User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Localized Pemphigus Foliaceus
To the Editor:
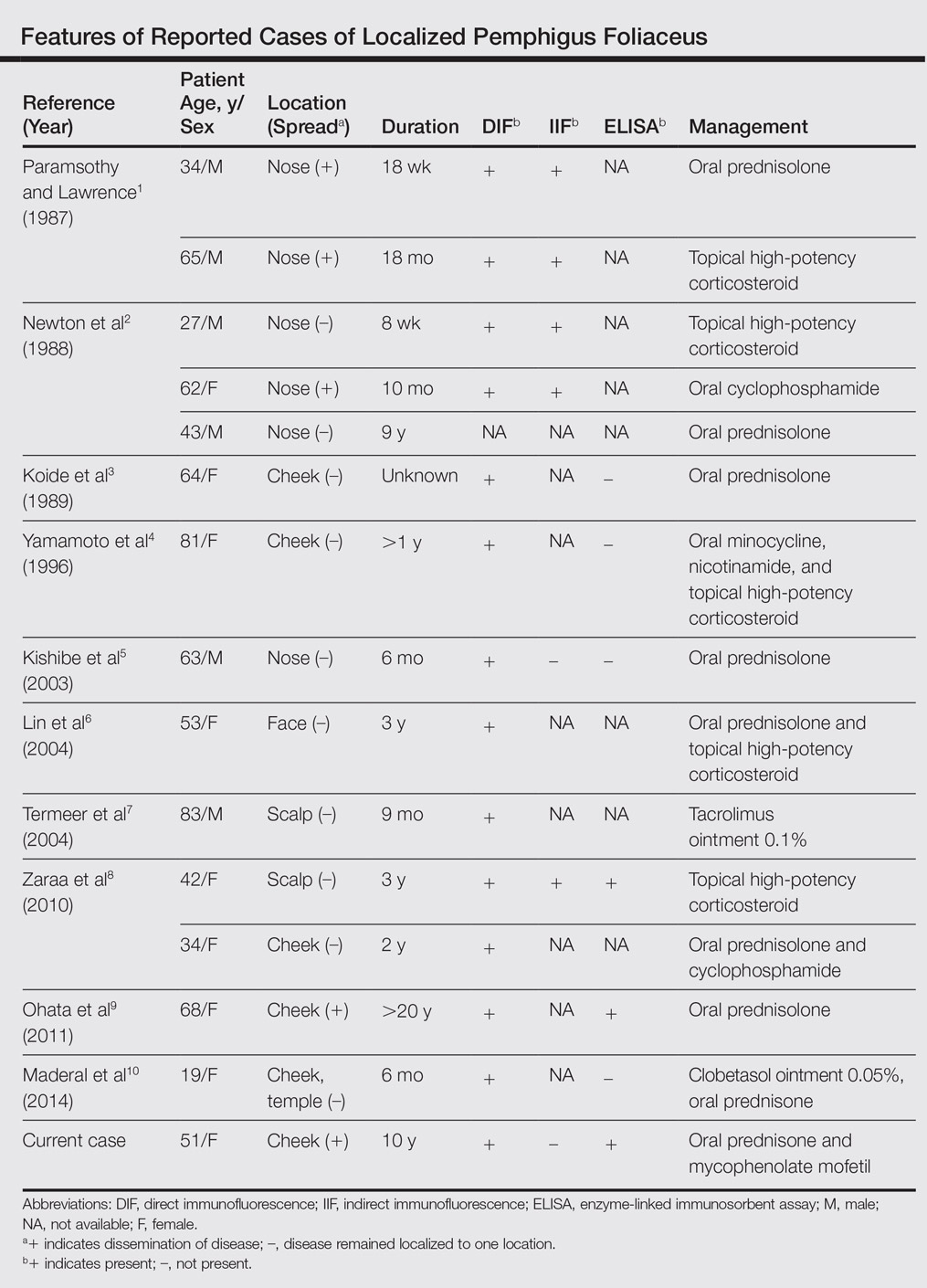
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
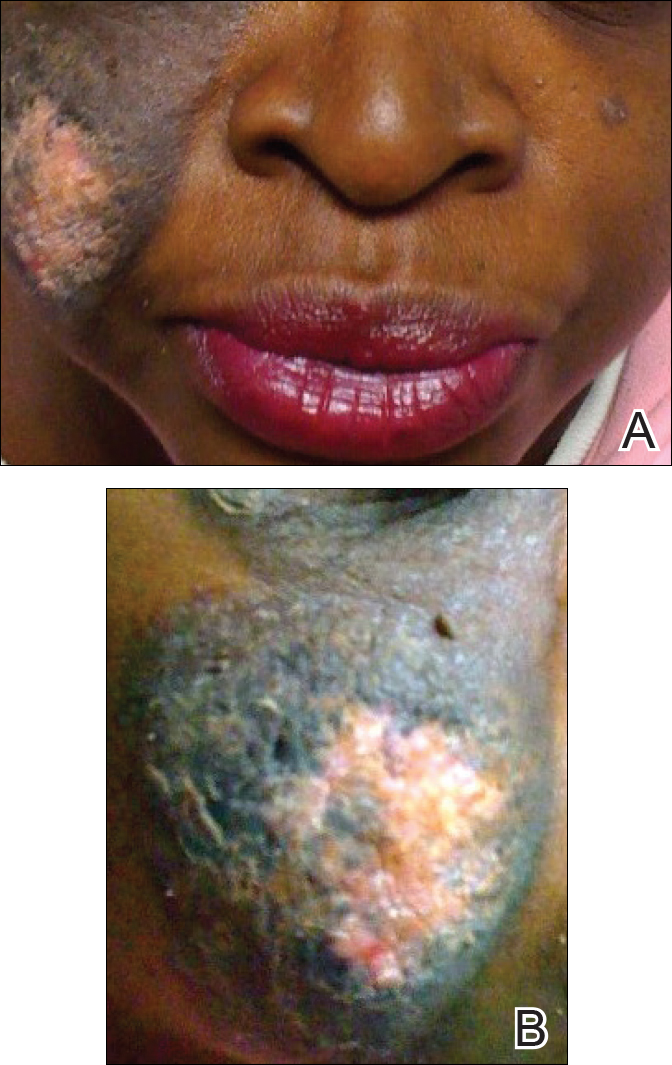
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
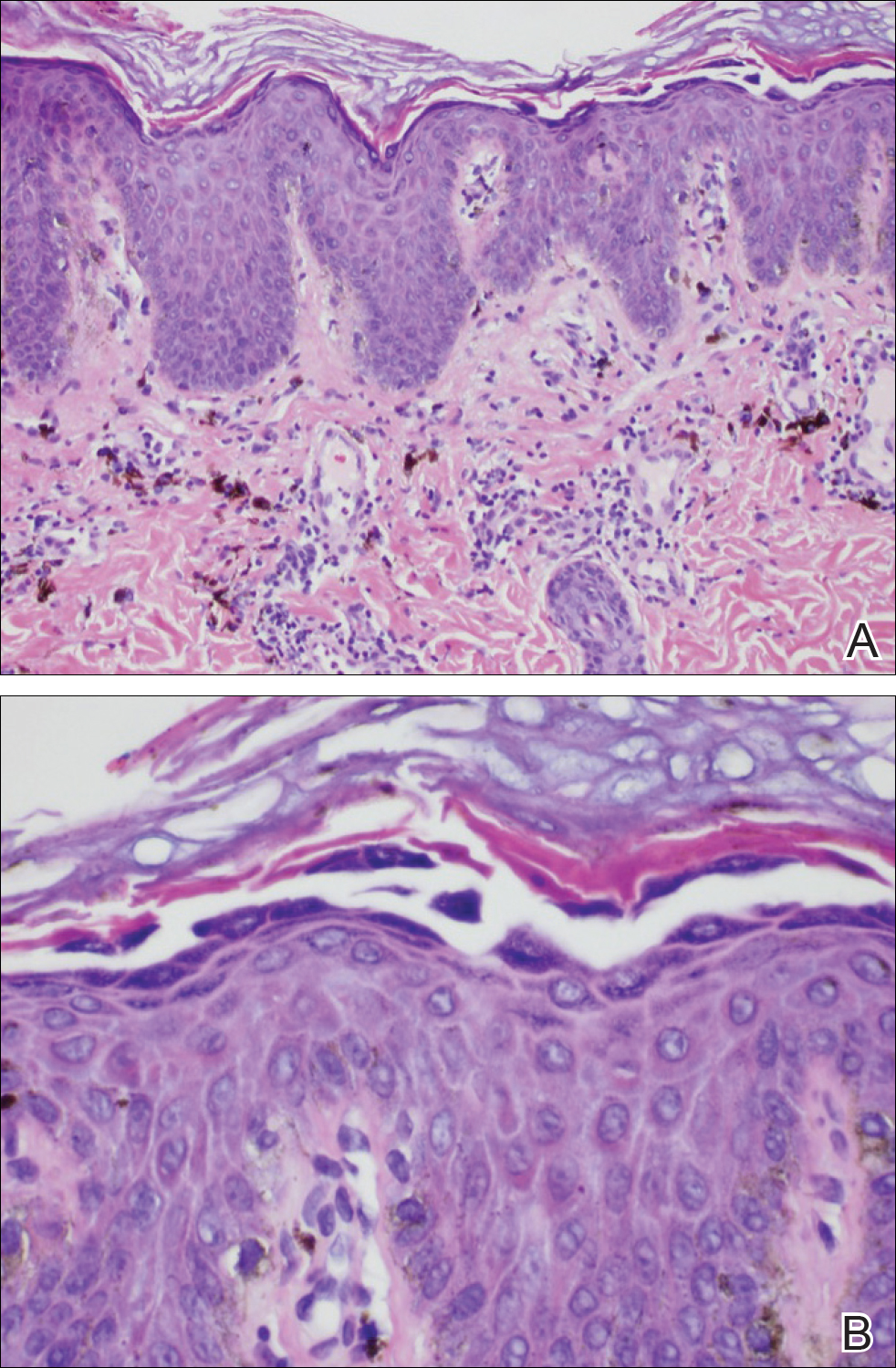
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.

The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.
- Paramsothy Y, Lawrence CM. “Tin-tack” sign in localized pemphigus foliaceus. Br J Dermatol. 1987;116:127-129.
- Newton JA, McGibbon DH, Monk B, et al. Pemphigus foliaceus localized to the nose. Br J Dermatol. 1988;118:303-312.
- Koide M, Kokura N, Takano N. Pemphigus foliaceus localized on the face [in Japanese]. Jpn J Dermatol. 1989;97:1262.
- Yamamoto S, Kanekura T, Gushi A, et al. A case of localized pemphigus foliaceus. J Dermatol. 1996;23:893-895.
- Kishibe M, Kinouchi M, Ishida-Yamamoto A, et al. Pemphigus foliaceus localized to the nose. Clin Exp Dermatol. 2003;28:560-562.
- Lin R, Ladd DJ, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
- Termeer CC, Technau K, Augustin M, et al. Topical tacrolimus (Protopic) for the treatment of a localized pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2004;18:636-637.
- Zaraa I, El Euch D, Kort R, et al. Localized pemphigus: a report of three cases. Int J Dermatol 2010;49:715-716.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- Maderal AD, Miner A, Nousari C, et al. Localized pemphigus foliaceus with unilateral facial involvement. Actas Dermosifiliogr. 2014;105:413-417.
- Cram DL, Winkelmann RK. Ultraviolet-induced acantholysis in pemphigus. Arch Dermatol. 1965;92:7-13.
- Kano Y, Shimosegawa M, Mizukawa Y, et al. Pemphigus foliaceus induced by exposure to sunlight. Dermatology. 2000;201:132-138.
- Lebe B, Gül Nıflıoğlu G, Seyrek S, et al. Evaluation of clinical and histopathologic/direct immunofluorescence diagnosis in autoimmune vesiculobullous dermatitis: utility of direct immunofluorescence. Turk Patoloji Derg. 2012;28:11-16.
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29:432-436.
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010-2017.
- Ng PP, Thng ST, Mohamed K, et al. Comparison of desmoglein ELISA and indirect immunofluorescence using two substrates (monkey esophagus and normal human skin) in the diagnosis of pemphigus. Australas J Dermatol. 2005;46:239-241.
To the Editor:
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.
The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.
To the Editor:
Pemphigus foliaceus is a rare autoimmune blistering disorder that typically presents with crusted scaly erosions in a seborrheic distribution. We describe a case of pemphigus foliaceus localized to the right cheek of 10 years’ duration that spread to other areas. With a PubMed search of articles indexed for MEDLINE yielding only 14 cases of localized pemphigus foliaceus (Table), it represents an extremely rare entity that often is a diagnostic challenge and may be a harbinger for disseminated disease months to years after the inciting lesion appears.
A 51-year-old woman presented with an asymptomatic cutaneous eruption that had remained localized to the right cheek for 10 years before it increased in size and new lesions developed on the left cheek, chest, and upper back. No inciting factors, such as contactants, insect bites, infections, medications, or recent travel were identified. On physical examination a well-demarcated, hypertrophic, verrucouslike plaque with central pink atrophy and exfoliative scale involved the right malar and submalar regions but spared the mucocutaneous junctions of the face (Figure 1). Subtle dark brown papules, some with overlying scale, speckled the left cheek, right jawline, chest, and upper back. The oral cavity was clear.
Leading differentials included hypertrophic discoid lupus erythematosus and pemphigus vegetans. Other considerations included sarcoidosis, granuloma faciale, lupus vulgaris, disseminated coccidioidomycosis or blastomycosis, and squamous cell carcinoma.
An initial biopsy revealed a lymphocytic lichenoid dermatitis with epidermal hyperplasia and scattered eosinophils for which the following differentials were provided: insect bite, hypertrophic lichen planus, prurigo nodularis superimposed on rosacea, and allergic contact dermatitis. Under these histologic diagnoses, tacrolimus ointment 0.03%, topical mid-potency corticosteroid, and a combination of oral doxycycline and metronidazole gel 1% were prescribed but failed to ameliorate her condition.
Because the clinical differentials were vast and noncorrelative with the original pathology, additional biopsies were performed: one from the edge of the large malar plaque, which was transected for hematoxylin and eosin (H&E) and tissue cultures; one perilesional to the large malar plaque for direct immunofluorescence (DIF); and one from the papule on the right jawline for H&E. Tissue cultures were negative for fungal and mycobacterial organisms. Both specimens submitted for H&E showed the prominent epidermal hyperplasia and lymphocytic dermal infiltrate noted on the original H&E but also demonstrated intragranular acantholysis (Figure 2). The DIF revealed intercellular IgG and C3 deposition throughout the epidermis (Figure 3). Indirect immunofluorescence was negative, but enzyme-linked immunosorbent assay detected circulating antidesmoglein-1 but not antidesmoglein-3 autoantibodies. Other serologies including antinuclear antibody, anti–double-stranded DNA, antihistone, anti–Sjögren syndrome A, and anti–Sjögren syndrome B antibodies were negative.
The diagnosis of localized pemphigus foliaceus was made and management with oral prednisone and mycophenolate mofetil resulted in improvement within weeks.
Localized pemphigus foliaceus is extremely rare with only 14 cases reported in the literature (Table).1-10 Its diagnosis is challenging, as the clinical presentation simulates various entities and the histological features and serological markers are difficult to capture.
Localized pemphigus foliaceus typically presents as an isolated, erythematous, scaly, crusted plaque involving the nose, cheek, or scalp and may mimic several conditions including contact dermatitis, seborrheic dermatitis, rosacea, cutaneous sarcoidosis, discoid lupus erythematosus, lupus vulgaris, impetigo contagiosa, solar keratosis, and nonmelanoma skin cancer.1-10
The predilection for sun-exposed areas suggests UV radiation may induce binding of antidesmoglein-1 autoantibodies with subsequent cytokine-mediated inflammation and acantholysis at these sites.11-13 Similarly, the immunomodulatory agent imiquimod has been reported to induce pemphigus foliaceus at its application sites.6
When pemphigus foliaceus is clinically discernible, the histology and DIF are in accordance with the clinical diagnosis 53.8% of the time.13 In cases of localized pemphigus foliaceus in which the diagnosis is more elusive, many biopsies often are needed to capture the characteristic intragranular acantholysis; this feature often is so subtle that unless the diagnosis is suspected, it is underappreciated or undetectable. In chronic lesions, it may be masked by secondary changes such as acanthosis, hyperkeratosis, and parakeratosis.14
In pemphigus foliaceus, detection of circulating antidesmoglein-1 autoantibodies by enzyme-linked immunosorbent assay is slightly more sensitive and specific compared to indirect immunofluorescence, but both correlate with disease activity.15,16 The low or absent autoantibody titers in localized pemphigus foliaceus may reflect its limited involvement, but dissemination of the disease with subsequent elevation of autoantibody titers may occur months to years after initial presentation,1,2,9 as was the case with our patient.
The majority of localized pemphigus foliaceus cases require systemic prednisone, sometimes in conjunction with nonsteroidal immunosuppressants or topical high-potency corticosteroids.1-3,5,6,8-10 One case was efficaciously managed with tacrolimus ointment 0.1%.7
Localized pemphigus foliaceus is a rare and challenging entity that must be a diagnostic consideration for any chronic focal plaque on the face or scalp, as it may herald disseminated disease.
- Paramsothy Y, Lawrence CM. “Tin-tack” sign in localized pemphigus foliaceus. Br J Dermatol. 1987;116:127-129.
- Newton JA, McGibbon DH, Monk B, et al. Pemphigus foliaceus localized to the nose. Br J Dermatol. 1988;118:303-312.
- Koide M, Kokura N, Takano N. Pemphigus foliaceus localized on the face [in Japanese]. Jpn J Dermatol. 1989;97:1262.
- Yamamoto S, Kanekura T, Gushi A, et al. A case of localized pemphigus foliaceus. J Dermatol. 1996;23:893-895.
- Kishibe M, Kinouchi M, Ishida-Yamamoto A, et al. Pemphigus foliaceus localized to the nose. Clin Exp Dermatol. 2003;28:560-562.
- Lin R, Ladd DJ, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
- Termeer CC, Technau K, Augustin M, et al. Topical tacrolimus (Protopic) for the treatment of a localized pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2004;18:636-637.
- Zaraa I, El Euch D, Kort R, et al. Localized pemphigus: a report of three cases. Int J Dermatol 2010;49:715-716.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- Maderal AD, Miner A, Nousari C, et al. Localized pemphigus foliaceus with unilateral facial involvement. Actas Dermosifiliogr. 2014;105:413-417.
- Cram DL, Winkelmann RK. Ultraviolet-induced acantholysis in pemphigus. Arch Dermatol. 1965;92:7-13.
- Kano Y, Shimosegawa M, Mizukawa Y, et al. Pemphigus foliaceus induced by exposure to sunlight. Dermatology. 2000;201:132-138.
- Lebe B, Gül Nıflıoğlu G, Seyrek S, et al. Evaluation of clinical and histopathologic/direct immunofluorescence diagnosis in autoimmune vesiculobullous dermatitis: utility of direct immunofluorescence. Turk Patoloji Derg. 2012;28:11-16.
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29:432-436.
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010-2017.
- Ng PP, Thng ST, Mohamed K, et al. Comparison of desmoglein ELISA and indirect immunofluorescence using two substrates (monkey esophagus and normal human skin) in the diagnosis of pemphigus. Australas J Dermatol. 2005;46:239-241.
- Paramsothy Y, Lawrence CM. “Tin-tack” sign in localized pemphigus foliaceus. Br J Dermatol. 1987;116:127-129.
- Newton JA, McGibbon DH, Monk B, et al. Pemphigus foliaceus localized to the nose. Br J Dermatol. 1988;118:303-312.
- Koide M, Kokura N, Takano N. Pemphigus foliaceus localized on the face [in Japanese]. Jpn J Dermatol. 1989;97:1262.
- Yamamoto S, Kanekura T, Gushi A, et al. A case of localized pemphigus foliaceus. J Dermatol. 1996;23:893-895.
- Kishibe M, Kinouchi M, Ishida-Yamamoto A, et al. Pemphigus foliaceus localized to the nose. Clin Exp Dermatol. 2003;28:560-562.
- Lin R, Ladd DJ, Powell DJ, et al. Localized pemphigus foliaceus induced by topical imiquimod treatment. Arch Dermatol. 2004;140:889-890.
- Termeer CC, Technau K, Augustin M, et al. Topical tacrolimus (Protopic) for the treatment of a localized pemphigus foliaceus. J Eur Acad Dermatol Venereol. 2004;18:636-637.
- Zaraa I, El Euch D, Kort R, et al. Localized pemphigus: a report of three cases. Int J Dermatol 2010;49:715-716.
- Ohata C, Akamatsu K, Imai N, et al. Localized pemphigus foliaceus exclusively involving the follicular infundibulum: a novel peau d’orange appearance. Eur J Dermatol. 2011;21:392-395.
- Maderal AD, Miner A, Nousari C, et al. Localized pemphigus foliaceus with unilateral facial involvement. Actas Dermosifiliogr. 2014;105:413-417.
- Cram DL, Winkelmann RK. Ultraviolet-induced acantholysis in pemphigus. Arch Dermatol. 1965;92:7-13.
- Kano Y, Shimosegawa M, Mizukawa Y, et al. Pemphigus foliaceus induced by exposure to sunlight. Dermatology. 2000;201:132-138.
- Lebe B, Gül Nıflıoğlu G, Seyrek S, et al. Evaluation of clinical and histopathologic/direct immunofluorescence diagnosis in autoimmune vesiculobullous dermatitis: utility of direct immunofluorescence. Turk Patoloji Derg. 2012;28:11-16.
- Joly P, Litrowski N. Pemphigus group (vulgaris, vegetans, foliaceus, herpetiformis, brasiliensis). Clin Dermatol. 2011;29:432-436.
- Ishii K, Amagai M, Hall RP, et al. Characterization of autoantibodies in pemphigus using antigen specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010-2017.
- Ng PP, Thng ST, Mohamed K, et al. Comparison of desmoglein ELISA and indirect immunofluorescence using two substrates (monkey esophagus and normal human skin) in the diagnosis of pemphigus. Australas J Dermatol. 2005;46:239-241.
Practice Points
- The diagnosis of pemphigus foliceus is challenging, as the clinical presentation simulates various entities.
- Clinicopathological correlation is important. If pathology and other diagnostics do not support clinical findings, trust your clinical assessment and consider repeating or adjusting the workup.
Severe Henoch-Schönlein Purpura Complicating Infliximab Therapy for Ulcerative Colitis
To the Editor:
Anti–tumor necrosis factor (TNF) α treatments have radically improved the management of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, and bowel diseases (eg, Crohn disease, ulcerative colitis [UC]). Because the number of patients treated with these agents has increased, uncommon adverse reactions have increasingly occurred. Cutaneous adverse reactions that have been reported with anti-TNF agents include immediate injection-site reaction, systemic infusion reactions, and delayed reactions.1 Among the delayed adverse reactions, psoriatic and eczematous eruptions as well as cutaneous infections are the most common, while cutaneous adverse effects related to an immune imbalance syndrome including vasculitis; lupuslike, lichenlike, and granulomatous eruptions; and skin cancer rarely are observed.1 Although most of the cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases. We report the case of severe Henoch-Schönlein purpura (HSP) following treatment with infliximab.
A 46-year-old man who was a nonsmoker with quiescent UC on infliximab for 30 months presented with palpable necrotic purpura on both legs (Figure) and arms as well as the abdomen of 10 days’ duration, along with diffuse joint pain and swelling. He had no history of infectious or gastrointestinal symptoms. The last infliximab infusion was performed 6 weeks prior to developing the purpura. His UC was diagnosed 10 years prior to the current presentation and was not associated with any extragastrointestinal manifestations. Since diagnosis, UC had failed to respond to therapies such as azathioprine, cyclosporine, and purinethol. The complete blood cell count was normal. The C-reactive protein level was 18.7 mg/L (reference range, <5 mg/L) and the erythrocyte sedimentation rate was 30 mm/h (reference range, 0–20 mm/h). Electrolytes, urea, creatinine clearance, and liver function were normal, and a chest radiograph and radiographs of the swollen joints were unremarkable. The total IgA level was elevated at 4 g/L (reference range, 0.7–4 g/L), with IgG and IgM levels within reference range. There was no hematuria or proteinuria on urinalysis. Tests for antinuclear antibodies, rheumatoid factor, circulating immune complexes, and antineutrophil cytoplasmic antibody were negative. Total complement, C3, and C4 levels also were normal. A skin biopsy confirmed a leukocytoclastic vasculitis of small vessels with C3 deposition. Serologic tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were negative. Based on these findings, the diagnosis of HSP was made. Systemic corticosteroids—120 mg daily of intravenous methylprednisolone for 3 days, followed by 1 mg/kg daily of oral prednisone for 2 weeks—were then introduced with rapid clinical improvement. Henoch-Schönlein purpura and joint symptoms completely resolved, but UC relapsed with bloody diarrhea and severe abdominal pain. Oral prednisone was maintained (1 mg/kg daily). Because of the severity of cutaneous vasculitis (HSP), a multidisciplinary decision was taken to definitively stop the anti-TNF agents and to first add azathioprine (2 mg/kg daily for 2 months), then subcutaneous methotrexate (25 mg weekly). Colonoscopy did not show any dysplasia or adenocarcinoma and confirmed the diagnosis of UC. After 6 months of combined therapy, UC was still active and we decided to perform a total colectomy with ileostomy formation. Complete remission of UC was obtained and maintained after 28 months of follow-up.
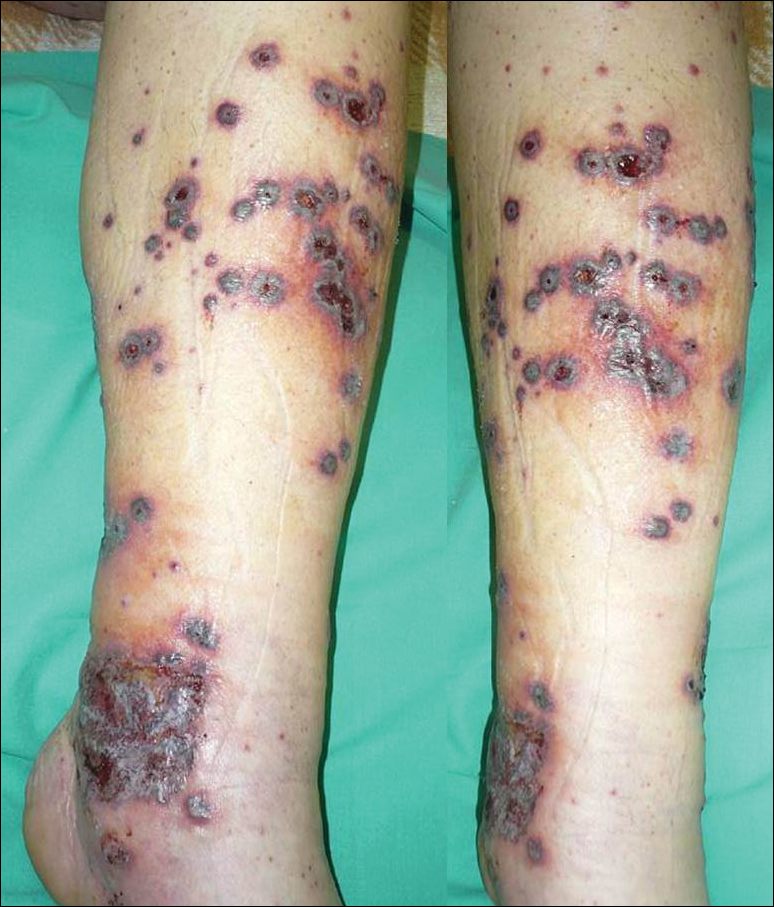
Henoch-Schönlein purpura is a multisystem small vessel leukocytoclastic vasculitis with the deposition of immune complexes containing IgA. Clinical manifestations may include palpable purpura, arthritis, enteritis, and nephritis. Henoch-Schönlein purpura usually affects children. Adult onset is rare but associated with more severe symptoms and a poor prognosis.2 The criteria for HSP, as defined by the American College of Rheumatology,3 include palpable purpura, 20 years or younger at disease onset, bowel angina, and presence of vascular wall granulocytes on biopsy. At least 2 of these criteria are required for HSP diagnosis. Various viral or bacterial infections and drugs can trigger HSP, which also can be associated with autoinflammatory or autoimmune diseases. The association of HSP and UC is a rare event, as demonstrated by de Oliveira et al.4 Although only 2 cases of cutaneous vasculitis mimicking HSP have been described in UC,4 we cannot exclude a possible association between HSP and UC. However, our patient had UC for 10 years and never had clinical manifestations of vasculitis.
There are 5 reports of HSP following etanercept5,6 or adalimumab7-9 therapy and 1 following infliximab therapy.10 In all cases, HSP occurred after several months of anti-TNF therapy. However, there also are reports of cutaneous vasculitis associated with arthralgia and glomerulonephritis that resolved after withdrawal of anti-TNF agents.11,12 It is possible that some of these reactions may have been manifestations of undiagnosed HSP. In a series of 113 patients who developed cutaneous vasculitis after anti-TNF agents, visceral vasculitis was observed in 24% of patients. Treatment of vasculitis involved withdrawal of the anti-TNF therapy in 101 cases (89%).13 In these UC patients with few therapeutic alternatives, the continuation of anti-TNF agents should be discussed. In the previous series,13 of 16 patients who were rechallenged with the same or a different TNF antagonist, 12 (75%) experienced vasculitis relapse, suggesting a class effect of TNF inhibition. Because of the severity of cutaneous vasculitis and as previously suggested in a recent analytical and comprehensive overview on paradoxical reactions under TNF blockers,1 we decided not to re-expose our patient to infliximab or to other anti-TNF agents.
In conclusion, HSP may occur during anti-TNF therapy and physicians need to be aware of this potentially serious complication.
- Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification ofHenoch-Schönlein purpura. an analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol. 2015;33(2, suppl 89):S44-S47.
