User login
Does the U.S. Food and Drug Administration control the price of drugs? Do other countries approve drugs faster than the United States? Can the FDA tell doctors how to prescribe drugs? There is a lot of misinformation about exactly what the FDA does and doesn’t do. Here are the top four FDA myths … BUSTED!
Myth 1: Patients cannot get access to drugs they need because the FDA is too slow in approving drugs.
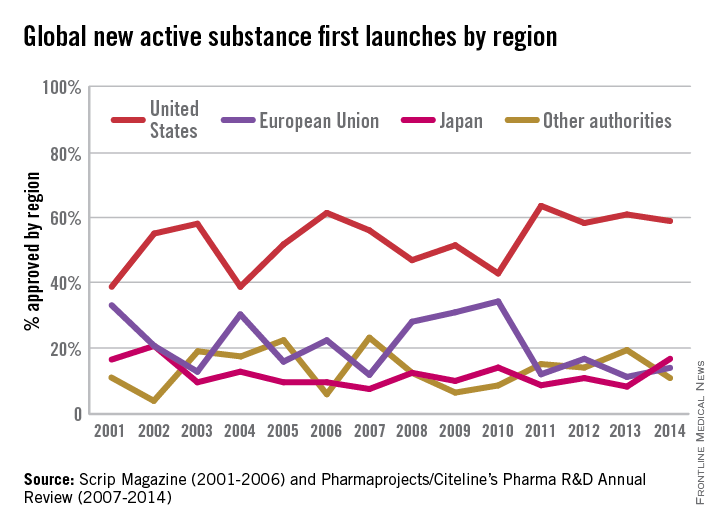
FACT: The FDA approves drugs faster on average than all other developed nations. That is 40 days faster than Japan; 70 days faster than Canada; and 174 days faster than the European Union (EU).1

People often say that drugs are being developed and made available to patients in other countries before they are marketed here in the United States. Physicians often gripe that their patients can’t get access to medicines that are available to others overseas. These anecdotes have led to the urban legend that the FDA evaluates and approves drugs at a slower rate than foreign agencies, especially the European Medicines Agency (EMA).
In a recent report by the British-based Centre for Innovation in Regulatory Science, “over 75% of the new drugs approved by Japan, EU, Canada, Australia, Switzerland, and FDA, from 2004 to 2013, were approved first by FDA.” The FDA continues to lead among regulatory agencies in drug approvals that ensure patients get access to the lifesaving medicines they need.
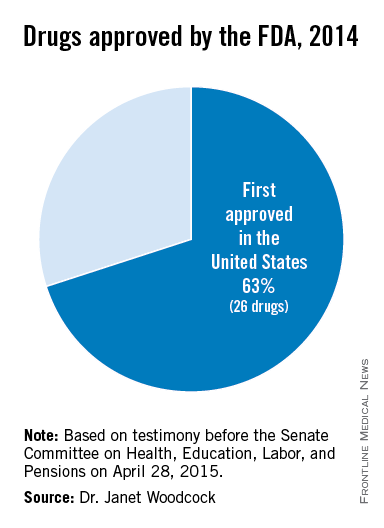
The U.S. Food and Drug Administration has significantly increased the speed by which new drugs reach patients in need. Between 2006 and 2010, three out of four new drugs approved in both Europe and the U.S. were first approved by the FDA. In the specific example of cancer, the FDA also approved 32 of the 35 prospective cancer drugs between 2003-2010, whereas the EMA only approved 26. Among those cancer drugs approved, over 90% were first approved by the FDA. Just last year, almost two-thirds of the novel drugs approved by the FDA (26 of 41 or about 63%) were approved in the United States before receiving approval in any other country.2
Myth 2: The FDA controls drug prices.
FACT: The FDA has no legal authority whatsoever to investigate or control the prices charged for marketed drugs. Manufacturers, distributors, and retailers establish these prices.
Drug prices do have a direct impact on the ability of people to cope with their illnesses as well as meet other expenses and the FDA recognizes that other factors beyond its purview, including insurance coverage and drug pricing, can determine patient access to drugs. However, the FDA has no legal authority whatsoever to investigate or control the prices charged for marketed drugs. Manufacturers, distributors, and retailers establish these prices.
