolis, Minnesota).
Factors that had the highest effect on the odds of a program graduating a chair/chief included the ratio of faculty to residents per year, presence of DF funding in 2008, number of years program was in existence, number of residents, number of full-time faculty, and number of full-time faculty on editorial boards of 6 major dermatology journals (Table 1). When controlling for each of these variables in the final multivariable analysis, programs with 4 or more faculty per resident had 3.31 times the odds of producing a chair/chief (95% confidence interval [CI], 1.14-9.66; P=.028).
Factors that had the highest effect on the odds of a program graduating a PD included status as department versus division, ratio of faculty to residents per year, presence of DF funding in 2008, number of lectures given by full-time faculty members at annual society meetings, number of residents, number of years program was in existence, number of full-time faculty, and number of publications from full-time faculty members (Table 2). The most significant factor associated with graduating PDs after controlling for other variables was the number of publications from full-time faculty members. The odds increased by 3.2% for every 1 additional publication and 32% for every 10 additional publications (95% CI, 1.01-1.06; P=.026).
Multiple linear regression demonstrated a positive relationship between the number of graduating chairs/chiefs and total full-time faculty members (R2=0.26; P=.034) and ratio of full-time faculty to residents (R2=0.29; P<.001). Marginally significant correlations were seen between the number of PDs and ratio of full-time faculty to residents (R2=0.32; P=.05) as well as the number of publications from full-time faculty members (R2=0.32; P=.05).
Comment
The ratio of full-time faculty to residents increased a program’s odds of graduating a chair/chief. More faculty members may lead to more opportunities for mentorship of residents and young faculty. Mentors are widely perceived to be integral to the learning and development of residents, not only in dermatology5 but across all specialties.6 Mentors also have been noted to play a key role in bolstering and maintaining interest in academics,7 which is true not only with regard to recruiting new residents but for retaining young faculty members. In a study (N=109) that examined factors associated with residents’ loss of interest in academic careers, half of the participants reported a lack of effective mentors, role models, and professional guidance.8 Mentors provide teaching, supervision, and advice, especially with regard to research and career paths.9 A large number of faculty members provides more opportunities for direct mentorship and offers residents more exposure to research, specialty clinics, and academic philosophies, which may positively influence and even inspire academic pursuits and leadership.3
Although the solution to producing future chairs/chiefs and PDs may lie in faculty guidance, finding and retaining faculty members as mentors amidst a shortage of academic dermatologists presents an underlying issue.3 In addition to a lack of mentorship, residents cite bureaucracy, salary differentials, and location to explain a loss of interest in academic careers.8 Several programs have been developed to address the recruitment of dermatology residents for academic careers, including combined medical-dermatology programs, 2+2 programs (2 years of clinical residency plus 2 additional research years), clinical research fellowships,10 and the Society for Investigative Dermatology’s Dermatology Resident Retreat for Future Academicians (http://www.sidnet.org/fortraineesandresidents).11 Perhaps recruitment should even start at the medical student level. In light of the academic strength of the current pool of dermatology residency applicants,12 training programs should continue to screen for applicants with sincere interests in academia.13 Students with more research and publications may be more likely to pursue academic careers, in accordance with prior studies of dermatology trainees.3,14 Studies also have shown that graduates of foreign dermatology residencies15 and individuals who hold both MD and PhD degrees may be more likely to enter into academic careers.16
For creating future chairs/chiefs and PDs, retention of young faculty in academics is as important as recruiting residents.17 At the mid-career level, the decline of funds for research has generated pressure for academic physicians to see increasing numbers of patients, leaving insufficient time for the many duties that accompany academic posts,2 including teaching and publishing. Other reasons that faculty members leave their posts before 40 years of age include financial and family concerns18 as well as the desire for more autonomy.4 Formalized training is seen with the American Academy of Dermatology’s Academic Dermatology Leadership Program (https://www.aad.org/members/leadership-institute/mentoring/aad-mentoring-opportunities/academic-dermatology-leadership-program-mentee), which promotes advanced leadership training to recent graduates.5 Other methods include support of young faculty with mentorship; grant applications; and administration at the department, hospital, and government levels.17 Recruitment of faculty from private practice may represent another potential source of faculty who wish to pursue more scholarly endeavors.4 Teaching has been cited as a primary reason for faculty members to remain in academia,18 and thus time for teaching must be protected. Such a strategy is in accordance with our findings that amount of annual DF funding received, number of full-time faculty publications, number of faculty members on editorial boards of major dermatology journals, and number of lectures given by full-time faculty factors at annual society meetings are positively associated with the odds of producing chairs/chiefs or PDs. In particular, the number of full-time faculty publications is directly related to increased odds of graduates becoming PDs. Residents and young faculty members who take part in research and attend national conferences may find inspiration or develop a passion for academic leadership.
A limitation to this study is that the ratio of faculty to graduated residents for some programs likely has fluctuated over the last 35 years. This study assumed that certain programs remained generally small or large during the course of their existence, which was controlled by using the ratio between faculty and residents. Additionally, the number of years that a program has been in existence influences the likelihood of that program to graduate higher numbers of residents who become chairs/chiefs or PDs. As a result, we used multiple linear regression to control for the number of residents and number of years that a program had been in existence. Finally, while the relationship between academic leaders and research may be explained by the increased likelihood of faculty with more funding, publications, or lectures to be selected for leadership roles, this finding supports the notion that research can contribute to leadership. This analysis identifies modifiable factors among residency training programs to improve the odds of graduating future academic leaders.
Conclusion
As the present study shows, the ratio of faculty to residents and the number of full-time faculty publications are key to graduating academic leaders in dermatology. Retention of faculty as leaders in academic dermatology is as crucial to the field as recruitment of residents into academic dermatology. Mentorship should be highly encouraged through the creation of formal programs and should not end at the resident level. Emphasizing the intellectually stimulating aspects of academia and providing administrative resources may help decrease the burden of academic duties, allowing the pursuit of teaching and research and ultimately the resources to become candidates for leadership positions in academia.
Leadership is widely recognized as a key component in the role of a physician,1 which is especially true in dermatology, a specialty that faces severe challenges in the recruitment and retention of academic faculty.2 A study of the dermatology workforce found that academic institutions are more likely to be seeking to hire new faculty2 and that many dermatology residency programs often are looking to replace chairpersons (chairs) and/or chiefs.3 Although fewer dermatology residents are pursuing academic careers than careers in private practice, full-time faculty members also are leaving their academic posts. This shift is demonstrated by the younger mean age of academic dermatologists2 and the increased rate of departure from academia prior to pursuing more formalized leadership roles.4
It has been suggested that the number of full-time faculty and number of faculty publications positively influence graduates of dermatology residency programs to pursue academic careers; however, variables affecting the likelihood of graduates of dermatology residency programs becoming academic leaders later in their career have not been well studied.3 The purpose of this study is to determine the factors that influence the development of program chairs/chiefs and program directors (PDs) of dermatology residency programs.
Methods
Data were collected from all accredited dermatology residency programs in the United States as of December 31, 2008. Residency programs that were started after 2004 were excluded from the study, as it was thought that these programs may not have graduated a sufficient number of residents for assessment. Military residency programs also were excluded, as graduates from these programs often do not freely choose their careers after residency.
Primary end points were the number of chairs/chiefs and PDs who had graduated from each dermatology residency program. Variables included the number of years the program had been in existence, status of the program as a department or division, number of full-time faculty members, number of residents, National Institutes of Health funding received in 2008 (http://www.report.nih.gov/award/index.cfm), Dermatology Foundation (DF) funding received (http://www.dermatologyfoundation.org/rap/), number of publications from full-time faculty members in 2008 (http://www.ncbi.nlm.nih.gov/pubmed/), number of faculty lectures given at annual meetings of 5 societies in 2008 (American Academy of Dermatology, the Society for Investigative Dermatology, the American Society of Dermatopathology, the Society for Pediatric Dermatology, and American Society for Dermatologic Surgery), and the number of faculty members on the editorial boards of 6 major dermatology journals (Journal of the American Academy of Dermatology, Journal of Investigative Dermatology, Archives of Dermatology [currently known as JAMA Dermatology], Dermatologic Surgery, Pediatric Dermatology, and Journal of Cutaneous Pathology). Data regarding faculty and residents were obtained from program Web sites and inquiries from individual programs. The year 1974 was used as a cutoff for the number of years a program had been in existence. Years of existence of a program was controlled for in the analysis. The ratio of faculty to residents was calculated per year and categorized as 4 or more or less than 4 to minimize the effect of changing program size over the years. For faculty members who split time between 2 residency programs, each program was given credit for the duration of time spent at that program. Faculty members who hold a PhD only and those who completed their residencies in non-US dermatology residency programs were excluded from the outcome variables. To avoid duplicate faculty publications, collections for each residency program were created within PubMed (ie, if 2 authors from the same program coauthored an article, it was only counted once toward the total number of faculty publications from that program).
Because the data were skewed (ie, there were a large number of programs with 0 graduating chairs/chiefs and PDs), nonparametric analyses were utilized. Logistic regression was used to calculate the odds of producing chairs/chiefs or PDs (yes vs no). Multiple logistic regression helped to determine those variables that were most closely associated with odds of graduating a chair/chief or PD. Variables with a significance level of P<.10 were considered in the multiple logistic regression, and backward selection was used to determine a model. Multiple linear regression was used to determine correlation coefficients for each of the variables and the number of chairs/chiefs or PDs graduated, controlling for the estimated number of graduates from the program and number of years the program had been in existence. Analyses for graduating chairs/chiefs and PDs were conducted separately. The final significance level used was P<.05. Data were analyzed using SAS version 9.3. This study was approved by the institutional review board at Kaiser Permanente Southern California.
Results
Data from 103 dermatology residency programs were included in the analysis. Of these programs, 47 had graduated at least 1 chair/chief and 55 had graduated at least 1 PD. Among the programs graduating any chairs/chiefs, they produced an average of 2.04 chairs/chiefs and 1.86 PDs. The 5 dermatology residency programs that graduated the highest total number of chairs/chiefs and PDs were Harvard University (Cambridge, Massachusetts), the University of Michigan (Ann Arbor, Michigan), New York University (New York, New York), Yale-New Haven Hospital (New Haven, Connecticut), and the University of Minnesota (Minneapolis, Minnesota).
Factors that had the highest effect on the odds of a program graduating a chair/chief included the ratio of faculty to residents per year, presence of DF funding in 2008, number of years program was in existence, number of residents, number of full-time faculty, and number of full-time faculty on editorial boards of 6 major dermatology journals (Table 1). When controlling for each of these variables in the final multivariable analysis, programs with 4 or more faculty per resident had 3.31 times the odds of producing a chair/chief (95% confidence interval [CI], 1.14-9.66; P=.028).
Factors that had the highest effect on the odds of a program graduating a PD included status as department versus division, ratio of faculty to residents per year, presence of DF funding in 2008, number of lectures given by full-time faculty members at annual society meetings, number of residents, number of years program was in existence, number of full-time faculty, and number of publications from full-time faculty members (Table 2). The most significant factor associated with graduating PDs after controlling for other variables was the number of publications from full-time faculty members. The odds increased by 3.2% for every 1 additional publication and 32% for every 10 additional publications (95% CI, 1.01-1.06; P=.026).
Multiple linear regression demonstrated a positive relationship between the number of graduating chairs/chiefs and total full-time faculty members (R2=0.26; P=.034) and ratio of full-time faculty to residents (R2=0.29; P<.001). Marginally significant correlations were seen between the number of PDs and ratio of full-time faculty to residents (R2=0.32; P=.05) as well as the number of publications from full-time faculty members (R2=0.32; P=.05).
Comment
The ratio of full-time faculty to residents increased a program’s odds of graduating a chair/chief. More faculty members may lead to more opportunities for mentorship of residents and young faculty. Mentors are widely perceived to be integral to the learning and development of residents, not only in dermatology5 but across all specialties.6 Mentors also have been noted to play a key role in bolstering and maintaining interest in academics,7 which is true not only with regard to recruiting new residents but for retaining young faculty members. In a study (N=109) that examined factors associated with residents’ loss of interest in academic careers, half of the participants reported a lack of effective mentors, role models, and professional guidance.8 Mentors provide teaching, supervision, and advice, especially with regard to research and career paths.9 A large number of faculty members provides more opportunities for direct mentorship and offers residents more exposure to research, specialty clinics, and academic philosophies, which may positively influence and even inspire academic pursuits and leadership.3
Although the solution to producing future chairs/chiefs and PDs may lie in faculty guidance, finding and retaining faculty members as mentors amidst a shortage of academic dermatologists presents an underlying issue.3 In addition to a lack of mentorship, residents cite bureaucracy, salary differentials, and location to explain a loss of interest in academic careers.8 Several programs have been developed to address the recruitment of dermatology residents for academic careers, including combined medical-dermatology programs, 2+2 programs (2 years of clinical residency plus 2 additional research years), clinical research fellowships,10 and the Society for Investigative Dermatology’s Dermatology Resident Retreat for Future Academicians (http://www.sidnet.org/fortraineesandresidents).11 Perhaps recruitment should even start at the medical student level. In light of the academic strength of the current pool of dermatology residency applicants,12 training programs should continue to screen for applicants with sincere interests in academia.13 Students with more research and publications may be more likely to pursue academic careers, in accordance with prior studies of dermatology trainees.3,14 Studies also have shown that graduates of foreign dermatology residencies15 and individuals who hold both MD and PhD degrees may be more likely to enter into academic careers.16
For creating future chairs/chiefs and PDs, retention of young faculty in academics is as important as recruiting residents.17 At the mid-career level, the decline of funds for research has generated pressure for academic physicians to see increasing numbers of patients, leaving insufficient time for the many duties that accompany academic posts,2 including teaching and publishing. Other reasons that faculty members leave their posts before 40 years of age include financial and family concerns18 as well as the desire for more autonomy.4 Formalized training is seen with the American Academy of Dermatology’s Academic Dermatology Leadership Program (https://www.aad.org/members/leadership-institute/mentoring/aad-mentoring-opportunities/academic-dermatology-leadership-program-mentee), which promotes advanced leadership training to recent graduates.5 Other methods include support of young faculty with mentorship; grant applications; and administration at the department, hospital, and government levels.17 Recruitment of faculty from private practice may represent another potential source of faculty who wish to pursue more scholarly endeavors.4 Teaching has been cited as a primary reason for faculty members to remain in academia,18 and thus time for teaching must be protected. Such a strategy is in accordance with our findings that amount of annual DF funding received, number of full-time faculty publications, number of faculty members on editorial boards of major dermatology journals, and number of lectures given by full-time faculty factors at annual society meetings are positively associated with the odds of producing chairs/chiefs or PDs. In particular, the number of full-time faculty publications is directly related to increased odds of graduates becoming PDs. Residents and young faculty members who take part in research and attend national conferences may find inspiration or develop a passion for academic leadership.
A limitation to this study is that the ratio of faculty to graduated residents for some programs likely has fluctuated over the last 35 years. This study assumed that certain programs remained generally small or large during the course of their existence, which was controlled by using the ratio between faculty and residents. Additionally, the number of years that a program has been in existence influences the likelihood of that program to graduate higher numbers of residents who become chairs/chiefs or PDs. As a result, we used multiple linear regression to control for the number of residents and number of years that a program had been in existence. Finally, while the relationship between academic leaders and research may be explained by the increased likelihood of faculty with more funding, publications, or lectures to be selected for leadership roles, this finding supports the notion that research can contribute to leadership. This analysis identifies modifiable factors among residency training programs to improve the odds of graduating future academic leaders.
Conclusion
As the present study shows, the ratio of faculty to residents and the number of full-time faculty publications are key to graduating academic leaders in dermatology. Retention of faculty as leaders in academic dermatology is as crucial to the field as recruitment of residents into academic dermatology. Mentorship should be highly encouraged through the creation of formal programs and should not end at the resident level. Emphasizing the intellectually stimulating aspects of academia and providing administrative resources may help decrease the burden of academic duties, allowing the pursuit of teaching and research and ultimately the resources to become candidates for leadership positions in academia.
1. Wood LD, Miller JJ, Marks JG Jr. The need for leadership: how can we better train the next generation of dermatologists? J Am Acad Dermatol. 2008;58:715-717.
2. Resneck JS Jr, Tierney EP, Kimball AB. Challenges facing academic dermatology: survey data on the faculty workforce. J Am Acad Dermatol. 2006;54:211-216.
3. Wu JJ, Ramirez CC, Alonso CA, et al. Dermatology residency program characteristics that correlate with graduates selecting an academic dermatology career. Arch Dermatol. 2006;142:845-850.
4. Loo DS, Liu CL, Geller AC, et al. Academic dermatology manpower: issues of recruitment and retention. Arch Dermatol. 2007;143:341-347.
5. Donovan JC. A survey of dermatology residency program directors’ views on mentorship. Dermatol Online J. 2009;15:1.
6. Sambunjak D, Straus SE, Marusi´c A. Mentoring in academic medicine: a systematic review. JAMA. 2006;296:1103-1115.
7. Rubenstein DS, Blauvelt A, Chen SC, et al. The future of academic dermatology in the United States: report on the resident retreat for future physician-scientists, June 15-17, 2001. J Am Acad Dermatol. 2002;47:300-303.
8. Reck SJ, Stratman EJ, Vogel C, et al. Assessment of residents’ loss of interest in academic careers and identification of correctable factors. Arch Dermatol. 2006;142:855-858.
9. Clark RA. Teacher, supervisor, adviser, or mentor? J Invest Dermatol. 2011;131:1779-1780.
10. Wu JJ. Current strategies to address the ongoing shortage of academic dermatologists. J Am Acad Dermatol. 2007;56:1065-1066.
11. Culton DA, Rubenstein DS, Diaz LA. The resident retreat for future academicians. J Invest Dermatol. 2010;130:1775-1777.
12. Wu JJ, Tyring SK. The academic strength of current dermatology residency applicants. Dermatol Online J. 2003;9:22.
13. Kia KF, Gielczyk RA, Ellis CN. Academia is the life for me, I’m sure. Arch Dermatol. 2006;142:911-913.
14. Miller CJ, Wood GC, Miller JJ, et al. Academics or private practice? the future of dermatologic surgery education. Dermatol Surg. 2006;32:70-75, discussion 76-78.
15. Wu JJ, Davis KF, Ramirez CC, et al. Graduates-of-foreign-dermatology residencies and military dermatology residencies and women in academic dermatology. Dermatol Online J. 2009;15:2.
16. Wu JJ, Davis KF, Ramirez CC, et al. MD/PhDs are more likely than MDs to choose a career in academic dermatology. Dermatol Online J. 2008;14:27.
17. Olerud JE. Academic workforce in dermatology. Arch Dermatol. 2007;143:409-410.
18. Turner E, Yoo J, Salter S, et al. Leadership workforce in academic dermatology. Arch Dermatol. 2007;143:948-949.
1. Wood LD, Miller JJ, Marks JG Jr. The need for leadership: how can we better train the next generation of dermatologists? J Am Acad Dermatol. 2008;58:715-717.
2. Resneck JS Jr, Tierney EP, Kimball AB. Challenges facing academic dermatology: survey data on the faculty workforce. J Am Acad Dermatol. 2006;54:211-216.
3. Wu JJ, Ramirez CC, Alonso CA, et al. Dermatology residency program characteristics that correlate with graduates selecting an academic dermatology career. Arch Dermatol. 2006;142:845-850.
4. Loo DS, Liu CL, Geller AC, et al. Academic dermatology manpower: issues of recruitment and retention. Arch Dermatol. 2007;143:341-347.
5. Donovan JC. A survey of dermatology residency program directors’ views on mentorship. Dermatol Online J. 2009;15:1.
6. Sambunjak D, Straus SE, Marusi´c A. Mentoring in academic medicine: a systematic review. JAMA. 2006;296:1103-1115.
7. Rubenstein DS, Blauvelt A, Chen SC, et al. The future of academic dermatology in the United States: report on the resident retreat for future physician-scientists, June 15-17, 2001. J Am Acad Dermatol. 2002;47:300-303.
8. Reck SJ, Stratman EJ, Vogel C, et al. Assessment of residents’ loss of interest in academic careers and identification of correctable factors. Arch Dermatol. 2006;142:855-858.
9. Clark RA. Teacher, supervisor, adviser, or mentor? J Invest Dermatol. 2011;131:1779-1780.
10. Wu JJ. Current strategies to address the ongoing shortage of academic dermatologists. J Am Acad Dermatol. 2007;56:1065-1066.
11. Culton DA, Rubenstein DS, Diaz LA. The resident retreat for future academicians. J Invest Dermatol. 2010;130:1775-1777.
12. Wu JJ, Tyring SK. The academic strength of current dermatology residency applicants. Dermatol Online J. 2003;9:22.
13. Kia KF, Gielczyk RA, Ellis CN. Academia is the life for me, I’m sure. Arch Dermatol. 2006;142:911-913.
14. Miller CJ, Wood GC, Miller JJ, et al. Academics or private practice? the future of dermatologic surgery education. Dermatol Surg. 2006;32:70-75, discussion 76-78.
15. Wu JJ, Davis KF, Ramirez CC, et al. Graduates-of-foreign-dermatology residencies and military dermatology residencies and women in academic dermatology. Dermatol Online J. 2009;15:2.
16. Wu JJ, Davis KF, Ramirez CC, et al. MD/PhDs are more likely than MDs to choose a career in academic dermatology. Dermatol Online J. 2008;14:27.
17. Olerud JE. Academic workforce in dermatology. Arch Dermatol. 2007;143:409-410.
18. Turner E, Yoo J, Salter S, et al. Leadership workforce in academic dermatology. Arch Dermatol. 2007;143:948-949.
Practice Points
- Leadership in dermatology is key to the future of academics.
- Opportunity for mentorship and research are the most important residency program factors leading to the graduation of future chairs/chiefs and program directors.
- The retention of residents and young faculty in academics can be aided by research and scholarly activity.

Cosmetic Corner: Dermatologists Weigh in on Aftershaves
To improve patient care and outcomes, leading dermatologists offered their recommendations on the top aftershaves. Consideration must be given to:
- The Art of Shaving Pre-Shave Oil
- CeraVe Facial Moisturizing Lotion AM
- Post Shave Cooling Gel
Cutis invites readers to send us their recommendations. Skin care products for babies, hand sanitizers, and cleansers for rosacea patients will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on the top aftershaves. Consideration must be given to:
- The Art of Shaving Pre-Shave Oil
- CeraVe Facial Moisturizing Lotion AM
- Post Shave Cooling Gel
Cutis invites readers to send us their recommendations. Skin care products for babies, hand sanitizers, and cleansers for rosacea patients will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on the top aftershaves. Consideration must be given to:
- The Art of Shaving Pre-Shave Oil
- CeraVe Facial Moisturizing Lotion AM
- Post Shave Cooling Gel
Cutis invites readers to send us their recommendations. Skin care products for babies, hand sanitizers, and cleansers for rosacea patients will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

Reflectance Confocal Microscopy: An Effective Diagnostic Tool for Dermatophytic Infections
There are a variety of well-established methods for diagnosing dermatophyte infections, including potassium hydroxide (KOH) preparations, fungal cultures, and skin biopsies. Each modality has its place in clinical practice, but they also have drawbacks. Reflectance confocal microscopy (RCM) is an emerging in vivo technology that could potentially serve as a sensitive, rapid, and noninvasive method of diagnosing dermatophytosis. Using near-infrared laser light scanning, RCM provides a quick noninvasive method of generating black-and-white, horizontal, quasipathology images that allow for the identification of cells and other structures similar to dermoscopy and histopathology.1 The images are obtained in a fully noninvasive fashion, as the device is placed in contact with the skin using a liquid medium. The process takes 5 to 15 minutes depending on the number of images obtained, and the images can then be displayed in real time on a computer screen or transmitted to a pathologist for evaluation.
Most initial applications of RCM focused on evaluating melanocytic lesions with the primary goal of differentiating between benign nevi and melanomas, thus reducing the need for skin biopsies.2-4 Efforts to develop RCM diagnostic criteria for identification of other skin cancers5,6 as well as to aid in the diagnosis of nonneoplastic skin conditions are ongoing.7 The potential applications of RCM are virtually limitless, as this modality can (at least partially) take the place of biopsies in a variety of clinical scenarios.2,8 Few reports have documented the utility of RCM as a diagnostic tool for onychomycosis9,10 and dermatophytic infections of the skin.11,12 Hui et al13 reported use for RCM for microscopic evaluation of mycelium features. Turan et al14 found that RCM could not replace the current diagnostic standards for tinea incognito but may be successfully used as an in vivo noninvasive screening tool to facilitate diagnosis. Because it provides high-resolution horizontal images extending from the surface of the stratum corneum to the superficial reticular dermis, RCM could be an effective tool in the diagnosis of cutaneous dermatophyte infections, as organisms usually are located in the stratum corneum of the epidermis in this infection. Branching hyphae are readily visible in the stratum corneum on RCM (Figure).