- de Oliveira GT, Martins SS, Deboni M, et al. Cutaneous vasculitis in ulcerative colitis mimicking Henoch-Schönlein purpura [published online May 22, 2012]. J Crohns Colitis. 2013;7:e69-e73.
- Marques I, Lagos A, Reis J, et al. Reversible Henoch-Schönlein purpura complicating adalimumab therapy. J Crohns Colitis. 2012;6:796-799.
- Rahman FZ, Takhar GK, Roy O, et al. Henoch-Schönlein purpura complicating adalimumab therapy for Crohn’s disease. World J Gastrointest Pharmacol Ther. 2010;1:119-122.
- Lee A, Kasama R, Evangelisto A, et al. Henoch-Schönlein purpura after etanercept therapy for psoriasis. J Clin Rheumatol. 2006;12:249-251.
- Duffy TN, Genta M, Moll S, et al. Henoch Schönlein purpura following etanercept treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(2, suppl 41):S106.
- LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature. Case Rep Rheumatol. 2016:2812980.
- Nobile S, Catassi C, Felici L. Herpes zoster infection followed by Henoch-Schönlein purpura in a girl receiving infliximab for ulcerative colitis. J Clin Rheumatol. 2009;15:101.
- Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol. 2004;31:1955-1958.
- Simms R, Kipgen D, Dahill S, et al. ANCA-associated renal vasculitis following anti-tumor necrosis factor alpha therapy. Am J Kidney Dis. 2008;51:e11-e14.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007;86:242-251.
To the Editor:
Anti–tumor necrosis factor (TNF) α treatments have radically improved the management of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, and bowel diseases (eg, Crohn disease, ulcerative colitis [UC]). Because the number of patients treated with these agents has increased, uncommon adverse reactions have increasingly occurred. Cutaneous adverse reactions that have been reported with anti-TNF agents include immediate injection-site reaction, systemic infusion reactions, and delayed reactions.1 Among the delayed adverse reactions, psoriatic and eczematous eruptions as well as cutaneous infections are the most common, while cutaneous adverse effects related to an immune imbalance syndrome including vasculitis; lupuslike, lichenlike, and granulomatous eruptions; and skin cancer rarely are observed.1 Although most of the cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases. We report the case of severe Henoch-Schönlein purpura (HSP) following treatment with infliximab.
A 46-year-old man who was a nonsmoker with quiescent UC on infliximab for 30 months presented with palpable necrotic purpura on both legs (Figure) and arms as well as the abdomen of 10 days’ duration, along with diffuse joint pain and swelling. He had no history of infectious or gastrointestinal symptoms. The last infliximab infusion was performed 6 weeks prior to developing the purpura. His UC was diagnosed 10 years prior to the current presentation and was not associated with any extragastrointestinal manifestations. Since diagnosis, UC had failed to respond to therapies such as azathioprine, cyclosporine, and purinethol. The complete blood cell count was normal. The C-reactive protein level was 18.7 mg/L (reference range, <5 mg/L) and the erythrocyte sedimentation rate was 30 mm/h (reference range, 0–20 mm/h). Electrolytes, urea, creatinine clearance, and liver function were normal, and a chest radiograph and radiographs of the swollen joints were unremarkable. The total IgA level was elevated at 4 g/L (reference range, 0.7–4 g/L), with IgG and IgM levels within reference range. There was no hematuria or proteinuria on urinalysis. Tests for antinuclear antibodies, rheumatoid factor, circulating immune complexes, and antineutrophil cytoplasmic antibody were negative. Total complement, C3, and C4 levels also were normal. A skin biopsy confirmed a leukocytoclastic vasculitis of small vessels with C3 deposition. Serologic tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were negative. Based on these findings, the diagnosis of HSP was made. Systemic corticosteroids—120 mg daily of intravenous methylprednisolone for 3 days, followed by 1 mg/kg daily of oral prednisone for 2 weeks—were then introduced with rapid clinical improvement. Henoch-Schönlein purpura and joint symptoms completely resolved, but UC relapsed with bloody diarrhea and severe abdominal pain. Oral prednisone was maintained (1 mg/kg daily). Because of the severity of cutaneous vasculitis (HSP), a multidisciplinary decision was taken to definitively stop the anti-TNF agents and to first add azathioprine (2 mg/kg daily for 2 months), then subcutaneous methotrexate (25 mg weekly). Colonoscopy did not show any dysplasia or adenocarcinoma and confirmed the diagnosis of UC. After 6 months of combined therapy, UC was still active and we decided to perform a total colectomy with ileostomy formation. Complete remission of UC was obtained and maintained after 28 months of follow-up.
Henoch-Schönlein purpura is a multisystem small vessel leukocytoclastic vasculitis with the deposition of immune complexes containing IgA. Clinical manifestations may include palpable purpura, arthritis, enteritis, and nephritis. Henoch-Schönlein purpura usually affects children. Adult onset is rare but associated with more severe symptoms and a poor prognosis.2 The criteria for HSP, as defined by the American College of Rheumatology,3 include palpable purpura, 20 years or younger at disease onset, bowel angina, and presence of vascular wall granulocytes on biopsy. At least 2 of these criteria are required for HSP diagnosis. Various viral or bacterial infections and drugs can trigger HSP, which also can be associated with autoinflammatory or autoimmune diseases. The association of HSP and UC is a rare event, as demonstrated by de Oliveira et al.4 Although only 2 cases of cutaneous vasculitis mimicking HSP have been described in UC,4 we cannot exclude a possible association between HSP and UC. However, our patient had UC for 10 years and never had clinical manifestations of vasculitis.
There are 5 reports of HSP following etanercept5,6 or adalimumab7-9 therapy and 1 following infliximab therapy.10 In all cases, HSP occurred after several months of anti-TNF therapy. However, there also are reports of cutaneous vasculitis associated with arthralgia and glomerulonephritis that resolved after withdrawal of anti-TNF agents.11,12 It is possible that some of these reactions may have been manifestations of undiagnosed HSP. In a series of 113 patients who developed cutaneous vasculitis after anti-TNF agents, visceral vasculitis was observed in 24% of patients. Treatment of vasculitis involved withdrawal of the anti-TNF therapy in 101 cases (89%).13 In these UC patients with few therapeutic alternatives, the continuation of anti-TNF agents should be discussed. In the previous series,13 of 16 patients who were rechallenged with the same or a different TNF antagonist, 12 (75%) experienced vasculitis relapse, suggesting a class effect of TNF inhibition. Because of the severity of cutaneous vasculitis and as previously suggested in a recent analytical and comprehensive overview on paradoxical reactions under TNF blockers,1 we decided not to re-expose our patient to infliximab or to other anti-TNF agents.
In conclusion, HSP may occur during anti-TNF therapy and physicians need to be aware of this potentially serious complication.
To the Editor:
Anti–tumor necrosis factor (TNF) α treatments have radically improved the management of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, and bowel diseases (eg, Crohn disease, ulcerative colitis [UC]). Because the number of patients treated with these agents has increased, uncommon adverse reactions have increasingly occurred. Cutaneous adverse reactions that have been reported with anti-TNF agents include immediate injection-site reaction, systemic infusion reactions, and delayed reactions.1 Among the delayed adverse reactions, psoriatic and eczematous eruptions as well as cutaneous infections are the most common, while cutaneous adverse effects related to an immune imbalance syndrome including vasculitis; lupuslike, lichenlike, and granulomatous eruptions; and skin cancer rarely are observed.1 Although most of the cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases. We report the case of severe Henoch-Schönlein purpura (HSP) following treatment with infliximab.
A 46-year-old man who was a nonsmoker with quiescent UC on infliximab for 30 months presented with palpable necrotic purpura on both legs (Figure) and arms as well as the abdomen of 10 days’ duration, along with diffuse joint pain and swelling. He had no history of infectious or gastrointestinal symptoms. The last infliximab infusion was performed 6 weeks prior to developing the purpura. His UC was diagnosed 10 years prior to the current presentation and was not associated with any extragastrointestinal manifestations. Since diagnosis, UC had failed to respond to therapies such as azathioprine, cyclosporine, and purinethol. The complete blood cell count was normal. The C-reactive protein level was 18.7 mg/L (reference range, <5 mg/L) and the erythrocyte sedimentation rate was 30 mm/h (reference range, 0–20 mm/h). Electrolytes, urea, creatinine clearance, and liver function were normal, and a chest radiograph and radiographs of the swollen joints were unremarkable. The total IgA level was elevated at 4 g/L (reference range, 0.7–4 g/L), with IgG and IgM levels within reference range. There was no hematuria or proteinuria on urinalysis. Tests for antinuclear antibodies, rheumatoid factor, circulating immune complexes, and antineutrophil cytoplasmic antibody were negative. Total complement, C3, and C4 levels also were normal. A skin biopsy confirmed a leukocytoclastic vasculitis of small vessels with C3 deposition. Serologic tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were negative. Based on these findings, the diagnosis of HSP was made. Systemic corticosteroids—120 mg daily of intravenous methylprednisolone for 3 days, followed by 1 mg/kg daily of oral prednisone for 2 weeks—were then introduced with rapid clinical improvement. Henoch-Schönlein purpura and joint symptoms completely resolved, but UC relapsed with bloody diarrhea and severe abdominal pain. Oral prednisone was maintained (1 mg/kg daily). Because of the severity of cutaneous vasculitis (HSP), a multidisciplinary decision was taken to definitively stop the anti-TNF agents and to first add azathioprine (2 mg/kg daily for 2 months), then subcutaneous methotrexate (25 mg weekly). Colonoscopy did not show any dysplasia or adenocarcinoma and confirmed the diagnosis of UC. After 6 months of combined therapy, UC was still active and we decided to perform a total colectomy with ileostomy formation. Complete remission of UC was obtained and maintained after 28 months of follow-up.
Henoch-Schönlein purpura is a multisystem small vessel leukocytoclastic vasculitis with the deposition of immune complexes containing IgA. Clinical manifestations may include palpable purpura, arthritis, enteritis, and nephritis. Henoch-Schönlein purpura usually affects children. Adult onset is rare but associated with more severe symptoms and a poor prognosis.2 The criteria for HSP, as defined by the American College of Rheumatology,3 include palpable purpura, 20 years or younger at disease onset, bowel angina, and presence of vascular wall granulocytes on biopsy. At least 2 of these criteria are required for HSP diagnosis. Various viral or bacterial infections and drugs can trigger HSP, which also can be associated with autoinflammatory or autoimmune diseases. The association of HSP and UC is a rare event, as demonstrated by de Oliveira et al.4 Although only 2 cases of cutaneous vasculitis mimicking HSP have been described in UC,4 we cannot exclude a possible association between HSP and UC. However, our patient had UC for 10 years and never had clinical manifestations of vasculitis.
There are 5 reports of HSP following etanercept5,6 or adalimumab7-9 therapy and 1 following infliximab therapy.10 In all cases, HSP occurred after several months of anti-TNF therapy. However, there also are reports of cutaneous vasculitis associated with arthralgia and glomerulonephritis that resolved after withdrawal of anti-TNF agents.11,12 It is possible that some of these reactions may have been manifestations of undiagnosed HSP. In a series of 113 patients who developed cutaneous vasculitis after anti-TNF agents, visceral vasculitis was observed in 24% of patients. Treatment of vasculitis involved withdrawal of the anti-TNF therapy in 101 cases (89%).13 In these UC patients with few therapeutic alternatives, the continuation of anti-TNF agents should be discussed. In the previous series,13 of 16 patients who were rechallenged with the same or a different TNF antagonist, 12 (75%) experienced vasculitis relapse, suggesting a class effect of TNF inhibition. Because of the severity of cutaneous vasculitis and as previously suggested in a recent analytical and comprehensive overview on paradoxical reactions under TNF blockers,1 we decided not to re-expose our patient to infliximab or to other anti-TNF agents.
In conclusion, HSP may occur during anti-TNF therapy and physicians need to be aware of this potentially serious complication.
- Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification ofHenoch-Schönlein purpura. an analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol. 2015;33(2, suppl 89):S44-S47.
- de Oliveira GT, Martins SS, Deboni M, et al. Cutaneous vasculitis in ulcerative colitis mimicking Henoch-Schönlein purpura [published online May 22, 2012]. J Crohns Colitis. 2013;7:e69-e73.
- Marques I, Lagos A, Reis J, et al. Reversible Henoch-Schönlein purpura complicating adalimumab therapy. J Crohns Colitis. 2012;6:796-799.
- Rahman FZ, Takhar GK, Roy O, et al. Henoch-Schönlein purpura complicating adalimumab therapy for Crohn’s disease. World J Gastrointest Pharmacol Ther. 2010;1:119-122.
- Lee A, Kasama R, Evangelisto A, et al. Henoch-Schönlein purpura after etanercept therapy for psoriasis. J Clin Rheumatol. 2006;12:249-251.
- Duffy TN, Genta M, Moll S, et al. Henoch Schönlein purpura following etanercept treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(2, suppl 41):S106.
- LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature. Case Rep Rheumatol. 2016:2812980.
- Nobile S, Catassi C, Felici L. Herpes zoster infection followed by Henoch-Schönlein purpura in a girl receiving infliximab for ulcerative colitis. J Clin Rheumatol. 2009;15:101.
- Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol. 2004;31:1955-1958.
- Simms R, Kipgen D, Dahill S, et al. ANCA-associated renal vasculitis following anti-tumor necrosis factor alpha therapy. Am J Kidney Dis. 2008;51:e11-e14.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007;86:242-251.
- Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification ofHenoch-Schönlein purpura. an analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol. 2015;33(2, suppl 89):S44-S47.
- de Oliveira GT, Martins SS, Deboni M, et al. Cutaneous vasculitis in ulcerative colitis mimicking Henoch-Schönlein purpura [published online May 22, 2012]. J Crohns Colitis. 2013;7:e69-e73.
- Marques I, Lagos A, Reis J, et al. Reversible Henoch-Schönlein purpura complicating adalimumab therapy. J Crohns Colitis. 2012;6:796-799.
- Rahman FZ, Takhar GK, Roy O, et al. Henoch-Schönlein purpura complicating adalimumab therapy for Crohn’s disease. World J Gastrointest Pharmacol Ther. 2010;1:119-122.
- Lee A, Kasama R, Evangelisto A, et al. Henoch-Schönlein purpura after etanercept therapy for psoriasis. J Clin Rheumatol. 2006;12:249-251.
- Duffy TN, Genta M, Moll S, et al. Henoch Schönlein purpura following etanercept treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(2, suppl 41):S106.
- LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature. Case Rep Rheumatol. 2016:2812980.
- Nobile S, Catassi C, Felici L. Herpes zoster infection followed by Henoch-Schönlein purpura in a girl receiving infliximab for ulcerative colitis. J Clin Rheumatol. 2009;15:101.
- Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol. 2004;31:1955-1958.
- Simms R, Kipgen D, Dahill S, et al. ANCA-associated renal vasculitis following anti-tumor necrosis factor alpha therapy. Am J Kidney Dis. 2008;51:e11-e14.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007;86:242-251.
Practice Points
- Cutaneous adverse effects may occur in approximately 20% of patients treated with anti–tumor necrosis factor (TNF) drugs.
- Henoch-Schönlein purpura (HSP), a small-vessel vasculitis, is an extremely rare complication of anti-TNF treatment.
- Although most cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases.
Questioning the Specificity and Sensitivity of ELISA for Bullous Pemphigoid Diagnosis
Bullous pemphigoid (BP) is the most common autoimmune blistering disease. The classic presentation of BP is a generalized, pruritic, bullous eruption in elderly patients, which is occasionally preceded by an urticarial prodrome. Immunopathologically, BP is characterized by IgG and sometimes IgE autoantibodies that target basement membrane zone proteins BP180 and BP230 of the epidermis.1
The diagnosis of BP should be suspected when an elderly patient presents with tense blisters and can be confirmed via diagnostic testing, including tissue histology and direct immunofluorescence (DIF) as the gold standard, as well as indirect immunofluorescence (IIF), enzyme-linked immunosorbent assay (ELISA), and most recently biochip technology as supportive tests.2 Since its advent, ELISA has gained popularity as a trustworthy diagnostic test for BP. The specificity of ELISA for BP diagnosis is reported to be 98% to 100%, which leads clinicians to believe that a positive ELISA equals certain diagnosis of BP; however, misdiagnosis of BP based on a positive ELISA result can occur.3-13 The treatment of BP often involves lifelong immunosuppressive therapy. Complications of immunosuppressive therapy contribute to morbidity and mortality in these patients, thus an accurate diagnosis is paramount before introducing therapy.14
We present the case of a 74-year-old man with a history of a pruritic nonbullous eruption who was diagnosed with BP and treated for 3 years based on positive ELISA results in the absence of confirmatory histology or DIF.
Case Report
A 74-year-old man with diabetes mellitus, hypertension, hyperlipidemia, benign prostatic hypertrophy, and obstructive sleep apnea presented for further evaluation and confirmation of a prior diagnosis of BP by an outside dermatologist. He reported a pruritic rash on the trunk, back, and extremities of 3 years’ duration. He denied occurrence of blisters at any time.
On presentation to an outside dermatologist 3 years ago, a biopsy was performed along with serologic studies due to the patient’s age and the possibility of an urticarial prodrome in BP. The biopsy revealed epidermal acanthosis, subepidermal separation, and a perivascular and interstitial infiltrate of lymphocytes and eosinophils in the papillary dermis. Direct immunofluorescence was nondiagnostic with a weak discontinuous pattern of IgG and IgA linearly along the basement membrane zone as well as few scattered and clumped cytoid bodies of IgM and IgA. Indirect immunofluoresence revealed a positive IgG titer of 1:40 on monkey esophagus substrate and a positive epidermal pattern on human split-skin substrate with a titer of 1:80. An ELISA for IgG autoantibodies against BP180 and BP230 yielded 15 U and 6 U, respectively (cut off value, 9 U). Based on the positive ELISA for IgG against BP180, a diagnosis of BP was made.
Over the following 3 years, the treatment included prednisone, tetracycline, nicotinamide, doxycycline, and dapsone. Therapy was suboptimal due to the patient’s comorbidities and socioeconomic status. Poorly controlled diabetes mellitus precluded consistent use of prednisone as recommended for BP. Tetracycline and nicotinamide were transiently effective in controlling the patient’s symptoms but were discontinued due to changes in his health insurance. Doxycycline and dapsone were ineffective. Throughout this 3-year period, the patient remained blister free, but the pruritic eruption was persistent.
The patient presented to our clinic due to his frustration with the lack of improvement and doubts about the BP diagnosis given the persistent absence of bullous lesions. Physical examination revealed numerous eroded, scaly, crusted papules on erythematous edematous plaques on all extremities, trunk, and back (Figure 1). The head, neck, face, and oral mucosa were spared. His history and clinical findings were atypical for BP and skin biopsies were performed. Histology revealed epidermal erosion with parakeratosis, spongiosis, and superficial perivascular lymphocytic inflammation with rare eosinophils without subepidermal split (Figure 2). Direct immunofluorescence was negative for IgG, IgA, IgM, C3, and C1q. Additionally, further review of the initial histology by another dermatopathologist revealed that the subepidermal separation reported was more likely artifactual clefts. These findings were not consistent with BP.
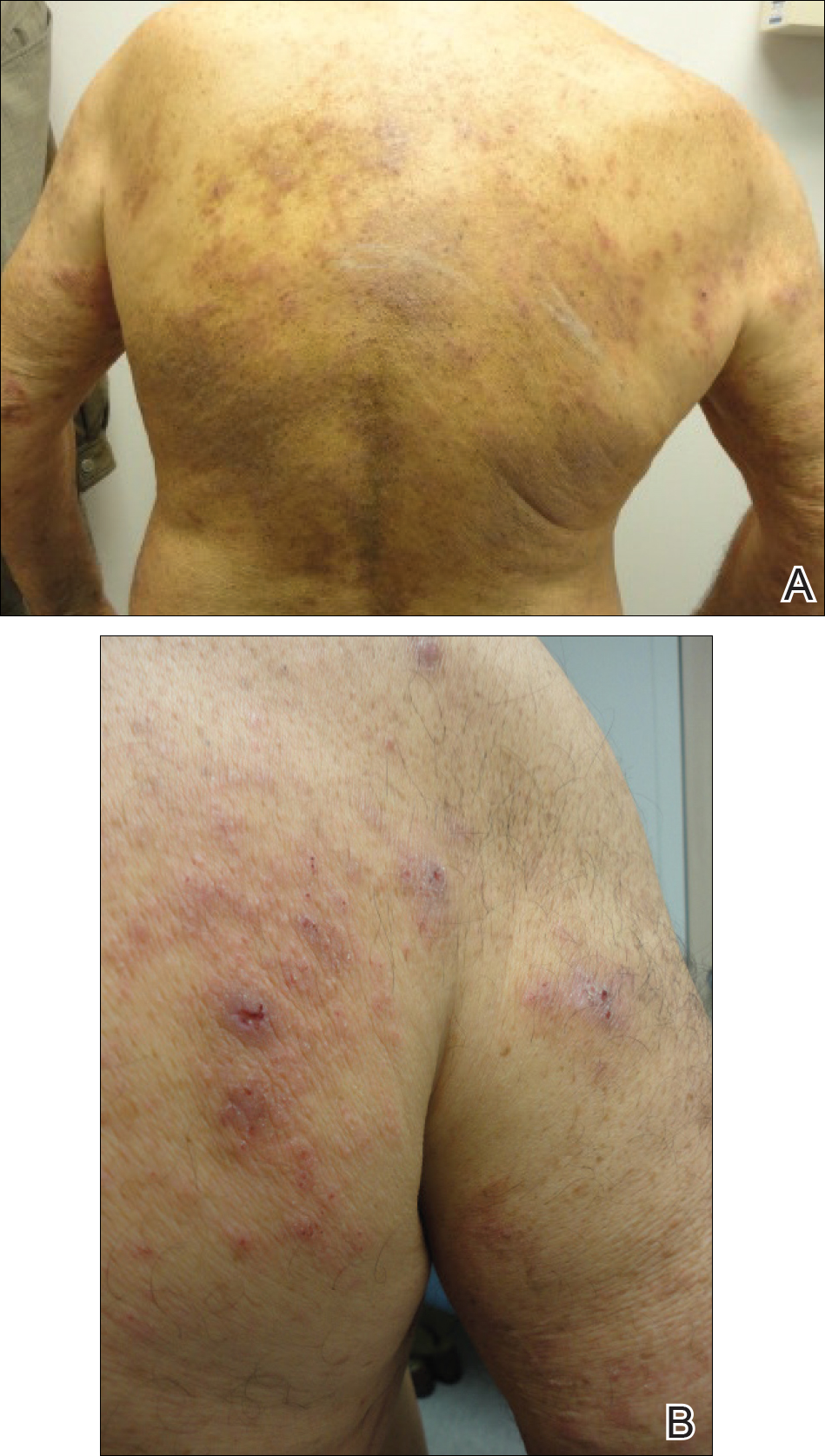

Given the patient’s clinical history, lack of bullae, and twice-negative DIF, the diagnosis was determined to be more consistent with eczematous spongiotic dermatitis. He refused a referral for phototherapy due to scheduling inconvenience. The patient was started on cyclosporine 0.5 mg/kg twice daily. After 10 days of treatment, he returned for follow-up and reported notable improvement in the pruritus. On physical examination, his dermatitis was improved with decreased erythema and inflammation.
The patient is being continued on extensive dry skin care with thick moisturizers and additional topical corticosteroid application on an as-needed basis.
Comment
Chronic immunosuppression contributes to morbidity and mortality in patients with BP; therefore, accurate diagnosis of BP is of utmost importance.14 A meta-analysis described ELISA as a test with high sensitivity and specificity (87% and 98%–100%, respectively) for diagnosis of BP.3 Nevertheless, there are opportunities for misdiagnosis using ELISA, as demonstrated in our case. To determine if the reported sensitivity and specificity of ELISA is accurate and reliable for clinical use, individual studies from the meta-analysis were reviewed.4,5,7-10,13,15 Issues identified in our review included dissimilar diagnostic procedures and patient populations among individual studies, several reports of positive ELISA in patients without BP, and a lack of explanation for these false-positive results.
There are notable differences in diagnostic procedures and patient populations among reports that establish the sensitivity and specificity of ELISA for BP diagnosis.3-13 Studies have detected IgG that targets the NC16A domain of the BP180 kD antigen, the C-terminal of the BP180 kD antigen, or the entire ectodomain of the BP180 kD antigen. Study patient populations varied in disease activity, stage, and treatment. Control patients included healthy patients as well as those with many dermatoses, including pemphigus vulgaris, systemic scleroderma, systemic lupus erythematosus, rheumatoid arthritis, lichen planus, and discoid lupus erythematosus.3-13 Due to these differences between individual studies, we believe the results that determine the overall sensitivity and specificity of ELISA for BP diagnosis must be interpreted with caution. For ELISA statistics to be clinically applicable to a specific patient, he/she should be similar to the patients studied. Therefore, we believe each study must be evaluated individually for applicability, given the differences that exist between them.
Furthermore, there have been several reports of false-positive ELISA results in patients with other dermatologic disorders, specifically in elderly patients with pruritus who do not fulfill clinical criteria for diagnosis with BP.16-18 In a population of elderly patients with pruritus for which no specific dermatological or systemic cause was identified, Hofmann et al18 found that 12% (3/25) of patients showed IgG reactivity to BP180 despite having negative DIF results. In another study of elderly patients with pruritic dermatoses, Feliciani et al17 found that 33% (5/15) of patients had IgG reactivity against BP230 or BP180, though they did not fulfill BP criteria based on clinical presentation and showed negative DIF and IIF results. These findings suggest that IgG reactivity against BP autoantibodies as determined by ELISA is not uncommon in pruritic diseases of the elderly.
Explanations for false-positive ELISA results were rare. A few authors suggested that false-positives could be attributed to an excessively low cutoff value,7-9 which was consistent with reports that the titer of autoantibodies to BP180 correlates with disease severity, suggesting that the higher titer of antibodies correlates with more severe disease and likely more accurate diagnosis.10,19,20 It is important to consider that patients who have low titers of BP180 autoantibodies with inconsistent clinical characteristics and DIF results may not truly have BP. Furthermore, to determine the clinical value of ELISA in identifying patients in the initial phase of BP, sera of BP patients should be compared with sera of elderly patients with pruritic skin disorders because they comprise the patient population that often requires diagnosis.18
Given the issues identified in our review of the literature, the published sensitivity and specificity of ELISA for BP diagnosis are likely overstated. In conclusion, ELISA should not be relied on as a single criterion adequate for diagnosis of BP.12,21 Rather, the diagnosis of BP can be obtained with a positive predictive value of 95% when a patient meets 3 of 4 clinical criteria (ie, absence of atrophic scars, absence of head and neck involvement, absence of mucosal involvement, and older than 70 years) and demonstrates linear deposits of predominantly IgG and/or C3 along the basement membrane zone of a perilesional biopsy on DIF.15 The gold standard for diagnosis of BP remains clinical presentation along with DIF, which can be supported by histology, IIF, and ELISA.22
- Delaporte E, Dubost-Brama A, Ghohestani R, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol. 1996;157:3642-3647.
- Schmidt E, Zillikens D. Diagnosis and clinical severity markers of bullous pemphigoid. F1000 Med Rep. 2009;1:15.
- Tampoia M, Giavarina D, Di Giorgio C, et al. Diagnostic accuracy of enzyme-linked immunosorbent assays (ELISA) to detect anti-skin autoantibodies in autoimmune blistering diseases: a systematic review and meta-analysis. Autoimmun Rev. 2012;12:121-126.
- Zillikens D, Mascaro JM, Rose PA, et al. A highly sensitive enzyme-linked immunosorbent assay for the detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J Invest Dermatol. 1997;109:679-683.
- Sitaru C, Dahnrich C, Probst C, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol. 2007;16:770-777.
- Yang B, Wang C, Chen S, et al. Evaluation of the combination of BP180-NC16a enzyme-linked immunosorbent assay and BP230 enzyme-linked immunosorbent assay in the diagnosis of bullous pemphigoid. Indian J Dermatol Venereol Leprol. 2012;78:722-727.
- Sakuma-Oyama Y, Powell AM, Oyama N, et al. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. Br J Dermatol. 2004;151:126-131.
- Tampoia M, Lattanzi V, Zucano A, et al. Evaluation of a new ELISA assay for detection of BP230 autoantibodies in bullous pemphigoid. Ann N Y Acad Sci. 2009;1173:15-20.
- Feng S, Lin L, Jin P, et al. Role of BP180NC16a-enzyme-linked immunosorbent assay (ELISA) in the diagnosis of bullous pemphigoid in China. Int J Dermatol. 2008;47:24-28.
- Kobayashi M, Amagai M, Kuroda-Kinoshita K, et al. BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci. 2002;30:224-232.
- Roussel A, Benichou J, Arivelo Randriamanantany Z, et al. Enzyme-linked immunosorbent assay for the combination of bullous pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:293-298.
- Chan, Lawrence S. ELISA instead of indirect IF in patients with BP. Arch Dermatol. 2011;147:291-292.
- Barnadas MA, Rubiales V, González J, et al. Enzyme-linked immunosorbent assay (ELISA) and indirect immunofluorescence testing in a bullous pemphigoid and pemphigoid gestationis. Int J Dermatol. 2008;47:1245-1249.
- Borradori L, Bernard P. Pemphigoid group. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2003:469.
- Vaillant L, Bernard P, Joly P, et al. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. Arch Dermatol. 1998;134:1075-1080.
- Fania L, Caldarola G, Muller R, et al. IgE recognition of bullous pemphigoid (BP)180 and BP230 in BP patients and elderly individuals with pruritic dermatoses. Clin Immunol. 2012;143:236-245.
- Feliciani C, Caldarola G, Kneisel A, et al. IgG autoantibody reactivity against bullous pemphigoid (BP) 180 and BP230 in elderly patients with pruritic dermatoses. Br J Dermatol. 2009;61:306-312.
