User login
More on ‘treatment resistance’
I wanted to thank Dr. Nasrallah for his bold article, “Treatment resistance is a myth!” (From the Editor,
Stanley N. Caroff, MD
Professor of Psychiatry
Perelman School of Medicine
University of Pennsylvania
Philadelphia, Pennsylvania
I thought Dr. Nasrallah’s editorial on treatment resistance was excellent. In my experience, bipolar depression often is not diagnosed in patients with long-standing depression. These patients do worse on antidepressants, which is interpreted by the clinician as treatment-resistant major depressive disorder. The other issue for me is that individuals with bipolar disorder with psychotic features are often diagnosed with schizophrenia or schizoaffective disorder and never receive a trial of lithium, which could alter the course of their illness in a dramatic fashion. For me, the underutilization of lithium is a real quality problem in our field. Keep up the good work!
Bruce J. Schwartz, MD
Deputy Chairman & Professor of PsychiatryMontefiore Medical Center and Albert Einstein College of Medicine
New York, New York
Are psychiatric advances still science fiction?
I read with great enthusiasm Dr. Nasrallah’s editorial “Today’s psychiatric neuroscience advances were science fiction during my residency” (From the Editor,
I have spent all my professional life serving in the public sector, mainly in New York, and can tell you that many of the brain exploration methods, methodologies, and clinical advances mentioned in this article unfortunately are still a dream for us. Still, we remain hopeful that someday those transformative advances will come to us, too, especially as the technology innovates and improves!
Vania Castillo, MD
New York, New York
Dr. Nasrallah responds
Thank you for your comments. Please remember that every single treatment you are currently using in the public mental health system was a research discovery at one point in the past, and it took many years to bring it to clinical practice. Translating basic neuroscience discoveries, such as the ones I mentioned in my editorial, into clinical practice not only takes time to develop and get approved for use, but also requires substantial funding and a cadre of psychiatric physician-scientists, both of which are in short supply.
“Warp speed” COVID-19 vaccine development was possible only because the deadly pandemic became such an urgent national crisis that the government opened its coffers and diverted billions of dollars to pharmaceutical companies, with a massive infrastructure of human talent and biotechnology, making this veritable “moonshot” a reality in 1 year instead of many. Regrettably, even though neuropsychiatric disorders are a serious societal plague that causes disability and early mortality from suicide, homicide, substance use, cardiovascular risk, and accelerated aging, they do not command the urgency of an infectious viral pandemic that rapidly killed millions and shut down societies all over the world.
You probably heard the saying “a journey of a thousand miles begins with a single step.” I believe we are more than one step—maybe more than 100 steps—toward the type of breakthroughs that we all crave for our long-suffering psychiatric patients. I am grateful for the medical advances we have made over the past 10 to 15 years, such as neuromodulation, rapid-acting parenteral antidepressants, nondopaminergic antipsychotics, therapeutic hallucinogens, early recognition and intervention, and many promising neurobiologic leads and novel therapeutic targets for the brain disorders we deal with every day.
The brain is the most complex, challenging, and physically inaccessible organ to explore and treat. In medicine, we can do heart, lung, liver, and kidney biopsies, but it is far too dangerous to do brain biopsies that would help uncover the molecular and cellular underpinnings of neuropsychiatric disorders. Yet thankfully, our knowledge of the brain structure and function in health and disease has grown by >100,000% over the past few decades compared to the preceding millennia of dark ignorance. Someday, we shall overcome.
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in their letters, or with manufacturers of competing products.
I wanted to thank Dr. Nasrallah for his bold article, “Treatment resistance is a myth!” (From the Editor,
Stanley N. Caroff, MD
Professor of Psychiatry
Perelman School of Medicine
University of Pennsylvania
Philadelphia, Pennsylvania
I thought Dr. Nasrallah’s editorial on treatment resistance was excellent. In my experience, bipolar depression often is not diagnosed in patients with long-standing depression. These patients do worse on antidepressants, which is interpreted by the clinician as treatment-resistant major depressive disorder. The other issue for me is that individuals with bipolar disorder with psychotic features are often diagnosed with schizophrenia or schizoaffective disorder and never receive a trial of lithium, which could alter the course of their illness in a dramatic fashion. For me, the underutilization of lithium is a real quality problem in our field. Keep up the good work!
Bruce J. Schwartz, MD
Deputy Chairman & Professor of PsychiatryMontefiore Medical Center and Albert Einstein College of Medicine
New York, New York
Are psychiatric advances still science fiction?
I read with great enthusiasm Dr. Nasrallah’s editorial “Today’s psychiatric neuroscience advances were science fiction during my residency” (From the Editor,
I have spent all my professional life serving in the public sector, mainly in New York, and can tell you that many of the brain exploration methods, methodologies, and clinical advances mentioned in this article unfortunately are still a dream for us. Still, we remain hopeful that someday those transformative advances will come to us, too, especially as the technology innovates and improves!
Vania Castillo, MD
New York, New York
Dr. Nasrallah responds
Thank you for your comments. Please remember that every single treatment you are currently using in the public mental health system was a research discovery at one point in the past, and it took many years to bring it to clinical practice. Translating basic neuroscience discoveries, such as the ones I mentioned in my editorial, into clinical practice not only takes time to develop and get approved for use, but also requires substantial funding and a cadre of psychiatric physician-scientists, both of which are in short supply.
“Warp speed” COVID-19 vaccine development was possible only because the deadly pandemic became such an urgent national crisis that the government opened its coffers and diverted billions of dollars to pharmaceutical companies, with a massive infrastructure of human talent and biotechnology, making this veritable “moonshot” a reality in 1 year instead of many. Regrettably, even though neuropsychiatric disorders are a serious societal plague that causes disability and early mortality from suicide, homicide, substance use, cardiovascular risk, and accelerated aging, they do not command the urgency of an infectious viral pandemic that rapidly killed millions and shut down societies all over the world.
You probably heard the saying “a journey of a thousand miles begins with a single step.” I believe we are more than one step—maybe more than 100 steps—toward the type of breakthroughs that we all crave for our long-suffering psychiatric patients. I am grateful for the medical advances we have made over the past 10 to 15 years, such as neuromodulation, rapid-acting parenteral antidepressants, nondopaminergic antipsychotics, therapeutic hallucinogens, early recognition and intervention, and many promising neurobiologic leads and novel therapeutic targets for the brain disorders we deal with every day.
The brain is the most complex, challenging, and physically inaccessible organ to explore and treat. In medicine, we can do heart, lung, liver, and kidney biopsies, but it is far too dangerous to do brain biopsies that would help uncover the molecular and cellular underpinnings of neuropsychiatric disorders. Yet thankfully, our knowledge of the brain structure and function in health and disease has grown by >100,000% over the past few decades compared to the preceding millennia of dark ignorance. Someday, we shall overcome.
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in their letters, or with manufacturers of competing products.
I wanted to thank Dr. Nasrallah for his bold article, “Treatment resistance is a myth!” (From the Editor,
Stanley N. Caroff, MD
Professor of Psychiatry
Perelman School of Medicine
University of Pennsylvania
Philadelphia, Pennsylvania
I thought Dr. Nasrallah’s editorial on treatment resistance was excellent. In my experience, bipolar depression often is not diagnosed in patients with long-standing depression. These patients do worse on antidepressants, which is interpreted by the clinician as treatment-resistant major depressive disorder. The other issue for me is that individuals with bipolar disorder with psychotic features are often diagnosed with schizophrenia or schizoaffective disorder and never receive a trial of lithium, which could alter the course of their illness in a dramatic fashion. For me, the underutilization of lithium is a real quality problem in our field. Keep up the good work!
Bruce J. Schwartz, MD
Deputy Chairman & Professor of PsychiatryMontefiore Medical Center and Albert Einstein College of Medicine
New York, New York
Are psychiatric advances still science fiction?
I read with great enthusiasm Dr. Nasrallah’s editorial “Today’s psychiatric neuroscience advances were science fiction during my residency” (From the Editor,
I have spent all my professional life serving in the public sector, mainly in New York, and can tell you that many of the brain exploration methods, methodologies, and clinical advances mentioned in this article unfortunately are still a dream for us. Still, we remain hopeful that someday those transformative advances will come to us, too, especially as the technology innovates and improves!
Vania Castillo, MD
New York, New York
Dr. Nasrallah responds
Thank you for your comments. Please remember that every single treatment you are currently using in the public mental health system was a research discovery at one point in the past, and it took many years to bring it to clinical practice. Translating basic neuroscience discoveries, such as the ones I mentioned in my editorial, into clinical practice not only takes time to develop and get approved for use, but also requires substantial funding and a cadre of psychiatric physician-scientists, both of which are in short supply.
“Warp speed” COVID-19 vaccine development was possible only because the deadly pandemic became such an urgent national crisis that the government opened its coffers and diverted billions of dollars to pharmaceutical companies, with a massive infrastructure of human talent and biotechnology, making this veritable “moonshot” a reality in 1 year instead of many. Regrettably, even though neuropsychiatric disorders are a serious societal plague that causes disability and early mortality from suicide, homicide, substance use, cardiovascular risk, and accelerated aging, they do not command the urgency of an infectious viral pandemic that rapidly killed millions and shut down societies all over the world.
You probably heard the saying “a journey of a thousand miles begins with a single step.” I believe we are more than one step—maybe more than 100 steps—toward the type of breakthroughs that we all crave for our long-suffering psychiatric patients. I am grateful for the medical advances we have made over the past 10 to 15 years, such as neuromodulation, rapid-acting parenteral antidepressants, nondopaminergic antipsychotics, therapeutic hallucinogens, early recognition and intervention, and many promising neurobiologic leads and novel therapeutic targets for the brain disorders we deal with every day.
The brain is the most complex, challenging, and physically inaccessible organ to explore and treat. In medicine, we can do heart, lung, liver, and kidney biopsies, but it is far too dangerous to do brain biopsies that would help uncover the molecular and cellular underpinnings of neuropsychiatric disorders. Yet thankfully, our knowledge of the brain structure and function in health and disease has grown by >100,000% over the past few decades compared to the preceding millennia of dark ignorance. Someday, we shall overcome.
Disclosures
The authors report no financial relationships with any companies whose products are mentioned in their letters, or with manufacturers of competing products.
Tardive dyskinesia is theme of awards competition for early career psychiatrists
Important advances in neuroscience and clinical psychiatry have been achieved in recent years, but there are significant gaps in knowledge and much that we don’t understand about the brain and behavior. Further advances depend on cultivating and supporting a new generation of dedicated basic science and clinical investigators. While there is a compelling need to attract, recruit, and encourage talented individuals to pursue scholarly interests, competing life and career demands often prove daunting.

The theme of the competition this year concerning tardive dyskinesia is timely and consistent with the mission of NMSIS to promote knowledge on neurologic side effects of antipsychotic drugs. Tardive dyskinesia can have a negative impact on the social, psychological, and physical well-being of patients; it remains a legacy of past treatment with antipsychotics; it is an increasing concern among an ever widening population of patients receiving even newer antipsychotics; and there are now two Food and Drug Administration–approved treatments for the disorder. Early career psychiatrists may have had limited instruction on tardive dyskinesia, which has not received prominent attention in curricular programs in recent years. Thus, in addition to supporting scholarly work and research experience, the 2018 Promising Scholars Award Program aims to promote knowledge and skills in managing patients with tardive dyskinesia.
Specific learning objectives are:
- Participants will learn the steps necessary to prepare a scientific manuscript for publication.
- Participants will review comments by expert referees and learn to incorporate and respond to the peer review process.
- Participants will review the evidence related to the diagnosis and treatment of tardive dyskinesia.
- Participants will be introduced to the spectrum of educational and networking opportunities at the Institute for Psychiatric Services conference.
In the past, this program was very popular and gained national recognition among psychiatric trainees. Numerous submitted papers were accepted for publication in peer-reviewed journals after the competition was completed.
Instructions for manuscript preparation are:
- First author must be a student, resident, or fellow.
- Papers should address specific issues related to the theme of tardive dyskinesia and be no longer than 15 double-spaced typed pages in length (excluding references and illustrations).
- Literature reviews, case reports, or studies that are original and newly developed or recently published are acceptable.
- Reviews and feedback will be provided by a panel of academic psychiatrists.
- Papers will be judged on relevance to tardive dyskinesia, originality, scholarship, scientific rigor, valid methodology, clinical significance, and organization.
To participate, papers and curriculum vitae of the first author must be submitted by July 1, 2018, to Dianne Daugherty by email at dianne@mhaus.org. Winners will be announced by Aug. 10, 2018. For additional information, write to dianne@mhaus.org or visit www.mhaus.org/nmsis/about-us/what-is-nmsis.
Dr. Caroff, professor of psychiatry, Corporal Michael J. Crescenz VA Medical Center and at the University of Pennsylvania, both in Philadelphia, is director of the NMSIS. He served as consultant to Neurocrine Biosciences and Teva Pharmaceutical Industries, and receives research grant funding from Neurocrine Biosciences.
Important advances in neuroscience and clinical psychiatry have been achieved in recent years, but there are significant gaps in knowledge and much that we don’t understand about the brain and behavior. Further advances depend on cultivating and supporting a new generation of dedicated basic science and clinical investigators. While there is a compelling need to attract, recruit, and encourage talented individuals to pursue scholarly interests, competing life and career demands often prove daunting.
The theme of the competition this year concerning tardive dyskinesia is timely and consistent with the mission of NMSIS to promote knowledge on neurologic side effects of antipsychotic drugs. Tardive dyskinesia can have a negative impact on the social, psychological, and physical well-being of patients; it remains a legacy of past treatment with antipsychotics; it is an increasing concern among an ever widening population of patients receiving even newer antipsychotics; and there are now two Food and Drug Administration–approved treatments for the disorder. Early career psychiatrists may have had limited instruction on tardive dyskinesia, which has not received prominent attention in curricular programs in recent years. Thus, in addition to supporting scholarly work and research experience, the 2018 Promising Scholars Award Program aims to promote knowledge and skills in managing patients with tardive dyskinesia.
Specific learning objectives are:
- Participants will learn the steps necessary to prepare a scientific manuscript for publication.
- Participants will review comments by expert referees and learn to incorporate and respond to the peer review process.
- Participants will review the evidence related to the diagnosis and treatment of tardive dyskinesia.
- Participants will be introduced to the spectrum of educational and networking opportunities at the Institute for Psychiatric Services conference.
In the past, this program was very popular and gained national recognition among psychiatric trainees. Numerous submitted papers were accepted for publication in peer-reviewed journals after the competition was completed.
Instructions for manuscript preparation are:
- First author must be a student, resident, or fellow.
- Papers should address specific issues related to the theme of tardive dyskinesia and be no longer than 15 double-spaced typed pages in length (excluding references and illustrations).
- Literature reviews, case reports, or studies that are original and newly developed or recently published are acceptable.
- Reviews and feedback will be provided by a panel of academic psychiatrists.
- Papers will be judged on relevance to tardive dyskinesia, originality, scholarship, scientific rigor, valid methodology, clinical significance, and organization.
To participate, papers and curriculum vitae of the first author must be submitted by July 1, 2018, to Dianne Daugherty by email at dianne@mhaus.org. Winners will be announced by Aug. 10, 2018. For additional information, write to dianne@mhaus.org or visit www.mhaus.org/nmsis/about-us/what-is-nmsis.
Dr. Caroff, professor of psychiatry, Corporal Michael J. Crescenz VA Medical Center and at the University of Pennsylvania, both in Philadelphia, is director of the NMSIS. He served as consultant to Neurocrine Biosciences and Teva Pharmaceutical Industries, and receives research grant funding from Neurocrine Biosciences.
Important advances in neuroscience and clinical psychiatry have been achieved in recent years, but there are significant gaps in knowledge and much that we don’t understand about the brain and behavior. Further advances depend on cultivating and supporting a new generation of dedicated basic science and clinical investigators. While there is a compelling need to attract, recruit, and encourage talented individuals to pursue scholarly interests, competing life and career demands often prove daunting.
The theme of the competition this year concerning tardive dyskinesia is timely and consistent with the mission of NMSIS to promote knowledge on neurologic side effects of antipsychotic drugs. Tardive dyskinesia can have a negative impact on the social, psychological, and physical well-being of patients; it remains a legacy of past treatment with antipsychotics; it is an increasing concern among an ever widening population of patients receiving even newer antipsychotics; and there are now two Food and Drug Administration–approved treatments for the disorder. Early career psychiatrists may have had limited instruction on tardive dyskinesia, which has not received prominent attention in curricular programs in recent years. Thus, in addition to supporting scholarly work and research experience, the 2018 Promising Scholars Award Program aims to promote knowledge and skills in managing patients with tardive dyskinesia.
Specific learning objectives are:
- Participants will learn the steps necessary to prepare a scientific manuscript for publication.
- Participants will review comments by expert referees and learn to incorporate and respond to the peer review process.
- Participants will review the evidence related to the diagnosis and treatment of tardive dyskinesia.
- Participants will be introduced to the spectrum of educational and networking opportunities at the Institute for Psychiatric Services conference.
In the past, this program was very popular and gained national recognition among psychiatric trainees. Numerous submitted papers were accepted for publication in peer-reviewed journals after the competition was completed.
Instructions for manuscript preparation are:
- First author must be a student, resident, or fellow.
- Papers should address specific issues related to the theme of tardive dyskinesia and be no longer than 15 double-spaced typed pages in length (excluding references and illustrations).
- Literature reviews, case reports, or studies that are original and newly developed or recently published are acceptable.
- Reviews and feedback will be provided by a panel of academic psychiatrists.
- Papers will be judged on relevance to tardive dyskinesia, originality, scholarship, scientific rigor, valid methodology, clinical significance, and organization.
To participate, papers and curriculum vitae of the first author must be submitted by July 1, 2018, to Dianne Daugherty by email at dianne@mhaus.org. Winners will be announced by Aug. 10, 2018. For additional information, write to dianne@mhaus.org or visit www.mhaus.org/nmsis/about-us/what-is-nmsis.
Dr. Caroff, professor of psychiatry, Corporal Michael J. Crescenz VA Medical Center and at the University of Pennsylvania, both in Philadelphia, is director of the NMSIS. He served as consultant to Neurocrine Biosciences and Teva Pharmaceutical Industries, and receives research grant funding from Neurocrine Biosciences.
Tardive dyskinesia is theme of awards competition for early career psychiatrists
Important advances in neuroscience and clinical psychiatry have been achieved in recent years, but there are significant gaps in knowledge and much that we don’t understand about the brain and behavior. Further advances depend on cultivating and supporting a new generation of dedicated basic science and clinical investigators. While there is a compelling need to attract, recruit, and encourage talented individuals to pursue scholarly interests, competing life and career demands often prove daunting.
The theme of the competition this year concerning tardive dyskinesia is timely and consistent with the mission of NMSIS to promote knowledge on neurologic side effects of antipsychotic drugs. Tardive dyskinesia can have a negative impact on the social, psychological, and physical well-being of patients; it remains a legacy of past treatment with antipsychotics; it is an increasing concern among an ever widening population of patients receiving even newer antipsychotics; and there are now two Food and Drug Administration–approved treatments for the disorder. Early career psychiatrists may have had limited instruction on tardive dyskinesia, which has not received prominent attention in curricular programs in recent years. Thus, in addition to supporting scholarly work and research experience, the 2018 Promising Scholars Award Program aims to promote knowledge and skills in managing patients with tardive dyskinesia.
Specific learning objectives are:
- Participants will learn the steps necessary to prepare a scientific manuscript for publication.
- Participants will review comments by expert referees and learn to incorporate and respond to the peer review process.
- Participants will review the evidence related to the diagnosis and treatment of tardive dyskinesia.
- Participants will be introduced to the spectrum of educational and networking opportunities at the Institute for Psychiatric Services conference.
In the past, this program was very popular and gained national recognition among psychiatric trainees. Numerous submitted papers were accepted for publication in peer-reviewed journals after the competition was completed.
Instructions for manuscript preparation are:
- First author must be a student, resident, or fellow.
- Papers should address specific issues related to the theme of tardive dyskinesia and be no longer than 15 double-spaced typed pages in length (excluding references and illustrations).
- Literature reviews, case reports, or studies that are original and newly developed or recently published are acceptable.
- Reviews and feedback will be provided by a panel of academic psychiatrists.
- Papers will be judged on relevance to tardive dyskinesia, originality, scholarship, scientific rigor, valid methodology, clinical significance, and organization.
To participate, papers and curriculum vitae of the first author must be submitted by July 1, 2018, to Dianne Daugherty by email at dianne@mhaus.org. Winners will be announced by Aug. 10, 2018. For additional information, write to dianne@mhaus.org or visit www.mhaus.org/nmsis/about-us/what-is-nmsis.
Dr. Caroff, professor of psychiatry, Corporal Michael J. Crescenz VA Medical Center and at the University of Pennsylvania, both in Philadelphia, is director of the NMSIS. He served as consultant to Neurocrine Biosciences and Teva Pharmaceutical Industries, and receives research grant funding from Neurocrine Biosciences.
Important advances in neuroscience and clinical psychiatry have been achieved in recent years, but there are significant gaps in knowledge and much that we don’t understand about the brain and behavior. Further advances depend on cultivating and supporting a new generation of dedicated basic science and clinical investigators. While there is a compelling need to attract, recruit, and encourage talented individuals to pursue scholarly interests, competing life and career demands often prove daunting.
The theme of the competition this year concerning tardive dyskinesia is timely and consistent with the mission of NMSIS to promote knowledge on neurologic side effects of antipsychotic drugs. Tardive dyskinesia can have a negative impact on the social, psychological, and physical well-being of patients; it remains a legacy of past treatment with antipsychotics; it is an increasing concern among an ever widening population of patients receiving even newer antipsychotics; and there are now two Food and Drug Administration–approved treatments for the disorder. Early career psychiatrists may have had limited instruction on tardive dyskinesia, which has not received prominent attention in curricular programs in recent years. Thus, in addition to supporting scholarly work and research experience, the 2018 Promising Scholars Award Program aims to promote knowledge and skills in managing patients with tardive dyskinesia.
Specific learning objectives are:
- Participants will learn the steps necessary to prepare a scientific manuscript for publication.
- Participants will review comments by expert referees and learn to incorporate and respond to the peer review process.
- Participants will review the evidence related to the diagnosis and treatment of tardive dyskinesia.
- Participants will be introduced to the spectrum of educational and networking opportunities at the Institute for Psychiatric Services conference.
In the past, this program was very popular and gained national recognition among psychiatric trainees. Numerous submitted papers were accepted for publication in peer-reviewed journals after the competition was completed.
Instructions for manuscript preparation are:
- First author must be a student, resident, or fellow.
- Papers should address specific issues related to the theme of tardive dyskinesia and be no longer than 15 double-spaced typed pages in length (excluding references and illustrations).
- Literature reviews, case reports, or studies that are original and newly developed or recently published are acceptable.
- Reviews and feedback will be provided by a panel of academic psychiatrists.
- Papers will be judged on relevance to tardive dyskinesia, originality, scholarship, scientific rigor, valid methodology, clinical significance, and organization.
To participate, papers and curriculum vitae of the first author must be submitted by July 1, 2018, to Dianne Daugherty by email at dianne@mhaus.org. Winners will be announced by Aug. 10, 2018. For additional information, write to dianne@mhaus.org or visit www.mhaus.org/nmsis/about-us/what-is-nmsis.
Dr. Caroff, professor of psychiatry, Corporal Michael J. Crescenz VA Medical Center and at the University of Pennsylvania, both in Philadelphia, is director of the NMSIS. He served as consultant to Neurocrine Biosciences and Teva Pharmaceutical Industries, and receives research grant funding from Neurocrine Biosciences.
Important advances in neuroscience and clinical psychiatry have been achieved in recent years, but there are significant gaps in knowledge and much that we don’t understand about the brain and behavior. Further advances depend on cultivating and supporting a new generation of dedicated basic science and clinical investigators. While there is a compelling need to attract, recruit, and encourage talented individuals to pursue scholarly interests, competing life and career demands often prove daunting.
The theme of the competition this year concerning tardive dyskinesia is timely and consistent with the mission of NMSIS to promote knowledge on neurologic side effects of antipsychotic drugs. Tardive dyskinesia can have a negative impact on the social, psychological, and physical well-being of patients; it remains a legacy of past treatment with antipsychotics; it is an increasing concern among an ever widening population of patients receiving even newer antipsychotics; and there are now two Food and Drug Administration–approved treatments for the disorder. Early career psychiatrists may have had limited instruction on tardive dyskinesia, which has not received prominent attention in curricular programs in recent years. Thus, in addition to supporting scholarly work and research experience, the 2018 Promising Scholars Award Program aims to promote knowledge and skills in managing patients with tardive dyskinesia.
Specific learning objectives are:
- Participants will learn the steps necessary to prepare a scientific manuscript for publication.
- Participants will review comments by expert referees and learn to incorporate and respond to the peer review process.
- Participants will review the evidence related to the diagnosis and treatment of tardive dyskinesia.
- Participants will be introduced to the spectrum of educational and networking opportunities at the Institute for Psychiatric Services conference.
In the past, this program was very popular and gained national recognition among psychiatric trainees. Numerous submitted papers were accepted for publication in peer-reviewed journals after the competition was completed.
Instructions for manuscript preparation are:
- First author must be a student, resident, or fellow.
- Papers should address specific issues related to the theme of tardive dyskinesia and be no longer than 15 double-spaced typed pages in length (excluding references and illustrations).
- Literature reviews, case reports, or studies that are original and newly developed or recently published are acceptable.
- Reviews and feedback will be provided by a panel of academic psychiatrists.
- Papers will be judged on relevance to tardive dyskinesia, originality, scholarship, scientific rigor, valid methodology, clinical significance, and organization.
To participate, papers and curriculum vitae of the first author must be submitted by July 1, 2018, to Dianne Daugherty by email at dianne@mhaus.org. Winners will be announced by Aug. 10, 2018. For additional information, write to dianne@mhaus.org or visit www.mhaus.org/nmsis/about-us/what-is-nmsis.
Dr. Caroff, professor of psychiatry, Corporal Michael J. Crescenz VA Medical Center and at the University of Pennsylvania, both in Philadelphia, is director of the NMSIS. He served as consultant to Neurocrine Biosciences and Teva Pharmaceutical Industries, and receives research grant funding from Neurocrine Biosciences.
Depression and deep brain stimulation: ‘Furor therapeuticus redux’
Looking back after a long and distinguished career, Leon Eisenberg, MD, invoked the term “furor therapeuticus” to describe overzealous treatment by doctors who became frustrated with therapeutic limitations or motivated by professional enthusiasm.1
With this in mind, Dr. Eisenberg criticized expansive marketing and prescribing of psychotropic drugs in an editorial published exactly 10 years ago. He might also have questioned the current interest in deep brain stimulation (DBS) as a treatment for depression and a growing list of behavioral disorders. Initial studies of DBS in depression were promising, but recent setbacks have brought research to a scientific and ethical crossroads that compels broader discussion.

Besides uncertainties over the right targets to stimulate, identification of the right candidates for DBS treatment can be difficult. Trials of DBS recruited highly selected depressed subjects with no consensus on symptoms or biomarkers that could be used to predict who might respond. Doctors still rely on clinical symptoms to distinguish patients with melancholic depression, who respond to medications or electroconvulsive therapy and might also respond to DBS, from patients with depressed mood because of psychosocial problems, who respond to psychotherapy or social interventions.
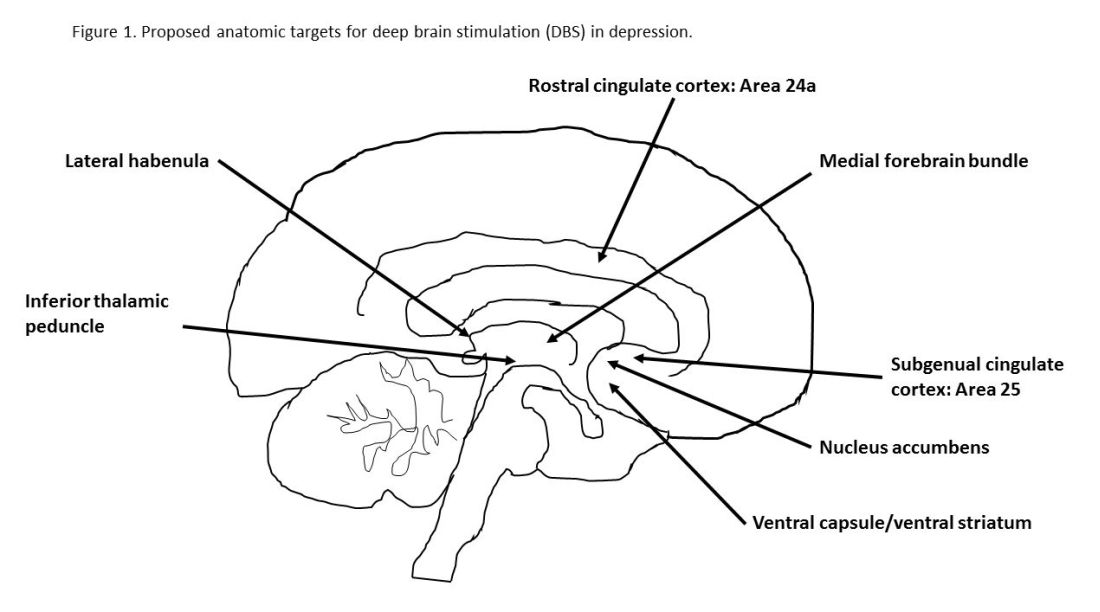
Evidence on the efficacy and safety of DBS in depression is mixed. Initial open trials were promising, with dramatic and sustained recovery in some patients, but they were limited by small numbers of subjects and a lack of randomized controls and standardized methods.4,5
DBS is not without serious side effects, and substantial maintenance costs are not always covered by insurance. So, two recent industry trials were eagerly anticipated but showed no significant differences between active and sham stimulations in depression.6,7 These disappointing results prompted soul-searching among investigators, who presented ingenious ideas for correcting shortcomings that could be tested in future trials but also raised doubts as to the prospects of DBS in depression.4,5
Given that DBS devices already are marketed for neurological disorders, regulation of practice is crucial to prevent off-label misuse in behavioral disorders.8 Federal agencies enforce rules governing DBS devices but rely on investigators and local review boards in research and on voluntary postmarketing reports by individual practitioners to monitor compliance and safety. Unscrupulous commercial interests could expand the market for these devices, as demonstrated by the proliferation of psychotropic drug prescribing decried by Dr. Eisenberg. DBS also must be restricted to specialized teams and medical centers to prevent inappropriate implantation by poorly trained providers.
