User login
Weight gain and excessive fatigue
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of Cushing syndrome (CS).
CS is a rare endocrine disease caused by prolonged exposure to high circulating cortisol levels. Exogenous hypercortisolism is the most common cause of CS. It is largely iatrogenic and results from the prolonged use of glucocorticoids. Less frequently, endogenous CS may occur as the result of excessive production of cortisol by adrenal glands. Endogenous CS can be ACTH-dependent or ACTH-independent. ACTH-dependent CS results from ACTH-secreting pituitary adenomas (Cushing disease) and ectopic ACTH secretion by neoplasms, whereas adrenal hyperplasia, adenoma, and carcinoma are the primary causes of ACTH-independent CS.
The annual incidence and prevalence of CS are unknown; the reported incidence of newly diagnosed cases has ranged from 1.2 to 2.4 per million people per year. Women are affected more often than are men, with a peak of incidence in the third to fourth decade of life. CS is associated with various metabolic, psychiatric, musculoskeletal, and cardiovascular comorbidities. Untreated, it is associated with increased mortality, typically as the result of cardiovascular and infectious complications; however, even in appropriately treated patients, mortality is elevated.
The chronic elevations of glucocorticoid concentrations in CS result in its characteristic phenotype, which includes weight gain, moon-shaped face, buffalo hump, muscle weakness, increased bruising, skin atrophy, red abdominal striae, menstrual irregularities, hirsutism, and acne. It is also associated with numerous comorbidities including diabetes, hypertension, hypercholesterolemia, and osteoporosis. Patients often experience mental health complications, such as depression, emotional lability, and cognitive dysfunction.
Given the rarity of CS and the fact that these symptoms overlap with other conditions, delayed diagnosis is common. The current obesity epidemic also poses diagnostic challenges because true CS can be difficult to differentiate from metabolic syndrome. The duration of hypercortisolism appears to be the most significant factor associated with the degree of morbidity and preterm mortality in CS; thus, an accurate diagnosis as early as possible is important.
Screening and diagnostic tests for CS evaluate cortisol secretion. Available options include late-night salivary cortisol (LNSC), impaired glucocorticoid feedback with overnight 1-mg DST or low-dose 2-day dexamethasone test (LDDT) and increased bioavailable cortisol with 24-hour UFC.
A 2021 consensus statement by Fleseriu and colleagues provides recommendations for the diagnosis of CS. If CS is suspected: begin with UFC, LNSC, or both; DST is an option if LNSC not feasible. If CS because of adrenal tumor is suspected: begin with DST because LNSC has lower specificity in these patients. To confirm CS, any of these tests can be used.
An individualized approach is recommended for the treatment of CS. The optimal approach for iatrogenic CS is to slowly taper exogenous steroids. Chronic exposure to steroids can suppress adrenal functioning; as such, recovery may take several months. Surgical resection is the first-line option for hypercortisolism because of Cushing disease, adrenal tumor, or ectopic tumor. Patients should be closely monitored after surgery to evaluate for possible recurrence. Radiotherapy may be recommended after failed transsphenoidal surgery or in Cushing disease with mass effect or invasion of surrounding structures. Medical therapy, such as pasireotide, cabergoline, and mifepristone, are also sometimes used. In addition, the treatment of comorbidities, such as obesity and type 2 diabetes, hypertension, osteoporosis, psychiatric issues, and electrolyte disorders, is critical.
Courtney Whittle, MD, MSW, Diplomate of ABOM, Pediatric Lead, Obesity Champion, TSPMG, Weight A Minute Clinic, Atlanta, Georgia.
Courtney Whittle, MD, MSW, Diplomate of ABOM, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of Cushing syndrome (CS).
CS is a rare endocrine disease caused by prolonged exposure to high circulating cortisol levels. Exogenous hypercortisolism is the most common cause of CS. It is largely iatrogenic and results from the prolonged use of glucocorticoids. Less frequently, endogenous CS may occur as the result of excessive production of cortisol by adrenal glands. Endogenous CS can be ACTH-dependent or ACTH-independent. ACTH-dependent CS results from ACTH-secreting pituitary adenomas (Cushing disease) and ectopic ACTH secretion by neoplasms, whereas adrenal hyperplasia, adenoma, and carcinoma are the primary causes of ACTH-independent CS.
The annual incidence and prevalence of CS are unknown; the reported incidence of newly diagnosed cases has ranged from 1.2 to 2.4 per million people per year. Women are affected more often than are men, with a peak of incidence in the third to fourth decade of life. CS is associated with various metabolic, psychiatric, musculoskeletal, and cardiovascular comorbidities. Untreated, it is associated with increased mortality, typically as the result of cardiovascular and infectious complications; however, even in appropriately treated patients, mortality is elevated.
The chronic elevations of glucocorticoid concentrations in CS result in its characteristic phenotype, which includes weight gain, moon-shaped face, buffalo hump, muscle weakness, increased bruising, skin atrophy, red abdominal striae, menstrual irregularities, hirsutism, and acne. It is also associated with numerous comorbidities including diabetes, hypertension, hypercholesterolemia, and osteoporosis. Patients often experience mental health complications, such as depression, emotional lability, and cognitive dysfunction.
Given the rarity of CS and the fact that these symptoms overlap with other conditions, delayed diagnosis is common. The current obesity epidemic also poses diagnostic challenges because true CS can be difficult to differentiate from metabolic syndrome. The duration of hypercortisolism appears to be the most significant factor associated with the degree of morbidity and preterm mortality in CS; thus, an accurate diagnosis as early as possible is important.
Screening and diagnostic tests for CS evaluate cortisol secretion. Available options include late-night salivary cortisol (LNSC), impaired glucocorticoid feedback with overnight 1-mg DST or low-dose 2-day dexamethasone test (LDDT) and increased bioavailable cortisol with 24-hour UFC.
A 2021 consensus statement by Fleseriu and colleagues provides recommendations for the diagnosis of CS. If CS is suspected: begin with UFC, LNSC, or both; DST is an option if LNSC not feasible. If CS because of adrenal tumor is suspected: begin with DST because LNSC has lower specificity in these patients. To confirm CS, any of these tests can be used.
An individualized approach is recommended for the treatment of CS. The optimal approach for iatrogenic CS is to slowly taper exogenous steroids. Chronic exposure to steroids can suppress adrenal functioning; as such, recovery may take several months. Surgical resection is the first-line option for hypercortisolism because of Cushing disease, adrenal tumor, or ectopic tumor. Patients should be closely monitored after surgery to evaluate for possible recurrence. Radiotherapy may be recommended after failed transsphenoidal surgery or in Cushing disease with mass effect or invasion of surrounding structures. Medical therapy, such as pasireotide, cabergoline, and mifepristone, are also sometimes used. In addition, the treatment of comorbidities, such as obesity and type 2 diabetes, hypertension, osteoporosis, psychiatric issues, and electrolyte disorders, is critical.
Courtney Whittle, MD, MSW, Diplomate of ABOM, Pediatric Lead, Obesity Champion, TSPMG, Weight A Minute Clinic, Atlanta, Georgia.
Courtney Whittle, MD, MSW, Diplomate of ABOM, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of Cushing syndrome (CS).
CS is a rare endocrine disease caused by prolonged exposure to high circulating cortisol levels. Exogenous hypercortisolism is the most common cause of CS. It is largely iatrogenic and results from the prolonged use of glucocorticoids. Less frequently, endogenous CS may occur as the result of excessive production of cortisol by adrenal glands. Endogenous CS can be ACTH-dependent or ACTH-independent. ACTH-dependent CS results from ACTH-secreting pituitary adenomas (Cushing disease) and ectopic ACTH secretion by neoplasms, whereas adrenal hyperplasia, adenoma, and carcinoma are the primary causes of ACTH-independent CS.
The annual incidence and prevalence of CS are unknown; the reported incidence of newly diagnosed cases has ranged from 1.2 to 2.4 per million people per year. Women are affected more often than are men, with a peak of incidence in the third to fourth decade of life. CS is associated with various metabolic, psychiatric, musculoskeletal, and cardiovascular comorbidities. Untreated, it is associated with increased mortality, typically as the result of cardiovascular and infectious complications; however, even in appropriately treated patients, mortality is elevated.
The chronic elevations of glucocorticoid concentrations in CS result in its characteristic phenotype, which includes weight gain, moon-shaped face, buffalo hump, muscle weakness, increased bruising, skin atrophy, red abdominal striae, menstrual irregularities, hirsutism, and acne. It is also associated with numerous comorbidities including diabetes, hypertension, hypercholesterolemia, and osteoporosis. Patients often experience mental health complications, such as depression, emotional lability, and cognitive dysfunction.
Given the rarity of CS and the fact that these symptoms overlap with other conditions, delayed diagnosis is common. The current obesity epidemic also poses diagnostic challenges because true CS can be difficult to differentiate from metabolic syndrome. The duration of hypercortisolism appears to be the most significant factor associated with the degree of morbidity and preterm mortality in CS; thus, an accurate diagnosis as early as possible is important.
Screening and diagnostic tests for CS evaluate cortisol secretion. Available options include late-night salivary cortisol (LNSC), impaired glucocorticoid feedback with overnight 1-mg DST or low-dose 2-day dexamethasone test (LDDT) and increased bioavailable cortisol with 24-hour UFC.
A 2021 consensus statement by Fleseriu and colleagues provides recommendations for the diagnosis of CS. If CS is suspected: begin with UFC, LNSC, or both; DST is an option if LNSC not feasible. If CS because of adrenal tumor is suspected: begin with DST because LNSC has lower specificity in these patients. To confirm CS, any of these tests can be used.
An individualized approach is recommended for the treatment of CS. The optimal approach for iatrogenic CS is to slowly taper exogenous steroids. Chronic exposure to steroids can suppress adrenal functioning; as such, recovery may take several months. Surgical resection is the first-line option for hypercortisolism because of Cushing disease, adrenal tumor, or ectopic tumor. Patients should be closely monitored after surgery to evaluate for possible recurrence. Radiotherapy may be recommended after failed transsphenoidal surgery or in Cushing disease with mass effect or invasion of surrounding structures. Medical therapy, such as pasireotide, cabergoline, and mifepristone, are also sometimes used. In addition, the treatment of comorbidities, such as obesity and type 2 diabetes, hypertension, osteoporosis, psychiatric issues, and electrolyte disorders, is critical.
Courtney Whittle, MD, MSW, Diplomate of ABOM, Pediatric Lead, Obesity Champion, TSPMG, Weight A Minute Clinic, Atlanta, Georgia.
Courtney Whittle, MD, MSW, Diplomate of ABOM, has disclosed no relevant financial relationships.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
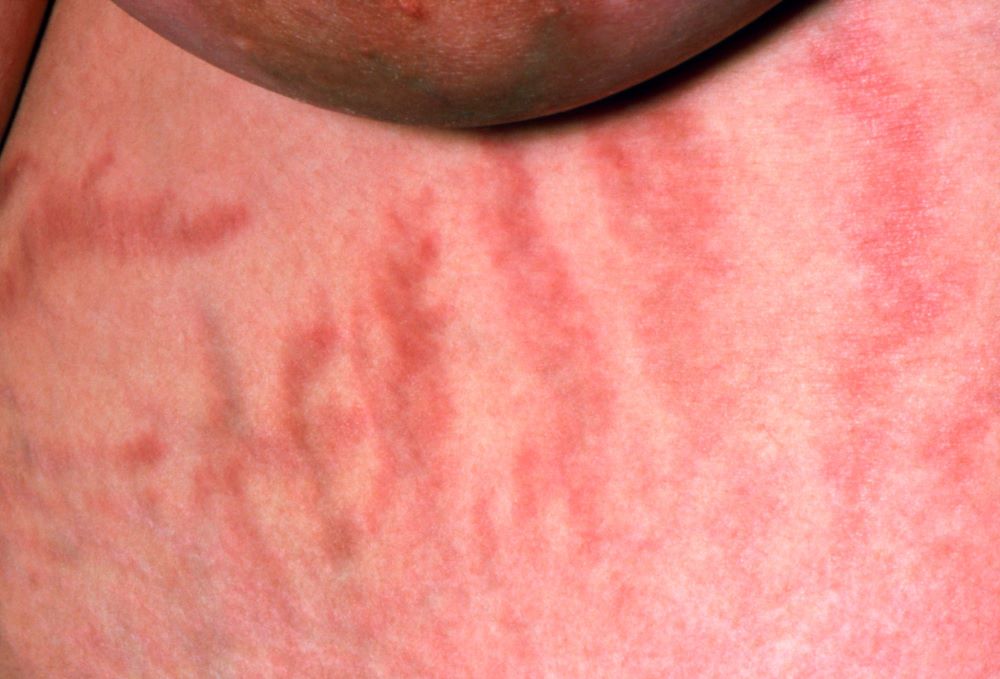
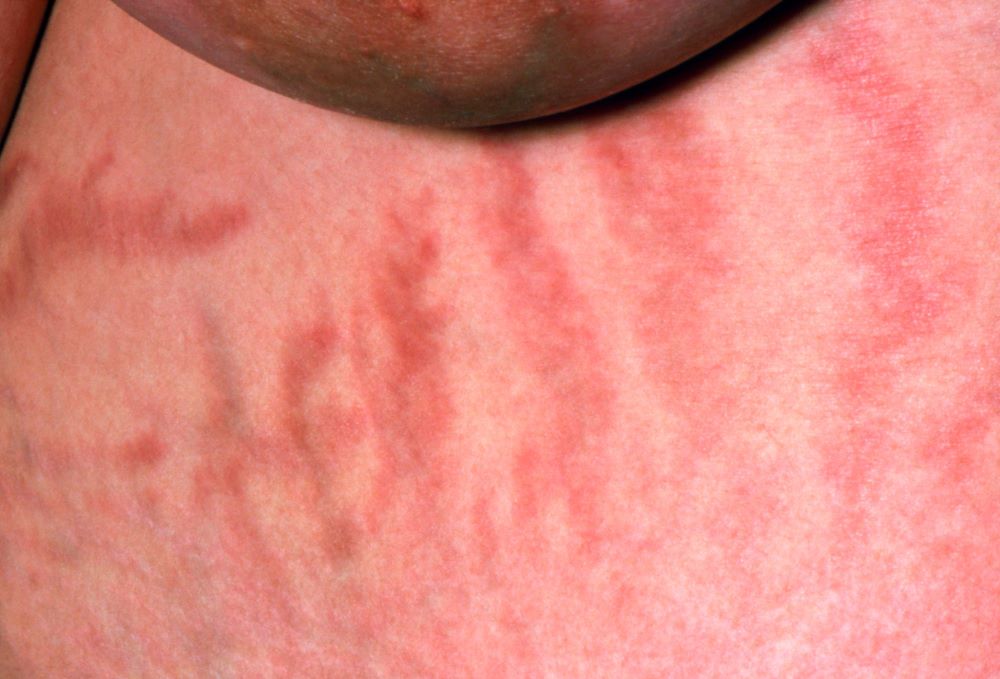
A 37-year-old woman presents with reports of insomnia, weight gain (approximately 12 lb over the last 9 months), and excessive fatigue. The patient's medical history is significant for hypertension (diagnosed 4 years earlier) and depression (diagnosed 7 years earlier). Her current medications include lisinopril 10 mg/d, bupropion 75 mg/d, and venlafaxine 75 mg/d. There is no history of alcohol or drug abuse; family history is unremarkable. The patient's height and weight are 5 ft 5 in and 182 lb (body mass index of 30.3).