- Hofmann SC, Tamm K, Hertl M, et al. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br J Dermatol. 2003;149:910-911.
- Schmidt E, Obe K, Brocker EB, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol. 2000;136:174-178.
- Feng S, Wu Q, Jin P, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Int J Dermatol. 2008;47:225-228.
- Di Zenzo G, Joly P, Zambruno G, et al. Sensitivity of immunofluorescence studies vs enzyme-linked immunosorbent assay for diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:1454-1456.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev. 2010;10:84-89.
Bullous pemphigoid (BP) is the most common autoimmune blistering disease. The classic presentation of BP is a generalized, pruritic, bullous eruption in elderly patients, which is occasionally preceded by an urticarial prodrome. Immunopathologically, BP is characterized by IgG and sometimes IgE autoantibodies that target basement membrane zone proteins BP180 and BP230 of the epidermis.1
The diagnosis of BP should be suspected when an elderly patient presents with tense blisters and can be confirmed via diagnostic testing, including tissue histology and direct immunofluorescence (DIF) as the gold standard, as well as indirect immunofluorescence (IIF), enzyme-linked immunosorbent assay (ELISA), and most recently biochip technology as supportive tests.2 Since its advent, ELISA has gained popularity as a trustworthy diagnostic test for BP. The specificity of ELISA for BP diagnosis is reported to be 98% to 100%, which leads clinicians to believe that a positive ELISA equals certain diagnosis of BP; however, misdiagnosis of BP based on a positive ELISA result can occur.3-13 The treatment of BP often involves lifelong immunosuppressive therapy. Complications of immunosuppressive therapy contribute to morbidity and mortality in these patients, thus an accurate diagnosis is paramount before introducing therapy.14
We present the case of a 74-year-old man with a history of a pruritic nonbullous eruption who was diagnosed with BP and treated for 3 years based on positive ELISA results in the absence of confirmatory histology or DIF.
Case Report
A 74-year-old man with diabetes mellitus, hypertension, hyperlipidemia, benign prostatic hypertrophy, and obstructive sleep apnea presented for further evaluation and confirmation of a prior diagnosis of BP by an outside dermatologist. He reported a pruritic rash on the trunk, back, and extremities of 3 years’ duration. He denied occurrence of blisters at any time.
On presentation to an outside dermatologist 3 years ago, a biopsy was performed along with serologic studies due to the patient’s age and the possibility of an urticarial prodrome in BP. The biopsy revealed epidermal acanthosis, subepidermal separation, and a perivascular and interstitial infiltrate of lymphocytes and eosinophils in the papillary dermis. Direct immunofluorescence was nondiagnostic with a weak discontinuous pattern of IgG and IgA linearly along the basement membrane zone as well as few scattered and clumped cytoid bodies of IgM and IgA. Indirect immunofluoresence revealed a positive IgG titer of 1:40 on monkey esophagus substrate and a positive epidermal pattern on human split-skin substrate with a titer of 1:80. An ELISA for IgG autoantibodies against BP180 and BP230 yielded 15 U and 6 U, respectively (cut off value, 9 U). Based on the positive ELISA for IgG against BP180, a diagnosis of BP was made.
Over the following 3 years, the treatment included prednisone, tetracycline, nicotinamide, doxycycline, and dapsone. Therapy was suboptimal due to the patient’s comorbidities and socioeconomic status. Poorly controlled diabetes mellitus precluded consistent use of prednisone as recommended for BP. Tetracycline and nicotinamide were transiently effective in controlling the patient’s symptoms but were discontinued due to changes in his health insurance. Doxycycline and dapsone were ineffective. Throughout this 3-year period, the patient remained blister free, but the pruritic eruption was persistent.
The patient presented to our clinic due to his frustration with the lack of improvement and doubts about the BP diagnosis given the persistent absence of bullous lesions. Physical examination revealed numerous eroded, scaly, crusted papules on erythematous edematous plaques on all extremities, trunk, and back (Figure 1). The head, neck, face, and oral mucosa were spared. His history and clinical findings were atypical for BP and skin biopsies were performed. Histology revealed epidermal erosion with parakeratosis, spongiosis, and superficial perivascular lymphocytic inflammation with rare eosinophils without subepidermal split (Figure 2). Direct immunofluorescence was negative for IgG, IgA, IgM, C3, and C1q. Additionally, further review of the initial histology by another dermatopathologist revealed that the subepidermal separation reported was more likely artifactual clefts. These findings were not consistent with BP.
Given the patient’s clinical history, lack of bullae, and twice-negative DIF, the diagnosis was determined to be more consistent with eczematous spongiotic dermatitis. He refused a referral for phototherapy due to scheduling inconvenience. The patient was started on cyclosporine 0.5 mg/kg twice daily. After 10 days of treatment, he returned for follow-up and reported notable improvement in the pruritus. On physical examination, his dermatitis was improved with decreased erythema and inflammation.
The patient is being continued on extensive dry skin care with thick moisturizers and additional topical corticosteroid application on an as-needed basis.
Comment
Chronic immunosuppression contributes to morbidity and mortality in patients with BP; therefore, accurate diagnosis of BP is of utmost importance.14 A meta-analysis described ELISA as a test with high sensitivity and specificity (87% and 98%–100%, respectively) for diagnosis of BP.3 Nevertheless, there are opportunities for misdiagnosis using ELISA, as demonstrated in our case. To determine if the reported sensitivity and specificity of ELISA is accurate and reliable for clinical use, individual studies from the meta-analysis were reviewed.4,5,7-10,13,15 Issues identified in our review included dissimilar diagnostic procedures and patient populations among individual studies, several reports of positive ELISA in patients without BP, and a lack of explanation for these false-positive results.
There are notable differences in diagnostic procedures and patient populations among reports that establish the sensitivity and specificity of ELISA for BP diagnosis.3-13 Studies have detected IgG that targets the NC16A domain of the BP180 kD antigen, the C-terminal of the BP180 kD antigen, or the entire ectodomain of the BP180 kD antigen. Study patient populations varied in disease activity, stage, and treatment. Control patients included healthy patients as well as those with many dermatoses, including pemphigus vulgaris, systemic scleroderma, systemic lupus erythematosus, rheumatoid arthritis, lichen planus, and discoid lupus erythematosus.3-13 Due to these differences between individual studies, we believe the results that determine the overall sensitivity and specificity of ELISA for BP diagnosis must be interpreted with caution. For ELISA statistics to be clinically applicable to a specific patient, he/she should be similar to the patients studied. Therefore, we believe each study must be evaluated individually for applicability, given the differences that exist between them.
Furthermore, there have been several reports of false-positive ELISA results in patients with other dermatologic disorders, specifically in elderly patients with pruritus who do not fulfill clinical criteria for diagnosis with BP.16-18 In a population of elderly patients with pruritus for which no specific dermatological or systemic cause was identified, Hofmann et al18 found that 12% (3/25) of patients showed IgG reactivity to BP180 despite having negative DIF results. In another study of elderly patients with pruritic dermatoses, Feliciani et al17 found that 33% (5/15) of patients had IgG reactivity against BP230 or BP180, though they did not fulfill BP criteria based on clinical presentation and showed negative DIF and IIF results. These findings suggest that IgG reactivity against BP autoantibodies as determined by ELISA is not uncommon in pruritic diseases of the elderly.
Explanations for false-positive ELISA results were rare. A few authors suggested that false-positives could be attributed to an excessively low cutoff value,7-9 which was consistent with reports that the titer of autoantibodies to BP180 correlates with disease severity, suggesting that the higher titer of antibodies correlates with more severe disease and likely more accurate diagnosis.10,19,20 It is important to consider that patients who have low titers of BP180 autoantibodies with inconsistent clinical characteristics and DIF results may not truly have BP. Furthermore, to determine the clinical value of ELISA in identifying patients in the initial phase of BP, sera of BP patients should be compared with sera of elderly patients with pruritic skin disorders because they comprise the patient population that often requires diagnosis.18
Given the issues identified in our review of the literature, the published sensitivity and specificity of ELISA for BP diagnosis are likely overstated. In conclusion, ELISA should not be relied on as a single criterion adequate for diagnosis of BP.12,21 Rather, the diagnosis of BP can be obtained with a positive predictive value of 95% when a patient meets 3 of 4 clinical criteria (ie, absence of atrophic scars, absence of head and neck involvement, absence of mucosal involvement, and older than 70 years) and demonstrates linear deposits of predominantly IgG and/or C3 along the basement membrane zone of a perilesional biopsy on DIF.15 The gold standard for diagnosis of BP remains clinical presentation along with DIF, which can be supported by histology, IIF, and ELISA.22
Bullous pemphigoid (BP) is the most common autoimmune blistering disease. The classic presentation of BP is a generalized, pruritic, bullous eruption in elderly patients, which is occasionally preceded by an urticarial prodrome. Immunopathologically, BP is characterized by IgG and sometimes IgE autoantibodies that target basement membrane zone proteins BP180 and BP230 of the epidermis.1
The diagnosis of BP should be suspected when an elderly patient presents with tense blisters and can be confirmed via diagnostic testing, including tissue histology and direct immunofluorescence (DIF) as the gold standard, as well as indirect immunofluorescence (IIF), enzyme-linked immunosorbent assay (ELISA), and most recently biochip technology as supportive tests.2 Since its advent, ELISA has gained popularity as a trustworthy diagnostic test for BP. The specificity of ELISA for BP diagnosis is reported to be 98% to 100%, which leads clinicians to believe that a positive ELISA equals certain diagnosis of BP; however, misdiagnosis of BP based on a positive ELISA result can occur.3-13 The treatment of BP often involves lifelong immunosuppressive therapy. Complications of immunosuppressive therapy contribute to morbidity and mortality in these patients, thus an accurate diagnosis is paramount before introducing therapy.14
We present the case of a 74-year-old man with a history of a pruritic nonbullous eruption who was diagnosed with BP and treated for 3 years based on positive ELISA results in the absence of confirmatory histology or DIF.
Case Report
A 74-year-old man with diabetes mellitus, hypertension, hyperlipidemia, benign prostatic hypertrophy, and obstructive sleep apnea presented for further evaluation and confirmation of a prior diagnosis of BP by an outside dermatologist. He reported a pruritic rash on the trunk, back, and extremities of 3 years’ duration. He denied occurrence of blisters at any time.
On presentation to an outside dermatologist 3 years ago, a biopsy was performed along with serologic studies due to the patient’s age and the possibility of an urticarial prodrome in BP. The biopsy revealed epidermal acanthosis, subepidermal separation, and a perivascular and interstitial infiltrate of lymphocytes and eosinophils in the papillary dermis. Direct immunofluorescence was nondiagnostic with a weak discontinuous pattern of IgG and IgA linearly along the basement membrane zone as well as few scattered and clumped cytoid bodies of IgM and IgA. Indirect immunofluoresence revealed a positive IgG titer of 1:40 on monkey esophagus substrate and a positive epidermal pattern on human split-skin substrate with a titer of 1:80. An ELISA for IgG autoantibodies against BP180 and BP230 yielded 15 U and 6 U, respectively (cut off value, 9 U). Based on the positive ELISA for IgG against BP180, a diagnosis of BP was made.
Over the following 3 years, the treatment included prednisone, tetracycline, nicotinamide, doxycycline, and dapsone. Therapy was suboptimal due to the patient’s comorbidities and socioeconomic status. Poorly controlled diabetes mellitus precluded consistent use of prednisone as recommended for BP. Tetracycline and nicotinamide were transiently effective in controlling the patient’s symptoms but were discontinued due to changes in his health insurance. Doxycycline and dapsone were ineffective. Throughout this 3-year period, the patient remained blister free, but the pruritic eruption was persistent.
The patient presented to our clinic due to his frustration with the lack of improvement and doubts about the BP diagnosis given the persistent absence of bullous lesions. Physical examination revealed numerous eroded, scaly, crusted papules on erythematous edematous plaques on all extremities, trunk, and back (Figure 1). The head, neck, face, and oral mucosa were spared. His history and clinical findings were atypical for BP and skin biopsies were performed. Histology revealed epidermal erosion with parakeratosis, spongiosis, and superficial perivascular lymphocytic inflammation with rare eosinophils without subepidermal split (Figure 2). Direct immunofluorescence was negative for IgG, IgA, IgM, C3, and C1q. Additionally, further review of the initial histology by another dermatopathologist revealed that the subepidermal separation reported was more likely artifactual clefts. These findings were not consistent with BP.
Given the patient’s clinical history, lack of bullae, and twice-negative DIF, the diagnosis was determined to be more consistent with eczematous spongiotic dermatitis. He refused a referral for phototherapy due to scheduling inconvenience. The patient was started on cyclosporine 0.5 mg/kg twice daily. After 10 days of treatment, he returned for follow-up and reported notable improvement in the pruritus. On physical examination, his dermatitis was improved with decreased erythema and inflammation.
The patient is being continued on extensive dry skin care with thick moisturizers and additional topical corticosteroid application on an as-needed basis.
Comment
Chronic immunosuppression contributes to morbidity and mortality in patients with BP; therefore, accurate diagnosis of BP is of utmost importance.14 A meta-analysis described ELISA as a test with high sensitivity and specificity (87% and 98%–100%, respectively) for diagnosis of BP.3 Nevertheless, there are opportunities for misdiagnosis using ELISA, as demonstrated in our case. To determine if the reported sensitivity and specificity of ELISA is accurate and reliable for clinical use, individual studies from the meta-analysis were reviewed.4,5,7-10,13,15 Issues identified in our review included dissimilar diagnostic procedures and patient populations among individual studies, several reports of positive ELISA in patients without BP, and a lack of explanation for these false-positive results.
There are notable differences in diagnostic procedures and patient populations among reports that establish the sensitivity and specificity of ELISA for BP diagnosis.3-13 Studies have detected IgG that targets the NC16A domain of the BP180 kD antigen, the C-terminal of the BP180 kD antigen, or the entire ectodomain of the BP180 kD antigen. Study patient populations varied in disease activity, stage, and treatment. Control patients included healthy patients as well as those with many dermatoses, including pemphigus vulgaris, systemic scleroderma, systemic lupus erythematosus, rheumatoid arthritis, lichen planus, and discoid lupus erythematosus.3-13 Due to these differences between individual studies, we believe the results that determine the overall sensitivity and specificity of ELISA for BP diagnosis must be interpreted with caution. For ELISA statistics to be clinically applicable to a specific patient, he/she should be similar to the patients studied. Therefore, we believe each study must be evaluated individually for applicability, given the differences that exist between them.
Furthermore, there have been several reports of false-positive ELISA results in patients with other dermatologic disorders, specifically in elderly patients with pruritus who do not fulfill clinical criteria for diagnosis with BP.16-18 In a population of elderly patients with pruritus for which no specific dermatological or systemic cause was identified, Hofmann et al18 found that 12% (3/25) of patients showed IgG reactivity to BP180 despite having negative DIF results. In another study of elderly patients with pruritic dermatoses, Feliciani et al17 found that 33% (5/15) of patients had IgG reactivity against BP230 or BP180, though they did not fulfill BP criteria based on clinical presentation and showed negative DIF and IIF results. These findings suggest that IgG reactivity against BP autoantibodies as determined by ELISA is not uncommon in pruritic diseases of the elderly.
Explanations for false-positive ELISA results were rare. A few authors suggested that false-positives could be attributed to an excessively low cutoff value,7-9 which was consistent with reports that the titer of autoantibodies to BP180 correlates with disease severity, suggesting that the higher titer of antibodies correlates with more severe disease and likely more accurate diagnosis.10,19,20 It is important to consider that patients who have low titers of BP180 autoantibodies with inconsistent clinical characteristics and DIF results may not truly have BP. Furthermore, to determine the clinical value of ELISA in identifying patients in the initial phase of BP, sera of BP patients should be compared with sera of elderly patients with pruritic skin disorders because they comprise the patient population that often requires diagnosis.18
Given the issues identified in our review of the literature, the published sensitivity and specificity of ELISA for BP diagnosis are likely overstated. In conclusion, ELISA should not be relied on as a single criterion adequate for diagnosis of BP.12,21 Rather, the diagnosis of BP can be obtained with a positive predictive value of 95% when a patient meets 3 of 4 clinical criteria (ie, absence of atrophic scars, absence of head and neck involvement, absence of mucosal involvement, and older than 70 years) and demonstrates linear deposits of predominantly IgG and/or C3 along the basement membrane zone of a perilesional biopsy on DIF.15 The gold standard for diagnosis of BP remains clinical presentation along with DIF, which can be supported by histology, IIF, and ELISA.22
- Delaporte E, Dubost-Brama A, Ghohestani R, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol. 1996;157:3642-3647.
- Schmidt E, Zillikens D. Diagnosis and clinical severity markers of bullous pemphigoid. F1000 Med Rep. 2009;1:15.
- Tampoia M, Giavarina D, Di Giorgio C, et al. Diagnostic accuracy of enzyme-linked immunosorbent assays (ELISA) to detect anti-skin autoantibodies in autoimmune blistering diseases: a systematic review and meta-analysis. Autoimmun Rev. 2012;12:121-126.
- Zillikens D, Mascaro JM, Rose PA, et al. A highly sensitive enzyme-linked immunosorbent assay for the detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J Invest Dermatol. 1997;109:679-683.
- Sitaru C, Dahnrich C, Probst C, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol. 2007;16:770-777.
- Yang B, Wang C, Chen S, et al. Evaluation of the combination of BP180-NC16a enzyme-linked immunosorbent assay and BP230 enzyme-linked immunosorbent assay in the diagnosis of bullous pemphigoid. Indian J Dermatol Venereol Leprol. 2012;78:722-727.
- Sakuma-Oyama Y, Powell AM, Oyama N, et al. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. Br J Dermatol. 2004;151:126-131.
- Tampoia M, Lattanzi V, Zucano A, et al. Evaluation of a new ELISA assay for detection of BP230 autoantibodies in bullous pemphigoid. Ann N Y Acad Sci. 2009;1173:15-20.
- Feng S, Lin L, Jin P, et al. Role of BP180NC16a-enzyme-linked immunosorbent assay (ELISA) in the diagnosis of bullous pemphigoid in China. Int J Dermatol. 2008;47:24-28.
- Kobayashi M, Amagai M, Kuroda-Kinoshita K, et al. BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci. 2002;30:224-232.
- Roussel A, Benichou J, Arivelo Randriamanantany Z, et al. Enzyme-linked immunosorbent assay for the combination of bullous pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:293-298.
- Chan, Lawrence S. ELISA instead of indirect IF in patients with BP. Arch Dermatol. 2011;147:291-292.
- Barnadas MA, Rubiales V, González J, et al. Enzyme-linked immunosorbent assay (ELISA) and indirect immunofluorescence testing in a bullous pemphigoid and pemphigoid gestationis. Int J Dermatol. 2008;47:1245-1249.
- Borradori L, Bernard P. Pemphigoid group. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2003:469.
- Vaillant L, Bernard P, Joly P, et al. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. Arch Dermatol. 1998;134:1075-1080.
- Fania L, Caldarola G, Muller R, et al. IgE recognition of bullous pemphigoid (BP)180 and BP230 in BP patients and elderly individuals with pruritic dermatoses. Clin Immunol. 2012;143:236-245.
- Feliciani C, Caldarola G, Kneisel A, et al. IgG autoantibody reactivity against bullous pemphigoid (BP) 180 and BP230 in elderly patients with pruritic dermatoses. Br J Dermatol. 2009;61:306-312.
- Hofmann SC, Tamm K, Hertl M, et al. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br J Dermatol. 2003;149:910-911.
- Schmidt E, Obe K, Brocker EB, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol. 2000;136:174-178.
- Feng S, Wu Q, Jin P, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Int J Dermatol. 2008;47:225-228.
- Di Zenzo G, Joly P, Zambruno G, et al. Sensitivity of immunofluorescence studies vs enzyme-linked immunosorbent assay for diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:1454-1456.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev. 2010;10:84-89.
- Delaporte E, Dubost-Brama A, Ghohestani R, et al. IgE autoantibodies directed against the major bullous pemphigoid antigen in patients with a severe form of pemphigoid. J Immunol. 1996;157:3642-3647.
- Schmidt E, Zillikens D. Diagnosis and clinical severity markers of bullous pemphigoid. F1000 Med Rep. 2009;1:15.
- Tampoia M, Giavarina D, Di Giorgio C, et al. Diagnostic accuracy of enzyme-linked immunosorbent assays (ELISA) to detect anti-skin autoantibodies in autoimmune blistering diseases: a systematic review and meta-analysis. Autoimmun Rev. 2012;12:121-126.
- Zillikens D, Mascaro JM, Rose PA, et al. A highly sensitive enzyme-linked immunosorbent assay for the detection of circulating anti-BP180 autoantibodies in patients with bullous pemphigoid. J Invest Dermatol. 1997;109:679-683.
- Sitaru C, Dahnrich C, Probst C, et al. Enzyme-linked immunosorbent assay using multimers of the 16th non-collagenous domain of the BP180 antigen for sensitive and specific detection of pemphigoid autoantibodies. Exp Dermatol. 2007;16:770-777.
- Yang B, Wang C, Chen S, et al. Evaluation of the combination of BP180-NC16a enzyme-linked immunosorbent assay and BP230 enzyme-linked immunosorbent assay in the diagnosis of bullous pemphigoid. Indian J Dermatol Venereol Leprol. 2012;78:722-727.
- Sakuma-Oyama Y, Powell AM, Oyama N, et al. Evaluation of a BP180-NC16a enzyme-linked immunosorbent assay in the initial diagnosis of bullous pemphigoid. Br J Dermatol. 2004;151:126-131.
- Tampoia M, Lattanzi V, Zucano A, et al. Evaluation of a new ELISA assay for detection of BP230 autoantibodies in bullous pemphigoid. Ann N Y Acad Sci. 2009;1173:15-20.
- Feng S, Lin L, Jin P, et al. Role of BP180NC16a-enzyme-linked immunosorbent assay (ELISA) in the diagnosis of bullous pemphigoid in China. Int J Dermatol. 2008;47:24-28.
- Kobayashi M, Amagai M, Kuroda-Kinoshita K, et al. BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci. 2002;30:224-232.
- Roussel A, Benichou J, Arivelo Randriamanantany Z, et al. Enzyme-linked immunosorbent assay for the combination of bullous pemphigoid antigens 1 and 2 in the diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:293-298.
- Chan, Lawrence S. ELISA instead of indirect IF in patients with BP. Arch Dermatol. 2011;147:291-292.
- Barnadas MA, Rubiales V, González J, et al. Enzyme-linked immunosorbent assay (ELISA) and indirect immunofluorescence testing in a bullous pemphigoid and pemphigoid gestationis. Int J Dermatol. 2008;47:1245-1249.
- Borradori L, Bernard P. Pemphigoid group. In: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatology. New York, NY: Mosby; 2003:469.
- Vaillant L, Bernard P, Joly P, et al. Evaluation of clinical criteria for diagnosis of bullous pemphigoid. Arch Dermatol. 1998;134:1075-1080.
- Fania L, Caldarola G, Muller R, et al. IgE recognition of bullous pemphigoid (BP)180 and BP230 in BP patients and elderly individuals with pruritic dermatoses. Clin Immunol. 2012;143:236-245.
- Feliciani C, Caldarola G, Kneisel A, et al. IgG autoantibody reactivity against bullous pemphigoid (BP) 180 and BP230 in elderly patients with pruritic dermatoses. Br J Dermatol. 2009;61:306-312.
- Hofmann SC, Tamm K, Hertl M, et al. Diagnostic value of an enzyme-linked immunosorbent assay using BP180 recombinant proteins in elderly patients with pruritic skin disorders. Br J Dermatol. 2003;149:910-911.
- Schmidt E, Obe K, Brocker EB, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Arch Dermatol. 2000;136:174-178.
- Feng S, Wu Q, Jin P, et al. Serum levels of autoantibodies to BP180 correlate with disease activity in patients with bullous pemphigoid. Int J Dermatol. 2008;47:225-228.
- Di Zenzo G, Joly P, Zambruno G, et al. Sensitivity of immunofluorescence studies vs enzyme-linked immunosorbent assay for diagnosis of bullous pemphigoid. Arch Dermatol. 2011;147:1454-1456.
- Schmidt E, Zillikens D. Modern diagnosis of autoimmune blistering skin diseases. Autoimmun Rev. 2010;10:84-89.
Practice Points
- A low serum level of autoantibodies to BP180 should be interpreted with caution because it is more likely to represent a false-positive than a high serum level.
- Rely on the gold standard for diagnosis of bullous pemphigoid: clinical presentation along with direct immunofluorescence, which can be supported by histology, indirect immunofluorescence, and enzyme-linked immunosorbent assay (ELISA) rather than ELISA alone.
Cosmetic Corner: Dermatologists Weigh in on Skin-Lightening Agents
To improve patient care and outcomes, leading dermatologists offered their recommendations on skin-lightening agents. Consideration must be given to:
- Even Better Clinical Dark Spot Corrector
Clinique Laboratories, LLC
Recommended by Gary Goldenberg, MD, New York, New York
- Lytera Skin Brightening Complex
SkinMedica, an Allergan compan
“It contains vitamin C, niacinamide, retinol, and licorice root extract to help lighten the skin and improve texture without hydroquinone.”—Anthony M. Rossi, MD, New York, New York
- Meladerm
Civant Skin Care
“This is an excellent hydroquinone-free cream for treating postinflammatory hyperpigmentation and melasma. It contains a combination of ingredients known to inhibit various steps along the melanogenesis pathway, such as retinyl palmitate, licorice extract, and arbutin, as well as lactic, kojic, and ascorbic acids.”—Cherise M. Levi, DO, New York, New York
Cutis invites readers to send us their recommendations. Cleansing devices, dry shampoos, athlete’s foot treatments, and face scrubs will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
[polldaddy:9711250]
To improve patient care and outcomes, leading dermatologists offered their recommendations on skin-lightening agents. Consideration must be given to:
- Even Better Clinical Dark Spot Corrector
Clinique Laboratories, LLC
Recommended by Gary Goldenberg, MD, New York, New York
- Lytera Skin Brightening Complex
SkinMedica, an Allergan compan
“It contains vitamin C, niacinamide, retinol, and licorice root extract to help lighten the skin and improve texture without hydroquinone.”—Anthony M. Rossi, MD, New York, New York
- Meladerm
Civant Skin Care
“This is an excellent hydroquinone-free cream for treating postinflammatory hyperpigmentation and melasma. It contains a combination of ingredients known to inhibit various steps along the melanogenesis pathway, such as retinyl palmitate, licorice extract, and arbutin, as well as lactic, kojic, and ascorbic acids.”—Cherise M. Levi, DO, New York, New York
Cutis invites readers to send us their recommendations. Cleansing devices, dry shampoos, athlete’s foot treatments, and face scrubs will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
[polldaddy:9711250]
To improve patient care and outcomes, leading dermatologists offered their recommendations on skin-lightening agents. Consideration must be given to:
- Even Better Clinical Dark Spot Corrector
Clinique Laboratories, LLC
Recommended by Gary Goldenberg, MD, New York, New York
- Lytera Skin Brightening Complex
SkinMedica, an Allergan compan
“It contains vitamin C, niacinamide, retinol, and licorice root extract to help lighten the skin and improve texture without hydroquinone.”—Anthony M. Rossi, MD, New York, New York
- Meladerm
Civant Skin Care
“This is an excellent hydroquinone-free cream for treating postinflammatory hyperpigmentation and melasma. It contains a combination of ingredients known to inhibit various steps along the melanogenesis pathway, such as retinyl palmitate, licorice extract, and arbutin, as well as lactic, kojic, and ascorbic acids.”—Cherise M. Levi, DO, New York, New York
Cutis invites readers to send us their recommendations. Cleansing devices, dry shampoos, athlete’s foot treatments, and face scrubs will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
[polldaddy:9711250]

Debunking Psoriasis Myths: Do Systemic Steroids Used in Psoriasis Patients Cause Pustular Psoriasis?
Myth: Systemic steroids cause pustular psoriasis
The advent of biologic therapy for psoriasis has changed the landscape of treatments offered to patients. Nevertheless, systemic therapies still play an important role, according to the American Academy of Dermatology psoriasis treatment guidelines, due to their oral route of administration and low cost compared to biologics. They are options for patients with moderate to severe psoriasis that is unresponsive to topical therapies or phototherapy. However, many dermatologists feel that it is inappropriate to prescribe oral steroids to psoriasis patients due to the risk for steroid-induced conversion to pustular psoriasis, the long-term side effects of steroids, and deterioration of psoriasis after withdrawal of steroids.
Pustular psoriasis appears clinically as white pustules (blisters of noninfectious pus) surrounded by red skin. The pus consists of white blood cells. There are a number of triggers in addition to systemic steroids, such as internal medications, irritating topical agents, overexposure to UV light, and pregnancy. Stopping an oral steroid abruptly can cause serious disease flares, fatigue, and joint pain.
Westphal et al described the case of a 70-year-old woman with palmoplantar psoriasis who was diagnosed with acute generalized exanthematous pustulosis that was treated with corticotherapy by injection and then oral prednisone. She experienced improvement, but her symptoms worsened when she was in the process of reducing the prednisone dose. The dose was increased again, and the same worsening of symptoms was experienced when the dose was reduced. After completely abandoning oral steroid therapy, she developed a severe case of generalized pustular psoriasis that was treated with acitretin. This case illustrates the dangerous consequences of abruptly discontinuing oral steroids.
However, dermatologists may be using oral steroids for psoriasis more often than treatment guidelines suggest. In 2014, Al-Dabagh et al evaluated how frequently systemic corticosteroids are prescribed for psoriasis in the United States. The researchers reported, "Despite the absence or discouragement of systemic corticosteroids in psoriasis management guidelines, systemic corticosteroids are among the most common systemic treatments used for psoriasis." They found that systemic corticosteroids were prescribed at 650,000 of 21,020,000 psoriasis visits, of which 93% were visits to dermatologists. Prednisone was the most commonly prescribed systemic corticosteroid, followed by methylprednisolone and dexamethasone. To prevent rebound flares, systemic corticosteroids were prescribed with a topical corticosteroid in 45% of the visits in patients with psoriasis as the sole diagnosis. They concluded, "The striking contrast between the guidelines for psoriasis management and actual practice suggests that there is an acute need to better understand the use of systemic corticosteroids for psoriasis."