Because behavioral disorders exact an enormous toll on patients, families, and society, better access to effective care and the search for better treatments must remain public health priorities.
Transformative, breakthrough discoveries in brain research will undoubtedly lead to improvements in treatment, including surgical devices in some cases, but, DBS is at risk of being exaggerated and oversold. Adverse consequences of misuse could provoke a public backlash that would have a chilling effect on vital brain research.
One possible way to prevent this is the risk evaluation and mitigation strategy established by the Food and Drug Administration to manage high-risk pharmaceuticals. The FDA mandates that certain high-risk drugs can be prescribed only if doctors are certified and only if patients are enrolled in a national registry where eligibility, course, and outcome are monitored. A similar mechanism should apply to high-risk surgical devices when used for behavioral disorders.9,10
People with behavioral disorders deserve the right to volunteer for experimental programs that offer hope of recovery for themselves and future generations, but they also deserve to be treated with the utmost scientific rigor and protection that society can provide.
Dr. Caroff is emeritus professor of psychiatry at the University of Pennsylvania, Philadelphia. He has received research grant funding from Sunovion Pharmaceuticals and serves as a consultant to Neurocrine Biosciences and TEVA.
References
1. Am J Psychiatry. 2007;164(4):552-5
2. Curr Behav Neurosci Rep. 2014;1(2):55-63
3. J Affect Disord. 2014;156:1-7
4. JAMA Psychiatry. 2016;739(5):439-40
5. Biol Psychiatry. 2016;79(4):e9-10
6. Stereotact Funct Neurosurg. 2015;93:366-9
7. Neurotherapeutics. 2014;11(3):475-84
8. J Neurol Neurosurg Psychiatry. 2014;85(9):1003-8
9. Brain Stimul. 2012;5(4):653-5
10. Fed Reg. 1977 May 23;42(99):26318-32
Looking back after a long and distinguished career, Leon Eisenberg, MD, invoked the term “furor therapeuticus” to describe overzealous treatment by doctors who became frustrated with therapeutic limitations or motivated by professional enthusiasm.1
With this in mind, Dr. Eisenberg criticized expansive marketing and prescribing of psychotropic drugs in an editorial published exactly 10 years ago. He might also have questioned the current interest in deep brain stimulation (DBS) as a treatment for depression and a growing list of behavioral disorders. Initial studies of DBS in depression were promising, but recent setbacks have brought research to a scientific and ethical crossroads that compels broader discussion.
Besides uncertainties over the right targets to stimulate, identification of the right candidates for DBS treatment can be difficult. Trials of DBS recruited highly selected depressed subjects with no consensus on symptoms or biomarkers that could be used to predict who might respond. Doctors still rely on clinical symptoms to distinguish patients with melancholic depression, who respond to medications or electroconvulsive therapy and might also respond to DBS, from patients with depressed mood because of psychosocial problems, who respond to psychotherapy or social interventions.
Evidence on the efficacy and safety of DBS in depression is mixed. Initial open trials were promising, with dramatic and sustained recovery in some patients, but they were limited by small numbers of subjects and a lack of randomized controls and standardized methods.4,5
DBS is not without serious side effects, and substantial maintenance costs are not always covered by insurance. So, two recent industry trials were eagerly anticipated but showed no significant differences between active and sham stimulations in depression.6,7 These disappointing results prompted soul-searching among investigators, who presented ingenious ideas for correcting shortcomings that could be tested in future trials but also raised doubts as to the prospects of DBS in depression.4,5
Given that DBS devices already are marketed for neurological disorders, regulation of practice is crucial to prevent off-label misuse in behavioral disorders.8 Federal agencies enforce rules governing DBS devices but rely on investigators and local review boards in research and on voluntary postmarketing reports by individual practitioners to monitor compliance and safety. Unscrupulous commercial interests could expand the market for these devices, as demonstrated by the proliferation of psychotropic drug prescribing decried by Dr. Eisenberg. DBS also must be restricted to specialized teams and medical centers to prevent inappropriate implantation by poorly trained providers.
Because behavioral disorders exact an enormous toll on patients, families, and society, better access to effective care and the search for better treatments must remain public health priorities.
Transformative, breakthrough discoveries in brain research will undoubtedly lead to improvements in treatment, including surgical devices in some cases, but, DBS is at risk of being exaggerated and oversold. Adverse consequences of misuse could provoke a public backlash that would have a chilling effect on vital brain research.
One possible way to prevent this is the risk evaluation and mitigation strategy established by the Food and Drug Administration to manage high-risk pharmaceuticals. The FDA mandates that certain high-risk drugs can be prescribed only if doctors are certified and only if patients are enrolled in a national registry where eligibility, course, and outcome are monitored. A similar mechanism should apply to high-risk surgical devices when used for behavioral disorders.9,10
People with behavioral disorders deserve the right to volunteer for experimental programs that offer hope of recovery for themselves and future generations, but they also deserve to be treated with the utmost scientific rigor and protection that society can provide.
Dr. Caroff is emeritus professor of psychiatry at the University of Pennsylvania, Philadelphia. He has received research grant funding from Sunovion Pharmaceuticals and serves as a consultant to Neurocrine Biosciences and TEVA.
References
1. Am J Psychiatry. 2007;164(4):552-5
2. Curr Behav Neurosci Rep. 2014;1(2):55-63
3. J Affect Disord. 2014;156:1-7
4. JAMA Psychiatry. 2016;739(5):439-40
5. Biol Psychiatry. 2016;79(4):e9-10
6. Stereotact Funct Neurosurg. 2015;93:366-9
7. Neurotherapeutics. 2014;11(3):475-84
8. J Neurol Neurosurg Psychiatry. 2014;85(9):1003-8
9. Brain Stimul. 2012;5(4):653-5
10. Fed Reg. 1977 May 23;42(99):26318-32
Looking back after a long and distinguished career, Leon Eisenberg, MD, invoked the term “furor therapeuticus” to describe overzealous treatment by doctors who became frustrated with therapeutic limitations or motivated by professional enthusiasm.1
With this in mind, Dr. Eisenberg criticized expansive marketing and prescribing of psychotropic drugs in an editorial published exactly 10 years ago. He might also have questioned the current interest in deep brain stimulation (DBS) as a treatment for depression and a growing list of behavioral disorders. Initial studies of DBS in depression were promising, but recent setbacks have brought research to a scientific and ethical crossroads that compels broader discussion.
Besides uncertainties over the right targets to stimulate, identification of the right candidates for DBS treatment can be difficult. Trials of DBS recruited highly selected depressed subjects with no consensus on symptoms or biomarkers that could be used to predict who might respond. Doctors still rely on clinical symptoms to distinguish patients with melancholic depression, who respond to medications or electroconvulsive therapy and might also respond to DBS, from patients with depressed mood because of psychosocial problems, who respond to psychotherapy or social interventions.
Evidence on the efficacy and safety of DBS in depression is mixed. Initial open trials were promising, with dramatic and sustained recovery in some patients, but they were limited by small numbers of subjects and a lack of randomized controls and standardized methods.4,5
DBS is not without serious side effects, and substantial maintenance costs are not always covered by insurance. So, two recent industry trials were eagerly anticipated but showed no significant differences between active and sham stimulations in depression.6,7 These disappointing results prompted soul-searching among investigators, who presented ingenious ideas for correcting shortcomings that could be tested in future trials but also raised doubts as to the prospects of DBS in depression.4,5
Given that DBS devices already are marketed for neurological disorders, regulation of practice is crucial to prevent off-label misuse in behavioral disorders.8 Federal agencies enforce rules governing DBS devices but rely on investigators and local review boards in research and on voluntary postmarketing reports by individual practitioners to monitor compliance and safety. Unscrupulous commercial interests could expand the market for these devices, as demonstrated by the proliferation of psychotropic drug prescribing decried by Dr. Eisenberg. DBS also must be restricted to specialized teams and medical centers to prevent inappropriate implantation by poorly trained providers.
Because behavioral disorders exact an enormous toll on patients, families, and society, better access to effective care and the search for better treatments must remain public health priorities.
Transformative, breakthrough discoveries in brain research will undoubtedly lead to improvements in treatment, including surgical devices in some cases, but, DBS is at risk of being exaggerated and oversold. Adverse consequences of misuse could provoke a public backlash that would have a chilling effect on vital brain research.
One possible way to prevent this is the risk evaluation and mitigation strategy established by the Food and Drug Administration to manage high-risk pharmaceuticals. The FDA mandates that certain high-risk drugs can be prescribed only if doctors are certified and only if patients are enrolled in a national registry where eligibility, course, and outcome are monitored. A similar mechanism should apply to high-risk surgical devices when used for behavioral disorders.9,10
People with behavioral disorders deserve the right to volunteer for experimental programs that offer hope of recovery for themselves and future generations, but they also deserve to be treated with the utmost scientific rigor and protection that society can provide.
Dr. Caroff is emeritus professor of psychiatry at the University of Pennsylvania, Philadelphia. He has received research grant funding from Sunovion Pharmaceuticals and serves as a consultant to Neurocrine Biosciences and TEVA.
References
1. Am J Psychiatry. 2007;164(4):552-5
2. Curr Behav Neurosci Rep. 2014;1(2):55-63
3. J Affect Disord. 2014;156:1-7
4. JAMA Psychiatry. 2016;739(5):439-40
5. Biol Psychiatry. 2016;79(4):e9-10
6. Stereotact Funct Neurosurg. 2015;93:366-9
7. Neurotherapeutics. 2014;11(3):475-84
8. J Neurol Neurosurg Psychiatry. 2014;85(9):1003-8
9. Brain Stimul. 2012;5(4):653-5
10. Fed Reg. 1977 May 23;42(99):26318-32
Steps to take when a patient develops tardive dyskinesia
Is there a rational management strategy for tardive dyskinesia?
Introduced into clinical practice more than a half century ago, antipsychotics are still the mainstay of schizophrenia treatment. However, from the earliest reports, antipsychotic efficacy was seemingly inseparable from extrapyramidal side effects (EPS) that manifested as acute and chronic involuntary movement disorders. Although acute extrapyramidal side effects could be prevented and treated, the late-arising symptoms of tardive dyskinesia (TD) seemed irreversible in most cases.
Concerns over TD stimulated extensive research and fueled efforts to develop new antipsychotics that spared the extrapyramidal motor system. Numerous industry-sponsored trials found a reduced risk of EPS—including TD—with newer, second-generation antipsychotics (SGAs), although this advantage diminished when modest doses of low- or mid-potency first-generation antipsychotics (FGAs) were used as the comparator.1-3 Nevertheless, in addition to the continued potential risk of introducing new cases of TD—even with SGAs—several other factors underscore the need to develop a rational strategy for clinical management of TD, including:
- thousands of patients are left with TD as a legacy of past treatment
- the neurophysiologic mechanisms underlying TD are not well understood
- there is no uniformly effective treatment to reverse TD
- TD may be irreversible in most cases.
Prevention
Because there is no “gold standard” treatment for TD, it is important to minimize the risk of TD by taking preventive measures and detecting incipient signs of the disorder. Preventive principles include:
- confirming and documenting the indication for antipsychotics
- using conservative maintenance doses and opting for lower potency or newer agents
- informing patients and caregivers of risk
- assessing for incipient signs of TD using the Abnormal Involuntary Movement Scale (AIMS),4 which should be administered at least every 3 to 6 months.
Confirming the diagnosis
TD presents as a polymorphous involuntary movement disorder,5-8 most often with nonrhythmic, repetitive, purposeless hyperkinetic symptoms. It usually affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing; bruxism; protrusion, curling, or twisting of the tongue; lip smacking, puckering, sucking, and pursing; retraction, grimacing or bridling of the mouth; bulging of the cheeks; or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, or upper or lower extremities also are common. Patients may experience axial symptoms affecting the neck, shoulders, spine, or pelvis. When severe, dyskinesias can affect breathing, swallowing, or speech, and interfere with walking and activities of daily living.
TD may present with nonchoreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but may represent separate subtypes with increased risk of progression, persistence, and severe disability. For example, tardive dystonia, which is estimated to occur in 1% to 4% of patients treated with antipsychotics,9 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia and other movement disorders also occur as tardive variants.10
Multiple diagnostic schemes for TD have been proposed; criteria proposed by Schooler and Kane have been widely accepted (Table 1).11 TD onset occurs insidiously over ≥3 months of antipsychotic treatment and may begin with tic-like movements or increased eye blinking. TD often is suppressed or masked by ongoing antipsychotic treatment and becomes apparent only when the drug is reduced, switched, or discontinued. Dyskinesias increase with emotional arousal, activation, or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, TD symptoms fluctuate over time; therefore, repeated measurements are necessary for reliable assessment of severity and persistence.
The differential diagnosis of TD necessitates conducting a careful medical and neurologic evaluation of all patients with new-onset movement disorders. Clues to neurologic causes include a family history of movement disorders, sudden onset or progressive course, associated medical or neurologic abnormalities, and asymmetry of symptoms. Some of the medical, neurologic, and psychiatric conditions to consider are listed in Table 2.12
Table 1
Schooler-Kane diagnostic criteria for TD
|
| Probable TD: meets criteria 1 through 3 Masked TD: meets criteria 1 through 3 but movements suppressed within 2 weeks by antipsychotic drugs Transient TD: movements not observed on subsequent examination within 3 months Withdrawal TD: movements observed within 2 weeks of antipsychotic drug discontinuation Persistent TD: movements persist for 3 months |
| TD: tardive dyskinesia Source: Reference 11 |
Table 2
Differential diagnosis of tardive dyskinesia
| Primary movement disorders |
|
| Secondary movement disorders |
|
| Source: Reference 12 |
Treatment decisions
If a patient develops TD, clinicians need to make several decisions (Algorithm). First, consider tapering any anticholinergic drugs unless acute EPS are prominent or tardive dystonia is present. Anticholinergic agents can worsen TD but not tardive dystonia; 60% of TD cases improve after discontinuing anticholinergics.13 Second, decide whether antipsychotics could be safely tapered or discontinued. If antipsychotics cannot be safely tapered, decide whether to maintain the patient’s present antipsychotic or switch to a more or less potent agent. Finally, decide whether a trial of an adjunctive antidyskinetic drug is warranted. All of these decisions require thorough discussion with patients and their families, accompanied by careful documentation.
Discontinuing, continuing, or switching antipsychotics. Discontinuing antipsychotics once TD becomes apparent is an option. However, the natural course of TD after drug withdrawal is unclear. Although drug withdrawal had been recommended to increase the odds of TD resolution, early studies showed withdrawing antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia).14 With long-term follow-up, 36% to 55% of patients eventually improved, which supports recommendations for drug reduction or withdrawal.14 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found only 2% of patients showed complete reversal of TD after drug discontinuation.15,16 In a meta-analysis, Soares and McGrath17 reported 37% of patients assigned to placebo across studies showed at least some improvement in TD, but concluded insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse after drug withdrawal in patients with schizophrenia (53% within 9 months).18
A second option for a stable patient with good control of psychotic symptoms but established or long-term TD is to continue the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision, and monitor carefully. In most cases, TD may not progress even with continued antipsychotic treatment, although symptoms may worsen in some cases. However, in a patient with new-onset or early signs of TD, the clinician may be obligated to switch to a lower-potency antipsychotic or newer SGA to improve the chance of resolution; switching is discussed below.
Data on the change in prevalence of TD within a population during continued antipsychotic treatment have been inconsistent. Some studies show an increase, while others show a decrease or no change at all.19 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients with TD have persistent symptoms, 10% to 30% have reduced symptoms, and 10% to 30% experience increased symptoms during treatment.13 Long-term studies estimated that up to 23% of patients may show loss of observable TD symptoms during treatment with FGAs in 1 year.19,20 Similarly, studies of SGAs have shown reduction of TD ratings; some found greater reductions, some found less reductions, and some no difference compared with FGAs.19,20 In some studies, improved TD outcomes were correlated with younger age, lower antipsychotic doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, there was a significant decline in TD severity ratings among 200 patients with TD at baseline who were randomized to receive 1 of 4 SGAs, but there were no significant differences among these SGAs in decline in AIMS scores (Figure).19 Fifty-five percent of these patients met criteria for TD at 2 consecutive post-baseline visits, 76% met criteria at some or all post-baseline visits, and 24% did not meet criteria at any subsequent visit. In addition, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS score. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients in this trial showed either persistence or fluctuation in observable TD symptoms.
Another alternative is to switch antipsychotics, keeping in mind the risk of destabilizing a patient and precipitating psychotic relapse. More potent antipsychotics—such as haloperidol—suppress TD in approximately 67% of patients and may be necessary to consider in patients with severe, disabling symptoms, although the safety of these drugs in relation to their impact on long-term TD outcome is unclear.13,21,22 On the other hand, lower-potency drugs and SGAs also have been associated with reduced TD symptoms23,24; this was confirmed by results of the CATIE trial cited above in which SGAs were associated with a significant reduction in TD severity ratings.19 Clozapine in particular has been recommended for suppressing TD, especially in cases of tardive dystonia.20 Surprisingly, data are limited and inconsistent in addressing whether high-potency FGAs suppress TD symptoms more than low-potency drugs or SGAs, and whether SGAs may suppress TD by mechanisms other than dopamine receptor blockade, which would enhance symptom remission.19,25,26
Apart from short-term suppression of TD symptoms, the advantage of switching to lower-potency antipsychotics or other SGAs would be to increase the odds of eventual TD resolution. Although there has been speculation that in contrast to high-potency FGAs, SGAs may increase the possibility of remission by actively reversing TD or by passively allowing time for TD to resolve, existing data are inconclusive as to whether treatment with SGAs or FGAs results in true recovery rather than symptom suppression. To distinguish remission from suppression, a few studies discontinued SGAs. Some reported continued absence of TD,27,28 but others found unmasking and reappearance of TD.29-31
Adjunctive antidyskinetic drugs. Agents that have been tested off-label for antidyskinetic effects could be considered if symptoms of TD remain problematic despite optimization of antipsychotic treatment, although none have been confirmed as uniformly effective in randomized controlled trials replicated by different investigators.13,17,22 These include dopamine-depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, antioxidants (vitamin E and B6), branched-chain amino acids, neuropeptides, cholinergic precursors, and cholinesterase inhibitors. Electroconvulsive therapy and botulinum toxin or surgical intervention (for tardive dystonia) also may be considered.
Hypotheses proposed to explain TD pathophysiology and thereby justify trials of specific antidyskinetic agents include dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production.32 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to loss of dopamine-mediated inhibition.33 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may suppress TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials that explored the use of cholinesterase inhibitors had mixed results.34-36 However, this hypothesis is supported by evidence from animal and human studies that correlated antipsychotic-induced changes in cholinergic activity with the delay in onset, irreversibility, and age-related risk of TD, the worsening of symptoms due to anticholinergic drugs, and the reduced liability of SGAs for causing TD. These findings suggest that further investigation of cholinergic mechanisms underlying TD may be worthwhile.35
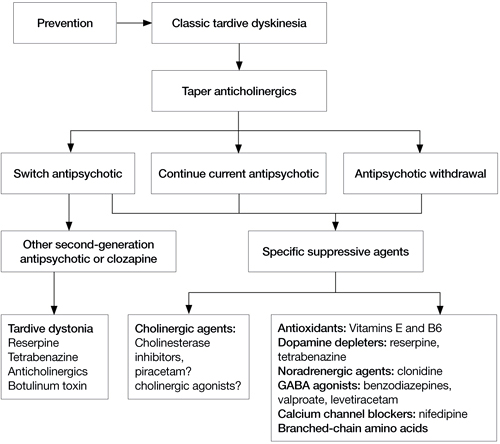
Algorithm: Proposed treatment algorithm for tardive dyskinesia
Source: Reprinted from Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia Trial. Neurol Clin. 2011;29:127-148 with permission from Elsevier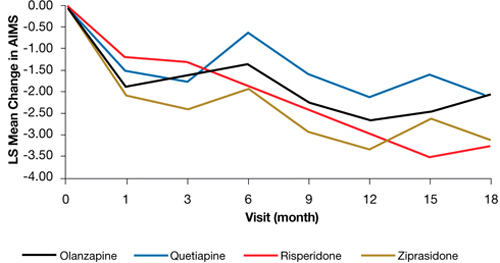
Figure: Adjusteda repeated measures model of change in total AIMS scores for patients with tardive dyskinesia at baseline in CATIE
a Model adjusted for baseline AIMS, baseline PANSS, and duration of illness. Adjusted P value for reduction in total AIMS score from baseline for all patients: P < .001. Treatment differences between the second-generation antipsychotics during the trial: P = .811
AIMS: Abnormal Involuntary Movement Scale; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; PANSS: Positive and Negative Syndrome Scale
Source: Reprinted from Caroff SN, Davis VG, Miller DD, et al; for the CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303 with permission from Physician Postgraduate Press, Inc.Related Resources
- National Institute of Neurological Disorders and Stroke. NINDS Tardive Dyskinesia Information Page. www.ninds.nih.gov/disorders/tardive/tardive.htm.
- WE MOVE (Worldwide Education and Awareness for Movement Disorders). www.wemove.org.
Drug Brand Names
- Botulinum toxin • Botox, Dysport, others
- Clonidine • Catapres
- Clozapine • Clozaril
- Haloperidol • Haldol
- Levetiracetam • Keppra
- Levodopa • Dopar, Larodopa
- Lithium • Lithobid, Eskalith, others
- Nifedipine • Adalat, Afeditab CR, others
- Olanzapine • Zyprexa
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Reserpine • Serpasil
- Risperidone • Risperdal
- Tetrabenazine • Xenazine
- Valproate • Depakote
- Ziprasidone • Geodon
Disclosures
Drs. Caroff, Dhopesh, and Campbell report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Miller receives research/grant support from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Ortho-McNeil-Janssen, and Pfizer Inc. and is a consultant to GlaxoSmithKline and Otsuka.
1. Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279-288.
2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
3. Leucht S, Wahlbeck K, Hamann J, et al. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361(9369):1581-1589.
4. Guy W. Abnormal involuntary movement scale (AIMS). In: Guy W ed. ECDEU assessment manual for psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
5. Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification description, and diagnosis. Clin Neuropharmacol. 1983;6(1):9-26.
6. Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York, NY: Raven Press; 1995:1485–1495.
7. Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991;4(2):109-120.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451-1462.
10. Burke RE, Kang UJ, Jankovic J, et al. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157-175.
11. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
12. American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Press, Inc; 1992.
13. Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583-609.
14. Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE Gardos G, eds. Tardive dyskinesia and neuroleptics: from dogma to reason. Washington, DC: American Psychiatric Press, Inc; 1986:76–97.
15. Glazer WM, Moore DC, Schooler NR, et al. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984;41(6):623-627.
16. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
17. Soares KV, McGrath JJ. The treatment of tardive dyskinesia—a systematic review and meta-analysis. Schizophr Res. 1999;39(1):1-16.
18. Gilbert PL, Harris MJ, McAdams LA, et al. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995;52(3):173-188.
19. Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303.
20. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
21. Jeste DV, Wyatt RJ. In search of treatment for tardive dyskinesia: review of the literature. Schizophr Bull. 1979;5(2):251-293.
22. Jeste DV, Lohr JB, Clark K, et al. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988;8(4 suppl):38S-48S.
23. Caroff SN, Mann SC, Campbell EC, et al. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(suppl 4):12-19.
24. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23-45.
25. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696-701.
26. Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res. 1990;3(5-6):315-320.
27. Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985-996.
28. Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102-106.
29. Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl). 1978;56(1):75-80.
30. Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472-477.
31. Small JG, Milstein V, Marhenke JD, et al. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263-267.
32. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(suppl 4):5-9.
33. Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993;34(10):713-738.
34. Caroff SN, Campbell EC, Havey J, et al. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001;62(10):772-775.
35. Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007;68(3):410-415.
36. Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006;12(4):261-263.
Introduced into clinical practice more than a half century ago, antipsychotics are still the mainstay of schizophrenia treatment. However, from the earliest reports, antipsychotic efficacy was seemingly inseparable from extrapyramidal side effects (EPS) that manifested as acute and chronic involuntary movement disorders. Although acute extrapyramidal side effects could be prevented and treated, the late-arising symptoms of tardive dyskinesia (TD) seemed irreversible in most cases.
Concerns over TD stimulated extensive research and fueled efforts to develop new antipsychotics that spared the extrapyramidal motor system. Numerous industry-sponsored trials found a reduced risk of EPS—including TD—with newer, second-generation antipsychotics (SGAs), although this advantage diminished when modest doses of low- or mid-potency first-generation antipsychotics (FGAs) were used as the comparator.1-3 Nevertheless, in addition to the continued potential risk of introducing new cases of TD—even with SGAs—several other factors underscore the need to develop a rational strategy for clinical management of TD, including:
- thousands of patients are left with TD as a legacy of past treatment
- the neurophysiologic mechanisms underlying TD are not well understood
- there is no uniformly effective treatment to reverse TD
- TD may be irreversible in most cases.
Prevention
Because there is no “gold standard” treatment for TD, it is important to minimize the risk of TD by taking preventive measures and detecting incipient signs of the disorder. Preventive principles include:
- confirming and documenting the indication for antipsychotics
- using conservative maintenance doses and opting for lower potency or newer agents
- informing patients and caregivers of risk
- assessing for incipient signs of TD using the Abnormal Involuntary Movement Scale (AIMS),4 which should be administered at least every 3 to 6 months.
Confirming the diagnosis
TD presents as a polymorphous involuntary movement disorder,5-8 most often with nonrhythmic, repetitive, purposeless hyperkinetic symptoms. It usually affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing; bruxism; protrusion, curling, or twisting of the tongue; lip smacking, puckering, sucking, and pursing; retraction, grimacing or bridling of the mouth; bulging of the cheeks; or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, or upper or lower extremities also are common. Patients may experience axial symptoms affecting the neck, shoulders, spine, or pelvis. When severe, dyskinesias can affect breathing, swallowing, or speech, and interfere with walking and activities of daily living.
TD may present with nonchoreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but may represent separate subtypes with increased risk of progression, persistence, and severe disability. For example, tardive dystonia, which is estimated to occur in 1% to 4% of patients treated with antipsychotics,9 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia and other movement disorders also occur as tardive variants.10
Multiple diagnostic schemes for TD have been proposed; criteria proposed by Schooler and Kane have been widely accepted (Table 1).11 TD onset occurs insidiously over ≥3 months of antipsychotic treatment and may begin with tic-like movements or increased eye blinking. TD often is suppressed or masked by ongoing antipsychotic treatment and becomes apparent only when the drug is reduced, switched, or discontinued. Dyskinesias increase with emotional arousal, activation, or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, TD symptoms fluctuate over time; therefore, repeated measurements are necessary for reliable assessment of severity and persistence.
The differential diagnosis of TD necessitates conducting a careful medical and neurologic evaluation of all patients with new-onset movement disorders. Clues to neurologic causes include a family history of movement disorders, sudden onset or progressive course, associated medical or neurologic abnormalities, and asymmetry of symptoms. Some of the medical, neurologic, and psychiatric conditions to consider are listed in Table 2.12
Table 1
Schooler-Kane diagnostic criteria for TD
|
| Probable TD: meets criteria 1 through 3 Masked TD: meets criteria 1 through 3 but movements suppressed within 2 weeks by antipsychotic drugs Transient TD: movements not observed on subsequent examination within 3 months Withdrawal TD: movements observed within 2 weeks of antipsychotic drug discontinuation Persistent TD: movements persist for 3 months |
| TD: tardive dyskinesia Source: Reference 11 |
Table 2
Differential diagnosis of tardive dyskinesia
| Primary movement disorders |
|
| Secondary movement disorders |
|
| Source: Reference 12 |
Treatment decisions
If a patient develops TD, clinicians need to make several decisions (Algorithm). First, consider tapering any anticholinergic drugs unless acute EPS are prominent or tardive dystonia is present. Anticholinergic agents can worsen TD but not tardive dystonia; 60% of TD cases improve after discontinuing anticholinergics.13 Second, decide whether antipsychotics could be safely tapered or discontinued. If antipsychotics cannot be safely tapered, decide whether to maintain the patient’s present antipsychotic or switch to a more or less potent agent. Finally, decide whether a trial of an adjunctive antidyskinetic drug is warranted. All of these decisions require thorough discussion with patients and their families, accompanied by careful documentation.
Discontinuing, continuing, or switching antipsychotics. Discontinuing antipsychotics once TD becomes apparent is an option. However, the natural course of TD after drug withdrawal is unclear. Although drug withdrawal had been recommended to increase the odds of TD resolution, early studies showed withdrawing antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia).14 With long-term follow-up, 36% to 55% of patients eventually improved, which supports recommendations for drug reduction or withdrawal.14 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found only 2% of patients showed complete reversal of TD after drug discontinuation.15,16 In a meta-analysis, Soares and McGrath17 reported 37% of patients assigned to placebo across studies showed at least some improvement in TD, but concluded insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse after drug withdrawal in patients with schizophrenia (53% within 9 months).18
A second option for a stable patient with good control of psychotic symptoms but established or long-term TD is to continue the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision, and monitor carefully. In most cases, TD may not progress even with continued antipsychotic treatment, although symptoms may worsen in some cases. However, in a patient with new-onset or early signs of TD, the clinician may be obligated to switch to a lower-potency antipsychotic or newer SGA to improve the chance of resolution; switching is discussed below.
