User login
High early recurrence rates with Merkel cell carcinoma
, and more than half of all patients with stage IV disease will have a recurrence within 1 year of definitive therapy, results of a new study show.
A study of 618 patients with MCC who were enrolled in a Seattle-based data repository shows that among all patients the 5-year recurrence rate was 40%. The risk of recurrence within the first year was 11% for patients with pathologic stage I disease, 33% for those with stage IIA/IIB disease, 45% for those with stage IIIB disease, and 58% for patients with pathologic stage IV MCC.

Approximately 95% of all recurrences happened within 3 years of the initial diagnosis, report Aubriana McEvoy, MD, from the University of Washington in Seattle, and colleagues.
“This cohort study indicates that the highest yield (and likely most cost-effective) time period for detecting MCC recurrence is 1-3 years after diagnosis,” they write in a study published online in JAMA Dermatology.
The estimated annual incidence of MCC in the United States in 2018 was 2,000 according to the American Cancer Society. The annual incidence rate is rising rapidly, however, and is estimated to reach 3,284 by 2025, McEvoy and colleagues write.
Although MCC is known to have high recurrence rates and is associated with a higher mortality rate than malignant melanoma, recurrence rate data are not captured by either the Surveillance, Epidemiology, and End Results (SEER) database or by the National Cancer Database. As a result, estimates of recurrence rates with MCC have been all over the map, ranging from 27% to 77%, depending on the population studied.
But as senior author Paul Nghiem, MD, PhD, professor and chair of dermatology at the University of Washington, Seattle, told this news organization, recurrence rates over time in their study were remarkably consistent.
“The biggest surprise to me was that, when we broke our nearly 20-year cohort into three 5- or 6-year chunks, every one of the groups had a 40% recurrence rate, within 1%. So we feel really confident that’s the right number,” he said.
Dr. Nghiem and colleagues report that, in contrast to patients with MCC, approximately 19% of patients with melanoma will have a recurrence, as will an estimated 5%-9% of patients with squamous cell carcinoma and 1%-10% of patients with basal cell carcinoma.
The fact that recurrence rates of MCC have remained stable over time despite presumed improvements in definitive therapy is disappointing, Dr. Nghiem acknowledged. He noted that it’s still unclear whether immunotherapy will have the same dramatic effect on survival rates for patients with MCC as it has for patients with malignant melanoma.
The high recurrence rates following definitive therapy for patients with early-stage disease was a novel finding, commented Shawn Demehri, MD, PhD, director of the high-risk skin cancer clinic at Massachusetts General Hospital in Boston.

“When you’re looking at patients with stage I or stage II, and they have definitive surgery but still have recurrences at a higher rate than melanoma, it brings home the point that these are among the most aggressive tumors of the skin,” he said in an interview.
The high recurrence rates seen with MCC are attributable to a variety of factors.
“This is a rare cancer of mostly older individuals with a lot of comorbidities, and also a cancer that, even though it is a primary cancer, might be detected a little later than even a melanoma primary tumor, just because of the nature of the neuroendocrine tumor cells,” he said.
Dr. Demehri was not involved in the study.
Prospective cohort
The study cohort consisted of 618 patients with MCC. The median age of the patients was 69, and 227 (37%) were women. The patients were enrolled within 6 months of their diagnosis in the prospective data repository from 2003 through 2019. Of this group, 223 had a recurrence of MCC.
As noted, there was a high risk of recurrence within 1 year, ranging from 11% for patients with pathologic stage I tumors to 58% for those with stage IV disease, and 95% of all recurrences occurred within 3 years of definitive therapy.
To get a better picture of the natural history of MCC recurrence, the investigators studied a cohort of patients with pathologically confirmed MCC who were prospectively enrolled from January 2003 through April 2019 in a data repository maintained at the University of Washington.
In addition to disease stage, factors associated with increased recurrence risk in univariable analyses include immunosuppression (hazard ratio, 2.4; P < .001), male sex (HR, 1.9; P < .001), known primary lesion among patients with clinically detectable nodal disease (HR, 2.3; P = .001), and older age (HR, 1.1, P = .06 for each 10-year increase).
Of the 187 patients in the cohort who died during the study, 121 died from MCC. At 4 years after diagnosis, MCC-specific survival rates were 95% for patients with pathologic stage I, 84% with stage IIA/IIB, 80% with stage IIIA, 58% with stage IIIB, and 41% with stage IV.
Evidence supports close monitoring within the first 3 years for patients with stage I-II MCC. Local recurrence within or adjacent to the primary tumor scar was associated with a 5-year MCC-specific survival rate of 85%, compared with 88% of patients with stage I or II disease who did not have recurrences.
“Because more than 90% of MCC recurrences arise within 3 years, it is appropriate to adjust surveillance intensity accordingly. Stage- and time-specific recurrence data can assist in appropriately focusing surveillance resources on patients and time intervals in which recurrence risk is highest,” the authors wrote.
“If you’re a patient who has not had your cancer come back for 3, 4, or 5 years, you can really cut down on the intensity of your follow-up and scans,” Dr. Nghiem said.
“We do now have two excellent blood tests that are working very well, and we have really good ways to detect the cancer coming back early, and that’s important, because we have potentially curative therapies that tend to work better if you catch the cancer early,” he said.
The study was supported by the National Institutes of Health. Dr. Nghiem reported personal fees and institutional support outside the study from several companies and patents for Merkel cell therapies with the University of Washington and University of Denmark. Dr. Demehri has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, and more than half of all patients with stage IV disease will have a recurrence within 1 year of definitive therapy, results of a new study show.
A study of 618 patients with MCC who were enrolled in a Seattle-based data repository shows that among all patients the 5-year recurrence rate was 40%. The risk of recurrence within the first year was 11% for patients with pathologic stage I disease, 33% for those with stage IIA/IIB disease, 45% for those with stage IIIB disease, and 58% for patients with pathologic stage IV MCC.
Approximately 95% of all recurrences happened within 3 years of the initial diagnosis, report Aubriana McEvoy, MD, from the University of Washington in Seattle, and colleagues.
“This cohort study indicates that the highest yield (and likely most cost-effective) time period for detecting MCC recurrence is 1-3 years after diagnosis,” they write in a study published online in JAMA Dermatology.
The estimated annual incidence of MCC in the United States in 2018 was 2,000 according to the American Cancer Society. The annual incidence rate is rising rapidly, however, and is estimated to reach 3,284 by 2025, McEvoy and colleagues write.
Although MCC is known to have high recurrence rates and is associated with a higher mortality rate than malignant melanoma, recurrence rate data are not captured by either the Surveillance, Epidemiology, and End Results (SEER) database or by the National Cancer Database. As a result, estimates of recurrence rates with MCC have been all over the map, ranging from 27% to 77%, depending on the population studied.
But as senior author Paul Nghiem, MD, PhD, professor and chair of dermatology at the University of Washington, Seattle, told this news organization, recurrence rates over time in their study were remarkably consistent.
“The biggest surprise to me was that, when we broke our nearly 20-year cohort into three 5- or 6-year chunks, every one of the groups had a 40% recurrence rate, within 1%. So we feel really confident that’s the right number,” he said.
Dr. Nghiem and colleagues report that, in contrast to patients with MCC, approximately 19% of patients with melanoma will have a recurrence, as will an estimated 5%-9% of patients with squamous cell carcinoma and 1%-10% of patients with basal cell carcinoma.
The fact that recurrence rates of MCC have remained stable over time despite presumed improvements in definitive therapy is disappointing, Dr. Nghiem acknowledged. He noted that it’s still unclear whether immunotherapy will have the same dramatic effect on survival rates for patients with MCC as it has for patients with malignant melanoma.
The high recurrence rates following definitive therapy for patients with early-stage disease was a novel finding, commented Shawn Demehri, MD, PhD, director of the high-risk skin cancer clinic at Massachusetts General Hospital in Boston.
“When you’re looking at patients with stage I or stage II, and they have definitive surgery but still have recurrences at a higher rate than melanoma, it brings home the point that these are among the most aggressive tumors of the skin,” he said in an interview.
The high recurrence rates seen with MCC are attributable to a variety of factors.
“This is a rare cancer of mostly older individuals with a lot of comorbidities, and also a cancer that, even though it is a primary cancer, might be detected a little later than even a melanoma primary tumor, just because of the nature of the neuroendocrine tumor cells,” he said.
Dr. Demehri was not involved in the study.
Prospective cohort
The study cohort consisted of 618 patients with MCC. The median age of the patients was 69, and 227 (37%) were women. The patients were enrolled within 6 months of their diagnosis in the prospective data repository from 2003 through 2019. Of this group, 223 had a recurrence of MCC.
As noted, there was a high risk of recurrence within 1 year, ranging from 11% for patients with pathologic stage I tumors to 58% for those with stage IV disease, and 95% of all recurrences occurred within 3 years of definitive therapy.
To get a better picture of the natural history of MCC recurrence, the investigators studied a cohort of patients with pathologically confirmed MCC who were prospectively enrolled from January 2003 through April 2019 in a data repository maintained at the University of Washington.
In addition to disease stage, factors associated with increased recurrence risk in univariable analyses include immunosuppression (hazard ratio, 2.4; P < .001), male sex (HR, 1.9; P < .001), known primary lesion among patients with clinically detectable nodal disease (HR, 2.3; P = .001), and older age (HR, 1.1, P = .06 for each 10-year increase).
Of the 187 patients in the cohort who died during the study, 121 died from MCC. At 4 years after diagnosis, MCC-specific survival rates were 95% for patients with pathologic stage I, 84% with stage IIA/IIB, 80% with stage IIIA, 58% with stage IIIB, and 41% with stage IV.
Evidence supports close monitoring within the first 3 years for patients with stage I-II MCC. Local recurrence within or adjacent to the primary tumor scar was associated with a 5-year MCC-specific survival rate of 85%, compared with 88% of patients with stage I or II disease who did not have recurrences.
“Because more than 90% of MCC recurrences arise within 3 years, it is appropriate to adjust surveillance intensity accordingly. Stage- and time-specific recurrence data can assist in appropriately focusing surveillance resources on patients and time intervals in which recurrence risk is highest,” the authors wrote.
“If you’re a patient who has not had your cancer come back for 3, 4, or 5 years, you can really cut down on the intensity of your follow-up and scans,” Dr. Nghiem said.
“We do now have two excellent blood tests that are working very well, and we have really good ways to detect the cancer coming back early, and that’s important, because we have potentially curative therapies that tend to work better if you catch the cancer early,” he said.
The study was supported by the National Institutes of Health. Dr. Nghiem reported personal fees and institutional support outside the study from several companies and patents for Merkel cell therapies with the University of Washington and University of Denmark. Dr. Demehri has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, and more than half of all patients with stage IV disease will have a recurrence within 1 year of definitive therapy, results of a new study show.
A study of 618 patients with MCC who were enrolled in a Seattle-based data repository shows that among all patients the 5-year recurrence rate was 40%. The risk of recurrence within the first year was 11% for patients with pathologic stage I disease, 33% for those with stage IIA/IIB disease, 45% for those with stage IIIB disease, and 58% for patients with pathologic stage IV MCC.
Approximately 95% of all recurrences happened within 3 years of the initial diagnosis, report Aubriana McEvoy, MD, from the University of Washington in Seattle, and colleagues.
“This cohort study indicates that the highest yield (and likely most cost-effective) time period for detecting MCC recurrence is 1-3 years after diagnosis,” they write in a study published online in JAMA Dermatology.
The estimated annual incidence of MCC in the United States in 2018 was 2,000 according to the American Cancer Society. The annual incidence rate is rising rapidly, however, and is estimated to reach 3,284 by 2025, McEvoy and colleagues write.
Although MCC is known to have high recurrence rates and is associated with a higher mortality rate than malignant melanoma, recurrence rate data are not captured by either the Surveillance, Epidemiology, and End Results (SEER) database or by the National Cancer Database. As a result, estimates of recurrence rates with MCC have been all over the map, ranging from 27% to 77%, depending on the population studied.
But as senior author Paul Nghiem, MD, PhD, professor and chair of dermatology at the University of Washington, Seattle, told this news organization, recurrence rates over time in their study were remarkably consistent.
“The biggest surprise to me was that, when we broke our nearly 20-year cohort into three 5- or 6-year chunks, every one of the groups had a 40% recurrence rate, within 1%. So we feel really confident that’s the right number,” he said.
Dr. Nghiem and colleagues report that, in contrast to patients with MCC, approximately 19% of patients with melanoma will have a recurrence, as will an estimated 5%-9% of patients with squamous cell carcinoma and 1%-10% of patients with basal cell carcinoma.
The fact that recurrence rates of MCC have remained stable over time despite presumed improvements in definitive therapy is disappointing, Dr. Nghiem acknowledged. He noted that it’s still unclear whether immunotherapy will have the same dramatic effect on survival rates for patients with MCC as it has for patients with malignant melanoma.
The high recurrence rates following definitive therapy for patients with early-stage disease was a novel finding, commented Shawn Demehri, MD, PhD, director of the high-risk skin cancer clinic at Massachusetts General Hospital in Boston.
“When you’re looking at patients with stage I or stage II, and they have definitive surgery but still have recurrences at a higher rate than melanoma, it brings home the point that these are among the most aggressive tumors of the skin,” he said in an interview.
The high recurrence rates seen with MCC are attributable to a variety of factors.
“This is a rare cancer of mostly older individuals with a lot of comorbidities, and also a cancer that, even though it is a primary cancer, might be detected a little later than even a melanoma primary tumor, just because of the nature of the neuroendocrine tumor cells,” he said.
Dr. Demehri was not involved in the study.
Prospective cohort
The study cohort consisted of 618 patients with MCC. The median age of the patients was 69, and 227 (37%) were women. The patients were enrolled within 6 months of their diagnosis in the prospective data repository from 2003 through 2019. Of this group, 223 had a recurrence of MCC.
As noted, there was a high risk of recurrence within 1 year, ranging from 11% for patients with pathologic stage I tumors to 58% for those with stage IV disease, and 95% of all recurrences occurred within 3 years of definitive therapy.
To get a better picture of the natural history of MCC recurrence, the investigators studied a cohort of patients with pathologically confirmed MCC who were prospectively enrolled from January 2003 through April 2019 in a data repository maintained at the University of Washington.
In addition to disease stage, factors associated with increased recurrence risk in univariable analyses include immunosuppression (hazard ratio, 2.4; P < .001), male sex (HR, 1.9; P < .001), known primary lesion among patients with clinically detectable nodal disease (HR, 2.3; P = .001), and older age (HR, 1.1, P = .06 for each 10-year increase).
Of the 187 patients in the cohort who died during the study, 121 died from MCC. At 4 years after diagnosis, MCC-specific survival rates were 95% for patients with pathologic stage I, 84% with stage IIA/IIB, 80% with stage IIIA, 58% with stage IIIB, and 41% with stage IV.
Evidence supports close monitoring within the first 3 years for patients with stage I-II MCC. Local recurrence within or adjacent to the primary tumor scar was associated with a 5-year MCC-specific survival rate of 85%, compared with 88% of patients with stage I or II disease who did not have recurrences.
“Because more than 90% of MCC recurrences arise within 3 years, it is appropriate to adjust surveillance intensity accordingly. Stage- and time-specific recurrence data can assist in appropriately focusing surveillance resources on patients and time intervals in which recurrence risk is highest,” the authors wrote.
“If you’re a patient who has not had your cancer come back for 3, 4, or 5 years, you can really cut down on the intensity of your follow-up and scans,” Dr. Nghiem said.
“We do now have two excellent blood tests that are working very well, and we have really good ways to detect the cancer coming back early, and that’s important, because we have potentially curative therapies that tend to work better if you catch the cancer early,” he said.
The study was supported by the National Institutes of Health. Dr. Nghiem reported personal fees and institutional support outside the study from several companies and patents for Merkel cell therapies with the University of Washington and University of Denmark. Dr. Demehri has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM JAMA DERMATOLOGY
New MIS-C guidance addresses diagnostic challenges, cardiac care
Updated guidance for health care providers on multisystem inflammatory syndrome in children (MIS-C) recognizes the evolving nature of the disease and offers strategies for pediatric rheumatologists, who also may be asked to recommend treatment for hyperinflammation in children with acute COVID-19.
Guidance is needed for many reasons, including the variable case definitions for MIS-C, the presence of MIS-C features in other infections and childhood rheumatic diseases, the extrapolation of treatment strategies from other conditions with similar presentations, and the issue of myocardial dysfunction, wrote Lauren A. Henderson, MD, MMSC, of Boston Children’s Hospital, and members of the American College of Rheumatology MIS-C and COVID-19–Related Hyperinflammation Task Force.

However, “modifications to treatment plans, particularly in patients with complex conditions, are highly disease, patient, geography, and time specific, and therefore must be individualized as part of a shared decision-making process,” the authors said. The updated guidance was published in Arthritis & Rheumatology.
Update needed in wake of Omicron
“We continue to see cases of MIS-C across the United States due to the spike in SARS-CoV-2 infections from the Omicron variant,” and therefore updated guidance is important at this time, Dr. Henderson told this news organization.
“MIS-C remains a serious complication of COVID-19 in children and the ACR wanted to continue to provide pediatricians with up-to-date recommendations for the management of MIS-C,” she said.
“Children began to present with MIS-C in April 2020. At that time, little was known about this entity. Most of the recommendations in the first version of the MIS-C guidance were based on expert opinion,” she explained. However, “over the last 2 years, pediatricians have worked very hard to conduct high-quality research studies to better understand MIS-C, so we now have more scientific evidence to guide our recommendations.
“In version three of the MIS-C guidance, there are new recommendations on treatment. Previously, it was unclear what medications should be used for first-line treatment in patients with MIS-C. Some children were given intravenous immunoglobulin while others were given IVIg and steroids together. Several new studies show that children with MIS-C who are treated with a combination of IVIg and steroids have better outcomes. Accordingly, the MIS-C guidance now recommends dual therapy with IVIg and steroids in children with MIS-C.”
Diagnostic evaluation
The guidance calls for maintaining a broad differential diagnosis of MIS-C, given that the condition remains rare, and that most children with COVID-19 present with mild symptoms and have excellent outcomes, the authors noted. The range of clinical features associated with MIS-C include fever, mucocutaneous findings, myocardial dysfunction, cardiac conduction abnormalities, shock, gastrointestinal symptoms, and lymphadenopathy.
Some patients also experience neurologic involvement in the form of severe headache, altered mental status, seizures, cranial nerve palsies, meningismus, cerebral edema, and ischemic or hemorrhagic stroke. Given the nonspecific nature of these symptoms, “it is imperative that a diagnostic evaluation for MIS-C include investigation for other possible causes, as deemed appropriate by the treating provider,” the authors emphasized. Other diagnostic considerations include the prevalence and chronology of COVID-19 in the community, which may change over time.
MIS-C and Kawasaki disease phenotypes
Earlier in the pandemic, when MIS-C first emerged, it was compared with Kawasaki disease (KD). “However, a closer examination of the literature shows that only about one-quarter to half of patients with a reported diagnosis of MIS-C meet the full diagnostic criteria for KD,” the authors wrote. Key features that separate MIS-C from KD include the greater incidence of KD among children in Japan and East Asia versus the higher incidence of MIS-C among non-Hispanic Black children. In addition, children with MIS-C have shown a wider age range, more prominent gastrointestinal and neurologic symptoms, and more frequent cardiac dysfunction, compared with those with KD.
Cardiac management
Close follow-up with cardiology is essential for children with MIS-C, according to the authors. The recommendations call for repeat echocardiograms for all children with MIS-C at a minimum of 7-14 days, then again at 4-6 weeks after the initial presentation. The authors also recommended additional echocardiograms for children with left ventricular dysfunction and cardiac aortic aneurysms.
MIS-C treatment
Current treatment recommendations emphasize that patients under investigation for MIS-C with life-threatening manifestations may need immunomodulatory therapy before a full diagnostic evaluation is complete, the authors said. However, patients without life-threatening manifestations should be evaluated before starting immunomodulatory treatment to avoid potentially harmful therapies for pediatric patients who don’t need them.
When MIS-C is refractory to initial immunomodulatory treatment, a second dose of IVIg is not recommended, but intensification therapy is advised with either high-dose (10-30 mg/kg per day) glucocorticoids, anakinra, or infliximab. However, there is little evidence available for selecting a specific agent for intensification therapy.
