User login
Post-MI beta-blockers: Less is more
WASHINGTON – Physicians seldom prescribe beta-blockers post MI in the full doses shown to improve survival in the classic randomized clinical trials conducted several decades ago, according to a large new study – and that, as it turns out, is a very good thing.
Results of the OBTAIN (Optimal Beta-Blocker Therapy After Myocardial Infarction) registry indicate that 2-year mortality post MI is lowest in patients discharged on a dose that’s roughly just 25% of target doses established in the landmark randomized trials, Dr. Jeffrey Goldberger reported at the annual meeting of the American College of Cardiology.

Two-year mortality post MI discharge followed a J-shaped curve: It was lowest, at 9.4%, in patients on about 25% of the target dose, and worst, at 14.9%, among those on more than 50% of the target dose. Mortality was intermediate in those whose beta-blocker dose hovered around either 12% or 50%. And patients on any of these four commonly prescribed dosing regimens fared significantly better than those who weren’t discharged on a beta-blocker, whose 2-year mortality was 21.7%, reported Dr. Goldberger, professor of medicine at Northwestern University in Chicago.
"Based on these data, we feel that following an MI it is important to treat patients with a beta-blocker. However, achieving the target doses used in prior randomized clinical trials is neither necessary nor desirable," he declared.
Quality improvement initiatives implemented in the past decade have resulted in greater than 90% utilization of beta-blockers after MI. However, practice guidelines don’t recommend specific beta-blockers or target doses. The OBTAIN registry was created in order to pin down the optimal dosing. The study hypothesis was that achieving target doses would be associated with better survival than with lower doses. Not so, investigators discovered.
The OBTAIN registry included 7,057 acute MI patients enrolled at 26 participating centers. In-hospital mortality was 4.7%. Of those patients who survived to discharge, 91.5% left the hospital on a beta-blocker. Metoprolol was prescribed in two-thirds of patients. The second most frequently prescribed beta-blocker post MI was carvedilol, utilized in one-quarter of patients.
The target daily doses normalized to those established in the landmark randomized trials as having a survival benefit are metoprolol 200 mg, carvedilol 50 mg, atenolol 100 mg, bisoprolol 10 mg, propranolol 180 mg, and timolol 20 mg.
Thirty-seven percent of patients discharged on a beta-blocker were on roughly 25% of the target dose. Roughly 25% of patients were on 12% of the target dose, a similar proportion got 50% of the target dose, and a mere 13% got more than 50% of the target dose. Three weeks post discharge, three-quarters of patients were on the same dose as when they left the hospital. Half of the rest had been titrated to a higher dose, the other half to a lower one.
In a multivariate analysis adjusted for demographics, diabetes and other comorbid conditions, ST-elevation MI, in-hospital therapies and length of stay, and other discharge medications, patients discharged on a beta-blocker had a 38%-51% lower mortality than those not on a beta-blocker. The most common reason that patients weren’t on a beta-blocker at discharge was hypotension.
Session cochair Dr. Yochai Birnbaum observed that the landmark randomized trials showing a mortality benefit for post-MI beta-blocker therapy were carried out in an era before statins and other potent secondary prevention therapies.
"It might be that when we add more and more medications, there are some interactions that are unclear. Maybe the previous recommendations are no longer valid," said Dr. Birnbaum, professor of medicine at Baylor College of Medicine, Houston.
The OBTAIN registry was sponsored by the National Heart, Lung, and Blood Institute. Dr. Goldberger reported having no relevant financial relationships.
WASHINGTON – Physicians seldom prescribe beta-blockers post MI in the full doses shown to improve survival in the classic randomized clinical trials conducted several decades ago, according to a large new study – and that, as it turns out, is a very good thing.
Results of the OBTAIN (Optimal Beta-Blocker Therapy After Myocardial Infarction) registry indicate that 2-year mortality post MI is lowest in patients discharged on a dose that’s roughly just 25% of target doses established in the landmark randomized trials, Dr. Jeffrey Goldberger reported at the annual meeting of the American College of Cardiology.
Two-year mortality post MI discharge followed a J-shaped curve: It was lowest, at 9.4%, in patients on about 25% of the target dose, and worst, at 14.9%, among those on more than 50% of the target dose. Mortality was intermediate in those whose beta-blocker dose hovered around either 12% or 50%. And patients on any of these four commonly prescribed dosing regimens fared significantly better than those who weren’t discharged on a beta-blocker, whose 2-year mortality was 21.7%, reported Dr. Goldberger, professor of medicine at Northwestern University in Chicago.
"Based on these data, we feel that following an MI it is important to treat patients with a beta-blocker. However, achieving the target doses used in prior randomized clinical trials is neither necessary nor desirable," he declared.
Quality improvement initiatives implemented in the past decade have resulted in greater than 90% utilization of beta-blockers after MI. However, practice guidelines don’t recommend specific beta-blockers or target doses. The OBTAIN registry was created in order to pin down the optimal dosing. The study hypothesis was that achieving target doses would be associated with better survival than with lower doses. Not so, investigators discovered.
The OBTAIN registry included 7,057 acute MI patients enrolled at 26 participating centers. In-hospital mortality was 4.7%. Of those patients who survived to discharge, 91.5% left the hospital on a beta-blocker. Metoprolol was prescribed in two-thirds of patients. The second most frequently prescribed beta-blocker post MI was carvedilol, utilized in one-quarter of patients.
The target daily doses normalized to those established in the landmark randomized trials as having a survival benefit are metoprolol 200 mg, carvedilol 50 mg, atenolol 100 mg, bisoprolol 10 mg, propranolol 180 mg, and timolol 20 mg.
Thirty-seven percent of patients discharged on a beta-blocker were on roughly 25% of the target dose. Roughly 25% of patients were on 12% of the target dose, a similar proportion got 50% of the target dose, and a mere 13% got more than 50% of the target dose. Three weeks post discharge, three-quarters of patients were on the same dose as when they left the hospital. Half of the rest had been titrated to a higher dose, the other half to a lower one.
In a multivariate analysis adjusted for demographics, diabetes and other comorbid conditions, ST-elevation MI, in-hospital therapies and length of stay, and other discharge medications, patients discharged on a beta-blocker had a 38%-51% lower mortality than those not on a beta-blocker. The most common reason that patients weren’t on a beta-blocker at discharge was hypotension.
Session cochair Dr. Yochai Birnbaum observed that the landmark randomized trials showing a mortality benefit for post-MI beta-blocker therapy were carried out in an era before statins and other potent secondary prevention therapies.
"It might be that when we add more and more medications, there are some interactions that are unclear. Maybe the previous recommendations are no longer valid," said Dr. Birnbaum, professor of medicine at Baylor College of Medicine, Houston.
The OBTAIN registry was sponsored by the National Heart, Lung, and Blood Institute. Dr. Goldberger reported having no relevant financial relationships.
WASHINGTON – Physicians seldom prescribe beta-blockers post MI in the full doses shown to improve survival in the classic randomized clinical trials conducted several decades ago, according to a large new study – and that, as it turns out, is a very good thing.
Results of the OBTAIN (Optimal Beta-Blocker Therapy After Myocardial Infarction) registry indicate that 2-year mortality post MI is lowest in patients discharged on a dose that’s roughly just 25% of target doses established in the landmark randomized trials, Dr. Jeffrey Goldberger reported at the annual meeting of the American College of Cardiology.
Two-year mortality post MI discharge followed a J-shaped curve: It was lowest, at 9.4%, in patients on about 25% of the target dose, and worst, at 14.9%, among those on more than 50% of the target dose. Mortality was intermediate in those whose beta-blocker dose hovered around either 12% or 50%. And patients on any of these four commonly prescribed dosing regimens fared significantly better than those who weren’t discharged on a beta-blocker, whose 2-year mortality was 21.7%, reported Dr. Goldberger, professor of medicine at Northwestern University in Chicago.
"Based on these data, we feel that following an MI it is important to treat patients with a beta-blocker. However, achieving the target doses used in prior randomized clinical trials is neither necessary nor desirable," he declared.
Quality improvement initiatives implemented in the past decade have resulted in greater than 90% utilization of beta-blockers after MI. However, practice guidelines don’t recommend specific beta-blockers or target doses. The OBTAIN registry was created in order to pin down the optimal dosing. The study hypothesis was that achieving target doses would be associated with better survival than with lower doses. Not so, investigators discovered.
The OBTAIN registry included 7,057 acute MI patients enrolled at 26 participating centers. In-hospital mortality was 4.7%. Of those patients who survived to discharge, 91.5% left the hospital on a beta-blocker. Metoprolol was prescribed in two-thirds of patients. The second most frequently prescribed beta-blocker post MI was carvedilol, utilized in one-quarter of patients.
The target daily doses normalized to those established in the landmark randomized trials as having a survival benefit are metoprolol 200 mg, carvedilol 50 mg, atenolol 100 mg, bisoprolol 10 mg, propranolol 180 mg, and timolol 20 mg.
Thirty-seven percent of patients discharged on a beta-blocker were on roughly 25% of the target dose. Roughly 25% of patients were on 12% of the target dose, a similar proportion got 50% of the target dose, and a mere 13% got more than 50% of the target dose. Three weeks post discharge, three-quarters of patients were on the same dose as when they left the hospital. Half of the rest had been titrated to a higher dose, the other half to a lower one.
In a multivariate analysis adjusted for demographics, diabetes and other comorbid conditions, ST-elevation MI, in-hospital therapies and length of stay, and other discharge medications, patients discharged on a beta-blocker had a 38%-51% lower mortality than those not on a beta-blocker. The most common reason that patients weren’t on a beta-blocker at discharge was hypotension.
Session cochair Dr. Yochai Birnbaum observed that the landmark randomized trials showing a mortality benefit for post-MI beta-blocker therapy were carried out in an era before statins and other potent secondary prevention therapies.
"It might be that when we add more and more medications, there are some interactions that are unclear. Maybe the previous recommendations are no longer valid," said Dr. Birnbaum, professor of medicine at Baylor College of Medicine, Houston.
The OBTAIN registry was sponsored by the National Heart, Lung, and Blood Institute. Dr. Goldberger reported having no relevant financial relationships.
AT ACC 14
Major finding: Patients discharged post MI on a beta-blocker at roughly 25% of the target dose – which was established as conferring a survival benefit in the earlier, classic randomized trials – had a significantly lower 2-year mortality than those discharged on higher or lower doses.
Data source: The observational OBTAIN registry included more than 7,000 acute MI patients at 26 sites.
Disclosures: Dr. Goldberger reported having no financial conflicts with regard to the National Heart, Lung, and Blood Institute–sponsored registry.

ACC/AHA Cardiovascular Risk Equations Get a Thumbs-up
WASHINGTON – The controversial cardiovascular risk estimator introduced in the current American College of Cardiology/American Heart Association risk-assessment guidelines demonstrated "moderate to good" predictive performance when applied to a large U.S. cohort for whom consideration of statin therapy is clinically relevant, Paul Muntner, Ph.D., reported at the annual meeting of the American College of Cardiology.
"We believe that the current study supports the validity of the pooled cohort risk equations to inform clinical management decisions," said Dr. Muntner, professor of epidemiology and of medicine at the University of Alabama at Birmingham.
The risk estimator has come under strong criticism since the guidelines were released last November. When critics applied the risk estimator to participants in the Women's Health Study, the Physicians' Health Study, and the Women's Health Initiative, they found a big discrepancy between the observed atherosclerotic cardiovascular disease (ASCVD) event rates during follow-up and the predicted rates based on the risk calculator, with the ACC/AHA risk estimator tending to markedly overestimate risk. But those analyses involved studies lacking surveillance mechanisms to identify ASCVD events that weren’t reported by participants, according to Dr. Muntner.
