User login
FDA grants "breakthrough" status to investigational T-cell therapy for relapsed/refractory ALL
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
A chimeric antigen receptor therapy in phase I studies for relapsed/refractory acute lymphoblastic leukemia has been granted "breakthrough therapy" status by the Food and Drug Administration, which will expedite the development and review of the product as a result, Novartis has announced.
The investigational T-cell therapy – CTL019 – is now being researched in phase I studies of pediatric and adult patients with relapsed/refractory ALL. CTL019 "uses CAR [chimeric antigen receptor] technology to reprogram a patient’s own T cells to ‘hunt’ cancer cells" that express specific proteins [CD19]. "After they have been reprogrammed, the T cells (now called CTL019) are reintroduced into the patient’s blood; they proliferate and bind to the targeted CD19+ cancer cells and destroy them," according to the statement issued by Novartis on July 7.
Researchers at the University of Pennsylvania, Philadelphia, are conducting phase I/II studies of CTL019 and are working with Novartis to study and develop CAR T-cell treatments as cancer treatments; they have submitted the filing for the breakthrough therapy status, the statement said.
If a drug is designated as breakthrough therapy, the FDA will expedite the development and review of such drug.
The breakthrough therapy designation was created by the 2012 FDA Safety and Innovation Act. The FDA defines a breakthrough therapy as a drug that is intended to treat a serious or life-threatening disease or condition, alone or in combination with other drugs, and where "preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development," according to the FDA website.
"Because of uncertainty of clinical trials, there is no guarantee that CTL019 will ever be commercially available anywhere in the world," the Novartis statement added.
FDA Approves Inhaled Insulin for Adults
A rapidly acting inhaled formulation of human insulin was approved by the Food and Drug Administration on June 27 for adults with type 1 and type 2 diabetes, with a boxed warning about the potential for bronchospasm and the requirement that the manufacturer conduct postmarketing studies.
The product, which will be marketed as Afrezza (insulin human) inhalation powder, is administered at the beginning of each meal or within 20 minutes of starting the meal, and "must be used in combination with long-acting insulin in patients with type 1 diabetes," according to the FDA.
Approval was based on studies of about 3,000 patients with type 1 or type 2 diabetes. Hypoglycemia, cough, and throat pain or irritation were the most common adverse effects associated with treatment.
At a meeting in April, an FDA advisory panel voted 13-1 that the data supported approval for treatment of adults with type 1 diabetes, and 14-0 that the data supported approval for treatment of adults with type 2 diabetes, although they had some concerns about the potential for lung cancer, acute bronchospasm, and other safety issues.
Panelists also noted that trials indicated that the inhaled insulin was not as effective as traditional insulin, and that it would not be appropriate for all patients with type 1 and type 2 diabetes, but agreed that more treatment options were needed.
The label includes a boxed warning about acute bronchospasm, which has been reported in patients with asthma and chronic obstructive pulmonary disease who have used Afrezza; it is contraindicated in patients with chronic lung disease.
To address the risks of bronchospasm, approval includes a Risk Evaluation and Mitigation Strategy (REMS), which will inform health care professionals about the risk of serious acute bronchospasm associated with treatment. The FDA is also requesting that the manufacturer, MannKind Corporation, conduct several postmarketing studies, including a study to evaluate the product in pediatric patients and a study to evaluate the potential risk of pulmonary malignancies, cardiovascular risk, and long-term pulmonary function effects.
A rapidly acting inhaled formulation of human insulin was approved by the Food and Drug Administration on June 27 for adults with type 1 and type 2 diabetes, with a boxed warning about the potential for bronchospasm and the requirement that the manufacturer conduct postmarketing studies.
The product, which will be marketed as Afrezza (insulin human) inhalation powder, is administered at the beginning of each meal or within 20 minutes of starting the meal, and "must be used in combination with long-acting insulin in patients with type 1 diabetes," according to the FDA.
Approval was based on studies of about 3,000 patients with type 1 or type 2 diabetes. Hypoglycemia, cough, and throat pain or irritation were the most common adverse effects associated with treatment.
At a meeting in April, an FDA advisory panel voted 13-1 that the data supported approval for treatment of adults with type 1 diabetes, and 14-0 that the data supported approval for treatment of adults with type 2 diabetes, although they had some concerns about the potential for lung cancer, acute bronchospasm, and other safety issues.
Panelists also noted that trials indicated that the inhaled insulin was not as effective as traditional insulin, and that it would not be appropriate for all patients with type 1 and type 2 diabetes, but agreed that more treatment options were needed.
The label includes a boxed warning about acute bronchospasm, which has been reported in patients with asthma and chronic obstructive pulmonary disease who have used Afrezza; it is contraindicated in patients with chronic lung disease.
To address the risks of bronchospasm, approval includes a Risk Evaluation and Mitigation Strategy (REMS), which will inform health care professionals about the risk of serious acute bronchospasm associated with treatment. The FDA is also requesting that the manufacturer, MannKind Corporation, conduct several postmarketing studies, including a study to evaluate the product in pediatric patients and a study to evaluate the potential risk of pulmonary malignancies, cardiovascular risk, and long-term pulmonary function effects.
A rapidly acting inhaled formulation of human insulin was approved by the Food and Drug Administration on June 27 for adults with type 1 and type 2 diabetes, with a boxed warning about the potential for bronchospasm and the requirement that the manufacturer conduct postmarketing studies.
The product, which will be marketed as Afrezza (insulin human) inhalation powder, is administered at the beginning of each meal or within 20 minutes of starting the meal, and "must be used in combination with long-acting insulin in patients with type 1 diabetes," according to the FDA.
Approval was based on studies of about 3,000 patients with type 1 or type 2 diabetes. Hypoglycemia, cough, and throat pain or irritation were the most common adverse effects associated with treatment.
At a meeting in April, an FDA advisory panel voted 13-1 that the data supported approval for treatment of adults with type 1 diabetes, and 14-0 that the data supported approval for treatment of adults with type 2 diabetes, although they had some concerns about the potential for lung cancer, acute bronchospasm, and other safety issues.
Panelists also noted that trials indicated that the inhaled insulin was not as effective as traditional insulin, and that it would not be appropriate for all patients with type 1 and type 2 diabetes, but agreed that more treatment options were needed.
The label includes a boxed warning about acute bronchospasm, which has been reported in patients with asthma and chronic obstructive pulmonary disease who have used Afrezza; it is contraindicated in patients with chronic lung disease.
To address the risks of bronchospasm, approval includes a Risk Evaluation and Mitigation Strategy (REMS), which will inform health care professionals about the risk of serious acute bronchospasm associated with treatment. The FDA is also requesting that the manufacturer, MannKind Corporation, conduct several postmarketing studies, including a study to evaluate the product in pediatric patients and a study to evaluate the potential risk of pulmonary malignancies, cardiovascular risk, and long-term pulmonary function effects.
FDA approves inhaled insulin for adults, with safety-monitoring plan
A rapidly acting inhaled formulation of human insulin was approved by the Food and Drug Administration on June 27 for adults with type 1 and type 2 diabetes, with a boxed warning about the potential for bronchospasm and the requirement that the manufacturer conduct postmarketing studies.
The product, which will be marketed as Afrezza (insulin human) inhalation powder, is administered at the beginning of each meal or within 20 minutes of starting the meal, and "must be used in combination with long-acting insulin in patients with type 1 diabetes," according to the FDA.
Approval was based on studies of about 3,000 patients with type 1 or type 2 diabetes. Hypoglycemia, cough, and throat pain or irritation were the most common adverse effects associated with treatment.
At a meeting in April, an FDA advisory panel voted 13-1 that the data supported approval for treatment of adults with type 1 diabetes, and 14-0 that the data supported approval for treatment of adults with type 2 diabetes, although they had some concerns about the potential for lung cancer, acute bronchospasm, and other safety issues.
Panelists also noted that trials indicated that the inhaled insulin was not as effective as traditional insulin, and that it would not be appropriate for all patients with type 1 and type 2 diabetes, but agreed that more treatment options were needed.
The label includes a boxed warning about acute bronchospasm, which has been reported in patients with asthma and chronic obstructive pulmonary disease who have used Afrezza; it is contraindicated in patients with chronic lung disease.
To address the risks of bronchospasm, approval includes a Risk Evaluation and Mitigation Strategy (REMS), which will inform health care professionals about the risk of serious acute bronchospasm associated with treatment. The FDA is also requesting that the manufacturer, MannKind Corporation, conduct several postmarketing studies, including a study to evaluate the product in pediatric patients and a study to evaluate the potential risk of pulmonary malignancies, cardiovascular risk, and long-term pulmonary function effects.
A rapidly acting inhaled formulation of human insulin was approved by the Food and Drug Administration on June 27 for adults with type 1 and type 2 diabetes, with a boxed warning about the potential for bronchospasm and the requirement that the manufacturer conduct postmarketing studies.
The product, which will be marketed as Afrezza (insulin human) inhalation powder, is administered at the beginning of each meal or within 20 minutes of starting the meal, and "must be used in combination with long-acting insulin in patients with type 1 diabetes," according to the FDA.
Approval was based on studies of about 3,000 patients with type 1 or type 2 diabetes. Hypoglycemia, cough, and throat pain or irritation were the most common adverse effects associated with treatment.
At a meeting in April, an FDA advisory panel voted 13-1 that the data supported approval for treatment of adults with type 1 diabetes, and 14-0 that the data supported approval for treatment of adults with type 2 diabetes, although they had some concerns about the potential for lung cancer, acute bronchospasm, and other safety issues.
Panelists also noted that trials indicated that the inhaled insulin was not as effective as traditional insulin, and that it would not be appropriate for all patients with type 1 and type 2 diabetes, but agreed that more treatment options were needed.
The label includes a boxed warning about acute bronchospasm, which has been reported in patients with asthma and chronic obstructive pulmonary disease who have used Afrezza; it is contraindicated in patients with chronic lung disease.
To address the risks of bronchospasm, approval includes a Risk Evaluation and Mitigation Strategy (REMS), which will inform health care professionals about the risk of serious acute bronchospasm associated with treatment. The FDA is also requesting that the manufacturer, MannKind Corporation, conduct several postmarketing studies, including a study to evaluate the product in pediatric patients and a study to evaluate the potential risk of pulmonary malignancies, cardiovascular risk, and long-term pulmonary function effects.
A rapidly acting inhaled formulation of human insulin was approved by the Food and Drug Administration on June 27 for adults with type 1 and type 2 diabetes, with a boxed warning about the potential for bronchospasm and the requirement that the manufacturer conduct postmarketing studies.
The product, which will be marketed as Afrezza (insulin human) inhalation powder, is administered at the beginning of each meal or within 20 minutes of starting the meal, and "must be used in combination with long-acting insulin in patients with type 1 diabetes," according to the FDA.
Approval was based on studies of about 3,000 patients with type 1 or type 2 diabetes. Hypoglycemia, cough, and throat pain or irritation were the most common adverse effects associated with treatment.
At a meeting in April, an FDA advisory panel voted 13-1 that the data supported approval for treatment of adults with type 1 diabetes, and 14-0 that the data supported approval for treatment of adults with type 2 diabetes, although they had some concerns about the potential for lung cancer, acute bronchospasm, and other safety issues.
