User login
Successful anti-TNF therapy may cancel excess coronary risk in RA
SAN DIEGO – Rheumatoid arthritis patients with a good response to tumor necrosis factor inhibitor therapy when assessed roughly 5 months into treatment had an acute coronary syndrome risk during the next 2 years that was no different than that of the matched general population in a large, Swedish national registry study.
"We could see that the risk of acute coronary syndrome in the TNF inhibitor–exposed population was doubled in the first year compared to the general population. But all this increased risk was carried by patients with a moderate or nonresponse to therapy. We saw no difference in risk between the general population and patients with a good response to treatment. My belief is that this benefit is not due to the TNF inhibitor as such, but rather it’s the control of inflammation that is crucial," Dr. Lotta Ljung said at the annual meeting of the American College of Rheumatology.

‘Good response’ was defined in this study via the EULAR response criteria: that is, a greater than 1.2-point improvement in the widely used Disease Activity Score 28 (DAS28) over baseline to a score of 3.2 or less at the 5-month evaluation.
Dr. Ljung, a senior consultant in rheumatology at Umea (Sweden) University Hospital, presented two analyses drawn from the Swedish Biologics Register, a national registry that captures 90% of all Swedes on biologic therapy for rheumatoid arthritis (RA). The study population included 7,704 RA patients with no history of ischemic heart disease when they started on their first TNF inhibitor during 2001-2010. They were matched by age, gender, and location to 23,112 RA patients who never took a biologic agent and to a second matched control group comprised of 38,520 individuals in the general population.
The crude incidence rate of acute coronary syndrome (ACS) in patients actively on TNF inhibitor therapy throughout follow-up was 5.7 events per 1,000 person-years, compared with 8.6 per 1,000 in biologic-naive RA patients and 3.3 per 1,000 in the matched general population.
In a fully adjusted Cox multivariate regression analysis factoring in socioeconomic variables, RA duration, joint surgery, and baseline atherosclerotic disease and other comorbid conditions, patients on anti-TNF therapy had a highly significant 27% reduction in ACS risk, compared with biologic-naive RA patients.
Nonetheless, patients on TNF inhibitor therapy remained at an adjusted 1.5-fold increased risk of ACS, compared with general population controls. However, this was significantly lower than the 2.3-fold elevated risk in biologic-naive RA patients.
In a separate analysis, the investigators took a closer look at the Swedish Biologics Register subgroup of the 4,931 RA patients on anti-TNF therapy for whom EULAR response data 5 months into treatment were available. Thirty-eight percent of these patients had a EULAR good response, 37% had a moderate response, and 25% had no response.
During 2 years of follow-up starting at the time of the EULAR response evaluation, the crude incidence rate of ACS among all TNF inhibitor–exposed RA patients, with close to 8,600 person-years of follow-up, was 6.9 cases per 1,000 person-years, compared with 3.4 per 1,000 among the matched general population controls. In an adjusted multivariate regression analysis, the ACS risk was 1.94-fold greater in moderate responders to anti-TNF therapy than in the general population and 2.53-fold greater in the nonresponders, but not significantly different between good responders and controls.
In addition, patients with an erythrocyte sedimentation rate (ESR) below 20 mm/hour at the time of their EULAR response evaluation had a subsequent 66% lower 1-year risk of ACS than did those with a higher ESR. And patients with a DAS28 remission at the 5-month evaluation – that is, a DAS28 below 2.6 – had a 79% lower ACS risk than did those with a DAS28 of 2.6 or above.
"This is dramatic," Dr. Ljung said in an interview. "I think it’s the first time we see a population in our RA cohorts that doesn’t have any proven cardiovascular risk increase compared with the general population. But it raises additional questions, of course, such as who are these patients who receive the good response: Is it due to factors related to their disease or background that gives them the opportunity to have the good response? We adjusted for a number of factors, but still ..."
She added that these studies contain two key take-home messages for rheumatologists: "I think the first thing for us to do is to treat our patients’ inflammation perfectly using traditional and biologic DMARDs. And the second thing is to be more aware of the traditional risk factors and start modifying those more aggressively for our patients."
The Swedish Biologics Register is funded by the Swedish Rheumatology Association, with support from half a dozen pharmaceutical companies. Dr. Ljung disclosed ties with AbbVie and Bristol-Myers Squibb.
SAN DIEGO – Rheumatoid arthritis patients with a good response to tumor necrosis factor inhibitor therapy when assessed roughly 5 months into treatment had an acute coronary syndrome risk during the next 2 years that was no different than that of the matched general population in a large, Swedish national registry study.
"We could see that the risk of acute coronary syndrome in the TNF inhibitor–exposed population was doubled in the first year compared to the general population. But all this increased risk was carried by patients with a moderate or nonresponse to therapy. We saw no difference in risk between the general population and patients with a good response to treatment. My belief is that this benefit is not due to the TNF inhibitor as such, but rather it’s the control of inflammation that is crucial," Dr. Lotta Ljung said at the annual meeting of the American College of Rheumatology.
‘Good response’ was defined in this study via the EULAR response criteria: that is, a greater than 1.2-point improvement in the widely used Disease Activity Score 28 (DAS28) over baseline to a score of 3.2 or less at the 5-month evaluation.
Dr. Ljung, a senior consultant in rheumatology at Umea (Sweden) University Hospital, presented two analyses drawn from the Swedish Biologics Register, a national registry that captures 90% of all Swedes on biologic therapy for rheumatoid arthritis (RA). The study population included 7,704 RA patients with no history of ischemic heart disease when they started on their first TNF inhibitor during 2001-2010. They were matched by age, gender, and location to 23,112 RA patients who never took a biologic agent and to a second matched control group comprised of 38,520 individuals in the general population.
The crude incidence rate of acute coronary syndrome (ACS) in patients actively on TNF inhibitor therapy throughout follow-up was 5.7 events per 1,000 person-years, compared with 8.6 per 1,000 in biologic-naive RA patients and 3.3 per 1,000 in the matched general population.
In a fully adjusted Cox multivariate regression analysis factoring in socioeconomic variables, RA duration, joint surgery, and baseline atherosclerotic disease and other comorbid conditions, patients on anti-TNF therapy had a highly significant 27% reduction in ACS risk, compared with biologic-naive RA patients.
Nonetheless, patients on TNF inhibitor therapy remained at an adjusted 1.5-fold increased risk of ACS, compared with general population controls. However, this was significantly lower than the 2.3-fold elevated risk in biologic-naive RA patients.
In a separate analysis, the investigators took a closer look at the Swedish Biologics Register subgroup of the 4,931 RA patients on anti-TNF therapy for whom EULAR response data 5 months into treatment were available. Thirty-eight percent of these patients had a EULAR good response, 37% had a moderate response, and 25% had no response.
During 2 years of follow-up starting at the time of the EULAR response evaluation, the crude incidence rate of ACS among all TNF inhibitor–exposed RA patients, with close to 8,600 person-years of follow-up, was 6.9 cases per 1,000 person-years, compared with 3.4 per 1,000 among the matched general population controls. In an adjusted multivariate regression analysis, the ACS risk was 1.94-fold greater in moderate responders to anti-TNF therapy than in the general population and 2.53-fold greater in the nonresponders, but not significantly different between good responders and controls.
In addition, patients with an erythrocyte sedimentation rate (ESR) below 20 mm/hour at the time of their EULAR response evaluation had a subsequent 66% lower 1-year risk of ACS than did those with a higher ESR. And patients with a DAS28 remission at the 5-month evaluation – that is, a DAS28 below 2.6 – had a 79% lower ACS risk than did those with a DAS28 of 2.6 or above.
"This is dramatic," Dr. Ljung said in an interview. "I think it’s the first time we see a population in our RA cohorts that doesn’t have any proven cardiovascular risk increase compared with the general population. But it raises additional questions, of course, such as who are these patients who receive the good response: Is it due to factors related to their disease or background that gives them the opportunity to have the good response? We adjusted for a number of factors, but still ..."
She added that these studies contain two key take-home messages for rheumatologists: "I think the first thing for us to do is to treat our patients’ inflammation perfectly using traditional and biologic DMARDs. And the second thing is to be more aware of the traditional risk factors and start modifying those more aggressively for our patients."
The Swedish Biologics Register is funded by the Swedish Rheumatology Association, with support from half a dozen pharmaceutical companies. Dr. Ljung disclosed ties with AbbVie and Bristol-Myers Squibb.
SAN DIEGO – Rheumatoid arthritis patients with a good response to tumor necrosis factor inhibitor therapy when assessed roughly 5 months into treatment had an acute coronary syndrome risk during the next 2 years that was no different than that of the matched general population in a large, Swedish national registry study.
"We could see that the risk of acute coronary syndrome in the TNF inhibitor–exposed population was doubled in the first year compared to the general population. But all this increased risk was carried by patients with a moderate or nonresponse to therapy. We saw no difference in risk between the general population and patients with a good response to treatment. My belief is that this benefit is not due to the TNF inhibitor as such, but rather it’s the control of inflammation that is crucial," Dr. Lotta Ljung said at the annual meeting of the American College of Rheumatology.
‘Good response’ was defined in this study via the EULAR response criteria: that is, a greater than 1.2-point improvement in the widely used Disease Activity Score 28 (DAS28) over baseline to a score of 3.2 or less at the 5-month evaluation.
Dr. Ljung, a senior consultant in rheumatology at Umea (Sweden) University Hospital, presented two analyses drawn from the Swedish Biologics Register, a national registry that captures 90% of all Swedes on biologic therapy for rheumatoid arthritis (RA). The study population included 7,704 RA patients with no history of ischemic heart disease when they started on their first TNF inhibitor during 2001-2010. They were matched by age, gender, and location to 23,112 RA patients who never took a biologic agent and to a second matched control group comprised of 38,520 individuals in the general population.
The crude incidence rate of acute coronary syndrome (ACS) in patients actively on TNF inhibitor therapy throughout follow-up was 5.7 events per 1,000 person-years, compared with 8.6 per 1,000 in biologic-naive RA patients and 3.3 per 1,000 in the matched general population.
In a fully adjusted Cox multivariate regression analysis factoring in socioeconomic variables, RA duration, joint surgery, and baseline atherosclerotic disease and other comorbid conditions, patients on anti-TNF therapy had a highly significant 27% reduction in ACS risk, compared with biologic-naive RA patients.
Nonetheless, patients on TNF inhibitor therapy remained at an adjusted 1.5-fold increased risk of ACS, compared with general population controls. However, this was significantly lower than the 2.3-fold elevated risk in biologic-naive RA patients.
In a separate analysis, the investigators took a closer look at the Swedish Biologics Register subgroup of the 4,931 RA patients on anti-TNF therapy for whom EULAR response data 5 months into treatment were available. Thirty-eight percent of these patients had a EULAR good response, 37% had a moderate response, and 25% had no response.
During 2 years of follow-up starting at the time of the EULAR response evaluation, the crude incidence rate of ACS among all TNF inhibitor–exposed RA patients, with close to 8,600 person-years of follow-up, was 6.9 cases per 1,000 person-years, compared with 3.4 per 1,000 among the matched general population controls. In an adjusted multivariate regression analysis, the ACS risk was 1.94-fold greater in moderate responders to anti-TNF therapy than in the general population and 2.53-fold greater in the nonresponders, but not significantly different between good responders and controls.
In addition, patients with an erythrocyte sedimentation rate (ESR) below 20 mm/hour at the time of their EULAR response evaluation had a subsequent 66% lower 1-year risk of ACS than did those with a higher ESR. And patients with a DAS28 remission at the 5-month evaluation – that is, a DAS28 below 2.6 – had a 79% lower ACS risk than did those with a DAS28 of 2.6 or above.
"This is dramatic," Dr. Ljung said in an interview. "I think it’s the first time we see a population in our RA cohorts that doesn’t have any proven cardiovascular risk increase compared with the general population. But it raises additional questions, of course, such as who are these patients who receive the good response: Is it due to factors related to their disease or background that gives them the opportunity to have the good response? We adjusted for a number of factors, but still ..."
She added that these studies contain two key take-home messages for rheumatologists: "I think the first thing for us to do is to treat our patients’ inflammation perfectly using traditional and biologic DMARDs. And the second thing is to be more aware of the traditional risk factors and start modifying those more aggressively for our patients."
The Swedish Biologics Register is funded by the Swedish Rheumatology Association, with support from half a dozen pharmaceutical companies. Dr. Ljung disclosed ties with AbbVie and Bristol-Myers Squibb.
AT THE ACR ANNUAL MEETING
Major finding: The acute coronary syndrome risk was 1.94-fold greater in rheumatoid arthritis patients with a moderate response to anti-TNF therapy than in the general population and 2.53-fold greater in those with no response, but not significantly different between controls and good responders (defined by the degree of improvement in the Disease Activity Score 28 when evaluated 5 months into treatment).
Data source: An observational study of 7,704 rheumatoid arthritis patients in the nationwide Swedish Biologics Register who started on a first tumor necrosis factor inhibitor during 2001-2010, together with 23,112 matched biologic-naive rheumatoid arthritis patients and 38,520 matched controls drawn from the general population.
Disclosures: The Swedish Biologics Register is funded by the Swedish Rheumatology Association, with support from half a dozen pharmaceutical companies. The presenter disclosed ties to AbbVie and Bristol-Myers Squibb.

TOPCAT: Spironolactone cuts hospitalizations for diastolic heart failure
DALLAS – Spironolactone did not hit a home run in the large international "treatment of preserved cardiac function heart failure with an aldosterone antagonist" (TOPCAT) trial, but it did knock out a solid single in the form of significantly reduced hospitalizations for this extremely common, chronic, high morbidity/mortality condition.