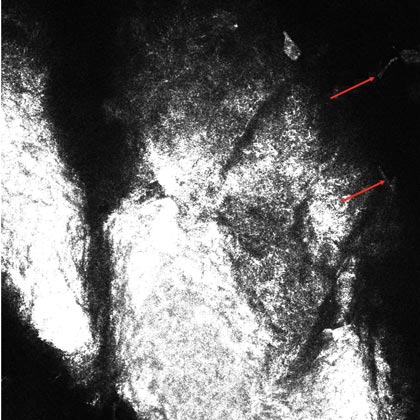
We reviewed a series of 9 cases from a private practice setting in which RCM was used to diagnose dermatophytosis. We compared the diagnostic accuracy of RCM to results from other diagnostic methods and the ultimate clinical outcome to determine the usefulness of this new technology.
Methods
Our retrospective chart review included all cases in which RCM was used and the clinical differential diagnosis included tinea corporis over a 4-month period in a private, single-specialty dermatology practice. All patients were treated by the same dermatologist. The RCM images were taken using an imaging system that had a horizontal optical resolution of less than 1.25 μm and a vertical optical resolution of less than 5.0 μm. The imaging was performed by medical assistants who were trained by the device manufacturer.
The sample sites were cleaned with isopropyl alcohol and a translucent contact ring was affixed to the skin using a liquid medium. The imaging head of the device was connected to the imaging ring and the images were taken. Identical imaging protocol was used in all patients. Multiple sets of horizontal images and one stack of vertical images were obtained. Patients reported no discomfort during the procedure, and the entire process was usually completed within 15 minutes. The images were sent to the pathologist for evaluation using the manufacturer’s telepathology system and were returned with a diagnosis within 24 hours. (On-site, real-time diagnosis also is possible if the dermatologist is trained in interpreting the images.)
In the chart review we looked for other diagnostic methods used as well as clinical outcomes. A case was considered to be positive for dermatophytic infection if any of the other diagnostic modalities yielded positive results or if a definitive resolution of the condition could be achieved using antifungal treatments alone.
Results
Ten patients (mean age, 43.1 years; age range, 16–76 years) with lesions that presented as possible dermatophytic infections underwent RCM analysis. In addition to RCM imaging, 5 patients underwent KOH testing of skin scrapings, 3 underwent analysis by fungal culture utilizing dermatophyte test medium (DTM), and 5 underwent biopsies. The findings are further summarized in the Table. One patient (patient 5) was excluded from the study because the RCM could not be evaluated due to the poor quality of the confocal images. Additionally, 2 patients (patients 2 and 7) had suboptimal imaging, which limited the evaluation.
Of the 9 evaluable cases, 4 (patients 1–4) were determined to be positive for the presence of dermatophytic infection through the fulfillment of criteria independent of RCM imaging. In each of those 4 cases, RCM images revealed the presence of hyphae, which indicated the presence of dermato-phytic infection. In these 4 cases, RCM and other diagnostic methods reached the same diagnosis.
In the other 5 cases (patients 6–10), the final diagnosis was not a dermatophytic infection. In 4 of those cases (patients 7–10), there were no signs of any structure resembling hyphae on the RCM images; however, in 1 case (patient 6), the RCM images showed structures that were consistent with the appearance of hyphae to the extent that the investigators, based solely on analysis of the RCM images, deemed a diagnosis indicating presence of a dermatophytic infection to be valid. In this case, a 38-year-old man presented with extensive scaly patches on the back of several months’ duration. Repeated skin biopsies showed hyperkeratosis and occasionally minimal spongiosis, while periodic acid–Schiff staining did not reveal fungal elements. Fungal cultures and KOH preparations were negative. Prior treatments with topical antifungals and steroids failed to improve the condition, which resolved rapidly with urea cream 40%. The interpretation of the RCM images in this patient did not match up with the results obtained from other methods of diagnosis and the clinical outcome; thus, we classified it as an incorrect diagnosis based on RCM analysis alone. In total, successful diagnosis using RCM imaging was achieved in 8 of 9 cases included in the analysis.
Comment
In this chart review, we evaluated the utility of using RCM in the diagnosis of dermatophytic infections of the skin by comparing findings noted on confocal imaging with those of other methods of diagnosis (Table). We included cases in which the clinical presentation raised the possibility of dermatophytic infection. Cases were considered positive for dermatophytes if KOH preparation, fungal culture, or skin biopsy (with or without periodic acid–Schiff staining) were positive or if there was a complete response to antifungal treatment alone. In this small number of cases, we found that RCM was 100% sensitive, as hyphae were readily seen in all cases of dermatophytic infections. In 1 RCM-positive case (patient 3), fungal culture with DTM was negative, but antifungal therapy was nonetheless given. Because the lesion resolved promptly with econazole, RCM proved to be true positive and DTM proved to be false negative (Table). Reflectance confocal microscopy imaging, however, was less specific. Of the 5 cases that showed no presence of dermatophytic infection, there was 1 case (patient 6) in which the pathologist could recognize structures that resembled fungal hyphae. There are various possible sources of structures masquerading as dermatophytes on confocal imaging, including the edges of nonnucleated loose keratinocytes, keratin fragments, and other foreign fibers. Evaluation by an experienced investigator can certainly help in limiting false-positive analyses, but a larger case study would be useful to develop a set of specific criteria to aid in the differentiation of fungal hyphae from other artifacts as well as to further define the sensitivity and specificity of RCM.
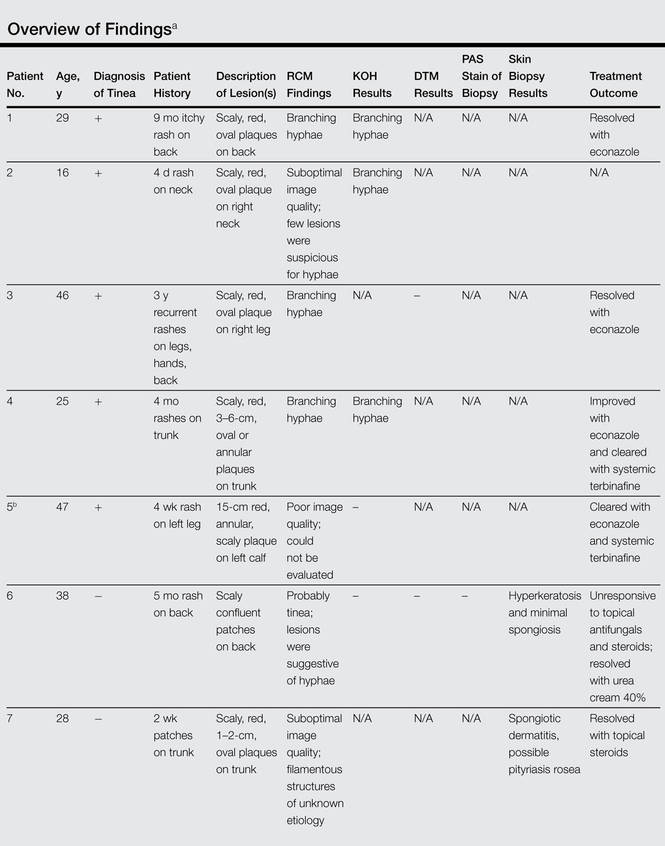
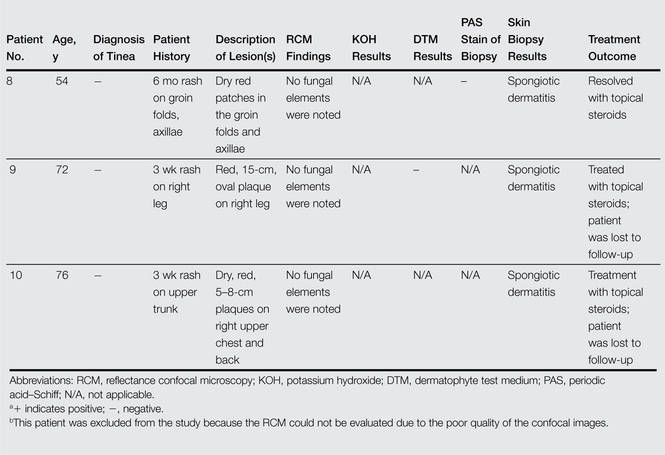
We also encountered difficulties with the technical aspects of RCM. One case (patient 5) was excluded from the analysis because the images were poor quality and could not be interpreted, and 2 cases (patients 2 and 7) had suboptimal images, in part due to operator error and in part due to equipment error that was recognized later on. The technical difficulties were problematic because no immediate review of image quality was available while patients were still present for possible reimaging. All of the images evaluated in this study were captured shortly after the RCM device was introduced to the practice. It is possible that with more training and a quick, on-site review of image quality, these technical problems could be avoided. Imaging protocols (ie, numbers and levels of scans taken by the confocal microscope) also could be adjusted so they include a large enough range to compensate for potential operator errors; however, these adjustments also could increase overall imaging time.
Conclusion
Based on our chart review of a small number of cases, we found that RCM can be a useful tool in diagnosing dermatophytic infections of the skin. With adequate training, dermatologists may be able to use RCM as an in-office tool to capture and evaluate images and subsequently diagnose or exclude dermatophytosis in a quick and noninvasive manner. However, further research and controlled studies of more cases will be required to develop accurate criteria for diagnosing fungal structures by RCM as well as to help determine the role of RCM in our diagnostic armamentarium.
1. Longo C, Farnetani F, Ciardo S, et al. Is confocal microscopy a valuable tool in diagnosing nodular lesions? a study of 140 cases. Br J Dermatol. 2013;169:58-67.
2. Debarbieux S, Dalle S, Depaepe L, et al. Second primary melanomas under BRAF blockers: study by reflectance confocal microscopy [published online a head of print April 1, 2013]. Br J Dermatol. 2013;168:1230-1235.
3. Schwartz RJ, Vera K, Navarrete N, et al. In vivo reflectance confocal microscopy of halo nevus. J Cutan Med Surg. 2013;17:33-38.
4. Pellacani G, Guitera P, Longo C, et al. The impact of in vivo reflectance confocal microscopy for the diagnostic accuracy of melanoma and equivocal melanocytic lesions. J Invest Dermatol. 2007;127:2759-2765.
5. Nori S, Rius-Diaz F, Cuevas J, et al. Sensitivity and specificity of reflectance-mode confocal micros-copy for in vivo diagnosis of basal cell carcinoma: a multicenter study. J Am Acad Dermatol. 2004;51:923-930.
6. Arzberger E, Komericki P, Ahlgrimm-Siess V, et al. Differentiation between balanitis and carcinoma in situ using reflectance confocal microscopy. JAMA Dermatol. 2013;149:440-445.
7. Ardigò M, Maliszewski I, Cota C, et al. Preliminary evaluation of in vivo reflectance confocal microscopy features of discoid lupus erythematosus. Br J Dermatol. 2007;156:1196-1203.
8. Longo C, Casari A, Pepe P, et al. Confocal microscopy insights into the treatment and cellular immune response of Basal cell carcinoma to photodynamic therapy. Dermatology. 2012;225:264-270.
9. Hongcharu W, Dwyer P, Gonzalez S, et al. Confirmation of onychomycosis by in vivo confocal microscopy. J Am Acad Dermatol. 2000;42:214-216.
10. Rothmund G, Sattler EC, Kaestle R, et al. Confocal laser scanning microscopy as a new valuable tool in the diagnosis of onychomycosis—comparison of six diagnostic methods [published online ahead of print April 23, 2012]. Mycoses. 2013;56:47-55.
11. Markus R, Huzaira M, Anderson RR, et al. A better potassium hydroxide preparation? Arch Dermatol. 2001;137:1076-1078.
12. Slutsky JB, Rabinovitz H, Grichnik JM, et al. Reflectance confocal microscopic features of dermatophytes, scabies, and Demodex. Arch Dermatol. 2011;147:1008.
13. Hui D, Xue-chang S, Ai-e X. Evaluation of reflectance confocal microscopy in dermatophytosis. Mycoses. 2013;56:130-133.
14. Turan E, Erdemir AT, Gurel MS, et al. A new diagnostic technique for tinea incognito: in vivo reflectance confocal microscopy. report of five cases. Skin Res Technol. 2013;19:e103-e107.
There are a variety of well-established methods for diagnosing dermatophyte infections, including potassium hydroxide (KOH) preparations, fungal cultures, and skin biopsies. Each modality has its place in clinical practice, but they also have drawbacks. Reflectance confocal microscopy (RCM) is an emerging in vivo technology that could potentially serve as a sensitive, rapid, and noninvasive method of diagnosing dermatophytosis. Using near-infrared laser light scanning, RCM provides a quick noninvasive method of generating black-and-white, horizontal, quasipathology images that allow for the identification of cells and other structures similar to dermoscopy and histopathology.1 The images are obtained in a fully noninvasive fashion, as the device is placed in contact with the skin using a liquid medium. The process takes 5 to 15 minutes depending on the number of images obtained, and the images can then be displayed in real time on a computer screen or transmitted to a pathologist for evaluation.
Most initial applications of RCM focused on evaluating melanocytic lesions with the primary goal of differentiating between benign nevi and melanomas, thus reducing the need for skin biopsies.2-4 Efforts to develop RCM diagnostic criteria for identification of other skin cancers5,6 as well as to aid in the diagnosis of nonneoplastic skin conditions are ongoing.7 The potential applications of RCM are virtually limitless, as this modality can (at least partially) take the place of biopsies in a variety of clinical scenarios.2,8 Few reports have documented the utility of RCM as a diagnostic tool for onychomycosis9,10 and dermatophytic infections of the skin.11,12 Hui et al13 reported use for RCM for microscopic evaluation of mycelium features. Turan et al14 found that RCM could not replace the current diagnostic standards for tinea incognito but may be successfully used as an in vivo noninvasive screening tool to facilitate diagnosis. Because it provides high-resolution horizontal images extending from the surface of the stratum corneum to the superficial reticular dermis, RCM could be an effective tool in the diagnosis of cutaneous dermatophyte infections, as organisms usually are located in the stratum corneum of the epidermis in this infection. Branching hyphae are readily visible in the stratum corneum on RCM (Figure).
We reviewed a series of 9 cases from a private practice setting in which RCM was used to diagnose dermatophytosis. We compared the diagnostic accuracy of RCM to results from other diagnostic methods and the ultimate clinical outcome to determine the usefulness of this new technology.
Methods
Our retrospective chart review included all cases in which RCM was used and the clinical differential diagnosis included tinea corporis over a 4-month period in a private, single-specialty dermatology practice. All patients were treated by the same dermatologist. The RCM images were taken using an imaging system that had a horizontal optical resolution of less than 1.25 μm and a vertical optical resolution of less than 5.0 μm. The imaging was performed by medical assistants who were trained by the device manufacturer.
The sample sites were cleaned with isopropyl alcohol and a translucent contact ring was affixed to the skin using a liquid medium. The imaging head of the device was connected to the imaging ring and the images were taken. Identical imaging protocol was used in all patients. Multiple sets of horizontal images and one stack of vertical images were obtained. Patients reported no discomfort during the procedure, and the entire process was usually completed within 15 minutes. The images were sent to the pathologist for evaluation using the manufacturer’s telepathology system and were returned with a diagnosis within 24 hours. (On-site, real-time diagnosis also is possible if the dermatologist is trained in interpreting the images.)
In the chart review we looked for other diagnostic methods used as well as clinical outcomes. A case was considered to be positive for dermatophytic infection if any of the other diagnostic modalities yielded positive results or if a definitive resolution of the condition could be achieved using antifungal treatments alone.
Results
Ten patients (mean age, 43.1 years; age range, 16–76 years) with lesions that presented as possible dermatophytic infections underwent RCM analysis. In addition to RCM imaging, 5 patients underwent KOH testing of skin scrapings, 3 underwent analysis by fungal culture utilizing dermatophyte test medium (DTM), and 5 underwent biopsies. The findings are further summarized in the Table. One patient (patient 5) was excluded from the study because the RCM could not be evaluated due to the poor quality of the confocal images. Additionally, 2 patients (patients 2 and 7) had suboptimal imaging, which limited the evaluation.
Of the 9 evaluable cases, 4 (patients 1–4) were determined to be positive for the presence of dermatophytic infection through the fulfillment of criteria independent of RCM imaging. In each of those 4 cases, RCM images revealed the presence of hyphae, which indicated the presence of dermato-phytic infection. In these 4 cases, RCM and other diagnostic methods reached the same diagnosis.
In the other 5 cases (patients 6–10), the final diagnosis was not a dermatophytic infection. In 4 of those cases (patients 7–10), there were no signs of any structure resembling hyphae on the RCM images; however, in 1 case (patient 6), the RCM images showed structures that were consistent with the appearance of hyphae to the extent that the investigators, based solely on analysis of the RCM images, deemed a diagnosis indicating presence of a dermatophytic infection to be valid. In this case, a 38-year-old man presented with extensive scaly patches on the back of several months’ duration. Repeated skin biopsies showed hyperkeratosis and occasionally minimal spongiosis, while periodic acid–Schiff staining did not reveal fungal elements. Fungal cultures and KOH preparations were negative. Prior treatments with topical antifungals and steroids failed to improve the condition, which resolved rapidly with urea cream 40%. The interpretation of the RCM images in this patient did not match up with the results obtained from other methods of diagnosis and the clinical outcome; thus, we classified it as an incorrect diagnosis based on RCM analysis alone. In total, successful diagnosis using RCM imaging was achieved in 8 of 9 cases included in the analysis.
Comment
In this chart review, we evaluated the utility of using RCM in the diagnosis of dermatophytic infections of the skin by comparing findings noted on confocal imaging with those of other methods of diagnosis (Table). We included cases in which the clinical presentation raised the possibility of dermatophytic infection. Cases were considered positive for dermatophytes if KOH preparation, fungal culture, or skin biopsy (with or without periodic acid–Schiff staining) were positive or if there was a complete response to antifungal treatment alone. In this small number of cases, we found that RCM was 100% sensitive, as hyphae were readily seen in all cases of dermatophytic infections. In 1 RCM-positive case (patient 3), fungal culture with DTM was negative, but antifungal therapy was nonetheless given. Because the lesion resolved promptly with econazole, RCM proved to be true positive and DTM proved to be false negative (Table). Reflectance confocal microscopy imaging, however, was less specific. Of the 5 cases that showed no presence of dermatophytic infection, there was 1 case (patient 6) in which the pathologist could recognize structures that resembled fungal hyphae. There are various possible sources of structures masquerading as dermatophytes on confocal imaging, including the edges of nonnucleated loose keratinocytes, keratin fragments, and other foreign fibers. Evaluation by an experienced investigator can certainly help in limiting false-positive analyses, but a larger case study would be useful to develop a set of specific criteria to aid in the differentiation of fungal hyphae from other artifacts as well as to further define the sensitivity and specificity of RCM.
We also encountered difficulties with the technical aspects of RCM. One case (patient 5) was excluded from the analysis because the images were poor quality and could not be interpreted, and 2 cases (patients 2 and 7) had suboptimal images, in part due to operator error and in part due to equipment error that was recognized later on. The technical difficulties were problematic because no immediate review of image quality was available while patients were still present for possible reimaging. All of the images evaluated in this study were captured shortly after the RCM device was introduced to the practice. It is possible that with more training and a quick, on-site review of image quality, these technical problems could be avoided. Imaging protocols (ie, numbers and levels of scans taken by the confocal microscope) also could be adjusted so they include a large enough range to compensate for potential operator errors; however, these adjustments also could increase overall imaging time.
Conclusion
Based on our chart review of a small number of cases, we found that RCM can be a useful tool in diagnosing dermatophytic infections of the skin. With adequate training, dermatologists may be able to use RCM as an in-office tool to capture and evaluate images and subsequently diagnose or exclude dermatophytosis in a quick and noninvasive manner. However, further research and controlled studies of more cases will be required to develop accurate criteria for diagnosing fungal structures by RCM as well as to help determine the role of RCM in our diagnostic armamentarium.
There are a variety of well-established methods for diagnosing dermatophyte infections, including potassium hydroxide (KOH) preparations, fungal cultures, and skin biopsies. Each modality has its place in clinical practice, but they also have drawbacks. Reflectance confocal microscopy (RCM) is an emerging in vivo technology that could potentially serve as a sensitive, rapid, and noninvasive method of diagnosing dermatophytosis. Using near-infrared laser light scanning, RCM provides a quick noninvasive method of generating black-and-white, horizontal, quasipathology images that allow for the identification of cells and other structures similar to dermoscopy and histopathology.1 The images are obtained in a fully noninvasive fashion, as the device is placed in contact with the skin using a liquid medium. The process takes 5 to 15 minutes depending on the number of images obtained, and the images can then be displayed in real time on a computer screen or transmitted to a pathologist for evaluation.
Most initial applications of RCM focused on evaluating melanocytic lesions with the primary goal of differentiating between benign nevi and melanomas, thus reducing the need for skin biopsies.2-4 Efforts to develop RCM diagnostic criteria for identification of other skin cancers5,6 as well as to aid in the diagnosis of nonneoplastic skin conditions are ongoing.7 The potential applications of RCM are virtually limitless, as this modality can (at least partially) take the place of biopsies in a variety of clinical scenarios.2,8 Few reports have documented the utility of RCM as a diagnostic tool for onychomycosis9,10 and dermatophytic infections of the skin.11,12 Hui et al13 reported use for RCM for microscopic evaluation of mycelium features. Turan et al14 found that RCM could not replace the current diagnostic standards for tinea incognito but may be successfully used as an in vivo noninvasive screening tool to facilitate diagnosis. Because it provides high-resolution horizontal images extending from the surface of the stratum corneum to the superficial reticular dermis, RCM could be an effective tool in the diagnosis of cutaneous dermatophyte infections, as organisms usually are located in the stratum corneum of the epidermis in this infection. Branching hyphae are readily visible in the stratum corneum on RCM (Figure).
We reviewed a series of 9 cases from a private practice setting in which RCM was used to diagnose dermatophytosis. We compared the diagnostic accuracy of RCM to results from other diagnostic methods and the ultimate clinical outcome to determine the usefulness of this new technology.
Methods
Our retrospective chart review included all cases in which RCM was used and the clinical differential diagnosis included tinea corporis over a 4-month period in a private, single-specialty dermatology practice. All patients were treated by the same dermatologist. The RCM images were taken using an imaging system that had a horizontal optical resolution of less than 1.25 μm and a vertical optical resolution of less than 5.0 μm. The imaging was performed by medical assistants who were trained by the device manufacturer.
The sample sites were cleaned with isopropyl alcohol and a translucent contact ring was affixed to the skin using a liquid medium. The imaging head of the device was connected to the imaging ring and the images were taken. Identical imaging protocol was used in all patients. Multiple sets of horizontal images and one stack of vertical images were obtained. Patients reported no discomfort during the procedure, and the entire process was usually completed within 15 minutes. The images were sent to the pathologist for evaluation using the manufacturer’s telepathology system and were returned with a diagnosis within 24 hours. (On-site, real-time diagnosis also is possible if the dermatologist is trained in interpreting the images.)
In the chart review we looked for other diagnostic methods used as well as clinical outcomes. A case was considered to be positive for dermatophytic infection if any of the other diagnostic modalities yielded positive results or if a definitive resolution of the condition could be achieved using antifungal treatments alone.
Results
Ten patients (mean age, 43.1 years; age range, 16–76 years) with lesions that presented as possible dermatophytic infections underwent RCM analysis. In addition to RCM imaging, 5 patients underwent KOH testing of skin scrapings, 3 underwent analysis by fungal culture utilizing dermatophyte test medium (DTM), and 5 underwent biopsies. The findings are further summarized in the Table. One patient (patient 5) was excluded from the study because the RCM could not be evaluated due to the poor quality of the confocal images. Additionally, 2 patients (patients 2 and 7) had suboptimal imaging, which limited the evaluation.
Of the 9 evaluable cases, 4 (patients 1–4) were determined to be positive for the presence of dermatophytic infection through the fulfillment of criteria independent of RCM imaging. In each of those 4 cases, RCM images revealed the presence of hyphae, which indicated the presence of dermato-phytic infection. In these 4 cases, RCM and other diagnostic methods reached the same diagnosis.
In the other 5 cases (patients 6–10), the final diagnosis was not a dermatophytic infection. In 4 of those cases (patients 7–10), there were no signs of any structure resembling hyphae on the RCM images; however, in 1 case (patient 6), the RCM images showed structures that were consistent with the appearance of hyphae to the extent that the investigators, based solely on analysis of the RCM images, deemed a diagnosis indicating presence of a dermatophytic infection to be valid. In this case, a 38-year-old man presented with extensive scaly patches on the back of several months’ duration. Repeated skin biopsies showed hyperkeratosis and occasionally minimal spongiosis, while periodic acid–Schiff staining did not reveal fungal elements. Fungal cultures and KOH preparations were negative. Prior treatments with topical antifungals and steroids failed to improve the condition, which resolved rapidly with urea cream 40%. The interpretation of the RCM images in this patient did not match up with the results obtained from other methods of diagnosis and the clinical outcome; thus, we classified it as an incorrect diagnosis based on RCM analysis alone. In total, successful diagnosis using RCM imaging was achieved in 8 of 9 cases included in the analysis.