Panelists also noted that trials indicated that the inhaled insulin was not as effective as traditional insulin, and that it would not be appropriate for all patients with type 1 and type 2 diabetes, but agreed that more treatment options were needed.
The label includes a boxed warning about acute bronchospasm, which has been reported in patients with asthma and chronic obstructive pulmonary disease who have used Afrezza; it is contraindicated in patients with chronic lung disease.
To address the risks of bronchospasm, approval includes a Risk Evaluation and Mitigation Strategy (REMS), which will inform health care professionals about the risk of serious acute bronchospasm associated with treatment. The FDA is also requesting that the manufacturer, MannKind Corporation, conduct several postmarketing studies, including a study to evaluate the product in pediatric patients and a study to evaluate the potential risk of pulmonary malignancies, cardiovascular risk, and long-term pulmonary function effects.
Phase III trial results needed before PARP inhibitor approved for ovarian cancer indication
SILVER SPRING, MD. – The decision on whether to approve the PARP inhibitor olaparib as maintenance treatment for a subgroup of women with platinum-sensitive relapsed ovarian cancer should be delayed until the results of a phase III study are available, according to the majority of a Food and Drug Administration advisory panel.
At a meeting of the FDA’s Oncologic Drugs Advisory Committee on June 25, it voted 11-2 that the effects of treatment – which included a median 7-month benefit in progression-free survival (PFS) over placebo in a subgroup of patients with ovarian cancer who have the germline BRCA (gBRCA) mutation in a phase II study – did not support an accelerated approval and that the decision should be delayed until the results of an ongoing phase III confirmatory study are available. The results of that study, the Study of Olaparib in Ovarian Cancer (SOLO) 2, are expected in mid-2016.
The FDA grants accelerated approval for drugs that treat serious or life-threatening conditions when there is evidence they provide a meaningful benefit over existing treatments, with endpoints considered "reasonably likely" to predict clinical benefit. The benefits, however, must be verified in a confirmatory trial. Otherwise, approval can be withdrawn. Because of multiple issues in the phase II study, including questions about the validity of the effects, statistical issues, and the potential for serious risks of the drug in a group of patients who otherwise may not be receiving treatment, the agency convened the panel meeting.
Olaparib – an inhibitor of poly ADP ribose polymerase (PARP), a key enzyme in one of the DNA repair pathways in human cells – "exploits tumor DNA repair pathway deficiencies to selectively induce cancer cell death," according to the manufacturer, AstraZeneca Pharmaceuticals. The proposed indication for olaparib, which is orally administered as monotherapy, is for "the maintenance treatment of adults with platinum-sensitive relapsed ovarian cancer (including fallopian tube or primary peritoneal) with germline BRCA (gBRCA) mutation as detected by an FDA-approved test, who are in response (complete response or partial response) to platinum-based chemotherapy."
The indication is based on a subgroup analysis of 96 patients with the gBRCA mutation who were enrolled in a phase II study of 265 women with platinum-sensitive relapsed high-grade ovarian cancer, randomized to treatment with olaparib or placebo as maintenance treatment after chemotherapy. In the study overall, the median PFS was 8.4 months for those on olaparib vs. 4.8 months among those on placebo, a statistically significant difference (hazard ratio, 0.35). In the 96 patients with the gBRCA mutation, however, the median PFS was 11.2 months among those on olaparib vs. 4.1 months among those on placebo, a significant difference (HR, 0.17).
Common adverse events included nausea, fatigue, abdominal pain, diarrhea and anemia – most were grade 1 or 2 but lasted a long time. Patients on olaparib experienced nausea for a median of 96 days, compared with 26 days among those on placebo. The median duration of other GI side effects also was higher among treated patients, including abdominal distension (147 vs. 34 days), constipation (44 vs. 4 days), and abdominal pain (75 vs. 18 days).
Among the issues raised by the FDA reviewers were the small number of patients with the mutation in the study, retrospective identification of most of those patients, and the possible imbalance in unknown prognostic factors between the treatment and placebo groups, as well as safety concerns – including the potentially increased risk of myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML) associated with treatment, considering the biology of the drug and cases reported in olaparib-treated patients.
In the study, there were four cases of MDS or AML, three in treated patients (2.2%). In a safety database of 2,618 patients treated with olaparib, there have been 22 cases of MDS or AML (0.8%).
Voting to delay approval, Dr. William McGuire III, medical director of gynecologic cancer outreach at Inova Fairfax Hospital in Falls Church, Va., said that he was conflicted, but was concerned because there is still no survival advantage evident. "This is a group of patients [with platinum-sensitive disease] that have generally good survival ... and I am most concerned about the possible signal that this drug may cause AML and MDS," which, he pointed out, "could actually lead to an early death in a patient who otherwise might live for a longer period of time."
Also voting with the majority, the panel chair, Dr. Mikkael Sekeres, director of the leukemia program at the Cleveland Clinic Taussig Cancer Institute, said that he was "extremely concerned about the risk of secondary cancers in ovarian cancer patients, who otherwise would receive no therapy at all," and that some of the secondary cancers may have been underreported. In addition, he said, "I was troubled about causing women months of nausea or other gastrointestinal side effects during a period of time they would otherwise be spending away from hospitals or clinics enjoying their lives."
One of the two panelists in favor of accelerated approval, Dr. Edward Trimble, director of the Center for Global Health at the National Cancer Institute, Rockville, Md., said that, in his view, the drug had a positive risk-benefit profile. It would be given to a limited number of ovarian cancer patients, was associated with a prolonged disease-free interval in the study, "and protected those patients from going on to intravenous cytotoxic therapy." The availability of an oral maintenance drug would make it possible for patients to avoid having to go to the hospital for chemotherapy for a period of time, which would be "a significant clinical benefit," he added.
SOLO 2 is an international, double-blind placebo-controlled randomized study comparing olaparib to placebo as maintenance therapy in 264* women with platinum-sensitive relapsed ovarian cancer and the gBRCA mutation. The study is evaluating a lower-dose, 300-mg-twice-a-day tablet formulation of olaparib.
The FDA is expected to make a decision by early October. The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, although occasionally a panelist is given a waiver.
emechcatie@frontlinemednews.com
*CORRECTION, 6/26/2014: An earlier version of this story misstated the number of patients enrolled in the SOLO 2 trial.
SILVER SPRING, MD. – The decision on whether to approve the PARP inhibitor olaparib as maintenance treatment for a subgroup of women with platinum-sensitive relapsed ovarian cancer should be delayed until the results of a phase III study are available, according to the majority of a Food and Drug Administration advisory panel.
At a meeting of the FDA’s Oncologic Drugs Advisory Committee on June 25, it voted 11-2 that the effects of treatment – which included a median 7-month benefit in progression-free survival (PFS) over placebo in a subgroup of patients with ovarian cancer who have the germline BRCA (gBRCA) mutation in a phase II study – did not support an accelerated approval and that the decision should be delayed until the results of an ongoing phase III confirmatory study are available. The results of that study, the Study of Olaparib in Ovarian Cancer (SOLO) 2, are expected in mid-2016.
The FDA grants accelerated approval for drugs that treat serious or life-threatening conditions when there is evidence they provide a meaningful benefit over existing treatments, with endpoints considered "reasonably likely" to predict clinical benefit. The benefits, however, must be verified in a confirmatory trial. Otherwise, approval can be withdrawn. Because of multiple issues in the phase II study, including questions about the validity of the effects, statistical issues, and the potential for serious risks of the drug in a group of patients who otherwise may not be receiving treatment, the agency convened the panel meeting.
Olaparib – an inhibitor of poly ADP ribose polymerase (PARP), a key enzyme in one of the DNA repair pathways in human cells – "exploits tumor DNA repair pathway deficiencies to selectively induce cancer cell death," according to the manufacturer, AstraZeneca Pharmaceuticals. The proposed indication for olaparib, which is orally administered as monotherapy, is for "the maintenance treatment of adults with platinum-sensitive relapsed ovarian cancer (including fallopian tube or primary peritoneal) with germline BRCA (gBRCA) mutation as detected by an FDA-approved test, who are in response (complete response or partial response) to platinum-based chemotherapy."
The indication is based on a subgroup analysis of 96 patients with the gBRCA mutation who were enrolled in a phase II study of 265 women with platinum-sensitive relapsed high-grade ovarian cancer, randomized to treatment with olaparib or placebo as maintenance treatment after chemotherapy. In the study overall, the median PFS was 8.4 months for those on olaparib vs. 4.8 months among those on placebo, a statistically significant difference (hazard ratio, 0.35). In the 96 patients with the gBRCA mutation, however, the median PFS was 11.2 months among those on olaparib vs. 4.1 months among those on placebo, a significant difference (HR, 0.17).
Common adverse events included nausea, fatigue, abdominal pain, diarrhea and anemia – most were grade 1 or 2 but lasted a long time. Patients on olaparib experienced nausea for a median of 96 days, compared with 26 days among those on placebo. The median duration of other GI side effects also was higher among treated patients, including abdominal distension (147 vs. 34 days), constipation (44 vs. 4 days), and abdominal pain (75 vs. 18 days).
Among the issues raised by the FDA reviewers were the small number of patients with the mutation in the study, retrospective identification of most of those patients, and the possible imbalance in unknown prognostic factors between the treatment and placebo groups, as well as safety concerns – including the potentially increased risk of myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML) associated with treatment, considering the biology of the drug and cases reported in olaparib-treated patients.
In the study, there were four cases of MDS or AML, three in treated patients (2.2%). In a safety database of 2,618 patients treated with olaparib, there have been 22 cases of MDS or AML (0.8%).
Voting to delay approval, Dr. William McGuire III, medical director of gynecologic cancer outreach at Inova Fairfax Hospital in Falls Church, Va., said that he was conflicted, but was concerned because there is still no survival advantage evident. "This is a group of patients [with platinum-sensitive disease] that have generally good survival ... and I am most concerned about the possible signal that this drug may cause AML and MDS," which, he pointed out, "could actually lead to an early death in a patient who otherwise might live for a longer period of time."
Also voting with the majority, the panel chair, Dr. Mikkael Sekeres, director of the leukemia program at the Cleveland Clinic Taussig Cancer Institute, said that he was "extremely concerned about the risk of secondary cancers in ovarian cancer patients, who otherwise would receive no therapy at all," and that some of the secondary cancers may have been underreported. In addition, he said, "I was troubled about causing women months of nausea or other gastrointestinal side effects during a period of time they would otherwise be spending away from hospitals or clinics enjoying their lives."
One of the two panelists in favor of accelerated approval, Dr. Edward Trimble, director of the Center for Global Health at the National Cancer Institute, Rockville, Md., said that, in his view, the drug had a positive risk-benefit profile. It would be given to a limited number of ovarian cancer patients, was associated with a prolonged disease-free interval in the study, "and protected those patients from going on to intravenous cytotoxic therapy." The availability of an oral maintenance drug would make it possible for patients to avoid having to go to the hospital for chemotherapy for a period of time, which would be "a significant clinical benefit," he added.