Data on the change in prevalence of TD within a population during continued antipsychotic treatment have been inconsistent. Some studies show an increase, while others show a decrease or no change at all.19 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients with TD have persistent symptoms, 10% to 30% have reduced symptoms, and 10% to 30% experience increased symptoms during treatment.13 Long-term studies estimated that up to 23% of patients may show loss of observable TD symptoms during treatment with FGAs in 1 year.19,20 Similarly, studies of SGAs have shown reduction of TD ratings; some found greater reductions, some found less reductions, and some no difference compared with FGAs.19,20 In some studies, improved TD outcomes were correlated with younger age, lower antipsychotic doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, there was a significant decline in TD severity ratings among 200 patients with TD at baseline who were randomized to receive 1 of 4 SGAs, but there were no significant differences among these SGAs in decline in AIMS scores (Figure).19 Fifty-five percent of these patients met criteria for TD at 2 consecutive post-baseline visits, 76% met criteria at some or all post-baseline visits, and 24% did not meet criteria at any subsequent visit. In addition, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS score. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients in this trial showed either persistence or fluctuation in observable TD symptoms.
Another alternative is to switch antipsychotics, keeping in mind the risk of destabilizing a patient and precipitating psychotic relapse. More potent antipsychotics—such as haloperidol—suppress TD in approximately 67% of patients and may be necessary to consider in patients with severe, disabling symptoms, although the safety of these drugs in relation to their impact on long-term TD outcome is unclear.13,21,22 On the other hand, lower-potency drugs and SGAs also have been associated with reduced TD symptoms23,24; this was confirmed by results of the CATIE trial cited above in which SGAs were associated with a significant reduction in TD severity ratings.19 Clozapine in particular has been recommended for suppressing TD, especially in cases of tardive dystonia.20 Surprisingly, data are limited and inconsistent in addressing whether high-potency FGAs suppress TD symptoms more than low-potency drugs or SGAs, and whether SGAs may suppress TD by mechanisms other than dopamine receptor blockade, which would enhance symptom remission.19,25,26
Apart from short-term suppression of TD symptoms, the advantage of switching to lower-potency antipsychotics or other SGAs would be to increase the odds of eventual TD resolution. Although there has been speculation that in contrast to high-potency FGAs, SGAs may increase the possibility of remission by actively reversing TD or by passively allowing time for TD to resolve, existing data are inconclusive as to whether treatment with SGAs or FGAs results in true recovery rather than symptom suppression. To distinguish remission from suppression, a few studies discontinued SGAs. Some reported continued absence of TD,27,28 but others found unmasking and reappearance of TD.29-31
Adjunctive antidyskinetic drugs. Agents that have been tested off-label for antidyskinetic effects could be considered if symptoms of TD remain problematic despite optimization of antipsychotic treatment, although none have been confirmed as uniformly effective in randomized controlled trials replicated by different investigators.13,17,22 These include dopamine-depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, antioxidants (vitamin E and B6), branched-chain amino acids, neuropeptides, cholinergic precursors, and cholinesterase inhibitors. Electroconvulsive therapy and botulinum toxin or surgical intervention (for tardive dystonia) also may be considered.
Hypotheses proposed to explain TD pathophysiology and thereby justify trials of specific antidyskinetic agents include dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production.32 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to loss of dopamine-mediated inhibition.33 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may suppress TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials that explored the use of cholinesterase inhibitors had mixed results.34-36 However, this hypothesis is supported by evidence from animal and human studies that correlated antipsychotic-induced changes in cholinergic activity with the delay in onset, irreversibility, and age-related risk of TD, the worsening of symptoms due to anticholinergic drugs, and the reduced liability of SGAs for causing TD. These findings suggest that further investigation of cholinergic mechanisms underlying TD may be worthwhile.35
Algorithm: Proposed treatment algorithm for tardive dyskinesia
Source: Reprinted from Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia Trial. Neurol Clin. 2011;29:127-148 with permission from Elsevier
Figure: Adjusteda repeated measures model of change in total AIMS scores for patients with tardive dyskinesia at baseline in CATIE
a Model adjusted for baseline AIMS, baseline PANSS, and duration of illness. Adjusted P value for reduction in total AIMS score from baseline for all patients: P < .001. Treatment differences between the second-generation antipsychotics during the trial: P = .811
AIMS: Abnormal Involuntary Movement Scale; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; PANSS: Positive and Negative Syndrome Scale
Source: Reprinted from Caroff SN, Davis VG, Miller DD, et al; for the CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303 with permission from Physician Postgraduate Press, Inc.Related Resources
- National Institute of Neurological Disorders and Stroke. NINDS Tardive Dyskinesia Information Page. www.ninds.nih.gov/disorders/tardive/tardive.htm.
- WE MOVE (Worldwide Education and Awareness for Movement Disorders). www.wemove.org.
Drug Brand Names
- Botulinum toxin • Botox, Dysport, others
- Clonidine • Catapres
- Clozapine • Clozaril
- Haloperidol • Haldol
- Levetiracetam • Keppra
- Levodopa • Dopar, Larodopa
- Lithium • Lithobid, Eskalith, others
- Nifedipine • Adalat, Afeditab CR, others
- Olanzapine • Zyprexa
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Reserpine • Serpasil
- Risperidone • Risperdal
- Tetrabenazine • Xenazine
- Valproate • Depakote
- Ziprasidone • Geodon
Disclosures
Drs. Caroff, Dhopesh, and Campbell report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Miller receives research/grant support from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Ortho-McNeil-Janssen, and Pfizer Inc. and is a consultant to GlaxoSmithKline and Otsuka.
Introduced into clinical practice more than a half century ago, antipsychotics are still the mainstay of schizophrenia treatment. However, from the earliest reports, antipsychotic efficacy was seemingly inseparable from extrapyramidal side effects (EPS) that manifested as acute and chronic involuntary movement disorders. Although acute extrapyramidal side effects could be prevented and treated, the late-arising symptoms of tardive dyskinesia (TD) seemed irreversible in most cases.
Concerns over TD stimulated extensive research and fueled efforts to develop new antipsychotics that spared the extrapyramidal motor system. Numerous industry-sponsored trials found a reduced risk of EPS—including TD—with newer, second-generation antipsychotics (SGAs), although this advantage diminished when modest doses of low- or mid-potency first-generation antipsychotics (FGAs) were used as the comparator.1-3 Nevertheless, in addition to the continued potential risk of introducing new cases of TD—even with SGAs—several other factors underscore the need to develop a rational strategy for clinical management of TD, including:
- thousands of patients are left with TD as a legacy of past treatment
- the neurophysiologic mechanisms underlying TD are not well understood
- there is no uniformly effective treatment to reverse TD
- TD may be irreversible in most cases.
Prevention
Because there is no “gold standard” treatment for TD, it is important to minimize the risk of TD by taking preventive measures and detecting incipient signs of the disorder. Preventive principles include:
- confirming and documenting the indication for antipsychotics
- using conservative maintenance doses and opting for lower potency or newer agents
- informing patients and caregivers of risk
- assessing for incipient signs of TD using the Abnormal Involuntary Movement Scale (AIMS),4 which should be administered at least every 3 to 6 months.
Confirming the diagnosis
TD presents as a polymorphous involuntary movement disorder,5-8 most often with nonrhythmic, repetitive, purposeless hyperkinetic symptoms. It usually affects orofacial and lingual musculature (“buccolinguomasticatory syndrome”) with chewing; bruxism; protrusion, curling, or twisting of the tongue; lip smacking, puckering, sucking, and pursing; retraction, grimacing or bridling of the mouth; bulging of the cheeks; or eye blinking and blepharospasm. Choreoathetoid movements of the fingers, hands, or upper or lower extremities also are common. Patients may experience axial symptoms affecting the neck, shoulders, spine, or pelvis. When severe, dyskinesias can affect breathing, swallowing, or speech, and interfere with walking and activities of daily living.
TD may present with nonchoreoathetoid symptoms that can be difficult to distinguish from acute EPS. These may co-exist with classic TD symptoms, but may represent separate subtypes with increased risk of progression, persistence, and severe disability. For example, tardive dystonia, which is estimated to occur in 1% to 4% of patients treated with antipsychotics,9 may be more generalized and disabling than TD, and may respond to anticholinergic agents. Akathisia and other movement disorders also occur as tardive variants.10
Multiple diagnostic schemes for TD have been proposed; criteria proposed by Schooler and Kane have been widely accepted (Table 1).11 TD onset occurs insidiously over ≥3 months of antipsychotic treatment and may begin with tic-like movements or increased eye blinking. TD often is suppressed or masked by ongoing antipsychotic treatment and becomes apparent only when the drug is reduced, switched, or discontinued. Dyskinesias increase with emotional arousal, activation, or distraction, and diminish with relaxation, sleep, or volitional effort. As a result, TD symptoms fluctuate over time; therefore, repeated measurements are necessary for reliable assessment of severity and persistence.
The differential diagnosis of TD necessitates conducting a careful medical and neurologic evaluation of all patients with new-onset movement disorders. Clues to neurologic causes include a family history of movement disorders, sudden onset or progressive course, associated medical or neurologic abnormalities, and asymmetry of symptoms. Some of the medical, neurologic, and psychiatric conditions to consider are listed in Table 2.12
Table 1
Schooler-Kane diagnostic criteria for TD
|
| Probable TD: meets criteria 1 through 3 Masked TD: meets criteria 1 through 3 but movements suppressed within 2 weeks by antipsychotic drugs Transient TD: movements not observed on subsequent examination within 3 months Withdrawal TD: movements observed within 2 weeks of antipsychotic drug discontinuation Persistent TD: movements persist for 3 months |
| TD: tardive dyskinesia Source: Reference 11 |
Table 2
Differential diagnosis of tardive dyskinesia
| Primary movement disorders |
|
| Secondary movement disorders |
|
| Source: Reference 12 |
Treatment decisions
If a patient develops TD, clinicians need to make several decisions (Algorithm). First, consider tapering any anticholinergic drugs unless acute EPS are prominent or tardive dystonia is present. Anticholinergic agents can worsen TD but not tardive dystonia; 60% of TD cases improve after discontinuing anticholinergics.13 Second, decide whether antipsychotics could be safely tapered or discontinued. If antipsychotics cannot be safely tapered, decide whether to maintain the patient’s present antipsychotic or switch to a more or less potent agent. Finally, decide whether a trial of an adjunctive antidyskinetic drug is warranted. All of these decisions require thorough discussion with patients and their families, accompanied by careful documentation.
Discontinuing, continuing, or switching antipsychotics. Discontinuing antipsychotics once TD becomes apparent is an option. However, the natural course of TD after drug withdrawal is unclear. Although drug withdrawal had been recommended to increase the odds of TD resolution, early studies showed withdrawing antipsychotics may lead to an initial worsening of TD in 33% to 53% of patients (unmasking or withdrawal dyskinesia).14 With long-term follow-up, 36% to 55% of patients eventually improved, which supports recommendations for drug reduction or withdrawal.14 However, complete and permanent reversibility beyond the withdrawal period is rare; Glazer et al found only 2% of patients showed complete reversal of TD after drug discontinuation.15,16 In a meta-analysis, Soares and McGrath17 reported 37% of patients assigned to placebo across studies showed at least some improvement in TD, but concluded insufficient evidence existed to support drug cessation or reduction as effective treatments for TD, especially when contrasted with robust evidence for the risk of psychotic relapse after drug withdrawal in patients with schizophrenia (53% within 9 months).18
A second option for a stable patient with good control of psychotic symptoms but established or long-term TD is to continue the antipsychotic, try to gradually reduce the dose, inform patients and caregivers of risks, document the decision, and monitor carefully. In most cases, TD may not progress even with continued antipsychotic treatment, although symptoms may worsen in some cases. However, in a patient with new-onset or early signs of TD, the clinician may be obligated to switch to a lower-potency antipsychotic or newer SGA to improve the chance of resolution; switching is discussed below.
Data on the change in prevalence of TD within a population during continued antipsychotic treatment have been inconsistent. Some studies show an increase, while others show a decrease or no change at all.19 However, prevalence rates obscure the dynamics of TD in individual patients. Roughly 50% of patients with TD have persistent symptoms, 10% to 30% have reduced symptoms, and 10% to 30% experience increased symptoms during treatment.13 Long-term studies estimated that up to 23% of patients may show loss of observable TD symptoms during treatment with FGAs in 1 year.19,20 Similarly, studies of SGAs have shown reduction of TD ratings; some found greater reductions, some found less reductions, and some no difference compared with FGAs.19,20 In some studies, improved TD outcomes were correlated with younger age, lower antipsychotic doses, reduced duration of drug treatment and dyskinesia, and increased length of follow-up.
In the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, there was a significant decline in TD severity ratings among 200 patients with TD at baseline who were randomized to receive 1 of 4 SGAs, but there were no significant differences among these SGAs in decline in AIMS scores (Figure).19 Fifty-five percent of these patients met criteria for TD at 2 consecutive post-baseline visits, 76% met criteria at some or all post-baseline visits, and 24% did not meet criteria at any subsequent visit. In addition, 32% showed ≥50% decrease and 7% showed ≥50% increase in AIMS score. Thus, similar to past evidence on the course of TD during treatment with FGAs or SGAs, most patients in this trial showed either persistence or fluctuation in observable TD symptoms.
Another alternative is to switch antipsychotics, keeping in mind the risk of destabilizing a patient and precipitating psychotic relapse. More potent antipsychotics—such as haloperidol—suppress TD in approximately 67% of patients and may be necessary to consider in patients with severe, disabling symptoms, although the safety of these drugs in relation to their impact on long-term TD outcome is unclear.13,21,22 On the other hand, lower-potency drugs and SGAs also have been associated with reduced TD symptoms23,24; this was confirmed by results of the CATIE trial cited above in which SGAs were associated with a significant reduction in TD severity ratings.19 Clozapine in particular has been recommended for suppressing TD, especially in cases of tardive dystonia.20 Surprisingly, data are limited and inconsistent in addressing whether high-potency FGAs suppress TD symptoms more than low-potency drugs or SGAs, and whether SGAs may suppress TD by mechanisms other than dopamine receptor blockade, which would enhance symptom remission.19,25,26
Apart from short-term suppression of TD symptoms, the advantage of switching to lower-potency antipsychotics or other SGAs would be to increase the odds of eventual TD resolution. Although there has been speculation that in contrast to high-potency FGAs, SGAs may increase the possibility of remission by actively reversing TD or by passively allowing time for TD to resolve, existing data are inconclusive as to whether treatment with SGAs or FGAs results in true recovery rather than symptom suppression. To distinguish remission from suppression, a few studies discontinued SGAs. Some reported continued absence of TD,27,28 but others found unmasking and reappearance of TD.29-31
Adjunctive antidyskinetic drugs. Agents that have been tested off-label for antidyskinetic effects could be considered if symptoms of TD remain problematic despite optimization of antipsychotic treatment, although none have been confirmed as uniformly effective in randomized controlled trials replicated by different investigators.13,17,22 These include dopamine-depleting agents, dopamine agonists, noradrenergic agonists and antagonists, GABAergic drugs (benzodiazepines, valproate, levetiracetam), lithium, calcium channel blockers, serotonergic drugs, antioxidants (vitamin E and B6), branched-chain amino acids, neuropeptides, cholinergic precursors, and cholinesterase inhibitors. Electroconvulsive therapy and botulinum toxin or surgical intervention (for tardive dystonia) also may be considered.
Hypotheses proposed to explain TD pathophysiology and thereby justify trials of specific antidyskinetic agents include dopamine receptor hypersensitivity, GABA insufficiency, and structural damage resulting from increased catecholamine metabolism and oxidative free radical production.32 Another hypothesis proposes that TD results from damage to striatal cholinergic interneurons due to loss of dopamine-mediated inhibition.33 If correct, this implies that cholinesterase inhibitors or cholinergic agonists may suppress TD by directly enhancing post-synaptic cholinergic activity, thereby compensating for the loss of pre-synaptic cholinergic neurons. Several preliminary trials that explored the use of cholinesterase inhibitors had mixed results.34-36 However, this hypothesis is supported by evidence from animal and human studies that correlated antipsychotic-induced changes in cholinergic activity with the delay in onset, irreversibility, and age-related risk of TD, the worsening of symptoms due to anticholinergic drugs, and the reduced liability of SGAs for causing TD. These findings suggest that further investigation of cholinergic mechanisms underlying TD may be worthwhile.35
Algorithm: Proposed treatment algorithm for tardive dyskinesia
Source: Reprinted from Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia Trial. Neurol Clin. 2011;29:127-148 with permission from Elsevier
Figure: Adjusteda repeated measures model of change in total AIMS scores for patients with tardive dyskinesia at baseline in CATIE
a Model adjusted for baseline AIMS, baseline PANSS, and duration of illness. Adjusted P value for reduction in total AIMS score from baseline for all patients: P < .001. Treatment differences between the second-generation antipsychotics during the trial: P = .811
AIMS: Abnormal Involuntary Movement Scale; CATIE: Clinical Antipsychotic Trials of Intervention Effectiveness; PANSS: Positive and Negative Syndrome Scale
Source: Reprinted from Caroff SN, Davis VG, Miller DD, et al; for the CATIE Investigators. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303 with permission from Physician Postgraduate Press, Inc.Related Resources
- National Institute of Neurological Disorders and Stroke. NINDS Tardive Dyskinesia Information Page. www.ninds.nih.gov/disorders/tardive/tardive.htm.
- WE MOVE (Worldwide Education and Awareness for Movement Disorders). www.wemove.org.
Drug Brand Names
- Botulinum toxin • Botox, Dysport, others
- Clonidine • Catapres
- Clozapine • Clozaril
- Haloperidol • Haldol
- Levetiracetam • Keppra
- Levodopa • Dopar, Larodopa
- Lithium • Lithobid, Eskalith, others
- Nifedipine • Adalat, Afeditab CR, others
- Olanzapine • Zyprexa
- Phenytoin • Dilantin
- Quetiapine • Seroquel
- Reserpine • Serpasil
- Risperidone • Risperdal
- Tetrabenazine • Xenazine
- Valproate • Depakote
- Ziprasidone • Geodon
Disclosures
Drs. Caroff, Dhopesh, and Campbell report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Miller receives research/grant support from AstraZeneca, Bristol-Myers Squibb, Eli Lilly and Company, Ortho-McNeil-Janssen, and Pfizer Inc. and is a consultant to GlaxoSmithKline and Otsuka.
1. Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279-288.
2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
3. Leucht S, Wahlbeck K, Hamann J, et al. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361(9369):1581-1589.
4. Guy W. Abnormal involuntary movement scale (AIMS). In: Guy W ed. ECDEU assessment manual for psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
5. Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification description, and diagnosis. Clin Neuropharmacol. 1983;6(1):9-26.
6. Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York, NY: Raven Press; 1995:1485–1495.
7. Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991;4(2):109-120.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451-1462.
10. Burke RE, Kang UJ, Jankovic J, et al. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157-175.
11. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
12. American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Press, Inc; 1992.
13. Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583-609.
14. Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE Gardos G, eds. Tardive dyskinesia and neuroleptics: from dogma to reason. Washington, DC: American Psychiatric Press, Inc; 1986:76–97.
15. Glazer WM, Moore DC, Schooler NR, et al. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984;41(6):623-627.
16. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
17. Soares KV, McGrath JJ. The treatment of tardive dyskinesia—a systematic review and meta-analysis. Schizophr Res. 1999;39(1):1-16.
18. Gilbert PL, Harris MJ, McAdams LA, et al. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995;52(3):173-188.
19. Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303.
20. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
21. Jeste DV, Wyatt RJ. In search of treatment for tardive dyskinesia: review of the literature. Schizophr Bull. 1979;5(2):251-293.
22. Jeste DV, Lohr JB, Clark K, et al. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988;8(4 suppl):38S-48S.
23. Caroff SN, Mann SC, Campbell EC, et al. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(suppl 4):12-19.
24. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23-45.
25. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696-701.
26. Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res. 1990;3(5-6):315-320.
27. Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985-996.
28. Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102-106.
29. Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl). 1978;56(1):75-80.
30. Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472-477.
31. Small JG, Milstein V, Marhenke JD, et al. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263-267.
32. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(suppl 4):5-9.
33. Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993;34(10):713-738.
34. Caroff SN, Campbell EC, Havey J, et al. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001;62(10):772-775.
35. Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007;68(3):410-415.
36. Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006;12(4):261-263.
1. Miller DD, Caroff SN, Davis SM, et al. Extrapyramidal side-effects of antipsychotics in a randomised trial. Br J Psychiatry. 2008;193(4):279-288.
2. Lieberman JA, Stroup TS, McEvoy JP, et al. Effectiveness of antipsychotic drugs in patients with chronic schizophrenia. N Engl J Med. 2005;353(12):1209-1223.
3. Leucht S, Wahlbeck K, Hamann J, et al. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361(9369):1581-1589.
4. Guy W. Abnormal involuntary movement scale (AIMS). In: Guy W ed. ECDEU assessment manual for psychopharmacology. Rockville, MD: U.S. Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, National Institute of Mental Health, Psychopharmacology Research Branch, Division of Extramural Research Programs; 1976:534–537.
5. Tarsy D. Neuroleptic-induced extrapyramidal reactions: classification description, and diagnosis. Clin Neuropharmacol. 1983;6(1):9-26.
6. Kane JM. Tardive dyskinesia: epidemiological and clinical presentation. In: Bloom FE Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York, NY: Raven Press; 1995:1485–1495.
7. Casey DE. Neuroleptic drug-induced extrapyramidal syndromes and tardive dyskinesia. Schizophr Res. 1991;4(2):109-120.
8. Caroff SN, Hurford I, Lybrand J, et al. Movement disorders induced by antipsychotic drugs: implications of the CATIE schizophrenia trial. Neurol Clin. 2011;29(1):127-148.
9. Dayalu P, Chou KL. Antipsychotic-induced extrapyramidal symptoms and their management. Expert Opin Pharmacother. 2008;9(9):1451-1462.
10. Burke RE, Kang UJ, Jankovic J, et al. Tardive akathisia: an analysis of clinical features and response to open therapeutic trials. Mov Disord. 1989;4(2):157-175.
11. Schooler NR, Kane JM. Research diagnoses for tardive dyskinesia. Arch Gen Psychiatry. 1982;39(4):486-487.
12. American Psychiatric Association. Tardive dyskinesia: a task force report of the American Psychiatric Association. Washington DC: American Psychiatric Press, Inc; 1992.
13. Egan MF, Apud J, Wyatt RJ. Treatment of tardive dyskinesia. Schizophr Bull. 1997;23(4):583-609.
14. Casey DE, Gerlach J. Tardive dyskinesia: what is the long-term outcome? In: Casey DE Gardos G, eds. Tardive dyskinesia and neuroleptics: from dogma to reason. Washington, DC: American Psychiatric Press, Inc; 1986:76–97.
15. Glazer WM, Moore DC, Schooler NR, et al. Tardive dyskinesia. A discontinuation study. Arch Gen Psychiatry. 1984;41(6):623-627.
16. Glazer WM, Morgenstern H, Schooler N, et al. Predictors of improvement in tardive dyskinesia following discontinuation of neuroleptic medication. Br J Psychiatry. 1990;157:585-592.
17. Soares KV, McGrath JJ. The treatment of tardive dyskinesia—a systematic review and meta-analysis. Schizophr Res. 1999;39(1):1-16.
18. Gilbert PL, Harris MJ, McAdams LA, et al. Neuroleptic withdrawal in schizophrenic patients. A review of the literature. Arch Gen Psychiatry. 1995;52(3):173-188.
19. Caroff SN, Davis VG, Miller DD, et al. Treatment outcomes of patients with tardive dyskinesia and chronic schizophrenia. J Clin Psychiatry. 2011;72(3):295-303.
20. Lieberman JA, Saltz BL, Johns CA, et al. The effects of clozapine on tardive dyskinesia. Br J Psychiatry. 1991;158:503-510.
21. Jeste DV, Wyatt RJ. In search of treatment for tardive dyskinesia: review of the literature. Schizophr Bull. 1979;5(2):251-293.
22. Jeste DV, Lohr JB, Clark K, et al. Pharmacological treatments of tardive dyskinesia in the 1980s. J Clin Psychopharmacol. 1988;8(4 suppl):38S-48S.
23. Caroff SN, Mann SC, Campbell EC, et al. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry. 2002;63(suppl 4):12-19.
24. Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23-45.
25. Emsley R, Turner HJ, Schronen J, et al. A single-blind, randomized trial comparing quetiapine and haloperidol in the treatment of tardive dyskinesia. J Clin Psychiatry. 2004;65(5):696-701.
26. Glazer WM, Hafez H. A comparison of masking effects of haloperidol versus molindone in tardive dyskinesia. Schizophr Res. 1990;3(5-6):315-320.
27. Kinon BJ, Jeste DV, Kollack-Walker S, et al. Olanzapine treatment for tardive dyskinesia in schizophrenia patients: a prospective clinical trial with patients randomized to blinded dose reduction periods. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(6):985-996.
28. Tamminga CA, Thaker GK, Moran M, et al. Clozapine in tardive dyskinesia: observations from human and animal model studies. J Clin Psychiatry. 1994;55(suppl B):102-106.
29. Simpson GM, Lee JH, Shrivastava RK. Clozapine in tardive dyskinesia. Psychopharmacology (Berl). 1978;56(1):75-80.
30. Ahmed S, Chengappa KN, Naidu VR, et al. Clozapine withdrawal-emergent dystonias and dyskinesias: a case series. J Clin Psychiatry. 1998;59(9):472-477.
31. Small JG, Milstein V, Marhenke JD, et al. Treatment outcome with clozapine in tardive dyskinesia, neuroleptic sensitivity, and treatment-resistant psychosis. J Clin Psychiatry. 1987;48(7):263-267.
32. Casey DE. Tardive dyskinesia: pathophysiology and animal models. J Clin Psychiatry. 2000;61(suppl 4):5-9.
33. Miller R, Chouinard G. Loss of striatal cholinergic neurons as a basis for tardive and L-dopa-induced dyskinesias neuroleptic-induced supersensitivity psychosis and refractory schizophrenia. Biol Psychiatry. 1993;34(10):713-738.
34. Caroff SN, Campbell EC, Havey J, et al. Treatment of tardive dyskinesia with donepezil: a pilot study. J Clin Psychiatry. 2001;62(10):772-775.
35. Caroff SN, Walker P, Campbell C, et al. Treatment of tardive dyskinesia with galantamine: a randomized controlled crossover trial. J Clin Psychiatry. 2007;68(3):410-415.
36. Caroff SN, Martine R, Kleiner-Fisman G, et al. Treatment of levodopa-induced dyskinesias with donepezil. Parkinsonism Relat Disord. 2006;12(4):261-263.
Neuroleptic malignant syndrome: Answers to 6 tough questions
Diagnosis and treatment of neuroleptic malignant syndrome (NMS) are controversial because this potentially life-threatening syndrome is rare and its presentation varies. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce. It may be possible, however, to develop rational treatment guidelines using empiric clinical data.1,2
This article examines the evidence related to 6 controversial aspects of NMS diagnosis and treatment:
- most-reliable risk factors
- NMS as a spectrum disorder
- what causes NMS
- NMS triggered by first-generation vs second-generation antipsychotics
- first-line interventions
- restarting antipsychotics after an NMS episode.
1. Are there reliable risk factors for NMS?
In small case-controlled studies, agitation, dehydration, and exhaustion were the most consistently found systemic factors believed to predispose patients taking antipsychotics to NMS (Table 1).3-5 Catatonia and organic brain syndromes may be separate risk factors.1,6
Preliminary studies also have implicated dopamine receptor abnormalities caused by genetic polymorphisms or effects of low serum iron.1,7,8 Pharmacologic studies have suggested that higher doses, rapid titration, and IM injections of antipsychotics are associated with increased NMS risk.3,5 Some studies suggest that 15% to 20% of NMS patients have a history of NMS episodes.1,2 In addition, high-potency first-generation antipsychotics (FGAs)—especially haloperidol—are assumed to carry higher risk than low-potency drugs and second-generation antipsychotics (SGAs), although this hypothesis remains difficult to prove.9-11
These risk factors, however, are not practical for estimating NMS risk in a given patient because they are relatively common compared with the low risk of NMS occurrence. For the vast majority of patients with psychotic symptoms, the benefits of properly indicated antipsychotic pharmacotherapy will outweigh the risks.
Table 1
| Systemic |
| Agitation |
| Dehydration |
| Exhaustion |
| Low serum iron concentrations (normal: 60 to 170 mcg/dL) |
| Diagnoses |
| History of NMS |
| Catatonia |
| Organic brain syndromes |
| Central nervous system |
| Dopamine receptor dysfunction |
| Basal ganglia dysfunction |
| Sympathetic nervous system dysfunction |
| Pharmacologic treatment* |
| Intramuscular or intravenous injections |
| High-potency dopamine antagonists |
| Rapid dose titration |
| High doses |
| FGAs compared with SGAs (?) |
*For individual patients, these common risk factors must be weighted again the benefits of antipsychotic therapy FGAs: first-generation antipsychotics; SGAs:second-generation antipsychotics; NMS: neuroleptic malignant syndromeSource: References 1-5
2. Is NMS related to parkinsonism, catatonia, or malignant hyperthermia?
Parkinsonsim. Some researchers have described NMS as an extreme parkinsonian crisis resulting from overwhelming blockade of dopamine pathways in the brain.1,2,12 In this view, NMS resembles the parkinsonian-hyperthermia syndrome that can occur in Parkinson's disease patients following abrupt discontinuation or loss of efficacy of dopaminergic therapy, which can be treated by reinstituting dopaminergic agents.13 Evidence to support this view includes:
- Parkinsonian signs are a cardinal feature of NMS.