"One of the challenges with those big studies is the underreporting of events. Let’s look at the Women’s Health Initiative. Roughly 25% of adjudicated events in that study were not detected because of the reliance on patient reporting. There were two reasons for this: Participants didn’t report a subsequently validated event, or hospital consent forms didn’t permit release of the chart to study investigators," he asserted.
Dr. Muntner presented a new analysis in which the ASCVD risk estimator was applied to participants in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) study, a prospective, observational, population-based study of more than 30,000 U.S. black and white patients. He and his coworkers compared the observed 5-year rates of the combined endpoint of death from coronary heart disease, nonfatal MI, or fatal or nonfatal stroke to rates projected by the risk equations.

The analysis was restricted to the 10,997 REGARDS participants who fell into the category of the population for whom the risk equations were designed as a guide in decision making regarding initiation of statin therapy: people aged 40-79 years without atherosclerotic cardiovascular disease or diabetes, not on a statin, and with an LDL cholesterol level of 70-189 mg/dL.
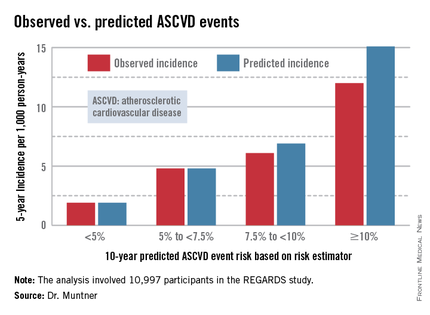
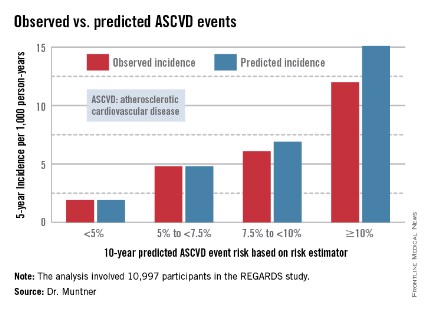
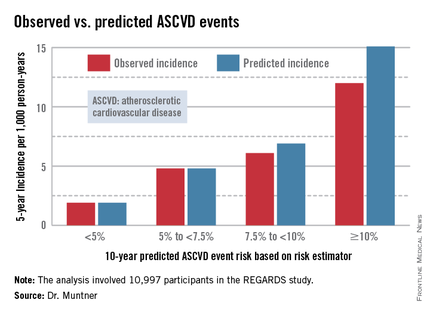
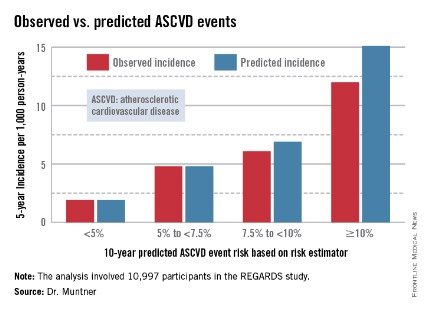
In participants in the lower 10-year ASCVD risk categories based on the equations, the predicted 5-year event rates were spot on with the observed rates. In patients at the higher end of the 10-year risk spectrum, the equations tended to overestimate the event risk (see chart). However, it should be noted that roughly 40% of the REGARDS cohort initiated statin therapy during the 5-year follow-up period, and that would have lowered their event rate, Dr. Muntner said.
The investigators also compared observed and predicted 5-year event rates in a separate REGARDS subgroup composed of 3,333 study participants with Medicare Part A insurance. In this older cohort, the risk equations tended to modestly underestimate the observed ASCVD event rate. "Overall, though, I would say this is pretty good calibration," the epidemiologist commented.
Simultaneous with Dr. Muntner’s presentation at ACC 14, the study results were published (JAMA 2014 April 9;311:1406-15).
The REGARDS study is funded by the National Institutes of Health, as was Dr. Muntner’s analysis. He reported having no relevant financial interests.
WASHINGTON – The controversial cardiovascular risk estimator introduced in the current American College of Cardiology/American Heart Association risk-assessment guidelines demonstrated "moderate to good" predictive performance when applied to a large U.S. cohort for whom consideration of statin therapy is clinically relevant, Paul Muntner, Ph.D., reported at the annual meeting of the American College of Cardiology.
"We believe that the current study supports the validity of the pooled cohort risk equations to inform clinical management decisions," said Dr. Muntner, professor of epidemiology and of medicine at the University of Alabama at Birmingham.
The risk estimator has come under strong criticism since the guidelines were released last November. When critics applied the risk estimator to participants in the Women's Health Study, the Physicians' Health Study, and the Women's Health Initiative, they found a big discrepancy between the observed atherosclerotic cardiovascular disease (ASCVD) event rates during follow-up and the predicted rates based on the risk calculator, with the ACC/AHA risk estimator tending to markedly overestimate risk. But those analyses involved studies lacking surveillance mechanisms to identify ASCVD events that weren’t reported by participants, according to Dr. Muntner.
"One of the challenges with those big studies is the underreporting of events. Let’s look at the Women’s Health Initiative. Roughly 25% of adjudicated events in that study were not detected because of the reliance on patient reporting. There were two reasons for this: Participants didn’t report a subsequently validated event, or hospital consent forms didn’t permit release of the chart to study investigators," he asserted.
Dr. Muntner presented a new analysis in which the ASCVD risk estimator was applied to participants in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) study, a prospective, observational, population-based study of more than 30,000 U.S. black and white patients. He and his coworkers compared the observed 5-year rates of the combined endpoint of death from coronary heart disease, nonfatal MI, or fatal or nonfatal stroke to rates projected by the risk equations.
The analysis was restricted to the 10,997 REGARDS participants who fell into the category of the population for whom the risk equations were designed as a guide in decision making regarding initiation of statin therapy: people aged 40-79 years without atherosclerotic cardiovascular disease or diabetes, not on a statin, and with an LDL cholesterol level of 70-189 mg/dL.
In participants in the lower 10-year ASCVD risk categories based on the equations, the predicted 5-year event rates were spot on with the observed rates. In patients at the higher end of the 10-year risk spectrum, the equations tended to overestimate the event risk (see chart). However, it should be noted that roughly 40% of the REGARDS cohort initiated statin therapy during the 5-year follow-up period, and that would have lowered their event rate, Dr. Muntner said.
The investigators also compared observed and predicted 5-year event rates in a separate REGARDS subgroup composed of 3,333 study participants with Medicare Part A insurance. In this older cohort, the risk equations tended to modestly underestimate the observed ASCVD event rate. "Overall, though, I would say this is pretty good calibration," the epidemiologist commented.
Simultaneous with Dr. Muntner’s presentation at ACC 14, the study results were published (JAMA 2014 April 9;311:1406-15).
The REGARDS study is funded by the National Institutes of Health, as was Dr. Muntner’s analysis. He reported having no relevant financial interests.
WASHINGTON – The controversial cardiovascular risk estimator introduced in the current American College of Cardiology/American Heart Association risk-assessment guidelines demonstrated "moderate to good" predictive performance when applied to a large U.S. cohort for whom consideration of statin therapy is clinically relevant, Paul Muntner, Ph.D., reported at the annual meeting of the American College of Cardiology.
"We believe that the current study supports the validity of the pooled cohort risk equations to inform clinical management decisions," said Dr. Muntner, professor of epidemiology and of medicine at the University of Alabama at Birmingham.
The risk estimator has come under strong criticism since the guidelines were released last November. When critics applied the risk estimator to participants in the Women's Health Study, the Physicians' Health Study, and the Women's Health Initiative, they found a big discrepancy between the observed atherosclerotic cardiovascular disease (ASCVD) event rates during follow-up and the predicted rates based on the risk calculator, with the ACC/AHA risk estimator tending to markedly overestimate risk. But those analyses involved studies lacking surveillance mechanisms to identify ASCVD events that weren’t reported by participants, according to Dr. Muntner.
"One of the challenges with those big studies is the underreporting of events. Let’s look at the Women’s Health Initiative. Roughly 25% of adjudicated events in that study were not detected because of the reliance on patient reporting. There were two reasons for this: Participants didn’t report a subsequently validated event, or hospital consent forms didn’t permit release of the chart to study investigators," he asserted.
Dr. Muntner presented a new analysis in which the ASCVD risk estimator was applied to participants in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) study, a prospective, observational, population-based study of more than 30,000 U.S. black and white patients. He and his coworkers compared the observed 5-year rates of the combined endpoint of death from coronary heart disease, nonfatal MI, or fatal or nonfatal stroke to rates projected by the risk equations.
The analysis was restricted to the 10,997 REGARDS participants who fell into the category of the population for whom the risk equations were designed as a guide in decision making regarding initiation of statin therapy: people aged 40-79 years without atherosclerotic cardiovascular disease or diabetes, not on a statin, and with an LDL cholesterol level of 70-189 mg/dL.
In participants in the lower 10-year ASCVD risk categories based on the equations, the predicted 5-year event rates were spot on with the observed rates. In patients at the higher end of the 10-year risk spectrum, the equations tended to overestimate the event risk (see chart). However, it should be noted that roughly 40% of the REGARDS cohort initiated statin therapy during the 5-year follow-up period, and that would have lowered their event rate, Dr. Muntner said.
The investigators also compared observed and predicted 5-year event rates in a separate REGARDS subgroup composed of 3,333 study participants with Medicare Part A insurance. In this older cohort, the risk equations tended to modestly underestimate the observed ASCVD event rate. "Overall, though, I would say this is pretty good calibration," the epidemiologist commented.
Simultaneous with Dr. Muntner’s presentation at ACC 14, the study results were published (JAMA 2014 April 9;311:1406-15).
The REGARDS study is funded by the National Institutes of Health, as was Dr. Muntner’s analysis. He reported having no relevant financial interests.
AT ACC 14
ACC/AHA cardiovascular risk equations get a thumbs-up
WASHINGTON – The controversial cardiovascular risk estimator introduced in the current American College of Cardiology/American Heart Association risk-assessment guidelines demonstrated "moderate to good" predictive performance when applied to a large U.S. cohort for whom consideration of statin therapy is clinically relevant, Paul Muntner, Ph.D., reported at the annual meeting of the American College of Cardiology.
"We believe that the current study supports the validity of the pooled cohort risk equations to inform clinical management decisions," said Dr. Muntner, professor of epidemiology and of medicine at the University of Alabama at Birmingham.
The risk estimator has come under strong criticism since the guidelines were released last November. When critics applied the risk estimator to participants in the Women's Health Study, the Physicians' Health Study, and the Women's Health Initiative, they found a big discrepancy between the observed atherosclerotic cardiovascular disease (ASCVD) event rates during follow-up and the predicted rates based on the risk calculator, with the ACC/AHA risk estimator tending to markedly overestimate risk. But those analyses involved studies lacking surveillance mechanisms to identify ASCVD events that weren’t reported by participants, according to Dr. Muntner.
"One of the challenges with those big studies is the underreporting of events. Let’s look at the Women’s Health Initiative. Roughly 25% of adjudicated events in that study were not detected because of the reliance on patient reporting. There were two reasons for this: Participants didn’t report a subsequently validated event, or hospital consent forms didn’t permit release of the chart to study investigators," he asserted.
Dr. Muntner presented a new analysis in which the ASCVD risk estimator was applied to participants in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) study, a prospective, observational, population-based study of more than 30,000 U.S. black and white patients. He and his coworkers compared the observed 5-year rates of the combined endpoint of death from coronary heart disease, nonfatal MI, or fatal or nonfatal stroke to rates projected by the risk equations.

