User login
Cost of Diagnosing Psoriasis and Rosacea for Dermatologists Versus Primary Care Physicians
Growing incentives to control health care costs may cause accountable care organizations (ACOs) to reconsider how diseases are best managed. Few studies have examined the cost difference between primary care providers (PCPs) and specialists in managing the same disease. Limited data have suggested that management of some diseases by a PCP may be less costly compared to a specialist1,2; however, it is not clear if this finding extends to skin disease. This study sought to assess the cost of seeing a dermatologist versus a PCP for diagnosis of the common skin diseases psoriasis and rosacea.
Methods
Patient data were obtained from the Humana database, a large commercial data set for claims and reimbursed costs encompassing 18,162,539 patients covered between January 2007 and December 2014. Our study population consisted of 3,944,465 patients with claims that included International Classification of Diseases, Ninth Revision (ICD-9), codes for dermatological diagnoses (680.0–709.9). We searched by ICD-9 code for US patients with primary diagnoses of psoriasis (696.1) and rosacea (695.3). We narrowed the search to include patients aged 30 to 64 years, as the diagnoses for these diseases are most common in patients older than 30 years. Patients who were older than 64 years were not included in the study, as most are covered by Medicare and therefore costs covered by Humana in this age group would not be as representative as in younger age groups. Total and average diagnosis-related costs per patient were compared between dermatologists and PCPs. Diagnosis-related costs encompassed physician reimbursement; laboratory and imaging costs, including skin biopsies; inpatient hospitalization cost; and any other charge that could be coded or billed by providers and reimbursed by the insurance company. To be eligible for reimbursement from Humana, dermatologists and PCPs must be registered with the insurer according to specialty board certification and practice credentialing, and they are reimbursed differently based on specialty. Drug costs, which would possibly skew the data toward providers using more expensive systemic medications (ie, dermatologists), were not included in this study, as the discussion is better reserved for long-term management of disease rather than diagnosis-related costs. All diagnoses of psoriasis were included in the study, which likely includes all severities of psoriasis, though we did not have the ability to further break down these diagnoses by severity.
Results
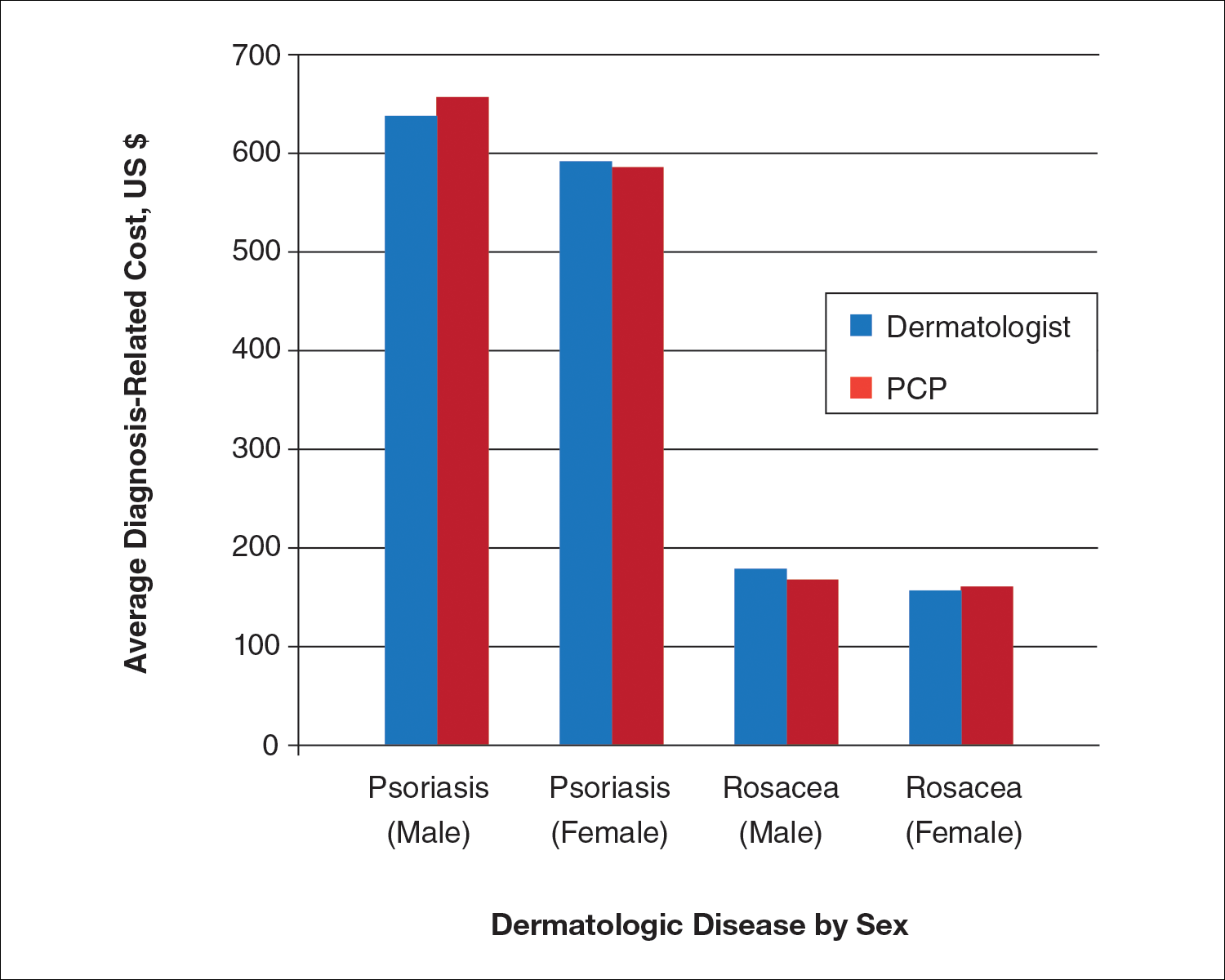
We identified 30,217 psoriasis patients and 37,561 rosacea patients. Of those patients with a primary diagnosis of psoriasis, 26,112 (86%) were seen by a dermatologist and 4105 (14%) were seen by a PCP (Table). Of those patients with a primary diagnosis of rosacea, 34,694 (92%) were seen by a dermatologist and 2867 (8%) were seen by a PCP (Table). There was little difference in the average diagnosis-related cost per patient for psoriasis in males (dermatologists, $638; PCPs, $657) versus females (dermatologists, $592; PCPs, $586) or between specialties (Figure). Findings were similar for rosacea in males (dermatologists, $179; PCPs, $168) versus females (dermatologists, $157; PCPs, $161). For these skin diseases, i

Comment
For the management of common skin disorders such as psoriasis and rosacea, there is little cost difference in seeing a dermatologist versus a PCP. Through extensive training and repeated exposure to many skin diseases, dermatologists are expected to be more comfortable in diagnosing and managing psoriasis and rosacea. Compared to PCPs, dermatologists have demonstrated increased diagnostic accuracy and efficiency when examining pigmented lesions and other dermatologic diseases in several studies.3-6 Although the current study shows that diagnosis-related costs for psoriasis and rosacea are essentially equal between dermatologists and PCPs, it actually may be less expensive for patients to see a dermatologist, as unnecessary tests, biopsies, or medications are more likely to be ordered/prescribed when there is less clinical diagnostic certainty.7,8 Additionally, seeing a PCP for diagnosis of a skin disease may be inefficient if subsequent referral to a dermatologist is needed, a common scenario that occurs when patients see a PCP for skin conditions.9
Our study had limitations, which is typical of a study using a claims database. We used ICD-9 codes recorded in patients’ medical claims to determine diagnosis of psoriasis and rosacea; therefore, our study and data are subject to coding errors. We could not assess the severity of disease, only the presence of disease. Further confirmation of diagnosis could have been made through searching for a second ICD-9 code in the patient’s history. Our data also are from a limited time period and may not represent costs from other time periods.
Conclusion
Given the lack of cost difference between both specialties, we conclude that ACOs should consider encouraging patients to seek care for dermatologic diseases by dermatologists who generally are more accurate and efficient skin diagnosticians, particularly if there is a shortage of PCPs within the ACO network.
- Wimo A, Religa D, Spångberg K, et al. Costs of diagnosing dementia: results from SveDem, the Swedish Dementia Registry. Int J Geriatr Psychiatry. 2013;28:1039-1044.
- Grunfeld E, Fitzpatrick R, Mant D, et al. Comparison of breast cancer patient satisfaction with follow-up in primary care versus specialist care: results from a randomized controlled trial. Br J Gen Pract. 1999;49:705-710.
- Chen SC, Pennie ML, Kolm P, et al. Diagnosing and managing cutaneous pigmented lesions: primary care physicians versus dermatologists. J Gen Intern Med. 2006;21:678-682.
- Federman D, Hogan D, Taylor JR, et al. A comparison of diagnosis, evaluation, and treatment of patients with dermatologic disorders. J Am Acad Dermatol. 1995;32:726-729.
- Feldman SR, Fleischer AB, Young AC, et al. Time-efficiency of nondermatologists compared with dermatologists in the care of skin disease. J Am Acad Dermatol. 1999;40:194-199.
- Feldman SR, Peterson SR, Fleischer AB Jr. Dermatologists meet the primary care standard for first contact management of skin disease. J Am Acad Dermatol. 1998;39(2, pt 1):182-186.
- Smith ES, Fleischer AB, Feldman SR. Nondermatologists are more likely than dermatologists to prescribe antifungal/corticosteroid products: an analysis of office visits for cutaneous fungal infections, 1990-1994. J Am Acad Dermatol. 1998;39:43-47.
- Shaffer MP, Feldman SR, Fleischer AB. Use of clotrimazole/betamethasone diproprionate by family physicians. Fam Med. 2000;32:561-565.
- Feldman SR, Fleischer AB, Chen JG. The gatekeeper model is inefficient for the delivery of dermatologic services. J Am Acad Dermatol. 1999;40:426-432.
Growing incentives to control health care costs may cause accountable care organizations (ACOs) to reconsider how diseases are best managed. Few studies have examined the cost difference between primary care providers (PCPs) and specialists in managing the same disease. Limited data have suggested that management of some diseases by a PCP may be less costly compared to a specialist1,2; however, it is not clear if this finding extends to skin disease. This study sought to assess the cost of seeing a dermatologist versus a PCP for diagnosis of the common skin diseases psoriasis and rosacea.
Methods
Patient data were obtained from the Humana database, a large commercial data set for claims and reimbursed costs encompassing 18,162,539 patients covered between January 2007 and December 2014. Our study population consisted of 3,944,465 patients with claims that included International Classification of Diseases, Ninth Revision (ICD-9), codes for dermatological diagnoses (680.0–709.9). We searched by ICD-9 code for US patients with primary diagnoses of psoriasis (696.1) and rosacea (695.3). We narrowed the search to include patients aged 30 to 64 years, as the diagnoses for these diseases are most common in patients older than 30 years. Patients who were older than 64 years were not included in the study, as most are covered by Medicare and therefore costs covered by Humana in this age group would not be as representative as in younger age groups. Total and average diagnosis-related costs per patient were compared between dermatologists and PCPs. Diagnosis-related costs encompassed physician reimbursement; laboratory and imaging costs, including skin biopsies; inpatient hospitalization cost; and any other charge that could be coded or billed by providers and reimbursed by the insurance company. To be eligible for reimbursement from Humana, dermatologists and PCPs must be registered with the insurer according to specialty board certification and practice credentialing, and they are reimbursed differently based on specialty. Drug costs, which would possibly skew the data toward providers using more expensive systemic medications (ie, dermatologists), were not included in this study, as the discussion is better reserved for long-term management of disease rather than diagnosis-related costs. All diagnoses of psoriasis were included in the study, which likely includes all severities of psoriasis, though we did not have the ability to further break down these diagnoses by severity.
Results
We identified 30,217 psoriasis patients and 37,561 rosacea patients. Of those patients with a primary diagnosis of psoriasis, 26,112 (86%) were seen by a dermatologist and 4105 (14%) were seen by a PCP (Table). Of those patients with a primary diagnosis of rosacea, 34,694 (92%) were seen by a dermatologist and 2867 (8%) were seen by a PCP (Table). There was little difference in the average diagnosis-related cost per patient for psoriasis in males (dermatologists, $638; PCPs, $657) versus females (dermatologists, $592; PCPs, $586) or between specialties (Figure). Findings were similar for rosacea in males (dermatologists, $179; PCPs, $168) versus females (dermatologists, $157; PCPs, $161). For these skin diseases, i
Comment
For the management of common skin disorders such as psoriasis and rosacea, there is little cost difference in seeing a dermatologist versus a PCP. Through extensive training and repeated exposure to many skin diseases, dermatologists are expected to be more comfortable in diagnosing and managing psoriasis and rosacea. Compared to PCPs, dermatologists have demonstrated increased diagnostic accuracy and efficiency when examining pigmented lesions and other dermatologic diseases in several studies.3-6 Although the current study shows that diagnosis-related costs for psoriasis and rosacea are essentially equal between dermatologists and PCPs, it actually may be less expensive for patients to see a dermatologist, as unnecessary tests, biopsies, or medications are more likely to be ordered/prescribed when there is less clinical diagnostic certainty.7,8 Additionally, seeing a PCP for diagnosis of a skin disease may be inefficient if subsequent referral to a dermatologist is needed, a common scenario that occurs when patients see a PCP for skin conditions.9
Our study had limitations, which is typical of a study using a claims database. We used ICD-9 codes recorded in patients’ medical claims to determine diagnosis of psoriasis and rosacea; therefore, our study and data are subject to coding errors. We could not assess the severity of disease, only the presence of disease. Further confirmation of diagnosis could have been made through searching for a second ICD-9 code in the patient’s history. Our data also are from a limited time period and may not represent costs from other time periods.
Conclusion
Given the lack of cost difference between both specialties, we conclude that ACOs should consider encouraging patients to seek care for dermatologic diseases by dermatologists who generally are more accurate and efficient skin diagnosticians, particularly if there is a shortage of PCPs within the ACO network.
Growing incentives to control health care costs may cause accountable care organizations (ACOs) to reconsider how diseases are best managed. Few studies have examined the cost difference between primary care providers (PCPs) and specialists in managing the same disease. Limited data have suggested that management of some diseases by a PCP may be less costly compared to a specialist1,2; however, it is not clear if this finding extends to skin disease. This study sought to assess the cost of seeing a dermatologist versus a PCP for diagnosis of the common skin diseases psoriasis and rosacea.
Methods
Patient data were obtained from the Humana database, a large commercial data set for claims and reimbursed costs encompassing 18,162,539 patients covered between January 2007 and December 2014. Our study population consisted of 3,944,465 patients with claims that included International Classification of Diseases, Ninth Revision (ICD-9), codes for dermatological diagnoses (680.0–709.9). We searched by ICD-9 code for US patients with primary diagnoses of psoriasis (696.1) and rosacea (695.3). We narrowed the search to include patients aged 30 to 64 years, as the diagnoses for these diseases are most common in patients older than 30 years. Patients who were older than 64 years were not included in the study, as most are covered by Medicare and therefore costs covered by Humana in this age group would not be as representative as in younger age groups. Total and average diagnosis-related costs per patient were compared between dermatologists and PCPs. Diagnosis-related costs encompassed physician reimbursement; laboratory and imaging costs, including skin biopsies; inpatient hospitalization cost; and any other charge that could be coded or billed by providers and reimbursed by the insurance company. To be eligible for reimbursement from Humana, dermatologists and PCPs must be registered with the insurer according to specialty board certification and practice credentialing, and they are reimbursed differently based on specialty. Drug costs, which would possibly skew the data toward providers using more expensive systemic medications (ie, dermatologists), were not included in this study, as the discussion is better reserved for long-term management of disease rather than diagnosis-related costs. All diagnoses of psoriasis were included in the study, which likely includes all severities of psoriasis, though we did not have the ability to further break down these diagnoses by severity.
Results
We identified 30,217 psoriasis patients and 37,561 rosacea patients. Of those patients with a primary diagnosis of psoriasis, 26,112 (86%) were seen by a dermatologist and 4105 (14%) were seen by a PCP (Table). Of those patients with a primary diagnosis of rosacea, 34,694 (92%) were seen by a dermatologist and 2867 (8%) were seen by a PCP (Table). There was little difference in the average diagnosis-related cost per patient for psoriasis in males (dermatologists, $638; PCPs, $657) versus females (dermatologists, $592; PCPs, $586) or between specialties (Figure). Findings were similar for rosacea in males (dermatologists, $179; PCPs, $168) versus females (dermatologists, $157; PCPs, $161). For these skin diseases, i
Comment
For the management of common skin disorders such as psoriasis and rosacea, there is little cost difference in seeing a dermatologist versus a PCP. Through extensive training and repeated exposure to many skin diseases, dermatologists are expected to be more comfortable in diagnosing and managing psoriasis and rosacea. Compared to PCPs, dermatologists have demonstrated increased diagnostic accuracy and efficiency when examining pigmented lesions and other dermatologic diseases in several studies.3-6 Although the current study shows that diagnosis-related costs for psoriasis and rosacea are essentially equal between dermatologists and PCPs, it actually may be less expensive for patients to see a dermatologist, as unnecessary tests, biopsies, or medications are more likely to be ordered/prescribed when there is less clinical diagnostic certainty.7,8 Additionally, seeing a PCP for diagnosis of a skin disease may be inefficient if subsequent referral to a dermatologist is needed, a common scenario that occurs when patients see a PCP for skin conditions.9
Our study had limitations, which is typical of a study using a claims database. We used ICD-9 codes recorded in patients’ medical claims to determine diagnosis of psoriasis and rosacea; therefore, our study and data are subject to coding errors. We could not assess the severity of disease, only the presence of disease. Further confirmation of diagnosis could have been made through searching for a second ICD-9 code in the patient’s history. Our data also are from a limited time period and may not represent costs from other time periods.
Conclusion
Given the lack of cost difference between both specialties, we conclude that ACOs should consider encouraging patients to seek care for dermatologic diseases by dermatologists who generally are more accurate and efficient skin diagnosticians, particularly if there is a shortage of PCPs within the ACO network.
- Wimo A, Religa D, Spångberg K, et al. Costs of diagnosing dementia: results from SveDem, the Swedish Dementia Registry. Int J Geriatr Psychiatry. 2013;28:1039-1044.
- Grunfeld E, Fitzpatrick R, Mant D, et al. Comparison of breast cancer patient satisfaction with follow-up in primary care versus specialist care: results from a randomized controlled trial. Br J Gen Pract. 1999;49:705-710.
- Chen SC, Pennie ML, Kolm P, et al. Diagnosing and managing cutaneous pigmented lesions: primary care physicians versus dermatologists. J Gen Intern Med. 2006;21:678-682.
- Federman D, Hogan D, Taylor JR, et al. A comparison of diagnosis, evaluation, and treatment of patients with dermatologic disorders. J Am Acad Dermatol. 1995;32:726-729.
- Feldman SR, Fleischer AB, Young AC, et al. Time-efficiency of nondermatologists compared with dermatologists in the care of skin disease. J Am Acad Dermatol. 1999;40:194-199.
- Feldman SR, Peterson SR, Fleischer AB Jr. Dermatologists meet the primary care standard for first contact management of skin disease. J Am Acad Dermatol. 1998;39(2, pt 1):182-186.
- Smith ES, Fleischer AB, Feldman SR. Nondermatologists are more likely than dermatologists to prescribe antifungal/corticosteroid products: an analysis of office visits for cutaneous fungal infections, 1990-1994. J Am Acad Dermatol. 1998;39:43-47.
- Shaffer MP, Feldman SR, Fleischer AB. Use of clotrimazole/betamethasone diproprionate by family physicians. Fam Med. 2000;32:561-565.
- Feldman SR, Fleischer AB, Chen JG. The gatekeeper model is inefficient for the delivery of dermatologic services. J Am Acad Dermatol. 1999;40:426-432.
