User login
Book review: “Sexual Citizens”
The Sexual Health Initiative to Foster Transformation (SHIFT)1 is a landmark study about sexual assault at college, which has generated 20 scientific articles and several chapters in books, but unfortunately, has not made its way into the psychiatric literature.
“Sexual Citizens: Sex, Power and Assault on Campus,” by Jennifer Hirsch and Shamus Khan, (available in audio book and paperback) was written as a follow up to the SHIFT study, so the rest of us can absorb the findings.2 This mixed-methods study included a survey of over 1,600 students aged 18-29 from Columbia University and Barnard College regarding their relationships and sexual histories, including assault. Data were collected using daily diaries, focus groups, and hundreds of hours of field work observation by young researchers. One- to 3-hour in-depth interviews exploring sexual experiences on campus were conducted with 151 students. These interviews are the focus of the book. It is a well-written, provocative story brimming with insights for those of us who lack the time to scour social science literature.

“Sexual Citizens” and the SHIFT study confirmed much of what we know. Sexual assault is common and has enduring effects. The study found that 36% of women and 15% of men had experienced unwanted, nonconsensual sexual contact by senior year. Twenty percent of women and 6% of men were rape survivors. Freshman, LGBTQ, and minority students were found at highest risk of assault. SHIFT reaffirmed that abstinence-only education is not a protective factor against college sexual assault, but neither was knowledge of affirmative consent (the practice of “ongoing and explicit” checking-in with partners) which few students ever employed. Encouragingly, students taught refusal skills were less likely to experience sexual assault.
Many of the book’s valuable lessons fall under the umbrella of failures of language and communication. For example, after drinking, they went to his room. She was expecting a social interaction, but with no other place to sit, they sat on his bed where she was coaxed or pressured into a sexual encounter. Afterward, she leaves, and it is never discussed again. One partner desires emotional intimacy, and the other, bragging rights in the fraternity or at the girls’ weekly brunch. Numerous personal stories like these, though at times heart wrenching, provide perspective on the barriers to addressing assault.
Subjects relayed experiences of assault by strangers or friends, and some provided details of their own actions as perpetrators. Stumbling around words and emotions, an avoidance of explicit language stemmed from shame, a fear of personal responsibility, the desire to maintain social cohesion, and concern for potential consequences for the perpetrator. Many subjects were resistant to calling nonconsensual sexual activity rape or even assault. Some who had perpetrated were unaware their behavior may have been experienced as assault, with recognition of this fact dawning during interviews.
This apparent limitation in self-reflective capacity may be in part due to the conceptualization of what assault is. Focus groups identified a discernible difference in how men and women understood assaults, with men believing rapes looked like a woman fighting back and screaming for help ... which is rarely what happens.
Notably absent among the interviewed are any flagrant perpetrators. The methodology section theorizes that individuals who intentionally harmed their peers were unlikely to choose to participate in this study. In addition, the characterization of assailants as “sociopathic predators” is based in a history of racialized imagery that leads us astray from the truth about campus sexual assault. Most assaults do not involve force, and SHIFT data showed 75% of victims knew their assailants. Ultimately, a major aim of the research was to study assault alongside healthy sex to “understand those pivotal moments when encounters change from being sex, to being assault.” Doing this requires understanding the where, how, and why students have sex, a more complicated undertaking than we may think.
In discussing their sexual lives, subjects frequently noted they did not have space to talk about their assaults. Though 81% of students discussed their experiences with someone, friend groups were often overburdened with stories, which minimized the victim’s experience. Furthermore, most had not sought help from the student counseling centers. Students navigating this complex field were frequently doing so in isolation. SHIFT found subjects to be eager to participate; they would often express thankfulness, and a sense of freedom in sharing with researchers. Commonly, students expressly did not want retribution for perpetrators, but simply a place to be heard without challenge. The current legal system precludes that possibility, leaving individuals without the option to confront perpetrators, and perpetrators often not knowing the extent of the damage they caused.
Where can psychiatrists have an impact right now? “Sexual Citizens” identifies four key areas for intervention to work toward a world with less sexual assault. These are:
- Improving diversity, inequality, and power distortions.
- Education about sex and sexual assault.
- Substance use.
- Mental health.
Substance use and mental health are especially relevant for psychiatrists (That substance use contributes to sexual assault is known by approximately ... everybody!). Unwanted sexual contact prior to college (20% of students) increased the odds of experiencing assault during college. Harm reduction strategies should be introduced before college, according to the SHIFT research, particularly in skills-based training on how to say “No” to unwanted sex. Psychiatrists are likely used to asking brief history questions related to sexual assault and rape. “Sexual Citizens” highlights the inadequacy of this blunt language and guides the reader toward a refined knowledge of the language needed to address sexual assault.
Dr. Whisler is a child and adolescent psychiatry fellow at the Stanford (Calif.) University. Dr. Higgins is affiliate associate professor of psychiatry and family medicine at the Medical University of South Carolina, Charleston.
References
1. Hirsch JS et al. Social dimensions of sexual consent among cisgender heterosexual college students: Insights from ethnographic research. J Adolesc Health. 2019 Jan;64(1):26-35. doi: 10.1016/j.jadohealth.2018.06.011.
2. Hirsch JS and Khan S. Sexual citizens: Sex, power, and assault on campus. New York: W.W. Norton & Company, 2020.
The Sexual Health Initiative to Foster Transformation (SHIFT)1 is a landmark study about sexual assault at college, which has generated 20 scientific articles and several chapters in books, but unfortunately, has not made its way into the psychiatric literature.
“Sexual Citizens: Sex, Power and Assault on Campus,” by Jennifer Hirsch and Shamus Khan, (available in audio book and paperback) was written as a follow up to the SHIFT study, so the rest of us can absorb the findings.2 This mixed-methods study included a survey of over 1,600 students aged 18-29 from Columbia University and Barnard College regarding their relationships and sexual histories, including assault. Data were collected using daily diaries, focus groups, and hundreds of hours of field work observation by young researchers. One- to 3-hour in-depth interviews exploring sexual experiences on campus were conducted with 151 students. These interviews are the focus of the book. It is a well-written, provocative story brimming with insights for those of us who lack the time to scour social science literature.
“Sexual Citizens” and the SHIFT study confirmed much of what we know. Sexual assault is common and has enduring effects. The study found that 36% of women and 15% of men had experienced unwanted, nonconsensual sexual contact by senior year. Twenty percent of women and 6% of men were rape survivors. Freshman, LGBTQ, and minority students were found at highest risk of assault. SHIFT reaffirmed that abstinence-only education is not a protective factor against college sexual assault, but neither was knowledge of affirmative consent (the practice of “ongoing and explicit” checking-in with partners) which few students ever employed. Encouragingly, students taught refusal skills were less likely to experience sexual assault.
Many of the book’s valuable lessons fall under the umbrella of failures of language and communication. For example, after drinking, they went to his room. She was expecting a social interaction, but with no other place to sit, they sat on his bed where she was coaxed or pressured into a sexual encounter. Afterward, she leaves, and it is never discussed again. One partner desires emotional intimacy, and the other, bragging rights in the fraternity or at the girls’ weekly brunch. Numerous personal stories like these, though at times heart wrenching, provide perspective on the barriers to addressing assault.
Subjects relayed experiences of assault by strangers or friends, and some provided details of their own actions as perpetrators. Stumbling around words and emotions, an avoidance of explicit language stemmed from shame, a fear of personal responsibility, the desire to maintain social cohesion, and concern for potential consequences for the perpetrator. Many subjects were resistant to calling nonconsensual sexual activity rape or even assault. Some who had perpetrated were unaware their behavior may have been experienced as assault, with recognition of this fact dawning during interviews.
This apparent limitation in self-reflective capacity may be in part due to the conceptualization of what assault is. Focus groups identified a discernible difference in how men and women understood assaults, with men believing rapes looked like a woman fighting back and screaming for help ... which is rarely what happens.
Notably absent among the interviewed are any flagrant perpetrators. The methodology section theorizes that individuals who intentionally harmed their peers were unlikely to choose to participate in this study. In addition, the characterization of assailants as “sociopathic predators” is based in a history of racialized imagery that leads us astray from the truth about campus sexual assault. Most assaults do not involve force, and SHIFT data showed 75% of victims knew their assailants. Ultimately, a major aim of the research was to study assault alongside healthy sex to “understand those pivotal moments when encounters change from being sex, to being assault.” Doing this requires understanding the where, how, and why students have sex, a more complicated undertaking than we may think.
In discussing their sexual lives, subjects frequently noted they did not have space to talk about their assaults. Though 81% of students discussed their experiences with someone, friend groups were often overburdened with stories, which minimized the victim’s experience. Furthermore, most had not sought help from the student counseling centers. Students navigating this complex field were frequently doing so in isolation. SHIFT found subjects to be eager to participate; they would often express thankfulness, and a sense of freedom in sharing with researchers. Commonly, students expressly did not want retribution for perpetrators, but simply a place to be heard without challenge. The current legal system precludes that possibility, leaving individuals without the option to confront perpetrators, and perpetrators often not knowing the extent of the damage they caused.
Where can psychiatrists have an impact right now? “Sexual Citizens” identifies four key areas for intervention to work toward a world with less sexual assault. These are:
- Improving diversity, inequality, and power distortions.
- Education about sex and sexual assault.
- Substance use.
- Mental health.
Substance use and mental health are especially relevant for psychiatrists (That substance use contributes to sexual assault is known by approximately ... everybody!). Unwanted sexual contact prior to college (20% of students) increased the odds of experiencing assault during college. Harm reduction strategies should be introduced before college, according to the SHIFT research, particularly in skills-based training on how to say “No” to unwanted sex. Psychiatrists are likely used to asking brief history questions related to sexual assault and rape. “Sexual Citizens” highlights the inadequacy of this blunt language and guides the reader toward a refined knowledge of the language needed to address sexual assault.
Dr. Whisler is a child and adolescent psychiatry fellow at the Stanford (Calif.) University. Dr. Higgins is affiliate associate professor of psychiatry and family medicine at the Medical University of South Carolina, Charleston.
References
1. Hirsch JS et al. Social dimensions of sexual consent among cisgender heterosexual college students: Insights from ethnographic research. J Adolesc Health. 2019 Jan;64(1):26-35. doi: 10.1016/j.jadohealth.2018.06.011.
2. Hirsch JS and Khan S. Sexual citizens: Sex, power, and assault on campus. New York: W.W. Norton & Company, 2020.
The Sexual Health Initiative to Foster Transformation (SHIFT)1 is a landmark study about sexual assault at college, which has generated 20 scientific articles and several chapters in books, but unfortunately, has not made its way into the psychiatric literature.
“Sexual Citizens: Sex, Power and Assault on Campus,” by Jennifer Hirsch and Shamus Khan, (available in audio book and paperback) was written as a follow up to the SHIFT study, so the rest of us can absorb the findings.2 This mixed-methods study included a survey of over 1,600 students aged 18-29 from Columbia University and Barnard College regarding their relationships and sexual histories, including assault. Data were collected using daily diaries, focus groups, and hundreds of hours of field work observation by young researchers. One- to 3-hour in-depth interviews exploring sexual experiences on campus were conducted with 151 students. These interviews are the focus of the book. It is a well-written, provocative story brimming with insights for those of us who lack the time to scour social science literature.
“Sexual Citizens” and the SHIFT study confirmed much of what we know. Sexual assault is common and has enduring effects. The study found that 36% of women and 15% of men had experienced unwanted, nonconsensual sexual contact by senior year. Twenty percent of women and 6% of men were rape survivors. Freshman, LGBTQ, and minority students were found at highest risk of assault. SHIFT reaffirmed that abstinence-only education is not a protective factor against college sexual assault, but neither was knowledge of affirmative consent (the practice of “ongoing and explicit” checking-in with partners) which few students ever employed. Encouragingly, students taught refusal skills were less likely to experience sexual assault.
Many of the book’s valuable lessons fall under the umbrella of failures of language and communication. For example, after drinking, they went to his room. She was expecting a social interaction, but with no other place to sit, they sat on his bed where she was coaxed or pressured into a sexual encounter. Afterward, she leaves, and it is never discussed again. One partner desires emotional intimacy, and the other, bragging rights in the fraternity or at the girls’ weekly brunch. Numerous personal stories like these, though at times heart wrenching, provide perspective on the barriers to addressing assault.
Subjects relayed experiences of assault by strangers or friends, and some provided details of their own actions as perpetrators. Stumbling around words and emotions, an avoidance of explicit language stemmed from shame, a fear of personal responsibility, the desire to maintain social cohesion, and concern for potential consequences for the perpetrator. Many subjects were resistant to calling nonconsensual sexual activity rape or even assault. Some who had perpetrated were unaware their behavior may have been experienced as assault, with recognition of this fact dawning during interviews.
This apparent limitation in self-reflective capacity may be in part due to the conceptualization of what assault is. Focus groups identified a discernible difference in how men and women understood assaults, with men believing rapes looked like a woman fighting back and screaming for help ... which is rarely what happens.
Notably absent among the interviewed are any flagrant perpetrators. The methodology section theorizes that individuals who intentionally harmed their peers were unlikely to choose to participate in this study. In addition, the characterization of assailants as “sociopathic predators” is based in a history of racialized imagery that leads us astray from the truth about campus sexual assault. Most assaults do not involve force, and SHIFT data showed 75% of victims knew their assailants. Ultimately, a major aim of the research was to study assault alongside healthy sex to “understand those pivotal moments when encounters change from being sex, to being assault.” Doing this requires understanding the where, how, and why students have sex, a more complicated undertaking than we may think.
In discussing their sexual lives, subjects frequently noted they did not have space to talk about their assaults. Though 81% of students discussed their experiences with someone, friend groups were often overburdened with stories, which minimized the victim’s experience. Furthermore, most had not sought help from the student counseling centers. Students navigating this complex field were frequently doing so in isolation. SHIFT found subjects to be eager to participate; they would often express thankfulness, and a sense of freedom in sharing with researchers. Commonly, students expressly did not want retribution for perpetrators, but simply a place to be heard without challenge. The current legal system precludes that possibility, leaving individuals without the option to confront perpetrators, and perpetrators often not knowing the extent of the damage they caused.
Where can psychiatrists have an impact right now? “Sexual Citizens” identifies four key areas for intervention to work toward a world with less sexual assault. These are:
- Improving diversity, inequality, and power distortions.
- Education about sex and sexual assault.
- Substance use.
- Mental health.
Substance use and mental health are especially relevant for psychiatrists (That substance use contributes to sexual assault is known by approximately ... everybody!). Unwanted sexual contact prior to college (20% of students) increased the odds of experiencing assault during college. Harm reduction strategies should be introduced before college, according to the SHIFT research, particularly in skills-based training on how to say “No” to unwanted sex. Psychiatrists are likely used to asking brief history questions related to sexual assault and rape. “Sexual Citizens” highlights the inadequacy of this blunt language and guides the reader toward a refined knowledge of the language needed to address sexual assault.
Dr. Whisler is a child and adolescent psychiatry fellow at the Stanford (Calif.) University. Dr. Higgins is affiliate associate professor of psychiatry and family medicine at the Medical University of South Carolina, Charleston.
References
1. Hirsch JS et al. Social dimensions of sexual consent among cisgender heterosexual college students: Insights from ethnographic research. J Adolesc Health. 2019 Jan;64(1):26-35. doi: 10.1016/j.jadohealth.2018.06.011.
2. Hirsch JS and Khan S. Sexual citizens: Sex, power, and assault on campus. New York: W.W. Norton & Company, 2020.
Do neural disconnects cause schizophrenia?
Advances in neuroimaging, cell biology, and post mortem analysis are starting to explain what happens in the brain of a person who develops schizophrenia. Schizophrenia appears to be a developmental disorder of disrupted neural connection within and between regions of the brain. These disruptions seem to result from genetic predispositions interacting with negative environmental events.
A matter of gray and white
Individuals with schizophrenia have deficits in gray matter and white matter, as illustrated by studies linking auditory hallucinations with brain regions associated with normal hearing (Box).
Gray matter. Magnetic resonance imaging (MRI) indicates that gray matter volume peaks in early adolescence and declines with age. The normal adolescent brain shrinks as inefficient neural connections are pruned away, a process that refines and matures gray matter. In individuals with schizophrenia, this reduction is more aggressive—perhaps because of excessive pruning—and occurs in the time frame when schizophrenia symptoms typically emerge.
Rapoport et al1 documented this process through sequential MRI scans in children with early-onset schizophrenia (mean age 14.5). Compared with age-matched healthy controls, youths with schizophrenia show greater and more rapid gray matter loss during late adolescence (Figure 1).2
Increased density. Reduced neuronal branching and spine formation also likely causes subtle reductions in gray matter volume (Figure 2). The resulting lack of dendritic connectivity may produce cognitive impairments and negative symptoms seen in schizophrenia.
Postmortem studies of gray matter cells show increased neuron density in patients with schizophrenia when compared with controls.3 Patients with schizophrenia have the same number of neurons as controls, but the neurons are more tightly packed because of reduced cell size, branching, and synapse formation.4
Research over the past decade has revealed schizophrenia to be a neurodegenerative disorder characterized by substantial brain tissue loss during first and subsequent psychotic episodes.5 Neuroimaging studies show that clinical and functional deterioration accompanies progressive loss of cortical gray matter volume and enlargement of cerebral ventricles. Thus, preventing relapses has come to be regarded as critical to long-term schizophrenia management.
Auditory hallucinations appear to emanate from the temporal lobe, the same brain region that processes external sound. Thus, it may be that patients experiencing hallucinations are misidentifying inner speech as coming from an outside source.
Using functional MRI to differentiate brain activity signals associated with hallucinating and nonhallucinating states, Dierks et al21 documented increased activity in auditory cortical gray matter during hallucinations in schizophrenia patients.
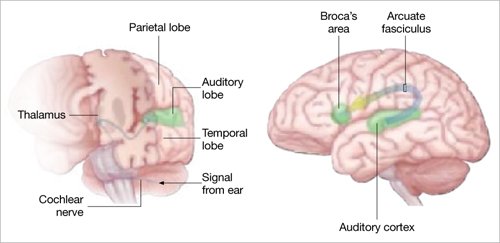
Auditory signals make synaptic connections in the thalamus (left) before reaching the auditory cortex. White matter fiber tracts called the arcuate fasciculus (right) connect the auditory cortex in the temporal lobe with Broca’s area in the frontal cortex.