The task force also advises giving low-dose aspirin (3-5 mg/kg per day, up to 81 mg once daily) to all MIS-C patients without active bleeding or significant bleeding risk until normalization of the platelet count and confirmed normal coronary arteries at least 4 weeks after diagnosis.
COVID-19 and hyperinflammation
The task force also noted a distinction between MIS-C and severe COVID-19 in children. Although many children with MIS-C are previously healthy, most children who develop severe COVID-19 during an initial infection have complex conditions or comorbidities such as developmental delay or genetic anomaly, or chronic conditions such as congenital heart disease, type 1 diabetes, or asthma, the authors said. They recommend that “hospitalized children with COVID-19 requiring supplemental oxygen or respiratory support should be considered for immunomodulatory therapy in addition to supportive care and antiviral medications.”
The authors acknowledged the limitations and evolving nature of the recommendations, which will continue to change and do not replace clinical judgment for the management of individual patients. In the meantime, the ACR will support the task force in reviewing new evidence and providing revised versions of the current document.
Many questions about MIS-C remain, Dr. Henderson said in an interview. “It can be very hard to diagnose children with MIS-C because many of the symptoms are similar to those seen in other febrile illness of childhood. We need to identify better biomarkers to help us make the diagnosis of MIS-C. In addition, we need studies to provide information about what treatments should be used if children fail to respond to IVIg and steroids. Finally, it appears that vaccination [against SARS-CoV-2] protects against severe forms of MIS-C, and studies are needed to see how vaccination protects children from MIS-C.”
The development of the guidance was supported by the American College of Rheumatology. Dr. Henderson disclosed relationships with companies including Sobi, Pfizer, and Adaptive Biotechnologies (less than $10,000) and research support from the Childhood Arthritis and Rheumatology Research Alliance and research grant support from Bristol-Myers Squibb.
A version of this article first appeared on Medscape.com.
Updated guidance for health care providers on multisystem inflammatory syndrome in children (MIS-C) recognizes the evolving nature of the disease and offers strategies for pediatric rheumatologists, who also may be asked to recommend treatment for hyperinflammation in children with acute COVID-19.
Guidance is needed for many reasons, including the variable case definitions for MIS-C, the presence of MIS-C features in other infections and childhood rheumatic diseases, the extrapolation of treatment strategies from other conditions with similar presentations, and the issue of myocardial dysfunction, wrote Lauren A. Henderson, MD, MMSC, of Boston Children’s Hospital, and members of the American College of Rheumatology MIS-C and COVID-19–Related Hyperinflammation Task Force.
However, “modifications to treatment plans, particularly in patients with complex conditions, are highly disease, patient, geography, and time specific, and therefore must be individualized as part of a shared decision-making process,” the authors said. The updated guidance was published in Arthritis & Rheumatology.
Update needed in wake of Omicron
“We continue to see cases of MIS-C across the United States due to the spike in SARS-CoV-2 infections from the Omicron variant,” and therefore updated guidance is important at this time, Dr. Henderson told this news organization.
“MIS-C remains a serious complication of COVID-19 in children and the ACR wanted to continue to provide pediatricians with up-to-date recommendations for the management of MIS-C,” she said.
“Children began to present with MIS-C in April 2020. At that time, little was known about this entity. Most of the recommendations in the first version of the MIS-C guidance were based on expert opinion,” she explained. However, “over the last 2 years, pediatricians have worked very hard to conduct high-quality research studies to better understand MIS-C, so we now have more scientific evidence to guide our recommendations.
“In version three of the MIS-C guidance, there are new recommendations on treatment. Previously, it was unclear what medications should be used for first-line treatment in patients with MIS-C. Some children were given intravenous immunoglobulin while others were given IVIg and steroids together. Several new studies show that children with MIS-C who are treated with a combination of IVIg and steroids have better outcomes. Accordingly, the MIS-C guidance now recommends dual therapy with IVIg and steroids in children with MIS-C.”
Diagnostic evaluation
The guidance calls for maintaining a broad differential diagnosis of MIS-C, given that the condition remains rare, and that most children with COVID-19 present with mild symptoms and have excellent outcomes, the authors noted. The range of clinical features associated with MIS-C include fever, mucocutaneous findings, myocardial dysfunction, cardiac conduction abnormalities, shock, gastrointestinal symptoms, and lymphadenopathy.
Some patients also experience neurologic involvement in the form of severe headache, altered mental status, seizures, cranial nerve palsies, meningismus, cerebral edema, and ischemic or hemorrhagic stroke. Given the nonspecific nature of these symptoms, “it is imperative that a diagnostic evaluation for MIS-C include investigation for other possible causes, as deemed appropriate by the treating provider,” the authors emphasized. Other diagnostic considerations include the prevalence and chronology of COVID-19 in the community, which may change over time.
MIS-C and Kawasaki disease phenotypes
Earlier in the pandemic, when MIS-C first emerged, it was compared with Kawasaki disease (KD). “However, a closer examination of the literature shows that only about one-quarter to half of patients with a reported diagnosis of MIS-C meet the full diagnostic criteria for KD,” the authors wrote. Key features that separate MIS-C from KD include the greater incidence of KD among children in Japan and East Asia versus the higher incidence of MIS-C among non-Hispanic Black children. In addition, children with MIS-C have shown a wider age range, more prominent gastrointestinal and neurologic symptoms, and more frequent cardiac dysfunction, compared with those with KD.
Cardiac management
Close follow-up with cardiology is essential for children with MIS-C, according to the authors. The recommendations call for repeat echocardiograms for all children with MIS-C at a minimum of 7-14 days, then again at 4-6 weeks after the initial presentation. The authors also recommended additional echocardiograms for children with left ventricular dysfunction and cardiac aortic aneurysms.
MIS-C treatment
Current treatment recommendations emphasize that patients under investigation for MIS-C with life-threatening manifestations may need immunomodulatory therapy before a full diagnostic evaluation is complete, the authors said. However, patients without life-threatening manifestations should be evaluated before starting immunomodulatory treatment to avoid potentially harmful therapies for pediatric patients who don’t need them.
When MIS-C is refractory to initial immunomodulatory treatment, a second dose of IVIg is not recommended, but intensification therapy is advised with either high-dose (10-30 mg/kg per day) glucocorticoids, anakinra, or infliximab. However, there is little evidence available for selecting a specific agent for intensification therapy.
The task force also advises giving low-dose aspirin (3-5 mg/kg per day, up to 81 mg once daily) to all MIS-C patients without active bleeding or significant bleeding risk until normalization of the platelet count and confirmed normal coronary arteries at least 4 weeks after diagnosis.
COVID-19 and hyperinflammation
The task force also noted a distinction between MIS-C and severe COVID-19 in children. Although many children with MIS-C are previously healthy, most children who develop severe COVID-19 during an initial infection have complex conditions or comorbidities such as developmental delay or genetic anomaly, or chronic conditions such as congenital heart disease, type 1 diabetes, or asthma, the authors said. They recommend that “hospitalized children with COVID-19 requiring supplemental oxygen or respiratory support should be considered for immunomodulatory therapy in addition to supportive care and antiviral medications.”
The authors acknowledged the limitations and evolving nature of the recommendations, which will continue to change and do not replace clinical judgment for the management of individual patients. In the meantime, the ACR will support the task force in reviewing new evidence and providing revised versions of the current document.
Many questions about MIS-C remain, Dr. Henderson said in an interview. “It can be very hard to diagnose children with MIS-C because many of the symptoms are similar to those seen in other febrile illness of childhood. We need to identify better biomarkers to help us make the diagnosis of MIS-C. In addition, we need studies to provide information about what treatments should be used if children fail to respond to IVIg and steroids. Finally, it appears that vaccination [against SARS-CoV-2] protects against severe forms of MIS-C, and studies are needed to see how vaccination protects children from MIS-C.”
The development of the guidance was supported by the American College of Rheumatology. Dr. Henderson disclosed relationships with companies including Sobi, Pfizer, and Adaptive Biotechnologies (less than $10,000) and research support from the Childhood Arthritis and Rheumatology Research Alliance and research grant support from Bristol-Myers Squibb.
A version of this article first appeared on Medscape.com.
Updated guidance for health care providers on multisystem inflammatory syndrome in children (MIS-C) recognizes the evolving nature of the disease and offers strategies for pediatric rheumatologists, who also may be asked to recommend treatment for hyperinflammation in children with acute COVID-19.
Guidance is needed for many reasons, including the variable case definitions for MIS-C, the presence of MIS-C features in other infections and childhood rheumatic diseases, the extrapolation of treatment strategies from other conditions with similar presentations, and the issue of myocardial dysfunction, wrote Lauren A. Henderson, MD, MMSC, of Boston Children’s Hospital, and members of the American College of Rheumatology MIS-C and COVID-19–Related Hyperinflammation Task Force.
However, “modifications to treatment plans, particularly in patients with complex conditions, are highly disease, patient, geography, and time specific, and therefore must be individualized as part of a shared decision-making process,” the authors said. The updated guidance was published in Arthritis & Rheumatology.
Update needed in wake of Omicron
“We continue to see cases of MIS-C across the United States due to the spike in SARS-CoV-2 infections from the Omicron variant,” and therefore updated guidance is important at this time, Dr. Henderson told this news organization.
“MIS-C remains a serious complication of COVID-19 in children and the ACR wanted to continue to provide pediatricians with up-to-date recommendations for the management of MIS-C,” she said.
“Children began to present with MIS-C in April 2020. At that time, little was known about this entity. Most of the recommendations in the first version of the MIS-C guidance were based on expert opinion,” she explained. However, “over the last 2 years, pediatricians have worked very hard to conduct high-quality research studies to better understand MIS-C, so we now have more scientific evidence to guide our recommendations.
“In version three of the MIS-C guidance, there are new recommendations on treatment. Previously, it was unclear what medications should be used for first-line treatment in patients with MIS-C. Some children were given intravenous immunoglobulin while others were given IVIg and steroids together. Several new studies show that children with MIS-C who are treated with a combination of IVIg and steroids have better outcomes. Accordingly, the MIS-C guidance now recommends dual therapy with IVIg and steroids in children with MIS-C.”
Diagnostic evaluation
The guidance calls for maintaining a broad differential diagnosis of MIS-C, given that the condition remains rare, and that most children with COVID-19 present with mild symptoms and have excellent outcomes, the authors noted. The range of clinical features associated with MIS-C include fever, mucocutaneous findings, myocardial dysfunction, cardiac conduction abnormalities, shock, gastrointestinal symptoms, and lymphadenopathy.
Some patients also experience neurologic involvement in the form of severe headache, altered mental status, seizures, cranial nerve palsies, meningismus, cerebral edema, and ischemic or hemorrhagic stroke. Given the nonspecific nature of these symptoms, “it is imperative that a diagnostic evaluation for MIS-C include investigation for other possible causes, as deemed appropriate by the treating provider,” the authors emphasized. Other diagnostic considerations include the prevalence and chronology of COVID-19 in the community, which may change over time.
MIS-C and Kawasaki disease phenotypes
Earlier in the pandemic, when MIS-C first emerged, it was compared with Kawasaki disease (KD). “However, a closer examination of the literature shows that only about one-quarter to half of patients with a reported diagnosis of MIS-C meet the full diagnostic criteria for KD,” the authors wrote. Key features that separate MIS-C from KD include the greater incidence of KD among children in Japan and East Asia versus the higher incidence of MIS-C among non-Hispanic Black children. In addition, children with MIS-C have shown a wider age range, more prominent gastrointestinal and neurologic symptoms, and more frequent cardiac dysfunction, compared with those with KD.
Cardiac management
Close follow-up with cardiology is essential for children with MIS-C, according to the authors. The recommendations call for repeat echocardiograms for all children with MIS-C at a minimum of 7-14 days, then again at 4-6 weeks after the initial presentation. The authors also recommended additional echocardiograms for children with left ventricular dysfunction and cardiac aortic aneurysms.
MIS-C treatment
Current treatment recommendations emphasize that patients under investigation for MIS-C with life-threatening manifestations may need immunomodulatory therapy before a full diagnostic evaluation is complete, the authors said. However, patients without life-threatening manifestations should be evaluated before starting immunomodulatory treatment to avoid potentially harmful therapies for pediatric patients who don’t need them.
When MIS-C is refractory to initial immunomodulatory treatment, a second dose of IVIg is not recommended, but intensification therapy is advised with either high-dose (10-30 mg/kg per day) glucocorticoids, anakinra, or infliximab. However, there is little evidence available for selecting a specific agent for intensification therapy.
The task force also advises giving low-dose aspirin (3-5 mg/kg per day, up to 81 mg once daily) to all MIS-C patients without active bleeding or significant bleeding risk until normalization of the platelet count and confirmed normal coronary arteries at least 4 weeks after diagnosis.
COVID-19 and hyperinflammation
The task force also noted a distinction between MIS-C and severe COVID-19 in children. Although many children with MIS-C are previously healthy, most children who develop severe COVID-19 during an initial infection have complex conditions or comorbidities such as developmental delay or genetic anomaly, or chronic conditions such as congenital heart disease, type 1 diabetes, or asthma, the authors said. They recommend that “hospitalized children with COVID-19 requiring supplemental oxygen or respiratory support should be considered for immunomodulatory therapy in addition to supportive care and antiviral medications.”
The authors acknowledged the limitations and evolving nature of the recommendations, which will continue to change and do not replace clinical judgment for the management of individual patients. In the meantime, the ACR will support the task force in reviewing new evidence and providing revised versions of the current document.
Many questions about MIS-C remain, Dr. Henderson said in an interview. “It can be very hard to diagnose children with MIS-C because many of the symptoms are similar to those seen in other febrile illness of childhood. We need to identify better biomarkers to help us make the diagnosis of MIS-C. In addition, we need studies to provide information about what treatments should be used if children fail to respond to IVIg and steroids. Finally, it appears that vaccination [against SARS-CoV-2] protects against severe forms of MIS-C, and studies are needed to see how vaccination protects children from MIS-C.”
The development of the guidance was supported by the American College of Rheumatology. Dr. Henderson disclosed relationships with companies including Sobi, Pfizer, and Adaptive Biotechnologies (less than $10,000) and research support from the Childhood Arthritis and Rheumatology Research Alliance and research grant support from Bristol-Myers Squibb.
A version of this article first appeared on Medscape.com.
FROM ARTHRITIS AND RHEUMATOLOGY
Growth hormone therapy for certain children may help them reach their potential
“Dr. Lilley, you’ll always be my favorite doctor; you helped me grow.”
These were the parting words from the last patient that I treated during my endocrinology fellowship. I had watched this young man grow from a prepubertal 17-year-old to a young man who had reached his predicted family height as I treated his delayed puberty caused by Kallmann syndrome, a problem that had been missed for years. It was the appropriate bookend for my chosen specialty.

Watching children grow and develop into who they were meant to be is one of my favorite things about endocrinology, as well as forming meaningful relationships with families. Treating detectable deficiencies in logical and measurable ways is also extremely satisfying.
Too little testosterone? That’s a problem I can solve. Too much thyroid hormone? There’s a blocker for that! Endocrinology can be a straightforward field, and when all goes well, everyone leaves happy.
Except when they don’t.
Gatekeepers for treatment for children’s growth
“Nice to meet you. We’re here to get growth hormone.”
“We’re here because his pediatrician made us come. We’ve already decided we’re not going to put hormones into his body.”
These are common statements I hear when I first meet new patients whose parents are concerned about their children’s growth. Pediatric endocrinologists, after all, are the usual gatekeepers for this treatment.
Growth hormone (GH) often makes the news for controversial reasons – most commonly for its abuse by elite athletes hoping to exploit its anabolic effects – causing parents to have varied opinions about its possible use in their children.
Some refuse endocrinology referrals at all owing to concerns that we will push daily injections on their children. Others demand referrals for their children of average height, hoping for every perceived advantage.
GH deficiency (GHD) – a condition where the pituitary gland fails to produce enough GH – can occur because of congenital pituitary malformations; anatomic, surgical, or traumatic interruptions to the gland; or enzyme deficiencies leading to faulty production.
GHD is just one reason for poor growth, however.
Growth is one of the most important indicators of health in children. A waning growth rate may be an early symptom of serious problems. In my clinic, I’ve diagnosed severe hypothyroidism in a marathon runner, a brain tumor, celiac disease in a teenager with no gastrointestinal complaints, autoimmune hepatitis, and several other diseases needing treatment in children who show no symptoms other than poor growth.
Barriers to normal growth
Sometimes, the die is cast for children to have barriers to normal growth. Several genetic conditions can lead to poor GH production or response, and GH treatment is often necessary to approximate normal height.
These may include:
- Turner syndrome (in females who are missing an X chromosome in whole or part) should be considered in every girl with abnormally short stature; mosaic forms of the condition may be subtle and lack classic features.
- Noonan syndrome is important to detect owing to the possibility of cardiac or renal malformations that may also occur in this condition, caused by a mutation in one of the genes in the RAS-MAPK pathway.
- Russell-Silver syndrome can cause intrauterine GH restriction and has been traced to uniparental disomy of chromosome 7 or duplications, mutations, or methylation defects in chromosome 11.
- Individuals with Prader-Willi syndrome, which is characterized by low muscle tone, hyperphagia, and hypogonadism, have demonstrated dramatic benefits from GH therapy, primarily in maintaining a normal body mass index.
Children who are small for their gestational age may be GH resistant, and those who do not catch up to their growth curve by the age of 2 years may require GH treatment to reach their height potential.
GH therapy isn’t entirely benign. Rare adverse effects of overtreatment can include slipped capital femoral epiphysis (a fracture to the growth plate) and pseudotumor cerebri (idiopathic intracranial hypertension).
Overtreatment can cause acromegaly, which results in coarsened features and large hands and feet.
When is GH therapy warranted?
“Growth hormone therapy has been denied by her insurer. They want you to fill out an appeal.”
Insurance approval in the United States can be a herculean effort because GH is expensive: Out-of-pocket costs are prohibitive for most people without insurance assistance, ranging from $7,000 to $30,000 annually.
Pediatric endocrinologists aren’t in the business of cosmetic endocrinology. Treatment of idiopathic short stature has been controversial since this became an indication for GH treatment.
GH isn’t always necessary. Diagnosing the underlying cause for poor growth is the most important step.
Often, we find constitutional delay of growth and puberty, or “late bloomers.” This condition is characterized by a delayed bone age (growth plates more open than expected for age) and delayed pubertal onset. These children will often reach a normal height despite starting as some of the smallest of their peers.
However, GH plays other roles in the body than simply propelling height. Children with congenital GHD will require GH treatment to prevent hypoglycemia, especially in infancy.
GH is needed even in adults with fused growth plates for normal lipid metabolism, bone accrual, and maintaining normal muscle mass.
I have noticed marked improvements in muscle tone in many children with developmental delays who are treated with GH, and research supports cognitive benefits for certain populations.
The most common regimens for GH focus on treatment via subcutaneous injection nightly, when GH is naturally produced; sometimes, injections are given six nights out of seven to provide a break or for splitting time between households.
Newer once-weekly formulations have recently received approval, as reported by this news organization, and are coming into use.
Pediatric endocrinologists measure height and follow growth factors closely with visits every 3-6 months. GH levels are not useful outside of provocative diagnostic (stimulation) testing.
Insulinlike growth factor 1 or insulinlike growth factor binding protein levels are analyzed per Tanner stage of puberty to assess appropriate response and to make dose adjustments.
Annual standardized films of the left hand help predict progress and anticipated adult height. Treatment usually persists through puberty until growth plates are closed; if true GHD is noticed, much smaller doses are continued through adulthood.
Regardless, conversations about GH happen with your friendly local pediatric endocrinologist.
We are thrilled to help shepherd patients through their growing age to meet their potential. For more information about GH treatment for children, the MAGIC Foundation is the perfect place to start.
Dr. Lilley is director of the pediatric diabetes and lipid program, Mississippi Center for Advanced Medicine, Madison. She disclosed no relevant conflicts of interest. A version of this article first appeared on Medscape.com.
“Dr. Lilley, you’ll always be my favorite doctor; you helped me grow.”
These were the parting words from the last patient that I treated during my endocrinology fellowship. I had watched this young man grow from a prepubertal 17-year-old to a young man who had reached his predicted family height as I treated his delayed puberty caused by Kallmann syndrome, a problem that had been missed for years. It was the appropriate bookend for my chosen specialty.