During physical examination, facial hirsutism is observed along with increased adipose tissue in the face (moon-shaped face), upper back at the base of the neck (buffalo hump), and abdomen. Vertical red abdominal striae are present. Several bruises are observed on the patient's thighs and arms; when questioned, she reports noting an increased tendency to bruise in recent months.
Pertinent laboratory findings include urinary free cortisol excretion (UFC) 324 mcg/24 h, 1-mg dexamethasone suppression test (DST) with a cortisol value of 3.64 mcg/dL (100.42 nmol/L), and adrenocorticotropic hormone (ACTH) level of 84.9 pg/mL.
Three-month history of fever
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of pleomorphic mantle cell lymphoma (MCL).
MCL is a rare, clinically and biologically heterogeneous B-cell non-Hodgkin lymphoma. It accounts for approximately 5%-7% of all lymphomas. In North America and Europe, its incidence is akin to that of noncutaneous, peripheral T-cell lymphomas. The typical age at diagnosis is between 60 and 70 years. Approximately 70% of all cases are seen in men.
Little is known about risk factors for the development of MCL. Factors that have been associated with the development of other lymphomas (eg, familial risk, immunosuppression, other immune disorders, chemical and occupational exposures, and infectious agents) have not been convincingly identified as predisposing factors for MCL, with the possible exception of family history.
MCL is usually associated with reciprocal chromosomal translocation between chromosomes 11 and 14, t(11;14)(q13:q32), resulting in overexpression of cyclin D1, which plays a key role in tumor cell proliferation through cell-cycle dysregulation, chromosomal instability, and epigenetic regulation. Tumor cells (monoclonal B cells) express surface immunoglobulin, immunoglobulin M, or immunoglobulin D. Cells are usually CD5+ and pan B-cell antigen positive (eg, CD19, CD20, CD22) with no expression of CD10 and CD23. Histologic features include small-to-medium lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Cytologic subtypes include classic MCL, the blastoid variant (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic variant (cells of varying size, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli). Blastoid and pleomorphic MCL typically have a more aggressive natural history and are associated with inferior clinical outcomes.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), an accurate pathologic diagnosis of the subtype is the most important initial step in the management of B-cell lymphomas, including pleomorphic MCL. The basic pathologic exam is the same for all subtypes, although additional testing may be needed in certain cases. An incisional or excisional lymph node biopsy is recommended. Fine-needle aspiration biopsy alone is typically not sufficient for the initial diagnosis of lymphoma; however, its diagnostic accuracy is significantly improved when it is used in combination with immunohistochemistry and flow cytometry. Immunohistochemistry is essential to differentiate MCL subtypes.
Essential workup procedures include a complete physical exam, with particular attention to node-bearing areas, including the Waldeyer ring, as well as the size of the liver and spleen, and assessment of performance status and B symptoms (fever, night sweats, unintentional weight loss). Laboratory studies should include complete blood count with differential, measurement of serum lactate dehydrogenase, hepatitis B virus testing, and a comprehensive metabolic panel. Required imaging studies include PET/CT (or chest/abdominal/pelvic CT with oral and intravenous contrast if PET/CT is not available) and multigated acquisition scanning or echocardiography when anthracyclines and anthracenedione-containing regimens are indicated.
A watch-and-wait approach may be appropriate for some patients with indolent MCL; however, patients with aggressive MCL, such as pleomorphic histology, require chemoimmunotherapy at diagnosis. For patients who relapse or achieve an incomplete response to first-line therapy, the NCCN guidelines recommend second-line treatment with a Bruton tyrosine kinase (BTK) inhibitor–containing regimen. Available BTK inhibitors include acalabrutinib, ibrutinib ± rituximab, zanubrutinib, and pirtobrutinib. Chemoimmunotherapy with lenalidomide + rituximab is another second-line option and may be particularly helpful for patients in whom a BTK inhibitor is contraindicated. Anti-CD19 CAR T-cell therapy is a recommended option for the third line and beyond.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of pleomorphic mantle cell lymphoma (MCL).
MCL is a rare, clinically and biologically heterogeneous B-cell non-Hodgkin lymphoma. It accounts for approximately 5%-7% of all lymphomas. In North America and Europe, its incidence is akin to that of noncutaneous, peripheral T-cell lymphomas. The typical age at diagnosis is between 60 and 70 years. Approximately 70% of all cases are seen in men.
Little is known about risk factors for the development of MCL. Factors that have been associated with the development of other lymphomas (eg, familial risk, immunosuppression, other immune disorders, chemical and occupational exposures, and infectious agents) have not been convincingly identified as predisposing factors for MCL, with the possible exception of family history.
MCL is usually associated with reciprocal chromosomal translocation between chromosomes 11 and 14, t(11;14)(q13:q32), resulting in overexpression of cyclin D1, which plays a key role in tumor cell proliferation through cell-cycle dysregulation, chromosomal instability, and epigenetic regulation. Tumor cells (monoclonal B cells) express surface immunoglobulin, immunoglobulin M, or immunoglobulin D. Cells are usually CD5+ and pan B-cell antigen positive (eg, CD19, CD20, CD22) with no expression of CD10 and CD23. Histologic features include small-to-medium lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Cytologic subtypes include classic MCL, the blastoid variant (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic variant (cells of varying size, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli). Blastoid and pleomorphic MCL typically have a more aggressive natural history and are associated with inferior clinical outcomes.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), an accurate pathologic diagnosis of the subtype is the most important initial step in the management of B-cell lymphomas, including pleomorphic MCL. The basic pathologic exam is the same for all subtypes, although additional testing may be needed in certain cases. An incisional or excisional lymph node biopsy is recommended. Fine-needle aspiration biopsy alone is typically not sufficient for the initial diagnosis of lymphoma; however, its diagnostic accuracy is significantly improved when it is used in combination with immunohistochemistry and flow cytometry. Immunohistochemistry is essential to differentiate MCL subtypes.
Essential workup procedures include a complete physical exam, with particular attention to node-bearing areas, including the Waldeyer ring, as well as the size of the liver and spleen, and assessment of performance status and B symptoms (fever, night sweats, unintentional weight loss). Laboratory studies should include complete blood count with differential, measurement of serum lactate dehydrogenase, hepatitis B virus testing, and a comprehensive metabolic panel. Required imaging studies include PET/CT (or chest/abdominal/pelvic CT with oral and intravenous contrast if PET/CT is not available) and multigated acquisition scanning or echocardiography when anthracyclines and anthracenedione-containing regimens are indicated.
A watch-and-wait approach may be appropriate for some patients with indolent MCL; however, patients with aggressive MCL, such as pleomorphic histology, require chemoimmunotherapy at diagnosis. For patients who relapse or achieve an incomplete response to first-line therapy, the NCCN guidelines recommend second-line treatment with a Bruton tyrosine kinase (BTK) inhibitor–containing regimen. Available BTK inhibitors include acalabrutinib, ibrutinib ± rituximab, zanubrutinib, and pirtobrutinib. Chemoimmunotherapy with lenalidomide + rituximab is another second-line option and may be particularly helpful for patients in whom a BTK inhibitor is contraindicated. Anti-CD19 CAR T-cell therapy is a recommended option for the third line and beyond.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
This patient's clinical presentation and laboratory findings are consistent with a diagnosis of pleomorphic mantle cell lymphoma (MCL).
MCL is a rare, clinically and biologically heterogeneous B-cell non-Hodgkin lymphoma. It accounts for approximately 5%-7% of all lymphomas. In North America and Europe, its incidence is akin to that of noncutaneous, peripheral T-cell lymphomas. The typical age at diagnosis is between 60 and 70 years. Approximately 70% of all cases are seen in men.
Little is known about risk factors for the development of MCL. Factors that have been associated with the development of other lymphomas (eg, familial risk, immunosuppression, other immune disorders, chemical and occupational exposures, and infectious agents) have not been convincingly identified as predisposing factors for MCL, with the possible exception of family history.
MCL is usually associated with reciprocal chromosomal translocation between chromosomes 11 and 14, t(11;14)(q13:q32), resulting in overexpression of cyclin D1, which plays a key role in tumor cell proliferation through cell-cycle dysregulation, chromosomal instability, and epigenetic regulation. Tumor cells (monoclonal B cells) express surface immunoglobulin, immunoglobulin M, or immunoglobulin D. Cells are usually CD5+ and pan B-cell antigen positive (eg, CD19, CD20, CD22) with no expression of CD10 and CD23. Histologic features include small-to-medium lymphocytes with scant cytoplasm, clumped chromatin, inconspicuous nucleoli, and prominent nuclear clefts. Cytologic subtypes include classic MCL, the blastoid variant (large cells, dispersed chromatin, and a high mitotic rate), and the pleomorphic variant (cells of varying size, although many are large, with pale cytoplasm, oval irregular nuclei, and prominent nucleoli). Blastoid and pleomorphic MCL typically have a more aggressive natural history and are associated with inferior clinical outcomes.
According to 2023 guidelines from the National Comprehensive Cancer Network (NCCN), an accurate pathologic diagnosis of the subtype is the most important initial step in the management of B-cell lymphomas, including pleomorphic MCL. The basic pathologic exam is the same for all subtypes, although additional testing may be needed in certain cases. An incisional or excisional lymph node biopsy is recommended. Fine-needle aspiration biopsy alone is typically not sufficient for the initial diagnosis of lymphoma; however, its diagnostic accuracy is significantly improved when it is used in combination with immunohistochemistry and flow cytometry. Immunohistochemistry is essential to differentiate MCL subtypes.
Essential workup procedures include a complete physical exam, with particular attention to node-bearing areas, including the Waldeyer ring, as well as the size of the liver and spleen, and assessment of performance status and B symptoms (fever, night sweats, unintentional weight loss). Laboratory studies should include complete blood count with differential, measurement of serum lactate dehydrogenase, hepatitis B virus testing, and a comprehensive metabolic panel. Required imaging studies include PET/CT (or chest/abdominal/pelvic CT with oral and intravenous contrast if PET/CT is not available) and multigated acquisition scanning or echocardiography when anthracyclines and anthracenedione-containing regimens are indicated.
A watch-and-wait approach may be appropriate for some patients with indolent MCL; however, patients with aggressive MCL, such as pleomorphic histology, require chemoimmunotherapy at diagnosis. For patients who relapse or achieve an incomplete response to first-line therapy, the NCCN guidelines recommend second-line treatment with a Bruton tyrosine kinase (BTK) inhibitor–containing regimen. Available BTK inhibitors include acalabrutinib, ibrutinib ± rituximab, zanubrutinib, and pirtobrutinib. Chemoimmunotherapy with lenalidomide + rituximab is another second-line option and may be particularly helpful for patients in whom a BTK inhibitor is contraindicated. Anti-CD19 CAR T-cell therapy is a recommended option for the third line and beyond.
Timothy J. Voorhees, MD, MSCR, Assistant Professor of Internal Medicine - Clinical, Division of Hematology, The Ohio State University James Comprehensive Cancer Center, Columbus, OH.
Timothy J. Voorhees, MD, MSCR, has disclosed the following relevant financial relationships:
Received research grant from: AstraZeneca; Morphosys; Incyte; Recordati.
Image Quizzes are fictional or fictionalized clinical scenarios intended to provide evidence-based educational takeaways.
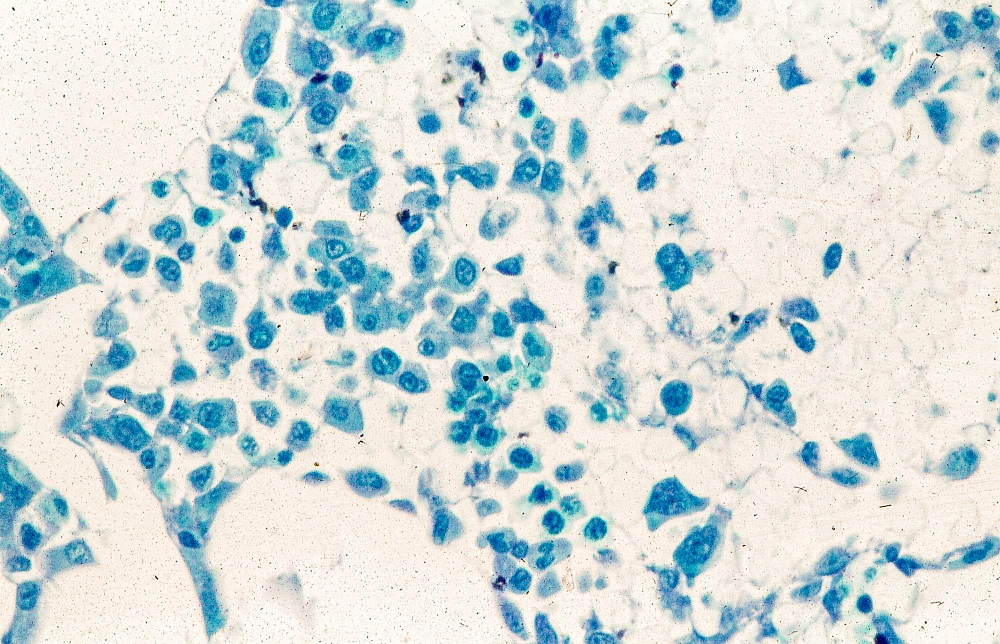
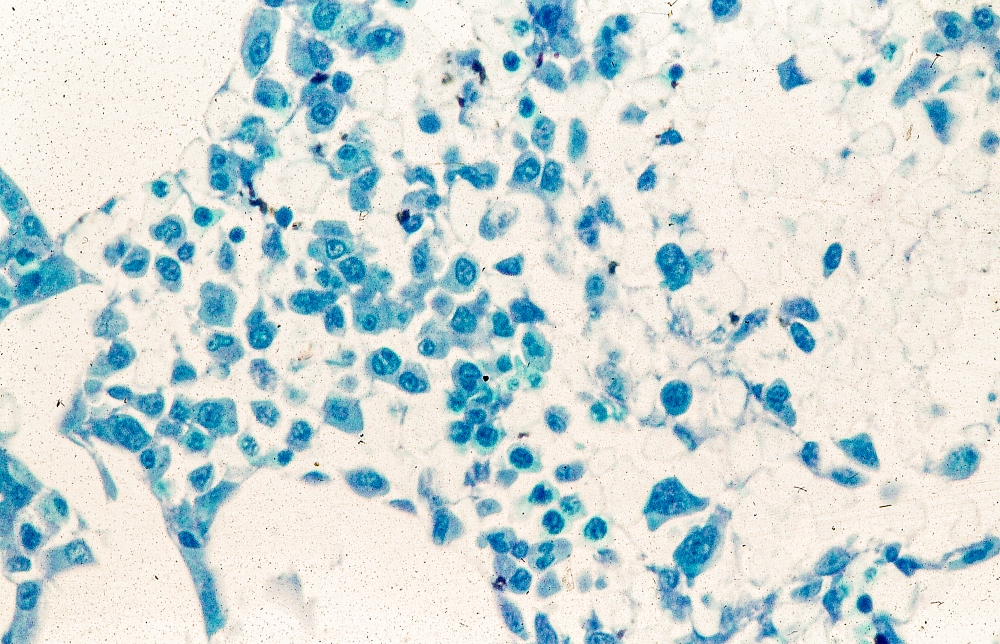
A 62-year-old man with no significant past medical history presents with a 3-month history of fever, night sweats, upper abdominal pain and bloating, and unintentional weight loss. He does not currently take any medications. His height and weight are 6 ft 2 in and 171 lb (BMI 22).