Source: Adapted from reference 2
Using MR diffusion tensor imaging, Hubl et al22 identified white matter changes in the arcuate fasciculus of schizophrenia patients prone to hallucinations, compared with healthy controls and patients who had schizophrenia but not hallucinations.
These findings support the understanding that auditory hallucinations originate from altered connectivity of the same regions that process normal hearing and speech. The schizophrenia patient may perceive external voices from aberrant internal signals.
Figure 1 Rates of gray matter volume loss during adolescence
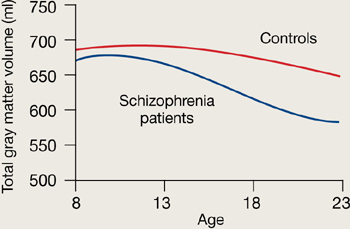
Youths with early-onset schizophrenia show greater gray matter volume loss during adolescence, compared with normal controls.
Source: Adapted from reference 2
Figure 2 Structural differences between neurons
in patients with schizophrenia and controls
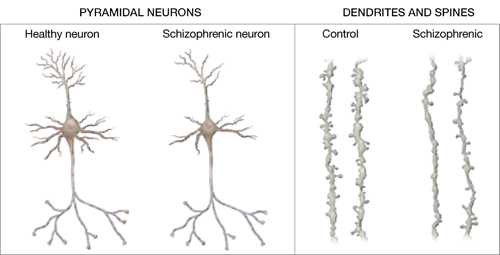
Schizophrenic neurons show reduced soma size, spine formation, and dendritic branching
Source: Adapted from reference 2White matter. Recent research suggests that white matter deficits also may be involved in schizophrenia’s pathophysiology. Studies using diffusion tensor imaging (DTI)—which measures the sum of vectors of water diffusion along axons—have documented white matter impairments in patients with schizophrenia.6
White matter tracks—myelinated axons that transport electrical signals among neurons—connect regions within the cortex and between the cortex and deeper brain structures. Disruption of white matter tracks may degrade signals and confuse neuronal communication.
Myelination. Genetic studies in patients with schizophrenia also have suggested that decreased neuron myelination may play a role in white matter deficits. Hakak et al8 examined more than 6,000 genes using microarray analysis and found only 17 genes were significantly down-regulated in patients with schizophrenia. Of those 17 genes, 6 were related to myelin and 11 showed no pattern.
Oligodendrocytes are glial cells that insulate axons with myelin and allow faster transmission of electrical impulses in the brain. In a postmortem study, Hof et al7 found 7 patients schizophrenia had 28% fewer oligodendrocytes per section of the superior frontal gyrus and 27% less white matter compared with 7 age-matched controls (Figure 3).
Figure 3 Reduced neuron myelination possible in schizophrenia
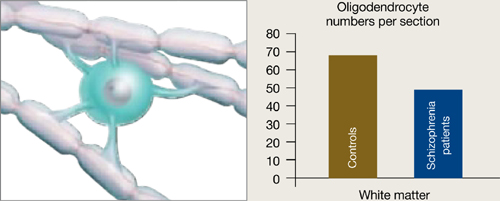
In a postmortem analysis, stained white matter sections taken from schizophrenia patients had fewer oligodendrocytes, cells that insulate axons with myelin and facilitate electrical transmission.
Source: Adapted from reference 2
Genes and the environment
Schizophrenia’s heritability is among the most repeated research findings in psychiatry.9 Other mechanisms besides genetics must be involved, however, as studies consistently show that monozygotic twins have a concordance rate of approximately 50% for the development of schizophrenia.
Environmental factors. Adverse environmental events may act in conjuction with genetic predisposition to trigger schizophrenia development. Ischemia or an impoverished diet, for example, have the potential to change DNA methylation.
Environmental factors associated with increased risk for schizophrenia include:
- maternal starvation during pregnancy10
- prenatal exposure to influenza11
- obstetrical complications with hypoxia12
- being born and raised in an urban environment13
- using marijuana during adolescence.14
Gene expression. Important genes may be silenced in individuals with increased DNA methylation and a susceptible genetic profile. Alterations in gene expression are the fundamental mechanism of behavioral change. Research shows that environmental events can alter gene expression without changing the genetic code, such as by adding methyl groups to DNA.15,16 The silencing of important developmental genes in this way can have devastating effects on development.
One explanation for the development of schizophrenia is that environmental events in susceptible individuals silence the production of proteins essential for maintaining neuronal connections through methylation of DNA. Postmortem analysis of brains of patients with schizophrenia show reduced mRNA of reelin,17 a protein produced in gamma-aminobutyric acid neurons involved in neuronal migration, axon branching, and synapse formation during brain development. Lowered production of proteins such as reelin may reduce connections between neurons and cause schizophrenia symptoms. Two research groups also have reported increased methylation of reelin DNA in postmortem studies of the brains of patients with schizophrenia.18,19 Increased methylation of DNA would silence production of this important protein.
Preventing neural disconnects? If schizophrenia is a developmental disorder resulting from failures in brain connectivity, then the ultimate treatment may be prevention. Recent research suggests that intervening with second-generation antipsychotics during the prodromal stage can prevent or delay the emergence of the disorder.20 Further research is needed to establish whether early intervention can prevent schizophrenia’s neuronal disruption.
1. Gogtay N, Sporn A, Rapoport J. Structural brain MRI studies in childhood-onset schizophrenia and childhood atypical psychosis. In: Lawrie S, Johnstone E, Weinberger D, eds. Schizophrenia: from neuroimaging to neuroscience. New York, NY: Oxford University Press; 2004.
2. Higgins ES, George MS. The neuroscience of clinical psychiatry. Philadelphia: Lippincott, Williams, and Wilkins; 2007.
3. Selemon LD. Increased cortical neuronal density in schizophrenia. Am J Psychiatry 2004;161(9):1564.-
4. Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 2000;57(1):65-73.
5. Csernansky JG. Neurodegeneration in schizophrenia: evidence from in vivo neuroimaging studies. Scientific World Journal 2007;7:135-43.
6. Kubicki M, McCarley R, Westin CF, et al. A review of diffusion tensor imaging studies in schizophrenia. J Psychiatr Res 2007;41(1-2):15-30.
7. Hof PR, Haroutunian V, Friedrich VL, Jr, et al. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol Psychiatry 2003;53(12):1075-85.
8. Hakak Y, Walker JR, Li C, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 2001;98(8):4746-51.
9. Shih RA, Belmonte PL, Zandi PP. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int Rev Psychiatr 2004;16(4):260-83.
10. McClellan JM, Susser E, King MC. Maternal famine, de novo mutations, and schizophrenia. JAMA 2006;296(5):582-4.
11. Limosin F, Rouillon F, Payan C, et al. Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta Psychiatr Scand 2003;107(5):331-5.
12. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 2002;159(7):1080-92.
13. Pedersen CB, Mortensen PB. Urbanization and traffic related exposures as risk factors for schizophrenia. BMC Psychiatry 2006;6:2.-
14. Arendt M, Rosenberg R, Foldager L, et al. Cannabis-induced psychosis and subsequent schizophrenia-spectrum disorders: follow-up study of 535 incident cases. Br J Psychiatry 2005;187:510-5.
15. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics 2003;33(suppl):245-54.
16. Abdolmaleky HM, Smith CL, Faraone SV, et al. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet 2004;127(1):51-9.
17. Fatemi SH, Stary JM, Earle JA, et al. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res 2005;72(2-3):109-22.
18. Abdolmaleky HM, Cheng KH, Russo A, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 2005;134(1):60-6.
19. Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA 2005;102(26):9341-6.
20. McGlashan TH, Zipursky RB, Perkins D, et al. Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am J Psychiatry 2006;163(5):790-799.
21. Dierks T, Linden DE, Jandl M, et al. Activation of Heschl’s gyrus during auditory hallucinations. Neuron 1999;22(3):615-21.
22. Hubl D, Koenig T, Strik W, et al. Pathways that make voices: white matter changes in auditory hallucinations. Arch Gen Psychiatry 2004;61(7):658-68.
Adapted from The neuroscience of clinical psychiatry, by Edmund S. Higgins and Mark S. George. Philadelphia: Lippincott, Williams, and Wilkins; 2007:251-63.
Advances in neuroimaging, cell biology, and post mortem analysis are starting to explain what happens in the brain of a person who develops schizophrenia. Schizophrenia appears to be a developmental disorder of disrupted neural connection within and between regions of the brain. These disruptions seem to result from genetic predispositions interacting with negative environmental events.
A matter of gray and white
Individuals with schizophrenia have deficits in gray matter and white matter, as illustrated by studies linking auditory hallucinations with brain regions associated with normal hearing (Box).
Gray matter. Magnetic resonance imaging (MRI) indicates that gray matter volume peaks in early adolescence and declines with age. The normal adolescent brain shrinks as inefficient neural connections are pruned away, a process that refines and matures gray matter. In individuals with schizophrenia, this reduction is more aggressive—perhaps because of excessive pruning—and occurs in the time frame when schizophrenia symptoms typically emerge.
Rapoport et al1 documented this process through sequential MRI scans in children with early-onset schizophrenia (mean age 14.5). Compared with age-matched healthy controls, youths with schizophrenia show greater and more rapid gray matter loss during late adolescence (Figure 1).2
Increased density. Reduced neuronal branching and spine formation also likely causes subtle reductions in gray matter volume (Figure 2). The resulting lack of dendritic connectivity may produce cognitive impairments and negative symptoms seen in schizophrenia.
Postmortem studies of gray matter cells show increased neuron density in patients with schizophrenia when compared with controls.3 Patients with schizophrenia have the same number of neurons as controls, but the neurons are more tightly packed because of reduced cell size, branching, and synapse formation.4
Research over the past decade has revealed schizophrenia to be a neurodegenerative disorder characterized by substantial brain tissue loss during first and subsequent psychotic episodes.5 Neuroimaging studies show that clinical and functional deterioration accompanies progressive loss of cortical gray matter volume and enlargement of cerebral ventricles. Thus, preventing relapses has come to be regarded as critical to long-term schizophrenia management.
Auditory hallucinations appear to emanate from the temporal lobe, the same brain region that processes external sound. Thus, it may be that patients experiencing hallucinations are misidentifying inner speech as coming from an outside source.
Using functional MRI to differentiate brain activity signals associated with hallucinating and nonhallucinating states, Dierks et al21 documented increased activity in auditory cortical gray matter during hallucinations in schizophrenia patients.
Auditory signals make synaptic connections in the thalamus (left) before reaching the auditory cortex. White matter fiber tracts called the arcuate fasciculus (right) connect the auditory cortex in the temporal lobe with Broca’s area in the frontal cortex.
Source: Adapted from reference 2
Using MR diffusion tensor imaging, Hubl et al22 identified white matter changes in the arcuate fasciculus of schizophrenia patients prone to hallucinations, compared with healthy controls and patients who had schizophrenia but not hallucinations.
These findings support the understanding that auditory hallucinations originate from altered connectivity of the same regions that process normal hearing and speech. The schizophrenia patient may perceive external voices from aberrant internal signals.
Figure 1 Rates of gray matter volume loss during adolescence
Youths with early-onset schizophrenia show greater gray matter volume loss during adolescence, compared with normal controls.
Source: Adapted from reference 2
Figure 2 Structural differences between neurons
in patients with schizophrenia and controls
Schizophrenic neurons show reduced soma size, spine formation, and dendritic branching
Source: Adapted from reference 2White matter. Recent research suggests that white matter deficits also may be involved in schizophrenia’s pathophysiology. Studies using diffusion tensor imaging (DTI)—which measures the sum of vectors of water diffusion along axons—have documented white matter impairments in patients with schizophrenia.6
White matter tracks—myelinated axons that transport electrical signals among neurons—connect regions within the cortex and between the cortex and deeper brain structures. Disruption of white matter tracks may degrade signals and confuse neuronal communication.
Myelination. Genetic studies in patients with schizophrenia also have suggested that decreased neuron myelination may play a role in white matter deficits. Hakak et al8 examined more than 6,000 genes using microarray analysis and found only 17 genes were significantly down-regulated in patients with schizophrenia. Of those 17 genes, 6 were related to myelin and 11 showed no pattern.
Oligodendrocytes are glial cells that insulate axons with myelin and allow faster transmission of electrical impulses in the brain. In a postmortem study, Hof et al7 found 7 patients schizophrenia had 28% fewer oligodendrocytes per section of the superior frontal gyrus and 27% less white matter compared with 7 age-matched controls (Figure 3).
Figure 3 Reduced neuron myelination possible in schizophrenia
In a postmortem analysis, stained white matter sections taken from schizophrenia patients had fewer oligodendrocytes, cells that insulate axons with myelin and facilitate electrical transmission.
Source: Adapted from reference 2
Genes and the environment
Schizophrenia’s heritability is among the most repeated research findings in psychiatry.9 Other mechanisms besides genetics must be involved, however, as studies consistently show that monozygotic twins have a concordance rate of approximately 50% for the development of schizophrenia.
Environmental factors. Adverse environmental events may act in conjuction with genetic predisposition to trigger schizophrenia development. Ischemia or an impoverished diet, for example, have the potential to change DNA methylation.
Environmental factors associated with increased risk for schizophrenia include:
- maternal starvation during pregnancy10
- prenatal exposure to influenza11
- obstetrical complications with hypoxia12
- being born and raised in an urban environment13
- using marijuana during adolescence.14
Gene expression. Important genes may be silenced in individuals with increased DNA methylation and a susceptible genetic profile. Alterations in gene expression are the fundamental mechanism of behavioral change. Research shows that environmental events can alter gene expression without changing the genetic code, such as by adding methyl groups to DNA.15,16 The silencing of important developmental genes in this way can have devastating effects on development.
One explanation for the development of schizophrenia is that environmental events in susceptible individuals silence the production of proteins essential for maintaining neuronal connections through methylation of DNA. Postmortem analysis of brains of patients with schizophrenia show reduced mRNA of reelin,17 a protein produced in gamma-aminobutyric acid neurons involved in neuronal migration, axon branching, and synapse formation during brain development. Lowered production of proteins such as reelin may reduce connections between neurons and cause schizophrenia symptoms. Two research groups also have reported increased methylation of reelin DNA in postmortem studies of the brains of patients with schizophrenia.18,19 Increased methylation of DNA would silence production of this important protein.
Preventing neural disconnects? If schizophrenia is a developmental disorder resulting from failures in brain connectivity, then the ultimate treatment may be prevention. Recent research suggests that intervening with second-generation antipsychotics during the prodromal stage can prevent or delay the emergence of the disorder.20 Further research is needed to establish whether early intervention can prevent schizophrenia’s neuronal disruption.
Advances in neuroimaging, cell biology, and post mortem analysis are starting to explain what happens in the brain of a person who develops schizophrenia. Schizophrenia appears to be a developmental disorder of disrupted neural connection within and between regions of the brain. These disruptions seem to result from genetic predispositions interacting with negative environmental events.
A matter of gray and white
Individuals with schizophrenia have deficits in gray matter and white matter, as illustrated by studies linking auditory hallucinations with brain regions associated with normal hearing (Box).
Gray matter. Magnetic resonance imaging (MRI) indicates that gray matter volume peaks in early adolescence and declines with age. The normal adolescent brain shrinks as inefficient neural connections are pruned away, a process that refines and matures gray matter. In individuals with schizophrenia, this reduction is more aggressive—perhaps because of excessive pruning—and occurs in the time frame when schizophrenia symptoms typically emerge.
Rapoport et al1 documented this process through sequential MRI scans in children with early-onset schizophrenia (mean age 14.5). Compared with age-matched healthy controls, youths with schizophrenia show greater and more rapid gray matter loss during late adolescence (Figure 1).2
Increased density. Reduced neuronal branching and spine formation also likely causes subtle reductions in gray matter volume (Figure 2). The resulting lack of dendritic connectivity may produce cognitive impairments and negative symptoms seen in schizophrenia.
Postmortem studies of gray matter cells show increased neuron density in patients with schizophrenia when compared with controls.3 Patients with schizophrenia have the same number of neurons as controls, but the neurons are more tightly packed because of reduced cell size, branching, and synapse formation.4
Research over the past decade has revealed schizophrenia to be a neurodegenerative disorder characterized by substantial brain tissue loss during first and subsequent psychotic episodes.5 Neuroimaging studies show that clinical and functional deterioration accompanies progressive loss of cortical gray matter volume and enlargement of cerebral ventricles. Thus, preventing relapses has come to be regarded as critical to long-term schizophrenia management.
Auditory hallucinations appear to emanate from the temporal lobe, the same brain region that processes external sound. Thus, it may be that patients experiencing hallucinations are misidentifying inner speech as coming from an outside source.
Using functional MRI to differentiate brain activity signals associated with hallucinating and nonhallucinating states, Dierks et al21 documented increased activity in auditory cortical gray matter during hallucinations in schizophrenia patients.
Auditory signals make synaptic connections in the thalamus (left) before reaching the auditory cortex. White matter fiber tracts called the arcuate fasciculus (right) connect the auditory cortex in the temporal lobe with Broca’s area in the frontal cortex.
Source: Adapted from reference 2
Using MR diffusion tensor imaging, Hubl et al22 identified white matter changes in the arcuate fasciculus of schizophrenia patients prone to hallucinations, compared with healthy controls and patients who had schizophrenia but not hallucinations.
These findings support the understanding that auditory hallucinations originate from altered connectivity of the same regions that process normal hearing and speech. The schizophrenia patient may perceive external voices from aberrant internal signals.
Figure 1 Rates of gray matter volume loss during adolescence
Youths with early-onset schizophrenia show greater gray matter volume loss during adolescence, compared with normal controls.
Source: Adapted from reference 2
Figure 2 Structural differences between neurons
in patients with schizophrenia and controls
Schizophrenic neurons show reduced soma size, spine formation, and dendritic branching
Source: Adapted from reference 2White matter. Recent research suggests that white matter deficits also may be involved in schizophrenia’s pathophysiology. Studies using diffusion tensor imaging (DTI)—which measures the sum of vectors of water diffusion along axons—have documented white matter impairments in patients with schizophrenia.6
White matter tracks—myelinated axons that transport electrical signals among neurons—connect regions within the cortex and between the cortex and deeper brain structures. Disruption of white matter tracks may degrade signals and confuse neuronal communication.