The analysis was restricted to the 10,997 REGARDS participants who fell into the category of the population for whom the risk equations were designed as a guide in decision making regarding initiation of statin therapy: people aged 40-79 years without atherosclerotic cardiovascular disease or diabetes, not on a statin, and with an LDL cholesterol level of 70-189 mg/dL.
In participants in the lower 10-year ASCVD risk categories based on the equations, the predicted 5-year event rates were spot on with the observed rates. In patients at the higher end of the 10-year risk spectrum, the equations tended to overestimate the event risk (see chart). However, it should be noted that roughly 40% of the REGARDS cohort initiated statin therapy during the 5-year follow-up period, and that would have lowered their event rate, Dr. Muntner said.
The investigators also compared observed and predicted 5-year event rates in a separate REGARDS subgroup composed of 3,333 study participants with Medicare Part A insurance. In this older cohort, the risk equations tended to modestly underestimate the observed ASCVD event rate. "Overall, though, I would say this is pretty good calibration," the epidemiologist commented.
Simultaneous with Dr. Muntner’s presentation at ACC 14, the study results were published (JAMA 2014 April 9;311:1406-15).
The REGARDS study is funded by the National Institutes of Health, as was Dr. Muntner’s analysis. He reported having no relevant financial interests.
WASHINGTON – The controversial cardiovascular risk estimator introduced in the current American College of Cardiology/American Heart Association risk-assessment guidelines demonstrated "moderate to good" predictive performance when applied to a large U.S. cohort for whom consideration of statin therapy is clinically relevant, Paul Muntner, Ph.D., reported at the annual meeting of the American College of Cardiology.
"We believe that the current study supports the validity of the pooled cohort risk equations to inform clinical management decisions," said Dr. Muntner, professor of epidemiology and of medicine at the University of Alabama at Birmingham.
The risk estimator has come under strong criticism since the guidelines were released last November. When critics applied the risk estimator to participants in the Women's Health Study, the Physicians' Health Study, and the Women's Health Initiative, they found a big discrepancy between the observed atherosclerotic cardiovascular disease (ASCVD) event rates during follow-up and the predicted rates based on the risk calculator, with the ACC/AHA risk estimator tending to markedly overestimate risk. But those analyses involved studies lacking surveillance mechanisms to identify ASCVD events that weren’t reported by participants, according to Dr. Muntner.
"One of the challenges with those big studies is the underreporting of events. Let’s look at the Women’s Health Initiative. Roughly 25% of adjudicated events in that study were not detected because of the reliance on patient reporting. There were two reasons for this: Participants didn’t report a subsequently validated event, or hospital consent forms didn’t permit release of the chart to study investigators," he asserted.
Dr. Muntner presented a new analysis in which the ASCVD risk estimator was applied to participants in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) study, a prospective, observational, population-based study of more than 30,000 U.S. black and white patients. He and his coworkers compared the observed 5-year rates of the combined endpoint of death from coronary heart disease, nonfatal MI, or fatal or nonfatal stroke to rates projected by the risk equations.
The analysis was restricted to the 10,997 REGARDS participants who fell into the category of the population for whom the risk equations were designed as a guide in decision making regarding initiation of statin therapy: people aged 40-79 years without atherosclerotic cardiovascular disease or diabetes, not on a statin, and with an LDL cholesterol level of 70-189 mg/dL.
In participants in the lower 10-year ASCVD risk categories based on the equations, the predicted 5-year event rates were spot on with the observed rates. In patients at the higher end of the 10-year risk spectrum, the equations tended to overestimate the event risk (see chart). However, it should be noted that roughly 40% of the REGARDS cohort initiated statin therapy during the 5-year follow-up period, and that would have lowered their event rate, Dr. Muntner said.
The investigators also compared observed and predicted 5-year event rates in a separate REGARDS subgroup composed of 3,333 study participants with Medicare Part A insurance. In this older cohort, the risk equations tended to modestly underestimate the observed ASCVD event rate. "Overall, though, I would say this is pretty good calibration," the epidemiologist commented.
Simultaneous with Dr. Muntner’s presentation at ACC 14, the study results were published (JAMA 2014 April 9;311:1406-15).
The REGARDS study is funded by the National Institutes of Health, as was Dr. Muntner’s analysis. He reported having no relevant financial interests.
WASHINGTON – The controversial cardiovascular risk estimator introduced in the current American College of Cardiology/American Heart Association risk-assessment guidelines demonstrated "moderate to good" predictive performance when applied to a large U.S. cohort for whom consideration of statin therapy is clinically relevant, Paul Muntner, Ph.D., reported at the annual meeting of the American College of Cardiology.
"We believe that the current study supports the validity of the pooled cohort risk equations to inform clinical management decisions," said Dr. Muntner, professor of epidemiology and of medicine at the University of Alabama at Birmingham.
The risk estimator has come under strong criticism since the guidelines were released last November. When critics applied the risk estimator to participants in the Women's Health Study, the Physicians' Health Study, and the Women's Health Initiative, they found a big discrepancy between the observed atherosclerotic cardiovascular disease (ASCVD) event rates during follow-up and the predicted rates based on the risk calculator, with the ACC/AHA risk estimator tending to markedly overestimate risk. But those analyses involved studies lacking surveillance mechanisms to identify ASCVD events that weren’t reported by participants, according to Dr. Muntner.
"One of the challenges with those big studies is the underreporting of events. Let’s look at the Women’s Health Initiative. Roughly 25% of adjudicated events in that study were not detected because of the reliance on patient reporting. There were two reasons for this: Participants didn’t report a subsequently validated event, or hospital consent forms didn’t permit release of the chart to study investigators," he asserted.
Dr. Muntner presented a new analysis in which the ASCVD risk estimator was applied to participants in the REGARDS (Reasons for Geographic and Racial Differences in Stroke) study, a prospective, observational, population-based study of more than 30,000 U.S. black and white patients. He and his coworkers compared the observed 5-year rates of the combined endpoint of death from coronary heart disease, nonfatal MI, or fatal or nonfatal stroke to rates projected by the risk equations.
The analysis was restricted to the 10,997 REGARDS participants who fell into the category of the population for whom the risk equations were designed as a guide in decision making regarding initiation of statin therapy: people aged 40-79 years without atherosclerotic cardiovascular disease or diabetes, not on a statin, and with an LDL cholesterol level of 70-189 mg/dL.
In participants in the lower 10-year ASCVD risk categories based on the equations, the predicted 5-year event rates were spot on with the observed rates. In patients at the higher end of the 10-year risk spectrum, the equations tended to overestimate the event risk (see chart). However, it should be noted that roughly 40% of the REGARDS cohort initiated statin therapy during the 5-year follow-up period, and that would have lowered their event rate, Dr. Muntner said.
The investigators also compared observed and predicted 5-year event rates in a separate REGARDS subgroup composed of 3,333 study participants with Medicare Part A insurance. In this older cohort, the risk equations tended to modestly underestimate the observed ASCVD event rate. "Overall, though, I would say this is pretty good calibration," the epidemiologist commented.
Simultaneous with Dr. Muntner’s presentation at ACC 14, the study results were published (JAMA 2014 April 9;311:1406-15).
The REGARDS study is funded by the National Institutes of Health, as was Dr. Muntner’s analysis. He reported having no relevant financial interests.
AT ACC 14
Major finding: The controversial cardiovascular risk equations at the heart of the latest ACC/AHA risk-assessment guidelines turned in a moderate to good performance in a validation study involving nearly 11,000 participants in a large, observational, prospective study.
Data source: The REGARDS study is a population-based study in which more than 30,000 U.S. black and white patients are being followed prospectively.
Disclosures: The analysis was funded by the National Institutes of Health. The presenter reported having no financial conflicts.
Crushing ticagrelor tablets sped platelet inhibition
WASHINGTON – Administration of crushed tablets of ticagrelor* may provide a way to dramatically accelerate the onset of platelet inhibition in patients with ST-elevation myocardial infarction.
In a proof-of-concept crossover study involving 36 healthy volunteers, crushing a whole 90-mg tablet of ticagrelor (Brilinta) and suspending it in 200 mL of water given orally resulted in a mean plasma ticagrelor concentration of 149 ng/mL 30 minutes post dose, compared with 34 ng/mL after a whole tablet was swallowed, Dr. Glenn F. Carlson reported at the annual meeting of the American College of Cardiology.
When the crushed tablet suspended in water was administered via nasogastric tube, the results were even more dramatic: a mean plasma ticagrelor concentration of 265 ng/mL 30 minutes post dose, or a ninefold greater mean plasma concentration compared with swallowing a whole tablet, said Dr. Carlson of AstraZeneca.
At 1 hour post dose, mean plasma concentrations of ticagrelor and its metabolite remained significantly greater with both of the crushed-tablet dosing strategies than with whole ticagrelor. However, 2 to 48 hours post dose, plasma concentrations were similar for all three drug administration strategies.
The clinical relevance of these findings lies in the well-established observation that in acute coronary syndromes – and especially in ST-elevation myocardial infarction – time is myocardium. That is, the more quickly that effective antiplatelet activity and reperfusion can be achieved, the less extensive the myocardial tissue damage, he noted.
The three modes of ticagrelor administration were found to be bioequivalent and equally safe.
The study was funded by AstraZeneca. Dr. Carlson is an employee of the company.
*CORRECTION, 4/29/2014: An earlier version of the article misstated the name of the drug ticagrelor.
WASHINGTON – Administration of crushed tablets of ticagrelor* may provide a way to dramatically accelerate the onset of platelet inhibition in patients with ST-elevation myocardial infarction.
In a proof-of-concept crossover study involving 36 healthy volunteers, crushing a whole 90-mg tablet of ticagrelor (Brilinta) and suspending it in 200 mL of water given orally resulted in a mean plasma ticagrelor concentration of 149 ng/mL 30 minutes post dose, compared with 34 ng/mL after a whole tablet was swallowed, Dr. Glenn F. Carlson reported at the annual meeting of the American College of Cardiology.
When the crushed tablet suspended in water was administered via nasogastric tube, the results were even more dramatic: a mean plasma ticagrelor concentration of 265 ng/mL 30 minutes post dose, or a ninefold greater mean plasma concentration compared with swallowing a whole tablet, said Dr. Carlson of AstraZeneca.
At 1 hour post dose, mean plasma concentrations of ticagrelor and its metabolite remained significantly greater with both of the crushed-tablet dosing strategies than with whole ticagrelor. However, 2 to 48 hours post dose, plasma concentrations were similar for all three drug administration strategies.
The clinical relevance of these findings lies in the well-established observation that in acute coronary syndromes – and especially in ST-elevation myocardial infarction – time is myocardium. That is, the more quickly that effective antiplatelet activity and reperfusion can be achieved, the less extensive the myocardial tissue damage, he noted.
The three modes of ticagrelor administration were found to be bioequivalent and equally safe.
The study was funded by AstraZeneca. Dr. Carlson is an employee of the company.
*CORRECTION, 4/29/2014: An earlier version of the article misstated the name of the drug ticagrelor.
WASHINGTON – Administration of crushed tablets of ticagrelor* may provide a way to dramatically accelerate the onset of platelet inhibition in patients with ST-elevation myocardial infarction.
In a proof-of-concept crossover study involving 36 healthy volunteers, crushing a whole 90-mg tablet of ticagrelor (Brilinta) and suspending it in 200 mL of water given orally resulted in a mean plasma ticagrelor concentration of 149 ng/mL 30 minutes post dose, compared with 34 ng/mL after a whole tablet was swallowed, Dr. Glenn F. Carlson reported at the annual meeting of the American College of Cardiology.
When the crushed tablet suspended in water was administered via nasogastric tube, the results were even more dramatic: a mean plasma ticagrelor concentration of 265 ng/mL 30 minutes post dose, or a ninefold greater mean plasma concentration compared with swallowing a whole tablet, said Dr. Carlson of AstraZeneca.
