User login
Adapting HCQ dose did not reduce SLE flare rates
A strategy to maintain hydroxychloroquine blood levels above 1,000 ng/mL in adult patients with systemic lupus erythematosus did not reduce the number of SLE flares during a 7-month follow-up period, results from a French study demonstrated.
"This study confirms the pharmacokinetic/pharmacodynamic relation for hydroxychloroquine (HCQ) in patients with SLE," investigators led by Prof. Nathalie Costedoat-Chalumeau reported in the November 2013 issue of Annals of the Rheumatic Diseases.
"Our results do not justify recommending a therapeutic adaptation of HCQ dose. However, we suggest that [HCQ] be measured to detect non-adherence, especially in patients with active disease, and to help patients with poor adherence reach [HCQ levels greater than or equal to] 1,000 ng/mL."
The purpose of the trial, known at the PLUS Study, was to determine the potential benefits of individualizing HCQ dosing schedules to reach a target of 1,000 ng/mL or greater and thereby decrease rates of SLE flare. It was carried out in 573 patients with SLE at 37 centers in France from June 2007 through August 2010.
Of the 573 patients, researchers randomized 171 to one of two treatment groups: 84 to no daily dose change (group A), and 87 to increased HCQ dose to achieve the target of 1,000 g/mL or greater (group B). The primary endpoint was the number of patients with flares during 7 months of follow-up (Ann. Rheum. Dis. 2013;72:1786-92).
At the time of randomization, the mean age of patients was 40 years, 87% were female, and their average HCQ dose was 400 mg/day. At 7 months of follow-up, the proportion of SLE flare rates was similar between the two groups (25% in group A vs. 27.6% in group B; P = .7). In a subset analysis of 57 patients from group A whose HCQ values were below 1,000 ng/mL after randomization and 39 patients from group B who maintained the therapeutic target dose of 1,000 ng/mL or higher after randomization, patients in group B tended to fewer flares compared with their counterparts in group A (20.5% vs. 35.1%, respectively; P = .12).
One reason that HCQ dosing above 1,000 ng/mL did not reduce the rate of SLE flares during the study period "may be that higher doses do not have an added therapeutic effect," the researchers speculated. "However, several factors may provide an alternative explanation of why our study did not provide its primary hypothesis." For one, the maintenance of HCQ above 1,000 ng/mL during the 7 months of follow up "was difficult to achieve." This could be explained by two factors, they continued. "The first is the pharmacokinetic variations of HCQ, but this explanation is unlikely because HCQ has a long terminal half-life and these patients were thought to be in a steady state. The second potential explanation might be adherence problems, even though known or suspected non-adherence was a major criterion for exclusion in our study. We found 10 patients with [HCQ] sufficiently low at inclusion to constitute an objective marker of lack of compliance."
The researchers acknowledged certain limitations of the study, including the potential for being underpowered (171 patients studied though the calculated sample size called for 200), and that the trend toward lower SLE flares in patients with higher HCQ "must be interpreted cautiously, since this analysis was not performed according to randomization group. The result might also be explained by better adherence to other medications, especially steroids."
The study was funded by a grant from the French PHRC 2005 Ministère de la Santé. Sanofi provided the HCQ and placebo tablets.
A strategy to maintain hydroxychloroquine blood levels above 1,000 ng/mL in adult patients with systemic lupus erythematosus did not reduce the number of SLE flares during a 7-month follow-up period, results from a French study demonstrated.
"This study confirms the pharmacokinetic/pharmacodynamic relation for hydroxychloroquine (HCQ) in patients with SLE," investigators led by Prof. Nathalie Costedoat-Chalumeau reported in the November 2013 issue of Annals of the Rheumatic Diseases.
"Our results do not justify recommending a therapeutic adaptation of HCQ dose. However, we suggest that [HCQ] be measured to detect non-adherence, especially in patients with active disease, and to help patients with poor adherence reach [HCQ levels greater than or equal to] 1,000 ng/mL."
The purpose of the trial, known at the PLUS Study, was to determine the potential benefits of individualizing HCQ dosing schedules to reach a target of 1,000 ng/mL or greater and thereby decrease rates of SLE flare. It was carried out in 573 patients with SLE at 37 centers in France from June 2007 through August 2010.
Of the 573 patients, researchers randomized 171 to one of two treatment groups: 84 to no daily dose change (group A), and 87 to increased HCQ dose to achieve the target of 1,000 g/mL or greater (group B). The primary endpoint was the number of patients with flares during 7 months of follow-up (Ann. Rheum. Dis. 2013;72:1786-92).
At the time of randomization, the mean age of patients was 40 years, 87% were female, and their average HCQ dose was 400 mg/day. At 7 months of follow-up, the proportion of SLE flare rates was similar between the two groups (25% in group A vs. 27.6% in group B; P = .7). In a subset analysis of 57 patients from group A whose HCQ values were below 1,000 ng/mL after randomization and 39 patients from group B who maintained the therapeutic target dose of 1,000 ng/mL or higher after randomization, patients in group B tended to fewer flares compared with their counterparts in group A (20.5% vs. 35.1%, respectively; P = .12).
One reason that HCQ dosing above 1,000 ng/mL did not reduce the rate of SLE flares during the study period "may be that higher doses do not have an added therapeutic effect," the researchers speculated. "However, several factors may provide an alternative explanation of why our study did not provide its primary hypothesis." For one, the maintenance of HCQ above 1,000 ng/mL during the 7 months of follow up "was difficult to achieve." This could be explained by two factors, they continued. "The first is the pharmacokinetic variations of HCQ, but this explanation is unlikely because HCQ has a long terminal half-life and these patients were thought to be in a steady state. The second potential explanation might be adherence problems, even though known or suspected non-adherence was a major criterion for exclusion in our study. We found 10 patients with [HCQ] sufficiently low at inclusion to constitute an objective marker of lack of compliance."
The researchers acknowledged certain limitations of the study, including the potential for being underpowered (171 patients studied though the calculated sample size called for 200), and that the trend toward lower SLE flares in patients with higher HCQ "must be interpreted cautiously, since this analysis was not performed according to randomization group. The result might also be explained by better adherence to other medications, especially steroids."
The study was funded by a grant from the French PHRC 2005 Ministère de la Santé. Sanofi provided the HCQ and placebo tablets.
A strategy to maintain hydroxychloroquine blood levels above 1,000 ng/mL in adult patients with systemic lupus erythematosus did not reduce the number of SLE flares during a 7-month follow-up period, results from a French study demonstrated.
"This study confirms the pharmacokinetic/pharmacodynamic relation for hydroxychloroquine (HCQ) in patients with SLE," investigators led by Prof. Nathalie Costedoat-Chalumeau reported in the November 2013 issue of Annals of the Rheumatic Diseases.
"Our results do not justify recommending a therapeutic adaptation of HCQ dose. However, we suggest that [HCQ] be measured to detect non-adherence, especially in patients with active disease, and to help patients with poor adherence reach [HCQ levels greater than or equal to] 1,000 ng/mL."
The purpose of the trial, known at the PLUS Study, was to determine the potential benefits of individualizing HCQ dosing schedules to reach a target of 1,000 ng/mL or greater and thereby decrease rates of SLE flare. It was carried out in 573 patients with SLE at 37 centers in France from June 2007 through August 2010.
Of the 573 patients, researchers randomized 171 to one of two treatment groups: 84 to no daily dose change (group A), and 87 to increased HCQ dose to achieve the target of 1,000 g/mL or greater (group B). The primary endpoint was the number of patients with flares during 7 months of follow-up (Ann. Rheum. Dis. 2013;72:1786-92).
At the time of randomization, the mean age of patients was 40 years, 87% were female, and their average HCQ dose was 400 mg/day. At 7 months of follow-up, the proportion of SLE flare rates was similar between the two groups (25% in group A vs. 27.6% in group B; P = .7). In a subset analysis of 57 patients from group A whose HCQ values were below 1,000 ng/mL after randomization and 39 patients from group B who maintained the therapeutic target dose of 1,000 ng/mL or higher after randomization, patients in group B tended to fewer flares compared with their counterparts in group A (20.5% vs. 35.1%, respectively; P = .12).
One reason that HCQ dosing above 1,000 ng/mL did not reduce the rate of SLE flares during the study period "may be that higher doses do not have an added therapeutic effect," the researchers speculated. "However, several factors may provide an alternative explanation of why our study did not provide its primary hypothesis." For one, the maintenance of HCQ above 1,000 ng/mL during the 7 months of follow up "was difficult to achieve." This could be explained by two factors, they continued. "The first is the pharmacokinetic variations of HCQ, but this explanation is unlikely because HCQ has a long terminal half-life and these patients were thought to be in a steady state. The second potential explanation might be adherence problems, even though known or suspected non-adherence was a major criterion for exclusion in our study. We found 10 patients with [HCQ] sufficiently low at inclusion to constitute an objective marker of lack of compliance."