Watching children grow and develop into who they were meant to be is one of my favorite things about endocrinology, as well as forming meaningful relationships with families. Treating detectable deficiencies in logical and measurable ways is also extremely satisfying.
Too little testosterone? That’s a problem I can solve. Too much thyroid hormone? There’s a blocker for that! Endocrinology can be a straightforward field, and when all goes well, everyone leaves happy.
Except when they don’t.
Gatekeepers for treatment for children’s growth
“Nice to meet you. We’re here to get growth hormone.”
“We’re here because his pediatrician made us come. We’ve already decided we’re not going to put hormones into his body.”
These are common statements I hear when I first meet new patients whose parents are concerned about their children’s growth. Pediatric endocrinologists, after all, are the usual gatekeepers for this treatment.
Growth hormone (GH) often makes the news for controversial reasons – most commonly for its abuse by elite athletes hoping to exploit its anabolic effects – causing parents to have varied opinions about its possible use in their children.
Some refuse endocrinology referrals at all owing to concerns that we will push daily injections on their children. Others demand referrals for their children of average height, hoping for every perceived advantage.
GH deficiency (GHD) – a condition where the pituitary gland fails to produce enough GH – can occur because of congenital pituitary malformations; anatomic, surgical, or traumatic interruptions to the gland; or enzyme deficiencies leading to faulty production.
GHD is just one reason for poor growth, however.
Growth is one of the most important indicators of health in children. A waning growth rate may be an early symptom of serious problems. In my clinic, I’ve diagnosed severe hypothyroidism in a marathon runner, a brain tumor, celiac disease in a teenager with no gastrointestinal complaints, autoimmune hepatitis, and several other diseases needing treatment in children who show no symptoms other than poor growth.
Barriers to normal growth
Sometimes, the die is cast for children to have barriers to normal growth. Several genetic conditions can lead to poor GH production or response, and GH treatment is often necessary to approximate normal height.
These may include:
- Turner syndrome (in females who are missing an X chromosome in whole or part) should be considered in every girl with abnormally short stature; mosaic forms of the condition may be subtle and lack classic features.
- Noonan syndrome is important to detect owing to the possibility of cardiac or renal malformations that may also occur in this condition, caused by a mutation in one of the genes in the RAS-MAPK pathway.
- Russell-Silver syndrome can cause intrauterine GH restriction and has been traced to uniparental disomy of chromosome 7 or duplications, mutations, or methylation defects in chromosome 11.
- Individuals with Prader-Willi syndrome, which is characterized by low muscle tone, hyperphagia, and hypogonadism, have demonstrated dramatic benefits from GH therapy, primarily in maintaining a normal body mass index.
Children who are small for their gestational age may be GH resistant, and those who do not catch up to their growth curve by the age of 2 years may require GH treatment to reach their height potential.
GH therapy isn’t entirely benign. Rare adverse effects of overtreatment can include slipped capital femoral epiphysis (a fracture to the growth plate) and pseudotumor cerebri (idiopathic intracranial hypertension).
Overtreatment can cause acromegaly, which results in coarsened features and large hands and feet.
When is GH therapy warranted?
“Growth hormone therapy has been denied by her insurer. They want you to fill out an appeal.”
Insurance approval in the United States can be a herculean effort because GH is expensive: Out-of-pocket costs are prohibitive for most people without insurance assistance, ranging from $7,000 to $30,000 annually.
Pediatric endocrinologists aren’t in the business of cosmetic endocrinology. Treatment of idiopathic short stature has been controversial since this became an indication for GH treatment.
GH isn’t always necessary. Diagnosing the underlying cause for poor growth is the most important step.
Often, we find constitutional delay of growth and puberty, or “late bloomers.” This condition is characterized by a delayed bone age (growth plates more open than expected for age) and delayed pubertal onset. These children will often reach a normal height despite starting as some of the smallest of their peers.
However, GH plays other roles in the body than simply propelling height. Children with congenital GHD will require GH treatment to prevent hypoglycemia, especially in infancy.
GH is needed even in adults with fused growth plates for normal lipid metabolism, bone accrual, and maintaining normal muscle mass.
I have noticed marked improvements in muscle tone in many children with developmental delays who are treated with GH, and research supports cognitive benefits for certain populations.
The most common regimens for GH focus on treatment via subcutaneous injection nightly, when GH is naturally produced; sometimes, injections are given six nights out of seven to provide a break or for splitting time between households.
Newer once-weekly formulations have recently received approval, as reported by this news organization, and are coming into use.
Pediatric endocrinologists measure height and follow growth factors closely with visits every 3-6 months. GH levels are not useful outside of provocative diagnostic (stimulation) testing.
Insulinlike growth factor 1 or insulinlike growth factor binding protein levels are analyzed per Tanner stage of puberty to assess appropriate response and to make dose adjustments.
Annual standardized films of the left hand help predict progress and anticipated adult height. Treatment usually persists through puberty until growth plates are closed; if true GHD is noticed, much smaller doses are continued through adulthood.
Regardless, conversations about GH happen with your friendly local pediatric endocrinologist.
We are thrilled to help shepherd patients through their growing age to meet their potential. For more information about GH treatment for children, the MAGIC Foundation is the perfect place to start.
Dr. Lilley is director of the pediatric diabetes and lipid program, Mississippi Center for Advanced Medicine, Madison. She disclosed no relevant conflicts of interest. A version of this article first appeared on Medscape.com.
“Dr. Lilley, you’ll always be my favorite doctor; you helped me grow.”
These were the parting words from the last patient that I treated during my endocrinology fellowship. I had watched this young man grow from a prepubertal 17-year-old to a young man who had reached his predicted family height as I treated his delayed puberty caused by Kallmann syndrome, a problem that had been missed for years. It was the appropriate bookend for my chosen specialty.
Watching children grow and develop into who they were meant to be is one of my favorite things about endocrinology, as well as forming meaningful relationships with families. Treating detectable deficiencies in logical and measurable ways is also extremely satisfying.
Too little testosterone? That’s a problem I can solve. Too much thyroid hormone? There’s a blocker for that! Endocrinology can be a straightforward field, and when all goes well, everyone leaves happy.
Except when they don’t.
Gatekeepers for treatment for children’s growth
“Nice to meet you. We’re here to get growth hormone.”
“We’re here because his pediatrician made us come. We’ve already decided we’re not going to put hormones into his body.”
These are common statements I hear when I first meet new patients whose parents are concerned about their children’s growth. Pediatric endocrinologists, after all, are the usual gatekeepers for this treatment.
Growth hormone (GH) often makes the news for controversial reasons – most commonly for its abuse by elite athletes hoping to exploit its anabolic effects – causing parents to have varied opinions about its possible use in their children.
Some refuse endocrinology referrals at all owing to concerns that we will push daily injections on their children. Others demand referrals for their children of average height, hoping for every perceived advantage.
GH deficiency (GHD) – a condition where the pituitary gland fails to produce enough GH – can occur because of congenital pituitary malformations; anatomic, surgical, or traumatic interruptions to the gland; or enzyme deficiencies leading to faulty production.
GHD is just one reason for poor growth, however.
Growth is one of the most important indicators of health in children. A waning growth rate may be an early symptom of serious problems. In my clinic, I’ve diagnosed severe hypothyroidism in a marathon runner, a brain tumor, celiac disease in a teenager with no gastrointestinal complaints, autoimmune hepatitis, and several other diseases needing treatment in children who show no symptoms other than poor growth.
Barriers to normal growth
Sometimes, the die is cast for children to have barriers to normal growth. Several genetic conditions can lead to poor GH production or response, and GH treatment is often necessary to approximate normal height.
These may include:
- Turner syndrome (in females who are missing an X chromosome in whole or part) should be considered in every girl with abnormally short stature; mosaic forms of the condition may be subtle and lack classic features.
- Noonan syndrome is important to detect owing to the possibility of cardiac or renal malformations that may also occur in this condition, caused by a mutation in one of the genes in the RAS-MAPK pathway.
- Russell-Silver syndrome can cause intrauterine GH restriction and has been traced to uniparental disomy of chromosome 7 or duplications, mutations, or methylation defects in chromosome 11.
- Individuals with Prader-Willi syndrome, which is characterized by low muscle tone, hyperphagia, and hypogonadism, have demonstrated dramatic benefits from GH therapy, primarily in maintaining a normal body mass index.
Children who are small for their gestational age may be GH resistant, and those who do not catch up to their growth curve by the age of 2 years may require GH treatment to reach their height potential.
GH therapy isn’t entirely benign. Rare adverse effects of overtreatment can include slipped capital femoral epiphysis (a fracture to the growth plate) and pseudotumor cerebri (idiopathic intracranial hypertension).
Overtreatment can cause acromegaly, which results in coarsened features and large hands and feet.
When is GH therapy warranted?
“Growth hormone therapy has been denied by her insurer. They want you to fill out an appeal.”
Insurance approval in the United States can be a herculean effort because GH is expensive: Out-of-pocket costs are prohibitive for most people without insurance assistance, ranging from $7,000 to $30,000 annually.
Pediatric endocrinologists aren’t in the business of cosmetic endocrinology. Treatment of idiopathic short stature has been controversial since this became an indication for GH treatment.
GH isn’t always necessary. Diagnosing the underlying cause for poor growth is the most important step.
Often, we find constitutional delay of growth and puberty, or “late bloomers.” This condition is characterized by a delayed bone age (growth plates more open than expected for age) and delayed pubertal onset. These children will often reach a normal height despite starting as some of the smallest of their peers.
However, GH plays other roles in the body than simply propelling height. Children with congenital GHD will require GH treatment to prevent hypoglycemia, especially in infancy.
GH is needed even in adults with fused growth plates for normal lipid metabolism, bone accrual, and maintaining normal muscle mass.
I have noticed marked improvements in muscle tone in many children with developmental delays who are treated with GH, and research supports cognitive benefits for certain populations.
The most common regimens for GH focus on treatment via subcutaneous injection nightly, when GH is naturally produced; sometimes, injections are given six nights out of seven to provide a break or for splitting time between households.
Newer once-weekly formulations have recently received approval, as reported by this news organization, and are coming into use.
Pediatric endocrinologists measure height and follow growth factors closely with visits every 3-6 months. GH levels are not useful outside of provocative diagnostic (stimulation) testing.
Insulinlike growth factor 1 or insulinlike growth factor binding protein levels are analyzed per Tanner stage of puberty to assess appropriate response and to make dose adjustments.
Annual standardized films of the left hand help predict progress and anticipated adult height. Treatment usually persists through puberty until growth plates are closed; if true GHD is noticed, much smaller doses are continued through adulthood.
Regardless, conversations about GH happen with your friendly local pediatric endocrinologist.
We are thrilled to help shepherd patients through their growing age to meet their potential. For more information about GH treatment for children, the MAGIC Foundation is the perfect place to start.
Dr. Lilley is director of the pediatric diabetes and lipid program, Mississippi Center for Advanced Medicine, Madison. She disclosed no relevant conflicts of interest. A version of this article first appeared on Medscape.com.
Cystic fibrosis in retreat, but still unbeaten
In 1938, the year that cystic fibrosis (CF) was first described clinically, four of five children born with the disease did not live past their first birthdays.
In 2019, the median age at death for patients enrolled in the Cystic Fibrosis Foundation (CFF) registry was 32 years, and the predicted life expectancy for patients with CF who were born from 2015 through 2019 was 46 years.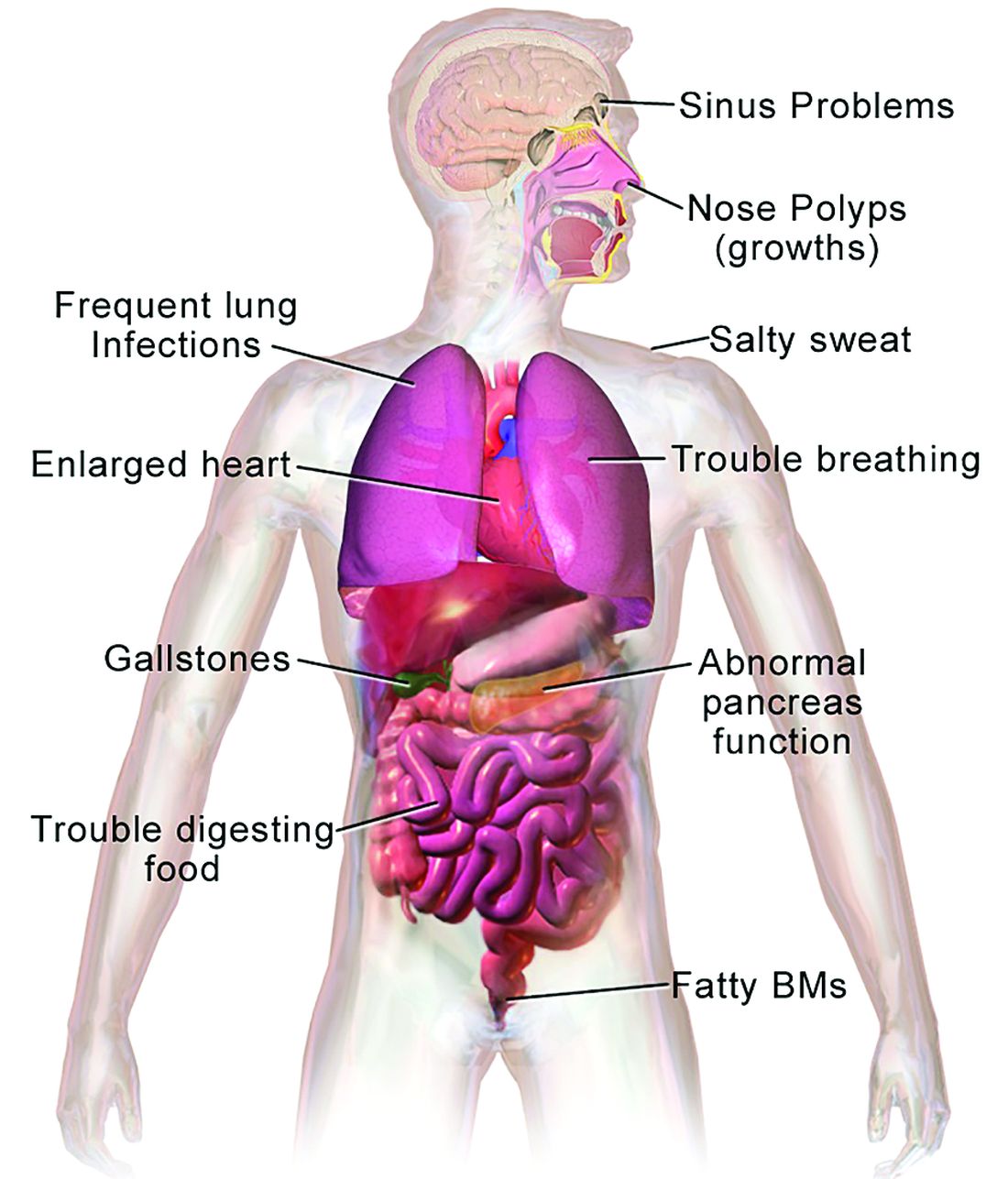
Those numbers reflect the remarkable progress made in the past 4 decades in the care of patients with CF, but also highlight the obstacles ahead, given that the predicted life expectancy for the overall U.S. population in 2019 (pre–COVID-19) was 78.9 years.
Julie Desch, MD, is a CF survivor who has beaten the odds and then some. At age 61, the retired surgical pathologist is a CF patient advocate, speaker, and a board member of the Cystic Fibrosis Research Institute, a not-for-profit organization that funds CF research and offers education, advocacy, and psychosocial support for persons with CF and their families and caregivers.
In an interview, Dr. Desch said that while there has been remarkable progress in her lifetime in the field of CF research and treatment, particularly in the development of drugs that modulate function of the underlying cause of approximately 90% of CF cases, there are still many CF patients who cannot benefit from these therapies.
“There are still 10% of people who don’t make a protein to be modified, so that’s a huge unmet need,” she said.
Genetic disorder
CF is a chronic autosomal recessive disorder with multiorgan and multisystem manifestations. It is caused by mutations in the CFTR gene, which codes for the protein CF transmembrane conductance regulator. CFTR controls transport of chloride ions across cell membranes, specifically the apical membrane of epithelial cells in tissues of the airways, intestinal tract, pancreas, kidneys, sweat glands, and the reproductive system, notably the vas deferens in males.
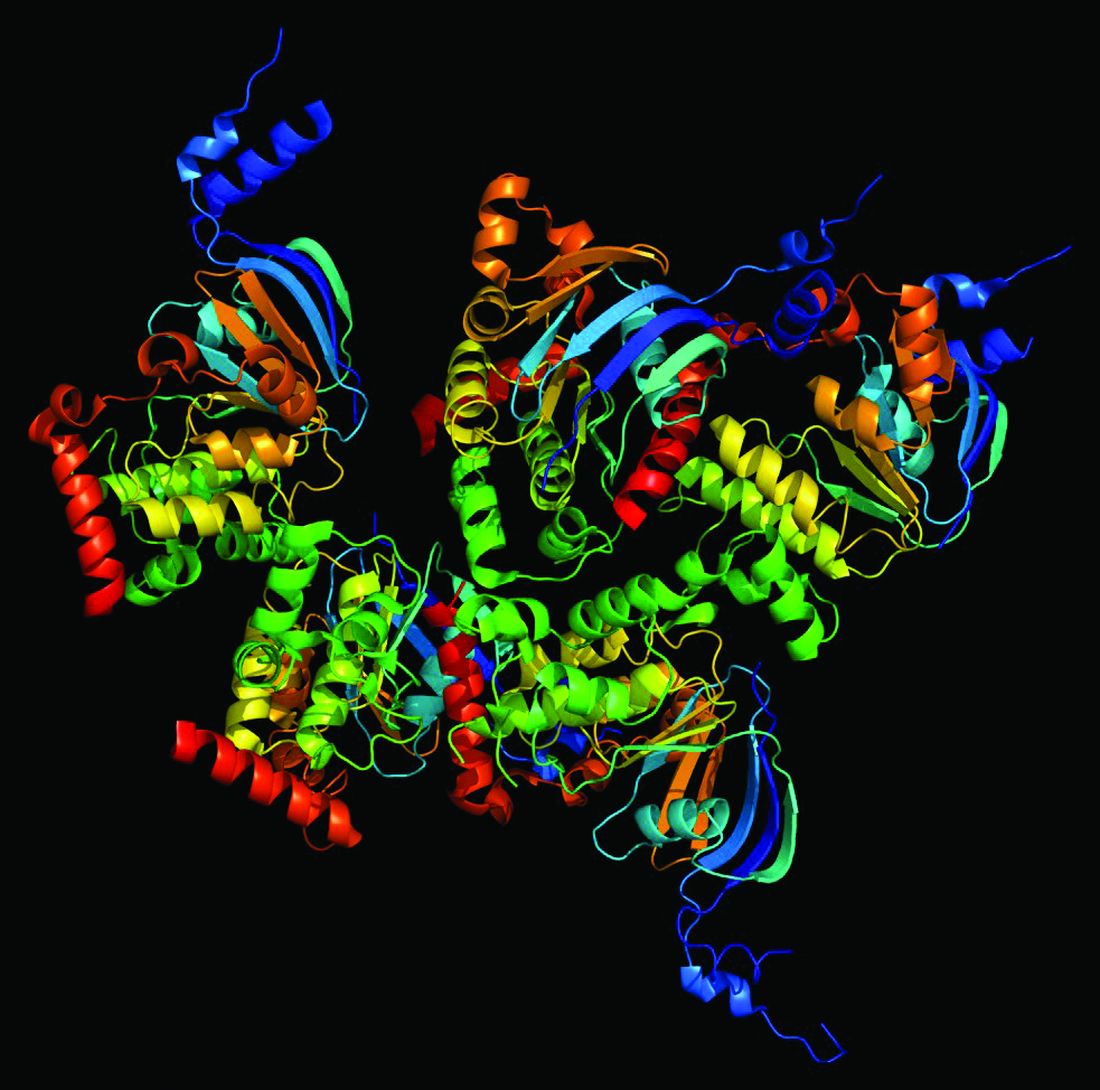
The F508 deletion (F508del) mutation is the most common, occurring in approximately 70% of persons with CF. It is a class 2-type protein processing mutation, leading to defects in cellular processing, protein stability, and chloride channel gating defects.