SOLO 2 is an international, double-blind placebo-controlled randomized study comparing olaparib to placebo as maintenance therapy in 264* women with platinum-sensitive relapsed ovarian cancer and the gBRCA mutation. The study is evaluating a lower-dose, 300-mg-twice-a-day tablet formulation of olaparib.
The FDA is expected to make a decision by early October. The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, although occasionally a panelist is given a waiver.
emechcatie@frontlinemednews.com
*CORRECTION, 6/26/2014: An earlier version of this story misstated the number of patients enrolled in the SOLO 2 trial.
SILVER SPRING, MD. – The decision on whether to approve the PARP inhibitor olaparib as maintenance treatment for a subgroup of women with platinum-sensitive relapsed ovarian cancer should be delayed until the results of a phase III study are available, according to the majority of a Food and Drug Administration advisory panel.
At a meeting of the FDA’s Oncologic Drugs Advisory Committee on June 25, it voted 11-2 that the effects of treatment – which included a median 7-month benefit in progression-free survival (PFS) over placebo in a subgroup of patients with ovarian cancer who have the germline BRCA (gBRCA) mutation in a phase II study – did not support an accelerated approval and that the decision should be delayed until the results of an ongoing phase III confirmatory study are available. The results of that study, the Study of Olaparib in Ovarian Cancer (SOLO) 2, are expected in mid-2016.
The FDA grants accelerated approval for drugs that treat serious or life-threatening conditions when there is evidence they provide a meaningful benefit over existing treatments, with endpoints considered "reasonably likely" to predict clinical benefit. The benefits, however, must be verified in a confirmatory trial. Otherwise, approval can be withdrawn. Because of multiple issues in the phase II study, including questions about the validity of the effects, statistical issues, and the potential for serious risks of the drug in a group of patients who otherwise may not be receiving treatment, the agency convened the panel meeting.
Olaparib – an inhibitor of poly ADP ribose polymerase (PARP), a key enzyme in one of the DNA repair pathways in human cells – "exploits tumor DNA repair pathway deficiencies to selectively induce cancer cell death," according to the manufacturer, AstraZeneca Pharmaceuticals. The proposed indication for olaparib, which is orally administered as monotherapy, is for "the maintenance treatment of adults with platinum-sensitive relapsed ovarian cancer (including fallopian tube or primary peritoneal) with germline BRCA (gBRCA) mutation as detected by an FDA-approved test, who are in response (complete response or partial response) to platinum-based chemotherapy."
The indication is based on a subgroup analysis of 96 patients with the gBRCA mutation who were enrolled in a phase II study of 265 women with platinum-sensitive relapsed high-grade ovarian cancer, randomized to treatment with olaparib or placebo as maintenance treatment after chemotherapy. In the study overall, the median PFS was 8.4 months for those on olaparib vs. 4.8 months among those on placebo, a statistically significant difference (hazard ratio, 0.35). In the 96 patients with the gBRCA mutation, however, the median PFS was 11.2 months among those on olaparib vs. 4.1 months among those on placebo, a significant difference (HR, 0.17).
Common adverse events included nausea, fatigue, abdominal pain, diarrhea and anemia – most were grade 1 or 2 but lasted a long time. Patients on olaparib experienced nausea for a median of 96 days, compared with 26 days among those on placebo. The median duration of other GI side effects also was higher among treated patients, including abdominal distension (147 vs. 34 days), constipation (44 vs. 4 days), and abdominal pain (75 vs. 18 days).
Among the issues raised by the FDA reviewers were the small number of patients with the mutation in the study, retrospective identification of most of those patients, and the possible imbalance in unknown prognostic factors between the treatment and placebo groups, as well as safety concerns – including the potentially increased risk of myelodysplastic syndrome (MDS) and acute myelogenous leukemia (AML) associated with treatment, considering the biology of the drug and cases reported in olaparib-treated patients.
In the study, there were four cases of MDS or AML, three in treated patients (2.2%). In a safety database of 2,618 patients treated with olaparib, there have been 22 cases of MDS or AML (0.8%).
Voting to delay approval, Dr. William McGuire III, medical director of gynecologic cancer outreach at Inova Fairfax Hospital in Falls Church, Va., said that he was conflicted, but was concerned because there is still no survival advantage evident. "This is a group of patients [with platinum-sensitive disease] that have generally good survival ... and I am most concerned about the possible signal that this drug may cause AML and MDS," which, he pointed out, "could actually lead to an early death in a patient who otherwise might live for a longer period of time."
Also voting with the majority, the panel chair, Dr. Mikkael Sekeres, director of the leukemia program at the Cleveland Clinic Taussig Cancer Institute, said that he was "extremely concerned about the risk of secondary cancers in ovarian cancer patients, who otherwise would receive no therapy at all," and that some of the secondary cancers may have been underreported. In addition, he said, "I was troubled about causing women months of nausea or other gastrointestinal side effects during a period of time they would otherwise be spending away from hospitals or clinics enjoying their lives."
One of the two panelists in favor of accelerated approval, Dr. Edward Trimble, director of the Center for Global Health at the National Cancer Institute, Rockville, Md., said that, in his view, the drug had a positive risk-benefit profile. It would be given to a limited number of ovarian cancer patients, was associated with a prolonged disease-free interval in the study, "and protected those patients from going on to intravenous cytotoxic therapy." The availability of an oral maintenance drug would make it possible for patients to avoid having to go to the hospital for chemotherapy for a period of time, which would be "a significant clinical benefit," he added.
SOLO 2 is an international, double-blind placebo-controlled randomized study comparing olaparib to placebo as maintenance therapy in 264* women with platinum-sensitive relapsed ovarian cancer and the gBRCA mutation. The study is evaluating a lower-dose, 300-mg-twice-a-day tablet formulation of olaparib.
The FDA is expected to make a decision by early October. The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, although occasionally a panelist is given a waiver.
emechcatie@frontlinemednews.com
*CORRECTION, 6/26/2014: An earlier version of this story misstated the number of patients enrolled in the SOLO 2 trial.
AT AN FDA ADVISORY COMMITTEE MEETING
FDA’s olmesartan probe showed no increased CV risk in diabetes
The Food and Drug Administration’s review of the safety of olmesartan has found "no clear evidence" of increased cardiovascular risks associated with the use of the drug in people with diabetes, but some concern remains about the possible risk with high doses, the agency reported on June 24.
The review, prompted by the ROADMAP (Randomized Olmesartan and Diabetes Microalbuminuria Prevention) study of patients with type 2 diabetes – which unexpectedly found an increased risk of cardiovascular death among patients on olmesartan, compared with those on placebo – has been completed, and recommendations for the use of the angiotensin receptor blocker (ARB) in people with diabetes will remain unchanged, according to the FDA.
Information about some of the studies reviewed by the FDA, however, including a large observational study of Medicare patients, will be included in the labels of olmesartan products, which include Benicar, Benicar HCT, AZO, Tribenzor, and generic formulations, the statement said.
"While data from the ROADMAP trial and the Medicare study have suggested that high-dose olmesartan may increase CV risk in diabetic patients, when considering the data from all trials and studies, they are not conclusive," the FDA statement said, adding, "Overall, we determined these studies do not clearly show an increased cardiovascular risk. Thus, the collective evidence available at this time does not support changing our recommendations for olmesartan use and does not support recommending that its use be avoided in patients with diabetes."
The risk of nonfatal MI, however, was lower among those on olmesartan in the ROADMAP study, which was designed to determine whether olmesartan could delay kidney damage in patients with type 2 diabetes (N. Engl. J. Med. 2011;364:907-17).
In the Medicare study of patients aged 65 years and older, the risk of death was increased among patients with diabetes, who received the highest (40 mg/day) dose of olmesartan for more than 6 months (hazard ratio, 2.0). Among patients who did not have diabetes, however, the highest dose was associated with a reduced risk of death (HR, 0.46). The conflicting results in these two patient groups in the study "are difficult to reconcile and raise uncertainty about the credibility of the findings in either group," the FDA said.
Other studies reviewed include a U.K. study of primary care medical records that compared outcomes of patients receiving high-dose olmesartan with those treated with high doses of other ARBs in over 58,000 patients. The study found an increased risk of overall death and of acute myocardial infarction associated with the use of high-dose olmesartan, which was not statistically significant (Pharmacoepidemiol. Drug Saf. 2014;23:340-7).
"Overall, these data raise concern of possible increased cardiovascular risk associated with the use of high-dose olmesartan in diabetic patients," the FDA concluded.
The FDA’s first safety communication on this issue was posted in June 2010, followed by an update in April 2011.
In 2013, about 1.8 million people received a dispensed prescription for olmesartan products from U.S. outpatient retail pharmacies, according to the FDA.
Serious adverse events in patients treated with olmesartan products should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
The Food and Drug Administration’s review of the safety of olmesartan has found "no clear evidence" of increased cardiovascular risks associated with the use of the drug in people with diabetes, but some concern remains about the possible risk with high doses, the agency reported on June 24.
The review, prompted by the ROADMAP (Randomized Olmesartan and Diabetes Microalbuminuria Prevention) study of patients with type 2 diabetes – which unexpectedly found an increased risk of cardiovascular death among patients on olmesartan, compared with those on placebo – has been completed, and recommendations for the use of the angiotensin receptor blocker (ARB) in people with diabetes will remain unchanged, according to the FDA.
Information about some of the studies reviewed by the FDA, however, including a large observational study of Medicare patients, will be included in the labels of olmesartan products, which include Benicar, Benicar HCT, AZO, Tribenzor, and generic formulations, the statement said.
"While data from the ROADMAP trial and the Medicare study have suggested that high-dose olmesartan may increase CV risk in diabetic patients, when considering the data from all trials and studies, they are not conclusive," the FDA statement said, adding, "Overall, we determined these studies do not clearly show an increased cardiovascular risk. Thus, the collective evidence available at this time does not support changing our recommendations for olmesartan use and does not support recommending that its use be avoided in patients with diabetes."
The risk of nonfatal MI, however, was lower among those on olmesartan in the ROADMAP study, which was designed to determine whether olmesartan could delay kidney damage in patients with type 2 diabetes (N. Engl. J. Med. 2011;364:907-17).
In the Medicare study of patients aged 65 years and older, the risk of death was increased among patients with diabetes, who received the highest (40 mg/day) dose of olmesartan for more than 6 months (hazard ratio, 2.0). Among patients who did not have diabetes, however, the highest dose was associated with a reduced risk of death (HR, 0.46). The conflicting results in these two patient groups in the study "are difficult to reconcile and raise uncertainty about the credibility of the findings in either group," the FDA said.
Other studies reviewed include a U.K. study of primary care medical records that compared outcomes of patients receiving high-dose olmesartan with those treated with high doses of other ARBs in over 58,000 patients. The study found an increased risk of overall death and of acute myocardial infarction associated with the use of high-dose olmesartan, which was not statistically significant (Pharmacoepidemiol. Drug Saf. 2014;23:340-7).
"Overall, these data raise concern of possible increased cardiovascular risk associated with the use of high-dose olmesartan in diabetic patients," the FDA concluded.
The FDA’s first safety communication on this issue was posted in June 2010, followed by an update in April 2011.