Physical examination reveals generalized lymphadenopathy and splenomegaly. Subsequently, an excisional lymph node biopsy is performed. Histologic examination of the specimen reveals sheets of mostly large cells of varying sizes, with nuclear overlap and extensive necrosis. Cytology findings include large lymphocytes with pale cytoplasm, clumped chromatin, oval irregular nuclei, and prominent nucleoli. Pertinent findings from immunohistochemical staining include the presence of t(11:14), Ki67 > 30%, CD5 and CD20 positivity, and CD10 and CD23 negativity. Centroblasts are absent.
Managing Type 2 Diabetes in Pediatric Patients
Type 2 diabetes (T2D) is associated with obesity and is increasing at an alarming rate in youth. Pediatric T2D disease progresses more rapidly than adult T2D and is harder to treat.
Dr Maria Redondo, a pediatric endocrinologist at Baylor College of Medicine in Houston, Texas, explains that insulin resistance is the major trigger of T2D, and beta-cell dysfunction is key to its development. In children, beta-cell dysfunction occurs more rapidly compared with adults. Children also have higher rates of complications and associated conditions, such as renal disease, cardiovascular disease, and nonalcoholic fatty liver disease.
Dr Redondo references evidence from the TODAY study, which indicates that treatment failure with first-line metformin is common in the pediatric population and affords minimal weight loss.
She then discusses GLP-1 receptor agonists as second-line therapy for children aged 10 years or older. There are currently two FDA-approved options: once-daily liraglutide and once-weekly exenatide. Both are given subcutaneously.
Finally, Dr Redondo highlights DPP-4 inhibitors, such as linagliptin, saxagliptin, and sitagliptin, as well as SGLT-2 inhibitors, such as dapagliflozin and canagliflozin, as emerging therapies currently in clinical trials.
--
Maria J. Redondo, MD, PhD, MPH, Professor, Department of Pediatrics, Baylor College of Medicine; Staff Physician, Texas Children's Hospital, Houston, Texas
Maria J. Redondo, MD, PhD, MPH, has disclosed the following relevant financial relationships:
Received research grant from: NIH; NIDDK
Type 2 diabetes (T2D) is associated with obesity and is increasing at an alarming rate in youth. Pediatric T2D disease progresses more rapidly than adult T2D and is harder to treat.
Dr Maria Redondo, a pediatric endocrinologist at Baylor College of Medicine in Houston, Texas, explains that insulin resistance is the major trigger of T2D, and beta-cell dysfunction is key to its development. In children, beta-cell dysfunction occurs more rapidly compared with adults. Children also have higher rates of complications and associated conditions, such as renal disease, cardiovascular disease, and nonalcoholic fatty liver disease.
Dr Redondo references evidence from the TODAY study, which indicates that treatment failure with first-line metformin is common in the pediatric population and affords minimal weight loss.
She then discusses GLP-1 receptor agonists as second-line therapy for children aged 10 years or older. There are currently two FDA-approved options: once-daily liraglutide and once-weekly exenatide. Both are given subcutaneously.
Finally, Dr Redondo highlights DPP-4 inhibitors, such as linagliptin, saxagliptin, and sitagliptin, as well as SGLT-2 inhibitors, such as dapagliflozin and canagliflozin, as emerging therapies currently in clinical trials.
--
Maria J. Redondo, MD, PhD, MPH, Professor, Department of Pediatrics, Baylor College of Medicine; Staff Physician, Texas Children's Hospital, Houston, Texas
Maria J. Redondo, MD, PhD, MPH, has disclosed the following relevant financial relationships:
Received research grant from: NIH; NIDDK
Type 2 diabetes (T2D) is associated with obesity and is increasing at an alarming rate in youth. Pediatric T2D disease progresses more rapidly than adult T2D and is harder to treat.
Dr Maria Redondo, a pediatric endocrinologist at Baylor College of Medicine in Houston, Texas, explains that insulin resistance is the major trigger of T2D, and beta-cell dysfunction is key to its development. In children, beta-cell dysfunction occurs more rapidly compared with adults. Children also have higher rates of complications and associated conditions, such as renal disease, cardiovascular disease, and nonalcoholic fatty liver disease.
Dr Redondo references evidence from the TODAY study, which indicates that treatment failure with first-line metformin is common in the pediatric population and affords minimal weight loss.
She then discusses GLP-1 receptor agonists as second-line therapy for children aged 10 years or older. There are currently two FDA-approved options: once-daily liraglutide and once-weekly exenatide. Both are given subcutaneously.
Finally, Dr Redondo highlights DPP-4 inhibitors, such as linagliptin, saxagliptin, and sitagliptin, as well as SGLT-2 inhibitors, such as dapagliflozin and canagliflozin, as emerging therapies currently in clinical trials.
--
Maria J. Redondo, MD, PhD, MPH, Professor, Department of Pediatrics, Baylor College of Medicine; Staff Physician, Texas Children's Hospital, Houston, Texas
Maria J. Redondo, MD, PhD, MPH, has disclosed the following relevant financial relationships:
Received research grant from: NIH; NIDDK

Endometriosis and Abnormal Uterine Bleeding

What is the link between endometriosis and abnormal uterine bleeding?
Dr. Lager: This is an important question because when people first learn about endometriosis, common symptoms include pain with periods, pelvic pain, but not necessarily abnormal uterine bleeding. However, many patients do complain of abnormal uterine bleeding when presenting with endometriosis.
There are a couple of reasons why abnormal uterine bleeding is important to consider. Within the spectrum of endometriosis, vaginal endometriosis can contribute to abnormal vaginal bleeding, most commonly cyclic or postcoital. The bleeding could be rectal due to deeply infiltrative endometriosis, although gastrointestinal etiologies should be included in the differential. Another link is coexisting diagnoses such as fibroids, adenomyosis, and endometrial polyps. In fact, the rates for coexisting conditions with endometriosis can be high and vary from study to study.
As an example, some studies show rates between 7% and 11%, where adenomyosis coexists with endometriosis. Other studies look at magnetic resonance imaging for adenomyosis and deep infiltrative endometriosis and find that women younger than 36 years have rates as high as 90% for coexisting diagnoses, and 79% for all women, regardless of the diagnosis.
The overlap is high. When I think particularly about adenomyosis and endometriosis, in some ways, the conditions are along a spectrum where adenomyosis involves ectopic endometrial glands in the myometrium, whereas endometriosis involves ectopic tissue outside of the uterus, predominantly in reproductive organs, but can be anywhere outside of the endometrium. So, when I think about abnormal uterine bleeding particularly associated with dysmenorrhea or pelvic pain, this can often be included in the constellation of symptoms for endometriosis.
Furthermore, it is important to rule out other causes of abnormal uterine bleeding because they would potentially change the treatment.
What are the current treatment options for endometriosis and abnormal uterine bleeding?
Dr. Lager: Treatments for endometriosis are inclusive of any overlapping conditions and we use a multidisciplinary approach to address symptoms. Medical treatments include hormonal management, including birth control pills, etonogestrel implants (Nexplanon), levonorgestrel-releasing intrauterine devices, progestin-only pills, gonadotropin-releasing hormone (GnRH) agonists, GnRH antagonists, and combination medications. Some medications do overlap and work for both, such as combined GnRH antagonists, estradiol, and progesterone.
Surgical management includes diagnostic laparoscopy with excision of endometriosis. If there is another coexisting diagnosis that is structural in nature, such as endometrial polyps, adenomyosis, or fibroids, surgical management may include hysteroscopy, myomectomy, or hysterectomy as indicated. When we consider surgical and nonsurgical approaches, it is important to be clear on the etiology of abnormal uterine bleeding to appropriately counsel patients for what the surgery could entail.
Have you found there to be any age or racial disparities in endometriosis treatment?
Dr. Lager: One of the things that is important about endometriosis, and in medicine in general, is to really think about how we approach race as a social construct. In the past, medicine has included race as a risk factor for certain medical conditions. And physicians in training were taught to use these risk factors to determine a differential diagnosis. However, this strategy has limited us in understanding how historical and structural racism affected patient diagnosis and treatment.
If we think back to literature that was published in the 1950s or the 1970s, Dr. Meeks was one of the physicians who described a set of characteristics of patients with endometriosis. He commented that typical patients were women who were goal-oriented, had private insurance, and experienced delayed marriage, among other traits.
The problem with this characterization was that patients would then present with symptoms of endometriosis who did not fit the original phenotype as historically described and they would be misdiagnosed and thus treated incorrectly. This incorrect treatment further reinforced incorrect stereotypes of patient presentations. These misdiagnoses could lead to unfortunate consequences in their activities of daily living as well as reproductive outcomes. We do not have data on how many patients may have been misdiagnosed and treated for pelvic inflammatory disease because they were not White, did not have private insurance, or had children early. This is an example of areas where we need to recognize systemic racism and classism and work hard to simply do better for our patients.
Although misdiagnosing based on stereotypes has decreased over time, I still think that original thinking can certainly affect patient referrals. When we look at the data of patients who are diagnosed with endometriosis, we find a higher rate of White patients (17%) compared to Black (10.1%), Asian (11.3%), and Hispanic patients (7.4%). Ensuring that all of our patients are getting appropriate referrals and diagnosis should be a priority.
When we think about the timing to initial diagnosis, globally, we know that there is a delay in diagnosis anywhere from 7 to 12 years, and then on top of that, those social constructs decrease the rate of diagnosis for certain patient populations. Misdiagnosis based on social constructs is unacceptable and one aspect that I think is very important to point out.
In a more recent study of 12,000 patients in 2022, the rate of surgical complications associated with endometriosis surgery was higher in women who were Black, Asian and Pacific Islander, and Native American/American Indian than in women who were White. These groups have a much higher rate of complications and higher rates of laparotomy—an open procedure—versus laparoscopy. In younger women, there is a higher rate of oophorectomy at the time of surgery for endometriosis than in older women.
Are there any best practices you would like to share with your peers?
Dr. Lager: For patients with abnormal uterine bleeding, it is important to consider other diagnoses and not assume that abnormal bleeding is solely related to endometriosis, while considering deeply infiltrative endometriosis in the differential.
When patients do present with cyclical bleeding, especially, for example, after hysterectomy, it is important to examine for either vaginal or vaginal cuff endometriosis because there can be other reasons that patients will have abnormal uterine bleeding related to atypical endometriosis.
It is important to know the patient’s history and focus on each patient’s level of pain, how it affects their day-to-day activities, and how they are experiencing that pain.
We all should be working to improve our understanding of social history and systemic racism as best as we can and make sure all patients are getting the right care that they deserve.
What is the link between endometriosis and abnormal uterine bleeding?
Dr. Lager: This is an important question because when people first learn about endometriosis, common symptoms include pain with periods, pelvic pain, but not necessarily abnormal uterine bleeding. However, many patients do complain of abnormal uterine bleeding when presenting with endometriosis.
There are a couple of reasons why abnormal uterine bleeding is important to consider. Within the spectrum of endometriosis, vaginal endometriosis can contribute to abnormal vaginal bleeding, most commonly cyclic or postcoital. The bleeding could be rectal due to deeply infiltrative endometriosis, although gastrointestinal etiologies should be included in the differential. Another link is coexisting diagnoses such as fibroids, adenomyosis, and endometrial polyps. In fact, the rates for coexisting conditions with endometriosis can be high and vary from study to study.
As an example, some studies show rates between 7% and 11%, where adenomyosis coexists with endometriosis. Other studies look at magnetic resonance imaging for adenomyosis and deep infiltrative endometriosis and find that women younger than 36 years have rates as high as 90% for coexisting diagnoses, and 79% for all women, regardless of the diagnosis.
The overlap is high. When I think particularly about adenomyosis and endometriosis, in some ways, the conditions are along a spectrum where adenomyosis involves ectopic endometrial glands in the myometrium, whereas endometriosis involves ectopic tissue outside of the uterus, predominantly in reproductive organs, but can be anywhere outside of the endometrium. So, when I think about abnormal uterine bleeding particularly associated with dysmenorrhea or pelvic pain, this can often be included in the constellation of symptoms for endometriosis.
Furthermore, it is important to rule out other causes of abnormal uterine bleeding because they would potentially change the treatment.
What are the current treatment options for endometriosis and abnormal uterine bleeding?
Dr. Lager: Treatments for endometriosis are inclusive of any overlapping conditions and we use a multidisciplinary approach to address symptoms. Medical treatments include hormonal management, including birth control pills, etonogestrel implants (Nexplanon), levonorgestrel-releasing intrauterine devices, progestin-only pills, gonadotropin-releasing hormone (GnRH) agonists, GnRH antagonists, and combination medications. Some medications do overlap and work for both, such as combined GnRH antagonists, estradiol, and progesterone.
Surgical management includes diagnostic laparoscopy with excision of endometriosis. If there is another coexisting diagnosis that is structural in nature, such as endometrial polyps, adenomyosis, or fibroids, surgical management may include hysteroscopy, myomectomy, or hysterectomy as indicated. When we consider surgical and nonsurgical approaches, it is important to be clear on the etiology of abnormal uterine bleeding to appropriately counsel patients for what the surgery could entail.
Have you found there to be any age or racial disparities in endometriosis treatment?
Dr. Lager: One of the things that is important about endometriosis, and in medicine in general, is to really think about how we approach race as a social construct. In the past, medicine has included race as a risk factor for certain medical conditions. And physicians in training were taught to use these risk factors to determine a differential diagnosis. However, this strategy has limited us in understanding how historical and structural racism affected patient diagnosis and treatment.
If we think back to literature that was published in the 1950s or the 1970s, Dr. Meeks was one of the physicians who described a set of characteristics of patients with endometriosis. He commented that typical patients were women who were goal-oriented, had private insurance, and experienced delayed marriage, among other traits.
The problem with this characterization was that patients would then present with symptoms of endometriosis who did not fit the original phenotype as historically described and they would be misdiagnosed and thus treated incorrectly. This incorrect treatment further reinforced incorrect stereotypes of patient presentations. These misdiagnoses could lead to unfortunate consequences in their activities of daily living as well as reproductive outcomes. We do not have data on how many patients may have been misdiagnosed and treated for pelvic inflammatory disease because they were not White, did not have private insurance, or had children early. This is an example of areas where we need to recognize systemic racism and classism and work hard to simply do better for our patients.