The benefits of systemic corticosteroids versus the frequency of adverse reactions should be weighed by dermatologists and patients to make evidence-based decisions about treatment. Patients should take oral steroids exactly as prescribed by physicians.
References
Al-Dabagh A, Al-Dabagh R, Davis SA, et al. Systemic corticosteroids are frequently prescribed for psoriasis. J Cutan Med Surg. 2014;18:195-199.
Delzell E. What you need to know about steroids. National Psoriasis Foundation website. https://www.psoriasis.org/advance/what-you-need-to-know-about-steroids. Published September 2, 2015. Accessed January 13, 2017.
Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
Pustular psoriasis. National Psoriasis Foundation website. https://www.psoriasis.org/about-psoriasis/types/pustular. Accessed January 13, 2017.
Westphal DC, Schettini APM, de Souza PP, et al. Generalized pustular psoriasis induced by systemic steroid dose reduction. An Bras Dermatol. 2016;91:664-666.
Expert Commentaries on next page
Expert Commentaries
When I was a resident, I was trained not to use systemic steroids in psoriasis patients for the reasons noted above, and I have faithfully followed these instructions 9 years into practice. However, I see many patients with severe psoriasis who are given systemic steroids by other physicians (ie, rheumatologists for psoriatic arthritis, pulmonologists for asthma). I often tell patients afterwards of the dangers of systemic steroids and to have them tell their other doctors to be cautious when giving another course of systemic steroids. However, I have yet to see a generalized pustular psoriasis outbreak or flare in psoriasis vulgaris after a course of systemic steroids. While I do not recommend systemic steroids for psoriasis patients since we have so many other systemic agents, I wonder if the risks that we were all trained about are really that high.
—Jashin J. Wu, MD (Los Angeles, California)
How bad is it to give patients with psoriasis systemic steroids? Are psoriasis patients treated with systemic steroids likely to get a pustular flare? Are patients with psoriasis who suddenly stop their corticosteroids more likely to get a pustular flare than psoriasis patients who suddenly stop other systemic psoriasis treatments? I don't have the answers to these questions. My sense is that we have a lot of dogma and strong opinions but very little hard evidence to answer these questions.
I don't typically prescribe systemic steroids to psoriasis patients, but systemic steroids are widely used. Sometimes there are problems. I have seen patients who received systemic steroids for psoriasis and who went on to have a pustular flare, but it's possible the systemic steroid was given because those patients were headed toward the pustular flare already.
I once had a psoriasis patient who came to see me with a suddenly inflamed tender joint. Not knowing what to do, I called a rheumatologist to see the patient. The rheumatologist, too busy to work the patient in, told me to give the patient a 2-week prednisone taper. I did, and nothing untoward happened with the psoriasis. This one anecdote doesn't give me much confidence that systemic steroids are safe for psoriasis patients.
Clearly, long-term steroids cause a host of problems (eg, osteoporosis, diabetes). But I'm not sure that the dogma that systemic steroids should be avoided in patients with psoriasis is well supported. Systemic steroids are being widely used, and I don't see an epidemic of pustular flares.
Is it a mistake to give systemic steroids to psoriasis patients? I just don't know.
—Steven R. Feldman, MD, PhD (Winston-Salem, North Carolina)
Myth: Systemic steroids cause pustular psoriasis
The advent of biologic therapy for psoriasis has changed the landscape of treatments offered to patients. Nevertheless, systemic therapies still play an important role, according to the American Academy of Dermatology psoriasis treatment guidelines, due to their oral route of administration and low cost compared to biologics. They are options for patients with moderate to severe psoriasis that is unresponsive to topical therapies or phototherapy. However, many dermatologists feel that it is inappropriate to prescribe oral steroids to psoriasis patients due to the risk for steroid-induced conversion to pustular psoriasis, the long-term side effects of steroids, and deterioration of psoriasis after withdrawal of steroids.
Pustular psoriasis appears clinically as white pustules (blisters of noninfectious pus) surrounded by red skin. The pus consists of white blood cells. There are a number of triggers in addition to systemic steroids, such as internal medications, irritating topical agents, overexposure to UV light, and pregnancy. Stopping an oral steroid abruptly can cause serious disease flares, fatigue, and joint pain.
Westphal et al described the case of a 70-year-old woman with palmoplantar psoriasis who was diagnosed with acute generalized exanthematous pustulosis that was treated with corticotherapy by injection and then oral prednisone. She experienced improvement, but her symptoms worsened when she was in the process of reducing the prednisone dose. The dose was increased again, and the same worsening of symptoms was experienced when the dose was reduced. After completely abandoning oral steroid therapy, she developed a severe case of generalized pustular psoriasis that was treated with acitretin. This case illustrates the dangerous consequences of abruptly discontinuing oral steroids.
However, dermatologists may be using oral steroids for psoriasis more often than treatment guidelines suggest. In 2014, Al-Dabagh et al evaluated how frequently systemic corticosteroids are prescribed for psoriasis in the United States. The researchers reported, "Despite the absence or discouragement of systemic corticosteroids in psoriasis management guidelines, systemic corticosteroids are among the most common systemic treatments used for psoriasis." They found that systemic corticosteroids were prescribed at 650,000 of 21,020,000 psoriasis visits, of which 93% were visits to dermatologists. Prednisone was the most commonly prescribed systemic corticosteroid, followed by methylprednisolone and dexamethasone. To prevent rebound flares, systemic corticosteroids were prescribed with a topical corticosteroid in 45% of the visits in patients with psoriasis as the sole diagnosis. They concluded, "The striking contrast between the guidelines for psoriasis management and actual practice suggests that there is an acute need to better understand the use of systemic corticosteroids for psoriasis."
The benefits of systemic corticosteroids versus the frequency of adverse reactions should be weighed by dermatologists and patients to make evidence-based decisions about treatment. Patients should take oral steroids exactly as prescribed by physicians.
References
Al-Dabagh A, Al-Dabagh R, Davis SA, et al. Systemic corticosteroids are frequently prescribed for psoriasis. J Cutan Med Surg. 2014;18:195-199.
Delzell E. What you need to know about steroids. National Psoriasis Foundation website. https://www.psoriasis.org/advance/what-you-need-to-know-about-steroids. Published September 2, 2015. Accessed January 13, 2017.
Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
Pustular psoriasis. National Psoriasis Foundation website. https://www.psoriasis.org/about-psoriasis/types/pustular. Accessed January 13, 2017.
Westphal DC, Schettini APM, de Souza PP, et al. Generalized pustular psoriasis induced by systemic steroid dose reduction. An Bras Dermatol. 2016;91:664-666.
Expert Commentaries on next page
Expert Commentaries
When I was a resident, I was trained not to use systemic steroids in psoriasis patients for the reasons noted above, and I have faithfully followed these instructions 9 years into practice. However, I see many patients with severe psoriasis who are given systemic steroids by other physicians (ie, rheumatologists for psoriatic arthritis, pulmonologists for asthma). I often tell patients afterwards of the dangers of systemic steroids and to have them tell their other doctors to be cautious when giving another course of systemic steroids. However, I have yet to see a generalized pustular psoriasis outbreak or flare in psoriasis vulgaris after a course of systemic steroids. While I do not recommend systemic steroids for psoriasis patients since we have so many other systemic agents, I wonder if the risks that we were all trained about are really that high.
—Jashin J. Wu, MD (Los Angeles, California)
How bad is it to give patients with psoriasis systemic steroids? Are psoriasis patients treated with systemic steroids likely to get a pustular flare? Are patients with psoriasis who suddenly stop their corticosteroids more likely to get a pustular flare than psoriasis patients who suddenly stop other systemic psoriasis treatments? I don't have the answers to these questions. My sense is that we have a lot of dogma and strong opinions but very little hard evidence to answer these questions.
I don't typically prescribe systemic steroids to psoriasis patients, but systemic steroids are widely used. Sometimes there are problems. I have seen patients who received systemic steroids for psoriasis and who went on to have a pustular flare, but it's possible the systemic steroid was given because those patients were headed toward the pustular flare already.
I once had a psoriasis patient who came to see me with a suddenly inflamed tender joint. Not knowing what to do, I called a rheumatologist to see the patient. The rheumatologist, too busy to work the patient in, told me to give the patient a 2-week prednisone taper. I did, and nothing untoward happened with the psoriasis. This one anecdote doesn't give me much confidence that systemic steroids are safe for psoriasis patients.
Clearly, long-term steroids cause a host of problems (eg, osteoporosis, diabetes). But I'm not sure that the dogma that systemic steroids should be avoided in patients with psoriasis is well supported. Systemic steroids are being widely used, and I don't see an epidemic of pustular flares.
Is it a mistake to give systemic steroids to psoriasis patients? I just don't know.
—Steven R. Feldman, MD, PhD (Winston-Salem, North Carolina)
Myth: Systemic steroids cause pustular psoriasis
The advent of biologic therapy for psoriasis has changed the landscape of treatments offered to patients. Nevertheless, systemic therapies still play an important role, according to the American Academy of Dermatology psoriasis treatment guidelines, due to their oral route of administration and low cost compared to biologics. They are options for patients with moderate to severe psoriasis that is unresponsive to topical therapies or phototherapy. However, many dermatologists feel that it is inappropriate to prescribe oral steroids to psoriasis patients due to the risk for steroid-induced conversion to pustular psoriasis, the long-term side effects of steroids, and deterioration of psoriasis after withdrawal of steroids.
Pustular psoriasis appears clinically as white pustules (blisters of noninfectious pus) surrounded by red skin. The pus consists of white blood cells. There are a number of triggers in addition to systemic steroids, such as internal medications, irritating topical agents, overexposure to UV light, and pregnancy. Stopping an oral steroid abruptly can cause serious disease flares, fatigue, and joint pain.
Westphal et al described the case of a 70-year-old woman with palmoplantar psoriasis who was diagnosed with acute generalized exanthematous pustulosis that was treated with corticotherapy by injection and then oral prednisone. She experienced improvement, but her symptoms worsened when she was in the process of reducing the prednisone dose. The dose was increased again, and the same worsening of symptoms was experienced when the dose was reduced. After completely abandoning oral steroid therapy, she developed a severe case of generalized pustular psoriasis that was treated with acitretin. This case illustrates the dangerous consequences of abruptly discontinuing oral steroids.
However, dermatologists may be using oral steroids for psoriasis more often than treatment guidelines suggest. In 2014, Al-Dabagh et al evaluated how frequently systemic corticosteroids are prescribed for psoriasis in the United States. The researchers reported, "Despite the absence or discouragement of systemic corticosteroids in psoriasis management guidelines, systemic corticosteroids are among the most common systemic treatments used for psoriasis." They found that systemic corticosteroids were prescribed at 650,000 of 21,020,000 psoriasis visits, of which 93% were visits to dermatologists. Prednisone was the most commonly prescribed systemic corticosteroid, followed by methylprednisolone and dexamethasone. To prevent rebound flares, systemic corticosteroids were prescribed with a topical corticosteroid in 45% of the visits in patients with psoriasis as the sole diagnosis. They concluded, "The striking contrast between the guidelines for psoriasis management and actual practice suggests that there is an acute need to better understand the use of systemic corticosteroids for psoriasis."
The benefits of systemic corticosteroids versus the frequency of adverse reactions should be weighed by dermatologists and patients to make evidence-based decisions about treatment. Patients should take oral steroids exactly as prescribed by physicians.
References
Al-Dabagh A, Al-Dabagh R, Davis SA, et al. Systemic corticosteroids are frequently prescribed for psoriasis. J Cutan Med Surg. 2014;18:195-199.
Delzell E. What you need to know about steroids. National Psoriasis Foundation website. https://www.psoriasis.org/advance/what-you-need-to-know-about-steroids. Published September 2, 2015. Accessed January 13, 2017.
Menter A, Korman NJ, Elmets CA, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: section 4. guidelines of care for the management and treatment of psoriasis with traditional systemic agents. J Am Acad Dermatol. 2009;61:451-485.
Pustular psoriasis. National Psoriasis Foundation website. https://www.psoriasis.org/about-psoriasis/types/pustular. Accessed January 13, 2017.
Westphal DC, Schettini APM, de Souza PP, et al. Generalized pustular psoriasis induced by systemic steroid dose reduction. An Bras Dermatol. 2016;91:664-666.
Expert Commentaries on next page
Expert Commentaries
When I was a resident, I was trained not to use systemic steroids in psoriasis patients for the reasons noted above, and I have faithfully followed these instructions 9 years into practice. However, I see many patients with severe psoriasis who are given systemic steroids by other physicians (ie, rheumatologists for psoriatic arthritis, pulmonologists for asthma). I often tell patients afterwards of the dangers of systemic steroids and to have them tell their other doctors to be cautious when giving another course of systemic steroids. However, I have yet to see a generalized pustular psoriasis outbreak or flare in psoriasis vulgaris after a course of systemic steroids. While I do not recommend systemic steroids for psoriasis patients since we have so many other systemic agents, I wonder if the risks that we were all trained about are really that high.
—Jashin J. Wu, MD (Los Angeles, California)
How bad is it to give patients with psoriasis systemic steroids? Are psoriasis patients treated with systemic steroids likely to get a pustular flare? Are patients with psoriasis who suddenly stop their corticosteroids more likely to get a pustular flare than psoriasis patients who suddenly stop other systemic psoriasis treatments? I don't have the answers to these questions. My sense is that we have a lot of dogma and strong opinions but very little hard evidence to answer these questions.
I don't typically prescribe systemic steroids to psoriasis patients, but systemic steroids are widely used. Sometimes there are problems. I have seen patients who received systemic steroids for psoriasis and who went on to have a pustular flare, but it's possible the systemic steroid was given because those patients were headed toward the pustular flare already.
I once had a psoriasis patient who came to see me with a suddenly inflamed tender joint. Not knowing what to do, I called a rheumatologist to see the patient. The rheumatologist, too busy to work the patient in, told me to give the patient a 2-week prednisone taper. I did, and nothing untoward happened with the psoriasis. This one anecdote doesn't give me much confidence that systemic steroids are safe for psoriasis patients.
Clearly, long-term steroids cause a host of problems (eg, osteoporosis, diabetes). But I'm not sure that the dogma that systemic steroids should be avoided in patients with psoriasis is well supported. Systemic steroids are being widely used, and I don't see an epidemic of pustular flares.
Is it a mistake to give systemic steroids to psoriasis patients? I just don't know.
—Steven R. Feldman, MD, PhD (Winston-Salem, North Carolina)

Telmisartan-Induced Lichen Planus Eruption Manifested on Vitiliginous Skin
To the Editor:
A 39-year-old man with a history of hypertension and vitiligo presented with a rapid-onset, generalized, pruritic rash covering the body of 4 weeks’ duration. He reported that the rash progressively worsened after developing mild sunburn. The patient stated that the rash was extremely pruritic with a burning sensation and was tender to touch. He was treated with betamethasone valerate cream 0.1% by an outside physician and an over-the-counter anti-itch lotion with no notable improvement. His only medication was telmisartan-hydrochlorothiazide (HCTZ) for hypertension. He denied any drug allergies.
Physical examination revealed multiple discrete and coalescent planar erythematous papules and plaques involving only the depigmented vitiliginous skin of the forehead, eyelids, and nape of the neck (Figure 1A), and confluent on the lateral aspect of the bilateral forearms (Figure 1B), dorsal aspect of the right hand, and bilateral dorsi of the feet. Wickham striae were noted on the lips (Figure 1C). A clinical diagnosis of lichen planus (LP) was made. The patient initially was prescribed halobetasol propionate ointment 0.05% twice daily. He reported notable relief of pruritus with reduction of overall symptoms and new lesion formation.
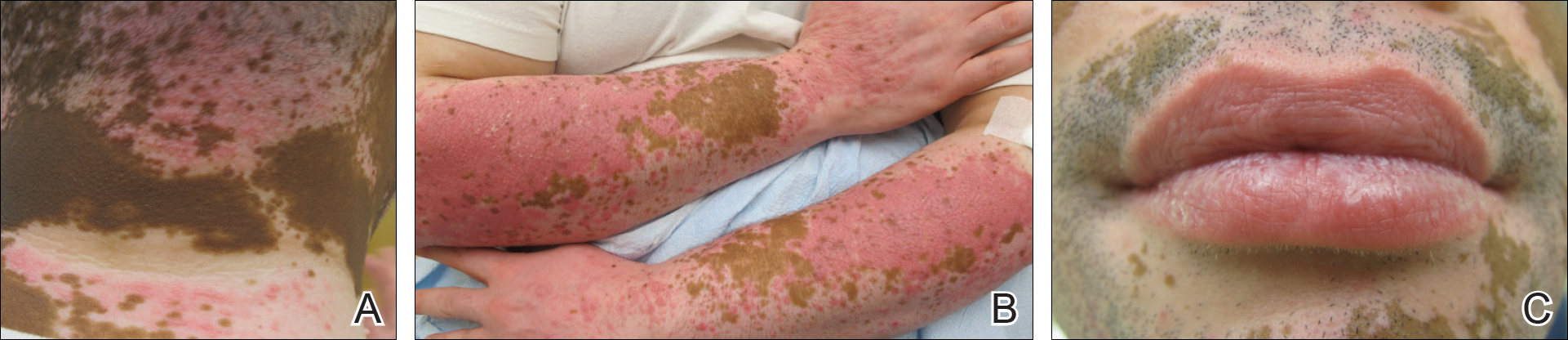
A 4-mm punch biopsy was performed on the left forearm. Histopathology revealed LP. Microscopic examination of the hematoxylin and eosin–stained specimen revealed a bandlike lymphohistiocytic infiltrate that extended across the papillary dermis, focally obscuring the dermoepidermal junction where there were vacuolar changes and colloid bodies. The epidermis showed sawtooth rete ridges, wedge-shaped foci of hypergranulosis, and compact hyperkeratosis (Figure 2).
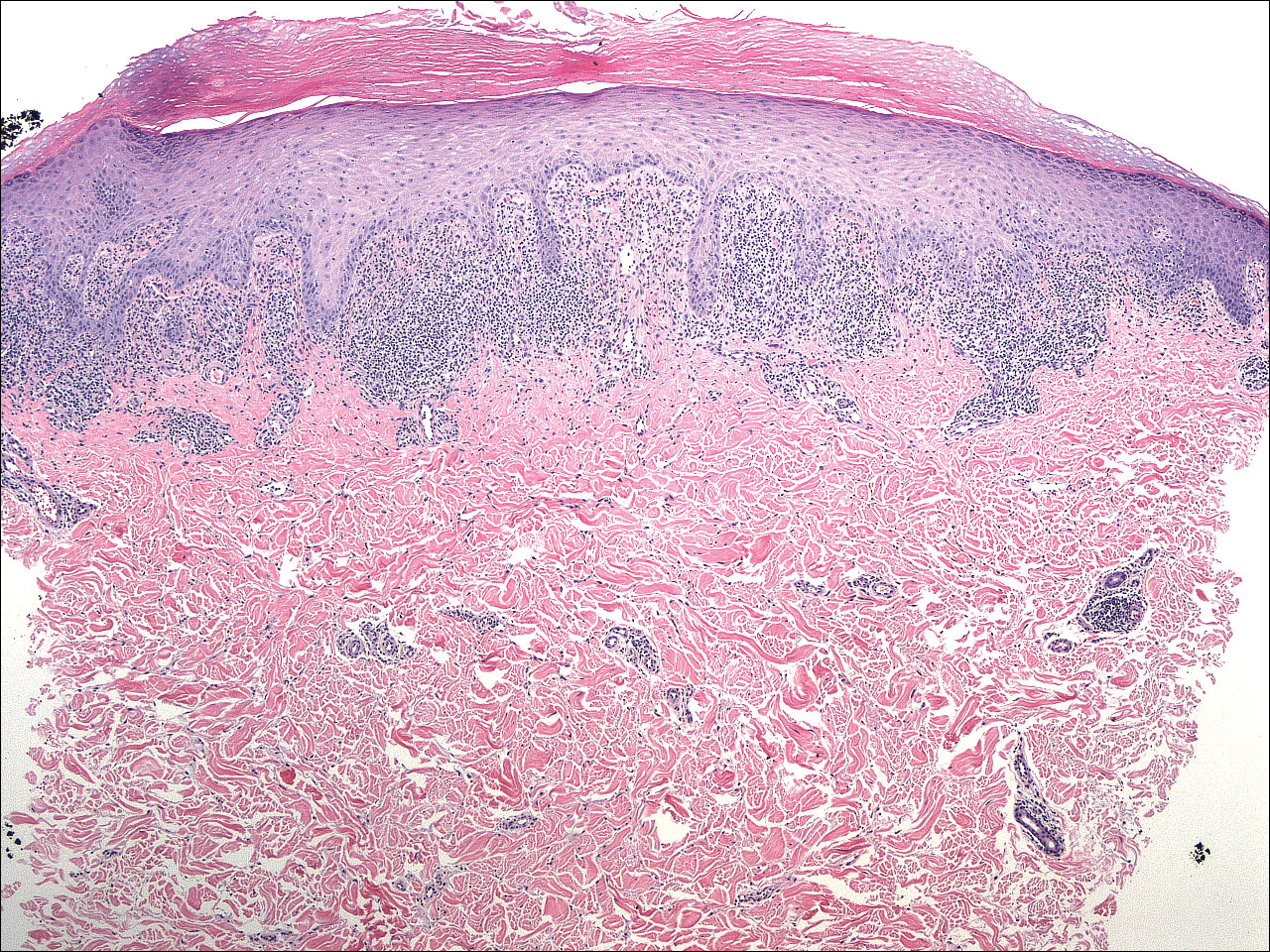
On further questioning during follow-up, the patient revealed that his hypertensive medication was changed from HCTZ, which he had been taking for the last 8 years, to the combination antihypertensive medication telmisartan-HCTZ before the onset of the skin eruption. Due to the temporal relationship between the new medication and onset of the eruption, the clinical impression was highly suspicious for drug-induced eruptive LP with Köbner phenomenon caused by the recent sunburn. Systemic workup for underlying causes of LP was negative. Laboratory tests revealed normal complete blood cell counts. The hepatitis panel included hepatitis A antibodies; hepatitis B surface, e antigen, and core antibodies; hepatitis B surface antigen and e antibodies; hepatitis C antibodies; and antinuclear antibodies, which were all negative.
The patient continued to develop new pruritic papules clinically consistent with LP. He was instructed to return to his primary care physician to change the telmisartan-HCTZ to a different class of antihypertensive medication. His medication was changed to atenolol. The patient also was instructed to continue the halobetasol propionate ointment 0.05% twice daily to the affected areas.
The patient returned for a follow-up visit 1 month later and reported notable improvement in pruritus and near-complete resolution of the LP after discontinuation of telmisartan-HCTZ. He also noted some degree of perifollicular repigmentation of the vitiliginous skin that had been unresponsive to prior therapy (Figure 3).
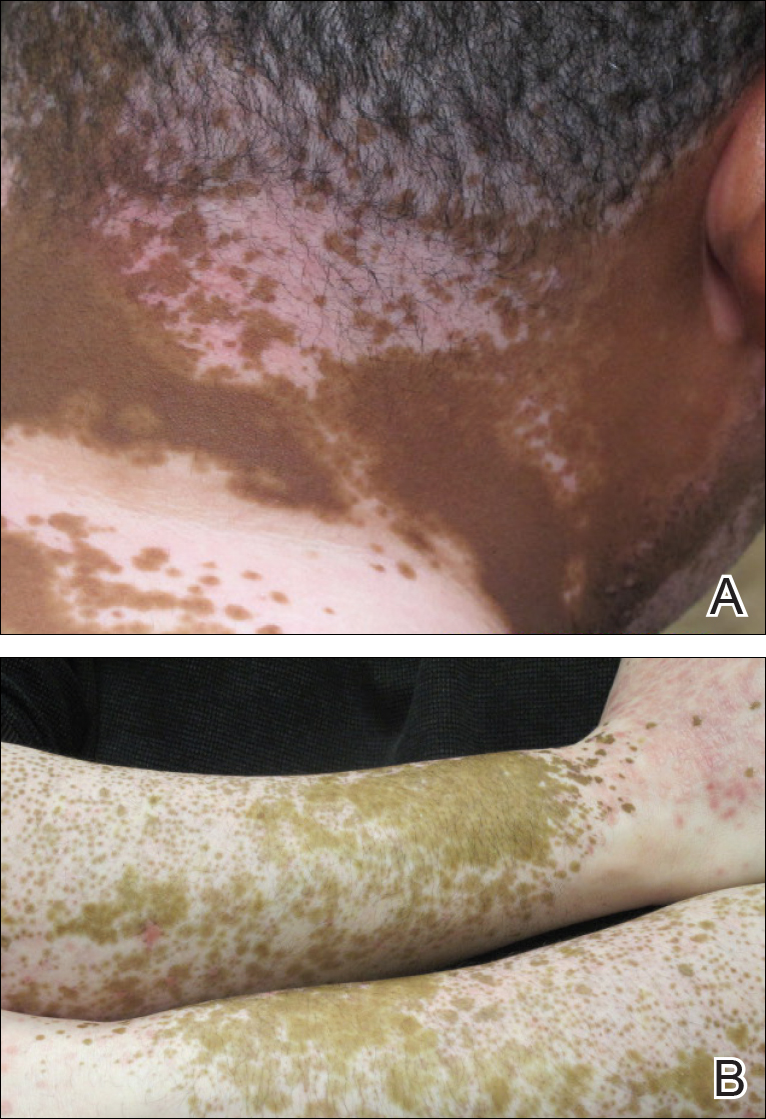
Lichen planus is a pruritic and inflammatory papulosquamous skin condition that presents as scaly, flat-topped, violaceous, polygonal-shaped papules commonly involving the flexor surface of the arms and legs, oral mucosa, scalp, nails, and genitalia. Clinically, LP can present in various forms including actinic, annular, atrophic, erosive, follicular, hypertrophic, linear, pigmented, and vesicular/bullous types. Koebnerization is common, especially in the linear form of LP. There are no specific laboratory findings or serologic markers seen in LP.
The exact cause of LP remains unknown. Clinical observations and anecdotal evidence have directed the cell-mediated immune response to insulting agents such as medications or contact allergy to metals triggering an abnormal cellular immune response. Various viral agents have been reported including hepatitis C virus, human herpesvirus, herpes simplex virus, and varicella-zoster virus.1-5 Other factors such as seasonal change and the environment may contribute to the development of LP and an increase in the incidence of LP eruption has been observed from January to July throughout the United States.6 Lichen planus also has been associated with other altered immune-related disease such as ulcerative colitis, alopecia areata, vitiligo, dermatomyositis, morphea, lichen sclerosis, and myasthenia gravis.7 Increased levels of emotional stress, particularly related to family members, often is related to the onset or aggravation of symptoms.8,9
Many drug-related LP-like and lichenoid eruptions have been reported with antihypertensive drugs, antimalarial drugs, diuretics, antidepressants, nonsteroidal anti-inflammatory drugs, antimicrobial drugs, and metals. In particular, medications such as captopril, enalapril, labetalol, propranolol, chlorothiazide, HCTZ, methyldopa, chloroquine, hydroxychloroquine, quinacrine, gold salts, penicillamine, and quinidine commonly are reported to induce lichenoid drug eruption.10
Several inflammatory papulosquamous skin conditions should be considered in the differential diagnosis before confirming the diagnosis of LP. It is important to rule out lupus erythematosus, especially if the oral mucosa and scalp are involved. In addition, erosive paraneoplastic pemphigus involving primarily the oral mucosa can resemble oral LP. Nail diseases such as psoriasis, onychomycosis, and alopecia areata should be considered as the differential diagnosis of nail disease. Genital involvement also can be seen in psoriasis and lichen sclerosus.
Treatment of LP is mainly symptomatic because of the benign nature of the disease and the high spontaneous remission rate with varying amount of time. If drugs, dental/metal implants, or underlying viral infections are the identifiable triggering factors of LP, the offending agents should be discontinued or removed. Additionally, topical or systemic treatments can be given depending on the severity of the disease, focusing mainly on symptomatic relief as well as the balance of risks and benefits associated with treatment.
Treatment options include topical and intralesional corticosteroids. Systemic medications such as oral corticosteroids and/or acitretin commonly are used in acute, severe, and disseminated cases, though treatment duration varies depending on the clinical response. Other systemic agents used to treat LP include griseofulvin, metronidazole, sulfasalazine, cyclosporine, and mycophenolate mofetil.
Phototherapy is considered an alternative therapy, especially for recalcitrant LP. UVA1 and narrowband UVB (wavelength, 311 nm) have been reported to effectively treat long-standing and therapy-resistant LP.11 In addition, a small study used the excimer laser (wavelength, 308 nm), which is well tolerated by patients, to treat focal recalcitrant oral lesions with excellent results.12 Photochemotherapy has been used with notable improvement, but the potential of carcinogenicity, especially in patients with Fitzpatrick skin types I and II, has limited its use.13
Our patient developed an unusual extensive LP eruption involving only vitiliginous skin shortly after initiation of the combined antihypertensive medication telmisartan-HCTZ, an angiotensin receptor blocker with a thiazide diuretic. Telmisartan and other angiotensin receptor blockers have not been reported to trigger LP; HCTZ is listed as one of the common drugs causing photosensitivity and LP.14,15 Although it is possible that our patient exhibited a delayed lichenoid drug eruption from the HCTZ, it is noteworthy that he did not experience a single episode of LP during his 8-year history of taking HCTZ. Instead, he developed the LP eruption shortly after the addition of telmisartan to his HCTZ antihypertensive regimen. The temporal relationship led us to direct the patient to the prescribing physician to discontinue telmisartan-HCTZ. After changing his antihypertensive medication to atenolol, the patient presented with improvement within the first month and near-complete resolution 2 months after the discontinuation of telmisartan-HCTZ.
