User login
Reports from the annual meeting of The Connective Tissue Oncology Society held in Rome, November 14-17, 2018 Sarcoma of the Year: Intimal Sarcoma
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
This year’s annual meeting of The Connective Tissue Oncology Society brought new insights on intimal sarcoma. Four studies in a featured session at the meeting examined both current and novel treatments for this rare and aggressive cancer, and emphasized the need for new therapies.
Anthracycline-based regimens as preferred first-line therapies
Anthracycline-based regimens were the preferred first-line therapies used in 83 adults with intimal sarcomas in a retrospective study of data from the World Sarcoma Network, reported by Anna Maria Frezza, MD, of the, Fondazione IRCCS Istituto Nazionale Tumori, Milan, Italy, and her colleagues. The researchers described the experience with anthracycline-based regimens as well as gemcitabine-based regimens and pazopanib among MDM2-positive patients with intimal sarcomas treated at 16 sarcoma reference centers in Europe, the United States, and Japan. Their findings speak to the need for new active drugs, which they said should target the MDM2 and CDK4 overexpression seen in patients with this rare sarcoma.Of the 83 patients studied, nearly all (76 patients) initially received an anthracycline-based regimen. Gemcitabine-based regimens were used in 29 patients and pazopanib in 10 patients; 20 of the 39 patients received more than one treatment.
Anthracycline-based regimens were associated with a 12-month progression-free survival rate of 38% in 76 patients with intimal sarcomas. All of the 76 patients received anthracycline regimens as their initial systemic therapy; 27 were treated for localized disease with a curative intent and the remaining 49 had advanced disease. The researchers also noted that anthracycline regimens were safely used in 22 patients with cardiac intimal sarcomas, as none of them died of cardiotoxicity.
Based on RECIST 1.1 measures, the overall response rate was 37% in 57 evaluable patients: 3 patients had a complete response, 18 had a partial response, 27 had stable disease, and 9 had progressive disease. For those with localized disease, the median time to progression was 14 months, and overall survival time was 51. For patients with advanced disease, the median time to progression was 8 months and overall survival was 22 months.
Outcomes were less favorable when patients were treated with gemcitabine regimens or pazopanib. In most of these cases, however, patients were either on their second (gemcitabine) or third (pazopanib) lines of therapy.
In the gemcitabine group, 2 patients were treated for localized disease with curative intent and 27 for advanced disease. Of 28 evaluable patients, best response was partial remission in 3, stable disease in 8, and progressive disease in 17. In the 27 patients with advanced disease, the median progression free survival time was 3 months and overall survival was 13 months.
All 10 patients in the pazopanib group had advanced disease and had undergone a median of two prior lines of therapy. One patient had a partial remission, 3 had stable disease, and 6 had progressive disease. The median progression free survival was 4 months and median overall survival was 12 months.
Rarest of the rare: Primary malignant sarcoma of the heart
Luke Smith, of the School of Clinical Medicine, University of Cambridge, U.K., detailed the experience of 28 patients diagnosed with sarcomas of the heart or great vessels at the university’s Royal Papworth Hospital and Addenbrooke’s Hospital between 2000 and 2018.
Based on this retrospective review, surgery offers the best chance for long-term survival for these patients, who would otherwise experience progressive heart failure and die. Adjuvant chemotherapy and radiation therapy might be able to extend their survival and improve symptomatic relief, he said, but these outcomes have not been prospectively studied.
Typically, the patients in this series, 20 with pulmonary artery sarcoma and 8 with cardiac sarcoma, presented with symptoms mimicking heart failure, pulmonary hypertension, or thromboembolic disease. Nearly all, 24 patients reported breathlessness. Eight patients had chest pain or tightness, 6 had cough, 6 had peripheral edema, 6 had constitutional symptoms, 3 had hemoptysis, and 1 had a TIA. Only 1 patient had a seriously impaired left ventricular ejection fraction of less than 30%. LVEF was normal at 55% or more in 16 patients, and moderately impaired at 30% or more in 10 patients.
Median overall survival was 17 months. The 19 patients who underwent surgical resection of their primary tumor survived much longer than the 10 patients who did not--median overall survival of 20 months vs. 9 months--but this finding may simply reflect more advanced disease in patients with inoperable disease. There were 3 perioperative deaths among the 19 patients who underwent surgery: 14 with pulmonary artery sarcomas had pulmonary endarterectomy and 4 with cardiac sarcomas underwent resection or maximal debulking of their tumors.
Based on the retrospective study, adjuvant chemotherapy and radiation were safe and may lead to better outcomes for these patients. Active chemotherapy regimens in the palliative setting included paclitaxel (angiosarcoma) and anthracycline ± ifosfamide.
Nine patients received post-surgical chemotherapy, and after completion five also had radiotherapy. The 3 cardiac sarcoma patients who had surgical resection with curative intent were treated with adjuvant ifosfamide-based chemotherapy (with close monitoring of fluid balance), and showed no evidence of disease on last follow-ups. One patient received post-operative paclitaxel following maximal debulking of a cardiac angiosarcoma.
Post-surgical anthracycline with and without ifosfamide were used in patients with pulmonary artery sarcomas with no clinical cardiotoxicity. Although the median overall survival for patients who received post-operative chemo- and radio-therapy was 28 months and the median overall survival with surgery alone was 9 months, the difference was not statistically significant.
In the palliative setting, partial responses were observed with paclitaxel and anthracycline (including liposomal doxorubicin) in patients with cardiac angiosarcoma. For pulmonary artery intimal sarcomas, partial responses were achieved with anthracycline with and without ifosfamide. Radiotherapy provided good local control.
The longest surviving pulmonary artery sarcoma patient, at 103 months, had pulmonary artery endarterectomy, followed by adjuvant epirubicin and radiotherapy. She developed lung metastases 7 years later and was treated with radiofrequency ablation. The longest surviving cardiac sarcoma patient, at 24 months, remains disease free. He had surgery to resect a high-grade undifferentiated sarcoma with involved margins, followed by adjuvant ifosfamide and radiotherapy to the right atrium.
Therapeutically exploitable genetic aberrations in intimal sarcomas
Imatinib and olaratumab might prove to be therapeutic approaches for some patients with intimal sarcomas, based on a retrospective evaluation of genetic aberrations in 11 patients with intimal sarcomas, Jason Roszik, PhD, MBA, reported at the meeting.
Dr. Roszik and his colleagues at the University of Texas MD Anderson Cancer Center, Houston, analyzed information on 11 patients with intimal sarcomas in the American Association for Cancer Research (AACR) project, Genomics Evidence Neoplasia Information Exchange (GENIE). Sampling was taken from the primary tumor in 8 patients and from the metastatic site in the other 3.
MDM2 amplifications were seen in 8 of 10 patients with available copy number alterations. Amplifications in the CDK pathway were present in 5, PDGFRA gain was seen in 4, and CDKN2A copy number loss was present in 3. Mutations that could be targeted with drugs included ALK, ATM/ATR, PTCH1 and PDGFRB, he said.
Unique genomic rearrangement events included PDE4DIP-NOTCH2 and MRPS30-ARID2 fusions. Co-occurring alterations included a NOTCH2 copy number gain in the PDE4DIP-NOTCH2 fusion tumor, and PDGFRB mutations in both fusion-positive cases.
The researchers also drew on the published findings of whole-exome sequencing and array-comparative genomic hybridization from an autopsy case of cardiac intimal sarcoma (Virchows Arch. 2017 Sep;471(3):423-428). That study identified concurrent PDGFRA amplification and PDGFRB mutation.
The researchers additionally examined clinical trial enrollments and could find no patient with intimal sarcoma among 406 sarcoma enrolled patients. Intimal sarcomas were not eligible for any clinical trial given the location of the tumors in major blood vessels.
“The somatic mutations and DNA copy number alterations in the PDGFR pathway relevant to the pathogenesis and potential targeted therapy of cardiac intimal sarcoma may be targeted by imatinib or olaratumab. Inclusion of such rare tumors in targeted therapy basket trials with a waiver for inclusion criteria is warranted,” Dr. Roszik and his colleagues concluded in the abstract of their presentation.
The promise of combination therapy
The “largest experience using multimodality therapy with proton based local therapy” for sarcomas involving the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries was reported by Yen-Lin E. Chen, MD, and her colleagues at Massachusetts General Hospital, Boston.
They examined an institutional sarcoma data repository of 13,950 patients and found 37 patients with sarcomas arising from the pericardium, myocardium, valves, pulmonary veins, or pulmonary arteries. These included 9 with unclassified pleomorphic sarcoma/malignant fibrous histiocytoma, 8 with angiosarcoma, 4 with spindle cell sarcoma, 4 with sarcoma not otherwise specified, 3 with leiomyosarcoma, 2 with osteosarcoma, 2 with Ewing sarcoma, and 1 each with chondrosarcoma, malignant peripheral nerve sheath tumor, rhabdomyosarcoma, synovial sarcoma, and intimal sarcoma.
Two-thirds of the patients had induction chemotherapy with or without maintenance therapy. Adriamycin, ifosfamide, and taxol therapies were most common. Two-thirds received proton based radiotherapy. Of the 23 patients who underwent resection, 11 were R2 (macroscopic positive margins), 3 were R1 (microscopic positive margins), and 9 were R0 (clear margins).
The 1-year overall survival rate was 64%, which fell to 37% at 3 years and to 28% at 5 years. Median survival was 28 months, twice that typically seen in the literature, Dr. Chen said.
For patients receiving proton based radiotherapy to a median dose of 64.8 GyRBE (range 63-72 GyRBE, 3 with additional intraoperative electrons), local failure free survivals were 80%, 64%, and 52% at 1, 3, and 5 years, respectively. For patients who did not receive radiotherapy, local failure free survival rates were 13%, 10%, 10%, respectively.
Overall, the 1, 3, and 5 year metastatic free survival rates were 25%, 14%, and 14%.
Survival rate was significantly better for patients with tumors smaller than 5 cm ( P =0.036), those over 40 years old ( P =0.028), those able to have surgery ( P =0.011), and those with non-angiosarcoma histologies ( P = 0.002).
Patterns of malignancies in patients with HIV-AIDS: a single institution observational study
India has the third largest HIV epidemic in the world because of its large population size, with 0.3% of the adult population infected with HIV. That translates to 2.1 million infected people, posing a significant challenge in the management of these individuals.1 In all, 43% of the infected are currently on highly active antiretroviral therapy (HAART).1 There has been a significant decrease in the number of HIV-AIDS–related deaths in recent years because of the remarkable increase in the use of antiretroviral therapy.2 However, the prolonged life expectancy in these patients has resulted in an increase in the risk of various new diseases such as cancers. With the complex interactions between altered immunity and infections, the risk of cancers is markedly increased in patients with HIV-AIDS.3 The spectrum of malignancies in this group of patients differs from that in the general population. In addition, the pattern and the magnitude of malignancies differ in different parts of the world.4 In this study, we have analyzed the pattern of malignancies in patients with HIV-AIDS in a regional cancer center in India. The aim of the study was to analyze the pattern of malignancies in patients with HIV-AIDS based on their age and sex and to document the CD4 counts at the time the malignancy was diagnosed.
Methods
We retrieved data from our institution’s medical records department on all patients who had HIV-AIDS and had been diagnosed with a malignancy. Data of all patients presenting with a malignancy and coexisting HIV-AIDS from January 2013 through December 2016 were analyzed initially. Only patients for whom there was a documented CD4 count were included in the final retrospective analysis. We analyzed the correlation between the patients’ CD4 counts and malignancies subclassified as AIDS-defining malignancies (ADMs; aggressive B-cell non-Hodgkin lymphoma [NHL] and cervical cancer) or non–AIDS-defining malignancies (NADMs; all other malignancies other than aggressive NHL and carcinoma cervix were defined as NADM). We also analyzed the correlation between the CD4 count and NHL and other malignancies. A statistical analysis was performed using SPSS Statistics for Windows, version 23 (IBM Corp, Armonk, NY). The independent sample Mann-Whitney U or Kruskal-Wallis tests were used for comparing the CD4 counts between the various subgroups of malignancies. The study was carried out in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines.
Results
A total of 370 patients who were diagnosed with malignancy and have coexisting HIV-AIDS were identified. In all, 85 patients were excluded because there were no CD4 counts available for them, and the remaining 285 patients were included in the final analysis. Of that total, 136 patients (48%) were men, and 149 (52%) were women.
The median age of the population was 44.8 years (5-80 years) at the time of diagnosis with malignancy. The mean CD4 count of the entire population was 235.4 cells/mm3 (50-734 cells/mm3). There were 104 patients with CD4 counts of ≤200 cells/mm3, and 181 patients had CD4 counts of >200 cells/mm3 (Table 1). All patients received the HAART regimen, efavirenz-lamuvidine-tenofovir (600 mg/300 mg/300 mg Telura).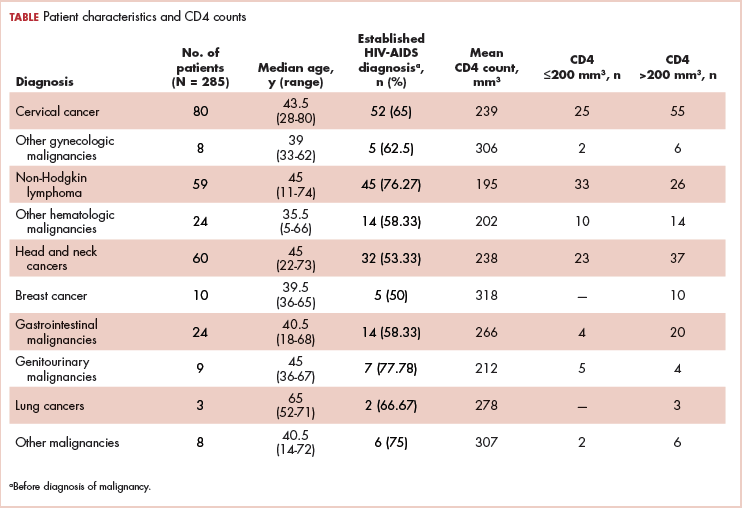
The most common malignancies in this population were gynecologic malignancies, followed by hematologic malignancies. Cervical cancer was the most common malignancy among women as well as in the overall study population. Among men, the most common malignancy was NHL. The second and third most common malignancies in men were carcinoma oral cavity and carcinoma oropharynx, respectively, whereas in women, they were NHL and breast cancer. The distribution of various hematologic, head and neck, and gastrointestinal malignancies in this group of patients is shown in Figures 1, 2, and 3.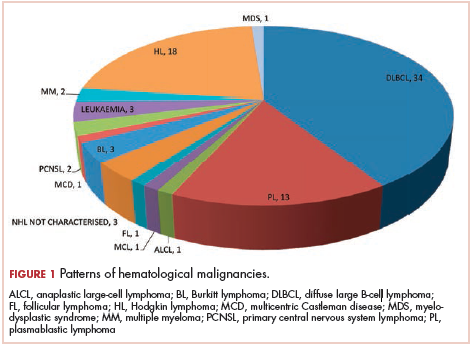
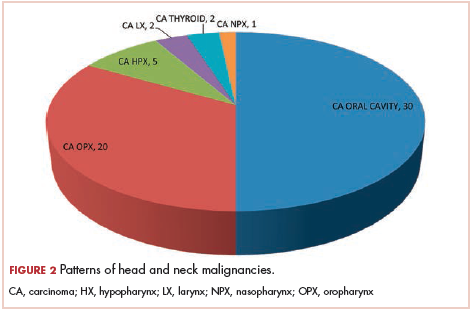
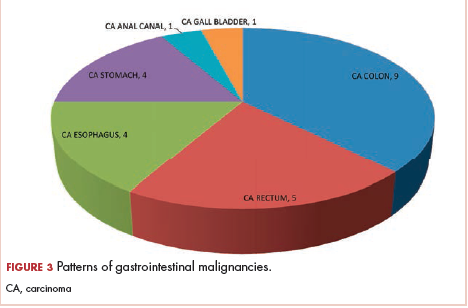
The ADMs in the study were NHL, including 2 patients diagnosed with primary central nervous system (CNS) lymphomas, and cervical cancer. No case of Kaposi sarcoma, also considered an ADM, was identified in this study. The common NADMs include head and neck malignancies (Figure 2), gastrointestinal malignancies (Figure 3), gynecological and genitourinary malignancies, and breast cancer. The mean CD4 count in the ADM subgroup was 221 cells/mm3, and in the NADM subgroup, it was 250 cells/mm3. There was a significant difference in the distribution of CD4 counts between the ADM and NADM subgroups (P = .03; Mann-Whitney U test). A statistical difference was also noted when the CD4 counts of the patients with NHL were compared with other malignancies (P = .0001; Mann-Whitney U test) There was no statistically significant difference noted when CD4 counts of patients with cervical cancer were compared with NADMs (P = .914).
Discussion
In 2015, a report from the Indian government estimated the prevalence of HIV in the country as 0.26% (0.22%-0.32%).5 The report also noted a decreasing trend in the number of new cases of HIV diagnosed and a decrease in the number of AIDS-related deaths.5 The decrease in deaths from AIDS is primarily attributed to the widespread use of HAART. With the introduction of HAART therapy, the survival of patients diagnosed with HIV-AIDS has increased markedly.6 However, newer challenges have emerged with improved survival, such as an increasing number of patients being diagnosed with malignancies. In the current HAART era, the pattern of malignancies in people living with HIV-AIDS has changed compared with the pre-HAART era.7 The literature suggests that worldwide, malignancies are encountered in about 30% patients with HIV-AIDS, but that percentage differs sharply from that encountered in India, where it is less than 5%.8 This may partly be explained by opportunistic infections such as tuberculosis in Indian patients, which remains the leading cause of death in the HIV-AIDS population. In our study, we retrospectively analyzed the pattern of malignancies in patients with HIV-AIDS.
Although few studies have quoted NHL as the predominant malignancy in their patients with HIV-AIDS, the predominant malignancy was cervical cancer in our patient population, as seen in few other studies.8-10 Head and neck malignancies also continue to be common malignancies in men with HIV-AIDS.10 Thus, an increase in malignancies induced by the human papillomavirus (HPV) can be seen in this group of patients. Only a few pediatric malignancies were noted in our study, and all of those patients had a vertical transmission of HIV.
Kaposi sarcoma is quite rare in the Indian population, and no case of Kaposi sarcoma was diagnosed in our study population. A similar finding was seen in several earlier publications from India. In the largest published series from India by Dhir and colleagues, evaluating 251 patients with HIV-AIDS and malignancy, no case of Kaposi sarcoma was reported.10 The authors mentioned that this finding might be because of the low seroprevalence of Kaposi sarcoma-associated herpesvirus in the Asian population.10 Three different studies from southern India have also not reported the incidence of Kaposi Sarcoma in their series of HIV-AIDS patients with malignancies,11-13 and similar findings were also reported in a study from northern India.9 The incidence of other immunodeficiency-related malignancies was identical to those reported in other studies in the literature.10,14
As seen in other studies, the CD4 counts in patients with ADM were significantly lower compared with those of patients with NADM, and that difference was not seen when CD4 counts of patients with cervical cancer were compared with patients in the NADM subgroup. The risk of NHL increases proportionally to the degree of immune suppression. The increased susceptibility to various infections in patients with low CD4 counts may also contribute to the occurrence of NHL in patients with low CD4 counts. The occurrence of various other rare cancers in patients with HIV-AIDS may be because of confounding rather than a direct HIV or immunosuppression effect.