- Withdrawal of dopamine agonists precipitates the syndrome.
- All triggering drugs are dopamine receptor antagonists.
- Risks of NMS correlates with drugs' dopamine receptor affinity.
- Dopaminergic agonists may be an effective treatment.
- Lesions in dopaminergic pathways produce a similar syndrome.
- Patients with NMS have demonstrated low cerebrospinal fluid concentrations of the dopamine metabolite homovanillic acid.14
Catatonia. Fink et al15 and others16-18 have persuasively argued that NMS represents a form of drug-induced malignant catatonia. Evidence supporting this includes:
- The 2 disorders share neuropsychiatric symptoms.
- Catalonic signs are common in NMS.19
- Malignant catatonia and NMS share physiologic and labratory signs.20
- Reintroduction of antipsychotics can acutely worsen both conditions.
- Benzodiazepines and electroconvulsive therapy (ECT) are effective treatments for both disorders.15-18
Lee21 examined the relationship between catatonic features and treatment response of 14 NMS patients. Most patients with catatonic symptoms responded to benzodiazepines, whereas none of those did who had an extrapyramidal-hyperthermic presentation without catatonia. Lee concluded that NMS is heterogeneous and may occur in catatonic and noncatatonic forms that differ in treatment response.
- similar clinical signs of rigidity, hyperthermia, and hypermetabolism
- similar psychologic and labratory signs, such as rhabdomyolysis
- hyperthermia in both responding to dantrolene.
Although the 2 are similar in presentation, malignant hyperthermia occurs intraoperatively and reflects a pharmacogenetic disorder of calcium regulation in skeletal muscle. Additionally, rigidity in malignant hyperthermia does not respond to peripheral-acting muscle relaxants.1,22 Evidence suggests that patients who have previously experienced an NMS episodes are not at risk for malignant hyperthermia.22
3. What is the pathophysiology of NMS?
NMS pathophysiology is complex and likely involves interplay between multiple central and systemic pathways and neurotransmitters. As described above, compelling evidence suggests that dopamine blockade plays a central role.12
Dopamine blockade in the hypothalamus is believed to contribute to thermoregulatory failure, and blockade in the nigrostriatal system likely contributes to muscle rigidity and hypermetabolism. The loss of dopaminergic input to the anterior cingulate-medial orbitofrontal circuit and the lateral orbitofrontal circuit likely con-tributes to the mental status changes and catatonic features seen in NMS.12
Some researchers have proposed competing or complementary hypotheses, however. For example, Gurrera23 proposed that patients who are prone to developing NMS have a vulnerability to a hyperactive and dysregulated sympathetic nervous system, and this trait—together with dopamine system disruption induced by dopamine-blocking agents—produces NMS. Other investigators have implicated serotonin, norepinephrine, gamma-aminobutyric acid and glutaminergic mechanisms.1,12,24,25
4. Are FGAs or SGAs more likely to cause NMS?
NMS is assumed to occur less frequently in patients treated with SGAs than in those receiving FGAs, although this hypothesisis unproven. Isolated reports of NMS have been associated with nearly every SGA.9-11 It is difficult to prove FGA vs SGA liabilities because:
- NMS is rare.
- Dosing practices may be more conser-vative now than in the past.
- Most clinicians are aware of the earlysigns of NMS.
In an epidemiological study of a large database, Stubner et al26 found that patients receiving SGAs had a lower risk of NMS than those treated with haloperidol.26 In this study, the overall rate of NMS was 0.02%.
NMS hotline data. We recently examined which medication classes were implicated in 111 NMS cases reported to the Neuroleptic Malignant Syndrome Information Service hotline (1-888-NMS-TEMP) between 1997 and 2006 (Figure). We included only cases of definite or probable NMS (as diagnosed by hotline consultants) in which a single antipsychotic was administered. Slightly more cases were attributed to FGAs (51%) than SGAs (45%). The remaining cases were attributed to neuroleptics used in medical settings (such as promethazineor prochlorperazine). Because they are now prescribed less often, FGAs accounted for a disproportionate number of NMS cases reported to the hotline. Haloperidol accounted for the majority of FGA cases and 44% of all cases. If we had excluded haloperidol and compared the NMS risk of SGAs to only intermediate- or low-potency FGAs, the relative advantage of SGAs would have been lost. On the other hand, it is clear that SGAs still carry a risk for NMS. Analyses suggest that the SGA-associated classic features of NMS—fever, muscle rigidity, and autonomic and mental status changes—are retained in patients receiving SGAs, although some may not develop the severe rigidity and extreme temperatures common in patients receiving FGAs.9-11 The milder clinical characteristics associated with SGAs may reflect more conservative prescribing patterns or increased awareness and earlier recognition of NMS, which would prevent fulminant presentations.
5. What is the evidence for specific NMS treatments?
NMS is rare, its presentation varies, and its progression is unpredictable. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce.
Even so, the notion that NMS represents an extreme variant of drug-induced parkinsonism or catatonia suggests that specific NMS treatments could be based on symptom severity or stage of presentation. We propose a treatment guideline basedon theoretical mechanisms and anecdotal data (Algorithm).2,27-29
Support. After immediate withdrawal of the offending medication, supportive therapy is the cornerstone of NMS treatment.1,2,27
For patients presenting with mild signs and symptoms, supportive care and careful clinical monitoring may be sufficient. Extreme hyperthermia demands volume resuscitation and cooling measures, intensive medical care, and careful monitoring for complications.
Treatment. Despite a lack of consensus on drug treatments for uncomplicated NMS, approximately 40% of patients with acute NMS receive pharmacologic treatments.2
Lorazepam, 1 to 2 mg parenterally, is a reasonable first-line therapy for NMS, especially in individuals with catatonic features.4,15-18,21,30,31 Some investigators recommend higher doses.15 Benzodiazepines are preferred if sedation is required in agitated NMS patients.4,15-18
Dopaminergic agents such as bromocriptine and amantadine enhance dopaminergic transmission to reverse parkinsonian symptoms and have been reported to reduce time to recovery and halve mortality rates when used alone or in conjunction with other treatments.13,27,32,33 Rapid discontinuation of these agents can result in rebound symptoms, although this may be true for any specific drug treatment of NMS.1,31,32
Dantrolene uncouples excitation-contraction coupling by enhancing calcium sequestration in sarcoplasmic reticulumin skeletal muscle and has been used to treat NMS hypermetabolic symptoms. Some reviews found improvement in up to 80% of NMS patients treated with dantrolene monotherapy.27,32-35 Compared with supportive care, time to recovery may be reduced—and mortality decreased by almost one-half—when dantrolene is used alone or in combination with other medications.
Not all case reports have shown that dantrolene, benzodiazepines, ordopaminergic agonists are effective in treating NMS.31,36 In our opinion, only advanced NMS cases—with extreme temperature elevations, severe rigidity, and evidence of systemic hypermetabolism—benefit from dantrolene treatment.1,2
ECT has been used successfully to reduce mortality from NMS and other catatonic-spectrum disorders. It is usually employed after supportive therapy and psychopharmacologic interventions fail.2,15,16,27,37 ECT for acute NMS typically consists of a series of 6 to 10 treatments with bilateral electrode placement. Daily ECT may be needed initially.15
6. Are antipsychotics contraindicated following an NMS episode?
The rate of NMS recurrence on retreatment with an antipsychotic has varied.38 We estimate that up to 30% of patients may be at risk of NMS recurrence when rechallenged with an antipsychotic.1 By following proper precautions (Table 2), however, you can safely treat most patients who require continued antipsychotic therapy.1,2 When you restart treatment, a lower-potency antipsychotic from a different chemical class may be a safer option than retrying the triggering agent, according to retrospective analyses of limited available data. A patient who develops NMS on a FGA might benefit from an SGA trial, although some risk of recurrence remains.1,10
Current Psychiatry 2007;6(8):89-95.
Drug brand names
- Amantadine • Symmetrel
- Bromocriptine • Parlodel
- Chlorpromazine • Thorazine
- Dantrolene • Dantrium
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Perphenazine • Trilafon
- Prochlorperazine • Compazine, Compro
- Promethazine • Phenergan
- Thioridazine • Mellaril
Disclosure
Dr. Strawn is an American Psychiatric Institute for Research and Education (APIRE)/Janssen Scholar.
Dr. Keck has received research support from or served as a consultant to Abbott Laboratories, American Diabetes Association, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly and Company, Janssen Pharmaceutica, National Institute of Mental Health, National Institute of Drug Abuse, Pfizer, Stanley Medical Research Institute, and UCB Pharma.
Dr. Caroff has received research support from Bristol-Myers Squibb, Ortho-McNeil Neurologics, and Pfizer.
1. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions 2nd ed. Washington, DC: American Psychiatric Publishing Inc; 2003; 1-44.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome Am J Psychiatry 2007;164:870-6.
3. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome Arch Gen Psychiatry 1989;46:914-18.
4. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome Am J Psychiatry 1989;146:717-25.
5. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry 1998;44:748-54.
6. White DA, Robins AH. Catatonia: harbinger of the neuroleptic malignant syndrome Br J Psychiatry 1991;158:419-21.
7. Rosebush PI, Mazurek MF. Serum iron and neuroleptic malignant syndrome. Lancet 1991;338:149-51.
8. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome Biol Psychiatry 1998;44:499-507.
9. Ananth J, Parameswaran S, Gunatilake S, et al. Neuroleptic malignant syndrome and atypical antipsychotic drugs J Clin Psychiatry 2004;65:464-70.
10. Caroff SN, Mann SC, Campbell EC. Atypical antipsychotics and neuroleptic malignant syndrome Psychiatr Ann 2000;30:314-21.
11. Hasan S, Buckley P. Novel antipsychotics and the neuroleptic malignant syndrome Am J Psychiatry 1998;155:1113-16.
12. Mann SC, Caroff SN, Fricchione G, Campbell EC. Central dopamine hypoactivity and the pathogenesis of neuroleptic malignant syndrome Psychiatr Ann 2000;30:363-74.
13. Factor SA, Santiago A. Parkinsonism-hyperpyrexia syndrome in Parkinson’s disease. In: Frucht SJ, Fahn S, eds. Movement disorder emergencies: diagnosis and treatment. Totowa, NJ: Humana Press; 2005; 29-40.
14. Nisijima K, Ishiguro T. Cerebrospinal fluid levels of monoamine metabolites and gamma-aminobutyric acid in neuroleptic malignant syndrome. J Psychiatr Res 1995;27:233-44.
15. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:1182-3.
16. Fricchione G, Bush G, Fozdar M, et al. Recognition and treatment of the catatonic syndrome. J Intensive Care Med 1997;12:135-47.
17. Philbrick KL, Rummans TA. Malignant catatonia. J Neuropsychiatry Clin Neurosci 1994;6:1-13.
18. Mann SC, Caroff SN, Bleier HR, et al. Lethal catatonia. Am J Psychiatry 1986;143:1374-81.
19. Koch M, Chandragiri S, Rizvi S, et al. Catatonic signs in neuroleptic malignant syndrome. Compr Psychiatry 2000;41:73-5.
20. Lee JW. Laboratory findings. In: Caroff SN, Mann SC, Francis A, Fricchoine GL, eds. Catatonia: from psychopathology to neurobiology Washington, DC: American Psychiatric Press, Inc; 2004; 65-75.
21. Lee JW. Catatonic variants, hyperthermic extrapyramidal reactions, and subtypes of neuroleptic malignant syndrome. Ann Clin Psychiatry 2007;19:9-16.
22. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiol 2001;28:387-93.
23. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry 1999;156:169-80.
24. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr 2000;5:26-33.
25. Weller M, Kornhuber J. A rationale for NMDA receptor antagonist therapy of the neuroleptic malignant syndrome. Med Hypotheses 1992;38:329-33.
26. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry 2004;37(suppl 1):S54-S64.
27. Davis JM, Caroff SN, Mann SC. Treatment of neuroleptic malignant syndrome. Psychiatr Ann 2000;30:325-31.
28. Adityanjee PA, Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988;153:107-11.
29. Woodbury MM, Woodbury MA. Neuroleptic-induced catatonia as a stage in the progression toward neuroleptic malignant syndrome. J Am Acad Child Adolesc Psychiatry 1992;31:1161-4.
30. Francis A, Chondragivi S, Rizvi S, et al. Is lorazepam a treatment for neuroleptic malignant syndrome? CNS Spectr 2000;5:54-7.
31. Rosebush PI, Stewart T, Mazurek MF. The treatment of neuroleptic malignant syndrome. Are dantrolene and bromocriptine useful adjuncts to supportive care? Br J Psychiatry 1991;159:709-12.
32. Sakkas P, Davis JM, Janicak PG, Wang ZY. Drug treatment of the neuroleptic malignant syndrome. Psychopharmacol Bull 1991;27:381-4.
33. Rosenberg MR, Green M. Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 1989;149:1927-31.
34. Yamawaki S, Morio M, Kazamutsuri G, et al. Clinical evaluation and effective usage of dantrolene sodium in neuroleptic malignant syndrome. Kiso to Rinsyou (Clinical Reports) 1993;27:1045-66.
35. Tsutsumi Y, Yamamoto K, Matsuura S, et al. The treatment of neuroleptic malignant syndrome using dantrolene sodium. Psychiatry Clin Neurosci 1998;52:433-8.
36. Reulbach U, Dutsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care 2007;11:R4.-
37. Troller JN, Sachdev PS. Electroconvulsive treatment of neuroleptic malignant syndrome: a review and report of cases. Aust N Z J Psychiatry 1999;33:650-9.
38. Pope HG, Aizley HG, Keck PE, Jr, McElroy SL. Neuroleptic malignant syndrome: long term follow-up of 20 cases. J Clin Psychiatry 1991;52:208-12.
Diagnosis and treatment of neuroleptic malignant syndrome (NMS) are controversial because this potentially life-threatening syndrome is rare and its presentation varies. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce. It may be possible, however, to develop rational treatment guidelines using empiric clinical data.1,2
This article examines the evidence related to 6 controversial aspects of NMS diagnosis and treatment:
- most-reliable risk factors
- NMS as a spectrum disorder
- what causes NMS
- NMS triggered by first-generation vs second-generation antipsychotics
- first-line interventions
- restarting antipsychotics after an NMS episode.
1. Are there reliable risk factors for NMS?
In small case-controlled studies, agitation, dehydration, and exhaustion were the most consistently found systemic factors believed to predispose patients taking antipsychotics to NMS (Table 1).3-5 Catatonia and organic brain syndromes may be separate risk factors.1,6
Preliminary studies also have implicated dopamine receptor abnormalities caused by genetic polymorphisms or effects of low serum iron.1,7,8 Pharmacologic studies have suggested that higher doses, rapid titration, and IM injections of antipsychotics are associated with increased NMS risk.3,5 Some studies suggest that 15% to 20% of NMS patients have a history of NMS episodes.1,2 In addition, high-potency first-generation antipsychotics (FGAs)—especially haloperidol—are assumed to carry higher risk than low-potency drugs and second-generation antipsychotics (SGAs), although this hypothesis remains difficult to prove.9-11
These risk factors, however, are not practical for estimating NMS risk in a given patient because they are relatively common compared with the low risk of NMS occurrence. For the vast majority of patients with psychotic symptoms, the benefits of properly indicated antipsychotic pharmacotherapy will outweigh the risks.
Table 1
| Systemic |
| Agitation |
| Dehydration |
| Exhaustion |
| Low serum iron concentrations (normal: 60 to 170 mcg/dL) |
| Diagnoses |
| History of NMS |
| Catatonia |
| Organic brain syndromes |
| Central nervous system |
| Dopamine receptor dysfunction |
| Basal ganglia dysfunction |
| Sympathetic nervous system dysfunction |
| Pharmacologic treatment* |
| Intramuscular or intravenous injections |
| High-potency dopamine antagonists |
| Rapid dose titration |
| High doses |
| FGAs compared with SGAs (?) |
*For individual patients, these common risk factors must be weighted again the benefits of antipsychotic therapy FGAs: first-generation antipsychotics; SGAs:second-generation antipsychotics; NMS: neuroleptic malignant syndromeSource: References 1-5
2. Is NMS related to parkinsonism, catatonia, or malignant hyperthermia?
Parkinsonsim. Some researchers have described NMS as an extreme parkinsonian crisis resulting from overwhelming blockade of dopamine pathways in the brain.1,2,12 In this view, NMS resembles the parkinsonian-hyperthermia syndrome that can occur in Parkinson's disease patients following abrupt discontinuation or loss of efficacy of dopaminergic therapy, which can be treated by reinstituting dopaminergic agents.13 Evidence to support this view includes:
- Parkinsonian signs are a cardinal feature of NMS.
- Withdrawal of dopamine agonists precipitates the syndrome.
- All triggering drugs are dopamine receptor antagonists.
- Risks of NMS correlates with drugs' dopamine receptor affinity.
- Dopaminergic agonists may be an effective treatment.
- Lesions in dopaminergic pathways produce a similar syndrome.
- Patients with NMS have demonstrated low cerebrospinal fluid concentrations of the dopamine metabolite homovanillic acid.14
Catatonia. Fink et al15 and others16-18 have persuasively argued that NMS represents a form of drug-induced malignant catatonia. Evidence supporting this includes:
- The 2 disorders share neuropsychiatric symptoms.
- Catalonic signs are common in NMS.19
- Malignant catatonia and NMS share physiologic and labratory signs.20
- Reintroduction of antipsychotics can acutely worsen both conditions.
- Benzodiazepines and electroconvulsive therapy (ECT) are effective treatments for both disorders.15-18
Lee21 examined the relationship between catatonic features and treatment response of 14 NMS patients. Most patients with catatonic symptoms responded to benzodiazepines, whereas none of those did who had an extrapyramidal-hyperthermic presentation without catatonia. Lee concluded that NMS is heterogeneous and may occur in catatonic and noncatatonic forms that differ in treatment response.
- similar clinical signs of rigidity, hyperthermia, and hypermetabolism
- similar psychologic and labratory signs, such as rhabdomyolysis
- hyperthermia in both responding to dantrolene.
Although the 2 are similar in presentation, malignant hyperthermia occurs intraoperatively and reflects a pharmacogenetic disorder of calcium regulation in skeletal muscle. Additionally, rigidity in malignant hyperthermia does not respond to peripheral-acting muscle relaxants.1,22 Evidence suggests that patients who have previously experienced an NMS episodes are not at risk for malignant hyperthermia.22
3. What is the pathophysiology of NMS?
NMS pathophysiology is complex and likely involves interplay between multiple central and systemic pathways and neurotransmitters. As described above, compelling evidence suggests that dopamine blockade plays a central role.12
Dopamine blockade in the hypothalamus is believed to contribute to thermoregulatory failure, and blockade in the nigrostriatal system likely contributes to muscle rigidity and hypermetabolism. The loss of dopaminergic input to the anterior cingulate-medial orbitofrontal circuit and the lateral orbitofrontal circuit likely con-tributes to the mental status changes and catatonic features seen in NMS.12
Some researchers have proposed competing or complementary hypotheses, however. For example, Gurrera23 proposed that patients who are prone to developing NMS have a vulnerability to a hyperactive and dysregulated sympathetic nervous system, and this trait—together with dopamine system disruption induced by dopamine-blocking agents—produces NMS. Other investigators have implicated serotonin, norepinephrine, gamma-aminobutyric acid and glutaminergic mechanisms.1,12,24,25
4. Are FGAs or SGAs more likely to cause NMS?
NMS is assumed to occur less frequently in patients treated with SGAs than in those receiving FGAs, although this hypothesisis unproven. Isolated reports of NMS have been associated with nearly every SGA.9-11 It is difficult to prove FGA vs SGA liabilities because:
- NMS is rare.
- Dosing practices may be more conser-vative now than in the past.
- Most clinicians are aware of the earlysigns of NMS.
In an epidemiological study of a large database, Stubner et al26 found that patients receiving SGAs had a lower risk of NMS than those treated with haloperidol.26 In this study, the overall rate of NMS was 0.02%.
NMS hotline data. We recently examined which medication classes were implicated in 111 NMS cases reported to the Neuroleptic Malignant Syndrome Information Service hotline (1-888-NMS-TEMP) between 1997 and 2006 (Figure). We included only cases of definite or probable NMS (as diagnosed by hotline consultants) in which a single antipsychotic was administered. Slightly more cases were attributed to FGAs (51%) than SGAs (45%). The remaining cases were attributed to neuroleptics used in medical settings (such as promethazineor prochlorperazine). Because they are now prescribed less often, FGAs accounted for a disproportionate number of NMS cases reported to the hotline. Haloperidol accounted for the majority of FGA cases and 44% of all cases. If we had excluded haloperidol and compared the NMS risk of SGAs to only intermediate- or low-potency FGAs, the relative advantage of SGAs would have been lost. On the other hand, it is clear that SGAs still carry a risk for NMS. Analyses suggest that the SGA-associated classic features of NMS—fever, muscle rigidity, and autonomic and mental status changes—are retained in patients receiving SGAs, although some may not develop the severe rigidity and extreme temperatures common in patients receiving FGAs.9-11 The milder clinical characteristics associated with SGAs may reflect more conservative prescribing patterns or increased awareness and earlier recognition of NMS, which would prevent fulminant presentations.
5. What is the evidence for specific NMS treatments?
NMS is rare, its presentation varies, and its progression is unpredictable. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce.
Even so, the notion that NMS represents an extreme variant of drug-induced parkinsonism or catatonia suggests that specific NMS treatments could be based on symptom severity or stage of presentation. We propose a treatment guideline basedon theoretical mechanisms and anecdotal data (Algorithm).2,27-29
Support. After immediate withdrawal of the offending medication, supportive therapy is the cornerstone of NMS treatment.1,2,27
For patients presenting with mild signs and symptoms, supportive care and careful clinical monitoring may be sufficient. Extreme hyperthermia demands volume resuscitation and cooling measures, intensive medical care, and careful monitoring for complications.
Treatment. Despite a lack of consensus on drug treatments for uncomplicated NMS, approximately 40% of patients with acute NMS receive pharmacologic treatments.2
Lorazepam, 1 to 2 mg parenterally, is a reasonable first-line therapy for NMS, especially in individuals with catatonic features.4,15-18,21,30,31 Some investigators recommend higher doses.15 Benzodiazepines are preferred if sedation is required in agitated NMS patients.4,15-18
Dopaminergic agents such as bromocriptine and amantadine enhance dopaminergic transmission to reverse parkinsonian symptoms and have been reported to reduce time to recovery and halve mortality rates when used alone or in conjunction with other treatments.13,27,32,33 Rapid discontinuation of these agents can result in rebound symptoms, although this may be true for any specific drug treatment of NMS.1,31,32
Dantrolene uncouples excitation-contraction coupling by enhancing calcium sequestration in sarcoplasmic reticulumin skeletal muscle and has been used to treat NMS hypermetabolic symptoms. Some reviews found improvement in up to 80% of NMS patients treated with dantrolene monotherapy.27,32-35 Compared with supportive care, time to recovery may be reduced—and mortality decreased by almost one-half—when dantrolene is used alone or in combination with other medications.
Not all case reports have shown that dantrolene, benzodiazepines, ordopaminergic agonists are effective in treating NMS.31,36 In our opinion, only advanced NMS cases—with extreme temperature elevations, severe rigidity, and evidence of systemic hypermetabolism—benefit from dantrolene treatment.1,2
ECT has been used successfully to reduce mortality from NMS and other catatonic-spectrum disorders. It is usually employed after supportive therapy and psychopharmacologic interventions fail.2,15,16,27,37 ECT for acute NMS typically consists of a series of 6 to 10 treatments with bilateral electrode placement. Daily ECT may be needed initially.15
6. Are antipsychotics contraindicated following an NMS episode?
The rate of NMS recurrence on retreatment with an antipsychotic has varied.38 We estimate that up to 30% of patients may be at risk of NMS recurrence when rechallenged with an antipsychotic.1 By following proper precautions (Table 2), however, you can safely treat most patients who require continued antipsychotic therapy.1,2 When you restart treatment, a lower-potency antipsychotic from a different chemical class may be a safer option than retrying the triggering agent, according to retrospective analyses of limited available data. A patient who develops NMS on a FGA might benefit from an SGA trial, although some risk of recurrence remains.1,10
Current Psychiatry 2007;6(8):89-95.
Drug brand names
- Amantadine • Symmetrel
- Bromocriptine • Parlodel
- Chlorpromazine • Thorazine
- Dantrolene • Dantrium
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Perphenazine • Trilafon
- Prochlorperazine • Compazine, Compro
- Promethazine • Phenergan
- Thioridazine • Mellaril
Disclosure
Dr. Strawn is an American Psychiatric Institute for Research and Education (APIRE)/Janssen Scholar.
Dr. Keck has received research support from or served as a consultant to Abbott Laboratories, American Diabetes Association, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly and Company, Janssen Pharmaceutica, National Institute of Mental Health, National Institute of Drug Abuse, Pfizer, Stanley Medical Research Institute, and UCB Pharma.
Dr. Caroff has received research support from Bristol-Myers Squibb, Ortho-McNeil Neurologics, and Pfizer.
Diagnosis and treatment of neuroleptic malignant syndrome (NMS) are controversial because this potentially life-threatening syndrome is rare and its presentation varies. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce. It may be possible, however, to develop rational treatment guidelines using empiric clinical data.1,2
This article examines the evidence related to 6 controversial aspects of NMS diagnosis and treatment:
- most-reliable risk factors
- NMS as a spectrum disorder
- what causes NMS
- NMS triggered by first-generation vs second-generation antipsychotics
- first-line interventions
- restarting antipsychotics after an NMS episode.
1. Are there reliable risk factors for NMS?
In small case-controlled studies, agitation, dehydration, and exhaustion were the most consistently found systemic factors believed to predispose patients taking antipsychotics to NMS (Table 1).3-5 Catatonia and organic brain syndromes may be separate risk factors.1,6
Preliminary studies also have implicated dopamine receptor abnormalities caused by genetic polymorphisms or effects of low serum iron.1,7,8 Pharmacologic studies have suggested that higher doses, rapid titration, and IM injections of antipsychotics are associated with increased NMS risk.3,5 Some studies suggest that 15% to 20% of NMS patients have a history of NMS episodes.1,2 In addition, high-potency first-generation antipsychotics (FGAs)—especially haloperidol—are assumed to carry higher risk than low-potency drugs and second-generation antipsychotics (SGAs), although this hypothesis remains difficult to prove.9-11
These risk factors, however, are not practical for estimating NMS risk in a given patient because they are relatively common compared with the low risk of NMS occurrence. For the vast majority of patients with psychotic symptoms, the benefits of properly indicated antipsychotic pharmacotherapy will outweigh the risks.
Table 1
| Systemic |
| Agitation |
| Dehydration |
| Exhaustion |
| Low serum iron concentrations (normal: 60 to 170 mcg/dL) |
| Diagnoses |
| History of NMS |
| Catatonia |
| Organic brain syndromes |
| Central nervous system |
| Dopamine receptor dysfunction |
| Basal ganglia dysfunction |
| Sympathetic nervous system dysfunction |
| Pharmacologic treatment* |
| Intramuscular or intravenous injections |
| High-potency dopamine antagonists |
| Rapid dose titration |
| High doses |
| FGAs compared with SGAs (?) |
*For individual patients, these common risk factors must be weighted again the benefits of antipsychotic therapy FGAs: first-generation antipsychotics; SGAs:second-generation antipsychotics; NMS: neuroleptic malignant syndromeSource: References 1-5
2. Is NMS related to parkinsonism, catatonia, or malignant hyperthermia?
Parkinsonsim. Some researchers have described NMS as an extreme parkinsonian crisis resulting from overwhelming blockade of dopamine pathways in the brain.1,2,12 In this view, NMS resembles the parkinsonian-hyperthermia syndrome that can occur in Parkinson's disease patients following abrupt discontinuation or loss of efficacy of dopaminergic therapy, which can be treated by reinstituting dopaminergic agents.13 Evidence to support this view includes:
- Parkinsonian signs are a cardinal feature of NMS.
- Withdrawal of dopamine agonists precipitates the syndrome.
- All triggering drugs are dopamine receptor antagonists.
- Risks of NMS correlates with drugs' dopamine receptor affinity.
- Dopaminergic agonists may be an effective treatment.
- Lesions in dopaminergic pathways produce a similar syndrome.
- Patients with NMS have demonstrated low cerebrospinal fluid concentrations of the dopamine metabolite homovanillic acid.14
Catatonia. Fink et al15 and others16-18 have persuasively argued that NMS represents a form of drug-induced malignant catatonia. Evidence supporting this includes:
- The 2 disorders share neuropsychiatric symptoms.
- Catalonic signs are common in NMS.19
- Malignant catatonia and NMS share physiologic and labratory signs.20
- Reintroduction of antipsychotics can acutely worsen both conditions.
- Benzodiazepines and electroconvulsive therapy (ECT) are effective treatments for both disorders.15-18
Lee21 examined the relationship between catatonic features and treatment response of 14 NMS patients. Most patients with catatonic symptoms responded to benzodiazepines, whereas none of those did who had an extrapyramidal-hyperthermic presentation without catatonia. Lee concluded that NMS is heterogeneous and may occur in catatonic and noncatatonic forms that differ in treatment response.
- similar clinical signs of rigidity, hyperthermia, and hypermetabolism
- similar psychologic and labratory signs, such as rhabdomyolysis
- hyperthermia in both responding to dantrolene.