It was this positive result for an important prespecified secondary outcome that enabled TOPCAT to avoid becoming roadkill. Technically, TOPCAT was a negative clinical trial in that spironolactone did not significantly outperform placebo on the primary composite outcome of cardiovascular mortality, heart failure hospitalization, or aborted cardiac arrest.
Yet that negative primary outcome was controversial: The aldosterone antagonist actually showed a significant positive result for the composite endpoint in North and South American participants, yet the results were resoundingly negative – and also considerably out of whack with the characteristic arc of progressive heart failure – among the nearly one-half of TOPCAT participants in Russia and the Republic of Georgia.
"What happened in Russia and Georgia we just don’t understand," Dr. Bertram Pitt, TOPCAT steering committee chair, said in an interview, shaking his head. "The event rate with placebo in Eastern Europe was so low it’s not compatible with anything we know about heart failure. The signs and symptoms of HFpEF [heart failure with preserved ejection fraction] are nonspecific; they can be due to obesity, lung disease, and other things. Clearly there are some people getting into the major trials of HFpEF that probably don’t have it."
TOPCAT was a randomized, double-blind clinical trial comprising 3,445 participants with symptomatic HFpEF at 250 sites in the United States and five other countries. They were randomized to spironolactone or placebo and followed prospectively for a mean of 3.3 years. The starting dose of the aldosterone antagonist was 15 mg/day, with a target of 30 mg/day. The drug could be titrated within the range of 15-45 mg/day. Eight months into the trial, the mean daily dose was 25 mg.

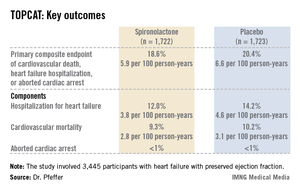
Presenting the TOPCAT results at the American Heart Association scientific sessions, Dr. Marc A. Pfeffer noted that the primary composite endpoint occurred in 20.4% of placebo-treated controls and 18.6% on spironolactone, a statistically nonsignificant difference. In contrast, the 17% reduction in the rate of hospitalization for HFpEF in the spironolactone group relative to controls was significant (P = .04). Moreover, the spironolactone-treated patients had a collective 394 HFpEF hospitalizations, markedly fewer than the 475 in controls. This translated to a hospitalization for heart failure with a preserved left ventricular ejection fraction occurring at a rate of 3.8 per 100 person-years in patients randomized to spironolactone, compared with the 4.6 per 100 person-years in placebo-treated controls.
Hyperkalemia in excess of 5.5 mmol/L occurred in 18.7% of the spironolactone group, twice the rate of controls (9.1%). And the incidence of a creatinine level more than double the upper limit of normal was 49% greater in the spironolactone group. That said, neither of these laboratory abnormalities resulted in any serious adverse consequences because investigators adjusted the dose in response, explained Dr. Pfeffer, professor of medicine at Harvard University, Boston.
He drew special attention to two points: The primary composite event rate in placebo-treated patients in the Americas was 31.8% consistent with what has been seen in other studies of HFpEF – compared to a mere 8.4% in Eastern Europe. And patients who qualified for TOPCAT on the basis of an elevated natriuretic peptide level had a primary endpoint rate of 15.9% with spironolactone, a highly significant 35% reduction compared with the 23.6% in controls, suggesting that an elevated baseline natriuretic peptide level may be a biomarker useful in identifying those HFpEF patients most likely to respond to an aldosterone antagonist.
Dr. Pfeffer said that because the pharmaceutical industry has zero interest in spironolactone and the National Heart, Lung, and Blood Institute (NHLBI), which sponsored TOPCAT, has finite resources, he doubts there will be any further large studies of the drug in HFpEF.
"One is going to have to make decisions based on this trial. I don’t see another trial behind us," the cardiologist said.
And while TOPCAT was flawed, he said it contains a compelling message for clinicians: "I think we have an important finding here. We’re very confident that with this generic medication, which costs pennies per day, we can reduce hospitalizations for heart failure, which are the major burden in patients with HFpEF."
Discussant Dr. Margaret M. Redfield was more cautious. While there was a sound rationale for studying spironolactone in HFpEF based upon its impressive benefits in systolic heart failure as shown in earlier landmark clinical trials, given that the drug didn’t result in a significant reduction in all-cause hospitalizations, she said she wants to see evidence of improved patient-centered outcomes, such as quality of life, before prescribing spironolactone for HFpEF.
"One has to worry that the problems with worsening renal function and hyperkalemia may be much more common in clinical practice than in the highly monitored environment of a clinical trial," added Dr. Redfield, professor of medicine at the Mayo Clinic, Rochester, Minn.

Other heart failure experts not involved in TOPCAT took a more positive view of the study.
Dr. Clyde W. Yancy, AHA spokesperson, said in an interview that HFpEF is probably even more common than systolic heart failure, and it is a disease for which physicians have had no proven-effective treatments.
"You can almost justifiably say, ‘Let’s just look at the Americas, where patients had event rates that are consistent with the disease we think we understand.’ And it looks like there’s a signal there. I think that there’s enough evidence in TOPCAT for the clinical pragmatist to be able to be comfortable that spironolactone is probably beneficial and can be given safely. If you are convinced you can give it safely in your own clinical scenario, I think you should use it," said Dr. Yancy, professor of medicine and chief of cardiology at Northwestern University, Chicago.
Dr. Marco Metra, a cardiologist at the University of Brescia (Italy), said he interprets the TOPCAT results as an indication for prescribing spironolactone, at least in HFpEF patients with high natriuretic peptide levels or multiple heart failure hospitalizations.
Dr. Pitt, emeritus professor of cardiovascular medicine at the University of Michigan, Ann Arbor, emphasized that in prescribing spironolactone, whether for HFpEF or systolic heart failure, it’s obligatory to measure potassium and creatinine levels at baseline, when changing the dose, and at each routine follow-up visit, titrating in response to the results. Failure to do so is akin to prescribing warfarin for a patient with atrial fibrillation and then never measuring the INR (international normalized ratio) he said.
TOPCAT was sponsored by the National, Heart, Lung, and Blood Institute. Dr. Pfeffer and Dr. Pitt serve as consultants to numerous pharmaceutical companies that had no involvement in the trial.
DALLAS – Spironolactone did not hit a home run in the large international "treatment of preserved cardiac function heart failure with an aldosterone antagonist" (TOPCAT) trial, but it did knock out a solid single in the form of significantly reduced hospitalizations for this extremely common, chronic, high morbidity/mortality condition.
It was this positive result for an important prespecified secondary outcome that enabled TOPCAT to avoid becoming roadkill. Technically, TOPCAT was a negative clinical trial in that spironolactone did not significantly outperform placebo on the primary composite outcome of cardiovascular mortality, heart failure hospitalization, or aborted cardiac arrest.
Yet that negative primary outcome was controversial: The aldosterone antagonist actually showed a significant positive result for the composite endpoint in North and South American participants, yet the results were resoundingly negative – and also considerably out of whack with the characteristic arc of progressive heart failure – among the nearly one-half of TOPCAT participants in Russia and the Republic of Georgia.
"What happened in Russia and Georgia we just don’t understand," Dr. Bertram Pitt, TOPCAT steering committee chair, said in an interview, shaking his head. "The event rate with placebo in Eastern Europe was so low it’s not compatible with anything we know about heart failure. The signs and symptoms of HFpEF [heart failure with preserved ejection fraction] are nonspecific; they can be due to obesity, lung disease, and other things. Clearly there are some people getting into the major trials of HFpEF that probably don’t have it."
TOPCAT was a randomized, double-blind clinical trial comprising 3,445 participants with symptomatic HFpEF at 250 sites in the United States and five other countries. They were randomized to spironolactone or placebo and followed prospectively for a mean of 3.3 years. The starting dose of the aldosterone antagonist was 15 mg/day, with a target of 30 mg/day. The drug could be titrated within the range of 15-45 mg/day. Eight months into the trial, the mean daily dose was 25 mg.
Presenting the TOPCAT results at the American Heart Association scientific sessions, Dr. Marc A. Pfeffer noted that the primary composite endpoint occurred in 20.4% of placebo-treated controls and 18.6% on spironolactone, a statistically nonsignificant difference. In contrast, the 17% reduction in the rate of hospitalization for HFpEF in the spironolactone group relative to controls was significant (P = .04). Moreover, the spironolactone-treated patients had a collective 394 HFpEF hospitalizations, markedly fewer than the 475 in controls. This translated to a hospitalization for heart failure with a preserved left ventricular ejection fraction occurring at a rate of 3.8 per 100 person-years in patients randomized to spironolactone, compared with the 4.6 per 100 person-years in placebo-treated controls.
Hyperkalemia in excess of 5.5 mmol/L occurred in 18.7% of the spironolactone group, twice the rate of controls (9.1%). And the incidence of a creatinine level more than double the upper limit of normal was 49% greater in the spironolactone group. That said, neither of these laboratory abnormalities resulted in any serious adverse consequences because investigators adjusted the dose in response, explained Dr. Pfeffer, professor of medicine at Harvard University, Boston.
He drew special attention to two points: The primary composite event rate in placebo-treated patients in the Americas was 31.8% consistent with what has been seen in other studies of HFpEF – compared to a mere 8.4% in Eastern Europe. And patients who qualified for TOPCAT on the basis of an elevated natriuretic peptide level had a primary endpoint rate of 15.9% with spironolactone, a highly significant 35% reduction compared with the 23.6% in controls, suggesting that an elevated baseline natriuretic peptide level may be a biomarker useful in identifying those HFpEF patients most likely to respond to an aldosterone antagonist.
Dr. Pfeffer said that because the pharmaceutical industry has zero interest in spironolactone and the National Heart, Lung, and Blood Institute (NHLBI), which sponsored TOPCAT, has finite resources, he doubts there will be any further large studies of the drug in HFpEF.
"One is going to have to make decisions based on this trial. I don’t see another trial behind us," the cardiologist said.
And while TOPCAT was flawed, he said it contains a compelling message for clinicians: "I think we have an important finding here. We’re very confident that with this generic medication, which costs pennies per day, we can reduce hospitalizations for heart failure, which are the major burden in patients with HFpEF."
Discussant Dr. Margaret M. Redfield was more cautious. While there was a sound rationale for studying spironolactone in HFpEF based upon its impressive benefits in systolic heart failure as shown in earlier landmark clinical trials, given that the drug didn’t result in a significant reduction in all-cause hospitalizations, she said she wants to see evidence of improved patient-centered outcomes, such as quality of life, before prescribing spironolactone for HFpEF.
"One has to worry that the problems with worsening renal function and hyperkalemia may be much more common in clinical practice than in the highly monitored environment of a clinical trial," added Dr. Redfield, professor of medicine at the Mayo Clinic, Rochester, Minn.
Other heart failure experts not involved in TOPCAT took a more positive view of the study.
Dr. Clyde W. Yancy, AHA spokesperson, said in an interview that HFpEF is probably even more common than systolic heart failure, and it is a disease for which physicians have had no proven-effective treatments.
"You can almost justifiably say, ‘Let’s just look at the Americas, where patients had event rates that are consistent with the disease we think we understand.’ And it looks like there’s a signal there. I think that there’s enough evidence in TOPCAT for the clinical pragmatist to be able to be comfortable that spironolactone is probably beneficial and can be given safely. If you are convinced you can give it safely in your own clinical scenario, I think you should use it," said Dr. Yancy, professor of medicine and chief of cardiology at Northwestern University, Chicago.
Dr. Marco Metra, a cardiologist at the University of Brescia (Italy), said he interprets the TOPCAT results as an indication for prescribing spironolactone, at least in HFpEF patients with high natriuretic peptide levels or multiple heart failure hospitalizations.
Dr. Pitt, emeritus professor of cardiovascular medicine at the University of Michigan, Ann Arbor, emphasized that in prescribing spironolactone, whether for HFpEF or systolic heart failure, it’s obligatory to measure potassium and creatinine levels at baseline, when changing the dose, and at each routine follow-up visit, titrating in response to the results. Failure to do so is akin to prescribing warfarin for a patient with atrial fibrillation and then never measuring the INR (international normalized ratio) he said.
TOPCAT was sponsored by the National, Heart, Lung, and Blood Institute. Dr. Pfeffer and Dr. Pitt serve as consultants to numerous pharmaceutical companies that had no involvement in the trial.
DALLAS – Spironolactone did not hit a home run in the large international "treatment of preserved cardiac function heart failure with an aldosterone antagonist" (TOPCAT) trial, but it did knock out a solid single in the form of significantly reduced hospitalizations for this extremely common, chronic, high morbidity/mortality condition.
It was this positive result for an important prespecified secondary outcome that enabled TOPCAT to avoid becoming roadkill. Technically, TOPCAT was a negative clinical trial in that spironolactone did not significantly outperform placebo on the primary composite outcome of cardiovascular mortality, heart failure hospitalization, or aborted cardiac arrest.
Yet that negative primary outcome was controversial: The aldosterone antagonist actually showed a significant positive result for the composite endpoint in North and South American participants, yet the results were resoundingly negative – and also considerably out of whack with the characteristic arc of progressive heart failure – among the nearly one-half of TOPCAT participants in Russia and the Republic of Georgia.
"What happened in Russia and Georgia we just don’t understand," Dr. Bertram Pitt, TOPCAT steering committee chair, said in an interview, shaking his head. "The event rate with placebo in Eastern Europe was so low it’s not compatible with anything we know about heart failure. The signs and symptoms of HFpEF [heart failure with preserved ejection fraction] are nonspecific; they can be due to obesity, lung disease, and other things. Clearly there are some people getting into the major trials of HFpEF that probably don’t have it."