Comment
In this chart review, we evaluated the utility of using RCM in the diagnosis of dermatophytic infections of the skin by comparing findings noted on confocal imaging with those of other methods of diagnosis (Table). We included cases in which the clinical presentation raised the possibility of dermatophytic infection. Cases were considered positive for dermatophytes if KOH preparation, fungal culture, or skin biopsy (with or without periodic acid–Schiff staining) were positive or if there was a complete response to antifungal treatment alone. In this small number of cases, we found that RCM was 100% sensitive, as hyphae were readily seen in all cases of dermatophytic infections. In 1 RCM-positive case (patient 3), fungal culture with DTM was negative, but antifungal therapy was nonetheless given. Because the lesion resolved promptly with econazole, RCM proved to be true positive and DTM proved to be false negative (Table). Reflectance confocal microscopy imaging, however, was less specific. Of the 5 cases that showed no presence of dermatophytic infection, there was 1 case (patient 6) in which the pathologist could recognize structures that resembled fungal hyphae. There are various possible sources of structures masquerading as dermatophytes on confocal imaging, including the edges of nonnucleated loose keratinocytes, keratin fragments, and other foreign fibers. Evaluation by an experienced investigator can certainly help in limiting false-positive analyses, but a larger case study would be useful to develop a set of specific criteria to aid in the differentiation of fungal hyphae from other artifacts as well as to further define the sensitivity and specificity of RCM.
We also encountered difficulties with the technical aspects of RCM. One case (patient 5) was excluded from the analysis because the images were poor quality and could not be interpreted, and 2 cases (patients 2 and 7) had suboptimal images, in part due to operator error and in part due to equipment error that was recognized later on. The technical difficulties were problematic because no immediate review of image quality was available while patients were still present for possible reimaging. All of the images evaluated in this study were captured shortly after the RCM device was introduced to the practice. It is possible that with more training and a quick, on-site review of image quality, these technical problems could be avoided. Imaging protocols (ie, numbers and levels of scans taken by the confocal microscope) also could be adjusted so they include a large enough range to compensate for potential operator errors; however, these adjustments also could increase overall imaging time.
Conclusion
Based on our chart review of a small number of cases, we found that RCM can be a useful tool in diagnosing dermatophytic infections of the skin. With adequate training, dermatologists may be able to use RCM as an in-office tool to capture and evaluate images and subsequently diagnose or exclude dermatophytosis in a quick and noninvasive manner. However, further research and controlled studies of more cases will be required to develop accurate criteria for diagnosing fungal structures by RCM as well as to help determine the role of RCM in our diagnostic armamentarium.
1. Longo C, Farnetani F, Ciardo S, et al. Is confocal microscopy a valuable tool in diagnosing nodular lesions? a study of 140 cases. Br J Dermatol. 2013;169:58-67.
2. Debarbieux S, Dalle S, Depaepe L, et al. Second primary melanomas under BRAF blockers: study by reflectance confocal microscopy [published online a head of print April 1, 2013]. Br J Dermatol. 2013;168:1230-1235.
3. Schwartz RJ, Vera K, Navarrete N, et al. In vivo reflectance confocal microscopy of halo nevus. J Cutan Med Surg. 2013;17:33-38.
4. Pellacani G, Guitera P, Longo C, et al. The impact of in vivo reflectance confocal microscopy for the diagnostic accuracy of melanoma and equivocal melanocytic lesions. J Invest Dermatol. 2007;127:2759-2765.
5. Nori S, Rius-Diaz F, Cuevas J, et al. Sensitivity and specificity of reflectance-mode confocal micros-copy for in vivo diagnosis of basal cell carcinoma: a multicenter study. J Am Acad Dermatol. 2004;51:923-930.
6. Arzberger E, Komericki P, Ahlgrimm-Siess V, et al. Differentiation between balanitis and carcinoma in situ using reflectance confocal microscopy. JAMA Dermatol. 2013;149:440-445.
7. Ardigò M, Maliszewski I, Cota C, et al. Preliminary evaluation of in vivo reflectance confocal microscopy features of discoid lupus erythematosus. Br J Dermatol. 2007;156:1196-1203.
8. Longo C, Casari A, Pepe P, et al. Confocal microscopy insights into the treatment and cellular immune response of Basal cell carcinoma to photodynamic therapy. Dermatology. 2012;225:264-270.
9. Hongcharu W, Dwyer P, Gonzalez S, et al. Confirmation of onychomycosis by in vivo confocal microscopy. J Am Acad Dermatol. 2000;42:214-216.
10. Rothmund G, Sattler EC, Kaestle R, et al. Confocal laser scanning microscopy as a new valuable tool in the diagnosis of onychomycosis—comparison of six diagnostic methods [published online ahead of print April 23, 2012]. Mycoses. 2013;56:47-55.
11. Markus R, Huzaira M, Anderson RR, et al. A better potassium hydroxide preparation? Arch Dermatol. 2001;137:1076-1078.
12. Slutsky JB, Rabinovitz H, Grichnik JM, et al. Reflectance confocal microscopic features of dermatophytes, scabies, and Demodex. Arch Dermatol. 2011;147:1008.
13. Hui D, Xue-chang S, Ai-e X. Evaluation of reflectance confocal microscopy in dermatophytosis. Mycoses. 2013;56:130-133.
14. Turan E, Erdemir AT, Gurel MS, et al. A new diagnostic technique for tinea incognito: in vivo reflectance confocal microscopy. report of five cases. Skin Res Technol. 2013;19:e103-e107.
1. Longo C, Farnetani F, Ciardo S, et al. Is confocal microscopy a valuable tool in diagnosing nodular lesions? a study of 140 cases. Br J Dermatol. 2013;169:58-67.
2. Debarbieux S, Dalle S, Depaepe L, et al. Second primary melanomas under BRAF blockers: study by reflectance confocal microscopy [published online a head of print April 1, 2013]. Br J Dermatol. 2013;168:1230-1235.
3. Schwartz RJ, Vera K, Navarrete N, et al. In vivo reflectance confocal microscopy of halo nevus. J Cutan Med Surg. 2013;17:33-38.
4. Pellacani G, Guitera P, Longo C, et al. The impact of in vivo reflectance confocal microscopy for the diagnostic accuracy of melanoma and equivocal melanocytic lesions. J Invest Dermatol. 2007;127:2759-2765.
5. Nori S, Rius-Diaz F, Cuevas J, et al. Sensitivity and specificity of reflectance-mode confocal micros-copy for in vivo diagnosis of basal cell carcinoma: a multicenter study. J Am Acad Dermatol. 2004;51:923-930.
6. Arzberger E, Komericki P, Ahlgrimm-Siess V, et al. Differentiation between balanitis and carcinoma in situ using reflectance confocal microscopy. JAMA Dermatol. 2013;149:440-445.
7. Ardigò M, Maliszewski I, Cota C, et al. Preliminary evaluation of in vivo reflectance confocal microscopy features of discoid lupus erythematosus. Br J Dermatol. 2007;156:1196-1203.
8. Longo C, Casari A, Pepe P, et al. Confocal microscopy insights into the treatment and cellular immune response of Basal cell carcinoma to photodynamic therapy. Dermatology. 2012;225:264-270.
9. Hongcharu W, Dwyer P, Gonzalez S, et al. Confirmation of onychomycosis by in vivo confocal microscopy. J Am Acad Dermatol. 2000;42:214-216.
10. Rothmund G, Sattler EC, Kaestle R, et al. Confocal laser scanning microscopy as a new valuable tool in the diagnosis of onychomycosis—comparison of six diagnostic methods [published online ahead of print April 23, 2012]. Mycoses. 2013;56:47-55.
11. Markus R, Huzaira M, Anderson RR, et al. A better potassium hydroxide preparation? Arch Dermatol. 2001;137:1076-1078.
12. Slutsky JB, Rabinovitz H, Grichnik JM, et al. Reflectance confocal microscopic features of dermatophytes, scabies, and Demodex. Arch Dermatol. 2011;147:1008.
13. Hui D, Xue-chang S, Ai-e X. Evaluation of reflectance confocal microscopy in dermatophytosis. Mycoses. 2013;56:130-133.
14. Turan E, Erdemir AT, Gurel MS, et al. A new diagnostic technique for tinea incognito: in vivo reflectance confocal microscopy. report of five cases. Skin Res Technol. 2013;19:e103-e107.
Practice Points
- Current methods for diagnosing dermatophytosis can be invasive, with variable sensitivity and/or slow turnaround time.
- Reflectance confocal microscopy is a promising option for rapid noninvasive diagnosis of dermatophytosis.
Epithelioid Sarcoma Resembling Benign Fibrous Histiocytoma
Epithelioid sarcoma (ES) is a rare malignant soft tissue neoplasm that is most often encountered on the distal extremities of young adults.1 Epithelioid sarcoma is notorious for its tendency to mimic palisading granulomatous processes such as granuloma annulare. We report a case of ES on the right hand of a 23-year-old man that resembled a benign fibrous histiocytoma (dermatofibroma) on incisional biopsy. The typical histopathologic features of ES were identified after amputation of the hand and evaluation of the deeper regions of the tumor. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.
|
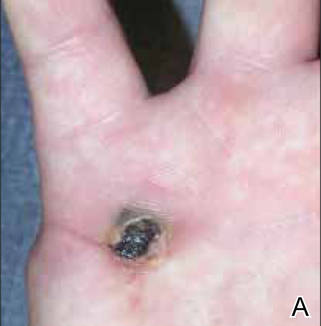
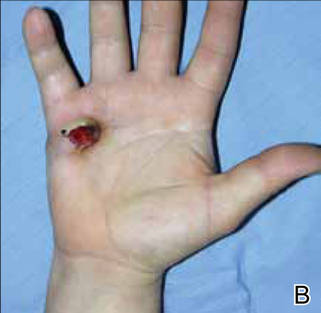
Case Report
A 23-year-old man presented with a nonhealing lesion on the right palm. His medical history was remarkable for a giant cell tumor of the tendon sheath involving the right fifth finger that had been treated via excision at an outside institution 2 years prior. Clinical examination revealed a 0.8×0.6-cm painful, firm, ulcerated dermal nodule with a hemorrhagic crust on the palmar surface of the right hand (Figure 1A). The clinical differential diagnosis included melanoma, traumatized verruca vulgaris, thrombosed pyogenic granuloma, and foreign body. A shave biopsy demonstrated verrucous epidermal hyperplasia, but the specimen did not include the dermis. Cultures of the lesion were positive for Staphylococcus aureus, and antibiotic therapy was initiated. In light of the clinical findings and the patient’s history of a giant cell tumor, imaging studies were performed. Magnetic resonance angiography showed abnormal masslike infiltrative enhancement throughout the soft tissues surrounding the right fifth metacarpal bone. The differential included a recurrent giant cell tumor, fibromatosis, and other soft tissue neoplasms.
After several missed appointments and surgery cancellations, the patient returned 4 months later for an incisional biopsy. Physical examination revealed a persistent palmar ulcer that had grown to 1.4×1 cm in size, along with an indurated purple plaque wrapping around the ulnar aspect of the right hand (Figure 1B). The biopsy demonstrated a proliferation of spindled and ovoid cells with scant cytoplasm that surrounded sclerotic collagen bundles resembling a dermatofibroma (Figure 2A). Cytologic atypia and mitotic activity were absent (Figure 2B). Glass slides of the original biopsy, which ultimately led to the diagnosis of the giant cell tumor of the tendon sheath more than 2 years earlier, were obtained and showed similar features. The proliferating cells were strongly and diffusely immunoreactive for vimentin, CD34, and cancer antigen 125 (CA 125). Scattered tumor cells strongly expressed cytokeratins (CKs) AE1/AE3 and cell adhesion molecule 5.2 (Figure 3). Staining for CD99 and epithelial membrane antigen was diffuse but weak. Factor XIIIa, S-100, CK7, smooth muscle actin, muscle-specific actin (HHF35), CD31, CD68, and B-cell lymphoma 2 were negative within the proliferating cells. Based on the clinical examination and results of the immunohistochemical staining, a diagnosis of ES was favored.
|
After a negative metastatic workup, amputation of the right hand was performed. The amputation specimen showed a tumor that extended through the entire hand with encasement of large vessels and tendons. Although the more superficial regions were cytologically bland, deep-seated regions of the tumor exhibited greater cellularity, nuclear pleomorphism, and mitotic activity (Figure 4). There was no bone involvement. Right axillary sentinel lymph nodes were negative for metastasis. Eighteen months later the patient developed chest and back pain with dyspnea. Thorascopic surgery was performed for a left pleural effusion and metastases to the left parietal pleura and adjacent soft tissue were identified. The patient was subsequently lost to follow-up.
Comment
First described by Enzinger1 in 1970, ES is a rare malignant soft tissue neoplasm that most frequently arises on the hands, forearms, and pretibial soft tissues of young adults.1-3 It is an aggressive tumor characterized by frequent recurrences and a high metastatic rate, with lung and regional lymph nodes being favored metastatic sites.1-5 Periods of several months or even years often pass between the initial presentation and establishment of a correct diagnosis, as ES frequently is mistaken for other benign conditions. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.6,7 In his original series of 62 cases, Enzinger1 noted that 17 patients were referred for treatment with a diagnosis of a benign fibrohistiocytic neoplasm, and other reports have described a resemblance to fibrous and fibrohistiocytic neoplasms.8-11 Mirra et al10 designated these tumors as fibromalike variants of ES. Additional subtypes of ES have subsequently been recognized, including those described as angiomatoid or angiosarcomalike, reflecting the potential of ES to resemble vascular tumors.12 A proximal type of ES also has been described. This lesion presents as a deep-seated tumor on the proximal limbs and is associated with more aggressive behavior. It lacks the granulomalike pattern and has more prominent epithelioid and rhabdoid histological presentation.13-15
Epithelioid sarcoma is a mesenchymal tumor that can display multidirectional differentiation that is primarily epithelial.16 The precise histogenesis of ES remains unclear, but studies have demonstrated a spectrum of differentiation that ranges from primitive myofibroblast or fibrohistiocytelike cells to those with well-developed epithelial properties.16,17 Epithelioid sarcoma characteristically coexpresses vimentin and low-molecular-weight CKs such as cell adhesion molecule 5.2. The tumor cells often are immunoreactive for epithelial membrane antigen and more than 50% of cases exhibit remarkable CD34 positivity.16 More recent studies have further refined the immunophenotype, demonstrating frequent expression of CK8 and CK19 but less commonly CK7, CK20, CK34bE12, and CK5/6.18-20 Additional studies reported that in 10 of 11 cases, ES was positive for CA 125 on immunohistochemical staining, and 3 of 5 patients also had elevated serum CA 125 levels.21,22 More recently, Hoshino et al23 showed elevated serum CA 125 levels in 5 of 7 patients with ES. Cancer antigen 125 is a high-molecular-weight glycoprotein commonly used in the identification of epithelial ovarian carcinomas; however, it also has been described in a number of other neoplasms including carcinomas of the breast, lungs, and colon and lymphoma.24-27 Although it appears that the addition of CA 125 to a panel of other immunohistochemical stains may be helpful in differentiating ES from other soft tissue sarcomas and serum CA 125 levels may help determine tumor burden, currently the number of cases studied is too small to definitively make that conclusion.21,23 In our case, the tumor cells were strongly and diffusely positive for CA 125. Serum CA 125 levels were not available.
Cytogenetic studies have failed to identify a consistent chromosomal abnormality in ES.5 Some analyses performed by comparative genomic hybridization on isolated cases and small case series indicate that the most frequent alterations involve 8q, 18q11, and 22q11.13,28,29 The tumor suppressor gene SMARCB1/INI1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1/integrase interactor 1) has been mapped to 22q11, and ES commonly shows absence of nuclear staining for this protein, indicating inactivation.13-15
Conclusion
Benign fibrohistiocytic proliferations should be included in the differential of histological mimickers of ES. Deep biopsies are essential to differentiate these benign tumors from fibrous histiocytomalike or fibromalike lesions of ES because superficial portions of ES may be well differentiated.
1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
2. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218-225.
3. Chase DR, Enzinger FM. Epithelioid sarcoma. diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241-263.
4. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
5. Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol. 1993;10:286-291.
6. Heenan PJ, Quirk CJ, Papadimitriou JM. Epithelioid sarcoma. a diagnostic problem. Am J Dermatopathol. 1986;8:95-104.
7. DiCaudo DJ, McCalmont TH, Wick MR. Selected diagnostic problems in neoplastic dermatopathology. Arch Pathol Lab Med. 2007;131:434-439.
8. Ormsby AH, Liou LS, Oriba HA, et al. Epithelioid sarcoma of the penis: report of an unusual case and review of the literature. Ann Diagn Pathol. 2000;4:88-94.
9. Lowentritt B, Parsons JK, Argani P, et al. Pediatric epithelioid sarcoma of the penis. J Urol. 2004;172:296-297.
10. Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
11. Tan SH, Ong BH. Spindle cell variant of epithelioid sarcoma: an easily misdiagnosed tumour. Australas J Dermatol. 2001;42:139-141.
12. von Hochstetter AR, Grant JW, Meyer VE, et al. Angiomatoid variant of epithelioid sarcoma. the value of immunohistochemistry in the differential diagnosis. Chir Organi Mov. 1990;75(suppl 1):158-162.
13. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012-4019.
14. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004;41:283-290.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35:891-897.
16. Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
17. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;19:265-275.
18. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
19. Humble SD, Prieto VG, Horenstein MG. Cytokeratin 7 and 20 expression in epithelioid sarcoma. J Cutan Pathol. 2003;30:242-246.
20. Lin L, Skacel M, Sigel JE, et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol. 2003;30:114-117.
21. Kato H, Hatori M, Kokubun S, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34:149-154.
22. Kato H, Hatori M, Watanabe M, et al. Epithelioid sarcomas with elevated serum CA125: report of two cases. Jpn J Clin Oncol. 2003;33:141-144.
23. Hoshino M, Kawashima H, Ogose A, et al. Serum CA 125 expression as a tumor marker for the diagnosis and monitoring the clinical course of epithelioid sarcoma [published online ahead of print September 16, 2009]. J Cancer Res Clin Oncol. 2010;136:457-464.
24. Lee AH, Paish EC, Marchio C, et al. The expression of Wilm’s tumour-1 and CA125 in invasive micropapillary carcinoma of the breast. Histopathology. 2007;51:824-828.
25. Homma S, Satoh H, Kagohashi K, et al. Production of CA125 by human lung cancer cell lines. Clin Exp Med. 2004;4:139-141.
26. Streppel MM, Vincent A, Mukherjee R, et al. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum Pathol. 2012;42:1755-1763.
27. Wei G, Yuping Z, Jun W, et al. CA125 expression in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006; 47:1322-1326.
28. Feely MG, Fidler ME, Nelson M, et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet. 2000;119:155-157.
29. Lushnikova T, Knuutila S, Miettinen M. DNA copy number changes in epithelioid sarcoma and its variants: a comparative genomic hybridization study. Mod Pathol. 2000;13:1092-1096.
Epithelioid sarcoma (ES) is a rare malignant soft tissue neoplasm that is most often encountered on the distal extremities of young adults.1 Epithelioid sarcoma is notorious for its tendency to mimic palisading granulomatous processes such as granuloma annulare. We report a case of ES on the right hand of a 23-year-old man that resembled a benign fibrous histiocytoma (dermatofibroma) on incisional biopsy. The typical histopathologic features of ES were identified after amputation of the hand and evaluation of the deeper regions of the tumor. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.
|
Case Report
A 23-year-old man presented with a nonhealing lesion on the right palm. His medical history was remarkable for a giant cell tumor of the tendon sheath involving the right fifth finger that had been treated via excision at an outside institution 2 years prior. Clinical examination revealed a 0.8×0.6-cm painful, firm, ulcerated dermal nodule with a hemorrhagic crust on the palmar surface of the right hand (Figure 1A). The clinical differential diagnosis included melanoma, traumatized verruca vulgaris, thrombosed pyogenic granuloma, and foreign body. A shave biopsy demonstrated verrucous epidermal hyperplasia, but the specimen did not include the dermis. Cultures of the lesion were positive for Staphylococcus aureus, and antibiotic therapy was initiated. In light of the clinical findings and the patient’s history of a giant cell tumor, imaging studies were performed. Magnetic resonance angiography showed abnormal masslike infiltrative enhancement throughout the soft tissues surrounding the right fifth metacarpal bone. The differential included a recurrent giant cell tumor, fibromatosis, and other soft tissue neoplasms.
After several missed appointments and surgery cancellations, the patient returned 4 months later for an incisional biopsy. Physical examination revealed a persistent palmar ulcer that had grown to 1.4×1 cm in size, along with an indurated purple plaque wrapping around the ulnar aspect of the right hand (Figure 1B). The biopsy demonstrated a proliferation of spindled and ovoid cells with scant cytoplasm that surrounded sclerotic collagen bundles resembling a dermatofibroma (Figure 2A). Cytologic atypia and mitotic activity were absent (Figure 2B). Glass slides of the original biopsy, which ultimately led to the diagnosis of the giant cell tumor of the tendon sheath more than 2 years earlier, were obtained and showed similar features. The proliferating cells were strongly and diffusely immunoreactive for vimentin, CD34, and cancer antigen 125 (CA 125). Scattered tumor cells strongly expressed cytokeratins (CKs) AE1/AE3 and cell adhesion molecule 5.2 (Figure 3). Staining for CD99 and epithelial membrane antigen was diffuse but weak. Factor XIIIa, S-100, CK7, smooth muscle actin, muscle-specific actin (HHF35), CD31, CD68, and B-cell lymphoma 2 were negative within the proliferating cells. Based on the clinical examination and results of the immunohistochemical staining, a diagnosis of ES was favored.
|
After a negative metastatic workup, amputation of the right hand was performed. The amputation specimen showed a tumor that extended through the entire hand with encasement of large vessels and tendons. Although the more superficial regions were cytologically bland, deep-seated regions of the tumor exhibited greater cellularity, nuclear pleomorphism, and mitotic activity (Figure 4). There was no bone involvement. Right axillary sentinel lymph nodes were negative for metastasis. Eighteen months later the patient developed chest and back pain with dyspnea. Thorascopic surgery was performed for a left pleural effusion and metastases to the left parietal pleura and adjacent soft tissue were identified. The patient was subsequently lost to follow-up.
Comment
First described by Enzinger1 in 1970, ES is a rare malignant soft tissue neoplasm that most frequently arises on the hands, forearms, and pretibial soft tissues of young adults.1-3 It is an aggressive tumor characterized by frequent recurrences and a high metastatic rate, with lung and regional lymph nodes being favored metastatic sites.1-5 Periods of several months or even years often pass between the initial presentation and establishment of a correct diagnosis, as ES frequently is mistaken for other benign conditions. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.6,7 In his original series of 62 cases, Enzinger1 noted that 17 patients were referred for treatment with a diagnosis of a benign fibrohistiocytic neoplasm, and other reports have described a resemblance to fibrous and fibrohistiocytic neoplasms.8-11 Mirra et al10 designated these tumors as fibromalike variants of ES. Additional subtypes of ES have subsequently been recognized, including those described as angiomatoid or angiosarcomalike, reflecting the potential of ES to resemble vascular tumors.12 A proximal type of ES also has been described. This lesion presents as a deep-seated tumor on the proximal limbs and is associated with more aggressive behavior. It lacks the granulomalike pattern and has more prominent epithelioid and rhabdoid histological presentation.13-15
Epithelioid sarcoma is a mesenchymal tumor that can display multidirectional differentiation that is primarily epithelial.16 The precise histogenesis of ES remains unclear, but studies have demonstrated a spectrum of differentiation that ranges from primitive myofibroblast or fibrohistiocytelike cells to those with well-developed epithelial properties.16,17 Epithelioid sarcoma characteristically coexpresses vimentin and low-molecular-weight CKs such as cell adhesion molecule 5.2. The tumor cells often are immunoreactive for epithelial membrane antigen and more than 50% of cases exhibit remarkable CD34 positivity.16 More recent studies have further refined the immunophenotype, demonstrating frequent expression of CK8 and CK19 but less commonly CK7, CK20, CK34bE12, and CK5/6.18-20 Additional studies reported that in 10 of 11 cases, ES was positive for CA 125 on immunohistochemical staining, and 3 of 5 patients also had elevated serum CA 125 levels.21,22 More recently, Hoshino et al23 showed elevated serum CA 125 levels in 5 of 7 patients with ES. Cancer antigen 125 is a high-molecular-weight glycoprotein commonly used in the identification of epithelial ovarian carcinomas; however, it also has been described in a number of other neoplasms including carcinomas of the breast, lungs, and colon and lymphoma.24-27 Although it appears that the addition of CA 125 to a panel of other immunohistochemical stains may be helpful in differentiating ES from other soft tissue sarcomas and serum CA 125 levels may help determine tumor burden, currently the number of cases studied is too small to definitively make that conclusion.21,23 In our case, the tumor cells were strongly and diffusely positive for CA 125. Serum CA 125 levels were not available.
