User login
Christopher Palmer has been an associate editor at MDedge News since 2017. When he's not tidying grammar, he writes short pieces about breaking FDA announcements and approvals, as well as journal articles. He proudly holds a BA in English and philosophy. Follow him on Twitter @cmacmpalm.
Liraglutide indication expanded to include children
according to a news release from the agency. The approval comes after granting the application Priority Review, which means the FDA aimed to take action on it within 6 months instead of the usual 10 months seen with a Standard Review.

Liraglutide injection has been approved for treatment of adults with type 2 diabetes since 2010, but this is the first noninsulin treatment for pediatric patients since metformin received approval for this population in 2000.
“The expanded indication provides an additional treatment option at a time when an increasing number of children are being diagnosed with [type 2 diabetes],” said Lisa Yanoff, MD, acting director of the division of metabolism and endocrinology products in the agency’s Center for Drug Evaluation and Research.
The approval is based on efficacy and safety results from several placebo-controlled trials in adults, and one trial in 134 pediatric patients aged 10 years or older. In the latter trial, which ran for more than 26 weeks, hemoglobin A1c levels fell below 7% in approximately 64% of patients treated with liraglutide injection, compared with 37% of those who received placebo. Those results were seen regardless of whether patients concomitantly used insulin.
Although an increase in hypoglycemia risk has sometimes been seen in adult patients taking liraglutide injection with insulin or other drugs that raise insulin production, this heightened risk was seen in the pediatric patients regardless of whether they took other therapies for diabetes.
Liraglutide injection carries a Boxed Warning, the FDA’s strongest warning, for heightened risk of thyroid C-cell tumors. Therefore, the agency recommends against patients with history or family members with history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2 using this treatment. Among its other warnings are those pertaining to patients with renal impairment or kidney failure, pancreatitis, and/or acute gallbladder disease.
The most common side effects are nausea, diarrhea, vomiting, decreased appetite, indigestion, and constipation. The full prescribing information can be found on the FDA website.
according to a news release from the agency. The approval comes after granting the application Priority Review, which means the FDA aimed to take action on it within 6 months instead of the usual 10 months seen with a Standard Review.
Liraglutide injection has been approved for treatment of adults with type 2 diabetes since 2010, but this is the first noninsulin treatment for pediatric patients since metformin received approval for this population in 2000.
“The expanded indication provides an additional treatment option at a time when an increasing number of children are being diagnosed with [type 2 diabetes],” said Lisa Yanoff, MD, acting director of the division of metabolism and endocrinology products in the agency’s Center for Drug Evaluation and Research.
The approval is based on efficacy and safety results from several placebo-controlled trials in adults, and one trial in 134 pediatric patients aged 10 years or older. In the latter trial, which ran for more than 26 weeks, hemoglobin A1c levels fell below 7% in approximately 64% of patients treated with liraglutide injection, compared with 37% of those who received placebo. Those results were seen regardless of whether patients concomitantly used insulin.
Although an increase in hypoglycemia risk has sometimes been seen in adult patients taking liraglutide injection with insulin or other drugs that raise insulin production, this heightened risk was seen in the pediatric patients regardless of whether they took other therapies for diabetes.
Liraglutide injection carries a Boxed Warning, the FDA’s strongest warning, for heightened risk of thyroid C-cell tumors. Therefore, the agency recommends against patients with history or family members with history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2 using this treatment. Among its other warnings are those pertaining to patients with renal impairment or kidney failure, pancreatitis, and/or acute gallbladder disease.
The most common side effects are nausea, diarrhea, vomiting, decreased appetite, indigestion, and constipation. The full prescribing information can be found on the FDA website.
according to a news release from the agency. The approval comes after granting the application Priority Review, which means the FDA aimed to take action on it within 6 months instead of the usual 10 months seen with a Standard Review.
Liraglutide injection has been approved for treatment of adults with type 2 diabetes since 2010, but this is the first noninsulin treatment for pediatric patients since metformin received approval for this population in 2000.
“The expanded indication provides an additional treatment option at a time when an increasing number of children are being diagnosed with [type 2 diabetes],” said Lisa Yanoff, MD, acting director of the division of metabolism and endocrinology products in the agency’s Center for Drug Evaluation and Research.
The approval is based on efficacy and safety results from several placebo-controlled trials in adults, and one trial in 134 pediatric patients aged 10 years or older. In the latter trial, which ran for more than 26 weeks, hemoglobin A1c levels fell below 7% in approximately 64% of patients treated with liraglutide injection, compared with 37% of those who received placebo. Those results were seen regardless of whether patients concomitantly used insulin.
Although an increase in hypoglycemia risk has sometimes been seen in adult patients taking liraglutide injection with insulin or other drugs that raise insulin production, this heightened risk was seen in the pediatric patients regardless of whether they took other therapies for diabetes.