In 2013, about 1.8 million people received a dispensed prescription for olmesartan products from U.S. outpatient retail pharmacies, according to the FDA.
Serious adverse events in patients treated with olmesartan products should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
The Food and Drug Administration’s review of the safety of olmesartan has found "no clear evidence" of increased cardiovascular risks associated with the use of the drug in people with diabetes, but some concern remains about the possible risk with high doses, the agency reported on June 24.
The review, prompted by the ROADMAP (Randomized Olmesartan and Diabetes Microalbuminuria Prevention) study of patients with type 2 diabetes – which unexpectedly found an increased risk of cardiovascular death among patients on olmesartan, compared with those on placebo – has been completed, and recommendations for the use of the angiotensin receptor blocker (ARB) in people with diabetes will remain unchanged, according to the FDA.
Information about some of the studies reviewed by the FDA, however, including a large observational study of Medicare patients, will be included in the labels of olmesartan products, which include Benicar, Benicar HCT, AZO, Tribenzor, and generic formulations, the statement said.
"While data from the ROADMAP trial and the Medicare study have suggested that high-dose olmesartan may increase CV risk in diabetic patients, when considering the data from all trials and studies, they are not conclusive," the FDA statement said, adding, "Overall, we determined these studies do not clearly show an increased cardiovascular risk. Thus, the collective evidence available at this time does not support changing our recommendations for olmesartan use and does not support recommending that its use be avoided in patients with diabetes."
The risk of nonfatal MI, however, was lower among those on olmesartan in the ROADMAP study, which was designed to determine whether olmesartan could delay kidney damage in patients with type 2 diabetes (N. Engl. J. Med. 2011;364:907-17).
In the Medicare study of patients aged 65 years and older, the risk of death was increased among patients with diabetes, who received the highest (40 mg/day) dose of olmesartan for more than 6 months (hazard ratio, 2.0). Among patients who did not have diabetes, however, the highest dose was associated with a reduced risk of death (HR, 0.46). The conflicting results in these two patient groups in the study "are difficult to reconcile and raise uncertainty about the credibility of the findings in either group," the FDA said.
Other studies reviewed include a U.K. study of primary care medical records that compared outcomes of patients receiving high-dose olmesartan with those treated with high doses of other ARBs in over 58,000 patients. The study found an increased risk of overall death and of acute myocardial infarction associated with the use of high-dose olmesartan, which was not statistically significant (Pharmacoepidemiol. Drug Saf. 2014;23:340-7).
"Overall, these data raise concern of possible increased cardiovascular risk associated with the use of high-dose olmesartan in diabetic patients," the FDA concluded.
The FDA’s first safety communication on this issue was posted in June 2010, followed by an update in April 2011.
In 2013, about 1.8 million people received a dispensed prescription for olmesartan products from U.S. outpatient retail pharmacies, according to the FDA.
Serious adverse events in patients treated with olmesartan products should be reported to the FDA’s MedWatch program at 800-332-1088 or www.fda.gov/medwatch.
FDA approves tedizolid for acute bacterial skin infections
Tedizolid, an oxazolidinone antibacterial drug, has been approved as an intravenous and oral treatment for acute bacterial skin and skin structure infections caused by gram positive bacteria, the Food and Drug Administration announced on June 20.
Tedizolid (Sivextro) is approved to treat patients with acute bacterial skin and skin structure infections (ABSSSI) caused by methicillin-resistant and methicillin-susceptible strains of Staphylococcus aureus, various streptococcus species, and Enterococcus faecalis.
At a meeting in March, the FDA’s Anti-infective Drugs Advisory Committee unanimously recommended that tedizolid be approved. The drug is the second new antibacterial drug to be approved by the FDA in the past month to treat ABSSSI. On May 23, the agency approved dalbavancin (Dalvance) to treat patients with ABSSSI caused by Staphylococcus aureus and various streptococcus species. Both drugs were designated as qualified infectious disease products, allowing them an additional 5 years of marketing exclusivity.
Tedizolid’s safety and efficacy were evaluated and shown to be as effective as linezolid in two clinical trials with 1,315 adults randomly assigned to receive tedizolid or linezolid.
One of the phase III studies, the ESTABLISH-2 study, an international, randomized study, compared 6 days of treatment with intravenous tedizolid to 10 days of treatment with IV linezolid in 666 patients with acute bacterial skin and skin structure infections, with the option of switching to oral treatment. The lesions were at least 75 cm2 in size, and were known to be or were suspected to be associated with gram-positive bacteria. Half the patients had cellulitis, 20% had a major cutaneous abscess, and about 30% had an infected wound; about 27% had MRSA.
The primary endpoint, an early clinical response (at least a 20% reduction in the lesion area 48-72 hours after starting treatment), was achieved by 85% of those treated with tedizolid and by 83% of those treated with linezolid, a difference that met the non-inferiority margin. Clinical responses were also similar at 7-14 days. At late follow-up, 18-25 days after the end of treatment, outcomes of MRSA infections "matched the overall results," according to the study, which was published online in the Lancet Infectious Diseases.
Fewer patients on tedizolid (16%) developed gastrointestinal-related adverse events than those on linezolid (20%); otherwise, treatment- associated adverse events were similar in the two groups.
Tedizolid has not been evaluated in neutropenic patients, so alternative therapies should be considered, the FDA said in its statement announcing the approval.
Sivextro is marketed by Cubist Pharmaceuticals.
Tedizolid, an oxazolidinone antibacterial drug, has been approved as an intravenous and oral treatment for acute bacterial skin and skin structure infections caused by gram positive bacteria, the Food and Drug Administration announced on June 20.
Tedizolid (Sivextro) is approved to treat patients with acute bacterial skin and skin structure infections (ABSSSI) caused by methicillin-resistant and methicillin-susceptible strains of Staphylococcus aureus, various streptococcus species, and Enterococcus faecalis.
At a meeting in March, the FDA’s Anti-infective Drugs Advisory Committee unanimously recommended that tedizolid be approved. The drug is the second new antibacterial drug to be approved by the FDA in the past month to treat ABSSSI. On May 23, the agency approved dalbavancin (Dalvance) to treat patients with ABSSSI caused by Staphylococcus aureus and various streptococcus species. Both drugs were designated as qualified infectious disease products, allowing them an additional 5 years of marketing exclusivity.
Tedizolid’s safety and efficacy were evaluated and shown to be as effective as linezolid in two clinical trials with 1,315 adults randomly assigned to receive tedizolid or linezolid.
One of the phase III studies, the ESTABLISH-2 study, an international, randomized study, compared 6 days of treatment with intravenous tedizolid to 10 days of treatment with IV linezolid in 666 patients with acute bacterial skin and skin structure infections, with the option of switching to oral treatment. The lesions were at least 75 cm2 in size, and were known to be or were suspected to be associated with gram-positive bacteria. Half the patients had cellulitis, 20% had a major cutaneous abscess, and about 30% had an infected wound; about 27% had MRSA.
The primary endpoint, an early clinical response (at least a 20% reduction in the lesion area 48-72 hours after starting treatment), was achieved by 85% of those treated with tedizolid and by 83% of those treated with linezolid, a difference that met the non-inferiority margin. Clinical responses were also similar at 7-14 days. At late follow-up, 18-25 days after the end of treatment, outcomes of MRSA infections "matched the overall results," according to the study, which was published online in the Lancet Infectious Diseases.
Fewer patients on tedizolid (16%) developed gastrointestinal-related adverse events than those on linezolid (20%); otherwise, treatment- associated adverse events were similar in the two groups.
Tedizolid has not been evaluated in neutropenic patients, so alternative therapies should be considered, the FDA said in its statement announcing the approval.
Sivextro is marketed by Cubist Pharmaceuticals.
Tedizolid, an oxazolidinone antibacterial drug, has been approved as an intravenous and oral treatment for acute bacterial skin and skin structure infections caused by gram positive bacteria, the Food and Drug Administration announced on June 20.
Tedizolid (Sivextro) is approved to treat patients with acute bacterial skin and skin structure infections (ABSSSI) caused by methicillin-resistant and methicillin-susceptible strains of Staphylococcus aureus, various streptococcus species, and Enterococcus faecalis.
At a meeting in March, the FDA’s Anti-infective Drugs Advisory Committee unanimously recommended that tedizolid be approved. The drug is the second new antibacterial drug to be approved by the FDA in the past month to treat ABSSSI. On May 23, the agency approved dalbavancin (Dalvance) to treat patients with ABSSSI caused by Staphylococcus aureus and various streptococcus species. Both drugs were designated as qualified infectious disease products, allowing them an additional 5 years of marketing exclusivity.
Tedizolid’s safety and efficacy were evaluated and shown to be as effective as linezolid in two clinical trials with 1,315 adults randomly assigned to receive tedizolid or linezolid.
One of the phase III studies, the ESTABLISH-2 study, an international, randomized study, compared 6 days of treatment with intravenous tedizolid to 10 days of treatment with IV linezolid in 666 patients with acute bacterial skin and skin structure infections, with the option of switching to oral treatment. The lesions were at least 75 cm2 in size, and were known to be or were suspected to be associated with gram-positive bacteria. Half the patients had cellulitis, 20% had a major cutaneous abscess, and about 30% had an infected wound; about 27% had MRSA.
The primary endpoint, an early clinical response (at least a 20% reduction in the lesion area 48-72 hours after starting treatment), was achieved by 85% of those treated with tedizolid and by 83% of those treated with linezolid, a difference that met the non-inferiority margin. Clinical responses were also similar at 7-14 days. At late follow-up, 18-25 days after the end of treatment, outcomes of MRSA infections "matched the overall results," according to the study, which was published online in the Lancet Infectious Diseases.
Fewer patients on tedizolid (16%) developed gastrointestinal-related adverse events than those on linezolid (20%); otherwise, treatment- associated adverse events were similar in the two groups.
Tedizolid has not been evaluated in neutropenic patients, so alternative therapies should be considered, the FDA said in its statement announcing the approval.
Sivextro is marketed by Cubist Pharmaceuticals.
Repeat biopsy and long-term surveillance key for rare Hodgkin’s lymphoma subtype
Time to progression was inferior in patients with advanced-stage nodular lymphocyte-predominant Hodgkin’s lymphoma, compared with patients with classical Hodgkin’s lymphoma, in a study that compared outcomes between the two groups of Hodgkin’s lymphoma patients enrolled in the British Columbia Cancer Agency database.
Over 10 years, time to progression was 63% in the nodular lymphocyte-predominant Hodgkin’s lymphoma (NLPHL) group, vs. 73% in the classical Hodgkin’s lymphoma (CHL) group (P =.040), reported Dr. Katharine Xing of the Centre for Lymphoid Cancer at the BCCA and the University of British Columbia, Vancouver, and her associates.
Transformation to an aggressive non–Hodgkin’s lymphoma (NHL) over 15 years occurred in 24% of those with NLPHL, but in none of those with CHL (P = .00018), and the median time to transformation among those with NLPHL was 5.45 years (Blood 2014;123:3567-73).