Although misdiagnosing based on stereotypes has decreased over time, I still think that original thinking can certainly affect patient referrals. When we look at the data of patients who are diagnosed with endometriosis, we find a higher rate of White patients (17%) compared to Black (10.1%), Asian (11.3%), and Hispanic patients (7.4%). Ensuring that all of our patients are getting appropriate referrals and diagnosis should be a priority.
When we think about the timing to initial diagnosis, globally, we know that there is a delay in diagnosis anywhere from 7 to 12 years, and then on top of that, those social constructs decrease the rate of diagnosis for certain patient populations. Misdiagnosis based on social constructs is unacceptable and one aspect that I think is very important to point out.
In a more recent study of 12,000 patients in 2022, the rate of surgical complications associated with endometriosis surgery was higher in women who were Black, Asian and Pacific Islander, and Native American/American Indian than in women who were White. These groups have a much higher rate of complications and higher rates of laparotomy—an open procedure—versus laparoscopy. In younger women, there is a higher rate of oophorectomy at the time of surgery for endometriosis than in older women.
Are there any best practices you would like to share with your peers?
Dr. Lager: For patients with abnormal uterine bleeding, it is important to consider other diagnoses and not assume that abnormal bleeding is solely related to endometriosis, while considering deeply infiltrative endometriosis in the differential.
When patients do present with cyclical bleeding, especially, for example, after hysterectomy, it is important to examine for either vaginal or vaginal cuff endometriosis because there can be other reasons that patients will have abnormal uterine bleeding related to atypical endometriosis.
It is important to know the patient’s history and focus on each patient’s level of pain, how it affects their day-to-day activities, and how they are experiencing that pain.
We all should be working to improve our understanding of social history and systemic racism as best as we can and make sure all patients are getting the right care that they deserve.
What is the link between endometriosis and abnormal uterine bleeding?
Dr. Lager: This is an important question because when people first learn about endometriosis, common symptoms include pain with periods, pelvic pain, but not necessarily abnormal uterine bleeding. However, many patients do complain of abnormal uterine bleeding when presenting with endometriosis.
There are a couple of reasons why abnormal uterine bleeding is important to consider. Within the spectrum of endometriosis, vaginal endometriosis can contribute to abnormal vaginal bleeding, most commonly cyclic or postcoital. The bleeding could be rectal due to deeply infiltrative endometriosis, although gastrointestinal etiologies should be included in the differential. Another link is coexisting diagnoses such as fibroids, adenomyosis, and endometrial polyps. In fact, the rates for coexisting conditions with endometriosis can be high and vary from study to study.
As an example, some studies show rates between 7% and 11%, where adenomyosis coexists with endometriosis. Other studies look at magnetic resonance imaging for adenomyosis and deep infiltrative endometriosis and find that women younger than 36 years have rates as high as 90% for coexisting diagnoses, and 79% for all women, regardless of the diagnosis.
The overlap is high. When I think particularly about adenomyosis and endometriosis, in some ways, the conditions are along a spectrum where adenomyosis involves ectopic endometrial glands in the myometrium, whereas endometriosis involves ectopic tissue outside of the uterus, predominantly in reproductive organs, but can be anywhere outside of the endometrium. So, when I think about abnormal uterine bleeding particularly associated with dysmenorrhea or pelvic pain, this can often be included in the constellation of symptoms for endometriosis.
Furthermore, it is important to rule out other causes of abnormal uterine bleeding because they would potentially change the treatment.
What are the current treatment options for endometriosis and abnormal uterine bleeding?
Dr. Lager: Treatments for endometriosis are inclusive of any overlapping conditions and we use a multidisciplinary approach to address symptoms. Medical treatments include hormonal management, including birth control pills, etonogestrel implants (Nexplanon), levonorgestrel-releasing intrauterine devices, progestin-only pills, gonadotropin-releasing hormone (GnRH) agonists, GnRH antagonists, and combination medications. Some medications do overlap and work for both, such as combined GnRH antagonists, estradiol, and progesterone.
Surgical management includes diagnostic laparoscopy with excision of endometriosis. If there is another coexisting diagnosis that is structural in nature, such as endometrial polyps, adenomyosis, or fibroids, surgical management may include hysteroscopy, myomectomy, or hysterectomy as indicated. When we consider surgical and nonsurgical approaches, it is important to be clear on the etiology of abnormal uterine bleeding to appropriately counsel patients for what the surgery could entail.
Have you found there to be any age or racial disparities in endometriosis treatment?
Dr. Lager: One of the things that is important about endometriosis, and in medicine in general, is to really think about how we approach race as a social construct. In the past, medicine has included race as a risk factor for certain medical conditions. And physicians in training were taught to use these risk factors to determine a differential diagnosis. However, this strategy has limited us in understanding how historical and structural racism affected patient diagnosis and treatment.
If we think back to literature that was published in the 1950s or the 1970s, Dr. Meeks was one of the physicians who described a set of characteristics of patients with endometriosis. He commented that typical patients were women who were goal-oriented, had private insurance, and experienced delayed marriage, among other traits.
The problem with this characterization was that patients would then present with symptoms of endometriosis who did not fit the original phenotype as historically described and they would be misdiagnosed and thus treated incorrectly. This incorrect treatment further reinforced incorrect stereotypes of patient presentations. These misdiagnoses could lead to unfortunate consequences in their activities of daily living as well as reproductive outcomes. We do not have data on how many patients may have been misdiagnosed and treated for pelvic inflammatory disease because they were not White, did not have private insurance, or had children early. This is an example of areas where we need to recognize systemic racism and classism and work hard to simply do better for our patients.
Although misdiagnosing based on stereotypes has decreased over time, I still think that original thinking can certainly affect patient referrals. When we look at the data of patients who are diagnosed with endometriosis, we find a higher rate of White patients (17%) compared to Black (10.1%), Asian (11.3%), and Hispanic patients (7.4%). Ensuring that all of our patients are getting appropriate referrals and diagnosis should be a priority.
When we think about the timing to initial diagnosis, globally, we know that there is a delay in diagnosis anywhere from 7 to 12 years, and then on top of that, those social constructs decrease the rate of diagnosis for certain patient populations. Misdiagnosis based on social constructs is unacceptable and one aspect that I think is very important to point out.
In a more recent study of 12,000 patients in 2022, the rate of surgical complications associated with endometriosis surgery was higher in women who were Black, Asian and Pacific Islander, and Native American/American Indian than in women who were White. These groups have a much higher rate of complications and higher rates of laparotomy—an open procedure—versus laparoscopy. In younger women, there is a higher rate of oophorectomy at the time of surgery for endometriosis than in older women.
Are there any best practices you would like to share with your peers?
Dr. Lager: For patients with abnormal uterine bleeding, it is important to consider other diagnoses and not assume that abnormal bleeding is solely related to endometriosis, while considering deeply infiltrative endometriosis in the differential.
When patients do present with cyclical bleeding, especially, for example, after hysterectomy, it is important to examine for either vaginal or vaginal cuff endometriosis because there can be other reasons that patients will have abnormal uterine bleeding related to atypical endometriosis.
It is important to know the patient’s history and focus on each patient’s level of pain, how it affects their day-to-day activities, and how they are experiencing that pain.
We all should be working to improve our understanding of social history and systemic racism as best as we can and make sure all patients are getting the right care that they deserve.
The Prediabetes Debate: Should the Diabetes Diagnostic Threshold Be Lowered, Allowing Clinicians to Intervene Earlier?

I believe that the diagnosis of type 2 diabetes mellitus (T2DM) should be broadened to match the glycemic thresholds currently used for prediabetes. This would eliminate the need for a separate category, the “prediabetes” nomenclature, and allow for earlier therapeutic intervention in patients. Current diabetes diagnostic thresholds do not reflect the latest advancements in T2DM understanding. The latest in T2DM research suggests that intervening and treating prediabetes earlier could potentially offer better clinical outcomes and enhance patients’ quality of life, as cell and tissue damage occurs early and leads to dysfunction prior to a diabetes diagnosis.1 T2DM is a highly complex disease with multifactorial causes beyond hyperglycemia that should be considered, such as hyperlipidemia, insulin resistance, hyperinsulinemia, and autoimmune inflammatory mechanisms that lead to β-cell dysfunction or failure, which results in hyperglycemia.
Prediabetes is associated with micro- and macrovascular complications that can occur early in the progression to frank disease state.1,2 This phase of diabetes also includes insulin resistance, impaired incretin action, insulin hypersecretion, increased lipolysis, and ectopic lipid storage—all of which damage β cells. These dysfunctions are also present in the frank diabetic disease state.3,4 Furthermore, diabetic retinopathy occurs in 8% to 12% of patients with prediabetes, and retinopathy begins earlier than previously thought, with neuroinflammation occurring even before vascular damage.5,6 Unfortunately, these neuro-inflammatory lesions cannot be detected with the typical instruments used in an ophthalmologist’s office.
It is believed that, through the principle of metabolic memory, even a moderate increase or episodic spikes in blood glucose can lead to negative effects in prediabetic patients who are susceptible to T2DM.1,5 Therefore, a lower diabetes diagnostic threshold could allow for earlier, more precise, and personalized therapies based on each patient’s individual risk factors and biomarkers. With a diagnosis of T2DM at the current prediabetes threshold, patients could receive treatment covered by health insurance while in the “prediabetic” state—treatment that would not have been previously approved. Patients should be treated earlier and on an individual basis with counseling on diet and lifestyle changes and antidiabetic agents to reduce glycemic levels, preserve β cells, and reduce cardiovascular [CV] or renal risk, among other complications.6,7
Moreover, the newer agents for treating diabetes such as glucagon-like pepetide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors, which are also associated with reduction in adverse CV and/or renal outcomes, could be beneficial if administered early in the prediabetic stage of disease.8,9
Early lifestyle and pharmacologic interventions can reduce the rate of progression from prediabetes to diabetes as well as complications and associated conditions, and even potentially result in remission or a full reversal of diabetes. These “side benefits” of lowering the diabetes diagnostic threshold (over and above glycemic control) make any cost-effectiveness calculations all the more advantageous to individual patients as well as to society.
Given the numerous benefits of earlier intervention for diabetes treatment, I believe a call to re-evaluate the current diabetes diagnostic threshold is in order, as it will do a great service for all patients who are currently at risk for developing T2DM.
Armato JP, DeFronzo RA, Abdul-Ghani M, Ruby RJ. Successful treatment of prediabetes in clinical practice using physiological assessment (STOP DIABETES). Lancet Diabetes Endocrinol. 2018;6:781-789.
Schwartz SS, Epstein S, Corkey BE, et al. A unified pathophysiological construct of diabetes and its complications. Trends Endocrinol Metab. 2017;28:645-655.
American Diabetes Association. Standards of Medical Care in Diabetes – 2021. Diabetes Care. 2021;44(S15–S39):S111-S124.
Brannick B, Wynn A, Dagogo-Jack S. Prediabetes as a toxic environment for the initiation of microvascular and macrovascular complications. Exp Biol Med (Maywood). 2016;241:1323-1331.
Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183-1197.
Sinclair SH, Schwartz SS. Diabetic retinopathy–an underdiagnosed and undertreated inflammatory, neuro-vascular complication of diabetes. Front Endocrinol (Lausanne). 2019;10:843.
Edwards CM, Cusi K. Prediabetes: a worldwide epidemic. Endocrinol Metab Clin N Am. 2016;45:751-764.
Kanat M, DeFronzo RA, Abdul-Ghani MA. Treatment of prediabetes. World J Diabetes. 2015;6:1207-1222.
Dankner R, Roth J. The personalized approach for detecting prediabetes and diabetes. Curr Diabetes Rev. 2016;12:58-65.
I believe that the diagnosis of type 2 diabetes mellitus (T2DM) should be broadened to match the glycemic thresholds currently used for prediabetes. This would eliminate the need for a separate category, the “prediabetes” nomenclature, and allow for earlier therapeutic intervention in patients. Current diabetes diagnostic thresholds do not reflect the latest advancements in T2DM understanding. The latest in T2DM research suggests that intervening and treating prediabetes earlier could potentially offer better clinical outcomes and enhance patients’ quality of life, as cell and tissue damage occurs early and leads to dysfunction prior to a diabetes diagnosis.1 T2DM is a highly complex disease with multifactorial causes beyond hyperglycemia that should be considered, such as hyperlipidemia, insulin resistance, hyperinsulinemia, and autoimmune inflammatory mechanisms that lead to β-cell dysfunction or failure, which results in hyperglycemia.
Prediabetes is associated with micro- and macrovascular complications that can occur early in the progression to frank disease state.1,2 This phase of diabetes also includes insulin resistance, impaired incretin action, insulin hypersecretion, increased lipolysis, and ectopic lipid storage—all of which damage β cells. These dysfunctions are also present in the frank diabetic disease state.3,4 Furthermore, diabetic retinopathy occurs in 8% to 12% of patients with prediabetes, and retinopathy begins earlier than previously thought, with neuroinflammation occurring even before vascular damage.5,6 Unfortunately, these neuro-inflammatory lesions cannot be detected with the typical instruments used in an ophthalmologist’s office.
It is believed that, through the principle of metabolic memory, even a moderate increase or episodic spikes in blood glucose can lead to negative effects in prediabetic patients who are susceptible to T2DM.1,5 Therefore, a lower diabetes diagnostic threshold could allow for earlier, more precise, and personalized therapies based on each patient’s individual risk factors and biomarkers. With a diagnosis of T2DM at the current prediabetes threshold, patients could receive treatment covered by health insurance while in the “prediabetic” state—treatment that would not have been previously approved. Patients should be treated earlier and on an individual basis with counseling on diet and lifestyle changes and antidiabetic agents to reduce glycemic levels, preserve β cells, and reduce cardiovascular [CV] or renal risk, among other complications.6,7
Moreover, the newer agents for treating diabetes such as glucagon-like pepetide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors, which are also associated with reduction in adverse CV and/or renal outcomes, could be beneficial if administered early in the prediabetic stage of disease.8,9
Early lifestyle and pharmacologic interventions can reduce the rate of progression from prediabetes to diabetes as well as complications and associated conditions, and even potentially result in remission or a full reversal of diabetes. These “side benefits” of lowering the diabetes diagnostic threshold (over and above glycemic control) make any cost-effectiveness calculations all the more advantageous to individual patients as well as to society.
Given the numerous benefits of earlier intervention for diabetes treatment, I believe a call to re-evaluate the current diabetes diagnostic threshold is in order, as it will do a great service for all patients who are currently at risk for developing T2DM.