Liraglutide injection carries a Boxed Warning, the FDA’s strongest warning, for heightened risk of thyroid C-cell tumors. Therefore, the agency recommends against patients with history or family members with history of medullary thyroid carcinoma or multiple endocrine neoplasia syndrome type 2 using this treatment. Among its other warnings are those pertaining to patients with renal impairment or kidney failure, pancreatitis, and/or acute gallbladder disease.
The most common side effects are nausea, diarrhea, vomiting, decreased appetite, indigestion, and constipation. The full prescribing information can be found on the FDA website.
Obesity and overweight declined among lower-income kids
The combined rate of , according to a study in JAMA.
Liping Pan, MD, MPH, of the Centers for Disease Control and Prevention and colleagues used data from the WIC Participant and Program Characteristics survey from 2010, 2012, 2014, and 2016 for 12,403,629 children aged 2-4 years from 50 states, Washington, D.C., and 5 territories. In addition to a –3.2% change (95% confidence interval, –3.3% to –3.2%) in adjusted prevalence difference for the combined rate of obesity and overweight seen between 2010 and 2016, the researchers found the crude prevalence decreased from 32.5% to 29.1%. A decrease was also seen for obesity alone (crude prevalence, 15.9% to 13.9%; adjusted prevalence difference, –1.9%; 95% CI, –1.9% to –1.8%).
One of the limitations of the study is that the characteristics of enrolled children might differ from those of children not enrolled in this WIC program; however, the researchers noted that they accounted for many demographic characteristics in the trend analyses.
“Reasons for the declines in obesity among young children in WIC remain undetermined but may include WIC food package revisions and local, state, and national initiatives,” they wrote.
SOURCE: Pan L et al. JAMA. 2019 Jun 18;321(23):2364-6.
The combined rate of , according to a study in JAMA.
Liping Pan, MD, MPH, of the Centers for Disease Control and Prevention and colleagues used data from the WIC Participant and Program Characteristics survey from 2010, 2012, 2014, and 2016 for 12,403,629 children aged 2-4 years from 50 states, Washington, D.C., and 5 territories. In addition to a –3.2% change (95% confidence interval, –3.3% to –3.2%) in adjusted prevalence difference for the combined rate of obesity and overweight seen between 2010 and 2016, the researchers found the crude prevalence decreased from 32.5% to 29.1%. A decrease was also seen for obesity alone (crude prevalence, 15.9% to 13.9%; adjusted prevalence difference, –1.9%; 95% CI, –1.9% to –1.8%).
One of the limitations of the study is that the characteristics of enrolled children might differ from those of children not enrolled in this WIC program; however, the researchers noted that they accounted for many demographic characteristics in the trend analyses.
“Reasons for the declines in obesity among young children in WIC remain undetermined but may include WIC food package revisions and local, state, and national initiatives,” they wrote.
SOURCE: Pan L et al. JAMA. 2019 Jun 18;321(23):2364-6.
The combined rate of , according to a study in JAMA.
Liping Pan, MD, MPH, of the Centers for Disease Control and Prevention and colleagues used data from the WIC Participant and Program Characteristics survey from 2010, 2012, 2014, and 2016 for 12,403,629 children aged 2-4 years from 50 states, Washington, D.C., and 5 territories. In addition to a –3.2% change (95% confidence interval, –3.3% to –3.2%) in adjusted prevalence difference for the combined rate of obesity and overweight seen between 2010 and 2016, the researchers found the crude prevalence decreased from 32.5% to 29.1%. A decrease was also seen for obesity alone (crude prevalence, 15.9% to 13.9%; adjusted prevalence difference, –1.9%; 95% CI, –1.9% to –1.8%).
One of the limitations of the study is that the characteristics of enrolled children might differ from those of children not enrolled in this WIC program; however, the researchers noted that they accounted for many demographic characteristics in the trend analyses.
“Reasons for the declines in obesity among young children in WIC remain undetermined but may include WIC food package revisions and local, state, and national initiatives,” they wrote.
SOURCE: Pan L et al. JAMA. 2019 Jun 18;321(23):2364-6.
FROM JAMA
Rosacea: Quality of life improves with treatment success
according to a survey from the National Rosacea Society.
The survey was conducted among 1,044 patients with rosacea, and 76% saw at least some improvement in their skin following treatment. Of all patients who responded to treatment, 40% reported improvement in psychological well-being, 35% reported improvement in social well-being, and 31% reported improvement in occupational well-being.
The percentages of patients reporting improvements were higher or lower depending on the degree of skin clearance. Among patients whose signs and symptoms of rosacea were virtually eliminated, 81% said psychological well-being improved, 71% said social well-being did so, and 62% said occupational well-being improved. Among those whose rosacea improved only slightly or moderately, the percentages of well-being improvement were only 24%, 21%, and 19%, respectively.
The National Rosacea Society serves as a resource to provide information on the disorder, as well as increase awareness and support research.
according to a survey from the National Rosacea Society.
The survey was conducted among 1,044 patients with rosacea, and 76% saw at least some improvement in their skin following treatment. Of all patients who responded to treatment, 40% reported improvement in psychological well-being, 35% reported improvement in social well-being, and 31% reported improvement in occupational well-being.