TOPCAT was a randomized, double-blind clinical trial comprising 3,445 participants with symptomatic HFpEF at 250 sites in the United States and five other countries. They were randomized to spironolactone or placebo and followed prospectively for a mean of 3.3 years. The starting dose of the aldosterone antagonist was 15 mg/day, with a target of 30 mg/day. The drug could be titrated within the range of 15-45 mg/day. Eight months into the trial, the mean daily dose was 25 mg.
Presenting the TOPCAT results at the American Heart Association scientific sessions, Dr. Marc A. Pfeffer noted that the primary composite endpoint occurred in 20.4% of placebo-treated controls and 18.6% on spironolactone, a statistically nonsignificant difference. In contrast, the 17% reduction in the rate of hospitalization for HFpEF in the spironolactone group relative to controls was significant (P = .04). Moreover, the spironolactone-treated patients had a collective 394 HFpEF hospitalizations, markedly fewer than the 475 in controls. This translated to a hospitalization for heart failure with a preserved left ventricular ejection fraction occurring at a rate of 3.8 per 100 person-years in patients randomized to spironolactone, compared with the 4.6 per 100 person-years in placebo-treated controls.
Hyperkalemia in excess of 5.5 mmol/L occurred in 18.7% of the spironolactone group, twice the rate of controls (9.1%). And the incidence of a creatinine level more than double the upper limit of normal was 49% greater in the spironolactone group. That said, neither of these laboratory abnormalities resulted in any serious adverse consequences because investigators adjusted the dose in response, explained Dr. Pfeffer, professor of medicine at Harvard University, Boston.
He drew special attention to two points: The primary composite event rate in placebo-treated patients in the Americas was 31.8% consistent with what has been seen in other studies of HFpEF – compared to a mere 8.4% in Eastern Europe. And patients who qualified for TOPCAT on the basis of an elevated natriuretic peptide level had a primary endpoint rate of 15.9% with spironolactone, a highly significant 35% reduction compared with the 23.6% in controls, suggesting that an elevated baseline natriuretic peptide level may be a biomarker useful in identifying those HFpEF patients most likely to respond to an aldosterone antagonist.
Dr. Pfeffer said that because the pharmaceutical industry has zero interest in spironolactone and the National Heart, Lung, and Blood Institute (NHLBI), which sponsored TOPCAT, has finite resources, he doubts there will be any further large studies of the drug in HFpEF.
"One is going to have to make decisions based on this trial. I don’t see another trial behind us," the cardiologist said.
And while TOPCAT was flawed, he said it contains a compelling message for clinicians: "I think we have an important finding here. We’re very confident that with this generic medication, which costs pennies per day, we can reduce hospitalizations for heart failure, which are the major burden in patients with HFpEF."
Discussant Dr. Margaret M. Redfield was more cautious. While there was a sound rationale for studying spironolactone in HFpEF based upon its impressive benefits in systolic heart failure as shown in earlier landmark clinical trials, given that the drug didn’t result in a significant reduction in all-cause hospitalizations, she said she wants to see evidence of improved patient-centered outcomes, such as quality of life, before prescribing spironolactone for HFpEF.
"One has to worry that the problems with worsening renal function and hyperkalemia may be much more common in clinical practice than in the highly monitored environment of a clinical trial," added Dr. Redfield, professor of medicine at the Mayo Clinic, Rochester, Minn.
Other heart failure experts not involved in TOPCAT took a more positive view of the study.
Dr. Clyde W. Yancy, AHA spokesperson, said in an interview that HFpEF is probably even more common than systolic heart failure, and it is a disease for which physicians have had no proven-effective treatments.
"You can almost justifiably say, ‘Let’s just look at the Americas, where patients had event rates that are consistent with the disease we think we understand.’ And it looks like there’s a signal there. I think that there’s enough evidence in TOPCAT for the clinical pragmatist to be able to be comfortable that spironolactone is probably beneficial and can be given safely. If you are convinced you can give it safely in your own clinical scenario, I think you should use it," said Dr. Yancy, professor of medicine and chief of cardiology at Northwestern University, Chicago.
Dr. Marco Metra, a cardiologist at the University of Brescia (Italy), said he interprets the TOPCAT results as an indication for prescribing spironolactone, at least in HFpEF patients with high natriuretic peptide levels or multiple heart failure hospitalizations.
Dr. Pitt, emeritus professor of cardiovascular medicine at the University of Michigan, Ann Arbor, emphasized that in prescribing spironolactone, whether for HFpEF or systolic heart failure, it’s obligatory to measure potassium and creatinine levels at baseline, when changing the dose, and at each routine follow-up visit, titrating in response to the results. Failure to do so is akin to prescribing warfarin for a patient with atrial fibrillation and then never measuring the INR (international normalized ratio) he said.
TOPCAT was sponsored by the National, Heart, Lung, and Blood Institute. Dr. Pfeffer and Dr. Pitt serve as consultants to numerous pharmaceutical companies that had no involvement in the trial.
AT THE AHA SCIENTIFIC SESSIONS
Major finding: Hospitalization for heart failure with a preserved left ventricular ejection fraction occurred at a rate of 3.8 per 100 person-years in patients randomized to spironolactone, a significant 17% risk reduction compared with the 4.6 per 100 person-years in placebo-treated controls.
Data source: TOPCAT, a randomized, double-blind, six-country clinical trial involving 3,445 patients with symptomatic heart failure and an ejection fraction of 45% or more.
Disclosures:. TOPCAT was sponsored by the National, Heart, Lung, and Blood Institute. Dr. Pfeffer and Dr. Pitt serve as consultants to pharmaceutical companies that had no involvement in the trial.

Gout cases jump sevenfold in half-century
SAN DIEGO – The number of Americans with gout has climbed sevenfold during the last 50 years, according to Dr. Eswar Krishnan.
The population burden of illness imposed by gout has risen both in men and women across all age groups, but most strikingly so in men older than 65 years, said Dr. Krishnan, director of clinical epidemiology in the division of immunology and rheumatology at Stanford (Calif.) University.
He turned to National Health and Nutrition Examination Survey (NHANES) data to gain a more precise picture of U.S. trends in gout prevalence over time than has previously been available. He accomplished this by comparing age- and sex-specific rates from the 1959-1962 and the 2009-2010 editions of the long-running Centers for Disease Control and Prevention–sponsored surveys.

The unadjusted population-based prevalence of self-reported gout jumped from 6 cases per 1,000 during 1959-1962 to 26 per 1,000 in 2009-2010. The estimated number of gout cases climbed from 1.1 million in 1960 to 8.1 million in 2010. Yet the proportion of gout patients who were women remained steady at 31% over time.
The mean age of Americans with gout rose over the half-century from 54 to 61 years among men and from 55 to 65 years among women, he reported at the annual meeting of the American College of Rheumatology.
Statistical analysis indicated that the increase in gout cases among women during the last 50 years could be accounted for entirely by the much-discussed societal growth in abdominal obesity. In contrast, the explanation for the increased prevalence of gout in men was multifactorial. The bulk of the increase was associated with an increased life span and the graying of America, coupled with higher rates of hypertension, diabetes, and abdominal obesity.
However, an immeasurable portion of the increase in gout during the past half-century is probably due to increased awareness of the disease among the U.S. population, Dr. Krishnan added.
NHANES is sponsored by the Centers for Disease Control and Prevention. Dr. Krishnan reported having received research grants from Takeda, which markets gout medication.
SAN DIEGO – The number of Americans with gout has climbed sevenfold during the last 50 years, according to Dr. Eswar Krishnan.
The population burden of illness imposed by gout has risen both in men and women across all age groups, but most strikingly so in men older than 65 years, said Dr. Krishnan, director of clinical epidemiology in the division of immunology and rheumatology at Stanford (Calif.) University.
He turned to National Health and Nutrition Examination Survey (NHANES) data to gain a more precise picture of U.S. trends in gout prevalence over time than has previously been available. He accomplished this by comparing age- and sex-specific rates from the 1959-1962 and the 2009-2010 editions of the long-running Centers for Disease Control and Prevention–sponsored surveys.
The unadjusted population-based prevalence of self-reported gout jumped from 6 cases per 1,000 during 1959-1962 to 26 per 1,000 in 2009-2010. The estimated number of gout cases climbed from 1.1 million in 1960 to 8.1 million in 2010. Yet the proportion of gout patients who were women remained steady at 31% over time.
The mean age of Americans with gout rose over the half-century from 54 to 61 years among men and from 55 to 65 years among women, he reported at the annual meeting of the American College of Rheumatology.
Statistical analysis indicated that the increase in gout cases among women during the last 50 years could be accounted for entirely by the much-discussed societal growth in abdominal obesity. In contrast, the explanation for the increased prevalence of gout in men was multifactorial. The bulk of the increase was associated with an increased life span and the graying of America, coupled with higher rates of hypertension, diabetes, and abdominal obesity.
However, an immeasurable portion of the increase in gout during the past half-century is probably due to increased awareness of the disease among the U.S. population, Dr. Krishnan added.
NHANES is sponsored by the Centers for Disease Control and Prevention. Dr. Krishnan reported having received research grants from Takeda, which markets gout medication.
SAN DIEGO – The number of Americans with gout has climbed sevenfold during the last 50 years, according to Dr. Eswar Krishnan.
The population burden of illness imposed by gout has risen both in men and women across all age groups, but most strikingly so in men older than 65 years, said Dr. Krishnan, director of clinical epidemiology in the division of immunology and rheumatology at Stanford (Calif.) University.
He turned to National Health and Nutrition Examination Survey (NHANES) data to gain a more precise picture of U.S. trends in gout prevalence over time than has previously been available. He accomplished this by comparing age- and sex-specific rates from the 1959-1962 and the 2009-2010 editions of the long-running Centers for Disease Control and Prevention–sponsored surveys.
The unadjusted population-based prevalence of self-reported gout jumped from 6 cases per 1,000 during 1959-1962 to 26 per 1,000 in 2009-2010. The estimated number of gout cases climbed from 1.1 million in 1960 to 8.1 million in 2010. Yet the proportion of gout patients who were women remained steady at 31% over time.
The mean age of Americans with gout rose over the half-century from 54 to 61 years among men and from 55 to 65 years among women, he reported at the annual meeting of the American College of Rheumatology.
Statistical analysis indicated that the increase in gout cases among women during the last 50 years could be accounted for entirely by the much-discussed societal growth in abdominal obesity. In contrast, the explanation for the increased prevalence of gout in men was multifactorial. The bulk of the increase was associated with an increased life span and the graying of America, coupled with higher rates of hypertension, diabetes, and abdominal obesity.
However, an immeasurable portion of the increase in gout during the past half-century is probably due to increased awareness of the disease among the U.S. population, Dr. Krishnan added.
NHANES is sponsored by the Centers for Disease Control and Prevention. Dr. Krishnan reported having received research grants from Takeda, which markets gout medication.
AT THE ACR ANNUAL MEETING
Major finding: The estimated number of Americans with gout in the United States increased more than sevenfold from 1.1 million in 1960 to 8.1 million in 2010.
Data source: The National Health and Nutrition Examination Survey is a program of periodic, large, government-funded, national cross-sectional surveys involving a representative sample of the U.S. population.
Disclosures: The National Health and Nutrition Examination Survey is sponsored by the Centers for Disease Control and Prevention. The presenter reported receiving research grants from Takeda, which markets gout medication.

Antimalarials prove protective against long-term lupus damage
SAN DIEGO – Potentially modifiable risk factors for future irreversible organ damage in lupus patients include hypertension, the use of corticosteroids, and higher levels of inflammation early on, according to findings from the SLICC (Systemic Lupus International Collaborating Clinics) Inception Cohort Study.
In addition, the study identified the use of antimalarial drugs as the one significant protective factor against steady accrual of irreversible organ damage in lupus patients.
"These findings help us pave the way to consider whether, firstly, one could use damage as a primary endpoint in future clinical trials in lupus – somewhat akin to how the erosion score is used in rheumatoid arthritis – and secondly, the results suggest particular interventions that might be important in reducing the risk of damage over time," Dr. Ian N. Bruce said at the annual meeting of the American College of Rheumatology.

The study also identified several fixed and unmodifiable risk factors for irreversible damage in lupus patients: older age at diagnosis, male gender, and being black or white Americans, added Dr. Bruce, professor of rheumatology at the University of Manchester (U.K.) and chair of the SLICC research group.
The SLICC Inception Cohort Study involves 1,722 patients at 31 centers in 11 countries in North America, Europe, and Asia who enrolled within 15 months after being formally diagnosed with systemic lupus erythematosus based upon the 1997 ACR criteria. They averaged 35 years of age and had an average of 4.25 comprehensive annual follow-up visits during the study period.
Irreversible organ damage was assessed using the SLICC/ACR Damage Index, or SDI. At baseline, 35% of patients had at least one item of damage as indicated by an SDI score of 1 or more. Over time, damage rates slowly and steadily increased such that by 6 years of follow-up 51% of participants had an SDI of at least 1.
In a multivariate analysis, patients with an SDI score of 1 at baseline had a highly significant 37% reduction in the risk of increasing their score during follow-up if they were taking antimalarials, compared with those not taking antimalarials.
On the other hand, patients with a baseline SDI of 1 were 61% more likely to experience an increase in their damage score during follow-up if they had hypertension and 43% more likely to do so if they were on corticosteroids than if they weren’t. Moreover, their risk of going from an SDI of 1 to a higher SDI indicative of mounting damage increased by 10% for every 3-point increase on the SLE Disease Activity Index (SLEDAI).