- Wimo A, Religa D, Spångberg K, et al. Costs of diagnosing dementia: results from SveDem, the Swedish Dementia Registry. Int J Geriatr Psychiatry. 2013;28:1039-1044.
- Grunfeld E, Fitzpatrick R, Mant D, et al. Comparison of breast cancer patient satisfaction with follow-up in primary care versus specialist care: results from a randomized controlled trial. Br J Gen Pract. 1999;49:705-710.
- Chen SC, Pennie ML, Kolm P, et al. Diagnosing and managing cutaneous pigmented lesions: primary care physicians versus dermatologists. J Gen Intern Med. 2006;21:678-682.
- Federman D, Hogan D, Taylor JR, et al. A comparison of diagnosis, evaluation, and treatment of patients with dermatologic disorders. J Am Acad Dermatol. 1995;32:726-729.
- Feldman SR, Fleischer AB, Young AC, et al. Time-efficiency of nondermatologists compared with dermatologists in the care of skin disease. J Am Acad Dermatol. 1999;40:194-199.
- Feldman SR, Peterson SR, Fleischer AB Jr. Dermatologists meet the primary care standard for first contact management of skin disease. J Am Acad Dermatol. 1998;39(2, pt 1):182-186.
- Smith ES, Fleischer AB, Feldman SR. Nondermatologists are more likely than dermatologists to prescribe antifungal/corticosteroid products: an analysis of office visits for cutaneous fungal infections, 1990-1994. J Am Acad Dermatol. 1998;39:43-47.
- Shaffer MP, Feldman SR, Fleischer AB. Use of clotrimazole/betamethasone diproprionate by family physicians. Fam Med. 2000;32:561-565.
- Feldman SR, Fleischer AB, Chen JG. The gatekeeper model is inefficient for the delivery of dermatologic services. J Am Acad Dermatol. 1999;40:426-432.
Practice Points
- Growing health care costs are causing accountable care organizations (ACOs) to reconsider how to best manage skin disease.
- There is little difference in average diagnosis-related cost between primary care physicians and dermatologists in diagnosing psoriasis or rosacea.
- With diagnosis costs essentially equal and increased dermatologist diagnostic accuracy, ACOs may encourage skin disease to be managed by dermatologists.
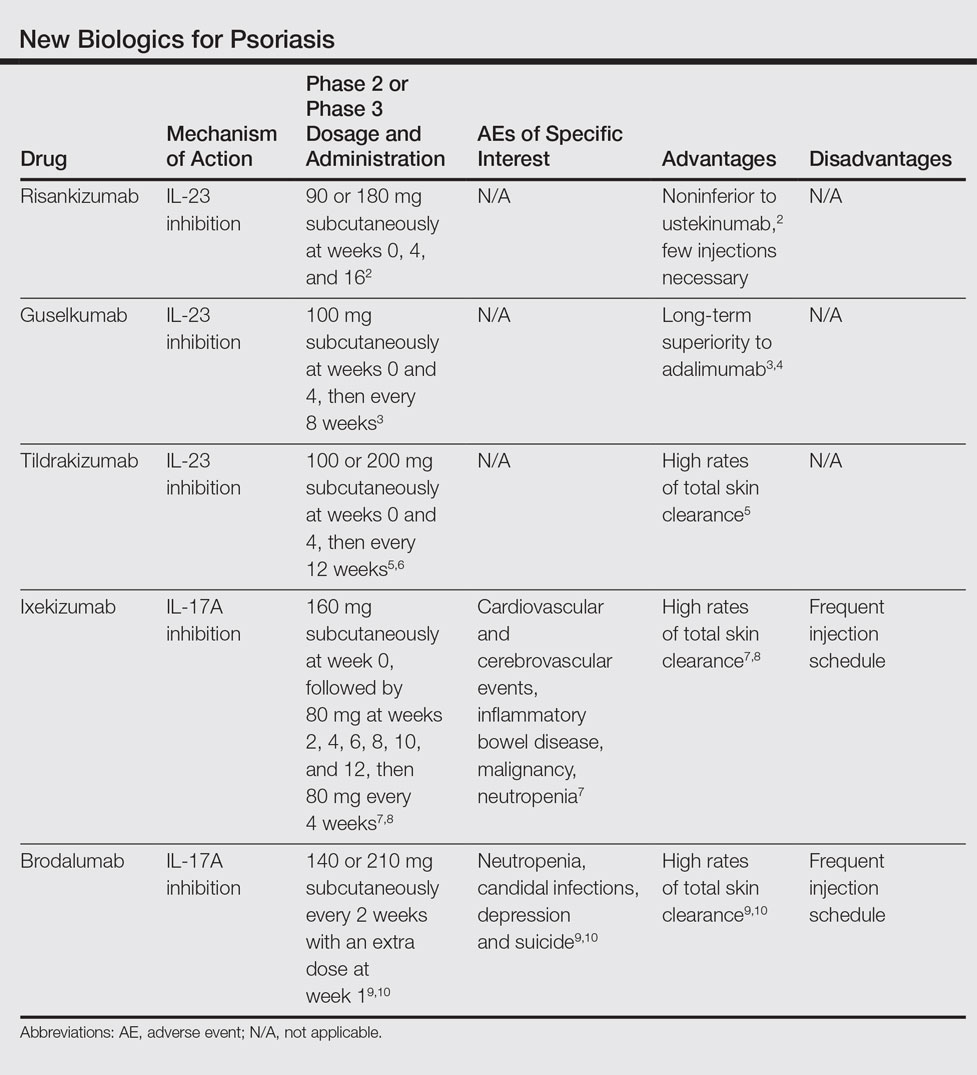
New Biologics in Psoriasis: An Update on IL-23 and IL-17 Inhibitors
The role of current biologic therapies in psoriasis predicates on the pathogenic role of upregulated, immune-related mechanisms that result in the activation of myeloid dendritic cells, which release IL-17, IL-23, and other cytokines to activate T cells, including helper T cell TH17. Along with other immune cells, TH17 produces IL-17. This proinflammatory cascade results in keratinocyte proliferation, angiogenesis, and migration of immune cells toward psoriatic lesions.1 Thus, the newest classes of biologics target IL-12, IL-23, and IL-17 to disrupt this inflammatory cascade.
We provide an updated review of the most recent clinical efficacy and safety data on the newest IL-23 and IL-17 inhibitors in the pipeline or approved for psoriasis, including risankizumab, guselkumab, tildrakizumab, ixekizumab, and brodalumab (Table). Ustekinumab and adalimumab, which have been previously approved by the US Food and Drug Administration (FDA), will be discussed here only as comparators.
IL-23 Inhibitors
Risankizumab
Risankizumab (formerly known as BI 655066)(Boehringer Ingelheim) is a selective human monoclonal antibody targeting the p19 subunit of IL-23 and currently is undergoing phase 3 trials for psoriasis. A proof-of-concept phase 1 study of 39 participants demonstrated efficacy after 12 weeks of treatment at varying subcutaneous and intravenous doses with placebo control.11 At week 12, 87% (27/31)(P<.001) of all risankizumab-treated participants achieved 75% reduction in psoriasis area and severity index (PASI) score compared to 0% of 8 placebo-treated participants. Common adverse effects (AEs) occurred in 65% (20/31) of risankizumab-treated participants, including non–dose-dependent upper respiratory tract infections, nasopharyngitis, and headache. Serious adverse events (SAEs) that occurred were considered unrelated to the study medication.11
A phase 2 trial of 166 participants compared 3 dosing regimens of subcutaneous risankizumab (single 18-mg dose at week 0; single 90-mg dose at weeks 0, 4, and 16; or single 180-mg dose at weeks 0, 4, and 16) and ustekinumab (weight-based single 45- or 90-mg dose at weeks 0, 4, and 16), demonstrating noninferiority at higher doses of risankizumab.2 Preliminary primary end point results at week 12 showed PASI 90 in 32.6% (P=.4667), 73.2% (P=.0013), 81.0% (P<.0001), and 40.0% of the treatment groups, respectively. Participants in the 180-mg risankizumab group achieved PASI 90 eight weeks faster than those on ustekinumab, lasting more than 2 months longer. Adverse effects were similar across all treatment groups and SAEs were unrelated to the study medications.2
Guselkumab
Guselkumab (Janssen Biotech, Inc) is a selective human monoclonal antibody against the p19 subunit of IL-23. The 52-week phase 2 X-PLORE trial compared dose-ranging subcutaneous guselkumab (5 mg at weeks 0 and 4, then every 12 weeks; 15 mg every 8 weeks; 50 mg at weeks 0 and 4, then every 12 weeks; 100 mg every 8 weeks; or 200 mg at weeks 0 and 4, then every 12 weeks), adalimumab (80-mg loading dose, followed by 40 mg at week 1, then every other week), and placebo in 293 randomized participants.4 At week 16, 34% (P=.002) of participants in the 5-mg guselkumab group, 61% (P<.001) in the 15-mg group, 79% (P<.001) in the 50-mg group, 86% (P<.001) in the 100-mg group, 83% (P<.001) in the 200-mg group, and 58% (P<.001) in the adalimumab group achieved physician global assessment (PGA) scores of 0 (clear) or 1 (minimal psoriasis) compared to 7% of the placebo group. Achievement of PASI 75 similarly favored the guselkumab (44% [P<.001]; 76% [no P value given]; 81% [P<.001]; 79% [P<.001]; and 81% [P<.001], respectively) and adalimumab treatment arms (70% [P<.001]) compared to 5% in the placebo group. In longer-term comparisons to week 40, participants in the 50-, 100-, and 200-mg guselkumab groups showed significantly greater remission of psoriatic lesions, measured by a PGA score of 0 or 1, than participants in the adalimumab group (71% [P=.05]; 77% [P=.005]; 81% [P=.01]; and 49%, respectively).4
Preliminary results from VOYAGE 1 (N=837), the first of several phase 3 trials, further demonstrate the superiority of guselkumab 100 mg at weeks 0 and 4 and then every 8 weeks over adalimumab (standard dosing) and placebo; at week 16, 73.3% (P<.001 for both comparisons) versus 49.7% and 2.9% of participants, respectively, achieved PASI 90, with sustained superiority of skin clearance in guselkumab-treated participants compared to adalimumab and placebo through week 48.3
Long-term safety data showed no dose dependence or trend from 0 to 16 weeks and 16 to 52 weeks of treatment regarding rates of AEs, SAEs, or serious infections.4 Between weeks 16 and 52, 48.9% of all guselkumab-treated participants exhibited AEs compared to 60.5% of adalimumab-treated participants and 51.3% of placebo participants. Overall infection rates also were lowest in the guselkumab group at 29.8% compared to 36.8% and 35.9%, respectively. Three participants treated with guselkumab had major cardiovascular events, including a fatal myocardial infarction. No cases of tuberculosis or serious opportunistic infections were reported.4
Tildrakizumab
Tildrakizumab (formerly known as MK-3222)(Sun Pharmaceutical Industries Ltd) is a human monoclonal antibody also targeting the p19 subunit of IL-23. In a phase 2 study of 355 participants with chronic plaque psoriasis, participants received 5-, 25-, 100-, or 200-mg subcutaneous tildrakizumab or placebo at weeks 0 and 4 and then every 12 weeks for a total of 52 weeks.6 At week 16, PASI 75 results were 33.3%, 64.4%, 66.3%, 74.4%, and 4.4%, respectively (P<.001 for each comparison). Improvement began within the first month of treatment, with median times to PASI 75 of 57 days at 200-mg dosing and 84 days at 100-mg dosing. Of those participants achieving PASI 75 by drug discontinuation at week 52, 96% of the 100-mg group and 93% of the 200-mg group maintained PASI 75 through week 72, suggesting low relapse rates after treatment cessation.6
In October 2016, the efficacy results of 2 pivotal phase 3 trials (reSURFACE 1 and reSURFACE 2) involving more than 1800 participants combined revealed PASI 90 achievement in an average of 54% of participants on tildrakizumab 100 mg and 59% of participants on tildrakizumab 200 mg at week 28.5 Achievement of PASI 100 occurred in 24% and 30% of participants at week 28, respectively. The second of these trials included an etanercept comparison group and demonstrated head-to-head superiority of 100 and 200 mg subcutaneous tildrakizumab at week 12 by end point measures.5
Treatment-related AEs occurred at rates of 25% in tildrakizumab-treated participants and 22% in placebo-treated participants, most frequently nasopharyngitis and headache.6 At least 1 AE occurred in 64% of tildrakizumab-treated participants without dose dependence compared to 69% of placebo-treated participants. Severe AEs thought to be drug treatment related were bacterial arthritis, lymphedema, melanoma, stroke, and epiglottitis.6
IL-17 Inhibitors
Ixekizumab
Ixekizumab (Eli Lilly and Company), a monoclonal inhibitor of IL-17A, is the most recently approved psoriasis biologic on the market and has been cleared for use in adults with moderate to severe plaque psoriasis. Recommended dosing is 160 mg (given in two 80-mg subcutaneous injections via an autoinjector or prefilled syringe) at week 0, followed by an 80-mg injection at weeks 2, 4, 6, 8, 10, and 12, and then 80 mg every 4 weeks thereafter. The FDA approved ixekizumab in March 2016 following favorable results of several phase 3 trials: UNCOVER-1, UNCOVER-2, and UNCOVER-3.7,8
In UNCOVER-1, 1296 participants were randomized to 1 of 2 ixekizumab treatment arms—160 mg starting dose at week 0, 80 mg every 2 or 4 weeks thereafter—or placebo.7 At week 12, 89.1%, 82.6%, and 3.9% achieved PASI 75, respectively (P<.001 for both). Importantly, high numbers of participants also achieved PASI 90 (70.9% in the 2-week group and 64.6% in the 4-week group vs 0.5% in the placebo group [P<.001]) and PASI 100 (35.3% and 33.6% vs 0%, respectively [P<.001]), suggesting high rates of disease clearance.7
UNCOVER-2 (N=1224) and UNCOVER-3 (N=1346) investigated the same 2 dosing regimens of ixekizumab compared to etanercept 50 mg biweekly and placebo.8 At week 12, the percentage of participants achieving PASI 90 in UNCOVER-2 was 70.7%, 59.7%, 18.7%, and 0.6%, respectively, and 68.1%, 65.3%, 25.7%, and 3.1%, respectively, in UNCOVER-3 (P<.0001 for all comparisons to placebo and etanercept). At week 12, PASI 100 results also showed striking superiority, with 40.5%, 30.8%, 5.3%, and 0.6% of participants, respectively, in UNCOVER-2, and 37.7%, 35%, 7.3%, and 0%, respectively, in UNCOVER-3, achieving complete clearance of disease (P<.0001 for all comparisons to placebo and etanercept). Responses to ixekizumab were observed as early as weeks 1 and 2, while no participants in the etanercept and placebo treatment groups achieved comparative efficiency.8
In an extension of UNCOVER-3, efficacy increased from week 12 to week 60 according to PASI 90 (68%–73% in the 2-week group; 65%–72% in the 4-week group) and PASI 100 measures (38%–55% in the 2-week group; 35%–52% in the 4-week group).7
The most common AEs associated with ixekizumab treatment from weeks 0 to 12 occurred at higher rates in the 2-week and 4-week ixekizumab groups compared to placebo, including nasopharyngitis (9.5% and 9% vs 8.7%, respectively), upper respiratory tract infection (4.4% and 3.9% vs 3.5%, respectively), injection-site reaction (10% and 7.7% vs 1%, respectively), arthralgia (4.4% and 4.3% vs 2.9%, respectively), and headache (2.5% and 1.9% vs 2.1%, respectively). Infections, including candidal, oral, vulvovaginal, and cutaneous, occurred in 27% of the 2-week dosing group and 27.4% of the 4-week dosing group compared to 22.9% of the placebo group during weeks 0 to 12, with candidal infections in particular occurring more frequently in the active treatment groups and exhibiting dose dependence. Other AEs of special interest that occurred among all ixekizumab-treated participants (n=3736) from weeks 0 to 60 were cardiovascular and cerebrovascular events (22 [0.6%]), inflammatory bowel disease (11 [0.3%]), non–skin cancer malignancy (14 [0.4%]), and nonmelanoma skin cancer (20 [0.5%]). Neutropenia occurred at higher rates in ixekizumab-treated participants (9.3% in the 2-week group and 8.6% in the 4-week group) compared to placebo (3.3%) and occurred in 11.5% of all ixekizumab participants over 60 weeks.7
Brodalumab
Brodalumab (Valeant Pharmaceuticals International, Inc) is a human monoclonal antibody targeting the IL-17A receptor currently under review for FDA approval after undergoing phase 3 trials. The first of these trials, AMAGINE-1, showed efficacy of subcutaneous brodalumab (140 or 210 mg administered every 2 weeks with an extra dose at week 1) compared to placebo in 661 participants.9 At week 12, 60%, 83%, and 3%, respectively, achieved PASI 75; 43%, 70%, and 1%, respectively, achieved PASI 90; and 23%, 42%, and 1%, respectively, achieved PASI 100 (P<.001 for all respective comparisons to placebo). These effects were retained through 52 weeks of treatment. The median time to complete disease clearance in participants reaching PASI 100 was 12 weeks. Conversely, participants who were re-randomized to placebo after week 12 of brodalumab treatment relapsed within weeks to months.9
AMAGINE-2 and AMAGINE-3 further demonstrated the efficacy of brodalumab (140 or 210 mg every 2 weeks with extra dose at week 1) compared to ustekinumab (45 or 90 mg weight-based standard dosing) and placebo in 1831 participants, respectively.10 In AMAGINE-2, 49% of participants in the 140-mg group (P<.001 vs placebo), 70% in the 210-mg group (P<.001 vs placebo), 47% in the ustekinumab group, and 3% in the placebo group achieved PASI 90 at week 12. Similarly, in AMAGINE-3, 52% of participants in the 140-mg group (P<.001), 69% in the 210-mg group (P<.001), 48% in the ustekinumab group, and 2% in the placebo group achieved PASI 90. Impressively, complete clearance (PASI 100) at week 12 occurred in 26% of the 140-mg group (P<.001 vs placebo), 44% of the 210-mg group (P<.001 vs placebo), and 22% of the ustekinumab group compared to 2% of the placebo group in AMAGINE-2, with similar rates in AMAGINE-3. Brodalumab was significantly superior to ustekinumab at the 210-mg dose by PASI 90 measures (P<.001) in both studies and at the 140-mg dose by PASI 100 measures (P=.007) in AMAGINE-3 only.10
Common AEs were nasopharyngitis, upper respiratory tract infection, headache, and arthralgia, all occurring at grossly similar rates (49%–60%) across all experimental groups in AMAGINE-1, AMAGINE-2, and AMAGINE-3 during the first 12-week treatment period.9,10 Brodalumab treatment groups had high rates of specific interest AEs compared to ustekinumab and placebo groups, including neutropenia (0.8%, 1.1%, 0.3%, and 0%, respectively) and candidal infections (0.8%, 1.3%, 0.3%, and 0.3%, respectively). Induction phase (weeks 0–12) depression rates were concerning, with 6 cases each in AMAGINE-2 (4 [0.7%] in the 140-mg group, 2 [0.3%] in the 210-mg group) and AMAGINE-3 (4 [0.6%] in the 140-mg group, 2 [0.3%] in the 210-mg group). Cases of neutropenia were mild, were not associated with major infection, and were transient or reversible. Depression rates after 52 weeks of treatment were 1.7% (23/1567) of brodalumab participants in AMAGINE-2 and 1.8% (21/1613) in AMAGINE-3. Three participants, all on constant 210-mg dosing through week 52, attempted suicide with 1 completion10; however, because no other IL-17 inhibitors were associated with depression or suicide in other trials, it has been suggested that these cases were incidental and not treatment related.12 An FDA advisory panel recommended approval of brodalumab in July 2016 despite ongoing concerns of depression and suicide.13
Conclusion
The robust investigation into IL-23 and IL-17 inhibitors to treat plaque psoriasis has yielded promising results, including the unprecedented rates of PASI 100 achievement with these new biologics. Risankizumab, ixekizumab, and brodalumab have demonstrated superior efficacy in trials compared to ustekinumab. Tildrakizumab has shown low disease relapse after drug cessation. Ixekizumab and brodalumab have shown high rates of total disease clearance. Thus far, safety findings for these pipeline biologics have been consistent with those of ustekinumab. With ixekizumab approved in 2016 and brodalumab under review, new options in biologic therapy will offer patients and clinicians greater choices in treating severe and recalcitrant psoriasis.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509.