I believe that the diagnosis of type 2 diabetes mellitus (T2DM) should be broadened to match the glycemic thresholds currently used for prediabetes. This would eliminate the need for a separate category, the “prediabetes” nomenclature, and allow for earlier therapeutic intervention in patients. Current diabetes diagnostic thresholds do not reflect the latest advancements in T2DM understanding. The latest in T2DM research suggests that intervening and treating prediabetes earlier could potentially offer better clinical outcomes and enhance patients’ quality of life, as cell and tissue damage occurs early and leads to dysfunction prior to a diabetes diagnosis.1 T2DM is a highly complex disease with multifactorial causes beyond hyperglycemia that should be considered, such as hyperlipidemia, insulin resistance, hyperinsulinemia, and autoimmune inflammatory mechanisms that lead to β-cell dysfunction or failure, which results in hyperglycemia.
Prediabetes is associated with micro- and macrovascular complications that can occur early in the progression to frank disease state.1,2 This phase of diabetes also includes insulin resistance, impaired incretin action, insulin hypersecretion, increased lipolysis, and ectopic lipid storage—all of which damage β cells. These dysfunctions are also present in the frank diabetic disease state.3,4 Furthermore, diabetic retinopathy occurs in 8% to 12% of patients with prediabetes, and retinopathy begins earlier than previously thought, with neuroinflammation occurring even before vascular damage.5,6 Unfortunately, these neuro-inflammatory lesions cannot be detected with the typical instruments used in an ophthalmologist’s office.
It is believed that, through the principle of metabolic memory, even a moderate increase or episodic spikes in blood glucose can lead to negative effects in prediabetic patients who are susceptible to T2DM.1,5 Therefore, a lower diabetes diagnostic threshold could allow for earlier, more precise, and personalized therapies based on each patient’s individual risk factors and biomarkers. With a diagnosis of T2DM at the current prediabetes threshold, patients could receive treatment covered by health insurance while in the “prediabetic” state—treatment that would not have been previously approved. Patients should be treated earlier and on an individual basis with counseling on diet and lifestyle changes and antidiabetic agents to reduce glycemic levels, preserve β cells, and reduce cardiovascular [CV] or renal risk, among other complications.6,7
Moreover, the newer agents for treating diabetes such as glucagon-like pepetide-1 receptor agonists and sodium-glucose cotransporter 2 inhibitors, which are also associated with reduction in adverse CV and/or renal outcomes, could be beneficial if administered early in the prediabetic stage of disease.8,9
Early lifestyle and pharmacologic interventions can reduce the rate of progression from prediabetes to diabetes as well as complications and associated conditions, and even potentially result in remission or a full reversal of diabetes. These “side benefits” of lowering the diabetes diagnostic threshold (over and above glycemic control) make any cost-effectiveness calculations all the more advantageous to individual patients as well as to society.
Given the numerous benefits of earlier intervention for diabetes treatment, I believe a call to re-evaluate the current diabetes diagnostic threshold is in order, as it will do a great service for all patients who are currently at risk for developing T2DM.
Armato JP, DeFronzo RA, Abdul-Ghani M, Ruby RJ. Successful treatment of prediabetes in clinical practice using physiological assessment (STOP DIABETES). Lancet Diabetes Endocrinol. 2018;6:781-789.
Schwartz SS, Epstein S, Corkey BE, et al. A unified pathophysiological construct of diabetes and its complications. Trends Endocrinol Metab. 2017;28:645-655.
American Diabetes Association. Standards of Medical Care in Diabetes – 2021. Diabetes Care. 2021;44(S15–S39):S111-S124.
Brannick B, Wynn A, Dagogo-Jack S. Prediabetes as a toxic environment for the initiation of microvascular and macrovascular complications. Exp Biol Med (Maywood). 2016;241:1323-1331.
Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183-1197.
Sinclair SH, Schwartz SS. Diabetic retinopathy–an underdiagnosed and undertreated inflammatory, neuro-vascular complication of diabetes. Front Endocrinol (Lausanne). 2019;10:843.
Edwards CM, Cusi K. Prediabetes: a worldwide epidemic. Endocrinol Metab Clin N Am. 2016;45:751-764.
Kanat M, DeFronzo RA, Abdul-Ghani MA. Treatment of prediabetes. World J Diabetes. 2015;6:1207-1222.
Dankner R, Roth J. The personalized approach for detecting prediabetes and diabetes. Curr Diabetes Rev. 2016;12:58-65.
Armato JP, DeFronzo RA, Abdul-Ghani M, Ruby RJ. Successful treatment of prediabetes in clinical practice using physiological assessment (STOP DIABETES). Lancet Diabetes Endocrinol. 2018;6:781-789.
Schwartz SS, Epstein S, Corkey BE, et al. A unified pathophysiological construct of diabetes and its complications. Trends Endocrinol Metab. 2017;28:645-655.
American Diabetes Association. Standards of Medical Care in Diabetes – 2021. Diabetes Care. 2021;44(S15–S39):S111-S124.
Brannick B, Wynn A, Dagogo-Jack S. Prediabetes as a toxic environment for the initiation of microvascular and macrovascular complications. Exp Biol Med (Maywood). 2016;241:1323-1331.
Report of the Expert Committee on the Diagnosis and Classification of Diabetes Mellitus. Diabetes Care. 1997;20:1183-1197.
Sinclair SH, Schwartz SS. Diabetic retinopathy–an underdiagnosed and undertreated inflammatory, neuro-vascular complication of diabetes. Front Endocrinol (Lausanne). 2019;10:843.
Edwards CM, Cusi K. Prediabetes: a worldwide epidemic. Endocrinol Metab Clin N Am. 2016;45:751-764.
Kanat M, DeFronzo RA, Abdul-Ghani MA. Treatment of prediabetes. World J Diabetes. 2015;6:1207-1222.
Dankner R, Roth J. The personalized approach for detecting prediabetes and diabetes. Curr Diabetes Rev. 2016;12:58-65.
PsA Pathophysiology and Etiology
Obesity Treatment
MCL Treatment
Acute Onset of Vitiligolike Depigmentation After Nivolumab Therapy for Systemic Melanoma
To the Editor:
Vitiligolike depigmentation has been known to develop around the sites of origin of melanoma or more rarely in patients treated with antimelanoma therapy.1 Vitiligo is characterized by white patchy depigmentation of the skin caused by the loss of functional melanocytes from the epidermis. The exact mechanisms of disease are unknown and multifactorial; however, autoimmunity plays a central role. Interferon gamma (IFN-γ), C-X-C chemokine ligand 10, and IL-22 have been identified as key mediators in an inflammatory cascade leading to the stimulation of the innate immune response against melanocyte antigens.2,3 Research suggests melanoma-associated vitiligolike leukoderma also results from an immune reaction directed against antigenic determinants shared by both normal and malignant melanocytes.3 Vitiligolike lesions have been associated with the use of immunomodulatory agents such as nivolumab, a fully humanized monoclonal IgG4 antibody, which blocks the programmed cell death protein 1 (PD-1) receptor that normally is expressed on T cells during the effector phase of T-cell activation.4,5 In the tumor microenvironment, the PD-1 receptor is stimulated, leading to downregulation of the T-cell effector function and destruction of T cells.5 Due to T-cell apoptosis and consequent suppression of the immune response, tumorigenesis continues. By inhibiting the PD-1 receptor, nivolumab increases the number of active T cells and antitumor response. However, the distressing side effect of vitiligolike depigmentation has been reported in 15% to 25% of treated patients.6
In a meta-analysis by Teulings et al,7 patients with new-onset vitiligo and malignant melanoma demonstrated a 2-fold decrease in cancer progression and a 4-fold decreased risk for death vs patients without vitiligo development. Thus, in patients with melanoma, vitiligolike depigmentation should be considered a good prognostic indicator as well as a visible sign of spontaneous or therapy-induced antihumoral immune response against melanocyte differentiation antigens, as it is associated with a notable survival benefit in patients receiving immunotherapy for metastatic melanoma.3 We describe a case of diffuse vitiligolike depigmentation that developed suddenly during nivolumab treatment, causing much distress to the patient.
A 75-year-old woman presented to the clinic with a chief concern of sudden diffuse skin discoloration primarily affecting the face, hands, and extremities of 3 weeks’ duration. She had a medical history of metastatic melanoma—the site of the primary melanoma was never identified—and she was undergoing immune-modulating therapy with nivolumab. She was on her fifth month of treatment and was experiencing a robust therapeutic response with a reported 100% clearance of the metastatic melanoma as observed on a positron emission tomography scan. The patchy depigmentation of skin was causing her much distress. Physical examination revealed diffuse patches of hypopigmentation on the trunk, face, and extremities (Figure). Shave biopsies of the right lateral arm demonstrated changes consistent with vitiligo, with an adjacent biopsy illustrating normal skin characteristics. Triamcinolone ointment 0.1% was initiated, with instruction to apply it to affected areas twice daily for 2 weeks. However, there was no improvement, and she discontinued use.

At 3-month follow-up, the depigmentation persisted, prompting a trial of hydroquinone cream 4% to be used sparingly in cosmetically sensitive areas such as the face and dorsal aspects of the hands. Additionally, diligent photoprotection was advised. Upon re-evaluation 9 months later, the patient remained in cancer remission, continued nivolumab therapy, and reported improvement in the hypopigmentation with a more even skin color with topical hydroquinone use. She no longer noticed starkly contrasting hypopigmented patches.
Vitiligo is a benign skin condition characterized by white depigmented macules and patches. The key feature of the disorder is loss of functional melanocytes from the cutaneous epidermis and sometimes from the hair follicles, with various theories on the cause. It has been suggested that the disease is multifactorial, involving both genetics and environmental factors.2 Regardless of the exact mechanism, the result is always the same: loss of melanin pigment in cells due to loss of melanocytes.
Autoimmunity plays a central role in the causation of vitiligo and was first suspected as a possible cause due to the association of vitiligo with several other autoimmune disorders, such as thyroiditis.8 An epidemiological survey from the United Kingdom and North America (N=2624) found that 19.4% of vitiligo patients aged 20 years or older also reported a clinical history of autoimmune thyroid disease compared with 2.4% of the overall White population of the same age.9 Interferon gamma, C-X-C chemokine ligand 10, and IL-22 receptors stimulate the innate immune response, resulting in an overactive danger signaling cascade, which leads to proinflammatory signals against melanocyte antigens.2,3 The adaptive immune system also participates in the progression of vitiligo by activating dermal dendritic cells to attack melanocytes along with melanocyte-specific cytotoxic T cells.
Immunomodulatory agents utilized in the treatment of metastatic melanoma have been linked to vitiligolike depigmentation. In those receiving PD-1 immunotherapy for metastatic melanoma, vitiligolike lesions have been reported in 15% to 25% of patients.6 Typically, the PD-1 molecule has a regulatory function on effector T cells. Interaction of the PD-1 receptor with its ligands occurs primarily in peripheral tissue causing apoptosis and downregulation of effector T cells with the goal of decreasing collateral damage to surrounding tissues by active T cells.5 In the tumor microenvironment, however, suppression of the host’s immune response is enhanced by aberrant stimulation of the PD-1 receptor, causing downregulation of the T-cell effector function, T-cell destruction, and apoptosis, which results in continued tumor growth. Nivolumab, a fully humanized monoclonal IgG4 antibody, selectively inhibits the PD-1 receptor, disrupting the regulator pathway that would typically end in T-cell destruction.5 Accordingly, the population of active T cells is increased along with the antitumor response.4,10 Nivolumab exhibits success as an immunotherapeutic agent, with an overall survival rate in patients with metastatic melanoma undergoing nivolumab therapy of 41% to 42% at 3 years and 35% at 5 years.11 However, therapeutic manipulation of the host’s immune response does not come without a cost. Vitiligolike lesions have been reported in up to a quarter of patients receiving PD-1 immunotherapy for metastatic melanoma.6
The relationship between vitiligolike depigmentation and melanoma can be explained by the immune activation against antigens associated with melanoma that also are expressed by normal melanocytes. In clinical observations of patients with melanoma and patients with vitiligo, antibodies to human melanocyte antigens were present in 80% (24/30) of patients vs 7% (2/28) in the control group.12 The autoimmune response results from a cross-reaction of melanoma cells that share the same antigens as normal melanocytes, such as melanoma antigen recognized by T cells 1 (MART-1), gp100, and tyrosinase.13,14
Development of vitiligolike depigmentation in patients with metastatic melanoma treated with nivolumab has been reported to occur between 2 and 15 months after the start of PD-1 therapy. This side effect of treatment correlates with favorable clinical outcomes.15,16 Enhancing immune recognition of melanocytes in patients with melanoma confers a survival advantage, as studies by Koh et al17 and Norlund et al18 involving patients who developed vitiligolike hypopigmentation associated with malignant melanoma indicated a better prognosis than for those without hypopigmentation. The 5-year survival rate of patients with both malignant melanoma and vitiligo was reported as 60% to 67% when it was estimated that only 30% to 50% of patients should have survived that duration of time.17,18 Similarly, a systematic review of patients with melanoma stages III and IV reported that those with associated hypopigmentation had a 2- to 4-fold decreased risk of disease progression and death compared to patients without depigmentation.7
Use of traditional treatment therapies for vitiligo is based on the ability of the therapy to suppress the immune system. However, in patients with metastatic melanoma undergoing immune-modulating cancer therapies, traditional treatment options may counter the antitumor effects of the targeted immunotherapies and should be used with caution. Our patient displayed improvement in the appearance of her starkly contrasting hypopigmented patches with the use of hydroquinone cream 4%, which induced necrotic death of melanocytes by inhibiting the conversion of L-3,4-dihydroxyphenylalanine to melanin by tyrosinase.19 The effect achieved by using topical hydroquinone 4% was a lighter skin appearance in areas of application.
There is no cure for vitiligo, and although it is a benign condition, it can negatively impact a patient's quality of life. In some countries, vitiligo is confused with leprosy, resulting in a social stigma attached to the diagnosis. Many patients are frightened or embarrassed by the diagnosis of vitiligo and its effects, and they often experience discrimination.2 Patients with vitiligo also experience more psychological difficulties such as depression.20 The unpredictability of vitiligo is associated with negative emotions including fear of spreading the lesions, shame, insecurity, and sadness.21 Supportive care measures, including psychological support and counseling, are recommended. Additionally, upon initiation of anti–PD-1 therapies, expectations should be discussed with patients concerning the possibilities of depigmentation and associated treatment results. Although the occurrence of vitiligo may cause the patient concern, it should be communicated that its presence is a positive indicator of a vigorous antimelanoma immunity and an increased survival rate.7
Vitiligolike depigmentation is a known rare adverse effect of nivolumab treatment. Although aesthetically unfavorable for the patient, the development of vitiligolike lesions while undergoing immunotherapy for melanoma may be a sign of a promising clinical outcome due to an effective immune response to melanoma antigens. Our patient remains in remission without any evidence of melanoma after 9 months of therapy, which offers support for a promising outcome for melanoma patients who experience vitiligolike depigmentation.
- de Golian E, Kwong BY, Swetter SM, et al. Cutaneous complications of targeted melanoma therapy. Curr Treat Options Oncol. 2016;17:57.