Patients with a baseline SDI of 0 were 64% more likely to progress to a score of 1 or more during follow-up if they were taking corticosteroids and 71% more likely to do so if they were hypertensive. Their risk also increased by 17% for each 3-point increase in SLEDAI. Men had a 48% greater risk of going from an SDI of 0 to 1 or more than women. Asians were 40% less likely to develop irreversible damage.
Each 1-point increase in SDI score was associated with a 46% increased risk of mortality, as well as with poorer health-related quality of life, especially as reflected in SF-36 physical component scores.
Session chair Dr. Roberto Caricchio of Temple University, Philadelphia, called the SLICC study "very important work."
"It teaches us to be aggressive up-front with our lupus patients, which we often aren’t. We tend to spare ourselves because it’s a chronic disease, and we know we’ll see these patients for the next 20 years, so we try to spare them from certain therapies," said Dr. Caricchio.
Dr. Bruce concurred. "I think a concerted effort to switch the disease off in almost a treat-to-target way, getting people into remission, may well be very important with regard to avoiding long-term damage. If we could do that without using steroids, that would be ideal," he commented.

"SLICC is interested in the fact that most clinical trials in lupus to date have taken a very small subsection of the population, those with high disease activity, and used a particular biologic agent or new molecule to show that it improved disease activity. But actually the majority of people with lupus – around 60% have low-grade, grumbling disease and are on low-dose steroids. And those are the ones who accumulate damage. I think we need to have a paradigm shift in how we do clinical trials in lupus and think about doing lupus trials against a damage endpoint," the rheumatologist continued.
Power calculations based upon the SLICC Inception Cohort Study suggest such trials could be relatively modest in size, he added.
SLICC receives financial support from GlaxoSmithKline, Bristol-Myers Squibb, and Human Genome Sciences. Dr. Bruce reported receiving research funding from GlaxoSmithKline, Bristol-Myers Squibb, Roche, and UCB.
SAN DIEGO – Potentially modifiable risk factors for future irreversible organ damage in lupus patients include hypertension, the use of corticosteroids, and higher levels of inflammation early on, according to findings from the SLICC (Systemic Lupus International Collaborating Clinics) Inception Cohort Study.
In addition, the study identified the use of antimalarial drugs as the one significant protective factor against steady accrual of irreversible organ damage in lupus patients.
"These findings help us pave the way to consider whether, firstly, one could use damage as a primary endpoint in future clinical trials in lupus – somewhat akin to how the erosion score is used in rheumatoid arthritis – and secondly, the results suggest particular interventions that might be important in reducing the risk of damage over time," Dr. Ian N. Bruce said at the annual meeting of the American College of Rheumatology.
The study also identified several fixed and unmodifiable risk factors for irreversible damage in lupus patients: older age at diagnosis, male gender, and being black or white Americans, added Dr. Bruce, professor of rheumatology at the University of Manchester (U.K.) and chair of the SLICC research group.
The SLICC Inception Cohort Study involves 1,722 patients at 31 centers in 11 countries in North America, Europe, and Asia who enrolled within 15 months after being formally diagnosed with systemic lupus erythematosus based upon the 1997 ACR criteria. They averaged 35 years of age and had an average of 4.25 comprehensive annual follow-up visits during the study period.
Irreversible organ damage was assessed using the SLICC/ACR Damage Index, or SDI. At baseline, 35% of patients had at least one item of damage as indicated by an SDI score of 1 or more. Over time, damage rates slowly and steadily increased such that by 6 years of follow-up 51% of participants had an SDI of at least 1.
In a multivariate analysis, patients with an SDI score of 1 at baseline had a highly significant 37% reduction in the risk of increasing their score during follow-up if they were taking antimalarials, compared with those not taking antimalarials.
On the other hand, patients with a baseline SDI of 1 were 61% more likely to experience an increase in their damage score during follow-up if they had hypertension and 43% more likely to do so if they were on corticosteroids than if they weren’t. Moreover, their risk of going from an SDI of 1 to a higher SDI indicative of mounting damage increased by 10% for every 3-point increase on the SLE Disease Activity Index (SLEDAI).
Patients with a baseline SDI of 0 were 64% more likely to progress to a score of 1 or more during follow-up if they were taking corticosteroids and 71% more likely to do so if they were hypertensive. Their risk also increased by 17% for each 3-point increase in SLEDAI. Men had a 48% greater risk of going from an SDI of 0 to 1 or more than women. Asians were 40% less likely to develop irreversible damage.
Each 1-point increase in SDI score was associated with a 46% increased risk of mortality, as well as with poorer health-related quality of life, especially as reflected in SF-36 physical component scores.
Session chair Dr. Roberto Caricchio of Temple University, Philadelphia, called the SLICC study "very important work."
"It teaches us to be aggressive up-front with our lupus patients, which we often aren’t. We tend to spare ourselves because it’s a chronic disease, and we know we’ll see these patients for the next 20 years, so we try to spare them from certain therapies," said Dr. Caricchio.
Dr. Bruce concurred. "I think a concerted effort to switch the disease off in almost a treat-to-target way, getting people into remission, may well be very important with regard to avoiding long-term damage. If we could do that without using steroids, that would be ideal," he commented.
"SLICC is interested in the fact that most clinical trials in lupus to date have taken a very small subsection of the population, those with high disease activity, and used a particular biologic agent or new molecule to show that it improved disease activity. But actually the majority of people with lupus – around 60% have low-grade, grumbling disease and are on low-dose steroids. And those are the ones who accumulate damage. I think we need to have a paradigm shift in how we do clinical trials in lupus and think about doing lupus trials against a damage endpoint," the rheumatologist continued.
Power calculations based upon the SLICC Inception Cohort Study suggest such trials could be relatively modest in size, he added.
SLICC receives financial support from GlaxoSmithKline, Bristol-Myers Squibb, and Human Genome Sciences. Dr. Bruce reported receiving research funding from GlaxoSmithKline, Bristol-Myers Squibb, Roche, and UCB.
SAN DIEGO – Potentially modifiable risk factors for future irreversible organ damage in lupus patients include hypertension, the use of corticosteroids, and higher levels of inflammation early on, according to findings from the SLICC (Systemic Lupus International Collaborating Clinics) Inception Cohort Study.
In addition, the study identified the use of antimalarial drugs as the one significant protective factor against steady accrual of irreversible organ damage in lupus patients.
"These findings help us pave the way to consider whether, firstly, one could use damage as a primary endpoint in future clinical trials in lupus – somewhat akin to how the erosion score is used in rheumatoid arthritis – and secondly, the results suggest particular interventions that might be important in reducing the risk of damage over time," Dr. Ian N. Bruce said at the annual meeting of the American College of Rheumatology.
The study also identified several fixed and unmodifiable risk factors for irreversible damage in lupus patients: older age at diagnosis, male gender, and being black or white Americans, added Dr. Bruce, professor of rheumatology at the University of Manchester (U.K.) and chair of the SLICC research group.
The SLICC Inception Cohort Study involves 1,722 patients at 31 centers in 11 countries in North America, Europe, and Asia who enrolled within 15 months after being formally diagnosed with systemic lupus erythematosus based upon the 1997 ACR criteria. They averaged 35 years of age and had an average of 4.25 comprehensive annual follow-up visits during the study period.
Irreversible organ damage was assessed using the SLICC/ACR Damage Index, or SDI. At baseline, 35% of patients had at least one item of damage as indicated by an SDI score of 1 or more. Over time, damage rates slowly and steadily increased such that by 6 years of follow-up 51% of participants had an SDI of at least 1.
In a multivariate analysis, patients with an SDI score of 1 at baseline had a highly significant 37% reduction in the risk of increasing their score during follow-up if they were taking antimalarials, compared with those not taking antimalarials.
On the other hand, patients with a baseline SDI of 1 were 61% more likely to experience an increase in their damage score during follow-up if they had hypertension and 43% more likely to do so if they were on corticosteroids than if they weren’t. Moreover, their risk of going from an SDI of 1 to a higher SDI indicative of mounting damage increased by 10% for every 3-point increase on the SLE Disease Activity Index (SLEDAI).
Patients with a baseline SDI of 0 were 64% more likely to progress to a score of 1 or more during follow-up if they were taking corticosteroids and 71% more likely to do so if they were hypertensive. Their risk also increased by 17% for each 3-point increase in SLEDAI. Men had a 48% greater risk of going from an SDI of 0 to 1 or more than women. Asians were 40% less likely to develop irreversible damage.
Each 1-point increase in SDI score was associated with a 46% increased risk of mortality, as well as with poorer health-related quality of life, especially as reflected in SF-36 physical component scores.
Session chair Dr. Roberto Caricchio of Temple University, Philadelphia, called the SLICC study "very important work."
"It teaches us to be aggressive up-front with our lupus patients, which we often aren’t. We tend to spare ourselves because it’s a chronic disease, and we know we’ll see these patients for the next 20 years, so we try to spare them from certain therapies," said Dr. Caricchio.
Dr. Bruce concurred. "I think a concerted effort to switch the disease off in almost a treat-to-target way, getting people into remission, may well be very important with regard to avoiding long-term damage. If we could do that without using steroids, that would be ideal," he commented.
"SLICC is interested in the fact that most clinical trials in lupus to date have taken a very small subsection of the population, those with high disease activity, and used a particular biologic agent or new molecule to show that it improved disease activity. But actually the majority of people with lupus – around 60% have low-grade, grumbling disease and are on low-dose steroids. And those are the ones who accumulate damage. I think we need to have a paradigm shift in how we do clinical trials in lupus and think about doing lupus trials against a damage endpoint," the rheumatologist continued.
Power calculations based upon the SLICC Inception Cohort Study suggest such trials could be relatively modest in size, he added.
SLICC receives financial support from GlaxoSmithKline, Bristol-Myers Squibb, and Human Genome Sciences. Dr. Bruce reported receiving research funding from GlaxoSmithKline, Bristol-Myers Squibb, Roche, and UCB.
AT THE ACR ANNUAL MEETING
Major finding: Patients with recently diagnosed SLE and no significant organ damage at baseline were 71% more likely to develop irreversible organ damage during follow-up if they were hypertensive and 64% more likely to do so if they were taking corticosteroids. Antimalarial drugs showed a protective effect against accrual of damage.
Data source: The Systemic Lupus International Collaborating Clinics Inception Cohort Study involves 1,722 patients at 31 centers in 11 countries, all recruited within 15 months after diagnosis.
Disclosures: The study group receives funding from GlaxoSmithKline, Bristol-Myers Squibb, and Human Genome Sciences. The presenter receives research grants from GlaxoSmithKline, Bristol-Myers Squibb, and several other pharmaceutical companies.

Minimally important differences defined for CDAI for rheumatoid arthritis
SAN DIEGO – The utility of the Clinical Disease Activity Index as a simple, practical tool to quantify rheumatoid arthritis activity in everyday practice has gotten a big boost as a result of formal determination of the absolute change in the index that constitutes a minimally important difference.
"Our hope and expectation is that these MIDs [minimally important differences] for absolute change in CDAI [Clinical Disease Activity Index] can be used to determine whether patients are improving, and to help manage the patients that we see in clinic in terms of what treatments might need to be altered," Dr. Jeffrey R. Curtis explained at the annual meeting of the American College of Rheumatology.
The Disease Activity Score 28 (DAS28) has a widely accepted MID of 1.2 units, making it highly useful in defining a patient’s magnitude of clinical improvement in clinical trial settings. For example, exceeding this MID early after going on tumor necrosis factor inhibitor therapy has been shown to predict high likelihood of low disease activity at 1 year (J. Rheumatol. 2012;39:1326-33). But the DAS28 is impractical except when a real-time measurement of acute phase reactants is available, which is often not the case in everyday practice. The CDAI could be a better option for clinicians who are willing to do a joint count. Until now, however, the MID for the CDAI in real-world settings has not been defined, noted Dr. Curtis of the University of Alabama, Birmingham.
He and his coinvestigators have rectified this situation. They accomplished this by analyzing data from the Canadian Early Arthritis Cohort (CATCH), a large observational cohort of patients with early rheumatoid arthritis. After exclusion of the 8% of CATCH participants with comorbid fibromyalgia, that left 1,191 patients with rheumatologist-diagnosed rheumatoid arthritis, a mean 5.8-month duration of symptoms at enrollment, and a baseline CDAI of 28.5.
The investigators compared changes in DAS28 and CDAI during a total of more than 3,200 pairs of rheumatology patient visits spaced at 3-month intervals during the first 12 months after enrollment.

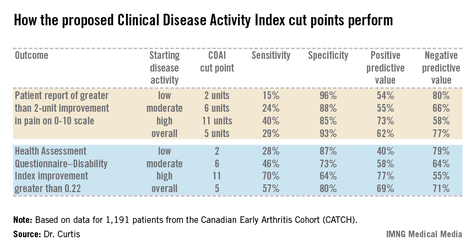
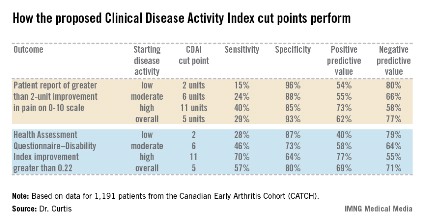
Overall, the best discriminator between a DAS28 MID indicative of significant clinical improvement as opposed to no improvement or worsening was a CDAI cut point of 5 units. That is, an absolute reduction of more than 5 units on the CDAI correlated well with a DAS28 improvement of at least 1.2 units. Indeed, the area under the receiver operator curve using a CDAI cut point of 5 units was 0.87. But there’s more to the story than that, according to Dr. Curtis.