The researchers acknowledged certain limitations of the study, including the potential for being underpowered (171 patients studied though the calculated sample size called for 200), and that the trend toward lower SLE flares in patients with higher HCQ "must be interpreted cautiously, since this analysis was not performed according to randomization group. The result might also be explained by better adherence to other medications, especially steroids."
The study was funded by a grant from the French PHRC 2005 Ministère de la Santé. Sanofi provided the HCQ and placebo tablets.
FROM ANNALS OF THE RHEUMATIC DISEASES
Major finding: At 7 months of follow-up, the flare rates in patients with systemic lupus erythematosus were similar between those who received no change in hydroxychloroquine (HCQ) dosing and those whose HCQ dose was increased to achieve a target level of 1,000 ng/mL or greater (25% vs. 27.6%, respectively).
Data source: A study of 171 adults with SLE who were randomized to one of the two treatment groups at 37 centers in France from June 2007 through August 2010.
Disclosures: The study was funded by a grant from the French PHRC 2005 Ministère de la Santé. Sanofi provided the study drug and the placebo tablets.
Investigational biologic lessened symptoms in some patients with lupus
An investigational biologic that targets T cells in patients with systemic lupus erythematosus significantly lessened symptoms in a subset of patients who completed a 24-week randomized trial.
In the overall analysis, the drug – rigerimod (Lupuzor) – did not perform significantly better than did placebo. But a post hoc analysis of patients with a higher clinical disease score found better results, with a response rate of 84% vs. 46% who took placebo, Dr. Robert Zimmer and his colleagues reported in the November issue of the Annals of the Rheumatic Diseases (2013 [doi:10.1136/annrheumdis-2012-202460]).
The drug was well tolerated, with 40% of the treatment group experiencing at least one side effect, compared with 49% of the placebo group, wrote Dr. Zimmer, president and chief science officer of ImmuPharma, the French company that is developing rigerimod.
Rigerimod is a made from a small nuclear ribonucleoprotein. Studies suggest that it inhibits T-cell reactivity, thereby reducing proteinuria, vasculitis, and dermatitis. It also prevented the production of antibodies to DNA from a lupus-model mouse.
The phase II study comprised an intent-to-treat group 149 patients who had baseline total scores of 6 or more on the SLEDAI-2K (Systemic Lupus Erythematosus Disease Activity Index-2K). They were randomized to three arms: subcutaneous injections of 200 mcg rigerimod every 4 weeks (group 1), every 2 weeks (group 2), or placebo injections.
All patients were treated for 12 weeks and then followed for an additional 12 weeks.
Most (96%) were women; their mean age at enrollment was about 38 years. The mean disease duration was about 8 years. The mean SLEDAI-2K total score was 11, and the mean physician’s global assessment (PGA), slightly more than 1. More than 80% were taking corticosteroids; about half were taking concomitant antimalarials and a quarter, concomitant azathioprine. Less than 5% were taking methotrexate.
Responders were considered those who had a reduction of at least 4 points in their baseline SLEDAI-2K score, with no increase in the PGA score. Treatment failures were those who had a severe disease flare, needed a steroid increase of at least 80 mg prednisone equivalent, or received biologics. Patients could continue their existing, stable medication regimens during the study.
The dropout rate was 9% (13 patients) – 8% of group 1, 2% of group 2 and 16% of the placebo group.
By the end of the 12-week treatment period, the response rate was significantly better in group 1 than in the placebo group (53% vs. 36%). Response in group 2 was not significantly different from group 1(45%).
By the end of week 24, the response rate had increased similarly in the entire study population (group 1, 59%: group 2, 59%; placebo, 53%). These rates were not significantly different from one another.
The target group analysis was conducted after the study’s end. The investigators restricted this group to patients whose SLEDAI-2K clinical –not overall – score was at least 6. "Despite the fact that this population had not been defined at the beginning of the study, the changes in the inclusion criteria led us to carefully analyze this population [clinical SLEDAI score of 6 and above] as this population will be the phase III population," they noted.
At week 12 in this population, there were significantly more responders in group 1 than in the placebo group (62% vs. 39%). The difference between group 2 and placebo was not statistically significant.
By week 24, groups 1 and 2 had similar response rates (69% and 62%), but these were not significantly higher than placebo (56%), with p values of 0.11 for group 1 vs. placebo and 0.28 for group 2 vs. placebo.
The investigators did not break down response by symptom, but did say "The apparent clinical benefit observed f or patients who received 200 mcg Lupuzor every 4 weeks, compared with those who received placebo every 2 weeks, were mainly due to an improvement in articular and cutaneous symptoms (arthritis and rash) at week 12."
The incidence of adverse events was low and similar between the groups; 40% of patients in each active group experienced at least one, compared with 49% of patients in the placebo group. The most common were injection site reactions, which occurred in 6% of group 1, 10% of group 2, and 2% of the placebo group.
One patient died during the trial. The cause of death was pneumonia, which was not deemed related to the study drug. The investigators noted that this patient had been on immunosuppressant therapy before entering the trial. Two other patients also developed pneumonia. "Thus," the investigators said, "while there did not appear to be an increased incidence of serious infections with Lupuzor in this short study, longer-term studies are needed to further characterise the overall tolerability profile of Lupuzor."
Finally, the investigators suggested that the clinical SLEDAI score, rather than the overall score, is the best way to assess this drug. "Comparing the number of responders in the target population and those of the overall population it appears that there is no change. Exactly the same number of responders was recorded in the three groups in the overall population and the target population. This indicates that the commonly used inclusion criterion [SLEDAI score of 6 and above] creates a bias in the evaluation of the study results in falsely reducing the response rate and therefore statistical analysis."
ImmuPharma is developing the drug and conducted the study.
An investigational biologic that targets T cells in patients with systemic lupus erythematosus significantly lessened symptoms in a subset of patients who completed a 24-week randomized trial.
In the overall analysis, the drug – rigerimod (Lupuzor) – did not perform significantly better than did placebo. But a post hoc analysis of patients with a higher clinical disease score found better results, with a response rate of 84% vs. 46% who took placebo, Dr. Robert Zimmer and his colleagues reported in the November issue of the Annals of the Rheumatic Diseases (2013 [doi:10.1136/annrheumdis-2012-202460]).
The drug was well tolerated, with 40% of the treatment group experiencing at least one side effect, compared with 49% of the placebo group, wrote Dr. Zimmer, president and chief science officer of ImmuPharma, the French company that is developing rigerimod.
Rigerimod is a made from a small nuclear ribonucleoprotein. Studies suggest that it inhibits T-cell reactivity, thereby reducing proteinuria, vasculitis, and dermatitis. It also prevented the production of antibodies to DNA from a lupus-model mouse.
The phase II study comprised an intent-to-treat group 149 patients who had baseline total scores of 6 or more on the SLEDAI-2K (Systemic Lupus Erythematosus Disease Activity Index-2K). They were randomized to three arms: subcutaneous injections of 200 mcg rigerimod every 4 weeks (group 1), every 2 weeks (group 2), or placebo injections.
All patients were treated for 12 weeks and then followed for an additional 12 weeks.
Most (96%) were women; their mean age at enrollment was about 38 years. The mean disease duration was about 8 years. The mean SLEDAI-2K total score was 11, and the mean physician’s global assessment (PGA), slightly more than 1. More than 80% were taking corticosteroids; about half were taking concomitant antimalarials and a quarter, concomitant azathioprine. Less than 5% were taking methotrexate.
Responders were considered those who had a reduction of at least 4 points in their baseline SLEDAI-2K score, with no increase in the PGA score. Treatment failures were those who had a severe disease flare, needed a steroid increase of at least 80 mg prednisone equivalent, or received biologics. Patients could continue their existing, stable medication regimens during the study.
The dropout rate was 9% (13 patients) – 8% of group 1, 2% of group 2 and 16% of the placebo group.
By the end of the 12-week treatment period, the response rate was significantly better in group 1 than in the placebo group (53% vs. 36%). Response in group 2 was not significantly different from group 1(45%).
By the end of week 24, the response rate had increased similarly in the entire study population (group 1, 59%: group 2, 59%; placebo, 53%). These rates were not significantly different from one another.
The target group analysis was conducted after the study’s end. The investigators restricted this group to patients whose SLEDAI-2K clinical –not overall – score was at least 6. "Despite the fact that this population had not been defined at the beginning of the study, the changes in the inclusion criteria led us to carefully analyze this population [clinical SLEDAI score of 6 and above] as this population will be the phase III population," they noted.
At week 12 in this population, there were significantly more responders in group 1 than in the placebo group (62% vs. 39%). The difference between group 2 and placebo was not statistically significant.
By week 24, groups 1 and 2 had similar response rates (69% and 62%), but these were not significantly higher than placebo (56%), with p values of 0.11 for group 1 vs. placebo and 0.28 for group 2 vs. placebo.
The investigators did not break down response by symptom, but did say "The apparent clinical benefit observed f or patients who received 200 mcg Lupuzor every 4 weeks, compared with those who received placebo every 2 weeks, were mainly due to an improvement in articular and cutaneous symptoms (arthritis and rash) at week 12."