Although the 2 are similar in presentation, malignant hyperthermia occurs intraoperatively and reflects a pharmacogenetic disorder of calcium regulation in skeletal muscle. Additionally, rigidity in malignant hyperthermia does not respond to peripheral-acting muscle relaxants.1,22 Evidence suggests that patients who have previously experienced an NMS episodes are not at risk for malignant hyperthermia.22
3. What is the pathophysiology of NMS?
NMS pathophysiology is complex and likely involves interplay between multiple central and systemic pathways and neurotransmitters. As described above, compelling evidence suggests that dopamine blockade plays a central role.12
Dopamine blockade in the hypothalamus is believed to contribute to thermoregulatory failure, and blockade in the nigrostriatal system likely contributes to muscle rigidity and hypermetabolism. The loss of dopaminergic input to the anterior cingulate-medial orbitofrontal circuit and the lateral orbitofrontal circuit likely con-tributes to the mental status changes and catatonic features seen in NMS.12
Some researchers have proposed competing or complementary hypotheses, however. For example, Gurrera23 proposed that patients who are prone to developing NMS have a vulnerability to a hyperactive and dysregulated sympathetic nervous system, and this trait—together with dopamine system disruption induced by dopamine-blocking agents—produces NMS. Other investigators have implicated serotonin, norepinephrine, gamma-aminobutyric acid and glutaminergic mechanisms.1,12,24,25
4. Are FGAs or SGAs more likely to cause NMS?
NMS is assumed to occur less frequently in patients treated with SGAs than in those receiving FGAs, although this hypothesisis unproven. Isolated reports of NMS have been associated with nearly every SGA.9-11 It is difficult to prove FGA vs SGA liabilities because:
- NMS is rare.
- Dosing practices may be more conser-vative now than in the past.
- Most clinicians are aware of the earlysigns of NMS.
In an epidemiological study of a large database, Stubner et al26 found that patients receiving SGAs had a lower risk of NMS than those treated with haloperidol.26 In this study, the overall rate of NMS was 0.02%.
NMS hotline data. We recently examined which medication classes were implicated in 111 NMS cases reported to the Neuroleptic Malignant Syndrome Information Service hotline (1-888-NMS-TEMP) between 1997 and 2006 (Figure). We included only cases of definite or probable NMS (as diagnosed by hotline consultants) in which a single antipsychotic was administered. Slightly more cases were attributed to FGAs (51%) than SGAs (45%). The remaining cases were attributed to neuroleptics used in medical settings (such as promethazineor prochlorperazine). Because they are now prescribed less often, FGAs accounted for a disproportionate number of NMS cases reported to the hotline. Haloperidol accounted for the majority of FGA cases and 44% of all cases. If we had excluded haloperidol and compared the NMS risk of SGAs to only intermediate- or low-potency FGAs, the relative advantage of SGAs would have been lost. On the other hand, it is clear that SGAs still carry a risk for NMS. Analyses suggest that the SGA-associated classic features of NMS—fever, muscle rigidity, and autonomic and mental status changes—are retained in patients receiving SGAs, although some may not develop the severe rigidity and extreme temperatures common in patients receiving FGAs.9-11 The milder clinical characteristics associated with SGAs may reflect more conservative prescribing patterns or increased awareness and earlier recognition of NMS, which would prevent fulminant presentations.
5. What is the evidence for specific NMS treatments?
NMS is rare, its presentation varies, and its progression is unpredictable. These factors make it difficult to evaluate treatments in controlled clinical trials, and data about the relative efficacy of specific interventions are scarce.
Even so, the notion that NMS represents an extreme variant of drug-induced parkinsonism or catatonia suggests that specific NMS treatments could be based on symptom severity or stage of presentation. We propose a treatment guideline basedon theoretical mechanisms and anecdotal data (Algorithm).2,27-29
Support. After immediate withdrawal of the offending medication, supportive therapy is the cornerstone of NMS treatment.1,2,27
For patients presenting with mild signs and symptoms, supportive care and careful clinical monitoring may be sufficient. Extreme hyperthermia demands volume resuscitation and cooling measures, intensive medical care, and careful monitoring for complications.
Treatment. Despite a lack of consensus on drug treatments for uncomplicated NMS, approximately 40% of patients with acute NMS receive pharmacologic treatments.2
Lorazepam, 1 to 2 mg parenterally, is a reasonable first-line therapy for NMS, especially in individuals with catatonic features.4,15-18,21,30,31 Some investigators recommend higher doses.15 Benzodiazepines are preferred if sedation is required in agitated NMS patients.4,15-18
Dopaminergic agents such as bromocriptine and amantadine enhance dopaminergic transmission to reverse parkinsonian symptoms and have been reported to reduce time to recovery and halve mortality rates when used alone or in conjunction with other treatments.13,27,32,33 Rapid discontinuation of these agents can result in rebound symptoms, although this may be true for any specific drug treatment of NMS.1,31,32
Dantrolene uncouples excitation-contraction coupling by enhancing calcium sequestration in sarcoplasmic reticulumin skeletal muscle and has been used to treat NMS hypermetabolic symptoms. Some reviews found improvement in up to 80% of NMS patients treated with dantrolene monotherapy.27,32-35 Compared with supportive care, time to recovery may be reduced—and mortality decreased by almost one-half—when dantrolene is used alone or in combination with other medications.
Not all case reports have shown that dantrolene, benzodiazepines, ordopaminergic agonists are effective in treating NMS.31,36 In our opinion, only advanced NMS cases—with extreme temperature elevations, severe rigidity, and evidence of systemic hypermetabolism—benefit from dantrolene treatment.1,2
ECT has been used successfully to reduce mortality from NMS and other catatonic-spectrum disorders. It is usually employed after supportive therapy and psychopharmacologic interventions fail.2,15,16,27,37 ECT for acute NMS typically consists of a series of 6 to 10 treatments with bilateral electrode placement. Daily ECT may be needed initially.15
6. Are antipsychotics contraindicated following an NMS episode?
The rate of NMS recurrence on retreatment with an antipsychotic has varied.38 We estimate that up to 30% of patients may be at risk of NMS recurrence when rechallenged with an antipsychotic.1 By following proper precautions (Table 2), however, you can safely treat most patients who require continued antipsychotic therapy.1,2 When you restart treatment, a lower-potency antipsychotic from a different chemical class may be a safer option than retrying the triggering agent, according to retrospective analyses of limited available data. A patient who develops NMS on a FGA might benefit from an SGA trial, although some risk of recurrence remains.1,10
Current Psychiatry 2007;6(8):89-95.
Drug brand names
- Amantadine • Symmetrel
- Bromocriptine • Parlodel
- Chlorpromazine • Thorazine
- Dantrolene • Dantrium
- Fluphenazine • Prolixin
- Haloperidol • Haldol
- Lorazepam • Ativan
- Loxapine • Loxitane
- Perphenazine • Trilafon
- Prochlorperazine • Compazine, Compro
- Promethazine • Phenergan
- Thioridazine • Mellaril
Disclosure
Dr. Strawn is an American Psychiatric Institute for Research and Education (APIRE)/Janssen Scholar.
Dr. Keck has received research support from or served as a consultant to Abbott Laboratories, American Diabetes Association, AstraZeneca Pharmaceuticals, Bristol-Myers Squibb, GlaxoSmithKline, Eli Lilly and Company, Janssen Pharmaceutica, National Institute of Mental Health, National Institute of Drug Abuse, Pfizer, Stanley Medical Research Institute, and UCB Pharma.
Dr. Caroff has received research support from Bristol-Myers Squibb, Ortho-McNeil Neurologics, and Pfizer.
1. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions 2nd ed. Washington, DC: American Psychiatric Publishing Inc; 2003; 1-44.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome Am J Psychiatry 2007;164:870-6.
3. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome Arch Gen Psychiatry 1989;46:914-18.
4. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome Am J Psychiatry 1989;146:717-25.
5. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry 1998;44:748-54.
6. White DA, Robins AH. Catatonia: harbinger of the neuroleptic malignant syndrome Br J Psychiatry 1991;158:419-21.
7. Rosebush PI, Mazurek MF. Serum iron and neuroleptic malignant syndrome. Lancet 1991;338:149-51.
8. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome Biol Psychiatry 1998;44:499-507.
9. Ananth J, Parameswaran S, Gunatilake S, et al. Neuroleptic malignant syndrome and atypical antipsychotic drugs J Clin Psychiatry 2004;65:464-70.
10. Caroff SN, Mann SC, Campbell EC. Atypical antipsychotics and neuroleptic malignant syndrome Psychiatr Ann 2000;30:314-21.
11. Hasan S, Buckley P. Novel antipsychotics and the neuroleptic malignant syndrome Am J Psychiatry 1998;155:1113-16.
12. Mann SC, Caroff SN, Fricchione G, Campbell EC. Central dopamine hypoactivity and the pathogenesis of neuroleptic malignant syndrome Psychiatr Ann 2000;30:363-74.
13. Factor SA, Santiago A. Parkinsonism-hyperpyrexia syndrome in Parkinson’s disease. In: Frucht SJ, Fahn S, eds. Movement disorder emergencies: diagnosis and treatment. Totowa, NJ: Humana Press; 2005; 29-40.
14. Nisijima K, Ishiguro T. Cerebrospinal fluid levels of monoamine metabolites and gamma-aminobutyric acid in neuroleptic malignant syndrome. J Psychiatr Res 1995;27:233-44.
15. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:1182-3.
16. Fricchione G, Bush G, Fozdar M, et al. Recognition and treatment of the catatonic syndrome. J Intensive Care Med 1997;12:135-47.
17. Philbrick KL, Rummans TA. Malignant catatonia. J Neuropsychiatry Clin Neurosci 1994;6:1-13.
18. Mann SC, Caroff SN, Bleier HR, et al. Lethal catatonia. Am J Psychiatry 1986;143:1374-81.
19. Koch M, Chandragiri S, Rizvi S, et al. Catatonic signs in neuroleptic malignant syndrome. Compr Psychiatry 2000;41:73-5.
20. Lee JW. Laboratory findings. In: Caroff SN, Mann SC, Francis A, Fricchoine GL, eds. Catatonia: from psychopathology to neurobiology Washington, DC: American Psychiatric Press, Inc; 2004; 65-75.
21. Lee JW. Catatonic variants, hyperthermic extrapyramidal reactions, and subtypes of neuroleptic malignant syndrome. Ann Clin Psychiatry 2007;19:9-16.
22. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiol 2001;28:387-93.
23. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry 1999;156:169-80.
24. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr 2000;5:26-33.
25. Weller M, Kornhuber J. A rationale for NMDA receptor antagonist therapy of the neuroleptic malignant syndrome. Med Hypotheses 1992;38:329-33.
26. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry 2004;37(suppl 1):S54-S64.
27. Davis JM, Caroff SN, Mann SC. Treatment of neuroleptic malignant syndrome. Psychiatr Ann 2000;30:325-31.
28. Adityanjee PA, Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988;153:107-11.
29. Woodbury MM, Woodbury MA. Neuroleptic-induced catatonia as a stage in the progression toward neuroleptic malignant syndrome. J Am Acad Child Adolesc Psychiatry 1992;31:1161-4.
30. Francis A, Chondragivi S, Rizvi S, et al. Is lorazepam a treatment for neuroleptic malignant syndrome? CNS Spectr 2000;5:54-7.
31. Rosebush PI, Stewart T, Mazurek MF. The treatment of neuroleptic malignant syndrome. Are dantrolene and bromocriptine useful adjuncts to supportive care? Br J Psychiatry 1991;159:709-12.
32. Sakkas P, Davis JM, Janicak PG, Wang ZY. Drug treatment of the neuroleptic malignant syndrome. Psychopharmacol Bull 1991;27:381-4.
33. Rosenberg MR, Green M. Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 1989;149:1927-31.
34. Yamawaki S, Morio M, Kazamutsuri G, et al. Clinical evaluation and effective usage of dantrolene sodium in neuroleptic malignant syndrome. Kiso to Rinsyou (Clinical Reports) 1993;27:1045-66.
35. Tsutsumi Y, Yamamoto K, Matsuura S, et al. The treatment of neuroleptic malignant syndrome using dantrolene sodium. Psychiatry Clin Neurosci 1998;52:433-8.
36. Reulbach U, Dutsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care 2007;11:R4.-
37. Troller JN, Sachdev PS. Electroconvulsive treatment of neuroleptic malignant syndrome: a review and report of cases. Aust N Z J Psychiatry 1999;33:650-9.
38. Pope HG, Aizley HG, Keck PE, Jr, McElroy SL. Neuroleptic malignant syndrome: long term follow-up of 20 cases. J Clin Psychiatry 1991;52:208-12.
1. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions 2nd ed. Washington, DC: American Psychiatric Publishing Inc; 2003; 1-44.
2. Strawn JR, Keck PE, Jr, Caroff SN. Neuroleptic malignant syndrome Am J Psychiatry 2007;164:870-6.
3. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome Arch Gen Psychiatry 1989;46:914-18.
4. Rosebush PI, Stewart TD. A prospective analysis of 24 episodes of neuroleptic malignant syndrome Am J Psychiatry 1989;146:717-25.
5. Berardi D, Amore M, Keck PE, Jr, et al. Clinical and pharmacologic risk factors for neuroleptic malignant syndrome: a case-control study. Biol Psychiatry 1998;44:748-54.
6. White DA, Robins AH. Catatonia: harbinger of the neuroleptic malignant syndrome Br J Psychiatry 1991;158:419-21.
7. Rosebush PI, Mazurek MF. Serum iron and neuroleptic malignant syndrome. Lancet 1991;338:149-51.
8. Lee JW. Serum iron in catatonia and neuroleptic malignant syndrome Biol Psychiatry 1998;44:499-507.
9. Ananth J, Parameswaran S, Gunatilake S, et al. Neuroleptic malignant syndrome and atypical antipsychotic drugs J Clin Psychiatry 2004;65:464-70.
10. Caroff SN, Mann SC, Campbell EC. Atypical antipsychotics and neuroleptic malignant syndrome Psychiatr Ann 2000;30:314-21.
11. Hasan S, Buckley P. Novel antipsychotics and the neuroleptic malignant syndrome Am J Psychiatry 1998;155:1113-16.
12. Mann SC, Caroff SN, Fricchione G, Campbell EC. Central dopamine hypoactivity and the pathogenesis of neuroleptic malignant syndrome Psychiatr Ann 2000;30:363-74.
13. Factor SA, Santiago A. Parkinsonism-hyperpyrexia syndrome in Parkinson’s disease. In: Frucht SJ, Fahn S, eds. Movement disorder emergencies: diagnosis and treatment. Totowa, NJ: Humana Press; 2005; 29-40.
14. Nisijima K, Ishiguro T. Cerebrospinal fluid levels of monoamine metabolites and gamma-aminobutyric acid in neuroleptic malignant syndrome. J Psychiatr Res 1995;27:233-44.
15. Fink M, Taylor MA. Neuroleptic malignant syndrome is malignant catatonia, warranting treatments efficacious for catatonia. Prog Neuropsychopharmacol Biol Psychiatry 2006;30:1182-3.
16. Fricchione G, Bush G, Fozdar M, et al. Recognition and treatment of the catatonic syndrome. J Intensive Care Med 1997;12:135-47.
17. Philbrick KL, Rummans TA. Malignant catatonia. J Neuropsychiatry Clin Neurosci 1994;6:1-13.
18. Mann SC, Caroff SN, Bleier HR, et al. Lethal catatonia. Am J Psychiatry 1986;143:1374-81.
19. Koch M, Chandragiri S, Rizvi S, et al. Catatonic signs in neuroleptic malignant syndrome. Compr Psychiatry 2000;41:73-5.
20. Lee JW. Laboratory findings. In: Caroff SN, Mann SC, Francis A, Fricchoine GL, eds. Catatonia: from psychopathology to neurobiology Washington, DC: American Psychiatric Press, Inc; 2004; 65-75.
21. Lee JW. Catatonic variants, hyperthermic extrapyramidal reactions, and subtypes of neuroleptic malignant syndrome. Ann Clin Psychiatry 2007;19:9-16.
22. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiol 2001;28:387-93.
23. Gurrera RJ. Sympathoadrenal hyperactivity and the etiology of neuroleptic malignant syndrome. Am J Psychiatry 1999;156:169-80.
24. Carroll BT. The universal field hypothesis of catatonia and neuroleptic malignant syndrome. CNS Spectr 2000;5:26-33.
25. Weller M, Kornhuber J. A rationale for NMDA receptor antagonist therapy of the neuroleptic malignant syndrome. Med Hypotheses 1992;38:329-33.
26. Stubner S, Rustenbeck E, Grohmann R, et al. Severe and uncommon involuntary movement disorders due to psychotropic drugs. Pharmacopsychiatry 2004;37(suppl 1):S54-S64.
27. Davis JM, Caroff SN, Mann SC. Treatment of neuroleptic malignant syndrome. Psychiatr Ann 2000;30:325-31.
28. Adityanjee PA, Singh S, Singh G, Ong S. Spectrum concept of neuroleptic malignant syndrome. Br J Psychiatry 1988;153:107-11.
29. Woodbury MM, Woodbury MA. Neuroleptic-induced catatonia as a stage in the progression toward neuroleptic malignant syndrome. J Am Acad Child Adolesc Psychiatry 1992;31:1161-4.
30. Francis A, Chondragivi S, Rizvi S, et al. Is lorazepam a treatment for neuroleptic malignant syndrome? CNS Spectr 2000;5:54-7.
31. Rosebush PI, Stewart T, Mazurek MF. The treatment of neuroleptic malignant syndrome. Are dantrolene and bromocriptine useful adjuncts to supportive care? Br J Psychiatry 1991;159:709-12.
32. Sakkas P, Davis JM, Janicak PG, Wang ZY. Drug treatment of the neuroleptic malignant syndrome. Psychopharmacol Bull 1991;27:381-4.
33. Rosenberg MR, Green M. Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 1989;149:1927-31.
34. Yamawaki S, Morio M, Kazamutsuri G, et al. Clinical evaluation and effective usage of dantrolene sodium in neuroleptic malignant syndrome. Kiso to Rinsyou (Clinical Reports) 1993;27:1045-66.
35. Tsutsumi Y, Yamamoto K, Matsuura S, et al. The treatment of neuroleptic malignant syndrome using dantrolene sodium. Psychiatry Clin Neurosci 1998;52:433-8.
36. Reulbach U, Dutsch C, Biermann T, et al. Managing an effective treatment for neuroleptic malignant syndrome. Crit Care 2007;11:R4.-
37. Troller JN, Sachdev PS. Electroconvulsive treatment of neuroleptic malignant syndrome: a review and report of cases. Aust N Z J Psychiatry 1999;33:650-9.
38. Pope HG, Aizley HG, Keck PE, Jr, McElroy SL. Neuroleptic malignant syndrome: long term follow-up of 20 cases. J Clin Psychiatry 1991;52:208-12.
Beware ictal activity that mimics psychiatric illness
Nonconvulsive status epilepticus (NCSE) is marked by neurobehavioral disturbances that resemble primary psychiatric disorders. Mistaken diagnosis and delayed treatment increase the risk of neurologic damage, so recognizing NCSE symptoms early is important.
To help you make a timely diagnosis, this article describes:
- neuropsychiatric manifestations of NCSE
- how to narrow the differential diagnosis by reviewing clinical symptoms and using electroencephalography (EEG)
- techniques used to rapidly halt ictal activity.
Status epilepticus (SE) is an acute medical emergency. Both forms—convulsive (CSE) and nonconvulsive (NCSE)—require early recognition and treatment. In the United States, 60 SE cases occur per 100,000 population/year, with mortality rates of 20% in adults and 38% in the elderly.1,2
Mortality risk. Data suggest patients with NCSE are unlikely to die unless NCSE co-occurs with CSE or severe medical illness such as delirium or acute complications. Mortality risk does not appear linked with a type of EEG discharge.3
Neurologic injury risk. Prolonged NCSE may cause permanent neurologic damage.4 Transient memory impairment has been reported after cessation of complex partial status epilepticus (CPSE).5 CPSE also has resulted in prolonged neurologic deficits, although concomitant medical illnesses might have contributed to the deficits.6 In one study, some patients gradually returned to baseline cognitive function after CPSE stopped, but they were not tested with standardized neuropsychological tools.7
No significant postictal memory impairment was observed on neuropsychological testing in patients with NCSE of frontal origin.8 A >5-year follow-up study of absence status epilepticus (ASE) found no evidence of long-term cognitive or behavioral decline, even though most patients had recurrent ASE.9 Similarly, no long-term sequelae were seen in patients with ASE.10,11
Triggers, neurologic symptoms
NCSE is an acute but treatable medical emergency that calls for assessing and supporting cardiac and respiratory function, monitoring vital signs, temperature reduction, and fluid replacement. Prognosis is usually good unless NCSE is associated with a serious medical illness (Box).1-11
Many metabolic, neurologic, pharmacologic, and medical abnormalities can precipitate NCSE (Table 1). The most common causes are hypoxia/anoxia, stroke, infection, subtherapeutic antiepileptic levels, alcohol and benzodiazepine intoxication/withdrawal, and metabolic abnormalities.4,7,10,12
NCSE manifests as absence status epilepticus (ASE) or complex partial status epilepticus (CPSE). A generally accepted diagnostic definition is ≥30 minutes of behavioral change from baseline, with diagnostic EEG findings.4,13 EEG is indispensable because the clinical manifestations of NCSE are predominantly behavioral, with minimal or no motor activity.
Table 1
Clinical factors that may precipitate NCSE
| Medical | Recent infection, hyperventilation, trauma, menstruation, pregnancy, renal dialysis, postoperative period, sleep deprivation |
| Metabolic | Hypoparathyroidism, renal failure, hyper/hyponatremia, hyper/hypoglycemia, hypocalcemia |
| Neurologic | Mental retardation, dementia, stroke |
| Pharmacologic | Low serum levels or abrupt discontinuation of anticonvulsants, alcohol intoxication/withdrawal, benzodiazepine withdrawal lithium and neuroleptic use, psychotropic overdose |
| Source : References 9,10,12,16 | |
ASE is reported primarily in children, although de novo cases have been described in elderly patients with no history of epilepsy.10,14
CPSE is usually associated with a history of focal epilepsy and vascular disease. CPSE has a focal onset, with subsequent secondary generalization. Onset is usually temporal in origin but also can be extratemporal.
Patients with CPSE often cycle between an “epileptic twilight state” with confusion and complete unresponsiveness with stereotyped automatisms. It can present with marked behavioral fluctuation or a change in mental status and is generally followed by a prolonged postictal state.4,7,13-15 Several NCSE cases have occurred in patients with no history of seizures.9,10,16
Historically, CPSE was reported to be less common than ASE, but this misconception was most likely caused by failure to recognize CPSE’s clinical presentation and rapid generalization on EEG.7,15
Neuropsychiatric features
Patients with NCSE may be referred for evaluation of an array of behavioral changes commonly seen in psychiatric practice. The differential diagnosis is extensive (Table 2) and includes neurologic and medical conditions often associated with catatonic syndrome.17,18
In a retrospective study, Kaplan12 assessed clinical presentations and reasons for diagnostic delay in 23 adults eventually diagnosed with NCSE. Presenting symptoms included:
- confusion, agitation, aggressive behavior
- lethargy, mutism, verbal perseveration, echolalia
- delirium, blinking, staring, chewing or picking behaviors
- tremulousness or myoclonus
- bizarre behavior (inappropriate laughing, crying, or singing)
- rigidity with waxy flexibility
- delusions, hallucinations.
A prospective study of 22 patients with NCSE found that 7 had a history of psychotic depression, schizophrenia, self-mutilation, bipolar disorder, or episodic severe aggression; 12 of 18 with ASE had a history of epilepsy, and 3 of 4 with CPSE had experienced seizures associated with cerebrovascular accident, right cerebral embolus, and thiazide-induced hyponatremia, respectively.16
Table 2
Differential diagnosis of NCSE
| Metabolic disorders | Hypo/hyperglycemia, hypercalcemia, Addison’s disease, Cushing’s disease, uremia |
| Neurologic disorders | Stroke, CNS tumors, closed head trauma, transient global amnesia, seizures, inflammatory and infectious encephalopathies |
| Psychiatric disorders | Schizophrenia, mood disorders, catatonia, malignant catatonia, somatoform disorders, conversion disorder, Asperger’s syndrome, malingering |
| Toxic disorders | Toxic encephalopathy, neuroleptic malignant syndrome, serotonin syndrome, alcohol and sedative-hypnotic withdrawal, drugs (lithium toxicity, tricyclics, baclofen, tiagabine, overdose) |
| Source: Reference 17,18 | |
Cerebrovascular disease, tumors, and trauma are the most common causes of late-life NCSE.4,19 De novo NCSE occasionally presents:
- after benzodiazepine withdrawal
- with neuroleptic, tricyclic antidepressant, or lithium treatment10,16
- with metabolic abnormalities and nonpsychotropic medications.10
Clinical symptoms
Clinical features of NCSE include cognitive changes, speech abnormalities, affective disturbances, psychosis, poor impulse control, and bizarre behaviors (Table 3). Some patients develop ictal phenomena resembling catatonia or clinical and EEG changes that mimic neuroleptic malignant syndrome (NMS).20-23
Table 3
Clinical features that raise suspicion of NCSE
| Domain | Features |
|---|---|
| Cognitive changes | Prolonged confusion, executive dysfunction, obtundation, attention/memory difficulties, lack of initiative, perseveration, stupor |
| Speech | Poverty of speech with monosyllabic answers, verbal perseveration, echolalia, palilalia, aphasia, paraphasic errors, confabulation, mutism |
| Affective | Prolonged fear, affective indifferent state with blank facial expression, hypomania, psychotic depression, inappropriate laughing and crying, anxiety states |
| Psychosis | Visual, auditory and cenesthetic hallucinations, delusions |
| Impulse control | Hostility, agitation, violence, groping, genital manipulation, picking, posturing |
| Others | Catatonic signs, autonomic disturbances |
| Source: References 5,7-9,12,15-17,20-23 | |
Among 29 patients with acute catatonic syndromes, epileptic activity was identified in 4. One patient with absence status was diagnosed with NMS during the catatonic period.26 Conversely, the commonality of clinical features has led to misdiagnosis of psychogenic catatonia as NCSE. EEG is necessary to exclude NCSE in these cases.
NMS. Yoshino et al27 described two patients taking neuroleptics who met criteria for NMS and had EEG changes consistent with NCSE. They later reported another patient with NCSE complicating NMS; the point at which NCSE developed was unknown, however, because EEG activity was not recorded at NMS onset.28 Based on NMS diagnostic criteria proposed by Caroff et al,29 these patients could have developed NCSE mimicking NMS.
EEG for diagnosis
Candidates. Because differentiating NCSE from similar conditions can be difficult, use EEG to confirm your clinical observations. No guidelines exist, but consider EEG when the patient’s history suggests NCSE. Ask the patient or family about:
- changes in mental status from baseline, especially new-onset catatonia or unexplained altered consciousness
- duration of events
- presence or absence of motor activity
- behavioral fluctuations
- presence or absence of automatisms or blinking.
EEG patterns. Table 4 summarized NCSE diagnostic criteria. NCSE shows characteristic patterns in ASE and CPSE,9,10,16,23 and EEG changes can be continuous or nearly continuous in both.
Table 4
EEG findings that support a clinical diagnosis of NCSE
| Clear-cut criteria |
|---|
| Frequent or continuous focal seizures, with ictal patterns that wax and wane with change in amplitude, frequency, and/or spatial distribution |
| Frequent or continuous generalized spike wave discharges: |
|
| Periodic lateralized epileptiform discharges (“PLEDs”) or bilateral periodic epileptiform discharges (“biPEDs") occurring in patients with coma from generalized tonic-clonic status epilepticus (subtle SE) |
| Probable (equivocal) criteria |
| Patients with acute cerebral damage who also show frequent or continuous EEG abnormalities without previous similar findings |
| Patients with epilepsy who show frequent or continuous generalized EEG abnormalities and similar interictal EEG patterns but whose clinical symptoms suggest NCSE |
| Source: References 4,12-14,17 |
31,32
In CPSE, less-synchronous epileptiform activity has been described, including rhythmical slow, rhythmic spikes, or rhythmic spike and slow waves. Two types of CPSE of frontal origin have been described:
- Type 1 presents clinically with mood disturbance and minimal confusion. EEG shows a frontal focus with a normal background.
- Type 2 presents clinically with confusion. EEG shows bilateral asymmetric frontal discharges.8
- generalized in 69%
- diffuse with focal predominance in 18%
- focal in 13%.
Distinguish between ictal and interictal EEG findings with epileptiform activity, because only the former is diagnostic for NCSE. Intravenous benzodiazepines might be necessary during EEG to verify the diagnosis.33
NCSE has developed after electroconvulsive therapy (ECT), but a cause-effect relationship is debatable. Interictal and abnormal EEG findings after ECT may be misdiagnosed as NCSE.34
Neuroimaging has limited clinical value because of the need for patient cooperation and specialized equipment.4 Head CT or MRI can exclude structural abnormalities. PET and SPECT show increased metabolism and blood flow, respectively, in NCSE. MR spectroscopy shows elevated lactate and decreased N-acetyl aspartate.
Halting ictal activity
To rapidly stop ictal activity—the main goal of treatment—recognizing and correcting precipitant factors is vital:
- Consider discontinuing medications that could lower the seizure threshold.
- Order a complete blood count, serum electrolytes, calcium, arterial-blood gas, liver and renal function tests, urine toxicology screen, and serum antiepileptic drug concentrations.