The FDA is responsible for assuring the American public that the drugs approved today are safe and effective. In carrying out this responsibility, the FDA also works under the law to make medicines accessible to doctors and their patients through strategies such as expanding access to generic medicines, reducing the time and cost of bringing new medicines to market, as well as ensuring safe use practices and the most up-to-date information for health care professionals and patients.
Myth 3: The FDA conducts clinical trials.
FACT: The FDA does not develop, manufacture, or test drugs. The FDA does oversee the testing of new drugs to evaluate the safety and effectiveness profile in clinical trials.
Drug manufacturers submit full reports on the drug so that the FDA can evaluate their data. These studies are conducted to answer whether the drug in study works for the proposed indication. By completing a full review and analysis of the submitted data, the FDA assesses the benefit and risk of each drug to determine if the drug will be approved.
Companies must submit an investigational new drug (IND) application to the FDA before testing can begin. The IND is the means through which the sponsor obtains an exemption from the FDA to begin testing and legally transporting or distributing a drug under investigation across state lines.
The FDA’s role in the development of a new drug begins when the drug sponsor (usually the manufacturer or potential marketer) wants to test the diagnostic or therapeutic potential in humans (after having screened the new molecule for pharmacological activity and acute toxicity potential in animals). It is at this point that the molecule changes in legal status under the Federal Food, Drug, and Cosmetic Act and becomes a new drug subject to specific requirements of the drug regulatory system.3
Myth 4: The FDA regulates the practice of medicine.
FACT: The FDA regulates the approval of a drug, but not how it is prescribed. Doctors are permitted to prescribe a drug for any reason they think is medically appropriate.
The FDA creates the “drug label” in conjunction with the drug company to ensure that promotional claims are not false or misleading. The drug label of FDA-approved drugs provides information about the drug, including the approved doses and how the drug should be given to treat the medical condition for which it was approved.
However, physicians often use drugs in a way that is different from that described in the FDA-approved drug label, an “off-label” use. This can mean that the drug is:
• Used for a different disease or medical condition.
• Given in a different way (such as by a different route).
• Given in a different dose.
Off-label is also called “nonapproved” or “unapproved” use of a drug. Because drug makers have not put their drugs through the studies required by the FDA to officially approve the drug for new uses, new uses for these drugs should be taken with caution as the FDA has not reviewed the clinical evidence to support the new use. Any off-label drug usage should include a detailed conversation between physician and patient.4
Dr. John J. Whyte is the director of Professional Affairs and Stakeholder Engagement (PASE) in the FDA’s Center for Drug Evaluation and Research (CDER).
Does the U.S. Food and Drug Administration control the price of drugs? Do other countries approve drugs faster than the United States? Can the FDA tell doctors how to prescribe drugs? There is a lot of misinformation about exactly what the FDA does and doesn’t do. Here are the top four FDA myths … BUSTED!
Myth 1: Patients cannot get access to drugs they need because the FDA is too slow in approving drugs.
FACT: The FDA approves drugs faster on average than all other developed nations. That is 40 days faster than Japan; 70 days faster than Canada; and 174 days faster than the European Union (EU).1
People often say that drugs are being developed and made available to patients in other countries before they are marketed here in the United States. Physicians often gripe that their patients can’t get access to medicines that are available to others overseas. These anecdotes have led to the urban legend that the FDA evaluates and approves drugs at a slower rate than foreign agencies, especially the European Medicines Agency (EMA).
In a recent report by the British-based Centre for Innovation in Regulatory Science, “over 75% of the new drugs approved by Japan, EU, Canada, Australia, Switzerland, and FDA, from 2004 to 2013, were approved first by FDA.” The FDA continues to lead among regulatory agencies in drug approvals that ensure patients get access to the lifesaving medicines they need.