The study compared 42 patients with advanced-stage NLPHL to 84 controls with advanced CHL, matched for age, sex, decade of diagnosis, stage, and chemotherapy type; all had been diagnosed between 1970 and 2011. Their mean age was 37 years, about two-thirds were men, most in both groups had stage III disease, and they were followed up for a median of about 11 years. Treatments included standard doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) and most received standard ABVD or ABVD-equivalent chemotherapy. The study was conducted to "highlight the distinct natural history of this rare HL subtype," which accounts for 5% of HL cases, the authors noted.
Over 10 years, "HL freedom from treatment failure," which reflects only relapses from HL, was 75% among those with NLPHL and 73% among those with CHL. Overall survival was also similar between the two groups (83.5% among those with NLPHL and 81% among those with CHL at 10 years).
Among their other findings was a significantly higher incidence of transformation over 10 years among those who had splenic involvement at the time of NLPHL diagnosis, compared with those who did not have splenic involvement (29% vs. 7.8%). When they looked at only those NLPHL patients who had received ABVD-like treatment, the incidence of transformation over 10 years was 34% among those with splenic involvement at diagnosis, vs. 9% among those who did not have splenic involvement (P = .014).
Since NLPHL is rare, information on the optimal treatment is limited, particularly for those with advanced disease, the authors pointed out. The analysis "highlights the distinct disease behavior of NLPHL, compared with CHL, and the need for repeat biopsy at relapse as well as long-term surveillance," the authors concluded. "Given the strong expression of CD20" on the lymphocyte predominant cells that distinguishes NLPHL from CHL, the results also provide "a rationale for further evaluation" of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) with rituximab, they added.
Time to progression was inferior in patients with advanced-stage nodular lymphocyte-predominant Hodgkin’s lymphoma, compared with patients with classical Hodgkin’s lymphoma, in a study that compared outcomes between the two groups of Hodgkin’s lymphoma patients enrolled in the British Columbia Cancer Agency database.
Over 10 years, time to progression was 63% in the nodular lymphocyte-predominant Hodgkin’s lymphoma (NLPHL) group, vs. 73% in the classical Hodgkin’s lymphoma (CHL) group (P =.040), reported Dr. Katharine Xing of the Centre for Lymphoid Cancer at the BCCA and the University of British Columbia, Vancouver, and her associates.
Transformation to an aggressive non–Hodgkin’s lymphoma (NHL) over 15 years occurred in 24% of those with NLPHL, but in none of those with CHL (P = .00018), and the median time to transformation among those with NLPHL was 5.45 years (Blood 2014;123:3567-73).
The study compared 42 patients with advanced-stage NLPHL to 84 controls with advanced CHL, matched for age, sex, decade of diagnosis, stage, and chemotherapy type; all had been diagnosed between 1970 and 2011. Their mean age was 37 years, about two-thirds were men, most in both groups had stage III disease, and they were followed up for a median of about 11 years. Treatments included standard doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) and most received standard ABVD or ABVD-equivalent chemotherapy. The study was conducted to "highlight the distinct natural history of this rare HL subtype," which accounts for 5% of HL cases, the authors noted.
Over 10 years, "HL freedom from treatment failure," which reflects only relapses from HL, was 75% among those with NLPHL and 73% among those with CHL. Overall survival was also similar between the two groups (83.5% among those with NLPHL and 81% among those with CHL at 10 years).
Among their other findings was a significantly higher incidence of transformation over 10 years among those who had splenic involvement at the time of NLPHL diagnosis, compared with those who did not have splenic involvement (29% vs. 7.8%). When they looked at only those NLPHL patients who had received ABVD-like treatment, the incidence of transformation over 10 years was 34% among those with splenic involvement at diagnosis, vs. 9% among those who did not have splenic involvement (P = .014).
Since NLPHL is rare, information on the optimal treatment is limited, particularly for those with advanced disease, the authors pointed out. The analysis "highlights the distinct disease behavior of NLPHL, compared with CHL, and the need for repeat biopsy at relapse as well as long-term surveillance," the authors concluded. "Given the strong expression of CD20" on the lymphocyte predominant cells that distinguishes NLPHL from CHL, the results also provide "a rationale for further evaluation" of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) with rituximab, they added.
Time to progression was inferior in patients with advanced-stage nodular lymphocyte-predominant Hodgkin’s lymphoma, compared with patients with classical Hodgkin’s lymphoma, in a study that compared outcomes between the two groups of Hodgkin’s lymphoma patients enrolled in the British Columbia Cancer Agency database.
Over 10 years, time to progression was 63% in the nodular lymphocyte-predominant Hodgkin’s lymphoma (NLPHL) group, vs. 73% in the classical Hodgkin’s lymphoma (CHL) group (P =.040), reported Dr. Katharine Xing of the Centre for Lymphoid Cancer at the BCCA and the University of British Columbia, Vancouver, and her associates.
Transformation to an aggressive non–Hodgkin’s lymphoma (NHL) over 15 years occurred in 24% of those with NLPHL, but in none of those with CHL (P = .00018), and the median time to transformation among those with NLPHL was 5.45 years (Blood 2014;123:3567-73).
The study compared 42 patients with advanced-stage NLPHL to 84 controls with advanced CHL, matched for age, sex, decade of diagnosis, stage, and chemotherapy type; all had been diagnosed between 1970 and 2011. Their mean age was 37 years, about two-thirds were men, most in both groups had stage III disease, and they were followed up for a median of about 11 years. Treatments included standard doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD) and most received standard ABVD or ABVD-equivalent chemotherapy. The study was conducted to "highlight the distinct natural history of this rare HL subtype," which accounts for 5% of HL cases, the authors noted.
Over 10 years, "HL freedom from treatment failure," which reflects only relapses from HL, was 75% among those with NLPHL and 73% among those with CHL. Overall survival was also similar between the two groups (83.5% among those with NLPHL and 81% among those with CHL at 10 years).
Among their other findings was a significantly higher incidence of transformation over 10 years among those who had splenic involvement at the time of NLPHL diagnosis, compared with those who did not have splenic involvement (29% vs. 7.8%). When they looked at only those NLPHL patients who had received ABVD-like treatment, the incidence of transformation over 10 years was 34% among those with splenic involvement at diagnosis, vs. 9% among those who did not have splenic involvement (P = .014).
Since NLPHL is rare, information on the optimal treatment is limited, particularly for those with advanced disease, the authors pointed out. The analysis "highlights the distinct disease behavior of NLPHL, compared with CHL, and the need for repeat biopsy at relapse as well as long-term surveillance," the authors concluded. "Given the strong expression of CD20" on the lymphocyte predominant cells that distinguishes NLPHL from CHL, the results also provide "a rationale for further evaluation" of CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) with rituximab, they added.
FROM BLOOD
Key clinical point: Repeat biopsy and long-term surveillance are necessary in nodular lymphocyte-predominant Hodgkin’s lymphoma.
Major finding: Overall survival was similar between patients with advanced-stage NLPHL and those with advanced-stage CHL, but differences between the two groups included an inferior time to progression among those with NLPHL over 10 years (63% vs 73%).
Data source: The study compared outcomes in 42 patients with advanced-stage NLPHL and 84 matched controls with advanced CHL, who were diagnosed between 1970 and 2011 and were enrolled in a Canadian cancer database.
Disclosures: Fourauthors received research funding from Roche; the remaining seven authors, including the lead author, had no relevant disclosures.
FDA panel votes favorably on implanted nerve-blocking device for obesity
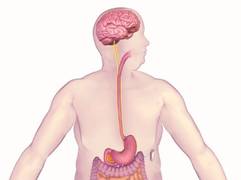
SILVER SPRING, MD. – A permanently implanted device thought to reduce hunger pangs by delivering electrical pulses to the intra-abdominal vagus nerve could be the next treatment approved for treating obesity, considering a Food and Drug Administration advisory panel’s positive vote on the device.
At a meeting on June 17, the FDA’s Gastroenterology-Urology Devices Panel voted 6-2, with one abstention, that the benefits of the Maestro Rechargeable System outweighed its risks for the indication under FDA review: for weight reduction in obese adults, with a body mass index of at least 40 kg/m2; or in those with a BMI of at least 35 kg/m2 with one or more obesity-related comorbid conditions, who have failed at least one supervised weight-management program within the past 5 years. Although the two primary effectiveness endpoints in the pivotal 12-month trial were not met, the safety endpoint was met and panelists voting in favor agreed that the study provided evidence that it was effective in helping some patients lose weight.
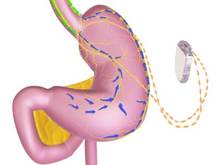
This is among the devices currently being studied that, if approved, could start to fill the large gap in obesity treatment options that currently exists between lifestyle modifications and pharmacologic treatment and bariatric surgery.
The main components of the device are a pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure under general anesthesia. External components include a mobile battery charger and a laptop computer used by clinicians to modify therapy and retrieve diagnostic information. By delivering intermittent electrical "blocking signals" to the anterior and posterior trunks of the intra-abdominal vagus nerve, the device "reduces sensations of hunger and produces satiety leading to weight loss," according to the manufacturer, EnteroMedics, which calls the treatment "VBLOC" therapy.
The ReCharge study, the main study submitted for approval, was a prospective, randomized, double-blind, sham-controlled study of 239 mostly female, white patients, whose mean age was about 47 years, and whose mean BMI at baseline was 41 (mean weight was about 250 lbs); about 5% had diabetes. A total of 162 had the device implanted and 77 had a nonfunctional neuroregulator implanted. At 12 months, almost 94% of the patients remained in the study.
One primary effectiveness endpoint was the mean percent excess weight loss (% EWL) among treated patients that was at least 10% greater than in the sham group (% EWL was calculated by dividing the weight lost by the excess weight). At 12 months, the mean % EWL was 24.4% in the treatment group, vs. 15.9% in the sham control group, an 8.5–percentage point difference. The second primary effectiveness endpoint, which applied only to those in the VBLOC group, had two components for the endpoint to be met: at least 55% of patients had least 20% EWL (which was met by 52.5% of patients); and at least 45% of patients had at least 25% EWL (which was met by 38.3% of patients).
Weight loss was maintained at 18 months, when the average % EWL in the VBLOC group was 25.2%, vs. 11.7% in the sham group, a 13.5% difference. However, 28% of the patients in the VBLOC group were lost to follow-up at that time, and the study was no longer blinded after 12 months.
With a 24% EWL after 1 year of treatment, an average patient – a 5’3"-tall woman weighing 225 lbs., with a BMI of 40 and 84 lbs. of excess weight – would lose 20 pounds, one of the FDA reviewers said at the meeting.
Over 12 months, among the 162 patients who received the device, there were 15 serious adverse events (9.3%): 9 related to the general surgery procedure (including 6 cases of nausea), 2 related to the implant or revision (one case of emesis and one case of atelectasis after the initial surgery), 3 device-related (pain at the neuroregulator site) and 1 treatment-related (gallbladder disease). Between 12 and 18 months, there was one gastric perforation during elective removal of the device.
Over 12 months, adverse events associated with the device included heartburn and dyspepsia, dysphagia, nausea, abdominal pain and pain at the neuroregulator site; nine surgical revisions were required in eight patients because of pain at the neuroregulator site (five cases), device malfunction (two cases), and readjustment of the neuroregulator because it was tilted (one case). In addition, five patients in the VBLOC group required surgical explantation of the device for jabbing pain during physical activity or heartburn symptoms.