- When possible, obtain neuroimaging and EEG in the emergency room for accurate diagnosis and prompt treatment.12
Response to benzodiazepines might be transient, lasting only hours or days. For instance, diazepam’s anticonvulsant effect may last
Newer antiepileptics—such as lamotrigine, levetiracetam, or topiramate—have been used with varying results, and their role in first-line treatment of NCSE is evolving. Rarely, the antiepileptic tiagabine precipitates or worsens NCSE.4,13,14
Related resources
- Epilepsy Foundation. www.epilepsyfoundation.org
- Neuroleptic Malignant Syndrome Information Service. Hotline for health professionals (888) 667-8367. www.nmsis.org
- Carbamazepine • Tegretol, Carbatrol
- Clonazepam • Klonopin
- Diazepam • Valium
- Lamotrigine • Lamictal
- Levetiracetam • Keppra
- Lithium carbonate • Lithobid, Eskalith CR
- Lorazepam • Ativan
- Phenobarbital • Luminal
- Phenytoin • Dilantin
- Tiagabine • Gabitril
- Topiramate • Topamax
- Valproic acid • Depakote
The authors report no financial relationship with any company whose products are mentioned in the article or with manufacturers of competing products.
Acknowledgment
Dr. Goveas was a geriatric psychiatry fellow, University of Pennsylvania, when he wrote this article in collaboration with his mentors, Drs. Caroff and Riggio.
1. DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46(4):1029-35.
2. Shorvon S. Status epilepticus: Its clinical features and treatment in children and adults Cambridge, UK: Cambridge University Press, 1994.
3. Shneker BF, Fountain NB. Assessment of acute morbidity and mortality in nonconvulsive status epilepticus. Neurology 2003;61:1066-73.
4. Walker M, Cross H, Smith S, et al. Nonconvulsive status epilepticus: Epilepsy research foundation workshop reports. Epileptic Disord 2005;7(3):53-296.
5. Engel J, Ludwig BI, Fetell M. Prolonged partial complex status epilepticus: EEG and behavioral observations. Neurology 1978;28:863-9.
6. Krumholz A, Sung GY, Fisher RS, et al. Complex partial status epilepticus accompanied by serious morbidity and mortality. Neurology 1995;45:1499-1504.
7. Ballenger CE, King DW, Gallagher BB. Partial complex status epilepticus. Neurology 1983;33:1545-52.
8. Thomas P, Zifkin B, Migneco O, et al. Nonconvulsive status epilepticus of frontal origin. Neurology 1999;52:1174-83.
9. Guberman A, Cantu-Reyna G, Stuss D, Broughton R. Nonconvulsive generalized status epilepticus: Clinical features, neuropsychological testing, and long-term follow-up. Neurology 1986;36:1284-91.
10. Thomas P, Beaumanoir A, Genton P, et al. ‘De novo’ absence status of late onset: Report of 11 cases. Neurology 1992;42:104-10.
11. Andermann F, Robb J. Absence status: a reappraisal following review of thirty-eight patients. Epilepsia 1972;13:177-87.
12. Kaplan PW. Nonconvulsive status epilepticus in the emergency room. Epilepsia 1996;37(7):643-50.
13. Riggio S. Nonconvulsive status epilepticus: Clinical features and diagnostic challenges. Psychiatr Clin N Am 2005;28(3):653-64.
14. Drislane FW. Presentation, evaluation, and treatment of nonconvulsive status epilepticus. Epilepsy Behav 2000;1(5):301-14.
15. Tomson T, Lindbom U, Nilsson BY. Nonconvulsive status epilepticus in adults: Thirty-two consecutive patients from a general hospital population. Epilepsia 1992;3(5):829-35.
16. Dunne JW, Summers QA, Stewart-Wynne EG. Non-convulsive status epilepticus: A prospective study in an adult general hospital. Q J Med 1987;62(238):117-26.
17. Kaplan PW. Behavioral manifestations of nonconvulsive status epilepticus. Epilepsy Behav 2002;3(2):122-39.
18. Mann SC. Malignant catatonia. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing Inc, 2003:121-43.
19. Sung CY, Chu NS. Status epilepticus in elderly: etiology, seizure type and outcome. Acta Neurol Scand 1989;80:51-6.
20. McLachlan RS, Blume WT. Isolated fear in complex partial status epilepticus. Ann Neurol 1980;8:639-41.
21. Walls MJ, Bowers TC, Dilsaver SC, Swann AC. Catatonia associated with depression secondary to complex partial epilepsy. J Clin Psychiatry 1993;54(2):73.-
22. Wells CE. Transient ictal psychosis. Arch Gen Psychiatry 1975;32:1201-3.
23. Agathonikou A, Panayiotopoulos CP, Giannakodimos S, Koutroumanidis M. Typical absence status in adults: Diagnostic and syndromic considerations. Epilepsia 1998;39(12):1265-76.
24. Lim J, Yagnik P, Schraeder P, Wheeler S. Ictal catatonia as a manifestation of nonconvulsive status epilepticus. J Neurol Neurosurg Psychiatry 1986;49:833-6.
25. Drury I, Klass DW, Westmoreland BF, Sharbrough FW. An acute syndrome with psychiatric symptoms and EEG abnormalities. Neurology 1985;35(6):911-14.
26. Primavera A, Fonti A, Novello P, et al. Epileptic seizures in patients with acute catatonic syndrome. J Neurol Neurosurg Psychiatry 1994;57(11):1419-22.
27. Yoshino A, Yoshimasu H, Tatsuzawa Y, et al. Nonconvulsive status epilepticus in two patients with neuroleptic malignant syndrome. J Clin Psychopharmacol 1998;18(4):347-9.
28. Yoshino A, Yoshimasu H. Nonconvulsive status epilepticus complicating neuroleptic malignant syndrome improved by intravenous diazepam. J Clin Psychopharmacol 2000;20(3):389-90.
29. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions, 2nd ed. Washington, DC: American Psychiatric Publishing; 2003:1-44.
30. Lob H, Roger J, Soulayrol R. Les etats de mal generalizes a expression confusionelle. In: Gastaut H, Roger J, Lob H, eds. Les etats de mal epileptiques. Paris: Masson; 1967:91-109.
31. Granner MA, Lee SI. Nonconvulsive status epilepticus: EEG analysis in a large series. Epilepsia 1994;35(1):42-7.
32. Niedermeyer E, Fineyre F, Riley T, Uematsu S. Absence status (petit mal status) with focal characteristics. Arch Neurol 1979;36:417-21.
33. Privitera M, Hoffman M, Moore JL, Jester D. EEG detection of nontonic-clonic status epilepticus in patients with altered consciousness. Epilepsy Res 1994;18:155-66.
34. Povlsen UJ, Wildschiodtz G, Hogenhaven H, Bolwig TG. Nonconvulsive status epilepticus after electroconvulsive therapy. J ECT 2003;19(3):164-9.
Nonconvulsive status epilepticus (NCSE) is marked by neurobehavioral disturbances that resemble primary psychiatric disorders. Mistaken diagnosis and delayed treatment increase the risk of neurologic damage, so recognizing NCSE symptoms early is important.
To help you make a timely diagnosis, this article describes:
- neuropsychiatric manifestations of NCSE
- how to narrow the differential diagnosis by reviewing clinical symptoms and using electroencephalography (EEG)
- techniques used to rapidly halt ictal activity.
Status epilepticus (SE) is an acute medical emergency. Both forms—convulsive (CSE) and nonconvulsive (NCSE)—require early recognition and treatment. In the United States, 60 SE cases occur per 100,000 population/year, with mortality rates of 20% in adults and 38% in the elderly.1,2
Mortality risk. Data suggest patients with NCSE are unlikely to die unless NCSE co-occurs with CSE or severe medical illness such as delirium or acute complications. Mortality risk does not appear linked with a type of EEG discharge.3
Neurologic injury risk. Prolonged NCSE may cause permanent neurologic damage.4 Transient memory impairment has been reported after cessation of complex partial status epilepticus (CPSE).5 CPSE also has resulted in prolonged neurologic deficits, although concomitant medical illnesses might have contributed to the deficits.6 In one study, some patients gradually returned to baseline cognitive function after CPSE stopped, but they were not tested with standardized neuropsychological tools.7
No significant postictal memory impairment was observed on neuropsychological testing in patients with NCSE of frontal origin.8 A >5-year follow-up study of absence status epilepticus (ASE) found no evidence of long-term cognitive or behavioral decline, even though most patients had recurrent ASE.9 Similarly, no long-term sequelae were seen in patients with ASE.10,11
Triggers, neurologic symptoms
NCSE is an acute but treatable medical emergency that calls for assessing and supporting cardiac and respiratory function, monitoring vital signs, temperature reduction, and fluid replacement. Prognosis is usually good unless NCSE is associated with a serious medical illness (Box).1-11
Many metabolic, neurologic, pharmacologic, and medical abnormalities can precipitate NCSE (Table 1). The most common causes are hypoxia/anoxia, stroke, infection, subtherapeutic antiepileptic levels, alcohol and benzodiazepine intoxication/withdrawal, and metabolic abnormalities.4,7,10,12
NCSE manifests as absence status epilepticus (ASE) or complex partial status epilepticus (CPSE). A generally accepted diagnostic definition is ≥30 minutes of behavioral change from baseline, with diagnostic EEG findings.4,13 EEG is indispensable because the clinical manifestations of NCSE are predominantly behavioral, with minimal or no motor activity.
Table 1
Clinical factors that may precipitate NCSE
| Medical | Recent infection, hyperventilation, trauma, menstruation, pregnancy, renal dialysis, postoperative period, sleep deprivation |
| Metabolic | Hypoparathyroidism, renal failure, hyper/hyponatremia, hyper/hypoglycemia, hypocalcemia |
| Neurologic | Mental retardation, dementia, stroke |
| Pharmacologic | Low serum levels or abrupt discontinuation of anticonvulsants, alcohol intoxication/withdrawal, benzodiazepine withdrawal lithium and neuroleptic use, psychotropic overdose |
| Source : References 9,10,12,16 | |
ASE is reported primarily in children, although de novo cases have been described in elderly patients with no history of epilepsy.10,14
CPSE is usually associated with a history of focal epilepsy and vascular disease. CPSE has a focal onset, with subsequent secondary generalization. Onset is usually temporal in origin but also can be extratemporal.
Patients with CPSE often cycle between an “epileptic twilight state” with confusion and complete unresponsiveness with stereotyped automatisms. It can present with marked behavioral fluctuation or a change in mental status and is generally followed by a prolonged postictal state.4,7,13-15 Several NCSE cases have occurred in patients with no history of seizures.9,10,16
Historically, CPSE was reported to be less common than ASE, but this misconception was most likely caused by failure to recognize CPSE’s clinical presentation and rapid generalization on EEG.7,15
Neuropsychiatric features
Patients with NCSE may be referred for evaluation of an array of behavioral changes commonly seen in psychiatric practice. The differential diagnosis is extensive (Table 2) and includes neurologic and medical conditions often associated with catatonic syndrome.17,18
In a retrospective study, Kaplan12 assessed clinical presentations and reasons for diagnostic delay in 23 adults eventually diagnosed with NCSE. Presenting symptoms included:
- confusion, agitation, aggressive behavior
- lethargy, mutism, verbal perseveration, echolalia
- delirium, blinking, staring, chewing or picking behaviors
- tremulousness or myoclonus
- bizarre behavior (inappropriate laughing, crying, or singing)
- rigidity with waxy flexibility
- delusions, hallucinations.
A prospective study of 22 patients with NCSE found that 7 had a history of psychotic depression, schizophrenia, self-mutilation, bipolar disorder, or episodic severe aggression; 12 of 18 with ASE had a history of epilepsy, and 3 of 4 with CPSE had experienced seizures associated with cerebrovascular accident, right cerebral embolus, and thiazide-induced hyponatremia, respectively.16
Table 2
Differential diagnosis of NCSE
| Metabolic disorders | Hypo/hyperglycemia, hypercalcemia, Addison’s disease, Cushing’s disease, uremia |
| Neurologic disorders | Stroke, CNS tumors, closed head trauma, transient global amnesia, seizures, inflammatory and infectious encephalopathies |
| Psychiatric disorders | Schizophrenia, mood disorders, catatonia, malignant catatonia, somatoform disorders, conversion disorder, Asperger’s syndrome, malingering |
| Toxic disorders | Toxic encephalopathy, neuroleptic malignant syndrome, serotonin syndrome, alcohol and sedative-hypnotic withdrawal, drugs (lithium toxicity, tricyclics, baclofen, tiagabine, overdose) |
| Source: Reference 17,18 | |
Cerebrovascular disease, tumors, and trauma are the most common causes of late-life NCSE.4,19 De novo NCSE occasionally presents:
- after benzodiazepine withdrawal
- with neuroleptic, tricyclic antidepressant, or lithium treatment10,16
- with metabolic abnormalities and nonpsychotropic medications.10
Clinical symptoms
Clinical features of NCSE include cognitive changes, speech abnormalities, affective disturbances, psychosis, poor impulse control, and bizarre behaviors (Table 3). Some patients develop ictal phenomena resembling catatonia or clinical and EEG changes that mimic neuroleptic malignant syndrome (NMS).20-23
Table 3
Clinical features that raise suspicion of NCSE
| Domain | Features |
|---|---|
| Cognitive changes | Prolonged confusion, executive dysfunction, obtundation, attention/memory difficulties, lack of initiative, perseveration, stupor |
| Speech | Poverty of speech with monosyllabic answers, verbal perseveration, echolalia, palilalia, aphasia, paraphasic errors, confabulation, mutism |
| Affective | Prolonged fear, affective indifferent state with blank facial expression, hypomania, psychotic depression, inappropriate laughing and crying, anxiety states |
| Psychosis | Visual, auditory and cenesthetic hallucinations, delusions |
| Impulse control | Hostility, agitation, violence, groping, genital manipulation, picking, posturing |
| Others | Catatonic signs, autonomic disturbances |
| Source: References 5,7-9,12,15-17,20-23 | |
Among 29 patients with acute catatonic syndromes, epileptic activity was identified in 4. One patient with absence status was diagnosed with NMS during the catatonic period.26 Conversely, the commonality of clinical features has led to misdiagnosis of psychogenic catatonia as NCSE. EEG is necessary to exclude NCSE in these cases.
NMS. Yoshino et al27 described two patients taking neuroleptics who met criteria for NMS and had EEG changes consistent with NCSE. They later reported another patient with NCSE complicating NMS; the point at which NCSE developed was unknown, however, because EEG activity was not recorded at NMS onset.28 Based on NMS diagnostic criteria proposed by Caroff et al,29 these patients could have developed NCSE mimicking NMS.
EEG for diagnosis
Candidates. Because differentiating NCSE from similar conditions can be difficult, use EEG to confirm your clinical observations. No guidelines exist, but consider EEG when the patient’s history suggests NCSE. Ask the patient or family about:
- changes in mental status from baseline, especially new-onset catatonia or unexplained altered consciousness
- duration of events
- presence or absence of motor activity
- behavioral fluctuations
- presence or absence of automatisms or blinking.
EEG patterns. Table 4 summarized NCSE diagnostic criteria. NCSE shows characteristic patterns in ASE and CPSE,9,10,16,23 and EEG changes can be continuous or nearly continuous in both.
Table 4
EEG findings that support a clinical diagnosis of NCSE
| Clear-cut criteria |
|---|
| Frequent or continuous focal seizures, with ictal patterns that wax and wane with change in amplitude, frequency, and/or spatial distribution |
| Frequent or continuous generalized spike wave discharges: |
|
| Periodic lateralized epileptiform discharges (“PLEDs”) or bilateral periodic epileptiform discharges (“biPEDs") occurring in patients with coma from generalized tonic-clonic status epilepticus (subtle SE) |
| Probable (equivocal) criteria |
| Patients with acute cerebral damage who also show frequent or continuous EEG abnormalities without previous similar findings |
| Patients with epilepsy who show frequent or continuous generalized EEG abnormalities and similar interictal EEG patterns but whose clinical symptoms suggest NCSE |
| Source: References 4,12-14,17 |
31,32
In CPSE, less-synchronous epileptiform activity has been described, including rhythmical slow, rhythmic spikes, or rhythmic spike and slow waves. Two types of CPSE of frontal origin have been described:
- Type 1 presents clinically with mood disturbance and minimal confusion. EEG shows a frontal focus with a normal background.
- Type 2 presents clinically with confusion. EEG shows bilateral asymmetric frontal discharges.8
- generalized in 69%
- diffuse with focal predominance in 18%
- focal in 13%.
Distinguish between ictal and interictal EEG findings with epileptiform activity, because only the former is diagnostic for NCSE. Intravenous benzodiazepines might be necessary during EEG to verify the diagnosis.33
NCSE has developed after electroconvulsive therapy (ECT), but a cause-effect relationship is debatable. Interictal and abnormal EEG findings after ECT may be misdiagnosed as NCSE.34
Neuroimaging has limited clinical value because of the need for patient cooperation and specialized equipment.4 Head CT or MRI can exclude structural abnormalities. PET and SPECT show increased metabolism and blood flow, respectively, in NCSE. MR spectroscopy shows elevated lactate and decreased N-acetyl aspartate.
Halting ictal activity
To rapidly stop ictal activity—the main goal of treatment—recognizing and correcting precipitant factors is vital:
- Consider discontinuing medications that could lower the seizure threshold.
- Order a complete blood count, serum electrolytes, calcium, arterial-blood gas, liver and renal function tests, urine toxicology screen, and serum antiepileptic drug concentrations.
- When possible, obtain neuroimaging and EEG in the emergency room for accurate diagnosis and prompt treatment.12
Response to benzodiazepines might be transient, lasting only hours or days. For instance, diazepam’s anticonvulsant effect may last
Newer antiepileptics—such as lamotrigine, levetiracetam, or topiramate—have been used with varying results, and their role in first-line treatment of NCSE is evolving. Rarely, the antiepileptic tiagabine precipitates or worsens NCSE.4,13,14
Related resources
- Epilepsy Foundation. www.epilepsyfoundation.org
- Neuroleptic Malignant Syndrome Information Service. Hotline for health professionals (888) 667-8367. www.nmsis.org
- Carbamazepine • Tegretol, Carbatrol
- Clonazepam • Klonopin
- Diazepam • Valium
- Lamotrigine • Lamictal
- Levetiracetam • Keppra
- Lithium carbonate • Lithobid, Eskalith CR
- Lorazepam • Ativan
- Phenobarbital • Luminal
- Phenytoin • Dilantin
- Tiagabine • Gabitril
- Topiramate • Topamax
- Valproic acid • Depakote
The authors report no financial relationship with any company whose products are mentioned in the article or with manufacturers of competing products.
Acknowledgment
Dr. Goveas was a geriatric psychiatry fellow, University of Pennsylvania, when he wrote this article in collaboration with his mentors, Drs. Caroff and Riggio.
Nonconvulsive status epilepticus (NCSE) is marked by neurobehavioral disturbances that resemble primary psychiatric disorders. Mistaken diagnosis and delayed treatment increase the risk of neurologic damage, so recognizing NCSE symptoms early is important.
To help you make a timely diagnosis, this article describes:
- neuropsychiatric manifestations of NCSE
- how to narrow the differential diagnosis by reviewing clinical symptoms and using electroencephalography (EEG)
- techniques used to rapidly halt ictal activity.
Status epilepticus (SE) is an acute medical emergency. Both forms—convulsive (CSE) and nonconvulsive (NCSE)—require early recognition and treatment. In the United States, 60 SE cases occur per 100,000 population/year, with mortality rates of 20% in adults and 38% in the elderly.1,2
Mortality risk. Data suggest patients with NCSE are unlikely to die unless NCSE co-occurs with CSE or severe medical illness such as delirium or acute complications. Mortality risk does not appear linked with a type of EEG discharge.3
Neurologic injury risk. Prolonged NCSE may cause permanent neurologic damage.4 Transient memory impairment has been reported after cessation of complex partial status epilepticus (CPSE).5 CPSE also has resulted in prolonged neurologic deficits, although concomitant medical illnesses might have contributed to the deficits.6 In one study, some patients gradually returned to baseline cognitive function after CPSE stopped, but they were not tested with standardized neuropsychological tools.7
No significant postictal memory impairment was observed on neuropsychological testing in patients with NCSE of frontal origin.8 A >5-year follow-up study of absence status epilepticus (ASE) found no evidence of long-term cognitive or behavioral decline, even though most patients had recurrent ASE.9 Similarly, no long-term sequelae were seen in patients with ASE.10,11
Triggers, neurologic symptoms
NCSE is an acute but treatable medical emergency that calls for assessing and supporting cardiac and respiratory function, monitoring vital signs, temperature reduction, and fluid replacement. Prognosis is usually good unless NCSE is associated with a serious medical illness (Box).1-11
Many metabolic, neurologic, pharmacologic, and medical abnormalities can precipitate NCSE (Table 1). The most common causes are hypoxia/anoxia, stroke, infection, subtherapeutic antiepileptic levels, alcohol and benzodiazepine intoxication/withdrawal, and metabolic abnormalities.4,7,10,12
NCSE manifests as absence status epilepticus (ASE) or complex partial status epilepticus (CPSE). A generally accepted diagnostic definition is ≥30 minutes of behavioral change from baseline, with diagnostic EEG findings.4,13 EEG is indispensable because the clinical manifestations of NCSE are predominantly behavioral, with minimal or no motor activity.
Table 1
Clinical factors that may precipitate NCSE
| Medical | Recent infection, hyperventilation, trauma, menstruation, pregnancy, renal dialysis, postoperative period, sleep deprivation |
| Metabolic | Hypoparathyroidism, renal failure, hyper/hyponatremia, hyper/hypoglycemia, hypocalcemia |
| Neurologic | Mental retardation, dementia, stroke |
| Pharmacologic | Low serum levels or abrupt discontinuation of anticonvulsants, alcohol intoxication/withdrawal, benzodiazepine withdrawal lithium and neuroleptic use, psychotropic overdose |
| Source : References 9,10,12,16 | |
ASE is reported primarily in children, although de novo cases have been described in elderly patients with no history of epilepsy.10,14
CPSE is usually associated with a history of focal epilepsy and vascular disease. CPSE has a focal onset, with subsequent secondary generalization. Onset is usually temporal in origin but also can be extratemporal.
Patients with CPSE often cycle between an “epileptic twilight state” with confusion and complete unresponsiveness with stereotyped automatisms. It can present with marked behavioral fluctuation or a change in mental status and is generally followed by a prolonged postictal state.4,7,13-15 Several NCSE cases have occurred in patients with no history of seizures.9,10,16
Historically, CPSE was reported to be less common than ASE, but this misconception was most likely caused by failure to recognize CPSE’s clinical presentation and rapid generalization on EEG.7,15
Neuropsychiatric features
Patients with NCSE may be referred for evaluation of an array of behavioral changes commonly seen in psychiatric practice. The differential diagnosis is extensive (Table 2) and includes neurologic and medical conditions often associated with catatonic syndrome.17,18
In a retrospective study, Kaplan12 assessed clinical presentations and reasons for diagnostic delay in 23 adults eventually diagnosed with NCSE. Presenting symptoms included:
- confusion, agitation, aggressive behavior
- lethargy, mutism, verbal perseveration, echolalia
- delirium, blinking, staring, chewing or picking behaviors
- tremulousness or myoclonus
- bizarre behavior (inappropriate laughing, crying, or singing)
- rigidity with waxy flexibility
- delusions, hallucinations.
A prospective study of 22 patients with NCSE found that 7 had a history of psychotic depression, schizophrenia, self-mutilation, bipolar disorder, or episodic severe aggression; 12 of 18 with ASE had a history of epilepsy, and 3 of 4 with CPSE had experienced seizures associated with cerebrovascular accident, right cerebral embolus, and thiazide-induced hyponatremia, respectively.16
Table 2
Differential diagnosis of NCSE
| Metabolic disorders | Hypo/hyperglycemia, hypercalcemia, Addison’s disease, Cushing’s disease, uremia |
| Neurologic disorders | Stroke, CNS tumors, closed head trauma, transient global amnesia, seizures, inflammatory and infectious encephalopathies |
| Psychiatric disorders | Schizophrenia, mood disorders, catatonia, malignant catatonia, somatoform disorders, conversion disorder, Asperger’s syndrome, malingering |
| Toxic disorders | Toxic encephalopathy, neuroleptic malignant syndrome, serotonin syndrome, alcohol and sedative-hypnotic withdrawal, drugs (lithium toxicity, tricyclics, baclofen, tiagabine, overdose) |
| Source: Reference 17,18 | |
Cerebrovascular disease, tumors, and trauma are the most common causes of late-life NCSE.4,19 De novo NCSE occasionally presents:
- after benzodiazepine withdrawal
- with neuroleptic, tricyclic antidepressant, or lithium treatment10,16
- with metabolic abnormalities and nonpsychotropic medications.10
Clinical symptoms
Clinical features of NCSE include cognitive changes, speech abnormalities, affective disturbances, psychosis, poor impulse control, and bizarre behaviors (Table 3). Some patients develop ictal phenomena resembling catatonia or clinical and EEG changes that mimic neuroleptic malignant syndrome (NMS).20-23
Table 3
Clinical features that raise suspicion of NCSE
| Domain | Features |
|---|---|
| Cognitive changes | Prolonged confusion, executive dysfunction, obtundation, attention/memory difficulties, lack of initiative, perseveration, stupor |
| Speech | Poverty of speech with monosyllabic answers, verbal perseveration, echolalia, palilalia, aphasia, paraphasic errors, confabulation, mutism |
| Affective | Prolonged fear, affective indifferent state with blank facial expression, hypomania, psychotic depression, inappropriate laughing and crying, anxiety states |
| Psychosis | Visual, auditory and cenesthetic hallucinations, delusions |
| Impulse control | Hostility, agitation, violence, groping, genital manipulation, picking, posturing |
| Others | Catatonic signs, autonomic disturbances |
| Source: References 5,7-9,12,15-17,20-23 | |
Among 29 patients with acute catatonic syndromes, epileptic activity was identified in 4. One patient with absence status was diagnosed with NMS during the catatonic period.26 Conversely, the commonality of clinical features has led to misdiagnosis of psychogenic catatonia as NCSE. EEG is necessary to exclude NCSE in these cases.
NMS. Yoshino et al27 described two patients taking neuroleptics who met criteria for NMS and had EEG changes consistent with NCSE. They later reported another patient with NCSE complicating NMS; the point at which NCSE developed was unknown, however, because EEG activity was not recorded at NMS onset.28 Based on NMS diagnostic criteria proposed by Caroff et al,29 these patients could have developed NCSE mimicking NMS.
EEG for diagnosis
Candidates. Because differentiating NCSE from similar conditions can be difficult, use EEG to confirm your clinical observations. No guidelines exist, but consider EEG when the patient’s history suggests NCSE. Ask the patient or family about:
- changes in mental status from baseline, especially new-onset catatonia or unexplained altered consciousness
- duration of events
- presence or absence of motor activity
- behavioral fluctuations
- presence or absence of automatisms or blinking.
EEG patterns. Table 4 summarized NCSE diagnostic criteria. NCSE shows characteristic patterns in ASE and CPSE,9,10,16,23 and EEG changes can be continuous or nearly continuous in both.
Table 4
EEG findings that support a clinical diagnosis of NCSE
| Clear-cut criteria |
|---|
| Frequent or continuous focal seizures, with ictal patterns that wax and wane with change in amplitude, frequency, and/or spatial distribution |
| Frequent or continuous generalized spike wave discharges: |
|
| Periodic lateralized epileptiform discharges (“PLEDs”) or bilateral periodic epileptiform discharges (“biPEDs") occurring in patients with coma from generalized tonic-clonic status epilepticus (subtle SE) |
| Probable (equivocal) criteria |
| Patients with acute cerebral damage who also show frequent or continuous EEG abnormalities without previous similar findings |
| Patients with epilepsy who show frequent or continuous generalized EEG abnormalities and similar interictal EEG patterns but whose clinical symptoms suggest NCSE |
| Source: References 4,12-14,17 |
31,32
In CPSE, less-synchronous epileptiform activity has been described, including rhythmical slow, rhythmic spikes, or rhythmic spike and slow waves. Two types of CPSE of frontal origin have been described:
- Type 1 presents clinically with mood disturbance and minimal confusion. EEG shows a frontal focus with a normal background.
- Type 2 presents clinically with confusion. EEG shows bilateral asymmetric frontal discharges.8
- generalized in 69%
- diffuse with focal predominance in 18%
- focal in 13%.
Distinguish between ictal and interictal EEG findings with epileptiform activity, because only the former is diagnostic for NCSE. Intravenous benzodiazepines might be necessary during EEG to verify the diagnosis.33
NCSE has developed after electroconvulsive therapy (ECT), but a cause-effect relationship is debatable. Interictal and abnormal EEG findings after ECT may be misdiagnosed as NCSE.34
Neuroimaging has limited clinical value because of the need for patient cooperation and specialized equipment.4 Head CT or MRI can exclude structural abnormalities. PET and SPECT show increased metabolism and blood flow, respectively, in NCSE. MR spectroscopy shows elevated lactate and decreased N-acetyl aspartate.
Halting ictal activity
To rapidly stop ictal activity—the main goal of treatment—recognizing and correcting precipitant factors is vital:
- Consider discontinuing medications that could lower the seizure threshold.
- Order a complete blood count, serum electrolytes, calcium, arterial-blood gas, liver and renal function tests, urine toxicology screen, and serum antiepileptic drug concentrations.
- When possible, obtain neuroimaging and EEG in the emergency room for accurate diagnosis and prompt treatment.12
Response to benzodiazepines might be transient, lasting only hours or days. For instance, diazepam’s anticonvulsant effect may last
Newer antiepileptics—such as lamotrigine, levetiracetam, or topiramate—have been used with varying results, and their role in first-line treatment of NCSE is evolving. Rarely, the antiepileptic tiagabine precipitates or worsens NCSE.4,13,14
Related resources
- Epilepsy Foundation. www.epilepsyfoundation.org
- Neuroleptic Malignant Syndrome Information Service. Hotline for health professionals (888) 667-8367. www.nmsis.org
- Carbamazepine • Tegretol, Carbatrol
- Clonazepam • Klonopin
- Diazepam • Valium
- Lamotrigine • Lamictal
- Levetiracetam • Keppra
- Lithium carbonate • Lithobid, Eskalith CR
- Lorazepam • Ativan
- Phenobarbital • Luminal
- Phenytoin • Dilantin
- Tiagabine • Gabitril
- Topiramate • Topamax
- Valproic acid • Depakote
The authors report no financial relationship with any company whose products are mentioned in the article or with manufacturers of competing products.