Our patient’s LP lesions only manifested on the skin affected by vitiligo, sparing the normal-pigmented skin. Studies have demonstrated an increased ratio of CD8+ T cells to CD4+ T cells as well as increased intercellular adhesion molecule 1 at the dermal level.10,16 Both vitiligo and LP share some common histopathologic features including highly populated CD8+ T cells and intercellular adhesion molecule 1. In our case, LP was triggered on the vitiliginous skin by telmisartan. Vitiligo in combination with trauma induced by sunburn may represent the trigger that altered the cellular immune response and created the telmisartan-induced LP. As a result, the LP eruption was confined to the vitiliginous skin lesions.
Perifollicular repigmentation was observed in our patient after the LP lesions resolved; the patient’s vitiligo was unresponsive to prior treatment. The inflammatory process occurring in LP may exert and interfere in the underlying autoimmune cytotoxic effect toward the melanocytes and the melanin synthesis. It may be of interest to find out if the inflammatory response of LP has a positive influence on the effect of melanogenesis pathways or on the underlying autoimmune-related inflammatory process in vitiligo. Further studies are needed to investigate the role of immunotherapy targeting specific inflammatory pathways and the impact on the repigmentation in vitiligo.
Acknowledgment—Special thanks to Paul Chu, MD (Port Chester, New York).
- Pilli M, Zerbini A, Vescovi P, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446-1452.
- De Vries HJ, van Marle J, Teunissen MB, et al. Lichen planus is associated with human herpesvirus type 7 replication and infiltration of plasmacytoid dendritic cells. Br J Dermatol. 2006;154:361-364.
- De Vries HJ, Teunissen MB, Zorgdrager F, et al. Lichen planus remission is associated with a decrease of human herpes virus type 7 protein expression in plasmacytoid dendritic cells. Arch Dermatol Res. 2007;299:213-219.
- Requena L, Kutzner H, Escalonilla P, et al. Cutaneous reactions at sites of herpes zoster scars: an expanded spectrum. Br J Dermatol. 1998;138:161-168.
- Al-Khenaizan S. Lichen planus occurring after hepatitis B vaccination: a new case. J Am Acad Dermatol. 2001;45:614-615.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Sadr-Ashkevari S. Familial actinic lichen planus: case reports in two brothers. Arch Int Med. 2001;4:204-206.
- Manolache L, Seceleanu-Petrescu D, Benea V. Lichen planus patients and stressful events. J Eur Acad Dermatol Venereol. 2008;22:437-441.
- Mahood JM. Familial lichen planus. Arch Dermatol. 1983;119:292-294.
- Shimizu M, Higaki Y, Higaki M, et al. The role of granzyme B-expressing CD8-positive T cells in apoptosis of keratinocytes in lichen planus. Arch Dermatol Res. 1997;289:527-532.
- Bécherel PA, Bussel A, Chosidow O, et al. Extracorporeal photochemotherapy for chronic erosive lichen planus. Lancet. 1998;351:805.
- Trehan M, Taylar CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol. 2004;140:415-420.
- Wackernagel A, Legat FJ, Hofer A, et al. Psoralen plus UVA vs. UVB-311 nm for the treatment of lichen planus. Photodermatol Photoimmunol Photomed. 2007;23:15-19.
- Fellner MJ. Lichen planus. Int J Dermatol. 1980;19:71-75.
- Moore DE. Drug-induced cutaneous photosensitivity: incidence, mechanism, prevention and management. Drug Saf. 2002;25:345-372.
- Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
To the Editor:
A 39-year-old man with a history of hypertension and vitiligo presented with a rapid-onset, generalized, pruritic rash covering the body of 4 weeks’ duration. He reported that the rash progressively worsened after developing mild sunburn. The patient stated that the rash was extremely pruritic with a burning sensation and was tender to touch. He was treated with betamethasone valerate cream 0.1% by an outside physician and an over-the-counter anti-itch lotion with no notable improvement. His only medication was telmisartan-hydrochlorothiazide (HCTZ) for hypertension. He denied any drug allergies.
Physical examination revealed multiple discrete and coalescent planar erythematous papules and plaques involving only the depigmented vitiliginous skin of the forehead, eyelids, and nape of the neck (Figure 1A), and confluent on the lateral aspect of the bilateral forearms (Figure 1B), dorsal aspect of the right hand, and bilateral dorsi of the feet. Wickham striae were noted on the lips (Figure 1C). A clinical diagnosis of lichen planus (LP) was made. The patient initially was prescribed halobetasol propionate ointment 0.05% twice daily. He reported notable relief of pruritus with reduction of overall symptoms and new lesion formation.
A 4-mm punch biopsy was performed on the left forearm. Histopathology revealed LP. Microscopic examination of the hematoxylin and eosin–stained specimen revealed a bandlike lymphohistiocytic infiltrate that extended across the papillary dermis, focally obscuring the dermoepidermal junction where there were vacuolar changes and colloid bodies. The epidermis showed sawtooth rete ridges, wedge-shaped foci of hypergranulosis, and compact hyperkeratosis (Figure 2).
On further questioning during follow-up, the patient revealed that his hypertensive medication was changed from HCTZ, which he had been taking for the last 8 years, to the combination antihypertensive medication telmisartan-HCTZ before the onset of the skin eruption. Due to the temporal relationship between the new medication and onset of the eruption, the clinical impression was highly suspicious for drug-induced eruptive LP with Köbner phenomenon caused by the recent sunburn. Systemic workup for underlying causes of LP was negative. Laboratory tests revealed normal complete blood cell counts. The hepatitis panel included hepatitis A antibodies; hepatitis B surface, e antigen, and core antibodies; hepatitis B surface antigen and e antibodies; hepatitis C antibodies; and antinuclear antibodies, which were all negative.
The patient continued to develop new pruritic papules clinically consistent with LP. He was instructed to return to his primary care physician to change the telmisartan-HCTZ to a different class of antihypertensive medication. His medication was changed to atenolol. The patient also was instructed to continue the halobetasol propionate ointment 0.05% twice daily to the affected areas.
The patient returned for a follow-up visit 1 month later and reported notable improvement in pruritus and near-complete resolution of the LP after discontinuation of telmisartan-HCTZ. He also noted some degree of perifollicular repigmentation of the vitiliginous skin that had been unresponsive to prior therapy (Figure 3).
Lichen planus is a pruritic and inflammatory papulosquamous skin condition that presents as scaly, flat-topped, violaceous, polygonal-shaped papules commonly involving the flexor surface of the arms and legs, oral mucosa, scalp, nails, and genitalia. Clinically, LP can present in various forms including actinic, annular, atrophic, erosive, follicular, hypertrophic, linear, pigmented, and vesicular/bullous types. Koebnerization is common, especially in the linear form of LP. There are no specific laboratory findings or serologic markers seen in LP.
The exact cause of LP remains unknown. Clinical observations and anecdotal evidence have directed the cell-mediated immune response to insulting agents such as medications or contact allergy to metals triggering an abnormal cellular immune response. Various viral agents have been reported including hepatitis C virus, human herpesvirus, herpes simplex virus, and varicella-zoster virus.1-5 Other factors such as seasonal change and the environment may contribute to the development of LP and an increase in the incidence of LP eruption has been observed from January to July throughout the United States.6 Lichen planus also has been associated with other altered immune-related disease such as ulcerative colitis, alopecia areata, vitiligo, dermatomyositis, morphea, lichen sclerosis, and myasthenia gravis.7 Increased levels of emotional stress, particularly related to family members, often is related to the onset or aggravation of symptoms.8,9
Many drug-related LP-like and lichenoid eruptions have been reported with antihypertensive drugs, antimalarial drugs, diuretics, antidepressants, nonsteroidal anti-inflammatory drugs, antimicrobial drugs, and metals. In particular, medications such as captopril, enalapril, labetalol, propranolol, chlorothiazide, HCTZ, methyldopa, chloroquine, hydroxychloroquine, quinacrine, gold salts, penicillamine, and quinidine commonly are reported to induce lichenoid drug eruption.10
Several inflammatory papulosquamous skin conditions should be considered in the differential diagnosis before confirming the diagnosis of LP. It is important to rule out lupus erythematosus, especially if the oral mucosa and scalp are involved. In addition, erosive paraneoplastic pemphigus involving primarily the oral mucosa can resemble oral LP. Nail diseases such as psoriasis, onychomycosis, and alopecia areata should be considered as the differential diagnosis of nail disease. Genital involvement also can be seen in psoriasis and lichen sclerosus.
Treatment of LP is mainly symptomatic because of the benign nature of the disease and the high spontaneous remission rate with varying amount of time. If drugs, dental/metal implants, or underlying viral infections are the identifiable triggering factors of LP, the offending agents should be discontinued or removed. Additionally, topical or systemic treatments can be given depending on the severity of the disease, focusing mainly on symptomatic relief as well as the balance of risks and benefits associated with treatment.
Treatment options include topical and intralesional corticosteroids. Systemic medications such as oral corticosteroids and/or acitretin commonly are used in acute, severe, and disseminated cases, though treatment duration varies depending on the clinical response. Other systemic agents used to treat LP include griseofulvin, metronidazole, sulfasalazine, cyclosporine, and mycophenolate mofetil.
Phototherapy is considered an alternative therapy, especially for recalcitrant LP. UVA1 and narrowband UVB (wavelength, 311 nm) have been reported to effectively treat long-standing and therapy-resistant LP.11 In addition, a small study used the excimer laser (wavelength, 308 nm), which is well tolerated by patients, to treat focal recalcitrant oral lesions with excellent results.12 Photochemotherapy has been used with notable improvement, but the potential of carcinogenicity, especially in patients with Fitzpatrick skin types I and II, has limited its use.13
Our patient developed an unusual extensive LP eruption involving only vitiliginous skin shortly after initiation of the combined antihypertensive medication telmisartan-HCTZ, an angiotensin receptor blocker with a thiazide diuretic. Telmisartan and other angiotensin receptor blockers have not been reported to trigger LP; HCTZ is listed as one of the common drugs causing photosensitivity and LP.14,15 Although it is possible that our patient exhibited a delayed lichenoid drug eruption from the HCTZ, it is noteworthy that he did not experience a single episode of LP during his 8-year history of taking HCTZ. Instead, he developed the LP eruption shortly after the addition of telmisartan to his HCTZ antihypertensive regimen. The temporal relationship led us to direct the patient to the prescribing physician to discontinue telmisartan-HCTZ. After changing his antihypertensive medication to atenolol, the patient presented with improvement within the first month and near-complete resolution 2 months after the discontinuation of telmisartan-HCTZ.
Our patient’s LP lesions only manifested on the skin affected by vitiligo, sparing the normal-pigmented skin. Studies have demonstrated an increased ratio of CD8+ T cells to CD4+ T cells as well as increased intercellular adhesion molecule 1 at the dermal level.10,16 Both vitiligo and LP share some common histopathologic features including highly populated CD8+ T cells and intercellular adhesion molecule 1. In our case, LP was triggered on the vitiliginous skin by telmisartan. Vitiligo in combination with trauma induced by sunburn may represent the trigger that altered the cellular immune response and created the telmisartan-induced LP. As a result, the LP eruption was confined to the vitiliginous skin lesions.
Perifollicular repigmentation was observed in our patient after the LP lesions resolved; the patient’s vitiligo was unresponsive to prior treatment. The inflammatory process occurring in LP may exert and interfere in the underlying autoimmune cytotoxic effect toward the melanocytes and the melanin synthesis. It may be of interest to find out if the inflammatory response of LP has a positive influence on the effect of melanogenesis pathways or on the underlying autoimmune-related inflammatory process in vitiligo. Further studies are needed to investigate the role of immunotherapy targeting specific inflammatory pathways and the impact on the repigmentation in vitiligo.
Acknowledgment—Special thanks to Paul Chu, MD (Port Chester, New York).
To the Editor:
A 39-year-old man with a history of hypertension and vitiligo presented with a rapid-onset, generalized, pruritic rash covering the body of 4 weeks’ duration. He reported that the rash progressively worsened after developing mild sunburn. The patient stated that the rash was extremely pruritic with a burning sensation and was tender to touch. He was treated with betamethasone valerate cream 0.1% by an outside physician and an over-the-counter anti-itch lotion with no notable improvement. His only medication was telmisartan-hydrochlorothiazide (HCTZ) for hypertension. He denied any drug allergies.
Physical examination revealed multiple discrete and coalescent planar erythematous papules and plaques involving only the depigmented vitiliginous skin of the forehead, eyelids, and nape of the neck (Figure 1A), and confluent on the lateral aspect of the bilateral forearms (Figure 1B), dorsal aspect of the right hand, and bilateral dorsi of the feet. Wickham striae were noted on the lips (Figure 1C). A clinical diagnosis of lichen planus (LP) was made. The patient initially was prescribed halobetasol propionate ointment 0.05% twice daily. He reported notable relief of pruritus with reduction of overall symptoms and new lesion formation.
A 4-mm punch biopsy was performed on the left forearm. Histopathology revealed LP. Microscopic examination of the hematoxylin and eosin–stained specimen revealed a bandlike lymphohistiocytic infiltrate that extended across the papillary dermis, focally obscuring the dermoepidermal junction where there were vacuolar changes and colloid bodies. The epidermis showed sawtooth rete ridges, wedge-shaped foci of hypergranulosis, and compact hyperkeratosis (Figure 2).
On further questioning during follow-up, the patient revealed that his hypertensive medication was changed from HCTZ, which he had been taking for the last 8 years, to the combination antihypertensive medication telmisartan-HCTZ before the onset of the skin eruption. Due to the temporal relationship between the new medication and onset of the eruption, the clinical impression was highly suspicious for drug-induced eruptive LP with Köbner phenomenon caused by the recent sunburn. Systemic workup for underlying causes of LP was negative. Laboratory tests revealed normal complete blood cell counts. The hepatitis panel included hepatitis A antibodies; hepatitis B surface, e antigen, and core antibodies; hepatitis B surface antigen and e antibodies; hepatitis C antibodies; and antinuclear antibodies, which were all negative.
The patient continued to develop new pruritic papules clinically consistent with LP. He was instructed to return to his primary care physician to change the telmisartan-HCTZ to a different class of antihypertensive medication. His medication was changed to atenolol. The patient also was instructed to continue the halobetasol propionate ointment 0.05% twice daily to the affected areas.
The patient returned for a follow-up visit 1 month later and reported notable improvement in pruritus and near-complete resolution of the LP after discontinuation of telmisartan-HCTZ. He also noted some degree of perifollicular repigmentation of the vitiliginous skin that had been unresponsive to prior therapy (Figure 3).
Lichen planus is a pruritic and inflammatory papulosquamous skin condition that presents as scaly, flat-topped, violaceous, polygonal-shaped papules commonly involving the flexor surface of the arms and legs, oral mucosa, scalp, nails, and genitalia. Clinically, LP can present in various forms including actinic, annular, atrophic, erosive, follicular, hypertrophic, linear, pigmented, and vesicular/bullous types. Koebnerization is common, especially in the linear form of LP. There are no specific laboratory findings or serologic markers seen in LP.
The exact cause of LP remains unknown. Clinical observations and anecdotal evidence have directed the cell-mediated immune response to insulting agents such as medications or contact allergy to metals triggering an abnormal cellular immune response. Various viral agents have been reported including hepatitis C virus, human herpesvirus, herpes simplex virus, and varicella-zoster virus.1-5 Other factors such as seasonal change and the environment may contribute to the development of LP and an increase in the incidence of LP eruption has been observed from January to July throughout the United States.6 Lichen planus also has been associated with other altered immune-related disease such as ulcerative colitis, alopecia areata, vitiligo, dermatomyositis, morphea, lichen sclerosis, and myasthenia gravis.7 Increased levels of emotional stress, particularly related to family members, often is related to the onset or aggravation of symptoms.8,9
Many drug-related LP-like and lichenoid eruptions have been reported with antihypertensive drugs, antimalarial drugs, diuretics, antidepressants, nonsteroidal anti-inflammatory drugs, antimicrobial drugs, and metals. In particular, medications such as captopril, enalapril, labetalol, propranolol, chlorothiazide, HCTZ, methyldopa, chloroquine, hydroxychloroquine, quinacrine, gold salts, penicillamine, and quinidine commonly are reported to induce lichenoid drug eruption.10
Several inflammatory papulosquamous skin conditions should be considered in the differential diagnosis before confirming the diagnosis of LP. It is important to rule out lupus erythematosus, especially if the oral mucosa and scalp are involved. In addition, erosive paraneoplastic pemphigus involving primarily the oral mucosa can resemble oral LP. Nail diseases such as psoriasis, onychomycosis, and alopecia areata should be considered as the differential diagnosis of nail disease. Genital involvement also can be seen in psoriasis and lichen sclerosus.
Treatment of LP is mainly symptomatic because of the benign nature of the disease and the high spontaneous remission rate with varying amount of time. If drugs, dental/metal implants, or underlying viral infections are the identifiable triggering factors of LP, the offending agents should be discontinued or removed. Additionally, topical or systemic treatments can be given depending on the severity of the disease, focusing mainly on symptomatic relief as well as the balance of risks and benefits associated with treatment.
Treatment options include topical and intralesional corticosteroids. Systemic medications such as oral corticosteroids and/or acitretin commonly are used in acute, severe, and disseminated cases, though treatment duration varies depending on the clinical response. Other systemic agents used to treat LP include griseofulvin, metronidazole, sulfasalazine, cyclosporine, and mycophenolate mofetil.
Phototherapy is considered an alternative therapy, especially for recalcitrant LP. UVA1 and narrowband UVB (wavelength, 311 nm) have been reported to effectively treat long-standing and therapy-resistant LP.11 In addition, a small study used the excimer laser (wavelength, 308 nm), which is well tolerated by patients, to treat focal recalcitrant oral lesions with excellent results.12 Photochemotherapy has been used with notable improvement, but the potential of carcinogenicity, especially in patients with Fitzpatrick skin types I and II, has limited its use.13
Our patient developed an unusual extensive LP eruption involving only vitiliginous skin shortly after initiation of the combined antihypertensive medication telmisartan-HCTZ, an angiotensin receptor blocker with a thiazide diuretic. Telmisartan and other angiotensin receptor blockers have not been reported to trigger LP; HCTZ is listed as one of the common drugs causing photosensitivity and LP.14,15 Although it is possible that our patient exhibited a delayed lichenoid drug eruption from the HCTZ, it is noteworthy that he did not experience a single episode of LP during his 8-year history of taking HCTZ. Instead, he developed the LP eruption shortly after the addition of telmisartan to his HCTZ antihypertensive regimen. The temporal relationship led us to direct the patient to the prescribing physician to discontinue telmisartan-HCTZ. After changing his antihypertensive medication to atenolol, the patient presented with improvement within the first month and near-complete resolution 2 months after the discontinuation of telmisartan-HCTZ.
Our patient’s LP lesions only manifested on the skin affected by vitiligo, sparing the normal-pigmented skin. Studies have demonstrated an increased ratio of CD8+ T cells to CD4+ T cells as well as increased intercellular adhesion molecule 1 at the dermal level.10,16 Both vitiligo and LP share some common histopathologic features including highly populated CD8+ T cells and intercellular adhesion molecule 1. In our case, LP was triggered on the vitiliginous skin by telmisartan. Vitiligo in combination with trauma induced by sunburn may represent the trigger that altered the cellular immune response and created the telmisartan-induced LP. As a result, the LP eruption was confined to the vitiliginous skin lesions.
Perifollicular repigmentation was observed in our patient after the LP lesions resolved; the patient’s vitiligo was unresponsive to prior treatment. The inflammatory process occurring in LP may exert and interfere in the underlying autoimmune cytotoxic effect toward the melanocytes and the melanin synthesis. It may be of interest to find out if the inflammatory response of LP has a positive influence on the effect of melanogenesis pathways or on the underlying autoimmune-related inflammatory process in vitiligo. Further studies are needed to investigate the role of immunotherapy targeting specific inflammatory pathways and the impact on the repigmentation in vitiligo.
Acknowledgment—Special thanks to Paul Chu, MD (Port Chester, New York).
- Pilli M, Zerbini A, Vescovi P, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446-1452.
- De Vries HJ, van Marle J, Teunissen MB, et al. Lichen planus is associated with human herpesvirus type 7 replication and infiltration of plasmacytoid dendritic cells. Br J Dermatol. 2006;154:361-364.
- De Vries HJ, Teunissen MB, Zorgdrager F, et al. Lichen planus remission is associated with a decrease of human herpes virus type 7 protein expression in plasmacytoid dendritic cells. Arch Dermatol Res. 2007;299:213-219.
- Requena L, Kutzner H, Escalonilla P, et al. Cutaneous reactions at sites of herpes zoster scars: an expanded spectrum. Br J Dermatol. 1998;138:161-168.
- Al-Khenaizan S. Lichen planus occurring after hepatitis B vaccination: a new case. J Am Acad Dermatol. 2001;45:614-615.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Sadr-Ashkevari S. Familial actinic lichen planus: case reports in two brothers. Arch Int Med. 2001;4:204-206.
- Manolache L, Seceleanu-Petrescu D, Benea V. Lichen planus patients and stressful events. J Eur Acad Dermatol Venereol. 2008;22:437-441.
- Mahood JM. Familial lichen planus. Arch Dermatol. 1983;119:292-294.
- Shimizu M, Higaki Y, Higaki M, et al. The role of granzyme B-expressing CD8-positive T cells in apoptosis of keratinocytes in lichen planus. Arch Dermatol Res. 1997;289:527-532.
- Bécherel PA, Bussel A, Chosidow O, et al. Extracorporeal photochemotherapy for chronic erosive lichen planus. Lancet. 1998;351:805.
- Trehan M, Taylar CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol. 2004;140:415-420.
- Wackernagel A, Legat FJ, Hofer A, et al. Psoralen plus UVA vs. UVB-311 nm for the treatment of lichen planus. Photodermatol Photoimmunol Photomed. 2007;23:15-19.
- Fellner MJ. Lichen planus. Int J Dermatol. 1980;19:71-75.
- Moore DE. Drug-induced cutaneous photosensitivity: incidence, mechanism, prevention and management. Drug Saf. 2002;25:345-372.
- Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
- Pilli M, Zerbini A, Vescovi P, et al. Oral lichen planus pathogenesis: a role for the HCV-specific cellular immune response. Hepatology. 2002;36:1446-1452.
- De Vries HJ, van Marle J, Teunissen MB, et al. Lichen planus is associated with human herpesvirus type 7 replication and infiltration of plasmacytoid dendritic cells. Br J Dermatol. 2006;154:361-364.
- De Vries HJ, Teunissen MB, Zorgdrager F, et al. Lichen planus remission is associated with a decrease of human herpes virus type 7 protein expression in plasmacytoid dendritic cells. Arch Dermatol Res. 2007;299:213-219.
- Requena L, Kutzner H, Escalonilla P, et al. Cutaneous reactions at sites of herpes zoster scars: an expanded spectrum. Br J Dermatol. 1998;138:161-168.
- Al-Khenaizan S. Lichen planus occurring after hepatitis B vaccination: a new case. J Am Acad Dermatol. 2001;45:614-615.
- Boyd AS, Neldner KH. Lichen planus. J Am Acad Dermatol. 1991;25:593-619.
- Sadr-Ashkevari S. Familial actinic lichen planus: case reports in two brothers. Arch Int Med. 2001;4:204-206.
- Manolache L, Seceleanu-Petrescu D, Benea V. Lichen planus patients and stressful events. J Eur Acad Dermatol Venereol. 2008;22:437-441.
- Mahood JM. Familial lichen planus. Arch Dermatol. 1983;119:292-294.
- Shimizu M, Higaki Y, Higaki M, et al. The role of granzyme B-expressing CD8-positive T cells in apoptosis of keratinocytes in lichen planus. Arch Dermatol Res. 1997;289:527-532.
- Bécherel PA, Bussel A, Chosidow O, et al. Extracorporeal photochemotherapy for chronic erosive lichen planus. Lancet. 1998;351:805.
- Trehan M, Taylar CR. Low-dose excimer 308-nm laser for the treatment of oral lichen planus. Arch Dermatol. 2004;140:415-420.
- Wackernagel A, Legat FJ, Hofer A, et al. Psoralen plus UVA vs. UVB-311 nm for the treatment of lichen planus. Photodermatol Photoimmunol Photomed. 2007;23:15-19.
- Fellner MJ. Lichen planus. Int J Dermatol. 1980;19:71-75.
- Moore DE. Drug-induced cutaneous photosensitivity: incidence, mechanism, prevention and management. Drug Saf. 2002;25:345-372.
- Ongenae K, Van Geel N, Naeyaert JM. Evidence for an autoimmune pathogenesis of vitiligo. Pigment Cell Res. 2003;16:90-100.
Practice Points
- Lichen planus (LP) is a T-cell–mediated autoimmune disease that affects the skin and often the mucosa, nails, and scalp.
- The etiology of LP is unknown. It can be induced by a variety of medications and may spread through the isomorphic phenomenon.
- Immune factors play a role in the development of LP, drug-induced LP, and vitiligo.
Bilateral Symmetric Onycholysis of Distal Fingernails
The Diagnosis: Allergic Contact Dermatitis
An allergic contact dermatitis (ACD) to acrylates was suspected and 4 patches were applied to the forearm (the North American Standard Series of the North American Contact Dermatitis Group). The patches were 2-hydroxyethyl methacrylate (2-HEMA) 2.0% permissible exposure limit (peL), ethyl acrylate 0.1% peL, tosylamide formaldehyde resin 10.0% peL, and methyl methacrylate 2.0% peL. A reading at 72 hours was performed and showed a positive reaction to hydroxyethyl methacrylate, ethyl acrylate, and methyl methacrylate, and a negative patch test to tosylamide formaldehyde resin (nail polish)(Figure). The patient was diagnosed with an allergic contact hypersensitivity to the aforementioned acrylates and instructed to avoid artificial nails and acrylate glues. She also was started on oral biotin supplements. On 6-month follow-up the patient had regrowth of all 10 fingernails without brittleness or splitting. She was able to use nail polishes but avoided all acrylic artificial nails and acrylate-containing personal care products.
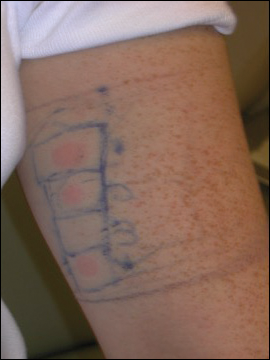
Acrylate Allergy and Artificial Nails
Acrylates are plastic materials formed by polymerization of acrylic or methacrylic acid monomers and have been cited as a major cause of occupational and nonoccupational contact dermatitis. Contact dermatitis to acrylates in artificial nails was first reported in the 1950s.1,2 Products containing 100% methyl methacrylate monomers in acrylic nails were banned by the US Food and Drug Administration in the early 1970s after receiving a number of complaints.3 However, no regulation prohibits the use of methyl methacrylate monomer in cosmetic products, and various methacrylate and acrylate monomers remain widely used.4 With a growing popularity in artificial nails, it is expected the number of sensitized persons will increase.
Acrylate allergy from sculptured nails concern self-curing resins made from a polymer powder and a liquid monomer solution. Advantages of new UV-cured products include the lack of unpleasant smell and simplified modeling. They also do not require an irritant, such as methacrylic acid, as a bonding agent. Instead, 2-HEMA and 2-hydroxypropyl methacrylate are added. These photobonded nails colloquially are called gel nails (acid free) as opposed to acrylic nails (using methacrylic acid as a primer). It is important to note that the esters of acrylic acid but not the acid itself sensitize patients, and sensitization is not caused by the uncured gel or the monomer solution but by the remaining monomers in the cured plastic nail and the dust filings that are produced during the finishing process.
Clinical Presentation
Symptoms of an ACD to nail acrylates include pruritus and fingertip dermatitis along with nail plate dystrophy. There may be pruritus at the nail base, with subsequent dryness, thickening, and onycholysis. The brittle nails may become split, discolored, and develop paronychia. Inadvertent contact with glue monomers or other acrylate-containing substances may cause eczematous lesions at distant sites. Avoidance of the allergen often results in complete restoration of the normal nail and fingertip within months.
Sensitization
Acrylates and methacrylates are ubiquitous materials used for both industrial and commercial applications. Due to their widespread industrial use, contact allergies to acrylates including 2-HEMA, 2-hydroxypropyl methacrylate, and triethyleneglycol diacrylate (TREGDA) are common. Cross-reaction of these compounds has been observed and is postulated to be due to reaction of the (meth)acrylate carboxyethyl group with the receptors of antigen-presenting cells.5 As a result, an individual with an acrylate allergy sensitized to one allergen often is allergic to its similar compounds and cross-reactors and must avoid the assortment of compounds containing these ingredients, which is important for individuals with occupational sensitization to a particular acrylate who is subsequently susceptible to other acrylate-containing compounds triggering allergic reactions when reexposure occurs in different settings.