Cytogenetic studies have failed to identify a consistent chromosomal abnormality in ES.5 Some analyses performed by comparative genomic hybridization on isolated cases and small case series indicate that the most frequent alterations involve 8q, 18q11, and 22q11.13,28,29 The tumor suppressor gene SMARCB1/INI1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1/integrase interactor 1) has been mapped to 22q11, and ES commonly shows absence of nuclear staining for this protein, indicating inactivation.13-15
Conclusion
Benign fibrohistiocytic proliferations should be included in the differential of histological mimickers of ES. Deep biopsies are essential to differentiate these benign tumors from fibrous histiocytomalike or fibromalike lesions of ES because superficial portions of ES may be well differentiated.
Epithelioid sarcoma (ES) is a rare malignant soft tissue neoplasm that is most often encountered on the distal extremities of young adults.1 Epithelioid sarcoma is notorious for its tendency to mimic palisading granulomatous processes such as granuloma annulare. We report a case of ES on the right hand of a 23-year-old man that resembled a benign fibrous histiocytoma (dermatofibroma) on incisional biopsy. The typical histopathologic features of ES were identified after amputation of the hand and evaluation of the deeper regions of the tumor. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.
|
Case Report
A 23-year-old man presented with a nonhealing lesion on the right palm. His medical history was remarkable for a giant cell tumor of the tendon sheath involving the right fifth finger that had been treated via excision at an outside institution 2 years prior. Clinical examination revealed a 0.8×0.6-cm painful, firm, ulcerated dermal nodule with a hemorrhagic crust on the palmar surface of the right hand (Figure 1A). The clinical differential diagnosis included melanoma, traumatized verruca vulgaris, thrombosed pyogenic granuloma, and foreign body. A shave biopsy demonstrated verrucous epidermal hyperplasia, but the specimen did not include the dermis. Cultures of the lesion were positive for Staphylococcus aureus, and antibiotic therapy was initiated. In light of the clinical findings and the patient’s history of a giant cell tumor, imaging studies were performed. Magnetic resonance angiography showed abnormal masslike infiltrative enhancement throughout the soft tissues surrounding the right fifth metacarpal bone. The differential included a recurrent giant cell tumor, fibromatosis, and other soft tissue neoplasms.
After several missed appointments and surgery cancellations, the patient returned 4 months later for an incisional biopsy. Physical examination revealed a persistent palmar ulcer that had grown to 1.4×1 cm in size, along with an indurated purple plaque wrapping around the ulnar aspect of the right hand (Figure 1B). The biopsy demonstrated a proliferation of spindled and ovoid cells with scant cytoplasm that surrounded sclerotic collagen bundles resembling a dermatofibroma (Figure 2A). Cytologic atypia and mitotic activity were absent (Figure 2B). Glass slides of the original biopsy, which ultimately led to the diagnosis of the giant cell tumor of the tendon sheath more than 2 years earlier, were obtained and showed similar features. The proliferating cells were strongly and diffusely immunoreactive for vimentin, CD34, and cancer antigen 125 (CA 125). Scattered tumor cells strongly expressed cytokeratins (CKs) AE1/AE3 and cell adhesion molecule 5.2 (Figure 3). Staining for CD99 and epithelial membrane antigen was diffuse but weak. Factor XIIIa, S-100, CK7, smooth muscle actin, muscle-specific actin (HHF35), CD31, CD68, and B-cell lymphoma 2 were negative within the proliferating cells. Based on the clinical examination and results of the immunohistochemical staining, a diagnosis of ES was favored.
|
After a negative metastatic workup, amputation of the right hand was performed. The amputation specimen showed a tumor that extended through the entire hand with encasement of large vessels and tendons. Although the more superficial regions were cytologically bland, deep-seated regions of the tumor exhibited greater cellularity, nuclear pleomorphism, and mitotic activity (Figure 4). There was no bone involvement. Right axillary sentinel lymph nodes were negative for metastasis. Eighteen months later the patient developed chest and back pain with dyspnea. Thorascopic surgery was performed for a left pleural effusion and metastases to the left parietal pleura and adjacent soft tissue were identified. The patient was subsequently lost to follow-up.
Comment
First described by Enzinger1 in 1970, ES is a rare malignant soft tissue neoplasm that most frequently arises on the hands, forearms, and pretibial soft tissues of young adults.1-3 It is an aggressive tumor characterized by frequent recurrences and a high metastatic rate, with lung and regional lymph nodes being favored metastatic sites.1-5 Periods of several months or even years often pass between the initial presentation and establishment of a correct diagnosis, as ES frequently is mistaken for other benign conditions. The tendency for ES to mimic granulomatous processes is a common diagnostic pitfall, but the potential for its close resemblance to benign fibrous histiocytoma is less recognized.6,7 In his original series of 62 cases, Enzinger1 noted that 17 patients were referred for treatment with a diagnosis of a benign fibrohistiocytic neoplasm, and other reports have described a resemblance to fibrous and fibrohistiocytic neoplasms.8-11 Mirra et al10 designated these tumors as fibromalike variants of ES. Additional subtypes of ES have subsequently been recognized, including those described as angiomatoid or angiosarcomalike, reflecting the potential of ES to resemble vascular tumors.12 A proximal type of ES also has been described. This lesion presents as a deep-seated tumor on the proximal limbs and is associated with more aggressive behavior. It lacks the granulomalike pattern and has more prominent epithelioid and rhabdoid histological presentation.13-15
Epithelioid sarcoma is a mesenchymal tumor that can display multidirectional differentiation that is primarily epithelial.16 The precise histogenesis of ES remains unclear, but studies have demonstrated a spectrum of differentiation that ranges from primitive myofibroblast or fibrohistiocytelike cells to those with well-developed epithelial properties.16,17 Epithelioid sarcoma characteristically coexpresses vimentin and low-molecular-weight CKs such as cell adhesion molecule 5.2. The tumor cells often are immunoreactive for epithelial membrane antigen and more than 50% of cases exhibit remarkable CD34 positivity.16 More recent studies have further refined the immunophenotype, demonstrating frequent expression of CK8 and CK19 but less commonly CK7, CK20, CK34bE12, and CK5/6.18-20 Additional studies reported that in 10 of 11 cases, ES was positive for CA 125 on immunohistochemical staining, and 3 of 5 patients also had elevated serum CA 125 levels.21,22 More recently, Hoshino et al23 showed elevated serum CA 125 levels in 5 of 7 patients with ES. Cancer antigen 125 is a high-molecular-weight glycoprotein commonly used in the identification of epithelial ovarian carcinomas; however, it also has been described in a number of other neoplasms including carcinomas of the breast, lungs, and colon and lymphoma.24-27 Although it appears that the addition of CA 125 to a panel of other immunohistochemical stains may be helpful in differentiating ES from other soft tissue sarcomas and serum CA 125 levels may help determine tumor burden, currently the number of cases studied is too small to definitively make that conclusion.21,23 In our case, the tumor cells were strongly and diffusely positive for CA 125. Serum CA 125 levels were not available.
Cytogenetic studies have failed to identify a consistent chromosomal abnormality in ES.5 Some analyses performed by comparative genomic hybridization on isolated cases and small case series indicate that the most frequent alterations involve 8q, 18q11, and 22q11.13,28,29 The tumor suppressor gene SMARCB1/INI1 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily B, member 1/integrase interactor 1) has been mapped to 22q11, and ES commonly shows absence of nuclear staining for this protein, indicating inactivation.13-15
Conclusion
Benign fibrohistiocytic proliferations should be included in the differential of histological mimickers of ES. Deep biopsies are essential to differentiate these benign tumors from fibrous histiocytomalike or fibromalike lesions of ES because superficial portions of ES may be well differentiated.
1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
2. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218-225.
3. Chase DR, Enzinger FM. Epithelioid sarcoma. diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241-263.
4. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
5. Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol. 1993;10:286-291.
6. Heenan PJ, Quirk CJ, Papadimitriou JM. Epithelioid sarcoma. a diagnostic problem. Am J Dermatopathol. 1986;8:95-104.
7. DiCaudo DJ, McCalmont TH, Wick MR. Selected diagnostic problems in neoplastic dermatopathology. Arch Pathol Lab Med. 2007;131:434-439.
8. Ormsby AH, Liou LS, Oriba HA, et al. Epithelioid sarcoma of the penis: report of an unusual case and review of the literature. Ann Diagn Pathol. 2000;4:88-94.
9. Lowentritt B, Parsons JK, Argani P, et al. Pediatric epithelioid sarcoma of the penis. J Urol. 2004;172:296-297.
10. Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
11. Tan SH, Ong BH. Spindle cell variant of epithelioid sarcoma: an easily misdiagnosed tumour. Australas J Dermatol. 2001;42:139-141.
12. von Hochstetter AR, Grant JW, Meyer VE, et al. Angiomatoid variant of epithelioid sarcoma. the value of immunohistochemistry in the differential diagnosis. Chir Organi Mov. 1990;75(suppl 1):158-162.
13. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012-4019.
14. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004;41:283-290.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35:891-897.
16. Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
17. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;19:265-275.
18. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
19. Humble SD, Prieto VG, Horenstein MG. Cytokeratin 7 and 20 expression in epithelioid sarcoma. J Cutan Pathol. 2003;30:242-246.
20. Lin L, Skacel M, Sigel JE, et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol. 2003;30:114-117.
21. Kato H, Hatori M, Kokubun S, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34:149-154.
22. Kato H, Hatori M, Watanabe M, et al. Epithelioid sarcomas with elevated serum CA125: report of two cases. Jpn J Clin Oncol. 2003;33:141-144.
23. Hoshino M, Kawashima H, Ogose A, et al. Serum CA 125 expression as a tumor marker for the diagnosis and monitoring the clinical course of epithelioid sarcoma [published online ahead of print September 16, 2009]. J Cancer Res Clin Oncol. 2010;136:457-464.
24. Lee AH, Paish EC, Marchio C, et al. The expression of Wilm’s tumour-1 and CA125 in invasive micropapillary carcinoma of the breast. Histopathology. 2007;51:824-828.
25. Homma S, Satoh H, Kagohashi K, et al. Production of CA125 by human lung cancer cell lines. Clin Exp Med. 2004;4:139-141.
26. Streppel MM, Vincent A, Mukherjee R, et al. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum Pathol. 2012;42:1755-1763.
27. Wei G, Yuping Z, Jun W, et al. CA125 expression in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006; 47:1322-1326.
28. Feely MG, Fidler ME, Nelson M, et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet. 2000;119:155-157.
29. Lushnikova T, Knuutila S, Miettinen M. DNA copy number changes in epithelioid sarcoma and its variants: a comparative genomic hybridization study. Mod Pathol. 2000;13:1092-1096.
1. Enzinger FM. Epitheloid sarcoma. a sarcoma simulating a granuloma or a carcinoma. Cancer. 1970;26:1029-1041.
2. Spillane AJ, Thomas JM, Fisher C. Epithelioid sarcoma: the clinicopathological complexities of this rare soft tissue sarcoma. Ann Surg Oncol. 2000;7:218-225.
3. Chase DR, Enzinger FM. Epithelioid sarcoma. diagnosis, prognostic indicators, and treatment. Am J Surg Pathol. 1985;9:241-263.
4. Fisher C. Epithelioid sarcoma of Enzinger. Adv Anat Pathol. 2006;13:114-121.
5. Evans HL, Baer SC. Epithelioid sarcoma: a clinicopathologic and prognostic study of 26 cases. Semin Diagn Pathol. 1993;10:286-291.
6. Heenan PJ, Quirk CJ, Papadimitriou JM. Epithelioid sarcoma. a diagnostic problem. Am J Dermatopathol. 1986;8:95-104.
7. DiCaudo DJ, McCalmont TH, Wick MR. Selected diagnostic problems in neoplastic dermatopathology. Arch Pathol Lab Med. 2007;131:434-439.
8. Ormsby AH, Liou LS, Oriba HA, et al. Epithelioid sarcoma of the penis: report of an unusual case and review of the literature. Ann Diagn Pathol. 2000;4:88-94.
9. Lowentritt B, Parsons JK, Argani P, et al. Pediatric epithelioid sarcoma of the penis. J Urol. 2004;172:296-297.
10. Mirra JM, Kessler S, Bhuta S, et al. The fibroma-like variant of epithelioid sarcoma. a fibrohistiocytic/myoid cell lesion often confused with benign and malignant spindle cell tumors. Cancer. 1992;69:1382-1395.
11. Tan SH, Ong BH. Spindle cell variant of epithelioid sarcoma: an easily misdiagnosed tumour. Australas J Dermatol. 2001;42:139-141.
12. von Hochstetter AR, Grant JW, Meyer VE, et al. Angiomatoid variant of epithelioid sarcoma. the value of immunohistochemistry in the differential diagnosis. Chir Organi Mov. 1990;75(suppl 1):158-162.
13. Modena P, Lualdi E, Facchinetti F, et al. SMARCB1/INI1 tumor suppressor gene is frequently inactivated in epithelioid sarcomas. Cancer Res. 2005;65:4012-4019.
14. Lualdi E, Modena P, Debiec-Rychter M, et al. Molecular cytogenetic characterization of proximal-type epithelioid sarcoma. Genes Chromosomes Cancer. 2004;41:283-290.
15. Kosemehmetoglu K, Kaygusuz G, Bahrami A, et al. Intra-articular epithelioid sarcoma showing mixed classic and proximal-type features: report of 2 cases, with immunohistochemical and molecular cytogenetic INI-1 study. Am J Surg Pathol. 2011;35:891-897.
16. Armah HB, Parwani AV. Epithelioid sarcoma. Arch Pathol Lab Med. 2009;133:814-819.
17. Fisher C. Epithelioid sarcoma: the spectrum of ultrastructural differentiation in seven immunohistochemically defined cases. Hum Pathol. 1988;19:265-275.
18. Miettinen M, Fanburg-Smith JC, Virolainen M, et al. Epithelioid sarcoma: an immunohistochemical analysis of 112 classical and variant cases and a discussion of the differential diagnosis. Hum Pathol. 1999;30:934-942.
19. Humble SD, Prieto VG, Horenstein MG. Cytokeratin 7 and 20 expression in epithelioid sarcoma. J Cutan Pathol. 2003;30:242-246.
20. Lin L, Skacel M, Sigel JE, et al. Epithelioid sarcoma: an immunohistochemical analysis evaluating the utility of cytokeratin 5/6 in distinguishing superficial epithelioid sarcoma from spindled squamous cell carcinoma. J Cutan Pathol. 2003;30:114-117.
21. Kato H, Hatori M, Kokubun S, et al. CA125 expression in epithelioid sarcoma. Jpn J Clin Oncol. 2004;34:149-154.
22. Kato H, Hatori M, Watanabe M, et al. Epithelioid sarcomas with elevated serum CA125: report of two cases. Jpn J Clin Oncol. 2003;33:141-144.
23. Hoshino M, Kawashima H, Ogose A, et al. Serum CA 125 expression as a tumor marker for the diagnosis and monitoring the clinical course of epithelioid sarcoma [published online ahead of print September 16, 2009]. J Cancer Res Clin Oncol. 2010;136:457-464.
24. Lee AH, Paish EC, Marchio C, et al. The expression of Wilm’s tumour-1 and CA125 in invasive micropapillary carcinoma of the breast. Histopathology. 2007;51:824-828.
25. Homma S, Satoh H, Kagohashi K, et al. Production of CA125 by human lung cancer cell lines. Clin Exp Med. 2004;4:139-141.
26. Streppel MM, Vincent A, Mukherjee R, et al. Mucin 16 (cancer antigen 125) expression in human tissues and cell lines and correlation with clinical outcome in adenocarcinomas of the pancreas, esophagus, stomach, and colon. Hum Pathol. 2012;42:1755-1763.
27. Wei G, Yuping Z, Jun W, et al. CA125 expression in patients with non-Hodgkin’s lymphoma. Leuk Lymphoma. 2006; 47:1322-1326.
28. Feely MG, Fidler ME, Nelson M, et al. Cytogenetic findings in a case of epithelioid sarcoma and a review of the literature. Cancer Genet Cytogenet. 2000;119:155-157.
29. Lushnikova T, Knuutila S, Miettinen M. DNA copy number changes in epithelioid sarcoma and its variants: a comparative genomic hybridization study. Mod Pathol. 2000;13:1092-1096.
Practice Points
- Epithelioid sarcoma should be considered in the clinical differential diagnosis of nonhealing recurrent lesions of the distal extremities in a young adult.
- Histological presentation of epithelioid sarcoma can mimic a number of benign granulomatous and fibrohistiocytic processes, including benign fibrous histiocytoma.
- Deeper biopsies may be needed to demonstrate the overtly malignant morphology characteristic of epithelioid sarcoma.
- Inactivation of SMARCB1/INI1 is a common molecular aberration identified in epithelioid sarcoma and can be demonstrated immunohistochemically by absence of nuclear staining in tumor cells.
Imiquimod Induces Sustained Remission of Actinic Damage: A Case Report Spanning One Decade of Observation
Sun damage and chronic exposure to UV radiation have been recognized as causative factors for the development of squamous cell carcinoma (SCC), its precursor actinic keratosis (AK), and basal cell carcinoma (BCC). Although surgical treatment is necessary for most advanced cases of skin cancer, several other therapeutic approaches have been described including the use of topical chemotherapy agents such as 5-fluorouracil (5-FU) and topical immunomodulators such as imiquimod. Unlike surgery, these agents provide the added benefit of treating larger fields of photodamaged skin. With the increasing prevalence of nonmelanoma skin cancers (NMSCs), the use of multiple topical agents for treatment will continue to become more common.
We present the case of a patient who underwent field therapy with topical 5-FU for diffuse actinic damage and AKs. There was no subsequent inflammatory response within the perimeter of a BCC that had been treated with imiquimod 10 years prior.
Case Report
An otherwise healthy 58-year-old man with a history of long-standing diffuse sun damage and multiple prior NMSCs presented for treatment of a recurrent BCC on the right cheek. The patient reported that the BCC had initially been biopsied and excised by his primary care physician. Two months later local recurrence was noted by the primary care physician and the patient was subsequently referred to our dermatology office. A 2-month treatment course with daily imiquimod cream 5% was initiated. This treatment caused extensive inflammation of the right cheek but was otherwise well tolerated (Figure 1).
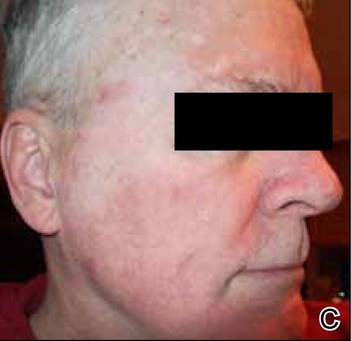 |
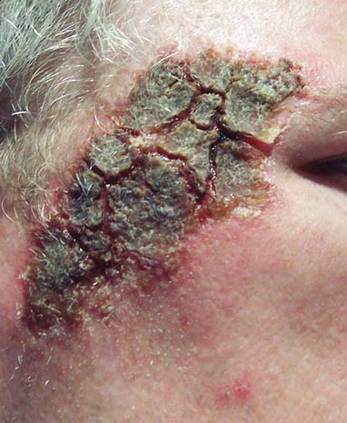
During a routine skin cancer screening 10 years later, no recurrence of the BCC was noted on the right cheek; however, the patient had developed multiple AKs on the face. Therapeutic options were discussed with the patient; he agreed to topical field therapy with 5-FU cream 0.5%. The patient applied the 5-FU cream to the entire face nightly for 1 month. During this time he experienced a brisk inflammatory response with painful cracking and redness of the skin. On follow-up, it was noted that the area on the right cheek that had been treated with imiquimod 10 years prior showed no inflammatory response despite nightly application of 5-FU cream to the area (Figure 2). The patient denied any routine use of sunscreen or other sun-protective practices.
Comment
Basal cell carcinoma is the most common skin cancer in the United States with an incidence of 1.4% to 2% per year. It has become more prevalent in recent decades, likely due to genetic predisposition and increasing cumulative sun exposure.1-4 A variety of treatment options are available. Surgical interventions include destruction via electrodesiccation and curettage, local excision, and Mohs micrographic surgery. One of the challenges in the management of BCC, as was the case in our patient, is the treatment of tumors that arise in cosmetically or functionally sensitive areas. Approaches that minimize the amount of tissue removed while ensuring the highest possible cure rate are favorable. In addition to surgery, topical imiquimod has been established as a potential treatment of BCC. Imiquimod, a nucleoside analogue of the imidazoquinoline family, is an agonist of toll-like receptors 7 and 8 that promotes cytokine-induced cell death via nuclear factor kB and a helper T cell TH1-weighted antitumor inflammatory response.5,6 Although clearance rates with imiquimod vary by drug regimen, success rates of 43% to 100% for superficial BCCs, 42% to 100% for nodular BCCs, and 56% to 63% for infiltrative BCCs have been reported.7 In a 2007 randomized study of imiquimod cream 5%, 5-FU ointment 5%, or cryosurgery for the treatment of AK, imiquimod resulted in superior and more reliable clearance with lower recurrence rates.8
Similar to BCC, AK is closely linked to lifetime cumulative sun exposure.9 Actinic keratoses have been well established as precursors to SCC, and some researchers advocate for their reclassification as early SCC in situ.10 The incidence of malignant conversion of AK to SCC has been estimated at 0.025% to 16% annually, with an estimated lifetime risk for malignant transformation of 8% per individual AK.11,12 Cryotherapy has been a mainstay for the treatment of isolated AK, and alternative therapies including curettage, photodynamic therapy, and laser therapy have been employed. Field-directed therapy has become a popular alternative that targets multiple lesions and field cancerization.8,13,14 Field cancerization implies that if one cell in the patient’s epidermis has been exposed to enough UV radiation to develop into a precancerous lesion or early skin cancer, then many other cells in the same environment likely have some degree of UV radiation–induced atypia.15 5-Fluorouacil is a pyrimidine analogue chemotherapeutic agent that inhibits thymidylate synthase and interferes with DNA synthesis.16 This mechanism of 5-FU commonly causes an inflammatory response characterized by burning, dryness, and redness, but these effects rarely force early discontinuation of treatment. A randomized controlled trial comparing 5-FU cream 0.5% to a placebo found that complete clearance rates at 4 weeks posttreatment were significantly higher in the treatment group (47.5%) versus placebo (3.4%)(P<.001).13 Additional trials have established no significant superiority of 5-FU cream 5% over 5-FU cream 0.5%, with a decrease in side effects noted in patients treated with the lower concentration.17
Our patient had a history of a recurrent BCC and was previously treated with imiquimod. He showed no inflammatory response to field therapy with 5-FU within the perimeter of prior immunomodulatory therapy. Although no frank scaling or crusting papules consistent with AK were observed in the previously treated area prior to 5-FU therapy, subclinical field damage in that area was expected because 10 years of additional sun exposure had accumulated since imiquimod therapy was completed. Several conclusions can be drawn from this observation. Primarily, no new clinically significant actinic lesions occurred on the previously treated skin. This observation is consistent with 12-month follow-up data on AKs treated with either 5-FU, imiquimod, or cryosurgery that identified imiquimod as having the lowest recurrence rate.8 Thus, a photoprotective effect may be ascribed to imiquimod therapy that extends beyond its drug effects on atypical keratinocytes. It has been one author’s personal experience (M.Q.) that patients treated with 5-FU experience recurrence of AKs within 3 to 5 years versus 10 years of remission with imiquimod. In our patient, imiquimod therapy seemed to reset the patient’s skin at the location of the prior BCC and surrounding field cancerization.
Studies with long-term follow-up are needed to investigate the need for re-treatment with imiquimod or 5-FU. The longevity of imiquimod treatment may be of importance beyond the treatment of AKs or NMSCs. For instance, during the treatment of lentigo maligna with imiquimod, Metcalf et al18 found a significant reduction in solar elastosis (P=.0036), normalization of epidermal thickness (P=.0073), and increased papillary dermal fibroplasia in pre- and posttreatment biopsies (P<.0001), which have been described as antiaging effects in the laypress. Some of these mechanisms appear to be implicated in the observations noted in our patient. The 10-year period between the 2 courses of therapy in our patient suggests that imiquimod may cause sustained healing of skin that was previously classified both clinically and microscopically as UV damaged.
Conclusion
Both topical immunomodulators such as imiquimod and topical chemotherapeutic agents such as 5-FU have a role in the field treatment of AK and the focal treatment of superficial BCC and SCC. As multiple topical immunomodulators continue to be evaluated, long-term studies assessing the need for re-treatment as well as the degree of sustained remission of sun damage will be necessary. We expect that their individual roles will continue to become more precisely defined and distinct in the coming years.
1. Flohil SC, de Vries E, Neumann HA, et al. Incidence, prevalence and future trends of primary basal cell carcinoma in the Netherlands. Acta Derm Venereol. 2011;91:24-30.