Acknowledgment
Dr. Goveas was a geriatric psychiatry fellow, University of Pennsylvania, when he wrote this article in collaboration with his mentors, Drs. Caroff and Riggio.
1. DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46(4):1029-35.
2. Shorvon S. Status epilepticus: Its clinical features and treatment in children and adults Cambridge, UK: Cambridge University Press, 1994.
3. Shneker BF, Fountain NB. Assessment of acute morbidity and mortality in nonconvulsive status epilepticus. Neurology 2003;61:1066-73.
4. Walker M, Cross H, Smith S, et al. Nonconvulsive status epilepticus: Epilepsy research foundation workshop reports. Epileptic Disord 2005;7(3):53-296.
5. Engel J, Ludwig BI, Fetell M. Prolonged partial complex status epilepticus: EEG and behavioral observations. Neurology 1978;28:863-9.
6. Krumholz A, Sung GY, Fisher RS, et al. Complex partial status epilepticus accompanied by serious morbidity and mortality. Neurology 1995;45:1499-1504.
7. Ballenger CE, King DW, Gallagher BB. Partial complex status epilepticus. Neurology 1983;33:1545-52.
8. Thomas P, Zifkin B, Migneco O, et al. Nonconvulsive status epilepticus of frontal origin. Neurology 1999;52:1174-83.
9. Guberman A, Cantu-Reyna G, Stuss D, Broughton R. Nonconvulsive generalized status epilepticus: Clinical features, neuropsychological testing, and long-term follow-up. Neurology 1986;36:1284-91.
10. Thomas P, Beaumanoir A, Genton P, et al. ‘De novo’ absence status of late onset: Report of 11 cases. Neurology 1992;42:104-10.
11. Andermann F, Robb J. Absence status: a reappraisal following review of thirty-eight patients. Epilepsia 1972;13:177-87.
12. Kaplan PW. Nonconvulsive status epilepticus in the emergency room. Epilepsia 1996;37(7):643-50.
13. Riggio S. Nonconvulsive status epilepticus: Clinical features and diagnostic challenges. Psychiatr Clin N Am 2005;28(3):653-64.
14. Drislane FW. Presentation, evaluation, and treatment of nonconvulsive status epilepticus. Epilepsy Behav 2000;1(5):301-14.
15. Tomson T, Lindbom U, Nilsson BY. Nonconvulsive status epilepticus in adults: Thirty-two consecutive patients from a general hospital population. Epilepsia 1992;3(5):829-35.
16. Dunne JW, Summers QA, Stewart-Wynne EG. Non-convulsive status epilepticus: A prospective study in an adult general hospital. Q J Med 1987;62(238):117-26.
17. Kaplan PW. Behavioral manifestations of nonconvulsive status epilepticus. Epilepsy Behav 2002;3(2):122-39.
18. Mann SC. Malignant catatonia. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing Inc, 2003:121-43.
19. Sung CY, Chu NS. Status epilepticus in elderly: etiology, seizure type and outcome. Acta Neurol Scand 1989;80:51-6.
20. McLachlan RS, Blume WT. Isolated fear in complex partial status epilepticus. Ann Neurol 1980;8:639-41.
21. Walls MJ, Bowers TC, Dilsaver SC, Swann AC. Catatonia associated with depression secondary to complex partial epilepsy. J Clin Psychiatry 1993;54(2):73.-
22. Wells CE. Transient ictal psychosis. Arch Gen Psychiatry 1975;32:1201-3.
23. Agathonikou A, Panayiotopoulos CP, Giannakodimos S, Koutroumanidis M. Typical absence status in adults: Diagnostic and syndromic considerations. Epilepsia 1998;39(12):1265-76.
24. Lim J, Yagnik P, Schraeder P, Wheeler S. Ictal catatonia as a manifestation of nonconvulsive status epilepticus. J Neurol Neurosurg Psychiatry 1986;49:833-6.
25. Drury I, Klass DW, Westmoreland BF, Sharbrough FW. An acute syndrome with psychiatric symptoms and EEG abnormalities. Neurology 1985;35(6):911-14.
26. Primavera A, Fonti A, Novello P, et al. Epileptic seizures in patients with acute catatonic syndrome. J Neurol Neurosurg Psychiatry 1994;57(11):1419-22.
27. Yoshino A, Yoshimasu H, Tatsuzawa Y, et al. Nonconvulsive status epilepticus in two patients with neuroleptic malignant syndrome. J Clin Psychopharmacol 1998;18(4):347-9.
28. Yoshino A, Yoshimasu H. Nonconvulsive status epilepticus complicating neuroleptic malignant syndrome improved by intravenous diazepam. J Clin Psychopharmacol 2000;20(3):389-90.
29. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions, 2nd ed. Washington, DC: American Psychiatric Publishing; 2003:1-44.
30. Lob H, Roger J, Soulayrol R. Les etats de mal generalizes a expression confusionelle. In: Gastaut H, Roger J, Lob H, eds. Les etats de mal epileptiques. Paris: Masson; 1967:91-109.
31. Granner MA, Lee SI. Nonconvulsive status epilepticus: EEG analysis in a large series. Epilepsia 1994;35(1):42-7.
32. Niedermeyer E, Fineyre F, Riley T, Uematsu S. Absence status (petit mal status) with focal characteristics. Arch Neurol 1979;36:417-21.
33. Privitera M, Hoffman M, Moore JL, Jester D. EEG detection of nontonic-clonic status epilepticus in patients with altered consciousness. Epilepsy Res 1994;18:155-66.
34. Povlsen UJ, Wildschiodtz G, Hogenhaven H, Bolwig TG. Nonconvulsive status epilepticus after electroconvulsive therapy. J ECT 2003;19(3):164-9.
1. DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996;46(4):1029-35.
2. Shorvon S. Status epilepticus: Its clinical features and treatment in children and adults Cambridge, UK: Cambridge University Press, 1994.
3. Shneker BF, Fountain NB. Assessment of acute morbidity and mortality in nonconvulsive status epilepticus. Neurology 2003;61:1066-73.
4. Walker M, Cross H, Smith S, et al. Nonconvulsive status epilepticus: Epilepsy research foundation workshop reports. Epileptic Disord 2005;7(3):53-296.
5. Engel J, Ludwig BI, Fetell M. Prolonged partial complex status epilepticus: EEG and behavioral observations. Neurology 1978;28:863-9.
6. Krumholz A, Sung GY, Fisher RS, et al. Complex partial status epilepticus accompanied by serious morbidity and mortality. Neurology 1995;45:1499-1504.
7. Ballenger CE, King DW, Gallagher BB. Partial complex status epilepticus. Neurology 1983;33:1545-52.
8. Thomas P, Zifkin B, Migneco O, et al. Nonconvulsive status epilepticus of frontal origin. Neurology 1999;52:1174-83.
9. Guberman A, Cantu-Reyna G, Stuss D, Broughton R. Nonconvulsive generalized status epilepticus: Clinical features, neuropsychological testing, and long-term follow-up. Neurology 1986;36:1284-91.
10. Thomas P, Beaumanoir A, Genton P, et al. ‘De novo’ absence status of late onset: Report of 11 cases. Neurology 1992;42:104-10.
11. Andermann F, Robb J. Absence status: a reappraisal following review of thirty-eight patients. Epilepsia 1972;13:177-87.
12. Kaplan PW. Nonconvulsive status epilepticus in the emergency room. Epilepsia 1996;37(7):643-50.
13. Riggio S. Nonconvulsive status epilepticus: Clinical features and diagnostic challenges. Psychiatr Clin N Am 2005;28(3):653-64.
14. Drislane FW. Presentation, evaluation, and treatment of nonconvulsive status epilepticus. Epilepsy Behav 2000;1(5):301-14.
15. Tomson T, Lindbom U, Nilsson BY. Nonconvulsive status epilepticus in adults: Thirty-two consecutive patients from a general hospital population. Epilepsia 1992;3(5):829-35.
16. Dunne JW, Summers QA, Stewart-Wynne EG. Non-convulsive status epilepticus: A prospective study in an adult general hospital. Q J Med 1987;62(238):117-26.
17. Kaplan PW. Behavioral manifestations of nonconvulsive status epilepticus. Epilepsy Behav 2002;3(2):122-39.
18. Mann SC. Malignant catatonia. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing Inc, 2003:121-43.
19. Sung CY, Chu NS. Status epilepticus in elderly: etiology, seizure type and outcome. Acta Neurol Scand 1989;80:51-6.
20. McLachlan RS, Blume WT. Isolated fear in complex partial status epilepticus. Ann Neurol 1980;8:639-41.
21. Walls MJ, Bowers TC, Dilsaver SC, Swann AC. Catatonia associated with depression secondary to complex partial epilepsy. J Clin Psychiatry 1993;54(2):73.-
22. Wells CE. Transient ictal psychosis. Arch Gen Psychiatry 1975;32:1201-3.
23. Agathonikou A, Panayiotopoulos CP, Giannakodimos S, Koutroumanidis M. Typical absence status in adults: Diagnostic and syndromic considerations. Epilepsia 1998;39(12):1265-76.
24. Lim J, Yagnik P, Schraeder P, Wheeler S. Ictal catatonia as a manifestation of nonconvulsive status epilepticus. J Neurol Neurosurg Psychiatry 1986;49:833-6.
25. Drury I, Klass DW, Westmoreland BF, Sharbrough FW. An acute syndrome with psychiatric symptoms and EEG abnormalities. Neurology 1985;35(6):911-14.
26. Primavera A, Fonti A, Novello P, et al. Epileptic seizures in patients with acute catatonic syndrome. J Neurol Neurosurg Psychiatry 1994;57(11):1419-22.
27. Yoshino A, Yoshimasu H, Tatsuzawa Y, et al. Nonconvulsive status epilepticus in two patients with neuroleptic malignant syndrome. J Clin Psychopharmacol 1998;18(4):347-9.
28. Yoshino A, Yoshimasu H. Nonconvulsive status epilepticus complicating neuroleptic malignant syndrome improved by intravenous diazepam. J Clin Psychopharmacol 2000;20(3):389-90.
29. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A, eds. Neuroleptic malignant syndrome and related conditions, 2nd ed. Washington, DC: American Psychiatric Publishing; 2003:1-44.
30. Lob H, Roger J, Soulayrol R. Les etats de mal generalizes a expression confusionelle. In: Gastaut H, Roger J, Lob H, eds. Les etats de mal epileptiques. Paris: Masson; 1967:91-109.
31. Granner MA, Lee SI. Nonconvulsive status epilepticus: EEG analysis in a large series. Epilepsia 1994;35(1):42-7.
32. Niedermeyer E, Fineyre F, Riley T, Uematsu S. Absence status (petit mal status) with focal characteristics. Arch Neurol 1979;36:417-21.
33. Privitera M, Hoffman M, Moore JL, Jester D. EEG detection of nontonic-clonic status epilepticus in patients with altered consciousness. Epilepsy Res 1994;18:155-66.
34. Povlsen UJ, Wildschiodtz G, Hogenhaven H, Bolwig TG. Nonconvulsive status epilepticus after electroconvulsive therapy. J ECT 2003;19(3):164-9.
Neuroleptic malignant syndrome: Still a risk, but which patients may be in danger?
Potentially fatal neuroleptic malignant syndrome (NMS)—though less common than in the past—can happen with either conventional or atypical antipsychotics.1,2 To help you protect patients when prescribing antipsychotics or consulting with other clinicians about these drugs, this article discusses:
- risk factors and clinical features that warn of NMS onset
- differential diagnosis of disease states most often confused with NMS
- management recommendations, including supportive measures and specific interventions such as benzodiazepines, dopamine agonists, dantrolene, and electroconvulsive therapy (ECT).
WHY NMS REMAINS RELEVANT
NMS remains a risk in susceptible patients receiving atypical antipsychotics, according to clinical reports and drug adverse event surveys (Figure).2
Moreover, NMS continues to be reported with conventional antipsychotics, which remain in widespread use. Patients who receive long-acting depot conventional antipsychotics are at risk for prolonged NMS episodes.
Figure 55 NMS cases reported with use of antipsychotics, 1998-2002
Probable or definite neuroleptic malignant syndrome cases associated with antipsychotic monotherapy reported to the Neuroleptic Malignant Syndrome Information Service.NMS in medical settings. Psychiatrists may be consulted when patients develop NMS while receiving conventional antipsychotics or other dopaminereceptor antagonists used in medical settings.3,4 Haloperidol remains the recommended drug of choice for treating agitation and delirium and continues to be the single most common trigger of NMS. Although often overlooked, antiemetics and sedatives with neuroleptic properties—such as prochlorperazine, metoclopramide, and promethazine—also have triggered NMS.
Other hyperthermic conditions. NMS is often considered in the differential diagnosis of patients who develop fever or encephalopathy while being treated with psychotropics. In these acute, complex, and often grave situations, psychiatrists may be consulted to recommend treatment for behavioral control or to distinguish NMS from other conditions.
NEWER VS. OLDER ANTIPSYCHOTICS
Has NMS incidence declined with the atypical agents? Probably, but providing proof is difficult:
- NMS is uncommon; its incidence in psychiatric patients treated with conventional antipsychotics is approximately 0.2%.5 To demonstrate reduced NMS incidence with atypicals, a very large sample of patients would be required to reach statistical significance.
- As doctors have used lower doses of conventional agents—which reduces the risk of NMS—any beneficial impact from atypicals has become more difficult to detect.5,6
- Reports of NMS frequency with atypicals may be inflated by bias in publishing adverse events with newer versus older agents.
- Patients switched to atypicals may represent a high-risk group that is intolerant or resistant to conventional antipsychotics.
So far, few unequivocal cases of NMS have been attributed to the use of quetiapine, ziprasidone, or aripiprazole, the most recently introduced atypicals. Moreover, case reports of NMS associated with clozapine, risperidone, or olanzapine2 are often difficult to interpret because of incomplete clinical details, varying diagnostic criteria, and concomitant use of more than one antipsychotic.
Milder NMS? Do the newer antipsychotics produce an “atypical” or milder form of NMS? Case reports indicate that extreme temperature elevations and extrapyramidal dysfunction are less frequent in NMS associated with atypical compared with conventional antipsychotics.2 However, case descriptions of NMS were heterogeneous even with conventional agents, and clinicians’ growing awareness of NMS even before atypicals were introduced allowed for earlier diagnosis of mild and partial NMS cases.7
CLINICAL FEATURES OF NMS
Regardless of drug selection, it is important to recognize early and mild signs of NMS. Any case can progress to a fulminant form that is more difficult to treat.
Patients at risk. NMS may be more likely to develop in patients with:5,6,8
- dehydration
- agitation
- low serum iron
- underlying brain damage
- catatonia.
Some patients may have genetic abnormalities in central dopamine systems that increase their susceptibility to NMS.6,9
Fifteen to 20% of patients who develop NMS have experienced a previous episode while taking antipsychotics, which is why taking a careful drug history is important.5,6 Although most often reported with therapeutic antipsychotic doses, NMS has been associated with rapid dose titration, especially when given parenterally.8
On the other hand, the practical value of these risk factors is often limited in individual cases and may lead one to overestimate NMS risk. NMS is rare and idiosyncratic. Risk factors may not outweigh antipsychotics’ benefits when these drugs are indicated for a patient with psychosis.
Incipient NMS. Identifying early signs of NMS may be impossible in fulminant cases, but patients with incipient NMS may show:
- unexpected mental status changes
- new-onset catatonia
- refractory extrapyramidal and bulbar signs such as rigidity, dysphagia, or dysarthria.5,7,10
Other clues to NMS onset include tachycardia, tachypnea, and elevated temperature or serum creatine phosphokinase (CPK). These signs, however, do not precede or progress to NMS in all cases. A high index of suspicion for NMS, tempered by sound clinical judgment, is called for when assessing all patients receiving antipsychotics.
Diagnostic criteria. Clinical signs of NMS as a fullblown hypermetabolic syndrome are distinctive and well described (Table 1).5,6,11,12 Elevated temperature is accompanied by profuse sweating. Extreme temperatures (>104° F), especially if prolonged or associated with hypoxia or hypotension, pose a high risk for brain damage, rhabdomyolysis, disseminated intravascular coagulation, multisystem organ failure, and death.
Muscle rigidity is a characteristic finding and may be accompanied by tremors, cogwheeling, myoclonus, or rhabdomyolysis. Changes in vital signs—such as tachycardia and tachypnea—are typical.
Mental status examination usually reveals catatonic signs of mutism and stupor, but delirium and coma also have been described. No laboratory findings are specific for NMS, but elevated white blood cell counts, low serum iron, metabolic acidosis, hypoxia, and elevated serum CPK and catecholamines have been reported.
Table 1
Common clinical features of NMS
| Signs and symptoms | Altered level of consciousness, catatonia, dysarthria, dysphagia, elevated temperature, labile blood pressure, muscle rigidity, mutism, myoclonus, tachycardia, tachypnea, tremor |
| Laboratory findings | Elevated catecholamines and serum creatine phosphokinase, hypoxia, leukocytosis, low serum iron, metabolic acidosis |
Resolution. If recognized promptly, NMS resolves within 1 to 2 weeks in two-thirds of patients after antipsychotics are discontinued. The average recovery time of 7 to 10 days may be prolonged in patients who were taking long-acting depot antipsychotics or in those with persistent residual catatonic symptoms.13
Risk of death. NMS remains potentially fatal, especially if high temperatures develop or episodes are prolonged. Causes of death include cardiorespiratory arrest, renal failure, pulmonary emboli, pneumonia, sepsis, disseminated intravascular coagulation, and multisystem organ failure.
DIFFERENTIAL DIAGNOSIS
Differential diagnosis of NMS encompasses disorders that present with fever and encephalopathy.5,6,12 Primary brain disorders that resemble NMS include:9,14-16
- infections
- acute psychotic disorders that progress to malignant catatonia or delirious mania
- midbrain structural lesions
- seizures.
Also exclude hormonal and autoimmune disorders and environmental heatstroke (Table 2).17,18 Similar hyperthermic syndromes have been reported with other toxins and drugs, including malignant hyperthermia of anesthesia, serotonin syndrome, and dopamine agonist withdrawal in patients with Parkinson’s disease (Table 3).5,6,19
Table 2
7 disease states most often confused with NMS
| Infections |
| Malignant catatonia secondary to psychotic disorders |
| Benign extrapyramidal side effects |
| Agitated delirium from diverse causes |
| Environmental heatstroke |
| Serotonin syndrome |
| Withdrawal from dopamine agonists, other drugs, or alcohol |
| Source: Neuroleptic Malignant Syndrome Information Service hotline |
Table 3
Drugs that can cause NMS-like hyperthermic syndromes
| Anticholinergics Dopamine antagonists Hallucinogens Inhalational anesthetics Monoamine oxidase inhibitors Psychostimulants Salicylates | Serotonergic drugs Succinylcholine Withdrawal from: • Dopamine agonists • Alcohol • Sedative/hypnotics • Baclofen |
MANAGING NMS
The standard approach to managing patients with NMS includes four steps:
- recognize the diagnosis early
- exclude alternate causes of symptoms
- discontinue suspected triggering drugs
- provide supportive care to reduce temperatures, ensure fluid balance, and detect complications.5,6,20
Beyond supportive care, several specific therapies have been proposed based on theoretical mechanisms of NMS and meta-analyses of offlabel use in anecdotal clinical reports (Table 4). If benzodiazepines, dopamine agonists, or dantrolene are effective, taper slowly after recovery to prevent rebound symptoms.
Benzodiazepines. Given the concept of NMS as a form of catatonia, benzodiazepines have been used effectively in some cases.20,21 A trial of lorazepam, 1 to 2 mg parenterally, is a reasonable first step. Higher doses may be required, with adequate monitoring of respiratory status. Oral lorazepam can maintain the therapeutic effect.
Dopamine agonists. To reverse the parkinsonism and dopamine antagonist properties of antipsychotics, dopamine agonists such as bromocriptine or amantadine have been tried and have reduced NMS duration and mortality.20,22,23 Newer drugs such as ropinorole and pramipexole may also be useful. Dopaminergic drugs, however, can worsen psychosis and cause hypotension and emesis.
Dantrolene may reduce hyperthermia related to skeletal muscle hypermetabolism of any cause and has been effective in rapidly reducing extreme temperatures in some NMS cases.20,22-26 Dantrolene is given IV, 1 to 2.5 mg/kg every 6 hours. An oral form can be substituted if a response is obtained. Dantrolene can impair respiratory and hepatic function and should not be combined with calcium channel blockers.
ECT is increasingly recognized as an effective NMS treatment and should not be overlooked for patients:
Standard ECT is given, although nondepolarizing muscle relaxants instead of succinylcholine are used in patients with serious rhabdomyolysis to avoid the risk of hyperkalemia.20
Recommendation. Although these modalities offer a spectrum of therapeutic options, it is premature to recommend any single remedy over others or over supportive care alone because:
- randomized, controlled trials have not been conducted
- NMS episodes are heterogeneous in presentation and outcome
- the syndrome is often self-limited after antipsychotics are discontinued.
I recommend that you choose therapies empirically, based on the character, severity, and duration of symptoms in a given case.5,6,20
Table 4
How to treat neuroleptic malignant syndrome
| General measures | Diagnose early, discontinue antipsychotic, provide supportive care |
| Specific interventions under investigation | |
| Benzodiazepines | Parenteral lorazepam, 1 to 2 mg or higher; monitor respiratory status |
| Dopamine agonists | Bromocriptine, 2.5 mg every 8 hours or amantadine, 100 mg every 8 hours; monitor psychosis, blood pressure, nausea |
| Dantrolene | 1 to 2.5 mg/kg IV every 6 hours; monitor respiratory and hepatic function; avoid calcium channel blockers |
| ECT | Standard administration; avoid succinylcholine in patients with rhabdomyolysis |
REDUCING RISK OF RECURRENCE
Patients vary in susceptibility to recurrence after they recover from NMS, but the risk approaches 30% with future exposure to antipsychotics.5,6 You may be able to minimize recurrence risk by:
- reducing risk factors, such as dehydration
- considering alternatives to antipsychotics, such as treating bipolar disorder with lithium or ECT
- using atypical instead of conventional antipsychotics, starting with low dosages and titrating slowly.
Before you reintroduce antipsychotics, carefully document informed consent and your rationale for treatment decisions in the patient’s chart.
Related resources
- Neuroleptic Malignant Syndrome Information Service. Hotline for health professionals. (888) 667-8367. www.nmsis.org
- Mann SC, Caroff SN, Keck PE Jr, Lazarus A. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003.
- Caroff SN, Mann SC, Francis A, Fricchione GL. Catatonia: from psychopathology to neurobiology. Washington, DC: American Psychiatric Publishing (in press).
Drug brand names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Haloperidol • Haldol
- Lorazepam • Ativan
- Metoclopramide • Reglan
- Olanzapine • Zyprexa
- Pramipexole • Mirapex
- Prochlorperazine • Compazine
- Promethazine • Phenergan
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ropinirole • Requip
- Ziprasidone • Geodon
Disclosure
Dr. Caroff receives research support from Janssen Pharmaceutica and Pfizer Inc., and is a consultant to Eli Lilly and Co. and Bristol-Myers Squibb Co.
1. Caroff SN, Mann SC, Campbell EC, Sullivan KA. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry 2002;63(suppl 4):12-19.
2. Caroff SN, Mann SC, Campbell EC. Atypical antipsychotics and neuroleptic malignant syndrome. Psychiatric Annals 2000;30(5):314-21.
3. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiology 2001;28(8):387-93.
4. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med 2002;30(11):2609.-
5. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am 1993;77(1):185-202.
6. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing Inc., 2003;1-44.
7. Velamoor VR, Swamy GN, Parmar RS, et al. Management of suspected neuroleptic malignant syndrome. Can J Psychiatry 1995;40(9):545-50.
8. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. Arch Gen Psychiatry 1989;46:914-18.
9. Mann SC. Malignant catatonia. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003;121-43.
10. Velamoor VR, Norman RMG, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994;182(3):168-73.
11. Delay J, Pichot P, Lemperiere T, et al. Un neuroleptique majeur non-phenothiazine et non reserpinique, l’haloperidol, dans le traitement des psychoses. Annales Medico-Psychologique 1960;118(1):145-52.
12. Caroff SN. The neuroleptic malignant syndrome. J Clin Psychiatry 1980;41(3):79-83.
13. Caroff SN, Mann SC, Keck PE, Jr, Francis A. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol 2000;20(2):257-9.
14. Caroff SN, Mann SC, McCarthy M, et al. Acute infectious encephalitis complicated by neuroleptic malignant syndrome. J Clin Psychopharmacol 1998;18(4):349-51.
15. Caroff SN, Mann SC, Gliatto MF, et al. Psychiatric manifestations of acute viral encephalitis. Psychiatric Annals 2001;31(3):193-204.
16. Caroff SN, Mann SC, Francis A, Fricchione GL (eds). Catatonia: from psychopathology to neurobiology. Washington, DC: American Psychiatric Publishing (in press).
17. Mann SC. Thermoregulatory mechanisms and antipsychotic drugrelated heatstroke. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003;45-74.
18. Caroff SN, Mann SC, Campbell EC. Risk of fatal heatstroke after hospitalization. Psychiatric Serv 2000;51(7):938.-
19. Keck PE, Jr. Serotonin syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003;75-92.
20. Davis JM, Caroff SN, Mann SC. Treatment of neuroleptic malignant syndrome. Psychiatric Annals 2000;30(5):325-31.
21. Francis A, Chandragiri S, Rizvi S, et al. Lorazepam a treatment for neuroleptic malignant syndrome? CNS Spectrums 2000;5(7):54-7.
22. Sakkas P, Davis JM, Hua J, Wang Z. Pharmacotherapy of neuroleptic malignant syndrome. Psychiatr Ann 1991;21:157-64.
23. Rosenberg MR, Green M. Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 1989;149:1927-31.
24. Henderson A, Longdon P. Fulminant metoclopramide-induced neuroleptic malignant syndrome rapidly responsive to intravenous dantrolene. Aust N Z J Med 1991;21:742-3.
25. Yamawaki S, Morio M, Kazamatsuri G, et al. Clinical evaluation and effective usage of dantrolene sodium in neuroleptic malignant syndrome. Kiso to Rinsyou (Clinical Reports) 1993;27(3):1045-66.
26. Tsutsumi Y, Yamamoto K, Matsuura S, et al. The treatment of neuroleptic malignant syndrome using dantrolene sodium. Psychiatry Clin Neurosci 1998;52:433-8.
27. Nisijima K, Ishiguro T. Electroconvulsive therapy for the treatment of neuroleptic malignant syndrome: a report of five cases. J ECT 1999;15:158-63.
28. Troller JN, Sachdev PS. Electroconvulsive treatment of neuroleptic malignant syndrome: a review and report of cases. Aust N Z J Psychiatry 1999;33:650-9.
Potentially fatal neuroleptic malignant syndrome (NMS)—though less common than in the past—can happen with either conventional or atypical antipsychotics.1,2 To help you protect patients when prescribing antipsychotics or consulting with other clinicians about these drugs, this article discusses:
- risk factors and clinical features that warn of NMS onset
- differential diagnosis of disease states most often confused with NMS
- management recommendations, including supportive measures and specific interventions such as benzodiazepines, dopamine agonists, dantrolene, and electroconvulsive therapy (ECT).
WHY NMS REMAINS RELEVANT
NMS remains a risk in susceptible patients receiving atypical antipsychotics, according to clinical reports and drug adverse event surveys (Figure).2
Moreover, NMS continues to be reported with conventional antipsychotics, which remain in widespread use. Patients who receive long-acting depot conventional antipsychotics are at risk for prolonged NMS episodes.
Figure 55 NMS cases reported with use of antipsychotics, 1998-2002
Probable or definite neuroleptic malignant syndrome cases associated with antipsychotic monotherapy reported to the Neuroleptic Malignant Syndrome Information Service.NMS in medical settings. Psychiatrists may be consulted when patients develop NMS while receiving conventional antipsychotics or other dopaminereceptor antagonists used in medical settings.3,4 Haloperidol remains the recommended drug of choice for treating agitation and delirium and continues to be the single most common trigger of NMS. Although often overlooked, antiemetics and sedatives with neuroleptic properties—such as prochlorperazine, metoclopramide, and promethazine—also have triggered NMS.
Other hyperthermic conditions. NMS is often considered in the differential diagnosis of patients who develop fever or encephalopathy while being treated with psychotropics. In these acute, complex, and often grave situations, psychiatrists may be consulted to recommend treatment for behavioral control or to distinguish NMS from other conditions.
NEWER VS. OLDER ANTIPSYCHOTICS
Has NMS incidence declined with the atypical agents? Probably, but providing proof is difficult:
- NMS is uncommon; its incidence in psychiatric patients treated with conventional antipsychotics is approximately 0.2%.5 To demonstrate reduced NMS incidence with atypicals, a very large sample of patients would be required to reach statistical significance.
- As doctors have used lower doses of conventional agents—which reduces the risk of NMS—any beneficial impact from atypicals has become more difficult to detect.5,6
- Reports of NMS frequency with atypicals may be inflated by bias in publishing adverse events with newer versus older agents.
- Patients switched to atypicals may represent a high-risk group that is intolerant or resistant to conventional antipsychotics.
So far, few unequivocal cases of NMS have been attributed to the use of quetiapine, ziprasidone, or aripiprazole, the most recently introduced atypicals. Moreover, case reports of NMS associated with clozapine, risperidone, or olanzapine2 are often difficult to interpret because of incomplete clinical details, varying diagnostic criteria, and concomitant use of more than one antipsychotic.