Allergens and Occupational Exposure
Acrylates in cosmetic nail products are a source of ACD for not only the customer but also the manicurist.6 The most frequently cited sources of ACD in beauticians are acrylate chemicals.7 However, acrylate compounds are an occupational hazard for a number of other specialists, including dentists and dental technicians, histology technicians, and individuals in the printing industry.8,9 Other individuals may be sensitized to acrylates through their inclusion in adhesives, dental bonding agents, hearing aids, electrocardiogram electrodes, artificial bone cement, and a myriad of other medical and nonmedical applications.4,10-12 For workers who cannot avoid occupational exposure to these allergens, polyvinyl alcohol and multilayer laminate gloves are recommended, as natural rubber latex gloves do not always provide adequate protection from many of these agents.10
Testing for Suspected Acrylate Allergy
Cross-reactivity among acrylates is widely considered in the literature but remains enigmatic and is an important consideration with regard to routine patch test screening.13 In the case of an acrylate allergy to nail products, using 2-HEMA and ethylene glycol dimethacrylate is effective in detecting sensitization by photobonded nails and in patients sensitized by powder liquid products.14 One study showed a patch test panel including 2-HEMA, ethylene glycol dimethacrylate, and TREGDA was effective in identifying the majority of individuals with an allergy to acrylates in nail products and nail technicians.15 Another study has shown the most commonly positive testing allergens to be HEMA, ethyl acrylate, and methyl methacrylate.16 If one is patch testing only one chemical, it appears 2-HEMA is preferred.17 However, broader panels of screening allergens are necessary to achieve an accurate diagnosis. Furthermore, different panels of test allergens have been shown to vary in their ability to detect an acrylate allergy in different occupational exposures.12
The time to patch test read also is important. A standard read at 72 hours is warranted; however, one study showed if only one read at day 3 was done without a subsequent day 7 read, then 25% of TREGDA and 50% of 2-HEMA allergies would have been missed in patients with occupational acrylate allergy.15 Other studies have reported late-appearing and long-lasting test reactions when testing for an acrylate allergy.18,19 Clinicians should be cognizant that an acrylate allergy may be present even if initial screening is negative but the history and clinical picture are suggestive.
- Canizares O. Contact dermatitis due to the acrylic materials used in artificial nails. AMA Arch Derm. 1956;74:141-143.
- Fisher AA, Franks A, Glick H. Allergic sensitization of the skin and nails to acrylic plastic nails. J Allergy. 1957;28:84-88.
- US Food and Drug Administration. Nail care products. http://www.fda.gov/Cosmetics/ProductsIngredients/Products/ucm127068.htm. Updated October 26, 2016. Accessed December 27, 2016.
- Haughton AM, Belsito DV. Acrylate allergy induced by acrylic nails resulting in prosthesis failure. J Am Acad Dermatol. 2008;59(5 suppl):S123-S124.
- Kanerva L. Cross-reactions of multifunctional methacrylates and acrylates. Acta Odontol Scand. 2001;59:320-329.
- Tammaro A, Narcisi A, Abruzzese C, et al. Fingertip dermatitis: occupational acrylate cross reaction. Allergol Int. 2014;63:609-610.
- Kwok C, Money A, Carder M, et al. Cases of occupational dermatitis and asthma in beauticians that were reported to The Health and Occupation Research (THOR) network from 1996 to 2011. Clin Exp Dermatol. 2014;39:590-595.
- Aalto-Korte K, Alanko K, Kuuliala O, et al. Methacrylate and acrylate allergy in dental personnel. Contact Dermatitis. 2007;57:324-330.
- Molina L, Amado A, Mattei PL 4th, et al. Contact dermatitis from acrylics in a histology laboratory assistant. Dermatitis. 2009;20:E11-E12.
- Prasad Hunasehally RY, Hughes TM, Stone NM. Atypical pattern of (meth)acrylate allergic contact dermatitis in dental professionals. Br Dent J. 2012;213:223-224.
- Stingeni L, Cerulli E, Spalletti A, et al. The role of acrylic acid impurity as a sensitizing component in electrocardiogram electrodes [published online January 27, 2015]. Contact Dermatitis. 2015;73:44-48.
- Sasseville D. Acrylates in contact dermatitis. Dermatitis. 2012;23:6-16.
- Fisher AA. Cross reactions between methyl methacrylate monomer and acrylic monomers presently used in acrylic nail preparations. Contact Dermatitis. 1980;6:345-347.
- Hemmer W, Focke M, Wantke F, et al. Allergic contact dermatitis to artificial fingernails prepared from UV light-cured acrylates. J Am Acad Dermatol. 1996;35(3, pt 1):377-380.
- Teik-Jin Goon A, Bruze M, Zimerson E, et al. Contact allergy to acrylates/methacrylates in the acrylate and nail acrylics series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2007;57:21-27.
- Drucker AM, Pratt MD. Acrylate contact allergy: patient characteristics and evaluation of screening allergens. Dermatitis. 2011;22:98-101.
- Ramos L, Cabral R, Goncalo M. Allergic contact dermatitis caused by acrylates and methacrylates--a 7-year study. Contact Dermatitis. 2014;71:102-107.
- Goon AT, Isaksson M, Zimerson E, et al. Contact allergy to (meth)acrylates in the dental series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2006;55:219-226.
- Isaksson M, Lindberg M, Sundberg K, et al. The development and course of patch-test reactions to 2-hydroxyethyl methacrylate and ethyleneglycol dimethacrylate. Contact Dermatitis. 2005;53:292-297.
The Diagnosis: Allergic Contact Dermatitis
An allergic contact dermatitis (ACD) to acrylates was suspected and 4 patches were applied to the forearm (the North American Standard Series of the North American Contact Dermatitis Group). The patches were 2-hydroxyethyl methacrylate (2-HEMA) 2.0% permissible exposure limit (peL), ethyl acrylate 0.1% peL, tosylamide formaldehyde resin 10.0% peL, and methyl methacrylate 2.0% peL. A reading at 72 hours was performed and showed a positive reaction to hydroxyethyl methacrylate, ethyl acrylate, and methyl methacrylate, and a negative patch test to tosylamide formaldehyde resin (nail polish)(Figure). The patient was diagnosed with an allergic contact hypersensitivity to the aforementioned acrylates and instructed to avoid artificial nails and acrylate glues. She also was started on oral biotin supplements. On 6-month follow-up the patient had regrowth of all 10 fingernails without brittleness or splitting. She was able to use nail polishes but avoided all acrylic artificial nails and acrylate-containing personal care products.
Acrylate Allergy and Artificial Nails
Acrylates are plastic materials formed by polymerization of acrylic or methacrylic acid monomers and have been cited as a major cause of occupational and nonoccupational contact dermatitis. Contact dermatitis to acrylates in artificial nails was first reported in the 1950s.1,2 Products containing 100% methyl methacrylate monomers in acrylic nails were banned by the US Food and Drug Administration in the early 1970s after receiving a number of complaints.3 However, no regulation prohibits the use of methyl methacrylate monomer in cosmetic products, and various methacrylate and acrylate monomers remain widely used.4 With a growing popularity in artificial nails, it is expected the number of sensitized persons will increase.
Acrylate allergy from sculptured nails concern self-curing resins made from a polymer powder and a liquid monomer solution. Advantages of new UV-cured products include the lack of unpleasant smell and simplified modeling. They also do not require an irritant, such as methacrylic acid, as a bonding agent. Instead, 2-HEMA and 2-hydroxypropyl methacrylate are added. These photobonded nails colloquially are called gel nails (acid free) as opposed to acrylic nails (using methacrylic acid as a primer). It is important to note that the esters of acrylic acid but not the acid itself sensitize patients, and sensitization is not caused by the uncured gel or the monomer solution but by the remaining monomers in the cured plastic nail and the dust filings that are produced during the finishing process.
Clinical Presentation
Symptoms of an ACD to nail acrylates include pruritus and fingertip dermatitis along with nail plate dystrophy. There may be pruritus at the nail base, with subsequent dryness, thickening, and onycholysis. The brittle nails may become split, discolored, and develop paronychia. Inadvertent contact with glue monomers or other acrylate-containing substances may cause eczematous lesions at distant sites. Avoidance of the allergen often results in complete restoration of the normal nail and fingertip within months.
Sensitization
Acrylates and methacrylates are ubiquitous materials used for both industrial and commercial applications. Due to their widespread industrial use, contact allergies to acrylates including 2-HEMA, 2-hydroxypropyl methacrylate, and triethyleneglycol diacrylate (TREGDA) are common. Cross-reaction of these compounds has been observed and is postulated to be due to reaction of the (meth)acrylate carboxyethyl group with the receptors of antigen-presenting cells.5 As a result, an individual with an acrylate allergy sensitized to one allergen often is allergic to its similar compounds and cross-reactors and must avoid the assortment of compounds containing these ingredients, which is important for individuals with occupational sensitization to a particular acrylate who is subsequently susceptible to other acrylate-containing compounds triggering allergic reactions when reexposure occurs in different settings.
Allergens and Occupational Exposure
Acrylates in cosmetic nail products are a source of ACD for not only the customer but also the manicurist.6 The most frequently cited sources of ACD in beauticians are acrylate chemicals.7 However, acrylate compounds are an occupational hazard for a number of other specialists, including dentists and dental technicians, histology technicians, and individuals in the printing industry.8,9 Other individuals may be sensitized to acrylates through their inclusion in adhesives, dental bonding agents, hearing aids, electrocardiogram electrodes, artificial bone cement, and a myriad of other medical and nonmedical applications.4,10-12 For workers who cannot avoid occupational exposure to these allergens, polyvinyl alcohol and multilayer laminate gloves are recommended, as natural rubber latex gloves do not always provide adequate protection from many of these agents.10
Testing for Suspected Acrylate Allergy
Cross-reactivity among acrylates is widely considered in the literature but remains enigmatic and is an important consideration with regard to routine patch test screening.13 In the case of an acrylate allergy to nail products, using 2-HEMA and ethylene glycol dimethacrylate is effective in detecting sensitization by photobonded nails and in patients sensitized by powder liquid products.14 One study showed a patch test panel including 2-HEMA, ethylene glycol dimethacrylate, and TREGDA was effective in identifying the majority of individuals with an allergy to acrylates in nail products and nail technicians.15 Another study has shown the most commonly positive testing allergens to be HEMA, ethyl acrylate, and methyl methacrylate.16 If one is patch testing only one chemical, it appears 2-HEMA is preferred.17 However, broader panels of screening allergens are necessary to achieve an accurate diagnosis. Furthermore, different panels of test allergens have been shown to vary in their ability to detect an acrylate allergy in different occupational exposures.12
The time to patch test read also is important. A standard read at 72 hours is warranted; however, one study showed if only one read at day 3 was done without a subsequent day 7 read, then 25% of TREGDA and 50% of 2-HEMA allergies would have been missed in patients with occupational acrylate allergy.15 Other studies have reported late-appearing and long-lasting test reactions when testing for an acrylate allergy.18,19 Clinicians should be cognizant that an acrylate allergy may be present even if initial screening is negative but the history and clinical picture are suggestive.
The Diagnosis: Allergic Contact Dermatitis
An allergic contact dermatitis (ACD) to acrylates was suspected and 4 patches were applied to the forearm (the North American Standard Series of the North American Contact Dermatitis Group). The patches were 2-hydroxyethyl methacrylate (2-HEMA) 2.0% permissible exposure limit (peL), ethyl acrylate 0.1% peL, tosylamide formaldehyde resin 10.0% peL, and methyl methacrylate 2.0% peL. A reading at 72 hours was performed and showed a positive reaction to hydroxyethyl methacrylate, ethyl acrylate, and methyl methacrylate, and a negative patch test to tosylamide formaldehyde resin (nail polish)(Figure). The patient was diagnosed with an allergic contact hypersensitivity to the aforementioned acrylates and instructed to avoid artificial nails and acrylate glues. She also was started on oral biotin supplements. On 6-month follow-up the patient had regrowth of all 10 fingernails without brittleness or splitting. She was able to use nail polishes but avoided all acrylic artificial nails and acrylate-containing personal care products.
Acrylate Allergy and Artificial Nails
Acrylates are plastic materials formed by polymerization of acrylic or methacrylic acid monomers and have been cited as a major cause of occupational and nonoccupational contact dermatitis. Contact dermatitis to acrylates in artificial nails was first reported in the 1950s.1,2 Products containing 100% methyl methacrylate monomers in acrylic nails were banned by the US Food and Drug Administration in the early 1970s after receiving a number of complaints.3 However, no regulation prohibits the use of methyl methacrylate monomer in cosmetic products, and various methacrylate and acrylate monomers remain widely used.4 With a growing popularity in artificial nails, it is expected the number of sensitized persons will increase.
Acrylate allergy from sculptured nails concern self-curing resins made from a polymer powder and a liquid monomer solution. Advantages of new UV-cured products include the lack of unpleasant smell and simplified modeling. They also do not require an irritant, such as methacrylic acid, as a bonding agent. Instead, 2-HEMA and 2-hydroxypropyl methacrylate are added. These photobonded nails colloquially are called gel nails (acid free) as opposed to acrylic nails (using methacrylic acid as a primer). It is important to note that the esters of acrylic acid but not the acid itself sensitize patients, and sensitization is not caused by the uncured gel or the monomer solution but by the remaining monomers in the cured plastic nail and the dust filings that are produced during the finishing process.
Clinical Presentation
Symptoms of an ACD to nail acrylates include pruritus and fingertip dermatitis along with nail plate dystrophy. There may be pruritus at the nail base, with subsequent dryness, thickening, and onycholysis. The brittle nails may become split, discolored, and develop paronychia. Inadvertent contact with glue monomers or other acrylate-containing substances may cause eczematous lesions at distant sites. Avoidance of the allergen often results in complete restoration of the normal nail and fingertip within months.
Sensitization
Acrylates and methacrylates are ubiquitous materials used for both industrial and commercial applications. Due to their widespread industrial use, contact allergies to acrylates including 2-HEMA, 2-hydroxypropyl methacrylate, and triethyleneglycol diacrylate (TREGDA) are common. Cross-reaction of these compounds has been observed and is postulated to be due to reaction of the (meth)acrylate carboxyethyl group with the receptors of antigen-presenting cells.5 As a result, an individual with an acrylate allergy sensitized to one allergen often is allergic to its similar compounds and cross-reactors and must avoid the assortment of compounds containing these ingredients, which is important for individuals with occupational sensitization to a particular acrylate who is subsequently susceptible to other acrylate-containing compounds triggering allergic reactions when reexposure occurs in different settings.
Allergens and Occupational Exposure
Acrylates in cosmetic nail products are a source of ACD for not only the customer but also the manicurist.6 The most frequently cited sources of ACD in beauticians are acrylate chemicals.7 However, acrylate compounds are an occupational hazard for a number of other specialists, including dentists and dental technicians, histology technicians, and individuals in the printing industry.8,9 Other individuals may be sensitized to acrylates through their inclusion in adhesives, dental bonding agents, hearing aids, electrocardiogram electrodes, artificial bone cement, and a myriad of other medical and nonmedical applications.4,10-12 For workers who cannot avoid occupational exposure to these allergens, polyvinyl alcohol and multilayer laminate gloves are recommended, as natural rubber latex gloves do not always provide adequate protection from many of these agents.10
Testing for Suspected Acrylate Allergy
Cross-reactivity among acrylates is widely considered in the literature but remains enigmatic and is an important consideration with regard to routine patch test screening.13 In the case of an acrylate allergy to nail products, using 2-HEMA and ethylene glycol dimethacrylate is effective in detecting sensitization by photobonded nails and in patients sensitized by powder liquid products.14 One study showed a patch test panel including 2-HEMA, ethylene glycol dimethacrylate, and TREGDA was effective in identifying the majority of individuals with an allergy to acrylates in nail products and nail technicians.15 Another study has shown the most commonly positive testing allergens to be HEMA, ethyl acrylate, and methyl methacrylate.16 If one is patch testing only one chemical, it appears 2-HEMA is preferred.17 However, broader panels of screening allergens are necessary to achieve an accurate diagnosis. Furthermore, different panels of test allergens have been shown to vary in their ability to detect an acrylate allergy in different occupational exposures.12
The time to patch test read also is important. A standard read at 72 hours is warranted; however, one study showed if only one read at day 3 was done without a subsequent day 7 read, then 25% of TREGDA and 50% of 2-HEMA allergies would have been missed in patients with occupational acrylate allergy.15 Other studies have reported late-appearing and long-lasting test reactions when testing for an acrylate allergy.18,19 Clinicians should be cognizant that an acrylate allergy may be present even if initial screening is negative but the history and clinical picture are suggestive.
- Canizares O. Contact dermatitis due to the acrylic materials used in artificial nails. AMA Arch Derm. 1956;74:141-143.
- Fisher AA, Franks A, Glick H. Allergic sensitization of the skin and nails to acrylic plastic nails. J Allergy. 1957;28:84-88.
- US Food and Drug Administration. Nail care products. http://www.fda.gov/Cosmetics/ProductsIngredients/Products/ucm127068.htm. Updated October 26, 2016. Accessed December 27, 2016.
- Haughton AM, Belsito DV. Acrylate allergy induced by acrylic nails resulting in prosthesis failure. J Am Acad Dermatol. 2008;59(5 suppl):S123-S124.
- Kanerva L. Cross-reactions of multifunctional methacrylates and acrylates. Acta Odontol Scand. 2001;59:320-329.
- Tammaro A, Narcisi A, Abruzzese C, et al. Fingertip dermatitis: occupational acrylate cross reaction. Allergol Int. 2014;63:609-610.
- Kwok C, Money A, Carder M, et al. Cases of occupational dermatitis and asthma in beauticians that were reported to The Health and Occupation Research (THOR) network from 1996 to 2011. Clin Exp Dermatol. 2014;39:590-595.
- Aalto-Korte K, Alanko K, Kuuliala O, et al. Methacrylate and acrylate allergy in dental personnel. Contact Dermatitis. 2007;57:324-330.
- Molina L, Amado A, Mattei PL 4th, et al. Contact dermatitis from acrylics in a histology laboratory assistant. Dermatitis. 2009;20:E11-E12.
- Prasad Hunasehally RY, Hughes TM, Stone NM. Atypical pattern of (meth)acrylate allergic contact dermatitis in dental professionals. Br Dent J. 2012;213:223-224.
- Stingeni L, Cerulli E, Spalletti A, et al. The role of acrylic acid impurity as a sensitizing component in electrocardiogram electrodes [published online January 27, 2015]. Contact Dermatitis. 2015;73:44-48.
- Sasseville D. Acrylates in contact dermatitis. Dermatitis. 2012;23:6-16.
- Fisher AA. Cross reactions between methyl methacrylate monomer and acrylic monomers presently used in acrylic nail preparations. Contact Dermatitis. 1980;6:345-347.
- Hemmer W, Focke M, Wantke F, et al. Allergic contact dermatitis to artificial fingernails prepared from UV light-cured acrylates. J Am Acad Dermatol. 1996;35(3, pt 1):377-380.
- Teik-Jin Goon A, Bruze M, Zimerson E, et al. Contact allergy to acrylates/methacrylates in the acrylate and nail acrylics series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2007;57:21-27.
- Drucker AM, Pratt MD. Acrylate contact allergy: patient characteristics and evaluation of screening allergens. Dermatitis. 2011;22:98-101.
- Ramos L, Cabral R, Goncalo M. Allergic contact dermatitis caused by acrylates and methacrylates--a 7-year study. Contact Dermatitis. 2014;71:102-107.
- Goon AT, Isaksson M, Zimerson E, et al. Contact allergy to (meth)acrylates in the dental series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2006;55:219-226.
- Isaksson M, Lindberg M, Sundberg K, et al. The development and course of patch-test reactions to 2-hydroxyethyl methacrylate and ethyleneglycol dimethacrylate. Contact Dermatitis. 2005;53:292-297.
- Canizares O. Contact dermatitis due to the acrylic materials used in artificial nails. AMA Arch Derm. 1956;74:141-143.
- Fisher AA, Franks A, Glick H. Allergic sensitization of the skin and nails to acrylic plastic nails. J Allergy. 1957;28:84-88.
- US Food and Drug Administration. Nail care products. http://www.fda.gov/Cosmetics/ProductsIngredients/Products/ucm127068.htm. Updated October 26, 2016. Accessed December 27, 2016.
- Haughton AM, Belsito DV. Acrylate allergy induced by acrylic nails resulting in prosthesis failure. J Am Acad Dermatol. 2008;59(5 suppl):S123-S124.
- Kanerva L. Cross-reactions of multifunctional methacrylates and acrylates. Acta Odontol Scand. 2001;59:320-329.
- Tammaro A, Narcisi A, Abruzzese C, et al. Fingertip dermatitis: occupational acrylate cross reaction. Allergol Int. 2014;63:609-610.
- Kwok C, Money A, Carder M, et al. Cases of occupational dermatitis and asthma in beauticians that were reported to The Health and Occupation Research (THOR) network from 1996 to 2011. Clin Exp Dermatol. 2014;39:590-595.
- Aalto-Korte K, Alanko K, Kuuliala O, et al. Methacrylate and acrylate allergy in dental personnel. Contact Dermatitis. 2007;57:324-330.
- Molina L, Amado A, Mattei PL 4th, et al. Contact dermatitis from acrylics in a histology laboratory assistant. Dermatitis. 2009;20:E11-E12.
- Prasad Hunasehally RY, Hughes TM, Stone NM. Atypical pattern of (meth)acrylate allergic contact dermatitis in dental professionals. Br Dent J. 2012;213:223-224.
- Stingeni L, Cerulli E, Spalletti A, et al. The role of acrylic acid impurity as a sensitizing component in electrocardiogram electrodes [published online January 27, 2015]. Contact Dermatitis. 2015;73:44-48.
- Sasseville D. Acrylates in contact dermatitis. Dermatitis. 2012;23:6-16.
- Fisher AA. Cross reactions between methyl methacrylate monomer and acrylic monomers presently used in acrylic nail preparations. Contact Dermatitis. 1980;6:345-347.
- Hemmer W, Focke M, Wantke F, et al. Allergic contact dermatitis to artificial fingernails prepared from UV light-cured acrylates. J Am Acad Dermatol. 1996;35(3, pt 1):377-380.
- Teik-Jin Goon A, Bruze M, Zimerson E, et al. Contact allergy to acrylates/methacrylates in the acrylate and nail acrylics series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2007;57:21-27.
- Drucker AM, Pratt MD. Acrylate contact allergy: patient characteristics and evaluation of screening allergens. Dermatitis. 2011;22:98-101.
- Ramos L, Cabral R, Goncalo M. Allergic contact dermatitis caused by acrylates and methacrylates--a 7-year study. Contact Dermatitis. 2014;71:102-107.
- Goon AT, Isaksson M, Zimerson E, et al. Contact allergy to (meth)acrylates in the dental series in southern Sweden: simultaneous positive patch test reaction patterns and possible screening allergens. Contact Dermatitis. 2006;55:219-226.
- Isaksson M, Lindberg M, Sundberg K, et al. The development and course of patch-test reactions to 2-hydroxyethyl methacrylate and ethyleneglycol dimethacrylate. Contact Dermatitis. 2005;53:292-297.
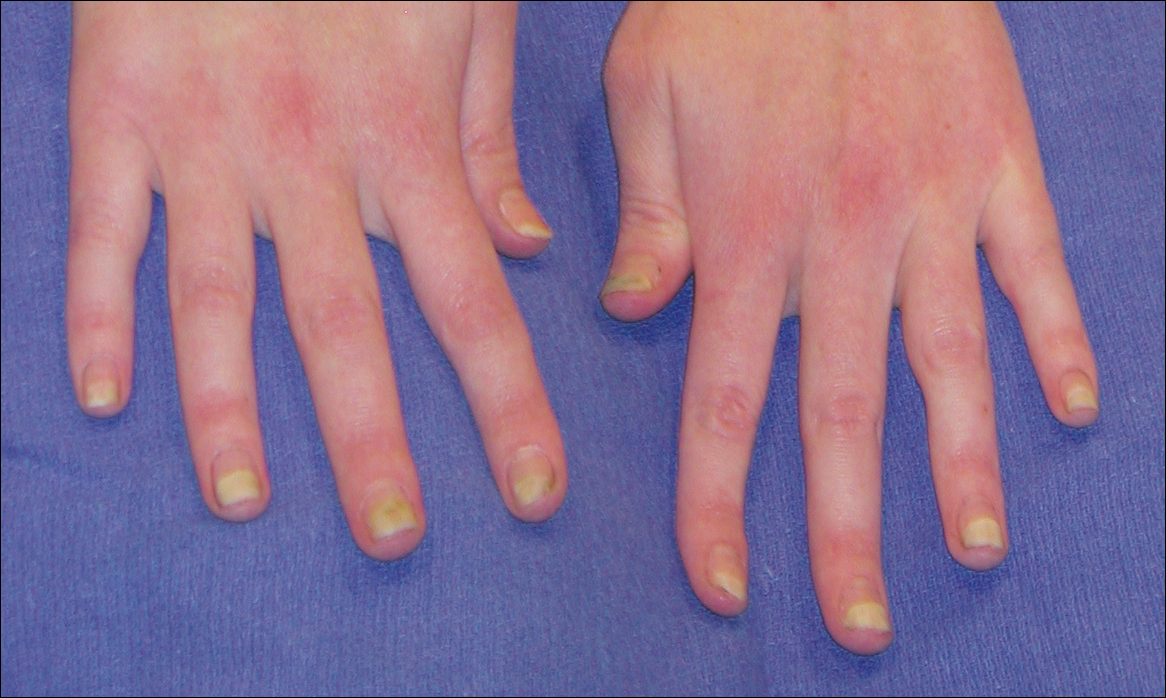
A 28-year-old woman presented with distal onycholysis of all 10 fingernails. The patient started to notice brittleness in the first, second, and third fingernails of the right hand 2 months prior. She had a 10-year history of wearing acrylic nails and reported a history of periungual eczema. On physical examination, all 10 fingernails had distal onycholysis and there was a green discoloration of the first fingernail on the left hand. On blood analysis, thyroid-stimulating hormone and free thyroxine were within reference range. A nail clipping showed onychodystrophy and a negative periodic acid-Schiff stain.
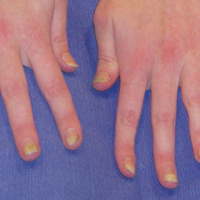
Healing of Leg Ulcers Associated With Granulomatosis With Polyangiitis (Wegener Granulomatosis) After Rituximab Therapy
To the Editor:
A 52-year-old woman with a history of arthralgia, rhinitis, sinusitis, and episodic epistaxis was admitted to the hospital with multiple nonhealing severe leg ulcerations. She noticed subcutaneous nodules on the legs 6 months prior to the development of ulcers. The lesions progressed from subcutaneous nodules to red-black skin discoloration, blister formation, and eventually ulceration. Over a period of months, the ulcers were treated with several courses of antibiotics and wound care including a single surgical debridement of one of the ulcers on the dorsum of the right foot. These interventions did not make a remarkable impact on ulcer healing.
On physical examination, the patient had scattered 4- to 5-mm palpable purpura on the knees, elbows, and feet bilaterally. She had multiple 1- to 8-cm indurated purple ulcerations with friable surfaces and raised irregular borders on the feet, toes, and lower legs bilaterally (Figure, A–C). One notably larger ulcer was found on the anterior aspect of the left thigh (Figure, A). Scattered 5- to 15-mm eschars were present on the legs bilaterally. She also had multiple large, firm, nonerythematous dermal plaques on the thighs bilaterally that measured several centimeters. There were no oral mucosal lesions and no ulcerations above the waist.
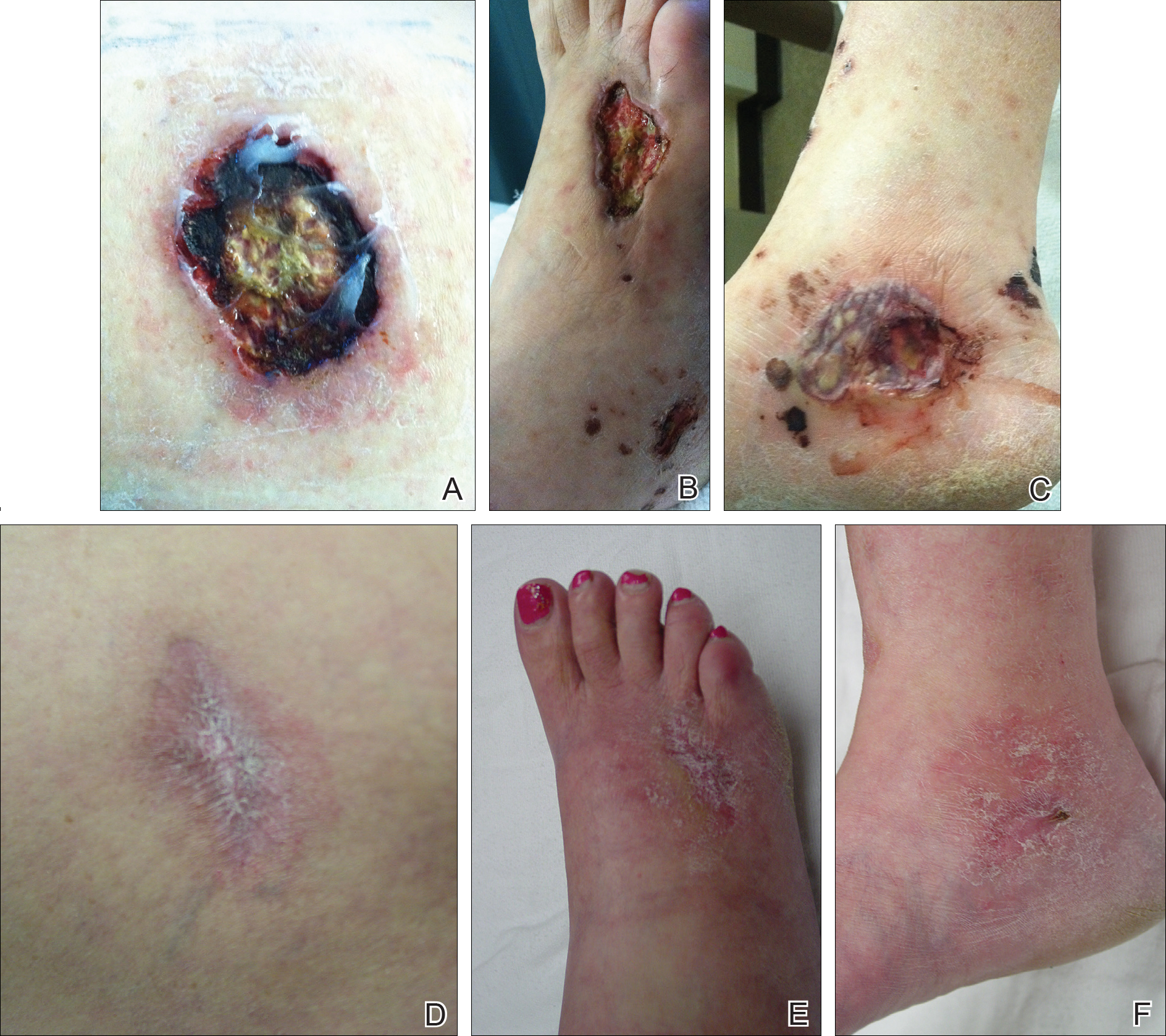
Magnetic resonance imaging of the foot showed some surrounding cellulitis but no osteomyelitis. Chest radiograph and computed tomography revealed bilateral apical nodules. Proteinase 3–antineutrophil cytoplasmic antibody (PR3-ANCA) testing was positive. Serum complement levels were normal. An antinuclear antibody test and rheumatoid factor were both negative. Skin biopsies were obtained from the thigh ulcer, foot ulcer, and purpuric lesions on the right knee. The results demonstrated leukocytoclastic vasculitis and neutrophilic small vessel vasculitis with necrotizing neutrophilic dermatitis and panniculitis. Granulomatosis with polyangiitis (GPA) was diagnosed based on these findings.