"If at all possible, it’s preferable to use more specific CDAI MID cut points conditional on where patients are starting in terms of disease activity. Patients need less of a change in order to say that they feel better if they’re already starting out doing pretty well, with low disease activity, than if they’re starting with high disease activity," the rheumatologist said.
The generally accepted CDAI threshold for low disease activity is a score below 10. Moderate disease activity is a score of 10-22, while high disease activity is a CDAI in excess of 22. Dr. Curtis and his coworkers determined that the MID for patients with low disease activity is an absolute difference greater than 2 units. For patients with moderately active disease, it’s greater than 6 units. And for those with high disease activity, an absolute reduction of greater than 11 units defines the MID.
The investigators also tested the utility of their CDAI cut points in comparison to changes in outcomes other than the DAS28, including patient self-reported pain and Health Assessment Questionnaire-Disability Index scores (see chart).
Dr. Curtis reported receiving grants from and serving as a consultant to Amgen and Pfizer, which sponsor the CATCH study. He also receives funding from other companies as well as the National Institutes of Health.
SAN DIEGO – The utility of the Clinical Disease Activity Index as a simple, practical tool to quantify rheumatoid arthritis activity in everyday practice has gotten a big boost as a result of formal determination of the absolute change in the index that constitutes a minimally important difference.
"Our hope and expectation is that these MIDs [minimally important differences] for absolute change in CDAI [Clinical Disease Activity Index] can be used to determine whether patients are improving, and to help manage the patients that we see in clinic in terms of what treatments might need to be altered," Dr. Jeffrey R. Curtis explained at the annual meeting of the American College of Rheumatology.
The Disease Activity Score 28 (DAS28) has a widely accepted MID of 1.2 units, making it highly useful in defining a patient’s magnitude of clinical improvement in clinical trial settings. For example, exceeding this MID early after going on tumor necrosis factor inhibitor therapy has been shown to predict high likelihood of low disease activity at 1 year (J. Rheumatol. 2012;39:1326-33). But the DAS28 is impractical except when a real-time measurement of acute phase reactants is available, which is often not the case in everyday practice. The CDAI could be a better option for clinicians who are willing to do a joint count. Until now, however, the MID for the CDAI in real-world settings has not been defined, noted Dr. Curtis of the University of Alabama, Birmingham.
He and his coinvestigators have rectified this situation. They accomplished this by analyzing data from the Canadian Early Arthritis Cohort (CATCH), a large observational cohort of patients with early rheumatoid arthritis. After exclusion of the 8% of CATCH participants with comorbid fibromyalgia, that left 1,191 patients with rheumatologist-diagnosed rheumatoid arthritis, a mean 5.8-month duration of symptoms at enrollment, and a baseline CDAI of 28.5.
The investigators compared changes in DAS28 and CDAI during a total of more than 3,200 pairs of rheumatology patient visits spaced at 3-month intervals during the first 12 months after enrollment.
Overall, the best discriminator between a DAS28 MID indicative of significant clinical improvement as opposed to no improvement or worsening was a CDAI cut point of 5 units. That is, an absolute reduction of more than 5 units on the CDAI correlated well with a DAS28 improvement of at least 1.2 units. Indeed, the area under the receiver operator curve using a CDAI cut point of 5 units was 0.87. But there’s more to the story than that, according to Dr. Curtis.
"If at all possible, it’s preferable to use more specific CDAI MID cut points conditional on where patients are starting in terms of disease activity. Patients need less of a change in order to say that they feel better if they’re already starting out doing pretty well, with low disease activity, than if they’re starting with high disease activity," the rheumatologist said.
The generally accepted CDAI threshold for low disease activity is a score below 10. Moderate disease activity is a score of 10-22, while high disease activity is a CDAI in excess of 22. Dr. Curtis and his coworkers determined that the MID for patients with low disease activity is an absolute difference greater than 2 units. For patients with moderately active disease, it’s greater than 6 units. And for those with high disease activity, an absolute reduction of greater than 11 units defines the MID.
The investigators also tested the utility of their CDAI cut points in comparison to changes in outcomes other than the DAS28, including patient self-reported pain and Health Assessment Questionnaire-Disability Index scores (see chart).
Dr. Curtis reported receiving grants from and serving as a consultant to Amgen and Pfizer, which sponsor the CATCH study. He also receives funding from other companies as well as the National Institutes of Health.
SAN DIEGO – The utility of the Clinical Disease Activity Index as a simple, practical tool to quantify rheumatoid arthritis activity in everyday practice has gotten a big boost as a result of formal determination of the absolute change in the index that constitutes a minimally important difference.
"Our hope and expectation is that these MIDs [minimally important differences] for absolute change in CDAI [Clinical Disease Activity Index] can be used to determine whether patients are improving, and to help manage the patients that we see in clinic in terms of what treatments might need to be altered," Dr. Jeffrey R. Curtis explained at the annual meeting of the American College of Rheumatology.
The Disease Activity Score 28 (DAS28) has a widely accepted MID of 1.2 units, making it highly useful in defining a patient’s magnitude of clinical improvement in clinical trial settings. For example, exceeding this MID early after going on tumor necrosis factor inhibitor therapy has been shown to predict high likelihood of low disease activity at 1 year (J. Rheumatol. 2012;39:1326-33). But the DAS28 is impractical except when a real-time measurement of acute phase reactants is available, which is often not the case in everyday practice. The CDAI could be a better option for clinicians who are willing to do a joint count. Until now, however, the MID for the CDAI in real-world settings has not been defined, noted Dr. Curtis of the University of Alabama, Birmingham.
He and his coinvestigators have rectified this situation. They accomplished this by analyzing data from the Canadian Early Arthritis Cohort (CATCH), a large observational cohort of patients with early rheumatoid arthritis. After exclusion of the 8% of CATCH participants with comorbid fibromyalgia, that left 1,191 patients with rheumatologist-diagnosed rheumatoid arthritis, a mean 5.8-month duration of symptoms at enrollment, and a baseline CDAI of 28.5.
The investigators compared changes in DAS28 and CDAI during a total of more than 3,200 pairs of rheumatology patient visits spaced at 3-month intervals during the first 12 months after enrollment.
Overall, the best discriminator between a DAS28 MID indicative of significant clinical improvement as opposed to no improvement or worsening was a CDAI cut point of 5 units. That is, an absolute reduction of more than 5 units on the CDAI correlated well with a DAS28 improvement of at least 1.2 units. Indeed, the area under the receiver operator curve using a CDAI cut point of 5 units was 0.87. But there’s more to the story than that, according to Dr. Curtis.
"If at all possible, it’s preferable to use more specific CDAI MID cut points conditional on where patients are starting in terms of disease activity. Patients need less of a change in order to say that they feel better if they’re already starting out doing pretty well, with low disease activity, than if they’re starting with high disease activity," the rheumatologist said.
The generally accepted CDAI threshold for low disease activity is a score below 10. Moderate disease activity is a score of 10-22, while high disease activity is a CDAI in excess of 22. Dr. Curtis and his coworkers determined that the MID for patients with low disease activity is an absolute difference greater than 2 units. For patients with moderately active disease, it’s greater than 6 units. And for those with high disease activity, an absolute reduction of greater than 11 units defines the MID.
The investigators also tested the utility of their CDAI cut points in comparison to changes in outcomes other than the DAS28, including patient self-reported pain and Health Assessment Questionnaire-Disability Index scores (see chart).
Dr. Curtis reported receiving grants from and serving as a consultant to Amgen and Pfizer, which sponsor the CATCH study. He also receives funding from other companies as well as the National Institutes of Health.
AT THE ACR ANNUAL MEETING
Major finding: The minimally important difference on the Clinical Disease Activity Index that defines clinically meaningful improvement in rheumatoid arthritis patients is an absolute reduction greater than 5 units.
Data source: This determination was made by correlating changes in the CDAI and the Disease Activity Score 28 in 1,191 patients with early rheumatoid arthritis in the CATCH cohort.
Disclosures: Dr. Curtis reported receiving grants from and serving as a consultant to Amgen and Pfizer, which sponsor the CATCH study. He also receives funding from other companies as well as the National Institutes of Health.
Relief for Brachioradial Pruritus
ISTANBUL, TURKEY – The capsaicin 8% patch known as Qutenza appears to be a novel and effective treatment for patients with brachioradial pruritus, according to a preliminary observational study.
"The big advantage of this new treatment strategy is the patient only needs one application of the patch instead of taking drugs daily. Most patients experience relief for 5-6 weeks afterward," Dr. Claudia Zeidler said at the annual congress of the European Academy of Dermatology and Venereology.
Brachioradial pruritus (BRP) is a neuropathic itch often accompanied by pain, burning, and stinging. The condition is localized to the dorsolateral forearms along the distribution of the C-5 or C-6 dermatomes. The condition is more common in women. Imaging studies of patients typically show spinal stenosis or other cervical spinal pathology entailing cervical nerve root compression, according to Dr. Zeidler of the University of Müenster, Germany.
She and her colleagues found that patients with BRP have significantly reduced intraepidermal nerve fiber density in lesional skin, compared with nonlesional skin on the ventral forearm. Patients also have reduced expression of transient receptor potential cation channel subfamily V, member 1 (TRPV1) in lesional skin, compared with nonlesional skin. TRPV1 affects the balance between keratinocyte differentiation and proliferation, and thus can influence epidermal barrier function.
Dr. Zeidler’s study enrolled 16 patients with an average BRP duration of 91 months who had undergone unsuccessful treatment attempts with various combinations of anticonvulsants, antidepressants, and antihistamines. Thirteen of the 16 patients were women, and ranged in age from 47 to 69 years. Affected skin areas were normal in appearance, aside from hyperpigmented scratch lesions that looked like prurigo nodularis in 12 patients. Magnetic resonance imaging showed 15 of the 16 patients had cervical nerve compression or degenerative changes.
Itching relief typically began the day after the in-office Qutenza treatment session. Three weeks post treatment, mean pruritus intensity scores on a 0-10 visual analog scale had dropped from 5.8 pretreatment to 1.1. Mean pain intensity scores fell from 4.2 to 0.8. Significant improvement in quality of life was documented via a reduction in Dermatology Life Quality Index scores from 8.9 pretreatment to 4.3. Levels of TRPV1 in lesional skin were significantly increased 3 weeks post treatment compared with baseline; however, intraepithelial nerve fiber density in lesional skin was not significantly different.
Side effects of the capsaicin 8% patch consisted of mild localized pain and burning lasting for 20 minutes to 2 days in 14 patients.
Additional patient follow-up at 2, 3, and 4 months indicated that the pruritus and pain relief typically lasted 5-6 weeks. Since the conclusion of the study, when symptoms return, patients come in for retreatment.
The 14-by-20-cm capsaicin 8% patch is approved in Europe for the treatment of nondiabetic neuropathic pain and in the United States for management of neuropathic pain associated with postherpetic neuralgia. The patch contains 179 mg of capsaicin. Dr. Zeidler and coworkers were careful to follow the detailed labeling instructions for the patch’s use.
The patch has to be administered by a physician or another health care professional. Self-treatment is not permitted. Removal of hair to promote patch adherence should be accomplished with the use of scissors, not by shaving. To reduce discomfort, the researchers applied topical lidocaine 2.5% for 1 hour prior to putting the patch directly on the itchy area. Upon removing the patch after 1 hour, a cleansing gel that comes with the patch was applied for 1 minute to remove any residual capsaicin.
Dr. Zeidler plans to conduct a randomized controlled trial in patients with BRP to confirm the observational study findings, and also to look at the long-term effects of repeated treatments. Her observational study was supported by Astellas Pharma. She reported having no financial conflicts.
ISTANBUL, TURKEY – The capsaicin 8% patch known as Qutenza appears to be a novel and effective treatment for patients with brachioradial pruritus, according to a preliminary observational study.
"The big advantage of this new treatment strategy is the patient only needs one application of the patch instead of taking drugs daily. Most patients experience relief for 5-6 weeks afterward," Dr. Claudia Zeidler said at the annual congress of the European Academy of Dermatology and Venereology.
Brachioradial pruritus (BRP) is a neuropathic itch often accompanied by pain, burning, and stinging. The condition is localized to the dorsolateral forearms along the distribution of the C-5 or C-6 dermatomes. The condition is more common in women. Imaging studies of patients typically show spinal stenosis or other cervical spinal pathology entailing cervical nerve root compression, according to Dr. Zeidler of the University of Müenster, Germany.
She and her colleagues found that patients with BRP have significantly reduced intraepidermal nerve fiber density in lesional skin, compared with nonlesional skin on the ventral forearm. Patients also have reduced expression of transient receptor potential cation channel subfamily V, member 1 (TRPV1) in lesional skin, compared with nonlesional skin. TRPV1 affects the balance between keratinocyte differentiation and proliferation, and thus can influence epidermal barrier function.
Dr. Zeidler’s study enrolled 16 patients with an average BRP duration of 91 months who had undergone unsuccessful treatment attempts with various combinations of anticonvulsants, antidepressants, and antihistamines. Thirteen of the 16 patients were women, and ranged in age from 47 to 69 years. Affected skin areas were normal in appearance, aside from hyperpigmented scratch lesions that looked like prurigo nodularis in 12 patients. Magnetic resonance imaging showed 15 of the 16 patients had cervical nerve compression or degenerative changes.