Despite the near unanimous vote on the risk-benefit question, the votes on safety and effectiveness were mixed. The panel voted 8-1 that there was reasonable assurance the device was safe for the indicated use, but voted 5-4 that there was not reasonable assurance that it was effective for that indication. Those voting in favor of risk-benefit were swayed by the evidence it was effective in some patients and was reasonably safe.
One of the panelists voting yes on the risk-benefit question, Dr. Karen Woods, a gastroenterologist at the Methodist Hospital, Houston, said that the data provided by the company indicated the device was beneficial and referred to the need for less-invasive devices to treat obesity. "I would say they have shown they have effectiveness, just not particularly meeting the exact criteria they created for the study," she noted. "I feel we’re filling in a little bit of an area of unmet need ... and it will either go on to be effective or it won’t be, but I don’t think we’re going to do a lot of harm."
The two panelists voting no on the risk-benefit question said they voted no because the effectiveness endpoints were not met, despite evidence of superior weight loss in the treated group. "It’s close, it’s approaching clinical and statistical significance, but it doesn’t reach it," said Dr. Gary Falk, a gastroenterologist and professor of medicine at the University of Pennsylvania, Philadelphia.
Concerns raised by the panelists included the lack of racial diversity and the low number of men enrolled in the study, which made it unclear whether the results could be applied to the general population of obese patients; possible detrimental effects on the vagus nerve after leads are explanted, and the lack of long-term follow-up data for a device that could remain in place for a lifetime. Another concern is that patients with the device cannot undergo magnetic resonance imaging.
The company expects an FDA decision on approval later this year, according to a statement released by EnteroMedics after the panel meeting. If the device is approved, the company plans postmarketing studies, a 5-year follow-up study of patients in the ReCharge trial and a registry study of 500 patients in the United States.
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, but occasionally, a panelist is given a waiver. At this meeting, a waiver was granted Dr. Woods, who holds stock in a company with a competing product, according to the FDA briefing documents.
SILVER SPRING, MD. – A permanently implanted device thought to reduce hunger pangs by delivering electrical pulses to the intra-abdominal vagus nerve could be the next treatment approved for treating obesity, considering a Food and Drug Administration advisory panel’s positive vote on the device.
At a meeting on June 17, the FDA’s Gastroenterology-Urology Devices Panel voted 6-2, with one abstention, that the benefits of the Maestro Rechargeable System outweighed its risks for the indication under FDA review: for weight reduction in obese adults, with a body mass index of at least 40 kg/m2; or in those with a BMI of at least 35 kg/m2 with one or more obesity-related comorbid conditions, who have failed at least one supervised weight-management program within the past 5 years. Although the two primary effectiveness endpoints in the pivotal 12-month trial were not met, the safety endpoint was met and panelists voting in favor agreed that the study provided evidence that it was effective in helping some patients lose weight.
This is among the devices currently being studied that, if approved, could start to fill the large gap in obesity treatment options that currently exists between lifestyle modifications and pharmacologic treatment and bariatric surgery.
The main components of the device are a pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure under general anesthesia. External components include a mobile battery charger and a laptop computer used by clinicians to modify therapy and retrieve diagnostic information. By delivering intermittent electrical "blocking signals" to the anterior and posterior trunks of the intra-abdominal vagus nerve, the device "reduces sensations of hunger and produces satiety leading to weight loss," according to the manufacturer, EnteroMedics, which calls the treatment "VBLOC" therapy.
The ReCharge study, the main study submitted for approval, was a prospective, randomized, double-blind, sham-controlled study of 239 mostly female, white patients, whose mean age was about 47 years, and whose mean BMI at baseline was 41 (mean weight was about 250 lbs); about 5% had diabetes. A total of 162 had the device implanted and 77 had a nonfunctional neuroregulator implanted. At 12 months, almost 94% of the patients remained in the study.
One primary effectiveness endpoint was the mean percent excess weight loss (% EWL) among treated patients that was at least 10% greater than in the sham group (% EWL was calculated by dividing the weight lost by the excess weight). At 12 months, the mean % EWL was 24.4% in the treatment group, vs. 15.9% in the sham control group, an 8.5–percentage point difference. The second primary effectiveness endpoint, which applied only to those in the VBLOC group, had two components for the endpoint to be met: at least 55% of patients had least 20% EWL (which was met by 52.5% of patients); and at least 45% of patients had at least 25% EWL (which was met by 38.3% of patients).
Weight loss was maintained at 18 months, when the average % EWL in the VBLOC group was 25.2%, vs. 11.7% in the sham group, a 13.5% difference. However, 28% of the patients in the VBLOC group were lost to follow-up at that time, and the study was no longer blinded after 12 months.
With a 24% EWL after 1 year of treatment, an average patient – a 5’3"-tall woman weighing 225 lbs., with a BMI of 40 and 84 lbs. of excess weight – would lose 20 pounds, one of the FDA reviewers said at the meeting.
Over 12 months, among the 162 patients who received the device, there were 15 serious adverse events (9.3%): 9 related to the general surgery procedure (including 6 cases of nausea), 2 related to the implant or revision (one case of emesis and one case of atelectasis after the initial surgery), 3 device-related (pain at the neuroregulator site) and 1 treatment-related (gallbladder disease). Between 12 and 18 months, there was one gastric perforation during elective removal of the device.
Over 12 months, adverse events associated with the device included heartburn and dyspepsia, dysphagia, nausea, abdominal pain and pain at the neuroregulator site; nine surgical revisions were required in eight patients because of pain at the neuroregulator site (five cases), device malfunction (two cases), and readjustment of the neuroregulator because it was tilted (one case). In addition, five patients in the VBLOC group required surgical explantation of the device for jabbing pain during physical activity or heartburn symptoms.
Despite the near unanimous vote on the risk-benefit question, the votes on safety and effectiveness were mixed. The panel voted 8-1 that there was reasonable assurance the device was safe for the indicated use, but voted 5-4 that there was not reasonable assurance that it was effective for that indication. Those voting in favor of risk-benefit were swayed by the evidence it was effective in some patients and was reasonably safe.
One of the panelists voting yes on the risk-benefit question, Dr. Karen Woods, a gastroenterologist at the Methodist Hospital, Houston, said that the data provided by the company indicated the device was beneficial and referred to the need for less-invasive devices to treat obesity. "I would say they have shown they have effectiveness, just not particularly meeting the exact criteria they created for the study," she noted. "I feel we’re filling in a little bit of an area of unmet need ... and it will either go on to be effective or it won’t be, but I don’t think we’re going to do a lot of harm."
The two panelists voting no on the risk-benefit question said they voted no because the effectiveness endpoints were not met, despite evidence of superior weight loss in the treated group. "It’s close, it’s approaching clinical and statistical significance, but it doesn’t reach it," said Dr. Gary Falk, a gastroenterologist and professor of medicine at the University of Pennsylvania, Philadelphia.
Concerns raised by the panelists included the lack of racial diversity and the low number of men enrolled in the study, which made it unclear whether the results could be applied to the general population of obese patients; possible detrimental effects on the vagus nerve after leads are explanted, and the lack of long-term follow-up data for a device that could remain in place for a lifetime. Another concern is that patients with the device cannot undergo magnetic resonance imaging.
The company expects an FDA decision on approval later this year, according to a statement released by EnteroMedics after the panel meeting. If the device is approved, the company plans postmarketing studies, a 5-year follow-up study of patients in the ReCharge trial and a registry study of 500 patients in the United States.
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, but occasionally, a panelist is given a waiver. At this meeting, a waiver was granted Dr. Woods, who holds stock in a company with a competing product, according to the FDA briefing documents.
SILVER SPRING, MD. – A permanently implanted device thought to reduce hunger pangs by delivering electrical pulses to the intra-abdominal vagus nerve could be the next treatment approved for treating obesity, considering a Food and Drug Administration advisory panel’s positive vote on the device.
At a meeting on June 17, the FDA’s Gastroenterology-Urology Devices Panel voted 6-2, with one abstention, that the benefits of the Maestro Rechargeable System outweighed its risks for the indication under FDA review: for weight reduction in obese adults, with a body mass index of at least 40 kg/m2; or in those with a BMI of at least 35 kg/m2 with one or more obesity-related comorbid conditions, who have failed at least one supervised weight-management program within the past 5 years. Although the two primary effectiveness endpoints in the pivotal 12-month trial were not met, the safety endpoint was met and panelists voting in favor agreed that the study provided evidence that it was effective in helping some patients lose weight.
This is among the devices currently being studied that, if approved, could start to fill the large gap in obesity treatment options that currently exists between lifestyle modifications and pharmacologic treatment and bariatric surgery.
The main components of the device are a pulse generator implanted into the lateral chest wall, connected to two electrical leads placed around the abdominal vagus nerve via a laparoscopic procedure under general anesthesia. External components include a mobile battery charger and a laptop computer used by clinicians to modify therapy and retrieve diagnostic information. By delivering intermittent electrical "blocking signals" to the anterior and posterior trunks of the intra-abdominal vagus nerve, the device "reduces sensations of hunger and produces satiety leading to weight loss," according to the manufacturer, EnteroMedics, which calls the treatment "VBLOC" therapy.
The ReCharge study, the main study submitted for approval, was a prospective, randomized, double-blind, sham-controlled study of 239 mostly female, white patients, whose mean age was about 47 years, and whose mean BMI at baseline was 41 (mean weight was about 250 lbs); about 5% had diabetes. A total of 162 had the device implanted and 77 had a nonfunctional neuroregulator implanted. At 12 months, almost 94% of the patients remained in the study.
One primary effectiveness endpoint was the mean percent excess weight loss (% EWL) among treated patients that was at least 10% greater than in the sham group (% EWL was calculated by dividing the weight lost by the excess weight). At 12 months, the mean % EWL was 24.4% in the treatment group, vs. 15.9% in the sham control group, an 8.5–percentage point difference. The second primary effectiveness endpoint, which applied only to those in the VBLOC group, had two components for the endpoint to be met: at least 55% of patients had least 20% EWL (which was met by 52.5% of patients); and at least 45% of patients had at least 25% EWL (which was met by 38.3% of patients).
Weight loss was maintained at 18 months, when the average % EWL in the VBLOC group was 25.2%, vs. 11.7% in the sham group, a 13.5% difference. However, 28% of the patients in the VBLOC group were lost to follow-up at that time, and the study was no longer blinded after 12 months.
With a 24% EWL after 1 year of treatment, an average patient – a 5’3"-tall woman weighing 225 lbs., with a BMI of 40 and 84 lbs. of excess weight – would lose 20 pounds, one of the FDA reviewers said at the meeting.
Over 12 months, among the 162 patients who received the device, there were 15 serious adverse events (9.3%): 9 related to the general surgery procedure (including 6 cases of nausea), 2 related to the implant or revision (one case of emesis and one case of atelectasis after the initial surgery), 3 device-related (pain at the neuroregulator site) and 1 treatment-related (gallbladder disease). Between 12 and 18 months, there was one gastric perforation during elective removal of the device.