Initial inpatient treatment included intravenous methylprednisolone (100 mg every 8 hours for 3 doses), followed by oral prednisone 60 mg daily. Two weeks later the ulcers were reevaluated and only mild improvement had occurred with the steroids. Therefore, rituximab (RTX) was initiated at 375 mg/m2 (700 mg) intravenously once weekly for 4 weeks. After 3 doses of RTX, the ulcerations were healing dramatically and the treatment was well tolerated. A rapid prednisone taper was started, and the patient received her fourth and final dose of RTX. Two months after the initial infusion, the thigh ulcer and most of the ulcerations on the feet and lower legs had almost completely resolved. Photographs were taken 5 months after initial RTX infusion (Figure, D–F). A chest radiograph 4 months after initial RTX infusion showed resolution of lung nodules. Two months after RTX induction therapy, azathioprine was added for maintenance but was stopped due to poor tolerance. Oral methotrexate 17.5 mg once weekly was added 5 months after RTX for maintenance and was well tolerated. At that time the prednisone dose was 10 mg daily and was successfully tapered to 5 mg by 9 months after RTX induction therapy.
Granulomatosis with polyangiitis (Wegener granulomatosis) is a granulomatous small- and medium-sized vessel vasculitis that traditionally affects the upper and lower respiratory tract and kidneys.1 Skin lesions also are quite common and include palpable purpura, ulcers, vesicles, papules, and subcutaneous nodules. Patients with active GPA also tend to have ANCAs directed against proteinase 3 (PR3-ANCA). Although GPA was once considered a fatal disease, treatment with cyclophosphamide combined with corticosteroids has been shown to substantially improve outcomes.1 Rituximab, a chimeric monoclonal anti-CD20 antibody, works by depleting B lymphocytes and has been used with success to treat diseases such as lymphoma and rheumatoid arthritis.2,3 The US Food and Drug Administration approved RTX for GPA and microscopic polyangiitis in 2011, with a number of trials supporting its efficacy.4
The success of RTX in treating GPA has been documented in case reports as well as several trials with extended follow-up. A single-center observational study of 53 patients showed that RTX was safe and effective for induction and maintenance of remission in patients with refractory GPA. This study also uncovered the potential for predicting relapse based on following B cell and ANCA levels and preventing relapse by initializing further treatment.5 Other small studies and case reports have shown similar success using RTX for refractory GPA.6-10 These studies included various combinations of concurrent therapies and various follow-up intervals. The Rituximab in ANCA-Associated Vasculitis (RAVE) trial compared RTX versus cyclophosphamide for ANCA-positive vasculitis.11 This multicenter, randomized, double-blind study found that RTX was as efficacious as cyclophosphamide for induction of remission in severe GPA.The data also suggested that RTX may be superior for relapsing disease.11 Another multicenter, open-label, randomized trial (RITUXVAS) compared RTX to cyclophosphamide in ANCA-associated renal vasculitis. This trial also found the 2 treatments to be similar in both efficacy in inducing remission and adverse events.12
Some conflicting reports have appeared on the effectiveness of using RTX for the granulomatous versus vasculitic manifestations of GPA. Aires et al13 showed failure of improvement in most patients with granulomatous manifestations of GPA in a study of 8 patients. A retrospective study including 59 patients who were treated with RTX also showed that complete remission was more common in patients with primarily vasculitic manifestations, not granulomatous manifestations.14 However, some case series that included patients with refractory ophthalmic GPA, a primarily granulomatous manifestation, have found success using RTX.15,16 More studies are needed to determine if there truly is a difference and whether this difference has an effect on when to use RTX. The skin lesions our patient demonstrated were due to the vasculitic component of the disease, and consequently, the rapid and complete response we observed would be consistent with the premise that the therapy works best for vasculitis.
Most of the trials assessing the efficacy of RTX utilize a tool such as the Wegener granulomatosis-specific Birmingham Vasculitis Activity Score.17 This measure of treatment response does include a skin item, but it is part of the composite response score. Consequently, a specific statement regarding skin improvement cannot be made. Additionally, little is reported pertaining to the treatment of skin-related findings in GPA. One report did specifically address the treatment of dermatologic manifestations of GPA utilizing systemic tacrolimus with oral prednisone successfully in 1 patient with GPA and a history of recurrent lower extremity nodules and ulcers.18 The efficacy of RTX in limited GPA was good in a small study of 8 patients. However, the study had only 1 patient with purpura and 1 patient with a subcutaneous nodule.19 Several other case series and studies have included patients with various cutaneous findings associated with GPA.5-7,9,11 However, they did not comment specifically on skin response to treatment, and the focus appeared to be on other organ system involvement. One case series did report improvement of lower extremity gangrene with RTX therapy for ANCA-associated vasculitis.8 Our report demonstrates a case of severe skin disease that responded well to RTX. It is common to have various skin findings in GPA, and our patient presented with notable skin disease. Although skin findings may not be the more life-threatening manifestations of the disease, they can be quite debilitating, as shown in our case report.
Our patient with notable leg ulcerations required hospitalization due to GPA and received RTX in addition to corticosteroids for treatment. We observed a rapid and dramatic improvement in the skin findings, which seemed to exceed expectations from steroids alone. The other manifestations of the disease including lung nodules also improved. Although cyclophosphamide and corticosteroids have been quite successful in induction of remission, cyclophosphamide is not without serious adverse effects. There also are some patients who have contraindications to cyclophosphamide or do not see successful results. In our brief review of the literature, RTX, a B cell–depleting antibody, has shown to have success in treating refractory and severe GPA. There is little reported specifically about treating the skin manifestations of GPA. A few studies and case reports mention skin findings but do not comment on the success of RTX in treating them. Although the severity of other organ involvement in GPA may take precedence, the skin findings can be quite debilitating, as in our patient. Patients with GPA and notable skin findings may benefit from RTX, and it would be beneficial to include these results in future studies using RTX to treat GPA.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116:488-498.
- Plosker GL, Figgitt DP. Rituximab: a review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs. 2003;63:803-843.
- Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793-2806.
- FDA approves Rituxan to treat two rare disorders [news release]. Silver Spring, MD: US Food and Drug Administration; April 19, 2011. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm251946.htm. Accessed January 6, 2017.
- Cartin-Ceba R, Golbin JM, Keogh KA, et al. Rituximab for remission induction and maintenance in refractory granulomatosis with polyangiitis (Wegener’s): ten-year experience at a single center. Arthritis Rheum. 2012;64:3770-3778.
- Keogh KA, Ytterberg SR, Fervenza FC, et al. Rituximab for refractory Wegener’s granulomatosis: report of a prospective, open-label pilot trial. Am J Respir Crit Care Med. 2006;173:180-187.
- Dalkilic E, Alkis N, Kamali S. Rituximab as a new therapeutic option in granulomatosis with polyangiitis: a report of two cases. Mod Rheumatol. 2012;22:463-466.
- Eriksson P. Nine patients with anti-neutrophil cytoplasmic antibody-positive vasculitis successfully treated with rituximab. J Intern Med. 2005;257:540-548.
- Oristrell J, Bejarano G, Jordana R, et al. Effectiveness of rituximab in severe Wegener’s granulomatosis: report of two cases and review of the literature. Open Respir Med J. 2009;3:94-99.
- Martinez Del Pero M, Chaudhry A, Jones RB, et al. B-cell depletion with rituximab for refractory head and neck Wegener’s granulomatosis: a cohort study. Clin Otolaryngol. 2009;34:328-335.
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221-232.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211-220.
- Aries PM, Hellmich B, Voswinkel J, et al. Lack of efficacy of rituximab in Wegener’s granulomatosis with refractory granulomatous manifestations. Ann Rheum Dis. 2006;65:853-858.
- Holle JU, Dubrau C, Herlyn K, et al. Rituximab for refractory granulomatosis with polyangiitis (Wegener’s granulomatosis): comparison of efficacy in granulomatous versus vasculitic manifestations. Ann Rheum Dis. 2012;71:327-333.
- Taylor SR, Salama AD, Joshi L, et al. Rituximab is effective in the treatment of refractory ophthalmic Wegener’s granulomatosis. Arthritis Rheum. 2009;60:1540-1547.
- Joshi L, Lightman SL, Salama AD, et al. Rituximab in refractory ophthalmic Wegener’s granulomatosis: PR3 titers may predict relapse, but repeat treatment can be effective. Ophthalmol. 2011;118:2498-2503.
- Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum. 2001;44:912-920.
- Wenzel J, Montag S, Wilsmann-Theis D, et al. Successful treatment of recalcitrant Wegener’s granulomatosis of the skin with tacrolimus (Prograf). Br J Dermatol. 2004;151:927-928.
- Seo P, Specks U, Keogh KA. Efficacy of rituximab in limited Wegener’s granulomatosis with refractory granulomatous manifestations. J Rheumatol. 2008;35:2017-2023.
To the Editor:
A 52-year-old woman with a history of arthralgia, rhinitis, sinusitis, and episodic epistaxis was admitted to the hospital with multiple nonhealing severe leg ulcerations. She noticed subcutaneous nodules on the legs 6 months prior to the development of ulcers. The lesions progressed from subcutaneous nodules to red-black skin discoloration, blister formation, and eventually ulceration. Over a period of months, the ulcers were treated with several courses of antibiotics and wound care including a single surgical debridement of one of the ulcers on the dorsum of the right foot. These interventions did not make a remarkable impact on ulcer healing.
On physical examination, the patient had scattered 4- to 5-mm palpable purpura on the knees, elbows, and feet bilaterally. She had multiple 1- to 8-cm indurated purple ulcerations with friable surfaces and raised irregular borders on the feet, toes, and lower legs bilaterally (Figure, A–C). One notably larger ulcer was found on the anterior aspect of the left thigh (Figure, A). Scattered 5- to 15-mm eschars were present on the legs bilaterally. She also had multiple large, firm, nonerythematous dermal plaques on the thighs bilaterally that measured several centimeters. There were no oral mucosal lesions and no ulcerations above the waist.
Magnetic resonance imaging of the foot showed some surrounding cellulitis but no osteomyelitis. Chest radiograph and computed tomography revealed bilateral apical nodules. Proteinase 3–antineutrophil cytoplasmic antibody (PR3-ANCA) testing was positive. Serum complement levels were normal. An antinuclear antibody test and rheumatoid factor were both negative. Skin biopsies were obtained from the thigh ulcer, foot ulcer, and purpuric lesions on the right knee. The results demonstrated leukocytoclastic vasculitis and neutrophilic small vessel vasculitis with necrotizing neutrophilic dermatitis and panniculitis. Granulomatosis with polyangiitis (GPA) was diagnosed based on these findings.
Initial inpatient treatment included intravenous methylprednisolone (100 mg every 8 hours for 3 doses), followed by oral prednisone 60 mg daily. Two weeks later the ulcers were reevaluated and only mild improvement had occurred with the steroids. Therefore, rituximab (RTX) was initiated at 375 mg/m2 (700 mg) intravenously once weekly for 4 weeks. After 3 doses of RTX, the ulcerations were healing dramatically and the treatment was well tolerated. A rapid prednisone taper was started, and the patient received her fourth and final dose of RTX. Two months after the initial infusion, the thigh ulcer and most of the ulcerations on the feet and lower legs had almost completely resolved. Photographs were taken 5 months after initial RTX infusion (Figure, D–F). A chest radiograph 4 months after initial RTX infusion showed resolution of lung nodules. Two months after RTX induction therapy, azathioprine was added for maintenance but was stopped due to poor tolerance. Oral methotrexate 17.5 mg once weekly was added 5 months after RTX for maintenance and was well tolerated. At that time the prednisone dose was 10 mg daily and was successfully tapered to 5 mg by 9 months after RTX induction therapy.
Granulomatosis with polyangiitis (Wegener granulomatosis) is a granulomatous small- and medium-sized vessel vasculitis that traditionally affects the upper and lower respiratory tract and kidneys.1 Skin lesions also are quite common and include palpable purpura, ulcers, vesicles, papules, and subcutaneous nodules. Patients with active GPA also tend to have ANCAs directed against proteinase 3 (PR3-ANCA). Although GPA was once considered a fatal disease, treatment with cyclophosphamide combined with corticosteroids has been shown to substantially improve outcomes.1 Rituximab, a chimeric monoclonal anti-CD20 antibody, works by depleting B lymphocytes and has been used with success to treat diseases such as lymphoma and rheumatoid arthritis.2,3 The US Food and Drug Administration approved RTX for GPA and microscopic polyangiitis in 2011, with a number of trials supporting its efficacy.4
The success of RTX in treating GPA has been documented in case reports as well as several trials with extended follow-up. A single-center observational study of 53 patients showed that RTX was safe and effective for induction and maintenance of remission in patients with refractory GPA. This study also uncovered the potential for predicting relapse based on following B cell and ANCA levels and preventing relapse by initializing further treatment.5 Other small studies and case reports have shown similar success using RTX for refractory GPA.6-10 These studies included various combinations of concurrent therapies and various follow-up intervals. The Rituximab in ANCA-Associated Vasculitis (RAVE) trial compared RTX versus cyclophosphamide for ANCA-positive vasculitis.11 This multicenter, randomized, double-blind study found that RTX was as efficacious as cyclophosphamide for induction of remission in severe GPA.The data also suggested that RTX may be superior for relapsing disease.11 Another multicenter, open-label, randomized trial (RITUXVAS) compared RTX to cyclophosphamide in ANCA-associated renal vasculitis. This trial also found the 2 treatments to be similar in both efficacy in inducing remission and adverse events.12
Some conflicting reports have appeared on the effectiveness of using RTX for the granulomatous versus vasculitic manifestations of GPA. Aires et al13 showed failure of improvement in most patients with granulomatous manifestations of GPA in a study of 8 patients. A retrospective study including 59 patients who were treated with RTX also showed that complete remission was more common in patients with primarily vasculitic manifestations, not granulomatous manifestations.14 However, some case series that included patients with refractory ophthalmic GPA, a primarily granulomatous manifestation, have found success using RTX.15,16 More studies are needed to determine if there truly is a difference and whether this difference has an effect on when to use RTX. The skin lesions our patient demonstrated were due to the vasculitic component of the disease, and consequently, the rapid and complete response we observed would be consistent with the premise that the therapy works best for vasculitis.
Most of the trials assessing the efficacy of RTX utilize a tool such as the Wegener granulomatosis-specific Birmingham Vasculitis Activity Score.17 This measure of treatment response does include a skin item, but it is part of the composite response score. Consequently, a specific statement regarding skin improvement cannot be made. Additionally, little is reported pertaining to the treatment of skin-related findings in GPA. One report did specifically address the treatment of dermatologic manifestations of GPA utilizing systemic tacrolimus with oral prednisone successfully in 1 patient with GPA and a history of recurrent lower extremity nodules and ulcers.18 The efficacy of RTX in limited GPA was good in a small study of 8 patients. However, the study had only 1 patient with purpura and 1 patient with a subcutaneous nodule.19 Several other case series and studies have included patients with various cutaneous findings associated with GPA.5-7,9,11 However, they did not comment specifically on skin response to treatment, and the focus appeared to be on other organ system involvement. One case series did report improvement of lower extremity gangrene with RTX therapy for ANCA-associated vasculitis.8 Our report demonstrates a case of severe skin disease that responded well to RTX. It is common to have various skin findings in GPA, and our patient presented with notable skin disease. Although skin findings may not be the more life-threatening manifestations of the disease, they can be quite debilitating, as shown in our case report.
Our patient with notable leg ulcerations required hospitalization due to GPA and received RTX in addition to corticosteroids for treatment. We observed a rapid and dramatic improvement in the skin findings, which seemed to exceed expectations from steroids alone. The other manifestations of the disease including lung nodules also improved. Although cyclophosphamide and corticosteroids have been quite successful in induction of remission, cyclophosphamide is not without serious adverse effects. There also are some patients who have contraindications to cyclophosphamide or do not see successful results. In our brief review of the literature, RTX, a B cell–depleting antibody, has shown to have success in treating refractory and severe GPA. There is little reported specifically about treating the skin manifestations of GPA. A few studies and case reports mention skin findings but do not comment on the success of RTX in treating them. Although the severity of other organ involvement in GPA may take precedence, the skin findings can be quite debilitating, as in our patient. Patients with GPA and notable skin findings may benefit from RTX, and it would be beneficial to include these results in future studies using RTX to treat GPA.
To the Editor:
A 52-year-old woman with a history of arthralgia, rhinitis, sinusitis, and episodic epistaxis was admitted to the hospital with multiple nonhealing severe leg ulcerations. She noticed subcutaneous nodules on the legs 6 months prior to the development of ulcers. The lesions progressed from subcutaneous nodules to red-black skin discoloration, blister formation, and eventually ulceration. Over a period of months, the ulcers were treated with several courses of antibiotics and wound care including a single surgical debridement of one of the ulcers on the dorsum of the right foot. These interventions did not make a remarkable impact on ulcer healing.
On physical examination, the patient had scattered 4- to 5-mm palpable purpura on the knees, elbows, and feet bilaterally. She had multiple 1- to 8-cm indurated purple ulcerations with friable surfaces and raised irregular borders on the feet, toes, and lower legs bilaterally (Figure, A–C). One notably larger ulcer was found on the anterior aspect of the left thigh (Figure, A). Scattered 5- to 15-mm eschars were present on the legs bilaterally. She also had multiple large, firm, nonerythematous dermal plaques on the thighs bilaterally that measured several centimeters. There were no oral mucosal lesions and no ulcerations above the waist.
Magnetic resonance imaging of the foot showed some surrounding cellulitis but no osteomyelitis. Chest radiograph and computed tomography revealed bilateral apical nodules. Proteinase 3–antineutrophil cytoplasmic antibody (PR3-ANCA) testing was positive. Serum complement levels were normal. An antinuclear antibody test and rheumatoid factor were both negative. Skin biopsies were obtained from the thigh ulcer, foot ulcer, and purpuric lesions on the right knee. The results demonstrated leukocytoclastic vasculitis and neutrophilic small vessel vasculitis with necrotizing neutrophilic dermatitis and panniculitis. Granulomatosis with polyangiitis (GPA) was diagnosed based on these findings.
Initial inpatient treatment included intravenous methylprednisolone (100 mg every 8 hours for 3 doses), followed by oral prednisone 60 mg daily. Two weeks later the ulcers were reevaluated and only mild improvement had occurred with the steroids. Therefore, rituximab (RTX) was initiated at 375 mg/m2 (700 mg) intravenously once weekly for 4 weeks. After 3 doses of RTX, the ulcerations were healing dramatically and the treatment was well tolerated. A rapid prednisone taper was started, and the patient received her fourth and final dose of RTX. Two months after the initial infusion, the thigh ulcer and most of the ulcerations on the feet and lower legs had almost completely resolved. Photographs were taken 5 months after initial RTX infusion (Figure, D–F). A chest radiograph 4 months after initial RTX infusion showed resolution of lung nodules. Two months after RTX induction therapy, azathioprine was added for maintenance but was stopped due to poor tolerance. Oral methotrexate 17.5 mg once weekly was added 5 months after RTX for maintenance and was well tolerated. At that time the prednisone dose was 10 mg daily and was successfully tapered to 5 mg by 9 months after RTX induction therapy.
Granulomatosis with polyangiitis (Wegener granulomatosis) is a granulomatous small- and medium-sized vessel vasculitis that traditionally affects the upper and lower respiratory tract and kidneys.1 Skin lesions also are quite common and include palpable purpura, ulcers, vesicles, papules, and subcutaneous nodules. Patients with active GPA also tend to have ANCAs directed against proteinase 3 (PR3-ANCA). Although GPA was once considered a fatal disease, treatment with cyclophosphamide combined with corticosteroids has been shown to substantially improve outcomes.1 Rituximab, a chimeric monoclonal anti-CD20 antibody, works by depleting B lymphocytes and has been used with success to treat diseases such as lymphoma and rheumatoid arthritis.2,3 The US Food and Drug Administration approved RTX for GPA and microscopic polyangiitis in 2011, with a number of trials supporting its efficacy.4
The success of RTX in treating GPA has been documented in case reports as well as several trials with extended follow-up. A single-center observational study of 53 patients showed that RTX was safe and effective for induction and maintenance of remission in patients with refractory GPA. This study also uncovered the potential for predicting relapse based on following B cell and ANCA levels and preventing relapse by initializing further treatment.5 Other small studies and case reports have shown similar success using RTX for refractory GPA.6-10 These studies included various combinations of concurrent therapies and various follow-up intervals. The Rituximab in ANCA-Associated Vasculitis (RAVE) trial compared RTX versus cyclophosphamide for ANCA-positive vasculitis.11 This multicenter, randomized, double-blind study found that RTX was as efficacious as cyclophosphamide for induction of remission in severe GPA.The data also suggested that RTX may be superior for relapsing disease.11 Another multicenter, open-label, randomized trial (RITUXVAS) compared RTX to cyclophosphamide in ANCA-associated renal vasculitis. This trial also found the 2 treatments to be similar in both efficacy in inducing remission and adverse events.12
Some conflicting reports have appeared on the effectiveness of using RTX for the granulomatous versus vasculitic manifestations of GPA. Aires et al13 showed failure of improvement in most patients with granulomatous manifestations of GPA in a study of 8 patients. A retrospective study including 59 patients who were treated with RTX also showed that complete remission was more common in patients with primarily vasculitic manifestations, not granulomatous manifestations.14 However, some case series that included patients with refractory ophthalmic GPA, a primarily granulomatous manifestation, have found success using RTX.15,16 More studies are needed to determine if there truly is a difference and whether this difference has an effect on when to use RTX. The skin lesions our patient demonstrated were due to the vasculitic component of the disease, and consequently, the rapid and complete response we observed would be consistent with the premise that the therapy works best for vasculitis.
Most of the trials assessing the efficacy of RTX utilize a tool such as the Wegener granulomatosis-specific Birmingham Vasculitis Activity Score.17 This measure of treatment response does include a skin item, but it is part of the composite response score. Consequently, a specific statement regarding skin improvement cannot be made. Additionally, little is reported pertaining to the treatment of skin-related findings in GPA. One report did specifically address the treatment of dermatologic manifestations of GPA utilizing systemic tacrolimus with oral prednisone successfully in 1 patient with GPA and a history of recurrent lower extremity nodules and ulcers.18 The efficacy of RTX in limited GPA was good in a small study of 8 patients. However, the study had only 1 patient with purpura and 1 patient with a subcutaneous nodule.19 Several other case series and studies have included patients with various cutaneous findings associated with GPA.5-7,9,11 However, they did not comment specifically on skin response to treatment, and the focus appeared to be on other organ system involvement. One case series did report improvement of lower extremity gangrene with RTX therapy for ANCA-associated vasculitis.8 Our report demonstrates a case of severe skin disease that responded well to RTX. It is common to have various skin findings in GPA, and our patient presented with notable skin disease. Although skin findings may not be the more life-threatening manifestations of the disease, they can be quite debilitating, as shown in our case report.
Our patient with notable leg ulcerations required hospitalization due to GPA and received RTX in addition to corticosteroids for treatment. We observed a rapid and dramatic improvement in the skin findings, which seemed to exceed expectations from steroids alone. The other manifestations of the disease including lung nodules also improved. Although cyclophosphamide and corticosteroids have been quite successful in induction of remission, cyclophosphamide is not without serious adverse effects. There also are some patients who have contraindications to cyclophosphamide or do not see successful results. In our brief review of the literature, RTX, a B cell–depleting antibody, has shown to have success in treating refractory and severe GPA. There is little reported specifically about treating the skin manifestations of GPA. A few studies and case reports mention skin findings but do not comment on the success of RTX in treating them. Although the severity of other organ involvement in GPA may take precedence, the skin findings can be quite debilitating, as in our patient. Patients with GPA and notable skin findings may benefit from RTX, and it would be beneficial to include these results in future studies using RTX to treat GPA.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116:488-498.
- Plosker GL, Figgitt DP. Rituximab: a review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs. 2003;63:803-843.
- Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793-2806.
- FDA approves Rituxan to treat two rare disorders [news release]. Silver Spring, MD: US Food and Drug Administration; April 19, 2011. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm251946.htm. Accessed January 6, 2017.
- Cartin-Ceba R, Golbin JM, Keogh KA, et al. Rituximab for remission induction and maintenance in refractory granulomatosis with polyangiitis (Wegener’s): ten-year experience at a single center. Arthritis Rheum. 2012;64:3770-3778.
- Keogh KA, Ytterberg SR, Fervenza FC, et al. Rituximab for refractory Wegener’s granulomatosis: report of a prospective, open-label pilot trial. Am J Respir Crit Care Med. 2006;173:180-187.
- Dalkilic E, Alkis N, Kamali S. Rituximab as a new therapeutic option in granulomatosis with polyangiitis: a report of two cases. Mod Rheumatol. 2012;22:463-466.
- Eriksson P. Nine patients with anti-neutrophil cytoplasmic antibody-positive vasculitis successfully treated with rituximab. J Intern Med. 2005;257:540-548.
- Oristrell J, Bejarano G, Jordana R, et al. Effectiveness of rituximab in severe Wegener’s granulomatosis: report of two cases and review of the literature. Open Respir Med J. 2009;3:94-99.
- Martinez Del Pero M, Chaudhry A, Jones RB, et al. B-cell depletion with rituximab for refractory head and neck Wegener’s granulomatosis: a cohort study. Clin Otolaryngol. 2009;34:328-335.
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221-232.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211-220.
- Aries PM, Hellmich B, Voswinkel J, et al. Lack of efficacy of rituximab in Wegener’s granulomatosis with refractory granulomatous manifestations. Ann Rheum Dis. 2006;65:853-858.
- Holle JU, Dubrau C, Herlyn K, et al. Rituximab for refractory granulomatosis with polyangiitis (Wegener’s granulomatosis): comparison of efficacy in granulomatous versus vasculitic manifestations. Ann Rheum Dis. 2012;71:327-333.
- Taylor SR, Salama AD, Joshi L, et al. Rituximab is effective in the treatment of refractory ophthalmic Wegener’s granulomatosis. Arthritis Rheum. 2009;60:1540-1547.
- Joshi L, Lightman SL, Salama AD, et al. Rituximab in refractory ophthalmic Wegener’s granulomatosis: PR3 titers may predict relapse, but repeat treatment can be effective. Ophthalmol. 2011;118:2498-2503.
- Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum. 2001;44:912-920.
- Wenzel J, Montag S, Wilsmann-Theis D, et al. Successful treatment of recalcitrant Wegener’s granulomatosis of the skin with tacrolimus (Prograf). Br J Dermatol. 2004;151:927-928.
- Seo P, Specks U, Keogh KA. Efficacy of rituximab in limited Wegener’s granulomatosis with refractory granulomatous manifestations. J Rheumatol. 2008;35:2017-2023.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med. 1992;116:488-498.
- Plosker GL, Figgitt DP. Rituximab: a review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs. 2003;63:803-843.
- Cohen SB, Emery P, Greenwald MW, et al. Rituximab for rheumatoid arthritis refractory to anti-tumor necrosis factor therapy: results of a multicenter, randomized, double-blind, placebo-controlled, phase III trial evaluating primary efficacy and safety at twenty-four weeks. Arthritis Rheum. 2006;54:2793-2806.
- FDA approves Rituxan to treat two rare disorders [news release]. Silver Spring, MD: US Food and Drug Administration; April 19, 2011. http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm251946.htm. Accessed January 6, 2017.
- Cartin-Ceba R, Golbin JM, Keogh KA, et al. Rituximab for remission induction and maintenance in refractory granulomatosis with polyangiitis (Wegener’s): ten-year experience at a single center. Arthritis Rheum. 2012;64:3770-3778.
- Keogh KA, Ytterberg SR, Fervenza FC, et al. Rituximab for refractory Wegener’s granulomatosis: report of a prospective, open-label pilot trial. Am J Respir Crit Care Med. 2006;173:180-187.
- Dalkilic E, Alkis N, Kamali S. Rituximab as a new therapeutic option in granulomatosis with polyangiitis: a report of two cases. Mod Rheumatol. 2012;22:463-466.
- Eriksson P. Nine patients with anti-neutrophil cytoplasmic antibody-positive vasculitis successfully treated with rituximab. J Intern Med. 2005;257:540-548.
- Oristrell J, Bejarano G, Jordana R, et al. Effectiveness of rituximab in severe Wegener’s granulomatosis: report of two cases and review of the literature. Open Respir Med J. 2009;3:94-99.
- Martinez Del Pero M, Chaudhry A, Jones RB, et al. B-cell depletion with rituximab for refractory head and neck Wegener’s granulomatosis: a cohort study. Clin Otolaryngol. 2009;34:328-335.
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221-232.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211-220.
- Aries PM, Hellmich B, Voswinkel J, et al. Lack of efficacy of rituximab in Wegener’s granulomatosis with refractory granulomatous manifestations. Ann Rheum Dis. 2006;65:853-858.