The percentages of patients reporting improvements were higher or lower depending on the degree of skin clearance. Among patients whose signs and symptoms of rosacea were virtually eliminated, 81% said psychological well-being improved, 71% said social well-being did so, and 62% said occupational well-being improved. Among those whose rosacea improved only slightly or moderately, the percentages of well-being improvement were only 24%, 21%, and 19%, respectively.
The National Rosacea Society serves as a resource to provide information on the disorder, as well as increase awareness and support research.
according to a survey from the National Rosacea Society.
The survey was conducted among 1,044 patients with rosacea, and 76% saw at least some improvement in their skin following treatment. Of all patients who responded to treatment, 40% reported improvement in psychological well-being, 35% reported improvement in social well-being, and 31% reported improvement in occupational well-being.
The percentages of patients reporting improvements were higher or lower depending on the degree of skin clearance. Among patients whose signs and symptoms of rosacea were virtually eliminated, 81% said psychological well-being improved, 71% said social well-being did so, and 62% said occupational well-being improved. Among those whose rosacea improved only slightly or moderately, the percentages of well-being improvement were only 24%, 21%, and 19%, respectively.
The National Rosacea Society serves as a resource to provide information on the disorder, as well as increase awareness and support research.
Teva expands its recall of losartan lots
The Food and Drug Administration has announced that , according to a release.
The recall for this and other angiotensin II receptor blockers was initiated by Teva on April 25, 2019, because of detection of unacceptable levels of the possibly cancer-causing impurity N-Nitroso-N-methyl-4-aminobutyric acid (NMBA). Teva expanded this recall on June 10, with another update issued on June 12.
Losartan is not the only ARB found to contain NMBA; a full list of all ARBs affected can be found on the FDA website and currently includes more than 1,100 lots being recalled. The list can be searched and sorted by such considerations as medicine in question, company involved, and lot number.
The Food and Drug Administration has announced that , according to a release.
The recall for this and other angiotensin II receptor blockers was initiated by Teva on April 25, 2019, because of detection of unacceptable levels of the possibly cancer-causing impurity N-Nitroso-N-methyl-4-aminobutyric acid (NMBA). Teva expanded this recall on June 10, with another update issued on June 12.
Losartan is not the only ARB found to contain NMBA; a full list of all ARBs affected can be found on the FDA website and currently includes more than 1,100 lots being recalled. The list can be searched and sorted by such considerations as medicine in question, company involved, and lot number.
The Food and Drug Administration has announced that , according to a release.
The recall for this and other angiotensin II receptor blockers was initiated by Teva on April 25, 2019, because of detection of unacceptable levels of the possibly cancer-causing impurity N-Nitroso-N-methyl-4-aminobutyric acid (NMBA). Teva expanded this recall on June 10, with another update issued on June 12.
Losartan is not the only ARB found to contain NMBA; a full list of all ARBs affected can be found on the FDA website and currently includes more than 1,100 lots being recalled. The list can be searched and sorted by such considerations as medicine in question, company involved, and lot number.
FDA invites sample submission for FDA-ARGOS database
which seeks to support research and regulatory decisions regarding DNA testing for pathogens with quality-controlled and curated genomic sequence data. Such testing and devices could be used as medical countermeasures against biothreats such as Ebola and Zika.
Infectious disease next-generation sequencing could use DNA analysis to help identify pathogens – from viruses to parasites – faster and more efficiently by, in theory, accomplishing with one test what was only possible before with many, according to the FDA. In order to not only further development of such tests and devices but also aid regulatory and scientific review of them, the FDA has collaborated with the Department of Defense, the National Center for Biotechnology Information, and Institute for Genome Sciences at the University of Maryland, Baltimore, to create FDA-ARGOS.
However, the FDA and its collaborators need samples of pathogens to continue developing the database, so they’ve invited health care professionals to submit samples for that purpose. More information, including preferred organism list and submission guidelines, can be found on the FDA-ARGOS website.
which seeks to support research and regulatory decisions regarding DNA testing for pathogens with quality-controlled and curated genomic sequence data. Such testing and devices could be used as medical countermeasures against biothreats such as Ebola and Zika.
Infectious disease next-generation sequencing could use DNA analysis to help identify pathogens – from viruses to parasites – faster and more efficiently by, in theory, accomplishing with one test what was only possible before with many, according to the FDA. In order to not only further development of such tests and devices but also aid regulatory and scientific review of them, the FDA has collaborated with the Department of Defense, the National Center for Biotechnology Information, and Institute for Genome Sciences at the University of Maryland, Baltimore, to create FDA-ARGOS.
However, the FDA and its collaborators need samples of pathogens to continue developing the database, so they’ve invited health care professionals to submit samples for that purpose. More information, including preferred organism list and submission guidelines, can be found on the FDA-ARGOS website.
which seeks to support research and regulatory decisions regarding DNA testing for pathogens with quality-controlled and curated genomic sequence data. Such testing and devices could be used as medical countermeasures against biothreats such as Ebola and Zika.