Over 12 months, adverse events associated with the device included heartburn and dyspepsia, dysphagia, nausea, abdominal pain and pain at the neuroregulator site; nine surgical revisions were required in eight patients because of pain at the neuroregulator site (five cases), device malfunction (two cases), and readjustment of the neuroregulator because it was tilted (one case). In addition, five patients in the VBLOC group required surgical explantation of the device for jabbing pain during physical activity or heartburn symptoms.
Despite the near unanimous vote on the risk-benefit question, the votes on safety and effectiveness were mixed. The panel voted 8-1 that there was reasonable assurance the device was safe for the indicated use, but voted 5-4 that there was not reasonable assurance that it was effective for that indication. Those voting in favor of risk-benefit were swayed by the evidence it was effective in some patients and was reasonably safe.
One of the panelists voting yes on the risk-benefit question, Dr. Karen Woods, a gastroenterologist at the Methodist Hospital, Houston, said that the data provided by the company indicated the device was beneficial and referred to the need for less-invasive devices to treat obesity. "I would say they have shown they have effectiveness, just not particularly meeting the exact criteria they created for the study," she noted. "I feel we’re filling in a little bit of an area of unmet need ... and it will either go on to be effective or it won’t be, but I don’t think we’re going to do a lot of harm."
The two panelists voting no on the risk-benefit question said they voted no because the effectiveness endpoints were not met, despite evidence of superior weight loss in the treated group. "It’s close, it’s approaching clinical and statistical significance, but it doesn’t reach it," said Dr. Gary Falk, a gastroenterologist and professor of medicine at the University of Pennsylvania, Philadelphia.
Concerns raised by the panelists included the lack of racial diversity and the low number of men enrolled in the study, which made it unclear whether the results could be applied to the general population of obese patients; possible detrimental effects on the vagus nerve after leads are explanted, and the lack of long-term follow-up data for a device that could remain in place for a lifetime. Another concern is that patients with the device cannot undergo magnetic resonance imaging.
The company expects an FDA decision on approval later this year, according to a statement released by EnteroMedics after the panel meeting. If the device is approved, the company plans postmarketing studies, a 5-year follow-up study of patients in the ReCharge trial and a registry study of 500 patients in the United States.
The FDA usually follows the recommendations of its advisory panels. Panel members have been cleared of potential conflicts, but occasionally, a panelist is given a waiver. At this meeting, a waiver was granted Dr. Woods, who holds stock in a company with a competing product, according to the FDA briefing documents.
AT AN FDA ADVISORY COMMITTEE MEETING
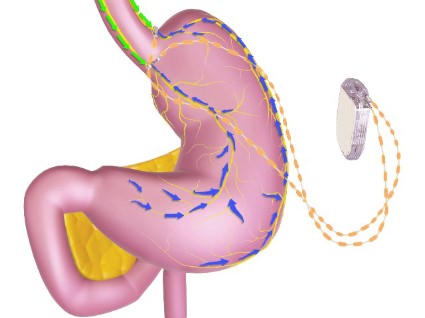
FDA approves vorapaxar, a new oral antiplatelet
The novel antiplatelet drug vorapaxar has been approved for reducing the risk of myocardial infarction, stroke, cardiovascular death, and need for coronary revascularization procedures in patients with a previous MI or peripheral arterial disease, the Food and Drug Administration announced on May 8.
Vorapaxar is an antagonist of protease-activated receptor-1 (PAR-1), which inhibits the action of thrombin on the platelet, and is the first drug in this class to be approved. Merck Sharp & Dohme will market the drug as Zontivity.
It comes in a tablet formulation.
At a meeting in January, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 10-1 to recommend approval of vorapaxar.
"In patients who have had a heart attack or who have peripheral arterial disease, this drug will lower the risk of heart attack, stroke, and cardiovascular death," Dr. Ellis F. Unger, director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research, reported in the FDA statement.
"In the study that supported the drug’s approval, Zontivity lowered this risk from 9.5% to 7.9% over a 3-year period – about 0.5% per year," he noted.
The FDA’s Cardiovascular and Renal Drugs Advisory Committee recommend approval of the novel antiplatelet drug for reducing atherothrombotic events in patients with a history of MI – the indication proposed by Merck. The recommended dose is one 2.5-mg tablet per day.
Because of the increased risk of bleeding, including life-threatening and fatal bleeding, the drug’s prescribing information includes a boxed warning about this risk and a medication guide informing patients about how to use the drug. A history of stroke, or transient ischemic attack, and a history of intracranial bleeding are contraindications.
The indication also includes the statement that treatment with vorapaxar has been shown to reduce the rate of the combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization.
The company conducted two phase III studies in two different groups of patients. In the TRACER (The Thrombin Receptor Antagonist for Clinical Event Reduction in ACS) study, which compared vorapaxar with placebo, added to standard therapy, as acute therapy (2.5 mg per day after a 40-mg loading dose, or placebo) in almost 13,000 hospitalized patients randomized within 24 hours of presenting with ACS (with non-ST elevation), it reduced the risk of atherothrombotic events (N. Engl. J. Med. 2012;366:20-33).
The study, however, was terminated early after an increased risk of major bleeding, including ICH, was detected and the company stopped the treatment in patients with a history of stroke or new stroke in the other phase III trial, the TRA 2P–TIMI 50 (Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Events) study. The company dropped plans to pursue the acute ACS indication.
The approved indication is based on the results of the TRA 2P–TIMI 50 study, which randomized 26,449 patients to placebo or 2.5 mg of vorapaxar a day, added to standard therapy (including other antiplatelet agents) in 26,449 outpatients with a previous MI, previous ischemic stroke, or peripheral arterial disease (N. Engl. J. Med. 2012;366:1404-13). (Almost 80% of the patients in the study who met the criteria in the proposed indication were on dual antiplatelet therapy with aspirin and a thienopyridine.)
In the overall cohort, including those who had to stop treatment early, compared with placebo, the risk of the primary composite endpoint of cardiovascular death, MI, stroke, or urgent coronary revascularization was reduced by 12%, and the risk of a secondary efficacy endpoint (CV death, MI, stroke) was reduced by 13% over 3 years among those treated with vorapaxar – effects that were statistically significant, Merck said.
The risk of severe or moderate bleeding was increased by 51% among those on vorapaxar over placebo.
The ICH rate over 3 years was 1% among those on vorapaxar, compared with 0.6% among those on placebo, but the absolute risk was higher in those with a history of stroke, according to Merck.
There is "clearly an unmet need" in this group of patients with a history of MI, "and a lack of therapies that have been shown to really be beneficial long term in this population," said the panel chair, Dr. Philip Sager, chair of the scientific programs committee, Cardiac Safety Research Consortium, San Francisco. "While there certainly is some bleeding risk, the benefits on the primary endpoint and secondary endpoint clearly outweigh those risks, and the benefit-risk relationship is positive," he added.
The panelist who voted against approval, Dr. Mori Krantz, director of the Colorado Prevention Center, Denver Health Medical Center, agreed that the primary efficacy endpoint had been met but was concerned about the size of the benefit and the large number of people needed to treat to benefit one patient.
"Although the intracranial hemorrhages and the major bleeds were potentially manageable, I worry about the amplification of that signal," because triple antiplatelet therapy is "unprecedented," he added.
Other safety concerns cited by panelists included a lack of an antidote and the lack of an effect in people weighing less than 60 kg (132 lb), an unresolved issue.
This is the first antiplatelet agent with a specific PAD indication. We now have multiple such agents to consider in our vascular patients. Time will tell which should be used in the management of the asymptomatic patient or those who have had either surgical or endovascular interventions. Currently, the other new agent, ticagrelor, is under investigation in the EUCLID study (Astra Zeneca). Hopefully there will ultimately be comparative studies of all antiplatelet agents. However, in my current practice I will prescribe clopidogrel for patients who do not have a contraindication and who can afford it. Otherwise aspirin will suffice in most patients. I reserve dual-antiplatelet agents for patients who demonstrate high risk or who have a history of graft or endovascular procedure thrombosis.
Dr. Russell Samson is the medical editor of Vascular Specialist.
This is the first antiplatelet agent with a specific PAD indication. We now have multiple such agents to consider in our vascular patients. Time will tell which should be used in the management of the asymptomatic patient or those who have had either surgical or endovascular interventions. Currently, the other new agent, ticagrelor, is under investigation in the EUCLID study (Astra Zeneca). Hopefully there will ultimately be comparative studies of all antiplatelet agents. However, in my current practice I will prescribe clopidogrel for patients who do not have a contraindication and who can afford it. Otherwise aspirin will suffice in most patients. I reserve dual-antiplatelet agents for patients who demonstrate high risk or who have a history of graft or endovascular procedure thrombosis.
Dr. Russell Samson is the medical editor of Vascular Specialist.
This is the first antiplatelet agent with a specific PAD indication. We now have multiple such agents to consider in our vascular patients. Time will tell which should be used in the management of the asymptomatic patient or those who have had either surgical or endovascular interventions. Currently, the other new agent, ticagrelor, is under investigation in the EUCLID study (Astra Zeneca). Hopefully there will ultimately be comparative studies of all antiplatelet agents. However, in my current practice I will prescribe clopidogrel for patients who do not have a contraindication and who can afford it. Otherwise aspirin will suffice in most patients. I reserve dual-antiplatelet agents for patients who demonstrate high risk or who have a history of graft or endovascular procedure thrombosis.
Dr. Russell Samson is the medical editor of Vascular Specialist.
The novel antiplatelet drug vorapaxar has been approved for reducing the risk of myocardial infarction, stroke, cardiovascular death, and need for coronary revascularization procedures in patients with a previous MI or peripheral arterial disease, the Food and Drug Administration announced on May 8.
Vorapaxar is an antagonist of protease-activated receptor-1 (PAR-1), which inhibits the action of thrombin on the platelet, and is the first drug in this class to be approved. Merck Sharp & Dohme will market the drug as Zontivity.
It comes in a tablet formulation.
At a meeting in January, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 10-1 to recommend approval of vorapaxar.
"In patients who have had a heart attack or who have peripheral arterial disease, this drug will lower the risk of heart attack, stroke, and cardiovascular death," Dr. Ellis F. Unger, director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research, reported in the FDA statement.
"In the study that supported the drug’s approval, Zontivity lowered this risk from 9.5% to 7.9% over a 3-year period – about 0.5% per year," he noted.
The FDA’s Cardiovascular and Renal Drugs Advisory Committee recommend approval of the novel antiplatelet drug for reducing atherothrombotic events in patients with a history of MI – the indication proposed by Merck. The recommended dose is one 2.5-mg tablet per day.
Because of the increased risk of bleeding, including life-threatening and fatal bleeding, the drug’s prescribing information includes a boxed warning about this risk and a medication guide informing patients about how to use the drug. A history of stroke, or transient ischemic attack, and a history of intracranial bleeding are contraindications.
The indication also includes the statement that treatment with vorapaxar has been shown to reduce the rate of the combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization.