The CFTR protein also secretes bicarbonate to regulate the pH of airway surface liquid, and inhibits the epithelial sodium channel, which mediates passive sodium transport across apical membranes of sodium-absorbing epithelial cells in the kidneys, intestine, and airways.
CF typically presents with the buildup in the lungs of abnormally viscous and sticky mucus leading to frequent, severe infections, particularly with Pseudomonas aeruginosa, progressive lung damage and, prior to the development of effective disease management, to premature death. The phenotype often includes malnutrition due to malabsorption, and failure to thrive.
Diagnosis
In all 50 U.S. states and the District of Columbia, newborns are screened for CF with an assay for immunoreactive trypsinogen (IRT) an indirect marker for pancreatic injury that is elevated in serum in most newborns with CF, but also detected in premature infants or those delivered under stressful circumstances. In some states newborns are tested only for IRT, with a diagnosis confirmed with a sweat chloride test and/or a CFTR mutation panel.
Treatment
There is no cure for CF, but the discovery of the gene in 1989 by Canadian and U.S. investigators has led to life-prolonging therapeutic interventions, specifically the development of CFTR modulators.
CFTR modulators include potentiators such as ivacaftor (Kalydeco), and correctors such as lumacaftor and tezacaftor (available in the combination Orkambi), and most recently in the triple combination of elexacaftor, tezacaftor, and ivacaftor (Trikafta; ETI).
Neil Sweezey, MD, FRCPC, a CF expert at The Hospital for Sick Children (SickKids) in Toronto, told this news organization that the ideal therapy for CF, genetic correction of the underlying mutations, is still not feasible, but that CFTR modulators are a close second.
“For 90% of patients, the three-drug combination Trikafta has been shown to be quite safe, quite tolerable, and quite remarkably beneficial,” he said.
In a study reported at CHEST 2021 by investigators from Nationwide Children’s Hospital in Columbus, Ohio, 32 adults who were started on the triple combination had significantly improved in forced expiratory volume in 1 second (FEV1), gain in body mass index, decreased sweat chloride and decreased colonization by Pseudomonas species. In addition, patients had significant improvements in blood inflammatory markers.
Christopher H. Goss, MD, FCCP, professor of pulmonary critical care and sleep medicine and professor of pediatrics at the University of Washington in Seattle, agreed that with the availability of the triple combination, “these are extraordinary times. An astounding fact is that most patients have complete resolution of cough, and the exacerbation rates have just plummeted,” he said in an interview.
Some of the reductions in exacerbations may be attributable to the COVID-19 pandemic, he noted, because patients in isolation have less exposure to circulating respiratory viruses.
“But it has been miraculous, and the clinical effect is certainly still more astounding than the effects of ivacaftor, which was the first truly breakthrough drug. Weight goes up, well-being increases, and the population lung function has shifted up to better grade lung function, in the entire population,” he said.
In addition, the need for lung and heart transplantation has sharply declined.
“I had a patient who had decided to forgo transplantation, despite absolutely horrible lung function, and he’s now bowling and leading a very productive life, when before he had been preparing for end of life,” Dr. Goss said.
Dr. Sweezey emphasized that as with all medications, patients being started on the triple combination require close monitoring for potential adverse events that might require dose modification or, for a small number of patients, withdrawal.
Burden of care
CFTR modulators have reduced but not eliminated the need for some patients to have mucolytic therapy, which may include dornase alfa, a recombinant human deoxyribonuclease (DNase) that reduces the viscosity of lung secretions, hypertonic saline inhaled twice daily (for patients 12 and older), mannitol, and physical manipulations to help patients clear mucus. This can include both manual percussion and the use of devices for high-frequency chest wall oscillation.
The complex nature of CF often requires a combination of other therapies to address comorbidities. These therapies may include infection prophylaxis and treatment with antibiotics and antifungals, nutrition support, and therapy for CF-related complications, including gastrointestinal issues, liver diseases, diabetes, and osteopenia that may be related to poor nutrient absorption, chronic inflammation, or other sequelae of CF.
In addition, patients often require frequent CF care center visits – ideally a minimum of every 3 months – which can result in significant loss of work or school time.
“Outcomes for patients in the long run have been absolutely proven to be best if they’re followed in big, established, multidisciplinary well-organized CF centers,” Dr. Sweezey said. “In the United States and Canada if you’re looked after on a regular basis, which means quarterly, every 3 months – whether you need it or not, you really do need it – and if the patients are seen and assessed and checked every 3 months all of their lives, they have small changes caught early, whether it’s an infection you can slap down with medication or a nutrition problem that may be affecting a child’s growth and development.”
“We’re really kind of at a pivotal moment in CF, where we realize things are changing,” said A. Whitney Brown, MD, senior director for clinical affairs at the Cystic Fibrosis Foundation, and an adult CF and lung transplant physician in the Inova Advanced Lung Disease Program in Falls Church, Va.
“Patient needs and interest have evolved, because of the pandemic and because of the highly effective modulator therapy, but we want to take great effort to study it in a rigorous way, to make sure that as we are agile and adapt the care model, that we can maintain the same quality outcomes that we have traditionally done,” she said in an interview.
The Lancet Respiratory Medicine Commission on the future of CF care states that models of care “need to consider management approaches (including disease monitoring) to maintain health and delay lung transplantation, while minimizing the burden of care for patients and their families.”
‘A great problem to have’
One of the most significant changes in CF care has been the growing population of CF patients like Dr. Desch who are living well into adulthood, with some approaching Medicare eligibility.
With the advent of triple therapy and CFTR modulators being started earlier in life, lung function can be preserved, damage to other organs can be minimized, and life expectancy for patients with CF will continue to improve.
“We’re anticipating that there may be some needs in the aging CF population that are different than what we have historically had,” Dr. Brown said. “Will there be geriatric providers that need to become experts in CF care? That’s a great problem to have,” she said.
Dr. Goss agreed, noting that CF is steadily shifting from a near uniformly fatal disease to a chronic disorder that in many cases can be managed “with a complex regimen of novel drugs, much like HIV.”
He noted that there are multiple drug interactions with the triple combination, “so it’s really important that people don’t start a CF patient on a drug without consulting a pharmacist, because you can totally inactivate ETI, or augment it dramatically, and we’ve seen both happen.”
Cost and access
All experts interviewed for this article agreed that while the care of patients with CF has improved exponentially over the last few decades, there are still troubling inequities in care.
One of the largest impediments is the cost of care, with the triple combination costing more than $300,000 per year.
“Clearly patients aren’t paying that, but insurance companies are, and that’s causing all kinds of trickle-down effects that definitely affect patients. The patients like myself who are able to have insurance that covers it benefit, but there are so many people that don’t,” Dr. Desch said.
Dr. Sweezey noted that prior to the advent of ETI, patients with CF in Canada had better outcomes and longer life expectancy than did similar patients in the United States because of universal access to care and coordinated services under Canada’s health care system, compared with the highly fragmented and inefficient U.S. system. He added that the wider availability of ETI in the United States vs. Canada may begin to narrow that gap, however.
As noted before, there is a substantial proportion of patients – an estimated 10% – who have CFTR mutations that are not correctable by currently available CFTR modulators, and these patients are at significant risk for irreversible airway complications and lung damage.
In addition, although CF occurs most frequently among people of White ancestry, the disease does not respect distinctions of race or ethnicity.
“It’s not just [Whites] – a lot of people from different racial backgrounds, ethnic backgrounds, are not being diagnosed or are not being diagnosed soon enough to have effective care early enough,” Dr. Desch said.
That statement is supported by the Lancet Respiratory Medicine Commission on the future of cystic fibrosis care, whose members noted in 2019 that “epidemiological studies in the past 2 decades have shown that cystic fibrosis occurs and is more frequent than was previously thought in populations of non-European descent, and the disease is now recognized in many regions of the world.”
The commission members noted that the costs of adequate CF care may be beyond the reach of many patients in developing nations.
Still, if the substantial barriers of cost and access can be overcome, the future will continue to look brighter for patients with CF. As Dr. Sweezey put it: “There are studies that are pushing lower age limits for using these modulators, and as the evidence builds for the efficacy and safety at younger ages, I think all of us are hoping that we’ll end up being able to use either the current or future modulators to actually prevent trouble in CF, rather than trying to come along and fix it after it’s been there.”
Dr. Brown disclosed advisory board activity for Vertex that ended prior to her joining the CF Foundation. Dr. Desch, Dr. Goss, and Dr. Sweezey reported no relevant conflicts of interest.
In 1938, the year that cystic fibrosis (CF) was first described clinically, four of five children born with the disease did not live past their first birthdays.
In 2019, the median age at death for patients enrolled in the Cystic Fibrosis Foundation (CFF) registry was 32 years, and the predicted life expectancy for patients with CF who were born from 2015 through 2019 was 46 years.
Those numbers reflect the remarkable progress made in the past 4 decades in the care of patients with CF, but also highlight the obstacles ahead, given that the predicted life expectancy for the overall U.S. population in 2019 (pre–COVID-19) was 78.9 years.
Julie Desch, MD, is a CF survivor who has beaten the odds and then some. At age 61, the retired surgical pathologist is a CF patient advocate, speaker, and a board member of the Cystic Fibrosis Research Institute, a not-for-profit organization that funds CF research and offers education, advocacy, and psychosocial support for persons with CF and their families and caregivers.
In an interview, Dr. Desch said that while there has been remarkable progress in her lifetime in the field of CF research and treatment, particularly in the development of drugs that modulate function of the underlying cause of approximately 90% of CF cases, there are still many CF patients who cannot benefit from these therapies.
“There are still 10% of people who don’t make a protein to be modified, so that’s a huge unmet need,” she said.
Genetic disorder
CF is a chronic autosomal recessive disorder with multiorgan and multisystem manifestations. It is caused by mutations in the CFTR gene, which codes for the protein CF transmembrane conductance regulator. CFTR controls transport of chloride ions across cell membranes, specifically the apical membrane of epithelial cells in tissues of the airways, intestinal tract, pancreas, kidneys, sweat glands, and the reproductive system, notably the vas deferens in males.
The F508 deletion (F508del) mutation is the most common, occurring in approximately 70% of persons with CF. It is a class 2-type protein processing mutation, leading to defects in cellular processing, protein stability, and chloride channel gating defects.
The CFTR protein also secretes bicarbonate to regulate the pH of airway surface liquid, and inhibits the epithelial sodium channel, which mediates passive sodium transport across apical membranes of sodium-absorbing epithelial cells in the kidneys, intestine, and airways.
CF typically presents with the buildup in the lungs of abnormally viscous and sticky mucus leading to frequent, severe infections, particularly with Pseudomonas aeruginosa, progressive lung damage and, prior to the development of effective disease management, to premature death. The phenotype often includes malnutrition due to malabsorption, and failure to thrive.
Diagnosis
In all 50 U.S. states and the District of Columbia, newborns are screened for CF with an assay for immunoreactive trypsinogen (IRT) an indirect marker for pancreatic injury that is elevated in serum in most newborns with CF, but also detected in premature infants or those delivered under stressful circumstances. In some states newborns are tested only for IRT, with a diagnosis confirmed with a sweat chloride test and/or a CFTR mutation panel.
Treatment
There is no cure for CF, but the discovery of the gene in 1989 by Canadian and U.S. investigators has led to life-prolonging therapeutic interventions, specifically the development of CFTR modulators.
CFTR modulators include potentiators such as ivacaftor (Kalydeco), and correctors such as lumacaftor and tezacaftor (available in the combination Orkambi), and most recently in the triple combination of elexacaftor, tezacaftor, and ivacaftor (Trikafta; ETI).
Neil Sweezey, MD, FRCPC, a CF expert at The Hospital for Sick Children (SickKids) in Toronto, told this news organization that the ideal therapy for CF, genetic correction of the underlying mutations, is still not feasible, but that CFTR modulators are a close second.
“For 90% of patients, the three-drug combination Trikafta has been shown to be quite safe, quite tolerable, and quite remarkably beneficial,” he said.
In a study reported at CHEST 2021 by investigators from Nationwide Children’s Hospital in Columbus, Ohio, 32 adults who were started on the triple combination had significantly improved in forced expiratory volume in 1 second (FEV1), gain in body mass index, decreased sweat chloride and decreased colonization by Pseudomonas species. In addition, patients had significant improvements in blood inflammatory markers.
Christopher H. Goss, MD, FCCP, professor of pulmonary critical care and sleep medicine and professor of pediatrics at the University of Washington in Seattle, agreed that with the availability of the triple combination, “these are extraordinary times. An astounding fact is that most patients have complete resolution of cough, and the exacerbation rates have just plummeted,” he said in an interview.
Some of the reductions in exacerbations may be attributable to the COVID-19 pandemic, he noted, because patients in isolation have less exposure to circulating respiratory viruses.
“But it has been miraculous, and the clinical effect is certainly still more astounding than the effects of ivacaftor, which was the first truly breakthrough drug. Weight goes up, well-being increases, and the population lung function has shifted up to better grade lung function, in the entire population,” he said.
In addition, the need for lung and heart transplantation has sharply declined.
“I had a patient who had decided to forgo transplantation, despite absolutely horrible lung function, and he’s now bowling and leading a very productive life, when before he had been preparing for end of life,” Dr. Goss said.
Dr. Sweezey emphasized that as with all medications, patients being started on the triple combination require close monitoring for potential adverse events that might require dose modification or, for a small number of patients, withdrawal.
Burden of care
CFTR modulators have reduced but not eliminated the need for some patients to have mucolytic therapy, which may include dornase alfa, a recombinant human deoxyribonuclease (DNase) that reduces the viscosity of lung secretions, hypertonic saline inhaled twice daily (for patients 12 and older), mannitol, and physical manipulations to help patients clear mucus. This can include both manual percussion and the use of devices for high-frequency chest wall oscillation.
The complex nature of CF often requires a combination of other therapies to address comorbidities. These therapies may include infection prophylaxis and treatment with antibiotics and antifungals, nutrition support, and therapy for CF-related complications, including gastrointestinal issues, liver diseases, diabetes, and osteopenia that may be related to poor nutrient absorption, chronic inflammation, or other sequelae of CF.
In addition, patients often require frequent CF care center visits – ideally a minimum of every 3 months – which can result in significant loss of work or school time.
“Outcomes for patients in the long run have been absolutely proven to be best if they’re followed in big, established, multidisciplinary well-organized CF centers,” Dr. Sweezey said. “In the United States and Canada if you’re looked after on a regular basis, which means quarterly, every 3 months – whether you need it or not, you really do need it – and if the patients are seen and assessed and checked every 3 months all of their lives, they have small changes caught early, whether it’s an infection you can slap down with medication or a nutrition problem that may be affecting a child’s growth and development.”
“We’re really kind of at a pivotal moment in CF, where we realize things are changing,” said A. Whitney Brown, MD, senior director for clinical affairs at the Cystic Fibrosis Foundation, and an adult CF and lung transplant physician in the Inova Advanced Lung Disease Program in Falls Church, Va.
“Patient needs and interest have evolved, because of the pandemic and because of the highly effective modulator therapy, but we want to take great effort to study it in a rigorous way, to make sure that as we are agile and adapt the care model, that we can maintain the same quality outcomes that we have traditionally done,” she said in an interview.
The Lancet Respiratory Medicine Commission on the future of CF care states that models of care “need to consider management approaches (including disease monitoring) to maintain health and delay lung transplantation, while minimizing the burden of care for patients and their families.”
‘A great problem to have’
One of the most significant changes in CF care has been the growing population of CF patients like Dr. Desch who are living well into adulthood, with some approaching Medicare eligibility.
With the advent of triple therapy and CFTR modulators being started earlier in life, lung function can be preserved, damage to other organs can be minimized, and life expectancy for patients with CF will continue to improve.
“We’re anticipating that there may be some needs in the aging CF population that are different than what we have historically had,” Dr. Brown said. “Will there be geriatric providers that need to become experts in CF care? That’s a great problem to have,” she said.
Dr. Goss agreed, noting that CF is steadily shifting from a near uniformly fatal disease to a chronic disorder that in many cases can be managed “with a complex regimen of novel drugs, much like HIV.”
He noted that there are multiple drug interactions with the triple combination, “so it’s really important that people don’t start a CF patient on a drug without consulting a pharmacist, because you can totally inactivate ETI, or augment it dramatically, and we’ve seen both happen.”
Cost and access
All experts interviewed for this article agreed that while the care of patients with CF has improved exponentially over the last few decades, there are still troubling inequities in care.
One of the largest impediments is the cost of care, with the triple combination costing more than $300,000 per year.
“Clearly patients aren’t paying that, but insurance companies are, and that’s causing all kinds of trickle-down effects that definitely affect patients. The patients like myself who are able to have insurance that covers it benefit, but there are so many people that don’t,” Dr. Desch said.
Dr. Sweezey noted that prior to the advent of ETI, patients with CF in Canada had better outcomes and longer life expectancy than did similar patients in the United States because of universal access to care and coordinated services under Canada’s health care system, compared with the highly fragmented and inefficient U.S. system. He added that the wider availability of ETI in the United States vs. Canada may begin to narrow that gap, however.
As noted before, there is a substantial proportion of patients – an estimated 10% – who have CFTR mutations that are not correctable by currently available CFTR modulators, and these patients are at significant risk for irreversible airway complications and lung damage.
In addition, although CF occurs most frequently among people of White ancestry, the disease does not respect distinctions of race or ethnicity.
“It’s not just [Whites] – a lot of people from different racial backgrounds, ethnic backgrounds, are not being diagnosed or are not being diagnosed soon enough to have effective care early enough,” Dr. Desch said.
That statement is supported by the Lancet Respiratory Medicine Commission on the future of cystic fibrosis care, whose members noted in 2019 that “epidemiological studies in the past 2 decades have shown that cystic fibrosis occurs and is more frequent than was previously thought in populations of non-European descent, and the disease is now recognized in many regions of the world.”
The commission members noted that the costs of adequate CF care may be beyond the reach of many patients in developing nations.
Still, if the substantial barriers of cost and access can be overcome, the future will continue to look brighter for patients with CF. As Dr. Sweezey put it: “There are studies that are pushing lower age limits for using these modulators, and as the evidence builds for the efficacy and safety at younger ages, I think all of us are hoping that we’ll end up being able to use either the current or future modulators to actually prevent trouble in CF, rather than trying to come along and fix it after it’s been there.”
Dr. Brown disclosed advisory board activity for Vertex that ended prior to her joining the CF Foundation. Dr. Desch, Dr. Goss, and Dr. Sweezey reported no relevant conflicts of interest.
In 1938, the year that cystic fibrosis (CF) was first described clinically, four of five children born with the disease did not live past their first birthdays.
In 2019, the median age at death for patients enrolled in the Cystic Fibrosis Foundation (CFF) registry was 32 years, and the predicted life expectancy for patients with CF who were born from 2015 through 2019 was 46 years.
Those numbers reflect the remarkable progress made in the past 4 decades in the care of patients with CF, but also highlight the obstacles ahead, given that the predicted life expectancy for the overall U.S. population in 2019 (pre–COVID-19) was 78.9 years.
Julie Desch, MD, is a CF survivor who has beaten the odds and then some. At age 61, the retired surgical pathologist is a CF patient advocate, speaker, and a board member of the Cystic Fibrosis Research Institute, a not-for-profit organization that funds CF research and offers education, advocacy, and psychosocial support for persons with CF and their families and caregivers.
In an interview, Dr. Desch said that while there has been remarkable progress in her lifetime in the field of CF research and treatment, particularly in the development of drugs that modulate function of the underlying cause of approximately 90% of CF cases, there are still many CF patients who cannot benefit from these therapies.
“There are still 10% of people who don’t make a protein to be modified, so that’s a huge unmet need,” she said.
Genetic disorder
CF is a chronic autosomal recessive disorder with multiorgan and multisystem manifestations. It is caused by mutations in the CFTR gene, which codes for the protein CF transmembrane conductance regulator. CFTR controls transport of chloride ions across cell membranes, specifically the apical membrane of epithelial cells in tissues of the airways, intestinal tract, pancreas, kidneys, sweat glands, and the reproductive system, notably the vas deferens in males.
The F508 deletion (F508del) mutation is the most common, occurring in approximately 70% of persons with CF. It is a class 2-type protein processing mutation, leading to defects in cellular processing, protein stability, and chloride channel gating defects.