- Holle JU, Dubrau C, Herlyn K, et al. Rituximab for refractory granulomatosis with polyangiitis (Wegener’s granulomatosis): comparison of efficacy in granulomatous versus vasculitic manifestations. Ann Rheum Dis. 2012;71:327-333.
- Taylor SR, Salama AD, Joshi L, et al. Rituximab is effective in the treatment of refractory ophthalmic Wegener’s granulomatosis. Arthritis Rheum. 2009;60:1540-1547.
- Joshi L, Lightman SL, Salama AD, et al. Rituximab in refractory ophthalmic Wegener’s granulomatosis: PR3 titers may predict relapse, but repeat treatment can be effective. Ophthalmol. 2011;118:2498-2503.
- Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS). Arthritis Rheum. 2001;44:912-920.
- Wenzel J, Montag S, Wilsmann-Theis D, et al. Successful treatment of recalcitrant Wegener’s granulomatosis of the skin with tacrolimus (Prograf). Br J Dermatol. 2004;151:927-928.
- Seo P, Specks U, Keogh KA. Efficacy of rituximab in limited Wegener’s granulomatosis with refractory granulomatous manifestations. J Rheumatol. 2008;35:2017-2023.
Practice Points
- Recognition of the dermatologic manifestations of granulomatosis with polyangiitis (GPA) may aid in an earlier diagnosis and appropriate treatment.
- Rituximab combined with corticosteroids may be a rapid and effective therapy for severe cutaneous ulcers related to GPA.
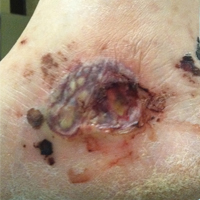
Cardiofaciocutaneous Syndrome and the Dermatologist’s Contribution to Diagnosis
To the Editor:
RASopathies, a class of developmental disorders, are caused by mutations in genes that encode protein components of the RAS/mitogen-activated protein kinase (MAPK) pathway. Each syndrome exhibits its phenotypic features; however, because all of them cause dysregulation of the RAS/MAPK pathway, there are numerous overlapping phenotypic features between the syndromes including cardiac defects, cutaneous abnormalities, characteristic facial features, neurocognitive impairment, and increased risk for developing some neoplastic disorders.
Cardiofaciocutaneous (CFC) syndrome is a RASopathy and is a genetic sporadic disease characterized by multiple congenital anomalies associated with mental retardation. It has a complex dermatological phenotype with many cutaneous features that can be helpful to differentiate CFC syndrome from Noonan and Costello syndromes, which also are classified as RASopathies.
A 3-year-old girl presented with skin xerosis and follicular hyperkeratosis of the face, neck, trunk, and limbs (Figure 1). Facial follicular hyperkeratotic papules on an erythematous base were associated with alopecia of the eyebrows (ulerythema ophryogenes). Hair was sparse and curly (Figure 2A). Facial dysmorphic features included a prominent forehead with bitemporal constriction, bilateral ptosis, a broad nasal base, lip contour in a Cupid’s bow, low-set earlobes with creases (Figure 2B), and a short and webbed neck.
Congenital heart disease, hypothyroidism, bilateral hydronephrosis, delayed motor development, and seizures were noted for the first 2 years. Brain computed tomography detected a dilated ventricular system with hydrocephalus. There was no family history of consanguinity.
Pregnancy was complicated by polyhydramnios and preeclampsia. The neonate was delivered at full-term and was readmitted at 6 days of age due to respiratory failure secondary to congenital chylothorax. Cardiac malformation was diagnosed as the ostium secundum atrial septal defect and interventricular and atrioventricular septal defects. Up to this point she was being treated for Turner syndrome.
The RASopathies are a class of human genetic syndromes that are caused by germ line mutations in genes that encode components of the RAS/MAPK pathway.1 There are many syndromes classified as RASopathies (Table).2,3
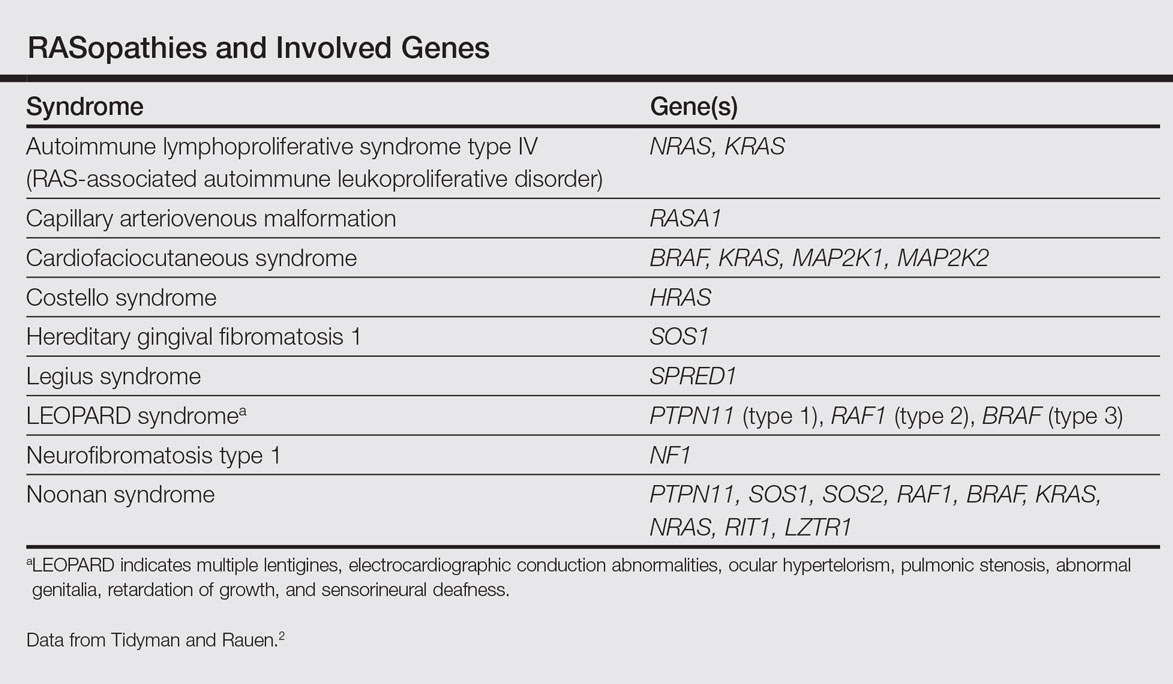
Cardiofaciocutaneous syndrome (Online Mendelian Inheritance in Man [OMIM] 115150) is a genetic disorder first described by Reynolds et al4 and is characterized by several cutaneous abnormalities, cardiac defects, dysmorphic craniofacial features, gastrointestinal dysmotility, and mental retardation. It occurs sporadically and is caused by functional activation of mutations in 4 different genes—BRAF, KRAS, MAP2K1, MAP2K2—of the RAS extracellular signal–regulated kinase molecular cascade that regulates cell differentiation, proliferation, and apoptosis.1
As a RASopathy, CFC syndrome is a member of a family of syndromes with similar phenotypes, which includes mainly Noonan and Costello syndromes. Psychomotor retardation and physical anomalies, the common denominator of all syndromes, may be explained by the effects of the mutations during early development.5,6
In CFC, relative macrocephaly, prominent forehead, bitemporal constriction, absence of eyebrows, palpebral ptosis, broad nasal root, bulbous nasal tip, and small chin commonly are found. The eyes are widely spaced and the palpebral fissures are downward slanting with epicanthic folds.1,4,7
Follicular keratosis of the arms, legs, and face occurs in 80% of cases of CFC and ulerythema ophryogenes with sparse eyebrows in 90% of cases. Sparse, curly, and slow-growing hair is found in 93% of patients. Xerotic scaly skin, hyperkeratosis of the palms and soles, infantile hemangiomas, and multiple melanocytic nevi also may occur.8
Cardiac abnormalities are seen in 75.7% of patients.1 Other features include mental retardation, delayed motor development, and structural abnormalities in the central nervous system, as well as seizures and electroencephalogram abnormalities. Unlike Noonan and Costello syndromes, it is unclear if patients with CFC syndrome are at an increased risk for cancer.1
Noonan syndrome (OMIM #163950) is a disorder characterized by congenital heart defects, short stature, skeletal abnormalities, distinctive facial dysmorphic features, and variable cognitive deficits. Other associated features include cryptorchidism, lymphatic dysplasia, bleeding tendency, and occasional hematologic malignancies during childhood. This syndrome is related to mutations in the PTPN11, SOS1, SOS2, RAF1, BRAF, KRAS, NRAS, RIT1, and LZTR1 genes.2,9-11 The typical ear shape and placement in Noonan syndrome is oval with an overfolded helix that is low set and posteriorly angulated, which is uncommon in CFC syndrome. Noonan syndrome is characterized by an inverted triangular face; hypertelorism; blue or blue-green iris color; webbed neck; limited skin involvement, mainly represented by multiple nevi; and a much milder developmental delay compared to CFC and Costello syndromes.1,11
Costello syndrome (OMIM #218040) is a rare condition comprised of severe postnatal feeding difficulties, mental retardation, coarse facial features, cardiovascular abnormalities (eg, pulmonic stenosis, hypertrophic cardiomyopathy, atrial tachycardia), tumor predisposition, and skin and musculoskeletal abnormalities.12 Costello syndrome is clinically diagnosed. This syndrome shows coarse facies with macrocephaly, downward-slanting palpebral fissures, epicanthal folds, bulbous nose with anteversed nostrils and low nasal bridge, full cheeks, large mouth, thick lips, large tongue, nasal papillomas, cutis laxa, low-set ears, short neck, diffuse skin hyperpigmentation, ulnar deviation of the hands, and nail dystrophy that are not observed in CFC. It is now accepted that the term Costello syndrome should be reserved for patients with HRAS mutation because of the specific risk profile of these patients.12 Remarkably, patients with Costello syndrome are at increased tumor risk (eg, rhabdomyosarcoma, neuroblastoma, bladder carcinoma).2,12
The diagnosis of CFC syndrome is purely clinical. There have been many attempts to delineate the syndrome, but none of the described traits are pathognomonic. In 2002, Kavamura et al7 created the CFC index, a useful diagnostic approach based on 82 clinical characteristics and their frequencies in the CFC population.
Skin abnormalities are helpful manifestations to differentiate CFC syndrome from Noonan and Costello syndromes. Patients with CFC syndrome present with follicular hyperkeratosis and absent eyebrows. Absent eyebrows, narrowed temples, and Cupid’s bow lip are hallmark features of CFC syndrome and are absent in Noonan and Costello syndromes. The presentation of palmoplantar hyperkeratosis also is a differentiating feature; in patients with Costello syndrome, it is found outside the pressure zones, whereas in those with CFC syndrome, it is present mainly in the pressure zones.1 Dermatologists can assist geneticists in the differential diagnosis of these syndromes.
The treatment of disorders with follicular plugging and xerosis is challenging. Emollients with urea, glycolic acid, and lactic acid could improve the appearance of the skin. Treatment with mutated MEK gene inhibitors is under investigation to restore normal development of affected embryos with CFC.2,13 This case and theoretical data show that skin manifestations can be helpful to differentiate CFC syndrome from other RASopathies such as Noonan and Costello syndromes.
- Roberts A, Allanson J, Jadico SK, et al. The cardiofaciocutaneous syndrome. J Med Genet. 2006;43:833-842.
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230-236.
- Stevenson D, Viskochil D, Mao R, et al. Legius syndrome. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK47312.
- Reynolds JF, Neri G, Herrmann JP, et al. New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement—the CFC syndrome. Am J Med Genet. 1986;25:413-427.
- Zenker M, Lehmann K, Schulz AL, et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet. 2007;44:131-135.
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287-1290.
- Kavamura MI, Peres CA, Alchorne MM, et al. CFC index for the diagnosis of cardiofaciocutaneous syndrome. Am J Med Genet. 2002;112:12-16.
- Siegel DH, McKenzie J, Frieden IJ, et al. Dermatological findings in 61 mutation-positive individuals with cardiofaciocutaneous syndrome. Br J Dermatol. 2011;164:521-529.
- Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol. 2010;1:2-26.
- Lo FS, Lin JL, Kuo MT, et al. Noonan syndrome caused by germline KRAS mutation in Taiwan: report of two patients and a review of the literature. Eur J Pediatr. 2009;168:919-923.
- Allanson JE, Roberts AE. Noonan syndrome. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1124/.
- Gripp KW, Lin AE. Costello syndrome. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1507/.
- Inoue S, Moriya M, Watanabe Y, et al. New BRAF knockin mice provide a pathogenetic mechanism of developmental defects and a therapeutic approach in cardio-facio-cutaneous syndrome. Hum Mol Genet. 2014;23:6553-6566.
To the Editor:
RASopathies, a class of developmental disorders, are caused by mutations in genes that encode protein components of the RAS/mitogen-activated protein kinase (MAPK) pathway. Each syndrome exhibits its phenotypic features; however, because all of them cause dysregulation of the RAS/MAPK pathway, there are numerous overlapping phenotypic features between the syndromes including cardiac defects, cutaneous abnormalities, characteristic facial features, neurocognitive impairment, and increased risk for developing some neoplastic disorders.
Cardiofaciocutaneous (CFC) syndrome is a RASopathy and is a genetic sporadic disease characterized by multiple congenital anomalies associated with mental retardation. It has a complex dermatological phenotype with many cutaneous features that can be helpful to differentiate CFC syndrome from Noonan and Costello syndromes, which also are classified as RASopathies.
A 3-year-old girl presented with skin xerosis and follicular hyperkeratosis of the face, neck, trunk, and limbs (Figure 1). Facial follicular hyperkeratotic papules on an erythematous base were associated with alopecia of the eyebrows (ulerythema ophryogenes). Hair was sparse and curly (Figure 2A). Facial dysmorphic features included a prominent forehead with bitemporal constriction, bilateral ptosis, a broad nasal base, lip contour in a Cupid’s bow, low-set earlobes with creases (Figure 2B), and a short and webbed neck.
Congenital heart disease, hypothyroidism, bilateral hydronephrosis, delayed motor development, and seizures were noted for the first 2 years. Brain computed tomography detected a dilated ventricular system with hydrocephalus. There was no family history of consanguinity.
Pregnancy was complicated by polyhydramnios and preeclampsia. The neonate was delivered at full-term and was readmitted at 6 days of age due to respiratory failure secondary to congenital chylothorax. Cardiac malformation was diagnosed as the ostium secundum atrial septal defect and interventricular and atrioventricular septal defects. Up to this point she was being treated for Turner syndrome.
The RASopathies are a class of human genetic syndromes that are caused by germ line mutations in genes that encode components of the RAS/MAPK pathway.1 There are many syndromes classified as RASopathies (Table).2,3
Cardiofaciocutaneous syndrome (Online Mendelian Inheritance in Man [OMIM] 115150) is a genetic disorder first described by Reynolds et al4 and is characterized by several cutaneous abnormalities, cardiac defects, dysmorphic craniofacial features, gastrointestinal dysmotility, and mental retardation. It occurs sporadically and is caused by functional activation of mutations in 4 different genes—BRAF, KRAS, MAP2K1, MAP2K2—of the RAS extracellular signal–regulated kinase molecular cascade that regulates cell differentiation, proliferation, and apoptosis.1
As a RASopathy, CFC syndrome is a member of a family of syndromes with similar phenotypes, which includes mainly Noonan and Costello syndromes. Psychomotor retardation and physical anomalies, the common denominator of all syndromes, may be explained by the effects of the mutations during early development.5,6
In CFC, relative macrocephaly, prominent forehead, bitemporal constriction, absence of eyebrows, palpebral ptosis, broad nasal root, bulbous nasal tip, and small chin commonly are found. The eyes are widely spaced and the palpebral fissures are downward slanting with epicanthic folds.1,4,7
Follicular keratosis of the arms, legs, and face occurs in 80% of cases of CFC and ulerythema ophryogenes with sparse eyebrows in 90% of cases. Sparse, curly, and slow-growing hair is found in 93% of patients. Xerotic scaly skin, hyperkeratosis of the palms and soles, infantile hemangiomas, and multiple melanocytic nevi also may occur.8
Cardiac abnormalities are seen in 75.7% of patients.1 Other features include mental retardation, delayed motor development, and structural abnormalities in the central nervous system, as well as seizures and electroencephalogram abnormalities. Unlike Noonan and Costello syndromes, it is unclear if patients with CFC syndrome are at an increased risk for cancer.1
Noonan syndrome (OMIM #163950) is a disorder characterized by congenital heart defects, short stature, skeletal abnormalities, distinctive facial dysmorphic features, and variable cognitive deficits. Other associated features include cryptorchidism, lymphatic dysplasia, bleeding tendency, and occasional hematologic malignancies during childhood. This syndrome is related to mutations in the PTPN11, SOS1, SOS2, RAF1, BRAF, KRAS, NRAS, RIT1, and LZTR1 genes.2,9-11 The typical ear shape and placement in Noonan syndrome is oval with an overfolded helix that is low set and posteriorly angulated, which is uncommon in CFC syndrome. Noonan syndrome is characterized by an inverted triangular face; hypertelorism; blue or blue-green iris color; webbed neck; limited skin involvement, mainly represented by multiple nevi; and a much milder developmental delay compared to CFC and Costello syndromes.1,11
Costello syndrome (OMIM #218040) is a rare condition comprised of severe postnatal feeding difficulties, mental retardation, coarse facial features, cardiovascular abnormalities (eg, pulmonic stenosis, hypertrophic cardiomyopathy, atrial tachycardia), tumor predisposition, and skin and musculoskeletal abnormalities.12 Costello syndrome is clinically diagnosed. This syndrome shows coarse facies with macrocephaly, downward-slanting palpebral fissures, epicanthal folds, bulbous nose with anteversed nostrils and low nasal bridge, full cheeks, large mouth, thick lips, large tongue, nasal papillomas, cutis laxa, low-set ears, short neck, diffuse skin hyperpigmentation, ulnar deviation of the hands, and nail dystrophy that are not observed in CFC. It is now accepted that the term Costello syndrome should be reserved for patients with HRAS mutation because of the specific risk profile of these patients.12 Remarkably, patients with Costello syndrome are at increased tumor risk (eg, rhabdomyosarcoma, neuroblastoma, bladder carcinoma).2,12
The diagnosis of CFC syndrome is purely clinical. There have been many attempts to delineate the syndrome, but none of the described traits are pathognomonic. In 2002, Kavamura et al7 created the CFC index, a useful diagnostic approach based on 82 clinical characteristics and their frequencies in the CFC population.
Skin abnormalities are helpful manifestations to differentiate CFC syndrome from Noonan and Costello syndromes. Patients with CFC syndrome present with follicular hyperkeratosis and absent eyebrows. Absent eyebrows, narrowed temples, and Cupid’s bow lip are hallmark features of CFC syndrome and are absent in Noonan and Costello syndromes. The presentation of palmoplantar hyperkeratosis also is a differentiating feature; in patients with Costello syndrome, it is found outside the pressure zones, whereas in those with CFC syndrome, it is present mainly in the pressure zones.1 Dermatologists can assist geneticists in the differential diagnosis of these syndromes.
The treatment of disorders with follicular plugging and xerosis is challenging. Emollients with urea, glycolic acid, and lactic acid could improve the appearance of the skin. Treatment with mutated MEK gene inhibitors is under investigation to restore normal development of affected embryos with CFC.2,13 This case and theoretical data show that skin manifestations can be helpful to differentiate CFC syndrome from other RASopathies such as Noonan and Costello syndromes.
To the Editor:
RASopathies, a class of developmental disorders, are caused by mutations in genes that encode protein components of the RAS/mitogen-activated protein kinase (MAPK) pathway. Each syndrome exhibits its phenotypic features; however, because all of them cause dysregulation of the RAS/MAPK pathway, there are numerous overlapping phenotypic features between the syndromes including cardiac defects, cutaneous abnormalities, characteristic facial features, neurocognitive impairment, and increased risk for developing some neoplastic disorders.
Cardiofaciocutaneous (CFC) syndrome is a RASopathy and is a genetic sporadic disease characterized by multiple congenital anomalies associated with mental retardation. It has a complex dermatological phenotype with many cutaneous features that can be helpful to differentiate CFC syndrome from Noonan and Costello syndromes, which also are classified as RASopathies.
A 3-year-old girl presented with skin xerosis and follicular hyperkeratosis of the face, neck, trunk, and limbs (Figure 1). Facial follicular hyperkeratotic papules on an erythematous base were associated with alopecia of the eyebrows (ulerythema ophryogenes). Hair was sparse and curly (Figure 2A). Facial dysmorphic features included a prominent forehead with bitemporal constriction, bilateral ptosis, a broad nasal base, lip contour in a Cupid’s bow, low-set earlobes with creases (Figure 2B), and a short and webbed neck.
Congenital heart disease, hypothyroidism, bilateral hydronephrosis, delayed motor development, and seizures were noted for the first 2 years. Brain computed tomography detected a dilated ventricular system with hydrocephalus. There was no family history of consanguinity.
Pregnancy was complicated by polyhydramnios and preeclampsia. The neonate was delivered at full-term and was readmitted at 6 days of age due to respiratory failure secondary to congenital chylothorax. Cardiac malformation was diagnosed as the ostium secundum atrial septal defect and interventricular and atrioventricular septal defects. Up to this point she was being treated for Turner syndrome.
The RASopathies are a class of human genetic syndromes that are caused by germ line mutations in genes that encode components of the RAS/MAPK pathway.1 There are many syndromes classified as RASopathies (Table).2,3
Cardiofaciocutaneous syndrome (Online Mendelian Inheritance in Man [OMIM] 115150) is a genetic disorder first described by Reynolds et al4 and is characterized by several cutaneous abnormalities, cardiac defects, dysmorphic craniofacial features, gastrointestinal dysmotility, and mental retardation. It occurs sporadically and is caused by functional activation of mutations in 4 different genes—BRAF, KRAS, MAP2K1, MAP2K2—of the RAS extracellular signal–regulated kinase molecular cascade that regulates cell differentiation, proliferation, and apoptosis.1
As a RASopathy, CFC syndrome is a member of a family of syndromes with similar phenotypes, which includes mainly Noonan and Costello syndromes. Psychomotor retardation and physical anomalies, the common denominator of all syndromes, may be explained by the effects of the mutations during early development.5,6
In CFC, relative macrocephaly, prominent forehead, bitemporal constriction, absence of eyebrows, palpebral ptosis, broad nasal root, bulbous nasal tip, and small chin commonly are found. The eyes are widely spaced and the palpebral fissures are downward slanting with epicanthic folds.1,4,7
Follicular keratosis of the arms, legs, and face occurs in 80% of cases of CFC and ulerythema ophryogenes with sparse eyebrows in 90% of cases. Sparse, curly, and slow-growing hair is found in 93% of patients. Xerotic scaly skin, hyperkeratosis of the palms and soles, infantile hemangiomas, and multiple melanocytic nevi also may occur.8
Cardiac abnormalities are seen in 75.7% of patients.1 Other features include mental retardation, delayed motor development, and structural abnormalities in the central nervous system, as well as seizures and electroencephalogram abnormalities. Unlike Noonan and Costello syndromes, it is unclear if patients with CFC syndrome are at an increased risk for cancer.1
Noonan syndrome (OMIM #163950) is a disorder characterized by congenital heart defects, short stature, skeletal abnormalities, distinctive facial dysmorphic features, and variable cognitive deficits. Other associated features include cryptorchidism, lymphatic dysplasia, bleeding tendency, and occasional hematologic malignancies during childhood. This syndrome is related to mutations in the PTPN11, SOS1, SOS2, RAF1, BRAF, KRAS, NRAS, RIT1, and LZTR1 genes.2,9-11 The typical ear shape and placement in Noonan syndrome is oval with an overfolded helix that is low set and posteriorly angulated, which is uncommon in CFC syndrome. Noonan syndrome is characterized by an inverted triangular face; hypertelorism; blue or blue-green iris color; webbed neck; limited skin involvement, mainly represented by multiple nevi; and a much milder developmental delay compared to CFC and Costello syndromes.1,11
Costello syndrome (OMIM #218040) is a rare condition comprised of severe postnatal feeding difficulties, mental retardation, coarse facial features, cardiovascular abnormalities (eg, pulmonic stenosis, hypertrophic cardiomyopathy, atrial tachycardia), tumor predisposition, and skin and musculoskeletal abnormalities.12 Costello syndrome is clinically diagnosed. This syndrome shows coarse facies with macrocephaly, downward-slanting palpebral fissures, epicanthal folds, bulbous nose with anteversed nostrils and low nasal bridge, full cheeks, large mouth, thick lips, large tongue, nasal papillomas, cutis laxa, low-set ears, short neck, diffuse skin hyperpigmentation, ulnar deviation of the hands, and nail dystrophy that are not observed in CFC. It is now accepted that the term Costello syndrome should be reserved for patients with HRAS mutation because of the specific risk profile of these patients.12 Remarkably, patients with Costello syndrome are at increased tumor risk (eg, rhabdomyosarcoma, neuroblastoma, bladder carcinoma).2,12
The diagnosis of CFC syndrome is purely clinical. There have been many attempts to delineate the syndrome, but none of the described traits are pathognomonic. In 2002, Kavamura et al7 created the CFC index, a useful diagnostic approach based on 82 clinical characteristics and their frequencies in the CFC population.
Skin abnormalities are helpful manifestations to differentiate CFC syndrome from Noonan and Costello syndromes. Patients with CFC syndrome present with follicular hyperkeratosis and absent eyebrows. Absent eyebrows, narrowed temples, and Cupid’s bow lip are hallmark features of CFC syndrome and are absent in Noonan and Costello syndromes. The presentation of palmoplantar hyperkeratosis also is a differentiating feature; in patients with Costello syndrome, it is found outside the pressure zones, whereas in those with CFC syndrome, it is present mainly in the pressure zones.1 Dermatologists can assist geneticists in the differential diagnosis of these syndromes.
The treatment of disorders with follicular plugging and xerosis is challenging. Emollients with urea, glycolic acid, and lactic acid could improve the appearance of the skin. Treatment with mutated MEK gene inhibitors is under investigation to restore normal development of affected embryos with CFC.2,13 This case and theoretical data show that skin manifestations can be helpful to differentiate CFC syndrome from other RASopathies such as Noonan and Costello syndromes.
- Roberts A, Allanson J, Jadico SK, et al. The cardiofaciocutaneous syndrome. J Med Genet. 2006;43:833-842.
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230-236.
- Stevenson D, Viskochil D, Mao R, et al. Legius syndrome. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK47312.
- Reynolds JF, Neri G, Herrmann JP, et al. New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement—the CFC syndrome. Am J Med Genet. 1986;25:413-427.
- Zenker M, Lehmann K, Schulz AL, et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet. 2007;44:131-135.
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287-1290.
- Kavamura MI, Peres CA, Alchorne MM, et al. CFC index for the diagnosis of cardiofaciocutaneous syndrome. Am J Med Genet. 2002;112:12-16.
- Siegel DH, McKenzie J, Frieden IJ, et al. Dermatological findings in 61 mutation-positive individuals with cardiofaciocutaneous syndrome. Br J Dermatol. 2011;164:521-529.
- Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol. 2010;1:2-26.
- Lo FS, Lin JL, Kuo MT, et al. Noonan syndrome caused by germline KRAS mutation in Taiwan: report of two patients and a review of the literature. Eur J Pediatr. 2009;168:919-923.
- Allanson JE, Roberts AE. Noonan syndrome. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1124/.
- Gripp KW, Lin AE. Costello syndrome. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1507/.
- Inoue S, Moriya M, Watanabe Y, et al. New BRAF knockin mice provide a pathogenetic mechanism of developmental defects and a therapeutic approach in cardio-facio-cutaneous syndrome. Hum Mol Genet. 2014;23:6553-6566.
- Roberts A, Allanson J, Jadico SK, et al. The cardiofaciocutaneous syndrome. J Med Genet. 2006;43:833-842.
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230-236.
- Stevenson D, Viskochil D, Mao R, et al. Legius syndrome. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK47312.
- Reynolds JF, Neri G, Herrmann JP, et al. New multiple congenital anomalies/mental retardation syndrome with cardio-facio-cutaneous involvement—the CFC syndrome. Am J Med Genet. 1986;25:413-427.
- Zenker M, Lehmann K, Schulz AL, et al. Expansion of the genotypic and phenotypic spectrum in patients with KRAS germline mutations. J Med Genet. 2007;44:131-135.
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287-1290.
- Kavamura MI, Peres CA, Alchorne MM, et al. CFC index for the diagnosis of cardiofaciocutaneous syndrome. Am J Med Genet. 2002;112:12-16.
- Siegel DH, McKenzie J, Frieden IJ, et al. Dermatological findings in 61 mutation-positive individuals with cardiofaciocutaneous syndrome. Br J Dermatol. 2011;164:521-529.
- Tartaglia M, Zampino G, Gelb BD. Noonan syndrome: clinical aspects and molecular pathogenesis. Mol Syndromol. 2010;1:2-26.
- Lo FS, Lin JL, Kuo MT, et al. Noonan syndrome caused by germline KRAS mutation in Taiwan: report of two patients and a review of the literature. Eur J Pediatr. 2009;168:919-923.
- Allanson JE, Roberts AE. Noonan syndrome. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1124/.
- Gripp KW, Lin AE. Costello syndrome. In: Pagon RA, Bird TD, Dolan CR, et al, eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/books/NBK1507/.
- Inoue S, Moriya M, Watanabe Y, et al. New BRAF knockin mice provide a pathogenetic mechanism of developmental defects and a therapeutic approach in cardio-facio-cutaneous syndrome. Hum Mol Genet. 2014;23:6553-6566.
Practice Points
- RASopathies, a class of developmental disorders, are caused by mutations in genes that encode protein components of the RAS/mitogen-activated protein kinase pathway. Cardiofaciocutaneous (CFC) syndrome is a RASopathy.
- Skin manifestations may help in differentiating CFC syndrome from other RASopathies.