The incidence of adverse events was low and similar between the groups; 40% of patients in each active group experienced at least one, compared with 49% of patients in the placebo group. The most common were injection site reactions, which occurred in 6% of group 1, 10% of group 2, and 2% of the placebo group.
One patient died during the trial. The cause of death was pneumonia, which was not deemed related to the study drug. The investigators noted that this patient had been on immunosuppressant therapy before entering the trial. Two other patients also developed pneumonia. "Thus," the investigators said, "while there did not appear to be an increased incidence of serious infections with Lupuzor in this short study, longer-term studies are needed to further characterise the overall tolerability profile of Lupuzor."
Finally, the investigators suggested that the clinical SLEDAI score, rather than the overall score, is the best way to assess this drug. "Comparing the number of responders in the target population and those of the overall population it appears that there is no change. Exactly the same number of responders was recorded in the three groups in the overall population and the target population. This indicates that the commonly used inclusion criterion [SLEDAI score of 6 and above] creates a bias in the evaluation of the study results in falsely reducing the response rate and therefore statistical analysis."
ImmuPharma is developing the drug and conducted the study.
An investigational biologic that targets T cells in patients with systemic lupus erythematosus significantly lessened symptoms in a subset of patients who completed a 24-week randomized trial.
In the overall analysis, the drug – rigerimod (Lupuzor) – did not perform significantly better than did placebo. But a post hoc analysis of patients with a higher clinical disease score found better results, with a response rate of 84% vs. 46% who took placebo, Dr. Robert Zimmer and his colleagues reported in the November issue of the Annals of the Rheumatic Diseases (2013 [doi:10.1136/annrheumdis-2012-202460]).
The drug was well tolerated, with 40% of the treatment group experiencing at least one side effect, compared with 49% of the placebo group, wrote Dr. Zimmer, president and chief science officer of ImmuPharma, the French company that is developing rigerimod.
Rigerimod is a made from a small nuclear ribonucleoprotein. Studies suggest that it inhibits T-cell reactivity, thereby reducing proteinuria, vasculitis, and dermatitis. It also prevented the production of antibodies to DNA from a lupus-model mouse.
The phase II study comprised an intent-to-treat group 149 patients who had baseline total scores of 6 or more on the SLEDAI-2K (Systemic Lupus Erythematosus Disease Activity Index-2K). They were randomized to three arms: subcutaneous injections of 200 mcg rigerimod every 4 weeks (group 1), every 2 weeks (group 2), or placebo injections.
All patients were treated for 12 weeks and then followed for an additional 12 weeks.
Most (96%) were women; their mean age at enrollment was about 38 years. The mean disease duration was about 8 years. The mean SLEDAI-2K total score was 11, and the mean physician’s global assessment (PGA), slightly more than 1. More than 80% were taking corticosteroids; about half were taking concomitant antimalarials and a quarter, concomitant azathioprine. Less than 5% were taking methotrexate.
Responders were considered those who had a reduction of at least 4 points in their baseline SLEDAI-2K score, with no increase in the PGA score. Treatment failures were those who had a severe disease flare, needed a steroid increase of at least 80 mg prednisone equivalent, or received biologics. Patients could continue their existing, stable medication regimens during the study.
The dropout rate was 9% (13 patients) – 8% of group 1, 2% of group 2 and 16% of the placebo group.
By the end of the 12-week treatment period, the response rate was significantly better in group 1 than in the placebo group (53% vs. 36%). Response in group 2 was not significantly different from group 1(45%).
By the end of week 24, the response rate had increased similarly in the entire study population (group 1, 59%: group 2, 59%; placebo, 53%). These rates were not significantly different from one another.
The target group analysis was conducted after the study’s end. The investigators restricted this group to patients whose SLEDAI-2K clinical –not overall – score was at least 6. "Despite the fact that this population had not been defined at the beginning of the study, the changes in the inclusion criteria led us to carefully analyze this population [clinical SLEDAI score of 6 and above] as this population will be the phase III population," they noted.
At week 12 in this population, there were significantly more responders in group 1 than in the placebo group (62% vs. 39%). The difference between group 2 and placebo was not statistically significant.
By week 24, groups 1 and 2 had similar response rates (69% and 62%), but these were not significantly higher than placebo (56%), with p values of 0.11 for group 1 vs. placebo and 0.28 for group 2 vs. placebo.
The investigators did not break down response by symptom, but did say "The apparent clinical benefit observed f or patients who received 200 mcg Lupuzor every 4 weeks, compared with those who received placebo every 2 weeks, were mainly due to an improvement in articular and cutaneous symptoms (arthritis and rash) at week 12."
The incidence of adverse events was low and similar between the groups; 40% of patients in each active group experienced at least one, compared with 49% of patients in the placebo group. The most common were injection site reactions, which occurred in 6% of group 1, 10% of group 2, and 2% of the placebo group.
One patient died during the trial. The cause of death was pneumonia, which was not deemed related to the study drug. The investigators noted that this patient had been on immunosuppressant therapy before entering the trial. Two other patients also developed pneumonia. "Thus," the investigators said, "while there did not appear to be an increased incidence of serious infections with Lupuzor in this short study, longer-term studies are needed to further characterise the overall tolerability profile of Lupuzor."
Finally, the investigators suggested that the clinical SLEDAI score, rather than the overall score, is the best way to assess this drug. "Comparing the number of responders in the target population and those of the overall population it appears that there is no change. Exactly the same number of responders was recorded in the three groups in the overall population and the target population. This indicates that the commonly used inclusion criterion [SLEDAI score of 6 and above] creates a bias in the evaluation of the study results in falsely reducing the response rate and therefore statistical analysis."
ImmuPharma is developing the drug and conducted the study.
Major finding: The investigational biologic rigerimod was superior to placebo in patients with a higher lupus clinical disease activity score, with a response rate of 84% vs. 46%.
Data source: The 24-week randomized, phase IIb trial involved 149 patients.
Disclosures: ImmuPharma is developing rigerimod and conducted the study. Dr. Zimmer is the company’s president and chief scientific officer.
Secukinumab soars in phase III psoriasis studies
ISTANBUL, TURKEY – The investigational interleukin-17A inhibitor secukinumab was the talk of Europe’s premier dermatology conference as a result of a series of world-premiere presentations of not one, but three, phase III clinical trials.
The entire secukinumab pivotal phase III program results were presented together at the annual congress of the European Academy of Dermatology and Venereology. Based on these promising results, Novartis plans to file for Food and Drug Administration and European regulatory approval before the end of 2013.
The three randomized, double-blind, multicenter pivotal trials of secukinumab in psoriasis collectively included 3,367 patients with moderate to severe chronic plaque psoriasis. All three studies were strongly positive. Efficacy rates were higher than with current biologics, clinical improvement was remarkably rapid, and the safety profile was reassuring.
"Those of us working in this area of research are really excited by this new data and what it says for the treatment of psoriasis patients," commented Dr. Richard Langley of Dalhousie University, Halifax, N.S.
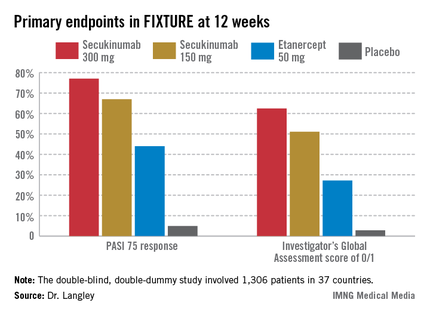
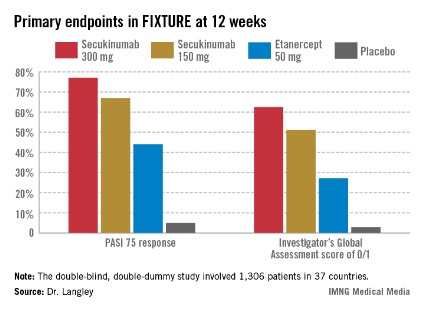
He presented the centerpiece of this trio of phase III trials, known as the FIXTURE trial. FIXTURE (Full Year Investigative Examination of Secukinumab vs. Etanercept Using 2 Dosing Regimens to Determine Efficacy in Psoriasis) was a 1,306-patient, double-blind, double-dummy trial conducted by 153 investigators in 37 countries. The trial featured a 1-year head-to-head comparison of secukinumab and the widely prescribed tumor necrosis factor (TNF) inhibitor etanercept (Enbrel). The IL-17A inhibitor outperformed etanercept in all of the prespecified primary and secondary outcome measures. Moreover, the two biologics proved equally safe.
"I think it’s interesting in comparing these two molecules that when we look at the data to date, we see comparable safety results. Etanercept is one of the most widely used biologics and has an excellent safety record. That’s reassuring for those of us who are investigating IL-17 signaling," observed Dr. Langley, also the current president of the Canadian Dermatology Association.