Myelination. Genetic studies in patients with schizophrenia also have suggested that decreased neuron myelination may play a role in white matter deficits. Hakak et al8 examined more than 6,000 genes using microarray analysis and found only 17 genes were significantly down-regulated in patients with schizophrenia. Of those 17 genes, 6 were related to myelin and 11 showed no pattern.
Oligodendrocytes are glial cells that insulate axons with myelin and allow faster transmission of electrical impulses in the brain. In a postmortem study, Hof et al7 found 7 patients schizophrenia had 28% fewer oligodendrocytes per section of the superior frontal gyrus and 27% less white matter compared with 7 age-matched controls (Figure 3).
Figure 3 Reduced neuron myelination possible in schizophrenia
In a postmortem analysis, stained white matter sections taken from schizophrenia patients had fewer oligodendrocytes, cells that insulate axons with myelin and facilitate electrical transmission.
Source: Adapted from reference 2
Genes and the environment
Schizophrenia’s heritability is among the most repeated research findings in psychiatry.9 Other mechanisms besides genetics must be involved, however, as studies consistently show that monozygotic twins have a concordance rate of approximately 50% for the development of schizophrenia.
Environmental factors. Adverse environmental events may act in conjuction with genetic predisposition to trigger schizophrenia development. Ischemia or an impoverished diet, for example, have the potential to change DNA methylation.
Environmental factors associated with increased risk for schizophrenia include:
- maternal starvation during pregnancy10
- prenatal exposure to influenza11
- obstetrical complications with hypoxia12
- being born and raised in an urban environment13
- using marijuana during adolescence.14
Gene expression. Important genes may be silenced in individuals with increased DNA methylation and a susceptible genetic profile. Alterations in gene expression are the fundamental mechanism of behavioral change. Research shows that environmental events can alter gene expression without changing the genetic code, such as by adding methyl groups to DNA.15,16 The silencing of important developmental genes in this way can have devastating effects on development.
One explanation for the development of schizophrenia is that environmental events in susceptible individuals silence the production of proteins essential for maintaining neuronal connections through methylation of DNA. Postmortem analysis of brains of patients with schizophrenia show reduced mRNA of reelin,17 a protein produced in gamma-aminobutyric acid neurons involved in neuronal migration, axon branching, and synapse formation during brain development. Lowered production of proteins such as reelin may reduce connections between neurons and cause schizophrenia symptoms. Two research groups also have reported increased methylation of reelin DNA in postmortem studies of the brains of patients with schizophrenia.18,19 Increased methylation of DNA would silence production of this important protein.
Preventing neural disconnects? If schizophrenia is a developmental disorder resulting from failures in brain connectivity, then the ultimate treatment may be prevention. Recent research suggests that intervening with second-generation antipsychotics during the prodromal stage can prevent or delay the emergence of the disorder.20 Further research is needed to establish whether early intervention can prevent schizophrenia’s neuronal disruption.
1. Gogtay N, Sporn A, Rapoport J. Structural brain MRI studies in childhood-onset schizophrenia and childhood atypical psychosis. In: Lawrie S, Johnstone E, Weinberger D, eds. Schizophrenia: from neuroimaging to neuroscience. New York, NY: Oxford University Press; 2004.
2. Higgins ES, George MS. The neuroscience of clinical psychiatry. Philadelphia: Lippincott, Williams, and Wilkins; 2007.
3. Selemon LD. Increased cortical neuronal density in schizophrenia. Am J Psychiatry 2004;161(9):1564.-
4. Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 2000;57(1):65-73.
5. Csernansky JG. Neurodegeneration in schizophrenia: evidence from in vivo neuroimaging studies. Scientific World Journal 2007;7:135-43.
6. Kubicki M, McCarley R, Westin CF, et al. A review of diffusion tensor imaging studies in schizophrenia. J Psychiatr Res 2007;41(1-2):15-30.
7. Hof PR, Haroutunian V, Friedrich VL, Jr, et al. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol Psychiatry 2003;53(12):1075-85.
8. Hakak Y, Walker JR, Li C, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 2001;98(8):4746-51.
9. Shih RA, Belmonte PL, Zandi PP. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int Rev Psychiatr 2004;16(4):260-83.
10. McClellan JM, Susser E, King MC. Maternal famine, de novo mutations, and schizophrenia. JAMA 2006;296(5):582-4.
11. Limosin F, Rouillon F, Payan C, et al. Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta Psychiatr Scand 2003;107(5):331-5.
12. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 2002;159(7):1080-92.
13. Pedersen CB, Mortensen PB. Urbanization and traffic related exposures as risk factors for schizophrenia. BMC Psychiatry 2006;6:2.-
14. Arendt M, Rosenberg R, Foldager L, et al. Cannabis-induced psychosis and subsequent schizophrenia-spectrum disorders: follow-up study of 535 incident cases. Br J Psychiatry 2005;187:510-5.
15. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics 2003;33(suppl):245-54.
16. Abdolmaleky HM, Smith CL, Faraone SV, et al. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet 2004;127(1):51-9.
17. Fatemi SH, Stary JM, Earle JA, et al. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res 2005;72(2-3):109-22.
18. Abdolmaleky HM, Cheng KH, Russo A, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 2005;134(1):60-6.
19. Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA 2005;102(26):9341-6.
20. McGlashan TH, Zipursky RB, Perkins D, et al. Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am J Psychiatry 2006;163(5):790-799.
21. Dierks T, Linden DE, Jandl M, et al. Activation of Heschl’s gyrus during auditory hallucinations. Neuron 1999;22(3):615-21.
22. Hubl D, Koenig T, Strik W, et al. Pathways that make voices: white matter changes in auditory hallucinations. Arch Gen Psychiatry 2004;61(7):658-68.
Adapted from The neuroscience of clinical psychiatry, by Edmund S. Higgins and Mark S. George. Philadelphia: Lippincott, Williams, and Wilkins; 2007:251-63.
1. Gogtay N, Sporn A, Rapoport J. Structural brain MRI studies in childhood-onset schizophrenia and childhood atypical psychosis. In: Lawrie S, Johnstone E, Weinberger D, eds. Schizophrenia: from neuroimaging to neuroscience. New York, NY: Oxford University Press; 2004.
2. Higgins ES, George MS. The neuroscience of clinical psychiatry. Philadelphia: Lippincott, Williams, and Wilkins; 2007.
3. Selemon LD. Increased cortical neuronal density in schizophrenia. Am J Psychiatry 2004;161(9):1564.-
4. Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 2000;57(1):65-73.
5. Csernansky JG. Neurodegeneration in schizophrenia: evidence from in vivo neuroimaging studies. Scientific World Journal 2007;7:135-43.
6. Kubicki M, McCarley R, Westin CF, et al. A review of diffusion tensor imaging studies in schizophrenia. J Psychiatr Res 2007;41(1-2):15-30.
7. Hof PR, Haroutunian V, Friedrich VL, Jr, et al. Loss and altered spatial distribution of oligodendrocytes in the superior frontal gyrus in schizophrenia. Biol Psychiatry 2003;53(12):1075-85.
8. Hakak Y, Walker JR, Li C, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci USA 2001;98(8):4746-51.
9. Shih RA, Belmonte PL, Zandi PP. A review of the evidence from family, twin and adoption studies for a genetic contribution to adult psychiatric disorders. Int Rev Psychiatr 2004;16(4):260-83.
10. McClellan JM, Susser E, King MC. Maternal famine, de novo mutations, and schizophrenia. JAMA 2006;296(5):582-4.
11. Limosin F, Rouillon F, Payan C, et al. Prenatal exposure to influenza as a risk factor for adult schizophrenia. Acta Psychiatr Scand 2003;107(5):331-5.
12. Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry 2002;159(7):1080-92.
13. Pedersen CB, Mortensen PB. Urbanization and traffic related exposures as risk factors for schizophrenia. BMC Psychiatry 2006;6:2.-
14. Arendt M, Rosenberg R, Foldager L, et al. Cannabis-induced psychosis and subsequent schizophrenia-spectrum disorders: follow-up study of 535 incident cases. Br J Psychiatry 2005;187:510-5.
15. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nature Genetics 2003;33(suppl):245-54.
16. Abdolmaleky HM, Smith CL, Faraone SV, et al. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet 2004;127(1):51-9.
17. Fatemi SH, Stary JM, Earle JA, et al. GABAergic dysfunction in schizophrenia and mood disorders as reflected by decreased levels of glutamic acid decarboxylase 65 and 67 kDa and Reelin proteins in cerebellum. Schizophr Res 2005;72(2-3):109-22.
18. Abdolmaleky HM, Cheng KH, Russo A, et al. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am J Med Genet B Neuropsychiatr Genet 2005;134(1):60-6.
19. Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA 2005;102(26):9341-6.
20. McGlashan TH, Zipursky RB, Perkins D, et al. Randomized, double-blind trial of olanzapine versus placebo in patients prodromally symptomatic for psychosis. Am J Psychiatry 2006;163(5):790-799.
21. Dierks T, Linden DE, Jandl M, et al. Activation of Heschl’s gyrus during auditory hallucinations. Neuron 1999;22(3):615-21.
22. Hubl D, Koenig T, Strik W, et al. Pathways that make voices: white matter changes in auditory hallucinations. Arch Gen Psychiatry 2004;61(7):658-68.
Adapted from The neuroscience of clinical psychiatry, by Edmund S. Higgins and Mark S. George. Philadelphia: Lippincott, Williams, and Wilkins; 2007:251-63.
How dopamine drives cocaine craving
Fighting cravings’ intense desire and obsessive thinking may be an addict’s most formidable challenge.1 Patients in recovery—desperate to stop abusing the substance—cannot control themselves after the craving is triggered. Remarkably, even after years of abstinence, cues reminding the addict of the substance—smells, sounds, or familiar surroundings—can ignite cravings and lead to relapse.
Dopamine and dope
A recent imaging study suggests that dopamine may be the culprit behind cravings. Research with cocaine and rodents suggests that dopamine released in the dorsal striatum is associated with drug-seeking behavior. Measuring craving in a rodent is impossible, but a recent imaging study examined how drug cues affect the brains of drug-addicted humans (Figure 1).2
Figure 1 Dopamine increase is associated with drug craving
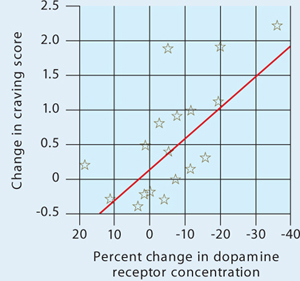
Changes in craving, as measured by Cocaine Craving Questionnaire scores, correlated with increased dopamine concentration in the putamen and caudate.
Source: Reference 2Volkow et al2 injected 18 cocaine-addicted patients with a dopamine D2 ligand that competes with endogenous dopamine and can be seen on positron emission tomography (PET). PET scans were then taken while each patient viewed a video of nature scenery (control) and then while watching scenes of drug preparation and simulated crack cocaine smoking.
When the control scan was subtracted from the cocaine-cued scan, the dorsal striatum—activated by the cocaine preparation cues—stood out (Figure 2), suggesting the neurobiological mechanisms responsible for craving.
The dorsal striatum is thought to be involved with selecting and initiating actions. In this study, the cocaine video caused a release of dopamine into the dorsal striatum and a desire for the drug. In an earlier study, hungry subjects who were shown food cues also showed increased dopamine activity in the dorsal striatum in association with a desire for food.3
Figure 2 Dopamine release in the dorsal striatum is linked with craving
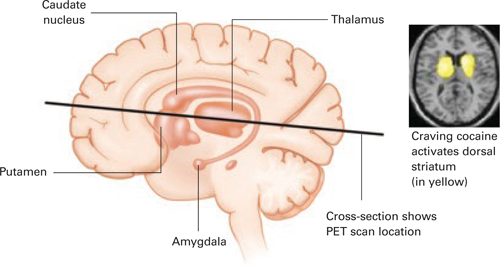
When cocaine-addicted patients watched a video depicting drug preparation and simulated crack cocaine smoking, PET scans of their brains showed dopamine release in the dorsal striatum.
Source: Reference 2Taken together, these studies suggest that dopamine in the dorsal striatum mediates craving for a desired object. The primary source of this neurotransmitter in the dorsal striatum is dopamine cells in the substantia nigra. The visual stimulus must activate these neurons in the substantia nigra to induce craving.
Caving into cravings
Desire precedes action and motivates behavior necessary for survival. Cocaine addiction apparently usurps the neurobiological mechanisms that motivate individuals to seek sustenance.
Developing an effective treatment for cocaine craving is a high priority at the National Institute on Drug Abuse.4 Medications including modafinil, propranolol, and disulfiram have been found effective for cocaine addiction in randomized, controlled trials, although none are FDA-approved for this use.4
One could speculate that antipsychotics—which are potent dopamine receptor blockers—might calm the cravings associated with cocaine addiction. Unfortunately, it is not that simple. Older antipsychotics might increase substance use in patients with schizophrenia and substance abuse.5 However, compelling evidence suggests that clozapine can reduce drug and alcohol use in dually diagnosed patients with schizophrenia.6 This provides some hope that the newer antipsychotic medications could provide a broad spectrum of pharmacologic activity that has the capacity to cool off cravings that stimulate drug-seeking behavior.
Drug brand names
- Clozapine • Clozaril
- Disulfiram • Antabuse
- Modafinil • Provigil
- Propranolol • Inderal
1. Weiss F. Neurobiology of craving, conditioned reward and relapse. Curr Opin Pharmacol 2005;5(1):9-19.
2. Volkow ND, Wang GJ, Telang F, et al. Cocaine cues and dopamine in dorsal striatum: mechanism of craving in cocaine addiction. J Neurosci 2006;26(24):6583-8.
3. Volkow ND, Wang GJ, Fowler JS, et al. “Nonhedonic” food motivation in humans involves dopamine in the dorsal striatum and methylphenidate amplifies this effect. Synapse 2002;44(3):175-80.
4. O’Brien CP. Anticraving medications for relapse prevention: a possible new class of psychoactive medications. Am J Psychiatry 2005;162(8):1423-31.
5. Green AI. Treatment of schizophrenia and comorbid substance abuse: pharmacologic approaches. J Clin Psychiatry 2006;67(suppl7):31-5.
6. Drake RE, Xie H, McHugo GJ, Green AI. The effects of clozapine on alcohol and drug use disorders among patients with schizophrenia. Schizophr Bull 2000;26(2):441-9.
Fighting cravings’ intense desire and obsessive thinking may be an addict’s most formidable challenge.1 Patients in recovery—desperate to stop abusing the substance—cannot control themselves after the craving is triggered. Remarkably, even after years of abstinence, cues reminding the addict of the substance—smells, sounds, or familiar surroundings—can ignite cravings and lead to relapse.
Dopamine and dope
A recent imaging study suggests that dopamine may be the culprit behind cravings. Research with cocaine and rodents suggests that dopamine released in the dorsal striatum is associated with drug-seeking behavior. Measuring craving in a rodent is impossible, but a recent imaging study examined how drug cues affect the brains of drug-addicted humans (Figure 1).2
Figure 1 Dopamine increase is associated with drug craving
Changes in craving, as measured by Cocaine Craving Questionnaire scores, correlated with increased dopamine concentration in the putamen and caudate.
Source: Reference 2Volkow et al2 injected 18 cocaine-addicted patients with a dopamine D2 ligand that competes with endogenous dopamine and can be seen on positron emission tomography (PET). PET scans were then taken while each patient viewed a video of nature scenery (control) and then while watching scenes of drug preparation and simulated crack cocaine smoking.
When the control scan was subtracted from the cocaine-cued scan, the dorsal striatum—activated by the cocaine preparation cues—stood out (Figure 2), suggesting the neurobiological mechanisms responsible for craving.
The dorsal striatum is thought to be involved with selecting and initiating actions. In this study, the cocaine video caused a release of dopamine into the dorsal striatum and a desire for the drug. In an earlier study, hungry subjects who were shown food cues also showed increased dopamine activity in the dorsal striatum in association with a desire for food.3
Figure 2 Dopamine release in the dorsal striatum is linked with craving
When cocaine-addicted patients watched a video depicting drug preparation and simulated crack cocaine smoking, PET scans of their brains showed dopamine release in the dorsal striatum.
Source: Reference 2Taken together, these studies suggest that dopamine in the dorsal striatum mediates craving for a desired object. The primary source of this neurotransmitter in the dorsal striatum is dopamine cells in the substantia nigra. The visual stimulus must activate these neurons in the substantia nigra to induce craving.
Caving into cravings
Desire precedes action and motivates behavior necessary for survival. Cocaine addiction apparently usurps the neurobiological mechanisms that motivate individuals to seek sustenance.
Developing an effective treatment for cocaine craving is a high priority at the National Institute on Drug Abuse.4 Medications including modafinil, propranolol, and disulfiram have been found effective for cocaine addiction in randomized, controlled trials, although none are FDA-approved for this use.4
One could speculate that antipsychotics—which are potent dopamine receptor blockers—might calm the cravings associated with cocaine addiction. Unfortunately, it is not that simple. Older antipsychotics might increase substance use in patients with schizophrenia and substance abuse.5 However, compelling evidence suggests that clozapine can reduce drug and alcohol use in dually diagnosed patients with schizophrenia.6 This provides some hope that the newer antipsychotic medications could provide a broad spectrum of pharmacologic activity that has the capacity to cool off cravings that stimulate drug-seeking behavior.
Drug brand names
- Clozapine • Clozaril
- Disulfiram • Antabuse
- Modafinil • Provigil
- Propranolol • Inderal
Fighting cravings’ intense desire and obsessive thinking may be an addict’s most formidable challenge.1 Patients in recovery—desperate to stop abusing the substance—cannot control themselves after the craving is triggered. Remarkably, even after years of abstinence, cues reminding the addict of the substance—smells, sounds, or familiar surroundings—can ignite cravings and lead to relapse.
Dopamine and dope
A recent imaging study suggests that dopamine may be the culprit behind cravings. Research with cocaine and rodents suggests that dopamine released in the dorsal striatum is associated with drug-seeking behavior. Measuring craving in a rodent is impossible, but a recent imaging study examined how drug cues affect the brains of drug-addicted humans (Figure 1).2
Figure 1 Dopamine increase is associated with drug craving
Changes in craving, as measured by Cocaine Craving Questionnaire scores, correlated with increased dopamine concentration in the putamen and caudate.