An increasing incidence of NADMs has been noted in the Western literature.7,14 ADMs remain the most common malignancies in the HIV-AIDS population, accounting for about 48% of all malignancies.8 This is in concordance with previous publications from India.8,10 With the widespread availability of generic HAART, the incidence of ADMs may decrease even more in the future. In developing countries where the screening procedures for malignancies in both the general population and patients with HIV-AIDS have not yet been implemented at a national level, premalignant lesions of the cervix are not detected.10 Cervical cancer is the most common malignancy in our study population, which underscores the importance of cervical cancer screening in patients with HIV-AIDS.
In the developed countries, following the introduction of HAART in HIV-AIDS management, the incidence of Kaposi sarcoma decreased by 60% to 70%, and the incidence of NHL decreased by 30% to 50%, whereas the rates of cervical cancer remained either stable or declined.15,16 Despite the declining trend, the incidence of these malignancies continues to be high among patients with HIV-AIDS compared with the general population.17 A study from the United States showed increasing trends in various NADMs (such as anal, lung, and liver cancers and Hodgkin lymphoma) from 2006 to 2010.17 In 2003, the number of patients with NADM were higher than the number of patients with ADM in the United States.14 In a population-based study from Brazil, ADMs were the most common malignancies diagnosed in patients with HIV-AIDS. A declining trend was noted in the incidence of ADMs in the population and an increasing trend in the incidence of NADMs. This increase in NADM incidence was contributed by anal and lung cancers.18 Studies from developing countries such as Uganda and Botswana have also shown a decrease in the incidence of Kaposi sarcoma after the introduction of HAART.19-21
Kaposi sarcoma, cervical cancer, NHL (including Burkitt lymphoma, immunoblastic lymphoma, and primary CNS lymphoma [PCNSL]) comprise ADMs. All 3 ADMs have an underlying viral infection as the causative agent.22 Kaposi sarcoma is caused by the Kaposi sarcoma herpes virus, for which seroprevalence varies worldwide.23 As already noted in this article, the incidence of Kaposi sarcoma among the HIV-AIDS population has decreased worldwide since the introduction of HAART. The preinvasive uterine cervix lesions and carcinoma cervix are caused by HPV. NHL in patients with HIV-AIDS is a predominantly aggressive B-cell neoplasm. Epstein-Barr virus is implicated for most of the ADM NHLs.24 PCNSL occurs in patients with low CD4 counts and poses a diagnostic challenge. The treatment outcomes for patients with PCNSL before the HAART era were dismal. With the widespread use of HAART, the treatment outcomes of patients with HIV-AIDS and NHL improved, and, currently, these patients are managed the same way as other patients with NHL.22
The increasing incidence of the NADM is partly attributed to the increasing incidence of these malignancies in the general population. An elevated risk of certain NADMs is also attributable to viral infections. The common NADMs in the United States are lung, anal, oropharyngeal, and hepatocellular cancers and Hodgkin lymphoma.14 The common NADMs in our study population were oral, oropharyngeal, colon, and breast cancers and Hodgkin lymphoma. One-third of head and neck cancers, including most oropharyngeal cancers, and cervical and anal cancers in patients with HIV-AIDS are related to HPV.25 Patients with HIV-AIDS are at increased risk for chronic HPV infection from immunosuppression. Chronic HPV infections and prolonged immunosuppression cause premalignant high-grade squamous intraepithelial lesions and invasive cancers.22 The initiation of and adherence to HAART leads to immune recovery and reduces high-risk HPV-associated morbidity.26 Findings from previous studies have demonstrated the benefits of screening for cervical cancer in patients with HIV-AIDS.27 The HPV vaccine is immunogenic in patients with HIV-AIDS and might help prevent HPV-associated malignancies.28
Conclusions
With the wide use of HAART by patients with HIV-AIDS, we can expect an increase in the survival of that population. The incidence of malignancies may also increase significantly in these patients, and further longitudinal studies are needed, as malignancies may emerge as the most common cause of death in patients with HIV-AIDS. In addition, the extensive use of HAART therapy and implementation of screening programs for cervical cancer in patients with HIV-AIDS could result in a decrease in the incidence of ADMs.
1. UNAIDS. Prevention gap report. http://www.unaids.org/sites/default/files/media_asset/2016-prevention-gap-report_en.pdf. Released 2016. Accessed December 27, 2017.
3. Dubrow R, Silverberg MJ, Park LS, Crothers K, Justice AC. HIV infection, aging, and immune function: implications for cancer risk and prevention. Curr Opin Oncol. 2012;24(5):506-516.
4. Biggar RJ, Chaturvedi AK, Bhatia K, Mbulaiteye SM. Cancer risk in persons with HIV-AIDS in India: a review and future directions for research. Infect Agent Cancer. 2009;4:4.
5. National AIDS Control Organisation & National Institute of Medical Statistics, ICMR, Ministry of Health & Family Welfare, Government of India. India HIV estimations 2015, technical report. http://www.naco.gov.in/sites/default/files/India%20HIV%20Estimations%202015.pdf. Published 2015. Accessed December 27, 2017.
6. Bonnet F, Lewden C, May T, et al. Malignancy-related causes of death in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Cancer. 2004;101(2):317-324.
7. Crum-Cianflone N, Hullsiek KH, Marconi V, et al. Trends in the incidence of cancers among HIV-infected persons and the impact of antiretroviral therapy: a 20-year cohort study. AIDS. 2009;23(1):41-50.
8. Sharma S, Soneja M, Ranjan S. Malignancies in human immunodeficiency virus infected patients in India: initial experience in the HAART era. Indian J Med Res. 2015;142(5):563-567.
9. Sachdeva RK, Sharma A, Singh S, Varma S. Spectrum of AIDS defining & non-AIDS defining malignancies in north India. In
10. Dhir AA, Sawant S, Dikshit RP, et al. Spectrum of HIV-AIDS related cancers in India. Cancer Causes Control. 2007;19(2):147-153.
11. Venkatesh KK, Saghayam S, Devaleenal B, et al. Spectrum of malignancies among HIV-infected patients in South India. Indian J Cancer. 2012;49(1):176-180.
12. Shruti P, Narayanan G, Puthuveettil J, Jayasree K, Vijayalakshmi K. Spectrum of HIV/AIDS-associated cancers in south India. J Clin Oncol. 2014;32(suppl):e12534.
13. Paul TR, Uppin MS, Uppin SG, et al. Spectrum of malignancies in human immunodeficiency virus–positive patients at a Tertiary Care Centre in South India. Indian J Cancer. 2014;51(4):459-463.
14. Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst. 2011;103(9):753-762.
15. Patel P, Hanson DL, Sullivan PS, et al. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992–2003. Ann Intern Med. 2008;148(10):728-736.
16. Engels EA, Biggar RJ, Hall HI, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123(1):187-194.
17. Robbins HA, Shiels MS, Pfeiffer RM, Engels EA. Epidemiologic contributions to recent cancer trends among HIV-infected people in the United States. AIDS. 2014;28(6):881-890.
18. Tanaka LF, Latorre MDRD, Gutierrez EB, Heumann C, Herbinger KH, Froeschl G. Trends in the incidence of AIDS-defining and non-AIDS-defining cancers in people living with AIDS: a population-based study from São Paulo, Brazil. Int J STD AIDS. 2017;28(12):1190-1198.
19. Mutyaba I, Phipps W, Krantz EM, et al. A population-level evaluation of the effect of antiretroviral therapy on cancer incidence in Kyadondo County, Uganda, 1999–2008. J Acquir Immune Defic Syndr. 2015;69(4):481-486.
20. Dryden-Peterson S, Medhin H, Kebabonye-Pusoentsi M, et al. Cancer incidence following expansion of HIV treatment in Botswana. PLoS ONE. 2015;10(8):e0135602.
21. Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12(1):6-11.
22. Yarchoan R, Uldrick TS. HIV-associated cancers and related diseases. N Engl J Med. 2018;378(11):1029-1041.
23. Gao SJ, Kingsley L, Li M, et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat Med. 1996;2(8):925-928.
24. Epstein-Barr virus and AIDS-associated lymphomas. Lancet. 1991;338(8773):979-981.
25. Picard A, Badoual C, Hourseau M, et al. Human papilloma virus prevalence in HIV patients with head and neck squamous cell carcinoma. AIDS. 2016;30(8):1257-1266.
26. Minkoff H, Zhong Y, Burk RD, et al. Influence of adherent and effective antiretroviral therapy use on human papillomavirus infection and squamous intraepithelial lesions in human immunodeficiency virus-positive women. J Infect Dis. 2010;201(5):681-690.
27. Ghebre RG, Grover S, Xu MJ, Chuang LT, Simonds H. Cervical cancer control in HIV-infected women: past, present and future. Gynecol Oncol Rep. 2017;21:101-108.
28. Kojic EM, Rana AI, Cu-Uvin S. Human papillomavirus vaccination in HIV-infected women: need for increased coverage. Expert Rev Vaccines. 2016;15(1):105-117.
India has the third largest HIV epidemic in the world because of its large population size, with 0.3% of the adult population infected with HIV. That translates to 2.1 million infected people, posing a significant challenge in the management of these individuals.1 In all, 43% of the infected are currently on highly active antiretroviral therapy (HAART).1 There has been a significant decrease in the number of HIV-AIDS–related deaths in recent years because of the remarkable increase in the use of antiretroviral therapy.2 However, the prolonged life expectancy in these patients has resulted in an increase in the risk of various new diseases such as cancers. With the complex interactions between altered immunity and infections, the risk of cancers is markedly increased in patients with HIV-AIDS.3 The spectrum of malignancies in this group of patients differs from that in the general population. In addition, the pattern and the magnitude of malignancies differ in different parts of the world.4 In this study, we have analyzed the pattern of malignancies in patients with HIV-AIDS in a regional cancer center in India. The aim of the study was to analyze the pattern of malignancies in patients with HIV-AIDS based on their age and sex and to document the CD4 counts at the time the malignancy was diagnosed.
Methods
We retrieved data from our institution’s medical records department on all patients who had HIV-AIDS and had been diagnosed with a malignancy. Data of all patients presenting with a malignancy and coexisting HIV-AIDS from January 2013 through December 2016 were analyzed initially. Only patients for whom there was a documented CD4 count were included in the final retrospective analysis. We analyzed the correlation between the patients’ CD4 counts and malignancies subclassified as AIDS-defining malignancies (ADMs; aggressive B-cell non-Hodgkin lymphoma [NHL] and cervical cancer) or non–AIDS-defining malignancies (NADMs; all other malignancies other than aggressive NHL and carcinoma cervix were defined as NADM). We also analyzed the correlation between the CD4 count and NHL and other malignancies. A statistical analysis was performed using SPSS Statistics for Windows, version 23 (IBM Corp, Armonk, NY). The independent sample Mann-Whitney U or Kruskal-Wallis tests were used for comparing the CD4 counts between the various subgroups of malignancies. The study was carried out in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines.
Results
A total of 370 patients who were diagnosed with malignancy and have coexisting HIV-AIDS were identified. In all, 85 patients were excluded because there were no CD4 counts available for them, and the remaining 285 patients were included in the final analysis. Of that total, 136 patients (48%) were men, and 149 (52%) were women.
The median age of the population was 44.8 years (5-80 years) at the time of diagnosis with malignancy. The mean CD4 count of the entire population was 235.4 cells/mm3 (50-734 cells/mm3). There were 104 patients with CD4 counts of ≤200 cells/mm3, and 181 patients had CD4 counts of >200 cells/mm3 (Table 1). All patients received the HAART regimen, efavirenz-lamuvidine-tenofovir (600 mg/300 mg/300 mg Telura).
The most common malignancies in this population were gynecologic malignancies, followed by hematologic malignancies. Cervical cancer was the most common malignancy among women as well as in the overall study population. Among men, the most common malignancy was NHL. The second and third most common malignancies in men were carcinoma oral cavity and carcinoma oropharynx, respectively, whereas in women, they were NHL and breast cancer. The distribution of various hematologic, head and neck, and gastrointestinal malignancies in this group of patients is shown in Figures 1, 2, and 3.
The ADMs in the study were NHL, including 2 patients diagnosed with primary central nervous system (CNS) lymphomas, and cervical cancer. No case of Kaposi sarcoma, also considered an ADM, was identified in this study. The common NADMs include head and neck malignancies (Figure 2), gastrointestinal malignancies (Figure 3), gynecological and genitourinary malignancies, and breast cancer. The mean CD4 count in the ADM subgroup was 221 cells/mm3, and in the NADM subgroup, it was 250 cells/mm3. There was a significant difference in the distribution of CD4 counts between the ADM and NADM subgroups (P = .03; Mann-Whitney U test). A statistical difference was also noted when the CD4 counts of the patients with NHL were compared with other malignancies (P = .0001; Mann-Whitney U test) There was no statistically significant difference noted when CD4 counts of patients with cervical cancer were compared with NADMs (P = .914).
Discussion
In 2015, a report from the Indian government estimated the prevalence of HIV in the country as 0.26% (0.22%-0.32%).5 The report also noted a decreasing trend in the number of new cases of HIV diagnosed and a decrease in the number of AIDS-related deaths.5 The decrease in deaths from AIDS is primarily attributed to the widespread use of HAART. With the introduction of HAART therapy, the survival of patients diagnosed with HIV-AIDS has increased markedly.6 However, newer challenges have emerged with improved survival, such as an increasing number of patients being diagnosed with malignancies. In the current HAART era, the pattern of malignancies in people living with HIV-AIDS has changed compared with the pre-HAART era.7 The literature suggests that worldwide, malignancies are encountered in about 30% patients with HIV-AIDS, but that percentage differs sharply from that encountered in India, where it is less than 5%.8 This may partly be explained by opportunistic infections such as tuberculosis in Indian patients, which remains the leading cause of death in the HIV-AIDS population. In our study, we retrospectively analyzed the pattern of malignancies in patients with HIV-AIDS.
Although few studies have quoted NHL as the predominant malignancy in their patients with HIV-AIDS, the predominant malignancy was cervical cancer in our patient population, as seen in few other studies.8-10 Head and neck malignancies also continue to be common malignancies in men with HIV-AIDS.10 Thus, an increase in malignancies induced by the human papillomavirus (HPV) can be seen in this group of patients. Only a few pediatric malignancies were noted in our study, and all of those patients had a vertical transmission of HIV.
Kaposi sarcoma is quite rare in the Indian population, and no case of Kaposi sarcoma was diagnosed in our study population. A similar finding was seen in several earlier publications from India. In the largest published series from India by Dhir and colleagues, evaluating 251 patients with HIV-AIDS and malignancy, no case of Kaposi sarcoma was reported.10 The authors mentioned that this finding might be because of the low seroprevalence of Kaposi sarcoma-associated herpesvirus in the Asian population.10 Three different studies from southern India have also not reported the incidence of Kaposi Sarcoma in their series of HIV-AIDS patients with malignancies,11-13 and similar findings were also reported in a study from northern India.9 The incidence of other immunodeficiency-related malignancies was identical to those reported in other studies in the literature.10,14
As seen in other studies, the CD4 counts in patients with ADM were significantly lower compared with those of patients with NADM, and that difference was not seen when CD4 counts of patients with cervical cancer were compared with patients in the NADM subgroup. The risk of NHL increases proportionally to the degree of immune suppression. The increased susceptibility to various infections in patients with low CD4 counts may also contribute to the occurrence of NHL in patients with low CD4 counts. The occurrence of various other rare cancers in patients with HIV-AIDS may be because of confounding rather than a direct HIV or immunosuppression effect.
An increasing incidence of NADMs has been noted in the Western literature.7,14 ADMs remain the most common malignancies in the HIV-AIDS population, accounting for about 48% of all malignancies.8 This is in concordance with previous publications from India.8,10 With the widespread availability of generic HAART, the incidence of ADMs may decrease even more in the future. In developing countries where the screening procedures for malignancies in both the general population and patients with HIV-AIDS have not yet been implemented at a national level, premalignant lesions of the cervix are not detected.10 Cervical cancer is the most common malignancy in our study population, which underscores the importance of cervical cancer screening in patients with HIV-AIDS.
In the developed countries, following the introduction of HAART in HIV-AIDS management, the incidence of Kaposi sarcoma decreased by 60% to 70%, and the incidence of NHL decreased by 30% to 50%, whereas the rates of cervical cancer remained either stable or declined.15,16 Despite the declining trend, the incidence of these malignancies continues to be high among patients with HIV-AIDS compared with the general population.17 A study from the United States showed increasing trends in various NADMs (such as anal, lung, and liver cancers and Hodgkin lymphoma) from 2006 to 2010.17 In 2003, the number of patients with NADM were higher than the number of patients with ADM in the United States.14 In a population-based study from Brazil, ADMs were the most common malignancies diagnosed in patients with HIV-AIDS. A declining trend was noted in the incidence of ADMs in the population and an increasing trend in the incidence of NADMs. This increase in NADM incidence was contributed by anal and lung cancers.18 Studies from developing countries such as Uganda and Botswana have also shown a decrease in the incidence of Kaposi sarcoma after the introduction of HAART.19-21
Kaposi sarcoma, cervical cancer, NHL (including Burkitt lymphoma, immunoblastic lymphoma, and primary CNS lymphoma [PCNSL]) comprise ADMs. All 3 ADMs have an underlying viral infection as the causative agent.22 Kaposi sarcoma is caused by the Kaposi sarcoma herpes virus, for which seroprevalence varies worldwide.23 As already noted in this article, the incidence of Kaposi sarcoma among the HIV-AIDS population has decreased worldwide since the introduction of HAART. The preinvasive uterine cervix lesions and carcinoma cervix are caused by HPV. NHL in patients with HIV-AIDS is a predominantly aggressive B-cell neoplasm. Epstein-Barr virus is implicated for most of the ADM NHLs.24 PCNSL occurs in patients with low CD4 counts and poses a diagnostic challenge. The treatment outcomes for patients with PCNSL before the HAART era were dismal. With the widespread use of HAART, the treatment outcomes of patients with HIV-AIDS and NHL improved, and, currently, these patients are managed the same way as other patients with NHL.22
The increasing incidence of the NADM is partly attributed to the increasing incidence of these malignancies in the general population. An elevated risk of certain NADMs is also attributable to viral infections. The common NADMs in the United States are lung, anal, oropharyngeal, and hepatocellular cancers and Hodgkin lymphoma.14 The common NADMs in our study population were oral, oropharyngeal, colon, and breast cancers and Hodgkin lymphoma. One-third of head and neck cancers, including most oropharyngeal cancers, and cervical and anal cancers in patients with HIV-AIDS are related to HPV.25 Patients with HIV-AIDS are at increased risk for chronic HPV infection from immunosuppression. Chronic HPV infections and prolonged immunosuppression cause premalignant high-grade squamous intraepithelial lesions and invasive cancers.22 The initiation of and adherence to HAART leads to immune recovery and reduces high-risk HPV-associated morbidity.26 Findings from previous studies have demonstrated the benefits of screening for cervical cancer in patients with HIV-AIDS.27 The HPV vaccine is immunogenic in patients with HIV-AIDS and might help prevent HPV-associated malignancies.28
Conclusions
With the wide use of HAART by patients with HIV-AIDS, we can expect an increase in the survival of that population. The incidence of malignancies may also increase significantly in these patients, and further longitudinal studies are needed, as malignancies may emerge as the most common cause of death in patients with HIV-AIDS. In addition, the extensive use of HAART therapy and implementation of screening programs for cervical cancer in patients with HIV-AIDS could result in a decrease in the incidence of ADMs.