FIXTURE participants had a mean baseline Psoriasis Area and Severity Index (PASI) score of 24, an average body mass index of 34 kg/m2, and a mean age in the mid-40s, and were previous inadequate responders to conventional systemic agents and/or biologics. The two co-primary endpoints were 12-week rates of at least a 75% reduction in PASI score, or PASI 75 response, and an Investigator’s Global Assessment (IGA) rating of 0/1, corresponding to clear or nearly clear, on a modified 5-point scale that provides more clinically meaningful information than the older 6-point IGA scale. Patients randomized to the secukinumab 300-mg or 150-mg groups did significantly better on both outcome measures than those assigned to etanercept.
The onset of benefit with secukinumab was swift; a statistically significant improvement compared with etanercept was seen within 2 weeks in terms of IGA scores and within 3 weeks for PASI 75. Secukinumab at 300 mg per subcutaneous injection – the most effective dose across all three pivotal trials – achieved roughly a 50% reduction in PASI scores after 3 weeks, compared with 8 weeks for etanercept, the dermatologist explained.
The PASI 75 response rate at 12 weeks was 77.1% in patients receiving secukinumab at 300 mg, 67% for those receiving secukinumab at 150 mg, 44% for etanercept 50 mg, and 4.9% for placebo.
The highest response rates with secukinumab were seen at week 16 rather than week 12. In the secukinumab 300-mg group, the PASI 75 rate at week 16 was 86.7%, with that high level of response being retained throughout the remainder of the 52-week study. In contrast, the PASI 75 rate at week 16 for etanercept was less than 60%. The IGA 0/1 rate at week 16 was 75.5% in the secukinumab 300-mg group and 60% with secukinumab 150 mg. The peak IGA 0/1 rate in the etanercept arm was only about 45%, and it came much later, at week 26.
Looking at more stringent secondary endpoints, the investigators found that a PASI 90 response was achieved by week 16 in 72.4% of the high-dose secukinumab group, compared with about 30% with etanercept. Twenty-four percent of patients on high-dose secukinumab had a PASI 100 response at week 12, compared with 4% on etanercept. By week 16, 36.8% of the high-dose secukinumab group had a PASI 100 response. At week 52, 65% of the secukinumab 300-mg group maintained a PASI 90 response, compared with 33% of the etanercept group.
The most common adverse event documented in FIXTURE was nasopharyngitis, which occurred in roughly one-third of patients in all four study arms. The serious infection rate was 1%-1.2% in all four study arms. No signals of an increase in malignancies or major adverse cardiovascular events were noted in any of the active treatment groups. Overall, adverse events were similar in both secukinumab arms and comparable to etanercept. Injection-site reactions occurred in 6.1% of the etanercept group and in none of the patients assigned to secukinumab or placebo.

The incidence of treatment-emergent antidrug antibodies in the secukinumab arms was 0.4%, and when it occurred, it had no impact on treatment efficacy or safety. Patients on etanercept weren’t tested for the emergence of antidrug antibodies.
Candida infections occurred in 4.7% of patients on secukinumab 300 mg, 2.3% of those on 150 mg, and 1.2% of the etanercept group. All cases in patients on secukinumab were mild or moderate and easily treated. Candida infections are a side effect of special interest because patients with a genetic defect in the IL-17 pathway can develop chronic mucocutaneous candidiasis. A theoretical concern that pharmacologic inhibition of IL-17 might create a big problem in this regard has not materialized.
The other two phase III secukinumab clinical trials presented in Istanbul – ERASURE and SCULPTURE – showed efficacy and safety results similar to those of FIXTURE. Unlike FIXTURE, neither of those trials featured a head-to-head comparison with another biologic.
These were the first completed phase III studies for any of the three biologics in this novel, highly promising class targeting IL-17 in the treatment of psoriasis and other immune-mediated inflammatory diseases. The other two agents in the pipeline targeting IL-17, brodalumab and ixekizumab, remain in ongoing phase III trials.
Because of the pivotal role IL-17 plays in generating inflammatory cytokines downstream, all three drugs are being studied for additional indications beyond psoriasis. For example, secukinumab, an IgG1 human monoclonal antibody, is in phase III clinical trials for psoriatic arthritis, ankylosing spondylitis, and rheumatoid arthritis, and in phase II for muscular sclerosis and asthma.
One audience member asked Dr. Langley for his thoughts regarding the absence of injection-site reactions in the secukinumab-treated patients. He replied that one obvious factor is that secukinumab injections are given much less frequently: once monthly during maintenance therapy as compared to once weekly with etanercept. In any event, he doesn’t consider injection-site reactions to be clinically relevant.
"Injection-site reactions don’t seem to be a major issue with any of the subcutaneous therapies. It has never in 15 years of using different biologics been an issue that’s made me stop treatment for a patient," he added.
Secukinumab was dosed at baseline, again weekly through week 4, then once monthly from week 8 through the study’s end. Etanercept was dosed at 50 mg twice weekly for 12 weeks, then once weekly in accord with the labeling instructions in most countries. Nonresponders at 12 weeks in the placebo arm were at that point randomized to secukinumab at 300 mg or 150 mg. The statistical analysis in FIXTURE was performed by nonresponder imputation.
"Those who are interested in statistics will know that that’s the most rigorous way that you can look at the data, because anybody who drops out of the study, whether they withdraw consent, have a protocol violation, or have an adverse event, even if they were responding at that time, are considered a failure or nonresponder," Dr. Langley explained.
Asked how he sees secukinumab fitting into patient management, Dr. Langley replied, "Most of us are forced to use conventional therapies and phototherapy to get approval for biologics, but after that I think the safety and efficacy of this drug put it right at the top. I could definitely use this drug as a first-line biologic for patients with plain psoriasis without psoriatic arthritis."
Coinvestigator Dr. Kristian Reich, professor of dermatology at Georg-August University in Göttingen, Germany, said he will probably continue to use TNF inhibitors in psoriasis patients with prominent arthritis, where he believes the TNF inhibitors are particularly beneficial, but he anticipates using secukinumab widely in others.
Dr. Bruce E. Strober, another active clinical trial researcher in psoriasis, said he likes what he sees in the IL-17 inhibitor data.
"I predict as a specialty we’ll become very confident in these drugs 5 years from now. A lot of us will be using them as first line in many patients. That’s my prediction. For now, though, I’m a big fan of TNF inhibition," added Dr. Strober, vice chair of dermatology at the University of Connecticut Medical Center, Farmington.
The secukinumab development program is sponsored by Novartis. Dr. Langley, Dr. Reich, and Dr. Strober disclosed having received research grants from and serving as consultants to Novartis and numerous other companies developing biologic agents for psoriasis.
ISTANBUL, TURKEY – The investigational interleukin-17A inhibitor secukinumab was the talk of Europe’s premier dermatology conference as a result of a series of world-premiere presentations of not one, but three, phase III clinical trials.
The entire secukinumab pivotal phase III program results were presented together at the annual congress of the European Academy of Dermatology and Venereology. Based on these promising results, Novartis plans to file for Food and Drug Administration and European regulatory approval before the end of 2013.
The three randomized, double-blind, multicenter pivotal trials of secukinumab in psoriasis collectively included 3,367 patients with moderate to severe chronic plaque psoriasis. All three studies were strongly positive. Efficacy rates were higher than with current biologics, clinical improvement was remarkably rapid, and the safety profile was reassuring.
"Those of us working in this area of research are really excited by this new data and what it says for the treatment of psoriasis patients," commented Dr. Richard Langley of Dalhousie University, Halifax, N.S.
He presented the centerpiece of this trio of phase III trials, known as the FIXTURE trial. FIXTURE (Full Year Investigative Examination of Secukinumab vs. Etanercept Using 2 Dosing Regimens to Determine Efficacy in Psoriasis) was a 1,306-patient, double-blind, double-dummy trial conducted by 153 investigators in 37 countries. The trial featured a 1-year head-to-head comparison of secukinumab and the widely prescribed tumor necrosis factor (TNF) inhibitor etanercept (Enbrel). The IL-17A inhibitor outperformed etanercept in all of the prespecified primary and secondary outcome measures. Moreover, the two biologics proved equally safe.
"I think it’s interesting in comparing these two molecules that when we look at the data to date, we see comparable safety results. Etanercept is one of the most widely used biologics and has an excellent safety record. That’s reassuring for those of us who are investigating IL-17 signaling," observed Dr. Langley, also the current president of the Canadian Dermatology Association.
FIXTURE participants had a mean baseline Psoriasis Area and Severity Index (PASI) score of 24, an average body mass index of 34 kg/m2, and a mean age in the mid-40s, and were previous inadequate responders to conventional systemic agents and/or biologics. The two co-primary endpoints were 12-week rates of at least a 75% reduction in PASI score, or PASI 75 response, and an Investigator’s Global Assessment (IGA) rating of 0/1, corresponding to clear or nearly clear, on a modified 5-point scale that provides more clinically meaningful information than the older 6-point IGA scale. Patients randomized to the secukinumab 300-mg or 150-mg groups did significantly better on both outcome measures than those assigned to etanercept.