- Papp K, Menter A, Sofen H, et al. Efficacy and safety of different dose regimens of a selective IL-23p19 inhibitor (BI 655066) compared with ustekinumab in patients with moderate-to-severe plaque psoriasis with and without psoriatic arthritis. Paper presented at: 2015 American College of Rheumatology/Association of Rheumatology Health Professionals Annual Meeting; November 6-11, 2015; San Francisco, CA.
- New phase 3 data show significant efficacy versus placebo and superiority of guselkumab versus Humira in treatment of moderate to severe plaque psoriasis [press release]. Vienna, Austria; Janssen Research & Development, LLC: October 1, 2016.
- Gordon KB, Duffin KC, Bissonnette R, et al. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N Engl J Med. 2015;373:136-144.
- Sun Pharma to announce late-breaking results for investigational IL-23p19 inhibitor, Tildrakizumab, achieves primary end point in both phase-3 studies in patients with moderate-to-severe plaque psoriasis [press release]. Mumbai, India; Sun Pharmaceutical Industries Ltd: October 1, 2016.
- Papp K, Thaci D, Reich K, et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol. 2015;173:930-939.
- Gordon KB, Blauvelt A, Papp KA, et al; UNCOVER-1 Study Group, UNCOVER-2 Study Group, UNCOVER-3 Study Group. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis [published online June 23, 2016]. Br J Dermatol. 2016;175:273-286.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial [published online March 1, 2015]. J Allergy Clin Immunol. 2015;136:116-124.e7.
- Chiricozzi A, Romanelli M, Saraceno R, et al. No meaningful association between suicidal behavior and the use of IL-17A-neutralizing or IL-17RA-blocking agents [published online August 31, 2016]. Expert Opin Drug Saf. 2016;15:1653-1659.
- FDA advisory committee recommends approval of brodalumab for treatment of moderate-to-severe plaque psoriasis [news release]. Laval, Quebec: Valeant Pharmaceuticals International, Inc; July 19, 2016.
The role of current biologic therapies in psoriasis predicates on the pathogenic role of upregulated, immune-related mechanisms that result in the activation of myeloid dendritic cells, which release IL-17, IL-23, and other cytokines to activate T cells, including helper T cell TH17. Along with other immune cells, TH17 produces IL-17. This proinflammatory cascade results in keratinocyte proliferation, angiogenesis, and migration of immune cells toward psoriatic lesions.1 Thus, the newest classes of biologics target IL-12, IL-23, and IL-17 to disrupt this inflammatory cascade.
We provide an updated review of the most recent clinical efficacy and safety data on the newest IL-23 and IL-17 inhibitors in the pipeline or approved for psoriasis, including risankizumab, guselkumab, tildrakizumab, ixekizumab, and brodalumab (Table). Ustekinumab and adalimumab, which have been previously approved by the US Food and Drug Administration (FDA), will be discussed here only as comparators.
IL-23 Inhibitors
Risankizumab
Risankizumab (formerly known as BI 655066)(Boehringer Ingelheim) is a selective human monoclonal antibody targeting the p19 subunit of IL-23 and currently is undergoing phase 3 trials for psoriasis. A proof-of-concept phase 1 study of 39 participants demonstrated efficacy after 12 weeks of treatment at varying subcutaneous and intravenous doses with placebo control.11 At week 12, 87% (27/31)(P<.001) of all risankizumab-treated participants achieved 75% reduction in psoriasis area and severity index (PASI) score compared to 0% of 8 placebo-treated participants. Common adverse effects (AEs) occurred in 65% (20/31) of risankizumab-treated participants, including non–dose-dependent upper respiratory tract infections, nasopharyngitis, and headache. Serious adverse events (SAEs) that occurred were considered unrelated to the study medication.11
A phase 2 trial of 166 participants compared 3 dosing regimens of subcutaneous risankizumab (single 18-mg dose at week 0; single 90-mg dose at weeks 0, 4, and 16; or single 180-mg dose at weeks 0, 4, and 16) and ustekinumab (weight-based single 45- or 90-mg dose at weeks 0, 4, and 16), demonstrating noninferiority at higher doses of risankizumab.2 Preliminary primary end point results at week 12 showed PASI 90 in 32.6% (P=.4667), 73.2% (P=.0013), 81.0% (P<.0001), and 40.0% of the treatment groups, respectively. Participants in the 180-mg risankizumab group achieved PASI 90 eight weeks faster than those on ustekinumab, lasting more than 2 months longer. Adverse effects were similar across all treatment groups and SAEs were unrelated to the study medications.2
Guselkumab
Guselkumab (Janssen Biotech, Inc) is a selective human monoclonal antibody against the p19 subunit of IL-23. The 52-week phase 2 X-PLORE trial compared dose-ranging subcutaneous guselkumab (5 mg at weeks 0 and 4, then every 12 weeks; 15 mg every 8 weeks; 50 mg at weeks 0 and 4, then every 12 weeks; 100 mg every 8 weeks; or 200 mg at weeks 0 and 4, then every 12 weeks), adalimumab (80-mg loading dose, followed by 40 mg at week 1, then every other week), and placebo in 293 randomized participants.4 At week 16, 34% (P=.002) of participants in the 5-mg guselkumab group, 61% (P<.001) in the 15-mg group, 79% (P<.001) in the 50-mg group, 86% (P<.001) in the 100-mg group, 83% (P<.001) in the 200-mg group, and 58% (P<.001) in the adalimumab group achieved physician global assessment (PGA) scores of 0 (clear) or 1 (minimal psoriasis) compared to 7% of the placebo group. Achievement of PASI 75 similarly favored the guselkumab (44% [P<.001]; 76% [no P value given]; 81% [P<.001]; 79% [P<.001]; and 81% [P<.001], respectively) and adalimumab treatment arms (70% [P<.001]) compared to 5% in the placebo group. In longer-term comparisons to week 40, participants in the 50-, 100-, and 200-mg guselkumab groups showed significantly greater remission of psoriatic lesions, measured by a PGA score of 0 or 1, than participants in the adalimumab group (71% [P=.05]; 77% [P=.005]; 81% [P=.01]; and 49%, respectively).4
Preliminary results from VOYAGE 1 (N=837), the first of several phase 3 trials, further demonstrate the superiority of guselkumab 100 mg at weeks 0 and 4 and then every 8 weeks over adalimumab (standard dosing) and placebo; at week 16, 73.3% (P<.001 for both comparisons) versus 49.7% and 2.9% of participants, respectively, achieved PASI 90, with sustained superiority of skin clearance in guselkumab-treated participants compared to adalimumab and placebo through week 48.3
Long-term safety data showed no dose dependence or trend from 0 to 16 weeks and 16 to 52 weeks of treatment regarding rates of AEs, SAEs, or serious infections.4 Between weeks 16 and 52, 48.9% of all guselkumab-treated participants exhibited AEs compared to 60.5% of adalimumab-treated participants and 51.3% of placebo participants. Overall infection rates also were lowest in the guselkumab group at 29.8% compared to 36.8% and 35.9%, respectively. Three participants treated with guselkumab had major cardiovascular events, including a fatal myocardial infarction. No cases of tuberculosis or serious opportunistic infections were reported.4
Tildrakizumab
Tildrakizumab (formerly known as MK-3222)(Sun Pharmaceutical Industries Ltd) is a human monoclonal antibody also targeting the p19 subunit of IL-23. In a phase 2 study of 355 participants with chronic plaque psoriasis, participants received 5-, 25-, 100-, or 200-mg subcutaneous tildrakizumab or placebo at weeks 0 and 4 and then every 12 weeks for a total of 52 weeks.6 At week 16, PASI 75 results were 33.3%, 64.4%, 66.3%, 74.4%, and 4.4%, respectively (P<.001 for each comparison). Improvement began within the first month of treatment, with median times to PASI 75 of 57 days at 200-mg dosing and 84 days at 100-mg dosing. Of those participants achieving PASI 75 by drug discontinuation at week 52, 96% of the 100-mg group and 93% of the 200-mg group maintained PASI 75 through week 72, suggesting low relapse rates after treatment cessation.6
In October 2016, the efficacy results of 2 pivotal phase 3 trials (reSURFACE 1 and reSURFACE 2) involving more than 1800 participants combined revealed PASI 90 achievement in an average of 54% of participants on tildrakizumab 100 mg and 59% of participants on tildrakizumab 200 mg at week 28.5 Achievement of PASI 100 occurred in 24% and 30% of participants at week 28, respectively. The second of these trials included an etanercept comparison group and demonstrated head-to-head superiority of 100 and 200 mg subcutaneous tildrakizumab at week 12 by end point measures.5
Treatment-related AEs occurred at rates of 25% in tildrakizumab-treated participants and 22% in placebo-treated participants, most frequently nasopharyngitis and headache.6 At least 1 AE occurred in 64% of tildrakizumab-treated participants without dose dependence compared to 69% of placebo-treated participants. Severe AEs thought to be drug treatment related were bacterial arthritis, lymphedema, melanoma, stroke, and epiglottitis.6
IL-17 Inhibitors
Ixekizumab
Ixekizumab (Eli Lilly and Company), a monoclonal inhibitor of IL-17A, is the most recently approved psoriasis biologic on the market and has been cleared for use in adults with moderate to severe plaque psoriasis. Recommended dosing is 160 mg (given in two 80-mg subcutaneous injections via an autoinjector or prefilled syringe) at week 0, followed by an 80-mg injection at weeks 2, 4, 6, 8, 10, and 12, and then 80 mg every 4 weeks thereafter. The FDA approved ixekizumab in March 2016 following favorable results of several phase 3 trials: UNCOVER-1, UNCOVER-2, and UNCOVER-3.7,8
In UNCOVER-1, 1296 participants were randomized to 1 of 2 ixekizumab treatment arms—160 mg starting dose at week 0, 80 mg every 2 or 4 weeks thereafter—or placebo.7 At week 12, 89.1%, 82.6%, and 3.9% achieved PASI 75, respectively (P<.001 for both). Importantly, high numbers of participants also achieved PASI 90 (70.9% in the 2-week group and 64.6% in the 4-week group vs 0.5% in the placebo group [P<.001]) and PASI 100 (35.3% and 33.6% vs 0%, respectively [P<.001]), suggesting high rates of disease clearance.7
UNCOVER-2 (N=1224) and UNCOVER-3 (N=1346) investigated the same 2 dosing regimens of ixekizumab compared to etanercept 50 mg biweekly and placebo.8 At week 12, the percentage of participants achieving PASI 90 in UNCOVER-2 was 70.7%, 59.7%, 18.7%, and 0.6%, respectively, and 68.1%, 65.3%, 25.7%, and 3.1%, respectively, in UNCOVER-3 (P<.0001 for all comparisons to placebo and etanercept). At week 12, PASI 100 results also showed striking superiority, with 40.5%, 30.8%, 5.3%, and 0.6% of participants, respectively, in UNCOVER-2, and 37.7%, 35%, 7.3%, and 0%, respectively, in UNCOVER-3, achieving complete clearance of disease (P<.0001 for all comparisons to placebo and etanercept). Responses to ixekizumab were observed as early as weeks 1 and 2, while no participants in the etanercept and placebo treatment groups achieved comparative efficiency.8
In an extension of UNCOVER-3, efficacy increased from week 12 to week 60 according to PASI 90 (68%–73% in the 2-week group; 65%–72% in the 4-week group) and PASI 100 measures (38%–55% in the 2-week group; 35%–52% in the 4-week group).7
The most common AEs associated with ixekizumab treatment from weeks 0 to 12 occurred at higher rates in the 2-week and 4-week ixekizumab groups compared to placebo, including nasopharyngitis (9.5% and 9% vs 8.7%, respectively), upper respiratory tract infection (4.4% and 3.9% vs 3.5%, respectively), injection-site reaction (10% and 7.7% vs 1%, respectively), arthralgia (4.4% and 4.3% vs 2.9%, respectively), and headache (2.5% and 1.9% vs 2.1%, respectively). Infections, including candidal, oral, vulvovaginal, and cutaneous, occurred in 27% of the 2-week dosing group and 27.4% of the 4-week dosing group compared to 22.9% of the placebo group during weeks 0 to 12, with candidal infections in particular occurring more frequently in the active treatment groups and exhibiting dose dependence. Other AEs of special interest that occurred among all ixekizumab-treated participants (n=3736) from weeks 0 to 60 were cardiovascular and cerebrovascular events (22 [0.6%]), inflammatory bowel disease (11 [0.3%]), non–skin cancer malignancy (14 [0.4%]), and nonmelanoma skin cancer (20 [0.5%]). Neutropenia occurred at higher rates in ixekizumab-treated participants (9.3% in the 2-week group and 8.6% in the 4-week group) compared to placebo (3.3%) and occurred in 11.5% of all ixekizumab participants over 60 weeks.7
Brodalumab
Brodalumab (Valeant Pharmaceuticals International, Inc) is a human monoclonal antibody targeting the IL-17A receptor currently under review for FDA approval after undergoing phase 3 trials. The first of these trials, AMAGINE-1, showed efficacy of subcutaneous brodalumab (140 or 210 mg administered every 2 weeks with an extra dose at week 1) compared to placebo in 661 participants.9 At week 12, 60%, 83%, and 3%, respectively, achieved PASI 75; 43%, 70%, and 1%, respectively, achieved PASI 90; and 23%, 42%, and 1%, respectively, achieved PASI 100 (P<.001 for all respective comparisons to placebo). These effects were retained through 52 weeks of treatment. The median time to complete disease clearance in participants reaching PASI 100 was 12 weeks. Conversely, participants who were re-randomized to placebo after week 12 of brodalumab treatment relapsed within weeks to months.9
AMAGINE-2 and AMAGINE-3 further demonstrated the efficacy of brodalumab (140 or 210 mg every 2 weeks with extra dose at week 1) compared to ustekinumab (45 or 90 mg weight-based standard dosing) and placebo in 1831 participants, respectively.10 In AMAGINE-2, 49% of participants in the 140-mg group (P<.001 vs placebo), 70% in the 210-mg group (P<.001 vs placebo), 47% in the ustekinumab group, and 3% in the placebo group achieved PASI 90 at week 12. Similarly, in AMAGINE-3, 52% of participants in the 140-mg group (P<.001), 69% in the 210-mg group (P<.001), 48% in the ustekinumab group, and 2% in the placebo group achieved PASI 90. Impressively, complete clearance (PASI 100) at week 12 occurred in 26% of the 140-mg group (P<.001 vs placebo), 44% of the 210-mg group (P<.001 vs placebo), and 22% of the ustekinumab group compared to 2% of the placebo group in AMAGINE-2, with similar rates in AMAGINE-3. Brodalumab was significantly superior to ustekinumab at the 210-mg dose by PASI 90 measures (P<.001) in both studies and at the 140-mg dose by PASI 100 measures (P=.007) in AMAGINE-3 only.10
Common AEs were nasopharyngitis, upper respiratory tract infection, headache, and arthralgia, all occurring at grossly similar rates (49%–60%) across all experimental groups in AMAGINE-1, AMAGINE-2, and AMAGINE-3 during the first 12-week treatment period.9,10 Brodalumab treatment groups had high rates of specific interest AEs compared to ustekinumab and placebo groups, including neutropenia (0.8%, 1.1%, 0.3%, and 0%, respectively) and candidal infections (0.8%, 1.3%, 0.3%, and 0.3%, respectively). Induction phase (weeks 0–12) depression rates were concerning, with 6 cases each in AMAGINE-2 (4 [0.7%] in the 140-mg group, 2 [0.3%] in the 210-mg group) and AMAGINE-3 (4 [0.6%] in the 140-mg group, 2 [0.3%] in the 210-mg group). Cases of neutropenia were mild, were not associated with major infection, and were transient or reversible. Depression rates after 52 weeks of treatment were 1.7% (23/1567) of brodalumab participants in AMAGINE-2 and 1.8% (21/1613) in AMAGINE-3. Three participants, all on constant 210-mg dosing through week 52, attempted suicide with 1 completion10; however, because no other IL-17 inhibitors were associated with depression or suicide in other trials, it has been suggested that these cases were incidental and not treatment related.12 An FDA advisory panel recommended approval of brodalumab in July 2016 despite ongoing concerns of depression and suicide.13
Conclusion
The robust investigation into IL-23 and IL-17 inhibitors to treat plaque psoriasis has yielded promising results, including the unprecedented rates of PASI 100 achievement with these new biologics. Risankizumab, ixekizumab, and brodalumab have demonstrated superior efficacy in trials compared to ustekinumab. Tildrakizumab has shown low disease relapse after drug cessation. Ixekizumab and brodalumab have shown high rates of total disease clearance. Thus far, safety findings for these pipeline biologics have been consistent with those of ustekinumab. With ixekizumab approved in 2016 and brodalumab under review, new options in biologic therapy will offer patients and clinicians greater choices in treating severe and recalcitrant psoriasis.
The role of current biologic therapies in psoriasis predicates on the pathogenic role of upregulated, immune-related mechanisms that result in the activation of myeloid dendritic cells, which release IL-17, IL-23, and other cytokines to activate T cells, including helper T cell TH17. Along with other immune cells, TH17 produces IL-17. This proinflammatory cascade results in keratinocyte proliferation, angiogenesis, and migration of immune cells toward psoriatic lesions.1 Thus, the newest classes of biologics target IL-12, IL-23, and IL-17 to disrupt this inflammatory cascade.
We provide an updated review of the most recent clinical efficacy and safety data on the newest IL-23 and IL-17 inhibitors in the pipeline or approved for psoriasis, including risankizumab, guselkumab, tildrakizumab, ixekizumab, and brodalumab (Table). Ustekinumab and adalimumab, which have been previously approved by the US Food and Drug Administration (FDA), will be discussed here only as comparators.