Source: Reference 2Volkow et al2 injected 18 cocaine-addicted patients with a dopamine D2 ligand that competes with endogenous dopamine and can be seen on positron emission tomography (PET). PET scans were then taken while each patient viewed a video of nature scenery (control) and then while watching scenes of drug preparation and simulated crack cocaine smoking.
When the control scan was subtracted from the cocaine-cued scan, the dorsal striatum—activated by the cocaine preparation cues—stood out (Figure 2), suggesting the neurobiological mechanisms responsible for craving.
The dorsal striatum is thought to be involved with selecting and initiating actions. In this study, the cocaine video caused a release of dopamine into the dorsal striatum and a desire for the drug. In an earlier study, hungry subjects who were shown food cues also showed increased dopamine activity in the dorsal striatum in association with a desire for food.3
Figure 2 Dopamine release in the dorsal striatum is linked with craving
When cocaine-addicted patients watched a video depicting drug preparation and simulated crack cocaine smoking, PET scans of their brains showed dopamine release in the dorsal striatum.
Source: Reference 2Taken together, these studies suggest that dopamine in the dorsal striatum mediates craving for a desired object. The primary source of this neurotransmitter in the dorsal striatum is dopamine cells in the substantia nigra. The visual stimulus must activate these neurons in the substantia nigra to induce craving.
Caving into cravings
Desire precedes action and motivates behavior necessary for survival. Cocaine addiction apparently usurps the neurobiological mechanisms that motivate individuals to seek sustenance.
Developing an effective treatment for cocaine craving is a high priority at the National Institute on Drug Abuse.4 Medications including modafinil, propranolol, and disulfiram have been found effective for cocaine addiction in randomized, controlled trials, although none are FDA-approved for this use.4
One could speculate that antipsychotics—which are potent dopamine receptor blockers—might calm the cravings associated with cocaine addiction. Unfortunately, it is not that simple. Older antipsychotics might increase substance use in patients with schizophrenia and substance abuse.5 However, compelling evidence suggests that clozapine can reduce drug and alcohol use in dually diagnosed patients with schizophrenia.6 This provides some hope that the newer antipsychotic medications could provide a broad spectrum of pharmacologic activity that has the capacity to cool off cravings that stimulate drug-seeking behavior.
Drug brand names
- Clozapine • Clozaril
- Disulfiram • Antabuse
- Modafinil • Provigil
- Propranolol • Inderal
1. Weiss F. Neurobiology of craving, conditioned reward and relapse. Curr Opin Pharmacol 2005;5(1):9-19.
2. Volkow ND, Wang GJ, Telang F, et al. Cocaine cues and dopamine in dorsal striatum: mechanism of craving in cocaine addiction. J Neurosci 2006;26(24):6583-8.
3. Volkow ND, Wang GJ, Fowler JS, et al. “Nonhedonic” food motivation in humans involves dopamine in the dorsal striatum and methylphenidate amplifies this effect. Synapse 2002;44(3):175-80.
4. O’Brien CP. Anticraving medications for relapse prevention: a possible new class of psychoactive medications. Am J Psychiatry 2005;162(8):1423-31.
5. Green AI. Treatment of schizophrenia and comorbid substance abuse: pharmacologic approaches. J Clin Psychiatry 2006;67(suppl7):31-5.
6. Drake RE, Xie H, McHugo GJ, Green AI. The effects of clozapine on alcohol and drug use disorders among patients with schizophrenia. Schizophr Bull 2000;26(2):441-9.
1. Weiss F. Neurobiology of craving, conditioned reward and relapse. Curr Opin Pharmacol 2005;5(1):9-19.
2. Volkow ND, Wang GJ, Telang F, et al. Cocaine cues and dopamine in dorsal striatum: mechanism of craving in cocaine addiction. J Neurosci 2006;26(24):6583-8.
3. Volkow ND, Wang GJ, Fowler JS, et al. “Nonhedonic” food motivation in humans involves dopamine in the dorsal striatum and methylphenidate amplifies this effect. Synapse 2002;44(3):175-80.
4. O’Brien CP. Anticraving medications for relapse prevention: a possible new class of psychoactive medications. Am J Psychiatry 2005;162(8):1423-31.
5. Green AI. Treatment of schizophrenia and comorbid substance abuse: pharmacologic approaches. J Clin Psychiatry 2006;67(suppl7):31-5.
6. Drake RE, Xie H, McHugo GJ, Green AI. The effects of clozapine on alcohol and drug use disorders among patients with schizophrenia. Schizophr Bull 2000;26(2):441-9.
Jump-starting depression treatment
The serendipitous discovery of medications that can improve mood transformed depression treatment more than 50 years ago.1 Most antidepressants produced since then could be called “me-too” medications because they all work by affecting the release of monoamines—serotonin, norepinephrine, and dopamine—which, in turn, modulate the activity of neurons that release glutamate.
Weeks may pass before antidepressants’ effect on monoamine-releasing neurons produces a therapeutic benefit, however, leaving many patients impaired or even suicidal. This delayed onset of action might be explained by antidepressants’ indirect blockade of glutamate. The route to more rapid results, therefore, might be to cut out the monoamine middlemen.
Glutamate clues
Glutamate, instead of monoamines, might offer a more direct means to affect mood and could be a new target for antidepressant treatment:2
- Positron-emission tomography of neuron function in depressed patients shows abnormal activity in neurons that release glutamate—so-called glutamate neurons.
- Approximately 60 % of neurons are glutamate neurons, the largest network of neurons in the brain.
- Increased glutamate activity is seen in depressed patients.
- Animal studies have shown that blocking the N-methyl-D-aspartate (NMDA) receptor—1 of 3 types of glutamate receptors—decreases depressive behavior.
- Chronic administration of antidepressant medication reduces the expression of NMDA receptors.
Rapid glutamate blockade
Prompted by evidence that glutamate may be involved in mood disorders, researchers at the National Institute of Mental Health designed a study to determine if blocking the NMDA receptor can produce a rapid antidepressant effect.3 They chose the agent ketamine for this study because of its potent affinity for the NMDA receptor.
Ketamine was developed in the 1960s as a general anaesthetic, and its use in the United States is limited almost exclusively to veterinary medicine. The drug’s propensity to induce perceptual disturbances limits its clinical use but enhances its illicit use.
Eighteen patients with treatment-resistant depression were enrolled in a randomized, placebo-controlled, double-blind, crossover study. After 2 weeks without medication, they received a single infusion of IV ketamine or placebo. One week later they received the alternate intervention. Changes in Hamilton Rating Scale for Depression scores were assessed after each infusion.
Figure Decrease in depressive symptoms with IV ketamine
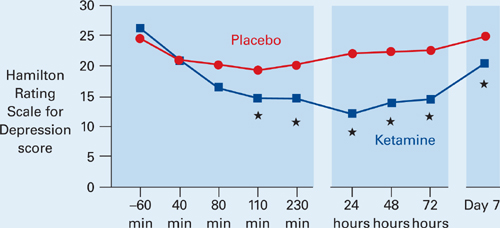
* = P <0.05
Patients who received ketamine (blue) showed a marked reduction of depressive symptoms within 2 hours compared with those who received placebo (red).
Source: Reference 3Patients receiving ketamine showed a robust and sustained reduction in depressive symptoms compared with placebo within 110 minutes (Figure). “To our knowledge,” the authors wrote, “there has never been a report of any other drug or somatic treatment—such as sleep deprivation, thyrotropin-releasing hormone, antidepressant, dexamethasone, or electroconvulsive therapy—that results in such a dramatic rapid and prolonged response with a single administration.”
Seventy-one percent of patients responded to IV ketamine within 24 hours, which is comparable to reported response rates after 8 weeks with oral antidepressants such as bupropion (62%), selective serotonin reuptake inhibitors (63%), and venlafaxine (65%).4,5
Caution and caveats
Despite these “dramatic” results, we must be cautious about extrapolating too much from this small study. Glutamate blockers such as ketamine can have serious adverse effects—including psychosis—and patients may not tolerate long-term interventions. Likewise, oral administration of memantine—another NMDA blocker—in a double-blind study did not effectively treat depression.6 Finally, subjects in the ketamine trial had chronic, treatment-resistant depression, and the results might not apply to other forms of depression.
The results suggest the possibility of a new option for depression treatment, however. Specifically, this option could expedite response and “jump-start” treatment through a novel mechanism so that persons with depression can get back on their feet more rapidly.
Drug brand names
- Bupropion • Wellbutrin
- Ketamine • Ketalar
- Memantine • Namenda
- Venlafaxine • Effexor
1. Higgins ES, George MS. Neuroscientific foundations of clinical psychiatry. Philadelphia, PA: Lippincott Williams and Wilkins; 2007. In press.
2. Kugaya A, Sanacora G. Beyond monoamines: glutamatergic function in mood disorders. CNS Spectrums 2005;10(10):808-19.
3. Zarate CA, Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006;63(8):856-64.
4. Thase ME, Haight BR, Richard N, et al. Remission rates following antidepressant therapy with bupropion or selective serotonin reuptake inhibitors: a meta-analysis of original data from 7 randomized controlled trials. J Clin Psychiatry 2005;66(8):974-81.
5. Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry 2001;62(11):869-77.
6. Zarate CA, Jr, Singh JB, Quiroz JA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry 2006;163(1):153-5.
The serendipitous discovery of medications that can improve mood transformed depression treatment more than 50 years ago.1 Most antidepressants produced since then could be called “me-too” medications because they all work by affecting the release of monoamines—serotonin, norepinephrine, and dopamine—which, in turn, modulate the activity of neurons that release glutamate.
Weeks may pass before antidepressants’ effect on monoamine-releasing neurons produces a therapeutic benefit, however, leaving many patients impaired or even suicidal. This delayed onset of action might be explained by antidepressants’ indirect blockade of glutamate. The route to more rapid results, therefore, might be to cut out the monoamine middlemen.
Glutamate clues
Glutamate, instead of monoamines, might offer a more direct means to affect mood and could be a new target for antidepressant treatment:2
- Positron-emission tomography of neuron function in depressed patients shows abnormal activity in neurons that release glutamate—so-called glutamate neurons.
- Approximately 60 % of neurons are glutamate neurons, the largest network of neurons in the brain.
- Increased glutamate activity is seen in depressed patients.
- Animal studies have shown that blocking the N-methyl-D-aspartate (NMDA) receptor—1 of 3 types of glutamate receptors—decreases depressive behavior.
- Chronic administration of antidepressant medication reduces the expression of NMDA receptors.
Rapid glutamate blockade
Prompted by evidence that glutamate may be involved in mood disorders, researchers at the National Institute of Mental Health designed a study to determine if blocking the NMDA receptor can produce a rapid antidepressant effect.3 They chose the agent ketamine for this study because of its potent affinity for the NMDA receptor.
Ketamine was developed in the 1960s as a general anaesthetic, and its use in the United States is limited almost exclusively to veterinary medicine. The drug’s propensity to induce perceptual disturbances limits its clinical use but enhances its illicit use.
Eighteen patients with treatment-resistant depression were enrolled in a randomized, placebo-controlled, double-blind, crossover study. After 2 weeks without medication, they received a single infusion of IV ketamine or placebo. One week later they received the alternate intervention. Changes in Hamilton Rating Scale for Depression scores were assessed after each infusion.
Figure Decrease in depressive symptoms with IV ketamine
* = P <0.05
Patients who received ketamine (blue) showed a marked reduction of depressive symptoms within 2 hours compared with those who received placebo (red).
Source: Reference 3Patients receiving ketamine showed a robust and sustained reduction in depressive symptoms compared with placebo within 110 minutes (Figure). “To our knowledge,” the authors wrote, “there has never been a report of any other drug or somatic treatment—such as sleep deprivation, thyrotropin-releasing hormone, antidepressant, dexamethasone, or electroconvulsive therapy—that results in such a dramatic rapid and prolonged response with a single administration.”
Seventy-one percent of patients responded to IV ketamine within 24 hours, which is comparable to reported response rates after 8 weeks with oral antidepressants such as bupropion (62%), selective serotonin reuptake inhibitors (63%), and venlafaxine (65%).4,5
Caution and caveats
Despite these “dramatic” results, we must be cautious about extrapolating too much from this small study. Glutamate blockers such as ketamine can have serious adverse effects—including psychosis—and patients may not tolerate long-term interventions. Likewise, oral administration of memantine—another NMDA blocker—in a double-blind study did not effectively treat depression.6 Finally, subjects in the ketamine trial had chronic, treatment-resistant depression, and the results might not apply to other forms of depression.
The results suggest the possibility of a new option for depression treatment, however. Specifically, this option could expedite response and “jump-start” treatment through a novel mechanism so that persons with depression can get back on their feet more rapidly.
Drug brand names
- Bupropion • Wellbutrin
- Ketamine • Ketalar
- Memantine • Namenda
- Venlafaxine • Effexor
The serendipitous discovery of medications that can improve mood transformed depression treatment more than 50 years ago.1 Most antidepressants produced since then could be called “me-too” medications because they all work by affecting the release of monoamines—serotonin, norepinephrine, and dopamine—which, in turn, modulate the activity of neurons that release glutamate.
Weeks may pass before antidepressants’ effect on monoamine-releasing neurons produces a therapeutic benefit, however, leaving many patients impaired or even suicidal. This delayed onset of action might be explained by antidepressants’ indirect blockade of glutamate. The route to more rapid results, therefore, might be to cut out the monoamine middlemen.
Glutamate clues
Glutamate, instead of monoamines, might offer a more direct means to affect mood and could be a new target for antidepressant treatment:2
- Positron-emission tomography of neuron function in depressed patients shows abnormal activity in neurons that release glutamate—so-called glutamate neurons.
- Approximately 60 % of neurons are glutamate neurons, the largest network of neurons in the brain.
- Increased glutamate activity is seen in depressed patients.
- Animal studies have shown that blocking the N-methyl-D-aspartate (NMDA) receptor—1 of 3 types of glutamate receptors—decreases depressive behavior.
- Chronic administration of antidepressant medication reduces the expression of NMDA receptors.
Rapid glutamate blockade
Prompted by evidence that glutamate may be involved in mood disorders, researchers at the National Institute of Mental Health designed a study to determine if blocking the NMDA receptor can produce a rapid antidepressant effect.3 They chose the agent ketamine for this study because of its potent affinity for the NMDA receptor.
Ketamine was developed in the 1960s as a general anaesthetic, and its use in the United States is limited almost exclusively to veterinary medicine. The drug’s propensity to induce perceptual disturbances limits its clinical use but enhances its illicit use.
Eighteen patients with treatment-resistant depression were enrolled in a randomized, placebo-controlled, double-blind, crossover study. After 2 weeks without medication, they received a single infusion of IV ketamine or placebo. One week later they received the alternate intervention. Changes in Hamilton Rating Scale for Depression scores were assessed after each infusion.
Figure Decrease in depressive symptoms with IV ketamine
* = P <0.05
Patients who received ketamine (blue) showed a marked reduction of depressive symptoms within 2 hours compared with those who received placebo (red).
Source: Reference 3Patients receiving ketamine showed a robust and sustained reduction in depressive symptoms compared with placebo within 110 minutes (Figure). “To our knowledge,” the authors wrote, “there has never been a report of any other drug or somatic treatment—such as sleep deprivation, thyrotropin-releasing hormone, antidepressant, dexamethasone, or electroconvulsive therapy—that results in such a dramatic rapid and prolonged response with a single administration.”
Seventy-one percent of patients responded to IV ketamine within 24 hours, which is comparable to reported response rates after 8 weeks with oral antidepressants such as bupropion (62%), selective serotonin reuptake inhibitors (63%), and venlafaxine (65%).4,5
Caution and caveats
Despite these “dramatic” results, we must be cautious about extrapolating too much from this small study. Glutamate blockers such as ketamine can have serious adverse effects—including psychosis—and patients may not tolerate long-term interventions. Likewise, oral administration of memantine—another NMDA blocker—in a double-blind study did not effectively treat depression.6 Finally, subjects in the ketamine trial had chronic, treatment-resistant depression, and the results might not apply to other forms of depression.
The results suggest the possibility of a new option for depression treatment, however. Specifically, this option could expedite response and “jump-start” treatment through a novel mechanism so that persons with depression can get back on their feet more rapidly.
Drug brand names
- Bupropion • Wellbutrin
- Ketamine • Ketalar
- Memantine • Namenda
- Venlafaxine • Effexor
1. Higgins ES, George MS. Neuroscientific foundations of clinical psychiatry. Philadelphia, PA: Lippincott Williams and Wilkins; 2007. In press.
2. Kugaya A, Sanacora G. Beyond monoamines: glutamatergic function in mood disorders. CNS Spectrums 2005;10(10):808-19.
3. Zarate CA, Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006;63(8):856-64.
4. Thase ME, Haight BR, Richard N, et al. Remission rates following antidepressant therapy with bupropion or selective serotonin reuptake inhibitors: a meta-analysis of original data from 7 randomized controlled trials. J Clin Psychiatry 2005;66(8):974-81.
5. Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry 2001;62(11):869-77.
6. Zarate CA, Jr, Singh JB, Quiroz JA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry 2006;163(1):153-5.
1. Higgins ES, George MS. Neuroscientific foundations of clinical psychiatry. Philadelphia, PA: Lippincott Williams and Wilkins; 2007. In press.
2. Kugaya A, Sanacora G. Beyond monoamines: glutamatergic function in mood disorders. CNS Spectrums 2005;10(10):808-19.
3. Zarate CA, Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006;63(8):856-64.
4. Thase ME, Haight BR, Richard N, et al. Remission rates following antidepressant therapy with bupropion or selective serotonin reuptake inhibitors: a meta-analysis of original data from 7 randomized controlled trials. J Clin Psychiatry 2005;66(8):974-81.
5. Entsuah AR, Huang H, Thase ME. Response and remission rates in different subpopulations with major depressive disorder administered venlafaxine, selective serotonin reuptake inhibitors, or placebo. J Clin Psychiatry 2001;62(11):869-77.
6. Zarate CA, Jr, Singh JB, Quiroz JA, et al. A double-blind, placebo-controlled study of memantine in the treatment of major depression. Am J Psychiatry 2006;163(1):153-5.