2. Donaldson MR, Coldiron BM. No end in sight: the skin cancer epidemic continues. Semin Cutan Med Surg. 2011;30:3-5.
3. Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer. I. Basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
4. Gailani MR, Leffell DJ, Ziegler A, et al. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J Natl Cancer Inst. 1996;88:349-354.
5. Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway [published online ahead of print January 22, 2002]. Nat Immunol. 2002;3:196-200.
6. Schön MP, Schön M. Imiquimod: mode of action. Br J Dermatol. 2007;157(suppl 2):8-13.
7. Love WE, Bernhard JD, Bordeaux JS. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch Dermatol. 2009;145:1431-1438.
8. Krawtchenko N, Roewert-Huber J, Ulrich M, et al. A randomised study of topical 5% imiquimod vs. topical5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(suppl 2):34-40.
9. Feldman SR, Fleischer AB Jr. Progression of actinic keratosis to squamous cell carcinoma revisited: clinical and treatment implications. Cutis. 2011;87:201-207.
10. Röwert-Huber J, Patel MJ, Forschner T, et al. Actinic keratosis is an early in situ squamous cell carcinoma: a proposal for reclassification. Br J Dermatol. 2007;156(suppl 3):8-12.
11. Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42(1 pt 2):23-24.
12. Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
13. Weiss J, Menter A, Hevia O, et al. Effective treatment of actinic keratosis with 0.5% fluorouracil cream for 1, 2, or 4 weeks. Cutis. 2002;70(2 suppl):22-29.
14. Almeida Gonçalves JC, De Noronha T. 5-fluouracil (5-FU) ointment in the treatment of skin tumours and keratoses. Dermatologica. 1970;140(suppl 1):97+.
15. Vanharanta S, Massagué J. Field cancerization: something new under the sun. Cell. 2012;149:1179-1181.
16. Robins P, Gupta AK. The use of topical fluorouracil to treat actinic keratosis. Cutis. 2002;70(2 suppl):4-7.
17. Kaur R, Alikhan A, Maibach H. Comparison of topical 5-fluorouracil formulations in actinic keratosis treatment. J Dermatolog Treat. 2010;2:267-271.
18. Metcalf S, Crowson AN, Naylor M, et al. Imiquimod as an antiaging agent [published online ahead of print December 20, 2006]. J Am Acad Dermatol. 2007;56:422-425.
Sun damage and chronic exposure to UV radiation have been recognized as causative factors for the development of squamous cell carcinoma (SCC), its precursor actinic keratosis (AK), and basal cell carcinoma (BCC). Although surgical treatment is necessary for most advanced cases of skin cancer, several other therapeutic approaches have been described including the use of topical chemotherapy agents such as 5-fluorouracil (5-FU) and topical immunomodulators such as imiquimod. Unlike surgery, these agents provide the added benefit of treating larger fields of photodamaged skin. With the increasing prevalence of nonmelanoma skin cancers (NMSCs), the use of multiple topical agents for treatment will continue to become more common.
We present the case of a patient who underwent field therapy with topical 5-FU for diffuse actinic damage and AKs. There was no subsequent inflammatory response within the perimeter of a BCC that had been treated with imiquimod 10 years prior.
Case Report
An otherwise healthy 58-year-old man with a history of long-standing diffuse sun damage and multiple prior NMSCs presented for treatment of a recurrent BCC on the right cheek. The patient reported that the BCC had initially been biopsied and excised by his primary care physician. Two months later local recurrence was noted by the primary care physician and the patient was subsequently referred to our dermatology office. A 2-month treatment course with daily imiquimod cream 5% was initiated. This treatment caused extensive inflammation of the right cheek but was otherwise well tolerated (Figure 1).
|
During a routine skin cancer screening 10 years later, no recurrence of the BCC was noted on the right cheek; however, the patient had developed multiple AKs on the face. Therapeutic options were discussed with the patient; he agreed to topical field therapy with 5-FU cream 0.5%. The patient applied the 5-FU cream to the entire face nightly for 1 month. During this time he experienced a brisk inflammatory response with painful cracking and redness of the skin. On follow-up, it was noted that the area on the right cheek that had been treated with imiquimod 10 years prior showed no inflammatory response despite nightly application of 5-FU cream to the area (Figure 2). The patient denied any routine use of sunscreen or other sun-protective practices.
Comment
Basal cell carcinoma is the most common skin cancer in the United States with an incidence of 1.4% to 2% per year. It has become more prevalent in recent decades, likely due to genetic predisposition and increasing cumulative sun exposure.1-4 A variety of treatment options are available. Surgical interventions include destruction via electrodesiccation and curettage, local excision, and Mohs micrographic surgery. One of the challenges in the management of BCC, as was the case in our patient, is the treatment of tumors that arise in cosmetically or functionally sensitive areas. Approaches that minimize the amount of tissue removed while ensuring the highest possible cure rate are favorable. In addition to surgery, topical imiquimod has been established as a potential treatment of BCC. Imiquimod, a nucleoside analogue of the imidazoquinoline family, is an agonist of toll-like receptors 7 and 8 that promotes cytokine-induced cell death via nuclear factor kB and a helper T cell TH1-weighted antitumor inflammatory response.5,6 Although clearance rates with imiquimod vary by drug regimen, success rates of 43% to 100% for superficial BCCs, 42% to 100% for nodular BCCs, and 56% to 63% for infiltrative BCCs have been reported.7 In a 2007 randomized study of imiquimod cream 5%, 5-FU ointment 5%, or cryosurgery for the treatment of AK, imiquimod resulted in superior and more reliable clearance with lower recurrence rates.8
Similar to BCC, AK is closely linked to lifetime cumulative sun exposure.9 Actinic keratoses have been well established as precursors to SCC, and some researchers advocate for their reclassification as early SCC in situ.10 The incidence of malignant conversion of AK to SCC has been estimated at 0.025% to 16% annually, with an estimated lifetime risk for malignant transformation of 8% per individual AK.11,12 Cryotherapy has been a mainstay for the treatment of isolated AK, and alternative therapies including curettage, photodynamic therapy, and laser therapy have been employed. Field-directed therapy has become a popular alternative that targets multiple lesions and field cancerization.8,13,14 Field cancerization implies that if one cell in the patient’s epidermis has been exposed to enough UV radiation to develop into a precancerous lesion or early skin cancer, then many other cells in the same environment likely have some degree of UV radiation–induced atypia.15 5-Fluorouacil is a pyrimidine analogue chemotherapeutic agent that inhibits thymidylate synthase and interferes with DNA synthesis.16 This mechanism of 5-FU commonly causes an inflammatory response characterized by burning, dryness, and redness, but these effects rarely force early discontinuation of treatment. A randomized controlled trial comparing 5-FU cream 0.5% to a placebo found that complete clearance rates at 4 weeks posttreatment were significantly higher in the treatment group (47.5%) versus placebo (3.4%)(P<.001).13 Additional trials have established no significant superiority of 5-FU cream 5% over 5-FU cream 0.5%, with a decrease in side effects noted in patients treated with the lower concentration.17
Our patient had a history of a recurrent BCC and was previously treated with imiquimod. He showed no inflammatory response to field therapy with 5-FU within the perimeter of prior immunomodulatory therapy. Although no frank scaling or crusting papules consistent with AK were observed in the previously treated area prior to 5-FU therapy, subclinical field damage in that area was expected because 10 years of additional sun exposure had accumulated since imiquimod therapy was completed. Several conclusions can be drawn from this observation. Primarily, no new clinically significant actinic lesions occurred on the previously treated skin. This observation is consistent with 12-month follow-up data on AKs treated with either 5-FU, imiquimod, or cryosurgery that identified imiquimod as having the lowest recurrence rate.8 Thus, a photoprotective effect may be ascribed to imiquimod therapy that extends beyond its drug effects on atypical keratinocytes. It has been one author’s personal experience (M.Q.) that patients treated with 5-FU experience recurrence of AKs within 3 to 5 years versus 10 years of remission with imiquimod. In our patient, imiquimod therapy seemed to reset the patient’s skin at the location of the prior BCC and surrounding field cancerization.
Studies with long-term follow-up are needed to investigate the need for re-treatment with imiquimod or 5-FU. The longevity of imiquimod treatment may be of importance beyond the treatment of AKs or NMSCs. For instance, during the treatment of lentigo maligna with imiquimod, Metcalf et al18 found a significant reduction in solar elastosis (P=.0036), normalization of epidermal thickness (P=.0073), and increased papillary dermal fibroplasia in pre- and posttreatment biopsies (P<.0001), which have been described as antiaging effects in the laypress. Some of these mechanisms appear to be implicated in the observations noted in our patient. The 10-year period between the 2 courses of therapy in our patient suggests that imiquimod may cause sustained healing of skin that was previously classified both clinically and microscopically as UV damaged.
Conclusion
Both topical immunomodulators such as imiquimod and topical chemotherapeutic agents such as 5-FU have a role in the field treatment of AK and the focal treatment of superficial BCC and SCC. As multiple topical immunomodulators continue to be evaluated, long-term studies assessing the need for re-treatment as well as the degree of sustained remission of sun damage will be necessary. We expect that their individual roles will continue to become more precisely defined and distinct in the coming years.
Sun damage and chronic exposure to UV radiation have been recognized as causative factors for the development of squamous cell carcinoma (SCC), its precursor actinic keratosis (AK), and basal cell carcinoma (BCC). Although surgical treatment is necessary for most advanced cases of skin cancer, several other therapeutic approaches have been described including the use of topical chemotherapy agents such as 5-fluorouracil (5-FU) and topical immunomodulators such as imiquimod. Unlike surgery, these agents provide the added benefit of treating larger fields of photodamaged skin. With the increasing prevalence of nonmelanoma skin cancers (NMSCs), the use of multiple topical agents for treatment will continue to become more common.
We present the case of a patient who underwent field therapy with topical 5-FU for diffuse actinic damage and AKs. There was no subsequent inflammatory response within the perimeter of a BCC that had been treated with imiquimod 10 years prior.
Case Report
An otherwise healthy 58-year-old man with a history of long-standing diffuse sun damage and multiple prior NMSCs presented for treatment of a recurrent BCC on the right cheek. The patient reported that the BCC had initially been biopsied and excised by his primary care physician. Two months later local recurrence was noted by the primary care physician and the patient was subsequently referred to our dermatology office. A 2-month treatment course with daily imiquimod cream 5% was initiated. This treatment caused extensive inflammation of the right cheek but was otherwise well tolerated (Figure 1).
|
During a routine skin cancer screening 10 years later, no recurrence of the BCC was noted on the right cheek; however, the patient had developed multiple AKs on the face. Therapeutic options were discussed with the patient; he agreed to topical field therapy with 5-FU cream 0.5%. The patient applied the 5-FU cream to the entire face nightly for 1 month. During this time he experienced a brisk inflammatory response with painful cracking and redness of the skin. On follow-up, it was noted that the area on the right cheek that had been treated with imiquimod 10 years prior showed no inflammatory response despite nightly application of 5-FU cream to the area (Figure 2). The patient denied any routine use of sunscreen or other sun-protective practices.
Comment
Basal cell carcinoma is the most common skin cancer in the United States with an incidence of 1.4% to 2% per year. It has become more prevalent in recent decades, likely due to genetic predisposition and increasing cumulative sun exposure.1-4 A variety of treatment options are available. Surgical interventions include destruction via electrodesiccation and curettage, local excision, and Mohs micrographic surgery. One of the challenges in the management of BCC, as was the case in our patient, is the treatment of tumors that arise in cosmetically or functionally sensitive areas. Approaches that minimize the amount of tissue removed while ensuring the highest possible cure rate are favorable. In addition to surgery, topical imiquimod has been established as a potential treatment of BCC. Imiquimod, a nucleoside analogue of the imidazoquinoline family, is an agonist of toll-like receptors 7 and 8 that promotes cytokine-induced cell death via nuclear factor kB and a helper T cell TH1-weighted antitumor inflammatory response.5,6 Although clearance rates with imiquimod vary by drug regimen, success rates of 43% to 100% for superficial BCCs, 42% to 100% for nodular BCCs, and 56% to 63% for infiltrative BCCs have been reported.7 In a 2007 randomized study of imiquimod cream 5%, 5-FU ointment 5%, or cryosurgery for the treatment of AK, imiquimod resulted in superior and more reliable clearance with lower recurrence rates.8
Similar to BCC, AK is closely linked to lifetime cumulative sun exposure.9 Actinic keratoses have been well established as precursors to SCC, and some researchers advocate for their reclassification as early SCC in situ.10 The incidence of malignant conversion of AK to SCC has been estimated at 0.025% to 16% annually, with an estimated lifetime risk for malignant transformation of 8% per individual AK.11,12 Cryotherapy has been a mainstay for the treatment of isolated AK, and alternative therapies including curettage, photodynamic therapy, and laser therapy have been employed. Field-directed therapy has become a popular alternative that targets multiple lesions and field cancerization.8,13,14 Field cancerization implies that if one cell in the patient’s epidermis has been exposed to enough UV radiation to develop into a precancerous lesion or early skin cancer, then many other cells in the same environment likely have some degree of UV radiation–induced atypia.15 5-Fluorouacil is a pyrimidine analogue chemotherapeutic agent that inhibits thymidylate synthase and interferes with DNA synthesis.16 This mechanism of 5-FU commonly causes an inflammatory response characterized by burning, dryness, and redness, but these effects rarely force early discontinuation of treatment. A randomized controlled trial comparing 5-FU cream 0.5% to a placebo found that complete clearance rates at 4 weeks posttreatment were significantly higher in the treatment group (47.5%) versus placebo (3.4%)(P<.001).13 Additional trials have established no significant superiority of 5-FU cream 5% over 5-FU cream 0.5%, with a decrease in side effects noted in patients treated with the lower concentration.17
Our patient had a history of a recurrent BCC and was previously treated with imiquimod. He showed no inflammatory response to field therapy with 5-FU within the perimeter of prior immunomodulatory therapy. Although no frank scaling or crusting papules consistent with AK were observed in the previously treated area prior to 5-FU therapy, subclinical field damage in that area was expected because 10 years of additional sun exposure had accumulated since imiquimod therapy was completed. Several conclusions can be drawn from this observation. Primarily, no new clinically significant actinic lesions occurred on the previously treated skin. This observation is consistent with 12-month follow-up data on AKs treated with either 5-FU, imiquimod, or cryosurgery that identified imiquimod as having the lowest recurrence rate.8 Thus, a photoprotective effect may be ascribed to imiquimod therapy that extends beyond its drug effects on atypical keratinocytes. It has been one author’s personal experience (M.Q.) that patients treated with 5-FU experience recurrence of AKs within 3 to 5 years versus 10 years of remission with imiquimod. In our patient, imiquimod therapy seemed to reset the patient’s skin at the location of the prior BCC and surrounding field cancerization.
Studies with long-term follow-up are needed to investigate the need for re-treatment with imiquimod or 5-FU. The longevity of imiquimod treatment may be of importance beyond the treatment of AKs or NMSCs. For instance, during the treatment of lentigo maligna with imiquimod, Metcalf et al18 found a significant reduction in solar elastosis (P=.0036), normalization of epidermal thickness (P=.0073), and increased papillary dermal fibroplasia in pre- and posttreatment biopsies (P<.0001), which have been described as antiaging effects in the laypress. Some of these mechanisms appear to be implicated in the observations noted in our patient. The 10-year period between the 2 courses of therapy in our patient suggests that imiquimod may cause sustained healing of skin that was previously classified both clinically and microscopically as UV damaged.
Conclusion
Both topical immunomodulators such as imiquimod and topical chemotherapeutic agents such as 5-FU have a role in the field treatment of AK and the focal treatment of superficial BCC and SCC. As multiple topical immunomodulators continue to be evaluated, long-term studies assessing the need for re-treatment as well as the degree of sustained remission of sun damage will be necessary. We expect that their individual roles will continue to become more precisely defined and distinct in the coming years.
1. Flohil SC, de Vries E, Neumann HA, et al. Incidence, prevalence and future trends of primary basal cell carcinoma in the Netherlands. Acta Derm Venereol. 2011;91:24-30.
2. Donaldson MR, Coldiron BM. No end in sight: the skin cancer epidemic continues. Semin Cutan Med Surg. 2011;30:3-5.
3. Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer. I. Basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
4. Gailani MR, Leffell DJ, Ziegler A, et al. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J Natl Cancer Inst. 1996;88:349-354.
5. Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway [published online ahead of print January 22, 2002]. Nat Immunol. 2002;3:196-200.
6. Schön MP, Schön M. Imiquimod: mode of action. Br J Dermatol. 2007;157(suppl 2):8-13.
7. Love WE, Bernhard JD, Bordeaux JS. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch Dermatol. 2009;145:1431-1438.
8. Krawtchenko N, Roewert-Huber J, Ulrich M, et al. A randomised study of topical 5% imiquimod vs. topical5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(suppl 2):34-40.
9. Feldman SR, Fleischer AB Jr. Progression of actinic keratosis to squamous cell carcinoma revisited: clinical and treatment implications. Cutis. 2011;87:201-207.
10. Röwert-Huber J, Patel MJ, Forschner T, et al. Actinic keratosis is an early in situ squamous cell carcinoma: a proposal for reclassification. Br J Dermatol. 2007;156(suppl 3):8-12.
11. Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42(1 pt 2):23-24.
12. Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
13. Weiss J, Menter A, Hevia O, et al. Effective treatment of actinic keratosis with 0.5% fluorouracil cream for 1, 2, or 4 weeks. Cutis. 2002;70(2 suppl):22-29.
14. Almeida Gonçalves JC, De Noronha T. 5-fluouracil (5-FU) ointment in the treatment of skin tumours and keratoses. Dermatologica. 1970;140(suppl 1):97+.
15. Vanharanta S, Massagué J. Field cancerization: something new under the sun. Cell. 2012;149:1179-1181.
16. Robins P, Gupta AK. The use of topical fluorouracil to treat actinic keratosis. Cutis. 2002;70(2 suppl):4-7.
17. Kaur R, Alikhan A, Maibach H. Comparison of topical 5-fluorouracil formulations in actinic keratosis treatment. J Dermatolog Treat. 2010;2:267-271.
18. Metcalf S, Crowson AN, Naylor M, et al. Imiquimod as an antiaging agent [published online ahead of print December 20, 2006]. J Am Acad Dermatol. 2007;56:422-425.
1. Flohil SC, de Vries E, Neumann HA, et al. Incidence, prevalence and future trends of primary basal cell carcinoma in the Netherlands. Acta Derm Venereol. 2011;91:24-30.
2. Donaldson MR, Coldiron BM. No end in sight: the skin cancer epidemic continues. Semin Cutan Med Surg. 2011;30:3-5.
3. Gallagher RP, Hill GB, Bajdik CD, et al. Sunlight exposure, pigmentary factors, and risk of nonmelanocytic skin cancer. I. Basal cell carcinoma. Arch Dermatol. 1995;131:157-163.
4. Gailani MR, Leffell DJ, Ziegler A, et al. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J Natl Cancer Inst. 1996;88:349-354.
5. Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway [published online ahead of print January 22, 2002]. Nat Immunol. 2002;3:196-200.
6. Schön MP, Schön M. Imiquimod: mode of action. Br J Dermatol. 2007;157(suppl 2):8-13.
7. Love WE, Bernhard JD, Bordeaux JS. Topical imiquimod or fluorouracil therapy for basal and squamous cell carcinoma: a systematic review. Arch Dermatol. 2009;145:1431-1438.
8. Krawtchenko N, Roewert-Huber J, Ulrich M, et al. A randomised study of topical 5% imiquimod vs. topical5-fluorouracil vs. cryosurgery in immunocompetent patients with actinic keratoses: a comparison of clinical and histological outcomes including 1-year follow-up. Br J Dermatol. 2007;157(suppl 2):34-40.
9. Feldman SR, Fleischer AB Jr. Progression of actinic keratosis to squamous cell carcinoma revisited: clinical and treatment implications. Cutis. 2011;87:201-207.
10. Röwert-Huber J, Patel MJ, Forschner T, et al. Actinic keratosis is an early in situ squamous cell carcinoma: a proposal for reclassification. Br J Dermatol. 2007;156(suppl 3):8-12.
11. Glogau RG. The risk of progression to invasive disease. J Am Acad Dermatol. 2000;42(1 pt 2):23-24.
12. Criscione VD, Weinstock MA, Naylor MF, et al. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523-2530.
13. Weiss J, Menter A, Hevia O, et al. Effective treatment of actinic keratosis with 0.5% fluorouracil cream for 1, 2, or 4 weeks. Cutis. 2002;70(2 suppl):22-29.
14. Almeida Gonçalves JC, De Noronha T. 5-fluouracil (5-FU) ointment in the treatment of skin tumours and keratoses. Dermatologica. 1970;140(suppl 1):97+.
15. Vanharanta S, Massagué J. Field cancerization: something new under the sun. Cell. 2012;149:1179-1181.
16. Robins P, Gupta AK. The use of topical fluorouracil to treat actinic keratosis. Cutis. 2002;70(2 suppl):4-7.
17. Kaur R, Alikhan A, Maibach H. Comparison of topical 5-fluorouracil formulations in actinic keratosis treatment. J Dermatolog Treat. 2010;2:267-271.
18. Metcalf S, Crowson AN, Naylor M, et al. Imiquimod as an antiaging agent [published online ahead of print December 20, 2006]. J Am Acad Dermatol. 2007;56:422-425.
Practice Points
- Topical immunomodulators such as imiquimod and topical chemotherapeutics such as 5-fluorouracil are effective in the field treatment of actinic keratoses.
- Prior topical immunomodulator use for nonmelanoma skin cancer may induce a sustained remission of actinic damage.
- The field effect of imiquimod treatment in actinically damaged skin may persist for several years.
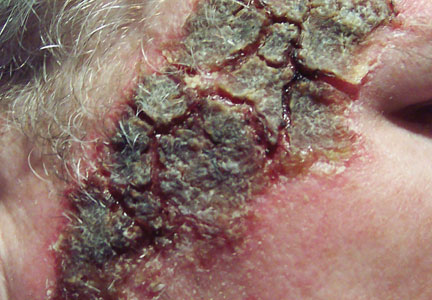
Superficial Acral Fibromyxoma and Other Slow-Growing Tumors in Acral Areas
First described by Fetsch et al1 in 2001, superficial acral fibromyxoma (SAFM) is a rare fibromyxoid mesenchymal tumor that typically affects the fingers and toes with frequent involvement of the nail unit. It is not widely recognized and remains poorly understood. We describe a series of 3 cases of SAFM encountered at our institution and provide a review of the literature on this unique tumor.
Case Reports
Patient 1
A 35-year-old man presented for treatment of a “wart” on the right fifth toe that had increased in size over the last year. He reported that the lesion was mildly painful and occasionally bled or drained clear fluid. He also noted cracking of the nail plate on the same toe. Physical examination revealed a firm, flesh-colored, 3-mm dermal papule on the proximal nail fold of the right fifth toe with subtle flattening of the underlying nail plate (Figure 1). The patient underwent biopsy of the involved proximal nail fold. Histopathology revealed a proliferation of small oval and spindle cells arranged in fascicles and bundles in the dermis (Figure 2). There was extensive mucin deposition associated with the spindle cell proliferation. Additionally, spindle cells and mucin surrounded and entrapped collagen bundles on the periphery of the lesion. Lesional cells were diffusely positive for CD34 and extended to the deep surgical margin (Figure 3). S-100 and factor XIIIa stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
|
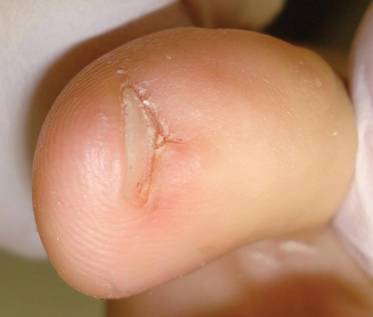
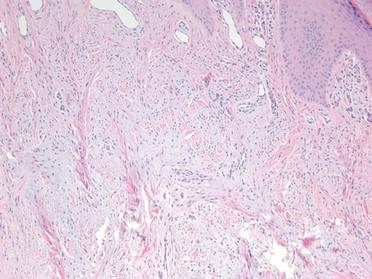

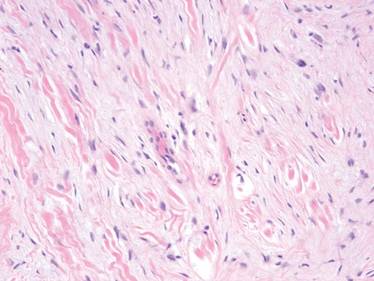
Patient 2
A 47-year-old man presented with an asymptomatic growth on the left fourth toe that had increased in size over the last year. Physical examination revealed an 8-mm, firm, fleshy, flesh-colored, smooth and slightly pedunculated papule on the distal aspect of the left fourth toe. The nail plate and periungual region were not involved. A shave biopsy of the papule was obtained. Histopathology demonstrated dermal stellate spindle cells arranged in a loose fascicular pattern with marked mucin deposition throughout the dermis (Figure 4). Lesional cells were positive for CD34. An S-100 stain highlighted dermal dendritic cells, but lesional cells were negative. No further excision was undertaken, and there was no evidence of recurrence at 1-year follow-up. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Patient 3
A 45-year-old woman presented with asymptomatic distal onycholysis of the right thumbnail of 1 year’s duration. She denied any history of trauma, and no bleeding or pigmentary changes were noted. Physical examination revealed a 5-mm flesh-colored papule on the hyponychium of the right thumb with focal onycholysis (Figure 5). A wedge biopsy of the lesion was performed. Histopathology showed an intradermal nodular proliferation of bland spindle cells arranged in loose fascicles and bundles and embedded in a myxoid stroma (Figure 6). CD34 staining strongly highlighted lesional cells. S-100 and neurofilament stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Comment
Clinically, SAFM typically presents as a slow-growing solitary nodule on the distal fingers or toes. The great toe is the most commonly affected digit, and the tumor may be subungual in up to two-thirds of cases.1 Unusual locations, such as the heel, also have been reported.2 Onset typically occurs in the fifth or sixth decade, and there is an approximately 2-fold higher incidence in men than women.1-3
Histopathologically, SAFM is a characteristically well-circumscribed but unencapsulated dermal tumor composed of spindle and stellate cells in a loose storiform or fascicular arrangement embedded in a myxoid, myxocollagenous, or collagenous stroma.4 The tumor often occupies the entire dermis and may extend into the subcutis or occasionally the underlying fascia and bone.4,5 Mast cells often are prominent, and microvascular accentuation also may be seen. Inflammatory infiltrates and multinucleated giant cells typically are not seen.6 Although 2 cases of atypical SAFM have been described,2 cellular atypia is not a characteristic feature of SAFM.