Infectious disease next-generation sequencing could use DNA analysis to help identify pathogens – from viruses to parasites – faster and more efficiently by, in theory, accomplishing with one test what was only possible before with many, according to the FDA. In order to not only further development of such tests and devices but also aid regulatory and scientific review of them, the FDA has collaborated with the Department of Defense, the National Center for Biotechnology Information, and Institute for Genome Sciences at the University of Maryland, Baltimore, to create FDA-ARGOS.
However, the FDA and its collaborators need samples of pathogens to continue developing the database, so they’ve invited health care professionals to submit samples for that purpose. More information, including preferred organism list and submission guidelines, can be found on the FDA-ARGOS website.
FDA approves Nucala’s new at-home formulations
, according to a press release from the drug’s developer. The biologic will now be available as an autoinjector and as a prefilled safety syringe.
The 100-mg subcutaneous mepolizumab injection is indicated as an add-on treatment for patients 12 years and older with severe eosinophilic asthma, and the three-dose 100-mg subcutaneous injections are indicated for the rare eosinophilic granulomatosis and polyangiitis, with the biologic administered every 4 weeks in either context. The release emphasizes that mepolizumab is not approved for acute bronchospasm or status asthmaticus. Health care professionals should first determine whether self-assisted administration or administration provided by a caregiver is appropriate, and then they should provide patients and/or caregivers with proper training in how to do so.
The approval is based on two open-label, single-arm, phase 3a studies that demonstrated successful administration was possible with these options among patients with severe eosinophilic asthma, at rates of 89%-95% in one study and 100% in the other. These results were followed by those of an open-label, parallel group, single-dose study that confirmed the pharmacokinetic and pharmacodynamic profiles of these new means of administration were comparable with those currently approved.
Mepolizumab is not indicated for those with a history of hypersensitivity to either mepolizumab or to the formulation’s excipients, such as anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, or rash. Any reductions of inhaled corticosteroids after initiation of mepolizumab should be gradual and under the supervision of a health care professional. Some infections by herpes zoster have been observed. The most common adverse reactions (occurring in 3% or more of patients and more often than with placebo) during the first 24 weeks of treatment were headache (19%), injection site reaction (8%), back pain (5%), fatigue (5%), influenza (3%), urinary tract infection (3%), abdominal pain upper (3%), pruritus (3%), eczema (3%), and muscle spasm (3%). Full prescribing information can be found on the FDA website.
, according to a press release from the drug’s developer. The biologic will now be available as an autoinjector and as a prefilled safety syringe.
The 100-mg subcutaneous mepolizumab injection is indicated as an add-on treatment for patients 12 years and older with severe eosinophilic asthma, and the three-dose 100-mg subcutaneous injections are indicated for the rare eosinophilic granulomatosis and polyangiitis, with the biologic administered every 4 weeks in either context. The release emphasizes that mepolizumab is not approved for acute bronchospasm or status asthmaticus. Health care professionals should first determine whether self-assisted administration or administration provided by a caregiver is appropriate, and then they should provide patients and/or caregivers with proper training in how to do so.
The approval is based on two open-label, single-arm, phase 3a studies that demonstrated successful administration was possible with these options among patients with severe eosinophilic asthma, at rates of 89%-95% in one study and 100% in the other. These results were followed by those of an open-label, parallel group, single-dose study that confirmed the pharmacokinetic and pharmacodynamic profiles of these new means of administration were comparable with those currently approved.
Mepolizumab is not indicated for those with a history of hypersensitivity to either mepolizumab or to the formulation’s excipients, such as anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, or rash. Any reductions of inhaled corticosteroids after initiation of mepolizumab should be gradual and under the supervision of a health care professional. Some infections by herpes zoster have been observed. The most common adverse reactions (occurring in 3% or more of patients and more often than with placebo) during the first 24 weeks of treatment were headache (19%), injection site reaction (8%), back pain (5%), fatigue (5%), influenza (3%), urinary tract infection (3%), abdominal pain upper (3%), pruritus (3%), eczema (3%), and muscle spasm (3%). Full prescribing information can be found on the FDA website.
, according to a press release from the drug’s developer. The biologic will now be available as an autoinjector and as a prefilled safety syringe.
The 100-mg subcutaneous mepolizumab injection is indicated as an add-on treatment for patients 12 years and older with severe eosinophilic asthma, and the three-dose 100-mg subcutaneous injections are indicated for the rare eosinophilic granulomatosis and polyangiitis, with the biologic administered every 4 weeks in either context. The release emphasizes that mepolizumab is not approved for acute bronchospasm or status asthmaticus. Health care professionals should first determine whether self-assisted administration or administration provided by a caregiver is appropriate, and then they should provide patients and/or caregivers with proper training in how to do so.