Brain workouts boost attention
Research over the past decade suggests that the brain, like muscles, might get stronger after a good workout. Some evidence suggests that patients with attention-deficit/hyperactivity disorder (ADHD) can improve focus and concentration with mental exercises.
Research has unveiled the brain’s remarkable capacity for structural change as the result of experience. Repeating a specific brain function enhances neural mechanisms by increasing synapse formation or generating new neurons.1 Increased use of neurons likely activates growth factor proteins that stimulate neural growth.
Healing the brain by exercising an impaired region is of great interest to physicians. For example, a group at Yale University showed that children with dyslexia who receive proper reading instruction read more fluently and show increased activity in the left hemisphere regions that decode words.2 Other research has shown that constraint-induced movement therapy—when a stroke patient is forced to use his or her impaired arm or leg by restraining the good one—can “reawaken” parts of the brain damaged by a stroke.3 Researchers also are studying whether intellectual exercises such as reading or learning new skills can forestall Alzheimer’s disease.
Neurofeedback basics
Can the brain’s malleability help children with ADHD? Imaging studies show that children with ADHD have structural and functional deficits in the prefrontal cortex, the area associated with attention. Stimulant medications prescribed for ADHD compensate for this defect by increasing dopaminergic neuron activity in the prefrontal cortex.
Neurofeedback—also called EEG biofeedback—also could alleviate ADHD symptoms. For 30 years, neurofeedback has been studied as an ADHD treatment with promising results.5 Improvement in attention, concentration, and working memory has been reported in up to 75% of cases in the literature, although randomized controlled trials have not been conducted.
Neurofeedback teaches an individual to regulate the electrical activity of his or her brain with mental exercises.4 EEG frequencies generally can be divided into four basic rhythms:
- beta rhythm is alert and focused
- alpha is relaxed
- theta is between awake and asleep
- delta is deep sleep (Figure 1).
The patient aims to spend more time in the alert beta rhythm and less time in the slower, more relaxed rhythms by thinking thoughts that generate the appropriate rhythm. More time spent in beta rhythm means better attention and concentration. A computer monitoring EEG frequencies helps the patient learn to regulate his or her brain rhythms.
Figure 1 EEG frequencies characterizing 4 basic brain rhythms
Adhd help
A University of Montreal research group recently completed an open, randomized, neurofeedback trial of 20 children ages 8 to 12 with ADHD who do not take psychostimulants or other medications for ADHD.6 Fifteen received neurofeedback therapy consisting of 40 one-hour sessions over 15 weeks. Five children who did not receive treatment served as the control group. All subjects took several neuropsychological tests measuring attention and hyperactivity before and after the study (Figure 2a). On average the neurofeedback group scored significantly higher on all measures of attention without side effects compared with the control group.
A comparison of functional brain imaging scans taken before and after the study showed increased anterior cingulate gyrus activity in the frontal cortex in the neurofeedback group but not the controls (Figure 2b). Subjects took the Counting Stroop Test—a mental exercise that involves the anterior cingulate gyrus—while in the scanner. The imaging studies were averaged within the groups, and the before and after scans were subtracted from each other. The increased activity translates into improved attention and concentration and decreased impulsivity, allowing children to perform better in school, get into less trouble, and have better relationships with parents.
Although we need more studies, this research suggests that neurofeedback might be a treatment option for patients with ADHD who cannot tolerate or do not wish to take medications.
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
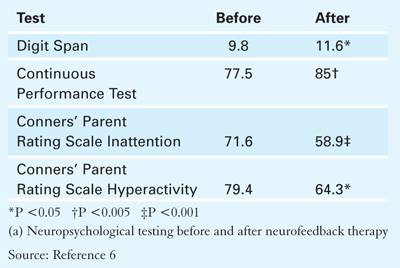
A. Neuropsychological testing
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
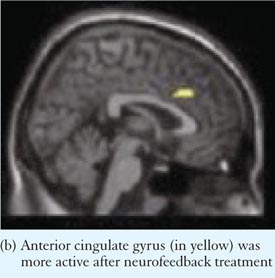
B. Imaging study
1. Gage FH. Brain, repair yourself. Sci Am 2003;289(3):46-53.
2. Shaywitz SE, Shaywitz BA. Dyslexia (specific reading disability). Biol Psychiatry 2005;57(11):1301-9.
3. Taub E, Uswatte G. Constraint-induced movement therapy: bridging from the primate laboratory to the stroke rehabilitation laboratory. J Rehabil Med 2003(41 suppl);34-40.
4. Kraft U. Train your brain. Sci Am Mind 2006;17(1):58-63.
5. Monastra VJ. Electroencephalographic biofeedback (neurotherapy) as a treatment for attention deficit hyperactivity disorder: rationale and empirical foundation. Child Adolesc Psychiatr Clin N Am 2005;14(1):55-82.
6. Levesque J, Beauregard M, Mensour B. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder: a functional magnetic resonance imaging study. Neurosci Lett 2006;394(3):216-21.
Research over the past decade suggests that the brain, like muscles, might get stronger after a good workout. Some evidence suggests that patients with attention-deficit/hyperactivity disorder (ADHD) can improve focus and concentration with mental exercises.
Research has unveiled the brain’s remarkable capacity for structural change as the result of experience. Repeating a specific brain function enhances neural mechanisms by increasing synapse formation or generating new neurons.1 Increased use of neurons likely activates growth factor proteins that stimulate neural growth.
Healing the brain by exercising an impaired region is of great interest to physicians. For example, a group at Yale University showed that children with dyslexia who receive proper reading instruction read more fluently and show increased activity in the left hemisphere regions that decode words.2 Other research has shown that constraint-induced movement therapy—when a stroke patient is forced to use his or her impaired arm or leg by restraining the good one—can “reawaken” parts of the brain damaged by a stroke.3 Researchers also are studying whether intellectual exercises such as reading or learning new skills can forestall Alzheimer’s disease.
Neurofeedback basics
Can the brain’s malleability help children with ADHD? Imaging studies show that children with ADHD have structural and functional deficits in the prefrontal cortex, the area associated with attention. Stimulant medications prescribed for ADHD compensate for this defect by increasing dopaminergic neuron activity in the prefrontal cortex.
Neurofeedback—also called EEG biofeedback—also could alleviate ADHD symptoms. For 30 years, neurofeedback has been studied as an ADHD treatment with promising results.5 Improvement in attention, concentration, and working memory has been reported in up to 75% of cases in the literature, although randomized controlled trials have not been conducted.
Neurofeedback teaches an individual to regulate the electrical activity of his or her brain with mental exercises.4 EEG frequencies generally can be divided into four basic rhythms:
- beta rhythm is alert and focused
- alpha is relaxed
- theta is between awake and asleep
- delta is deep sleep (Figure 1).
The patient aims to spend more time in the alert beta rhythm and less time in the slower, more relaxed rhythms by thinking thoughts that generate the appropriate rhythm. More time spent in beta rhythm means better attention and concentration. A computer monitoring EEG frequencies helps the patient learn to regulate his or her brain rhythms.
Figure 1 EEG frequencies characterizing 4 basic brain rhythms
Adhd help
A University of Montreal research group recently completed an open, randomized, neurofeedback trial of 20 children ages 8 to 12 with ADHD who do not take psychostimulants or other medications for ADHD.6 Fifteen received neurofeedback therapy consisting of 40 one-hour sessions over 15 weeks. Five children who did not receive treatment served as the control group. All subjects took several neuropsychological tests measuring attention and hyperactivity before and after the study (Figure 2a). On average the neurofeedback group scored significantly higher on all measures of attention without side effects compared with the control group.
A comparison of functional brain imaging scans taken before and after the study showed increased anterior cingulate gyrus activity in the frontal cortex in the neurofeedback group but not the controls (Figure 2b). Subjects took the Counting Stroop Test—a mental exercise that involves the anterior cingulate gyrus—while in the scanner. The imaging studies were averaged within the groups, and the before and after scans were subtracted from each other. The increased activity translates into improved attention and concentration and decreased impulsivity, allowing children to perform better in school, get into less trouble, and have better relationships with parents.
Although we need more studies, this research suggests that neurofeedback might be a treatment option for patients with ADHD who cannot tolerate or do not wish to take medications.
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
A. Neuropsychological testing
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
B. Imaging study
Research over the past decade suggests that the brain, like muscles, might get stronger after a good workout. Some evidence suggests that patients with attention-deficit/hyperactivity disorder (ADHD) can improve focus and concentration with mental exercises.
Research has unveiled the brain’s remarkable capacity for structural change as the result of experience. Repeating a specific brain function enhances neural mechanisms by increasing synapse formation or generating new neurons.1 Increased use of neurons likely activates growth factor proteins that stimulate neural growth.
Healing the brain by exercising an impaired region is of great interest to physicians. For example, a group at Yale University showed that children with dyslexia who receive proper reading instruction read more fluently and show increased activity in the left hemisphere regions that decode words.2 Other research has shown that constraint-induced movement therapy—when a stroke patient is forced to use his or her impaired arm or leg by restraining the good one—can “reawaken” parts of the brain damaged by a stroke.3 Researchers also are studying whether intellectual exercises such as reading or learning new skills can forestall Alzheimer’s disease.
Neurofeedback basics
Can the brain’s malleability help children with ADHD? Imaging studies show that children with ADHD have structural and functional deficits in the prefrontal cortex, the area associated with attention. Stimulant medications prescribed for ADHD compensate for this defect by increasing dopaminergic neuron activity in the prefrontal cortex.
Neurofeedback—also called EEG biofeedback—also could alleviate ADHD symptoms. For 30 years, neurofeedback has been studied as an ADHD treatment with promising results.5 Improvement in attention, concentration, and working memory has been reported in up to 75% of cases in the literature, although randomized controlled trials have not been conducted.
Neurofeedback teaches an individual to regulate the electrical activity of his or her brain with mental exercises.4 EEG frequencies generally can be divided into four basic rhythms:
- beta rhythm is alert and focused
- alpha is relaxed
- theta is between awake and asleep
- delta is deep sleep (Figure 1).
The patient aims to spend more time in the alert beta rhythm and less time in the slower, more relaxed rhythms by thinking thoughts that generate the appropriate rhythm. More time spent in beta rhythm means better attention and concentration. A computer monitoring EEG frequencies helps the patient learn to regulate his or her brain rhythms.
Figure 1 EEG frequencies characterizing 4 basic brain rhythms
Adhd help
A University of Montreal research group recently completed an open, randomized, neurofeedback trial of 20 children ages 8 to 12 with ADHD who do not take psychostimulants or other medications for ADHD.6 Fifteen received neurofeedback therapy consisting of 40 one-hour sessions over 15 weeks. Five children who did not receive treatment served as the control group. All subjects took several neuropsychological tests measuring attention and hyperactivity before and after the study (Figure 2a). On average the neurofeedback group scored significantly higher on all measures of attention without side effects compared with the control group.
A comparison of functional brain imaging scans taken before and after the study showed increased anterior cingulate gyrus activity in the frontal cortex in the neurofeedback group but not the controls (Figure 2b). Subjects took the Counting Stroop Test—a mental exercise that involves the anterior cingulate gyrus—while in the scanner. The imaging studies were averaged within the groups, and the before and after scans were subtracted from each other. The increased activity translates into improved attention and concentration and decreased impulsivity, allowing children to perform better in school, get into less trouble, and have better relationships with parents.
Although we need more studies, this research suggests that neurofeedback might be a treatment option for patients with ADHD who cannot tolerate or do not wish to take medications.
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
A. Neuropsychological testing
Figure 2 Neurofeedback training: Improvement in neuropsychological and imaging studies
B. Imaging study
1. Gage FH. Brain, repair yourself. Sci Am 2003;289(3):46-53.
2. Shaywitz SE, Shaywitz BA. Dyslexia (specific reading disability). Biol Psychiatry 2005;57(11):1301-9.
3. Taub E, Uswatte G. Constraint-induced movement therapy: bridging from the primate laboratory to the stroke rehabilitation laboratory. J Rehabil Med 2003(41 suppl);34-40.
4. Kraft U. Train your brain. Sci Am Mind 2006;17(1):58-63.
5. Monastra VJ. Electroencephalographic biofeedback (neurotherapy) as a treatment for attention deficit hyperactivity disorder: rationale and empirical foundation. Child Adolesc Psychiatr Clin N Am 2005;14(1):55-82.
6. Levesque J, Beauregard M, Mensour B. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder: a functional magnetic resonance imaging study. Neurosci Lett 2006;394(3):216-21.
1. Gage FH. Brain, repair yourself. Sci Am 2003;289(3):46-53.
2. Shaywitz SE, Shaywitz BA. Dyslexia (specific reading disability). Biol Psychiatry 2005;57(11):1301-9.
3. Taub E, Uswatte G. Constraint-induced movement therapy: bridging from the primate laboratory to the stroke rehabilitation laboratory. J Rehabil Med 2003(41 suppl);34-40.
4. Kraft U. Train your brain. Sci Am Mind 2006;17(1):58-63.
5. Monastra VJ. Electroencephalographic biofeedback (neurotherapy) as a treatment for attention deficit hyperactivity disorder: rationale and empirical foundation. Child Adolesc Psychiatr Clin N Am 2005;14(1):55-82.
6. Levesque J, Beauregard M, Mensour B. Effect of neurofeedback training on the neural substrates of selective attention in children with attention-deficit/hyperactivity disorder: a functional magnetic resonance imaging study. Neurosci Lett 2006;394(3):216-21.
Learning from lab-rat love
What’s a woman to do when sexual encounters become difficult or less satisfying?
- Vascularly directed sexual dysfunction treatments for men are not the answer; phosphodiesterase inhibitors such as sildenafil did no better than placebo among 800 women with various sexual problems.1
- The testosterone patch increases sexual interest for some women but failed to win FDA approval because of long-term safety concerns.
Even more frustrating, libido loss is a common side effect with some widely prescribed psychotropics—such as selective serotonin reuptake inhibitors. But an agent shown to boost libido in female rats may give women with sexual dysfunction a pharmaceutical option.
From sunscreen to aphrodisiac
Approximately 10 years ago, University of Arizona researchers studied a melanocortin-stimulating hormone analogue while trying to develop a product to allow light-skinned persons to tan without ultraviolet ray exposure. The product indeed darkened skin, but it also triggered spontaneous erections in all 3 men in the pilot study.2 A modified version of the erection-inducing peptide—now called bremelanotide—is being tested in phase 3 clinical trials as a prospective erectile dysfunction treatment.3
Unlike its predecessors, bremelanotide works directly on neural pathways that control sexual function, rather than on the vascular system. But its highest-interest feature may be its effect on female sexual desire—at least in rats.
Behavioral neurologist James Pfaus, PhD, identified behaviors female rats use to arouse males—what might be called the rodent equivalent of flirtation. These behaviors include solicitations (meeting head to head with males, then abruptly fleeing), pacing, hops, and darts.
His group then tested the effect of various doses of subcutaneous bremelanotide on 40 ovariectomized female rats primed with estradiol and progesterone.4 Researchers paired each female with a male for 30 minutes and recorded their behaviors. Compared with females who received placebo or low-dose bremelanotide, high-dose bremelanotide females performed significantly more “flirtatious” behaviors (Figure). The researchers attributed this difference to increased sexual desire.
The drug’s mechanism of action remains in question, but it appears to act centrally. Injecting it directly into female rats’ ventricles produced similar behaviors when they were paired with receptive males. Also, markers of neuronal activity have been found in the anterior aspect of the hypothalamus and nucleus accumbens—areas associated with sexual activity and pleasure.
FigureBremelanotide increases sexually solicitous behaviors in female rats
Source: Reference 4. Reprinted with permission. Copyright 2004, National Academy of Sciences, USA.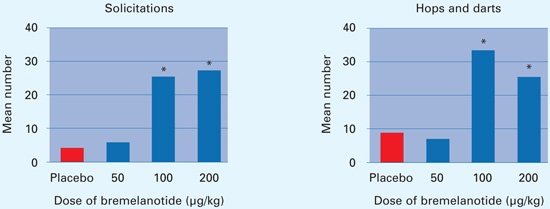
Pilot study in women
A randomized, double-blind, placebo-controlled, crossover trial suggests that bremelanotide as a nasal spray might increase desire in women with female sexual dysfunction (FSD).5 When 18 perimenopausal women with FSD were given bremelanotide or placebo, the bremelanotide group reported greater sexual desire and genital arousal while watching a sexually explicit video.
Specifically, this pilot study—part of the drug’s phase 2 trials in women—found that:
- 72% of women who took bremelanotide reported feelings of genital arousal, compared with 39% in the placebo group
- 67% of treated women experienced sexual desire versus 22% of controls.
Is fsd a pathology?
Some clinicians worry that FSD is a “disease” made up by pharmaceutical industry marketers. They argue that hypoactive sexual desire is not a disorder but an adaptive response to physical changes or relationship difficulties.6
On the other hand, a national survey of 1,749 women and 1,410 men ages 18 to 59 found sexual difficulties to be more prevalent in women than in men:7
- 43% of women reported sexual dysfunction, compared with 31% of men.
- 26% of women reported inability to achieve orgasm compared with 8% of men.
An effective and safe medication targeted at improving women’s sexual desire might help some patients, such as peri- or postmenopausal women, in otherwise healthy relationships.
1. Basson R, McInnes R, Smith MD, et al. Efficacy and safety of sildenafil citrate in women with sexual dysfunction associated with female sexual arousal disorder. J Womens Health Gen Based Med 2002;11(4):367-77.
2. Dorr RT, Lines R, Levine N, et al. Evaluation of melanotan-II, a superpotent cyclic melanotropic peptide in a pilot phase-I clinical study. Life Sci 1996;58(20):1777-84.
3. Molinoff PB, Shadiack AM, Earle D, et al. PT-141: A melanocortin agonist for the treatment of sexual dysfunction. Ann NY Acad Sci 2003;994:96-102.
4. Pfaus JG, Shadiack A, Van Soest T, et al. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci USA 2004;101(27):10201-4.
5. Perelman MA, Diamond LE, Earle DC, et al. The potential role of bremelanotide (PT-141) as a pharmacologic intervention for FSD. Poster presented at: International Society for the Study of Women’s Sexual Health annual meeting; March 10, 2006; Lisbon, Portugal.
6. Moynihan R. The marketing of a disease: female sexual dysfunction. BMJ 2005;330(7484):192-4.
7. Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999;281(6):537-44.
What’s a woman to do when sexual encounters become difficult or less satisfying?