The company conducted two phase III studies in two different groups of patients. In the TRACER (The Thrombin Receptor Antagonist for Clinical Event Reduction in ACS) study, which compared vorapaxar with placebo, added to standard therapy, as acute therapy (2.5 mg per day after a 40-mg loading dose, or placebo) in almost 13,000 hospitalized patients randomized within 24 hours of presenting with ACS (with non-ST elevation), it reduced the risk of atherothrombotic events (N. Engl. J. Med. 2012;366:20-33).
The study, however, was terminated early after an increased risk of major bleeding, including ICH, was detected and the company stopped the treatment in patients with a history of stroke or new stroke in the other phase III trial, the TRA 2P–TIMI 50 (Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Events) study. The company dropped plans to pursue the acute ACS indication.
The approved indication is based on the results of the TRA 2P–TIMI 50 study, which randomized 26,449 patients to placebo or 2.5 mg of vorapaxar a day, added to standard therapy (including other antiplatelet agents) in 26,449 outpatients with a previous MI, previous ischemic stroke, or peripheral arterial disease (N. Engl. J. Med. 2012;366:1404-13). (Almost 80% of the patients in the study who met the criteria in the proposed indication were on dual antiplatelet therapy with aspirin and a thienopyridine.)
In the overall cohort, including those who had to stop treatment early, compared with placebo, the risk of the primary composite endpoint of cardiovascular death, MI, stroke, or urgent coronary revascularization was reduced by 12%, and the risk of a secondary efficacy endpoint (CV death, MI, stroke) was reduced by 13% over 3 years among those treated with vorapaxar – effects that were statistically significant, Merck said.
The risk of severe or moderate bleeding was increased by 51% among those on vorapaxar over placebo.
The ICH rate over 3 years was 1% among those on vorapaxar, compared with 0.6% among those on placebo, but the absolute risk was higher in those with a history of stroke, according to Merck.
There is "clearly an unmet need" in this group of patients with a history of MI, "and a lack of therapies that have been shown to really be beneficial long term in this population," said the panel chair, Dr. Philip Sager, chair of the scientific programs committee, Cardiac Safety Research Consortium, San Francisco. "While there certainly is some bleeding risk, the benefits on the primary endpoint and secondary endpoint clearly outweigh those risks, and the benefit-risk relationship is positive," he added.
The panelist who voted against approval, Dr. Mori Krantz, director of the Colorado Prevention Center, Denver Health Medical Center, agreed that the primary efficacy endpoint had been met but was concerned about the size of the benefit and the large number of people needed to treat to benefit one patient.
"Although the intracranial hemorrhages and the major bleeds were potentially manageable, I worry about the amplification of that signal," because triple antiplatelet therapy is "unprecedented," he added.
Other safety concerns cited by panelists included a lack of an antidote and the lack of an effect in people weighing less than 60 kg (132 lb), an unresolved issue.
The novel antiplatelet drug vorapaxar has been approved for reducing the risk of myocardial infarction, stroke, cardiovascular death, and need for coronary revascularization procedures in patients with a previous MI or peripheral arterial disease, the Food and Drug Administration announced on May 8.
Vorapaxar is an antagonist of protease-activated receptor-1 (PAR-1), which inhibits the action of thrombin on the platelet, and is the first drug in this class to be approved. Merck Sharp & Dohme will market the drug as Zontivity.
It comes in a tablet formulation.
At a meeting in January, the FDA’s Cardiovascular and Renal Drugs Advisory Committee voted 10-1 to recommend approval of vorapaxar.
"In patients who have had a heart attack or who have peripheral arterial disease, this drug will lower the risk of heart attack, stroke, and cardiovascular death," Dr. Ellis F. Unger, director of the Office of Drug Evaluation I in the FDA’s Center for Drug Evaluation and Research, reported in the FDA statement.
"In the study that supported the drug’s approval, Zontivity lowered this risk from 9.5% to 7.9% over a 3-year period – about 0.5% per year," he noted.
The FDA’s Cardiovascular and Renal Drugs Advisory Committee recommend approval of the novel antiplatelet drug for reducing atherothrombotic events in patients with a history of MI – the indication proposed by Merck. The recommended dose is one 2.5-mg tablet per day.
Because of the increased risk of bleeding, including life-threatening and fatal bleeding, the drug’s prescribing information includes a boxed warning about this risk and a medication guide informing patients about how to use the drug. A history of stroke, or transient ischemic attack, and a history of intracranial bleeding are contraindications.
The indication also includes the statement that treatment with vorapaxar has been shown to reduce the rate of the combined endpoint of cardiovascular death, MI, stroke, and urgent coronary revascularization.
The company conducted two phase III studies in two different groups of patients. In the TRACER (The Thrombin Receptor Antagonist for Clinical Event Reduction in ACS) study, which compared vorapaxar with placebo, added to standard therapy, as acute therapy (2.5 mg per day after a 40-mg loading dose, or placebo) in almost 13,000 hospitalized patients randomized within 24 hours of presenting with ACS (with non-ST elevation), it reduced the risk of atherothrombotic events (N. Engl. J. Med. 2012;366:20-33).
The study, however, was terminated early after an increased risk of major bleeding, including ICH, was detected and the company stopped the treatment in patients with a history of stroke or new stroke in the other phase III trial, the TRA 2P–TIMI 50 (Thrombin-Receptor Antagonist in Secondary Prevention of Atherothrombotic Events) study. The company dropped plans to pursue the acute ACS indication.
The approved indication is based on the results of the TRA 2P–TIMI 50 study, which randomized 26,449 patients to placebo or 2.5 mg of vorapaxar a day, added to standard therapy (including other antiplatelet agents) in 26,449 outpatients with a previous MI, previous ischemic stroke, or peripheral arterial disease (N. Engl. J. Med. 2012;366:1404-13). (Almost 80% of the patients in the study who met the criteria in the proposed indication were on dual antiplatelet therapy with aspirin and a thienopyridine.)
In the overall cohort, including those who had to stop treatment early, compared with placebo, the risk of the primary composite endpoint of cardiovascular death, MI, stroke, or urgent coronary revascularization was reduced by 12%, and the risk of a secondary efficacy endpoint (CV death, MI, stroke) was reduced by 13% over 3 years among those treated with vorapaxar – effects that were statistically significant, Merck said.
The risk of severe or moderate bleeding was increased by 51% among those on vorapaxar over placebo.
The ICH rate over 3 years was 1% among those on vorapaxar, compared with 0.6% among those on placebo, but the absolute risk was higher in those with a history of stroke, according to Merck.
There is "clearly an unmet need" in this group of patients with a history of MI, "and a lack of therapies that have been shown to really be beneficial long term in this population," said the panel chair, Dr. Philip Sager, chair of the scientific programs committee, Cardiac Safety Research Consortium, San Francisco. "While there certainly is some bleeding risk, the benefits on the primary endpoint and secondary endpoint clearly outweigh those risks, and the benefit-risk relationship is positive," he added.
The panelist who voted against approval, Dr. Mori Krantz, director of the Colorado Prevention Center, Denver Health Medical Center, agreed that the primary efficacy endpoint had been met but was concerned about the size of the benefit and the large number of people needed to treat to benefit one patient.
"Although the intracranial hemorrhages and the major bleeds were potentially manageable, I worry about the amplification of that signal," because triple antiplatelet therapy is "unprecedented," he added.
Other safety concerns cited by panelists included a lack of an antidote and the lack of an effect in people weighing less than 60 kg (132 lb), an unresolved issue.
Lymph node mapping agent approval expanded to include head and neck cancer evaluation
Lymphoseek, a diagnostic imaging agent, has been approved for helping evaluate the spread of squamous cell carcinoma in patients with head and neck cancer, the Food and Drug Administration announced.
In a statement issued on June 13, the FDA said that with this approval, Lymphoseek (technetium 99m tilmanocept) injection "can now be used to guide" sentinel lymph node biopsies in patients with cancer of the head and neck, which "will allow for the option of more limited lymph node surgery in patients with sentinel nodes negative for cancer."
To use Lymphoseek, "doctors inject the drug into the tumor area and later, using a handheld radiation detector, find the sentinel lymph nodes that have taken up Lymphoseek’s radioactivity," Dr. Libero Marzella, director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
This is a new indication for Lymphoseek, marketed by Navidea Biopharmaceuticals. In March 2013, it was approved for assisting in the localization of lymph nodes draining a primary tumor in patients with breast cancer or melanoma.
Approval of the new indication is based on a study that used Lymphoseek in the evaluation of 85 patents with squamous cell carcinoma of the lip, oral cavity, and skin, which "showed that Lymphoseek-guided sentinel lymph node biopsy accurately determined if the cancer had spread through the lymphatic system," according to the FDA. Pain and irritation at the injection site were the most common adverse effects associated with Lymphoseek in the study.
Lymphoseek, a diagnostic imaging agent, has been approved for helping evaluate the spread of squamous cell carcinoma in patients with head and neck cancer, the Food and Drug Administration announced.
In a statement issued on June 13, the FDA said that with this approval, Lymphoseek (technetium 99m tilmanocept) injection "can now be used to guide" sentinel lymph node biopsies in patients with cancer of the head and neck, which "will allow for the option of more limited lymph node surgery in patients with sentinel nodes negative for cancer."
To use Lymphoseek, "doctors inject the drug into the tumor area and later, using a handheld radiation detector, find the sentinel lymph nodes that have taken up Lymphoseek’s radioactivity," Dr. Libero Marzella, director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
This is a new indication for Lymphoseek, marketed by Navidea Biopharmaceuticals. In March 2013, it was approved for assisting in the localization of lymph nodes draining a primary tumor in patients with breast cancer or melanoma.
Approval of the new indication is based on a study that used Lymphoseek in the evaluation of 85 patents with squamous cell carcinoma of the lip, oral cavity, and skin, which "showed that Lymphoseek-guided sentinel lymph node biopsy accurately determined if the cancer had spread through the lymphatic system," according to the FDA. Pain and irritation at the injection site were the most common adverse effects associated with Lymphoseek in the study.
Lymphoseek, a diagnostic imaging agent, has been approved for helping evaluate the spread of squamous cell carcinoma in patients with head and neck cancer, the Food and Drug Administration announced.
In a statement issued on June 13, the FDA said that with this approval, Lymphoseek (technetium 99m tilmanocept) injection "can now be used to guide" sentinel lymph node biopsies in patients with cancer of the head and neck, which "will allow for the option of more limited lymph node surgery in patients with sentinel nodes negative for cancer."
To use Lymphoseek, "doctors inject the drug into the tumor area and later, using a handheld radiation detector, find the sentinel lymph nodes that have taken up Lymphoseek’s radioactivity," Dr. Libero Marzella, director of the Division of Medical Imaging Products in the FDA’s Center for Drug Evaluation and Research, said in the statement.
This is a new indication for Lymphoseek, marketed by Navidea Biopharmaceuticals. In March 2013, it was approved for assisting in the localization of lymph nodes draining a primary tumor in patients with breast cancer or melanoma.
Approval of the new indication is based on a study that used Lymphoseek in the evaluation of 85 patents with squamous cell carcinoma of the lip, oral cavity, and skin, which "showed that Lymphoseek-guided sentinel lymph node biopsy accurately determined if the cancer had spread through the lymphatic system," according to the FDA. Pain and irritation at the injection site were the most common adverse effects associated with Lymphoseek in the study.