At 1 hour post dose, mean plasma concentrations of ticagrelor and its metabolite remained significantly greater with both of the crushed-tablet dosing strategies than with whole ticagrelor. However, 2 to 48 hours post dose, plasma concentrations were similar for all three drug administration strategies.
The clinical relevance of these findings lies in the well-established observation that in acute coronary syndromes – and especially in ST-elevation myocardial infarction – time is myocardium. That is, the more quickly that effective antiplatelet activity and reperfusion can be achieved, the less extensive the myocardial tissue damage, he noted.
The three modes of ticagrelor administration were found to be bioequivalent and equally safe.
The study was funded by AstraZeneca. Dr. Carlson is an employee of the company.
*CORRECTION, 4/29/2014: An earlier version of the article misstated the name of the drug ticagrelor.
AT ACC 14
Major finding: Mean plasma concentration of ticagralor was roughly fivefold greater 30 minutes post dose via a crushed tablet in water vs. a tablet swallowed whole, and ninefold greater when the medication was delivered in a suspension via nasogastric tube.
Data source: This was a single-center, randomized, crossover study involving 36 healthy volunteers who on separate occasions swallowed one 90-mg tablet of the antiplatelet agent ticagralor whole, drank a crushed tablet suspended in 200 mL of water, or received the suspension via nasogastric tube.
Disclosures: The study was funded by AstraZeneca. Dr. Carlson is an employee of the company.
Ultrasound-facilitated endovascular thrombolysis 'a game changer' in acute pulmonary embolism
WASHINGTON – Ultrasound-facilitated, catheter-directed, low-dose fibrinolysis for acute massive or submassive pulmonary embolism significantly improves right ventricular function, reduces pulmonary hypertension and angiographic evidence of obstruction, and lessens the risk of fibrinolysis-associated intracranial hemorrhage, according to a prospective multicenter clinical trial.
"By minimizing the risk of intracranial bleeding, ultrasound-facilitated, catheter-directed, low-dose fibrinolysis represents a potential game changer in the treatment of high-risk pulmonary embolism patients," Dr. Gregory Piazza said in presenting the results of the SEATTLE II study at the annual meeting of the American College of Cardiology.

Full-dose systemic fibrinolysis has long been the go-to advanced therapy for high-risk pulmonary embolism (PE), but physicians are leery of the associated 2%-3% risk of catastrophic intracranial hemorrhage, noted Dr. Piazza, a cardiologist at Brigham and Women’s Hospital and Harvard University, Boston.
SEATTLE II was a single-arm, 21-site, prospective study in which 150 patients with high-risk PE underwent treatment using the commercially available EKOS EkoSonic Endovascular System.
Twenty-one percent of patients had massive PE, defined as presentation with syncope, cardiogenic shock, resuscitated cardiac arrest, or persistent hypotension. The remaining 79% had submassive PE, with normal blood pressure but evidence of right ventricular dysfunction. All participants had to have a right ventricular/left ventricular ratio (RV/LV) of 0.9 or greater on the same chest CT scan used in diagnosing the PE. This CT documentation of right ventricular dysfunction has been associated in a meta-analysis of patients with submassive PE with a 7.4-fold increased risk of death due to PE compared to normotensive PE patients with normal right ventricular function (J. Thromb. Haemost. 2013;11:1823-32).
The primary endpoint was change in RV/LV on chest CT from baseline to 48 hours after initiation of fibrinolysis. This ratio improved from 1.55 to 1.13, for a statistically and clinically significant 27% reduction. A similar-size improvement was seen in pulmonary artery systolic pressure – a secondary efficacy endpoint – which decreased from 51.4 mm Hg before treatment to 37.5 mm Hg post procedure and 36.9 mm Hg at 48 hours. Both efficacy endpoints improved to a similar extent regardless of whether patients had massive or submassive PE.
The mean Modified Miller Pulmonary Artery Angiographic Obstruction Score improved by 30%, from 22.5 pretreatment to 15.8 at 48 hours.
Three in-hospital deaths occurred. One was due to massive PE which occurred before the fibrinolysis procedure could be completed. The others were not directly attributable to PE: One involved overwhelming sepsis and the other was due to progressive respiratory failure. Major bleeding occurred in 11% of patients; however, 16 of the 17 events were classified as GUSTO moderate bleeds, with only a single GUSTO severe hemorrhage.
There were no intracranial hemorrhages.
The fibrinolytic agent used in SEATTLE II was tissue plasminogen activator, delivered at 1 mg/hr for a total dose of 24 mg. Patients with unilateral PE received a single device and 24 hours of infusion time. The 86% of patients who had bilateral disease got two devices and 12 hours of therapy.
The proprietary EKOS system consists of two catheters: an outer infusion catheter with side holes that elute the fibrinolytic agent, and an inner core catheter with ultrasound transducers placed at regular intervals. These transducers produce low-intensity ultrasound which serves two purposes. Through a process called acoustic streaming, the low-intensity ultrasound helps push the fibrinolytic agent closer to the thrombus. Plus, the ultrasound energy causes the clot fibrin to reconfigure from a tight lattice to a more porous structure that promotes deeper penetration of the fibrinolytic, Dr. Piazza explained.
Audience members greeted the study results enthusiastically.
"Most of us really do believe that for safety reasons this is the way to go, rather than systemic fibrinolysis," one said.
Dr. Piazza said the next step in defining the EKOS system’s role in clinical practice will be to study briefer infusion times as a means of achieving faster patient improvement with reduced use of hospital resources, including ICU and intermediate-care unit time.
The EKOS system has been approved in the United States since 2005 for treatment of blood clots in the arms and legs. In Europe it gained an additional indication for treatment of massive and submassive PE in 2011.
The SEATTLE II study was sponsored by EKOS Corp. Dr. Piazza reported receiving a research grant from the company.
WASHINGTON – Ultrasound-facilitated, catheter-directed, low-dose fibrinolysis for acute massive or submassive pulmonary embolism significantly improves right ventricular function, reduces pulmonary hypertension and angiographic evidence of obstruction, and lessens the risk of fibrinolysis-associated intracranial hemorrhage, according to a prospective multicenter clinical trial.
"By minimizing the risk of intracranial bleeding, ultrasound-facilitated, catheter-directed, low-dose fibrinolysis represents a potential game changer in the treatment of high-risk pulmonary embolism patients," Dr. Gregory Piazza said in presenting the results of the SEATTLE II study at the annual meeting of the American College of Cardiology.
Full-dose systemic fibrinolysis has long been the go-to advanced therapy for high-risk pulmonary embolism (PE), but physicians are leery of the associated 2%-3% risk of catastrophic intracranial hemorrhage, noted Dr. Piazza, a cardiologist at Brigham and Women’s Hospital and Harvard University, Boston.
SEATTLE II was a single-arm, 21-site, prospective study in which 150 patients with high-risk PE underwent treatment using the commercially available EKOS EkoSonic Endovascular System.
Twenty-one percent of patients had massive PE, defined as presentation with syncope, cardiogenic shock, resuscitated cardiac arrest, or persistent hypotension. The remaining 79% had submassive PE, with normal blood pressure but evidence of right ventricular dysfunction. All participants had to have a right ventricular/left ventricular ratio (RV/LV) of 0.9 or greater on the same chest CT scan used in diagnosing the PE. This CT documentation of right ventricular dysfunction has been associated in a meta-analysis of patients with submassive PE with a 7.4-fold increased risk of death due to PE compared to normotensive PE patients with normal right ventricular function (J. Thromb. Haemost. 2013;11:1823-32).
The primary endpoint was change in RV/LV on chest CT from baseline to 48 hours after initiation of fibrinolysis. This ratio improved from 1.55 to 1.13, for a statistically and clinically significant 27% reduction. A similar-size improvement was seen in pulmonary artery systolic pressure – a secondary efficacy endpoint – which decreased from 51.4 mm Hg before treatment to 37.5 mm Hg post procedure and 36.9 mm Hg at 48 hours. Both efficacy endpoints improved to a similar extent regardless of whether patients had massive or submassive PE.
The mean Modified Miller Pulmonary Artery Angiographic Obstruction Score improved by 30%, from 22.5 pretreatment to 15.8 at 48 hours.
Three in-hospital deaths occurred. One was due to massive PE which occurred before the fibrinolysis procedure could be completed. The others were not directly attributable to PE: One involved overwhelming sepsis and the other was due to progressive respiratory failure. Major bleeding occurred in 11% of patients; however, 16 of the 17 events were classified as GUSTO moderate bleeds, with only a single GUSTO severe hemorrhage.
There were no intracranial hemorrhages.
The fibrinolytic agent used in SEATTLE II was tissue plasminogen activator, delivered at 1 mg/hr for a total dose of 24 mg. Patients with unilateral PE received a single device and 24 hours of infusion time. The 86% of patients who had bilateral disease got two devices and 12 hours of therapy.
The proprietary EKOS system consists of two catheters: an outer infusion catheter with side holes that elute the fibrinolytic agent, and an inner core catheter with ultrasound transducers placed at regular intervals. These transducers produce low-intensity ultrasound which serves two purposes. Through a process called acoustic streaming, the low-intensity ultrasound helps push the fibrinolytic agent closer to the thrombus. Plus, the ultrasound energy causes the clot fibrin to reconfigure from a tight lattice to a more porous structure that promotes deeper penetration of the fibrinolytic, Dr. Piazza explained.
Audience members greeted the study results enthusiastically.
"Most of us really do believe that for safety reasons this is the way to go, rather than systemic fibrinolysis," one said.
Dr. Piazza said the next step in defining the EKOS system’s role in clinical practice will be to study briefer infusion times as a means of achieving faster patient improvement with reduced use of hospital resources, including ICU and intermediate-care unit time.
The EKOS system has been approved in the United States since 2005 for treatment of blood clots in the arms and legs. In Europe it gained an additional indication for treatment of massive and submassive PE in 2011.
The SEATTLE II study was sponsored by EKOS Corp. Dr. Piazza reported receiving a research grant from the company.
WASHINGTON – Ultrasound-facilitated, catheter-directed, low-dose fibrinolysis for acute massive or submassive pulmonary embolism significantly improves right ventricular function, reduces pulmonary hypertension and angiographic evidence of obstruction, and lessens the risk of fibrinolysis-associated intracranial hemorrhage, according to a prospective multicenter clinical trial.
"By minimizing the risk of intracranial bleeding, ultrasound-facilitated, catheter-directed, low-dose fibrinolysis represents a potential game changer in the treatment of high-risk pulmonary embolism patients," Dr. Gregory Piazza said in presenting the results of the SEATTLE II study at the annual meeting of the American College of Cardiology.
Full-dose systemic fibrinolysis has long been the go-to advanced therapy for high-risk pulmonary embolism (PE), but physicians are leery of the associated 2%-3% risk of catastrophic intracranial hemorrhage, noted Dr. Piazza, a cardiologist at Brigham and Women’s Hospital and Harvard University, Boston.
SEATTLE II was a single-arm, 21-site, prospective study in which 150 patients with high-risk PE underwent treatment using the commercially available EKOS EkoSonic Endovascular System.
Twenty-one percent of patients had massive PE, defined as presentation with syncope, cardiogenic shock, resuscitated cardiac arrest, or persistent hypotension. The remaining 79% had submassive PE, with normal blood pressure but evidence of right ventricular dysfunction. All participants had to have a right ventricular/left ventricular ratio (RV/LV) of 0.9 or greater on the same chest CT scan used in diagnosing the PE. This CT documentation of right ventricular dysfunction has been associated in a meta-analysis of patients with submassive PE with a 7.4-fold increased risk of death due to PE compared to normotensive PE patients with normal right ventricular function (J. Thromb. Haemost. 2013;11:1823-32).