- Vascularly directed sexual dysfunction treatments for men are not the answer; phosphodiesterase inhibitors such as sildenafil did no better than placebo among 800 women with various sexual problems.1
- The testosterone patch increases sexual interest for some women but failed to win FDA approval because of long-term safety concerns.
Even more frustrating, libido loss is a common side effect with some widely prescribed psychotropics—such as selective serotonin reuptake inhibitors. But an agent shown to boost libido in female rats may give women with sexual dysfunction a pharmaceutical option.
From sunscreen to aphrodisiac
Approximately 10 years ago, University of Arizona researchers studied a melanocortin-stimulating hormone analogue while trying to develop a product to allow light-skinned persons to tan without ultraviolet ray exposure. The product indeed darkened skin, but it also triggered spontaneous erections in all 3 men in the pilot study.2 A modified version of the erection-inducing peptide—now called bremelanotide—is being tested in phase 3 clinical trials as a prospective erectile dysfunction treatment.3
Unlike its predecessors, bremelanotide works directly on neural pathways that control sexual function, rather than on the vascular system. But its highest-interest feature may be its effect on female sexual desire—at least in rats.
Behavioral neurologist James Pfaus, PhD, identified behaviors female rats use to arouse males—what might be called the rodent equivalent of flirtation. These behaviors include solicitations (meeting head to head with males, then abruptly fleeing), pacing, hops, and darts.
His group then tested the effect of various doses of subcutaneous bremelanotide on 40 ovariectomized female rats primed with estradiol and progesterone.4 Researchers paired each female with a male for 30 minutes and recorded their behaviors. Compared with females who received placebo or low-dose bremelanotide, high-dose bremelanotide females performed significantly more “flirtatious” behaviors (Figure). The researchers attributed this difference to increased sexual desire.
The drug’s mechanism of action remains in question, but it appears to act centrally. Injecting it directly into female rats’ ventricles produced similar behaviors when they were paired with receptive males. Also, markers of neuronal activity have been found in the anterior aspect of the hypothalamus and nucleus accumbens—areas associated with sexual activity and pleasure.
FigureBremelanotide increases sexually solicitous behaviors in female rats
Source: Reference 4. Reprinted with permission. Copyright 2004, National Academy of Sciences, USA.
Pilot study in women
A randomized, double-blind, placebo-controlled, crossover trial suggests that bremelanotide as a nasal spray might increase desire in women with female sexual dysfunction (FSD).5 When 18 perimenopausal women with FSD were given bremelanotide or placebo, the bremelanotide group reported greater sexual desire and genital arousal while watching a sexually explicit video.
Specifically, this pilot study—part of the drug’s phase 2 trials in women—found that:
- 72% of women who took bremelanotide reported feelings of genital arousal, compared with 39% in the placebo group
- 67% of treated women experienced sexual desire versus 22% of controls.
Is fsd a pathology?
Some clinicians worry that FSD is a “disease” made up by pharmaceutical industry marketers. They argue that hypoactive sexual desire is not a disorder but an adaptive response to physical changes or relationship difficulties.6
On the other hand, a national survey of 1,749 women and 1,410 men ages 18 to 59 found sexual difficulties to be more prevalent in women than in men:7
- 43% of women reported sexual dysfunction, compared with 31% of men.
- 26% of women reported inability to achieve orgasm compared with 8% of men.
An effective and safe medication targeted at improving women’s sexual desire might help some patients, such as peri- or postmenopausal women, in otherwise healthy relationships.
What’s a woman to do when sexual encounters become difficult or less satisfying?
- Vascularly directed sexual dysfunction treatments for men are not the answer; phosphodiesterase inhibitors such as sildenafil did no better than placebo among 800 women with various sexual problems.1
- The testosterone patch increases sexual interest for some women but failed to win FDA approval because of long-term safety concerns.
Even more frustrating, libido loss is a common side effect with some widely prescribed psychotropics—such as selective serotonin reuptake inhibitors. But an agent shown to boost libido in female rats may give women with sexual dysfunction a pharmaceutical option.
From sunscreen to aphrodisiac
Approximately 10 years ago, University of Arizona researchers studied a melanocortin-stimulating hormone analogue while trying to develop a product to allow light-skinned persons to tan without ultraviolet ray exposure. The product indeed darkened skin, but it also triggered spontaneous erections in all 3 men in the pilot study.2 A modified version of the erection-inducing peptide—now called bremelanotide—is being tested in phase 3 clinical trials as a prospective erectile dysfunction treatment.3
Unlike its predecessors, bremelanotide works directly on neural pathways that control sexual function, rather than on the vascular system. But its highest-interest feature may be its effect on female sexual desire—at least in rats.
Behavioral neurologist James Pfaus, PhD, identified behaviors female rats use to arouse males—what might be called the rodent equivalent of flirtation. These behaviors include solicitations (meeting head to head with males, then abruptly fleeing), pacing, hops, and darts.
His group then tested the effect of various doses of subcutaneous bremelanotide on 40 ovariectomized female rats primed with estradiol and progesterone.4 Researchers paired each female with a male for 30 minutes and recorded their behaviors. Compared with females who received placebo or low-dose bremelanotide, high-dose bremelanotide females performed significantly more “flirtatious” behaviors (Figure). The researchers attributed this difference to increased sexual desire.
The drug’s mechanism of action remains in question, but it appears to act centrally. Injecting it directly into female rats’ ventricles produced similar behaviors when they were paired with receptive males. Also, markers of neuronal activity have been found in the anterior aspect of the hypothalamus and nucleus accumbens—areas associated with sexual activity and pleasure.
FigureBremelanotide increases sexually solicitous behaviors in female rats
Source: Reference 4. Reprinted with permission. Copyright 2004, National Academy of Sciences, USA.
Pilot study in women
A randomized, double-blind, placebo-controlled, crossover trial suggests that bremelanotide as a nasal spray might increase desire in women with female sexual dysfunction (FSD).5 When 18 perimenopausal women with FSD were given bremelanotide or placebo, the bremelanotide group reported greater sexual desire and genital arousal while watching a sexually explicit video.
Specifically, this pilot study—part of the drug’s phase 2 trials in women—found that:
- 72% of women who took bremelanotide reported feelings of genital arousal, compared with 39% in the placebo group
- 67% of treated women experienced sexual desire versus 22% of controls.
Is fsd a pathology?
Some clinicians worry that FSD is a “disease” made up by pharmaceutical industry marketers. They argue that hypoactive sexual desire is not a disorder but an adaptive response to physical changes or relationship difficulties.6
On the other hand, a national survey of 1,749 women and 1,410 men ages 18 to 59 found sexual difficulties to be more prevalent in women than in men:7
- 43% of women reported sexual dysfunction, compared with 31% of men.
- 26% of women reported inability to achieve orgasm compared with 8% of men.
An effective and safe medication targeted at improving women’s sexual desire might help some patients, such as peri- or postmenopausal women, in otherwise healthy relationships.
1. Basson R, McInnes R, Smith MD, et al. Efficacy and safety of sildenafil citrate in women with sexual dysfunction associated with female sexual arousal disorder. J Womens Health Gen Based Med 2002;11(4):367-77.
2. Dorr RT, Lines R, Levine N, et al. Evaluation of melanotan-II, a superpotent cyclic melanotropic peptide in a pilot phase-I clinical study. Life Sci 1996;58(20):1777-84.
3. Molinoff PB, Shadiack AM, Earle D, et al. PT-141: A melanocortin agonist for the treatment of sexual dysfunction. Ann NY Acad Sci 2003;994:96-102.
4. Pfaus JG, Shadiack A, Van Soest T, et al. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci USA 2004;101(27):10201-4.
5. Perelman MA, Diamond LE, Earle DC, et al. The potential role of bremelanotide (PT-141) as a pharmacologic intervention for FSD. Poster presented at: International Society for the Study of Women’s Sexual Health annual meeting; March 10, 2006; Lisbon, Portugal.
6. Moynihan R. The marketing of a disease: female sexual dysfunction. BMJ 2005;330(7484):192-4.
7. Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999;281(6):537-44.
1. Basson R, McInnes R, Smith MD, et al. Efficacy and safety of sildenafil citrate in women with sexual dysfunction associated with female sexual arousal disorder. J Womens Health Gen Based Med 2002;11(4):367-77.
2. Dorr RT, Lines R, Levine N, et al. Evaluation of melanotan-II, a superpotent cyclic melanotropic peptide in a pilot phase-I clinical study. Life Sci 1996;58(20):1777-84.
3. Molinoff PB, Shadiack AM, Earle D, et al. PT-141: A melanocortin agonist for the treatment of sexual dysfunction. Ann NY Acad Sci 2003;994:96-102.
4. Pfaus JG, Shadiack A, Van Soest T, et al. Selective facilitation of sexual solicitation in the female rat by a melanocortin receptor agonist. Proc Natl Acad Sci USA 2004;101(27):10201-4.
5. Perelman MA, Diamond LE, Earle DC, et al. The potential role of bremelanotide (PT-141) as a pharmacologic intervention for FSD. Poster presented at: International Society for the Study of Women’s Sexual Health annual meeting; March 10, 2006; Lisbon, Portugal.
6. Moynihan R. The marketing of a disease: female sexual dysfunction. BMJ 2005;330(7484):192-4.
7. Laumann EO, Paik A, Rosen RC. Sexual dysfunction in the United States: prevalence and predictors. JAMA 1999;281(6):537-44.
Glutamatergic TB drug ‘cools off’ anxiety disorders
Think of anxiety disorders as an overactive brain alarm that psychotropics and exposure therapy quiet via separate mechanisms. Psychotropics “cool off” the alarm by curtailing excitability of the amygdala, brainstem nuclei, and hypothalamus.1 Psychological treatments, particularly exposure therapy, seek to teach the brain not to fear the dreaded object.2
One would assume that combining medication and exposure therapy for anxiety would be beneficial, but results have been disappointing.3 Anxiolytics do not enhance—and many impede—learning that occurs during psychotherapy. When the medications are tapered, patients who receive psychotherapy plus placebo typically experience more-enduring benefit than those receiving psychotherapy plus active medication.4-6
An unlikely candidate—a glutamatergic tuberculosis (TB) drug—may offer a solution. The drug and others in its class may potentiate psychotherapy’s effects by enhancing learning rather than relief.7
Glutamate/learning link
Glutamate neurons have 3 types of glutamatergic receptors, with the NMDA and AMPA types perceived as most important because of their possible role in memory development. Creating new memories may involve strengthening signals between glutamate neurons. The exact cellular mechanism is unknown, but it may involve greater release of neurotransmitters or formation of new synapses.
Stronger signaling—and hence learning—may depend on opening the NMDA receptor to enhance postsynaptic potential.8 Opening both NMDA and AMPA receptors generates a stronger signal and allows calcium influx, compared with opening the AMPA receptor alone (Figure). This combination can activate genes that control protein synthesis and result in structural changes necessary for developing long-term memories.
D-cycloserine—a partial agonist at the NMDA receptor—is usually used as an antibiotic to treat TB. The drug also has been shown to enhance the learning process that underlies fear extinction in rats. A group at Emory University studied the effect of adding the medication to exposure therapy in humans with acrophobia.7
FigureNMDA receptor agonists may enhance learning in psychotherapy
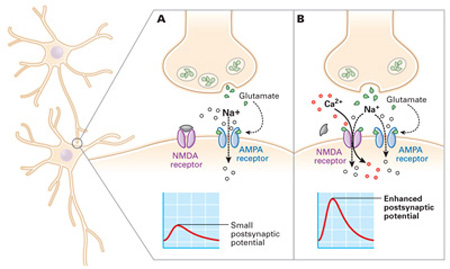
Normally, only glutamate neurons’ AMPA receptors activate in response to glutamate release (A). Both AMPA and NMDA receptors open in response to NMDA receptor agonists like D-cycloserine (B). Consequent stronger signaling may improve memory.
Going ‘up’
The researchers developed a virtual reality exposure system in which participants felt as if they were standing in an elevator, watching the floors recede as they rose 19 stories. Exposure therapy—seven weekly 35- to 45-minute sessions—has been shown to reduce the fear patients with acrophobia experience in virtual elevators.
Of 27 subjects, 10 received placebo and 17 received D-cycloserine, 50 mg or 500 mg. Subjects took their pills 2 to 4 hours before an exposure session. All participants experienced 2 virtual exposure sessions 1 to 2 weeks apart, which is considered suboptimal treatment for acrophobia.
Three months after the study, the D-cycloserine groups showed a markedly reduced fear of heights on the virtual elevator, while the placebo group showed no change from baseline. Fear levels were measured by subjective report of discomfort at each “floor.” D-cycloserine subjects also reported significantly greater reductions in measures of acrophobia in their daily lives.
Interestingly, both the D-cycloserine and placebo groups were equally frightened during virtual reality exposure. Only later did the D-cycloserine groups report less fear when exposed to heights, indicating that D-cycloserine enhanced learning that occurred during exposure therapy.
It’s exciting to think that medications could enhance and accelerate healing by activating the appropriate receptors during psychotherapy and give patients enduring benefits without the need for continued treatment.
If shown to be effective in larger studies, glutamatergic medications plus psychotherapy could provide more effective therapy for anxiety disorder. This approach is reported to be under investigation for treating anorexia nervosa, social phobia, panic disorder, and obsessive-compulsive disorder.9
1. Lydiard RB. Break the ‘fear circuit’ in resistant panic disorder. Current Psychiatry 2003;2(11):12-22.
2. Tynes LL, Tynes SF. Panic attacks: help sufferers recover with cognitive-behavioral therapy. Current Psychiatry 2005;4(11):51-60.
3. Otto MW, Smits JAJ, Reese HE. Combined psychotherapy and pharmacotherapy for mood and anxiety disorders in adults: review and analysis. Clinical Psychology: Science & Practice 2005;12(1):72-86.
4. Barlow DH, Gorman JM, Shear MK, Woods SW. Cognitive-behavioral therapy, imipramine, or their combination for panic disorder: A randomized controlled trial. JAMA 2000;283(19):2529-36.
5. Haug TT, Blomhoff S, Hellstrom K, et al. Exposure therapy and sertraline in social phobia: I-year follow-up of a randomised controlled trial. Br J Psychiatry 2003;182:312-8.
6. Marks IM, Swinson RP, Basoglu M, et al. Alprazolam and exposure alone and combined in panic disorder with agoraphobia. A controlled study in London and Toronto. Br J Psychiatry 1993;162:776-87.
7. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry 2004;61(11):1136-44.
8. Purves D, Augustine GJ, Fitzpatrick D, et al. Plasticity of mature synapses and circuits. In: Neuroscience (3rd ed). Sunderland, MA: Sinauer; 2004:575-610.
9. O’Connor A. A pill that helps ease grip of irrational fears. New York Times March 22, 2005.
Think of anxiety disorders as an overactive brain alarm that psychotropics and exposure therapy quiet via separate mechanisms. Psychotropics “cool off” the alarm by curtailing excitability of the amygdala, brainstem nuclei, and hypothalamus.1 Psychological treatments, particularly exposure therapy, seek to teach the brain not to fear the dreaded object.2
One would assume that combining medication and exposure therapy for anxiety would be beneficial, but results have been disappointing.3 Anxiolytics do not enhance—and many impede—learning that occurs during psychotherapy. When the medications are tapered, patients who receive psychotherapy plus placebo typically experience more-enduring benefit than those receiving psychotherapy plus active medication.4-6
An unlikely candidate—a glutamatergic tuberculosis (TB) drug—may offer a solution. The drug and others in its class may potentiate psychotherapy’s effects by enhancing learning rather than relief.7
Glutamate/learning link
Glutamate neurons have 3 types of glutamatergic receptors, with the NMDA and AMPA types perceived as most important because of their possible role in memory development. Creating new memories may involve strengthening signals between glutamate neurons. The exact cellular mechanism is unknown, but it may involve greater release of neurotransmitters or formation of new synapses.
Stronger signaling—and hence learning—may depend on opening the NMDA receptor to enhance postsynaptic potential.8 Opening both NMDA and AMPA receptors generates a stronger signal and allows calcium influx, compared with opening the AMPA receptor alone (Figure). This combination can activate genes that control protein synthesis and result in structural changes necessary for developing long-term memories.
D-cycloserine—a partial agonist at the NMDA receptor—is usually used as an antibiotic to treat TB. The drug also has been shown to enhance the learning process that underlies fear extinction in rats. A group at Emory University studied the effect of adding the medication to exposure therapy in humans with acrophobia.7
FigureNMDA receptor agonists may enhance learning in psychotherapy
Normally, only glutamate neurons’ AMPA receptors activate in response to glutamate release (A). Both AMPA and NMDA receptors open in response to NMDA receptor agonists like D-cycloserine (B). Consequent stronger signaling may improve memory.
Going ‘up’
The researchers developed a virtual reality exposure system in which participants felt as if they were standing in an elevator, watching the floors recede as they rose 19 stories. Exposure therapy—seven weekly 35- to 45-minute sessions—has been shown to reduce the fear patients with acrophobia experience in virtual elevators.
Of 27 subjects, 10 received placebo and 17 received D-cycloserine, 50 mg or 500 mg. Subjects took their pills 2 to 4 hours before an exposure session. All participants experienced 2 virtual exposure sessions 1 to 2 weeks apart, which is considered suboptimal treatment for acrophobia.
Three months after the study, the D-cycloserine groups showed a markedly reduced fear of heights on the virtual elevator, while the placebo group showed no change from baseline. Fear levels were measured by subjective report of discomfort at each “floor.” D-cycloserine subjects also reported significantly greater reductions in measures of acrophobia in their daily lives.
Interestingly, both the D-cycloserine and placebo groups were equally frightened during virtual reality exposure. Only later did the D-cycloserine groups report less fear when exposed to heights, indicating that D-cycloserine enhanced learning that occurred during exposure therapy.
It’s exciting to think that medications could enhance and accelerate healing by activating the appropriate receptors during psychotherapy and give patients enduring benefits without the need for continued treatment.