IL-23 Inhibitors
Risankizumab
Risankizumab (formerly known as BI 655066)(Boehringer Ingelheim) is a selective human monoclonal antibody targeting the p19 subunit of IL-23 and currently is undergoing phase 3 trials for psoriasis. A proof-of-concept phase 1 study of 39 participants demonstrated efficacy after 12 weeks of treatment at varying subcutaneous and intravenous doses with placebo control.11 At week 12, 87% (27/31)(P<.001) of all risankizumab-treated participants achieved 75% reduction in psoriasis area and severity index (PASI) score compared to 0% of 8 placebo-treated participants. Common adverse effects (AEs) occurred in 65% (20/31) of risankizumab-treated participants, including non–dose-dependent upper respiratory tract infections, nasopharyngitis, and headache. Serious adverse events (SAEs) that occurred were considered unrelated to the study medication.11
A phase 2 trial of 166 participants compared 3 dosing regimens of subcutaneous risankizumab (single 18-mg dose at week 0; single 90-mg dose at weeks 0, 4, and 16; or single 180-mg dose at weeks 0, 4, and 16) and ustekinumab (weight-based single 45- or 90-mg dose at weeks 0, 4, and 16), demonstrating noninferiority at higher doses of risankizumab.2 Preliminary primary end point results at week 12 showed PASI 90 in 32.6% (P=.4667), 73.2% (P=.0013), 81.0% (P<.0001), and 40.0% of the treatment groups, respectively. Participants in the 180-mg risankizumab group achieved PASI 90 eight weeks faster than those on ustekinumab, lasting more than 2 months longer. Adverse effects were similar across all treatment groups and SAEs were unrelated to the study medications.2
Guselkumab
Guselkumab (Janssen Biotech, Inc) is a selective human monoclonal antibody against the p19 subunit of IL-23. The 52-week phase 2 X-PLORE trial compared dose-ranging subcutaneous guselkumab (5 mg at weeks 0 and 4, then every 12 weeks; 15 mg every 8 weeks; 50 mg at weeks 0 and 4, then every 12 weeks; 100 mg every 8 weeks; or 200 mg at weeks 0 and 4, then every 12 weeks), adalimumab (80-mg loading dose, followed by 40 mg at week 1, then every other week), and placebo in 293 randomized participants.4 At week 16, 34% (P=.002) of participants in the 5-mg guselkumab group, 61% (P<.001) in the 15-mg group, 79% (P<.001) in the 50-mg group, 86% (P<.001) in the 100-mg group, 83% (P<.001) in the 200-mg group, and 58% (P<.001) in the adalimumab group achieved physician global assessment (PGA) scores of 0 (clear) or 1 (minimal psoriasis) compared to 7% of the placebo group. Achievement of PASI 75 similarly favored the guselkumab (44% [P<.001]; 76% [no P value given]; 81% [P<.001]; 79% [P<.001]; and 81% [P<.001], respectively) and adalimumab treatment arms (70% [P<.001]) compared to 5% in the placebo group. In longer-term comparisons to week 40, participants in the 50-, 100-, and 200-mg guselkumab groups showed significantly greater remission of psoriatic lesions, measured by a PGA score of 0 or 1, than participants in the adalimumab group (71% [P=.05]; 77% [P=.005]; 81% [P=.01]; and 49%, respectively).4
Preliminary results from VOYAGE 1 (N=837), the first of several phase 3 trials, further demonstrate the superiority of guselkumab 100 mg at weeks 0 and 4 and then every 8 weeks over adalimumab (standard dosing) and placebo; at week 16, 73.3% (P<.001 for both comparisons) versus 49.7% and 2.9% of participants, respectively, achieved PASI 90, with sustained superiority of skin clearance in guselkumab-treated participants compared to adalimumab and placebo through week 48.3
Long-term safety data showed no dose dependence or trend from 0 to 16 weeks and 16 to 52 weeks of treatment regarding rates of AEs, SAEs, or serious infections.4 Between weeks 16 and 52, 48.9% of all guselkumab-treated participants exhibited AEs compared to 60.5% of adalimumab-treated participants and 51.3% of placebo participants. Overall infection rates also were lowest in the guselkumab group at 29.8% compared to 36.8% and 35.9%, respectively. Three participants treated with guselkumab had major cardiovascular events, including a fatal myocardial infarction. No cases of tuberculosis or serious opportunistic infections were reported.4
Tildrakizumab
Tildrakizumab (formerly known as MK-3222)(Sun Pharmaceutical Industries Ltd) is a human monoclonal antibody also targeting the p19 subunit of IL-23. In a phase 2 study of 355 participants with chronic plaque psoriasis, participants received 5-, 25-, 100-, or 200-mg subcutaneous tildrakizumab or placebo at weeks 0 and 4 and then every 12 weeks for a total of 52 weeks.6 At week 16, PASI 75 results were 33.3%, 64.4%, 66.3%, 74.4%, and 4.4%, respectively (P<.001 for each comparison). Improvement began within the first month of treatment, with median times to PASI 75 of 57 days at 200-mg dosing and 84 days at 100-mg dosing. Of those participants achieving PASI 75 by drug discontinuation at week 52, 96% of the 100-mg group and 93% of the 200-mg group maintained PASI 75 through week 72, suggesting low relapse rates after treatment cessation.6
In October 2016, the efficacy results of 2 pivotal phase 3 trials (reSURFACE 1 and reSURFACE 2) involving more than 1800 participants combined revealed PASI 90 achievement in an average of 54% of participants on tildrakizumab 100 mg and 59% of participants on tildrakizumab 200 mg at week 28.5 Achievement of PASI 100 occurred in 24% and 30% of participants at week 28, respectively. The second of these trials included an etanercept comparison group and demonstrated head-to-head superiority of 100 and 200 mg subcutaneous tildrakizumab at week 12 by end point measures.5
Treatment-related AEs occurred at rates of 25% in tildrakizumab-treated participants and 22% in placebo-treated participants, most frequently nasopharyngitis and headache.6 At least 1 AE occurred in 64% of tildrakizumab-treated participants without dose dependence compared to 69% of placebo-treated participants. Severe AEs thought to be drug treatment related were bacterial arthritis, lymphedema, melanoma, stroke, and epiglottitis.6
IL-17 Inhibitors
Ixekizumab
Ixekizumab (Eli Lilly and Company), a monoclonal inhibitor of IL-17A, is the most recently approved psoriasis biologic on the market and has been cleared for use in adults with moderate to severe plaque psoriasis. Recommended dosing is 160 mg (given in two 80-mg subcutaneous injections via an autoinjector or prefilled syringe) at week 0, followed by an 80-mg injection at weeks 2, 4, 6, 8, 10, and 12, and then 80 mg every 4 weeks thereafter. The FDA approved ixekizumab in March 2016 following favorable results of several phase 3 trials: UNCOVER-1, UNCOVER-2, and UNCOVER-3.7,8
In UNCOVER-1, 1296 participants were randomized to 1 of 2 ixekizumab treatment arms—160 mg starting dose at week 0, 80 mg every 2 or 4 weeks thereafter—or placebo.7 At week 12, 89.1%, 82.6%, and 3.9% achieved PASI 75, respectively (P<.001 for both). Importantly, high numbers of participants also achieved PASI 90 (70.9% in the 2-week group and 64.6% in the 4-week group vs 0.5% in the placebo group [P<.001]) and PASI 100 (35.3% and 33.6% vs 0%, respectively [P<.001]), suggesting high rates of disease clearance.7
UNCOVER-2 (N=1224) and UNCOVER-3 (N=1346) investigated the same 2 dosing regimens of ixekizumab compared to etanercept 50 mg biweekly and placebo.8 At week 12, the percentage of participants achieving PASI 90 in UNCOVER-2 was 70.7%, 59.7%, 18.7%, and 0.6%, respectively, and 68.1%, 65.3%, 25.7%, and 3.1%, respectively, in UNCOVER-3 (P<.0001 for all comparisons to placebo and etanercept). At week 12, PASI 100 results also showed striking superiority, with 40.5%, 30.8%, 5.3%, and 0.6% of participants, respectively, in UNCOVER-2, and 37.7%, 35%, 7.3%, and 0%, respectively, in UNCOVER-3, achieving complete clearance of disease (P<.0001 for all comparisons to placebo and etanercept). Responses to ixekizumab were observed as early as weeks 1 and 2, while no participants in the etanercept and placebo treatment groups achieved comparative efficiency.8
In an extension of UNCOVER-3, efficacy increased from week 12 to week 60 according to PASI 90 (68%–73% in the 2-week group; 65%–72% in the 4-week group) and PASI 100 measures (38%–55% in the 2-week group; 35%–52% in the 4-week group).7
The most common AEs associated with ixekizumab treatment from weeks 0 to 12 occurred at higher rates in the 2-week and 4-week ixekizumab groups compared to placebo, including nasopharyngitis (9.5% and 9% vs 8.7%, respectively), upper respiratory tract infection (4.4% and 3.9% vs 3.5%, respectively), injection-site reaction (10% and 7.7% vs 1%, respectively), arthralgia (4.4% and 4.3% vs 2.9%, respectively), and headache (2.5% and 1.9% vs 2.1%, respectively). Infections, including candidal, oral, vulvovaginal, and cutaneous, occurred in 27% of the 2-week dosing group and 27.4% of the 4-week dosing group compared to 22.9% of the placebo group during weeks 0 to 12, with candidal infections in particular occurring more frequently in the active treatment groups and exhibiting dose dependence. Other AEs of special interest that occurred among all ixekizumab-treated participants (n=3736) from weeks 0 to 60 were cardiovascular and cerebrovascular events (22 [0.6%]), inflammatory bowel disease (11 [0.3%]), non–skin cancer malignancy (14 [0.4%]), and nonmelanoma skin cancer (20 [0.5%]). Neutropenia occurred at higher rates in ixekizumab-treated participants (9.3% in the 2-week group and 8.6% in the 4-week group) compared to placebo (3.3%) and occurred in 11.5% of all ixekizumab participants over 60 weeks.7
Brodalumab
Brodalumab (Valeant Pharmaceuticals International, Inc) is a human monoclonal antibody targeting the IL-17A receptor currently under review for FDA approval after undergoing phase 3 trials. The first of these trials, AMAGINE-1, showed efficacy of subcutaneous brodalumab (140 or 210 mg administered every 2 weeks with an extra dose at week 1) compared to placebo in 661 participants.9 At week 12, 60%, 83%, and 3%, respectively, achieved PASI 75; 43%, 70%, and 1%, respectively, achieved PASI 90; and 23%, 42%, and 1%, respectively, achieved PASI 100 (P<.001 for all respective comparisons to placebo). These effects were retained through 52 weeks of treatment. The median time to complete disease clearance in participants reaching PASI 100 was 12 weeks. Conversely, participants who were re-randomized to placebo after week 12 of brodalumab treatment relapsed within weeks to months.9
AMAGINE-2 and AMAGINE-3 further demonstrated the efficacy of brodalumab (140 or 210 mg every 2 weeks with extra dose at week 1) compared to ustekinumab (45 or 90 mg weight-based standard dosing) and placebo in 1831 participants, respectively.10 In AMAGINE-2, 49% of participants in the 140-mg group (P<.001 vs placebo), 70% in the 210-mg group (P<.001 vs placebo), 47% in the ustekinumab group, and 3% in the placebo group achieved PASI 90 at week 12. Similarly, in AMAGINE-3, 52% of participants in the 140-mg group (P<.001), 69% in the 210-mg group (P<.001), 48% in the ustekinumab group, and 2% in the placebo group achieved PASI 90. Impressively, complete clearance (PASI 100) at week 12 occurred in 26% of the 140-mg group (P<.001 vs placebo), 44% of the 210-mg group (P<.001 vs placebo), and 22% of the ustekinumab group compared to 2% of the placebo group in AMAGINE-2, with similar rates in AMAGINE-3. Brodalumab was significantly superior to ustekinumab at the 210-mg dose by PASI 90 measures (P<.001) in both studies and at the 140-mg dose by PASI 100 measures (P=.007) in AMAGINE-3 only.10
Common AEs were nasopharyngitis, upper respiratory tract infection, headache, and arthralgia, all occurring at grossly similar rates (49%–60%) across all experimental groups in AMAGINE-1, AMAGINE-2, and AMAGINE-3 during the first 12-week treatment period.9,10 Brodalumab treatment groups had high rates of specific interest AEs compared to ustekinumab and placebo groups, including neutropenia (0.8%, 1.1%, 0.3%, and 0%, respectively) and candidal infections (0.8%, 1.3%, 0.3%, and 0.3%, respectively). Induction phase (weeks 0–12) depression rates were concerning, with 6 cases each in AMAGINE-2 (4 [0.7%] in the 140-mg group, 2 [0.3%] in the 210-mg group) and AMAGINE-3 (4 [0.6%] in the 140-mg group, 2 [0.3%] in the 210-mg group). Cases of neutropenia were mild, were not associated with major infection, and were transient or reversible. Depression rates after 52 weeks of treatment were 1.7% (23/1567) of brodalumab participants in AMAGINE-2 and 1.8% (21/1613) in AMAGINE-3. Three participants, all on constant 210-mg dosing through week 52, attempted suicide with 1 completion10; however, because no other IL-17 inhibitors were associated with depression or suicide in other trials, it has been suggested that these cases were incidental and not treatment related.12 An FDA advisory panel recommended approval of brodalumab in July 2016 despite ongoing concerns of depression and suicide.13
Conclusion
The robust investigation into IL-23 and IL-17 inhibitors to treat plaque psoriasis has yielded promising results, including the unprecedented rates of PASI 100 achievement with these new biologics. Risankizumab, ixekizumab, and brodalumab have demonstrated superior efficacy in trials compared to ustekinumab. Tildrakizumab has shown low disease relapse after drug cessation. Ixekizumab and brodalumab have shown high rates of total disease clearance. Thus far, safety findings for these pipeline biologics have been consistent with those of ustekinumab. With ixekizumab approved in 2016 and brodalumab under review, new options in biologic therapy will offer patients and clinicians greater choices in treating severe and recalcitrant psoriasis.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509.
- Papp K, Menter A, Sofen H, et al. Efficacy and safety of different dose regimens of a selective IL-23p19 inhibitor (BI 655066) compared with ustekinumab in patients with moderate-to-severe plaque psoriasis with and without psoriatic arthritis. Paper presented at: 2015 American College of Rheumatology/Association of Rheumatology Health Professionals Annual Meeting; November 6-11, 2015; San Francisco, CA.
- New phase 3 data show significant efficacy versus placebo and superiority of guselkumab versus Humira in treatment of moderate to severe plaque psoriasis [press release]. Vienna, Austria; Janssen Research & Development, LLC: October 1, 2016.
- Gordon KB, Duffin KC, Bissonnette R, et al. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N Engl J Med. 2015;373:136-144.
- Sun Pharma to announce late-breaking results for investigational IL-23p19 inhibitor, Tildrakizumab, achieves primary end point in both phase-3 studies in patients with moderate-to-severe plaque psoriasis [press release]. Mumbai, India; Sun Pharmaceutical Industries Ltd: October 1, 2016.
- Papp K, Thaci D, Reich K, et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol. 2015;173:930-939.
- Gordon KB, Blauvelt A, Papp KA, et al; UNCOVER-1 Study Group, UNCOVER-2 Study Group, UNCOVER-3 Study Group. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis [published online June 23, 2016]. Br J Dermatol. 2016;175:273-286.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial [published online March 1, 2015]. J Allergy Clin Immunol. 2015;136:116-124.e7.
- Chiricozzi A, Romanelli M, Saraceno R, et al. No meaningful association between suicidal behavior and the use of IL-17A-neutralizing or IL-17RA-blocking agents [published online August 31, 2016]. Expert Opin Drug Saf. 2016;15:1653-1659.
- FDA advisory committee recommends approval of brodalumab for treatment of moderate-to-severe plaque psoriasis [news release]. Laval, Quebec: Valeant Pharmaceuticals International, Inc; July 19, 2016.
- Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med. 2009;361:496-509.
- Papp K, Menter A, Sofen H, et al. Efficacy and safety of different dose regimens of a selective IL-23p19 inhibitor (BI 655066) compared with ustekinumab in patients with moderate-to-severe plaque psoriasis with and without psoriatic arthritis. Paper presented at: 2015 American College of Rheumatology/Association of Rheumatology Health Professionals Annual Meeting; November 6-11, 2015; San Francisco, CA.
- New phase 3 data show significant efficacy versus placebo and superiority of guselkumab versus Humira in treatment of moderate to severe plaque psoriasis [press release]. Vienna, Austria; Janssen Research & Development, LLC: October 1, 2016.
- Gordon KB, Duffin KC, Bissonnette R, et al. A phase 2 trial of guselkumab versus adalimumab for plaque psoriasis. N Engl J Med. 2015;373:136-144.
- Sun Pharma to announce late-breaking results for investigational IL-23p19 inhibitor, Tildrakizumab, achieves primary end point in both phase-3 studies in patients with moderate-to-severe plaque psoriasis [press release]. Mumbai, India; Sun Pharmaceutical Industries Ltd: October 1, 2016.
- Papp K, Thaci D, Reich K, et al. Tildrakizumab (MK-3222), an anti-interleukin-23p19 monoclonal antibody, improves psoriasis in a phase IIb randomized placebo-controlled trial. Br J Dermatol. 2015;173:930-939.
- Gordon KB, Blauvelt A, Papp KA, et al; UNCOVER-1 Study Group, UNCOVER-2 Study Group, UNCOVER-3 Study Group. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375:345-356.
- Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet. 2015;386:541-551.
- Papp KA, Reich K, Paul C, et al. A prospective phase III, randomized, double-blind, placebo-controlled study of brodalumab in patients with moderate-to-severe plaque psoriasis [published online June 23, 2016]. Br J Dermatol. 2016;175:273-286.
- Lebwohl M, Strober B, Menter A, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318-1328.
- Krueger JG, Ferris LK, Menter A, et al. Anti-IL-23A mAb BI 655066 for treatment of moderate-to-severe psoriasis: safety, efficacy, pharmacokinetics, and biomarker results of a single-rising-dose, randomized, double-blind, placebo-controlled trial [published online March 1, 2015]. J Allergy Clin Immunol. 2015;136:116-124.e7.
- Chiricozzi A, Romanelli M, Saraceno R, et al. No meaningful association between suicidal behavior and the use of IL-17A-neutralizing or IL-17RA-blocking agents [published online August 31, 2016]. Expert Opin Drug Saf. 2016;15:1653-1659.
- FDA advisory committee recommends approval of brodalumab for treatment of moderate-to-severe plaque psoriasis [news release]. Laval, Quebec: Valeant Pharmaceuticals International, Inc; July 19, 2016.
Practice Points
- The newest biologics for treatment of moderate to severe plaque psoriasis are IL-23 and IL-17 inhibitors with unprecedented efficacy of complete skin clearance compared to older biologics.
- Risankizumab, guselkumab, and tildrakizumab are new IL-23 inhibitors currently in phase 3 trials with promising early efficacy and safety results.
- Ixekizumab, which recently was approved, and brodalumab, which is pending US Food and Drug Administration review, are new IL-17 inhibitors that achieved total skin clearance in more than one-quarter of phase 3 participants after 12 weeks of treatment.

VIDEO: Pediatric psoriasis patients prepare for biologics
WAILEA, Hawaii – The first approval of a biologic for pediatric psoriasis, and ongoing clinical trials of other biologics in children with psoriasis, are among the encouraging therapeutic developments for this patient population, Wynnis Tom, MD, said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
“We are incredibly excited ... as pediatric dermatologists that we’re finally seeing breakthroughs” in terms of Food and Drug Administration activity regarding the use of biologics for treating psoriasis in children, Dr. Tom said in a video interview at the seminar.
Etanercept (Enbrel) is now approved for children with psoriasis, the first biologic indicated for pediatric psoriasis, and clinical trials of other biologics that have been available for adults and nonbiologic products for pediatric psoriasis are underway, she said.
However, getting insurance coverage can still be a challenge, although having long-term efficacy and safety data helps, noted Dr. Tom of the department of dermatology and pediatrics, University of California, San Diego, and Rady Children’s Hospital, San Diego. Also helpful is sending letters to insurers on behalf of the patient, describing the patient’s quality of life, descriptions of treatments that have been unsuccessful, and even photos documenting the disease in the child, she added.
Dr. Tom disclosed ties with Promius, Celgene, and Janssen.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, Hawaii – The first approval of a biologic for pediatric psoriasis, and ongoing clinical trials of other biologics in children with psoriasis, are among the encouraging therapeutic developments for this patient population, Wynnis Tom, MD, said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
“We are incredibly excited ... as pediatric dermatologists that we’re finally seeing breakthroughs” in terms of Food and Drug Administration activity regarding the use of biologics for treating psoriasis in children, Dr. Tom said in a video interview at the seminar.
Etanercept (Enbrel) is now approved for children with psoriasis, the first biologic indicated for pediatric psoriasis, and clinical trials of other biologics that have been available for adults and nonbiologic products for pediatric psoriasis are underway, she said.
However, getting insurance coverage can still be a challenge, although having long-term efficacy and safety data helps, noted Dr. Tom of the department of dermatology and pediatrics, University of California, San Diego, and Rady Children’s Hospital, San Diego. Also helpful is sending letters to insurers on behalf of the patient, describing the patient’s quality of life, descriptions of treatments that have been unsuccessful, and even photos documenting the disease in the child, she added.
Dr. Tom disclosed ties with Promius, Celgene, and Janssen.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, Hawaii – The first approval of a biologic for pediatric psoriasis, and ongoing clinical trials of other biologics in children with psoriasis, are among the encouraging therapeutic developments for this patient population, Wynnis Tom, MD, said at the Hawaii Dermatology Seminar provided by Global Academy for Medical Education/Skin Disease Education Foundation.