Itching relief typically began the day after the in-office Qutenza treatment session. Three weeks post treatment, mean pruritus intensity scores on a 0-10 visual analog scale had dropped from 5.8 pretreatment to 1.1. Mean pain intensity scores fell from 4.2 to 0.8. Significant improvement in quality of life was documented via a reduction in Dermatology Life Quality Index scores from 8.9 pretreatment to 4.3. Levels of TRPV1 in lesional skin were significantly increased 3 weeks post treatment compared with baseline; however, intraepithelial nerve fiber density in lesional skin was not significantly different.
Side effects of the capsaicin 8% patch consisted of mild localized pain and burning lasting for 20 minutes to 2 days in 14 patients.
Additional patient follow-up at 2, 3, and 4 months indicated that the pruritus and pain relief typically lasted 5-6 weeks. Since the conclusion of the study, when symptoms return, patients come in for retreatment.
The 14-by-20-cm capsaicin 8% patch is approved in Europe for the treatment of nondiabetic neuropathic pain and in the United States for management of neuropathic pain associated with postherpetic neuralgia. The patch contains 179 mg of capsaicin. Dr. Zeidler and coworkers were careful to follow the detailed labeling instructions for the patch’s use.
The patch has to be administered by a physician or another health care professional. Self-treatment is not permitted. Removal of hair to promote patch adherence should be accomplished with the use of scissors, not by shaving. To reduce discomfort, the researchers applied topical lidocaine 2.5% for 1 hour prior to putting the patch directly on the itchy area. Upon removing the patch after 1 hour, a cleansing gel that comes with the patch was applied for 1 minute to remove any residual capsaicin.
Dr. Zeidler plans to conduct a randomized controlled trial in patients with BRP to confirm the observational study findings, and also to look at the long-term effects of repeated treatments. Her observational study was supported by Astellas Pharma. She reported having no financial conflicts.
ISTANBUL, TURKEY – The capsaicin 8% patch known as Qutenza appears to be a novel and effective treatment for patients with brachioradial pruritus, according to a preliminary observational study.
"The big advantage of this new treatment strategy is the patient only needs one application of the patch instead of taking drugs daily. Most patients experience relief for 5-6 weeks afterward," Dr. Claudia Zeidler said at the annual congress of the European Academy of Dermatology and Venereology.
Brachioradial pruritus (BRP) is a neuropathic itch often accompanied by pain, burning, and stinging. The condition is localized to the dorsolateral forearms along the distribution of the C-5 or C-6 dermatomes. The condition is more common in women. Imaging studies of patients typically show spinal stenosis or other cervical spinal pathology entailing cervical nerve root compression, according to Dr. Zeidler of the University of Müenster, Germany.
She and her colleagues found that patients with BRP have significantly reduced intraepidermal nerve fiber density in lesional skin, compared with nonlesional skin on the ventral forearm. Patients also have reduced expression of transient receptor potential cation channel subfamily V, member 1 (TRPV1) in lesional skin, compared with nonlesional skin. TRPV1 affects the balance between keratinocyte differentiation and proliferation, and thus can influence epidermal barrier function.
Dr. Zeidler’s study enrolled 16 patients with an average BRP duration of 91 months who had undergone unsuccessful treatment attempts with various combinations of anticonvulsants, antidepressants, and antihistamines. Thirteen of the 16 patients were women, and ranged in age from 47 to 69 years. Affected skin areas were normal in appearance, aside from hyperpigmented scratch lesions that looked like prurigo nodularis in 12 patients. Magnetic resonance imaging showed 15 of the 16 patients had cervical nerve compression or degenerative changes.
Itching relief typically began the day after the in-office Qutenza treatment session. Three weeks post treatment, mean pruritus intensity scores on a 0-10 visual analog scale had dropped from 5.8 pretreatment to 1.1. Mean pain intensity scores fell from 4.2 to 0.8. Significant improvement in quality of life was documented via a reduction in Dermatology Life Quality Index scores from 8.9 pretreatment to 4.3. Levels of TRPV1 in lesional skin were significantly increased 3 weeks post treatment compared with baseline; however, intraepithelial nerve fiber density in lesional skin was not significantly different.
Side effects of the capsaicin 8% patch consisted of mild localized pain and burning lasting for 20 minutes to 2 days in 14 patients.
Additional patient follow-up at 2, 3, and 4 months indicated that the pruritus and pain relief typically lasted 5-6 weeks. Since the conclusion of the study, when symptoms return, patients come in for retreatment.
The 14-by-20-cm capsaicin 8% patch is approved in Europe for the treatment of nondiabetic neuropathic pain and in the United States for management of neuropathic pain associated with postherpetic neuralgia. The patch contains 179 mg of capsaicin. Dr. Zeidler and coworkers were careful to follow the detailed labeling instructions for the patch’s use.
The patch has to be administered by a physician or another health care professional. Self-treatment is not permitted. Removal of hair to promote patch adherence should be accomplished with the use of scissors, not by shaving. To reduce discomfort, the researchers applied topical lidocaine 2.5% for 1 hour prior to putting the patch directly on the itchy area. Upon removing the patch after 1 hour, a cleansing gel that comes with the patch was applied for 1 minute to remove any residual capsaicin.
Dr. Zeidler plans to conduct a randomized controlled trial in patients with BRP to confirm the observational study findings, and also to look at the long-term effects of repeated treatments. Her observational study was supported by Astellas Pharma. She reported having no financial conflicts.
AT THE EADV CONGRESS
Capsaicin 8% patch offers relief for brachioradial pruritus
ISTANBUL, TURKEY – The capsaicin 8% patch known as Qutenza appears to be a novel and effective treatment for patients with brachioradial pruritus, according to a preliminary observational study.
"The big advantage of this new treatment strategy is the patient only needs one application of the patch instead of taking drugs daily. Most patients experience relief for 5-6 weeks afterward," Dr. Claudia Zeidler said at the annual congress of the European Academy of Dermatology and Venereology.
Brachioradial pruritus (BRP) is a neuropathic itch often accompanied by pain, burning, and stinging. The condition is localized to the dorsolateral forearms along the distribution of the C-5 or C-6 dermatomes. The condition is more common in women. Imaging studies of patients typically show spinal stenosis or other cervical spinal pathology entailing cervical nerve root compression, according to Dr. Zeidler of the University of Müenster, Germany.
She and her colleagues found that patients with BRP have significantly reduced intraepidermal nerve fiber density in lesional skin, compared with nonlesional skin on the ventral forearm. Patients also have reduced expression of transient receptor potential cation channel subfamily V, member 1 (TRPV1) in lesional skin, compared with nonlesional skin. TRPV1 affects the balance between keratinocyte differentiation and proliferation, and thus can influence epidermal barrier function.
Dr. Zeidler’s study enrolled 16 patients with an average BRP duration of 91 months who had undergone unsuccessful treatment attempts with various combinations of anticonvulsants, antidepressants, and antihistamines. Thirteen of the 16 patients were women, and ranged in age from 47 to 69 years. Affected skin areas were normal in appearance, aside from hyperpigmented scratch lesions that looked like prurigo nodularis in 12 patients. Magnetic resonance imaging showed 15 of the 16 patients had cervical nerve compression or degenerative changes.
Itching relief typically began the day after the in-office Qutenza treatment session. Three weeks post treatment, mean pruritus intensity scores on a 0-10 visual analog scale had dropped from 5.8 pretreatment to 1.1. Mean pain intensity scores fell from 4.2 to 0.8. Significant improvement in quality of life was documented via a reduction in Dermatology Life Quality Index scores from 8.9 pretreatment to 4.3. Levels of TRPV1 in lesional skin were significantly increased 3 weeks post treatment compared with baseline; however, intraepithelial nerve fiber density in lesional skin was not significantly different.
Side effects of the capsaicin 8% patch consisted of mild localized pain and burning lasting for 20 minutes to 2 days in 14 patients.
Additional patient follow-up at 2, 3, and 4 months indicated that the pruritus and pain relief typically lasted 5-6 weeks. Since the conclusion of the study, when symptoms return, patients come in for retreatment.
The 14-by-20-cm capsaicin 8% patch is approved in Europe for the treatment of nondiabetic neuropathic pain and in the United States for management of neuropathic pain associated with postherpetic neuralgia. The patch contains 179 mg of capsaicin. Dr. Zeidler and coworkers were careful to follow the detailed labeling instructions for the patch’s use.
The patch has to be administered by a physician or another health care professional. Self-treatment is not permitted. Removal of hair to promote patch adherence should be accomplished with the use of scissors, not by shaving. To reduce discomfort, the researchers applied topical lidocaine 2.5% for 1 hour prior to putting the patch directly on the itchy area. Upon removing the patch after 1 hour, a cleansing gel that comes with the patch was applied for 1 minute to remove any residual capsaicin.
Dr. Zeidler plans to conduct a randomized controlled trial in patients with BRP to confirm the observational study findings, and also to look at the long-term effects of repeated treatments. Her observational study was supported by Astellas Pharma. She reported having no financial conflicts.
ISTANBUL, TURKEY – The capsaicin 8% patch known as Qutenza appears to be a novel and effective treatment for patients with brachioradial pruritus, according to a preliminary observational study.
"The big advantage of this new treatment strategy is the patient only needs one application of the patch instead of taking drugs daily. Most patients experience relief for 5-6 weeks afterward," Dr. Claudia Zeidler said at the annual congress of the European Academy of Dermatology and Venereology.
Brachioradial pruritus (BRP) is a neuropathic itch often accompanied by pain, burning, and stinging. The condition is localized to the dorsolateral forearms along the distribution of the C-5 or C-6 dermatomes. The condition is more common in women. Imaging studies of patients typically show spinal stenosis or other cervical spinal pathology entailing cervical nerve root compression, according to Dr. Zeidler of the University of Müenster, Germany.
She and her colleagues found that patients with BRP have significantly reduced intraepidermal nerve fiber density in lesional skin, compared with nonlesional skin on the ventral forearm. Patients also have reduced expression of transient receptor potential cation channel subfamily V, member 1 (TRPV1) in lesional skin, compared with nonlesional skin. TRPV1 affects the balance between keratinocyte differentiation and proliferation, and thus can influence epidermal barrier function.
Dr. Zeidler’s study enrolled 16 patients with an average BRP duration of 91 months who had undergone unsuccessful treatment attempts with various combinations of anticonvulsants, antidepressants, and antihistamines. Thirteen of the 16 patients were women, and ranged in age from 47 to 69 years. Affected skin areas were normal in appearance, aside from hyperpigmented scratch lesions that looked like prurigo nodularis in 12 patients. Magnetic resonance imaging showed 15 of the 16 patients had cervical nerve compression or degenerative changes.
Itching relief typically began the day after the in-office Qutenza treatment session. Three weeks post treatment, mean pruritus intensity scores on a 0-10 visual analog scale had dropped from 5.8 pretreatment to 1.1. Mean pain intensity scores fell from 4.2 to 0.8. Significant improvement in quality of life was documented via a reduction in Dermatology Life Quality Index scores from 8.9 pretreatment to 4.3. Levels of TRPV1 in lesional skin were significantly increased 3 weeks post treatment compared with baseline; however, intraepithelial nerve fiber density in lesional skin was not significantly different.
Side effects of the capsaicin 8% patch consisted of mild localized pain and burning lasting for 20 minutes to 2 days in 14 patients.
Additional patient follow-up at 2, 3, and 4 months indicated that the pruritus and pain relief typically lasted 5-6 weeks. Since the conclusion of the study, when symptoms return, patients come in for retreatment.
The 14-by-20-cm capsaicin 8% patch is approved in Europe for the treatment of nondiabetic neuropathic pain and in the United States for management of neuropathic pain associated with postherpetic neuralgia. The patch contains 179 mg of capsaicin. Dr. Zeidler and coworkers were careful to follow the detailed labeling instructions for the patch’s use.
The patch has to be administered by a physician or another health care professional. Self-treatment is not permitted. Removal of hair to promote patch adherence should be accomplished with the use of scissors, not by shaving. To reduce discomfort, the researchers applied topical lidocaine 2.5% for 1 hour prior to putting the patch directly on the itchy area. Upon removing the patch after 1 hour, a cleansing gel that comes with the patch was applied for 1 minute to remove any residual capsaicin.
Dr. Zeidler plans to conduct a randomized controlled trial in patients with BRP to confirm the observational study findings, and also to look at the long-term effects of repeated treatments. Her observational study was supported by Astellas Pharma. She reported having no financial conflicts.
ISTANBUL, TURKEY – The capsaicin 8% patch known as Qutenza appears to be a novel and effective treatment for patients with brachioradial pruritus, according to a preliminary observational study.
"The big advantage of this new treatment strategy is the patient only needs one application of the patch instead of taking drugs daily. Most patients experience relief for 5-6 weeks afterward," Dr. Claudia Zeidler said at the annual congress of the European Academy of Dermatology and Venereology.
Brachioradial pruritus (BRP) is a neuropathic itch often accompanied by pain, burning, and stinging. The condition is localized to the dorsolateral forearms along the distribution of the C-5 or C-6 dermatomes. The condition is more common in women. Imaging studies of patients typically show spinal stenosis or other cervical spinal pathology entailing cervical nerve root compression, according to Dr. Zeidler of the University of Müenster, Germany.
She and her colleagues found that patients with BRP have significantly reduced intraepidermal nerve fiber density in lesional skin, compared with nonlesional skin on the ventral forearm. Patients also have reduced expression of transient receptor potential cation channel subfamily V, member 1 (TRPV1) in lesional skin, compared with nonlesional skin. TRPV1 affects the balance between keratinocyte differentiation and proliferation, and thus can influence epidermal barrier function.