The primary endpoint was change in RV/LV on chest CT from baseline to 48 hours after initiation of fibrinolysis. This ratio improved from 1.55 to 1.13, for a statistically and clinically significant 27% reduction. A similar-size improvement was seen in pulmonary artery systolic pressure – a secondary efficacy endpoint – which decreased from 51.4 mm Hg before treatment to 37.5 mm Hg post procedure and 36.9 mm Hg at 48 hours. Both efficacy endpoints improved to a similar extent regardless of whether patients had massive or submassive PE.
The mean Modified Miller Pulmonary Artery Angiographic Obstruction Score improved by 30%, from 22.5 pretreatment to 15.8 at 48 hours.
Three in-hospital deaths occurred. One was due to massive PE which occurred before the fibrinolysis procedure could be completed. The others were not directly attributable to PE: One involved overwhelming sepsis and the other was due to progressive respiratory failure. Major bleeding occurred in 11% of patients; however, 16 of the 17 events were classified as GUSTO moderate bleeds, with only a single GUSTO severe hemorrhage.
There were no intracranial hemorrhages.
The fibrinolytic agent used in SEATTLE II was tissue plasminogen activator, delivered at 1 mg/hr for a total dose of 24 mg. Patients with unilateral PE received a single device and 24 hours of infusion time. The 86% of patients who had bilateral disease got two devices and 12 hours of therapy.
The proprietary EKOS system consists of two catheters: an outer infusion catheter with side holes that elute the fibrinolytic agent, and an inner core catheter with ultrasound transducers placed at regular intervals. These transducers produce low-intensity ultrasound which serves two purposes. Through a process called acoustic streaming, the low-intensity ultrasound helps push the fibrinolytic agent closer to the thrombus. Plus, the ultrasound energy causes the clot fibrin to reconfigure from a tight lattice to a more porous structure that promotes deeper penetration of the fibrinolytic, Dr. Piazza explained.
Audience members greeted the study results enthusiastically.
"Most of us really do believe that for safety reasons this is the way to go, rather than systemic fibrinolysis," one said.
Dr. Piazza said the next step in defining the EKOS system’s role in clinical practice will be to study briefer infusion times as a means of achieving faster patient improvement with reduced use of hospital resources, including ICU and intermediate-care unit time.
The EKOS system has been approved in the United States since 2005 for treatment of blood clots in the arms and legs. In Europe it gained an additional indication for treatment of massive and submassive PE in 2011.
The SEATTLE II study was sponsored by EKOS Corp. Dr. Piazza reported receiving a research grant from the company.
AT ACC 14
Major finding: Ultrasound-facilitated, catheter-directed, low-dose fibrinolysis in patients with high-risk pulmonary embolism improved right ventricular function and reduced pulmonary hypertension with no intracranial hemorrhages or fatal bleeding events.
Data source: SEATTLE II, a multicenter, single-arm, prospective study of 150 patients with massive or submassive pulmonary embolism.
Disclosures: The study was sponsored by EKOS Corp. The presenter received a research grant from the company.

Off-label use of novel anticoagulants accelerates
WASHINGTON – The off-label use of novel oral anticoagulants for stroke prevention in patients with valvular atrial fibrillation has climbed steeply since the drugs reached the marketplace, mirroring the medications’ rapid adoption for the approved indication of preventing strokes in nonvalvular AF, according to Dr. Sandeep Mahrendra Jani*.
An analysis of 190,227 nonvalvular atrial fibrillation (NVAF) patients in 95 practices participating in the American College of Cardiology’s National Cardiovascular Data Registry – PINNACLE Registry – showed that during the first quarter of 2011, just 4.8% were on dabigatran, the sole novel oral anticoagulant then available. By the fourth quarter of 2012, however, 14.9% of NVAF patients were on a novel oral anticoagulant, either dabigatran or the subsequently approved rivaroxiban, he reported at the annual meeting of the ACC.
Similarly, among 2,142 registry participants with valvular atrial fibrillation (AF), the use of any novel oral anticoagulant shot up from 2.7% in the first quarter of 2011 to 13.8% in the fourth quarter of 2012, noted Dr. Jani of Medstar Washington (D.C.) Hospital Center.
During this time – prior to the arrival of apixiban on the market – the use of warfarin for stroke prevention in patients with NVAF declined from 47.9% to 44.3%. Among patients with valvular atrial fibrillation, the prevalence of warfarin therapy fell from 65.8% in the first quarter of 2011 to 60.1% in fourth quarter 2012.
During the first quarter of 2011, 51.2% of all patients with NVAF and 66.4% with valvular AF were on any oral anticoagulant. By fourth quarter 2012, these rates had increased to 56.9% and 66.8%, respectively.
The use of dabigatran in patients with valvular AF took a hit in late 2012 in response to the premature halt of the RE-ALIGN (Dabigatran Etexilate in Patients With Mechanical Heart Valves) trial, followed by the Food and Drug Administration’s warning against using dabigatran in patients with mechanical heart valves. Dabigatran was used by 2.7% of valvular AF patients in the first quarter of 2011, rising steadily to 12.1% by the third quarter of 2012, then plunging to just 1.4% in the year’s final quarter.
In light of the rapidly accelerating use of novel oral anticoagulants in patients with valvular AF, despite a lack of evidence of efficacy for stroke prevention in this setting, further studies are a priority, Dr. Jani said.
The PINNACLE registry is funded by the ACC, with founding sponsorship provided by Bristol-Myers Squibb and Pfizer. Dr. Jani reported having no relevant financial conflicts.
*Correction, 4/29/2014: An earlier version of this article misstated the name of study author Dr. Sandeep Mahrendra Jani.
WASHINGTON – The off-label use of novel oral anticoagulants for stroke prevention in patients with valvular atrial fibrillation has climbed steeply since the drugs reached the marketplace, mirroring the medications’ rapid adoption for the approved indication of preventing strokes in nonvalvular AF, according to Dr. Sandeep Mahrendra Jani*.
An analysis of 190,227 nonvalvular atrial fibrillation (NVAF) patients in 95 practices participating in the American College of Cardiology’s National Cardiovascular Data Registry – PINNACLE Registry – showed that during the first quarter of 2011, just 4.8% were on dabigatran, the sole novel oral anticoagulant then available. By the fourth quarter of 2012, however, 14.9% of NVAF patients were on a novel oral anticoagulant, either dabigatran or the subsequently approved rivaroxiban, he reported at the annual meeting of the ACC.
Similarly, among 2,142 registry participants with valvular atrial fibrillation (AF), the use of any novel oral anticoagulant shot up from 2.7% in the first quarter of 2011 to 13.8% in the fourth quarter of 2012, noted Dr. Jani of Medstar Washington (D.C.) Hospital Center.
During this time – prior to the arrival of apixiban on the market – the use of warfarin for stroke prevention in patients with NVAF declined from 47.9% to 44.3%. Among patients with valvular atrial fibrillation, the prevalence of warfarin therapy fell from 65.8% in the first quarter of 2011 to 60.1% in fourth quarter 2012.
During the first quarter of 2011, 51.2% of all patients with NVAF and 66.4% with valvular AF were on any oral anticoagulant. By fourth quarter 2012, these rates had increased to 56.9% and 66.8%, respectively.
The use of dabigatran in patients with valvular AF took a hit in late 2012 in response to the premature halt of the RE-ALIGN (Dabigatran Etexilate in Patients With Mechanical Heart Valves) trial, followed by the Food and Drug Administration’s warning against using dabigatran in patients with mechanical heart valves. Dabigatran was used by 2.7% of valvular AF patients in the first quarter of 2011, rising steadily to 12.1% by the third quarter of 2012, then plunging to just 1.4% in the year’s final quarter.
In light of the rapidly accelerating use of novel oral anticoagulants in patients with valvular AF, despite a lack of evidence of efficacy for stroke prevention in this setting, further studies are a priority, Dr. Jani said.
The PINNACLE registry is funded by the ACC, with founding sponsorship provided by Bristol-Myers Squibb and Pfizer. Dr. Jani reported having no relevant financial conflicts.
*Correction, 4/29/2014: An earlier version of this article misstated the name of study author Dr. Sandeep Mahrendra Jani.
WASHINGTON – The off-label use of novel oral anticoagulants for stroke prevention in patients with valvular atrial fibrillation has climbed steeply since the drugs reached the marketplace, mirroring the medications’ rapid adoption for the approved indication of preventing strokes in nonvalvular AF, according to Dr. Sandeep Mahrendra Jani*.
An analysis of 190,227 nonvalvular atrial fibrillation (NVAF) patients in 95 practices participating in the American College of Cardiology’s National Cardiovascular Data Registry – PINNACLE Registry – showed that during the first quarter of 2011, just 4.8% were on dabigatran, the sole novel oral anticoagulant then available. By the fourth quarter of 2012, however, 14.9% of NVAF patients were on a novel oral anticoagulant, either dabigatran or the subsequently approved rivaroxiban, he reported at the annual meeting of the ACC.
Similarly, among 2,142 registry participants with valvular atrial fibrillation (AF), the use of any novel oral anticoagulant shot up from 2.7% in the first quarter of 2011 to 13.8% in the fourth quarter of 2012, noted Dr. Jani of Medstar Washington (D.C.) Hospital Center.
During this time – prior to the arrival of apixiban on the market – the use of warfarin for stroke prevention in patients with NVAF declined from 47.9% to 44.3%. Among patients with valvular atrial fibrillation, the prevalence of warfarin therapy fell from 65.8% in the first quarter of 2011 to 60.1% in fourth quarter 2012.
During the first quarter of 2011, 51.2% of all patients with NVAF and 66.4% with valvular AF were on any oral anticoagulant. By fourth quarter 2012, these rates had increased to 56.9% and 66.8%, respectively.
The use of dabigatran in patients with valvular AF took a hit in late 2012 in response to the premature halt of the RE-ALIGN (Dabigatran Etexilate in Patients With Mechanical Heart Valves) trial, followed by the Food and Drug Administration’s warning against using dabigatran in patients with mechanical heart valves. Dabigatran was used by 2.7% of valvular AF patients in the first quarter of 2011, rising steadily to 12.1% by the third quarter of 2012, then plunging to just 1.4% in the year’s final quarter.
In light of the rapidly accelerating use of novel oral anticoagulants in patients with valvular AF, despite a lack of evidence of efficacy for stroke prevention in this setting, further studies are a priority, Dr. Jani said.
The PINNACLE registry is funded by the ACC, with founding sponsorship provided by Bristol-Myers Squibb and Pfizer. Dr. Jani reported having no relevant financial conflicts.
*Correction, 4/29/2014: An earlier version of this article misstated the name of study author Dr. Sandeep Mahrendra Jani.
AT ACC 14
Major finding: By the fourth quarter of 2012, 14.9% of patients with nonvalvular AF were on a novel anticoagulant. So were 13.8% of those with valvular AF, even though this is an off-label use of these drugs.
Data source: This study involved more than 190,000 patients with nonvalvular AF and 2,142 with valvular AF in 95 practices participating in the PINNACLE Registry.
Disclosures The PINNACLE Registry is funded by the ACC’s National Cardiovascular Data Registry. The presenter reported having no relevant financial conflicts.
'Favorable signal' for secondary coronary prevention in STABILITY
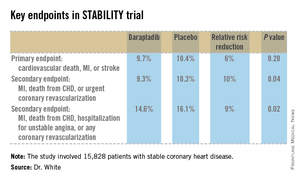
WASHINGTON – A novel pharmacologic approach to the prevention of ischemic events in patients with stable coronary heart disease failed to reduce the combined risk of cardiovascular death, acute myocardial infarction, or stroke in a nearly 16,000-patient megatrial known as STABILITY.