India has the third largest HIV epidemic in the world because of its large population size, with 0.3% of the adult population infected with HIV. That translates to 2.1 million infected people, posing a significant challenge in the management of these individuals.1 In all, 43% of the infected are currently on highly active antiretroviral therapy (HAART).1 There has been a significant decrease in the number of HIV-AIDS–related deaths in recent years because of the remarkable increase in the use of antiretroviral therapy.2 However, the prolonged life expectancy in these patients has resulted in an increase in the risk of various new diseases such as cancers. With the complex interactions between altered immunity and infections, the risk of cancers is markedly increased in patients with HIV-AIDS.3 The spectrum of malignancies in this group of patients differs from that in the general population. In addition, the pattern and the magnitude of malignancies differ in different parts of the world.4 In this study, we have analyzed the pattern of malignancies in patients with HIV-AIDS in a regional cancer center in India. The aim of the study was to analyze the pattern of malignancies in patients with HIV-AIDS based on their age and sex and to document the CD4 counts at the time the malignancy was diagnosed.
Methods
We retrieved data from our institution’s medical records department on all patients who had HIV-AIDS and had been diagnosed with a malignancy. Data of all patients presenting with a malignancy and coexisting HIV-AIDS from January 2013 through December 2016 were analyzed initially. Only patients for whom there was a documented CD4 count were included in the final retrospective analysis. We analyzed the correlation between the patients’ CD4 counts and malignancies subclassified as AIDS-defining malignancies (ADMs; aggressive B-cell non-Hodgkin lymphoma [NHL] and cervical cancer) or non–AIDS-defining malignancies (NADMs; all other malignancies other than aggressive NHL and carcinoma cervix were defined as NADM). We also analyzed the correlation between the CD4 count and NHL and other malignancies. A statistical analysis was performed using SPSS Statistics for Windows, version 23 (IBM Corp, Armonk, NY). The independent sample Mann-Whitney U or Kruskal-Wallis tests were used for comparing the CD4 counts between the various subgroups of malignancies. The study was carried out in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines.
Results
A total of 370 patients who were diagnosed with malignancy and have coexisting HIV-AIDS were identified. In all, 85 patients were excluded because there were no CD4 counts available for them, and the remaining 285 patients were included in the final analysis. Of that total, 136 patients (48%) were men, and 149 (52%) were women.
The median age of the population was 44.8 years (5-80 years) at the time of diagnosis with malignancy. The mean CD4 count of the entire population was 235.4 cells/mm3 (50-734 cells/mm3). There were 104 patients with CD4 counts of ≤200 cells/mm3, and 181 patients had CD4 counts of >200 cells/mm3 (Table 1). All patients received the HAART regimen, efavirenz-lamuvidine-tenofovir (600 mg/300 mg/300 mg Telura).
The most common malignancies in this population were gynecologic malignancies, followed by hematologic malignancies. Cervical cancer was the most common malignancy among women as well as in the overall study population. Among men, the most common malignancy was NHL. The second and third most common malignancies in men were carcinoma oral cavity and carcinoma oropharynx, respectively, whereas in women, they were NHL and breast cancer. The distribution of various hematologic, head and neck, and gastrointestinal malignancies in this group of patients is shown in Figures 1, 2, and 3.
The ADMs in the study were NHL, including 2 patients diagnosed with primary central nervous system (CNS) lymphomas, and cervical cancer. No case of Kaposi sarcoma, also considered an ADM, was identified in this study. The common NADMs include head and neck malignancies (Figure 2), gastrointestinal malignancies (Figure 3), gynecological and genitourinary malignancies, and breast cancer. The mean CD4 count in the ADM subgroup was 221 cells/mm3, and in the NADM subgroup, it was 250 cells/mm3. There was a significant difference in the distribution of CD4 counts between the ADM and NADM subgroups (P = .03; Mann-Whitney U test). A statistical difference was also noted when the CD4 counts of the patients with NHL were compared with other malignancies (P = .0001; Mann-Whitney U test) There was no statistically significant difference noted when CD4 counts of patients with cervical cancer were compared with NADMs (P = .914).
Discussion
In 2015, a report from the Indian government estimated the prevalence of HIV in the country as 0.26% (0.22%-0.32%).5 The report also noted a decreasing trend in the number of new cases of HIV diagnosed and a decrease in the number of AIDS-related deaths.5 The decrease in deaths from AIDS is primarily attributed to the widespread use of HAART. With the introduction of HAART therapy, the survival of patients diagnosed with HIV-AIDS has increased markedly.6 However, newer challenges have emerged with improved survival, such as an increasing number of patients being diagnosed with malignancies. In the current HAART era, the pattern of malignancies in people living with HIV-AIDS has changed compared with the pre-HAART era.7 The literature suggests that worldwide, malignancies are encountered in about 30% patients with HIV-AIDS, but that percentage differs sharply from that encountered in India, where it is less than 5%.8 This may partly be explained by opportunistic infections such as tuberculosis in Indian patients, which remains the leading cause of death in the HIV-AIDS population. In our study, we retrospectively analyzed the pattern of malignancies in patients with HIV-AIDS.
Although few studies have quoted NHL as the predominant malignancy in their patients with HIV-AIDS, the predominant malignancy was cervical cancer in our patient population, as seen in few other studies.8-10 Head and neck malignancies also continue to be common malignancies in men with HIV-AIDS.10 Thus, an increase in malignancies induced by the human papillomavirus (HPV) can be seen in this group of patients. Only a few pediatric malignancies were noted in our study, and all of those patients had a vertical transmission of HIV.
Kaposi sarcoma is quite rare in the Indian population, and no case of Kaposi sarcoma was diagnosed in our study population. A similar finding was seen in several earlier publications from India. In the largest published series from India by Dhir and colleagues, evaluating 251 patients with HIV-AIDS and malignancy, no case of Kaposi sarcoma was reported.10 The authors mentioned that this finding might be because of the low seroprevalence of Kaposi sarcoma-associated herpesvirus in the Asian population.10 Three different studies from southern India have also not reported the incidence of Kaposi Sarcoma in their series of HIV-AIDS patients with malignancies,11-13 and similar findings were also reported in a study from northern India.9 The incidence of other immunodeficiency-related malignancies was identical to those reported in other studies in the literature.10,14
As seen in other studies, the CD4 counts in patients with ADM were significantly lower compared with those of patients with NADM, and that difference was not seen when CD4 counts of patients with cervical cancer were compared with patients in the NADM subgroup. The risk of NHL increases proportionally to the degree of immune suppression. The increased susceptibility to various infections in patients with low CD4 counts may also contribute to the occurrence of NHL in patients with low CD4 counts. The occurrence of various other rare cancers in patients with HIV-AIDS may be because of confounding rather than a direct HIV or immunosuppression effect.
An increasing incidence of NADMs has been noted in the Western literature.7,14 ADMs remain the most common malignancies in the HIV-AIDS population, accounting for about 48% of all malignancies.8 This is in concordance with previous publications from India.8,10 With the widespread availability of generic HAART, the incidence of ADMs may decrease even more in the future. In developing countries where the screening procedures for malignancies in both the general population and patients with HIV-AIDS have not yet been implemented at a national level, premalignant lesions of the cervix are not detected.10 Cervical cancer is the most common malignancy in our study population, which underscores the importance of cervical cancer screening in patients with HIV-AIDS.
In the developed countries, following the introduction of HAART in HIV-AIDS management, the incidence of Kaposi sarcoma decreased by 60% to 70%, and the incidence of NHL decreased by 30% to 50%, whereas the rates of cervical cancer remained either stable or declined.15,16 Despite the declining trend, the incidence of these malignancies continues to be high among patients with HIV-AIDS compared with the general population.17 A study from the United States showed increasing trends in various NADMs (such as anal, lung, and liver cancers and Hodgkin lymphoma) from 2006 to 2010.17 In 2003, the number of patients with NADM were higher than the number of patients with ADM in the United States.14 In a population-based study from Brazil, ADMs were the most common malignancies diagnosed in patients with HIV-AIDS. A declining trend was noted in the incidence of ADMs in the population and an increasing trend in the incidence of NADMs. This increase in NADM incidence was contributed by anal and lung cancers.18 Studies from developing countries such as Uganda and Botswana have also shown a decrease in the incidence of Kaposi sarcoma after the introduction of HAART.19-21
Kaposi sarcoma, cervical cancer, NHL (including Burkitt lymphoma, immunoblastic lymphoma, and primary CNS lymphoma [PCNSL]) comprise ADMs. All 3 ADMs have an underlying viral infection as the causative agent.22 Kaposi sarcoma is caused by the Kaposi sarcoma herpes virus, for which seroprevalence varies worldwide.23 As already noted in this article, the incidence of Kaposi sarcoma among the HIV-AIDS population has decreased worldwide since the introduction of HAART. The preinvasive uterine cervix lesions and carcinoma cervix are caused by HPV. NHL in patients with HIV-AIDS is a predominantly aggressive B-cell neoplasm. Epstein-Barr virus is implicated for most of the ADM NHLs.24 PCNSL occurs in patients with low CD4 counts and poses a diagnostic challenge. The treatment outcomes for patients with PCNSL before the HAART era were dismal. With the widespread use of HAART, the treatment outcomes of patients with HIV-AIDS and NHL improved, and, currently, these patients are managed the same way as other patients with NHL.22
The increasing incidence of the NADM is partly attributed to the increasing incidence of these malignancies in the general population. An elevated risk of certain NADMs is also attributable to viral infections. The common NADMs in the United States are lung, anal, oropharyngeal, and hepatocellular cancers and Hodgkin lymphoma.14 The common NADMs in our study population were oral, oropharyngeal, colon, and breast cancers and Hodgkin lymphoma. One-third of head and neck cancers, including most oropharyngeal cancers, and cervical and anal cancers in patients with HIV-AIDS are related to HPV.25 Patients with HIV-AIDS are at increased risk for chronic HPV infection from immunosuppression. Chronic HPV infections and prolonged immunosuppression cause premalignant high-grade squamous intraepithelial lesions and invasive cancers.22 The initiation of and adherence to HAART leads to immune recovery and reduces high-risk HPV-associated morbidity.26 Findings from previous studies have demonstrated the benefits of screening for cervical cancer in patients with HIV-AIDS.27 The HPV vaccine is immunogenic in patients with HIV-AIDS and might help prevent HPV-associated malignancies.28
Conclusions
With the wide use of HAART by patients with HIV-AIDS, we can expect an increase in the survival of that population. The incidence of malignancies may also increase significantly in these patients, and further longitudinal studies are needed, as malignancies may emerge as the most common cause of death in patients with HIV-AIDS. In addition, the extensive use of HAART therapy and implementation of screening programs for cervical cancer in patients with HIV-AIDS could result in a decrease in the incidence of ADMs.
1. UNAIDS. Prevention gap report. http://www.unaids.org/sites/default/files/media_asset/2016-prevention-gap-report_en.pdf. Released 2016. Accessed December 27, 2017.
3. Dubrow R, Silverberg MJ, Park LS, Crothers K, Justice AC. HIV infection, aging, and immune function: implications for cancer risk and prevention. Curr Opin Oncol. 2012;24(5):506-516.
4. Biggar RJ, Chaturvedi AK, Bhatia K, Mbulaiteye SM. Cancer risk in persons with HIV-AIDS in India: a review and future directions for research. Infect Agent Cancer. 2009;4:4.
5. National AIDS Control Organisation & National Institute of Medical Statistics, ICMR, Ministry of Health & Family Welfare, Government of India. India HIV estimations 2015, technical report. http://www.naco.gov.in/sites/default/files/India%20HIV%20Estimations%202015.pdf. Published 2015. Accessed December 27, 2017.
6. Bonnet F, Lewden C, May T, et al. Malignancy-related causes of death in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Cancer. 2004;101(2):317-324.
7. Crum-Cianflone N, Hullsiek KH, Marconi V, et al. Trends in the incidence of cancers among HIV-infected persons and the impact of antiretroviral therapy: a 20-year cohort study. AIDS. 2009;23(1):41-50.
8. Sharma S, Soneja M, Ranjan S. Malignancies in human immunodeficiency virus infected patients in India: initial experience in the HAART era. Indian J Med Res. 2015;142(5):563-567.
9. Sachdeva RK, Sharma A, Singh S, Varma S. Spectrum of AIDS defining & non-AIDS defining malignancies in north India. In
10. Dhir AA, Sawant S, Dikshit RP, et al. Spectrum of HIV-AIDS related cancers in India. Cancer Causes Control. 2007;19(2):147-153.
11. Venkatesh KK, Saghayam S, Devaleenal B, et al. Spectrum of malignancies among HIV-infected patients in South India. Indian J Cancer. 2012;49(1):176-180.
12. Shruti P, Narayanan G, Puthuveettil J, Jayasree K, Vijayalakshmi K. Spectrum of HIV/AIDS-associated cancers in south India. J Clin Oncol. 2014;32(suppl):e12534.
13. Paul TR, Uppin MS, Uppin SG, et al. Spectrum of malignancies in human immunodeficiency virus–positive patients at a Tertiary Care Centre in South India. Indian J Cancer. 2014;51(4):459-463.
14. Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst. 2011;103(9):753-762.
15. Patel P, Hanson DL, Sullivan PS, et al. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992–2003. Ann Intern Med. 2008;148(10):728-736.
16. Engels EA, Biggar RJ, Hall HI, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123(1):187-194.
17. Robbins HA, Shiels MS, Pfeiffer RM, Engels EA. Epidemiologic contributions to recent cancer trends among HIV-infected people in the United States. AIDS. 2014;28(6):881-890.
18. Tanaka LF, Latorre MDRD, Gutierrez EB, Heumann C, Herbinger KH, Froeschl G. Trends in the incidence of AIDS-defining and non-AIDS-defining cancers in people living with AIDS: a population-based study from São Paulo, Brazil. Int J STD AIDS. 2017;28(12):1190-1198.
19. Mutyaba I, Phipps W, Krantz EM, et al. A population-level evaluation of the effect of antiretroviral therapy on cancer incidence in Kyadondo County, Uganda, 1999–2008. J Acquir Immune Defic Syndr. 2015;69(4):481-486.
20. Dryden-Peterson S, Medhin H, Kebabonye-Pusoentsi M, et al. Cancer incidence following expansion of HIV treatment in Botswana. PLoS ONE. 2015;10(8):e0135602.
21. Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12(1):6-11.
22. Yarchoan R, Uldrick TS. HIV-associated cancers and related diseases. N Engl J Med. 2018;378(11):1029-1041.
23. Gao SJ, Kingsley L, Li M, et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat Med. 1996;2(8):925-928.
24. Epstein-Barr virus and AIDS-associated lymphomas. Lancet. 1991;338(8773):979-981.
25. Picard A, Badoual C, Hourseau M, et al. Human papilloma virus prevalence in HIV patients with head and neck squamous cell carcinoma. AIDS. 2016;30(8):1257-1266.
26. Minkoff H, Zhong Y, Burk RD, et al. Influence of adherent and effective antiretroviral therapy use on human papillomavirus infection and squamous intraepithelial lesions in human immunodeficiency virus-positive women. J Infect Dis. 2010;201(5):681-690.
27. Ghebre RG, Grover S, Xu MJ, Chuang LT, Simonds H. Cervical cancer control in HIV-infected women: past, present and future. Gynecol Oncol Rep. 2017;21:101-108.
28. Kojic EM, Rana AI, Cu-Uvin S. Human papillomavirus vaccination in HIV-infected women: need for increased coverage. Expert Rev Vaccines. 2016;15(1):105-117.
1. UNAIDS. Prevention gap report. http://www.unaids.org/sites/default/files/media_asset/2016-prevention-gap-report_en.pdf. Released 2016. Accessed December 27, 2017.
3. Dubrow R, Silverberg MJ, Park LS, Crothers K, Justice AC. HIV infection, aging, and immune function: implications for cancer risk and prevention. Curr Opin Oncol. 2012;24(5):506-516.
4. Biggar RJ, Chaturvedi AK, Bhatia K, Mbulaiteye SM. Cancer risk in persons with HIV-AIDS in India: a review and future directions for research. Infect Agent Cancer. 2009;4:4.
5. National AIDS Control Organisation & National Institute of Medical Statistics, ICMR, Ministry of Health & Family Welfare, Government of India. India HIV estimations 2015, technical report. http://www.naco.gov.in/sites/default/files/India%20HIV%20Estimations%202015.pdf. Published 2015. Accessed December 27, 2017.
6. Bonnet F, Lewden C, May T, et al. Malignancy-related causes of death in human immunodeficiency virus-infected patients in the era of highly active antiretroviral therapy. Cancer. 2004;101(2):317-324.
7. Crum-Cianflone N, Hullsiek KH, Marconi V, et al. Trends in the incidence of cancers among HIV-infected persons and the impact of antiretroviral therapy: a 20-year cohort study. AIDS. 2009;23(1):41-50.
8. Sharma S, Soneja M, Ranjan S. Malignancies in human immunodeficiency virus infected patients in India: initial experience in the HAART era. Indian J Med Res. 2015;142(5):563-567.
9. Sachdeva RK, Sharma A, Singh S, Varma S. Spectrum of AIDS defining & non-AIDS defining malignancies in north India. In
10. Dhir AA, Sawant S, Dikshit RP, et al. Spectrum of HIV-AIDS related cancers in India. Cancer Causes Control. 2007;19(2):147-153.
11. Venkatesh KK, Saghayam S, Devaleenal B, et al. Spectrum of malignancies among HIV-infected patients in South India. Indian J Cancer. 2012;49(1):176-180.
12. Shruti P, Narayanan G, Puthuveettil J, Jayasree K, Vijayalakshmi K. Spectrum of HIV/AIDS-associated cancers in south India. J Clin Oncol. 2014;32(suppl):e12534.
13. Paul TR, Uppin MS, Uppin SG, et al. Spectrum of malignancies in human immunodeficiency virus–positive patients at a Tertiary Care Centre in South India. Indian J Cancer. 2014;51(4):459-463.
14. Shiels MS, Pfeiffer RM, Gail MH, et al. Cancer burden in the HIV-infected population in the United States. J Natl Cancer Inst. 2011;103(9):753-762.
15. Patel P, Hanson DL, Sullivan PS, et al. Incidence of types of cancer among HIV-infected persons compared with the general population in the United States, 1992–2003. Ann Intern Med. 2008;148(10):728-736.
16. Engels EA, Biggar RJ, Hall HI, et al. Cancer risk in people infected with human immunodeficiency virus in the United States. Int J Cancer. 2008;123(1):187-194.
17. Robbins HA, Shiels MS, Pfeiffer RM, Engels EA. Epidemiologic contributions to recent cancer trends among HIV-infected people in the United States. AIDS. 2014;28(6):881-890.
18. Tanaka LF, Latorre MDRD, Gutierrez EB, Heumann C, Herbinger KH, Froeschl G. Trends in the incidence of AIDS-defining and non-AIDS-defining cancers in people living with AIDS: a population-based study from São Paulo, Brazil. Int J STD AIDS. 2017;28(12):1190-1198.
19. Mutyaba I, Phipps W, Krantz EM, et al. A population-level evaluation of the effect of antiretroviral therapy on cancer incidence in Kyadondo County, Uganda, 1999–2008. J Acquir Immune Defic Syndr. 2015;69(4):481-486.
20. Dryden-Peterson S, Medhin H, Kebabonye-Pusoentsi M, et al. Cancer incidence following expansion of HIV treatment in Botswana. PLoS ONE. 2015;10(8):e0135602.