The approval is based on two open-label, single-arm, phase 3a studies that demonstrated successful administration was possible with these options among patients with severe eosinophilic asthma, at rates of 89%-95% in one study and 100% in the other. These results were followed by those of an open-label, parallel group, single-dose study that confirmed the pharmacokinetic and pharmacodynamic profiles of these new means of administration were comparable with those currently approved.
Mepolizumab is not indicated for those with a history of hypersensitivity to either mepolizumab or to the formulation’s excipients, such as anaphylaxis, angioedema, bronchospasm, hypotension, urticaria, or rash. Any reductions of inhaled corticosteroids after initiation of mepolizumab should be gradual and under the supervision of a health care professional. Some infections by herpes zoster have been observed. The most common adverse reactions (occurring in 3% or more of patients and more often than with placebo) during the first 24 weeks of treatment were headache (19%), injection site reaction (8%), back pain (5%), fatigue (5%), influenza (3%), urinary tract infection (3%), abdominal pain upper (3%), pruritus (3%), eczema (3%), and muscle spasm (3%). Full prescribing information can be found on the FDA website.
FDA: Vinpocetine associated with fetal harms, miscarriage
according to a statement from the agency.
This warning is based on data reviewed by the FDA, including a report from the National Institutes of Health’s National Toxicology Program, that show associations between vinpocetine use and decreased fetal weight and increased risk of miscarriage in animals. The agency is particularly concerned because products containing this ingredient, including those marketed as improving energy and memory, are widely available to women of childbearing age. As a result, the agency has recommended these women not take vinpocetine.
Vinpocetine is a synthetically produced compound used in dietary supplements either on its own or in combination and may be referred to as Vinca minor extract, lesser periwinkle extract, or common periwinkle extract on product labels. Although vinpocetine is regulated in some countries as a prescription drug, when it’s sold in dietary supplements in the United States, the FDA does not usually review those products or their labeling before they become available to consumers under the same safety and effectiveness standards used to evaluate drug products.
“Today’s safety warning is just one of many steps the FDA is taking to adapt to the realities of the evolving dietary supplement industry,” according to the agency’s statement. “Protecting the public from unsafe dietary supplements remains a top priority for the FDA.”
The full statement regarding vinpocetine and its risks can be found on the FDA website.
according to a statement from the agency.
This warning is based on data reviewed by the FDA, including a report from the National Institutes of Health’s National Toxicology Program, that show associations between vinpocetine use and decreased fetal weight and increased risk of miscarriage in animals. The agency is particularly concerned because products containing this ingredient, including those marketed as improving energy and memory, are widely available to women of childbearing age. As a result, the agency has recommended these women not take vinpocetine.
Vinpocetine is a synthetically produced compound used in dietary supplements either on its own or in combination and may be referred to as Vinca minor extract, lesser periwinkle extract, or common periwinkle extract on product labels. Although vinpocetine is regulated in some countries as a prescription drug, when it’s sold in dietary supplements in the United States, the FDA does not usually review those products or their labeling before they become available to consumers under the same safety and effectiveness standards used to evaluate drug products.
“Today’s safety warning is just one of many steps the FDA is taking to adapt to the realities of the evolving dietary supplement industry,” according to the agency’s statement. “Protecting the public from unsafe dietary supplements remains a top priority for the FDA.”
The full statement regarding vinpocetine and its risks can be found on the FDA website.
according to a statement from the agency.
This warning is based on data reviewed by the FDA, including a report from the National Institutes of Health’s National Toxicology Program, that show associations between vinpocetine use and decreased fetal weight and increased risk of miscarriage in animals. The agency is particularly concerned because products containing this ingredient, including those marketed as improving energy and memory, are widely available to women of childbearing age. As a result, the agency has recommended these women not take vinpocetine.
Vinpocetine is a synthetically produced compound used in dietary supplements either on its own or in combination and may be referred to as Vinca minor extract, lesser periwinkle extract, or common periwinkle extract on product labels. Although vinpocetine is regulated in some countries as a prescription drug, when it’s sold in dietary supplements in the United States, the FDA does not usually review those products or their labeling before they become available to consumers under the same safety and effectiveness standards used to evaluate drug products.
“Today’s safety warning is just one of many steps the FDA is taking to adapt to the realities of the evolving dietary supplement industry,” according to the agency’s statement. “Protecting the public from unsafe dietary supplements remains a top priority for the FDA.”
The full statement regarding vinpocetine and its risks can be found on the FDA website.
Timolol shortens propranolol use in infantile hemangioma
according to a study published in Pediatric Dermatology.