The CFTR protein also secretes bicarbonate to regulate the pH of airway surface liquid, and inhibits the epithelial sodium channel, which mediates passive sodium transport across apical membranes of sodium-absorbing epithelial cells in the kidneys, intestine, and airways.
CF typically presents with the buildup in the lungs of abnormally viscous and sticky mucus leading to frequent, severe infections, particularly with Pseudomonas aeruginosa, progressive lung damage and, prior to the development of effective disease management, to premature death. The phenotype often includes malnutrition due to malabsorption, and failure to thrive.
Diagnosis
In all 50 U.S. states and the District of Columbia, newborns are screened for CF with an assay for immunoreactive trypsinogen (IRT) an indirect marker for pancreatic injury that is elevated in serum in most newborns with CF, but also detected in premature infants or those delivered under stressful circumstances. In some states newborns are tested only for IRT, with a diagnosis confirmed with a sweat chloride test and/or a CFTR mutation panel.
Treatment
There is no cure for CF, but the discovery of the gene in 1989 by Canadian and U.S. investigators has led to life-prolonging therapeutic interventions, specifically the development of CFTR modulators.
CFTR modulators include potentiators such as ivacaftor (Kalydeco), and correctors such as lumacaftor and tezacaftor (available in the combination Orkambi), and most recently in the triple combination of elexacaftor, tezacaftor, and ivacaftor (Trikafta; ETI).
Neil Sweezey, MD, FRCPC, a CF expert at The Hospital for Sick Children (SickKids) in Toronto, told this news organization that the ideal therapy for CF, genetic correction of the underlying mutations, is still not feasible, but that CFTR modulators are a close second.
“For 90% of patients, the three-drug combination Trikafta has been shown to be quite safe, quite tolerable, and quite remarkably beneficial,” he said.
In a study reported at CHEST 2021 by investigators from Nationwide Children’s Hospital in Columbus, Ohio, 32 adults who were started on the triple combination had significantly improved in forced expiratory volume in 1 second (FEV1), gain in body mass index, decreased sweat chloride and decreased colonization by Pseudomonas species. In addition, patients had significant improvements in blood inflammatory markers.
Christopher H. Goss, MD, FCCP, professor of pulmonary critical care and sleep medicine and professor of pediatrics at the University of Washington in Seattle, agreed that with the availability of the triple combination, “these are extraordinary times. An astounding fact is that most patients have complete resolution of cough, and the exacerbation rates have just plummeted,” he said in an interview.
Some of the reductions in exacerbations may be attributable to the COVID-19 pandemic, he noted, because patients in isolation have less exposure to circulating respiratory viruses.
“But it has been miraculous, and the clinical effect is certainly still more astounding than the effects of ivacaftor, which was the first truly breakthrough drug. Weight goes up, well-being increases, and the population lung function has shifted up to better grade lung function, in the entire population,” he said.
In addition, the need for lung and heart transplantation has sharply declined.
“I had a patient who had decided to forgo transplantation, despite absolutely horrible lung function, and he’s now bowling and leading a very productive life, when before he had been preparing for end of life,” Dr. Goss said.
Dr. Sweezey emphasized that as with all medications, patients being started on the triple combination require close monitoring for potential adverse events that might require dose modification or, for a small number of patients, withdrawal.
Burden of care
CFTR modulators have reduced but not eliminated the need for some patients to have mucolytic therapy, which may include dornase alfa, a recombinant human deoxyribonuclease (DNase) that reduces the viscosity of lung secretions, hypertonic saline inhaled twice daily (for patients 12 and older), mannitol, and physical manipulations to help patients clear mucus. This can include both manual percussion and the use of devices for high-frequency chest wall oscillation.
The complex nature of CF often requires a combination of other therapies to address comorbidities. These therapies may include infection prophylaxis and treatment with antibiotics and antifungals, nutrition support, and therapy for CF-related complications, including gastrointestinal issues, liver diseases, diabetes, and osteopenia that may be related to poor nutrient absorption, chronic inflammation, or other sequelae of CF.
In addition, patients often require frequent CF care center visits – ideally a minimum of every 3 months – which can result in significant loss of work or school time.
“Outcomes for patients in the long run have been absolutely proven to be best if they’re followed in big, established, multidisciplinary well-organized CF centers,” Dr. Sweezey said. “In the United States and Canada if you’re looked after on a regular basis, which means quarterly, every 3 months – whether you need it or not, you really do need it – and if the patients are seen and assessed and checked every 3 months all of their lives, they have small changes caught early, whether it’s an infection you can slap down with medication or a nutrition problem that may be affecting a child’s growth and development.”
“We’re really kind of at a pivotal moment in CF, where we realize things are changing,” said A. Whitney Brown, MD, senior director for clinical affairs at the Cystic Fibrosis Foundation, and an adult CF and lung transplant physician in the Inova Advanced Lung Disease Program in Falls Church, Va.
“Patient needs and interest have evolved, because of the pandemic and because of the highly effective modulator therapy, but we want to take great effort to study it in a rigorous way, to make sure that as we are agile and adapt the care model, that we can maintain the same quality outcomes that we have traditionally done,” she said in an interview.
The Lancet Respiratory Medicine Commission on the future of CF care states that models of care “need to consider management approaches (including disease monitoring) to maintain health and delay lung transplantation, while minimizing the burden of care for patients and their families.”
‘A great problem to have’
One of the most significant changes in CF care has been the growing population of CF patients like Dr. Desch who are living well into adulthood, with some approaching Medicare eligibility.
With the advent of triple therapy and CFTR modulators being started earlier in life, lung function can be preserved, damage to other organs can be minimized, and life expectancy for patients with CF will continue to improve.
“We’re anticipating that there may be some needs in the aging CF population that are different than what we have historically had,” Dr. Brown said. “Will there be geriatric providers that need to become experts in CF care? That’s a great problem to have,” she said.
Dr. Goss agreed, noting that CF is steadily shifting from a near uniformly fatal disease to a chronic disorder that in many cases can be managed “with a complex regimen of novel drugs, much like HIV.”
He noted that there are multiple drug interactions with the triple combination, “so it’s really important that people don’t start a CF patient on a drug without consulting a pharmacist, because you can totally inactivate ETI, or augment it dramatically, and we’ve seen both happen.”
Cost and access
All experts interviewed for this article agreed that while the care of patients with CF has improved exponentially over the last few decades, there are still troubling inequities in care.
One of the largest impediments is the cost of care, with the triple combination costing more than $300,000 per year.
“Clearly patients aren’t paying that, but insurance companies are, and that’s causing all kinds of trickle-down effects that definitely affect patients. The patients like myself who are able to have insurance that covers it benefit, but there are so many people that don’t,” Dr. Desch said.
Dr. Sweezey noted that prior to the advent of ETI, patients with CF in Canada had better outcomes and longer life expectancy than did similar patients in the United States because of universal access to care and coordinated services under Canada’s health care system, compared with the highly fragmented and inefficient U.S. system. He added that the wider availability of ETI in the United States vs. Canada may begin to narrow that gap, however.
As noted before, there is a substantial proportion of patients – an estimated 10% – who have CFTR mutations that are not correctable by currently available CFTR modulators, and these patients are at significant risk for irreversible airway complications and lung damage.
In addition, although CF occurs most frequently among people of White ancestry, the disease does not respect distinctions of race or ethnicity.
“It’s not just [Whites] – a lot of people from different racial backgrounds, ethnic backgrounds, are not being diagnosed or are not being diagnosed soon enough to have effective care early enough,” Dr. Desch said.
That statement is supported by the Lancet Respiratory Medicine Commission on the future of cystic fibrosis care, whose members noted in 2019 that “epidemiological studies in the past 2 decades have shown that cystic fibrosis occurs and is more frequent than was previously thought in populations of non-European descent, and the disease is now recognized in many regions of the world.”
The commission members noted that the costs of adequate CF care may be beyond the reach of many patients in developing nations.
Still, if the substantial barriers of cost and access can be overcome, the future will continue to look brighter for patients with CF. As Dr. Sweezey put it: “There are studies that are pushing lower age limits for using these modulators, and as the evidence builds for the efficacy and safety at younger ages, I think all of us are hoping that we’ll end up being able to use either the current or future modulators to actually prevent trouble in CF, rather than trying to come along and fix it after it’s been there.”
Dr. Brown disclosed advisory board activity for Vertex that ended prior to her joining the CF Foundation. Dr. Desch, Dr. Goss, and Dr. Sweezey reported no relevant conflicts of interest.
Lipedema: A potentially devastating, often unrecognized disease
” according to C. William Hanke, MD, MPH.
“This disease is well known in Europe, especially in the Netherlands, Germany, and Austria, but in this country, I believe most dermatologists have never heard of it,” Dr. Hanke said at the ODAC Dermatology, Aesthetic & Surgical Conference.

Clinically, patients with lipedema – also known as “two-body syndrome” – present with a symmetric, bilateral increase in subcutaneous fat, with “cuffs of fat” around the ankles. It usually affects the legs and thighs; the hands and feet are not affected.
“From the waist on up, the body looks like one person, and from the waist on down, it looks like an entirely different person,” said Dr. Hanke, a dermatologist who is program director for the micrographic surgery and dermatologic oncology fellowship training program at Ascension St. Vincent Hospital in Indianapolis. “Just think of the difficulty that the person has with their life in terms of buying clothes or social interactions. This is a devastating problem.”
Lipedema almost always affects women and is progressive from puberty. “Characteristically, patients have pain and bruise easily in the areas of lipedema,” said Dr. Hanke, who has served as president of the American Academy of Dermatology, the American Society for Dermatologic Surgery, the American College of Mohs Surgery, and the International Society for Dermatologic Surgery. The affected areas are painful to touch, making exercise uncomfortable for patients, he said.
Lipedema can be masked by obesity, “so, if you superimpose generalized obesity on lipedema, you have an even more difficult problem,” he added. “A physician who doesn’t understand the disease may perform standard nontumescent liposuction under general anesthesia, with cannulas, which traumatize lipedematous fat. Thereby, a patient with lipedema can then be inadvertently transformed into a patient with lympholipedema. Then you’ve got even an even worse problem.”
One might think that the rate of diabetes would be high among lipedema patients, “but diabetes is essentially nonexistent in this group,” he continued. However, patients with lipedema “may develop hypothyroidism, venous disease, joint pain, and fibrosis in the fat as the disease progresses.”
Lipedema stages, treatment
Lipedema is defined by three clinical stages: Stage one is characterized by an enlarged subcutaneous fat department, but the skin surface is smooth. In stage 2, the skin surface becomes wavy with irregularities and dents, and in stage 3, patients develop large deforming nodules and hanging flaps.
“If we can diagnose lipedema in the early stages and perform tumescent liposuction using tumescent local anesthesia, we can prevent the progression of the disease,” Dr. Hanke said. For patients who meet criteria for tumescent liposuction, three to six treatments may be required for stage 3 disease. “Tumescent local anesthesia should be used, because liposuction using tumescent local anesthesia is atraumatic to fat,” he said. “Usually, the most painful areas are treated first.”
In a single-center study from Germany that followed 85 patients who underwent tumescent liposuction for lipedema, researchers found that improvements in pain, bruising, and mobility were sustained at 4 and 8 years following the procedure. Patient quality of life and cosmetic appearance were also sustained.
In terms of liposuction’s cosmetic effects, “the goal of liposuction in lipedema patients is different,” Dr. Hanke said. “The goal is to get these people moving again, stabilize their weight, and minimize progression of the disease. Cosmetic improvement is secondary.”
A more recent follow-up study of 60 patients from the same single-center German study showed that the positive effects of liposuction lasted 12 years postoperatively without relevant progression of disease.
Following the first International Consensus Conference on Lipedema in Vienna in 2017, Dr. Hanke and colleagues published guidelines on preventing progression of lipedema with liposuction using tumescent local anesthesia.
“If patients with lipedema gain weight, the problem becomes even worse,” he said. “A sensible diet and nontraumatic exercise like water aerobics is ideal. If patients pursue yo-yo dieting, more and more fat stays in the legs after each cycle. Sometimes I’ll refer overweight patients with lipedema for a bariatric surgery consult.”
Dr. Hanke noted that Karen Herbst, MD, PhD, an endocrinologist at the University of Arizona, Tucson, who is widely considered an expert on the medical management of lipedema, has a website on lipedema care.
Dr. Hanke reported having no financial conflicts related to his presentation.
” according to C. William Hanke, MD, MPH.
“This disease is well known in Europe, especially in the Netherlands, Germany, and Austria, but in this country, I believe most dermatologists have never heard of it,” Dr. Hanke said at the ODAC Dermatology, Aesthetic & Surgical Conference.
Clinically, patients with lipedema – also known as “two-body syndrome” – present with a symmetric, bilateral increase in subcutaneous fat, with “cuffs of fat” around the ankles. It usually affects the legs and thighs; the hands and feet are not affected.
“From the waist on up, the body looks like one person, and from the waist on down, it looks like an entirely different person,” said Dr. Hanke, a dermatologist who is program director for the micrographic surgery and dermatologic oncology fellowship training program at Ascension St. Vincent Hospital in Indianapolis. “Just think of the difficulty that the person has with their life in terms of buying clothes or social interactions. This is a devastating problem.”
Lipedema almost always affects women and is progressive from puberty. “Characteristically, patients have pain and bruise easily in the areas of lipedema,” said Dr. Hanke, who has served as president of the American Academy of Dermatology, the American Society for Dermatologic Surgery, the American College of Mohs Surgery, and the International Society for Dermatologic Surgery. The affected areas are painful to touch, making exercise uncomfortable for patients, he said.
Lipedema can be masked by obesity, “so, if you superimpose generalized obesity on lipedema, you have an even more difficult problem,” he added. “A physician who doesn’t understand the disease may perform standard nontumescent liposuction under general anesthesia, with cannulas, which traumatize lipedematous fat. Thereby, a patient with lipedema can then be inadvertently transformed into a patient with lympholipedema. Then you’ve got even an even worse problem.”
One might think that the rate of diabetes would be high among lipedema patients, “but diabetes is essentially nonexistent in this group,” he continued. However, patients with lipedema “may develop hypothyroidism, venous disease, joint pain, and fibrosis in the fat as the disease progresses.”
Lipedema stages, treatment
Lipedema is defined by three clinical stages: Stage one is characterized by an enlarged subcutaneous fat department, but the skin surface is smooth. In stage 2, the skin surface becomes wavy with irregularities and dents, and in stage 3, patients develop large deforming nodules and hanging flaps.
“If we can diagnose lipedema in the early stages and perform tumescent liposuction using tumescent local anesthesia, we can prevent the progression of the disease,” Dr. Hanke said. For patients who meet criteria for tumescent liposuction, three to six treatments may be required for stage 3 disease. “Tumescent local anesthesia should be used, because liposuction using tumescent local anesthesia is atraumatic to fat,” he said. “Usually, the most painful areas are treated first.”
In a single-center study from Germany that followed 85 patients who underwent tumescent liposuction for lipedema, researchers found that improvements in pain, bruising, and mobility were sustained at 4 and 8 years following the procedure. Patient quality of life and cosmetic appearance were also sustained.
In terms of liposuction’s cosmetic effects, “the goal of liposuction in lipedema patients is different,” Dr. Hanke said. “The goal is to get these people moving again, stabilize their weight, and minimize progression of the disease. Cosmetic improvement is secondary.”
A more recent follow-up study of 60 patients from the same single-center German study showed that the positive effects of liposuction lasted 12 years postoperatively without relevant progression of disease.
Following the first International Consensus Conference on Lipedema in Vienna in 2017, Dr. Hanke and colleagues published guidelines on preventing progression of lipedema with liposuction using tumescent local anesthesia.
“If patients with lipedema gain weight, the problem becomes even worse,” he said. “A sensible diet and nontraumatic exercise like water aerobics is ideal. If patients pursue yo-yo dieting, more and more fat stays in the legs after each cycle. Sometimes I’ll refer overweight patients with lipedema for a bariatric surgery consult.”
Dr. Hanke noted that Karen Herbst, MD, PhD, an endocrinologist at the University of Arizona, Tucson, who is widely considered an expert on the medical management of lipedema, has a website on lipedema care.
Dr. Hanke reported having no financial conflicts related to his presentation.
” according to C. William Hanke, MD, MPH.
“This disease is well known in Europe, especially in the Netherlands, Germany, and Austria, but in this country, I believe most dermatologists have never heard of it,” Dr. Hanke said at the ODAC Dermatology, Aesthetic & Surgical Conference.
Clinically, patients with lipedema – also known as “two-body syndrome” – present with a symmetric, bilateral increase in subcutaneous fat, with “cuffs of fat” around the ankles. It usually affects the legs and thighs; the hands and feet are not affected.
“From the waist on up, the body looks like one person, and from the waist on down, it looks like an entirely different person,” said Dr. Hanke, a dermatologist who is program director for the micrographic surgery and dermatologic oncology fellowship training program at Ascension St. Vincent Hospital in Indianapolis. “Just think of the difficulty that the person has with their life in terms of buying clothes or social interactions. This is a devastating problem.”
Lipedema almost always affects women and is progressive from puberty. “Characteristically, patients have pain and bruise easily in the areas of lipedema,” said Dr. Hanke, who has served as president of the American Academy of Dermatology, the American Society for Dermatologic Surgery, the American College of Mohs Surgery, and the International Society for Dermatologic Surgery. The affected areas are painful to touch, making exercise uncomfortable for patients, he said.
Lipedema can be masked by obesity, “so, if you superimpose generalized obesity on lipedema, you have an even more difficult problem,” he added. “A physician who doesn’t understand the disease may perform standard nontumescent liposuction under general anesthesia, with cannulas, which traumatize lipedematous fat. Thereby, a patient with lipedema can then be inadvertently transformed into a patient with lympholipedema. Then you’ve got even an even worse problem.”
One might think that the rate of diabetes would be high among lipedema patients, “but diabetes is essentially nonexistent in this group,” he continued. However, patients with lipedema “may develop hypothyroidism, venous disease, joint pain, and fibrosis in the fat as the disease progresses.”
Lipedema stages, treatment
Lipedema is defined by three clinical stages: Stage one is characterized by an enlarged subcutaneous fat department, but the skin surface is smooth. In stage 2, the skin surface becomes wavy with irregularities and dents, and in stage 3, patients develop large deforming nodules and hanging flaps.
“If we can diagnose lipedema in the early stages and perform tumescent liposuction using tumescent local anesthesia, we can prevent the progression of the disease,” Dr. Hanke said. For patients who meet criteria for tumescent liposuction, three to six treatments may be required for stage 3 disease. “Tumescent local anesthesia should be used, because liposuction using tumescent local anesthesia is atraumatic to fat,” he said. “Usually, the most painful areas are treated first.”
In a single-center study from Germany that followed 85 patients who underwent tumescent liposuction for lipedema, researchers found that improvements in pain, bruising, and mobility were sustained at 4 and 8 years following the procedure. Patient quality of life and cosmetic appearance were also sustained.
In terms of liposuction’s cosmetic effects, “the goal of liposuction in lipedema patients is different,” Dr. Hanke said. “The goal is to get these people moving again, stabilize their weight, and minimize progression of the disease. Cosmetic improvement is secondary.”
A more recent follow-up study of 60 patients from the same single-center German study showed that the positive effects of liposuction lasted 12 years postoperatively without relevant progression of disease.
Following the first International Consensus Conference on Lipedema in Vienna in 2017, Dr. Hanke and colleagues published guidelines on preventing progression of lipedema with liposuction using tumescent local anesthesia.
“If patients with lipedema gain weight, the problem becomes even worse,” he said. “A sensible diet and nontraumatic exercise like water aerobics is ideal. If patients pursue yo-yo dieting, more and more fat stays in the legs after each cycle. Sometimes I’ll refer overweight patients with lipedema for a bariatric surgery consult.”
Dr. Hanke noted that Karen Herbst, MD, PhD, an endocrinologist at the University of Arizona, Tucson, who is widely considered an expert on the medical management of lipedema, has a website on lipedema care.
Dr. Hanke reported having no financial conflicts related to his presentation.
FROM ODAC 2022
FDA approves levoketoconazole for Cushing syndrome
The Food and Drug Administration has approved levoketoconazole (Recorlev, Xeris Biopharma) for the treatment of endogenous hypercortisolemia in adults with Cushing syndrome for whom surgery is not possible or was not curative.