- Ezzedine K, Eleftheriadou V, Whitton M, et al. Vitiligo. Lancet. 2015;386:74-84.
- Ortonne, JP, Passeron, T. Vitiligo and other disorders of hypopigmentation. In: Bolognia J, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1087-1114.
- Opdivo. Package insert. Bristol-Myers Squibb Company; 2023.
- Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19:5300-5309.
- Hwang SJE, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74:455-461.e1.
- Teulings HE, Limpens J, Jansen SN, et al. Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta-analysis. J Clin Oncol. 2015;33:773-781.
- Gey A, Diallo A, Seneschal J, et al. Autoimmune thyroid disease in vitiligo: multivariate analysis indicates intricate pathomechanisms. Br J Dermatol. 2013;168:756-761.
- Alkhateeb A, Fain PR, Thody A, et al. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. 2003;16:208-214.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
- Hodi FS, Kluger H, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Cancer Res. 2016;76(14 suppl):CT001.
- Cui J, Bystryn JC. Melanoma and vitiligo are associated with antibody responses to similar antigens on pigment cells. Arch Dermatol. 1995;131:314-318.
- Lynch SA, Bouchard BN, Vijayasaradhi S, et al. Antigens of melanocytes and melanoma. Cancer Metastasis Rev. 1991;10:141-150.
- Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab cutaneous adverse events and their association with disease progression. JAMA Dermatol. 2015;15:1206-1212.
- Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45-51.
- Nakamura Y, Tanaka R, Asami Y, et al. Correlation between vitiligo occurrence and clinical benefit in advanced melanoma patients treated with nivolumab: a multi-institutional retrospective study. J Dermatol. 2017;44:117-122.
- Koh HK, Sober AJ, Nakagawa H, et al. Malignant melanoma and vitiligo-like leukoderma: an electron microscope study. J Am Acad Dermatol. 1983;9:696-708.
- Nordlund JJ, Kirkwood JM, Forget BM, et al. Vitiligo in patients with metastatic melanoma: a good prognostic sign. J Am Acad Dermatol. 1983;9:689-696.
- Palumbo A, d’Ischia M, Misuraca G, et al. Mechanism of inhibition of melanogenesis by hydroquinone. Biochim Biophys Acta. 1991;1073:85-90.
- Lai YC, Yew YW, Kennedy C, et al. Vitiligo and depression: a systematic review and meta-analysis of observational studies. Br J Dermatol. 2017;177:708-718.
- Nogueira LSC, Zancanaro PCQ, Azambuja RD. Vitiligo and emotions. An Bras Dermatol. 2009;84:41-45.
To the Editor:
Vitiligolike depigmentation has been known to develop around the sites of origin of melanoma or more rarely in patients treated with antimelanoma therapy.1 Vitiligo is characterized by white patchy depigmentation of the skin caused by the loss of functional melanocytes from the epidermis. The exact mechanisms of disease are unknown and multifactorial; however, autoimmunity plays a central role. Interferon gamma (IFN-γ), C-X-C chemokine ligand 10, and IL-22 have been identified as key mediators in an inflammatory cascade leading to the stimulation of the innate immune response against melanocyte antigens.2,3 Research suggests melanoma-associated vitiligolike leukoderma also results from an immune reaction directed against antigenic determinants shared by both normal and malignant melanocytes.3 Vitiligolike lesions have been associated with the use of immunomodulatory agents such as nivolumab, a fully humanized monoclonal IgG4 antibody, which blocks the programmed cell death protein 1 (PD-1) receptor that normally is expressed on T cells during the effector phase of T-cell activation.4,5 In the tumor microenvironment, the PD-1 receptor is stimulated, leading to downregulation of the T-cell effector function and destruction of T cells.5 Due to T-cell apoptosis and consequent suppression of the immune response, tumorigenesis continues. By inhibiting the PD-1 receptor, nivolumab increases the number of active T cells and antitumor response. However, the distressing side effect of vitiligolike depigmentation has been reported in 15% to 25% of treated patients.6
In a meta-analysis by Teulings et al,7 patients with new-onset vitiligo and malignant melanoma demonstrated a 2-fold decrease in cancer progression and a 4-fold decreased risk for death vs patients without vitiligo development. Thus, in patients with melanoma, vitiligolike depigmentation should be considered a good prognostic indicator as well as a visible sign of spontaneous or therapy-induced antihumoral immune response against melanocyte differentiation antigens, as it is associated with a notable survival benefit in patients receiving immunotherapy for metastatic melanoma.3 We describe a case of diffuse vitiligolike depigmentation that developed suddenly during nivolumab treatment, causing much distress to the patient.
A 75-year-old woman presented to the clinic with a chief concern of sudden diffuse skin discoloration primarily affecting the face, hands, and extremities of 3 weeks’ duration. She had a medical history of metastatic melanoma—the site of the primary melanoma was never identified—and she was undergoing immune-modulating therapy with nivolumab. She was on her fifth month of treatment and was experiencing a robust therapeutic response with a reported 100% clearance of the metastatic melanoma as observed on a positron emission tomography scan. The patchy depigmentation of skin was causing her much distress. Physical examination revealed diffuse patches of hypopigmentation on the trunk, face, and extremities (Figure). Shave biopsies of the right lateral arm demonstrated changes consistent with vitiligo, with an adjacent biopsy illustrating normal skin characteristics. Triamcinolone ointment 0.1% was initiated, with instruction to apply it to affected areas twice daily for 2 weeks. However, there was no improvement, and she discontinued use.
At 3-month follow-up, the depigmentation persisted, prompting a trial of hydroquinone cream 4% to be used sparingly in cosmetically sensitive areas such as the face and dorsal aspects of the hands. Additionally, diligent photoprotection was advised. Upon re-evaluation 9 months later, the patient remained in cancer remission, continued nivolumab therapy, and reported improvement in the hypopigmentation with a more even skin color with topical hydroquinone use. She no longer noticed starkly contrasting hypopigmented patches.
Vitiligo is a benign skin condition characterized by white depigmented macules and patches. The key feature of the disorder is loss of functional melanocytes from the cutaneous epidermis and sometimes from the hair follicles, with various theories on the cause. It has been suggested that the disease is multifactorial, involving both genetics and environmental factors.2 Regardless of the exact mechanism, the result is always the same: loss of melanin pigment in cells due to loss of melanocytes.
Autoimmunity plays a central role in the causation of vitiligo and was first suspected as a possible cause due to the association of vitiligo with several other autoimmune disorders, such as thyroiditis.8 An epidemiological survey from the United Kingdom and North America (N=2624) found that 19.4% of vitiligo patients aged 20 years or older also reported a clinical history of autoimmune thyroid disease compared with 2.4% of the overall White population of the same age.9 Interferon gamma, C-X-C chemokine ligand 10, and IL-22 receptors stimulate the innate immune response, resulting in an overactive danger signaling cascade, which leads to proinflammatory signals against melanocyte antigens.2,3 The adaptive immune system also participates in the progression of vitiligo by activating dermal dendritic cells to attack melanocytes along with melanocyte-specific cytotoxic T cells.
Immunomodulatory agents utilized in the treatment of metastatic melanoma have been linked to vitiligolike depigmentation. In those receiving PD-1 immunotherapy for metastatic melanoma, vitiligolike lesions have been reported in 15% to 25% of patients.6 Typically, the PD-1 molecule has a regulatory function on effector T cells. Interaction of the PD-1 receptor with its ligands occurs primarily in peripheral tissue causing apoptosis and downregulation of effector T cells with the goal of decreasing collateral damage to surrounding tissues by active T cells.5 In the tumor microenvironment, however, suppression of the host’s immune response is enhanced by aberrant stimulation of the PD-1 receptor, causing downregulation of the T-cell effector function, T-cell destruction, and apoptosis, which results in continued tumor growth. Nivolumab, a fully humanized monoclonal IgG4 antibody, selectively inhibits the PD-1 receptor, disrupting the regulator pathway that would typically end in T-cell destruction.5 Accordingly, the population of active T cells is increased along with the antitumor response.4,10 Nivolumab exhibits success as an immunotherapeutic agent, with an overall survival rate in patients with metastatic melanoma undergoing nivolumab therapy of 41% to 42% at 3 years and 35% at 5 years.11 However, therapeutic manipulation of the host’s immune response does not come without a cost. Vitiligolike lesions have been reported in up to a quarter of patients receiving PD-1 immunotherapy for metastatic melanoma.6
The relationship between vitiligolike depigmentation and melanoma can be explained by the immune activation against antigens associated with melanoma that also are expressed by normal melanocytes. In clinical observations of patients with melanoma and patients with vitiligo, antibodies to human melanocyte antigens were present in 80% (24/30) of patients vs 7% (2/28) in the control group.12 The autoimmune response results from a cross-reaction of melanoma cells that share the same antigens as normal melanocytes, such as melanoma antigen recognized by T cells 1 (MART-1), gp100, and tyrosinase.13,14
Development of vitiligolike depigmentation in patients with metastatic melanoma treated with nivolumab has been reported to occur between 2 and 15 months after the start of PD-1 therapy. This side effect of treatment correlates with favorable clinical outcomes.15,16 Enhancing immune recognition of melanocytes in patients with melanoma confers a survival advantage, as studies by Koh et al17 and Norlund et al18 involving patients who developed vitiligolike hypopigmentation associated with malignant melanoma indicated a better prognosis than for those without hypopigmentation. The 5-year survival rate of patients with both malignant melanoma and vitiligo was reported as 60% to 67% when it was estimated that only 30% to 50% of patients should have survived that duration of time.17,18 Similarly, a systematic review of patients with melanoma stages III and IV reported that those with associated hypopigmentation had a 2- to 4-fold decreased risk of disease progression and death compared to patients without depigmentation.7
Use of traditional treatment therapies for vitiligo is based on the ability of the therapy to suppress the immune system. However, in patients with metastatic melanoma undergoing immune-modulating cancer therapies, traditional treatment options may counter the antitumor effects of the targeted immunotherapies and should be used with caution. Our patient displayed improvement in the appearance of her starkly contrasting hypopigmented patches with the use of hydroquinone cream 4%, which induced necrotic death of melanocytes by inhibiting the conversion of L-3,4-dihydroxyphenylalanine to melanin by tyrosinase.19 The effect achieved by using topical hydroquinone 4% was a lighter skin appearance in areas of application.
There is no cure for vitiligo, and although it is a benign condition, it can negatively impact a patient's quality of life. In some countries, vitiligo is confused with leprosy, resulting in a social stigma attached to the diagnosis. Many patients are frightened or embarrassed by the diagnosis of vitiligo and its effects, and they often experience discrimination.2 Patients with vitiligo also experience more psychological difficulties such as depression.20 The unpredictability of vitiligo is associated with negative emotions including fear of spreading the lesions, shame, insecurity, and sadness.21 Supportive care measures, including psychological support and counseling, are recommended. Additionally, upon initiation of anti–PD-1 therapies, expectations should be discussed with patients concerning the possibilities of depigmentation and associated treatment results. Although the occurrence of vitiligo may cause the patient concern, it should be communicated that its presence is a positive indicator of a vigorous antimelanoma immunity and an increased survival rate.7
Vitiligolike depigmentation is a known rare adverse effect of nivolumab treatment. Although aesthetically unfavorable for the patient, the development of vitiligolike lesions while undergoing immunotherapy for melanoma may be a sign of a promising clinical outcome due to an effective immune response to melanoma antigens. Our patient remains in remission without any evidence of melanoma after 9 months of therapy, which offers support for a promising outcome for melanoma patients who experience vitiligolike depigmentation.
To the Editor:
Vitiligolike depigmentation has been known to develop around the sites of origin of melanoma or more rarely in patients treated with antimelanoma therapy.1 Vitiligo is characterized by white patchy depigmentation of the skin caused by the loss of functional melanocytes from the epidermis. The exact mechanisms of disease are unknown and multifactorial; however, autoimmunity plays a central role. Interferon gamma (IFN-γ), C-X-C chemokine ligand 10, and IL-22 have been identified as key mediators in an inflammatory cascade leading to the stimulation of the innate immune response against melanocyte antigens.2,3 Research suggests melanoma-associated vitiligolike leukoderma also results from an immune reaction directed against antigenic determinants shared by both normal and malignant melanocytes.3 Vitiligolike lesions have been associated with the use of immunomodulatory agents such as nivolumab, a fully humanized monoclonal IgG4 antibody, which blocks the programmed cell death protein 1 (PD-1) receptor that normally is expressed on T cells during the effector phase of T-cell activation.4,5 In the tumor microenvironment, the PD-1 receptor is stimulated, leading to downregulation of the T-cell effector function and destruction of T cells.5 Due to T-cell apoptosis and consequent suppression of the immune response, tumorigenesis continues. By inhibiting the PD-1 receptor, nivolumab increases the number of active T cells and antitumor response. However, the distressing side effect of vitiligolike depigmentation has been reported in 15% to 25% of treated patients.6
In a meta-analysis by Teulings et al,7 patients with new-onset vitiligo and malignant melanoma demonstrated a 2-fold decrease in cancer progression and a 4-fold decreased risk for death vs patients without vitiligo development. Thus, in patients with melanoma, vitiligolike depigmentation should be considered a good prognostic indicator as well as a visible sign of spontaneous or therapy-induced antihumoral immune response against melanocyte differentiation antigens, as it is associated with a notable survival benefit in patients receiving immunotherapy for metastatic melanoma.3 We describe a case of diffuse vitiligolike depigmentation that developed suddenly during nivolumab treatment, causing much distress to the patient.
A 75-year-old woman presented to the clinic with a chief concern of sudden diffuse skin discoloration primarily affecting the face, hands, and extremities of 3 weeks’ duration. She had a medical history of metastatic melanoma—the site of the primary melanoma was never identified—and she was undergoing immune-modulating therapy with nivolumab. She was on her fifth month of treatment and was experiencing a robust therapeutic response with a reported 100% clearance of the metastatic melanoma as observed on a positron emission tomography scan. The patchy depigmentation of skin was causing her much distress. Physical examination revealed diffuse patches of hypopigmentation on the trunk, face, and extremities (Figure). Shave biopsies of the right lateral arm demonstrated changes consistent with vitiligo, with an adjacent biopsy illustrating normal skin characteristics. Triamcinolone ointment 0.1% was initiated, with instruction to apply it to affected areas twice daily for 2 weeks. However, there was no improvement, and she discontinued use.
At 3-month follow-up, the depigmentation persisted, prompting a trial of hydroquinone cream 4% to be used sparingly in cosmetically sensitive areas such as the face and dorsal aspects of the hands. Additionally, diligent photoprotection was advised. Upon re-evaluation 9 months later, the patient remained in cancer remission, continued nivolumab therapy, and reported improvement in the hypopigmentation with a more even skin color with topical hydroquinone use. She no longer noticed starkly contrasting hypopigmented patches.
Vitiligo is a benign skin condition characterized by white depigmented macules and patches. The key feature of the disorder is loss of functional melanocytes from the cutaneous epidermis and sometimes from the hair follicles, with various theories on the cause. It has been suggested that the disease is multifactorial, involving both genetics and environmental factors.2 Regardless of the exact mechanism, the result is always the same: loss of melanin pigment in cells due to loss of melanocytes.