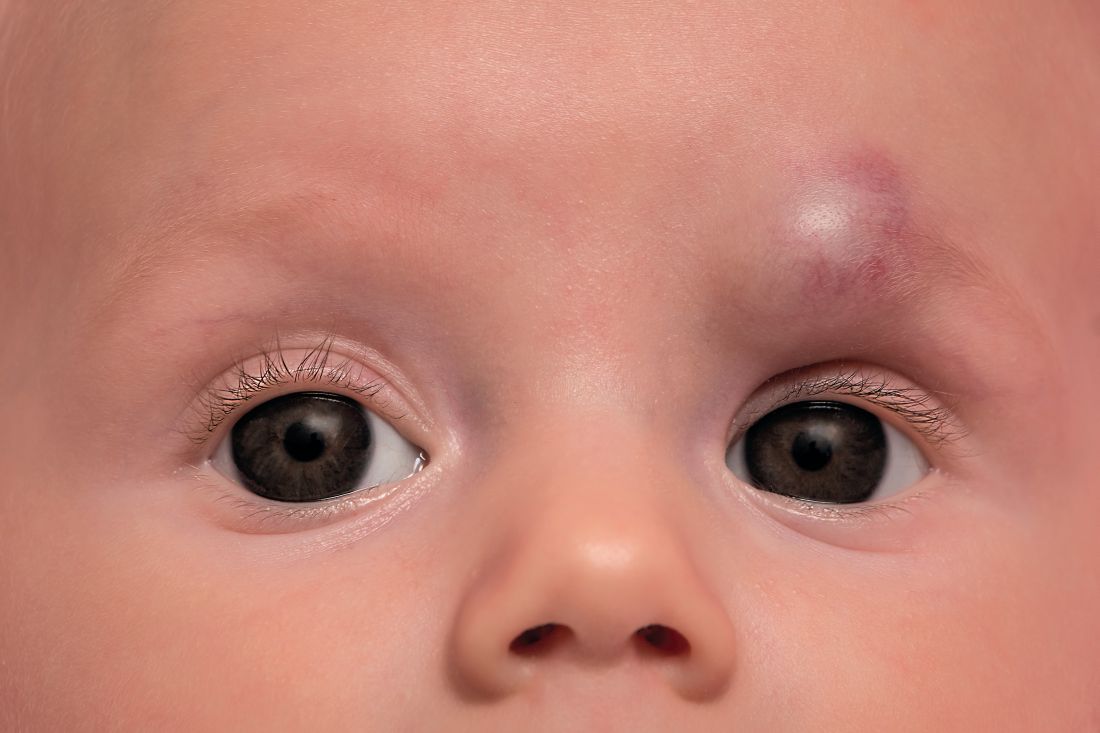
Diana B. Mannschreck, BSN, of Johns Hopkins University, Baltimore, and colleagues performed a retrospective chart review of 559 patients with infantile hemangioma seen in the dermatology clinic at Johns Hopkins between December 2008 and January 2018. Patients received any of five courses of treatment, including oral propranolol followed by topical timolol, propranolol only, and timolol only. Of the courses evaluated, propranolol followed by timolol had the shortest duration of propranolol therapy – a median of 2.2 months shorter than propranolol-only therapy (P = .0006). This sequential regimen also was associated with no reinitiations of propranolol therapy following tapering, whereas 13% of those receiving propranolol alone had to reinitiate it after tapering.
This is of interest because oral beta-blockers, including propranolol, have been associated with rare but serious adverse events, such as bronchospasm, hypotension, and hypoglycemia.
Limitations of the study include its retrospective and single-center nature. There was no funding or disclosure information given.
SOURCE: Mannschreck DB et al. Pediatr Dermatol. 2019 Apr 9. doi: 10.1111/pde.13816.
according to a study published in Pediatric Dermatology.
Diana B. Mannschreck, BSN, of Johns Hopkins University, Baltimore, and colleagues performed a retrospective chart review of 559 patients with infantile hemangioma seen in the dermatology clinic at Johns Hopkins between December 2008 and January 2018. Patients received any of five courses of treatment, including oral propranolol followed by topical timolol, propranolol only, and timolol only. Of the courses evaluated, propranolol followed by timolol had the shortest duration of propranolol therapy – a median of 2.2 months shorter than propranolol-only therapy (P = .0006). This sequential regimen also was associated with no reinitiations of propranolol therapy following tapering, whereas 13% of those receiving propranolol alone had to reinitiate it after tapering.
This is of interest because oral beta-blockers, including propranolol, have been associated with rare but serious adverse events, such as bronchospasm, hypotension, and hypoglycemia.
Limitations of the study include its retrospective and single-center nature. There was no funding or disclosure information given.
SOURCE: Mannschreck DB et al. Pediatr Dermatol. 2019 Apr 9. doi: 10.1111/pde.13816.
according to a study published in Pediatric Dermatology.
Diana B. Mannschreck, BSN, of Johns Hopkins University, Baltimore, and colleagues performed a retrospective chart review of 559 patients with infantile hemangioma seen in the dermatology clinic at Johns Hopkins between December 2008 and January 2018. Patients received any of five courses of treatment, including oral propranolol followed by topical timolol, propranolol only, and timolol only. Of the courses evaluated, propranolol followed by timolol had the shortest duration of propranolol therapy – a median of 2.2 months shorter than propranolol-only therapy (P = .0006). This sequential regimen also was associated with no reinitiations of propranolol therapy following tapering, whereas 13% of those receiving propranolol alone had to reinitiate it after tapering.