21. Shiels MS, Engels EA. Evolving epidemiology of HIV-associated malignancies. Curr Opin HIV AIDS. 2017;12(1):6-11.
22. Yarchoan R, Uldrick TS. HIV-associated cancers and related diseases. N Engl J Med. 2018;378(11):1029-1041.
23. Gao SJ, Kingsley L, Li M, et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nat Med. 1996;2(8):925-928.
24. Epstein-Barr virus and AIDS-associated lymphomas. Lancet. 1991;338(8773):979-981.
25. Picard A, Badoual C, Hourseau M, et al. Human papilloma virus prevalence in HIV patients with head and neck squamous cell carcinoma. AIDS. 2016;30(8):1257-1266.
26. Minkoff H, Zhong Y, Burk RD, et al. Influence of adherent and effective antiretroviral therapy use on human papillomavirus infection and squamous intraepithelial lesions in human immunodeficiency virus-positive women. J Infect Dis. 2010;201(5):681-690.
27. Ghebre RG, Grover S, Xu MJ, Chuang LT, Simonds H. Cervical cancer control in HIV-infected women: past, present and future. Gynecol Oncol Rep. 2017;21:101-108.
28. Kojic EM, Rana AI, Cu-Uvin S. Human papillomavirus vaccination in HIV-infected women: need for increased coverage. Expert Rev Vaccines. 2016;15(1):105-117.
FDA approves larotrectinib for cancers with NTRK gene fusions
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
for adult and pediatric patients with solid tumors that harbor a genetic aberration known as an neurotrophic receptor tyrosine kinase (NTRK) fusion.
Specifically, the oral inhibitor of tropomyosin receptor kinase is approved for patients with solid tumors that have a NTRK gene fusion without a known acquired resistance mutation and have metastatic disease, are likely to experience severe morbidity from surgical resection, have no satisfactory alternative treatments, or have cancer that has progressed following treatment, the FDA said in a press release.
NTRK fusions are found in both children and adults with dozens of different cancer types. The genetic aberration tends to be rare in common cancers and nearly universal in certain uncommon cancers.
Approval of larotrectinib (Vitrakvi) was based on overall response rate and response duration in the first 55 patients with unresectable or metastatic solid tumors harboring an NTRK gene fusion enrolled across three multicenter, open-label, single-arm clinical trials. The ORR was 75% (95% confidence interval, 61%-85%), including 22% complete responses and 53% partial responses. At the time of database lock, median duration of response had not been reached, the FDA said.
The most common tumors were salivary gland tumors (22%), soft tissue sarcomas (20%), infantile fibrosarcomas (13%), and thyroid cancers (9%). Identification of positive NTRK gene fusion status was prospectively determined in local laboratories using next-generation sequencing or fluorescence in situ hybridization.
Results of the three trials, a phase 1 trial among 8 adult patients (LOXO-TRK-14001), a phase 1/2 trial among 12 pediatric patients (SCOUT), and a phase 2 basket trial among 35 adult and adolescent patients (NAVIGATE), were presented at the annual meeting of the American Society of Clinical Oncology in 2017.
The safety of larotrectinib was evaluated in 176 patients enrolled across the three clinical trials, including 44 pediatric patients. The most common adverse reactions with larotrectinib were fatigue, nausea, dizziness, vomiting, increased AST, cough, increased ALT, constipation, and diarrhea, the FDA said.
The recommended larotrectinib doses are 100 mg orally twice daily for adults and 100 mg/m2 orally twice daily (maximum of 100 mg per dose) for pediatric patients.
The approval of larotrectinib follows the approval of pembrolizumab for certain solid tumors with the MSI-H biomarker, as the first approval for the treatment of cancer based on a biomarker rather than the particular body part or organ system affected by the tumor.
The FDA granted the accelerated approval of Vitrakvi to Loxo Oncology and Bayer.
Immunotherapy may hold the key to defeating virally associated cancers
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.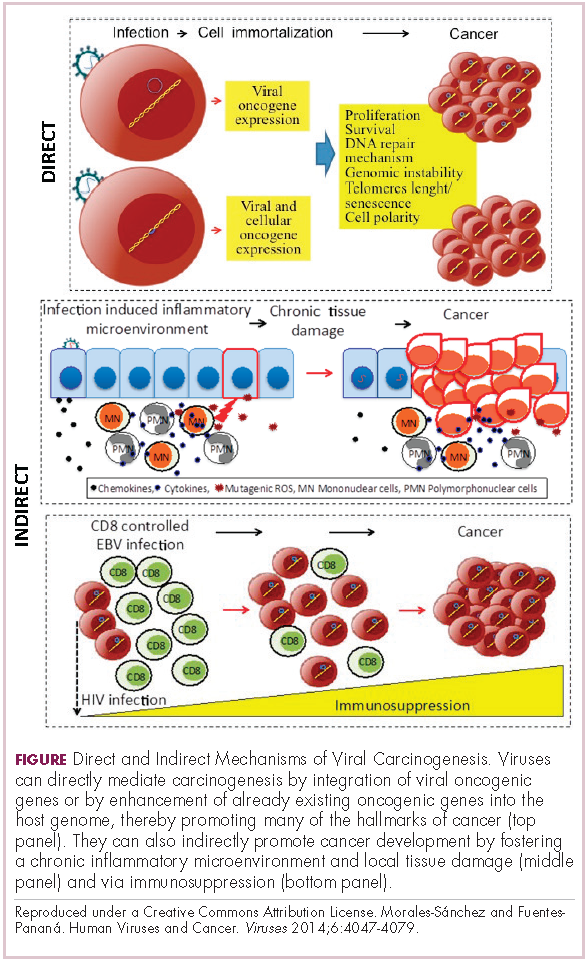
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).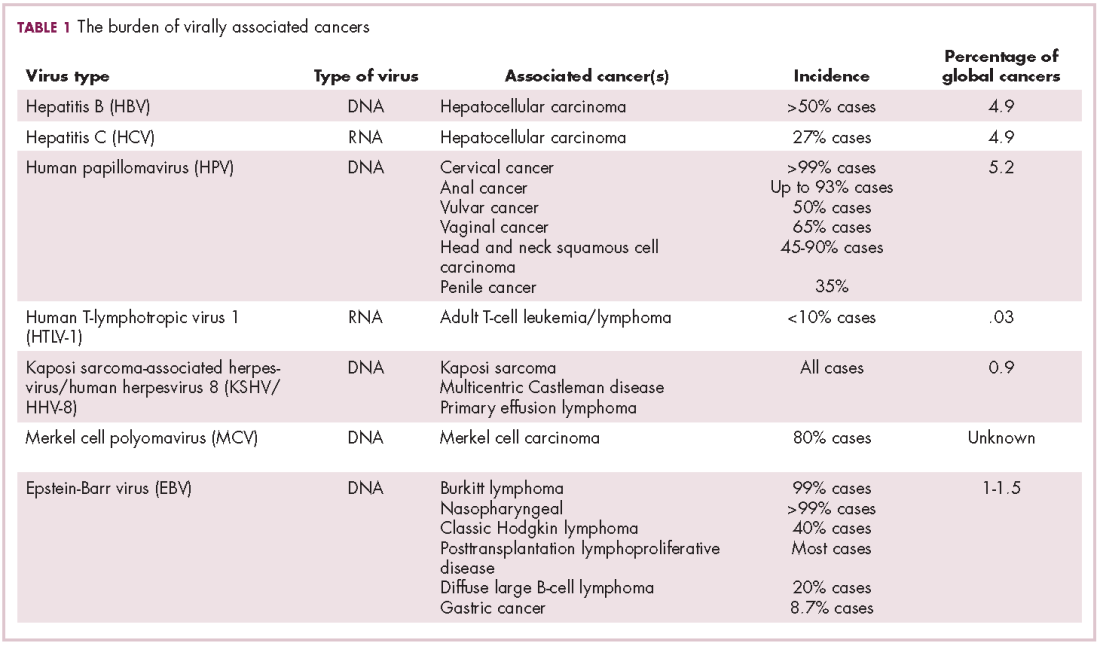
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9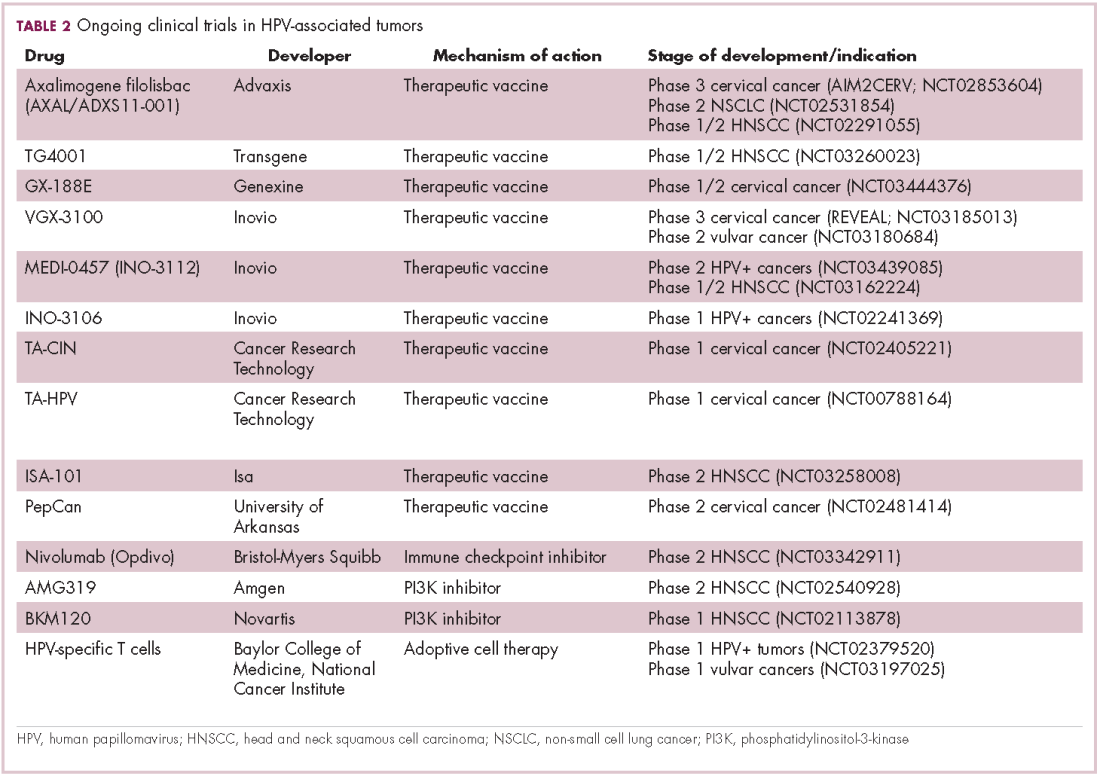
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).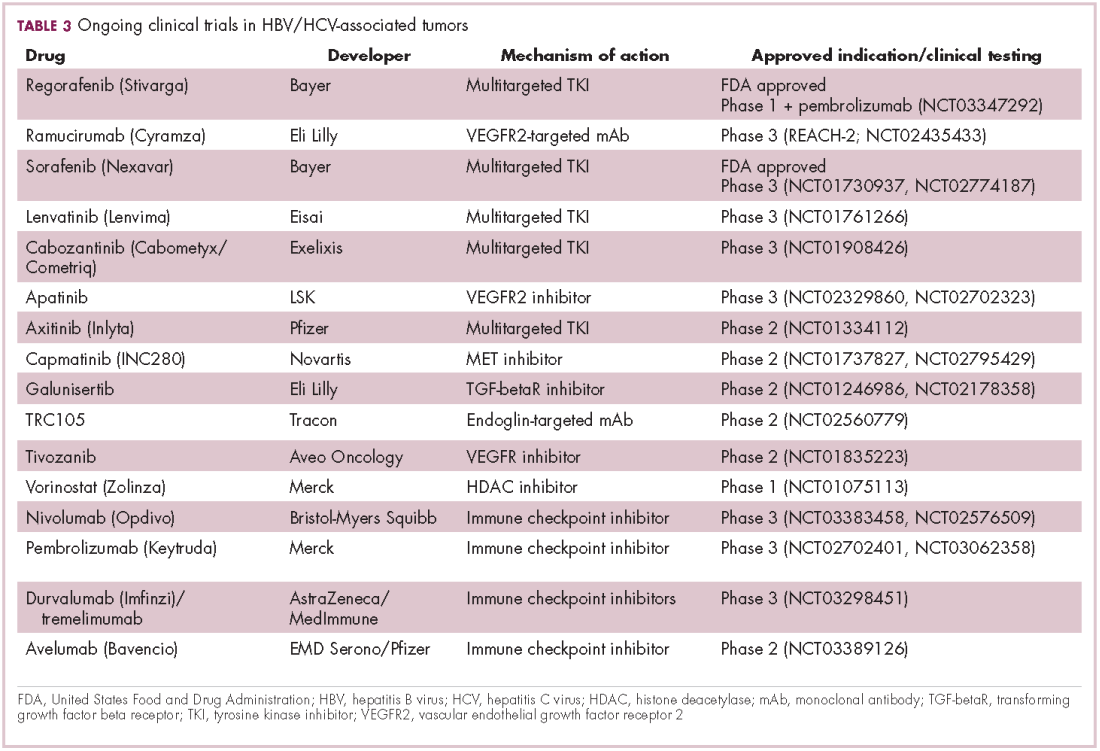
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).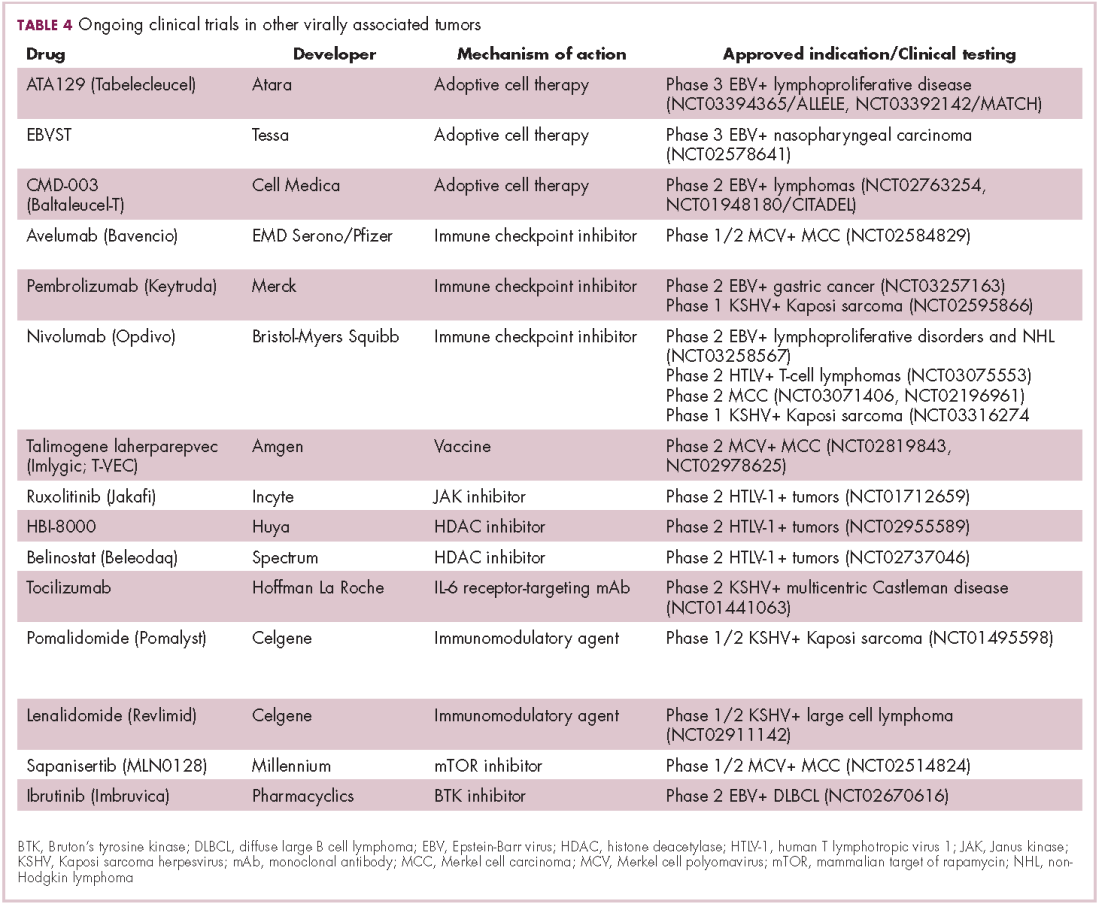
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
Infection with certain viruses has been causally linked to the development of cancer. In recent years, an improved understanding of the unique pathology and molecular underpinnings of these virally associated cancers has prompted the development of more personalized treatment strategies, with a particular focus on immunotherapy. Here, we describe some of the latest developments.
The link between viruses and cancer
Suspicions about a possible role of viral infections in the development of cancer were first aroused in the early 1900s. The seminal discovery is traced back to Peyton Rous, who showed that a malignant tumor growing in a chicken could be transferred to a healthy bird by injecting it with tumor extracts that contained no actual tumor cells.1
The infectious etiology of human cancer, however, remained controversial until many years later when the first cancer-causing virus, Epstein-Barr virus (EBV), was identified in cell cultures from patients with Burkitt lymphoma. Shortly afterward, the Rous sarcoma virus was unveiled as the oncogenic agent behind Rous’ observations.2Seven viruses have now been linked to the development of cancers and are thought to be responsible for around 12% of all cancer cases worldwide. The burden is likely to increase as technological advancements make it easier to establish a causal link between viruses and cancer development.3
In addition to making these links, researchers have also made significant headway in understanding how viruses cause cancer. Cancerous transformation of host cells occurs in only a minority of those who are infected with oncogenic viruses and often occurs in the setting of chronic infection.
Viruses can mediate carcinogenesis by direct and/or indirect mechanisms (Figure 1). Many of the hallmarks of cancer, the key attributes that drive the transformation from a normal cell to a malignant one, are compatible with the virus’s needs, such as needing to avoid cell death, increasing cell proliferation, and avoiding detection by the immune system.
Viruses hijack the cellular machinery to meet those needs and they can do this either by producing viral proteins that have an oncogenic effect or by integrating their genetic material into the host cell genome. When the latter occurs, the process of integration can also cause damage to the DNA, which further increases the risk of cancer-promoting changes occurring in the host genome.
Viruses can indirectly contribute to carcinogenesis by fostering a microenvironment of chronic inflammation, causing oxidative stress and local tissue damage, and by suppressing the antitumor immune response.4,5
Screening and prevention efforts have helped to reduce the burden of several different virally associated cancers. However, for the substantial proportion of patients who are still affected by these cancers, there is a pressing need for new therapeutic options, particularly since genome sequencing studies have revealed that these cancers can often have distinct underlying molecular mechanisms.
Vaccines lead the charge in HPV-driven cancers
German virologist Harald zur Hausen received the Nobel Prize in 2008 for his discovery of the oncogenic role of human papillomaviruses (HPVs), a large family of more than 100 DNA viruses that infect the epithelial cells of the skin and mucous membranes. They are responsible for the largest number of virally associated cancer cases globally – around 5% (Table 1).
A number of different cancer types are linked to HPV infection, but it is best known as the cause of cervical cancer. The development of diagnostic blood tests and prophylactic vaccines for prevention and early intervention in HPV infection has helped to reduce the incidence of cervical cancer. Conversely, another type of HPV-associated cancer, head and neck squamous cell carcinoma (HNSCC), has seen increased incidence in recent years.