Dr. Zeidler’s study enrolled 16 patients with an average BRP duration of 91 months who had undergone unsuccessful treatment attempts with various combinations of anticonvulsants, antidepressants, and antihistamines. Thirteen of the 16 patients were women, and ranged in age from 47 to 69 years. Affected skin areas were normal in appearance, aside from hyperpigmented scratch lesions that looked like prurigo nodularis in 12 patients. Magnetic resonance imaging showed 15 of the 16 patients had cervical nerve compression or degenerative changes.
Itching relief typically began the day after the in-office Qutenza treatment session. Three weeks post treatment, mean pruritus intensity scores on a 0-10 visual analog scale had dropped from 5.8 pretreatment to 1.1. Mean pain intensity scores fell from 4.2 to 0.8. Significant improvement in quality of life was documented via a reduction in Dermatology Life Quality Index scores from 8.9 pretreatment to 4.3. Levels of TRPV1 in lesional skin were significantly increased 3 weeks post treatment compared with baseline; however, intraepithelial nerve fiber density in lesional skin was not significantly different.
Side effects of the capsaicin 8% patch consisted of mild localized pain and burning lasting for 20 minutes to 2 days in 14 patients.
Additional patient follow-up at 2, 3, and 4 months indicated that the pruritus and pain relief typically lasted 5-6 weeks. Since the conclusion of the study, when symptoms return, patients come in for retreatment.
The 14-by-20-cm capsaicin 8% patch is approved in Europe for the treatment of nondiabetic neuropathic pain and in the United States for management of neuropathic pain associated with postherpetic neuralgia. The patch contains 179 mg of capsaicin. Dr. Zeidler and coworkers were careful to follow the detailed labeling instructions for the patch’s use.
The patch has to be administered by a physician or another health care professional. Self-treatment is not permitted. Removal of hair to promote patch adherence should be accomplished with the use of scissors, not by shaving. To reduce discomfort, the researchers applied topical lidocaine 2.5% for 1 hour prior to putting the patch directly on the itchy area. Upon removing the patch after 1 hour, a cleansing gel that comes with the patch was applied for 1 minute to remove any residual capsaicin.
Dr. Zeidler plans to conduct a randomized controlled trial in patients with BRP to confirm the observational study findings, and also to look at the long-term effects of repeated treatments. Her observational study was supported by Astellas Pharma. She reported having no financial conflicts.
AT THE EADV CONGRESS
Major finding: Three weeks after a single hour-long application of the capsaicin 8% patch known as Qutenza, mean itch intensity on a 0-10 visual analog scale in patients with brachioradial pruritus was 1.1, compared with 5.8 pretreatment.
Data source: An uncontrolled prospective observational study involving 16 treated patients with longstanding brachioradial pruritus refractory to various combinations of anticonvulsants, antihistamines, and antidepressants.
Disclosures: The investigator-initiated study was supported by Astellas Pharma. The presenter reported having no financial conflicts.
Depression accounts for psoriatics’ increased MI risk
SAN DIEGO – Depression is an independent risk factor for acute myocardial infarction in patients with psoriasis or psoriatic arthritis, a large population-based cohort study indicates.
In this study of more than 10,000 British Columbians with psoriasis and/or psoriatic arthritis, the increased risk of MI was confined to the patient subset having comorbid depression, Lindsay C. Burns, Ph.D., reported at the annual meeting of the American College of Rheumatology.
"These data underscore the need to actively screen for depression among psoriasis and psoriatic arthritis patients and closely monitor cardiovascular health in this high-risk group to improve long-term survival," declared Dr. Burns of the University of British Columbia, Vancouver.
She and her coinvestigators mined the comprehensive health records available for 4.1 million adults through British Columbia’s universal medical insurance coverage system in order to identify all 10,041 patients who were diagnosed with psoriasis by a dermatologist or psoriatic arthritis by a rheumatologist during 1996-2006, and who at that time had no history of MI. The patients were matched by age, gender, and years of follow-up with 47,415 controls.
Acute MI occurred in 268 patients with psoriasis or psoriatic arthritis, for an incidence rate of 5.8 cases per 1,000 person-years. The incidence rate of physician-diagnosed depression was 3.4 per 1,000 person-years in the psoriatic group, with a 10-year prevalence of 21.6%.
In a multivariate regression analysis adjusted for comorbid conditions, socioeconomic status, health resource utilization, age, and gender, individuals with psoriasis or psoriatic arthritis were 26% more likely to be depressed than controls. Diagnosis of depression in psoriatic patients during the follow-up period increased their risk of having an MI by an adjusted 80% compared with psoriatic patients without the psychiatric diagnosis.
Psoriatic subjects without depression had a statistically nonsignificant 10% increased risk of MI compared with nonpsoriatic controls. In contrast, psoriatic patients with diagnosed depression had a 60% greater MI risk than controls, according to Dr. Burns.
The study was funded by the Arthritis Research Center of Canada. Dr. Burns reported having no financial conflicts of interest.
SAN DIEGO – Depression is an independent risk factor for acute myocardial infarction in patients with psoriasis or psoriatic arthritis, a large population-based cohort study indicates.
In this study of more than 10,000 British Columbians with psoriasis and/or psoriatic arthritis, the increased risk of MI was confined to the patient subset having comorbid depression, Lindsay C. Burns, Ph.D., reported at the annual meeting of the American College of Rheumatology.
"These data underscore the need to actively screen for depression among psoriasis and psoriatic arthritis patients and closely monitor cardiovascular health in this high-risk group to improve long-term survival," declared Dr. Burns of the University of British Columbia, Vancouver.
She and her coinvestigators mined the comprehensive health records available for 4.1 million adults through British Columbia’s universal medical insurance coverage system in order to identify all 10,041 patients who were diagnosed with psoriasis by a dermatologist or psoriatic arthritis by a rheumatologist during 1996-2006, and who at that time had no history of MI. The patients were matched by age, gender, and years of follow-up with 47,415 controls.
Acute MI occurred in 268 patients with psoriasis or psoriatic arthritis, for an incidence rate of 5.8 cases per 1,000 person-years. The incidence rate of physician-diagnosed depression was 3.4 per 1,000 person-years in the psoriatic group, with a 10-year prevalence of 21.6%.
In a multivariate regression analysis adjusted for comorbid conditions, socioeconomic status, health resource utilization, age, and gender, individuals with psoriasis or psoriatic arthritis were 26% more likely to be depressed than controls. Diagnosis of depression in psoriatic patients during the follow-up period increased their risk of having an MI by an adjusted 80% compared with psoriatic patients without the psychiatric diagnosis.
Psoriatic subjects without depression had a statistically nonsignificant 10% increased risk of MI compared with nonpsoriatic controls. In contrast, psoriatic patients with diagnosed depression had a 60% greater MI risk than controls, according to Dr. Burns.
The study was funded by the Arthritis Research Center of Canada. Dr. Burns reported having no financial conflicts of interest.
SAN DIEGO – Depression is an independent risk factor for acute myocardial infarction in patients with psoriasis or psoriatic arthritis, a large population-based cohort study indicates.
In this study of more than 10,000 British Columbians with psoriasis and/or psoriatic arthritis, the increased risk of MI was confined to the patient subset having comorbid depression, Lindsay C. Burns, Ph.D., reported at the annual meeting of the American College of Rheumatology.
"These data underscore the need to actively screen for depression among psoriasis and psoriatic arthritis patients and closely monitor cardiovascular health in this high-risk group to improve long-term survival," declared Dr. Burns of the University of British Columbia, Vancouver.
She and her coinvestigators mined the comprehensive health records available for 4.1 million adults through British Columbia’s universal medical insurance coverage system in order to identify all 10,041 patients who were diagnosed with psoriasis by a dermatologist or psoriatic arthritis by a rheumatologist during 1996-2006, and who at that time had no history of MI. The patients were matched by age, gender, and years of follow-up with 47,415 controls.
Acute MI occurred in 268 patients with psoriasis or psoriatic arthritis, for an incidence rate of 5.8 cases per 1,000 person-years. The incidence rate of physician-diagnosed depression was 3.4 per 1,000 person-years in the psoriatic group, with a 10-year prevalence of 21.6%.
In a multivariate regression analysis adjusted for comorbid conditions, socioeconomic status, health resource utilization, age, and gender, individuals with psoriasis or psoriatic arthritis were 26% more likely to be depressed than controls. Diagnosis of depression in psoriatic patients during the follow-up period increased their risk of having an MI by an adjusted 80% compared with psoriatic patients without the psychiatric diagnosis.
Psoriatic subjects without depression had a statistically nonsignificant 10% increased risk of MI compared with nonpsoriatic controls. In contrast, psoriatic patients with diagnosed depression had a 60% greater MI risk than controls, according to Dr. Burns.
The study was funded by the Arthritis Research Center of Canada. Dr. Burns reported having no financial conflicts of interest.
AT THE ACR ANNUAL MEETING
Major finding: Patients with psoriasis or psoriatic arthritis who developed comorbid depression had a highly significant 80% increased risk of acute MI during follow-up, compared with those without depression.
Data source: This was a population-based cohort study involving 10,041 adults diagnosed with psoriasis or psoriatic arthritis in British Columbia during 1996-2006 and more than 47,000 controls.
Disclosures: The study was funded by the Arthritis Research Center of Canada. Dr. Burns reported having no financial conflicts of interest.
Apremilast’s positive study results made it the talk of ACR 2013
SAN DIEGO – The novel oral phosphodiesterase-4 inhibitor apremilast cut an impressively wide swath through the annual meeting of the American College of Rheumatology on the strength of positive results in three separate pivotal phase III trials for psoriatic arthritis and a favorable phase II study in Behçet’s syndrome.
Dr. Alvin F. Wells reported that apremilast resulted in clinically meaningful improvement in psoriatic arthritis symptoms, physical function, and associated skin psoriasis at 16 weeks in the PALACE 4 trial. Moreover, the improvement remained durable through 52 weeks in the phase III clinical trial.
PALACE 4 compared apremilast against placebo as a first-line treatment in 527 patients with psoriatic arthritis not previously treated with a disease-modifying antirheumatic drug. This was a population with active disease: a mean baseline 11 swollen and 20 tender joints; a Health Assessment Questionnaire Disability Index score averaging 1.07; and a 50% prevalence of dactylitis, with a mean severity score of 2.0. Sixty-five percent of patients had enthesitis, with a median Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) of 3.0. Participants had a mean 16-year history of psoriasis and a 3.4-year duration of psoriatic arthritis.
The primary study endpoint was attainment of an American College of Rheumatology 20% improvement (ACR20) response at 16 weeks. This was achieved in 29.2% of patients randomized to apremilast at 20 mg twice daily, 32.3% on 30 mg twice daily, and 16.9% on placebo. At week 16, patients on placebo were re-randomized to apremilast at 20 mg or 30 mg twice weekly. By week 52, an ACR20 response was achieved by 53% of patients in the apremilast 20 mg twice-daily group and 59% of those on the higher dose, according to Dr. Wells, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
Apremilast also scored well on numerous secondary endpoints (see chart). For example, the swollen joint count decreased by a median of 89% over the course of 52 weeks in patients on apremilast at 20 mg twice daily.
"The way I like to think of that result is at least 50% of patients had an 89% improvement in their swollen joint count," he explained.
During the first 16 weeks of PALACE 4, there were fewer serious adverse events in apremilast-treated patients than with placebo-treated patients. The most frequent side effects of apremilast were nausea, diarrhea, and headache, affecting 16%, 12%, and 8%, respectively, of patients in the higher-dose arm. These adverse events were almost exclusively mild or moderate, they began within the first 2 weeks of treatment, and they resolved in 4 weeks despite continued treatment. Less than 2% of apremilast-treated patients dropped out of the study due to nausea or diarrhea through week 52.
PALACE 4 differed from the PALACE 2 and 3 trials, also presented at the ACR meeting, in that PALACE 2 and 3 involved only patients with psoriatic arthritis previously treated with biologic agents or other disease-modifying antirheumatic drugs. All three studies had the same design, and the results in terms of both efficacy and safety were consistent across the full clinical trial program. No clinically meaningful changes in laboratory values were seen in any of the PALACE trials, suggesting ongoing lab monitoring may not be necessary.
Also at the ACR annual meeting, Dr. Gülen Hatemi of Istanbul University, Turkey, in a reprise of her report several weeks earlier at the annual congress of the European Academy of Dermatology and Venereology, presented a phase II study showing apremilast to be highly effective in treating the oral ulcers that are the cardinal feature of Behçet’s syndrome.
Celgene filed for approval of apremilast for psoriatic arthritis with the Food and Drug Administration earlier this year and anticipates a decision in March 2014. The company also plans to apply to the European regulatory agency for the same indication before year’s end, and to petition the FDA for approval in psoriasis within the same time frame. A 500-patient phase III study of apremilast in ankylosing spondylitis, known as POSTURE, is well underway.
PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
SAN DIEGO – The novel oral phosphodiesterase-4 inhibitor apremilast cut an impressively wide swath through the annual meeting of the American College of Rheumatology on the strength of positive results in three separate pivotal phase III trials for psoriatic arthritis and a favorable phase II study in Behçet’s syndrome.
Dr. Alvin F. Wells reported that apremilast resulted in clinically meaningful improvement in psoriatic arthritis symptoms, physical function, and associated skin psoriasis at 16 weeks in the PALACE 4 trial. Moreover, the improvement remained durable through 52 weeks in the phase III clinical trial.
PALACE 4 compared apremilast against placebo as a first-line treatment in 527 patients with psoriatic arthritis not previously treated with a disease-modifying antirheumatic drug. This was a population with active disease: a mean baseline 11 swollen and 20 tender joints; a Health Assessment Questionnaire Disability Index score averaging 1.07; and a 50% prevalence of dactylitis, with a mean severity score of 2.0. Sixty-five percent of patients had enthesitis, with a median Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) of 3.0. Participants had a mean 16-year history of psoriasis and a 3.4-year duration of psoriatic arthritis.