Endogenous Cushing syndrome is a relatively rare condition characterized by chronically elevated cortisol levels, typically arising from a benign pituitary tumor. Left untreated, it can lead to reproductive problems and hirsutism in women, as well as serious complications, including diabetes, hypertension, tissue fragility, and mood disorders. Half of patients will die within 5 years if left untreated.

Levoketoconazole inhibits cortisol synthesis. The FDA approval was based on efficacy and safety data from two phase 3 studies involving a total of 166 patients with endogenous Cushing syndrome. In both the open-label, single-arm SONICS study and the randomized, placebo-controlled LOGICS trial, the drug significantly reduced and normalized mean urinary free cortisol levels and improved several secondary endpoints. The ongoing open-label OPTICS study will gather long-term data.
The Recorlev label includes boxed warnings about the potential for life-threatening hepatotoxicity and QT prolongation. Prior to and during treatment, patients should undergo liver enzyme testing, ECG, and correction of hypokalemia and hypomagnesemia.
The most common adverse reactions (occurring in less than 20%) include nausea/vomiting, hypokalemia, hemorrhage/contusion, systemic hypertension, headache, hepatic injury, abnormal uterine bleeding, erythema, fatigue, abdominal pain/dyspepsia, arthritis, upper respiratory infection, myalgia, arrhythmia, back pain, insomnia/sleep disturbances, and peripheral edema.
“Cushing syndrome is a rare disease that can be physically and emotionally devastating to the patient. Most patients endure years of symptoms prior to obtaining a diagnosis and are then faced with limited effective treatment options ... We are excited to see that the long and complicated path of rare drug development has reached FDA approval on a new therapeutic option for our underserved Cushing’s community,” Leslie Edwin, president of the Cushing’s Support & Research Foundation, said in a Xeris statement.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved levoketoconazole (Recorlev, Xeris Biopharma) for the treatment of endogenous hypercortisolemia in adults with Cushing syndrome for whom surgery is not possible or was not curative.
Endogenous Cushing syndrome is a relatively rare condition characterized by chronically elevated cortisol levels, typically arising from a benign pituitary tumor. Left untreated, it can lead to reproductive problems and hirsutism in women, as well as serious complications, including diabetes, hypertension, tissue fragility, and mood disorders. Half of patients will die within 5 years if left untreated.
Levoketoconazole inhibits cortisol synthesis. The FDA approval was based on efficacy and safety data from two phase 3 studies involving a total of 166 patients with endogenous Cushing syndrome. In both the open-label, single-arm SONICS study and the randomized, placebo-controlled LOGICS trial, the drug significantly reduced and normalized mean urinary free cortisol levels and improved several secondary endpoints. The ongoing open-label OPTICS study will gather long-term data.
The Recorlev label includes boxed warnings about the potential for life-threatening hepatotoxicity and QT prolongation. Prior to and during treatment, patients should undergo liver enzyme testing, ECG, and correction of hypokalemia and hypomagnesemia.
The most common adverse reactions (occurring in less than 20%) include nausea/vomiting, hypokalemia, hemorrhage/contusion, systemic hypertension, headache, hepatic injury, abnormal uterine bleeding, erythema, fatigue, abdominal pain/dyspepsia, arthritis, upper respiratory infection, myalgia, arrhythmia, back pain, insomnia/sleep disturbances, and peripheral edema.
“Cushing syndrome is a rare disease that can be physically and emotionally devastating to the patient. Most patients endure years of symptoms prior to obtaining a diagnosis and are then faced with limited effective treatment options ... We are excited to see that the long and complicated path of rare drug development has reached FDA approval on a new therapeutic option for our underserved Cushing’s community,” Leslie Edwin, president of the Cushing’s Support & Research Foundation, said in a Xeris statement.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration has approved levoketoconazole (Recorlev, Xeris Biopharma) for the treatment of endogenous hypercortisolemia in adults with Cushing syndrome for whom surgery is not possible or was not curative.
Endogenous Cushing syndrome is a relatively rare condition characterized by chronically elevated cortisol levels, typically arising from a benign pituitary tumor. Left untreated, it can lead to reproductive problems and hirsutism in women, as well as serious complications, including diabetes, hypertension, tissue fragility, and mood disorders. Half of patients will die within 5 years if left untreated.
Levoketoconazole inhibits cortisol synthesis. The FDA approval was based on efficacy and safety data from two phase 3 studies involving a total of 166 patients with endogenous Cushing syndrome. In both the open-label, single-arm SONICS study and the randomized, placebo-controlled LOGICS trial, the drug significantly reduced and normalized mean urinary free cortisol levels and improved several secondary endpoints. The ongoing open-label OPTICS study will gather long-term data.
The Recorlev label includes boxed warnings about the potential for life-threatening hepatotoxicity and QT prolongation. Prior to and during treatment, patients should undergo liver enzyme testing, ECG, and correction of hypokalemia and hypomagnesemia.
The most common adverse reactions (occurring in less than 20%) include nausea/vomiting, hypokalemia, hemorrhage/contusion, systemic hypertension, headache, hepatic injury, abnormal uterine bleeding, erythema, fatigue, abdominal pain/dyspepsia, arthritis, upper respiratory infection, myalgia, arrhythmia, back pain, insomnia/sleep disturbances, and peripheral edema.
“Cushing syndrome is a rare disease that can be physically and emotionally devastating to the patient. Most patients endure years of symptoms prior to obtaining a diagnosis and are then faced with limited effective treatment options ... We are excited to see that the long and complicated path of rare drug development has reached FDA approval on a new therapeutic option for our underserved Cushing’s community,” Leslie Edwin, president of the Cushing’s Support & Research Foundation, said in a Xeris statement.
A version of this article first appeared on Medscape.com.
Case series show no consensus on treatment for palmoplantar pustulosis, generalized pustular psoriasis
that evaluated the characteristics and course of the disease in patients diagnosed with PPP or GPP.
“These case series confirm the rarity of both generalized pustular psoriasis and palmoplantar pustulosis (PPP) and highlight the persistence of symptoms over time and the lack of effective treatment options available to patients,” Megan H. Noe, MD, MPH, MSCE, first author of both case series and assistant professor of dermatology, Harvard Medical School, and a dermatologist at Brigham and Women’s Hospital, both in Boston, said in an interview. In both studies, she added, “more than 20 different therapies were utilized, demonstrating a lack of consensus regarding effective treatment.”
The two case series were published in JAMA Dermatology.
Palmoplantar pustulosis
In the case series of 197 patients with PPP , data were obtained from a retrospective review at 20 academic dermatology practices in the United States between January 2007 and December 2018. The patients were mostly women (73.6%) who were White (60.9%), with a mean age of 53 years; 38.1% were current smokers, and 27.4% were former smokers, and the mean follow-up time was 22.1 months. About half (48.2%) of patients who presented to their respective centers had skin pain, 19.8% had problems using their hands and feet, 12.7% had arthralgias, and 2% had myalgias. Clinicians who examined these patients found pustules on the palms (80.2%), soles (76.7%), and both palms and soles (59.9%); some nail unit involvement was reported in 10.2%.
Patients were treated with a variety of topical therapies, systemic steroids, systemic anti-infectives, and systemic psoriasis therapies, Dr. Noe and colleagues said. The most common initial treatments included a topical steroid (84.8%), with the vast majority of clinicians using a high-potency topical steroid (153 of 167 patients; 91.6%), or topical therapy only (64.5%).
Other initial treatments used were other types of topical medications in 34 of the patients in the series (17.3%), such as a vitamin D analogue in 27 patients (79.4%); oral systemic treatments such as acitretin in 27 patients (13.7%) or methotrexate in 22 patients (11.2%); narrowband UVB phototherapy in 15 patients (7.7%); systemic steroids in 10 patients (5.1%); or systemic antibiotics in 9 patients (4.6%). Less commonly used were biologic agents like adalimumab, used in 6 patients (3.1%).
The researchers also examined health care utilization in 128 patients and found that 82% had at least one follow-up visit, 31.3% required two to three follow-up visits, and 18.8% had five or more follow-up visits. When adjusted to account for age and sex, there was a decreased risk of requiring five or more healthcare visits per year for women (odds ratio, 0.49; 95% confidence interval, 0.25-0.95)
Generalized pustular psoriasis
Dr. Noe and colleagues also evaluated 95 patients with GPP in a retrospective longitudinal case series of patients treated at 20 academic dermatology practices in the United States between January 2007 and December 2018. As in the PPP group, most patients in the GPP case series were women (70.5%), and over half were White (53.7%); the mean age was 50.3 years old, and the mean follow-up time was 19.8 months. A majority of patients with GPP were never-smokers (52.6%) or former smokers (20%). When patients with GPP initially presented to the study sites, 36.8% were admitted as inpatients, 9.5% presented in the emergency department, and 53.7% presented in an outpatient or ambulatory dermatology setting.
GPP commonly appeared on the trunk and extremities, but was “also reported on the scalp, face, genitals, nail unit, and mucous membranes in a minority of patients,” the researchers said. Overall, 62.1% of patients had skin pain, 26.2% had joint pain, 16.8% reported tachycardia, and 9.5% reported fever. Hypertension, depression, diabetes, chronic kidney disease, and hypothyroidism were common comorbidities of GPP, the researchers noted.
Clinicians reported treating GPP with topical steroids (86.3%) and topical treatments alone (32.3%). Oral systemic treatments such as acitretin (24.2%), cyclosporine (22.1%), and methotrexate (13.7%) were also used, as well as systemic steroids (20%). Other treatments used were narrowband UVB phototherapy (5.3%) and biologic agents like adalimumab (4.2%) and infliximab (4.2%).
For 53 patients with follow-up data of at least 6 months, 19 (35.8%) had been hospitalized because of their symptoms, and 8 patients were hospitalized for further GPP-specific concerns. Patients with GPP had a median 3.2 dermatology visits per year and a maximum of 18 visits. A model that was adjusted for age and sex showed women were at a decreased risk for being admitted to the hospital or emergency department in the follow-up period (odds ratio, 0.19; 95% confidence interval, 0.04-0.83).
PPP and GPP in practice
Sylvia Hsu, MD, professor and chair of the department of dermatology at Temple University, Philadelphia, who was not involved with the research, noted that most dermatologists will see few, if any, cases of PPP and GPP in a year. At her center, she estimated that she sees about one PPP case per week, and one or two cases of GPP a year. In general, she said that her clinical experience matched what was found by the authors of both case series.

For patients with PPP, “I would say the average dermatologist would probably start out with a superpotent topical steroid like clobetasol or halobetasol ointment,” Dr. Hsu said.
If they are not of childbearing age, she added, she would also prescribe acitretin, which she avoids giving to patients of childbearing age because of its teratogenicity. “Acitretin has the reputation that it doesn’t work well or fast for psoriasis. It doesn’t work well or fast for plaque-type psoriasis, but it works well and fast for pustular psoriasis,” she said.
In place of acitretin, Dr. Hsu recommended cyclosporine for a patient of childbearing age as a short-term solution to resolve symptoms before transitioning them to another therapy. “A woman of childbearing age, you put on cyclosporine, you’ve got to transition to something else,” she said. “And so many times you wean them off, the pustular psoriasis comes back because the topical steroid doesn’t work that well.”
One possible option is the interluekin-23 inhibitor guselkumab (approved by the Food and Drug Administration for treating moderate to severe plaque psoriasis and psoriatic arthritis) but cost and effectiveness can be a factor. Although studies have shown efficacy, biologics as treatments for PPP are “hit or miss,” Dr. Hsu said.
Regarding use of systemic therapies, Dr. Hsu cautioned against using them to treat plaque-type psoriasis. “We always learn, don’t use a systemic steroid like prednisone to treat psoriasis because it helps, but it comes back with a vengeance,” she said. “Sometimes when you treat plaque-type psoriasis with prednisone, it could come back with a vengeance, and it can come back as generalized pustular psoriasis.”
For patients with GPP, “you need a quick fix” because of the painful symptoms associated with the disease, Dr. Hsu said. In this case, she recommended cyclosporine and said she would avoid prescribing topical medications. “You’re going to have to give an oral drug because usually when we’re seeing somebody with GPP, they’re either a hospital consult or they just walked in the door,” she said. After prescribing cyclosporine, you would transition to another treatment like a biologic “as quickly as you can” with the knowledge that the biologic “may or may not work.”
New treatment options needed
Commenting on both case series in a related editorial, Edward W. Cowen, MD, MHSc, senior clinician and head of the dermatology consultation service in the dermatology branch of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Md., said that “much of the clinical presentation of pustular disease remains a mystery,” including why tobacco use is a risk factor for developing pustular psoriasis, and why tumor necrosis factor inhibitors “induce pustular disease in a small number of patients” with psoriasis vulgaris.

“Most importantly, we still do not know if localized and generalized pustular psoriasis all truly represent different variants of the same disease process, and if not, which biologic treatment represents the best option for a given clinical variant,” he wrote.
Dr. Cowen noted that the multi-institutional approach to collecting the retrospective data in these case series could be used as a “basic framework to build on for future clinical trials for rare skin diseases such as pustular psoriasis.”
In the interview, Dr. Noe said that she hoped that the “Pustular Psoriasis in the US Research Group” she and her coauthors created for the case series could help with the development of prospective clinical trials. “For pustular psoriasis and other rare diseases in dermatology, multi-institutional collaborations are necessary to conduct prospective research,” she said.
“While not directly studied in our research, I think it is important to consider the negative impact on quality of life, experienced by patients with pustular psoriasis. In our study, many patients experienced exacerbations of their disease over time, and it is important to consider the impact this has on patients,” she said in the interview. “Continued research on pustular psoriasis is necessary to decrease the negative impact of these diseases on the lives of our patients.”
The case series were funded in part by an institutional grant from Boehringer Ingelheim. The authors report relationships with various pharmaceutical and biopharmaceutical companies, technology companies, medical publishing companies, medical journals, and medical societies with connections to the topic area in the form of serving in roles as a chief medical editor, consultant, data safety monitoring board member, deputy editor, principal investigator, research investigator, scientific adviser, or speaker; or having received grants, honoraria, personal fees, or research funding. Dr. Cowen has no disclosures. Dr. Hsu reports serving on a Boehringer Ingelheim advisory board for a product being evaluated as a potential treatment for GPP.
that evaluated the characteristics and course of the disease in patients diagnosed with PPP or GPP.
“These case series confirm the rarity of both generalized pustular psoriasis and palmoplantar pustulosis (PPP) and highlight the persistence of symptoms over time and the lack of effective treatment options available to patients,” Megan H. Noe, MD, MPH, MSCE, first author of both case series and assistant professor of dermatology, Harvard Medical School, and a dermatologist at Brigham and Women’s Hospital, both in Boston, said in an interview. In both studies, she added, “more than 20 different therapies were utilized, demonstrating a lack of consensus regarding effective treatment.”
The two case series were published in JAMA Dermatology.
Palmoplantar pustulosis
In the case series of 197 patients with PPP , data were obtained from a retrospective review at 20 academic dermatology practices in the United States between January 2007 and December 2018. The patients were mostly women (73.6%) who were White (60.9%), with a mean age of 53 years; 38.1% were current smokers, and 27.4% were former smokers, and the mean follow-up time was 22.1 months. About half (48.2%) of patients who presented to their respective centers had skin pain, 19.8% had problems using their hands and feet, 12.7% had arthralgias, and 2% had myalgias. Clinicians who examined these patients found pustules on the palms (80.2%), soles (76.7%), and both palms and soles (59.9%); some nail unit involvement was reported in 10.2%.
Patients were treated with a variety of topical therapies, systemic steroids, systemic anti-infectives, and systemic psoriasis therapies, Dr. Noe and colleagues said. The most common initial treatments included a topical steroid (84.8%), with the vast majority of clinicians using a high-potency topical steroid (153 of 167 patients; 91.6%), or topical therapy only (64.5%).
Other initial treatments used were other types of topical medications in 34 of the patients in the series (17.3%), such as a vitamin D analogue in 27 patients (79.4%); oral systemic treatments such as acitretin in 27 patients (13.7%) or methotrexate in 22 patients (11.2%); narrowband UVB phototherapy in 15 patients (7.7%); systemic steroids in 10 patients (5.1%); or systemic antibiotics in 9 patients (4.6%). Less commonly used were biologic agents like adalimumab, used in 6 patients (3.1%).
The researchers also examined health care utilization in 128 patients and found that 82% had at least one follow-up visit, 31.3% required two to three follow-up visits, and 18.8% had five or more follow-up visits. When adjusted to account for age and sex, there was a decreased risk of requiring five or more healthcare visits per year for women (odds ratio, 0.49; 95% confidence interval, 0.25-0.95)
Generalized pustular psoriasis
Dr. Noe and colleagues also evaluated 95 patients with GPP in a retrospective longitudinal case series of patients treated at 20 academic dermatology practices in the United States between January 2007 and December 2018. As in the PPP group, most patients in the GPP case series were women (70.5%), and over half were White (53.7%); the mean age was 50.3 years old, and the mean follow-up time was 19.8 months. A majority of patients with GPP were never-smokers (52.6%) or former smokers (20%). When patients with GPP initially presented to the study sites, 36.8% were admitted as inpatients, 9.5% presented in the emergency department, and 53.7% presented in an outpatient or ambulatory dermatology setting.
GPP commonly appeared on the trunk and extremities, but was “also reported on the scalp, face, genitals, nail unit, and mucous membranes in a minority of patients,” the researchers said. Overall, 62.1% of patients had skin pain, 26.2% had joint pain, 16.8% reported tachycardia, and 9.5% reported fever. Hypertension, depression, diabetes, chronic kidney disease, and hypothyroidism were common comorbidities of GPP, the researchers noted.
Clinicians reported treating GPP with topical steroids (86.3%) and topical treatments alone (32.3%). Oral systemic treatments such as acitretin (24.2%), cyclosporine (22.1%), and methotrexate (13.7%) were also used, as well as systemic steroids (20%). Other treatments used were narrowband UVB phototherapy (5.3%) and biologic agents like adalimumab (4.2%) and infliximab (4.2%).
For 53 patients with follow-up data of at least 6 months, 19 (35.8%) had been hospitalized because of their symptoms, and 8 patients were hospitalized for further GPP-specific concerns. Patients with GPP had a median 3.2 dermatology visits per year and a maximum of 18 visits. A model that was adjusted for age and sex showed women were at a decreased risk for being admitted to the hospital or emergency department in the follow-up period (odds ratio, 0.19; 95% confidence interval, 0.04-0.83).
PPP and GPP in practice
Sylvia Hsu, MD, professor and chair of the department of dermatology at Temple University, Philadelphia, who was not involved with the research, noted that most dermatologists will see few, if any, cases of PPP and GPP in a year. At her center, she estimated that she sees about one PPP case per week, and one or two cases of GPP a year. In general, she said that her clinical experience matched what was found by the authors of both case series.
For patients with PPP, “I would say the average dermatologist would probably start out with a superpotent topical steroid like clobetasol or halobetasol ointment,” Dr. Hsu said.
If they are not of childbearing age, she added, she would also prescribe acitretin, which she avoids giving to patients of childbearing age because of its teratogenicity. “Acitretin has the reputation that it doesn’t work well or fast for psoriasis. It doesn’t work well or fast for plaque-type psoriasis, but it works well and fast for pustular psoriasis,” she said.
In place of acitretin, Dr. Hsu recommended cyclosporine for a patient of childbearing age as a short-term solution to resolve symptoms before transitioning them to another therapy. “A woman of childbearing age, you put on cyclosporine, you’ve got to transition to something else,” she said. “And so many times you wean them off, the pustular psoriasis comes back because the topical steroid doesn’t work that well.”
One possible option is the interluekin-23 inhibitor guselkumab (approved by the Food and Drug Administration for treating moderate to severe plaque psoriasis and psoriatic arthritis) but cost and effectiveness can be a factor. Although studies have shown efficacy, biologics as treatments for PPP are “hit or miss,” Dr. Hsu said.
Regarding use of systemic therapies, Dr. Hsu cautioned against using them to treat plaque-type psoriasis. “We always learn, don’t use a systemic steroid like prednisone to treat psoriasis because it helps, but it comes back with a vengeance,” she said. “Sometimes when you treat plaque-type psoriasis with prednisone, it could come back with a vengeance, and it can come back as generalized pustular psoriasis.”