The onset of benefit with secukinumab was swift; a statistically significant improvement compared with etanercept was seen within 2 weeks in terms of IGA scores and within 3 weeks for PASI 75. Secukinumab at 300 mg per subcutaneous injection – the most effective dose across all three pivotal trials – achieved roughly a 50% reduction in PASI scores after 3 weeks, compared with 8 weeks for etanercept, the dermatologist explained.
The PASI 75 response rate at 12 weeks was 77.1% in patients receiving secukinumab at 300 mg, 67% for those receiving secukinumab at 150 mg, 44% for etanercept 50 mg, and 4.9% for placebo.
The highest response rates with secukinumab were seen at week 16 rather than week 12. In the secukinumab 300-mg group, the PASI 75 rate at week 16 was 86.7%, with that high level of response being retained throughout the remainder of the 52-week study. In contrast, the PASI 75 rate at week 16 for etanercept was less than 60%. The IGA 0/1 rate at week 16 was 75.5% in the secukinumab 300-mg group and 60% with secukinumab 150 mg. The peak IGA 0/1 rate in the etanercept arm was only about 45%, and it came much later, at week 26.
Looking at more stringent secondary endpoints, the investigators found that a PASI 90 response was achieved by week 16 in 72.4% of the high-dose secukinumab group, compared with about 30% with etanercept. Twenty-four percent of patients on high-dose secukinumab had a PASI 100 response at week 12, compared with 4% on etanercept. By week 16, 36.8% of the high-dose secukinumab group had a PASI 100 response. At week 52, 65% of the secukinumab 300-mg group maintained a PASI 90 response, compared with 33% of the etanercept group.
The most common adverse event documented in FIXTURE was nasopharyngitis, which occurred in roughly one-third of patients in all four study arms. The serious infection rate was 1%-1.2% in all four study arms. No signals of an increase in malignancies or major adverse cardiovascular events were noted in any of the active treatment groups. Overall, adverse events were similar in both secukinumab arms and comparable to etanercept. Injection-site reactions occurred in 6.1% of the etanercept group and in none of the patients assigned to secukinumab or placebo.
The incidence of treatment-emergent antidrug antibodies in the secukinumab arms was 0.4%, and when it occurred, it had no impact on treatment efficacy or safety. Patients on etanercept weren’t tested for the emergence of antidrug antibodies.
Candida infections occurred in 4.7% of patients on secukinumab 300 mg, 2.3% of those on 150 mg, and 1.2% of the etanercept group. All cases in patients on secukinumab were mild or moderate and easily treated. Candida infections are a side effect of special interest because patients with a genetic defect in the IL-17 pathway can develop chronic mucocutaneous candidiasis. A theoretical concern that pharmacologic inhibition of IL-17 might create a big problem in this regard has not materialized.
The other two phase III secukinumab clinical trials presented in Istanbul – ERASURE and SCULPTURE – showed efficacy and safety results similar to those of FIXTURE. Unlike FIXTURE, neither of those trials featured a head-to-head comparison with another biologic.
These were the first completed phase III studies for any of the three biologics in this novel, highly promising class targeting IL-17 in the treatment of psoriasis and other immune-mediated inflammatory diseases. The other two agents in the pipeline targeting IL-17, brodalumab and ixekizumab, remain in ongoing phase III trials.
Because of the pivotal role IL-17 plays in generating inflammatory cytokines downstream, all three drugs are being studied for additional indications beyond psoriasis. For example, secukinumab, an IgG1 human monoclonal antibody, is in phase III clinical trials for psoriatic arthritis, ankylosing spondylitis, and rheumatoid arthritis, and in phase II for muscular sclerosis and asthma.
One audience member asked Dr. Langley for his thoughts regarding the absence of injection-site reactions in the secukinumab-treated patients. He replied that one obvious factor is that secukinumab injections are given much less frequently: once monthly during maintenance therapy as compared to once weekly with etanercept. In any event, he doesn’t consider injection-site reactions to be clinically relevant.
"Injection-site reactions don’t seem to be a major issue with any of the subcutaneous therapies. It has never in 15 years of using different biologics been an issue that’s made me stop treatment for a patient," he added.
Secukinumab was dosed at baseline, again weekly through week 4, then once monthly from week 8 through the study’s end. Etanercept was dosed at 50 mg twice weekly for 12 weeks, then once weekly in accord with the labeling instructions in most countries. Nonresponders at 12 weeks in the placebo arm were at that point randomized to secukinumab at 300 mg or 150 mg. The statistical analysis in FIXTURE was performed by nonresponder imputation.
"Those who are interested in statistics will know that that’s the most rigorous way that you can look at the data, because anybody who drops out of the study, whether they withdraw consent, have a protocol violation, or have an adverse event, even if they were responding at that time, are considered a failure or nonresponder," Dr. Langley explained.
Asked how he sees secukinumab fitting into patient management, Dr. Langley replied, "Most of us are forced to use conventional therapies and phototherapy to get approval for biologics, but after that I think the safety and efficacy of this drug put it right at the top. I could definitely use this drug as a first-line biologic for patients with plain psoriasis without psoriatic arthritis."
Coinvestigator Dr. Kristian Reich, professor of dermatology at Georg-August University in Göttingen, Germany, said he will probably continue to use TNF inhibitors in psoriasis patients with prominent arthritis, where he believes the TNF inhibitors are particularly beneficial, but he anticipates using secukinumab widely in others.
Dr. Bruce E. Strober, another active clinical trial researcher in psoriasis, said he likes what he sees in the IL-17 inhibitor data.
"I predict as a specialty we’ll become very confident in these drugs 5 years from now. A lot of us will be using them as first line in many patients. That’s my prediction. For now, though, I’m a big fan of TNF inhibition," added Dr. Strober, vice chair of dermatology at the University of Connecticut Medical Center, Farmington.
The secukinumab development program is sponsored by Novartis. Dr. Langley, Dr. Reich, and Dr. Strober disclosed having received research grants from and serving as consultants to Novartis and numerous other companies developing biologic agents for psoriasis.
ISTANBUL, TURKEY – The investigational interleukin-17A inhibitor secukinumab was the talk of Europe’s premier dermatology conference as a result of a series of world-premiere presentations of not one, but three, phase III clinical trials.
The entire secukinumab pivotal phase III program results were presented together at the annual congress of the European Academy of Dermatology and Venereology. Based on these promising results, Novartis plans to file for Food and Drug Administration and European regulatory approval before the end of 2013.
The three randomized, double-blind, multicenter pivotal trials of secukinumab in psoriasis collectively included 3,367 patients with moderate to severe chronic plaque psoriasis. All three studies were strongly positive. Efficacy rates were higher than with current biologics, clinical improvement was remarkably rapid, and the safety profile was reassuring.
"Those of us working in this area of research are really excited by this new data and what it says for the treatment of psoriasis patients," commented Dr. Richard Langley of Dalhousie University, Halifax, N.S.
He presented the centerpiece of this trio of phase III trials, known as the FIXTURE trial. FIXTURE (Full Year Investigative Examination of Secukinumab vs. Etanercept Using 2 Dosing Regimens to Determine Efficacy in Psoriasis) was a 1,306-patient, double-blind, double-dummy trial conducted by 153 investigators in 37 countries. The trial featured a 1-year head-to-head comparison of secukinumab and the widely prescribed tumor necrosis factor (TNF) inhibitor etanercept (Enbrel). The IL-17A inhibitor outperformed etanercept in all of the prespecified primary and secondary outcome measures. Moreover, the two biologics proved equally safe.
"I think it’s interesting in comparing these two molecules that when we look at the data to date, we see comparable safety results. Etanercept is one of the most widely used biologics and has an excellent safety record. That’s reassuring for those of us who are investigating IL-17 signaling," observed Dr. Langley, also the current president of the Canadian Dermatology Association.
FIXTURE participants had a mean baseline Psoriasis Area and Severity Index (PASI) score of 24, an average body mass index of 34 kg/m2, and a mean age in the mid-40s, and were previous inadequate responders to conventional systemic agents and/or biologics. The two co-primary endpoints were 12-week rates of at least a 75% reduction in PASI score, or PASI 75 response, and an Investigator’s Global Assessment (IGA) rating of 0/1, corresponding to clear or nearly clear, on a modified 5-point scale that provides more clinically meaningful information than the older 6-point IGA scale. Patients randomized to the secukinumab 300-mg or 150-mg groups did significantly better on both outcome measures than those assigned to etanercept.
The onset of benefit with secukinumab was swift; a statistically significant improvement compared with etanercept was seen within 2 weeks in terms of IGA scores and within 3 weeks for PASI 75. Secukinumab at 300 mg per subcutaneous injection – the most effective dose across all three pivotal trials – achieved roughly a 50% reduction in PASI scores after 3 weeks, compared with 8 weeks for etanercept, the dermatologist explained.