HPVs are categorized according to their oncogenic potential as high, intermediate, or low risk. The high-risk HPV16 and HPV18 strains are most commonly associated with cancer. They are thought to cause cancer predominantly through integration into the host genome. The HPV genome is composed of 8 genes encoding proteins that regulate viral replication and assembly. The E6 and E7 genes are the most highly oncogenic; as the HPV DNA is inserted into the host genome, the transcriptional regulator of E6/E7 is lost, leading to their increased expression. These genes have significant oncogenic potential because of their interaction with 2 tumor suppressor proteins, p53 and pRb.6,7
The largest investment in therapeutic development for HPV-positive cancers has been in the realm of immunotherapy in an effort to boost the anti-tumor immune response. In particular, there has been a focus on the development of therapeutic vaccines, designed to prime the anti-tumor immune response to recognize viral antigens. A variety of different types of vaccines are being developed, including live, attenuated and inactivated vaccines that are protein, DNA, or peptide based. Most developed to date target the E6/E7 proteins from the HPV16/18 strains (Table 2).8,9
Other immunotherapies are also being evaluated, including immune checkpoint inhibitors, antibodies designed to target one of the principal mechanisms of immune evasion exploited by cancer cells. The combination of immune checkpoint inhibitors with vaccines is a particularly promising strategy in HPV-associated cancers. At the European Society for Medical Oncology Congress in 2017, the results of a phase 2 trial of nivolumab in combination with ISA-101 were presented.
Among 24 patients with HPV-positive tumors, the majority oropharyngeal cancers, the combination elicited an overall response rate (ORR) of 33%, including 2 complete responses (CRs). Most adverse events (AEs) were mild to moderate in severity and included fever, injection site reactions, fatigue and nausea.14
Hepatocellular carcinoma: a tale of two viruses
The hepatitis viruses are a group of 5 unrelated viruses that causes inflammation of the liver. Hepatitis B (HBV), a DNA virus, and hepatitis C (HCV), an RNA virus, are also oncoviruses; HBV in particular is one of the main causes of hepatocellular carcinoma (HCC), the most common type of liver cancer.
The highly inflammatory environment fostered by HBV and HCV infection causes liver damage that often leads to cirrhosis. Continued infection can drive permanent damage to the hepatocytes, leading to genetic and epigenetic damage and driving oncogenesis. As an RNA virus, HCV doesn’t integrate into the genome and no confirmed viral oncoproteins have been identified to date, therefore it mostly drives cancer through these indirect mechanisms, which is also reflected in the fact that HCV-associated HCC predominantly occurs against a backdrop of liver cirrhosis.
HBV does integrate into the host genome. Genome sequencing studies revealed hundreds of integration sites, but most commonly they disrupted host genes involved in telomere stability and cell cycle regulation, providing some insight into the mechanisms by which HBV-associated HCC develops. In addition, HBV produces several oncoproteins, including HBx, which disrupts gene transcription, cell signaling pathways, cell cycle progress, apoptosis and other cellular processes.15,16
Multitargeted tyrosine kinase inhibitors (TKIs) have been the focal point of therapeutic development in HCC. However, following the approval of sorafenib in 2008, there was a dearth of effective new treatment options despite substantial efforts and numerous phase 3 trials. More recently, immunotherapy has also come to the forefront, especially immune checkpoint inhibitors.
Last year marked the first new drug approvals in nearly a decade – the TKI regorafenib (Stivarga) and immune checkpoint inhibitor nivolumab (Opdivo), both in the second-line setting after failure of sorafenib. Treatment options in this setting may continue to expand, with the TKIs cabozantinib and lenvatinib and the immune checkpoint inhibitor pembrolizumab and the combination of durvalumab and tremelimumab hot on their heels.17-20 Many of these drugs are also being evaluated in the front-line setting in comparison with sorafenib (Table 3).
At the current time, the treatment strategy for patients with HCC is independent of etiology, however, there are significant ongoing efforts to try to tease out the implications of infection for treatment efficacy. A recent meta-analysis of patients treated with sorafenib in 3 randomized phase 3 trials (n = 3,526) suggested that it improved overall survival (OS) among patients who were HCV-positive, but HBV-negative.21
Studies of the vascular endothelial growth factor receptor 2-targeting monoclonal antibody ramucirumab, on the other hand, suggested that it may have a greater OS benefit in patients with HBV, while regorafenib seemed to have a comparable OS benefit in both subgroups.22-25 The immune checkpoint inhibitors studied thus far seem to elicit responses irrespective of infection status.
A phase 2 trial of the immune checkpoint inhibitor tremelimumab was conducted specifically in patients with advanced HCC and chronic HCV infection. The disease control rate (DCR) was 76.4%, with 17.6% partial response (PR) rate. There was also a significant drop in viral load, suggesting that tremelimumab may have antiviral effects.26,27,28
Adoptive cell therapy promising in EBV-positive cancers
More than 90% of the global population is infected with EBV, making it one of the most common human viruses. It is a member of the herpesvirus family that is probably best known as the cause of infectious mononucleosis. On rare occasions, however, EBV can cause tumor development, though our understanding of its exact pathogenic role in cancer is still incomplete.
EBV is a DNA virus that doesn’t tend to integrate into the host genome, but instead remains in the nucleus in the form of episomes and produces several oncoproteins, including latent membrane protein-1. It is associated with a range of different cancer types, including Burkitt lymphoma and other B-cell malignancies. It also infects epithelial cells and can cause nasopharyngeal carcinoma and gastric cancer, however, much less is known about the molecular underpinnings of these EBV-positive cancer types.26,27Gastric cancers actually comprise the largest group of EBV-associated tumors because of the global incidence of this cancer type. The Cancer Genome Atlas Research Network recently characterized gastric cancer on a molecular level and identified an EBV-positive subgroup as a distinct clinical entity with unique molecular characteristics.29
The focus of therapeutic development has again been on immunotherapy, however in this case the idea of collecting the patients T cells, engineering them to recognize EBV, and then reinfusing them into the patient – adoptive cell therapy – has gained the most traction (Table 4).
Two presentations at the American Society of Hematology annual meeting in 2017 detailed ongoing clinical trials of Atara Biotherapeutics’ ATA129 and Cell Medica’s CMD-003. ATA129 was associated with a high response rate and a low rate of serious AEs in patients with posttransplant lymphoproliferative disorder; ORR was 80% in 6 patients treated after hematopoietic stem cell transplantation, and 83% in 6 patients after solid organ transplant.30
CMD-003, meanwhile, demonstrated preliminary signs of activity and safety in patients with relapsed extranodal NK/T-cell lymphoma, according to early results from the phase 2 CITADEL trial. Among 6 evaluable patients, the ORR was 50% and the DCR was 67%.31
Newest oncovirus on the block
The most recently discovered cancer-associated virus is Merkel cell polyomavirus (MCV), a DNA virus that was identified in 2008. Like EBV, virtually the whole global adult population is infected with MCV. It is linked to the development of a highly aggressive and lethal, though rare, form of skin cancer – Merkel cell carcinoma.
MCV is found in around 80% of MCC cases and in fewer than 10% of melanomas and other skin cancers. Thus far, several direct mechanisms of oncogenesis have been described, including integration of MCV into the host genome and the production of viral oncogenes, though their precise function is as yet unclear.32-34
The American Cancer Society estimates that only 1500 cases of MCC are diagnosed each year in the United States.35 Its rarity makes it difficult to conduct clinical trials with sufficient power, yet some headway has still been made.
Around half of MCCs express the programmed cell death ligand 1 (PD-L1) on their surface, making them a logical candidate for immune checkpoint inhibition. In 2017, avelumab became the first FDA-approved drug for the treatment of MCC. Approval was based on the JAVELIN Merkel 200 study in which 88 patients received avelumab. After 1 year of follow-up the ORR was 31.8%, with a CR rate of 9%.36
Genome sequencing studies suggest that the mutational profile of MCV-positive tumors is quite different to those that are MCV-negative, which could have therapeutic implications. To date, these implications have not been delineated, given the challenge of small patient numbers, however an ongoing phase 1/2 trial is evaluating the combination of avelumab and radiation therapy or recombinant interferon beta, with or without MCV-specific cytotoxic T cells in patients with MCC and MCV infection.
The 2 other known cancer-causing viruses are human T-lymphotropic virus 1 (HTLV-1), a retrovirus associated with adult T-cell leukemia/lymphoma (ATL) and Kaposi sarcoma herpesvirus (KSHV). The latter is the causative agent of Kaposi sarcoma, often in combination with human immunodeficiency virus (HIV), a rare skin tumor that became renowned in the 1980s as an AIDS-defining illness.
The incidence of HTLV-1- and KSHV-positive tumors is substantially lower than the other virally associated cancers and, like MCC, this makes studying them and conducting clinical trials of novel therapeutic options a challenge. Nonetheless, several trials of targeted therapies and immunotherapies are underway.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
1. Rous PA. Transmissible avain neoplasm. (Sarcoma of the common fowl). J Exp Med. 1910;12(5):696-705.
2. Epstein MA, Achong BG, Barr YM. Virus particles in cultured lymphoblasts from Burkitt's lymphoma. Lancet. 1964;1(7335):702-703.
3. Mesri Enrique A, Feitelson MA, Munger K. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host & Microbe. 2014;15(3):266-282.
4. Santana-Davila R, Bhatia S, Chow LQ. Harnessing the immune system as a therapeutic tool in virus-associated cancers. JAMA Oncol. 2017;3(1):106-112.
5. Tashiro H, Brenner MK. Immunotherapy against cancer-related viruses. Cell Res. 2017;27(1):59-73.
6. Brianti P, De Flammineis E, Mercuri SR. Review of HPV-related diseases and cancers. New Microbiol. 2017;40(2):80-85.
7. Tulay P, Serakinci N. The route to HPV-associated neoplastic transformation: a review of the literature. Crit Rev Eukaryot Gene Expr. 2016;26(1):27-39.
8. Smola S. Immunopathogenesis of HPV-associated cancers and prospects for immunotherapy. Viruses. 2017;9(9).
9. Rosales R, Rosales C. Immune therapy for human papillomaviruses-related cancers. World Journal of Clinical Oncology. 2014;5(5):1002-1019.
10. Miles B, Safran HP, Monk BJ. Therapeutic options for treatment of human papillomavirus-associated cancers - novel immunologic vaccines: ADXS11-001. Gynecol Oncol Res Pract. 2017;4:10.
11. Miles BA, Monk BJ, Safran HP. Mechanistic insights into ADXS11-001 human papillomavirus-associated cancer immunotherapy. Gynecol Oncol Res Pract. 2017;4:9.
12. Huh W, Dizon D, Powell M, Landrum L, Leath C. A prospective phase II trial of the listeria-based human papillomavirus immunotherapy axalimogene filolisbac in second and third-line metastatic cervical cancer: A NRG oncology group trial. Paper presented at: Annual Meeting on Women's Cancer; March 12-15, 2017, 2017; National Harbor, MD.
13. Petit RG, Mehta A, Jain M, et al. ADXS11-001 immunotherapy targeting HPV-E7: final results from a Phase II study in Indian women with recurrent cervical cancer. Journal for Immunotherapy of Cancer. 2014;2(Suppl 3):P92-P92.
14. Glisson B, Massarelli E, William W, et al. Nivolumab and ISA 101 HPV vaccine in incurable HPV-16+ cancer. Ann Oncol. 2017;28(suppl_5):v403-v427.
15. Ding X-X, Zhu Q-G, Zhang S-M, et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget. 2017;8(33):55715-55730.
16. Ringehan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017;372(1732):20160274.
17. Abou-Alfa G, Meyer T, Cheng AL, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: results from the randomized phase III CELESTIAL trial. J Clin Oncol. 2017;36(Suppl 4S):abstr 207.
18. Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet. 2018.
19. Zhu AX, Finn RS, Cattan S, et al. KEYNOTE-224: Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. J Clin Oncol. 2018;36(Suppl 4S):Abstr 209.
20. Kelley RK, Abou-Alfa GK, Bendell JC, et al. Phase I/II study of durvalumab and tremelimumab in patients with unresectable hepatocellular carcinoma (HCC): Phase I safety and efficacy analyses. Journal of Clinical Oncology. 2017;35(15_suppl):4073-4073.
21. Jackson R, Psarelli E-E, Berhane S, Khan H, Johnson P. Impact of Viral Status on Survival in Patients Receiving Sorafenib for Advanced Hepatocellular Cancer: A Meta-Analysis of Randomized Phase III Trials. Journal of Clinical Oncology. 2017;35(6):622-628.
22. Kudo M. Molecular Targeted Agents for Hepatocellular Carcinoma: Current Status and Future Perspectives. Liver Cancer. 2017;6(2):101-112.
23. zur Hausen H, Meinhof W, Scheiber W, Bornkamm GW. Attempts to detect virus-secific DNA in human tumors. I. Nucleic acid hybridizations with complementary RNA of human wart virus. Int J Cancer. 1974;13(5):650-656.
24. Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56-66.
25. Bruix J, Tak WY, Gasbarrini A, et al. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013;49(16):3412-3419.
26. Neparidze N, Lacy J. Malignancies associated with epstein-barr virus: pathobiology, clinical features, and evolving treatments. Clin Adv Hematol Oncol. 2014;12(6):358-371.
27. Ozoya OO, Sokol L, Dalia S. EBV-Related Malignancies, Outcomes and Novel Prevention Strategies. Infect Disord Drug Targets. 2016;16(1):4-21.
28. Sangro B, Gomez-Martin C, de la Mata M, et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol. 2013;59(1):81-88.
29. The Cancer Genome Atlas Research N. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513:202.
30. Prockop S, Li A, Baiocchi R, et al. Efficacy and safety of ATA129, partially matched allogeneic third-party Epstein-Barr virus-targeted cytotoxic T lymphocytes in a multicenter study for post-transplant lymphoproliferative disorder. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
31. Kim W, Ardeshna K, Lin Y, et al. Autologous EBV-specific T cells (CMD-003): Early results from a multicenter, multinational Phase 2 trial for treatment of EBV-associated NK/T-cell lymphoma. Paper presented at: 59th Annual Meeting of the American Society of Hematology; December 9-12, 2017, 2017; Atlanta, GA.
32. Schadendorf D, Lebbé C, zur Hausen A, et al. Merkel cell carcinoma: Epidemiology, prognosis, therapy and unmet medical needs. European Journal of Cancer. 2017;71:53-69.
33. Spurgeon ME, Lambert PF. Merkel cell polyomavirus: a newly discovered human virus with oncogenic potential. Virology. 2013;435(1):118-130.
34. Tello TL, Coggshall K, Yom SS, Yu SS. Merkel cell carcinoma: An update and review: Current and future therapy. J Am Acad Dermatol. 2018;78(3):445-454.
35. American Cancer Society. Key Statistics for Merkel Cell Carcinoma. 2015; https://www.cancer.org/cancer/merkel-cell-skin-cancer/about/key-statistics.html#written_by. Accessed March 7th, 2017.
36. Kaufman HL, Russell J, Hamid O, et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet Oncology.17(10):1374-1385.
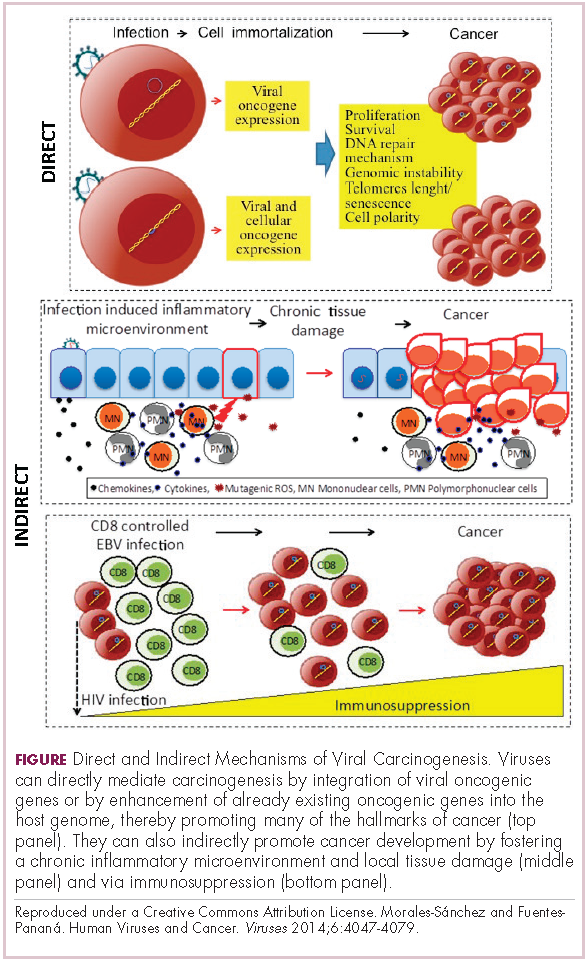
Primary renal synovial sarcoma – a diagnostic dilemma
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).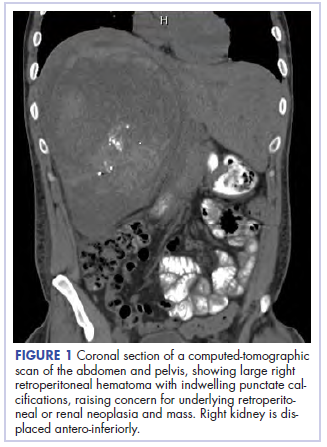
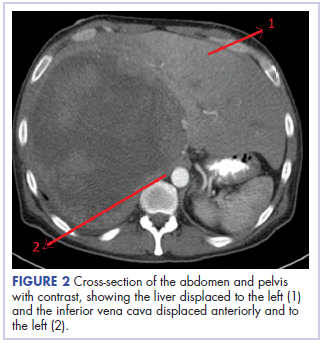
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
Soft tissue sarcomas are rare mesenchymal tumors that comprise 1% of all malignancies. Synovial sarcoma accounts for 5% to 10% of adult soft tissue sarcomas and usually occurs in close association with joint capsules, tendon sheaths, and bursa in the extremities of young and middle-aged adults.1 Synovial sarcomas have been reported in other unusual sites, including the head and neck, thoracic and abdominal wall, retroperitoneum, bone, pleura, and visceral organs such as the lung, prostate, or kidney.2 Primary renal synovial sarcoma is an extremely rare tumor accounting for <2% of all malignant renal tumors.3 To the best of our knowledge, fewer than 50 cases of primary renal synovial sarcoma have been described in the English literature.4 It presents as a diagnostic dilemma because of the dearth of specific clinical and imaging findings and is often confused with benign and malignant tumors. The differential diagnosis includes angiomyolipoma, renal cell carcinoma with sarcomatoid differentiation, metastatic sarcoma, hemangiopericytoma, malignant solitary fibrous tumor, Wilms tumor, and malignant peripheral nerve sheath tumor. Hence, a combination of histomorphologic, immunohistochemical, cytogenetic, and molecular studies that show a unique chromosomal translocation t(X;18) (p11;q11) is imperative in the diagnosis of primary renal synovial sarcoma.4 In the present report, we present the case of a 38-year-old man who was diagnosed with primary renal synovial sarcoma.
Case presentation and summary
A 38-year-old man with a medical history of gastroesophageal reflux disease and Barrett’s esophagus presented to our hospital for the first time with persistent and progressive right-sided flank and abdominal pain that was aggravated after a minor trauma to the back. There was no associated hematuria or dysuria.
Of note is that he had experienced intermittent flank pain for 2 years before this transfer. He had initially been diagnosed at his local hospital close to his home by ultrasound with an angiomyolipoma of 2 × 3 cm arising from the upper pole of his right kidney, which remained stable on repeat sonograms. About 22 months after his initial presentation at his local hospital, the flank pain increased, and a computed-tomographic (CT) scan revealed a perinephric hematoma that was thought to originate from a ruptured angiomyolipoma. He subsequently underwent embolization, but his symptoms recurred soon after. He presented again to his local hospital where CT imaging revealed a significant increase in the size of the retroperitoneal mass, and findings were suggestive of a hematoma. Subsequent angiogram did not reveal active extravasation, so a biopsy was performed.