Autoimmunity plays a central role in the causation of vitiligo and was first suspected as a possible cause due to the association of vitiligo with several other autoimmune disorders, such as thyroiditis.8 An epidemiological survey from the United Kingdom and North America (N=2624) found that 19.4% of vitiligo patients aged 20 years or older also reported a clinical history of autoimmune thyroid disease compared with 2.4% of the overall White population of the same age.9 Interferon gamma, C-X-C chemokine ligand 10, and IL-22 receptors stimulate the innate immune response, resulting in an overactive danger signaling cascade, which leads to proinflammatory signals against melanocyte antigens.2,3 The adaptive immune system also participates in the progression of vitiligo by activating dermal dendritic cells to attack melanocytes along with melanocyte-specific cytotoxic T cells.
Immunomodulatory agents utilized in the treatment of metastatic melanoma have been linked to vitiligolike depigmentation. In those receiving PD-1 immunotherapy for metastatic melanoma, vitiligolike lesions have been reported in 15% to 25% of patients.6 Typically, the PD-1 molecule has a regulatory function on effector T cells. Interaction of the PD-1 receptor with its ligands occurs primarily in peripheral tissue causing apoptosis and downregulation of effector T cells with the goal of decreasing collateral damage to surrounding tissues by active T cells.5 In the tumor microenvironment, however, suppression of the host’s immune response is enhanced by aberrant stimulation of the PD-1 receptor, causing downregulation of the T-cell effector function, T-cell destruction, and apoptosis, which results in continued tumor growth. Nivolumab, a fully humanized monoclonal IgG4 antibody, selectively inhibits the PD-1 receptor, disrupting the regulator pathway that would typically end in T-cell destruction.5 Accordingly, the population of active T cells is increased along with the antitumor response.4,10 Nivolumab exhibits success as an immunotherapeutic agent, with an overall survival rate in patients with metastatic melanoma undergoing nivolumab therapy of 41% to 42% at 3 years and 35% at 5 years.11 However, therapeutic manipulation of the host’s immune response does not come without a cost. Vitiligolike lesions have been reported in up to a quarter of patients receiving PD-1 immunotherapy for metastatic melanoma.6
The relationship between vitiligolike depigmentation and melanoma can be explained by the immune activation against antigens associated with melanoma that also are expressed by normal melanocytes. In clinical observations of patients with melanoma and patients with vitiligo, antibodies to human melanocyte antigens were present in 80% (24/30) of patients vs 7% (2/28) in the control group.12 The autoimmune response results from a cross-reaction of melanoma cells that share the same antigens as normal melanocytes, such as melanoma antigen recognized by T cells 1 (MART-1), gp100, and tyrosinase.13,14
Development of vitiligolike depigmentation in patients with metastatic melanoma treated with nivolumab has been reported to occur between 2 and 15 months after the start of PD-1 therapy. This side effect of treatment correlates with favorable clinical outcomes.15,16 Enhancing immune recognition of melanocytes in patients with melanoma confers a survival advantage, as studies by Koh et al17 and Norlund et al18 involving patients who developed vitiligolike hypopigmentation associated with malignant melanoma indicated a better prognosis than for those without hypopigmentation. The 5-year survival rate of patients with both malignant melanoma and vitiligo was reported as 60% to 67% when it was estimated that only 30% to 50% of patients should have survived that duration of time.17,18 Similarly, a systematic review of patients with melanoma stages III and IV reported that those with associated hypopigmentation had a 2- to 4-fold decreased risk of disease progression and death compared to patients without depigmentation.7
Use of traditional treatment therapies for vitiligo is based on the ability of the therapy to suppress the immune system. However, in patients with metastatic melanoma undergoing immune-modulating cancer therapies, traditional treatment options may counter the antitumor effects of the targeted immunotherapies and should be used with caution. Our patient displayed improvement in the appearance of her starkly contrasting hypopigmented patches with the use of hydroquinone cream 4%, which induced necrotic death of melanocytes by inhibiting the conversion of L-3,4-dihydroxyphenylalanine to melanin by tyrosinase.19 The effect achieved by using topical hydroquinone 4% was a lighter skin appearance in areas of application.
There is no cure for vitiligo, and although it is a benign condition, it can negatively impact a patient's quality of life. In some countries, vitiligo is confused with leprosy, resulting in a social stigma attached to the diagnosis. Many patients are frightened or embarrassed by the diagnosis of vitiligo and its effects, and they often experience discrimination.2 Patients with vitiligo also experience more psychological difficulties such as depression.20 The unpredictability of vitiligo is associated with negative emotions including fear of spreading the lesions, shame, insecurity, and sadness.21 Supportive care measures, including psychological support and counseling, are recommended. Additionally, upon initiation of anti–PD-1 therapies, expectations should be discussed with patients concerning the possibilities of depigmentation and associated treatment results. Although the occurrence of vitiligo may cause the patient concern, it should be communicated that its presence is a positive indicator of a vigorous antimelanoma immunity and an increased survival rate.7
Vitiligolike depigmentation is a known rare adverse effect of nivolumab treatment. Although aesthetically unfavorable for the patient, the development of vitiligolike lesions while undergoing immunotherapy for melanoma may be a sign of a promising clinical outcome due to an effective immune response to melanoma antigens. Our patient remains in remission without any evidence of melanoma after 9 months of therapy, which offers support for a promising outcome for melanoma patients who experience vitiligolike depigmentation.
- de Golian E, Kwong BY, Swetter SM, et al. Cutaneous complications of targeted melanoma therapy. Curr Treat Options Oncol. 2016;17:57.
- Ezzedine K, Eleftheriadou V, Whitton M, et al. Vitiligo. Lancet. 2015;386:74-84.
- Ortonne, JP, Passeron, T. Vitiligo and other disorders of hypopigmentation. In: Bolognia J, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1087-1114.
- Opdivo. Package insert. Bristol-Myers Squibb Company; 2023.
- Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19:5300-5309.
- Hwang SJE, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74:455-461.e1.
- Teulings HE, Limpens J, Jansen SN, et al. Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta-analysis. J Clin Oncol. 2015;33:773-781.
- Gey A, Diallo A, Seneschal J, et al. Autoimmune thyroid disease in vitiligo: multivariate analysis indicates intricate pathomechanisms. Br J Dermatol. 2013;168:756-761.
- Alkhateeb A, Fain PR, Thody A, et al. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. 2003;16:208-214.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
- Hodi FS, Kluger H, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Cancer Res. 2016;76(14 suppl):CT001.
- Cui J, Bystryn JC. Melanoma and vitiligo are associated with antibody responses to similar antigens on pigment cells. Arch Dermatol. 1995;131:314-318.
- Lynch SA, Bouchard BN, Vijayasaradhi S, et al. Antigens of melanocytes and melanoma. Cancer Metastasis Rev. 1991;10:141-150.
- Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab cutaneous adverse events and their association with disease progression. JAMA Dermatol. 2015;15:1206-1212.
- Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45-51.
- Nakamura Y, Tanaka R, Asami Y, et al. Correlation between vitiligo occurrence and clinical benefit in advanced melanoma patients treated with nivolumab: a multi-institutional retrospective study. J Dermatol. 2017;44:117-122.
- Koh HK, Sober AJ, Nakagawa H, et al. Malignant melanoma and vitiligo-like leukoderma: an electron microscope study. J Am Acad Dermatol. 1983;9:696-708.
- Nordlund JJ, Kirkwood JM, Forget BM, et al. Vitiligo in patients with metastatic melanoma: a good prognostic sign. J Am Acad Dermatol. 1983;9:689-696.
- Palumbo A, d’Ischia M, Misuraca G, et al. Mechanism of inhibition of melanogenesis by hydroquinone. Biochim Biophys Acta. 1991;1073:85-90.
- Lai YC, Yew YW, Kennedy C, et al. Vitiligo and depression: a systematic review and meta-analysis of observational studies. Br J Dermatol. 2017;177:708-718.
- Nogueira LSC, Zancanaro PCQ, Azambuja RD. Vitiligo and emotions. An Bras Dermatol. 2009;84:41-45.
- de Golian E, Kwong BY, Swetter SM, et al. Cutaneous complications of targeted melanoma therapy. Curr Treat Options Oncol. 2016;17:57.
- Ezzedine K, Eleftheriadou V, Whitton M, et al. Vitiligo. Lancet. 2015;386:74-84.
- Ortonne, JP, Passeron, T. Vitiligo and other disorders of hypopigmentation. In: Bolognia J, Schaffer JV, Cerroni L, eds. Dermatology. 4th ed. Elsevier; 2018:1087-1114.
- Opdivo. Package insert. Bristol-Myers Squibb Company; 2023.
- Ott PA, Hodi FS, Robert C. CTLA-4 and PD-1/PD-L1 blockade: new immunotherapeutic modalities with durable clinical benefit in melanoma patients. Clin Cancer Res. 2013;19:5300-5309.
- Hwang SJE, Carlos G, Wakade D, et al. Cutaneous adverse events (AEs) of anti-programmed cell death (PD)-1 therapy in patients with metastatic melanoma: a single-institution cohort. J Am Acad Dermatol. 2016;74:455-461.e1.
- Teulings HE, Limpens J, Jansen SN, et al. Vitiligo-like depigmentation in patients with stage III-IV melanoma receiving immunotherapy and its association with survival: a systematic review and meta-analysis. J Clin Oncol. 2015;33:773-781.
- Gey A, Diallo A, Seneschal J, et al. Autoimmune thyroid disease in vitiligo: multivariate analysis indicates intricate pathomechanisms. Br J Dermatol. 2013;168:756-761.
- Alkhateeb A, Fain PR, Thody A, et al. Epidemiology of vitiligo and associated autoimmune diseases in Caucasian probands and their families. Pigment Cell Res. 2003;16:208-214.
- Robert C, Long GV, Brady B, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320-330.
- Hodi FS, Kluger H, Sznol M, et al. Durable, long-term survival in previously treated patients with advanced melanoma who received nivolumab monotherapy in a phase I trial. Cancer Res. 2016;76(14 suppl):CT001.
- Cui J, Bystryn JC. Melanoma and vitiligo are associated with antibody responses to similar antigens on pigment cells. Arch Dermatol. 1995;131:314-318.
- Lynch SA, Bouchard BN, Vijayasaradhi S, et al. Antigens of melanocytes and melanoma. Cancer Metastasis Rev. 1991;10:141-150.
- Sanlorenzo M, Vujic I, Daud A, et al. Pembrolizumab cutaneous adverse events and their association with disease progression. JAMA Dermatol. 2015;15:1206-1212.
- Hua C, Boussemart L, Mateus C, et al. Association of vitiligo with tumor response in patients with metastatic melanoma treated with pembrolizumab. JAMA Dermatol. 2016;152:45-51.
- Nakamura Y, Tanaka R, Asami Y, et al. Correlation between vitiligo occurrence and clinical benefit in advanced melanoma patients treated with nivolumab: a multi-institutional retrospective study. J Dermatol. 2017;44:117-122.
- Koh HK, Sober AJ, Nakagawa H, et al. Malignant melanoma and vitiligo-like leukoderma: an electron microscope study. J Am Acad Dermatol. 1983;9:696-708.
- Nordlund JJ, Kirkwood JM, Forget BM, et al. Vitiligo in patients with metastatic melanoma: a good prognostic sign. J Am Acad Dermatol. 1983;9:689-696.
- Palumbo A, d’Ischia M, Misuraca G, et al. Mechanism of inhibition of melanogenesis by hydroquinone. Biochim Biophys Acta. 1991;1073:85-90.
- Lai YC, Yew YW, Kennedy C, et al. Vitiligo and depression: a systematic review and meta-analysis of observational studies. Br J Dermatol. 2017;177:708-718.
- Nogueira LSC, Zancanaro PCQ, Azambuja RD. Vitiligo and emotions. An Bras Dermatol. 2009;84:41-45.
Practice Points
- New-onset vitiligo coinciding with malignant melanoma should be considered a good prognostic indicator.
- Daily use of hydroquinone cream 4% in conjunction with diligent photoprotection was shown to even overall skin tone in a patient experiencing leukoderma from nivolumab therapy.

Collision Course of a Basal Cell Carcinoma and Apocrine Hidrocystoma on the Scalp
To the Editor:
A collision tumor is the coexistence of 2 discrete tumors in the same neoplasm, possibly comprising a malignant tumor and a benign tumor, and thereby complicating appropriate diagnosis and treatment. We present a case of a basal cell carcinoma (BCC) of the scalp that was later found to be in collision with an apocrine hidrocystoma that might have arisen from a nevus sebaceus. Although rare, BCC can coexist with apocrine hidrocystoma. Jayaprakasam and Rene1 reported a case of a collision tumor containing BCC and hidrocystoma on the eyelid.1 We present a case of a BCC on the scalp that was later found to be in collision with an apocrine hidrocystoma that possibly arose from a nevus sebaceus.
.")
A 92-year-old Black woman with a biopsy-confirmed primary BCC of the left parietal scalp presented for Mohs micrographic surgery. The pathology report from an outside facility was reviewed. The initial diagnosis had been made with 2 punch biopsies from separate areas of the large nodule—one consistent with nodular and pigmented BCC (Figure 1), and the other revealed nodular ulcerated BCC. Physical examination prior to Mohs surgery revealed a mobile, flesh-colored, 6.2×6.0-cm nodule with minimal overlying hair on the left parietal scalp (Figure 2). During stage-I processing by the histopathology laboratory, large cystic structures were encountered; en face frozen sections showed a cystic tumor. Excised tissue was submitted for permanent processing to aid in diagnosis; the initial diagnostic biopsy slides were requested from the outside facility for review.

The initial diagnostic biopsy slides were reviewed and found to be consistent with nodular and pigmented BCC, as previously reported. Findings from hematoxylin and eosin staining of tissue obtained from Mohs sections were consistent with a combined neoplasm comprising BCC (Figure 3A) and apocrine hidrocystoma (Figure 3B). In addition, one section was characterized by acanthosis, papillomatosis, and sebaceous glands—similar to findings that are seen in a nevus sebaceus (Figure 3C).
")
The BCC was cleared after stage I; the final wound size was 7×6.6 cm. Although benign apocrine hidrocystoma was still evident at the margin, further excision was not performed at the request of the patient and her family. Partial primary wound closure was performed with pulley sutures. A xenograft was placed over the unclosed central portion. The wound was permitted to heal by second intention.