The PASI 75 response rate at 12 weeks was 77.1% in patients receiving secukinumab at 300 mg, 67% for those receiving secukinumab at 150 mg, 44% for etanercept 50 mg, and 4.9% for placebo.
The highest response rates with secukinumab were seen at week 16 rather than week 12. In the secukinumab 300-mg group, the PASI 75 rate at week 16 was 86.7%, with that high level of response being retained throughout the remainder of the 52-week study. In contrast, the PASI 75 rate at week 16 for etanercept was less than 60%. The IGA 0/1 rate at week 16 was 75.5% in the secukinumab 300-mg group and 60% with secukinumab 150 mg. The peak IGA 0/1 rate in the etanercept arm was only about 45%, and it came much later, at week 26.
Looking at more stringent secondary endpoints, the investigators found that a PASI 90 response was achieved by week 16 in 72.4% of the high-dose secukinumab group, compared with about 30% with etanercept. Twenty-four percent of patients on high-dose secukinumab had a PASI 100 response at week 12, compared with 4% on etanercept. By week 16, 36.8% of the high-dose secukinumab group had a PASI 100 response. At week 52, 65% of the secukinumab 300-mg group maintained a PASI 90 response, compared with 33% of the etanercept group.
The most common adverse event documented in FIXTURE was nasopharyngitis, which occurred in roughly one-third of patients in all four study arms. The serious infection rate was 1%-1.2% in all four study arms. No signals of an increase in malignancies or major adverse cardiovascular events were noted in any of the active treatment groups. Overall, adverse events were similar in both secukinumab arms and comparable to etanercept. Injection-site reactions occurred in 6.1% of the etanercept group and in none of the patients assigned to secukinumab or placebo.
The incidence of treatment-emergent antidrug antibodies in the secukinumab arms was 0.4%, and when it occurred, it had no impact on treatment efficacy or safety. Patients on etanercept weren’t tested for the emergence of antidrug antibodies.
Candida infections occurred in 4.7% of patients on secukinumab 300 mg, 2.3% of those on 150 mg, and 1.2% of the etanercept group. All cases in patients on secukinumab were mild or moderate and easily treated. Candida infections are a side effect of special interest because patients with a genetic defect in the IL-17 pathway can develop chronic mucocutaneous candidiasis. A theoretical concern that pharmacologic inhibition of IL-17 might create a big problem in this regard has not materialized.
The other two phase III secukinumab clinical trials presented in Istanbul – ERASURE and SCULPTURE – showed efficacy and safety results similar to those of FIXTURE. Unlike FIXTURE, neither of those trials featured a head-to-head comparison with another biologic.
These were the first completed phase III studies for any of the three biologics in this novel, highly promising class targeting IL-17 in the treatment of psoriasis and other immune-mediated inflammatory diseases. The other two agents in the pipeline targeting IL-17, brodalumab and ixekizumab, remain in ongoing phase III trials.
Because of the pivotal role IL-17 plays in generating inflammatory cytokines downstream, all three drugs are being studied for additional indications beyond psoriasis. For example, secukinumab, an IgG1 human monoclonal antibody, is in phase III clinical trials for psoriatic arthritis, ankylosing spondylitis, and rheumatoid arthritis, and in phase II for muscular sclerosis and asthma.
One audience member asked Dr. Langley for his thoughts regarding the absence of injection-site reactions in the secukinumab-treated patients. He replied that one obvious factor is that secukinumab injections are given much less frequently: once monthly during maintenance therapy as compared to once weekly with etanercept. In any event, he doesn’t consider injection-site reactions to be clinically relevant.
"Injection-site reactions don’t seem to be a major issue with any of the subcutaneous therapies. It has never in 15 years of using different biologics been an issue that’s made me stop treatment for a patient," he added.
Secukinumab was dosed at baseline, again weekly through week 4, then once monthly from week 8 through the study’s end. Etanercept was dosed at 50 mg twice weekly for 12 weeks, then once weekly in accord with the labeling instructions in most countries. Nonresponders at 12 weeks in the placebo arm were at that point randomized to secukinumab at 300 mg or 150 mg. The statistical analysis in FIXTURE was performed by nonresponder imputation.
"Those who are interested in statistics will know that that’s the most rigorous way that you can look at the data, because anybody who drops out of the study, whether they withdraw consent, have a protocol violation, or have an adverse event, even if they were responding at that time, are considered a failure or nonresponder," Dr. Langley explained.
Asked how he sees secukinumab fitting into patient management, Dr. Langley replied, "Most of us are forced to use conventional therapies and phototherapy to get approval for biologics, but after that I think the safety and efficacy of this drug put it right at the top. I could definitely use this drug as a first-line biologic for patients with plain psoriasis without psoriatic arthritis."
Coinvestigator Dr. Kristian Reich, professor of dermatology at Georg-August University in Göttingen, Germany, said he will probably continue to use TNF inhibitors in psoriasis patients with prominent arthritis, where he believes the TNF inhibitors are particularly beneficial, but he anticipates using secukinumab widely in others.
Dr. Bruce E. Strober, another active clinical trial researcher in psoriasis, said he likes what he sees in the IL-17 inhibitor data.
"I predict as a specialty we’ll become very confident in these drugs 5 years from now. A lot of us will be using them as first line in many patients. That’s my prediction. For now, though, I’m a big fan of TNF inhibition," added Dr. Strober, vice chair of dermatology at the University of Connecticut Medical Center, Farmington.
The secukinumab development program is sponsored by Novartis. Dr. Langley, Dr. Reich, and Dr. Strober disclosed having received research grants from and serving as consultants to Novartis and numerous other companies developing biologic agents for psoriasis.
AT THE EADV CONGRESS
Major finding: The PASI 75 response rate at 12 weeks in patients with moderate to severe chronic plaque psoriasis was 77.1% in those randomized to the investigational interleukin-17A inhibitor secukinumab at 300 mg, 67% for secukinumab at 150 mg, 44% for etanercept 50 mg, and 4.9% for placebo.
Data source: The FIXTURE trial was a randomized, double-blind, double-dummy, year-long, phase III trial involving 1,306 patients with chronic plaque psoriasis not adequately responsive to prior systemic therapies.
Disclosures: The secukinumab development program is sponsored by Novartis. Dr. Langley, Dr. Reich, and Dr. Strober disclosed having received research grants from and serving as consultants to Novartis and numerous other companies developing biologic agents for psoriasis.
Psoriasis linked to nonalcoholic fatty liver disease
ISTANBUL, TURKEY – The prevalence of psoriasis was significantly greater in subjects with nonalcoholic fatty liver disease by a margin of 7% to 4.2%, according to findings from a long-term, population-based cohort study including nearly 4,000 individuals.
This newly identified link between two common diseases – one of which manifests as visible, angry skin lesions; the other silent, insidious, and often unrecognized until its late stages – contains an important message for clinicians, said Dr. Ella van der Voort of Erasmus University in Rotterdam, the Netherlands.
"Physicians should be aware of this increased risk before prescribing for psoriasis patients medications with potential for liver toxicity – and I’m not talking about just psoriasis medications, but also medications in general, like statins," she said at the annual congress of the European Academy of Dermatology and Venereology.

Psoriasis was present in 5.1% of the study group. Most cases were mild, with affected patients having a mean psoriasis area severity index of 2.9 out of a possible 72 and no use of systemic psoriasis therapies. Nevertheless, the prevalence of nonalcoholic fatty liver disease (NAFLD) was 46.2% among the 118 psoriasis patients, compared with 33% in the 2,174 subjects without psoriasis.
NAFLD is now the most common form of chronic liver disease in Western countries. By 2020, when new well-tolerated and highly effective medications for hepatitis C are expected to have made a mark, NAFLD may be the leading reason for liver transplantation in developed countries, Dr. van der Voort observed.
The Rotterdam Study is designed to examine contributors to chronic diseases in the elderly, including cardiovascular, endocrinologic, psychiatric, neurologic, and eye diseases. The first wave of participants enrolled in 1989-93. Dr. van der Voort’s study focused on the third cohort, consisting of 3,932 elderly Rotterdam-area residents enrolled in 2006-2008. The mean age at enrollment was 76 years, and the ongoing routine evaluation of the cohort includes liver ultrasound, lab tests for liver enzyme levels and serologic evidence of hepatitis, and a skin examination.
Dr. van der Voort’s analysis was restricted to the 2,292 third-wave subjects who were not heavy drinkers and didn’t have chronic hepatitis, either of which rules out the diagnosis of NAFLD. The participants were not taking hepatotoxic medications, and there was no uncertainty as to the skin examination findings.
The hallmark of NAFLD is the accumulation of triglycerides in the hepatocytes. The previously well-described risk factors for NAFLD include the metabolic syndrome and its individual components, as well as sedentary lifestyle, smoking, and a poor-quality diet. There is also a genetic component: individuals with a mutation in the phospholipase domain-containing protein 3 (PNPLA3) gene, which codes for an enzyme functioning in hydrolyzation of triglycerides, are predisposed to NAFLD.