“We are incredibly excited ... as pediatric dermatologists that we’re finally seeing breakthroughs” in terms of Food and Drug Administration activity regarding the use of biologics for treating psoriasis in children, Dr. Tom said in a video interview at the seminar.
Etanercept (Enbrel) is now approved for children with psoriasis, the first biologic indicated for pediatric psoriasis, and clinical trials of other biologics that have been available for adults and nonbiologic products for pediatric psoriasis are underway, she said.
However, getting insurance coverage can still be a challenge, although having long-term efficacy and safety data helps, noted Dr. Tom of the department of dermatology and pediatrics, University of California, San Diego, and Rady Children’s Hospital, San Diego. Also helpful is sending letters to insurers on behalf of the patient, describing the patient’s quality of life, descriptions of treatments that have been unsuccessful, and even photos documenting the disease in the child, she added.
Dr. Tom disclosed ties with Promius, Celgene, and Janssen.
SDEF and this news organization are owned by the same parent company.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT SDEF HAWAII DERMATOLOGY SEMINAR

The Role of Biologic Therapy for Psoriasis in Cardiovascular Risk Reduction
The cardiovascular comorbidities associated with psoriasis have been well documented; however, the mechanism by which psoriasis increases the risk for cardiovascular disease (CVD) remains unclear. Elevated systemic inflammatory cytokines and mediators may play a key role in their association, which prompts the questions: Do systemic medications have a protective effect? Do patients on systemic antipsoriatic treatment have a decreased risk for major adverse cardiovascular events (MACEs) compared with untreated patients?
We believe the shared inflammatory processes involved in psoriasis and atherosclerosis formation are potential targets for therapy in reducing the incidence of CVD and its associated complications. A growing amount of evidence suggests cardioprotective effects associated with antipsoriatic treatments such as tumor necrosis factor (TNF) inhibitors and methotrexate. Gkalpakiotis et al1 demonstrated a reduction in serum E-selectin (mean [standard deviation], 53.04 [23.54] ng/mL vs 35.32 [8.70] ng/mL; P<.001) and IL-22 (25.11 [19.9] pg/mL vs 12.83 [8.42] pg/mL; P<.001) after 3 months of adalimumab administration in patients with moderate to severe psoriasis. Both E-selectin and IL-22 are associated with the development of atherosclerosis, endothelial dysfunction, and an increased incidence of CVD. Similarly, Wu et al2 demonstrated a statistically significant reduction (–5.04 mg/dL [95% confidence interval [CI], –8.24 to –2.12; P<.01) in C-reactive protein in patients with psoriasis, psoriatic arthritis, and rheumatoid arthritis after concurrent use of methotrexate and TNF inhibitors.
Solomon et al3 compared the rate of newly diagnosed diabetes mellitus among psoriasis and rheumatoid arthritis patients treated with TNF inhibitors, methotrexate, hydroxychloroquine, and other nonbiologic disease-modifying antirheumatic drugs. The authors’ findings suggest that those who take a TNF inhibitor (hazard ratio [HR], 0.62; 95% CI, 0.42-0.91) and hydroxychloroquine (HR, 0.54; 95% CI, 0.36-0.80) are at lower risk for diabetes mellitus compared to those treated with nonbiologic disease-modifying antirheumatic drugs. Conversely, the methotrexate (HR, 0.77; 95% CI, 0.53-1.13) cohort did not show a statistically significant reduction in diabetes risk.3
Pina et al4 revealed improvement in endothelial function after 6 months of adalimumab use in patients with moderate to severe psoriasis. To evaluate the presence of subclinical endothelial dysfunction, the authors assessed brachial artery reactivity by measuring flow-mediated dilation and carotid artery stiffness by pulse wave velocity. Patients showed an increase in flow-mediated dilation (mean [SD], 6.19% [2.44%] vs 7.46% [2.43%]; P=.008) and reduction in pulse wave velocity (6.28 [1.04] m/s vs 5.69 [1.31] m/s; P=.03) compared to baseline measurements, indicating an improvement of endothelial function.4
Ahlehoff et al5 observed for improvements in subclinical left ventricular dysfunction in psoriasis patients after treatment with biologics. Using echocardiography, they assessed for changes in diastolic function and left ventricular systolic deformation (defined by global longitudinal strain). Of patients who received 3 months of biologic therapy (TNF inhibitor orIL-12/23 inhibitor) and maintained at minimum a psoriasis area and severity index 50 response, all demonstrated an improvement in diastolic function (mean [SD], 8.1 [2.1] vs 6.7 [1.9]; P<.001) and global longitudinal strain (mean [SD], –16.8% [2.1%] vs –18.3% [2.3%]; P<.001). Of note, patients who did not achieve a psoriasis area and severity index 50 response at follow-up did not exhibit an improvement in subclinical myocardial function.5
Moreover, a Danish nationwide study with up to 5-year follow-up evaluated the risk for MACE (ie, cardiovascular death, myocardial infarction, stroke) in patients with severe psoriasis receiving systemic anti-inflammatory medications and nonsystemic therapies including topical treatments, phototherapy, and climate therapy.6 Compared to nonsystemic therapies, methotrexate use (HR, 0.53; 95% CI, 0.34-0.83) was associated with a decreased risk for cardiovascular events. However, a protective decreased risk was not found among patients who used systemic cyclosporine (HR, 1.06; 95% CI, 0.26-4.27) or retinoids (HR, 1.80; 95% CI, 1.03-2.96). Any biological drug use had a comparable but nonsignificant reduction of cardiovascular events (HR, 0.58; 95% CI, 0.30-1.10). After multivariable adjustment, TNF inhibitors were associated with a statistically significant decreased risk for cardiovascular events (HR, 0.46; 95% CI, 0.22-0.98; P=.04) compared to nonsystemic therapies. The IL-12/23 inhibitor did not demonstrate this relationship (HR, 1.52; 95% CI, 0.47-4.94).6
Lastly, Wu et al7 compared the risk for MACE (ie, myocardial infarction, stroke, unstable angina, transient ischemic attack) between patients with psoriasis who received TNF inhibitors or methotrexate. The TNF inhibitor and methotrexate cohorts were observed for a median of 12 months and 9 months, respectively. After adjusting for potential confounding factors, they found a 45% reduction (HR, 0.55; 95% CI, 0.45-0.67) in cardiovascular event risk in the TNF inhibitor cohort compared with the methotrexate cohort. Notably, analyses also showed comparatively fewer cardiovascular events in the TNF inhibitor cohort throughout all time points—6, 12, 18, 24, 60 months—in the observation period. Regression analysis revealed an 11% reduction in cardiovascular events (HR, 0.89; 95% CI, 0.80-0.98) with each additional 6 months of cumulative TNF inhibitor exposure.
The current sum of evidence suggests cardioprotective effects of TNF inhibitor and methotrexate use. However, given the cumulative systemic toxicity and inferior cutaneous efficacy of methotrexate, TNF inhibitors will likely play a more significant role going forward. The role of methotrexate may be for its simultaneous use with biologic therapies to limit immunogenicity. Newer biologic agents such as IL-12/23 and IL-17 inhibitors have not yet been as extensively studied for their effects on cardiovascular risk as their TNF inhibitor counterparts. However, because of their shared ability to target specific immunological pathways, it is plausible that IL-12/23 and IL-17 agents may exhibit cardioprotective effects.8
Patients with psoriasis should be counseled and educated about the increased risk for CVD and its associated morbidity and mortality risk. Screening for modifiable risk factors and recommending therapeutic lifestyle changes also is appropriate. Future studies should help define the role of specific systemic drugs in reducing the risk for CVD in patients with psoriasis.
- Gkalpakiotis S, Arenbergerova M, Gkalpakioti P, et al. Impact of adalimumab treatment on cardiovascular risk biomarkers in psoriasis: results of a pilot study [published online October 24, 2016]. J Dermatol. doi:10.1111/1346-8138.13661.
- Wu JJ, Rowan CG, Bebchuk JD, et al. Association between tumor necrosis factor inhibitor (TNFi) therapy and changes in C-reactive protein (CRP), blood pressure, and alanine aminotransferase (ALT) among patients with psoriasis, psoriatic arthritis, or rheumatoid arthritis [published online March 5, 2015]. J Am Acad Dermatol. 2015;72:917-919.
- Solomon DH, Massarotti E, Garg R, et al. Association between disease-modifying antirheumatic drugs and diabetes risk in patients with rheumatoid arthritis and psoriasis. JAMA. 2011;305:2525-2531.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-alpha therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6-month prospective study. J Dermatol. 2016;43:1267-1272.
- Ahlehoff O, Hansen PR, Gislason GH, et al. Myocardial function and effects of biologic therapy in patients with severe psoriasis: a prospective echocardiographic study [published online April 6, 2015]. J Eur Acad Dermatol Venereol. 2016;30:819-823.
- Ahlehoff O, Skov L, Gislason G, et al. Cardiovascular outcomes and systemic anti-inflammatory drugs in patients with severe psoriasis: 5-year follow-up of a Danish nationwide cohort [published online October 10, 2014]. J Eur Acad Dermatol Venereol. 2015;29:1128-1134.
- Wu JJ, Guérin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-α inhibitors versus methotrexate [published online October 26, 2016]. J Am Acad Dermatol. 2017;76:81-90.
- Egeberg A, Skov L. Management of cardiovascular disease in patients with psoriasis. Expert Opin Pharmacother. 2016;17:1509-1516.
The cardiovascular comorbidities associated with psoriasis have been well documented; however, the mechanism by which psoriasis increases the risk for cardiovascular disease (CVD) remains unclear. Elevated systemic inflammatory cytokines and mediators may play a key role in their association, which prompts the questions: Do systemic medications have a protective effect? Do patients on systemic antipsoriatic treatment have a decreased risk for major adverse cardiovascular events (MACEs) compared with untreated patients?
We believe the shared inflammatory processes involved in psoriasis and atherosclerosis formation are potential targets for therapy in reducing the incidence of CVD and its associated complications. A growing amount of evidence suggests cardioprotective effects associated with antipsoriatic treatments such as tumor necrosis factor (TNF) inhibitors and methotrexate. Gkalpakiotis et al1 demonstrated a reduction in serum E-selectin (mean [standard deviation], 53.04 [23.54] ng/mL vs 35.32 [8.70] ng/mL; P<.001) and IL-22 (25.11 [19.9] pg/mL vs 12.83 [8.42] pg/mL; P<.001) after 3 months of adalimumab administration in patients with moderate to severe psoriasis. Both E-selectin and IL-22 are associated with the development of atherosclerosis, endothelial dysfunction, and an increased incidence of CVD. Similarly, Wu et al2 demonstrated a statistically significant reduction (–5.04 mg/dL [95% confidence interval [CI], –8.24 to –2.12; P<.01) in C-reactive protein in patients with psoriasis, psoriatic arthritis, and rheumatoid arthritis after concurrent use of methotrexate and TNF inhibitors.
Solomon et al3 compared the rate of newly diagnosed diabetes mellitus among psoriasis and rheumatoid arthritis patients treated with TNF inhibitors, methotrexate, hydroxychloroquine, and other nonbiologic disease-modifying antirheumatic drugs. The authors’ findings suggest that those who take a TNF inhibitor (hazard ratio [HR], 0.62; 95% CI, 0.42-0.91) and hydroxychloroquine (HR, 0.54; 95% CI, 0.36-0.80) are at lower risk for diabetes mellitus compared to those treated with nonbiologic disease-modifying antirheumatic drugs. Conversely, the methotrexate (HR, 0.77; 95% CI, 0.53-1.13) cohort did not show a statistically significant reduction in diabetes risk.3
Pina et al4 revealed improvement in endothelial function after 6 months of adalimumab use in patients with moderate to severe psoriasis. To evaluate the presence of subclinical endothelial dysfunction, the authors assessed brachial artery reactivity by measuring flow-mediated dilation and carotid artery stiffness by pulse wave velocity. Patients showed an increase in flow-mediated dilation (mean [SD], 6.19% [2.44%] vs 7.46% [2.43%]; P=.008) and reduction in pulse wave velocity (6.28 [1.04] m/s vs 5.69 [1.31] m/s; P=.03) compared to baseline measurements, indicating an improvement of endothelial function.4
Ahlehoff et al5 observed for improvements in subclinical left ventricular dysfunction in psoriasis patients after treatment with biologics. Using echocardiography, they assessed for changes in diastolic function and left ventricular systolic deformation (defined by global longitudinal strain). Of patients who received 3 months of biologic therapy (TNF inhibitor orIL-12/23 inhibitor) and maintained at minimum a psoriasis area and severity index 50 response, all demonstrated an improvement in diastolic function (mean [SD], 8.1 [2.1] vs 6.7 [1.9]; P<.001) and global longitudinal strain (mean [SD], –16.8% [2.1%] vs –18.3% [2.3%]; P<.001). Of note, patients who did not achieve a psoriasis area and severity index 50 response at follow-up did not exhibit an improvement in subclinical myocardial function.5
Moreover, a Danish nationwide study with up to 5-year follow-up evaluated the risk for MACE (ie, cardiovascular death, myocardial infarction, stroke) in patients with severe psoriasis receiving systemic anti-inflammatory medications and nonsystemic therapies including topical treatments, phototherapy, and climate therapy.6 Compared to nonsystemic therapies, methotrexate use (HR, 0.53; 95% CI, 0.34-0.83) was associated with a decreased risk for cardiovascular events. However, a protective decreased risk was not found among patients who used systemic cyclosporine (HR, 1.06; 95% CI, 0.26-4.27) or retinoids (HR, 1.80; 95% CI, 1.03-2.96). Any biological drug use had a comparable but nonsignificant reduction of cardiovascular events (HR, 0.58; 95% CI, 0.30-1.10). After multivariable adjustment, TNF inhibitors were associated with a statistically significant decreased risk for cardiovascular events (HR, 0.46; 95% CI, 0.22-0.98; P=.04) compared to nonsystemic therapies. The IL-12/23 inhibitor did not demonstrate this relationship (HR, 1.52; 95% CI, 0.47-4.94).6
Lastly, Wu et al7 compared the risk for MACE (ie, myocardial infarction, stroke, unstable angina, transient ischemic attack) between patients with psoriasis who received TNF inhibitors or methotrexate. The TNF inhibitor and methotrexate cohorts were observed for a median of 12 months and 9 months, respectively. After adjusting for potential confounding factors, they found a 45% reduction (HR, 0.55; 95% CI, 0.45-0.67) in cardiovascular event risk in the TNF inhibitor cohort compared with the methotrexate cohort. Notably, analyses also showed comparatively fewer cardiovascular events in the TNF inhibitor cohort throughout all time points—6, 12, 18, 24, 60 months—in the observation period. Regression analysis revealed an 11% reduction in cardiovascular events (HR, 0.89; 95% CI, 0.80-0.98) with each additional 6 months of cumulative TNF inhibitor exposure.
The current sum of evidence suggests cardioprotective effects of TNF inhibitor and methotrexate use. However, given the cumulative systemic toxicity and inferior cutaneous efficacy of methotrexate, TNF inhibitors will likely play a more significant role going forward. The role of methotrexate may be for its simultaneous use with biologic therapies to limit immunogenicity. Newer biologic agents such as IL-12/23 and IL-17 inhibitors have not yet been as extensively studied for their effects on cardiovascular risk as their TNF inhibitor counterparts. However, because of their shared ability to target specific immunological pathways, it is plausible that IL-12/23 and IL-17 agents may exhibit cardioprotective effects.8
Patients with psoriasis should be counseled and educated about the increased risk for CVD and its associated morbidity and mortality risk. Screening for modifiable risk factors and recommending therapeutic lifestyle changes also is appropriate. Future studies should help define the role of specific systemic drugs in reducing the risk for CVD in patients with psoriasis.
The cardiovascular comorbidities associated with psoriasis have been well documented; however, the mechanism by which psoriasis increases the risk for cardiovascular disease (CVD) remains unclear. Elevated systemic inflammatory cytokines and mediators may play a key role in their association, which prompts the questions: Do systemic medications have a protective effect? Do patients on systemic antipsoriatic treatment have a decreased risk for major adverse cardiovascular events (MACEs) compared with untreated patients?
We believe the shared inflammatory processes involved in psoriasis and atherosclerosis formation are potential targets for therapy in reducing the incidence of CVD and its associated complications. A growing amount of evidence suggests cardioprotective effects associated with antipsoriatic treatments such as tumor necrosis factor (TNF) inhibitors and methotrexate. Gkalpakiotis et al1 demonstrated a reduction in serum E-selectin (mean [standard deviation], 53.04 [23.54] ng/mL vs 35.32 [8.70] ng/mL; P<.001) and IL-22 (25.11 [19.9] pg/mL vs 12.83 [8.42] pg/mL; P<.001) after 3 months of adalimumab administration in patients with moderate to severe psoriasis. Both E-selectin and IL-22 are associated with the development of atherosclerosis, endothelial dysfunction, and an increased incidence of CVD. Similarly, Wu et al2 demonstrated a statistically significant reduction (–5.04 mg/dL [95% confidence interval [CI], –8.24 to –2.12; P<.01) in C-reactive protein in patients with psoriasis, psoriatic arthritis, and rheumatoid arthritis after concurrent use of methotrexate and TNF inhibitors.
Solomon et al3 compared the rate of newly diagnosed diabetes mellitus among psoriasis and rheumatoid arthritis patients treated with TNF inhibitors, methotrexate, hydroxychloroquine, and other nonbiologic disease-modifying antirheumatic drugs. The authors’ findings suggest that those who take a TNF inhibitor (hazard ratio [HR], 0.62; 95% CI, 0.42-0.91) and hydroxychloroquine (HR, 0.54; 95% CI, 0.36-0.80) are at lower risk for diabetes mellitus compared to those treated with nonbiologic disease-modifying antirheumatic drugs. Conversely, the methotrexate (HR, 0.77; 95% CI, 0.53-1.13) cohort did not show a statistically significant reduction in diabetes risk.3
Pina et al4 revealed improvement in endothelial function after 6 months of adalimumab use in patients with moderate to severe psoriasis. To evaluate the presence of subclinical endothelial dysfunction, the authors assessed brachial artery reactivity by measuring flow-mediated dilation and carotid artery stiffness by pulse wave velocity. Patients showed an increase in flow-mediated dilation (mean [SD], 6.19% [2.44%] vs 7.46% [2.43%]; P=.008) and reduction in pulse wave velocity (6.28 [1.04] m/s vs 5.69 [1.31] m/s; P=.03) compared to baseline measurements, indicating an improvement of endothelial function.4
Ahlehoff et al5 observed for improvements in subclinical left ventricular dysfunction in psoriasis patients after treatment with biologics. Using echocardiography, they assessed for changes in diastolic function and left ventricular systolic deformation (defined by global longitudinal strain). Of patients who received 3 months of biologic therapy (TNF inhibitor orIL-12/23 inhibitor) and maintained at minimum a psoriasis area and severity index 50 response, all demonstrated an improvement in diastolic function (mean [SD], 8.1 [2.1] vs 6.7 [1.9]; P<.001) and global longitudinal strain (mean [SD], –16.8% [2.1%] vs –18.3% [2.3%]; P<.001). Of note, patients who did not achieve a psoriasis area and severity index 50 response at follow-up did not exhibit an improvement in subclinical myocardial function.5
Moreover, a Danish nationwide study with up to 5-year follow-up evaluated the risk for MACE (ie, cardiovascular death, myocardial infarction, stroke) in patients with severe psoriasis receiving systemic anti-inflammatory medications and nonsystemic therapies including topical treatments, phototherapy, and climate therapy.6 Compared to nonsystemic therapies, methotrexate use (HR, 0.53; 95% CI, 0.34-0.83) was associated with a decreased risk for cardiovascular events. However, a protective decreased risk was not found among patients who used systemic cyclosporine (HR, 1.06; 95% CI, 0.26-4.27) or retinoids (HR, 1.80; 95% CI, 1.03-2.96). Any biological drug use had a comparable but nonsignificant reduction of cardiovascular events (HR, 0.58; 95% CI, 0.30-1.10). After multivariable adjustment, TNF inhibitors were associated with a statistically significant decreased risk for cardiovascular events (HR, 0.46; 95% CI, 0.22-0.98; P=.04) compared to nonsystemic therapies. The IL-12/23 inhibitor did not demonstrate this relationship (HR, 1.52; 95% CI, 0.47-4.94).6
Lastly, Wu et al7 compared the risk for MACE (ie, myocardial infarction, stroke, unstable angina, transient ischemic attack) between patients with psoriasis who received TNF inhibitors or methotrexate. The TNF inhibitor and methotrexate cohorts were observed for a median of 12 months and 9 months, respectively. After adjusting for potential confounding factors, they found a 45% reduction (HR, 0.55; 95% CI, 0.45-0.67) in cardiovascular event risk in the TNF inhibitor cohort compared with the methotrexate cohort. Notably, analyses also showed comparatively fewer cardiovascular events in the TNF inhibitor cohort throughout all time points—6, 12, 18, 24, 60 months—in the observation period. Regression analysis revealed an 11% reduction in cardiovascular events (HR, 0.89; 95% CI, 0.80-0.98) with each additional 6 months of cumulative TNF inhibitor exposure.