Before confirmatory pathologic evaluation could be completed, the patient presented to his local hospital again in excruciating pain. A CT scan of his abdomen and pelvis demonstrated a massive subacute on chronic hematoma in the right retroperitoneum measuring 22 × 19 × 18 cm, with calcifications originating from an upper pole right renal neoplasm. The right kidney was displaced antero-inferiorly, and the inferior vena cava was displaced anteriorly and to the left. The preliminary pathology returned with findings suggestive of sarcoma (Figures 1 and 2).
The patient was then transferred to our institution, where he was evaluated by medical and surgical oncology. A CT scan of the chest and magnetic-resonance imaging (MRI) of the brain did not reveal metastatic disease. He underwent exploratory laparotomy that involved the resection of a 22-cm retroperitoneal mass, right nephrectomy, right adrenalectomy, partial right hepatectomy, and a full thickness resection of the right postero-inferior diaphragm followed by mesh repair because of involvement by the tumor.
In its entirety, the specimen was a mass of 26 × 24 × 14 cm. It was sectioned to show extensively necrotic and hemorrhagic variegated white to tan-red parenchyma (Figure 3). Histology revealed a poorly differentiated malignant neoplasm composed of round cells with scant amphophilic cytoplasm arranged in solid, variably sized nests separated by prominent thin-walled branching vascular channels (Figure 4). The mitotic rate was high. It was determined to be a histologically ungraded sarcoma according to the French Federation of Comprehensive Cancer Centers system of grading soft tissue sarcomas; the margins were indeterminate. Immunohistochemistry was positive for EMA, TLE1, and negative for AE1/AE3, S100, STAT6, and Nkx2.2. Molecular pathology fluorescent in situ hybridization (FISH) analysis demonstrated positivity for SS18 gene rearrangement (SS18-SSX1 fusion).
After recovering from surgery, the patient received adjuvant chemotherapy with doxorubicin and ifosfamide. It has been almost 16 months since we first saw this patient. He was started on doxorubicin 20 mg/m2 on days 1 to 4, ifosfamide 2,500 mg on days 1 to 4, and mesna 800 mg on days 1 to 4, for a total of 6 cycles. He did well for the first 5 months, after which he developed disease recurrence in the postoperative nephrectomy bed (a biopsy showed it to be recurrent synovial sarcoma) as well as pulmonary nodules, for which he was started on trabectedin 1.5 mg/m2 every 3 weeks. Two months later, a CT scan showed an increase in the size of his retroperitoneal mass, and the treatment was changed to pazopanib 400 mg daily orally, on which he remained at the time of publication.
Discussion
Synovial sarcoma is the fourth most common type of soft tissue sarcoma, accounting for 2.5% to 10.5% of all primary soft tissue malignancies worldwide. It occurs most frequently in adolescents and young adults, with most patients presenting between the ages of 15 and 40 years. Median age of presentation is 36 years. Despite the nomenclature, synovial sarcoma does not arise in intra-articular locations but typically occurs in proximity to joints in the extremities. Synovial sarcomas are less commonly described in other sites, including the head and neck, mediastinum, intraperitoneum, retroperitoneum, lung, pleura, and kidney.4,5 Renal synovial sarcoma was first described in a published article by Argani and colleagues in 2000.5
Adult renal mesenchymal tumors are classified into benign and malignant tumors on the basis of the histologic features and clinicobiologic behavior.6,7 The benign esenchymal renal tumors include angiomyolipoma, leiomyoma, hemangioma, lymphangioma, juxtaglomerular cell tumor, renomedullary interstitial cell tumor (medullary fibroma), lipoma, solitary fibrous tumor, and schwannoma. Malignant renal tumors of mesenchymal origin include leiomyosarcoma, rhabdomyosarcoma, angiosarcoma, osteosarcoma, fibrosarcoma, malignant fibrous histiocytoma, solitary fibrous tumor, and synovial sarcoma.
Most of these tumor types cause the same nonspecific symptoms in patients – abdominal pain, flank pain, abdominal fullness, a palpable mass, and hematuria – although they can be clinically silent. The average duration of symptoms in synovial sarcoma is 2 to 4 years.8 The long duration of symptoms and initial slow growth of synovial sarcomas may give a false impression of a benign process.
A preoperative radiological diagnosis of primary renal synovial sarcoma may be suspected by analyzing the tumor’s growth patterns on CT scans.9 Renal synovial sarcomas often appear as large, well-defined soft tissue masses that can extend into the renal pelvis or into the perinephric region.9 A CT scan may identify soft tissue calcifications, especially subtle ones in areas where the tumor anatomy is complex. A CT scan may also reveal areas of hemorrhage, necrosis, or cyst formation within the tumor, and can easily confirm bone involvement. Intravenous contrast may help in differentiating the mass from adjacent muscle and neurovascular complex.9,10 On MRI, renal synovial sarcomas are often described as nonspecific heterogeneous masses, although they may also exhibit heterogeneous enhancement of hemorrhagic areas, calcifications, and air-fluid levels (known as “triple sign”) as well as septae. The triple sign may be identified as areas of low, intermediate, and high signal intensity, correlating with areas of hemorrhage, calcification, and air-fluid level.9,10 Signal intensity is about equal to that of skeletal muscle on T1-weighted MRI and higher than that of subcutaneous fat on T2-weighted MRI.
In the present case, the tumor was initially misdiagnosed as an angiomyolipoma, the most common benign tumor of the kidney. Angiomyolipomas are usually solid triphasic tumors arising from the renal cortex and are composed of 3 major elements: dysmorphic blood vessels, smooth muscle components, and adipose tissue. When angiomyolipomas are large enough, they are readily recognized by the identification of macroscopic fat within the tumor, either by CT scan or MRI.11 When they are small, they may be difficult to distinguish from a small cyst on CT because of volume averaging.
On pathology, synovial sarcoma has dual epithelial and mesenchymal differentiation. They are frequently multi-lobulated, and areas of necrosis, hemorrhage, and cyst formation are also common. There are 3 main histologic subtypes of synovial sarcoma: biphasic (20%-30%), monophasic (50%-60%), and poorly differentiated (15%-25%). Poorly differentiated synovial sarcomas are generally epithelioid in morphology, have high mitotic activity (usually 10-20 mitoses/10 high-power field; range is <5 for well differentiated, low-grade tumors), and can be confused with round cell tumors such as Ewing sarcoma. Poorly differentiated synovial sarcomas are high-grade tumors.
Immunohistochemical studies can confirm the pathological diagnosis. Synovial sarcomas usually stain positive for Bcl2, CD99/Mic2, CD56, Vim, and focally for EMA but negatively for desmin, actin, WT1, S-100, CD34, and CD31.5 Currently, the gold standard for diagnosis and hallmark for synovial sarcomas are the t (X;18) translocation and SYT-SSX gene fusion products (SYT-SSX1 in 67% and SYT-SSX2 in 33% of cases). These can be detected either by FISH or reverse-transcription polymerase chain reaction. This genetic alteration is identified in more than 90% of synovial sarcomas and is highly specific.
The role of SYT-SSX gene fusion in the pathogenesis of synovial sarcoma is an active area of investigation. The fusion of SYT with SSX translates into a fusion protein that binds to the transcription activator SMARCA4 that is involved in chromatin remodeling, thus displacing both the wildtype SYT and the tumor suppressor gene SMARCB1. The modified protein complex then binds at several super-enhancer loci, unlocking suppressed genes such as Sox2, which is known to be necessary for synovial sarcoma proliferation. Alterations in SMARCB1 are involved in several cancer types, implicating this event as a driver of these malignancies.12 This results in a global alteration in chromatin remodeling that needs to be better understood to design targeted therapies.
The clinical course of synovial sarcoma, regardless of the tissue of origin, is typically poor. Multiple clinical and pathologic factors, including tumor size, location, patient age, and presence of poorly differentiated areas, are thought to have prognostic significance. A tumor size of more than 5 cm at presentation has the greatest impact on prognosis, with studies showing 5-year survival rates of 64% for patients with tumors smaller than 5 cm and 26% for patients with masses greater than 5 cm.13,14 High-grade synovial sarcoma is favored in tumors that have cystic components, hemorrhage, and fluid levels and the triple sign.
Patients with tumors in the extremities have a more favorable prognosis than those with lesions in the head and neck area or axially, a feature that likely reflects better surgical control available for extremity lesions. Patient age of less than 15 to 20 years is also associated with a better long-term prognosis.15,16 Varela-Duran and Enzinger17 reported that the presence of extensive calcifications suggests improved long-term survival, with 5-year survival rates of 82% and decreased rates of local recurrence (32%) and metastatic disease (29%). The poorly differentiated subtype is associated with a worsened prognosis, with a 5-year survival rate of 20% through 30%.18,19 Other pathologic factors associated with worsened prognosis include presence of rhabdoid cells, extensive tumor necrosis, high nuclear grade, p53 mutations, and high mitotic rate (>10 mitoses/10 high-power field). More recently, the gene fusion type SYT-SSX2 (more common in monophasic lesions) has been associated with an improved prognosis, compared with that for SYT-SSX1, and an 89% metastasis-free survival.20
Although there are no guidelines for the treatment of primary renal synovial sarcoma because of the limited number of cases reported, surgery is considered the first choice. Adjuvant chemotherapy with an anthracycline (doxorubicin or epirubicin) combined with ifosfamide has been the most frequently used regimen in published cases, especially in those in which patients have poor prognostic factors as mentioned above.
Overall, the 5-year survival rate ranges from 36% to 76%.14 The clinical course of synovial sarcoma is characterized by a high rate of local recurrence (30%-50%) and metastatic disease (41%). Most metastases occur within the first 2 to 5 years after treatment cessation. Metastases are present in 16% to 25% of patients at their initial presentation, with the most frequent metastatic site being the lung, followed by the lymph nodes (4%-18%) and bone (8%-11%).
Conclusion
Primary renal synovial sarcoma is extremely rare, and preoperative diagnosis is difficult in the absence of specific clinical or imaging findings. A high index of suspicion combined with pathologic, immunohistochemical, cytogenetic, and molecular studies is essential for accurate diagnosis and subsequent treatment planning. The differential diagnosis of renal synovial sarcoma can be extensive, and our experience with this patient illustrates the diagnostic dilemma associated with renal synovial sarcoma.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
1. Majumder A, Dey S, Khandakar B, Medda S, Chandra Paul P. Primary renal synovial sarcoma: a rare tumor with an atypical presentation. Arch Iran Med. 2014;17(10):726-728.
2. Fetsch JF, Meis JM. Synovial sarcoma of the abdominal wall. Cancer. 1993;72(2):469 477.
3. Wang Z, Zhong Z, Zhu L, et al. Primary synovial sarcoma of the kidney: a case report. Oncol Lett. 2015;10(6):3542-3544.
4. Abbas M, Dämmrich ME, Braubach P, et al. Synovial sarcoma of the kidney in a young patient with a review of the literature. Rare tumors. 2014;6(2):5393
5. Argani P, Faria PA, Epstein JI, et al. Primary renal synovial sarcoma: molecular and morphologic delineation of an entity previously included among embryonal sarcomas of the kidney. Am J Surg Pathol. 2000;24(8):1087-1096.
6. Eble JN, Sauter G, Epstein JI, Sesterhenn IA, eds. World Health Organization classification of tumours: pathology and genetics of tumours of the urinary system and male genital organs. Lyon, France: IARC; 2004.
7. Tamboli P, Ro JY, Amin MB, Ligato S, Ayala AG. Benign tumors and tumor-like lesions of the adult kidney. Part II: benign mesenchymal and mixed neoplasms, and tumor-like lesions. Adv Anat Pathol. 2000;7(1):47-66.
8. Weiss SW, Goldblum JR. Malignant soft tissue tumors of uncertain type. In: Weiss SW, Goldblum JR, eds. Enzinger and Weiss’s soft tissue tumors. 4th ed. St. Louis, MO: Mosby, 2001; 1483-1565.
9. Lacovelli R, Altavilla A, Ciardi A, et al. Clinical and pathological features of primary renal synovial sarcoma: analysis of 64 cases from 11 years of medical literature. BJU Int. 2012;110(10):1449-1454.
10. Alhazzani AR, El-Sharkawy MS, Hassan H. Primary retroperitoneal synovial sarcoma in CT and MRI. Urol Ann. 2010;2(1):39-41.
11. Katabathina VS, Vikram R, Nagar AM, Tamboli P, Menias CO, Prasad SR. Mesenchymal neoplasms of the kidney in adults: imaging spectrum with radiologic-pathologic correlation. Radiographics. 2010;30(6):1525-1540.
12. Sápi Z, Papp G, Szendrői M, et al. Epigenetic regulation of SMARCB1 by miR-206, -381 and -671- 5p is evident in a variety of SMARCB1 immunonegative soft tissue sarcomas, while miR-765 appears specific for epithelioid sarcoma. A miRNA study of 223 soft tissue sarcomas. Genes Chromosomes Cancer. 2016;55(10):786-802.
13. Ferrari A, Gronchi A, Casanova M, et al. Synovial sarcoma: a retrospective analysis of 271 patients of all ages treated at a single institution. Cancer. 2004;101(3):627-634.
14. Rangheard AS, Vanel D, Viala J, Schwaab G, Casiraghi O, Sigal R. Synovial sarcomas of the head and neck: CT and MR imaging findings of eight patients. Am J Neuroradiol. 2001;22(5):851-857.
15. Oda Y, Hashimoto H, Tsuneyoshi M, Takeshita S. Survival in synovial sarcoma: a multivariate study of prognostic factors with special emphasis on the comparison between early death and long-term survival. Am J Surg Pathol. 1993;17(1):35-44.
16. Raney RB. Synovial sarcoma in young people: background, prognostic factors and therapeutic questions. J Pediatr Hematol Oncol. 2005;27(4):207-211.
17. Varela-Duran J, Enzinger FM. Calcifying synovial sarcoma. Cancer. 1982;50(2):345-352.
18. Cagle LA, Mirra JM, Storm FK, Roe DJ, Eilber FR. Histologic features relating to prognosis in synovial sarcoma. Cancer. 1987;59(10):1810-1814.
19. Skytting B, Meis-Kindblom JM, Larsson O, et al. Synovial sarcoma – identification of favorable and unfavorable histologic types: a Scandinavian sarcoma group study of 104 cases. Acta Orthop Scand. 1999:70(6):543-554.
20. Murphey MD, Gibson MS, Jennings BT, Crespo-Rodríguez AM, Fanburg-Smith J, Gajewski DA. Imaging of synovial sarcoma with radiologic-pathologic correlation. Radiographics. 2006;26(5):1543-1565.
TKIs and immunotherapy hold promise for alveolar soft part sarcoma
Alveolar soft part sarcoma (ASPS) has often proven to be resistant to conventional doxorubicin-based chemotherapy, but tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs) may provide new treatment strategies for this rare type of sarcoma, according to a literature review.
A rare, translocation-driven sarcoma of the soft tissues, ASPS often affects young adults and is characterized by indolent behavior and early metastasis. Despite its resistance to chemotherapy, studies indicate that survival is often prolonged in patients with metastatic disease. The literature has shown 5-year survival rates at about 60%, and this percentage has remained fairly consistent for the past 3 decades.
Luca Paoluzzi, MD, of New York University, and Robert G. Maki, MD, PhD, of Hofstra University, Hempstead, N.Y., reviewed the literature from 1952 to March 2018, in order to gain a better understanding of ASPS and the opportunities “for the translation of such knowledge into clinical practice,” they wrote in JAMA.
From a therapeutic standpoint, ASPS is characterized by sensitivity to vascular endothelial growth factor receptor–predominant TKIs, compared with other soft tissue sarcomas (STS), and recent data have emphasized that it is responsive to new immunotherapy regimens including ICIs. Pazopanib is currently the only agent that has received regulatory approval for use in STS refractory to other treatments and it appears to have consistent activity in metastatic ASPS. Management of ASPS generally also involves surgical resection and/or systemic treatment for metastatic disease. Conventional agents such as anthracycline-based chemotherapy have demonstrated a poor response rate lower than 10%, and while a complete resection may be curative, metastases are common and can occur years after resection of the primary tumor.
Conversely, ICIs “represent a promising area of drug development in ASPS; the data to date are limited but encouraging,” wrote Dr. Paoluzzi and Dr. Maki.
They pointed to one study that included 50 patients with sarcoma with 14 different subtypes of STS who were enrolled in immunotherapy trials conducted at the University of Texas MD Anderson Cancer Center, Houston. There were two pretreated patients with ASPS (two to four prior lines) in the cohort who received antiprogrammed death-ligand 1–based therapy, and achieved a partial response bordering on a complete response that lasted 8 and 12 months. An additional two patients achieved stable disease.
Another paper, presented at the 2017 Connective Tissue Oncology Society annual meeting, presented preliminary data from a phase 2 study that showed four of nine evaluable patients with ASPS treated with the TKI axitinib, combined with pembrolizumab, achieved a partial response. Three others had stable disease.
“Pathway-driven basket trials facilitate the enrollment of patients with such uncommon cancers and should provide valuable information regarding a second type of immune responsiveness to ICIs, one that is not a function of high tumor mutational burden,” the authors concluded.
No outside funding sources were reported. Dr. Maki reported receiving consultant fees from numerous sources and research support to New York University from Immune Design, Immunocore, Eli Lilly, Presage Biosciences, TRACON Pharmaceuticals, SARC, Regeneron, and Genentech. No other conflicts were reported.
SOURCE: doi: 10.1001/jamaoncol.2018.4490.
Alveolar soft part sarcoma (ASPS) has often proven to be resistant to conventional doxorubicin-based chemotherapy, but tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs) may provide new treatment strategies for this rare type of sarcoma, according to a literature review.
A rare, translocation-driven sarcoma of the soft tissues, ASPS often affects young adults and is characterized by indolent behavior and early metastasis. Despite its resistance to chemotherapy, studies indicate that survival is often prolonged in patients with metastatic disease. The literature has shown 5-year survival rates at about 60%, and this percentage has remained fairly consistent for the past 3 decades.
Luca Paoluzzi, MD, of New York University, and Robert G. Maki, MD, PhD, of Hofstra University, Hempstead, N.Y., reviewed the literature from 1952 to March 2018, in order to gain a better understanding of ASPS and the opportunities “for the translation of such knowledge into clinical practice,” they wrote in JAMA.
From a therapeutic standpoint, ASPS is characterized by sensitivity to vascular endothelial growth factor receptor–predominant TKIs, compared with other soft tissue sarcomas (STS), and recent data have emphasized that it is responsive to new immunotherapy regimens including ICIs. Pazopanib is currently the only agent that has received regulatory approval for use in STS refractory to other treatments and it appears to have consistent activity in metastatic ASPS. Management of ASPS generally also involves surgical resection and/or systemic treatment for metastatic disease. Conventional agents such as anthracycline-based chemotherapy have demonstrated a poor response rate lower than 10%, and while a complete resection may be curative, metastases are common and can occur years after resection of the primary tumor.
Conversely, ICIs “represent a promising area of drug development in ASPS; the data to date are limited but encouraging,” wrote Dr. Paoluzzi and Dr. Maki.
They pointed to one study that included 50 patients with sarcoma with 14 different subtypes of STS who were enrolled in immunotherapy trials conducted at the University of Texas MD Anderson Cancer Center, Houston. There were two pretreated patients with ASPS (two to four prior lines) in the cohort who received antiprogrammed death-ligand 1–based therapy, and achieved a partial response bordering on a complete response that lasted 8 and 12 months. An additional two patients achieved stable disease.