The prevalence of metabolic syndrome was 62% in subjects with psoriasis and 52% in those without psoriasis. Individuals with psoriasis also were more likely to be smokers by a margin of 15% to 8%. After adjusting for those variables, as well as alcohol intake, age, gender, liver enzyme levels, and other potential confounders, two strong independent predictors of NAFLD were metabolic syndrome, with an adjusted odds ratio of 3.5, and psoriasis, with an odds ratio of 1.7.
In a multivariate analysis, psoriasis was not only independently associated with a 70% increased likelihood of NAFLD, but psoriasis patients with NAFLD were also 60% more likely to have the more severe forms of NAFLD.
The Rotterdam study is funded chiefly by Dutch scientific research foundations, with secondary pharmaceutical industry support. Dr. van der Voort reported having no financial conflicts of interest.
ISTANBUL, TURKEY – The prevalence of psoriasis was significantly greater in subjects with nonalcoholic fatty liver disease by a margin of 7% to 4.2%, according to findings from a long-term, population-based cohort study including nearly 4,000 individuals.
This newly identified link between two common diseases – one of which manifests as visible, angry skin lesions; the other silent, insidious, and often unrecognized until its late stages – contains an important message for clinicians, said Dr. Ella van der Voort of Erasmus University in Rotterdam, the Netherlands.
"Physicians should be aware of this increased risk before prescribing for psoriasis patients medications with potential for liver toxicity – and I’m not talking about just psoriasis medications, but also medications in general, like statins," she said at the annual congress of the European Academy of Dermatology and Venereology.
Psoriasis was present in 5.1% of the study group. Most cases were mild, with affected patients having a mean psoriasis area severity index of 2.9 out of a possible 72 and no use of systemic psoriasis therapies. Nevertheless, the prevalence of nonalcoholic fatty liver disease (NAFLD) was 46.2% among the 118 psoriasis patients, compared with 33% in the 2,174 subjects without psoriasis.
NAFLD is now the most common form of chronic liver disease in Western countries. By 2020, when new well-tolerated and highly effective medications for hepatitis C are expected to have made a mark, NAFLD may be the leading reason for liver transplantation in developed countries, Dr. van der Voort observed.
The Rotterdam Study is designed to examine contributors to chronic diseases in the elderly, including cardiovascular, endocrinologic, psychiatric, neurologic, and eye diseases. The first wave of participants enrolled in 1989-93. Dr. van der Voort’s study focused on the third cohort, consisting of 3,932 elderly Rotterdam-area residents enrolled in 2006-2008. The mean age at enrollment was 76 years, and the ongoing routine evaluation of the cohort includes liver ultrasound, lab tests for liver enzyme levels and serologic evidence of hepatitis, and a skin examination.
Dr. van der Voort’s analysis was restricted to the 2,292 third-wave subjects who were not heavy drinkers and didn’t have chronic hepatitis, either of which rules out the diagnosis of NAFLD. The participants were not taking hepatotoxic medications, and there was no uncertainty as to the skin examination findings.
The hallmark of NAFLD is the accumulation of triglycerides in the hepatocytes. The previously well-described risk factors for NAFLD include the metabolic syndrome and its individual components, as well as sedentary lifestyle, smoking, and a poor-quality diet. There is also a genetic component: individuals with a mutation in the phospholipase domain-containing protein 3 (PNPLA3) gene, which codes for an enzyme functioning in hydrolyzation of triglycerides, are predisposed to NAFLD.
The prevalence of metabolic syndrome was 62% in subjects with psoriasis and 52% in those without psoriasis. Individuals with psoriasis also were more likely to be smokers by a margin of 15% to 8%. After adjusting for those variables, as well as alcohol intake, age, gender, liver enzyme levels, and other potential confounders, two strong independent predictors of NAFLD were metabolic syndrome, with an adjusted odds ratio of 3.5, and psoriasis, with an odds ratio of 1.7.
In a multivariate analysis, psoriasis was not only independently associated with a 70% increased likelihood of NAFLD, but psoriasis patients with NAFLD were also 60% more likely to have the more severe forms of NAFLD.
The Rotterdam study is funded chiefly by Dutch scientific research foundations, with secondary pharmaceutical industry support. Dr. van der Voort reported having no financial conflicts of interest.
ISTANBUL, TURKEY – The prevalence of psoriasis was significantly greater in subjects with nonalcoholic fatty liver disease by a margin of 7% to 4.2%, according to findings from a long-term, population-based cohort study including nearly 4,000 individuals.
This newly identified link between two common diseases – one of which manifests as visible, angry skin lesions; the other silent, insidious, and often unrecognized until its late stages – contains an important message for clinicians, said Dr. Ella van der Voort of Erasmus University in Rotterdam, the Netherlands.
"Physicians should be aware of this increased risk before prescribing for psoriasis patients medications with potential for liver toxicity – and I’m not talking about just psoriasis medications, but also medications in general, like statins," she said at the annual congress of the European Academy of Dermatology and Venereology.
Psoriasis was present in 5.1% of the study group. Most cases were mild, with affected patients having a mean psoriasis area severity index of 2.9 out of a possible 72 and no use of systemic psoriasis therapies. Nevertheless, the prevalence of nonalcoholic fatty liver disease (NAFLD) was 46.2% among the 118 psoriasis patients, compared with 33% in the 2,174 subjects without psoriasis.
NAFLD is now the most common form of chronic liver disease in Western countries. By 2020, when new well-tolerated and highly effective medications for hepatitis C are expected to have made a mark, NAFLD may be the leading reason for liver transplantation in developed countries, Dr. van der Voort observed.
The Rotterdam Study is designed to examine contributors to chronic diseases in the elderly, including cardiovascular, endocrinologic, psychiatric, neurologic, and eye diseases. The first wave of participants enrolled in 1989-93. Dr. van der Voort’s study focused on the third cohort, consisting of 3,932 elderly Rotterdam-area residents enrolled in 2006-2008. The mean age at enrollment was 76 years, and the ongoing routine evaluation of the cohort includes liver ultrasound, lab tests for liver enzyme levels and serologic evidence of hepatitis, and a skin examination.
Dr. van der Voort’s analysis was restricted to the 2,292 third-wave subjects who were not heavy drinkers and didn’t have chronic hepatitis, either of which rules out the diagnosis of NAFLD. The participants were not taking hepatotoxic medications, and there was no uncertainty as to the skin examination findings.
The hallmark of NAFLD is the accumulation of triglycerides in the hepatocytes. The previously well-described risk factors for NAFLD include the metabolic syndrome and its individual components, as well as sedentary lifestyle, smoking, and a poor-quality diet. There is also a genetic component: individuals with a mutation in the phospholipase domain-containing protein 3 (PNPLA3) gene, which codes for an enzyme functioning in hydrolyzation of triglycerides, are predisposed to NAFLD.
The prevalence of metabolic syndrome was 62% in subjects with psoriasis and 52% in those without psoriasis. Individuals with psoriasis also were more likely to be smokers by a margin of 15% to 8%. After adjusting for those variables, as well as alcohol intake, age, gender, liver enzyme levels, and other potential confounders, two strong independent predictors of NAFLD were metabolic syndrome, with an adjusted odds ratio of 3.5, and psoriasis, with an odds ratio of 1.7.
In a multivariate analysis, psoriasis was not only independently associated with a 70% increased likelihood of NAFLD, but psoriasis patients with NAFLD were also 60% more likely to have the more severe forms of NAFLD.
The Rotterdam study is funded chiefly by Dutch scientific research foundations, with secondary pharmaceutical industry support. Dr. van der Voort reported having no financial conflicts of interest.
AT THE EADV CONGRESS
Major finding: The prevalence of nonalcoholic fatty liver disease in a large cohort of elderly subjects was 46.2% among those with psoriasis and 33% in those without. In a multivariate regression analysis, psoriasis was independently associated with a 70% increased likelihood of the hepatic disease.
Data source: The Rotterdam Study is a long-standing prospective, population-based cohort study. The aim is to determine key factors in the development of a range of chronic diseases common in the elderly.
Disclosures: The Rotterdam Study is funded primarily by Dutch scientific research foundations. The presenter reported having no financial conflicts.

Most data reassure regarding TNF inhibitors and cancer
LAS VEGAS – Concerns about use of a tumor necrosis factor inhibitor in a rheumatoid arthritis patient with a history of cancer are common, but the data are largely reassuring, according to Dr. Iain McInnes.
He used as an example a case involving a 56-year-old woman who had erosive RA that was anti-citrullinated peptide antibody–positive (ACPA+). The woman failed triple therapy, responded well to etanercept, and achieved clinical remission after 9 months. The patient then developed breast cancer and her TNF inhibitor was stopped, as recommended by current American College of Rheumatology guidelines.

The patient returned a year later after undergoing "the whole gamut from the oncological armamentarium," including chemotherapy, radiation therapy, and surgery.
"So what do you do? She wants her TNF blocker back – she did very well on it," said Dr. McInnes, who is Muirhead Chair of Medicine and Director of Institute of Infection, Immunity, and Inflammation at the University of Glasgow, Scotland.