If shown to be effective in larger studies, glutamatergic medications plus psychotherapy could provide more effective therapy for anxiety disorder. This approach is reported to be under investigation for treating anorexia nervosa, social phobia, panic disorder, and obsessive-compulsive disorder.9
Think of anxiety disorders as an overactive brain alarm that psychotropics and exposure therapy quiet via separate mechanisms. Psychotropics “cool off” the alarm by curtailing excitability of the amygdala, brainstem nuclei, and hypothalamus.1 Psychological treatments, particularly exposure therapy, seek to teach the brain not to fear the dreaded object.2
One would assume that combining medication and exposure therapy for anxiety would be beneficial, but results have been disappointing.3 Anxiolytics do not enhance—and many impede—learning that occurs during psychotherapy. When the medications are tapered, patients who receive psychotherapy plus placebo typically experience more-enduring benefit than those receiving psychotherapy plus active medication.4-6
An unlikely candidate—a glutamatergic tuberculosis (TB) drug—may offer a solution. The drug and others in its class may potentiate psychotherapy’s effects by enhancing learning rather than relief.7
Glutamate/learning link
Glutamate neurons have 3 types of glutamatergic receptors, with the NMDA and AMPA types perceived as most important because of their possible role in memory development. Creating new memories may involve strengthening signals between glutamate neurons. The exact cellular mechanism is unknown, but it may involve greater release of neurotransmitters or formation of new synapses.
Stronger signaling—and hence learning—may depend on opening the NMDA receptor to enhance postsynaptic potential.8 Opening both NMDA and AMPA receptors generates a stronger signal and allows calcium influx, compared with opening the AMPA receptor alone (Figure). This combination can activate genes that control protein synthesis and result in structural changes necessary for developing long-term memories.
D-cycloserine—a partial agonist at the NMDA receptor—is usually used as an antibiotic to treat TB. The drug also has been shown to enhance the learning process that underlies fear extinction in rats. A group at Emory University studied the effect of adding the medication to exposure therapy in humans with acrophobia.7
FigureNMDA receptor agonists may enhance learning in psychotherapy
Normally, only glutamate neurons’ AMPA receptors activate in response to glutamate release (A). Both AMPA and NMDA receptors open in response to NMDA receptor agonists like D-cycloserine (B). Consequent stronger signaling may improve memory.
Going ‘up’
The researchers developed a virtual reality exposure system in which participants felt as if they were standing in an elevator, watching the floors recede as they rose 19 stories. Exposure therapy—seven weekly 35- to 45-minute sessions—has been shown to reduce the fear patients with acrophobia experience in virtual elevators.
Of 27 subjects, 10 received placebo and 17 received D-cycloserine, 50 mg or 500 mg. Subjects took their pills 2 to 4 hours before an exposure session. All participants experienced 2 virtual exposure sessions 1 to 2 weeks apart, which is considered suboptimal treatment for acrophobia.
Three months after the study, the D-cycloserine groups showed a markedly reduced fear of heights on the virtual elevator, while the placebo group showed no change from baseline. Fear levels were measured by subjective report of discomfort at each “floor.” D-cycloserine subjects also reported significantly greater reductions in measures of acrophobia in their daily lives.
Interestingly, both the D-cycloserine and placebo groups were equally frightened during virtual reality exposure. Only later did the D-cycloserine groups report less fear when exposed to heights, indicating that D-cycloserine enhanced learning that occurred during exposure therapy.
It’s exciting to think that medications could enhance and accelerate healing by activating the appropriate receptors during psychotherapy and give patients enduring benefits without the need for continued treatment.
If shown to be effective in larger studies, glutamatergic medications plus psychotherapy could provide more effective therapy for anxiety disorder. This approach is reported to be under investigation for treating anorexia nervosa, social phobia, panic disorder, and obsessive-compulsive disorder.9
1. Lydiard RB. Break the ‘fear circuit’ in resistant panic disorder. Current Psychiatry 2003;2(11):12-22.
2. Tynes LL, Tynes SF. Panic attacks: help sufferers recover with cognitive-behavioral therapy. Current Psychiatry 2005;4(11):51-60.
3. Otto MW, Smits JAJ, Reese HE. Combined psychotherapy and pharmacotherapy for mood and anxiety disorders in adults: review and analysis. Clinical Psychology: Science & Practice 2005;12(1):72-86.
4. Barlow DH, Gorman JM, Shear MK, Woods SW. Cognitive-behavioral therapy, imipramine, or their combination for panic disorder: A randomized controlled trial. JAMA 2000;283(19):2529-36.
5. Haug TT, Blomhoff S, Hellstrom K, et al. Exposure therapy and sertraline in social phobia: I-year follow-up of a randomised controlled trial. Br J Psychiatry 2003;182:312-8.
6. Marks IM, Swinson RP, Basoglu M, et al. Alprazolam and exposure alone and combined in panic disorder with agoraphobia. A controlled study in London and Toronto. Br J Psychiatry 1993;162:776-87.
7. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry 2004;61(11):1136-44.
8. Purves D, Augustine GJ, Fitzpatrick D, et al. Plasticity of mature synapses and circuits. In: Neuroscience (3rd ed). Sunderland, MA: Sinauer; 2004:575-610.
9. O’Connor A. A pill that helps ease grip of irrational fears. New York Times March 22, 2005.
1. Lydiard RB. Break the ‘fear circuit’ in resistant panic disorder. Current Psychiatry 2003;2(11):12-22.
2. Tynes LL, Tynes SF. Panic attacks: help sufferers recover with cognitive-behavioral therapy. Current Psychiatry 2005;4(11):51-60.
3. Otto MW, Smits JAJ, Reese HE. Combined psychotherapy and pharmacotherapy for mood and anxiety disorders in adults: review and analysis. Clinical Psychology: Science & Practice 2005;12(1):72-86.
4. Barlow DH, Gorman JM, Shear MK, Woods SW. Cognitive-behavioral therapy, imipramine, or their combination for panic disorder: A randomized controlled trial. JAMA 2000;283(19):2529-36.
5. Haug TT, Blomhoff S, Hellstrom K, et al. Exposure therapy and sertraline in social phobia: I-year follow-up of a randomised controlled trial. Br J Psychiatry 2003;182:312-8.
6. Marks IM, Swinson RP, Basoglu M, et al. Alprazolam and exposure alone and combined in panic disorder with agoraphobia. A controlled study in London and Toronto. Br J Psychiatry 1993;162:776-87.
7. Ressler KJ, Rothbaum BO, Tannenbaum L, et al. Cognitive enhancers as adjuncts to psychotherapy: use of D-cycloserine in phobic individuals to facilitate extinction of fear. Arch Gen Psychiatry 2004;61(11):1136-44.
8. Purves D, Augustine GJ, Fitzpatrick D, et al. Plasticity of mature synapses and circuits. In: Neuroscience (3rd ed). Sunderland, MA: Sinauer; 2004:575-610.
9. O’Connor A. A pill that helps ease grip of irrational fears. New York Times March 22, 2005.
Can a vaccine prevent Alzheimer’s disease?
Deposition of amyloid-β peptide (Aβ) is believed to contribute to Alzheimer’s disease (AD) pathogenesis. Derived from a larger precursor protein, Aβ aggregates into plaques, and may promote neuronal death and, ultimately, dementia.
Current treatments alleviate symptoms without slowing underlying neurodegeneration. The prospect of harnessing the immune system to target the Aβ peptide offers an intriguing option for preventing this devastating, increasingly common disease.
Anti-a BETA antibodies
Transgenic mice bred to overexpress AD genes have responded remarkably in studies using the immune system to target the amyloid-β peptide.3 Several mouse groups have shown plaque reduction (Figure 1) and improved cognitive performance. These findings substantiate the amyloid hypothesis in AD pathogenesis.
A host could acquire anti-Aβ antibodies though two basic approaches (Figure 2):2
Figure 1 Differences in amyloid deposition between control and immunized mice
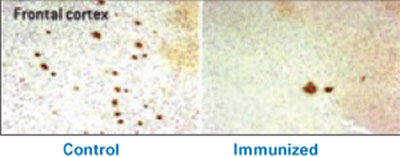
Frontal cortex of an unvaccinated mouse (left) shows more amyloid deposits (dark spots) than that of a mouse producing antibodies against the amyloid-β peptide (right).
Source: Image by Cynthia A. Lemere, PhD. Used with permission.Active immunization exposes the subject to the antigen (in this case the Aβ peptide) and allows T cells and B cells to produce anti-Aβ antibodies. This approach has been studied in humans, but adverse effects have stymied its development.
Passive immunization, which involves developing anti-Aβ antibodies in a separate source, aims to clear Aβ peptide without requiring an immunologic response from the host. Large doses of antibodies administered weekly or monthly would be needed to build adequate plasma levels in the CNS, and large quantities of circulating antibodies could cause hemorrhagic stroke.
A troublesome trial
After successful preclinical and phase 1 testing of a vaccine against the Aβ peptide (called AN-1792), a phase 2a placebo-controlled trial in 2001 followed patients with mild to moderate AD. Drug administration was halted after 18 patients (6%) developed meningoencephalitis after several months.4 However, 300 patients with AD and 72 control patients had received at least one injection, and double-blind assessments were maintained for 12 months.
Figure 2 Methods for immunizing against Aβ peptide
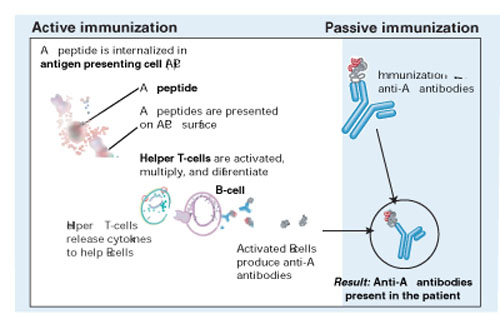
Active immunization produces anti-Aβ antibodies via immunologic response to vaccination. With passive immunization, anti-Aβ antibodies are administered directly.
Illustration by Rich LaRocco.All patients with meningoencephalitis had received the vaccine but not all developed an immune response, suggesting that something other than the antibodies—such as T cells—caused the encephalitis. Twelve patients recovered, but six had persistent cognitive and neurologic deficits.
More-optimistic news
Of the 300 patients who received an active vaccine, 20% developed an adequate antibody response.5
The responders showed no significant difference from the placebo group in most outcome measures but showed less worsening in the nine-component Neuropsychological Test Battery (NTB) (P=0.02). Of particular interest, antibody responders showed significant improvement in the NTB—s memory domain (P=0.03). Further, subjects with higher IgG antibody titers showed greater improvement than did other responders.
More work ahead
Although the outcome of this initial AN-1792 trial is disappointing because of its discontinuation and mixed results, T cell infiltration and amyloid depletion were found during postmortem examinations of two vaccine recipients.6
Pharmaceutical companies are testing two compounds for AD immunotherapy:7
- AAB-001, a human monoclonal antibody, targets all 42 Aβ amino acids via passive immunization and has entered phase 2 trials.
- ACC-001, an Aβ immuno-conjugate designed to elicit an active antibody response, began phase 1 testing last fall.
These efforts suggest that an “Alzheimer’s vaccine” could be produced, provided it could attack the Aβ peptide without inducing a significant cellular reaction.
1. Neugroschl JA, Kolevzor A, Samuels SC, Marir DB. Dementia. In: Sadock BJ, Sadock VA (eds). Kaplan & Sadock’s comprehensive text-book of psychiatry (8th ed). Philadelphia: Lippincott Williams & Wilkins; 2005:1068-93.
2. Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci 2002;3:824-8.
3. Schenk D, Hagen M, Seubert P. Current progress in beta-amyloid immunotherapy. Current Opin Immunol 2004;16:599-606.
4. Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoen-cephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003;61:46-54.
5. Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005;64:1553-62.
6. Ferrer I, Boada Rovira M, Sanchez Guerra ML, et al. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathology 2004;14(1):11-20.
7. Sullivan MG. Immunotherapy studies for AD back on track. Psychiatry News 2005;33(11):69.-
Deposition of amyloid-β peptide (Aβ) is believed to contribute to Alzheimer’s disease (AD) pathogenesis. Derived from a larger precursor protein, Aβ aggregates into plaques, and may promote neuronal death and, ultimately, dementia.
Current treatments alleviate symptoms without slowing underlying neurodegeneration. The prospect of harnessing the immune system to target the Aβ peptide offers an intriguing option for preventing this devastating, increasingly common disease.
Anti-a BETA antibodies
Transgenic mice bred to overexpress AD genes have responded remarkably in studies using the immune system to target the amyloid-β peptide.3 Several mouse groups have shown plaque reduction (Figure 1) and improved cognitive performance. These findings substantiate the amyloid hypothesis in AD pathogenesis.
A host could acquire anti-Aβ antibodies though two basic approaches (Figure 2):2
Figure 1 Differences in amyloid deposition between control and immunized mice
Frontal cortex of an unvaccinated mouse (left) shows more amyloid deposits (dark spots) than that of a mouse producing antibodies against the amyloid-β peptide (right).
Source: Image by Cynthia A. Lemere, PhD. Used with permission.Active immunization exposes the subject to the antigen (in this case the Aβ peptide) and allows T cells and B cells to produce anti-Aβ antibodies. This approach has been studied in humans, but adverse effects have stymied its development.
Passive immunization, which involves developing anti-Aβ antibodies in a separate source, aims to clear Aβ peptide without requiring an immunologic response from the host. Large doses of antibodies administered weekly or monthly would be needed to build adequate plasma levels in the CNS, and large quantities of circulating antibodies could cause hemorrhagic stroke.
A troublesome trial
After successful preclinical and phase 1 testing of a vaccine against the Aβ peptide (called AN-1792), a phase 2a placebo-controlled trial in 2001 followed patients with mild to moderate AD. Drug administration was halted after 18 patients (6%) developed meningoencephalitis after several months.4 However, 300 patients with AD and 72 control patients had received at least one injection, and double-blind assessments were maintained for 12 months.
Figure 2 Methods for immunizing against Aβ peptide
Active immunization produces anti-Aβ antibodies via immunologic response to vaccination. With passive immunization, anti-Aβ antibodies are administered directly.
Illustration by Rich LaRocco.All patients with meningoencephalitis had received the vaccine but not all developed an immune response, suggesting that something other than the antibodies—such as T cells—caused the encephalitis. Twelve patients recovered, but six had persistent cognitive and neurologic deficits.
More-optimistic news
Of the 300 patients who received an active vaccine, 20% developed an adequate antibody response.5
The responders showed no significant difference from the placebo group in most outcome measures but showed less worsening in the nine-component Neuropsychological Test Battery (NTB) (P=0.02). Of particular interest, antibody responders showed significant improvement in the NTB—s memory domain (P=0.03). Further, subjects with higher IgG antibody titers showed greater improvement than did other responders.
More work ahead
Although the outcome of this initial AN-1792 trial is disappointing because of its discontinuation and mixed results, T cell infiltration and amyloid depletion were found during postmortem examinations of two vaccine recipients.6
Pharmaceutical companies are testing two compounds for AD immunotherapy:7
- AAB-001, a human monoclonal antibody, targets all 42 Aβ amino acids via passive immunization and has entered phase 2 trials.
- ACC-001, an Aβ immuno-conjugate designed to elicit an active antibody response, began phase 1 testing last fall.
These efforts suggest that an “Alzheimer’s vaccine” could be produced, provided it could attack the Aβ peptide without inducing a significant cellular reaction.
Deposition of amyloid-β peptide (Aβ) is believed to contribute to Alzheimer’s disease (AD) pathogenesis. Derived from a larger precursor protein, Aβ aggregates into plaques, and may promote neuronal death and, ultimately, dementia.
Current treatments alleviate symptoms without slowing underlying neurodegeneration. The prospect of harnessing the immune system to target the Aβ peptide offers an intriguing option for preventing this devastating, increasingly common disease.
Anti-a BETA antibodies
Transgenic mice bred to overexpress AD genes have responded remarkably in studies using the immune system to target the amyloid-β peptide.3 Several mouse groups have shown plaque reduction (Figure 1) and improved cognitive performance. These findings substantiate the amyloid hypothesis in AD pathogenesis.
A host could acquire anti-Aβ antibodies though two basic approaches (Figure 2):2
Figure 1 Differences in amyloid deposition between control and immunized mice
Frontal cortex of an unvaccinated mouse (left) shows more amyloid deposits (dark spots) than that of a mouse producing antibodies against the amyloid-β peptide (right).
Source: Image by Cynthia A. Lemere, PhD. Used with permission.Active immunization exposes the subject to the antigen (in this case the Aβ peptide) and allows T cells and B cells to produce anti-Aβ antibodies. This approach has been studied in humans, but adverse effects have stymied its development.
Passive immunization, which involves developing anti-Aβ antibodies in a separate source, aims to clear Aβ peptide without requiring an immunologic response from the host. Large doses of antibodies administered weekly or monthly would be needed to build adequate plasma levels in the CNS, and large quantities of circulating antibodies could cause hemorrhagic stroke.
A troublesome trial
After successful preclinical and phase 1 testing of a vaccine against the Aβ peptide (called AN-1792), a phase 2a placebo-controlled trial in 2001 followed patients with mild to moderate AD. Drug administration was halted after 18 patients (6%) developed meningoencephalitis after several months.4 However, 300 patients with AD and 72 control patients had received at least one injection, and double-blind assessments were maintained for 12 months.
Figure 2 Methods for immunizing against Aβ peptide
Active immunization produces anti-Aβ antibodies via immunologic response to vaccination. With passive immunization, anti-Aβ antibodies are administered directly.
Illustration by Rich LaRocco.All patients with meningoencephalitis had received the vaccine but not all developed an immune response, suggesting that something other than the antibodies—such as T cells—caused the encephalitis. Twelve patients recovered, but six had persistent cognitive and neurologic deficits.
More-optimistic news
Of the 300 patients who received an active vaccine, 20% developed an adequate antibody response.5
The responders showed no significant difference from the placebo group in most outcome measures but showed less worsening in the nine-component Neuropsychological Test Battery (NTB) (P=0.02). Of particular interest, antibody responders showed significant improvement in the NTB—s memory domain (P=0.03). Further, subjects with higher IgG antibody titers showed greater improvement than did other responders.
More work ahead
Although the outcome of this initial AN-1792 trial is disappointing because of its discontinuation and mixed results, T cell infiltration and amyloid depletion were found during postmortem examinations of two vaccine recipients.6
Pharmaceutical companies are testing two compounds for AD immunotherapy:7
- AAB-001, a human monoclonal antibody, targets all 42 Aβ amino acids via passive immunization and has entered phase 2 trials.
- ACC-001, an Aβ immuno-conjugate designed to elicit an active antibody response, began phase 1 testing last fall.