That being said, darapladib, the oral inhibitor of lipoprotein-associated phospholipase A2 (Lp-PLA2) under study, did significantly reduce the rate of the prespecified secondary endpoint of major coronary events, compared with placebo. And that’s an encouraging finding, Dr. Harvey D. White said at the annual meeting of the American College of Cardiology.
"We believe that there is a signal here on coronary events. This is the first large trial of a new treatment addressing a novel mechanism of inhibition of inflammation in the atherosclerotic plaque, and we believe this signal may be very important for patient care," said Dr. White, director of coronary care and cardiovascular research at Auckland (New Zealand) City Hospital.
Patients with stable coronary heart disease (CHD) constitute a population with an unmet need, as evidenced by the substantial clinical event rate in the placebo group, he added.
Lp-PLA2 is produced by white blood cells and macrophages and carried by low-density lipoprotein (LDL) cholesterol. Within an atheroma, Lp-PLA2 increases production of inflammatory products associated with necrotic core expansion. Stable atheromatous plaques contain little Lp-PLA2, but vulnerable or ruptured plaques contain it in high concentrations. A strong association between Lp-PLA2 activity and the risk of CHD has been shown in several dozen studies (Lancet 2010;375:1536-44). And darapladib reduces Lp-PLA2 levels by roughly 60%. In coronary imaging studies, the drug halts progression of coronary artery necrotic plaque core volume, the cardiologist noted.
STABILITY (the Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) was a 39-nation clinical trial that included 15,828 patients with stable CHD who were randomized to receive once-daily darapladib at 160 mg or placebo and then followed prospectively for a median of 3.7 years. The combined rate of cardiovascular death, myocardial infarction, or stroke in patients randomized to darapladib was 9.7%, yielding a nonsignificant 6% relative risk reduction, compared with placebo. There was a consistent benefit favoring darapladib across all coronary-specific endpoints (see chart).
Diarrhea was significantly more common in the darapladib group. Three percent of patients randomized to darapladib discontinued treatment due to diarrhea, another 2% because of abnormal feces, 2% due to abnormal skin odor, and 1% for abnormal-smelling urine. This is believed to be a consequence of the sulfhydryl group in the drug molecule.
The STABILITY trial was hampered in that the study was designed before epidemiologic evidence showed Lp-PLA2 levels are unrelated to stroke risk. Yet stroke was a component of the primary endpoint. Also, the rate of background guideline-directed medical therapy in STABILITY participants was greater than in any previous secondary prevention trial: 93% of subjects were on aspirin and 97% on a statin – and statin therapy in and of itself has been shown to reduce Lp-PLA2 by up to 35%. One-third of STABILITY patients had a baseline LDL below 70 mg/dL, and only one-quarter had an LDL above 100 mg/dL.
In addition, three-quarters of subjects had previously undergone coronary revascularization, with one-third of participants having had coronary artery bypass surgery.
"This high rate of optimized background therapy may have made it hard to modulate events with darapladib," Dr. White observed.
Baseline levels of Lp-PLA2 are still being collected and will be reported later. Planned secondary analyses will assess whether darapladib has a more pronounced benefit in patients with high baseline Lp-PLA2.
A sister study known as SOLID, being led by the Harvard University–based TIMI group, will report results later this year. SOLID is evaluating darapladib for the prevention of ischemic events in patients started on the drug within 1 month after an acute coronary syndrome rather than in patients with stable coronary disease as in STABILITY.
"We can’t guess the results, but you could hypothesize that since there’s more inflammation in the cardiac core when the plaque is disrupted, SOLID will provide very interesting data," the cardiologist said.
Simultaneous with Dr. White’s presentation at ACC 14, the STABILITY results were published online at NEJM.org (doi: 10.1056/NEJMoa1315878).
STABILITY was sponsored by GlaxoSmithKline. Dr. White reported receiving research grant and travel support from the company.
WASHINGTON – A novel pharmacologic approach to the prevention of ischemic events in patients with stable coronary heart disease failed to reduce the combined risk of cardiovascular death, acute myocardial infarction, or stroke in a nearly 16,000-patient megatrial known as STABILITY.
That being said, darapladib, the oral inhibitor of lipoprotein-associated phospholipase A2 (Lp-PLA2) under study, did significantly reduce the rate of the prespecified secondary endpoint of major coronary events, compared with placebo. And that’s an encouraging finding, Dr. Harvey D. White said at the annual meeting of the American College of Cardiology.
"We believe that there is a signal here on coronary events. This is the first large trial of a new treatment addressing a novel mechanism of inhibition of inflammation in the atherosclerotic plaque, and we believe this signal may be very important for patient care," said Dr. White, director of coronary care and cardiovascular research at Auckland (New Zealand) City Hospital.
Patients with stable coronary heart disease (CHD) constitute a population with an unmet need, as evidenced by the substantial clinical event rate in the placebo group, he added.
Lp-PLA2 is produced by white blood cells and macrophages and carried by low-density lipoprotein (LDL) cholesterol. Within an atheroma, Lp-PLA2 increases production of inflammatory products associated with necrotic core expansion. Stable atheromatous plaques contain little Lp-PLA2, but vulnerable or ruptured plaques contain it in high concentrations. A strong association between Lp-PLA2 activity and the risk of CHD has been shown in several dozen studies (Lancet 2010;375:1536-44). And darapladib reduces Lp-PLA2 levels by roughly 60%. In coronary imaging studies, the drug halts progression of coronary artery necrotic plaque core volume, the cardiologist noted.
STABILITY (the Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) was a 39-nation clinical trial that included 15,828 patients with stable CHD who were randomized to receive once-daily darapladib at 160 mg or placebo and then followed prospectively for a median of 3.7 years. The combined rate of cardiovascular death, myocardial infarction, or stroke in patients randomized to darapladib was 9.7%, yielding a nonsignificant 6% relative risk reduction, compared with placebo. There was a consistent benefit favoring darapladib across all coronary-specific endpoints (see chart).
Diarrhea was significantly more common in the darapladib group. Three percent of patients randomized to darapladib discontinued treatment due to diarrhea, another 2% because of abnormal feces, 2% due to abnormal skin odor, and 1% for abnormal-smelling urine. This is believed to be a consequence of the sulfhydryl group in the drug molecule.
The STABILITY trial was hampered in that the study was designed before epidemiologic evidence showed Lp-PLA2 levels are unrelated to stroke risk. Yet stroke was a component of the primary endpoint. Also, the rate of background guideline-directed medical therapy in STABILITY participants was greater than in any previous secondary prevention trial: 93% of subjects were on aspirin and 97% on a statin – and statin therapy in and of itself has been shown to reduce Lp-PLA2 by up to 35%. One-third of STABILITY patients had a baseline LDL below 70 mg/dL, and only one-quarter had an LDL above 100 mg/dL.
In addition, three-quarters of subjects had previously undergone coronary revascularization, with one-third of participants having had coronary artery bypass surgery.
"This high rate of optimized background therapy may have made it hard to modulate events with darapladib," Dr. White observed.
Baseline levels of Lp-PLA2 are still being collected and will be reported later. Planned secondary analyses will assess whether darapladib has a more pronounced benefit in patients with high baseline Lp-PLA2.
A sister study known as SOLID, being led by the Harvard University–based TIMI group, will report results later this year. SOLID is evaluating darapladib for the prevention of ischemic events in patients started on the drug within 1 month after an acute coronary syndrome rather than in patients with stable coronary disease as in STABILITY.
"We can’t guess the results, but you could hypothesize that since there’s more inflammation in the cardiac core when the plaque is disrupted, SOLID will provide very interesting data," the cardiologist said.
Simultaneous with Dr. White’s presentation at ACC 14, the STABILITY results were published online at NEJM.org (doi: 10.1056/NEJMoa1315878).
STABILITY was sponsored by GlaxoSmithKline. Dr. White reported receiving research grant and travel support from the company.
WASHINGTON – A novel pharmacologic approach to the prevention of ischemic events in patients with stable coronary heart disease failed to reduce the combined risk of cardiovascular death, acute myocardial infarction, or stroke in a nearly 16,000-patient megatrial known as STABILITY.
That being said, darapladib, the oral inhibitor of lipoprotein-associated phospholipase A2 (Lp-PLA2) under study, did significantly reduce the rate of the prespecified secondary endpoint of major coronary events, compared with placebo. And that’s an encouraging finding, Dr. Harvey D. White said at the annual meeting of the American College of Cardiology.
"We believe that there is a signal here on coronary events. This is the first large trial of a new treatment addressing a novel mechanism of inhibition of inflammation in the atherosclerotic plaque, and we believe this signal may be very important for patient care," said Dr. White, director of coronary care and cardiovascular research at Auckland (New Zealand) City Hospital.
Patients with stable coronary heart disease (CHD) constitute a population with an unmet need, as evidenced by the substantial clinical event rate in the placebo group, he added.
Lp-PLA2 is produced by white blood cells and macrophages and carried by low-density lipoprotein (LDL) cholesterol. Within an atheroma, Lp-PLA2 increases production of inflammatory products associated with necrotic core expansion. Stable atheromatous plaques contain little Lp-PLA2, but vulnerable or ruptured plaques contain it in high concentrations. A strong association between Lp-PLA2 activity and the risk of CHD has been shown in several dozen studies (Lancet 2010;375:1536-44). And darapladib reduces Lp-PLA2 levels by roughly 60%. In coronary imaging studies, the drug halts progression of coronary artery necrotic plaque core volume, the cardiologist noted.
STABILITY (the Stabilization of Atherosclerotic Plaque by Initiation of Darapladib Therapy) was a 39-nation clinical trial that included 15,828 patients with stable CHD who were randomized to receive once-daily darapladib at 160 mg or placebo and then followed prospectively for a median of 3.7 years. The combined rate of cardiovascular death, myocardial infarction, or stroke in patients randomized to darapladib was 9.7%, yielding a nonsignificant 6% relative risk reduction, compared with placebo. There was a consistent benefit favoring darapladib across all coronary-specific endpoints (see chart).
Diarrhea was significantly more common in the darapladib group. Three percent of patients randomized to darapladib discontinued treatment due to diarrhea, another 2% because of abnormal feces, 2% due to abnormal skin odor, and 1% for abnormal-smelling urine. This is believed to be a consequence of the sulfhydryl group in the drug molecule.
The STABILITY trial was hampered in that the study was designed before epidemiologic evidence showed Lp-PLA2 levels are unrelated to stroke risk. Yet stroke was a component of the primary endpoint. Also, the rate of background guideline-directed medical therapy in STABILITY participants was greater than in any previous secondary prevention trial: 93% of subjects were on aspirin and 97% on a statin – and statin therapy in and of itself has been shown to reduce Lp-PLA2 by up to 35%. One-third of STABILITY patients had a baseline LDL below 70 mg/dL, and only one-quarter had an LDL above 100 mg/dL.
In addition, three-quarters of subjects had previously undergone coronary revascularization, with one-third of participants having had coronary artery bypass surgery.
"This high rate of optimized background therapy may have made it hard to modulate events with darapladib," Dr. White observed.
Baseline levels of Lp-PLA2 are still being collected and will be reported later. Planned secondary analyses will assess whether darapladib has a more pronounced benefit in patients with high baseline Lp-PLA2.
A sister study known as SOLID, being led by the Harvard University–based TIMI group, will report results later this year. SOLID is evaluating darapladib for the prevention of ischemic events in patients started on the drug within 1 month after an acute coronary syndrome rather than in patients with stable coronary disease as in STABILITY.
"We can’t guess the results, but you could hypothesize that since there’s more inflammation in the cardiac core when the plaque is disrupted, SOLID will provide very interesting data," the cardiologist said.