The immunohistochemical profile of SAFM is characterized by diffuse or focal expression of CD34, focal expression of epithelial membrane antigen (EMA), CD99 expression, and varying numbers of factor XIIIa–positive histiocytes.2,3 Positive staining for vimentin also is common. Staining typically is negative for S-100, human melanoma black 45, keratin, smooth muscle actin, and desmin.
The standard treatment of SAFM is complete local resection of the tumor, though some patients have been treated with partial excision or biopsy and partial or complete digital amputation.1 Local recurrence may occur in up to 20% of cases; however, approximately two-thirds of the reported recurrences in the literature occurred after incomplete tumor excision.1,2 It may be more appropriate to consider these cases as persistent rather than recurrent tumors. Superficial acral fibromyxoma is considered a benign tumor, with no known cases of metastases.4
|
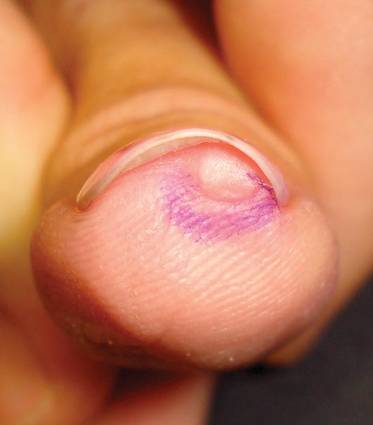
A broad differential diagnosis exists for SAFM and it can be difficult to differentiate it from a wide variety of benign and malignant tumors that may be seen on the nail unit and distal extremities (Table). Myxoid neurofibromas typically present as solitary lesions on the hands and feet. Similar to SAFM, myxoid neurofibromas are unencapsulated dermal tumors composed of spindle-shaped cells in which mast cells often are conspicuous.2,7 However, tumor cells in myxoid neurofibromas are S-100 positive, and the lesions typically do not show vasculature accentuation.4,7
Sclerosing perineuriomas are benign fibrous tumors of the fingers and palms. Histopathologically, bland spindle cells arranged in fascicles and whorls are observed in a hyalinized collagen matrix.8 Immunohistochemically, sclerosing perineuriomas are positive for EMA and negative for S-100, but unlike SAFM, these tumors usually are CD34 negative.8
Superficial angiomyxomas typically are located on the head and neck but also may be found in other locations such as the trunk. They present as cutaneous papules or polypoid lesions. Histopathologically, superficial angiomyxomas are poorly circumscribed with a lobular pattern. Spindle-shaped fibroblasts exist in a myxoid matrix with neutrophils and thin-walled capillaries. The fibroblasts are variably positive for CD34 but also are S-100 positive.1,9
Myxoid dermatofibrosarcoma protuberans is a rare, locally aggressive, mesenchymal tumor of the skin and subcutis2 that typically presents on the trunk, proximal extremities, or head and neck; occurrence on the fingers or toes is exceedingly rare.2,10 Histopathologically, a myxoid stroma contains sheets of bland spindle-shaped cells with minimal to no atypia, sometimes arranged in a storiform pattern. The tumor characteristically invades deeply into the subcutaneous tissues. CD34 is characteristically positive and S-100 is negative.2,10
Low-grade myxofibrosarcoma is a soft tissue sarcoma easily confused with other spindle cell tumors. It is one of the most common sarcomas in adults but rarely arises in acral areas.2 It is characterized by a nodular growth pattern with marked nuclear atypia and perivascular clustering of tumor cells. CD34 staining may be positive in some cases.11
Similar to SAFM, myxoinflammatory fibroblastic sarcoma has a predilection for the extremities.4 However, it typically presents as a subcutaneous mass and has no documented tendency for nail bed involvement. Also unlike SAFM, it has a remarkable inflammatory infiltrate and characteristic virocyte or Reed-Sternberg cells.12
Acquired digital fibrokeratomas are benign neoplasms that occur on fingers and toes; the classic clinical presentation is a solitary smooth nodule or dome, often with a characteristic projecting configuration and horn shape.1 Histopathologically, these tumors are paucicellular with thick, vertically oriented, interwoven collagen bundles; cells may be positive for CD34 but are negative for EMA.1,13 Related to acquired digital fibrokeratomas are Koenen tumors, which share a similar histology but are distinguished by their clinical characteristics. For example, Koenen tumors tend to be multifocal and are strongly associated with tuberous sclerosis. These tumors also have a tendency to recur.1
Conclusion
Our report of 3 typical cases of SAFM highlights the need to keep this increasingly recognized and well-defined clinicopathological entity in the differential for slow-growing tumors in acral locations, particularly those in the periungual and subungual regions.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
2. Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
3. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
4. André J, Theunis A, Richert B, et al. Superficial acral fibromyxoma: clinical and pathological features. Am J Dermatopathol. 2004;26:472-474.
5. Kazakov DV, Mentzel T, Burg G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
6. Meyerle JH, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
7. Graadt van Roggen JF, Hogendoorn PC, Fletcher CD. Myxoid tumours of soft tissue. Histopathology. 1999;35:291-312.
8. Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
9. Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23:910-917.
10. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. a study of 115 cases. Cancer. 1962;15:717-725.
11. Wada T, Hasegawa T, Nagoya S, et al. Myxofibrosarcoma with an infiltrative growth pattern: a case report. Jpn J Clin Oncol. 2000;30:458-462.
12. Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998;22:911-924.
13. Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;97:120-129.
First described by Fetsch et al1 in 2001, superficial acral fibromyxoma (SAFM) is a rare fibromyxoid mesenchymal tumor that typically affects the fingers and toes with frequent involvement of the nail unit. It is not widely recognized and remains poorly understood. We describe a series of 3 cases of SAFM encountered at our institution and provide a review of the literature on this unique tumor.
Case Reports
Patient 1
A 35-year-old man presented for treatment of a “wart” on the right fifth toe that had increased in size over the last year. He reported that the lesion was mildly painful and occasionally bled or drained clear fluid. He also noted cracking of the nail plate on the same toe. Physical examination revealed a firm, flesh-colored, 3-mm dermal papule on the proximal nail fold of the right fifth toe with subtle flattening of the underlying nail plate (Figure 1). The patient underwent biopsy of the involved proximal nail fold. Histopathology revealed a proliferation of small oval and spindle cells arranged in fascicles and bundles in the dermis (Figure 2). There was extensive mucin deposition associated with the spindle cell proliferation. Additionally, spindle cells and mucin surrounded and entrapped collagen bundles on the periphery of the lesion. Lesional cells were diffusely positive for CD34 and extended to the deep surgical margin (Figure 3). S-100 and factor XIIIa stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
|
Patient 2
A 47-year-old man presented with an asymptomatic growth on the left fourth toe that had increased in size over the last year. Physical examination revealed an 8-mm, firm, fleshy, flesh-colored, smooth and slightly pedunculated papule on the distal aspect of the left fourth toe. The nail plate and periungual region were not involved. A shave biopsy of the papule was obtained. Histopathology demonstrated dermal stellate spindle cells arranged in a loose fascicular pattern with marked mucin deposition throughout the dermis (Figure 4). Lesional cells were positive for CD34. An S-100 stain highlighted dermal dendritic cells, but lesional cells were negative. No further excision was undertaken, and there was no evidence of recurrence at 1-year follow-up. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Patient 3
A 45-year-old woman presented with asymptomatic distal onycholysis of the right thumbnail of 1 year’s duration. She denied any history of trauma, and no bleeding or pigmentary changes were noted. Physical examination revealed a 5-mm flesh-colored papule on the hyponychium of the right thumb with focal onycholysis (Figure 5). A wedge biopsy of the lesion was performed. Histopathology showed an intradermal nodular proliferation of bland spindle cells arranged in loose fascicles and bundles and embedded in a myxoid stroma (Figure 6). CD34 staining strongly highlighted lesional cells. S-100 and neurofilament stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Comment
Clinically, SAFM typically presents as a slow-growing solitary nodule on the distal fingers or toes. The great toe is the most commonly affected digit, and the tumor may be subungual in up to two-thirds of cases.1 Unusual locations, such as the heel, also have been reported.2 Onset typically occurs in the fifth or sixth decade, and there is an approximately 2-fold higher incidence in men than women.1-3
Histopathologically, SAFM is a characteristically well-circumscribed but unencapsulated dermal tumor composed of spindle and stellate cells in a loose storiform or fascicular arrangement embedded in a myxoid, myxocollagenous, or collagenous stroma.4 The tumor often occupies the entire dermis and may extend into the subcutis or occasionally the underlying fascia and bone.4,5 Mast cells often are prominent, and microvascular accentuation also may be seen. Inflammatory infiltrates and multinucleated giant cells typically are not seen.6 Although 2 cases of atypical SAFM have been described,2 cellular atypia is not a characteristic feature of SAFM.
The immunohistochemical profile of SAFM is characterized by diffuse or focal expression of CD34, focal expression of epithelial membrane antigen (EMA), CD99 expression, and varying numbers of factor XIIIa–positive histiocytes.2,3 Positive staining for vimentin also is common. Staining typically is negative for S-100, human melanoma black 45, keratin, smooth muscle actin, and desmin.
The standard treatment of SAFM is complete local resection of the tumor, though some patients have been treated with partial excision or biopsy and partial or complete digital amputation.1 Local recurrence may occur in up to 20% of cases; however, approximately two-thirds of the reported recurrences in the literature occurred after incomplete tumor excision.1,2 It may be more appropriate to consider these cases as persistent rather than recurrent tumors. Superficial acral fibromyxoma is considered a benign tumor, with no known cases of metastases.4
|
A broad differential diagnosis exists for SAFM and it can be difficult to differentiate it from a wide variety of benign and malignant tumors that may be seen on the nail unit and distal extremities (Table). Myxoid neurofibromas typically present as solitary lesions on the hands and feet. Similar to SAFM, myxoid neurofibromas are unencapsulated dermal tumors composed of spindle-shaped cells in which mast cells often are conspicuous.2,7 However, tumor cells in myxoid neurofibromas are S-100 positive, and the lesions typically do not show vasculature accentuation.4,7
Sclerosing perineuriomas are benign fibrous tumors of the fingers and palms. Histopathologically, bland spindle cells arranged in fascicles and whorls are observed in a hyalinized collagen matrix.8 Immunohistochemically, sclerosing perineuriomas are positive for EMA and negative for S-100, but unlike SAFM, these tumors usually are CD34 negative.8
Superficial angiomyxomas typically are located on the head and neck but also may be found in other locations such as the trunk. They present as cutaneous papules or polypoid lesions. Histopathologically, superficial angiomyxomas are poorly circumscribed with a lobular pattern. Spindle-shaped fibroblasts exist in a myxoid matrix with neutrophils and thin-walled capillaries. The fibroblasts are variably positive for CD34 but also are S-100 positive.1,9
Myxoid dermatofibrosarcoma protuberans is a rare, locally aggressive, mesenchymal tumor of the skin and subcutis2 that typically presents on the trunk, proximal extremities, or head and neck; occurrence on the fingers or toes is exceedingly rare.2,10 Histopathologically, a myxoid stroma contains sheets of bland spindle-shaped cells with minimal to no atypia, sometimes arranged in a storiform pattern. The tumor characteristically invades deeply into the subcutaneous tissues. CD34 is characteristically positive and S-100 is negative.2,10
Low-grade myxofibrosarcoma is a soft tissue sarcoma easily confused with other spindle cell tumors. It is one of the most common sarcomas in adults but rarely arises in acral areas.2 It is characterized by a nodular growth pattern with marked nuclear atypia and perivascular clustering of tumor cells. CD34 staining may be positive in some cases.11
Similar to SAFM, myxoinflammatory fibroblastic sarcoma has a predilection for the extremities.4 However, it typically presents as a subcutaneous mass and has no documented tendency for nail bed involvement. Also unlike SAFM, it has a remarkable inflammatory infiltrate and characteristic virocyte or Reed-Sternberg cells.12
Acquired digital fibrokeratomas are benign neoplasms that occur on fingers and toes; the classic clinical presentation is a solitary smooth nodule or dome, often with a characteristic projecting configuration and horn shape.1 Histopathologically, these tumors are paucicellular with thick, vertically oriented, interwoven collagen bundles; cells may be positive for CD34 but are negative for EMA.1,13 Related to acquired digital fibrokeratomas are Koenen tumors, which share a similar histology but are distinguished by their clinical characteristics. For example, Koenen tumors tend to be multifocal and are strongly associated with tuberous sclerosis. These tumors also have a tendency to recur.1
Conclusion
Our report of 3 typical cases of SAFM highlights the need to keep this increasingly recognized and well-defined clinicopathological entity in the differential for slow-growing tumors in acral locations, particularly those in the periungual and subungual regions.
First described by Fetsch et al1 in 2001, superficial acral fibromyxoma (SAFM) is a rare fibromyxoid mesenchymal tumor that typically affects the fingers and toes with frequent involvement of the nail unit. It is not widely recognized and remains poorly understood. We describe a series of 3 cases of SAFM encountered at our institution and provide a review of the literature on this unique tumor.
Case Reports
Patient 1
A 35-year-old man presented for treatment of a “wart” on the right fifth toe that had increased in size over the last year. He reported that the lesion was mildly painful and occasionally bled or drained clear fluid. He also noted cracking of the nail plate on the same toe. Physical examination revealed a firm, flesh-colored, 3-mm dermal papule on the proximal nail fold of the right fifth toe with subtle flattening of the underlying nail plate (Figure 1). The patient underwent biopsy of the involved proximal nail fold. Histopathology revealed a proliferation of small oval and spindle cells arranged in fascicles and bundles in the dermis (Figure 2). There was extensive mucin deposition associated with the spindle cell proliferation. Additionally, spindle cells and mucin surrounded and entrapped collagen bundles on the periphery of the lesion. Lesional cells were diffusely positive for CD34 and extended to the deep surgical margin (Figure 3). S-100 and factor XIIIa stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
|
Patient 2
A 47-year-old man presented with an asymptomatic growth on the left fourth toe that had increased in size over the last year. Physical examination revealed an 8-mm, firm, fleshy, flesh-colored, smooth and slightly pedunculated papule on the distal aspect of the left fourth toe. The nail plate and periungual region were not involved. A shave biopsy of the papule was obtained. Histopathology demonstrated dermal stellate spindle cells arranged in a loose fascicular pattern with marked mucin deposition throughout the dermis (Figure 4). Lesional cells were positive for CD34. An S-100 stain highlighted dermal dendritic cells, but lesional cells were negative. No further excision was undertaken, and there was no evidence of recurrence at 1-year follow-up. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Patient 3
A 45-year-old woman presented with asymptomatic distal onycholysis of the right thumbnail of 1 year’s duration. She denied any history of trauma, and no bleeding or pigmentary changes were noted. Physical examination revealed a 5-mm flesh-colored papule on the hyponychium of the right thumb with focal onycholysis (Figure 5). A wedge biopsy of the lesion was performed. Histopathology showed an intradermal nodular proliferation of bland spindle cells arranged in loose fascicles and bundles and embedded in a myxoid stroma (Figure 6). CD34 staining strongly highlighted lesional cells. S-100 and neurofilament stains were negative. The diagnosis of SAFM was made based on the acral location, histopathologic appearance, and immunohistochemical profile of the tumor.
Comment
Clinically, SAFM typically presents as a slow-growing solitary nodule on the distal fingers or toes. The great toe is the most commonly affected digit, and the tumor may be subungual in up to two-thirds of cases.1 Unusual locations, such as the heel, also have been reported.2 Onset typically occurs in the fifth or sixth decade, and there is an approximately 2-fold higher incidence in men than women.1-3
Histopathologically, SAFM is a characteristically well-circumscribed but unencapsulated dermal tumor composed of spindle and stellate cells in a loose storiform or fascicular arrangement embedded in a myxoid, myxocollagenous, or collagenous stroma.4 The tumor often occupies the entire dermis and may extend into the subcutis or occasionally the underlying fascia and bone.4,5 Mast cells often are prominent, and microvascular accentuation also may be seen. Inflammatory infiltrates and multinucleated giant cells typically are not seen.6 Although 2 cases of atypical SAFM have been described,2 cellular atypia is not a characteristic feature of SAFM.
The immunohistochemical profile of SAFM is characterized by diffuse or focal expression of CD34, focal expression of epithelial membrane antigen (EMA), CD99 expression, and varying numbers of factor XIIIa–positive histiocytes.2,3 Positive staining for vimentin also is common. Staining typically is negative for S-100, human melanoma black 45, keratin, smooth muscle actin, and desmin.
The standard treatment of SAFM is complete local resection of the tumor, though some patients have been treated with partial excision or biopsy and partial or complete digital amputation.1 Local recurrence may occur in up to 20% of cases; however, approximately two-thirds of the reported recurrences in the literature occurred after incomplete tumor excision.1,2 It may be more appropriate to consider these cases as persistent rather than recurrent tumors. Superficial acral fibromyxoma is considered a benign tumor, with no known cases of metastases.4
|
A broad differential diagnosis exists for SAFM and it can be difficult to differentiate it from a wide variety of benign and malignant tumors that may be seen on the nail unit and distal extremities (Table). Myxoid neurofibromas typically present as solitary lesions on the hands and feet. Similar to SAFM, myxoid neurofibromas are unencapsulated dermal tumors composed of spindle-shaped cells in which mast cells often are conspicuous.2,7 However, tumor cells in myxoid neurofibromas are S-100 positive, and the lesions typically do not show vasculature accentuation.4,7
Sclerosing perineuriomas are benign fibrous tumors of the fingers and palms. Histopathologically, bland spindle cells arranged in fascicles and whorls are observed in a hyalinized collagen matrix.8 Immunohistochemically, sclerosing perineuriomas are positive for EMA and negative for S-100, but unlike SAFM, these tumors usually are CD34 negative.8
Superficial angiomyxomas typically are located on the head and neck but also may be found in other locations such as the trunk. They present as cutaneous papules or polypoid lesions. Histopathologically, superficial angiomyxomas are poorly circumscribed with a lobular pattern. Spindle-shaped fibroblasts exist in a myxoid matrix with neutrophils and thin-walled capillaries. The fibroblasts are variably positive for CD34 but also are S-100 positive.1,9
Myxoid dermatofibrosarcoma protuberans is a rare, locally aggressive, mesenchymal tumor of the skin and subcutis2 that typically presents on the trunk, proximal extremities, or head and neck; occurrence on the fingers or toes is exceedingly rare.2,10 Histopathologically, a myxoid stroma contains sheets of bland spindle-shaped cells with minimal to no atypia, sometimes arranged in a storiform pattern. The tumor characteristically invades deeply into the subcutaneous tissues. CD34 is characteristically positive and S-100 is negative.2,10
Low-grade myxofibrosarcoma is a soft tissue sarcoma easily confused with other spindle cell tumors. It is one of the most common sarcomas in adults but rarely arises in acral areas.2 It is characterized by a nodular growth pattern with marked nuclear atypia and perivascular clustering of tumor cells. CD34 staining may be positive in some cases.11
Similar to SAFM, myxoinflammatory fibroblastic sarcoma has a predilection for the extremities.4 However, it typically presents as a subcutaneous mass and has no documented tendency for nail bed involvement. Also unlike SAFM, it has a remarkable inflammatory infiltrate and characteristic virocyte or Reed-Sternberg cells.12
Acquired digital fibrokeratomas are benign neoplasms that occur on fingers and toes; the classic clinical presentation is a solitary smooth nodule or dome, often with a characteristic projecting configuration and horn shape.1 Histopathologically, these tumors are paucicellular with thick, vertically oriented, interwoven collagen bundles; cells may be positive for CD34 but are negative for EMA.1,13 Related to acquired digital fibrokeratomas are Koenen tumors, which share a similar histology but are distinguished by their clinical characteristics. For example, Koenen tumors tend to be multifocal and are strongly associated with tuberous sclerosis. These tumors also have a tendency to recur.1
Conclusion
Our report of 3 typical cases of SAFM highlights the need to keep this increasingly recognized and well-defined clinicopathological entity in the differential for slow-growing tumors in acral locations, particularly those in the periungual and subungual regions.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
2. Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
3. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
4. André J, Theunis A, Richert B, et al. Superficial acral fibromyxoma: clinical and pathological features. Am J Dermatopathol. 2004;26:472-474.
5. Kazakov DV, Mentzel T, Burg G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
6. Meyerle JH, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
7. Graadt van Roggen JF, Hogendoorn PC, Fletcher CD. Myxoid tumours of soft tissue. Histopathology. 1999;35:291-312.
8. Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
9. Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23:910-917.
10. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. a study of 115 cases. Cancer. 1962;15:717-725.
11. Wada T, Hasegawa T, Nagoya S, et al. Myxofibrosarcoma with an infiltrative growth pattern: a case report. Jpn J Clin Oncol. 2000;30:458-462.
12. Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998;22:911-924.
13. Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;97:120-129.
1. Fetsch JF, Laskin WB, Miettinen M. Superficial acral fibromyxoma: a clinicopathologic and immunohistochemical analysis of 37 cases of a distinctive soft tissue tumor with a predilection for the fingers and toes. Hum Pathol. 2001;32:704-714.
2. Al-Daraji WI, Miettinen M. Superficial acral fibromyxoma: a clinicopathological analysis of 32 tumors including 4 in the heel. J Cutan Pathol. 2008;35:1020-1026.
3. Hollmann TJ, Bovée JV, Fletcher CD. Digital fibromyxoma (superficial acral fibromyxoma): a detailed characterization of 124 cases. Am J Surg Pathol. 2012;36:789-798.
4. André J, Theunis A, Richert B, et al. Superficial acral fibromyxoma: clinical and pathological features. Am J Dermatopathol. 2004;26:472-474.
5. Kazakov DV, Mentzel T, Burg G, et al. Superficial acral fibromyxoma: report of two cases. Dermatology. 2002;205:285-288.
6. Meyerle JH, Keller RA, Krivda SJ. Superficial acral fibromyxoma of the index finger. J Am Acad Dermatol. 2004;50:134-136.
7. Graadt van Roggen JF, Hogendoorn PC, Fletcher CD. Myxoid tumours of soft tissue. Histopathology. 1999;35:291-312.
8. Fetsch JF, Miettinen M. Sclerosing perineurioma: a clinicopathologic study of 19 cases of a distinctive soft tissue lesion with a predilection for the fingers and palms of young adults. Am J Surg Pathol. 1997;21:1433-1442.
9. Calonje E, Guerin D, McCormick D, et al. Superficial angiomyxoma: clinicopathologic analysis of a series of distinctive but poorly recognized cutaneous tumors with tendency for recurrence. Am J Surg Pathol. 1999;23:910-917.
10. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. a study of 115 cases. Cancer. 1962;15:717-725.
11. Wada T, Hasegawa T, Nagoya S, et al. Myxofibrosarcoma with an infiltrative growth pattern: a case report. Jpn J Clin Oncol. 2000;30:458-462.
12. Meis-Kindblom JM, Kindblom LG. Acral myxoinflammatory fibroblastic sarcoma: a low-grade tumor of the hands and feet. Am J Surg Pathol. 1998;22:911-924.
13. Bart RS, Andrade R, Kopf AW, et al. Acquired digital fibrokeratomas. Arch Dermatol. 1968;97:120-129.
Practice Points
- Superficial acral fibromyxoma (SAFM) is a rare but distinct tumor that may affect the nail bed and nail plate, and it may clinically or histopathologically mimic other tumors of the distal extremities.