The current sum of evidence suggests cardioprotective effects of TNF inhibitor and methotrexate use. However, given the cumulative systemic toxicity and inferior cutaneous efficacy of methotrexate, TNF inhibitors will likely play a more significant role going forward. The role of methotrexate may be for its simultaneous use with biologic therapies to limit immunogenicity. Newer biologic agents such as IL-12/23 and IL-17 inhibitors have not yet been as extensively studied for their effects on cardiovascular risk as their TNF inhibitor counterparts. However, because of their shared ability to target specific immunological pathways, it is plausible that IL-12/23 and IL-17 agents may exhibit cardioprotective effects.8
Patients with psoriasis should be counseled and educated about the increased risk for CVD and its associated morbidity and mortality risk. Screening for modifiable risk factors and recommending therapeutic lifestyle changes also is appropriate. Future studies should help define the role of specific systemic drugs in reducing the risk for CVD in patients with psoriasis.
- Gkalpakiotis S, Arenbergerova M, Gkalpakioti P, et al. Impact of adalimumab treatment on cardiovascular risk biomarkers in psoriasis: results of a pilot study [published online October 24, 2016]. J Dermatol. doi:10.1111/1346-8138.13661.
- Wu JJ, Rowan CG, Bebchuk JD, et al. Association between tumor necrosis factor inhibitor (TNFi) therapy and changes in C-reactive protein (CRP), blood pressure, and alanine aminotransferase (ALT) among patients with psoriasis, psoriatic arthritis, or rheumatoid arthritis [published online March 5, 2015]. J Am Acad Dermatol. 2015;72:917-919.
- Solomon DH, Massarotti E, Garg R, et al. Association between disease-modifying antirheumatic drugs and diabetes risk in patients with rheumatoid arthritis and psoriasis. JAMA. 2011;305:2525-2531.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-alpha therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6-month prospective study. J Dermatol. 2016;43:1267-1272.
- Ahlehoff O, Hansen PR, Gislason GH, et al. Myocardial function and effects of biologic therapy in patients with severe psoriasis: a prospective echocardiographic study [published online April 6, 2015]. J Eur Acad Dermatol Venereol. 2016;30:819-823.
- Ahlehoff O, Skov L, Gislason G, et al. Cardiovascular outcomes and systemic anti-inflammatory drugs in patients with severe psoriasis: 5-year follow-up of a Danish nationwide cohort [published online October 10, 2014]. J Eur Acad Dermatol Venereol. 2015;29:1128-1134.
- Wu JJ, Guérin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-α inhibitors versus methotrexate [published online October 26, 2016]. J Am Acad Dermatol. 2017;76:81-90.
- Egeberg A, Skov L. Management of cardiovascular disease in patients with psoriasis. Expert Opin Pharmacother. 2016;17:1509-1516.
- Gkalpakiotis S, Arenbergerova M, Gkalpakioti P, et al. Impact of adalimumab treatment on cardiovascular risk biomarkers in psoriasis: results of a pilot study [published online October 24, 2016]. J Dermatol. doi:10.1111/1346-8138.13661.
- Wu JJ, Rowan CG, Bebchuk JD, et al. Association between tumor necrosis factor inhibitor (TNFi) therapy and changes in C-reactive protein (CRP), blood pressure, and alanine aminotransferase (ALT) among patients with psoriasis, psoriatic arthritis, or rheumatoid arthritis [published online March 5, 2015]. J Am Acad Dermatol. 2015;72:917-919.
- Solomon DH, Massarotti E, Garg R, et al. Association between disease-modifying antirheumatic drugs and diabetes risk in patients with rheumatoid arthritis and psoriasis. JAMA. 2011;305:2525-2531.
- Pina T, Corrales A, Lopez-Mejias R, et al. Anti-tumor necrosis factor-alpha therapy improves endothelial function and arterial stiffness in patients with moderate to severe psoriasis: a 6-month prospective study. J Dermatol. 2016;43:1267-1272.
- Ahlehoff O, Hansen PR, Gislason GH, et al. Myocardial function and effects of biologic therapy in patients with severe psoriasis: a prospective echocardiographic study [published online April 6, 2015]. J Eur Acad Dermatol Venereol. 2016;30:819-823.
- Ahlehoff O, Skov L, Gislason G, et al. Cardiovascular outcomes and systemic anti-inflammatory drugs in patients with severe psoriasis: 5-year follow-up of a Danish nationwide cohort [published online October 10, 2014]. J Eur Acad Dermatol Venereol. 2015;29:1128-1134.
- Wu JJ, Guérin A, Sundaram M, et al. Cardiovascular event risk assessment in psoriasis patients treated with tumor necrosis factor-α inhibitors versus methotrexate [published online October 26, 2016]. J Am Acad Dermatol. 2017;76:81-90.
- Egeberg A, Skov L. Management of cardiovascular disease in patients with psoriasis. Expert Opin Pharmacother. 2016;17:1509-1516.

VIDEO: Adding methotrexate to a biologic may help achieve treatment goal
WAILEA, Hawaii – Combining methotrexate with a biologic is an off-label use for psoriasis patients but is supported by information from the psoriatic arthritis literature, said J. Mark Jackson, MD, of the University of Louisville (Ky.).
In fact, the combination treatment may improve symptoms in patients with both psoriasis and arthritis not well controlled with one drug, Dr. Jackson explained in a video interview at the meeting, provided by Global Academy for Medical Education/Skin Disease Education Foundation.
As for dosage, “I do think we can use less methotrexate in combination” with a biologic for psoriasis when using methotrexate alone, he noted. “It’s like the addition of a spice to the right dish,” he added. Patients may need “just a little bit to get them over the edge and really get them to that efficacy point that creates the success that you need.”
Dr. Jackson disclosed financial relationships with companies including AbbVie, Amgen, Celgene, Dermira, Galderma, Genentech, Janssen, Lilly, Medimetriks, Merck, Novartis, Pfizer, Promius, and Top MD.
SDEF and this news organization are owned by the same parent organization.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, Hawaii – Combining methotrexate with a biologic is an off-label use for psoriasis patients but is supported by information from the psoriatic arthritis literature, said J. Mark Jackson, MD, of the University of Louisville (Ky.).
In fact, the combination treatment may improve symptoms in patients with both psoriasis and arthritis not well controlled with one drug, Dr. Jackson explained in a video interview at the meeting, provided by Global Academy for Medical Education/Skin Disease Education Foundation.
As for dosage, “I do think we can use less methotrexate in combination” with a biologic for psoriasis when using methotrexate alone, he noted. “It’s like the addition of a spice to the right dish,” he added. Patients may need “just a little bit to get them over the edge and really get them to that efficacy point that creates the success that you need.”
Dr. Jackson disclosed financial relationships with companies including AbbVie, Amgen, Celgene, Dermira, Galderma, Genentech, Janssen, Lilly, Medimetriks, Merck, Novartis, Pfizer, Promius, and Top MD.
SDEF and this news organization are owned by the same parent organization.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, Hawaii – Combining methotrexate with a biologic is an off-label use for psoriasis patients but is supported by information from the psoriatic arthritis literature, said J. Mark Jackson, MD, of the University of Louisville (Ky.).
In fact, the combination treatment may improve symptoms in patients with both psoriasis and arthritis not well controlled with one drug, Dr. Jackson explained in a video interview at the meeting, provided by Global Academy for Medical Education/Skin Disease Education Foundation.
As for dosage, “I do think we can use less methotrexate in combination” with a biologic for psoriasis when using methotrexate alone, he noted. “It’s like the addition of a spice to the right dish,” he added. Patients may need “just a little bit to get them over the edge and really get them to that efficacy point that creates the success that you need.”
Dr. Jackson disclosed financial relationships with companies including AbbVie, Amgen, Celgene, Dermira, Galderma, Genentech, Janssen, Lilly, Medimetriks, Merck, Novartis, Pfizer, Promius, and Top MD.
SDEF and this news organization are owned by the same parent organization.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT SDEF HAWAII DERMATOLOGY SEMINAR

VIDEO: Interchangeability of biosimilars and parent compounds raise potential efficacy issues
WAILEA, Hawaii – Biosimilars are coming to dermatology, but their impact from a clinical and insurance standpoint has yet to be determined, according to Kenneth B. Gordon, MD, professor and chair of the department of dermatology at the Medical College of Wisconsin, Milwaukee.
Biosimilars seem to have reasonably good efficacy, and safety to date has been “pretty consistent” with safety associated with the parent compounds, but a key issue will be how they are used in patients on anti-TNF agents who have been doing well over time, Dr. Gordon said in a video interview. Because these medicines cross react in terms of immunogenicity and are not quite the same, “trying to use them interchangeably, as many insurance companies will ask us to do, is going to be difficult,” he noted.
His concern does not apply to a new patient starting on a biosimilar. “The problem I see is when insurance companies and pharmacies start mandating us going back and forth between medicines, and running into difficulty with loss of effect of those drugs,” he explained. “It’s not a safety issue, it’s more of an issue of losing efficacy of a formerly active drug,” he said at the meeting provided by Global Academy for Medical Education/Skin Disease Education Foundation.
Dr. Gordon disclosed financial relationships with AbbVie, Amgen, Boehringer Ingelheim, Janssen, Lilly, Celgene, Novartis, and Sun.
SDEF and this news organization are owned by the same parent organization.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, Hawaii – Biosimilars are coming to dermatology, but their impact from a clinical and insurance standpoint has yet to be determined, according to Kenneth B. Gordon, MD, professor and chair of the department of dermatology at the Medical College of Wisconsin, Milwaukee.
Biosimilars seem to have reasonably good efficacy, and safety to date has been “pretty consistent” with safety associated with the parent compounds, but a key issue will be how they are used in patients on anti-TNF agents who have been doing well over time, Dr. Gordon said in a video interview. Because these medicines cross react in terms of immunogenicity and are not quite the same, “trying to use them interchangeably, as many insurance companies will ask us to do, is going to be difficult,” he noted.
His concern does not apply to a new patient starting on a biosimilar. “The problem I see is when insurance companies and pharmacies start mandating us going back and forth between medicines, and running into difficulty with loss of effect of those drugs,” he explained. “It’s not a safety issue, it’s more of an issue of losing efficacy of a formerly active drug,” he said at the meeting provided by Global Academy for Medical Education/Skin Disease Education Foundation.
Dr. Gordon disclosed financial relationships with AbbVie, Amgen, Boehringer Ingelheim, Janssen, Lilly, Celgene, Novartis, and Sun.
SDEF and this news organization are owned by the same parent organization.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
WAILEA, Hawaii – Biosimilars are coming to dermatology, but their impact from a clinical and insurance standpoint has yet to be determined, according to Kenneth B. Gordon, MD, professor and chair of the department of dermatology at the Medical College of Wisconsin, Milwaukee.
Biosimilars seem to have reasonably good efficacy, and safety to date has been “pretty consistent” with safety associated with the parent compounds, but a key issue will be how they are used in patients on anti-TNF agents who have been doing well over time, Dr. Gordon said in a video interview. Because these medicines cross react in terms of immunogenicity and are not quite the same, “trying to use them interchangeably, as many insurance companies will ask us to do, is going to be difficult,” he noted.
His concern does not apply to a new patient starting on a biosimilar. “The problem I see is when insurance companies and pharmacies start mandating us going back and forth between medicines, and running into difficulty with loss of effect of those drugs,” he explained. “It’s not a safety issue, it’s more of an issue of losing efficacy of a formerly active drug,” he said at the meeting provided by Global Academy for Medical Education/Skin Disease Education Foundation.
Dr. Gordon disclosed financial relationships with AbbVie, Amgen, Boehringer Ingelheim, Janssen, Lilly, Celgene, Novartis, and Sun.
SDEF and this news organization are owned by the same parent organization.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
AT SDEF HAWAII DERMATOLOGY SEMINAR

MTX side effects limit patient use
Side effects in conjunction with inadequate disease control lead patients with plaque psoriasis to discontinue methotrexate (MTX) treatment, Dr. Marisol Otero and her colleagues reported.
The investigators identified 85 adult patients with plaque psoriasis from the Continuous Assessment of Psoriasis Treatment Use Registry With Methotrexate (MTX-CAPTURE) who had been treated with MTX for up to of 5.2 years. All had been started on MTX in accordance with Dutch and European guidelines.
Dose adjustments during treatment were made at physicians’ discretion and most patients (84) received folic acid supplements to protect against gastrointestinal side effects. Patients were required to have at least one follow up session with their physician during the study, according to Dr. Otero of the department of dermatology at Radboud University, Nijmegen, the Netherlands, and her colleagues.
At the end of 5 years, 55 patients (64.7%) had discontinued MTX, defined as cessation of MTX for more than 90 days or addition of another systemic psoriasis medication (Br J Dermatol. doi: 10.1111/bjd.15305).
Of the patients who discontinued treatment, 19 (34.5%) did so solely because of side effects, 14 (25.5%) discontinued because of lack of efficacy, and 7 (12.7%) cited the combination of side effects and ineffectiveness. Nine (16.4%) decided to end treatment for other reasons including personal decision, desire for pregnancy, and clinically inactive disease. Six (10.9%) were lost to for follow-up.
Side effects alone were the primary determinant in drug survival, with an overall drug survival rate for MTX of 1.8 years, Dr. Otero noted.
“It was remarkable that discontinuation due to side effects and ineffectiveness, were both common, while our hypothesis was that drug survival of MTX would be mainly limited by side effects,” the investigators said. “Side effects alone or in combination with inadequate disease control were more important in the context of treatment discontinuation than inadequate disease control solely.”
Side effects in conjunction with inadequate disease control lead patients with plaque psoriasis to discontinue methotrexate (MTX) treatment, Dr. Marisol Otero and her colleagues reported.
The investigators identified 85 adult patients with plaque psoriasis from the Continuous Assessment of Psoriasis Treatment Use Registry With Methotrexate (MTX-CAPTURE) who had been treated with MTX for up to of 5.2 years. All had been started on MTX in accordance with Dutch and European guidelines.
Dose adjustments during treatment were made at physicians’ discretion and most patients (84) received folic acid supplements to protect against gastrointestinal side effects. Patients were required to have at least one follow up session with their physician during the study, according to Dr. Otero of the department of dermatology at Radboud University, Nijmegen, the Netherlands, and her colleagues.
At the end of 5 years, 55 patients (64.7%) had discontinued MTX, defined as cessation of MTX for more than 90 days or addition of another systemic psoriasis medication (Br J Dermatol. doi: 10.1111/bjd.15305).
Of the patients who discontinued treatment, 19 (34.5%) did so solely because of side effects, 14 (25.5%) discontinued because of lack of efficacy, and 7 (12.7%) cited the combination of side effects and ineffectiveness. Nine (16.4%) decided to end treatment for other reasons including personal decision, desire for pregnancy, and clinically inactive disease. Six (10.9%) were lost to for follow-up.
Side effects alone were the primary determinant in drug survival, with an overall drug survival rate for MTX of 1.8 years, Dr. Otero noted.
“It was remarkable that discontinuation due to side effects and ineffectiveness, were both common, while our hypothesis was that drug survival of MTX would be mainly limited by side effects,” the investigators said. “Side effects alone or in combination with inadequate disease control were more important in the context of treatment discontinuation than inadequate disease control solely.”
Side effects in conjunction with inadequate disease control lead patients with plaque psoriasis to discontinue methotrexate (MTX) treatment, Dr. Marisol Otero and her colleagues reported.
The investigators identified 85 adult patients with plaque psoriasis from the Continuous Assessment of Psoriasis Treatment Use Registry With Methotrexate (MTX-CAPTURE) who had been treated with MTX for up to of 5.2 years. All had been started on MTX in accordance with Dutch and European guidelines.
Dose adjustments during treatment were made at physicians’ discretion and most patients (84) received folic acid supplements to protect against gastrointestinal side effects. Patients were required to have at least one follow up session with their physician during the study, according to Dr. Otero of the department of dermatology at Radboud University, Nijmegen, the Netherlands, and her colleagues.
At the end of 5 years, 55 patients (64.7%) had discontinued MTX, defined as cessation of MTX for more than 90 days or addition of another systemic psoriasis medication (Br J Dermatol. doi: 10.1111/bjd.15305).
Of the patients who discontinued treatment, 19 (34.5%) did so solely because of side effects, 14 (25.5%) discontinued because of lack of efficacy, and 7 (12.7%) cited the combination of side effects and ineffectiveness. Nine (16.4%) decided to end treatment for other reasons including personal decision, desire for pregnancy, and clinically inactive disease. Six (10.9%) were lost to for follow-up.
Side effects alone were the primary determinant in drug survival, with an overall drug survival rate for MTX of 1.8 years, Dr. Otero noted.
“It was remarkable that discontinuation due to side effects and ineffectiveness, were both common, while our hypothesis was that drug survival of MTX would be mainly limited by side effects,” the investigators said. “Side effects alone or in combination with inadequate disease control were more important in the context of treatment discontinuation than inadequate disease control solely.”
FROM THE BRITISH JOURNAL OF DERMATOLOGY
Key clinical point:
Major finding: More than one-third (34.5%) of patients stopped MTX because of side effects while more than a quarter (25.5%) did so due to lack of efficacy.
Data source: Analysis of 85 patients from a prospective noninterventional daily practice registry.
Disclosures: The study received no external funding. Dr. Otero has worked as a consultant for Eli Lilly. Other investigators reported consultancies and/or clinical trial work with multiple major pharmaceutical companies.
FDA opens abbreviated approval pathway for interchangeable biosimilars
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.

“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).

“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
The Food and Drug Administration has proposed a regulatory path for biosimilar biologics that are interchangeable with the reference product, paving the way for a new generation of less-expensive versions of these unique drugs.
But bringing an interchangeable biosimilar to market won’t be easy. The bar for interchangeability will be high, requiring that manufacturers prove switching between the new and older products is safe. And clinicians, while cautiously optimistic, aren’t thrilled with the industry payoff that could come with the designation: freedom for insurance companies and pharmacies to switch products at the dispensing level without requiring a new prescription.
The draft FDA guidance for industry, “Considerations in Demonstrating Interchangeability With a Reference Product,” arises from the Biologics Price Competition and Innovation Act of 2009. That section of the Affordable Care Act provides for abbreviated approval pathways for biological products that are demonstrated to be “highly similar” (biosimilar) to or “interchangeable” with an FDA-approved biological product.
The difference between these appellations is subtle but critical to the regulatory process – and perhaps to patient safety. Regulators recognize that the structure of these large, highly complex molecules can never precisely replicate the reference product. But to be labeled a “biosimilar,” developers must prove that the new product functions essentially the same; there can be no clinically meaningful differences in terms of safety, purity, and potency. Unlike a generic medication, a biosimilar can’t be substituted for its reference product at the pharmacy level. If a physician wants the patient on that biosimilar, the script must specify it.