Simultaneous with Dr. White’s presentation at ACC 14, the STABILITY results were published online at NEJM.org (doi: 10.1056/NEJMoa1315878).
STABILITY was sponsored by GlaxoSmithKline. Dr. White reported receiving research grant and travel support from the company.
AT ACC 14
Major finding: The combined rate of cardiovascular death, myocardial infarction, or stroke in patients randomized to darapladib was 9.7%, yielding a nonsignificant 6% relative risk reduction, compared with placebo. But a significant reduction was seen in major coronary events.
Data source: STABILITY, a randomized, double-blind study in which 15,828 patients with stable coronary heart disease were followed on darapladib or placebo for a median of 3.7 years.
Disclosures: STABILITY was sponsored by GlaxoSmithKline. Dr. White reported receiving research grant and travel support from the company.
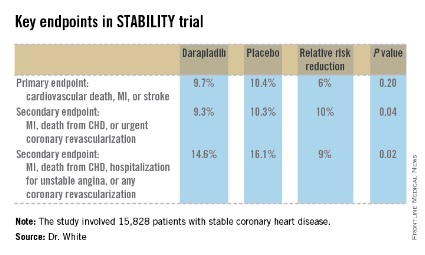
Antipsychotics’ risk in dementia revised upward
ORLANDO – The mortality risk associated with prescription of antipsychotics or valproic acid for behavioral disturbances in patients with dementia is considerably greater than previously estimated, according to a massive national study.
This retrospective study included 45,669 matched pairs of Veterans Affairs patients over age 65 with dementia diagnosed during 1998-2009. Thus, there were 45,669 VA patients over age 65 with dementia diagnosed during 1998-2009 who were on one of the medications under scrutiny: haloperidol, olanzapine, quetiapine, risperidone, valproic acid, or a nontricyclic antidepressant. Each of these patients was matched to an older VA patient with dementia who was not on any of those medications and had not been for at least the past 6 months.
Investigators defined the number needed to harm (NNH) as the number of dementia patients who would need to be on one of the medications for 180 days in order to result in one additional death compared with nonuser matched controls. The NNH ranged from a worst-case scenario of 15 for haloperidol to 50 for quetiapine, Dr. Donovan T. Maust reported at the annual meeting of the American Association for Geriatric Psychiatry.

The patient pairs were matched based on demographics; comorbid medical and psychiatric diagnoses, including delirium within the prior 12 months; and a history of psychiatric hospitalization, noted Dr. Maust of the section on geriatric psychiatry at the University of Michigan, Ann Arbor.
The mortality NNHs found in this large study are considerably lower – that is, more unfavorable than in the two earlier meta-analyses conducted by other investigators. In the first meta-analysis, the estimated NNH for second-generation antipsychotics versus placebo was 100 (JAMA 2005;294:1934-43). More recently, a 2011 Agency for Healthcare Research Quality comparative effectiveness review found an NNH of 87 for second-generation antipsychotics when used in treating behavioral disturbances in elderly patients with dementia. The explanation for the larger, more favorable NNHs found in these meta-analyses is that they relied mainly upon studies of 8-12 weeks’ duration and so failed to capture the additional drug-related deaths that occurred over the course of the 180-day study in VA patients, the psychiatrist said.
In addition to providing a truer picture of the mortality risks of antipsychotics and valproic acid, the other advantage of the VA study is that it delineates the mortality risks for individual medications compared with no use at all. Earlier studies lumped antipsychotics together as a class, he noted.
"You can see that the risk is basically doubled with risperidone compared to quetiapine. So clinically I would use quetiapine as my first-line agent if I had to use anything. And I’ve stopped using haloperidol altogether," Dr. Maust said in an interview.
After the Food and Drug Administration issued its 2005 black box warning about the increased mortality risk of atypical antipsychotics in elderly patients with dementia, prescription of those drugs for the treatment of agitation and other behavioral symptoms in dementia outpatients dropped off, while the use of conventional antipsychotics climbed. With the 2008 black box warning regarding conventional antipsychotics, outpatient prescriptions for those agents also tailed off, but the use of valproic acid increased. However, the NNH of 29 for valproic acid seen in the new study shows this drug is problematic, too.
"It’s a bit like Whac-a-Mole where once one drug goes off the list, then people try something else. They’re such frustrating behaviors, and it’s such a distressing situation for the patient, the family, and the providers that it makes sense that people are trying alternatives," Dr. Maust commented.
The NNH of 158 for patients on antidepressant therapy seen in this study is probably not clinically meaningful in that it approaches the background mortality risk in this elderly, sick population, he added.
While the use of antipsychotics to treat behavioral disturbances in patients with dementia may have dropped off in outpatient settings since the black box warnings, that’s not the case in the inpatient setting – at least, not in the geriatric psychiatry inpatient unit at the Mayo Clinic in Rochester, Minn., Dr. Jung In "Kristin" Lee reported in a separate presentation at the meeting.
She presented a retrospective study of all patients hospitalized in the unit with a discharge diagnosis of dementia over the course of two study years: 2002 and 2012. In 2002, well before the black box warnings, 58% of 52 dementia patients were discharged on an antipsychotic agent, with quetiapine being the most commonly prescribed. Similarly, 65% of the 43 dementia patients discharged in 2012 were on an antipsychotic, with quetiapine once again being No. 1. In both years only a single dementia patient was discharged on a conventional antipsychotic, according to Dr. Lee of the Mayo Clinic.
Dr. Maust’s study was sponsored by the National Institute of Mental Health and the University of Michigan Program for Positive Aging. He reported having no financial conflicts, as did Dr. Lee, whose study was unfunded.
ORLANDO – The mortality risk associated with prescription of antipsychotics or valproic acid for behavioral disturbances in patients with dementia is considerably greater than previously estimated, according to a massive national study.
This retrospective study included 45,669 matched pairs of Veterans Affairs patients over age 65 with dementia diagnosed during 1998-2009. Thus, there were 45,669 VA patients over age 65 with dementia diagnosed during 1998-2009 who were on one of the medications under scrutiny: haloperidol, olanzapine, quetiapine, risperidone, valproic acid, or a nontricyclic antidepressant. Each of these patients was matched to an older VA patient with dementia who was not on any of those medications and had not been for at least the past 6 months.
Investigators defined the number needed to harm (NNH) as the number of dementia patients who would need to be on one of the medications for 180 days in order to result in one additional death compared with nonuser matched controls. The NNH ranged from a worst-case scenario of 15 for haloperidol to 50 for quetiapine, Dr. Donovan T. Maust reported at the annual meeting of the American Association for Geriatric Psychiatry.
The patient pairs were matched based on demographics; comorbid medical and psychiatric diagnoses, including delirium within the prior 12 months; and a history of psychiatric hospitalization, noted Dr. Maust of the section on geriatric psychiatry at the University of Michigan, Ann Arbor.
The mortality NNHs found in this large study are considerably lower – that is, more unfavorable than in the two earlier meta-analyses conducted by other investigators. In the first meta-analysis, the estimated NNH for second-generation antipsychotics versus placebo was 100 (JAMA 2005;294:1934-43). More recently, a 2011 Agency for Healthcare Research Quality comparative effectiveness review found an NNH of 87 for second-generation antipsychotics when used in treating behavioral disturbances in elderly patients with dementia. The explanation for the larger, more favorable NNHs found in these meta-analyses is that they relied mainly upon studies of 8-12 weeks’ duration and so failed to capture the additional drug-related deaths that occurred over the course of the 180-day study in VA patients, the psychiatrist said.
In addition to providing a truer picture of the mortality risks of antipsychotics and valproic acid, the other advantage of the VA study is that it delineates the mortality risks for individual medications compared with no use at all. Earlier studies lumped antipsychotics together as a class, he noted.
"You can see that the risk is basically doubled with risperidone compared to quetiapine. So clinically I would use quetiapine as my first-line agent if I had to use anything. And I’ve stopped using haloperidol altogether," Dr. Maust said in an interview.
After the Food and Drug Administration issued its 2005 black box warning about the increased mortality risk of atypical antipsychotics in elderly patients with dementia, prescription of those drugs for the treatment of agitation and other behavioral symptoms in dementia outpatients dropped off, while the use of conventional antipsychotics climbed. With the 2008 black box warning regarding conventional antipsychotics, outpatient prescriptions for those agents also tailed off, but the use of valproic acid increased. However, the NNH of 29 for valproic acid seen in the new study shows this drug is problematic, too.
"It’s a bit like Whac-a-Mole where once one drug goes off the list, then people try something else. They’re such frustrating behaviors, and it’s such a distressing situation for the patient, the family, and the providers that it makes sense that people are trying alternatives," Dr. Maust commented.
The NNH of 158 for patients on antidepressant therapy seen in this study is probably not clinically meaningful in that it approaches the background mortality risk in this elderly, sick population, he added.
While the use of antipsychotics to treat behavioral disturbances in patients with dementia may have dropped off in outpatient settings since the black box warnings, that’s not the case in the inpatient setting – at least, not in the geriatric psychiatry inpatient unit at the Mayo Clinic in Rochester, Minn., Dr. Jung In "Kristin" Lee reported in a separate presentation at the meeting.
She presented a retrospective study of all patients hospitalized in the unit with a discharge diagnosis of dementia over the course of two study years: 2002 and 2012. In 2002, well before the black box warnings, 58% of 52 dementia patients were discharged on an antipsychotic agent, with quetiapine being the most commonly prescribed. Similarly, 65% of the 43 dementia patients discharged in 2012 were on an antipsychotic, with quetiapine once again being No. 1. In both years only a single dementia patient was discharged on a conventional antipsychotic, according to Dr. Lee of the Mayo Clinic.
Dr. Maust’s study was sponsored by the National Institute of Mental Health and the University of Michigan Program for Positive Aging. He reported having no financial conflicts, as did Dr. Lee, whose study was unfunded.
ORLANDO – The mortality risk associated with prescription of antipsychotics or valproic acid for behavioral disturbances in patients with dementia is considerably greater than previously estimated, according to a massive national study.
This retrospective study included 45,669 matched pairs of Veterans Affairs patients over age 65 with dementia diagnosed during 1998-2009. Thus, there were 45,669 VA patients over age 65 with dementia diagnosed during 1998-2009 who were on one of the medications under scrutiny: haloperidol, olanzapine, quetiapine, risperidone, valproic acid, or a nontricyclic antidepressant. Each of these patients was matched to an older VA patient with dementia who was not on any of those medications and had not been for at least the past 6 months.
Investigators defined the number needed to harm (NNH) as the number of dementia patients who would need to be on one of the medications for 180 days in order to result in one additional death compared with nonuser matched controls. The NNH ranged from a worst-case scenario of 15 for haloperidol to 50 for quetiapine, Dr. Donovan T. Maust reported at the annual meeting of the American Association for Geriatric Psychiatry.
The patient pairs were matched based on demographics; comorbid medical and psychiatric diagnoses, including delirium within the prior 12 months; and a history of psychiatric hospitalization, noted Dr. Maust of the section on geriatric psychiatry at the University of Michigan, Ann Arbor.
The mortality NNHs found in this large study are considerably lower – that is, more unfavorable than in the two earlier meta-analyses conducted by other investigators. In the first meta-analysis, the estimated NNH for second-generation antipsychotics versus placebo was 100 (JAMA 2005;294:1934-43). More recently, a 2011 Agency for Healthcare Research Quality comparative effectiveness review found an NNH of 87 for second-generation antipsychotics when used in treating behavioral disturbances in elderly patients with dementia. The explanation for the larger, more favorable NNHs found in these meta-analyses is that they relied mainly upon studies of 8-12 weeks’ duration and so failed to capture the additional drug-related deaths that occurred over the course of the 180-day study in VA patients, the psychiatrist said.