- Although SAFM is considered a benign tumor, it frequently persists or recurs after incomplete excision, and therefore complete local resection may be recommended, particularly for symptomatic lesions.
Dermatologic Toxicity in a Patient Receiving Liposomal Doxorubicin
To the Editor:
Liposomal doxorubicin hydrochloride is an anthracycline topoisomerase inhibitor indicated for ovarian cancer, AIDS-related Kaposi sarcoma, and multiple myeloma.1 It also has been used with limited success in a clinical trial of previously treated patients with endometrial cancer.2 The most common adverse reactions include asthenia, fatigue, fever, anorexia, nausea, vomiting, stomatitis, diarrhea, constipation, hand-and-foot syndrome, rash, neutropenia, thrombocytopenia, and anemia.1
A 58-year-old woman with a history of stage IIIA endometrial cancer underwent a total abdominal hysterectomy and bilateral salpingo-oophorectomy soon after diagnosis. She then completed 5 high-dose-rate brachytherapy treatments and 6 cycles of paclitaxel and carboplatin. Follow-up imaging revealed pulmonary metastasis. The patient was then enrolled in a clinical trial but was switched to 40 mg/m2 liposomal doxorubicin given once every 28 days for 5 cycles after progression of disease.
After each dose of doxorubicin, she developed redness of the palms and soles. Following the third cycle of doxorubicin, a painful rash involving the thighs and axilla appeared with some desquamation in the left axilla. Three weeks after the fourth dose of doxorubicin, she presented with severe worsening of the rash to involve the extensor elbows (Figure 1), back, and lower legs with bilateral axillary desquamation. The bilateral medial thighs were erythematous with maceration that was tender and blanchable (Figure 2). The total affected body surface area was 10% to 15%. There was no involvement of the mucosa. She was treated with hydrogel sheet dressings and silver sulfadiazine cream 1%.
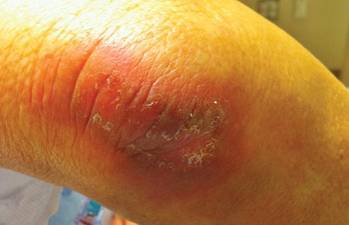 |
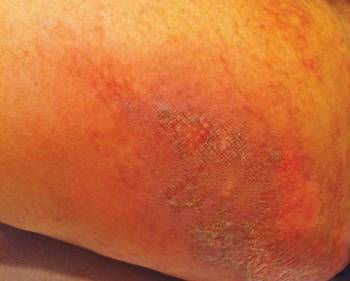 |
The patient’s rash was thought to be due to doxorubicin toxicity; however, a 4-mm punch biopsy specimen from the left thigh was taken for culture and hemotoxylin and eosin stain to rule out other possibilities. Biopsy was consistent with a drug reaction, revealing superficial perivascular dermatitis with keratinocyte atypia of the epidermis. Doxorubicin was discontinued and the rash resolved completely within 2 weeks, except for some thickening of the skin on the palms, soles, and thighs. After a delay of approximately 1 week, doxorubicin was resumed at a lower dose of 30 mg/m2. No dermatologic symptoms followed treatment at this dose.
Four clinical patterns of doxorubicin toxicity are recognized. The most common pattern is acral erythema, also known as hand-and-foot syndrome, which is followed by desquamation of the palms and soles, occurring in approximately 50% of patients. Ten percent of patients experience a diffuse follicular rash with mild, diffuse, scaly erythema and follicular accentuation that often occurs over the lateral limbs but also may occur over the trunk. New melanotic macules may appear on the trunk or extremities including palms and soles.3 Finally, an intertrigolike eruption exacerbated by friction with erythematous patches over skin folds or in areas of friction also has been described.3-5 Our patient presented with a combination of dermatologic toxicities including acral erythema and intertrigolike eruption. Acral erythema occurred in 24 of 60 patients and intertrigolike eruption occurred in 5 of 60 patients in one study.3 Another report documented both occurring together.5
Treatment of doxorubicin skin toxicity consists of reduction of the dose of doxorubicin, supportive care, and patient education. Specific treatments include topical wound care, emollient creams, and pain management with analgesics. Other interventions include wearing loose clothing, avoiding vigorous exercise, and sitting on padded surfaces.6
Doxorubicin skin toxicity presents in several clinical patterns. Although acral erythema is the most common pattern, severe intertrigolike eruptions similar to our case may occur. Physicians caring for patients receiving doxorubicin should be aware of the variety of presentations of skin toxicity and the possible need for dose reduction to decrease symptoms.
1. Doxil [package insert]. Horsham, PA: Janssen Products, LP; 2014.
2. Muggia FM, Blessing JA, Sorosky J, et al. Phase II trial of the pegylated liposomal doxorubicin in previously treated metastatic endometrial cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2002;20:2360-2364.
3. Lotem M, Hubert A, Lyass O, et al. Skin toxic effects of polyethylene glycol-coated liposomal doxorubicin. Arch Dermatol. 2000;136:1475-1480.
4. Korver GE, Ronald H, Petersen MJ. An intertrigo-like eruption from pegylated liposomal doxorubicin. J Drugs Dermatol. 2006;5:901-902.
5. Sánchez Henarejos P, Ros Martinez S, Marín Zafra GR,
et al. Intertrigo-like eruption caused by pegylated liposomal doxorubicin (PLD). Clin Transl Oncol. 2009;11:486-487.
6. von Moos R, Thuerlimann BJ, Aapro M, et al. Pegylated liposomal doxorubicin-associated hand-foot syndrome: recommendations of an international panel of experts [published online ahead of print March 10, 2008]. Eur J Cancer. 2008;44:781-790.
To the Editor:
Liposomal doxorubicin hydrochloride is an anthracycline topoisomerase inhibitor indicated for ovarian cancer, AIDS-related Kaposi sarcoma, and multiple myeloma.1 It also has been used with limited success in a clinical trial of previously treated patients with endometrial cancer.2 The most common adverse reactions include asthenia, fatigue, fever, anorexia, nausea, vomiting, stomatitis, diarrhea, constipation, hand-and-foot syndrome, rash, neutropenia, thrombocytopenia, and anemia.1
A 58-year-old woman with a history of stage IIIA endometrial cancer underwent a total abdominal hysterectomy and bilateral salpingo-oophorectomy soon after diagnosis. She then completed 5 high-dose-rate brachytherapy treatments and 6 cycles of paclitaxel and carboplatin. Follow-up imaging revealed pulmonary metastasis. The patient was then enrolled in a clinical trial but was switched to 40 mg/m2 liposomal doxorubicin given once every 28 days for 5 cycles after progression of disease.
After each dose of doxorubicin, she developed redness of the palms and soles. Following the third cycle of doxorubicin, a painful rash involving the thighs and axilla appeared with some desquamation in the left axilla. Three weeks after the fourth dose of doxorubicin, she presented with severe worsening of the rash to involve the extensor elbows (Figure 1), back, and lower legs with bilateral axillary desquamation. The bilateral medial thighs were erythematous with maceration that was tender and blanchable (Figure 2). The total affected body surface area was 10% to 15%. There was no involvement of the mucosa. She was treated with hydrogel sheet dressings and silver sulfadiazine cream 1%.
|
|
The patient’s rash was thought to be due to doxorubicin toxicity; however, a 4-mm punch biopsy specimen from the left thigh was taken for culture and hemotoxylin and eosin stain to rule out other possibilities. Biopsy was consistent with a drug reaction, revealing superficial perivascular dermatitis with keratinocyte atypia of the epidermis. Doxorubicin was discontinued and the rash resolved completely within 2 weeks, except for some thickening of the skin on the palms, soles, and thighs. After a delay of approximately 1 week, doxorubicin was resumed at a lower dose of 30 mg/m2. No dermatologic symptoms followed treatment at this dose.
Four clinical patterns of doxorubicin toxicity are recognized. The most common pattern is acral erythema, also known as hand-and-foot syndrome, which is followed by desquamation of the palms and soles, occurring in approximately 50% of patients. Ten percent of patients experience a diffuse follicular rash with mild, diffuse, scaly erythema and follicular accentuation that often occurs over the lateral limbs but also may occur over the trunk. New melanotic macules may appear on the trunk or extremities including palms and soles.3 Finally, an intertrigolike eruption exacerbated by friction with erythematous patches over skin folds or in areas of friction also has been described.3-5 Our patient presented with a combination of dermatologic toxicities including acral erythema and intertrigolike eruption. Acral erythema occurred in 24 of 60 patients and intertrigolike eruption occurred in 5 of 60 patients in one study.3 Another report documented both occurring together.5
Treatment of doxorubicin skin toxicity consists of reduction of the dose of doxorubicin, supportive care, and patient education. Specific treatments include topical wound care, emollient creams, and pain management with analgesics. Other interventions include wearing loose clothing, avoiding vigorous exercise, and sitting on padded surfaces.6
Doxorubicin skin toxicity presents in several clinical patterns. Although acral erythema is the most common pattern, severe intertrigolike eruptions similar to our case may occur. Physicians caring for patients receiving doxorubicin should be aware of the variety of presentations of skin toxicity and the possible need for dose reduction to decrease symptoms.
To the Editor:
Liposomal doxorubicin hydrochloride is an anthracycline topoisomerase inhibitor indicated for ovarian cancer, AIDS-related Kaposi sarcoma, and multiple myeloma.1 It also has been used with limited success in a clinical trial of previously treated patients with endometrial cancer.2 The most common adverse reactions include asthenia, fatigue, fever, anorexia, nausea, vomiting, stomatitis, diarrhea, constipation, hand-and-foot syndrome, rash, neutropenia, thrombocytopenia, and anemia.1
A 58-year-old woman with a history of stage IIIA endometrial cancer underwent a total abdominal hysterectomy and bilateral salpingo-oophorectomy soon after diagnosis. She then completed 5 high-dose-rate brachytherapy treatments and 6 cycles of paclitaxel and carboplatin. Follow-up imaging revealed pulmonary metastasis. The patient was then enrolled in a clinical trial but was switched to 40 mg/m2 liposomal doxorubicin given once every 28 days for 5 cycles after progression of disease.
After each dose of doxorubicin, she developed redness of the palms and soles. Following the third cycle of doxorubicin, a painful rash involving the thighs and axilla appeared with some desquamation in the left axilla. Three weeks after the fourth dose of doxorubicin, she presented with severe worsening of the rash to involve the extensor elbows (Figure 1), back, and lower legs with bilateral axillary desquamation. The bilateral medial thighs were erythematous with maceration that was tender and blanchable (Figure 2). The total affected body surface area was 10% to 15%. There was no involvement of the mucosa. She was treated with hydrogel sheet dressings and silver sulfadiazine cream 1%.
|
|
The patient’s rash was thought to be due to doxorubicin toxicity; however, a 4-mm punch biopsy specimen from the left thigh was taken for culture and hemotoxylin and eosin stain to rule out other possibilities. Biopsy was consistent with a drug reaction, revealing superficial perivascular dermatitis with keratinocyte atypia of the epidermis. Doxorubicin was discontinued and the rash resolved completely within 2 weeks, except for some thickening of the skin on the palms, soles, and thighs. After a delay of approximately 1 week, doxorubicin was resumed at a lower dose of 30 mg/m2. No dermatologic symptoms followed treatment at this dose.
Four clinical patterns of doxorubicin toxicity are recognized. The most common pattern is acral erythema, also known as hand-and-foot syndrome, which is followed by desquamation of the palms and soles, occurring in approximately 50% of patients. Ten percent of patients experience a diffuse follicular rash with mild, diffuse, scaly erythema and follicular accentuation that often occurs over the lateral limbs but also may occur over the trunk. New melanotic macules may appear on the trunk or extremities including palms and soles.3 Finally, an intertrigolike eruption exacerbated by friction with erythematous patches over skin folds or in areas of friction also has been described.3-5 Our patient presented with a combination of dermatologic toxicities including acral erythema and intertrigolike eruption. Acral erythema occurred in 24 of 60 patients and intertrigolike eruption occurred in 5 of 60 patients in one study.3 Another report documented both occurring together.5
Treatment of doxorubicin skin toxicity consists of reduction of the dose of doxorubicin, supportive care, and patient education. Specific treatments include topical wound care, emollient creams, and pain management with analgesics. Other interventions include wearing loose clothing, avoiding vigorous exercise, and sitting on padded surfaces.6
Doxorubicin skin toxicity presents in several clinical patterns. Although acral erythema is the most common pattern, severe intertrigolike eruptions similar to our case may occur. Physicians caring for patients receiving doxorubicin should be aware of the variety of presentations of skin toxicity and the possible need for dose reduction to decrease symptoms.
1. Doxil [package insert]. Horsham, PA: Janssen Products, LP; 2014.
2. Muggia FM, Blessing JA, Sorosky J, et al. Phase II trial of the pegylated liposomal doxorubicin in previously treated metastatic endometrial cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2002;20:2360-2364.
3. Lotem M, Hubert A, Lyass O, et al. Skin toxic effects of polyethylene glycol-coated liposomal doxorubicin. Arch Dermatol. 2000;136:1475-1480.
4. Korver GE, Ronald H, Petersen MJ. An intertrigo-like eruption from pegylated liposomal doxorubicin. J Drugs Dermatol. 2006;5:901-902.
5. Sánchez Henarejos P, Ros Martinez S, Marín Zafra GR,
et al. Intertrigo-like eruption caused by pegylated liposomal doxorubicin (PLD). Clin Transl Oncol. 2009;11:486-487.
6. von Moos R, Thuerlimann BJ, Aapro M, et al. Pegylated liposomal doxorubicin-associated hand-foot syndrome: recommendations of an international panel of experts [published online ahead of print March 10, 2008]. Eur J Cancer. 2008;44:781-790.
1. Doxil [package insert]. Horsham, PA: Janssen Products, LP; 2014.
2. Muggia FM, Blessing JA, Sorosky J, et al. Phase II trial of the pegylated liposomal doxorubicin in previously treated metastatic endometrial cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2002;20:2360-2364.
3. Lotem M, Hubert A, Lyass O, et al. Skin toxic effects of polyethylene glycol-coated liposomal doxorubicin. Arch Dermatol. 2000;136:1475-1480.
4. Korver GE, Ronald H, Petersen MJ. An intertrigo-like eruption from pegylated liposomal doxorubicin. J Drugs Dermatol. 2006;5:901-902.
5. Sánchez Henarejos P, Ros Martinez S, Marín Zafra GR,
et al. Intertrigo-like eruption caused by pegylated liposomal doxorubicin (PLD). Clin Transl Oncol. 2009;11:486-487.
6. von Moos R, Thuerlimann BJ, Aapro M, et al. Pegylated liposomal doxorubicin-associated hand-foot syndrome: recommendations of an international panel of experts [published online ahead of print March 10, 2008]. Eur J Cancer. 2008;44:781-790.
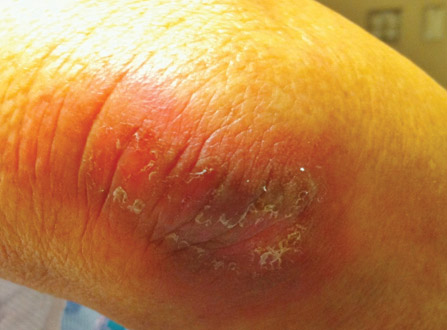
An Unusual Case of Sporadic Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome
To the Editor:
Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCCS) is a rare, highly penetrant, autosomal-dominant disorder that has been reported in approximately 200 families worldwide.1,2 More than 90% of patients with HLRCCS develop multiple cutaneous leiomyomata, frequently in a segmental distribution, that increase in number and size with age. The extent of skin lesions is variable, even within the same family. Approximately 90% of female family members also have symptomatic uterine leiomyomata; 10% to 16% of these patients develop aggressive renal cell carcinomas,3 with more than 50% dying of metastatic disease within 5 years of diagnosis. Clinical diagnosis is established by the presence of multiple cutaneous leiomyomata, at least 1 of which should be histologically confirmed, or by a single leiomyoma in the presence of a positive family history.4
Mutations of fumarate hydratase (FH), a Krebs cycle enzyme that interconverts fumarate and malate, have been implicated in this syndrome.5 The homotetrameric 50 kDa protein exists in the mitochondrial matrix and the cytoplasm. Diagnosis is confirmed by molecular genetic testing for FH mutations or rarely by demonstrating reduced activity of FH enzyme. So far, at least 155 variations in DNA sequence of FH have been identified in HLRCCS. However, no definite genotype-phenotype correlations have been established yet. We present the case of a sporadic form of HLRCCS, which is rare.
A 27-year-old man presented with multiple slowly growing, painful lesions on the chest and back of 11 years’ duration. Physical examination revealed approximately twenty 2- to 4-mm pink-tan papules on the left side of the chest and several 2- to 7-mm tan-pink papules on the upper back (Figure 1A). The lesions were tender to touch, pressure, and cold temperatures. Microscopic examination of one of the lesions on the back showed benign smooth muscle proliferation expanding the reticular dermis, consistent with a cutaneous leiomyoma (Figure 1B).
  Figure 1. Cluster of slow-growing, 2- to 7-mm, slightly erythematous papules on the upper back (A). Shave biopsy showed an unencapsulated dermal proliferation composed of interlacing fascicles of smooth muscle bundles with bland morphology, cigar-shaped nuclei, and lack of mitotic activity, compatible with cutaneous leiomyoma (B)(H&E, original magnification ×40). |
Based on the clinical presentation, the possibility of HLRCCS was raised. Subsequently, the FH gene was sequenced from the peripheral blood revealing a heterozygous 4-base pair frameshift deletion mutation (TGAA deleted at positions 1083 through 1086 [complementary DNA][c.1083_1086delTGAA]), confirming the diagnosis (Figure 2). There was no family history of leiomyomata of the skin or uterus or renal tumors. Therefore, this case represents sporadic HLRCCS. Magnetic resonance imaging revealed only a 0.4-cm renal cortical cyst for which he was monitored for approximately a year but was lost to follow-up.

The molecular mechanism of tumorigenesis in HLRCCS is poorly understood.6 Under normal circumstances, hypoxia-inducible factor (HIF) is hydroxylated by HIF prolyl hydroxylase after which it is targeted for an ubiquitin-mediated degradation (Figure 3 [top panel]). In the absence of FH, there is accumulation of fumarate, an inhibitor of HIF prolyl hydroxylase, leading to an increase in intracellular levels of unhydroxylated and undegradable HIF (Figure 3 [bottom panel]). Because of insufficient malate levels, the glucose metabolism through Krebs cycle shifts toward anaerobic glycolysis, even when sufficient oxygen is present to support respiration, creating a pseudohypoxic milieu that is similar to the Warburg effect. This environment leads to further stabilization of HIF, which is a transcription factor, that upregulates the expression of angiogenic factors (eg, vascular endothelial growth factor), growth factors (eg, erythropoietin, transforming growth factor a, platelet-derived growth factor), glucose transporters (eg, glucose transporter 1), and glycolytic enzymes (eg, phosphokinase mutase 1, lactate dehydrogenase A). These alterations may favor tumor growth by increasing the availability of biosynthetic intermediates needed for cellular proliferation and survival.
Patients with renal tumor–associated hereditary syndromes may present initially to dermatologists; therefore, it is important to recognize the cutaneous manifestations of these conditions because early diagnosis of renal cancer may prove to be lifesaving.
1. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825-829.
2. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer [published online ahead of print February 27, 2001]. Proc Natl Acad Sci U S A. 2001;98:3387-3392.
3. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America [published online ahead of print May 22, 2003]. Am J Hum Genet. 2003;73:95-106.
4. Ferzli PG, Millett CR, Newman MD, et al. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis. 2008;81:41-48.
5. Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;25:20.
6. Sudarshan S, Pinto PA, Neckers L, et al. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104-110.
To the Editor:
Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCCS) is a rare, highly penetrant, autosomal-dominant disorder that has been reported in approximately 200 families worldwide.1,2 More than 90% of patients with HLRCCS develop multiple cutaneous leiomyomata, frequently in a segmental distribution, that increase in number and size with age. The extent of skin lesions is variable, even within the same family. Approximately 90% of female family members also have symptomatic uterine leiomyomata; 10% to 16% of these patients develop aggressive renal cell carcinomas,3 with more than 50% dying of metastatic disease within 5 years of diagnosis. Clinical diagnosis is established by the presence of multiple cutaneous leiomyomata, at least 1 of which should be histologically confirmed, or by a single leiomyoma in the presence of a positive family history.4
Mutations of fumarate hydratase (FH), a Krebs cycle enzyme that interconverts fumarate and malate, have been implicated in this syndrome.5 The homotetrameric 50 kDa protein exists in the mitochondrial matrix and the cytoplasm. Diagnosis is confirmed by molecular genetic testing for FH mutations or rarely by demonstrating reduced activity of FH enzyme. So far, at least 155 variations in DNA sequence of FH have been identified in HLRCCS. However, no definite genotype-phenotype correlations have been established yet. We present the case of a sporadic form of HLRCCS, which is rare.
A 27-year-old man presented with multiple slowly growing, painful lesions on the chest and back of 11 years’ duration. Physical examination revealed approximately twenty 2- to 4-mm pink-tan papules on the left side of the chest and several 2- to 7-mm tan-pink papules on the upper back (Figure 1A). The lesions were tender to touch, pressure, and cold temperatures. Microscopic examination of one of the lesions on the back showed benign smooth muscle proliferation expanding the reticular dermis, consistent with a cutaneous leiomyoma (Figure 1B).
Figure 1. Cluster of slow-growing, 2- to 7-mm, slightly erythematous papules on the upper back (A). Shave biopsy showed an unencapsulated dermal proliferation composed of interlacing fascicles of smooth muscle bundles with bland morphology, cigar-shaped nuclei, and lack of mitotic activity, compatible with cutaneous leiomyoma (B)(H&E, original magnification ×40). |
Based on the clinical presentation, the possibility of HLRCCS was raised. Subsequently, the FH gene was sequenced from the peripheral blood revealing a heterozygous 4-base pair frameshift deletion mutation (TGAA deleted at positions 1083 through 1086 [complementary DNA][c.1083_1086delTGAA]), confirming the diagnosis (Figure 2). There was no family history of leiomyomata of the skin or uterus or renal tumors. Therefore, this case represents sporadic HLRCCS. Magnetic resonance imaging revealed only a 0.4-cm renal cortical cyst for which he was monitored for approximately a year but was lost to follow-up.
The molecular mechanism of tumorigenesis in HLRCCS is poorly understood.6 Under normal circumstances, hypoxia-inducible factor (HIF) is hydroxylated by HIF prolyl hydroxylase after which it is targeted for an ubiquitin-mediated degradation (Figure 3 [top panel]). In the absence of FH, there is accumulation of fumarate, an inhibitor of HIF prolyl hydroxylase, leading to an increase in intracellular levels of unhydroxylated and undegradable HIF (Figure 3 [bottom panel]). Because of insufficient malate levels, the glucose metabolism through Krebs cycle shifts toward anaerobic glycolysis, even when sufficient oxygen is present to support respiration, creating a pseudohypoxic milieu that is similar to the Warburg effect. This environment leads to further stabilization of HIF, which is a transcription factor, that upregulates the expression of angiogenic factors (eg, vascular endothelial growth factor), growth factors (eg, erythropoietin, transforming growth factor a, platelet-derived growth factor), glucose transporters (eg, glucose transporter 1), and glycolytic enzymes (eg, phosphokinase mutase 1, lactate dehydrogenase A). These alterations may favor tumor growth by increasing the availability of biosynthetic intermediates needed for cellular proliferation and survival.
Patients with renal tumor–associated hereditary syndromes may present initially to dermatologists; therefore, it is important to recognize the cutaneous manifestations of these conditions because early diagnosis of renal cancer may prove to be lifesaving.
To the Editor:
Hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCCS) is a rare, highly penetrant, autosomal-dominant disorder that has been reported in approximately 200 families worldwide.1,2 More than 90% of patients with HLRCCS develop multiple cutaneous leiomyomata, frequently in a segmental distribution, that increase in number and size with age. The extent of skin lesions is variable, even within the same family. Approximately 90% of female family members also have symptomatic uterine leiomyomata; 10% to 16% of these patients develop aggressive renal cell carcinomas,3 with more than 50% dying of metastatic disease within 5 years of diagnosis. Clinical diagnosis is established by the presence of multiple cutaneous leiomyomata, at least 1 of which should be histologically confirmed, or by a single leiomyoma in the presence of a positive family history.4
Mutations of fumarate hydratase (FH), a Krebs cycle enzyme that interconverts fumarate and malate, have been implicated in this syndrome.5 The homotetrameric 50 kDa protein exists in the mitochondrial matrix and the cytoplasm. Diagnosis is confirmed by molecular genetic testing for FH mutations or rarely by demonstrating reduced activity of FH enzyme. So far, at least 155 variations in DNA sequence of FH have been identified in HLRCCS. However, no definite genotype-phenotype correlations have been established yet. We present the case of a sporadic form of HLRCCS, which is rare.