Another paper, presented at the 2017 Connective Tissue Oncology Society annual meeting, presented preliminary data from a phase 2 study that showed four of nine evaluable patients with ASPS treated with the TKI axitinib, combined with pembrolizumab, achieved a partial response. Three others had stable disease.
“Pathway-driven basket trials facilitate the enrollment of patients with such uncommon cancers and should provide valuable information regarding a second type of immune responsiveness to ICIs, one that is not a function of high tumor mutational burden,” the authors concluded.
No outside funding sources were reported. Dr. Maki reported receiving consultant fees from numerous sources and research support to New York University from Immune Design, Immunocore, Eli Lilly, Presage Biosciences, TRACON Pharmaceuticals, SARC, Regeneron, and Genentech. No other conflicts were reported.
SOURCE: doi: 10.1001/jamaoncol.2018.4490.
Alveolar soft part sarcoma (ASPS) has often proven to be resistant to conventional doxorubicin-based chemotherapy, but tyrosine kinase inhibitors (TKIs) and immune checkpoint inhibitors (ICIs) may provide new treatment strategies for this rare type of sarcoma, according to a literature review.
A rare, translocation-driven sarcoma of the soft tissues, ASPS often affects young adults and is characterized by indolent behavior and early metastasis. Despite its resistance to chemotherapy, studies indicate that survival is often prolonged in patients with metastatic disease. The literature has shown 5-year survival rates at about 60%, and this percentage has remained fairly consistent for the past 3 decades.
Luca Paoluzzi, MD, of New York University, and Robert G. Maki, MD, PhD, of Hofstra University, Hempstead, N.Y., reviewed the literature from 1952 to March 2018, in order to gain a better understanding of ASPS and the opportunities “for the translation of such knowledge into clinical practice,” they wrote in JAMA.
From a therapeutic standpoint, ASPS is characterized by sensitivity to vascular endothelial growth factor receptor–predominant TKIs, compared with other soft tissue sarcomas (STS), and recent data have emphasized that it is responsive to new immunotherapy regimens including ICIs. Pazopanib is currently the only agent that has received regulatory approval for use in STS refractory to other treatments and it appears to have consistent activity in metastatic ASPS. Management of ASPS generally also involves surgical resection and/or systemic treatment for metastatic disease. Conventional agents such as anthracycline-based chemotherapy have demonstrated a poor response rate lower than 10%, and while a complete resection may be curative, metastases are common and can occur years after resection of the primary tumor.
Conversely, ICIs “represent a promising area of drug development in ASPS; the data to date are limited but encouraging,” wrote Dr. Paoluzzi and Dr. Maki.
They pointed to one study that included 50 patients with sarcoma with 14 different subtypes of STS who were enrolled in immunotherapy trials conducted at the University of Texas MD Anderson Cancer Center, Houston. There were two pretreated patients with ASPS (two to four prior lines) in the cohort who received antiprogrammed death-ligand 1–based therapy, and achieved a partial response bordering on a complete response that lasted 8 and 12 months. An additional two patients achieved stable disease.
Another paper, presented at the 2017 Connective Tissue Oncology Society annual meeting, presented preliminary data from a phase 2 study that showed four of nine evaluable patients with ASPS treated with the TKI axitinib, combined with pembrolizumab, achieved a partial response. Three others had stable disease.
“Pathway-driven basket trials facilitate the enrollment of patients with such uncommon cancers and should provide valuable information regarding a second type of immune responsiveness to ICIs, one that is not a function of high tumor mutational burden,” the authors concluded.
No outside funding sources were reported. Dr. Maki reported receiving consultant fees from numerous sources and research support to New York University from Immune Design, Immunocore, Eli Lilly, Presage Biosciences, TRACON Pharmaceuticals, SARC, Regeneron, and Genentech. No other conflicts were reported.
SOURCE: doi: 10.1001/jamaoncol.2018.4490.
FROM JAMA
Key clinical point: Alveolar soft part sarcoma has often proven to be resistant to conventional doxorubicin-based chemotherapy, tyrosine kinase inhibitors and immune checkpoint inhibitors may provide new treatment strategies.
Major finding: In one study of sarcoma patients enrolled in immunotherapy trials, two pretreated patients with alveolar soft part sarcoma (two to four prior lines) who received antiprogrammed death-ligand 1–based therapy achieved partial responses, bordering on a complete response, that lasted 8 and 12 months.
Study details: A review of literature concerning treatment for alveolar soft part sarcoma.
Disclosures: No outside funding sources were reported. Dr. Maki reported receiving consultant fees from numerous sources and research support to New York University from Immune Design, Immunocore, Eli Lilly, Presage Biosciences, TRACON Pharmaceuticals, SARC, Regeneron, and Genentech. No other conflicts were reported.
Source:
Predicting treatment response in leiomyosarcoma, liposarcoma
Aberrations in oncogenic pathways and immune modulation influence treatment response in patients with metastatic leiomyosarcoma or liposarcoma, based on an analysis of whole-exome sequencing of tumor samples from patients in a completed phase 3 randomized trial comparing trabectedin and dacarbazine.
In that trial, trabectedin benefit was mostly seen in patients with leiomyosarcoma, as well as in patients with myxoid/round cell sarcomas, and less so in those with dedifferentiated and pleomorphic liposarcomas.
Gurpreet Kapoor, PhD, of LabConnect, Seattle, and colleagues examined aberrations in oncogenic pathways (DNA damage response, PI3K, MDM2-p53) and in immune modulation and then correlated the genomic aberrations with prospective data on clinical outcomes in the trial.
For the study, presented at the annual meeting of the American Society of Clinical Oncology in Chicago, archival tumor samples were collected from 456 of the 518 patients; 180 had uterine leiomyosarcomas, 149 had nonuterine leiomyosarcomas, 66 had dedifferentiated liposarcomas, 46 had myxoid liposarcomas, and 15 had pleomorphic liposarcomas.
Peripheral blood samples from a subset of 346 patients were also analyzed as matched normal to filter noise from nonpathogenic variants in the whole-exome sequencing.
Consistent with sarcoma data from The Cancer Genome Atlas, frequent homozygous gene deletions with relatively low mutational load were noted in these leiomyosarcoma and liposarcoma samples. TP53 and RB1 alterations were more frequent in leiomyosarcomas than in liposarcomas and were not associated with clinical outcomes. Analyses of 103 DNA damage-response genes found somatic alterations exceeded 20% across subtypes and correlated with improved progression-free survival in only uterine leiomyosarcomas (hazard ratio, 0.63; P = .03).
Genomic alterations in PI3K pathway genes were noted in 30% of myxoid liposarcomas and were associated with a worse rate of progression-free survival (HR, 3.0; P = .045).
A trend towards better overall survival was noted in dedifferentiated liposarcoma patients with MDM2 amplification as compared with normal MDM2 copy number.
Certain subtype-specific genomic aberrations in immune modulation pathways were associated with worse clinical outcomes in patients with uterine leiomyosarcoma or dedifferentiated liposarcoma. Alterations in immune suppressors were associated with improved clinical outcomes in nonuterine leiomyosarcomas and alterations in lipid metabolism were associated with improved clinical outcomes in dedifferentiated liposarcomas.
The invited discussant for the study, Mark Andrew Dickson, MD, of Memorial Sloan Kettering Cancer Center, New York, noted that “the real take-home here is that the TMBs (tumor mutation burdens) are relatively low across all of the L-type sarcomas.
“The pattern and prevalence of genomic aberrations that we’re seeing in this cohort of patients prospectively analyzed on a clinical trial are consistent with prior reports. ... including CDK4 and MDM2 in dedifferentiated liposarcoma, PI3-kinase in some myxoid/round cells, p53 in leiomyosarcoma and liposarcoma, and so on.”
Generally, tumor mutation burden is low in L-type sarcomas, and there are some intriguing associations with benefit to therapies, such as PI3-kinase pathway and potential resistance to trabectedin and high tumor mutation burden and potential sensitivity to trabectedin, that need to be explored and validated in another larger cohort, he said.
“I also am increasingly coming to terms with the fact that the tumors like leiomyosarcoma, which have low tumor mutation burden, and which so far have proven fairly immune to immunotherapy, based on all of the negative PD-1 data that we’ve seen, and that also have recurrent, relatively unactionable mutations, like p53 and Rb, remain very difficult to treat,” Dr. Dickson concluded.
SOURCE: Kapoor G et al. ASCO 2018, Abstract 11513.
Aberrations in oncogenic pathways and immune modulation influence treatment response in patients with metastatic leiomyosarcoma or liposarcoma, based on an analysis of whole-exome sequencing of tumor samples from patients in a completed phase 3 randomized trial comparing trabectedin and dacarbazine.
In that trial, trabectedin benefit was mostly seen in patients with leiomyosarcoma, as well as in patients with myxoid/round cell sarcomas, and less so in those with dedifferentiated and pleomorphic liposarcomas.
Gurpreet Kapoor, PhD, of LabConnect, Seattle, and colleagues examined aberrations in oncogenic pathways (DNA damage response, PI3K, MDM2-p53) and in immune modulation and then correlated the genomic aberrations with prospective data on clinical outcomes in the trial.
For the study, presented at the annual meeting of the American Society of Clinical Oncology in Chicago, archival tumor samples were collected from 456 of the 518 patients; 180 had uterine leiomyosarcomas, 149 had nonuterine leiomyosarcomas, 66 had dedifferentiated liposarcomas, 46 had myxoid liposarcomas, and 15 had pleomorphic liposarcomas.
Peripheral blood samples from a subset of 346 patients were also analyzed as matched normal to filter noise from nonpathogenic variants in the whole-exome sequencing.
Consistent with sarcoma data from The Cancer Genome Atlas, frequent homozygous gene deletions with relatively low mutational load were noted in these leiomyosarcoma and liposarcoma samples. TP53 and RB1 alterations were more frequent in leiomyosarcomas than in liposarcomas and were not associated with clinical outcomes. Analyses of 103 DNA damage-response genes found somatic alterations exceeded 20% across subtypes and correlated with improved progression-free survival in only uterine leiomyosarcomas (hazard ratio, 0.63; P = .03).
Genomic alterations in PI3K pathway genes were noted in 30% of myxoid liposarcomas and were associated with a worse rate of progression-free survival (HR, 3.0; P = .045).
A trend towards better overall survival was noted in dedifferentiated liposarcoma patients with MDM2 amplification as compared with normal MDM2 copy number.
Certain subtype-specific genomic aberrations in immune modulation pathways were associated with worse clinical outcomes in patients with uterine leiomyosarcoma or dedifferentiated liposarcoma. Alterations in immune suppressors were associated with improved clinical outcomes in nonuterine leiomyosarcomas and alterations in lipid metabolism were associated with improved clinical outcomes in dedifferentiated liposarcomas.
The invited discussant for the study, Mark Andrew Dickson, MD, of Memorial Sloan Kettering Cancer Center, New York, noted that “the real take-home here is that the TMBs (tumor mutation burdens) are relatively low across all of the L-type sarcomas.
“The pattern and prevalence of genomic aberrations that we’re seeing in this cohort of patients prospectively analyzed on a clinical trial are consistent with prior reports. ... including CDK4 and MDM2 in dedifferentiated liposarcoma, PI3-kinase in some myxoid/round cells, p53 in leiomyosarcoma and liposarcoma, and so on.”
Generally, tumor mutation burden is low in L-type sarcomas, and there are some intriguing associations with benefit to therapies, such as PI3-kinase pathway and potential resistance to trabectedin and high tumor mutation burden and potential sensitivity to trabectedin, that need to be explored and validated in another larger cohort, he said.
“I also am increasingly coming to terms with the fact that the tumors like leiomyosarcoma, which have low tumor mutation burden, and which so far have proven fairly immune to immunotherapy, based on all of the negative PD-1 data that we’ve seen, and that also have recurrent, relatively unactionable mutations, like p53 and Rb, remain very difficult to treat,” Dr. Dickson concluded.
SOURCE: Kapoor G et al. ASCO 2018, Abstract 11513.
Aberrations in oncogenic pathways and immune modulation influence treatment response in patients with metastatic leiomyosarcoma or liposarcoma, based on an analysis of whole-exome sequencing of tumor samples from patients in a completed phase 3 randomized trial comparing trabectedin and dacarbazine.
In that trial, trabectedin benefit was mostly seen in patients with leiomyosarcoma, as well as in patients with myxoid/round cell sarcomas, and less so in those with dedifferentiated and pleomorphic liposarcomas.
Gurpreet Kapoor, PhD, of LabConnect, Seattle, and colleagues examined aberrations in oncogenic pathways (DNA damage response, PI3K, MDM2-p53) and in immune modulation and then correlated the genomic aberrations with prospective data on clinical outcomes in the trial.
For the study, presented at the annual meeting of the American Society of Clinical Oncology in Chicago, archival tumor samples were collected from 456 of the 518 patients; 180 had uterine leiomyosarcomas, 149 had nonuterine leiomyosarcomas, 66 had dedifferentiated liposarcomas, 46 had myxoid liposarcomas, and 15 had pleomorphic liposarcomas.
Peripheral blood samples from a subset of 346 patients were also analyzed as matched normal to filter noise from nonpathogenic variants in the whole-exome sequencing.
Consistent with sarcoma data from The Cancer Genome Atlas, frequent homozygous gene deletions with relatively low mutational load were noted in these leiomyosarcoma and liposarcoma samples. TP53 and RB1 alterations were more frequent in leiomyosarcomas than in liposarcomas and were not associated with clinical outcomes. Analyses of 103 DNA damage-response genes found somatic alterations exceeded 20% across subtypes and correlated with improved progression-free survival in only uterine leiomyosarcomas (hazard ratio, 0.63; P = .03).
Genomic alterations in PI3K pathway genes were noted in 30% of myxoid liposarcomas and were associated with a worse rate of progression-free survival (HR, 3.0; P = .045).
A trend towards better overall survival was noted in dedifferentiated liposarcoma patients with MDM2 amplification as compared with normal MDM2 copy number.
Certain subtype-specific genomic aberrations in immune modulation pathways were associated with worse clinical outcomes in patients with uterine leiomyosarcoma or dedifferentiated liposarcoma. Alterations in immune suppressors were associated with improved clinical outcomes in nonuterine leiomyosarcomas and alterations in lipid metabolism were associated with improved clinical outcomes in dedifferentiated liposarcomas.
The invited discussant for the study, Mark Andrew Dickson, MD, of Memorial Sloan Kettering Cancer Center, New York, noted that “the real take-home here is that the TMBs (tumor mutation burdens) are relatively low across all of the L-type sarcomas.
“The pattern and prevalence of genomic aberrations that we’re seeing in this cohort of patients prospectively analyzed on a clinical trial are consistent with prior reports. ... including CDK4 and MDM2 in dedifferentiated liposarcoma, PI3-kinase in some myxoid/round cells, p53 in leiomyosarcoma and liposarcoma, and so on.”
Generally, tumor mutation burden is low in L-type sarcomas, and there are some intriguing associations with benefit to therapies, such as PI3-kinase pathway and potential resistance to trabectedin and high tumor mutation burden and potential sensitivity to trabectedin, that need to be explored and validated in another larger cohort, he said.
“I also am increasingly coming to terms with the fact that the tumors like leiomyosarcoma, which have low tumor mutation burden, and which so far have proven fairly immune to immunotherapy, based on all of the negative PD-1 data that we’ve seen, and that also have recurrent, relatively unactionable mutations, like p53 and Rb, remain very difficult to treat,” Dr. Dickson concluded.
SOURCE: Kapoor G et al. ASCO 2018, Abstract 11513.
REPORTING FROM ASCO 2018
Key clinical point: Aberrations in oncogenic pathways and immune modulation influence treatment response in patients with metastatic leiomyosarcoma or liposarcoma.
Major finding: Genomic alterations in PI3K pathway genes were noted in 30% of myxoid liposarcomas and were associated with a worse rate of progression-free survival (HR, 3.0; P = .045).
Study details: Archival tumor samples were collected from 456 of the 518 patients; 180 had uterine leiomyosarcomas, 149 had nonuterine leiomyosarcomas, 66 had dedifferentiated liposarcomas, 46 had myxoid liposarcomas, and 15 had pleomorphic liposarcomas in the completed phase 3 randomized trial comparing trabectedin and dacarbazine.
Disclosures: Dr. Kapoor is employed by LabConnect, Seattle. Research funding was supplied by Janssen Research & Development.
Source: Kapoor G et al. ASCO 2018, Abstract 11513.
FDA lifts partial hold on tazemetostat trials
The U.S. Food and Drug Administration has lifted the partial clinical hold on trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas, according to a press release from the drug’s developer Epizyme.

The patient had been on study for approximately 15 months and had achieved a confirmed partial response. The patient has since discontinued tazemetostat and responded to treatment for T-LBL.
“This remains the only case of T-LBL we’ve seen in more than 750 patients treated with tazemetostat,” Robert Bazemore, president and chief executive officer of Epizyme, said in a webcast on Sept. 24.
Epizyme assessed the risk of secondary malignancies, including T-LBL, as well as the overall risks and benefits of tazemetostat treatment, conducting a review of the published literature and an examination of efficacy and safety data across all of its tazemetostat trials. A panel of external scientific and medical experts who reviewed the findings concluded that T-LBL risks appear to be confined to pediatric patients who received higher doses of the drug. The phase 1 pediatric study in which the patient developed T-LBL included higher doses of tazemetostat than those used in the phase 2 adult studies.
“The team at Epizyme has worked diligently in collaboration with external experts and the FDA over the past several months,” Mr. Bazemore said.
The company is not making any substantial changes to trial designs or the patient populations involved in tazemetostat trials. However, Epizyme is modifying dosing in the pediatric studies, improving patient monitoring, and making changes to exclusion criteria to reduce the potential risk of T-LBL and other secondary malignancies. Mr. Bazemore said Epizyme hopes to submit a New Drug Application for tazemetostat in the treatment of epithelioid sarcoma.
Tazemetostat is under investigation as monotherapy in phase 2 trials of follicular lymphoma and solid-tumor malignancies. The drug is also being studied as part of combination therapy for non–small cell lung cancer and diffuse large B-cell lymphoma (DLBCL).
In August, Epizyme announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with DLBCL. However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Epizyme is now working to resolve partial clinical holds placed on tazemetostat in France and Germany in order to resume trial enrollment in those countries.
The U.S. Food and Drug Administration has lifted the partial clinical hold on trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas, according to a press release from the drug’s developer Epizyme.
The patient had been on study for approximately 15 months and had achieved a confirmed partial response. The patient has since discontinued tazemetostat and responded to treatment for T-LBL.
“This remains the only case of T-LBL we’ve seen in more than 750 patients treated with tazemetostat,” Robert Bazemore, president and chief executive officer of Epizyme, said in a webcast on Sept. 24.
Epizyme assessed the risk of secondary malignancies, including T-LBL, as well as the overall risks and benefits of tazemetostat treatment, conducting a review of the published literature and an examination of efficacy and safety data across all of its tazemetostat trials. A panel of external scientific and medical experts who reviewed the findings concluded that T-LBL risks appear to be confined to pediatric patients who received higher doses of the drug. The phase 1 pediatric study in which the patient developed T-LBL included higher doses of tazemetostat than those used in the phase 2 adult studies.
“The team at Epizyme has worked diligently in collaboration with external experts and the FDA over the past several months,” Mr. Bazemore said.