The primary study endpoint was attainment of an American College of Rheumatology 20% improvement (ACR20) response at 16 weeks. This was achieved in 29.2% of patients randomized to apremilast at 20 mg twice daily, 32.3% on 30 mg twice daily, and 16.9% on placebo. At week 16, patients on placebo were re-randomized to apremilast at 20 mg or 30 mg twice weekly. By week 52, an ACR20 response was achieved by 53% of patients in the apremilast 20 mg twice-daily group and 59% of those on the higher dose, according to Dr. Wells, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
Apremilast also scored well on numerous secondary endpoints (see chart). For example, the swollen joint count decreased by a median of 89% over the course of 52 weeks in patients on apremilast at 20 mg twice daily.
"The way I like to think of that result is at least 50% of patients had an 89% improvement in their swollen joint count," he explained.
During the first 16 weeks of PALACE 4, there were fewer serious adverse events in apremilast-treated patients than with placebo-treated patients. The most frequent side effects of apremilast were nausea, diarrhea, and headache, affecting 16%, 12%, and 8%, respectively, of patients in the higher-dose arm. These adverse events were almost exclusively mild or moderate, they began within the first 2 weeks of treatment, and they resolved in 4 weeks despite continued treatment. Less than 2% of apremilast-treated patients dropped out of the study due to nausea or diarrhea through week 52.
PALACE 4 differed from the PALACE 2 and 3 trials, also presented at the ACR meeting, in that PALACE 2 and 3 involved only patients with psoriatic arthritis previously treated with biologic agents or other disease-modifying antirheumatic drugs. All three studies had the same design, and the results in terms of both efficacy and safety were consistent across the full clinical trial program. No clinically meaningful changes in laboratory values were seen in any of the PALACE trials, suggesting ongoing lab monitoring may not be necessary.
Also at the ACR annual meeting, Dr. Gülen Hatemi of Istanbul University, Turkey, in a reprise of her report several weeks earlier at the annual congress of the European Academy of Dermatology and Venereology, presented a phase II study showing apremilast to be highly effective in treating the oral ulcers that are the cardinal feature of Behçet’s syndrome.
Celgene filed for approval of apremilast for psoriatic arthritis with the Food and Drug Administration earlier this year and anticipates a decision in March 2014. The company also plans to apply to the European regulatory agency for the same indication before year’s end, and to petition the FDA for approval in psoriasis within the same time frame. A 500-patient phase III study of apremilast in ankylosing spondylitis, known as POSTURE, is well underway.
PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
SAN DIEGO – The novel oral phosphodiesterase-4 inhibitor apremilast cut an impressively wide swath through the annual meeting of the American College of Rheumatology on the strength of positive results in three separate pivotal phase III trials for psoriatic arthritis and a favorable phase II study in Behçet’s syndrome.
Dr. Alvin F. Wells reported that apremilast resulted in clinically meaningful improvement in psoriatic arthritis symptoms, physical function, and associated skin psoriasis at 16 weeks in the PALACE 4 trial. Moreover, the improvement remained durable through 52 weeks in the phase III clinical trial.
PALACE 4 compared apremilast against placebo as a first-line treatment in 527 patients with psoriatic arthritis not previously treated with a disease-modifying antirheumatic drug. This was a population with active disease: a mean baseline 11 swollen and 20 tender joints; a Health Assessment Questionnaire Disability Index score averaging 1.07; and a 50% prevalence of dactylitis, with a mean severity score of 2.0. Sixty-five percent of patients had enthesitis, with a median Maastricht Ankylosing Spondylitis Enthesitis Score (MASES) of 3.0. Participants had a mean 16-year history of psoriasis and a 3.4-year duration of psoriatic arthritis.
The primary study endpoint was attainment of an American College of Rheumatology 20% improvement (ACR20) response at 16 weeks. This was achieved in 29.2% of patients randomized to apremilast at 20 mg twice daily, 32.3% on 30 mg twice daily, and 16.9% on placebo. At week 16, patients on placebo were re-randomized to apremilast at 20 mg or 30 mg twice weekly. By week 52, an ACR20 response was achieved by 53% of patients in the apremilast 20 mg twice-daily group and 59% of those on the higher dose, according to Dr. Wells, director of the Rheumatology and Immunotherapy Center in Franklin, Wisc.
Apremilast also scored well on numerous secondary endpoints (see chart). For example, the swollen joint count decreased by a median of 89% over the course of 52 weeks in patients on apremilast at 20 mg twice daily.
"The way I like to think of that result is at least 50% of patients had an 89% improvement in their swollen joint count," he explained.
During the first 16 weeks of PALACE 4, there were fewer serious adverse events in apremilast-treated patients than with placebo-treated patients. The most frequent side effects of apremilast were nausea, diarrhea, and headache, affecting 16%, 12%, and 8%, respectively, of patients in the higher-dose arm. These adverse events were almost exclusively mild or moderate, they began within the first 2 weeks of treatment, and they resolved in 4 weeks despite continued treatment. Less than 2% of apremilast-treated patients dropped out of the study due to nausea or diarrhea through week 52.
PALACE 4 differed from the PALACE 2 and 3 trials, also presented at the ACR meeting, in that PALACE 2 and 3 involved only patients with psoriatic arthritis previously treated with biologic agents or other disease-modifying antirheumatic drugs. All three studies had the same design, and the results in terms of both efficacy and safety were consistent across the full clinical trial program. No clinically meaningful changes in laboratory values were seen in any of the PALACE trials, suggesting ongoing lab monitoring may not be necessary.
Also at the ACR annual meeting, Dr. Gülen Hatemi of Istanbul University, Turkey, in a reprise of her report several weeks earlier at the annual congress of the European Academy of Dermatology and Venereology, presented a phase II study showing apremilast to be highly effective in treating the oral ulcers that are the cardinal feature of Behçet’s syndrome.
Celgene filed for approval of apremilast for psoriatic arthritis with the Food and Drug Administration earlier this year and anticipates a decision in March 2014. The company also plans to apply to the European regulatory agency for the same indication before year’s end, and to petition the FDA for approval in psoriasis within the same time frame. A 500-patient phase III study of apremilast in ankylosing spondylitis, known as POSTURE, is well underway.
PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
AT THE ACR ANNUAL MEETING
Major finding: After 16 weeks of treatment, the investigational drug apremilast achieved an ACR20 response of 29.2% at 20 mg twice daily as first-line therapy in patients with psoriatic arthritis and 32.3% at 30 mg twice daily, whereas placebo-treated controls achieved a rate of 16.9%.
Data source: The PALACE 4 study was a pivotal phase III, randomized, placebo-controlled trial including 527 patients with active psoriatic arthritis.
Disclosures: PALACE 4 was sponsored by Celgene. Dr. Wells reported receiving a research grant from the company.
Cyclophosphamide and rituximab combo reduced severe lupus flares
SAN DIEGO – The combination of intravenous cyclophosphamide and rituximab shows promise for the reduction of lupus flares, both renal and nonrenal, in patients with severe systemic lupus erythematosus, according to Dr. Ali Shahzad.
Moreover, this benefit did not come at the cost of a significant increase in infections, as compared with intravenous cyclophosphamide monotherapy, Dr. Shahzad reported at the annual meeting of the American College of Rheumatology.
He reported on 43 patients with severe, recurrent SLE. Thirty-one were placed on intravenous cyclophosphamide monotherapy administered according to the National Institutes of Health standard protocol for the treatment of lupus nephritis. The other 12 got cyclophosphamide plus two 1,000-mg doses of rituximab (Rituxan) given 15 days apart. The combination regimen was given at the physician’s discretion for recalcitrant or recurrent flares of lupus nephritis in 10 of 12 cases, and for treatment-resistant CNS lupus or other extrarenal lupus in the other 2. Eight of the 12 recipients of combination therapy had previously been treated with intravenous cyclophosphamide, in most cases for lupus nephritis.
In the combination therapy group, the median duration of follow-up prior to dual therapy was 36 months, with an additional 21 months of follow-up after receiving the combination. Prior to combination therapy, these 12 patients collectively had 38 lupus flares, 26 of which featured renal involvement. In contrast, post treatment they developed just 13 flares, only 3 of which were renal, according to Dr. Shahzad of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Md.
The 43 SLE patients had a total of 15 bacterial, 6 fungal, and 18 viral infections. And although there was a consistent trend toward higher rates in the combination therapy group, the differences fell far short of statistical significance.
Dr. Shahzad acknowledged the small sample size and retrospective design as important study limitations. However, based upon these encouraging, albeit preliminary, study findings, he and his NIH colleagues said they are planning a prospective clinical trial examining the efficacy and tolerability of the intravenous cyclophosphamide/rituximab combination in patients with severe, recurrent SLE.
Dr. Shahzad reported having no financial conflicts regarding this NIH-sponsored study.
SAN DIEGO – The combination of intravenous cyclophosphamide and rituximab shows promise for the reduction of lupus flares, both renal and nonrenal, in patients with severe systemic lupus erythematosus, according to Dr. Ali Shahzad.
Moreover, this benefit did not come at the cost of a significant increase in infections, as compared with intravenous cyclophosphamide monotherapy, Dr. Shahzad reported at the annual meeting of the American College of Rheumatology.
He reported on 43 patients with severe, recurrent SLE. Thirty-one were placed on intravenous cyclophosphamide monotherapy administered according to the National Institutes of Health standard protocol for the treatment of lupus nephritis. The other 12 got cyclophosphamide plus two 1,000-mg doses of rituximab (Rituxan) given 15 days apart. The combination regimen was given at the physician’s discretion for recalcitrant or recurrent flares of lupus nephritis in 10 of 12 cases, and for treatment-resistant CNS lupus or other extrarenal lupus in the other 2. Eight of the 12 recipients of combination therapy had previously been treated with intravenous cyclophosphamide, in most cases for lupus nephritis.
In the combination therapy group, the median duration of follow-up prior to dual therapy was 36 months, with an additional 21 months of follow-up after receiving the combination. Prior to combination therapy, these 12 patients collectively had 38 lupus flares, 26 of which featured renal involvement. In contrast, post treatment they developed just 13 flares, only 3 of which were renal, according to Dr. Shahzad of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Md.
The 43 SLE patients had a total of 15 bacterial, 6 fungal, and 18 viral infections. And although there was a consistent trend toward higher rates in the combination therapy group, the differences fell far short of statistical significance.
Dr. Shahzad acknowledged the small sample size and retrospective design as important study limitations. However, based upon these encouraging, albeit preliminary, study findings, he and his NIH colleagues said they are planning a prospective clinical trial examining the efficacy and tolerability of the intravenous cyclophosphamide/rituximab combination in patients with severe, recurrent SLE.
Dr. Shahzad reported having no financial conflicts regarding this NIH-sponsored study.
SAN DIEGO – The combination of intravenous cyclophosphamide and rituximab shows promise for the reduction of lupus flares, both renal and nonrenal, in patients with severe systemic lupus erythematosus, according to Dr. Ali Shahzad.
Moreover, this benefit did not come at the cost of a significant increase in infections, as compared with intravenous cyclophosphamide monotherapy, Dr. Shahzad reported at the annual meeting of the American College of Rheumatology.
He reported on 43 patients with severe, recurrent SLE. Thirty-one were placed on intravenous cyclophosphamide monotherapy administered according to the National Institutes of Health standard protocol for the treatment of lupus nephritis. The other 12 got cyclophosphamide plus two 1,000-mg doses of rituximab (Rituxan) given 15 days apart. The combination regimen was given at the physician’s discretion for recalcitrant or recurrent flares of lupus nephritis in 10 of 12 cases, and for treatment-resistant CNS lupus or other extrarenal lupus in the other 2. Eight of the 12 recipients of combination therapy had previously been treated with intravenous cyclophosphamide, in most cases for lupus nephritis.
In the combination therapy group, the median duration of follow-up prior to dual therapy was 36 months, with an additional 21 months of follow-up after receiving the combination. Prior to combination therapy, these 12 patients collectively had 38 lupus flares, 26 of which featured renal involvement. In contrast, post treatment they developed just 13 flares, only 3 of which were renal, according to Dr. Shahzad of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Md.
The 43 SLE patients had a total of 15 bacterial, 6 fungal, and 18 viral infections. And although there was a consistent trend toward higher rates in the combination therapy group, the differences fell far short of statistical significance.
Dr. Shahzad acknowledged the small sample size and retrospective design as important study limitations. However, based upon these encouraging, albeit preliminary, study findings, he and his NIH colleagues said they are planning a prospective clinical trial examining the efficacy and tolerability of the intravenous cyclophosphamide/rituximab combination in patients with severe, recurrent SLE.
Dr. Shahzad reported having no financial conflicts regarding this NIH-sponsored study.
AT THE ACR ANNUAL MEETING
Major finding: Patients with severe recurrent systemic lupus erythematosus collectively had 38 lupus flares, including 26 with renal involvement, during a median 36-month period before undergoing combination therapy with intravenous cyclophosphamide and rituximab; they had 13 flares, only 3 of which featured renal involvement, during 21 months afterward.
Data source: A retrospective study including 43 patients with severe recurrent SLE, 31 of whom were placed on intravenous cyclophosphamide monotherapy, while 12 received combination therapy with cyclophosphamide plus rituximab.
Disclosures: This study was sponsored by the National Institutes of Health. The presenter, an employee of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, reported having no relevant financial conflicts.