The U.S. Food and Drug Administration has significantly increased the speed by which new drugs reach patients in need. Between 2006 and 2010, three out of four new drugs approved in both Europe and the U.S. were first approved by the FDA. In the specific example of cancer, the FDA also approved 32 of the 35 prospective cancer drugs between 2003-2010, whereas the EMA only approved 26. Among those cancer drugs approved, over 90% were first approved by the FDA. Just last year, almost two-thirds of the novel drugs approved by the FDA (26 of 41 or about 63%) were approved in the United States before receiving approval in any other country.2
Myth 2: The FDA controls drug prices.
FACT: The FDA has no legal authority whatsoever to investigate or control the prices charged for marketed drugs. Manufacturers, distributors, and retailers establish these prices.
Drug prices do have a direct impact on the ability of people to cope with their illnesses as well as meet other expenses and the FDA recognizes that other factors beyond its purview, including insurance coverage and drug pricing, can determine patient access to drugs. However, the FDA has no legal authority whatsoever to investigate or control the prices charged for marketed drugs. Manufacturers, distributors, and retailers establish these prices.
The FDA is responsible for assuring the American public that the drugs approved today are safe and effective. In carrying out this responsibility, the FDA also works under the law to make medicines accessible to doctors and their patients through strategies such as expanding access to generic medicines, reducing the time and cost of bringing new medicines to market, as well as ensuring safe use practices and the most up-to-date information for health care professionals and patients.
Myth 3: The FDA conducts clinical trials.
FACT: The FDA does not develop, manufacture, or test drugs. The FDA does oversee the testing of new drugs to evaluate the safety and effectiveness profile in clinical trials.
Drug manufacturers submit full reports on the drug so that the FDA can evaluate their data. These studies are conducted to answer whether the drug in study works for the proposed indication. By completing a full review and analysis of the submitted data, the FDA assesses the benefit and risk of each drug to determine if the drug will be approved.
Companies must submit an investigational new drug (IND) application to the FDA before testing can begin. The IND is the means through which the sponsor obtains an exemption from the FDA to begin testing and legally transporting or distributing a drug under investigation across state lines.
The FDA’s role in the development of a new drug begins when the drug sponsor (usually the manufacturer or potential marketer) wants to test the diagnostic or therapeutic potential in humans (after having screened the new molecule for pharmacological activity and acute toxicity potential in animals). It is at this point that the molecule changes in legal status under the Federal Food, Drug, and Cosmetic Act and becomes a new drug subject to specific requirements of the drug regulatory system.3
Myth 4: The FDA regulates the practice of medicine.
FACT: The FDA regulates the approval of a drug, but not how it is prescribed. Doctors are permitted to prescribe a drug for any reason they think is medically appropriate.
The FDA creates the “drug label” in conjunction with the drug company to ensure that promotional claims are not false or misleading. The drug label of FDA-approved drugs provides information about the drug, including the approved doses and how the drug should be given to treat the medical condition for which it was approved.
However, physicians often use drugs in a way that is different from that described in the FDA-approved drug label, an “off-label” use. This can mean that the drug is:
• Used for a different disease or medical condition.
• Given in a different way (such as by a different route).
• Given in a different dose.
Off-label is also called “nonapproved” or “unapproved” use of a drug. Because drug makers have not put their drugs through the studies required by the FDA to officially approve the drug for new uses, new uses for these drugs should be taken with caution as the FDA has not reviewed the clinical evidence to support the new use. Any off-label drug usage should include a detailed conversation between physician and patient.4
Dr. John J. Whyte is the director of Professional Affairs and Stakeholder Engagement (PASE) in the FDA’s Center for Drug Evaluation and Research (CDER).
Does the U.S. Food and Drug Administration control the price of drugs? Do other countries approve drugs faster than the United States? Can the FDA tell doctors how to prescribe drugs? There is a lot of misinformation about exactly what the FDA does and doesn’t do. Here are the top four FDA myths … BUSTED!
Myth 1: Patients cannot get access to drugs they need because the FDA is too slow in approving drugs.
FACT: The FDA approves drugs faster on average than all other developed nations. That is 40 days faster than Japan; 70 days faster than Canada; and 174 days faster than the European Union (EU).1
People often say that drugs are being developed and made available to patients in other countries before they are marketed here in the United States. Physicians often gripe that their patients can’t get access to medicines that are available to others overseas. These anecdotes have led to the urban legend that the FDA evaluates and approves drugs at a slower rate than foreign agencies, especially the European Medicines Agency (EMA).