These efforts suggest that an “Alzheimer’s vaccine” could be produced, provided it could attack the Aβ peptide without inducing a significant cellular reaction.
1. Neugroschl JA, Kolevzor A, Samuels SC, Marir DB. Dementia. In: Sadock BJ, Sadock VA (eds). Kaplan & Sadock’s comprehensive text-book of psychiatry (8th ed). Philadelphia: Lippincott Williams & Wilkins; 2005:1068-93.
2. Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci 2002;3:824-8.
3. Schenk D, Hagen M, Seubert P. Current progress in beta-amyloid immunotherapy. Current Opin Immunol 2004;16:599-606.
4. Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoen-cephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003;61:46-54.
5. Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005;64:1553-62.
6. Ferrer I, Boada Rovira M, Sanchez Guerra ML, et al. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathology 2004;14(1):11-20.
7. Sullivan MG. Immunotherapy studies for AD back on track. Psychiatry News 2005;33(11):69.-
1. Neugroschl JA, Kolevzor A, Samuels SC, Marir DB. Dementia. In: Sadock BJ, Sadock VA (eds). Kaplan & Sadock’s comprehensive text-book of psychiatry (8th ed). Philadelphia: Lippincott Williams & Wilkins; 2005:1068-93.
2. Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci 2002;3:824-8.
3. Schenk D, Hagen M, Seubert P. Current progress in beta-amyloid immunotherapy. Current Opin Immunol 2004;16:599-606.
4. Orgogozo JM, Gilman S, Dartigues JF, et al. Subacute meningoen-cephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 2003;61:46-54.
5. Gilman S, Koller M, Black RS, et al. Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 2005;64:1553-62.
6. Ferrer I, Boada Rovira M, Sanchez Guerra ML, et al. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathology 2004;14(1):11-20.
7. Sullivan MG. Immunotherapy studies for AD back on track. Psychiatry News 2005;33(11):69.-
Does chronic pain shrink the brain?
Chronic pain—particularly lower back pain—is frustrating to both patient and clinician. Because most cases lack an obvious physical explanation, the doctor may wonder if the patient is faking or exaggerating—that the pain is “in the patient’s head.” Studies suggest this cerebral component may exist—but not in ways you might expect.
How the brain processes pain
According to traditional belief, the brain passively receives noxious signals from injured tissue (nociceptive) or damaged nerve (neuropathic). Extensive—some would say excessive—tests are often conducted in search of a bone or muscle injury that might explain the pain.
Functional imaging studies across 15 years have shown activity in various brain regions when subjects feel pain. In addition to the somatosensory cortex, pain also activates brain areas involved with mood, attention, and anxiety. More important, the brain does not passively receive signals from the periphery but can inhibit ascending signals with endogenous opioids, such as endorphins and enkephalins.
Apkarian et alPosttraumatic stress disorder: Nature and nurture?, May 2004.)
Drugs. Medications and other substances taken to alleviate pain might also reduce gray matter. Excessive alcohol and opioid use have long-term adverse effects on the CNS.3 Is treatment or self-medication mildly toxic to the brain?
Overuse atrophy. Apkarian et al1 propose that cortical loss may be secondary to overuse. They suggest that persistent pain perception— and the resultant negative affect and stress—causes an excitotoxic and inflammatory state that wears out portions of the brain circuitry. If this is true, then chronic pain itself causes cerebral atrophy.
Whatever the explanation, this study indicates that chronic lower back pain pathology extends beyond the lower back.
Related resources
- Deyo RA, Weinstein JN. Low back pain. N Engl J Med 2001;344:363-70.
- International Association for the Study of Pain. www.iasp-pain.org.
1. Apkarian AV, Sosa Y, Sonty S, et al. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci 2004;24:10410-5.
2. Lorenz J, Minoshima S, Casey KL. Keeping pain out of mind: the role of the dorsolateral prefrontal cortex in pain modulation. Brain 2003;126:1079-91.
3. Goldman D, Barr CS. Restoring the addicted brain. N Engl J Med 2002;347:843-5.
Dr. Higgins is clinical associate professor of family medicine and psychiatry, Medical University of South Carolina, Charleston (higginse@musc.edu).
Chronic pain—particularly lower back pain—is frustrating to both patient and clinician. Because most cases lack an obvious physical explanation, the doctor may wonder if the patient is faking or exaggerating—that the pain is “in the patient’s head.” Studies suggest this cerebral component may exist—but not in ways you might expect.
How the brain processes pain
According to traditional belief, the brain passively receives noxious signals from injured tissue (nociceptive) or damaged nerve (neuropathic). Extensive—some would say excessive—tests are often conducted in search of a bone or muscle injury that might explain the pain.
Functional imaging studies across 15 years have shown activity in various brain regions when subjects feel pain. In addition to the somatosensory cortex, pain also activates brain areas involved with mood, attention, and anxiety. More important, the brain does not passively receive signals from the periphery but can inhibit ascending signals with endogenous opioids, such as endorphins and enkephalins.
Apkarian et alPosttraumatic stress disorder: Nature and nurture?, May 2004.)
Drugs. Medications and other substances taken to alleviate pain might also reduce gray matter. Excessive alcohol and opioid use have long-term adverse effects on the CNS.3 Is treatment or self-medication mildly toxic to the brain?
Overuse atrophy. Apkarian et al1 propose that cortical loss may be secondary to overuse. They suggest that persistent pain perception— and the resultant negative affect and stress—causes an excitotoxic and inflammatory state that wears out portions of the brain circuitry. If this is true, then chronic pain itself causes cerebral atrophy.
Whatever the explanation, this study indicates that chronic lower back pain pathology extends beyond the lower back.
Related resources
- Deyo RA, Weinstein JN. Low back pain. N Engl J Med 2001;344:363-70.
- International Association for the Study of Pain. www.iasp-pain.org.
Chronic pain—particularly lower back pain—is frustrating to both patient and clinician. Because most cases lack an obvious physical explanation, the doctor may wonder if the patient is faking or exaggerating—that the pain is “in the patient’s head.” Studies suggest this cerebral component may exist—but not in ways you might expect.
How the brain processes pain
According to traditional belief, the brain passively receives noxious signals from injured tissue (nociceptive) or damaged nerve (neuropathic). Extensive—some would say excessive—tests are often conducted in search of a bone or muscle injury that might explain the pain.
Functional imaging studies across 15 years have shown activity in various brain regions when subjects feel pain. In addition to the somatosensory cortex, pain also activates brain areas involved with mood, attention, and anxiety. More important, the brain does not passively receive signals from the periphery but can inhibit ascending signals with endogenous opioids, such as endorphins and enkephalins.
Apkarian et alPosttraumatic stress disorder: Nature and nurture?, May 2004.)
Drugs. Medications and other substances taken to alleviate pain might also reduce gray matter. Excessive alcohol and opioid use have long-term adverse effects on the CNS.3 Is treatment or self-medication mildly toxic to the brain?
Overuse atrophy. Apkarian et al1 propose that cortical loss may be secondary to overuse. They suggest that persistent pain perception— and the resultant negative affect and stress—causes an excitotoxic and inflammatory state that wears out portions of the brain circuitry. If this is true, then chronic pain itself causes cerebral atrophy.
Whatever the explanation, this study indicates that chronic lower back pain pathology extends beyond the lower back.
Related resources
- Deyo RA, Weinstein JN. Low back pain. N Engl J Med 2001;344:363-70.
- International Association for the Study of Pain. www.iasp-pain.org.
1. Apkarian AV, Sosa Y, Sonty S, et al. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci 2004;24:10410-5.
2. Lorenz J, Minoshima S, Casey KL. Keeping pain out of mind: the role of the dorsolateral prefrontal cortex in pain modulation. Brain 2003;126:1079-91.
3. Goldman D, Barr CS. Restoring the addicted brain. N Engl J Med 2002;347:843-5.
Dr. Higgins is clinical associate professor of family medicine and psychiatry, Medical University of South Carolina, Charleston (higginse@musc.edu).
1. Apkarian AV, Sosa Y, Sonty S, et al. Chronic back pain is associated with decreased prefrontal and thalamic gray matter density. J Neurosci 2004;24:10410-5.
2. Lorenz J, Minoshima S, Casey KL. Keeping pain out of mind: the role of the dorsolateral prefrontal cortex in pain modulation. Brain 2003;126:1079-91.
3. Goldman D, Barr CS. Restoring the addicted brain. N Engl J Med 2002;347:843-5.
Dr. Higgins is clinical associate professor of family medicine and psychiatry, Medical University of South Carolina, Charleston (higginse@musc.edu).
Blame the brain for promiscuity?
Why do some people stay married for decades while others jump from one relationship to the next? If humans are like rodents, recent research suggests that neuropeptides may help forge the ties that bind.
FOLLOWING VOLE BEHAVIOR
The vole, which inhabits many grasslands in the United States, is a rodent resembling a mouse but related to the lemming. Males in some vole species (such as meadow voles) show no partner preference whereas males in other species prefer one partner, share the same nest with her, and help care for their offspring.1
The neuropeptides oxytocin and arginine vasopressin are mediators of pair bonding. The brain of a prairie vole—a species in which males tend to bond with female partners—has more vasopressin receptors than that of the solitary meadow vole (Figure 1).
Figure 1 More vasopressin in bonding voles
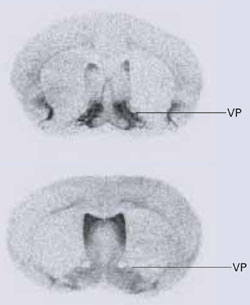
Ventral forebrain autoradiograms show greater vasopressin (VP) expression in prairie voles (top) than meadow voles.
Reprinted with permission. © 2004, Nature Publishing Group.Lim et al, however, found that meadow voles were more likely to bond with a single female after the males’ vasopressin receptors were increased.2 The researchers isolated and replicated the gene sequence responsible for the vasopressin receptor in the prairie vole. Then, using a viral vector, they injected the gene into the ventral pallidum of 11 male meadow voles. Eleven other voles received placebo.
Two weeks later, each sexually naïve male vole was housed for 24 hours with a sexually receptive female vole. Then, each male was placed for 3 hours in a three-chamber apparatus with the partner female in one chamber and a novel female in another. The time spent huddling with each female was recorded.
Across 3 hours, the placebo group voles spent 10 to 15 minutes with either female—normal behavior for this species. By contrast, the voles that received the vasopressin receptors spent approximately 40 minutes with their partners but only about 5 minutes with the novel voles, thus showing more affiliative behavior and a clear preference for their mates (Figure 2). The findings suggest that the researchers may have produced a profound change in social behavior by altering one gene.
IMPLICATIONS FOR HUMANS
How this research relates to humans is unknown, as there is no sound evidence of a link between vole and human pair bonding. Likewise, the influence of the higher cortical areas in orchestrating human behaviors cannot be underestimated.
Neuroimaging, however, has shown that brain regions rich with oxytocin and vasopressin receptors are activated while a person views pictures of loved ones.3 Additionally, mens’ vasopressin levels have been shown to increase when they are sexually aroused.4 Whether these findings one day lead to a medicine that promotes monogamous behavior in men remains to be seen.5
Figure 2 Male meadow voles’ interactions with females after vasopressin or placebo treatment
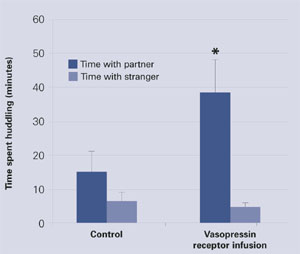
Source: Adapted from reference 2
1. Young LJ, Wang Z. The neurobiology of pair bonding. Nat Neurosci 2004;7:1048-54.
2. Lim MM, Wang Z, Olazabal DE, et al. Enhanced partner preference in a promiscuous species by manipulating the expression of a single gene. Nature 2004;429(6993):754-7.
3. Bartels A, Zeki S. The neural correlates of maternal and romantic love. Neuroimage 2004;21:1155-66.
4. Murphy MR, Seckl JR, Burton S, et al. Changes in oxytocin and vasopressin secretion during sexual activity in men. J Clin Endocrinol Metab 1987;65:738-41.
5. Konner M. The ties that bind. Nature 2004;429(6993):705.-
Why do some people stay married for decades while others jump from one relationship to the next? If humans are like rodents, recent research suggests that neuropeptides may help forge the ties that bind.
FOLLOWING VOLE BEHAVIOR
The vole, which inhabits many grasslands in the United States, is a rodent resembling a mouse but related to the lemming. Males in some vole species (such as meadow voles) show no partner preference whereas males in other species prefer one partner, share the same nest with her, and help care for their offspring.1
The neuropeptides oxytocin and arginine vasopressin are mediators of pair bonding. The brain of a prairie vole—a species in which males tend to bond with female partners—has more vasopressin receptors than that of the solitary meadow vole (Figure 1).
Figure 1 More vasopressin in bonding voles
Ventral forebrain autoradiograms show greater vasopressin (VP) expression in prairie voles (top) than meadow voles.
Reprinted with permission. © 2004, Nature Publishing Group.Lim et al, however, found that meadow voles were more likely to bond with a single female after the males’ vasopressin receptors were increased.2 The researchers isolated and replicated the gene sequence responsible for the vasopressin receptor in the prairie vole. Then, using a viral vector, they injected the gene into the ventral pallidum of 11 male meadow voles. Eleven other voles received placebo.
Two weeks later, each sexually naïve male vole was housed for 24 hours with a sexually receptive female vole. Then, each male was placed for 3 hours in a three-chamber apparatus with the partner female in one chamber and a novel female in another. The time spent huddling with each female was recorded.
Across 3 hours, the placebo group voles spent 10 to 15 minutes with either female—normal behavior for this species. By contrast, the voles that received the vasopressin receptors spent approximately 40 minutes with their partners but only about 5 minutes with the novel voles, thus showing more affiliative behavior and a clear preference for their mates (Figure 2). The findings suggest that the researchers may have produced a profound change in social behavior by altering one gene.
IMPLICATIONS FOR HUMANS
How this research relates to humans is unknown, as there is no sound evidence of a link between vole and human pair bonding. Likewise, the influence of the higher cortical areas in orchestrating human behaviors cannot be underestimated.
Neuroimaging, however, has shown that brain regions rich with oxytocin and vasopressin receptors are activated while a person views pictures of loved ones.3 Additionally, mens’ vasopressin levels have been shown to increase when they are sexually aroused.4 Whether these findings one day lead to a medicine that promotes monogamous behavior in men remains to be seen.5
Figure 2 Male meadow voles’ interactions with females after vasopressin or placebo treatment
Source: Adapted from reference 2
Why do some people stay married for decades while others jump from one relationship to the next? If humans are like rodents, recent research suggests that neuropeptides may help forge the ties that bind.
FOLLOWING VOLE BEHAVIOR
The vole, which inhabits many grasslands in the United States, is a rodent resembling a mouse but related to the lemming. Males in some vole species (such as meadow voles) show no partner preference whereas males in other species prefer one partner, share the same nest with her, and help care for their offspring.1
The neuropeptides oxytocin and arginine vasopressin are mediators of pair bonding. The brain of a prairie vole—a species in which males tend to bond with female partners—has more vasopressin receptors than that of the solitary meadow vole (Figure 1).
Figure 1 More vasopressin in bonding voles
Ventral forebrain autoradiograms show greater vasopressin (VP) expression in prairie voles (top) than meadow voles.
Reprinted with permission. © 2004, Nature Publishing Group.Lim et al, however, found that meadow voles were more likely to bond with a single female after the males’ vasopressin receptors were increased.2 The researchers isolated and replicated the gene sequence responsible for the vasopressin receptor in the prairie vole. Then, using a viral vector, they injected the gene into the ventral pallidum of 11 male meadow voles. Eleven other voles received placebo.
Two weeks later, each sexually naïve male vole was housed for 24 hours with a sexually receptive female vole. Then, each male was placed for 3 hours in a three-chamber apparatus with the partner female in one chamber and a novel female in another. The time spent huddling with each female was recorded.
Across 3 hours, the placebo group voles spent 10 to 15 minutes with either female—normal behavior for this species. By contrast, the voles that received the vasopressin receptors spent approximately 40 minutes with their partners but only about 5 minutes with the novel voles, thus showing more affiliative behavior and a clear preference for their mates (Figure 2). The findings suggest that the researchers may have produced a profound change in social behavior by altering one gene.
IMPLICATIONS FOR HUMANS
How this research relates to humans is unknown, as there is no sound evidence of a link between vole and human pair bonding. Likewise, the influence of the higher cortical areas in orchestrating human behaviors cannot be underestimated.
Neuroimaging, however, has shown that brain regions rich with oxytocin and vasopressin receptors are activated while a person views pictures of loved ones.3 Additionally, mens’ vasopressin levels have been shown to increase when they are sexually aroused.4 Whether these findings one day lead to a medicine that promotes monogamous behavior in men remains to be seen.5
Figure 2 Male meadow voles’ interactions with females after vasopressin or placebo treatment
Source: Adapted from reference 2
1. Young LJ, Wang Z. The neurobiology of pair bonding. Nat Neurosci 2004;7:1048-54.
2. Lim MM, Wang Z, Olazabal DE, et al. Enhanced partner preference in a promiscuous species by manipulating the expression of a single gene. Nature 2004;429(6993):754-7.
3. Bartels A, Zeki S. The neural correlates of maternal and romantic love. Neuroimage 2004;21:1155-66.
4. Murphy MR, Seckl JR, Burton S, et al. Changes in oxytocin and vasopressin secretion during sexual activity in men. J Clin Endocrinol Metab 1987;65:738-41.
5. Konner M. The ties that bind. Nature 2004;429(6993):705.-
1. Young LJ, Wang Z. The neurobiology of pair bonding. Nat Neurosci 2004;7:1048-54.
2. Lim MM, Wang Z, Olazabal DE, et al. Enhanced partner preference in a promiscuous species by manipulating the expression of a single gene. Nature 2004;429(6993):754-7.
3. Bartels A, Zeki S. The neural correlates of maternal and romantic love. Neuroimage 2004;21:1155-66.
4. Murphy MR, Seckl JR, Burton S, et al. Changes in oxytocin and vasopressin secretion during sexual activity in men. J Clin Endocrinol Metab 1987;65:738-41.
5. Konner M. The ties that bind. Nature 2004;429(6993):705.-