This is of interest because oral beta-blockers, including propranolol, have been associated with rare but serious adverse events, such as bronchospasm, hypotension, and hypoglycemia.
Limitations of the study include its retrospective and single-center nature. There was no funding or disclosure information given.
SOURCE: Mannschreck DB et al. Pediatr Dermatol. 2019 Apr 9. doi: 10.1111/pde.13816.
FROM PEDIATRIC DERMATOLOGY
Some “slime”-related contact dermatitis is allergic
The viscous homemade children’s plaything known as “slime” has been associated with allergic, as well as irritant, contact dermatitis of the hands thanks to an array of possible compounds with which it can be made, according to a case report in Pediatric Dermatology. The report details many possible compounds causing the dermatitis reactions seen by health care professionals.

In the case, which was reported by L. Elizabeth Anderson, MD, of the Children’s Hospital of Philadelphia and colleagues, an 11-year-old girl with a history of atopic dermatitis presented with hand dermatitis that was suspected to be related to playing with slime. After her dermatitis failed to respond to strong topical steroids, she was referred for patch testing, with positivity for methylchloroisothiazolinone/methylisothiazolinone (MCI/MI). After all contact with any products containing MCI/MI was eliminated, her hand dermatitis cleared, and bodywide atopic dermatitis improved some as well.
MCI/MI and MI are among the most commonly suspected culprits in cases of slime-related contact dermatitis. Although most cases are irritant contact dermatitis, some are allergic and can be detected using patch tests. MCI/MI is included in the T.R.U.E. Test, but according to the case report, 37% of patients with allergy to MI alone will not have positive response with the T.R.U.E. Test because of the low concentrations of MI in that test. The authors of this case report also listed many other the potential allergens in popular slime recipes; however, many are not included in the T.R.U.E. Test.
“While the T.R.U.E. Test does not capture most of the potential allergens in popular slime recipes, the recently published Pediatric Baseline Patch Test Series by Yu et al. [Dermatitis. 2018;29:206-12] does and is recommended for use in patients suspected of having dermatitis secondary to slime,” Dr. Anderson and associates wrote.
SOURCE: Anderson LE et al. Pediatr Dermatol. 2019 Mar 13. doi: 10.1111/pde.13792.
The viscous homemade children’s plaything known as “slime” has been associated with allergic, as well as irritant, contact dermatitis of the hands thanks to an array of possible compounds with which it can be made, according to a case report in Pediatric Dermatology. The report details many possible compounds causing the dermatitis reactions seen by health care professionals.
In the case, which was reported by L. Elizabeth Anderson, MD, of the Children’s Hospital of Philadelphia and colleagues, an 11-year-old girl with a history of atopic dermatitis presented with hand dermatitis that was suspected to be related to playing with slime. After her dermatitis failed to respond to strong topical steroids, she was referred for patch testing, with positivity for methylchloroisothiazolinone/methylisothiazolinone (MCI/MI). After all contact with any products containing MCI/MI was eliminated, her hand dermatitis cleared, and bodywide atopic dermatitis improved some as well.
MCI/MI and MI are among the most commonly suspected culprits in cases of slime-related contact dermatitis. Although most cases are irritant contact dermatitis, some are allergic and can be detected using patch tests. MCI/MI is included in the T.R.U.E. Test, but according to the case report, 37% of patients with allergy to MI alone will not have positive response with the T.R.U.E. Test because of the low concentrations of MI in that test. The authors of this case report also listed many other the potential allergens in popular slime recipes; however, many are not included in the T.R.U.E. Test.
“While the T.R.U.E. Test does not capture most of the potential allergens in popular slime recipes, the recently published Pediatric Baseline Patch Test Series by Yu et al. [Dermatitis. 2018;29:206-12] does and is recommended for use in patients suspected of having dermatitis secondary to slime,” Dr. Anderson and associates wrote.
SOURCE: Anderson LE et al. Pediatr Dermatol. 2019 Mar 13. doi: 10.1111/pde.13792.
The viscous homemade children’s plaything known as “slime” has been associated with allergic, as well as irritant, contact dermatitis of the hands thanks to an array of possible compounds with which it can be made, according to a case report in Pediatric Dermatology. The report details many possible compounds causing the dermatitis reactions seen by health care professionals.
In the case, which was reported by L. Elizabeth Anderson, MD, of the Children’s Hospital of Philadelphia and colleagues, an 11-year-old girl with a history of atopic dermatitis presented with hand dermatitis that was suspected to be related to playing with slime. After her dermatitis failed to respond to strong topical steroids, she was referred for patch testing, with positivity for methylchloroisothiazolinone/methylisothiazolinone (MCI/MI). After all contact with any products containing MCI/MI was eliminated, her hand dermatitis cleared, and bodywide atopic dermatitis improved some as well.