In a recent report by the British-based Centre for Innovation in Regulatory Science, “over 75% of the new drugs approved by Japan, EU, Canada, Australia, Switzerland, and FDA, from 2004 to 2013, were approved first by FDA.” The FDA continues to lead among regulatory agencies in drug approvals that ensure patients get access to the lifesaving medicines they need.
The U.S. Food and Drug Administration has significantly increased the speed by which new drugs reach patients in need. Between 2006 and 2010, three out of four new drugs approved in both Europe and the U.S. were first approved by the FDA. In the specific example of cancer, the FDA also approved 32 of the 35 prospective cancer drugs between 2003-2010, whereas the EMA only approved 26. Among those cancer drugs approved, over 90% were first approved by the FDA. Just last year, almost two-thirds of the novel drugs approved by the FDA (26 of 41 or about 63%) were approved in the United States before receiving approval in any other country.2
Myth 2: The FDA controls drug prices.
FACT: The FDA has no legal authority whatsoever to investigate or control the prices charged for marketed drugs. Manufacturers, distributors, and retailers establish these prices.
Drug prices do have a direct impact on the ability of people to cope with their illnesses as well as meet other expenses and the FDA recognizes that other factors beyond its purview, including insurance coverage and drug pricing, can determine patient access to drugs. However, the FDA has no legal authority whatsoever to investigate or control the prices charged for marketed drugs. Manufacturers, distributors, and retailers establish these prices.
The FDA is responsible for assuring the American public that the drugs approved today are safe and effective. In carrying out this responsibility, the FDA also works under the law to make medicines accessible to doctors and their patients through strategies such as expanding access to generic medicines, reducing the time and cost of bringing new medicines to market, as well as ensuring safe use practices and the most up-to-date information for health care professionals and patients.
Myth 3: The FDA conducts clinical trials.
FACT: The FDA does not develop, manufacture, or test drugs. The FDA does oversee the testing of new drugs to evaluate the safety and effectiveness profile in clinical trials.
Drug manufacturers submit full reports on the drug so that the FDA can evaluate their data. These studies are conducted to answer whether the drug in study works for the proposed indication. By completing a full review and analysis of the submitted data, the FDA assesses the benefit and risk of each drug to determine if the drug will be approved.
Companies must submit an investigational new drug (IND) application to the FDA before testing can begin. The IND is the means through which the sponsor obtains an exemption from the FDA to begin testing and legally transporting or distributing a drug under investigation across state lines.
The FDA’s role in the development of a new drug begins when the drug sponsor (usually the manufacturer or potential marketer) wants to test the diagnostic or therapeutic potential in humans (after having screened the new molecule for pharmacological activity and acute toxicity potential in animals). It is at this point that the molecule changes in legal status under the Federal Food, Drug, and Cosmetic Act and becomes a new drug subject to specific requirements of the drug regulatory system.3
Myth 4: The FDA regulates the practice of medicine.
FACT: The FDA regulates the approval of a drug, but not how it is prescribed. Doctors are permitted to prescribe a drug for any reason they think is medically appropriate.
The FDA creates the “drug label” in conjunction with the drug company to ensure that promotional claims are not false or misleading. The drug label of FDA-approved drugs provides information about the drug, including the approved doses and how the drug should be given to treat the medical condition for which it was approved.
However, physicians often use drugs in a way that is different from that described in the FDA-approved drug label, an “off-label” use. This can mean that the drug is:
• Used for a different disease or medical condition.
• Given in a different way (such as by a different route).
• Given in a different dose.
Off-label is also called “nonapproved” or “unapproved” use of a drug. Because drug makers have not put their drugs through the studies required by the FDA to officially approve the drug for new uses, new uses for these drugs should be taken with caution as the FDA has not reviewed the clinical evidence to support the new use. Any off-label drug usage should include a detailed conversation between physician and patient.4
Dr. John J. Whyte is the director of Professional Affairs and Stakeholder Engagement (PASE) in the FDA’s Center for Drug Evaluation and Research (CDER).