A 27-year-old man presented with multiple slowly growing, painful lesions on the chest and back of 11 years’ duration. Physical examination revealed approximately twenty 2- to 4-mm pink-tan papules on the left side of the chest and several 2- to 7-mm tan-pink papules on the upper back (Figure 1A). The lesions were tender to touch, pressure, and cold temperatures. Microscopic examination of one of the lesions on the back showed benign smooth muscle proliferation expanding the reticular dermis, consistent with a cutaneous leiomyoma (Figure 1B).
Figure 1. Cluster of slow-growing, 2- to 7-mm, slightly erythematous papules on the upper back (A). Shave biopsy showed an unencapsulated dermal proliferation composed of interlacing fascicles of smooth muscle bundles with bland morphology, cigar-shaped nuclei, and lack of mitotic activity, compatible with cutaneous leiomyoma (B)(H&E, original magnification ×40). |
Based on the clinical presentation, the possibility of HLRCCS was raised. Subsequently, the FH gene was sequenced from the peripheral blood revealing a heterozygous 4-base pair frameshift deletion mutation (TGAA deleted at positions 1083 through 1086 [complementary DNA][c.1083_1086delTGAA]), confirming the diagnosis (Figure 2). There was no family history of leiomyomata of the skin or uterus or renal tumors. Therefore, this case represents sporadic HLRCCS. Magnetic resonance imaging revealed only a 0.4-cm renal cortical cyst for which he was monitored for approximately a year but was lost to follow-up.
The molecular mechanism of tumorigenesis in HLRCCS is poorly understood.6 Under normal circumstances, hypoxia-inducible factor (HIF) is hydroxylated by HIF prolyl hydroxylase after which it is targeted for an ubiquitin-mediated degradation (Figure 3 [top panel]). In the absence of FH, there is accumulation of fumarate, an inhibitor of HIF prolyl hydroxylase, leading to an increase in intracellular levels of unhydroxylated and undegradable HIF (Figure 3 [bottom panel]). Because of insufficient malate levels, the glucose metabolism through Krebs cycle shifts toward anaerobic glycolysis, even when sufficient oxygen is present to support respiration, creating a pseudohypoxic milieu that is similar to the Warburg effect. This environment leads to further stabilization of HIF, which is a transcription factor, that upregulates the expression of angiogenic factors (eg, vascular endothelial growth factor), growth factors (eg, erythropoietin, transforming growth factor a, platelet-derived growth factor), glucose transporters (eg, glucose transporter 1), and glycolytic enzymes (eg, phosphokinase mutase 1, lactate dehydrogenase A). These alterations may favor tumor growth by increasing the availability of biosynthetic intermediates needed for cellular proliferation and survival.
Patients with renal tumor–associated hereditary syndromes may present initially to dermatologists; therefore, it is important to recognize the cutaneous manifestations of these conditions because early diagnosis of renal cancer may prove to be lifesaving.
1. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825-829.
2. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer [published online ahead of print February 27, 2001]. Proc Natl Acad Sci U S A. 2001;98:3387-3392.
3. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America [published online ahead of print May 22, 2003]. Am J Hum Genet. 2003;73:95-106.
4. Ferzli PG, Millett CR, Newman MD, et al. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis. 2008;81:41-48.
5. Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;25:20.
6. Sudarshan S, Pinto PA, Neckers L, et al. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104-110.
1. Kiuru M, Launonen V, Hietala M, et al. Familial cutaneous leiomyomatosis is a two-hit condition associated with renal cell cancer of characteristic histopathology. Am J Pathol. 2001;159:825-829.
2. Launonen V, Vierimaa O, Kiuru M, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer [published online ahead of print February 27, 2001]. Proc Natl Acad Sci U S A. 2001;98:3387-3392.
3. Toro JR, Nickerson ML, Wei MH, et al. Mutations in the fumarate hydratase gene cause hereditary leiomyomatosis and renal cell cancer in families in North America [published online ahead of print May 22, 2003]. Am J Hum Genet. 2003;73:95-106.
4. Ferzli PG, Millett CR, Newman MD, et al. The dermatologist’s guide to hereditary syndromes with renal tumors. Cutis. 2008;81:41-48.
5. Bayley JP, Launonen V, Tomlinson IP. The FH mutation database: an online database of fumarate hydratase mutations involved in the MCUL (HLRCC) tumor syndrome and congenital fumarase deficiency. BMC Med Genet. 2008;25:20.
6. Sudarshan S, Pinto PA, Neckers L, et al. Mechanisms of disease: hereditary leiomyomatosis and renal cell cancer—a distinct form of hereditary kidney cancer. Nat Clin Pract Urol. 2007;4:104-110.
Tuberculosis Cutis Orificialis in an Immunocompetent Patient
To the Editor:
Orificial tuberculosis (OT) constitutes 2% of cutaneous tuberculosis cases and 0.01% to 1% of all clinical presentations of tuberculosis.1 It is clinically classified as primary or secondary OT. In primary OT, the oral mucosa is the initial site of the infection without any internal organ involvement.2 This form is more prevalent among men and young adults.1,3 Secondary OT is the cutaneous tuberculosis type that occurs in patients with internal organ tuberculosis from autoinoculation of bacilli to the orificial area. It is more common and usually affects elderly patients.2-4 We present the development of primary OT in an immunocompetent woman.
A 51-year-old woman was admitted with painful enlarging oral ulcers of 1 year’s duration. There was no history of tuberculosis infection, dental trauma, or smoking habit prior to the development of oral ulcers, and no family history of tuberculosis. On dermatological examination white-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate and gingiva were observed (Figures 1A and 1B). Oral hygiene was good. There was no regional lym-phadenopathy on palpation. Physical findings were normal. The histopathology of the biopsy from the gingival ulcer revealed noncaseating granulomatous inflammation in the dermis (Figure 2). Ziehl-Neelsen and periodic acid–Schiff stains were negative for acid-fast bacilli and fungi, respectively.
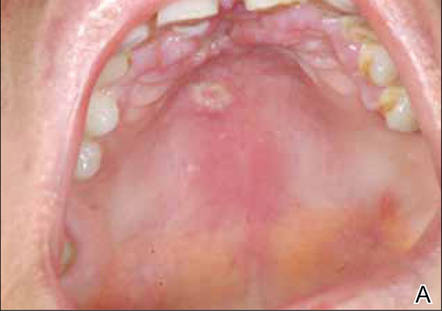 | 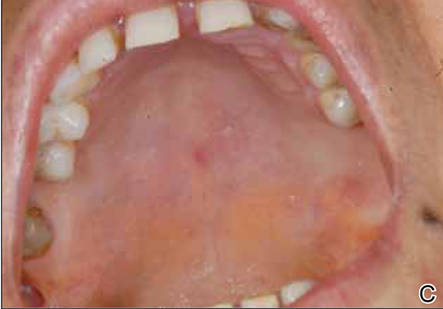 | |
 | 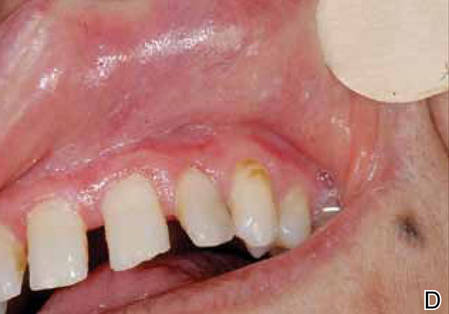 | |
| Figure 1. White-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate (A) and gingiva (B). Resolution of lesions on the hard palate (C) and the gingiva (D) after 9 months of antituberculosis therapy. | ||
Laboratory results from blood chemistry, complete blood cell count, erythrocyte sedimentation rate, C-reactive protein level, and urine analysis, as well as titers of serum immunoglobulin, antineutrophil cytoplasmic antibodies, and antinuclear antibodies, were within reference range. Human immunodeficiency virus serology was negative. Chest radiography and ultrasonography of the abdomen revealed no abnormalities. A purified protein derivative (tuberculin) test showed an induration of 20 mm. Mycobacterium tuberculosis grew on the culture of the tissue specimen.
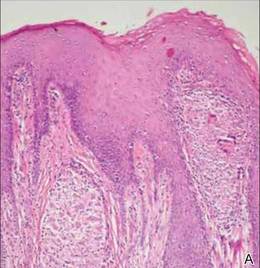 |
|
The patient was diagnosed with primary OT and treated with isoniazid (300 mg daily), rifampin (600 mg daily), ethambutol (1500 mg daily), and pyrazinamide (2000 mg daily). At 2 months of therapy the lesions started to heal and showed complete resolution at the end of 9 months of treatment (Figures 1C and 1D). There was no recurrence at 2-year follow-up.
Orificial tuberculosis is a rare form of cutaneous tuberculosis. Therefore, it is regarded as a “forgotten disease” in the literature.5 The pathogenesis of OT has not been clearly defined. The intact mucous membrane, thickened squamous epithelium, cleansing and antibacterial function of saliva, and presence of saprophytes act as protective mechanisms against penetration of mycobacteria. Some factors such as poor oral hygiene, smoking, local trauma, or presence of a dental cyst or an abscess may predispose the direct mucosal inoculation of the tuberculosis bacilli.1,2,6 Orificial tuberculosis may develop as an opportunistic infection in 1.33% of human immunodeficiency virus patients.7 However, our patient had no prior trauma or known predisposing factors.
The common presentation of OT is an ulcerative lesion with irregular and well-delineated margins with a yellowish granular base. A vesicle, papule, or nodule may precede the ulcers. The presence of yellow satellite nodules around the lesion is characteristic for OT.1,2,6 Although there were no satellite nodules around the ulcerations in our case, the lesions had a yellowish granular base and irregular, well-delineated margins.
The tongue, gingiva, lips, tonsils, and epiglottis are the most common sites of involvement.2,5,8 Hard palate involvement rarely has been reported, even in immunosuppressed patients.7 Enlarged painful cervical lymph nodes may accompany the disease. Most patients with OT have been reported to have active pulmonary tuberculosis at the time of diagnosis.6,8 Thus, internal organ involvement, particularly the pulmonary system, should be checked in patients with OT. In our patient, the hard palate was involved together with the gingiva despite the absence of immunosuppression. No internal organ involvement was found in the systemic evaluation.
Orificial tuberculosis is a form of cutaneous tuberculosis that is difficult to diagnose because of the varying nature of clinical features, failure of growth of M tuberculosis on culture, and rarity of the disease.1,2,6 As in our case, biopsies may not always exhibit a caseous necrosis, which is specific to tuberculosis.6,7 Thus, it may be difficult to distinguish oral cavity tuberculosis from conditions demonstrating oral ulcers such as bullous diseases, trauma, fungal diseases, syphilis, sarcoidosis, or squamous cell carcinoma by evaluating only signs and symptoms.6 Clinical suspicion is the first and foremost step in the diagnostic process of OT. In our patient, OT was suspected in the differential diagnosis because the resistant oral ulcerations showed the most common presentation of OT: irregular and well-delineated margins and a yellowish granular base. By considering tuberculosis within the differential diagnosis in our patient, microbiologic cultivation was performed from the oral mucosa and accurate diagnosis was established by determination of the pathogen’s growth in the culture.
Because of the increased incidence of tuberculosis and unusual manifestations, clinicians may easily overlook OT. It should be considered in the differential diagnosis of resistant nodules or ulcers of the oral cavity.
1. Kiliç A, Gül U, Gönül M, et al. Orificial tuberculosis of the lip: a case report and review of the literature. Int J Dermatol. 2009;48:178-180.
2. Ito FA, de Andrade CR, Vargas PA, et al. Primary tuberculosis of the oral cavity. Oral Dis. 2005;11:50-53.
3. Dixit R, Sharma S, Nuwal P. Tuberculosis of oral cavity. Indian J Tuberc. 2008;55:51-53.
4. Smolka W, Burger H, Iizuka T, et al. Primary tuberculosis of the oral cavity in an elderly nonimmunosuppressed patient: case report and review of the literature. Arch Otolaryngol Head Neck Surg. 2008;134:1107-1109.
5. Rodrigues G, Carnelio S, Valliathan M. Primary isolated gingival tuberculosis. Braz J Infect Dis. 2007;11:172-173.
6. Vilar FC, de Souza A, Moya MJ, et al. Atypical oral lesion in a patient with pulmonary tuberculosis. Int J Dermatol. 2009;48:910-912.
7. Kakisi OK, Kechagia AS, Kakisis IK, et al. Tuberculosis of the oral cavity: a systematic review. Eur J Oral Sci. 2010;118:103-109.
8. Eng HL, Lu SY, Yang CH, et al. Oral tuberculosis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:415-420.
To the Editor:
Orificial tuberculosis (OT) constitutes 2% of cutaneous tuberculosis cases and 0.01% to 1% of all clinical presentations of tuberculosis.1 It is clinically classified as primary or secondary OT. In primary OT, the oral mucosa is the initial site of the infection without any internal organ involvement.2 This form is more prevalent among men and young adults.1,3 Secondary OT is the cutaneous tuberculosis type that occurs in patients with internal organ tuberculosis from autoinoculation of bacilli to the orificial area. It is more common and usually affects elderly patients.2-4 We present the development of primary OT in an immunocompetent woman.
A 51-year-old woman was admitted with painful enlarging oral ulcers of 1 year’s duration. There was no history of tuberculosis infection, dental trauma, or smoking habit prior to the development of oral ulcers, and no family history of tuberculosis. On dermatological examination white-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate and gingiva were observed (Figures 1A and 1B). Oral hygiene was good. There was no regional lym-phadenopathy on palpation. Physical findings were normal. The histopathology of the biopsy from the gingival ulcer revealed noncaseating granulomatous inflammation in the dermis (Figure 2). Ziehl-Neelsen and periodic acid–Schiff stains were negative for acid-fast bacilli and fungi, respectively.
| | |
| | |
| Figure 1. White-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate (A) and gingiva (B). Resolution of lesions on the hard palate (C) and the gingiva (D) after 9 months of antituberculosis therapy. | ||
Laboratory results from blood chemistry, complete blood cell count, erythrocyte sedimentation rate, C-reactive protein level, and urine analysis, as well as titers of serum immunoglobulin, antineutrophil cytoplasmic antibodies, and antinuclear antibodies, were within reference range. Human immunodeficiency virus serology was negative. Chest radiography and ultrasonography of the abdomen revealed no abnormalities. A purified protein derivative (tuberculin) test showed an induration of 20 mm. Mycobacterium tuberculosis grew on the culture of the tissue specimen.
|
|
The patient was diagnosed with primary OT and treated with isoniazid (300 mg daily), rifampin (600 mg daily), ethambutol (1500 mg daily), and pyrazinamide (2000 mg daily). At 2 months of therapy the lesions started to heal and showed complete resolution at the end of 9 months of treatment (Figures 1C and 1D). There was no recurrence at 2-year follow-up.
Orificial tuberculosis is a rare form of cutaneous tuberculosis. Therefore, it is regarded as a “forgotten disease” in the literature.5 The pathogenesis of OT has not been clearly defined. The intact mucous membrane, thickened squamous epithelium, cleansing and antibacterial function of saliva, and presence of saprophytes act as protective mechanisms against penetration of mycobacteria. Some factors such as poor oral hygiene, smoking, local trauma, or presence of a dental cyst or an abscess may predispose the direct mucosal inoculation of the tuberculosis bacilli.1,2,6 Orificial tuberculosis may develop as an opportunistic infection in 1.33% of human immunodeficiency virus patients.7 However, our patient had no prior trauma or known predisposing factors.
The common presentation of OT is an ulcerative lesion with irregular and well-delineated margins with a yellowish granular base. A vesicle, papule, or nodule may precede the ulcers. The presence of yellow satellite nodules around the lesion is characteristic for OT.1,2,6 Although there were no satellite nodules around the ulcerations in our case, the lesions had a yellowish granular base and irregular, well-delineated margins.
The tongue, gingiva, lips, tonsils, and epiglottis are the most common sites of involvement.2,5,8 Hard palate involvement rarely has been reported, even in immunosuppressed patients.7 Enlarged painful cervical lymph nodes may accompany the disease. Most patients with OT have been reported to have active pulmonary tuberculosis at the time of diagnosis.6,8 Thus, internal organ involvement, particularly the pulmonary system, should be checked in patients with OT. In our patient, the hard palate was involved together with the gingiva despite the absence of immunosuppression. No internal organ involvement was found in the systemic evaluation.
Orificial tuberculosis is a form of cutaneous tuberculosis that is difficult to diagnose because of the varying nature of clinical features, failure of growth of M tuberculosis on culture, and rarity of the disease.1,2,6 As in our case, biopsies may not always exhibit a caseous necrosis, which is specific to tuberculosis.6,7 Thus, it may be difficult to distinguish oral cavity tuberculosis from conditions demonstrating oral ulcers such as bullous diseases, trauma, fungal diseases, syphilis, sarcoidosis, or squamous cell carcinoma by evaluating only signs and symptoms.6 Clinical suspicion is the first and foremost step in the diagnostic process of OT. In our patient, OT was suspected in the differential diagnosis because the resistant oral ulcerations showed the most common presentation of OT: irregular and well-delineated margins and a yellowish granular base. By considering tuberculosis within the differential diagnosis in our patient, microbiologic cultivation was performed from the oral mucosa and accurate diagnosis was established by determination of the pathogen’s growth in the culture.
Because of the increased incidence of tuberculosis and unusual manifestations, clinicians may easily overlook OT. It should be considered in the differential diagnosis of resistant nodules or ulcers of the oral cavity.
To the Editor:
Orificial tuberculosis (OT) constitutes 2% of cutaneous tuberculosis cases and 0.01% to 1% of all clinical presentations of tuberculosis.1 It is clinically classified as primary or secondary OT. In primary OT, the oral mucosa is the initial site of the infection without any internal organ involvement.2 This form is more prevalent among men and young adults.1,3 Secondary OT is the cutaneous tuberculosis type that occurs in patients with internal organ tuberculosis from autoinoculation of bacilli to the orificial area. It is more common and usually affects elderly patients.2-4 We present the development of primary OT in an immunocompetent woman.
A 51-year-old woman was admitted with painful enlarging oral ulcers of 1 year’s duration. There was no history of tuberculosis infection, dental trauma, or smoking habit prior to the development of oral ulcers, and no family history of tuberculosis. On dermatological examination white-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate and gingiva were observed (Figures 1A and 1B). Oral hygiene was good. There was no regional lym-phadenopathy on palpation. Physical findings were normal. The histopathology of the biopsy from the gingival ulcer revealed noncaseating granulomatous inflammation in the dermis (Figure 2). Ziehl-Neelsen and periodic acid–Schiff stains were negative for acid-fast bacilli and fungi, respectively.
| | |
| | |
| Figure 1. White-yellow indurated ulcers with 1×1.5-cm irregular margins located on the hard palate (A) and gingiva (B). Resolution of lesions on the hard palate (C) and the gingiva (D) after 9 months of antituberculosis therapy. | ||
Laboratory results from blood chemistry, complete blood cell count, erythrocyte sedimentation rate, C-reactive protein level, and urine analysis, as well as titers of serum immunoglobulin, antineutrophil cytoplasmic antibodies, and antinuclear antibodies, were within reference range. Human immunodeficiency virus serology was negative. Chest radiography and ultrasonography of the abdomen revealed no abnormalities. A purified protein derivative (tuberculin) test showed an induration of 20 mm. Mycobacterium tuberculosis grew on the culture of the tissue specimen.
|
|
The patient was diagnosed with primary OT and treated with isoniazid (300 mg daily), rifampin (600 mg daily), ethambutol (1500 mg daily), and pyrazinamide (2000 mg daily). At 2 months of therapy the lesions started to heal and showed complete resolution at the end of 9 months of treatment (Figures 1C and 1D). There was no recurrence at 2-year follow-up.
Orificial tuberculosis is a rare form of cutaneous tuberculosis. Therefore, it is regarded as a “forgotten disease” in the literature.5 The pathogenesis of OT has not been clearly defined. The intact mucous membrane, thickened squamous epithelium, cleansing and antibacterial function of saliva, and presence of saprophytes act as protective mechanisms against penetration of mycobacteria. Some factors such as poor oral hygiene, smoking, local trauma, or presence of a dental cyst or an abscess may predispose the direct mucosal inoculation of the tuberculosis bacilli.1,2,6 Orificial tuberculosis may develop as an opportunistic infection in 1.33% of human immunodeficiency virus patients.7 However, our patient had no prior trauma or known predisposing factors.
The common presentation of OT is an ulcerative lesion with irregular and well-delineated margins with a yellowish granular base. A vesicle, papule, or nodule may precede the ulcers. The presence of yellow satellite nodules around the lesion is characteristic for OT.1,2,6 Although there were no satellite nodules around the ulcerations in our case, the lesions had a yellowish granular base and irregular, well-delineated margins.
The tongue, gingiva, lips, tonsils, and epiglottis are the most common sites of involvement.2,5,8 Hard palate involvement rarely has been reported, even in immunosuppressed patients.7 Enlarged painful cervical lymph nodes may accompany the disease. Most patients with OT have been reported to have active pulmonary tuberculosis at the time of diagnosis.6,8 Thus, internal organ involvement, particularly the pulmonary system, should be checked in patients with OT. In our patient, the hard palate was involved together with the gingiva despite the absence of immunosuppression. No internal organ involvement was found in the systemic evaluation.
Orificial tuberculosis is a form of cutaneous tuberculosis that is difficult to diagnose because of the varying nature of clinical features, failure of growth of M tuberculosis on culture, and rarity of the disease.1,2,6 As in our case, biopsies may not always exhibit a caseous necrosis, which is specific to tuberculosis.6,7 Thus, it may be difficult to distinguish oral cavity tuberculosis from conditions demonstrating oral ulcers such as bullous diseases, trauma, fungal diseases, syphilis, sarcoidosis, or squamous cell carcinoma by evaluating only signs and symptoms.6 Clinical suspicion is the first and foremost step in the diagnostic process of OT. In our patient, OT was suspected in the differential diagnosis because the resistant oral ulcerations showed the most common presentation of OT: irregular and well-delineated margins and a yellowish granular base. By considering tuberculosis within the differential diagnosis in our patient, microbiologic cultivation was performed from the oral mucosa and accurate diagnosis was established by determination of the pathogen’s growth in the culture.
Because of the increased incidence of tuberculosis and unusual manifestations, clinicians may easily overlook OT. It should be considered in the differential diagnosis of resistant nodules or ulcers of the oral cavity.
1. Kiliç A, Gül U, Gönül M, et al. Orificial tuberculosis of the lip: a case report and review of the literature. Int J Dermatol. 2009;48:178-180.
2. Ito FA, de Andrade CR, Vargas PA, et al. Primary tuberculosis of the oral cavity. Oral Dis. 2005;11:50-53.
3. Dixit R, Sharma S, Nuwal P. Tuberculosis of oral cavity. Indian J Tuberc. 2008;55:51-53.
4. Smolka W, Burger H, Iizuka T, et al. Primary tuberculosis of the oral cavity in an elderly nonimmunosuppressed patient: case report and review of the literature. Arch Otolaryngol Head Neck Surg. 2008;134:1107-1109.
5. Rodrigues G, Carnelio S, Valliathan M. Primary isolated gingival tuberculosis. Braz J Infect Dis. 2007;11:172-173.
6. Vilar FC, de Souza A, Moya MJ, et al. Atypical oral lesion in a patient with pulmonary tuberculosis. Int J Dermatol. 2009;48:910-912.
7. Kakisi OK, Kechagia AS, Kakisis IK, et al. Tuberculosis of the oral cavity: a systematic review. Eur J Oral Sci. 2010;118:103-109.
8. Eng HL, Lu SY, Yang CH, et al. Oral tuberculosis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:415-420.
1. Kiliç A, Gül U, Gönül M, et al. Orificial tuberculosis of the lip: a case report and review of the literature. Int J Dermatol. 2009;48:178-180.
2. Ito FA, de Andrade CR, Vargas PA, et al. Primary tuberculosis of the oral cavity. Oral Dis. 2005;11:50-53.
3. Dixit R, Sharma S, Nuwal P. Tuberculosis of oral cavity. Indian J Tuberc. 2008;55:51-53.
4. Smolka W, Burger H, Iizuka T, et al. Primary tuberculosis of the oral cavity in an elderly nonimmunosuppressed patient: case report and review of the literature. Arch Otolaryngol Head Neck Surg. 2008;134:1107-1109.
5. Rodrigues G, Carnelio S, Valliathan M. Primary isolated gingival tuberculosis. Braz J Infect Dis. 2007;11:172-173.
6. Vilar FC, de Souza A, Moya MJ, et al. Atypical oral lesion in a patient with pulmonary tuberculosis. Int J Dermatol. 2009;48:910-912.
7. Kakisi OK, Kechagia AS, Kakisis IK, et al. Tuberculosis of the oral cavity: a systematic review. Eur J Oral Sci. 2010;118:103-109.
8. Eng HL, Lu SY, Yang CH, et al. Oral tuberculosis. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 1996;81:415-420.