The clinical differential diagnosis of a scalp nodule includes a pilar cyst, BCC, squamous cell carcinoma, melanoma, cutaneous metastasis, adnexal tumor, atypical fibroxanthoma, and collision tumor. A collision tumor—the association of 2 or more benign or malignant neoplasms—represents a well-known pitfall in making a correct clinical and pathologic diagnosis.2 Many theories have been proposed to explain the pathophysiology of collision tumors. Some authors have speculated that they arise from involvement of related cell types.1 Other theories include induction by cytokines and growth factors secreted from one tumor that provides an ideal environment for proliferation of other cell types, a field cancerization effect of sun-damaged skin, or a coincidence.2
In our case, it is possible that the 2 tumors arose from a nevus sebaceus. One retrospective study of 706 cases of nevus sebaceus (707 specimens) found that 22.5% of cases developed secondary proliferation; of those cases, 18.9% were benign.3 Additionally, in 4.2% of cases of nevus sebaceus, proliferation of 2 or more tumors developed. The most common malignant neoplasm to develop from nevus sebaceus was BCC, followed by squamous cell carcinoma and sebaceous carcinoma. The most common benign neoplasm to develop from nevus sebaceus was trichoblastoma, followed by syringocystadenoma papilliferum.3
Our case highlights the possibility of a sampling error when performing a biopsy of any large neoplasm. Additionally, Mohs surgeons should maintain high clinical suspicion for collision tumors when encountering a large tumor with pathology inconsistent with the original biopsy. Apocrine hidrocystoma should be considered in the differential diagnosis of a large cystic mass of the scalp. Also, it is important to recognize that malignant lesions, such as BCC, can coexist with another benign tumor. Basal cell carcinoma is rare in Black patients, supporting our belief that our patient’s tumors arose from a nevus sebaceus.
It also is important for Mohs surgeons to consider any potential discrepancy between the initial pathology report and Mohs intraoperative pathology that can impact diagnosis, the aggressiveness of the tumors identified, and how such aggressiveness may affect management options.4,5 Some dermatology practices request biopsy slides from patients who are referred for Mohs micrographic surgery for internal review by a dermatopathologist before surgery is performed; however, this protocol requires additional time and adds costs for the overall health care system.4 One study found that internal review of outside biopsy slides resulted in a change in diagnosis in 2.2% of patients (N=3345)—affecting management in 61% of cases in which the diagnosis was changed.4 Another study (N=163) found that the reported aggressiveness of 50.5% of nonmelanoma cases in an initial biopsy report was changed during Mohs micrographic surgery.5 Mohs surgeons should be aware that discrepancies can occur, and if a discrepancy is discovered, the procedure may be paused until the initial biopsy slide is reviewed and further information is collected.
- Jayaprakasam A, Rene C. A benign or malignant eyelid lump—can you tell? an unusual collision tumour highlighting the difficulty differentiating a hidrocystoma from a basal cell carcinoma. BMJ Case Reports. 2012;2012:bcr1220115307. doi:10.1136/bcr.12.2011.5307
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603. doi:10.1097/DAD.0b013e3181a88116
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337. doi:10.1016/j.jaad.2013.10.004
- Butler ST, Youker SR, Mandrell J, et al. The importance of reviewing pathology specimens before Mohs surgery. Dermatol Surg. 2009;35:407-412. doi:10.1111/j.1524-4725.2008.01056.x
- Stiegel E, Lam C, Schowalter M, et al. Correlation between original biopsy pathology and Mohs intraoperative pathology. Dermatol Surg. 2018;44:193-197. doi:10.1097/DSS.0000000000001276
To the Editor:
A collision tumor is the coexistence of 2 discrete tumors in the same neoplasm, possibly comprising a malignant tumor and a benign tumor, and thereby complicating appropriate diagnosis and treatment. We present a case of a basal cell carcinoma (BCC) of the scalp that was later found to be in collision with an apocrine hidrocystoma that might have arisen from a nevus sebaceus. Although rare, BCC can coexist with apocrine hidrocystoma. Jayaprakasam and Rene1 reported a case of a collision tumor containing BCC and hidrocystoma on the eyelid.1 We present a case of a BCC on the scalp that was later found to be in collision with an apocrine hidrocystoma that possibly arose from a nevus sebaceus.
A 92-year-old Black woman with a biopsy-confirmed primary BCC of the left parietal scalp presented for Mohs micrographic surgery. The pathology report from an outside facility was reviewed. The initial diagnosis had been made with 2 punch biopsies from separate areas of the large nodule—one consistent with nodular and pigmented BCC (Figure 1), and the other revealed nodular ulcerated BCC. Physical examination prior to Mohs surgery revealed a mobile, flesh-colored, 6.2×6.0-cm nodule with minimal overlying hair on the left parietal scalp (Figure 2). During stage-I processing by the histopathology laboratory, large cystic structures were encountered; en face frozen sections showed a cystic tumor. Excised tissue was submitted for permanent processing to aid in diagnosis; the initial diagnostic biopsy slides were requested from the outside facility for review.
The initial diagnostic biopsy slides were reviewed and found to be consistent with nodular and pigmented BCC, as previously reported. Findings from hematoxylin and eosin staining of tissue obtained from Mohs sections were consistent with a combined neoplasm comprising BCC (Figure 3A) and apocrine hidrocystoma (Figure 3B). In addition, one section was characterized by acanthosis, papillomatosis, and sebaceous glands—similar to findings that are seen in a nevus sebaceus (Figure 3C).
The BCC was cleared after stage I; the final wound size was 7×6.6 cm. Although benign apocrine hidrocystoma was still evident at the margin, further excision was not performed at the request of the patient and her family. Partial primary wound closure was performed with pulley sutures. A xenograft was placed over the unclosed central portion. The wound was permitted to heal by second intention.
The clinical differential diagnosis of a scalp nodule includes a pilar cyst, BCC, squamous cell carcinoma, melanoma, cutaneous metastasis, adnexal tumor, atypical fibroxanthoma, and collision tumor. A collision tumor—the association of 2 or more benign or malignant neoplasms—represents a well-known pitfall in making a correct clinical and pathologic diagnosis.2 Many theories have been proposed to explain the pathophysiology of collision tumors. Some authors have speculated that they arise from involvement of related cell types.1 Other theories include induction by cytokines and growth factors secreted from one tumor that provides an ideal environment for proliferation of other cell types, a field cancerization effect of sun-damaged skin, or a coincidence.2
In our case, it is possible that the 2 tumors arose from a nevus sebaceus. One retrospective study of 706 cases of nevus sebaceus (707 specimens) found that 22.5% of cases developed secondary proliferation; of those cases, 18.9% were benign.3 Additionally, in 4.2% of cases of nevus sebaceus, proliferation of 2 or more tumors developed. The most common malignant neoplasm to develop from nevus sebaceus was BCC, followed by squamous cell carcinoma and sebaceous carcinoma. The most common benign neoplasm to develop from nevus sebaceus was trichoblastoma, followed by syringocystadenoma papilliferum.3
Our case highlights the possibility of a sampling error when performing a biopsy of any large neoplasm. Additionally, Mohs surgeons should maintain high clinical suspicion for collision tumors when encountering a large tumor with pathology inconsistent with the original biopsy. Apocrine hidrocystoma should be considered in the differential diagnosis of a large cystic mass of the scalp. Also, it is important to recognize that malignant lesions, such as BCC, can coexist with another benign tumor. Basal cell carcinoma is rare in Black patients, supporting our belief that our patient’s tumors arose from a nevus sebaceus.
It also is important for Mohs surgeons to consider any potential discrepancy between the initial pathology report and Mohs intraoperative pathology that can impact diagnosis, the aggressiveness of the tumors identified, and how such aggressiveness may affect management options.4,5 Some dermatology practices request biopsy slides from patients who are referred for Mohs micrographic surgery for internal review by a dermatopathologist before surgery is performed; however, this protocol requires additional time and adds costs for the overall health care system.4 One study found that internal review of outside biopsy slides resulted in a change in diagnosis in 2.2% of patients (N=3345)—affecting management in 61% of cases in which the diagnosis was changed.4 Another study (N=163) found that the reported aggressiveness of 50.5% of nonmelanoma cases in an initial biopsy report was changed during Mohs micrographic surgery.5 Mohs surgeons should be aware that discrepancies can occur, and if a discrepancy is discovered, the procedure may be paused until the initial biopsy slide is reviewed and further information is collected.
To the Editor:
A collision tumor is the coexistence of 2 discrete tumors in the same neoplasm, possibly comprising a malignant tumor and a benign tumor, and thereby complicating appropriate diagnosis and treatment. We present a case of a basal cell carcinoma (BCC) of the scalp that was later found to be in collision with an apocrine hidrocystoma that might have arisen from a nevus sebaceus. Although rare, BCC can coexist with apocrine hidrocystoma. Jayaprakasam and Rene1 reported a case of a collision tumor containing BCC and hidrocystoma on the eyelid.1 We present a case of a BCC on the scalp that was later found to be in collision with an apocrine hidrocystoma that possibly arose from a nevus sebaceus.
A 92-year-old Black woman with a biopsy-confirmed primary BCC of the left parietal scalp presented for Mohs micrographic surgery. The pathology report from an outside facility was reviewed. The initial diagnosis had been made with 2 punch biopsies from separate areas of the large nodule—one consistent with nodular and pigmented BCC (Figure 1), and the other revealed nodular ulcerated BCC. Physical examination prior to Mohs surgery revealed a mobile, flesh-colored, 6.2×6.0-cm nodule with minimal overlying hair on the left parietal scalp (Figure 2). During stage-I processing by the histopathology laboratory, large cystic structures were encountered; en face frozen sections showed a cystic tumor. Excised tissue was submitted for permanent processing to aid in diagnosis; the initial diagnostic biopsy slides were requested from the outside facility for review.
The initial diagnostic biopsy slides were reviewed and found to be consistent with nodular and pigmented BCC, as previously reported. Findings from hematoxylin and eosin staining of tissue obtained from Mohs sections were consistent with a combined neoplasm comprising BCC (Figure 3A) and apocrine hidrocystoma (Figure 3B). In addition, one section was characterized by acanthosis, papillomatosis, and sebaceous glands—similar to findings that are seen in a nevus sebaceus (Figure 3C).
The BCC was cleared after stage I; the final wound size was 7×6.6 cm. Although benign apocrine hidrocystoma was still evident at the margin, further excision was not performed at the request of the patient and her family. Partial primary wound closure was performed with pulley sutures. A xenograft was placed over the unclosed central portion. The wound was permitted to heal by second intention.
The clinical differential diagnosis of a scalp nodule includes a pilar cyst, BCC, squamous cell carcinoma, melanoma, cutaneous metastasis, adnexal tumor, atypical fibroxanthoma, and collision tumor. A collision tumor—the association of 2 or more benign or malignant neoplasms—represents a well-known pitfall in making a correct clinical and pathologic diagnosis.2 Many theories have been proposed to explain the pathophysiology of collision tumors. Some authors have speculated that they arise from involvement of related cell types.1 Other theories include induction by cytokines and growth factors secreted from one tumor that provides an ideal environment for proliferation of other cell types, a field cancerization effect of sun-damaged skin, or a coincidence.2
In our case, it is possible that the 2 tumors arose from a nevus sebaceus. One retrospective study of 706 cases of nevus sebaceus (707 specimens) found that 22.5% of cases developed secondary proliferation; of those cases, 18.9% were benign.3 Additionally, in 4.2% of cases of nevus sebaceus, proliferation of 2 or more tumors developed. The most common malignant neoplasm to develop from nevus sebaceus was BCC, followed by squamous cell carcinoma and sebaceous carcinoma. The most common benign neoplasm to develop from nevus sebaceus was trichoblastoma, followed by syringocystadenoma papilliferum.3
Our case highlights the possibility of a sampling error when performing a biopsy of any large neoplasm. Additionally, Mohs surgeons should maintain high clinical suspicion for collision tumors when encountering a large tumor with pathology inconsistent with the original biopsy. Apocrine hidrocystoma should be considered in the differential diagnosis of a large cystic mass of the scalp. Also, it is important to recognize that malignant lesions, such as BCC, can coexist with another benign tumor. Basal cell carcinoma is rare in Black patients, supporting our belief that our patient’s tumors arose from a nevus sebaceus.
It also is important for Mohs surgeons to consider any potential discrepancy between the initial pathology report and Mohs intraoperative pathology that can impact diagnosis, the aggressiveness of the tumors identified, and how such aggressiveness may affect management options.4,5 Some dermatology practices request biopsy slides from patients who are referred for Mohs micrographic surgery for internal review by a dermatopathologist before surgery is performed; however, this protocol requires additional time and adds costs for the overall health care system.4 One study found that internal review of outside biopsy slides resulted in a change in diagnosis in 2.2% of patients (N=3345)—affecting management in 61% of cases in which the diagnosis was changed.4 Another study (N=163) found that the reported aggressiveness of 50.5% of nonmelanoma cases in an initial biopsy report was changed during Mohs micrographic surgery.5 Mohs surgeons should be aware that discrepancies can occur, and if a discrepancy is discovered, the procedure may be paused until the initial biopsy slide is reviewed and further information is collected.
- Jayaprakasam A, Rene C. A benign or malignant eyelid lump—can you tell? an unusual collision tumour highlighting the difficulty differentiating a hidrocystoma from a basal cell carcinoma. BMJ Case Reports. 2012;2012:bcr1220115307. doi:10.1136/bcr.12.2011.5307
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603. doi:10.1097/DAD.0b013e3181a88116
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337. doi:10.1016/j.jaad.2013.10.004
- Butler ST, Youker SR, Mandrell J, et al. The importance of reviewing pathology specimens before Mohs surgery. Dermatol Surg. 2009;35:407-412. doi:10.1111/j.1524-4725.2008.01056.x
- Stiegel E, Lam C, Schowalter M, et al. Correlation between original biopsy pathology and Mohs intraoperative pathology. Dermatol Surg. 2018;44:193-197. doi:10.1097/DSS.0000000000001276
- Jayaprakasam A, Rene C. A benign or malignant eyelid lump—can you tell? an unusual collision tumour highlighting the difficulty differentiating a hidrocystoma from a basal cell carcinoma. BMJ Case Reports. 2012;2012:bcr1220115307. doi:10.1136/bcr.12.2011.5307
- Miteva M, Herschthal D, Ricotti C, et al. A rare case of a cutaneous squamomelanocytic tumor: revisiting the histogenesis of combined neoplasms. Am J Dermatopathol. 2009;31:599-603. doi:10.1097/DAD.0b013e3181a88116
- Idriss MH, Elston DM. Secondary neoplasms associated with nevus sebaceus of Jadassohn: a study of 707 cases. J Am Acad Dermatol. 2014;70:332-337. doi:10.1016/j.jaad.2013.10.004
- Butler ST, Youker SR, Mandrell J, et al. The importance of reviewing pathology specimens before Mohs surgery. Dermatol Surg. 2009;35:407-412. doi:10.1111/j.1524-4725.2008.01056.x
- Stiegel E, Lam C, Schowalter M, et al. Correlation between original biopsy pathology and Mohs intraoperative pathology. Dermatol Surg. 2018;44:193-197. doi:10.1097/DSS.0000000000001276
PRACTICE POINTS
- When collision tumors are encountered during Mohs micrographic surgery, review of the initial diagnostic material is recommended.
- Permanent processing of Mohs excisions may be helpful in determining the diagnosis of the occult second tumor diagnosis.