MCI/MI and MI are among the most commonly suspected culprits in cases of slime-related contact dermatitis. Although most cases are irritant contact dermatitis, some are allergic and can be detected using patch tests. MCI/MI is included in the T.R.U.E. Test, but according to the case report, 37% of patients with allergy to MI alone will not have positive response with the T.R.U.E. Test because of the low concentrations of MI in that test. The authors of this case report also listed many other the potential allergens in popular slime recipes; however, many are not included in the T.R.U.E. Test.
“While the T.R.U.E. Test does not capture most of the potential allergens in popular slime recipes, the recently published Pediatric Baseline Patch Test Series by Yu et al. [Dermatitis. 2018;29:206-12] does and is recommended for use in patients suspected of having dermatitis secondary to slime,” Dr. Anderson and associates wrote.
SOURCE: Anderson LE et al. Pediatr Dermatol. 2019 Mar 13. doi: 10.1111/pde.13792.
FROM PEDIATRIC DERMATOLOGY
FDA approves first drug for steroid-refractory acute GVHD
The Food and Drug Administration has approved Jafaki (ruxolitinib) for treatment of steroid-refractory acute graft-versus-host disease (GVHD) in adult and pediatric patients 12 years and older.
Ruxolitinib will be made available to appropriate patients immediately, according to a statement from Incyte, which markets the drug. The company noted that ruxolitinib is the first FDA-approved treatment for this indication.
The approval is based on data from the open-label, single-arm, multicenter REACH1 trial, which studied ruxolitinib in combination with corticosteroids. The 71 patients in the trial had grade 2-4 acute GVHD after allogeneic hematopoietic stem cell transplant; of these patients, 49 were refractory to steroids alone, 12 had received at least two prior therapies for GVHD, and 10 did not otherwise meet the FDA definition of steroid refractory.
The trial’s primary endpoints were day-28 overall response rate and response duration. Among the 49 patients with steroid only–refractory GVHD, the overall response rate was 100% for grade 2 GVHD, 40.7% for grade 3, and 44.4% for grade 4. Median response duration was 16 days. For all 49 of these patients, the overall response rate was 57%, and the complete response rate was 31%.
Among all 71 participants, the most frequently reported adverse reactions were infections (55%) and edema (51%); anemia (71%), thrombocytopenia (75%), and neutropenia (58%) were the most common laboratory abnormalities.
The Food and Drug Administration has approved Jafaki (ruxolitinib) for treatment of steroid-refractory acute graft-versus-host disease (GVHD) in adult and pediatric patients 12 years and older.
Ruxolitinib will be made available to appropriate patients immediately, according to a statement from Incyte, which markets the drug. The company noted that ruxolitinib is the first FDA-approved treatment for this indication.
The approval is based on data from the open-label, single-arm, multicenter REACH1 trial, which studied ruxolitinib in combination with corticosteroids. The 71 patients in the trial had grade 2-4 acute GVHD after allogeneic hematopoietic stem cell transplant; of these patients, 49 were refractory to steroids alone, 12 had received at least two prior therapies for GVHD, and 10 did not otherwise meet the FDA definition of steroid refractory.
The trial’s primary endpoints were day-28 overall response rate and response duration. Among the 49 patients with steroid only–refractory GVHD, the overall response rate was 100% for grade 2 GVHD, 40.7% for grade 3, and 44.4% for grade 4. Median response duration was 16 days. For all 49 of these patients, the overall response rate was 57%, and the complete response rate was 31%.
Among all 71 participants, the most frequently reported adverse reactions were infections (55%) and edema (51%); anemia (71%), thrombocytopenia (75%), and neutropenia (58%) were the most common laboratory abnormalities.
The Food and Drug Administration has approved Jafaki (ruxolitinib) for treatment of steroid-refractory acute graft-versus-host disease (GVHD) in adult and pediatric patients 12 years and older.
Ruxolitinib will be made available to appropriate patients immediately, according to a statement from Incyte, which markets the drug. The company noted that ruxolitinib is the first FDA-approved treatment for this indication.
The approval is based on data from the open-label, single-arm, multicenter REACH1 trial, which studied ruxolitinib in combination with corticosteroids. The 71 patients in the trial had grade 2-4 acute GVHD after allogeneic hematopoietic stem cell transplant; of these patients, 49 were refractory to steroids alone, 12 had received at least two prior therapies for GVHD, and 10 did not otherwise meet the FDA definition of steroid refractory.
The trial’s primary endpoints were day-28 overall response rate and response duration. Among the 49 patients with steroid only–refractory GVHD, the overall response rate was 100% for grade 2 GVHD, 40.7% for grade 3, and 44.4% for grade 4. Median response duration was 16 days. For all 49 of these patients, the overall response rate was 57%, and the complete response rate was 31%.
Among all 71 participants, the most frequently reported adverse reactions were infections (55%) and edema (51%); anemia (71%), thrombocytopenia (75%), and neutropenia (58%) were the most common laboratory abnormalities.