Interchangeables jump a higher regulatory bar
An “interchangeable biosimilar,” though, would have to jump a higher regulatory bar. Not only must it produce the same clinical result as the reference product, it also must be benignly interchangeable with it, conferring no additional risk if a patient switches from the reference to the biosimilar and back again. A pharmacist could, if permitted by state law, substitute an interchangeable product for the reference product without going through the prescriber.
Like biosimilars, interchangeable products need not be tested in every disease for which the reference drug is approved, according to the document. Once they are proved safe for one indication, those data can be extrapolated to allow approval for the other indications as well. Nor do biosimilars need to prove efficacy per se, as their molecular similarity to the reference product ensures that they bind to the same receptor and exert the same therapeutic effect.
The biosimilar/interchangeable market has been slow to take off in the United States. There are no approved interchangeable biosimilars, and only four biosimilars – three of which were approved in 2016:
• Sandoz’ filgrastim-sndz (Zarxio).
• Pfizer’s and Celltrion’s infliximab-dyyb (Inflectra).
• Sandoz’ etanercept-szzs (Erelzi).
• Amgen’s adalimumab-atto (Amjevita).
Switching studies is the key to achieving the interchangeable designation, according to the FDA document. They must include at least two full switches between the candidate product and the reference product, which must be licensed in the United States.
But because these products are so structurally diverse, the FDA isn’t imposing a one-size-fits-all process on them. Instead, the molecular complexity and immunogenicity of each product will dictate its approval requirements.
Those with relatively low structural complexity, high molecular similarity to the reference product, and a low incidence of immunogenic adverse events may only need a single switching study to achieve the “interchangeability” designation.
The bar will be higher for a product with high structural complexity that is not as similar to the reference product, or which has been associated with immunogenic adverse events. For this product, FDA might also require extensive safety postmarketing data for the product as a licensed biosimilar, as well as a switching study.
Pharmacokinetics, pharmacodynamics, immunogenicity, and safety will be the primary endpoints of a switching study. Efficacy data are not necessary but can be used as supportive endpoints. Any safety signals in a switching study would raise regulatory eyebrows whether they came from the candidate product or the reference product. Since the study replicates what could happen if the two were used sequentially, it makes little difference from which product the event might arise.
“If an apparent difference in immune response or adverse events is noticed between the switching and nonswitching arms of the study ... it would raise concerns as to whether the proposed interchangeable product is interchangeable, regardless of whether the proposed interchangeable product or the reference product or the switching of the two products actually caused the event,” the document notes.
The E.U. vs. U.S. experience
The United States is only now getting a taste of what has become common fare in the European Union, said Angus Worthing, MD, chair of the American College of Rheumatology’s Government Affairs Committee. The European Medicines Agency approved its first biosimilar in 2006. Since then, 23 such drugs have come on the market, at an average price of about 30% less than the reference drug. Prices for some drugs have dropped as much as 70% in countries in which national health care systems abandoned the reference product in favor of the competing biosimilar, Dr. Worthing said in an interview.
“But the U.S. doesn’t have a national health care system, so it won’t work like that here.” In fact, he noted, brand-new data show that Medicare actually paid 22% more for the infliximab biosimilar Inflectra than it did for Remicade in the last quarter of 2016.
It’s not immediately apparent why this is the case, but it’s probably related to company discounts and rebates on these very expensive drugs. According to the report in Inside Health Policy, Janssen Biotech may have increased its discount on the drug to compete with Inflectra’s launch price of 15% below Remicade’s wholesale cost. Prices won’t moderate as much in the United States as in the European Union until several biosimilars of the same class appear, Dr. Worthing said.
There have already been allegations that big pharma manipulates international and national pricing to reduce biosimilar competition.
In June, Russian biotech company Biocad filed a lawsuit in New York charging Roche/Genentech with price fixing. The suit alleges that the companies cut the cost of three cancer drugs (Avastin, Herceptin, and Rituxan/MabThera) in Russia, where Biocad markets biosimilars for each. At the same time, Biocad alleges, the companies raised U.S. prices on those drugs to make up for the money they were losing on the Russian market.
“I think most of the cost benefits will accrue to insurance plans and pharmacy managers, but maybe not to the patients themselves,” he said in an interview. “The most important beneficiaries may not see a single penny of benefit.”
It may be difficult to extrapolate the European economic experience into the U.S. health care market, but the safety record of its biosimilar armamentarium is solid. None of the biosimilars approved in the E.U. have ever been recalled or removed from the European market because of regulatory or safety concerns.
Nonmedical switching raises concerns
Academic medical societies and clinicians interviewed for this article view the proposed approval pathway with cautious optimism. While acknowledging the potential benefit of reducing the costs of prohibitively expensive drugs, they uniformly insist that patient safety – not economic pressure – should be the driving force here.
“I was initially skeptical, and I do believe that we need very close pharmacovigilance in monitoring these for safety,” said Gideon Smith, MD, PhD, a dermatologist at Massachusetts General Hospital, Boston. “But there has been huge uptake of these products in the E.U., and the data are so extensive that we can be reasonably confident these drugs are effective, and no good reason to believe the safety will be any different.”
He is not as comfortable with the prospect of pharmacy-level substitution of an interchangeable biosimilar with the reference product – a feeling that other clinicians echoed.
“I think this is a fundamental issue that should have been dealt with on a federal level. Physicians should always be involved in the decision,” said Dr. Smith, who spoke at an FDA advisory committee meeting last summer on behalf of the American Academy of Dermatology (AAD).
“In general, the GI field is OK with the idea of starting someone on a new prescription [of an interchangeable biosimilar], but not so much with the idea of switching around,” said Dr. Hanauer, who is the Clifford Joseph Barborka Professor of Gastroenterology at Northwestern University, Chicago. “In these biologic compounds, very small differences can be amplified” and alter therapeutic response.
The possibility of switching from the reference to the biosimilar and maybe back again worries him. He hearkened back to the approval of Remicade, when patients who had taken it during clinical trials only were finally able to obtain it on the market. Dr. Hanauer explained that, “20% of them developed serum sickness reactions after the reexposure.”
He also expressed some concern about quality control in international manufacturing plants, citing a 2005 epidemic of immune-mediated pure red cell anemia in patients who received an epoetin alfa biosimilar manufactured in Thailand. The prefilled syringes had an uncoated rubber stopper that apparently reacted with polysorbate 60 in the solution – an interaction that increased immunogenicity when the drug was administered subcutaneously.
Dr. Smith concurred. “We know that some patients produce antibodies to biologics if they come on and off, and so we discourage that. The concern is that switching may lead to an increased rate of medication failure, if you have to switch back. This is especially troubling in the case of a hard-to-control patient with severe flares. If they’re being well controlled on a medication, the last thing you want to do is change it for no good clinical reason. And we may well be forced to do that.”
Neither the AAD nor the American College of Gastroenterology has a published stand on the FDA’s proposed guidance for interchangeable biosimilars. The preliminary view of the American College of Rheumatology is a positive one, Dr. Worthing said. However, ACR feels pharmacy-level switching should be a joint, not unilateral, decision.
“Our position statement on biosimilars has been that if it’s legal for a pharmacy to make that switch then we want the doctor and the patient to know, so we can track for safety signals.”
Bringing any biosimilar to market, though, takes a lot of money and a lot of time. And while companies are growing cell lines and producing new molecules that mimic existing drugs, science marches on, said Dr. Smith.
“If we keep dragging our feet on this issue, it might end up being a moot point,” he said. Newer drugs are achieving better results, raising the bar for therapeutic success. An example is the monoclonal antibody secukinumab (Cosentyx), an inhibitor of interleukin 17A. In October 2016, late-breaking data released at the annual meeting of the European Academy of Dermatology and Venereology impressed the dermatology community. In psoriasis patients, the drug maintained 90% skin clearance for 4 years in 66% of patients, and 100% clearance for 4 years in 43%.
Not only does this kind of efficacy provide symptomatic relief, it also prevents the expensive long-term morbidity associated with psoriasis, Dr. Smith said.
“Even if these new medications are considerably more expensive upfront than a biosimilar for an older drug, they may end up being less expensive in the long run.”
Dr. Krant and Dr. Worthing had no financial disclosures. Dr. Smith has received grants from Allergan and Cipher Pharmaceuticals. Dr. Hanauer has received grants from numerous pharmaceutical companies that manufacture biologics.
*This article was updated 1/31/2017.
msullivan@frontlinemedcom.com
On Twitter @alz_gal
Severe Henoch-Schönlein Purpura Complicating Infliximab Therapy for Ulcerative Colitis
To the Editor:
Anti–tumor necrosis factor (TNF) α treatments have radically improved the management of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, and bowel diseases (eg, Crohn disease, ulcerative colitis [UC]). Because the number of patients treated with these agents has increased, uncommon adverse reactions have increasingly occurred. Cutaneous adverse reactions that have been reported with anti-TNF agents include immediate injection-site reaction, systemic infusion reactions, and delayed reactions.1 Among the delayed adverse reactions, psoriatic and eczematous eruptions as well as cutaneous infections are the most common, while cutaneous adverse effects related to an immune imbalance syndrome including vasculitis; lupuslike, lichenlike, and granulomatous eruptions; and skin cancer rarely are observed.1 Although most of the cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases. We report the case of severe Henoch-Schönlein purpura (HSP) following treatment with infliximab.
A 46-year-old man who was a nonsmoker with quiescent UC on infliximab for 30 months presented with palpable necrotic purpura on both legs (Figure) and arms as well as the abdomen of 10 days’ duration, along with diffuse joint pain and swelling. He had no history of infectious or gastrointestinal symptoms. The last infliximab infusion was performed 6 weeks prior to developing the purpura. His UC was diagnosed 10 years prior to the current presentation and was not associated with any extragastrointestinal manifestations. Since diagnosis, UC had failed to respond to therapies such as azathioprine, cyclosporine, and purinethol. The complete blood cell count was normal. The C-reactive protein level was 18.7 mg/L (reference range, <5 mg/L) and the erythrocyte sedimentation rate was 30 mm/h (reference range, 0–20 mm/h). Electrolytes, urea, creatinine clearance, and liver function were normal, and a chest radiograph and radiographs of the swollen joints were unremarkable. The total IgA level was elevated at 4 g/L (reference range, 0.7–4 g/L), with IgG and IgM levels within reference range. There was no hematuria or proteinuria on urinalysis. Tests for antinuclear antibodies, rheumatoid factor, circulating immune complexes, and antineutrophil cytoplasmic antibody were negative. Total complement, C3, and C4 levels also were normal. A skin biopsy confirmed a leukocytoclastic vasculitis of small vessels with C3 deposition. Serologic tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were negative. Based on these findings, the diagnosis of HSP was made. Systemic corticosteroids—120 mg daily of intravenous methylprednisolone for 3 days, followed by 1 mg/kg daily of oral prednisone for 2 weeks—were then introduced with rapid clinical improvement. Henoch-Schönlein purpura and joint symptoms completely resolved, but UC relapsed with bloody diarrhea and severe abdominal pain. Oral prednisone was maintained (1 mg/kg daily). Because of the severity of cutaneous vasculitis (HSP), a multidisciplinary decision was taken to definitively stop the anti-TNF agents and to first add azathioprine (2 mg/kg daily for 2 months), then subcutaneous methotrexate (25 mg weekly). Colonoscopy did not show any dysplasia or adenocarcinoma and confirmed the diagnosis of UC. After 6 months of combined therapy, UC was still active and we decided to perform a total colectomy with ileostomy formation. Complete remission of UC was obtained and maintained after 28 months of follow-up.
Henoch-Schönlein purpura is a multisystem small vessel leukocytoclastic vasculitis with the deposition of immune complexes containing IgA. Clinical manifestations may include palpable purpura, arthritis, enteritis, and nephritis. Henoch-Schönlein purpura usually affects children. Adult onset is rare but associated with more severe symptoms and a poor prognosis.2 The criteria for HSP, as defined by the American College of Rheumatology,3 include palpable purpura, 20 years or younger at disease onset, bowel angina, and presence of vascular wall granulocytes on biopsy. At least 2 of these criteria are required for HSP diagnosis. Various viral or bacterial infections and drugs can trigger HSP, which also can be associated with autoinflammatory or autoimmune diseases. The association of HSP and UC is a rare event, as demonstrated by de Oliveira et al.4 Although only 2 cases of cutaneous vasculitis mimicking HSP have been described in UC,4 we cannot exclude a possible association between HSP and UC. However, our patient had UC for 10 years and never had clinical manifestations of vasculitis.
There are 5 reports of HSP following etanercept5,6 or adalimumab7-9 therapy and 1 following infliximab therapy.10 In all cases, HSP occurred after several months of anti-TNF therapy. However, there also are reports of cutaneous vasculitis associated with arthralgia and glomerulonephritis that resolved after withdrawal of anti-TNF agents.11,12 It is possible that some of these reactions may have been manifestations of undiagnosed HSP. In a series of 113 patients who developed cutaneous vasculitis after anti-TNF agents, visceral vasculitis was observed in 24% of patients. Treatment of vasculitis involved withdrawal of the anti-TNF therapy in 101 cases (89%).13 In these UC patients with few therapeutic alternatives, the continuation of anti-TNF agents should be discussed. In the previous series,13 of 16 patients who were rechallenged with the same or a different TNF antagonist, 12 (75%) experienced vasculitis relapse, suggesting a class effect of TNF inhibition. Because of the severity of cutaneous vasculitis and as previously suggested in a recent analytical and comprehensive overview on paradoxical reactions under TNF blockers,1 we decided not to re-expose our patient to infliximab or to other anti-TNF agents.
In conclusion, HSP may occur during anti-TNF therapy and physicians need to be aware of this potentially serious complication.
- Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification ofHenoch-Schönlein purpura. an analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol. 2015;33(2, suppl 89):S44-S47.
- de Oliveira GT, Martins SS, Deboni M, et al. Cutaneous vasculitis in ulcerative colitis mimicking Henoch-Schönlein purpura [published online May 22, 2012]. J Crohns Colitis. 2013;7:e69-e73.
- Marques I, Lagos A, Reis J, et al. Reversible Henoch-Schönlein purpura complicating adalimumab therapy. J Crohns Colitis. 2012;6:796-799.
- Rahman FZ, Takhar GK, Roy O, et al. Henoch-Schönlein purpura complicating adalimumab therapy for Crohn’s disease. World J Gastrointest Pharmacol Ther. 2010;1:119-122.
- Lee A, Kasama R, Evangelisto A, et al. Henoch-Schönlein purpura after etanercept therapy for psoriasis. J Clin Rheumatol. 2006;12:249-251.
- Duffy TN, Genta M, Moll S, et al. Henoch Schönlein purpura following etanercept treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(2, suppl 41):S106.
- LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature. Case Rep Rheumatol. 2016:2812980.
- Nobile S, Catassi C, Felici L. Herpes zoster infection followed by Henoch-Schönlein purpura in a girl receiving infliximab for ulcerative colitis. J Clin Rheumatol. 2009;15:101.
- Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol. 2004;31:1955-1958.
- Simms R, Kipgen D, Dahill S, et al. ANCA-associated renal vasculitis following anti-tumor necrosis factor alpha therapy. Am J Kidney Dis. 2008;51:e11-e14.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007;86:242-251.
To the Editor:
Anti–tumor necrosis factor (TNF) α treatments have radically improved the management of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, and bowel diseases (eg, Crohn disease, ulcerative colitis [UC]). Because the number of patients treated with these agents has increased, uncommon adverse reactions have increasingly occurred. Cutaneous adverse reactions that have been reported with anti-TNF agents include immediate injection-site reaction, systemic infusion reactions, and delayed reactions.1 Among the delayed adverse reactions, psoriatic and eczematous eruptions as well as cutaneous infections are the most common, while cutaneous adverse effects related to an immune imbalance syndrome including vasculitis; lupuslike, lichenlike, and granulomatous eruptions; and skin cancer rarely are observed.1 Although most of the cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases. We report the case of severe Henoch-Schönlein purpura (HSP) following treatment with infliximab.
A 46-year-old man who was a nonsmoker with quiescent UC on infliximab for 30 months presented with palpable necrotic purpura on both legs (Figure) and arms as well as the abdomen of 10 days’ duration, along with diffuse joint pain and swelling. He had no history of infectious or gastrointestinal symptoms. The last infliximab infusion was performed 6 weeks prior to developing the purpura. His UC was diagnosed 10 years prior to the current presentation and was not associated with any extragastrointestinal manifestations. Since diagnosis, UC had failed to respond to therapies such as azathioprine, cyclosporine, and purinethol. The complete blood cell count was normal. The C-reactive protein level was 18.7 mg/L (reference range, <5 mg/L) and the erythrocyte sedimentation rate was 30 mm/h (reference range, 0–20 mm/h). Electrolytes, urea, creatinine clearance, and liver function were normal, and a chest radiograph and radiographs of the swollen joints were unremarkable. The total IgA level was elevated at 4 g/L (reference range, 0.7–4 g/L), with IgG and IgM levels within reference range. There was no hematuria or proteinuria on urinalysis. Tests for antinuclear antibodies, rheumatoid factor, circulating immune complexes, and antineutrophil cytoplasmic antibody were negative. Total complement, C3, and C4 levels also were normal. A skin biopsy confirmed a leukocytoclastic vasculitis of small vessels with C3 deposition. Serologic tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were negative. Based on these findings, the diagnosis of HSP was made. Systemic corticosteroids—120 mg daily of intravenous methylprednisolone for 3 days, followed by 1 mg/kg daily of oral prednisone for 2 weeks—were then introduced with rapid clinical improvement. Henoch-Schönlein purpura and joint symptoms completely resolved, but UC relapsed with bloody diarrhea and severe abdominal pain. Oral prednisone was maintained (1 mg/kg daily). Because of the severity of cutaneous vasculitis (HSP), a multidisciplinary decision was taken to definitively stop the anti-TNF agents and to first add azathioprine (2 mg/kg daily for 2 months), then subcutaneous methotrexate (25 mg weekly). Colonoscopy did not show any dysplasia or adenocarcinoma and confirmed the diagnosis of UC. After 6 months of combined therapy, UC was still active and we decided to perform a total colectomy with ileostomy formation. Complete remission of UC was obtained and maintained after 28 months of follow-up.
Henoch-Schönlein purpura is a multisystem small vessel leukocytoclastic vasculitis with the deposition of immune complexes containing IgA. Clinical manifestations may include palpable purpura, arthritis, enteritis, and nephritis. Henoch-Schönlein purpura usually affects children. Adult onset is rare but associated with more severe symptoms and a poor prognosis.2 The criteria for HSP, as defined by the American College of Rheumatology,3 include palpable purpura, 20 years or younger at disease onset, bowel angina, and presence of vascular wall granulocytes on biopsy. At least 2 of these criteria are required for HSP diagnosis. Various viral or bacterial infections and drugs can trigger HSP, which also can be associated with autoinflammatory or autoimmune diseases. The association of HSP and UC is a rare event, as demonstrated by de Oliveira et al.4 Although only 2 cases of cutaneous vasculitis mimicking HSP have been described in UC,4 we cannot exclude a possible association between HSP and UC. However, our patient had UC for 10 years and never had clinical manifestations of vasculitis.
There are 5 reports of HSP following etanercept5,6 or adalimumab7-9 therapy and 1 following infliximab therapy.10 In all cases, HSP occurred after several months of anti-TNF therapy. However, there also are reports of cutaneous vasculitis associated with arthralgia and glomerulonephritis that resolved after withdrawal of anti-TNF agents.11,12 It is possible that some of these reactions may have been manifestations of undiagnosed HSP. In a series of 113 patients who developed cutaneous vasculitis after anti-TNF agents, visceral vasculitis was observed in 24% of patients. Treatment of vasculitis involved withdrawal of the anti-TNF therapy in 101 cases (89%).13 In these UC patients with few therapeutic alternatives, the continuation of anti-TNF agents should be discussed. In the previous series,13 of 16 patients who were rechallenged with the same or a different TNF antagonist, 12 (75%) experienced vasculitis relapse, suggesting a class effect of TNF inhibition. Because of the severity of cutaneous vasculitis and as previously suggested in a recent analytical and comprehensive overview on paradoxical reactions under TNF blockers,1 we decided not to re-expose our patient to infliximab or to other anti-TNF agents.