In addition to providing a truer picture of the mortality risks of antipsychotics and valproic acid, the other advantage of the VA study is that it delineates the mortality risks for individual medications compared with no use at all. Earlier studies lumped antipsychotics together as a class, he noted.
"You can see that the risk is basically doubled with risperidone compared to quetiapine. So clinically I would use quetiapine as my first-line agent if I had to use anything. And I’ve stopped using haloperidol altogether," Dr. Maust said in an interview.
After the Food and Drug Administration issued its 2005 black box warning about the increased mortality risk of atypical antipsychotics in elderly patients with dementia, prescription of those drugs for the treatment of agitation and other behavioral symptoms in dementia outpatients dropped off, while the use of conventional antipsychotics climbed. With the 2008 black box warning regarding conventional antipsychotics, outpatient prescriptions for those agents also tailed off, but the use of valproic acid increased. However, the NNH of 29 for valproic acid seen in the new study shows this drug is problematic, too.
"It’s a bit like Whac-a-Mole where once one drug goes off the list, then people try something else. They’re such frustrating behaviors, and it’s such a distressing situation for the patient, the family, and the providers that it makes sense that people are trying alternatives," Dr. Maust commented.
The NNH of 158 for patients on antidepressant therapy seen in this study is probably not clinically meaningful in that it approaches the background mortality risk in this elderly, sick population, he added.
While the use of antipsychotics to treat behavioral disturbances in patients with dementia may have dropped off in outpatient settings since the black box warnings, that’s not the case in the inpatient setting – at least, not in the geriatric psychiatry inpatient unit at the Mayo Clinic in Rochester, Minn., Dr. Jung In "Kristin" Lee reported in a separate presentation at the meeting.
She presented a retrospective study of all patients hospitalized in the unit with a discharge diagnosis of dementia over the course of two study years: 2002 and 2012. In 2002, well before the black box warnings, 58% of 52 dementia patients were discharged on an antipsychotic agent, with quetiapine being the most commonly prescribed. Similarly, 65% of the 43 dementia patients discharged in 2012 were on an antipsychotic, with quetiapine once again being No. 1. In both years only a single dementia patient was discharged on a conventional antipsychotic, according to Dr. Lee of the Mayo Clinic.
Dr. Maust’s study was sponsored by the National Institute of Mental Health and the University of Michigan Program for Positive Aging. He reported having no financial conflicts, as did Dr. Lee, whose study was unfunded.
AT THE AAGP ANNUAL MEETING
Major finding: Among elderly patients with dementia placed on an antipsychotic to treat behavioral problems, the number needed to harm – that is, the number of patients that needed to be on the medication for 180 days in order to result in one additional death compared with nontreatment – ranged from 15 with haloperidol to a best-case scenario of 50 with quetiapine.
Data source: A retrospective study involving more than 90,000 Veterans Affairs patients over age 65 with dementia: 45,669 who were placed on an antipsychotic agent, valproic acid, or an antidepressant to manage behavioral problems and an equal number of closely matched patients not on any of the medications under scrutiny.
Disclosures: The study was supported by the National Institute of Mental Health and the University of Michigan Program for Positive Aging. The presenter reported having no financial conflicts.

Asenapine often beneficial in geriatric bipolar disorder
ORLANDO – Low-dose asenapine provided global improvement in symptoms and mood in older nondemented patients with bipolar disorder with prior suboptimal response to other therapies, according to a pilot study.
This atypical antipsychotic agent, which was used in the study at a mean daily dose of just 7.4 mg, is a novel therapy worth considering in selected geriatric patients with bipolar disorder, Dr. Martha N. Sajatovic said at the annual meeting of the American Association for Geriatric Psychiatry.

She presented a 12-week, prospective, open-label pilot study of asenapine (Saphris) in 15 nondemented patients over age 60 years with type 1 or 2 bipolar disorder who had a poor response to previous treatments. Patients started on asenapine at 5 mg/day and were titrated as indicated to a maximum of 20 mg/day, although the mean dose used was 7.4 mg/day.
The 11 subjects who completed the study showed significant improvements on the Brief Psychiatric Rating Scale, on the Clinical Global Impression, Bipolar version, and on measures of mood symptoms. No changes were found from baseline in terms of mean body mass index, abnormal movement scores, blood glucose levels, lipid levels, or functional status as measured by the Short Form–12, or on the WHO-Disability Assessment Scale II.

Of the four study dropouts, one left because of elevated liver function test results, another because of the emergence of manic symptoms while on asenapine, and two others for reasons unrelated to the study medication, according to Dr. Sajatovic, professor of psychiatry and director of the geropsychiatry program at Case Western Reserve University, Cleveland.
The most common treatment-emergent adverse event was mild, transient GI discomfort, which affected one-third of participants.
The addition of asenapine to the armamentarium for bipolar disorder in later life is a welcome development, she said. Of the 6 million American adults with bipolar disorder, roughly 1 million are over the age of 60. Traditional mood stabilizers such as lithium often have limiting side effects in the older population, and treatment failures are common, she noted.
This pilot study was sponsored by NV Organon/Merck. Dr. Satajovic reported receiving research grants from Merck and other companies, as well as serving as a consultant to Prophase, Otsuka, Pfizer, Amgen, and United BioSource.
ORLANDO – Low-dose asenapine provided global improvement in symptoms and mood in older nondemented patients with bipolar disorder with prior suboptimal response to other therapies, according to a pilot study.
This atypical antipsychotic agent, which was used in the study at a mean daily dose of just 7.4 mg, is a novel therapy worth considering in selected geriatric patients with bipolar disorder, Dr. Martha N. Sajatovic said at the annual meeting of the American Association for Geriatric Psychiatry.
She presented a 12-week, prospective, open-label pilot study of asenapine (Saphris) in 15 nondemented patients over age 60 years with type 1 or 2 bipolar disorder who had a poor response to previous treatments. Patients started on asenapine at 5 mg/day and were titrated as indicated to a maximum of 20 mg/day, although the mean dose used was 7.4 mg/day.
The 11 subjects who completed the study showed significant improvements on the Brief Psychiatric Rating Scale, on the Clinical Global Impression, Bipolar version, and on measures of mood symptoms. No changes were found from baseline in terms of mean body mass index, abnormal movement scores, blood glucose levels, lipid levels, or functional status as measured by the Short Form–12, or on the WHO-Disability Assessment Scale II.
Of the four study dropouts, one left because of elevated liver function test results, another because of the emergence of manic symptoms while on asenapine, and two others for reasons unrelated to the study medication, according to Dr. Sajatovic, professor of psychiatry and director of the geropsychiatry program at Case Western Reserve University, Cleveland.
The most common treatment-emergent adverse event was mild, transient GI discomfort, which affected one-third of participants.
The addition of asenapine to the armamentarium for bipolar disorder in later life is a welcome development, she said. Of the 6 million American adults with bipolar disorder, roughly 1 million are over the age of 60. Traditional mood stabilizers such as lithium often have limiting side effects in the older population, and treatment failures are common, she noted.
This pilot study was sponsored by NV Organon/Merck. Dr. Satajovic reported receiving research grants from Merck and other companies, as well as serving as a consultant to Prophase, Otsuka, Pfizer, Amgen, and United BioSource.
ORLANDO – Low-dose asenapine provided global improvement in symptoms and mood in older nondemented patients with bipolar disorder with prior suboptimal response to other therapies, according to a pilot study.
This atypical antipsychotic agent, which was used in the study at a mean daily dose of just 7.4 mg, is a novel therapy worth considering in selected geriatric patients with bipolar disorder, Dr. Martha N. Sajatovic said at the annual meeting of the American Association for Geriatric Psychiatry.
She presented a 12-week, prospective, open-label pilot study of asenapine (Saphris) in 15 nondemented patients over age 60 years with type 1 or 2 bipolar disorder who had a poor response to previous treatments. Patients started on asenapine at 5 mg/day and were titrated as indicated to a maximum of 20 mg/day, although the mean dose used was 7.4 mg/day.
The 11 subjects who completed the study showed significant improvements on the Brief Psychiatric Rating Scale, on the Clinical Global Impression, Bipolar version, and on measures of mood symptoms. No changes were found from baseline in terms of mean body mass index, abnormal movement scores, blood glucose levels, lipid levels, or functional status as measured by the Short Form–12, or on the WHO-Disability Assessment Scale II.
Of the four study dropouts, one left because of elevated liver function test results, another because of the emergence of manic symptoms while on asenapine, and two others for reasons unrelated to the study medication, according to Dr. Sajatovic, professor of psychiatry and director of the geropsychiatry program at Case Western Reserve University, Cleveland.
The most common treatment-emergent adverse event was mild, transient GI discomfort, which affected one-third of participants.
The addition of asenapine to the armamentarium for bipolar disorder in later life is a welcome development, she said. Of the 6 million American adults with bipolar disorder, roughly 1 million are over the age of 60. Traditional mood stabilizers such as lithium often have limiting side effects in the older population, and treatment failures are common, she noted.
This pilot study was sponsored by NV Organon/Merck. Dr. Satajovic reported receiving research grants from Merck and other companies, as well as serving as a consultant to Prophase, Otsuka, Pfizer, Amgen, and United BioSource.
AT THE AAGP ANNUAL MEETING
Major finding: The atypical antipsychotic asenapine produced significant improvement in symptoms and mood in patients with later-life bipolar disorder that previously had shown a poor response to other therapies.
Data source: A prospective, 12-week, open-label pilot study in which 15 nondemented patients over age 60 with bipolar disorder suboptimally responsive to other therapies were treated with asenapine starting at 5 mg/day.
Disclosures: This pilot study was sponsored by NV Organon/Merck. Dr. Satajovic reported receiving research grants from Merck and other companies, as well as serving as a consultant to Prophase, Otsuka, Pfizer, Amgen, and United BioSource.
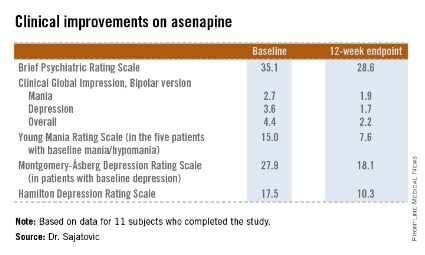
VIDEO: PCSK9 inhibitors placed in perspective
WASHINGTON – "We’re at the dawn of an era of biological therapeutics in cardiology," Dr. Peter Libby, chief of cardiovascular medicine at the Brigham and Women’s Hospital in Boston, told us at the annual meeting of the American College of Cardiology, as he provided the lowdown on the potentially game-changing novel class of LDL cholesterol–lowering medications known as PCSK9 inhibitors.
One such agent, evolocumab, was the focus of five highly positive phase III clinical trials presented at ACC 14.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WASHINGTON – "We’re at the dawn of an era of biological therapeutics in cardiology," Dr. Peter Libby, chief of cardiovascular medicine at the Brigham and Women’s Hospital in Boston, told us at the annual meeting of the American College of Cardiology, as he provided the lowdown on the potentially game-changing novel class of LDL cholesterol–lowering medications known as PCSK9 inhibitors.
One such agent, evolocumab, was the focus of five highly positive phase III clinical trials presented at ACC 14.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WASHINGTON – "We’re at the dawn of an era of biological therapeutics in cardiology," Dr. Peter Libby, chief of cardiovascular medicine at the Brigham and Women’s Hospital in Boston, told us at the annual meeting of the American College of Cardiology, as he provided the lowdown on the potentially game-changing novel class of LDL cholesterol–lowering medications known as PCSK9 inhibitors.
One such agent, evolocumab, was the focus of five highly positive phase III clinical trials presented at ACC 14.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT ACC 14