For patients with GPP, “you need a quick fix” because of the painful symptoms associated with the disease, Dr. Hsu said. In this case, she recommended cyclosporine and said she would avoid prescribing topical medications. “You’re going to have to give an oral drug because usually when we’re seeing somebody with GPP, they’re either a hospital consult or they just walked in the door,” she said. After prescribing cyclosporine, you would transition to another treatment like a biologic “as quickly as you can” with the knowledge that the biologic “may or may not work.”
New treatment options needed
Commenting on both case series in a related editorial, Edward W. Cowen, MD, MHSc, senior clinician and head of the dermatology consultation service in the dermatology branch of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Md., said that “much of the clinical presentation of pustular disease remains a mystery,” including why tobacco use is a risk factor for developing pustular psoriasis, and why tumor necrosis factor inhibitors “induce pustular disease in a small number of patients” with psoriasis vulgaris.
“Most importantly, we still do not know if localized and generalized pustular psoriasis all truly represent different variants of the same disease process, and if not, which biologic treatment represents the best option for a given clinical variant,” he wrote.
Dr. Cowen noted that the multi-institutional approach to collecting the retrospective data in these case series could be used as a “basic framework to build on for future clinical trials for rare skin diseases such as pustular psoriasis.”
In the interview, Dr. Noe said that she hoped that the “Pustular Psoriasis in the US Research Group” she and her coauthors created for the case series could help with the development of prospective clinical trials. “For pustular psoriasis and other rare diseases in dermatology, multi-institutional collaborations are necessary to conduct prospective research,” she said.
“While not directly studied in our research, I think it is important to consider the negative impact on quality of life, experienced by patients with pustular psoriasis. In our study, many patients experienced exacerbations of their disease over time, and it is important to consider the impact this has on patients,” she said in the interview. “Continued research on pustular psoriasis is necessary to decrease the negative impact of these diseases on the lives of our patients.”
The case series were funded in part by an institutional grant from Boehringer Ingelheim. The authors report relationships with various pharmaceutical and biopharmaceutical companies, technology companies, medical publishing companies, medical journals, and medical societies with connections to the topic area in the form of serving in roles as a chief medical editor, consultant, data safety monitoring board member, deputy editor, principal investigator, research investigator, scientific adviser, or speaker; or having received grants, honoraria, personal fees, or research funding. Dr. Cowen has no disclosures. Dr. Hsu reports serving on a Boehringer Ingelheim advisory board for a product being evaluated as a potential treatment for GPP.
that evaluated the characteristics and course of the disease in patients diagnosed with PPP or GPP.
“These case series confirm the rarity of both generalized pustular psoriasis and palmoplantar pustulosis (PPP) and highlight the persistence of symptoms over time and the lack of effective treatment options available to patients,” Megan H. Noe, MD, MPH, MSCE, first author of both case series and assistant professor of dermatology, Harvard Medical School, and a dermatologist at Brigham and Women’s Hospital, both in Boston, said in an interview. In both studies, she added, “more than 20 different therapies were utilized, demonstrating a lack of consensus regarding effective treatment.”
The two case series were published in JAMA Dermatology.
Palmoplantar pustulosis
In the case series of 197 patients with PPP , data were obtained from a retrospective review at 20 academic dermatology practices in the United States between January 2007 and December 2018. The patients were mostly women (73.6%) who were White (60.9%), with a mean age of 53 years; 38.1% were current smokers, and 27.4% were former smokers, and the mean follow-up time was 22.1 months. About half (48.2%) of patients who presented to their respective centers had skin pain, 19.8% had problems using their hands and feet, 12.7% had arthralgias, and 2% had myalgias. Clinicians who examined these patients found pustules on the palms (80.2%), soles (76.7%), and both palms and soles (59.9%); some nail unit involvement was reported in 10.2%.
Patients were treated with a variety of topical therapies, systemic steroids, systemic anti-infectives, and systemic psoriasis therapies, Dr. Noe and colleagues said. The most common initial treatments included a topical steroid (84.8%), with the vast majority of clinicians using a high-potency topical steroid (153 of 167 patients; 91.6%), or topical therapy only (64.5%).
Other initial treatments used were other types of topical medications in 34 of the patients in the series (17.3%), such as a vitamin D analogue in 27 patients (79.4%); oral systemic treatments such as acitretin in 27 patients (13.7%) or methotrexate in 22 patients (11.2%); narrowband UVB phototherapy in 15 patients (7.7%); systemic steroids in 10 patients (5.1%); or systemic antibiotics in 9 patients (4.6%). Less commonly used were biologic agents like adalimumab, used in 6 patients (3.1%).
The researchers also examined health care utilization in 128 patients and found that 82% had at least one follow-up visit, 31.3% required two to three follow-up visits, and 18.8% had five or more follow-up visits. When adjusted to account for age and sex, there was a decreased risk of requiring five or more healthcare visits per year for women (odds ratio, 0.49; 95% confidence interval, 0.25-0.95)
Generalized pustular psoriasis
Dr. Noe and colleagues also evaluated 95 patients with GPP in a retrospective longitudinal case series of patients treated at 20 academic dermatology practices in the United States between January 2007 and December 2018. As in the PPP group, most patients in the GPP case series were women (70.5%), and over half were White (53.7%); the mean age was 50.3 years old, and the mean follow-up time was 19.8 months. A majority of patients with GPP were never-smokers (52.6%) or former smokers (20%). When patients with GPP initially presented to the study sites, 36.8% were admitted as inpatients, 9.5% presented in the emergency department, and 53.7% presented in an outpatient or ambulatory dermatology setting.
GPP commonly appeared on the trunk and extremities, but was “also reported on the scalp, face, genitals, nail unit, and mucous membranes in a minority of patients,” the researchers said. Overall, 62.1% of patients had skin pain, 26.2% had joint pain, 16.8% reported tachycardia, and 9.5% reported fever. Hypertension, depression, diabetes, chronic kidney disease, and hypothyroidism were common comorbidities of GPP, the researchers noted.
Clinicians reported treating GPP with topical steroids (86.3%) and topical treatments alone (32.3%). Oral systemic treatments such as acitretin (24.2%), cyclosporine (22.1%), and methotrexate (13.7%) were also used, as well as systemic steroids (20%). Other treatments used were narrowband UVB phototherapy (5.3%) and biologic agents like adalimumab (4.2%) and infliximab (4.2%).
For 53 patients with follow-up data of at least 6 months, 19 (35.8%) had been hospitalized because of their symptoms, and 8 patients were hospitalized for further GPP-specific concerns. Patients with GPP had a median 3.2 dermatology visits per year and a maximum of 18 visits. A model that was adjusted for age and sex showed women were at a decreased risk for being admitted to the hospital or emergency department in the follow-up period (odds ratio, 0.19; 95% confidence interval, 0.04-0.83).
PPP and GPP in practice
Sylvia Hsu, MD, professor and chair of the department of dermatology at Temple University, Philadelphia, who was not involved with the research, noted that most dermatologists will see few, if any, cases of PPP and GPP in a year. At her center, she estimated that she sees about one PPP case per week, and one or two cases of GPP a year. In general, she said that her clinical experience matched what was found by the authors of both case series.
For patients with PPP, “I would say the average dermatologist would probably start out with a superpotent topical steroid like clobetasol or halobetasol ointment,” Dr. Hsu said.
If they are not of childbearing age, she added, she would also prescribe acitretin, which she avoids giving to patients of childbearing age because of its teratogenicity. “Acitretin has the reputation that it doesn’t work well or fast for psoriasis. It doesn’t work well or fast for plaque-type psoriasis, but it works well and fast for pustular psoriasis,” she said.
In place of acitretin, Dr. Hsu recommended cyclosporine for a patient of childbearing age as a short-term solution to resolve symptoms before transitioning them to another therapy. “A woman of childbearing age, you put on cyclosporine, you’ve got to transition to something else,” she said. “And so many times you wean them off, the pustular psoriasis comes back because the topical steroid doesn’t work that well.”
One possible option is the interluekin-23 inhibitor guselkumab (approved by the Food and Drug Administration for treating moderate to severe plaque psoriasis and psoriatic arthritis) but cost and effectiveness can be a factor. Although studies have shown efficacy, biologics as treatments for PPP are “hit or miss,” Dr. Hsu said.
Regarding use of systemic therapies, Dr. Hsu cautioned against using them to treat plaque-type psoriasis. “We always learn, don’t use a systemic steroid like prednisone to treat psoriasis because it helps, but it comes back with a vengeance,” she said. “Sometimes when you treat plaque-type psoriasis with prednisone, it could come back with a vengeance, and it can come back as generalized pustular psoriasis.”
For patients with GPP, “you need a quick fix” because of the painful symptoms associated with the disease, Dr. Hsu said. In this case, she recommended cyclosporine and said she would avoid prescribing topical medications. “You’re going to have to give an oral drug because usually when we’re seeing somebody with GPP, they’re either a hospital consult or they just walked in the door,” she said. After prescribing cyclosporine, you would transition to another treatment like a biologic “as quickly as you can” with the knowledge that the biologic “may or may not work.”
New treatment options needed
Commenting on both case series in a related editorial, Edward W. Cowen, MD, MHSc, senior clinician and head of the dermatology consultation service in the dermatology branch of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, Bethesda, Md., said that “much of the clinical presentation of pustular disease remains a mystery,” including why tobacco use is a risk factor for developing pustular psoriasis, and why tumor necrosis factor inhibitors “induce pustular disease in a small number of patients” with psoriasis vulgaris.
“Most importantly, we still do not know if localized and generalized pustular psoriasis all truly represent different variants of the same disease process, and if not, which biologic treatment represents the best option for a given clinical variant,” he wrote.
Dr. Cowen noted that the multi-institutional approach to collecting the retrospective data in these case series could be used as a “basic framework to build on for future clinical trials for rare skin diseases such as pustular psoriasis.”
In the interview, Dr. Noe said that she hoped that the “Pustular Psoriasis in the US Research Group” she and her coauthors created for the case series could help with the development of prospective clinical trials. “For pustular psoriasis and other rare diseases in dermatology, multi-institutional collaborations are necessary to conduct prospective research,” she said.
“While not directly studied in our research, I think it is important to consider the negative impact on quality of life, experienced by patients with pustular psoriasis. In our study, many patients experienced exacerbations of their disease over time, and it is important to consider the impact this has on patients,” she said in the interview. “Continued research on pustular psoriasis is necessary to decrease the negative impact of these diseases on the lives of our patients.”
The case series were funded in part by an institutional grant from Boehringer Ingelheim. The authors report relationships with various pharmaceutical and biopharmaceutical companies, technology companies, medical publishing companies, medical journals, and medical societies with connections to the topic area in the form of serving in roles as a chief medical editor, consultant, data safety monitoring board member, deputy editor, principal investigator, research investigator, scientific adviser, or speaker; or having received grants, honoraria, personal fees, or research funding. Dr. Cowen has no disclosures. Dr. Hsu reports serving on a Boehringer Ingelheim advisory board for a product being evaluated as a potential treatment for GPP.
FROM JAMA DERMATOLOGY
Spesolimab speeds lesion clearance in generalized pustular psoriasis
GPP is a life-threatening skin condition involving the widespread eruption of sterile pustules, with a clinical course that “can be relapsing with recurrent flares or persistent with intermittent flares,” Hervé Bachelez, MD, of the Université de Paris and coauthors wrote. GPP patients are often hospitalized, and mortality ranges from 2% to 16% from causes that include sepsis and cardiorespiratory failure.
“The role of the interleukin-36 pathway in GPP is supported by the finding of loss-of-function mutations in the interleukin-36 receptor antagonist gene (IL36RN) and associated genes (CARD14, AP1S3, SERPINA3, and MPO) and by the overexpression of interleukin-36 cytokines in GPP skin lesions,” therefore, IL-36 is a potential treatment target to manage flares, they explained.
In the multicenter, double-blind trial, published in the New England Journal of Medicine, the researchers randomized 35 adults with GPP flares to a single 900-mg intravenous dose of spesolimab and 18 to placebo. Patients in both groups could receive an open-label dose of spesolimab after day 8; all patients were followed for 12 weeks.
The primary study endpoint was the Generalized Pustular Psoriasis Physician Global Assessment (GPPGA) pustulation subscore of 0 at 1 week after treatment. The GPPGA ranges from 0 (no visible pustules) to 4 (severe pustules). At baseline, 46% spesolimab patients and 39% placebo patients had a GPPGA pustulation subscore of 3, and 37% and 33%, respectively, had a pustulation subscore of 4.
After 1 week, 54% of the spesolimab patients had no visible pustules, compared with 6% of placebo patients; the difference was statistically significant (P < .001). The main secondary endpoint was a score of 0 or 1 (clear or almost clear skin) on the GPPGA total score after 1 week. Significantly more spesolimab patients had GPPGA total scores of 0 or 1, compared with placebo patients (43% vs. 11%, respectively; P = .02).
Overall, 6 of 35 spesolimab patients (17%) and 6% of those in the placebo groups developed infections during the first week, and 24 of 51 patients (47%) who had received spesolimab at any point during the study developed infections by week 12. Infections included urinary tract infections (three cases), influenza (three), otitis externa (two), folliculitis (two), upper respiratory tract infection (two), and pustule (two).
In the first week, 6% of spesolimab patients and none of the placebo patients reported serious adverse events; at week 12, 12% of patients who had received at least one spesolimab dose reported a serious adverse event. In addition, antidrug antibodies were identified in 23 (46%) of the 50 patients who received at least one dose of spesolimab.
“Symptoms that were observed in two patients who received spesolimab were reported as a drug reaction with eosinophilia and systemic symptoms (DRESS),” the authors noted. One patient had a RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions) score and the other had a score of 3; a score below 2 indicates no DRESS, and a score of 2 or 3 indicates “possible DRESS,” they added.
“Because 15 of the 18 patients who were assigned to the placebo group received open-label spesolimab, the effect of spesolimab as compared with that of placebo could not be determined after week 1,” the researchers noted.
The study findings were limited by several factors including the short randomization period and small study population, the researchers noted. However, the effect sizes for both the primary and secondary endpoints were large, which strengthened the results.
The results support data from previous studies suggesting a role for IL-36 in the pathogenesis of GPP, and support the need for longer and larger studies of the safety and effectiveness of spesolimab for GPP patients, they concluded.
No FDA-approved therapy
“GPP is a very rare but devastating life-threatening disease that presents with the sudden onset of pustules throughout the skin,” Joel Gelfand, MD, professor of dermatology and director of the psoriasis and phototherapy center at the University of Pennsylvania, Philadelphia, said in an interview. “Without rapid treatment, GPP can result in death. Currently there are no [Food and Drug Administration]–approved treatments for this orphan disease.”
Dr. Gelfand said he was surprised by the degree of efficacy and the speed of the patient response to spesolimab, compared with placebo, which he described as “truly remarkable.” Based on the current study results, “spesolimab offers a tremendous step forward for our patients,” he added.
Looking ahead, Dr. Gelfand noted that “longer-term studies with a comparator, such as a biologic that targets IL-17, would be helpful to more fully understand the safety, efficacy, and role that spesolimab will have in real-world patients.”
On Dec. 15, Boehringer Ingelheim announced that the FDA had granted priority review for spesolimab for treating GPP flares.
The study was supported by Boehringer Ingelheim. Lead author Dr. Bachelez had no financial conflicts to disclose. Several authors are employees of Boehringer Ingelheim. Dr. Gelfand is a consultant for the study sponsor Boehringer Ingelheim and has received research grants from Boehringer Ingelheim to his institution to support an investigator-initiated study. He also disclosed serving as a consultant and receiving research grants from other manufacturers of psoriasis products.
GPP is a life-threatening skin condition involving the widespread eruption of sterile pustules, with a clinical course that “can be relapsing with recurrent flares or persistent with intermittent flares,” Hervé Bachelez, MD, of the Université de Paris and coauthors wrote. GPP patients are often hospitalized, and mortality ranges from 2% to 16% from causes that include sepsis and cardiorespiratory failure.
“The role of the interleukin-36 pathway in GPP is supported by the finding of loss-of-function mutations in the interleukin-36 receptor antagonist gene (IL36RN) and associated genes (CARD14, AP1S3, SERPINA3, and MPO) and by the overexpression of interleukin-36 cytokines in GPP skin lesions,” therefore, IL-36 is a potential treatment target to manage flares, they explained.
In the multicenter, double-blind trial, published in the New England Journal of Medicine, the researchers randomized 35 adults with GPP flares to a single 900-mg intravenous dose of spesolimab and 18 to placebo. Patients in both groups could receive an open-label dose of spesolimab after day 8; all patients were followed for 12 weeks.
The primary study endpoint was the Generalized Pustular Psoriasis Physician Global Assessment (GPPGA) pustulation subscore of 0 at 1 week after treatment. The GPPGA ranges from 0 (no visible pustules) to 4 (severe pustules). At baseline, 46% spesolimab patients and 39% placebo patients had a GPPGA pustulation subscore of 3, and 37% and 33%, respectively, had a pustulation subscore of 4.
After 1 week, 54% of the spesolimab patients had no visible pustules, compared with 6% of placebo patients; the difference was statistically significant (P < .001). The main secondary endpoint was a score of 0 or 1 (clear or almost clear skin) on the GPPGA total score after 1 week. Significantly more spesolimab patients had GPPGA total scores of 0 or 1, compared with placebo patients (43% vs. 11%, respectively; P = .02).
Overall, 6 of 35 spesolimab patients (17%) and 6% of those in the placebo groups developed infections during the first week, and 24 of 51 patients (47%) who had received spesolimab at any point during the study developed infections by week 12. Infections included urinary tract infections (three cases), influenza (three), otitis externa (two), folliculitis (two), upper respiratory tract infection (two), and pustule (two).
In the first week, 6% of spesolimab patients and none of the placebo patients reported serious adverse events; at week 12, 12% of patients who had received at least one spesolimab dose reported a serious adverse event. In addition, antidrug antibodies were identified in 23 (46%) of the 50 patients who received at least one dose of spesolimab.
“Symptoms that were observed in two patients who received spesolimab were reported as a drug reaction with eosinophilia and systemic symptoms (DRESS),” the authors noted. One patient had a RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions) score and the other had a score of 3; a score below 2 indicates no DRESS, and a score of 2 or 3 indicates “possible DRESS,” they added.
“Because 15 of the 18 patients who were assigned to the placebo group received open-label spesolimab, the effect of spesolimab as compared with that of placebo could not be determined after week 1,” the researchers noted.
The study findings were limited by several factors including the short randomization period and small study population, the researchers noted. However, the effect sizes for both the primary and secondary endpoints were large, which strengthened the results.
The results support data from previous studies suggesting a role for IL-36 in the pathogenesis of GPP, and support the need for longer and larger studies of the safety and effectiveness of spesolimab for GPP patients, they concluded.
No FDA-approved therapy
“GPP is a very rare but devastating life-threatening disease that presents with the sudden onset of pustules throughout the skin,” Joel Gelfand, MD, professor of dermatology and director of the psoriasis and phototherapy center at the University of Pennsylvania, Philadelphia, said in an interview. “Without rapid treatment, GPP can result in death. Currently there are no [Food and Drug Administration]–approved treatments for this orphan disease.”
Dr. Gelfand said he was surprised by the degree of efficacy and the speed of the patient response to spesolimab, compared with placebo, which he described as “truly remarkable.” Based on the current study results, “spesolimab offers a tremendous step forward for our patients,” he added.
Looking ahead, Dr. Gelfand noted that “longer-term studies with a comparator, such as a biologic that targets IL-17, would be helpful to more fully understand the safety, efficacy, and role that spesolimab will have in real-world patients.”
On Dec. 15, Boehringer Ingelheim announced that the FDA had granted priority review for spesolimab for treating GPP flares.
The study was supported by Boehringer Ingelheim. Lead author Dr. Bachelez had no financial conflicts to disclose. Several authors are employees of Boehringer Ingelheim. Dr. Gelfand is a consultant for the study sponsor Boehringer Ingelheim and has received research grants from Boehringer Ingelheim to his institution to support an investigator-initiated study. He also disclosed serving as a consultant and receiving research grants from other manufacturers of psoriasis products.
GPP is a life-threatening skin condition involving the widespread eruption of sterile pustules, with a clinical course that “can be relapsing with recurrent flares or persistent with intermittent flares,” Hervé Bachelez, MD, of the Université de Paris and coauthors wrote. GPP patients are often hospitalized, and mortality ranges from 2% to 16% from causes that include sepsis and cardiorespiratory failure.