Milder NMS? Do the newer antipsychotics produce an “atypical” or milder form of NMS? Case reports indicate that extreme temperature elevations and extrapyramidal dysfunction are less frequent in NMS associated with atypical compared with conventional antipsychotics.2 However, case descriptions of NMS were heterogeneous even with conventional agents, and clinicians’ growing awareness of NMS even before atypicals were introduced allowed for earlier diagnosis of mild and partial NMS cases.7
CLINICAL FEATURES OF NMS
Regardless of drug selection, it is important to recognize early and mild signs of NMS. Any case can progress to a fulminant form that is more difficult to treat.
Patients at risk. NMS may be more likely to develop in patients with:5,6,8
- dehydration
- agitation
- low serum iron
- underlying brain damage
- catatonia.
Some patients may have genetic abnormalities in central dopamine systems that increase their susceptibility to NMS.6,9
Fifteen to 20% of patients who develop NMS have experienced a previous episode while taking antipsychotics, which is why taking a careful drug history is important.5,6 Although most often reported with therapeutic antipsychotic doses, NMS has been associated with rapid dose titration, especially when given parenterally.8
On the other hand, the practical value of these risk factors is often limited in individual cases and may lead one to overestimate NMS risk. NMS is rare and idiosyncratic. Risk factors may not outweigh antipsychotics’ benefits when these drugs are indicated for a patient with psychosis.
Incipient NMS. Identifying early signs of NMS may be impossible in fulminant cases, but patients with incipient NMS may show:
- unexpected mental status changes
- new-onset catatonia
- refractory extrapyramidal and bulbar signs such as rigidity, dysphagia, or dysarthria.5,7,10
Other clues to NMS onset include tachycardia, tachypnea, and elevated temperature or serum creatine phosphokinase (CPK). These signs, however, do not precede or progress to NMS in all cases. A high index of suspicion for NMS, tempered by sound clinical judgment, is called for when assessing all patients receiving antipsychotics.
Diagnostic criteria. Clinical signs of NMS as a fullblown hypermetabolic syndrome are distinctive and well described (Table 1).5,6,11,12 Elevated temperature is accompanied by profuse sweating. Extreme temperatures (>104° F), especially if prolonged or associated with hypoxia or hypotension, pose a high risk for brain damage, rhabdomyolysis, disseminated intravascular coagulation, multisystem organ failure, and death.
Muscle rigidity is a characteristic finding and may be accompanied by tremors, cogwheeling, myoclonus, or rhabdomyolysis. Changes in vital signs—such as tachycardia and tachypnea—are typical.
Mental status examination usually reveals catatonic signs of mutism and stupor, but delirium and coma also have been described. No laboratory findings are specific for NMS, but elevated white blood cell counts, low serum iron, metabolic acidosis, hypoxia, and elevated serum CPK and catecholamines have been reported.
Table 1
Common clinical features of NMS
| Signs and symptoms | Altered level of consciousness, catatonia, dysarthria, dysphagia, elevated temperature, labile blood pressure, muscle rigidity, mutism, myoclonus, tachycardia, tachypnea, tremor |
| Laboratory findings | Elevated catecholamines and serum creatine phosphokinase, hypoxia, leukocytosis, low serum iron, metabolic acidosis |
Resolution. If recognized promptly, NMS resolves within 1 to 2 weeks in two-thirds of patients after antipsychotics are discontinued. The average recovery time of 7 to 10 days may be prolonged in patients who were taking long-acting depot antipsychotics or in those with persistent residual catatonic symptoms.13
Risk of death. NMS remains potentially fatal, especially if high temperatures develop or episodes are prolonged. Causes of death include cardiorespiratory arrest, renal failure, pulmonary emboli, pneumonia, sepsis, disseminated intravascular coagulation, and multisystem organ failure.
DIFFERENTIAL DIAGNOSIS
Differential diagnosis of NMS encompasses disorders that present with fever and encephalopathy.5,6,12 Primary brain disorders that resemble NMS include:9,14-16
- infections
- acute psychotic disorders that progress to malignant catatonia or delirious mania
- midbrain structural lesions
- seizures.
Also exclude hormonal and autoimmune disorders and environmental heatstroke (Table 2).17,18 Similar hyperthermic syndromes have been reported with other toxins and drugs, including malignant hyperthermia of anesthesia, serotonin syndrome, and dopamine agonist withdrawal in patients with Parkinson’s disease (Table 3).5,6,19
Table 2
7 disease states most often confused with NMS
| Infections |
| Malignant catatonia secondary to psychotic disorders |
| Benign extrapyramidal side effects |
| Agitated delirium from diverse causes |
| Environmental heatstroke |
| Serotonin syndrome |
| Withdrawal from dopamine agonists, other drugs, or alcohol |
| Source: Neuroleptic Malignant Syndrome Information Service hotline |
Table 3
Drugs that can cause NMS-like hyperthermic syndromes
| Anticholinergics Dopamine antagonists Hallucinogens Inhalational anesthetics Monoamine oxidase inhibitors Psychostimulants Salicylates | Serotonergic drugs Succinylcholine Withdrawal from: • Dopamine agonists • Alcohol • Sedative/hypnotics • Baclofen |
MANAGING NMS
The standard approach to managing patients with NMS includes four steps:
- recognize the diagnosis early
- exclude alternate causes of symptoms
- discontinue suspected triggering drugs
- provide supportive care to reduce temperatures, ensure fluid balance, and detect complications.5,6,20
Beyond supportive care, several specific therapies have been proposed based on theoretical mechanisms of NMS and meta-analyses of offlabel use in anecdotal clinical reports (Table 4). If benzodiazepines, dopamine agonists, or dantrolene are effective, taper slowly after recovery to prevent rebound symptoms.
Benzodiazepines. Given the concept of NMS as a form of catatonia, benzodiazepines have been used effectively in some cases.20,21 A trial of lorazepam, 1 to 2 mg parenterally, is a reasonable first step. Higher doses may be required, with adequate monitoring of respiratory status. Oral lorazepam can maintain the therapeutic effect.
Dopamine agonists. To reverse the parkinsonism and dopamine antagonist properties of antipsychotics, dopamine agonists such as bromocriptine or amantadine have been tried and have reduced NMS duration and mortality.20,22,23 Newer drugs such as ropinorole and pramipexole may also be useful. Dopaminergic drugs, however, can worsen psychosis and cause hypotension and emesis.
Dantrolene may reduce hyperthermia related to skeletal muscle hypermetabolism of any cause and has been effective in rapidly reducing extreme temperatures in some NMS cases.20,22-26 Dantrolene is given IV, 1 to 2.5 mg/kg every 6 hours. An oral form can be substituted if a response is obtained. Dantrolene can impair respiratory and hepatic function and should not be combined with calcium channel blockers.
ECT is increasingly recognized as an effective NMS treatment and should not be overlooked for patients:
Standard ECT is given, although nondepolarizing muscle relaxants instead of succinylcholine are used in patients with serious rhabdomyolysis to avoid the risk of hyperkalemia.20
Recommendation. Although these modalities offer a spectrum of therapeutic options, it is premature to recommend any single remedy over others or over supportive care alone because:
- randomized, controlled trials have not been conducted
- NMS episodes are heterogeneous in presentation and outcome
- the syndrome is often self-limited after antipsychotics are discontinued.
I recommend that you choose therapies empirically, based on the character, severity, and duration of symptoms in a given case.5,6,20
Table 4
How to treat neuroleptic malignant syndrome
| General measures | Diagnose early, discontinue antipsychotic, provide supportive care |
| Specific interventions under investigation | |
| Benzodiazepines | Parenteral lorazepam, 1 to 2 mg or higher; monitor respiratory status |
| Dopamine agonists | Bromocriptine, 2.5 mg every 8 hours or amantadine, 100 mg every 8 hours; monitor psychosis, blood pressure, nausea |
| Dantrolene | 1 to 2.5 mg/kg IV every 6 hours; monitor respiratory and hepatic function; avoid calcium channel blockers |
| ECT | Standard administration; avoid succinylcholine in patients with rhabdomyolysis |
REDUCING RISK OF RECURRENCE
Patients vary in susceptibility to recurrence after they recover from NMS, but the risk approaches 30% with future exposure to antipsychotics.5,6 You may be able to minimize recurrence risk by:
- reducing risk factors, such as dehydration
- considering alternatives to antipsychotics, such as treating bipolar disorder with lithium or ECT
- using atypical instead of conventional antipsychotics, starting with low dosages and titrating slowly.
Before you reintroduce antipsychotics, carefully document informed consent and your rationale for treatment decisions in the patient’s chart.
Related resources
- Neuroleptic Malignant Syndrome Information Service. Hotline for health professionals. (888) 667-8367. www.nmsis.org
- Mann SC, Caroff SN, Keck PE Jr, Lazarus A. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003.
- Caroff SN, Mann SC, Francis A, Fricchione GL. Catatonia: from psychopathology to neurobiology. Washington, DC: American Psychiatric Publishing (in press).
Drug brand names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Haloperidol • Haldol
- Lorazepam • Ativan
- Metoclopramide • Reglan
- Olanzapine • Zyprexa
- Pramipexole • Mirapex
- Prochlorperazine • Compazine
- Promethazine • Phenergan
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ropinirole • Requip
- Ziprasidone • Geodon
Disclosure
Dr. Caroff receives research support from Janssen Pharmaceutica and Pfizer Inc., and is a consultant to Eli Lilly and Co. and Bristol-Myers Squibb Co.
Potentially fatal neuroleptic malignant syndrome (NMS)—though less common than in the past—can happen with either conventional or atypical antipsychotics.1,2 To help you protect patients when prescribing antipsychotics or consulting with other clinicians about these drugs, this article discusses:
- risk factors and clinical features that warn of NMS onset
- differential diagnosis of disease states most often confused with NMS
- management recommendations, including supportive measures and specific interventions such as benzodiazepines, dopamine agonists, dantrolene, and electroconvulsive therapy (ECT).
WHY NMS REMAINS RELEVANT
NMS remains a risk in susceptible patients receiving atypical antipsychotics, according to clinical reports and drug adverse event surveys (Figure).2
Moreover, NMS continues to be reported with conventional antipsychotics, which remain in widespread use. Patients who receive long-acting depot conventional antipsychotics are at risk for prolonged NMS episodes.
Figure 55 NMS cases reported with use of antipsychotics, 1998-2002
Probable or definite neuroleptic malignant syndrome cases associated with antipsychotic monotherapy reported to the Neuroleptic Malignant Syndrome Information Service.NMS in medical settings. Psychiatrists may be consulted when patients develop NMS while receiving conventional antipsychotics or other dopaminereceptor antagonists used in medical settings.3,4 Haloperidol remains the recommended drug of choice for treating agitation and delirium and continues to be the single most common trigger of NMS. Although often overlooked, antiemetics and sedatives with neuroleptic properties—such as prochlorperazine, metoclopramide, and promethazine—also have triggered NMS.
Other hyperthermic conditions. NMS is often considered in the differential diagnosis of patients who develop fever or encephalopathy while being treated with psychotropics. In these acute, complex, and often grave situations, psychiatrists may be consulted to recommend treatment for behavioral control or to distinguish NMS from other conditions.
NEWER VS. OLDER ANTIPSYCHOTICS
Has NMS incidence declined with the atypical agents? Probably, but providing proof is difficult:
- NMS is uncommon; its incidence in psychiatric patients treated with conventional antipsychotics is approximately 0.2%.5 To demonstrate reduced NMS incidence with atypicals, a very large sample of patients would be required to reach statistical significance.
- As doctors have used lower doses of conventional agents—which reduces the risk of NMS—any beneficial impact from atypicals has become more difficult to detect.5,6
- Reports of NMS frequency with atypicals may be inflated by bias in publishing adverse events with newer versus older agents.
- Patients switched to atypicals may represent a high-risk group that is intolerant or resistant to conventional antipsychotics.
So far, few unequivocal cases of NMS have been attributed to the use of quetiapine, ziprasidone, or aripiprazole, the most recently introduced atypicals. Moreover, case reports of NMS associated with clozapine, risperidone, or olanzapine2 are often difficult to interpret because of incomplete clinical details, varying diagnostic criteria, and concomitant use of more than one antipsychotic.
Milder NMS? Do the newer antipsychotics produce an “atypical” or milder form of NMS? Case reports indicate that extreme temperature elevations and extrapyramidal dysfunction are less frequent in NMS associated with atypical compared with conventional antipsychotics.2 However, case descriptions of NMS were heterogeneous even with conventional agents, and clinicians’ growing awareness of NMS even before atypicals were introduced allowed for earlier diagnosis of mild and partial NMS cases.7
CLINICAL FEATURES OF NMS
Regardless of drug selection, it is important to recognize early and mild signs of NMS. Any case can progress to a fulminant form that is more difficult to treat.
Patients at risk. NMS may be more likely to develop in patients with:5,6,8
- dehydration
- agitation
- low serum iron
- underlying brain damage
- catatonia.
Some patients may have genetic abnormalities in central dopamine systems that increase their susceptibility to NMS.6,9
Fifteen to 20% of patients who develop NMS have experienced a previous episode while taking antipsychotics, which is why taking a careful drug history is important.5,6 Although most often reported with therapeutic antipsychotic doses, NMS has been associated with rapid dose titration, especially when given parenterally.8
On the other hand, the practical value of these risk factors is often limited in individual cases and may lead one to overestimate NMS risk. NMS is rare and idiosyncratic. Risk factors may not outweigh antipsychotics’ benefits when these drugs are indicated for a patient with psychosis.
Incipient NMS. Identifying early signs of NMS may be impossible in fulminant cases, but patients with incipient NMS may show:
- unexpected mental status changes
- new-onset catatonia
- refractory extrapyramidal and bulbar signs such as rigidity, dysphagia, or dysarthria.5,7,10
Other clues to NMS onset include tachycardia, tachypnea, and elevated temperature or serum creatine phosphokinase (CPK). These signs, however, do not precede or progress to NMS in all cases. A high index of suspicion for NMS, tempered by sound clinical judgment, is called for when assessing all patients receiving antipsychotics.
Diagnostic criteria. Clinical signs of NMS as a fullblown hypermetabolic syndrome are distinctive and well described (Table 1).5,6,11,12 Elevated temperature is accompanied by profuse sweating. Extreme temperatures (>104° F), especially if prolonged or associated with hypoxia or hypotension, pose a high risk for brain damage, rhabdomyolysis, disseminated intravascular coagulation, multisystem organ failure, and death.
Muscle rigidity is a characteristic finding and may be accompanied by tremors, cogwheeling, myoclonus, or rhabdomyolysis. Changes in vital signs—such as tachycardia and tachypnea—are typical.
Mental status examination usually reveals catatonic signs of mutism and stupor, but delirium and coma also have been described. No laboratory findings are specific for NMS, but elevated white blood cell counts, low serum iron, metabolic acidosis, hypoxia, and elevated serum CPK and catecholamines have been reported.
Table 1
Common clinical features of NMS
| Signs and symptoms | Altered level of consciousness, catatonia, dysarthria, dysphagia, elevated temperature, labile blood pressure, muscle rigidity, mutism, myoclonus, tachycardia, tachypnea, tremor |
| Laboratory findings | Elevated catecholamines and serum creatine phosphokinase, hypoxia, leukocytosis, low serum iron, metabolic acidosis |
Resolution. If recognized promptly, NMS resolves within 1 to 2 weeks in two-thirds of patients after antipsychotics are discontinued. The average recovery time of 7 to 10 days may be prolonged in patients who were taking long-acting depot antipsychotics or in those with persistent residual catatonic symptoms.13
Risk of death. NMS remains potentially fatal, especially if high temperatures develop or episodes are prolonged. Causes of death include cardiorespiratory arrest, renal failure, pulmonary emboli, pneumonia, sepsis, disseminated intravascular coagulation, and multisystem organ failure.
DIFFERENTIAL DIAGNOSIS
Differential diagnosis of NMS encompasses disorders that present with fever and encephalopathy.5,6,12 Primary brain disorders that resemble NMS include:9,14-16
- infections
- acute psychotic disorders that progress to malignant catatonia or delirious mania
- midbrain structural lesions
- seizures.
Also exclude hormonal and autoimmune disorders and environmental heatstroke (Table 2).17,18 Similar hyperthermic syndromes have been reported with other toxins and drugs, including malignant hyperthermia of anesthesia, serotonin syndrome, and dopamine agonist withdrawal in patients with Parkinson’s disease (Table 3).5,6,19
Table 2
7 disease states most often confused with NMS
| Infections |
| Malignant catatonia secondary to psychotic disorders |
| Benign extrapyramidal side effects |
| Agitated delirium from diverse causes |
| Environmental heatstroke |
| Serotonin syndrome |
| Withdrawal from dopamine agonists, other drugs, or alcohol |
| Source: Neuroleptic Malignant Syndrome Information Service hotline |
Table 3
Drugs that can cause NMS-like hyperthermic syndromes
| Anticholinergics Dopamine antagonists Hallucinogens Inhalational anesthetics Monoamine oxidase inhibitors Psychostimulants Salicylates | Serotonergic drugs Succinylcholine Withdrawal from: • Dopamine agonists • Alcohol • Sedative/hypnotics • Baclofen |
MANAGING NMS
The standard approach to managing patients with NMS includes four steps:
- recognize the diagnosis early
- exclude alternate causes of symptoms
- discontinue suspected triggering drugs
- provide supportive care to reduce temperatures, ensure fluid balance, and detect complications.5,6,20
Beyond supportive care, several specific therapies have been proposed based on theoretical mechanisms of NMS and meta-analyses of offlabel use in anecdotal clinical reports (Table 4). If benzodiazepines, dopamine agonists, or dantrolene are effective, taper slowly after recovery to prevent rebound symptoms.
Benzodiazepines. Given the concept of NMS as a form of catatonia, benzodiazepines have been used effectively in some cases.20,21 A trial of lorazepam, 1 to 2 mg parenterally, is a reasonable first step. Higher doses may be required, with adequate monitoring of respiratory status. Oral lorazepam can maintain the therapeutic effect.
Dopamine agonists. To reverse the parkinsonism and dopamine antagonist properties of antipsychotics, dopamine agonists such as bromocriptine or amantadine have been tried and have reduced NMS duration and mortality.20,22,23 Newer drugs such as ropinorole and pramipexole may also be useful. Dopaminergic drugs, however, can worsen psychosis and cause hypotension and emesis.
Dantrolene may reduce hyperthermia related to skeletal muscle hypermetabolism of any cause and has been effective in rapidly reducing extreme temperatures in some NMS cases.20,22-26 Dantrolene is given IV, 1 to 2.5 mg/kg every 6 hours. An oral form can be substituted if a response is obtained. Dantrolene can impair respiratory and hepatic function and should not be combined with calcium channel blockers.
ECT is increasingly recognized as an effective NMS treatment and should not be overlooked for patients:
Standard ECT is given, although nondepolarizing muscle relaxants instead of succinylcholine are used in patients with serious rhabdomyolysis to avoid the risk of hyperkalemia.20
Recommendation. Although these modalities offer a spectrum of therapeutic options, it is premature to recommend any single remedy over others or over supportive care alone because:
- randomized, controlled trials have not been conducted
- NMS episodes are heterogeneous in presentation and outcome
- the syndrome is often self-limited after antipsychotics are discontinued.
I recommend that you choose therapies empirically, based on the character, severity, and duration of symptoms in a given case.5,6,20
Table 4
How to treat neuroleptic malignant syndrome
| General measures | Diagnose early, discontinue antipsychotic, provide supportive care |
| Specific interventions under investigation | |
| Benzodiazepines | Parenteral lorazepam, 1 to 2 mg or higher; monitor respiratory status |
| Dopamine agonists | Bromocriptine, 2.5 mg every 8 hours or amantadine, 100 mg every 8 hours; monitor psychosis, blood pressure, nausea |
| Dantrolene | 1 to 2.5 mg/kg IV every 6 hours; monitor respiratory and hepatic function; avoid calcium channel blockers |
| ECT | Standard administration; avoid succinylcholine in patients with rhabdomyolysis |
REDUCING RISK OF RECURRENCE
Patients vary in susceptibility to recurrence after they recover from NMS, but the risk approaches 30% with future exposure to antipsychotics.5,6 You may be able to minimize recurrence risk by:
- reducing risk factors, such as dehydration
- considering alternatives to antipsychotics, such as treating bipolar disorder with lithium or ECT
- using atypical instead of conventional antipsychotics, starting with low dosages and titrating slowly.
Before you reintroduce antipsychotics, carefully document informed consent and your rationale for treatment decisions in the patient’s chart.
Related resources
- Neuroleptic Malignant Syndrome Information Service. Hotline for health professionals. (888) 667-8367. www.nmsis.org
- Mann SC, Caroff SN, Keck PE Jr, Lazarus A. Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003.
- Caroff SN, Mann SC, Francis A, Fricchione GL. Catatonia: from psychopathology to neurobiology. Washington, DC: American Psychiatric Publishing (in press).
Drug brand names
- Amantadine • Symmetrel
- Aripiprazole • Abilify
- Bromocriptine • Parlodel
- Clozapine • Clozaril
- Dantrolene • Dantrium
- Haloperidol • Haldol
- Lorazepam • Ativan
- Metoclopramide • Reglan
- Olanzapine • Zyprexa
- Pramipexole • Mirapex
- Prochlorperazine • Compazine
- Promethazine • Phenergan
- Quetiapine • Seroquel
- Risperidone • Risperdal
- Ropinirole • Requip
- Ziprasidone • Geodon
Disclosure
Dr. Caroff receives research support from Janssen Pharmaceutica and Pfizer Inc., and is a consultant to Eli Lilly and Co. and Bristol-Myers Squibb Co.
1. Caroff SN, Mann SC, Campbell EC, Sullivan KA. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry 2002;63(suppl 4):12-19.
2. Caroff SN, Mann SC, Campbell EC. Atypical antipsychotics and neuroleptic malignant syndrome. Psychiatric Annals 2000;30(5):314-21.
3. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiology 2001;28(8):387-93.
4. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med 2002;30(11):2609.-
5. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am 1993;77(1):185-202.
6. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing Inc., 2003;1-44.
7. Velamoor VR, Swamy GN, Parmar RS, et al. Management of suspected neuroleptic malignant syndrome. Can J Psychiatry 1995;40(9):545-50.
8. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. Arch Gen Psychiatry 1989;46:914-18.
9. Mann SC. Malignant catatonia. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003;121-43.
10. Velamoor VR, Norman RMG, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994;182(3):168-73.
11. Delay J, Pichot P, Lemperiere T, et al. Un neuroleptique majeur non-phenothiazine et non reserpinique, l’haloperidol, dans le traitement des psychoses. Annales Medico-Psychologique 1960;118(1):145-52.
12. Caroff SN. The neuroleptic malignant syndrome. J Clin Psychiatry 1980;41(3):79-83.
13. Caroff SN, Mann SC, Keck PE, Jr, Francis A. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol 2000;20(2):257-9.
14. Caroff SN, Mann SC, McCarthy M, et al. Acute infectious encephalitis complicated by neuroleptic malignant syndrome. J Clin Psychopharmacol 1998;18(4):349-51.
15. Caroff SN, Mann SC, Gliatto MF, et al. Psychiatric manifestations of acute viral encephalitis. Psychiatric Annals 2001;31(3):193-204.
16. Caroff SN, Mann SC, Francis A, Fricchione GL (eds). Catatonia: from psychopathology to neurobiology. Washington, DC: American Psychiatric Publishing (in press).
17. Mann SC. Thermoregulatory mechanisms and antipsychotic drugrelated heatstroke. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003;45-74.
18. Caroff SN, Mann SC, Campbell EC. Risk of fatal heatstroke after hospitalization. Psychiatric Serv 2000;51(7):938.-
19. Keck PE, Jr. Serotonin syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003;75-92.
20. Davis JM, Caroff SN, Mann SC. Treatment of neuroleptic malignant syndrome. Psychiatric Annals 2000;30(5):325-31.
21. Francis A, Chandragiri S, Rizvi S, et al. Lorazepam a treatment for neuroleptic malignant syndrome? CNS Spectrums 2000;5(7):54-7.
22. Sakkas P, Davis JM, Hua J, Wang Z. Pharmacotherapy of neuroleptic malignant syndrome. Psychiatr Ann 1991;21:157-64.
23. Rosenberg MR, Green M. Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 1989;149:1927-31.
24. Henderson A, Longdon P. Fulminant metoclopramide-induced neuroleptic malignant syndrome rapidly responsive to intravenous dantrolene. Aust N Z J Med 1991;21:742-3.
25. Yamawaki S, Morio M, Kazamatsuri G, et al. Clinical evaluation and effective usage of dantrolene sodium in neuroleptic malignant syndrome. Kiso to Rinsyou (Clinical Reports) 1993;27(3):1045-66.
26. Tsutsumi Y, Yamamoto K, Matsuura S, et al. The treatment of neuroleptic malignant syndrome using dantrolene sodium. Psychiatry Clin Neurosci 1998;52:433-8.
27. Nisijima K, Ishiguro T. Electroconvulsive therapy for the treatment of neuroleptic malignant syndrome: a report of five cases. J ECT 1999;15:158-63.
28. Troller JN, Sachdev PS. Electroconvulsive treatment of neuroleptic malignant syndrome: a review and report of cases. Aust N Z J Psychiatry 1999;33:650-9.
1. Caroff SN, Mann SC, Campbell EC, Sullivan KA. Movement disorders associated with atypical antipsychotic drugs. J Clin Psychiatry 2002;63(suppl 4):12-19.
2. Caroff SN, Mann SC, Campbell EC. Atypical antipsychotics and neuroleptic malignant syndrome. Psychiatric Annals 2000;30(5):314-21.
3. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the perioperative setting. Am J Anesthesiology 2001;28(8):387-93.
4. Caroff SN, Rosenberg H, Mann SC, et al. Neuroleptic malignant syndrome in the critical care unit. Crit Care Med 2002;30(11):2609.-
5. Caroff SN, Mann SC. Neuroleptic malignant syndrome. Med Clin North Am 1993;77(1):185-202.
6. Caroff SN. Neuroleptic malignant syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing Inc., 2003;1-44.
7. Velamoor VR, Swamy GN, Parmar RS, et al. Management of suspected neuroleptic malignant syndrome. Can J Psychiatry 1995;40(9):545-50.
8. Keck PE, Jr, Pope HG, Jr, Cohen BM, et al. Risk factors for neuroleptic malignant syndrome. Arch Gen Psychiatry 1989;46:914-18.
9. Mann SC. Malignant catatonia. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003;121-43.
10. Velamoor VR, Norman RMG, Caroff SN, et al. Progression of symptoms in neuroleptic malignant syndrome. J Nerv Ment Dis 1994;182(3):168-73.
11. Delay J, Pichot P, Lemperiere T, et al. Un neuroleptique majeur non-phenothiazine et non reserpinique, l’haloperidol, dans le traitement des psychoses. Annales Medico-Psychologique 1960;118(1):145-52.
12. Caroff SN. The neuroleptic malignant syndrome. J Clin Psychiatry 1980;41(3):79-83.
13. Caroff SN, Mann SC, Keck PE, Jr, Francis A. Residual catatonic state following neuroleptic malignant syndrome. J Clin Psychopharmacol 2000;20(2):257-9.
14. Caroff SN, Mann SC, McCarthy M, et al. Acute infectious encephalitis complicated by neuroleptic malignant syndrome. J Clin Psychopharmacol 1998;18(4):349-51.
15. Caroff SN, Mann SC, Gliatto MF, et al. Psychiatric manifestations of acute viral encephalitis. Psychiatric Annals 2001;31(3):193-204.
16. Caroff SN, Mann SC, Francis A, Fricchione GL (eds). Catatonia: from psychopathology to neurobiology. Washington, DC: American Psychiatric Publishing (in press).
17. Mann SC. Thermoregulatory mechanisms and antipsychotic drugrelated heatstroke. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003;45-74.
18. Caroff SN, Mann SC, Campbell EC. Risk of fatal heatstroke after hospitalization. Psychiatric Serv 2000;51(7):938.-
19. Keck PE, Jr. Serotonin syndrome. In: Mann SC, Caroff SN, Keck PE Jr, Lazarus A (eds). Neuroleptic malignant syndrome and related conditions (2nd ed). Washington, DC: American Psychiatric Publishing, 2003;75-92.
20. Davis JM, Caroff SN, Mann SC. Treatment of neuroleptic malignant syndrome. Psychiatric Annals 2000;30(5):325-31.
21. Francis A, Chandragiri S, Rizvi S, et al. Lorazepam a treatment for neuroleptic malignant syndrome? CNS Spectrums 2000;5(7):54-7.
22. Sakkas P, Davis JM, Hua J, Wang Z. Pharmacotherapy of neuroleptic malignant syndrome. Psychiatr Ann 1991;21:157-64.
23. Rosenberg MR, Green M. Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 1989;149:1927-31.
24. Henderson A, Longdon P. Fulminant metoclopramide-induced neuroleptic malignant syndrome rapidly responsive to intravenous dantrolene. Aust N Z J Med 1991;21:742-3.
25. Yamawaki S, Morio M, Kazamatsuri G, et al. Clinical evaluation and effective usage of dantrolene sodium in neuroleptic malignant syndrome. Kiso to Rinsyou (Clinical Reports) 1993;27(3):1045-66.
26. Tsutsumi Y, Yamamoto K, Matsuura S, et al. The treatment of neuroleptic malignant syndrome using dantrolene sodium. Psychiatry Clin Neurosci 1998;52:433-8.
27. Nisijima K, Ishiguro T. Electroconvulsive therapy for the treatment of neuroleptic malignant syndrome: a report of five cases. J ECT 1999;15:158-63.
28. Troller JN, Sachdev PS. Electroconvulsive treatment of neuroleptic malignant syndrome: a review and report of cases. Aust N Z J Psychiatry 1999;33:650-9.