In conclusion, HSP may occur during anti-TNF therapy and physicians need to be aware of this potentially serious complication.
To the Editor:
Anti–tumor necrosis factor (TNF) α treatments have radically improved the management of chronic inflammatory conditions, including rheumatoid arthritis, ankylosing spondylitis, psoriasis and psoriatic arthritis, and bowel diseases (eg, Crohn disease, ulcerative colitis [UC]). Because the number of patients treated with these agents has increased, uncommon adverse reactions have increasingly occurred. Cutaneous adverse reactions that have been reported with anti-TNF agents include immediate injection-site reaction, systemic infusion reactions, and delayed reactions.1 Among the delayed adverse reactions, psoriatic and eczematous eruptions as well as cutaneous infections are the most common, while cutaneous adverse effects related to an immune imbalance syndrome including vasculitis; lupuslike, lichenlike, and granulomatous eruptions; and skin cancer rarely are observed.1 Although most of the cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases. We report the case of severe Henoch-Schönlein purpura (HSP) following treatment with infliximab.
A 46-year-old man who was a nonsmoker with quiescent UC on infliximab for 30 months presented with palpable necrotic purpura on both legs (Figure) and arms as well as the abdomen of 10 days’ duration, along with diffuse joint pain and swelling. He had no history of infectious or gastrointestinal symptoms. The last infliximab infusion was performed 6 weeks prior to developing the purpura. His UC was diagnosed 10 years prior to the current presentation and was not associated with any extragastrointestinal manifestations. Since diagnosis, UC had failed to respond to therapies such as azathioprine, cyclosporine, and purinethol. The complete blood cell count was normal. The C-reactive protein level was 18.7 mg/L (reference range, <5 mg/L) and the erythrocyte sedimentation rate was 30 mm/h (reference range, 0–20 mm/h). Electrolytes, urea, creatinine clearance, and liver function were normal, and a chest radiograph and radiographs of the swollen joints were unremarkable. The total IgA level was elevated at 4 g/L (reference range, 0.7–4 g/L), with IgG and IgM levels within reference range. There was no hematuria or proteinuria on urinalysis. Tests for antinuclear antibodies, rheumatoid factor, circulating immune complexes, and antineutrophil cytoplasmic antibody were negative. Total complement, C3, and C4 levels also were normal. A skin biopsy confirmed a leukocytoclastic vasculitis of small vessels with C3 deposition. Serologic tests for hepatitis B virus, hepatitis C virus, and human immunodeficiency virus were negative. Based on these findings, the diagnosis of HSP was made. Systemic corticosteroids—120 mg daily of intravenous methylprednisolone for 3 days, followed by 1 mg/kg daily of oral prednisone for 2 weeks—were then introduced with rapid clinical improvement. Henoch-Schönlein purpura and joint symptoms completely resolved, but UC relapsed with bloody diarrhea and severe abdominal pain. Oral prednisone was maintained (1 mg/kg daily). Because of the severity of cutaneous vasculitis (HSP), a multidisciplinary decision was taken to definitively stop the anti-TNF agents and to first add azathioprine (2 mg/kg daily for 2 months), then subcutaneous methotrexate (25 mg weekly). Colonoscopy did not show any dysplasia or adenocarcinoma and confirmed the diagnosis of UC. After 6 months of combined therapy, UC was still active and we decided to perform a total colectomy with ileostomy formation. Complete remission of UC was obtained and maintained after 28 months of follow-up.
Henoch-Schönlein purpura is a multisystem small vessel leukocytoclastic vasculitis with the deposition of immune complexes containing IgA. Clinical manifestations may include palpable purpura, arthritis, enteritis, and nephritis. Henoch-Schönlein purpura usually affects children. Adult onset is rare but associated with more severe symptoms and a poor prognosis.2 The criteria for HSP, as defined by the American College of Rheumatology,3 include palpable purpura, 20 years or younger at disease onset, bowel angina, and presence of vascular wall granulocytes on biopsy. At least 2 of these criteria are required for HSP diagnosis. Various viral or bacterial infections and drugs can trigger HSP, which also can be associated with autoinflammatory or autoimmune diseases. The association of HSP and UC is a rare event, as demonstrated by de Oliveira et al.4 Although only 2 cases of cutaneous vasculitis mimicking HSP have been described in UC,4 we cannot exclude a possible association between HSP and UC. However, our patient had UC for 10 years and never had clinical manifestations of vasculitis.
There are 5 reports of HSP following etanercept5,6 or adalimumab7-9 therapy and 1 following infliximab therapy.10 In all cases, HSP occurred after several months of anti-TNF therapy. However, there also are reports of cutaneous vasculitis associated with arthralgia and glomerulonephritis that resolved after withdrawal of anti-TNF agents.11,12 It is possible that some of these reactions may have been manifestations of undiagnosed HSP. In a series of 113 patients who developed cutaneous vasculitis after anti-TNF agents, visceral vasculitis was observed in 24% of patients. Treatment of vasculitis involved withdrawal of the anti-TNF therapy in 101 cases (89%).13 In these UC patients with few therapeutic alternatives, the continuation of anti-TNF agents should be discussed. In the previous series,13 of 16 patients who were rechallenged with the same or a different TNF antagonist, 12 (75%) experienced vasculitis relapse, suggesting a class effect of TNF inhibition. Because of the severity of cutaneous vasculitis and as previously suggested in a recent analytical and comprehensive overview on paradoxical reactions under TNF blockers,1 we decided not to re-expose our patient to infliximab or to other anti-TNF agents.
In conclusion, HSP may occur during anti-TNF therapy and physicians need to be aware of this potentially serious complication.
- Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification ofHenoch-Schönlein purpura. an analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol. 2015;33(2, suppl 89):S44-S47.
- de Oliveira GT, Martins SS, Deboni M, et al. Cutaneous vasculitis in ulcerative colitis mimicking Henoch-Schönlein purpura [published online May 22, 2012]. J Crohns Colitis. 2013;7:e69-e73.
- Marques I, Lagos A, Reis J, et al. Reversible Henoch-Schönlein purpura complicating adalimumab therapy. J Crohns Colitis. 2012;6:796-799.
- Rahman FZ, Takhar GK, Roy O, et al. Henoch-Schönlein purpura complicating adalimumab therapy for Crohn’s disease. World J Gastrointest Pharmacol Ther. 2010;1:119-122.
- Lee A, Kasama R, Evangelisto A, et al. Henoch-Schönlein purpura after etanercept therapy for psoriasis. J Clin Rheumatol. 2006;12:249-251.
- Duffy TN, Genta M, Moll S, et al. Henoch Schönlein purpura following etanercept treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(2, suppl 41):S106.
- LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature. Case Rep Rheumatol. 2016:2812980.
- Nobile S, Catassi C, Felici L. Herpes zoster infection followed by Henoch-Schönlein purpura in a girl receiving infliximab for ulcerative colitis. J Clin Rheumatol. 2009;15:101.
- Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol. 2004;31:1955-1958.
- Simms R, Kipgen D, Dahill S, et al. ANCA-associated renal vasculitis following anti-tumor necrosis factor alpha therapy. Am J Kidney Dis. 2008;51:e11-e14.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007;86:242-251.
- Toussirot É, Aubin F. Paradoxical reactions under TNF-α blocking agents and other biological agents given for chronic immune-mediated diseases: an analytical and comprehensive overview. RMD Open. 2016;2:e000239.
- Saulsbury FT. Henoch-Schönlein purpura. Curr Opin Rheumatol. 2001;13:35-40.
- Ortiz-Sanjuán F, Blanco R, Hernández JL, et al. Applicability of the 2006 European League Against Rheumatism (EULAR) criteria for the classification ofHenoch-Schönlein purpura. an analysis based on 766 patients with cutaneous vasculitis. Clin Exp Rheumatol. 2015;33(2, suppl 89):S44-S47.
- de Oliveira GT, Martins SS, Deboni M, et al. Cutaneous vasculitis in ulcerative colitis mimicking Henoch-Schönlein purpura [published online May 22, 2012]. J Crohns Colitis. 2013;7:e69-e73.
- Marques I, Lagos A, Reis J, et al. Reversible Henoch-Schönlein purpura complicating adalimumab therapy. J Crohns Colitis. 2012;6:796-799.
- Rahman FZ, Takhar GK, Roy O, et al. Henoch-Schönlein purpura complicating adalimumab therapy for Crohn’s disease. World J Gastrointest Pharmacol Ther. 2010;1:119-122.
- Lee A, Kasama R, Evangelisto A, et al. Henoch-Schönlein purpura after etanercept therapy for psoriasis. J Clin Rheumatol. 2006;12:249-251.
- Duffy TN, Genta M, Moll S, et al. Henoch Schönlein purpura following etanercept treatment of rheumatoid arthritis. Clin Exp Rheumatol. 2006;24(2, suppl 41):S106.
- LaConti JJ, Donet JA, Cho-Vega JH, et al. Henoch-Schönlein purpura with adalimumab therapy for ulcerative colitis: a case report and review of the literature. Case Rep Rheumatol. 2016:2812980.
- Nobile S, Catassi C, Felici L. Herpes zoster infection followed by Henoch-Schönlein purpura in a girl receiving infliximab for ulcerative colitis. J Clin Rheumatol. 2009;15:101.
- Mohan N, Edwards ET, Cupps TR, et al. Leukocytoclastic vasculitis associated with tumor necrosis factor-alpha blocking agents. J Rheumatol. 2004;31:1955-1958.
- Simms R, Kipgen D, Dahill S, et al. ANCA-associated renal vasculitis following anti-tumor necrosis factor alpha therapy. Am J Kidney Dis. 2008;51:e11-e14.
- Ramos-Casals M, Brito-Zerón P, Muñoz S, et al. Autoimmune diseases induced by TNF-targeted therapies: analysis of 233 cases. Medicine (Baltimore). 2007;86:242-251.
Practice Points
- Cutaneous adverse effects may occur in approximately 20% of patients treated with anti–tumor necrosis factor (TNF) drugs.
- Henoch-Schönlein purpura (HSP), a small-vessel vasculitis, is an extremely rare complication of anti-TNF treatment.
- Although most cutaneous adverse effects do not require anti-TNF treatment discontinuation and are resolved with symptomatic treatment, anti-TNF therapy must be stopped in more severe cases.
Bone fractures more likely to occur in psoriasis, PsA patients
Individuals who have psoriasis or psoriatic arthritis are at a significantly higher risk of also suffering bone fractures, particularly in their hip and vertebrae, according to a new study published in the Annals of the Rheumatic Diseases.
“To our knowledge, these are the first population-based estimates of the risk for incident fracture and osteoporosis in patients with psoriasis and/or PsA [psoriatic arthritis] and the first longitudinal cohort study to address this issue,” wrote the authors of the study, led by Alexis Ogdie-Beatty, MD, of the University of Pennsylvania in Philadelphia (Ann Rheum Dis. 2017 Jan 16. doi: 10.1136/annrheumdis-2016-210441).

A total of 9,788 PsA and 158,323 psoriasis patients were included in the study, along with 39,306 RA patients and 821,834 individuals from the general population. Psoriasis patients were divided into groups classified as mild (n = 149,809) or severe (n = 8,514). The average age of each cohort ranged from nearly 47 years to almost 59 years, with all cohorts comprising mostly females, ranging from about 51% to 69%.
“We found that the risk for any fracture in patients with PsA and severe psoriasis was similar to RA [but] patients with PsA and psoriasis had an increased incidence of fracture compared with the general population by 7%-26%,” the authors explained. “The incidence of vertebral fracture was also increased in patients with severe psoriasis and while hip fracture was elevated in both psoriasis groups, it was only statistically significant in patients with mild psoriasis relative to matched controls after adjusting for risk factors for osteoporosis.”
Dr. Ogdie-Beatty and her colleagues found that all of the conditions conferred an elevated risk for fractures anywhere in the body when compared with the general population, reaching hazard ratios of 1.16 (95% confidence interval, 1.06-1.27) for people with PsA, 1.07 (95% CI, 1.05-1.10) for mild psoriasis, 1.26 for severe psoriasis (95% CI, 1.15-1.39), and 1.23 for RA (95% CI, 1.18-1.28). The risk for hip fractures was only significantly higher for mild (hazard ratio, 1.13; 95% CI, 1.04-1.22) and severe psoriasis (HR, 1.21; 95% CI, 0.88-1.66), and RA (HR, 1.55; 95% CI, 1.40-1.72). Individuals with PsA did not have a significantly higher risk for vertebral fractures (HR, 1.07; 95% CI, 0.66-1.72), whereas those with mild psoriasis (HR, 1.17; 95% CI, 1.03-1.33), severe psoriasis (HR, 2.23; 95% CI, 1.54-3.22), or RA did (HR, 1.53; 95% CI, 1.30-1.80). Each of these models were fully adjusted for multiple different osteoporosis risk factors, although they were all commonly adjusted for age, sex, atrial fibrillation, diabetes, chronic obstructive pulmonary disease, stroke, SSRI use, tricyclic antidepressant use, oral steroids, smoking, and categorical body mass index.
Individual coauthors disclosed receiving funding for their work from the National Institutes of Health, as well as grants from the Department of Veterans Affairs and the Rheumatology Research Foundation. Three of the authors reported receiving payment for continuing medical education work related to psoriatic arthritis or psoriasis.
Individuals who have psoriasis or psoriatic arthritis are at a significantly higher risk of also suffering bone fractures, particularly in their hip and vertebrae, according to a new study published in the Annals of the Rheumatic Diseases.
“To our knowledge, these are the first population-based estimates of the risk for incident fracture and osteoporosis in patients with psoriasis and/or PsA [psoriatic arthritis] and the first longitudinal cohort study to address this issue,” wrote the authors of the study, led by Alexis Ogdie-Beatty, MD, of the University of Pennsylvania in Philadelphia (Ann Rheum Dis. 2017 Jan 16. doi: 10.1136/annrheumdis-2016-210441).
A total of 9,788 PsA and 158,323 psoriasis patients were included in the study, along with 39,306 RA patients and 821,834 individuals from the general population. Psoriasis patients were divided into groups classified as mild (n = 149,809) or severe (n = 8,514). The average age of each cohort ranged from nearly 47 years to almost 59 years, with all cohorts comprising mostly females, ranging from about 51% to 69%.
“We found that the risk for any fracture in patients with PsA and severe psoriasis was similar to RA [but] patients with PsA and psoriasis had an increased incidence of fracture compared with the general population by 7%-26%,” the authors explained. “The incidence of vertebral fracture was also increased in patients with severe psoriasis and while hip fracture was elevated in both psoriasis groups, it was only statistically significant in patients with mild psoriasis relative to matched controls after adjusting for risk factors for osteoporosis.”
Dr. Ogdie-Beatty and her colleagues found that all of the conditions conferred an elevated risk for fractures anywhere in the body when compared with the general population, reaching hazard ratios of 1.16 (95% confidence interval, 1.06-1.27) for people with PsA, 1.07 (95% CI, 1.05-1.10) for mild psoriasis, 1.26 for severe psoriasis (95% CI, 1.15-1.39), and 1.23 for RA (95% CI, 1.18-1.28). The risk for hip fractures was only significantly higher for mild (hazard ratio, 1.13; 95% CI, 1.04-1.22) and severe psoriasis (HR, 1.21; 95% CI, 0.88-1.66), and RA (HR, 1.55; 95% CI, 1.40-1.72). Individuals with PsA did not have a significantly higher risk for vertebral fractures (HR, 1.07; 95% CI, 0.66-1.72), whereas those with mild psoriasis (HR, 1.17; 95% CI, 1.03-1.33), severe psoriasis (HR, 2.23; 95% CI, 1.54-3.22), or RA did (HR, 1.53; 95% CI, 1.30-1.80). Each of these models were fully adjusted for multiple different osteoporosis risk factors, although they were all commonly adjusted for age, sex, atrial fibrillation, diabetes, chronic obstructive pulmonary disease, stroke, SSRI use, tricyclic antidepressant use, oral steroids, smoking, and categorical body mass index.
Individual coauthors disclosed receiving funding for their work from the National Institutes of Health, as well as grants from the Department of Veterans Affairs and the Rheumatology Research Foundation. Three of the authors reported receiving payment for continuing medical education work related to psoriatic arthritis or psoriasis.
Individuals who have psoriasis or psoriatic arthritis are at a significantly higher risk of also suffering bone fractures, particularly in their hip and vertebrae, according to a new study published in the Annals of the Rheumatic Diseases.
“To our knowledge, these are the first population-based estimates of the risk for incident fracture and osteoporosis in patients with psoriasis and/or PsA [psoriatic arthritis] and the first longitudinal cohort study to address this issue,” wrote the authors of the study, led by Alexis Ogdie-Beatty, MD, of the University of Pennsylvania in Philadelphia (Ann Rheum Dis. 2017 Jan 16. doi: 10.1136/annrheumdis-2016-210441).
A total of 9,788 PsA and 158,323 psoriasis patients were included in the study, along with 39,306 RA patients and 821,834 individuals from the general population. Psoriasis patients were divided into groups classified as mild (n = 149,809) or severe (n = 8,514). The average age of each cohort ranged from nearly 47 years to almost 59 years, with all cohorts comprising mostly females, ranging from about 51% to 69%.
“We found that the risk for any fracture in patients with PsA and severe psoriasis was similar to RA [but] patients with PsA and psoriasis had an increased incidence of fracture compared with the general population by 7%-26%,” the authors explained. “The incidence of vertebral fracture was also increased in patients with severe psoriasis and while hip fracture was elevated in both psoriasis groups, it was only statistically significant in patients with mild psoriasis relative to matched controls after adjusting for risk factors for osteoporosis.”
Dr. Ogdie-Beatty and her colleagues found that all of the conditions conferred an elevated risk for fractures anywhere in the body when compared with the general population, reaching hazard ratios of 1.16 (95% confidence interval, 1.06-1.27) for people with PsA, 1.07 (95% CI, 1.05-1.10) for mild psoriasis, 1.26 for severe psoriasis (95% CI, 1.15-1.39), and 1.23 for RA (95% CI, 1.18-1.28). The risk for hip fractures was only significantly higher for mild (hazard ratio, 1.13; 95% CI, 1.04-1.22) and severe psoriasis (HR, 1.21; 95% CI, 0.88-1.66), and RA (HR, 1.55; 95% CI, 1.40-1.72). Individuals with PsA did not have a significantly higher risk for vertebral fractures (HR, 1.07; 95% CI, 0.66-1.72), whereas those with mild psoriasis (HR, 1.17; 95% CI, 1.03-1.33), severe psoriasis (HR, 2.23; 95% CI, 1.54-3.22), or RA did (HR, 1.53; 95% CI, 1.30-1.80). Each of these models were fully adjusted for multiple different osteoporosis risk factors, although they were all commonly adjusted for age, sex, atrial fibrillation, diabetes, chronic obstructive pulmonary disease, stroke, SSRI use, tricyclic antidepressant use, oral steroids, smoking, and categorical body mass index.
Individual coauthors disclosed receiving funding for their work from the National Institutes of Health, as well as grants from the Department of Veterans Affairs and the Rheumatology Research Foundation. Three of the authors reported receiving payment for continuing medical education work related to psoriatic arthritis or psoriasis.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point:
Major finding: The risk of fracture for patients with PsA and psoriasis had a risk of fracture that was increased by 7%-26% in comparison with the general U.K. population.
Data source: Population-based, longitudinal cohort study of 9,788 PsA patients and 158,323 psoriasis patients in the United Kingdom during 1994-2014.
Disclosures: Individual coauthors disclosed receiving funding for their work from the National Institutes of Health, as well as grants from the Department of Veterans Affairs and the Rheumatology Research Foundation. Three of the authors reported receiving payment for continuing medical education work related to psoriatic arthritis or psoriasis.