“The role of the interleukin-36 pathway in GPP is supported by the finding of loss-of-function mutations in the interleukin-36 receptor antagonist gene (IL36RN) and associated genes (CARD14, AP1S3, SERPINA3, and MPO) and by the overexpression of interleukin-36 cytokines in GPP skin lesions,” therefore, IL-36 is a potential treatment target to manage flares, they explained.
In the multicenter, double-blind trial, published in the New England Journal of Medicine, the researchers randomized 35 adults with GPP flares to a single 900-mg intravenous dose of spesolimab and 18 to placebo. Patients in both groups could receive an open-label dose of spesolimab after day 8; all patients were followed for 12 weeks.
The primary study endpoint was the Generalized Pustular Psoriasis Physician Global Assessment (GPPGA) pustulation subscore of 0 at 1 week after treatment. The GPPGA ranges from 0 (no visible pustules) to 4 (severe pustules). At baseline, 46% spesolimab patients and 39% placebo patients had a GPPGA pustulation subscore of 3, and 37% and 33%, respectively, had a pustulation subscore of 4.
After 1 week, 54% of the spesolimab patients had no visible pustules, compared with 6% of placebo patients; the difference was statistically significant (P < .001). The main secondary endpoint was a score of 0 or 1 (clear or almost clear skin) on the GPPGA total score after 1 week. Significantly more spesolimab patients had GPPGA total scores of 0 or 1, compared with placebo patients (43% vs. 11%, respectively; P = .02).
Overall, 6 of 35 spesolimab patients (17%) and 6% of those in the placebo groups developed infections during the first week, and 24 of 51 patients (47%) who had received spesolimab at any point during the study developed infections by week 12. Infections included urinary tract infections (three cases), influenza (three), otitis externa (two), folliculitis (two), upper respiratory tract infection (two), and pustule (two).
In the first week, 6% of spesolimab patients and none of the placebo patients reported serious adverse events; at week 12, 12% of patients who had received at least one spesolimab dose reported a serious adverse event. In addition, antidrug antibodies were identified in 23 (46%) of the 50 patients who received at least one dose of spesolimab.
“Symptoms that were observed in two patients who received spesolimab were reported as a drug reaction with eosinophilia and systemic symptoms (DRESS),” the authors noted. One patient had a RegiSCAR (European Registry of Severe Cutaneous Adverse Reactions) score and the other had a score of 3; a score below 2 indicates no DRESS, and a score of 2 or 3 indicates “possible DRESS,” they added.
“Because 15 of the 18 patients who were assigned to the placebo group received open-label spesolimab, the effect of spesolimab as compared with that of placebo could not be determined after week 1,” the researchers noted.
The study findings were limited by several factors including the short randomization period and small study population, the researchers noted. However, the effect sizes for both the primary and secondary endpoints were large, which strengthened the results.
The results support data from previous studies suggesting a role for IL-36 in the pathogenesis of GPP, and support the need for longer and larger studies of the safety and effectiveness of spesolimab for GPP patients, they concluded.
No FDA-approved therapy
“GPP is a very rare but devastating life-threatening disease that presents with the sudden onset of pustules throughout the skin,” Joel Gelfand, MD, professor of dermatology and director of the psoriasis and phototherapy center at the University of Pennsylvania, Philadelphia, said in an interview. “Without rapid treatment, GPP can result in death. Currently there are no [Food and Drug Administration]–approved treatments for this orphan disease.”
Dr. Gelfand said he was surprised by the degree of efficacy and the speed of the patient response to spesolimab, compared with placebo, which he described as “truly remarkable.” Based on the current study results, “spesolimab offers a tremendous step forward for our patients,” he added.
Looking ahead, Dr. Gelfand noted that “longer-term studies with a comparator, such as a biologic that targets IL-17, would be helpful to more fully understand the safety, efficacy, and role that spesolimab will have in real-world patients.”
On Dec. 15, Boehringer Ingelheim announced that the FDA had granted priority review for spesolimab for treating GPP flares.
The study was supported by Boehringer Ingelheim. Lead author Dr. Bachelez had no financial conflicts to disclose. Several authors are employees of Boehringer Ingelheim. Dr. Gelfand is a consultant for the study sponsor Boehringer Ingelheim and has received research grants from Boehringer Ingelheim to his institution to support an investigator-initiated study. He also disclosed serving as a consultant and receiving research grants from other manufacturers of psoriasis products.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Prurigo nodularis has two disease endotypes, a cluster analysis shows
A cluster analysis of circulating plasma biomarkers in prurigo nodularis (PN) has identified two disease endotypes with inflammatory and noninflammatory biomarker profiles.

said senior author Shawn G. Kwatra, MD, of the department of dermatology at Johns Hopkins University, Baltimore. “This is the beginning of personalized medicine in prurigo nodularis.”
He and others have long observed significant clinical heterogeneity both in the presentation of PN – with the nodules in African American patients, for instance, appearing larger, thicker, and more fibrotic – and in patients’ response to immunomodulating and neuromodulating therapies.
To avoid the introduction of bias, the researchers used an unsupervised machine-learning approach to analyze the levels of 12 inflammatory biomarkers in 20 patients with PN and in matched healthy controls. The biomarkers were chosen based on their demonstrated dysregulation in PN and other inflammatory dermatoses.
The researchers then conducted a population-level analysis using multicenter electronic medical record data to explore inflammatory markers and verify findings from the cluster analysis. The study was published online Oct. 27, 2021, in the Journal of Investigative Dermatology.
One cluster of the 20 patients had higher levels of nine inflammatory biomarkers representing multiple immune axes: Higher interleukin-1 alpha, IL-4, IL-5, IL-6, IL-10, IL-17A, IL-22, and IL-25. This cluster had a higher percentage of Black patients, a higher severity of itch, and lower quality of life scores, the authors report in the preprint.
The other cluster – without such an inflammatory profile – had fewer Black patients and a higher percentage of patients with myelopathy (e.g. spinal stenosis, spinal trauma, degenerative disc disease). The rates of inflammatory comorbidities and of immune- and neuromodulating treatments at the time of blood draw were relatively equivalent between the two clusters.
In the subsequent population-level analysis, using data from a global health research network of EMRs from almost 50 health care organizations, Black patients with PN were found to have higher erythrocyte sedimentation rate, C-reactive protein, ferritin, and eosinophils, and lower transferrin, than White patients with PN. (The analysis included only Black and White patients.)
There are no Food and Drug Administration–approved therapies for PN, and “clinicians need to be really creative in managing these patients,” Dr. Kwatra said.
“There may be suggestions at the bedside that patients have more immune dysregulation, or maybe I’ll see increased circulating blood eosinophils,” he said. “And there are those who don’t seem to have any immune dysregulation and have more features of neurosensitization ... who may have a history of neck pain or back injury.”
The existence of endotypes in PN suggests that patients may benefit from personalized therapies with either immunomodulating or neuromodulating treatments, he and his colleagues wrote. “Further neuroimmune phenotyping studies of PN may pave the way for a future precision medicine management approach.”
Studies of PN conducted in Europe have been almost exclusively in White patients, Dr. Kwatra noted, even though PN has been shown to disproportionately affect Black and other racial/ethnic-minority patients.
Black patients with PN were found to have the highest all-cause mortality over 20 years post diagnosis in a separate analysis of over 22,000 patients with PN. Using data from the same health research network, Dr. Kwatra and coinvestigators stratified patients by race/ethnicity and compared each subgroup with a corresponding subgroup of similar race/ethnicity to control for inherent differences in mortality.
Overall, patients with PN had higher all-cause mortality than controls (hazard ratio, 1.70), likely because of a high comorbidity burden, they wrote in their research letter. Black patients with PN had the highest mortality (HR, 2.07), followed by White (HR, 1.74) and Hispanic (HR, 1.62) patients.
PN may exacerbate existing racial disparities in the social determinants of health, and Black patients may suffer from greater systemic inflammation, Dr. Kwatra and coauthors wrote. Certainly, he said, these findings, as well as the finding of a distinct inflammatory signature in Black patients with PN, support “that the disease burden is much higher” in these patients.
Dr Kwatra disclosed that he is an advisory board member/consultant for Celldex Therapeutics, Galderma, Incyte, Pfizer, Regeneron, and Kiniksa Pharmaceuticals and has received grant funding from several companies. His research is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Grants from the Dermatology Foundation and the Skin of Color Society also helped fund the cluster analysis.
A cluster analysis of circulating plasma biomarkers in prurigo nodularis (PN) has identified two disease endotypes with inflammatory and noninflammatory biomarker profiles.
said senior author Shawn G. Kwatra, MD, of the department of dermatology at Johns Hopkins University, Baltimore. “This is the beginning of personalized medicine in prurigo nodularis.”
He and others have long observed significant clinical heterogeneity both in the presentation of PN – with the nodules in African American patients, for instance, appearing larger, thicker, and more fibrotic – and in patients’ response to immunomodulating and neuromodulating therapies.
To avoid the introduction of bias, the researchers used an unsupervised machine-learning approach to analyze the levels of 12 inflammatory biomarkers in 20 patients with PN and in matched healthy controls. The biomarkers were chosen based on their demonstrated dysregulation in PN and other inflammatory dermatoses.
The researchers then conducted a population-level analysis using multicenter electronic medical record data to explore inflammatory markers and verify findings from the cluster analysis. The study was published online Oct. 27, 2021, in the Journal of Investigative Dermatology.
One cluster of the 20 patients had higher levels of nine inflammatory biomarkers representing multiple immune axes: Higher interleukin-1 alpha, IL-4, IL-5, IL-6, IL-10, IL-17A, IL-22, and IL-25. This cluster had a higher percentage of Black patients, a higher severity of itch, and lower quality of life scores, the authors report in the preprint.
The other cluster – without such an inflammatory profile – had fewer Black patients and a higher percentage of patients with myelopathy (e.g. spinal stenosis, spinal trauma, degenerative disc disease). The rates of inflammatory comorbidities and of immune- and neuromodulating treatments at the time of blood draw were relatively equivalent between the two clusters.
In the subsequent population-level analysis, using data from a global health research network of EMRs from almost 50 health care organizations, Black patients with PN were found to have higher erythrocyte sedimentation rate, C-reactive protein, ferritin, and eosinophils, and lower transferrin, than White patients with PN. (The analysis included only Black and White patients.)
There are no Food and Drug Administration–approved therapies for PN, and “clinicians need to be really creative in managing these patients,” Dr. Kwatra said.
“There may be suggestions at the bedside that patients have more immune dysregulation, or maybe I’ll see increased circulating blood eosinophils,” he said. “And there are those who don’t seem to have any immune dysregulation and have more features of neurosensitization ... who may have a history of neck pain or back injury.”
The existence of endotypes in PN suggests that patients may benefit from personalized therapies with either immunomodulating or neuromodulating treatments, he and his colleagues wrote. “Further neuroimmune phenotyping studies of PN may pave the way for a future precision medicine management approach.”
Studies of PN conducted in Europe have been almost exclusively in White patients, Dr. Kwatra noted, even though PN has been shown to disproportionately affect Black and other racial/ethnic-minority patients.
Black patients with PN were found to have the highest all-cause mortality over 20 years post diagnosis in a separate analysis of over 22,000 patients with PN. Using data from the same health research network, Dr. Kwatra and coinvestigators stratified patients by race/ethnicity and compared each subgroup with a corresponding subgroup of similar race/ethnicity to control for inherent differences in mortality.
Overall, patients with PN had higher all-cause mortality than controls (hazard ratio, 1.70), likely because of a high comorbidity burden, they wrote in their research letter. Black patients with PN had the highest mortality (HR, 2.07), followed by White (HR, 1.74) and Hispanic (HR, 1.62) patients.
PN may exacerbate existing racial disparities in the social determinants of health, and Black patients may suffer from greater systemic inflammation, Dr. Kwatra and coauthors wrote. Certainly, he said, these findings, as well as the finding of a distinct inflammatory signature in Black patients with PN, support “that the disease burden is much higher” in these patients.
Dr Kwatra disclosed that he is an advisory board member/consultant for Celldex Therapeutics, Galderma, Incyte, Pfizer, Regeneron, and Kiniksa Pharmaceuticals and has received grant funding from several companies. His research is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Grants from the Dermatology Foundation and the Skin of Color Society also helped fund the cluster analysis.
A cluster analysis of circulating plasma biomarkers in prurigo nodularis (PN) has identified two disease endotypes with inflammatory and noninflammatory biomarker profiles.
said senior author Shawn G. Kwatra, MD, of the department of dermatology at Johns Hopkins University, Baltimore. “This is the beginning of personalized medicine in prurigo nodularis.”
He and others have long observed significant clinical heterogeneity both in the presentation of PN – with the nodules in African American patients, for instance, appearing larger, thicker, and more fibrotic – and in patients’ response to immunomodulating and neuromodulating therapies.
To avoid the introduction of bias, the researchers used an unsupervised machine-learning approach to analyze the levels of 12 inflammatory biomarkers in 20 patients with PN and in matched healthy controls. The biomarkers were chosen based on their demonstrated dysregulation in PN and other inflammatory dermatoses.
The researchers then conducted a population-level analysis using multicenter electronic medical record data to explore inflammatory markers and verify findings from the cluster analysis. The study was published online Oct. 27, 2021, in the Journal of Investigative Dermatology.
One cluster of the 20 patients had higher levels of nine inflammatory biomarkers representing multiple immune axes: Higher interleukin-1 alpha, IL-4, IL-5, IL-6, IL-10, IL-17A, IL-22, and IL-25. This cluster had a higher percentage of Black patients, a higher severity of itch, and lower quality of life scores, the authors report in the preprint.
The other cluster – without such an inflammatory profile – had fewer Black patients and a higher percentage of patients with myelopathy (e.g. spinal stenosis, spinal trauma, degenerative disc disease). The rates of inflammatory comorbidities and of immune- and neuromodulating treatments at the time of blood draw were relatively equivalent between the two clusters.
In the subsequent population-level analysis, using data from a global health research network of EMRs from almost 50 health care organizations, Black patients with PN were found to have higher erythrocyte sedimentation rate, C-reactive protein, ferritin, and eosinophils, and lower transferrin, than White patients with PN. (The analysis included only Black and White patients.)
There are no Food and Drug Administration–approved therapies for PN, and “clinicians need to be really creative in managing these patients,” Dr. Kwatra said.
“There may be suggestions at the bedside that patients have more immune dysregulation, or maybe I’ll see increased circulating blood eosinophils,” he said. “And there are those who don’t seem to have any immune dysregulation and have more features of neurosensitization ... who may have a history of neck pain or back injury.”
The existence of endotypes in PN suggests that patients may benefit from personalized therapies with either immunomodulating or neuromodulating treatments, he and his colleagues wrote. “Further neuroimmune phenotyping studies of PN may pave the way for a future precision medicine management approach.”
Studies of PN conducted in Europe have been almost exclusively in White patients, Dr. Kwatra noted, even though PN has been shown to disproportionately affect Black and other racial/ethnic-minority patients.
Black patients with PN were found to have the highest all-cause mortality over 20 years post diagnosis in a separate analysis of over 22,000 patients with PN. Using data from the same health research network, Dr. Kwatra and coinvestigators stratified patients by race/ethnicity and compared each subgroup with a corresponding subgroup of similar race/ethnicity to control for inherent differences in mortality.
Overall, patients with PN had higher all-cause mortality than controls (hazard ratio, 1.70), likely because of a high comorbidity burden, they wrote in their research letter. Black patients with PN had the highest mortality (HR, 2.07), followed by White (HR, 1.74) and Hispanic (HR, 1.62) patients.
PN may exacerbate existing racial disparities in the social determinants of health, and Black patients may suffer from greater systemic inflammation, Dr. Kwatra and coauthors wrote. Certainly, he said, these findings, as well as the finding of a distinct inflammatory signature in Black patients with PN, support “that the disease burden is much higher” in these patients.
Dr Kwatra disclosed that he is an advisory board member/consultant for Celldex Therapeutics, Galderma, Incyte, Pfizer, Regeneron, and Kiniksa Pharmaceuticals and has received grant funding from several companies. His research is supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Grants from the Dermatology Foundation and the Skin of Color Society also helped fund the cluster analysis.
FROM THE JOURNAL OF INVESTIGATIVE DERMATOLOGY
New option for flares in pustular psoriasis
according to a statement from manufacturer Boehringer Ingelheim. The FDA also granted Priority Review to spesolimab. Priority Review is a designation granted to medications that would offer significant improvement over the currently available treatments.
Generalized pustular psoriasis (GPP), though rare, is a potentially life-threatening condition that is distinct from plaque psoriasis. Throughout the course of the disease, which is caused by the accumulation of neutrophils in the skin, patients may experience persistent disease with intermittent flares or relapsing disease with recurrent flares. The neutrophil accumulation results in the eruption of sterile, yet painful pustules across all parts of the body.
“While the severity of GPP flares can vary, if left untreated they can be life threatening due to complications such as sepsis and multisystem organ failure,” and have a significant impact on quality of life, according to the company statement.
The FDA also has granted spesolimab an Orphan Drug Designation for the treatment of GPP, and a Breakthrough Therapy Designation for the treatment of GPP flares in adults.
A marketing authorization application for spesolimab for the treatment of GPP was accepted for evaluation by the European Medicines Agency in October 2021, according to a company press release issued at that time.
A protocol for a phase 2 study of spesolimab versus placebo for treating acute flares in GPP patients was published in October in BMJ Open, after a phase 1 proof-of-concept study published in 2019 showed the potential of an IL-36 receptor antagonist to improve disease scores in adults with GPP.
More information is available on the Boehringer Ingelheim website.
according to a statement from manufacturer Boehringer Ingelheim. The FDA also granted Priority Review to spesolimab. Priority Review is a designation granted to medications that would offer significant improvement over the currently available treatments.
Generalized pustular psoriasis (GPP), though rare, is a potentially life-threatening condition that is distinct from plaque psoriasis. Throughout the course of the disease, which is caused by the accumulation of neutrophils in the skin, patients may experience persistent disease with intermittent flares or relapsing disease with recurrent flares. The neutrophil accumulation results in the eruption of sterile, yet painful pustules across all parts of the body.
“While the severity of GPP flares can vary, if left untreated they can be life threatening due to complications such as sepsis and multisystem organ failure,” and have a significant impact on quality of life, according to the company statement.
The FDA also has granted spesolimab an Orphan Drug Designation for the treatment of GPP, and a Breakthrough Therapy Designation for the treatment of GPP flares in adults.
A marketing authorization application for spesolimab for the treatment of GPP was accepted for evaluation by the European Medicines Agency in October 2021, according to a company press release issued at that time.
A protocol for a phase 2 study of spesolimab versus placebo for treating acute flares in GPP patients was published in October in BMJ Open, after a phase 1 proof-of-concept study published in 2019 showed the potential of an IL-36 receptor antagonist to improve disease scores in adults with GPP.
More information is available on the Boehringer Ingelheim website.
according to a statement from manufacturer Boehringer Ingelheim. The FDA also granted Priority Review to spesolimab. Priority Review is a designation granted to medications that would offer significant improvement over the currently available treatments.
Generalized pustular psoriasis (GPP), though rare, is a potentially life-threatening condition that is distinct from plaque psoriasis. Throughout the course of the disease, which is caused by the accumulation of neutrophils in the skin, patients may experience persistent disease with intermittent flares or relapsing disease with recurrent flares. The neutrophil accumulation results in the eruption of sterile, yet painful pustules across all parts of the body.
“While the severity of GPP flares can vary, if left untreated they can be life threatening due to complications such as sepsis and multisystem organ failure,” and have a significant impact on quality of life, according to the company statement.
The FDA also has granted spesolimab an Orphan Drug Designation for the treatment of GPP, and a Breakthrough Therapy Designation for the treatment of GPP flares in adults.
A marketing authorization application for spesolimab for the treatment of GPP was accepted for evaluation by the European Medicines Agency in October 2021, according to a company press release issued at that time.
A protocol for a phase 2 study of spesolimab versus placebo for treating acute flares in GPP patients was published in October in BMJ Open, after a phase 1 proof-of-concept study published in 2019 showed the potential of an IL-36 receptor antagonist to improve disease scores in adults with GPP.
More information is available on the Boehringer Ingelheim website.