The company is not making any substantial changes to trial designs or the patient populations involved in tazemetostat trials. However, Epizyme is modifying dosing in the pediatric studies, improving patient monitoring, and making changes to exclusion criteria to reduce the potential risk of T-LBL and other secondary malignancies. Mr. Bazemore said Epizyme hopes to submit a New Drug Application for tazemetostat in the treatment of epithelioid sarcoma.
Tazemetostat is under investigation as monotherapy in phase 2 trials of follicular lymphoma and solid-tumor malignancies. The drug is also being studied as part of combination therapy for non–small cell lung cancer and diffuse large B-cell lymphoma (DLBCL).
In August, Epizyme announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with DLBCL. However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Epizyme is now working to resolve partial clinical holds placed on tazemetostat in France and Germany in order to resume trial enrollment in those countries.
The U.S. Food and Drug Administration has lifted the partial clinical hold on trials of tazemetostat, an EZH2 inhibitor being developed to treat solid tumors and lymphomas, according to a press release from the drug’s developer Epizyme.
The patient had been on study for approximately 15 months and had achieved a confirmed partial response. The patient has since discontinued tazemetostat and responded to treatment for T-LBL.
“This remains the only case of T-LBL we’ve seen in more than 750 patients treated with tazemetostat,” Robert Bazemore, president and chief executive officer of Epizyme, said in a webcast on Sept. 24.
Epizyme assessed the risk of secondary malignancies, including T-LBL, as well as the overall risks and benefits of tazemetostat treatment, conducting a review of the published literature and an examination of efficacy and safety data across all of its tazemetostat trials. A panel of external scientific and medical experts who reviewed the findings concluded that T-LBL risks appear to be confined to pediatric patients who received higher doses of the drug. The phase 1 pediatric study in which the patient developed T-LBL included higher doses of tazemetostat than those used in the phase 2 adult studies.
“The team at Epizyme has worked diligently in collaboration with external experts and the FDA over the past several months,” Mr. Bazemore said.
The company is not making any substantial changes to trial designs or the patient populations involved in tazemetostat trials. However, Epizyme is modifying dosing in the pediatric studies, improving patient monitoring, and making changes to exclusion criteria to reduce the potential risk of T-LBL and other secondary malignancies. Mr. Bazemore said Epizyme hopes to submit a New Drug Application for tazemetostat in the treatment of epithelioid sarcoma.
Tazemetostat is under investigation as monotherapy in phase 2 trials of follicular lymphoma and solid-tumor malignancies. The drug is also being studied as part of combination therapy for non–small cell lung cancer and diffuse large B-cell lymphoma (DLBCL).
In August, Epizyme announced its decision to stop developing tazemetostat for use as monotherapy or in combination with prednisolone for patients with DLBCL. However, tazemetostat is still under investigation as a potential treatment for DLBCL as part of other combination regimens.
Epizyme is now working to resolve partial clinical holds placed on tazemetostat in France and Germany in order to resume trial enrollment in those countries.
Improved survival in liposarcoma
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
PARP inhibitor plus trabectedin shows promise for sarcoma
A combination of trabectedin and the PARP inhibitor olaparib may be a safe and effective therapy for patients with sarcoma, the recent TOMAS trial found.
High PARP1 expression was associated with treatment response, reported Giovanni Grignani, MD, of the Medical Oncology_Sarcoma Unit at Istituto di Ricovero e Cura a Carattere Scientifico in Candiolo, Italy, and his colleagues.
PARP inhibitors prevent repair of DNA damage, suggesting potential synergisms with DNA-damaging anticancer agents. Preclinical models support this strategy; however, clinical trials have found that toxicities restrict doses below antitumor activity levels.
“In view of these findings, trabectedin could be an ideal drug to use in combination with PARP1/2 inhibitors for two reasons: its favourable haemopoietic toxicity profile and its unique mechanisms of action,” the authors wrote in The Lancet Oncology. Trabectedin bends the minor groove of DNA toward the major groove, which activates PARP1 in an attempt to repair the damage. Preclinical trials showed that a PARP inhibitor such as olaparib would block this PARP1 activation, ultimately resulting in a more robust response than with either drug alone.
The phase 1b, open-label TOMAS trial involved 50 patients with sarcoma who had experienced disease progression after standard therapy. The study was divided into two cohorts: dose-escalation and dose-expansion. Patients received a median of four cycles of therapy with a median follow-up of 10 months (some patients are still undergoing treatment). The primary endpoint was maximum tolerated dose. The investigators also evaluated pharmacokinetics, pharmacodynamics, and various response measures.
Although adverse events occurred, these were manageable, and the authors concluded that the combination is safe for further investigation. The most common grade 3 or higher adverse events were lymphopenia (64%), neutropenia (62%), thrombocytopenia (28%), anemia (26%), hypophosphatemia (40%), and alanine aminotransferase elevation (18%). The maximum tolerated dose (recommended phase 2 dose) was olaparib 150 mg twice daily and trabectedin 1.1 mg/m2 every 3 weeks.
“These doses allowed us to minimize the need for dose reductions and continue treatment for as long as tumour control was maintained,” the authors wrote. Previous treatments impacted tolerability. The researchers noted that “patients who had received more than two lines of therapy had a higher risk of developing dose-limiting toxicities than those patients who had been treated with only one line of therapy.”
Overall, 14% of patients responded to therapy. Six-month progression-free survival was more common in patients with soft tissue sarcoma (38%) than other tumor types. More patients with high PARP1 expression achieved 6-month PFS compared with patients who had low PARP1 expression (59% vs. 8%; P = .01).
“The combination of olaparib and trabectedin exploits the potential of two different first-in-class drugs and shows tolerability and activity in homologous repair-proficient tumors,” the authors concluded.
They are planning two phase 2 studies in the future; one “comparing trabectedin alone versus the combination of trabectedin and olaparib, stratifying patients according to PARP1 expression,” and an “after-platinum-failure study of patients with ovarian cancer regardless of patients’ BRCA1/2 and BRCAness status.”
The TOMAS trial was funded by the Italian Association for Cancer Research, the Foundation for Research on Musculoskeletal and Rare Tumors, the Italian Ministry of Health, and PharmaMar. The authors reported compensation from Lilly, Novartis, Bayer, Eisai, Amgen, and others.
SOURCE: Grignani et al. Lancet Oncol. 2018 Sep 11. doi: 10.1016/S1470-2045(18)30438-8.
The phase 1b TOMAS trial by Grignani et al. showed that PARP inhibitor combination therapy may be a safe and effective option for patients with sarcoma, and a phase 2 study is warranted, according to Benjamin A. Nacev, MD, and William D. Tap, MD.
PARP inhibitors mitigate DNA damage repair, suggesting potential for synergistic combinations with DNA-damaging anticancer agents. Unfortunately, previous combinations have revealed toxicity issues.
“The first clinical example of this approach was the combination of the alkylating drug temozolomide and the PARP inhibitor rucaparib, which was hampered by dose-limiting myelosuppression,” Dr. Nacev and Dr. Tap wrote in an editorial in The Lancet Oncology.
In the TOMAS trial, Grignani et al. assessed a combination of trabectedin and the PARP inhibitor olaparib. Preclinical data showed synergistic activity in sarcoma cell lines, and the authors predicted tolerable myelosuppression with trabectedin.
Their predictions yielded promising results: Approximately one-third of patients with soft-tissue sarcoma were progression free at 6 months. Although myelosuppression did occur, the adverse event profile was tolerable.
As drug synergisms are biologically complex, “a key success of the TOMAS trial is the effective use of exploratory pharmacodynamic endpoints including PARP1 expression, PARylation, and mutational status of the DNA damage repair pathway.”
“For example, efficacy in the TOMAS trial correlated with PARP1 expression, with greater 6-month progression-free survival in the high PARP1 expression group than the low expression group.”
“The TOMAS investigators should be commended for doing the important bench-to-bedside approach of rationally designing and testing a drug combination to leverage available active drugs. We agree with the authors’ call for further investigation of trabectedin and olaparib in a randomised phase 2 trial in soft tissue sarcoma.”
William D. Tap, MD is chief of the Sarcoma Medical Oncology Service and Benjamin A. Nacev, MD is a third-year medical oncology/hematology fellow at Memorial Sloan Kettering Cancer Center in New York. Dr. Tap reported personal fees from Eli Lilly, Novartis, Eisai, and others. These comments are adapted from their accompanying editorial .
The phase 1b TOMAS trial by Grignani et al. showed that PARP inhibitor combination therapy may be a safe and effective option for patients with sarcoma, and a phase 2 study is warranted, according to Benjamin A. Nacev, MD, and William D. Tap, MD.
PARP inhibitors mitigate DNA damage repair, suggesting potential for synergistic combinations with DNA-damaging anticancer agents. Unfortunately, previous combinations have revealed toxicity issues.
“The first clinical example of this approach was the combination of the alkylating drug temozolomide and the PARP inhibitor rucaparib, which was hampered by dose-limiting myelosuppression,” Dr. Nacev and Dr. Tap wrote in an editorial in The Lancet Oncology.
In the TOMAS trial, Grignani et al. assessed a combination of trabectedin and the PARP inhibitor olaparib. Preclinical data showed synergistic activity in sarcoma cell lines, and the authors predicted tolerable myelosuppression with trabectedin.
Their predictions yielded promising results: Approximately one-third of patients with soft-tissue sarcoma were progression free at 6 months. Although myelosuppression did occur, the adverse event profile was tolerable.
As drug synergisms are biologically complex, “a key success of the TOMAS trial is the effective use of exploratory pharmacodynamic endpoints including PARP1 expression, PARylation, and mutational status of the DNA damage repair pathway.”
“For example, efficacy in the TOMAS trial correlated with PARP1 expression, with greater 6-month progression-free survival in the high PARP1 expression group than the low expression group.”
“The TOMAS investigators should be commended for doing the important bench-to-bedside approach of rationally designing and testing a drug combination to leverage available active drugs. We agree with the authors’ call for further investigation of trabectedin and olaparib in a randomised phase 2 trial in soft tissue sarcoma.”
William D. Tap, MD is chief of the Sarcoma Medical Oncology Service and Benjamin A. Nacev, MD is a third-year medical oncology/hematology fellow at Memorial Sloan Kettering Cancer Center in New York. Dr. Tap reported personal fees from Eli Lilly, Novartis, Eisai, and others. These comments are adapted from their accompanying editorial .
The phase 1b TOMAS trial by Grignani et al. showed that PARP inhibitor combination therapy may be a safe and effective option for patients with sarcoma, and a phase 2 study is warranted, according to Benjamin A. Nacev, MD, and William D. Tap, MD.
PARP inhibitors mitigate DNA damage repair, suggesting potential for synergistic combinations with DNA-damaging anticancer agents. Unfortunately, previous combinations have revealed toxicity issues.
“The first clinical example of this approach was the combination of the alkylating drug temozolomide and the PARP inhibitor rucaparib, which was hampered by dose-limiting myelosuppression,” Dr. Nacev and Dr. Tap wrote in an editorial in The Lancet Oncology.
In the TOMAS trial, Grignani et al. assessed a combination of trabectedin and the PARP inhibitor olaparib. Preclinical data showed synergistic activity in sarcoma cell lines, and the authors predicted tolerable myelosuppression with trabectedin.
Their predictions yielded promising results: Approximately one-third of patients with soft-tissue sarcoma were progression free at 6 months. Although myelosuppression did occur, the adverse event profile was tolerable.
As drug synergisms are biologically complex, “a key success of the TOMAS trial is the effective use of exploratory pharmacodynamic endpoints including PARP1 expression, PARylation, and mutational status of the DNA damage repair pathway.”
“For example, efficacy in the TOMAS trial correlated with PARP1 expression, with greater 6-month progression-free survival in the high PARP1 expression group than the low expression group.”
“The TOMAS investigators should be commended for doing the important bench-to-bedside approach of rationally designing and testing a drug combination to leverage available active drugs. We agree with the authors’ call for further investigation of trabectedin and olaparib in a randomised phase 2 trial in soft tissue sarcoma.”
William D. Tap, MD is chief of the Sarcoma Medical Oncology Service and Benjamin A. Nacev, MD is a third-year medical oncology/hematology fellow at Memorial Sloan Kettering Cancer Center in New York. Dr. Tap reported personal fees from Eli Lilly, Novartis, Eisai, and others. These comments are adapted from their accompanying editorial .
A combination of trabectedin and the PARP inhibitor olaparib may be a safe and effective therapy for patients with sarcoma, the recent TOMAS trial found.
High PARP1 expression was associated with treatment response, reported Giovanni Grignani, MD, of the Medical Oncology_Sarcoma Unit at Istituto di Ricovero e Cura a Carattere Scientifico in Candiolo, Italy, and his colleagues.
PARP inhibitors prevent repair of DNA damage, suggesting potential synergisms with DNA-damaging anticancer agents. Preclinical models support this strategy; however, clinical trials have found that toxicities restrict doses below antitumor activity levels.
“In view of these findings, trabectedin could be an ideal drug to use in combination with PARP1/2 inhibitors for two reasons: its favourable haemopoietic toxicity profile and its unique mechanisms of action,” the authors wrote in The Lancet Oncology. Trabectedin bends the minor groove of DNA toward the major groove, which activates PARP1 in an attempt to repair the damage. Preclinical trials showed that a PARP inhibitor such as olaparib would block this PARP1 activation, ultimately resulting in a more robust response than with either drug alone.
The phase 1b, open-label TOMAS trial involved 50 patients with sarcoma who had experienced disease progression after standard therapy. The study was divided into two cohorts: dose-escalation and dose-expansion. Patients received a median of four cycles of therapy with a median follow-up of 10 months (some patients are still undergoing treatment). The primary endpoint was maximum tolerated dose. The investigators also evaluated pharmacokinetics, pharmacodynamics, and various response measures.
Although adverse events occurred, these were manageable, and the authors concluded that the combination is safe for further investigation. The most common grade 3 or higher adverse events were lymphopenia (64%), neutropenia (62%), thrombocytopenia (28%), anemia (26%), hypophosphatemia (40%), and alanine aminotransferase elevation (18%). The maximum tolerated dose (recommended phase 2 dose) was olaparib 150 mg twice daily and trabectedin 1.1 mg/m2 every 3 weeks.
“These doses allowed us to minimize the need for dose reductions and continue treatment for as long as tumour control was maintained,” the authors wrote. Previous treatments impacted tolerability. The researchers noted that “patients who had received more than two lines of therapy had a higher risk of developing dose-limiting toxicities than those patients who had been treated with only one line of therapy.”
Overall, 14% of patients responded to therapy. Six-month progression-free survival was more common in patients with soft tissue sarcoma (38%) than other tumor types. More patients with high PARP1 expression achieved 6-month PFS compared with patients who had low PARP1 expression (59% vs. 8%; P = .01).
“The combination of olaparib and trabectedin exploits the potential of two different first-in-class drugs and shows tolerability and activity in homologous repair-proficient tumors,” the authors concluded.
They are planning two phase 2 studies in the future; one “comparing trabectedin alone versus the combination of trabectedin and olaparib, stratifying patients according to PARP1 expression,” and an “after-platinum-failure study of patients with ovarian cancer regardless of patients’ BRCA1/2 and BRCAness status.”
The TOMAS trial was funded by the Italian Association for Cancer Research, the Foundation for Research on Musculoskeletal and Rare Tumors, the Italian Ministry of Health, and PharmaMar. The authors reported compensation from Lilly, Novartis, Bayer, Eisai, Amgen, and others.
SOURCE: Grignani et al. Lancet Oncol. 2018 Sep 11. doi: 10.1016/S1470-2045(18)30438-8.
A combination of trabectedin and the PARP inhibitor olaparib may be a safe and effective therapy for patients with sarcoma, the recent TOMAS trial found.
High PARP1 expression was associated with treatment response, reported Giovanni Grignani, MD, of the Medical Oncology_Sarcoma Unit at Istituto di Ricovero e Cura a Carattere Scientifico in Candiolo, Italy, and his colleagues.
PARP inhibitors prevent repair of DNA damage, suggesting potential synergisms with DNA-damaging anticancer agents. Preclinical models support this strategy; however, clinical trials have found that toxicities restrict doses below antitumor activity levels.
“In view of these findings, trabectedin could be an ideal drug to use in combination with PARP1/2 inhibitors for two reasons: its favourable haemopoietic toxicity profile and its unique mechanisms of action,” the authors wrote in The Lancet Oncology. Trabectedin bends the minor groove of DNA toward the major groove, which activates PARP1 in an attempt to repair the damage. Preclinical trials showed that a PARP inhibitor such as olaparib would block this PARP1 activation, ultimately resulting in a more robust response than with either drug alone.
The phase 1b, open-label TOMAS trial involved 50 patients with sarcoma who had experienced disease progression after standard therapy. The study was divided into two cohorts: dose-escalation and dose-expansion. Patients received a median of four cycles of therapy with a median follow-up of 10 months (some patients are still undergoing treatment). The primary endpoint was maximum tolerated dose. The investigators also evaluated pharmacokinetics, pharmacodynamics, and various response measures.
Although adverse events occurred, these were manageable, and the authors concluded that the combination is safe for further investigation. The most common grade 3 or higher adverse events were lymphopenia (64%), neutropenia (62%), thrombocytopenia (28%), anemia (26%), hypophosphatemia (40%), and alanine aminotransferase elevation (18%). The maximum tolerated dose (recommended phase 2 dose) was olaparib 150 mg twice daily and trabectedin 1.1 mg/m2 every 3 weeks.
“These doses allowed us to minimize the need for dose reductions and continue treatment for as long as tumour control was maintained,” the authors wrote. Previous treatments impacted tolerability. The researchers noted that “patients who had received more than two lines of therapy had a higher risk of developing dose-limiting toxicities than those patients who had been treated with only one line of therapy.”
Overall, 14% of patients responded to therapy. Six-month progression-free survival was more common in patients with soft tissue sarcoma (38%) than other tumor types. More patients with high PARP1 expression achieved 6-month PFS compared with patients who had low PARP1 expression (59% vs. 8%; P = .01).
“The combination of olaparib and trabectedin exploits the potential of two different first-in-class drugs and shows tolerability and activity in homologous repair-proficient tumors,” the authors concluded.
They are planning two phase 2 studies in the future; one “comparing trabectedin alone versus the combination of trabectedin and olaparib, stratifying patients according to PARP1 expression,” and an “after-platinum-failure study of patients with ovarian cancer regardless of patients’ BRCA1/2 and BRCAness status.”
The TOMAS trial was funded by the Italian Association for Cancer Research, the Foundation for Research on Musculoskeletal and Rare Tumors, the Italian Ministry of Health, and PharmaMar. The authors reported compensation from Lilly, Novartis, Bayer, Eisai, Amgen, and others.
SOURCE: Grignani et al. Lancet Oncol. 2018 Sep 11. doi: 10.1016/S1470-2045(18)30438-8.
FROM THE LANCET ONCOLOGY
Key clinical point: A combination of trabectedin and the PARP inhibitor olaparib may be a safe and effective therapy for patients with sarcoma.
Major finding: Of those with high PARP1 expression, 59% were progression free 6 months after treatment.
Study details: TOMAS was an open-label phase 1b trial involving 50 patients with sarcoma who had disease progression after standard therapy.
Disclosures: The study was funded by the Italian Association for Cancer Research, the Foundation for Research on Musculoskeletal and Rare Tumors, the Italian Ministry of Health, and PharmaMar. The authors reported compensation from Lilly, Novartis, Bayer, Eisai, Amgen, and others.
Source: Grignani et al. Lancet Oncol. 2018 Sep 11. doi: 10.1016/S1470-2045(18)30438-8.