Prescribe a conventional disease-modifying antirheumatic drug (DMARD)? Try a different mode of action?
What if the patient is a 56-year-old female with ACPA+ erosive RA who failed triple therapy and leflunomide; is doing poorly; has a history of breast cancer treated successfully 3 years ago by lumpectomy, radiation therapy and chemotherapy; and asks for a change in therapy?
According to the ACR guidelines, any biologic agent can be used in a patient with solid malignancy or nonmelanoma skin cancer that was treated more than 5 years ago, while rituximab should be considered in those treated for a solid malignancy within the last 5 years, those treated with nonmelanoma skin cancer within the last 5 years, those treated for skin melanoma, and those treated for lymphoproliferative malignancy.
The British Society of Rheumatology guidelines are "broadly similar but less restrictive on the use of rituximab, and less restrictive of the time duration from solid malignancy to decision making," Dr. McInnes said at Perspectives in Rheumatic Diseases 2013.
A number of studies looking at whether TNF inhibitors are associated with increased cancer risk have been published and, basically, "the more one looks at the data, the less concerned one becomes," he said.
In a large meta-analysis that included 74 randomized controlled studies, more than 15,400 patients treated with TNF inhibitors, and nearly 7,500 controls, investigators found no evidence to support or refute a link between TNF inhibitors treatment and short-term risk of all cancers except nonmelanoma skin cancer. The risk of nonmelanoma skin cancer, however, was nearly more than doubled in the treatment group (Pharmacoedpidemiol. Drug Saf. 2011;20:119-30).
A 2012 study demonstrated a very low risk of malignancy among patients on TNF inhibitors across several registries (Rheum. Dis. Clin. North Am. 2012;38:761-70).
Also, a study published earlier this year showed that, in nearly 40,000 patients with various types of rheumatic diseases, treatment with anti-TNF drugs was not associated with a short-term increase in the risk of cancer, compared with commonly used therapies for immune-mediated chronic inflammatory diseases (Arthritis Rheum. 2013;65:48-58).
Another large population-based cohort study published this year (BMJ 2013;346:f1939), showed that RA patients treated with TNF inhibitors are not at increased overall risk for cancer, but they do have a 50% increased relative risk of invasive melanoma. The increase in absolute melanoma risk is small, however, and "may not markedly shift the overall risk-benefit balance of TNF inhibitors as used in clinical practice but might do so in patients at high risk of melanoma for other reasons."
Notably, another large study published in 2011 linked data from multiple Swedish clinical registries of RA with nationwide data on hospitalization and outpatient visits for RA, showing that cancers that occur in patients taking TNF inhibitors are not characterized by any distinction, such as a later stage at presentation or worse survival.
"This is true whether you look at overall cancer or lung, colorectal, or hematologic cancers. You can reassure your patient," Dr. McInnes said.
As for RA patients with a history of prior malignancy, findings from a 2010 analysis of data from the British Society for Rheumatology Biologics Register showed that, in 117 patients on a DMARD and 177 on an anti-TNF drug, the risk of cancer was actually greater among those on a DMARD, with a rate of incident malignancy per 1,000 person-years of 25.3 in the anti-TNF patients, compared with 38.3 in the DMARD patients (Arthritis Care Res. 2010;62:755-63).
"When you look at the numbers they are, by and large, reassuring, and this is the message you should give to patients, but make decisions on an individual-patient basis," Dr. McInnes said.
Dr. McInnes disclosed that he has been a speaker, adviser, and/or investigator for Janssen, Roche, Pfizer, BMS, and Novartis. The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
LAS VEGAS – Concerns about use of a tumor necrosis factor inhibitor in a rheumatoid arthritis patient with a history of cancer are common, but the data are largely reassuring, according to Dr. Iain McInnes.
He used as an example a case involving a 56-year-old woman who had erosive RA that was anti-citrullinated peptide antibody–positive (ACPA+). The woman failed triple therapy, responded well to etanercept, and achieved clinical remission after 9 months. The patient then developed breast cancer and her TNF inhibitor was stopped, as recommended by current American College of Rheumatology guidelines.
The patient returned a year later after undergoing "the whole gamut from the oncological armamentarium," including chemotherapy, radiation therapy, and surgery.
"So what do you do? She wants her TNF blocker back – she did very well on it," said Dr. McInnes, who is Muirhead Chair of Medicine and Director of Institute of Infection, Immunity, and Inflammation at the University of Glasgow, Scotland.
Prescribe a conventional disease-modifying antirheumatic drug (DMARD)? Try a different mode of action?
What if the patient is a 56-year-old female with ACPA+ erosive RA who failed triple therapy and leflunomide; is doing poorly; has a history of breast cancer treated successfully 3 years ago by lumpectomy, radiation therapy and chemotherapy; and asks for a change in therapy?
According to the ACR guidelines, any biologic agent can be used in a patient with solid malignancy or nonmelanoma skin cancer that was treated more than 5 years ago, while rituximab should be considered in those treated for a solid malignancy within the last 5 years, those treated with nonmelanoma skin cancer within the last 5 years, those treated for skin melanoma, and those treated for lymphoproliferative malignancy.
The British Society of Rheumatology guidelines are "broadly similar but less restrictive on the use of rituximab, and less restrictive of the time duration from solid malignancy to decision making," Dr. McInnes said at Perspectives in Rheumatic Diseases 2013.
A number of studies looking at whether TNF inhibitors are associated with increased cancer risk have been published and, basically, "the more one looks at the data, the less concerned one becomes," he said.
In a large meta-analysis that included 74 randomized controlled studies, more than 15,400 patients treated with TNF inhibitors, and nearly 7,500 controls, investigators found no evidence to support or refute a link between TNF inhibitors treatment and short-term risk of all cancers except nonmelanoma skin cancer. The risk of nonmelanoma skin cancer, however, was nearly more than doubled in the treatment group (Pharmacoedpidemiol. Drug Saf. 2011;20:119-30).
A 2012 study demonstrated a very low risk of malignancy among patients on TNF inhibitors across several registries (Rheum. Dis. Clin. North Am. 2012;38:761-70).
Also, a study published earlier this year showed that, in nearly 40,000 patients with various types of rheumatic diseases, treatment with anti-TNF drugs was not associated with a short-term increase in the risk of cancer, compared with commonly used therapies for immune-mediated chronic inflammatory diseases (Arthritis Rheum. 2013;65:48-58).
Another large population-based cohort study published this year (BMJ 2013;346:f1939), showed that RA patients treated with TNF inhibitors are not at increased overall risk for cancer, but they do have a 50% increased relative risk of invasive melanoma. The increase in absolute melanoma risk is small, however, and "may not markedly shift the overall risk-benefit balance of TNF inhibitors as used in clinical practice but might do so in patients at high risk of melanoma for other reasons."
Notably, another large study published in 2011 linked data from multiple Swedish clinical registries of RA with nationwide data on hospitalization and outpatient visits for RA, showing that cancers that occur in patients taking TNF inhibitors are not characterized by any distinction, such as a later stage at presentation or worse survival.
"This is true whether you look at overall cancer or lung, colorectal, or hematologic cancers. You can reassure your patient," Dr. McInnes said.
As for RA patients with a history of prior malignancy, findings from a 2010 analysis of data from the British Society for Rheumatology Biologics Register showed that, in 117 patients on a DMARD and 177 on an anti-TNF drug, the risk of cancer was actually greater among those on a DMARD, with a rate of incident malignancy per 1,000 person-years of 25.3 in the anti-TNF patients, compared with 38.3 in the DMARD patients (Arthritis Care Res. 2010;62:755-63).
"When you look at the numbers they are, by and large, reassuring, and this is the message you should give to patients, but make decisions on an individual-patient basis," Dr. McInnes said.
Dr. McInnes disclosed that he has been a speaker, adviser, and/or investigator for Janssen, Roche, Pfizer, BMS, and Novartis. The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
LAS VEGAS – Concerns about use of a tumor necrosis factor inhibitor in a rheumatoid arthritis patient with a history of cancer are common, but the data are largely reassuring, according to Dr. Iain McInnes.
He used as an example a case involving a 56-year-old woman who had erosive RA that was anti-citrullinated peptide antibody–positive (ACPA+). The woman failed triple therapy, responded well to etanercept, and achieved clinical remission after 9 months. The patient then developed breast cancer and her TNF inhibitor was stopped, as recommended by current American College of Rheumatology guidelines.
The patient returned a year later after undergoing "the whole gamut from the oncological armamentarium," including chemotherapy, radiation therapy, and surgery.
"So what do you do? She wants her TNF blocker back – she did very well on it," said Dr. McInnes, who is Muirhead Chair of Medicine and Director of Institute of Infection, Immunity, and Inflammation at the University of Glasgow, Scotland.
Prescribe a conventional disease-modifying antirheumatic drug (DMARD)? Try a different mode of action?
What if the patient is a 56-year-old female with ACPA+ erosive RA who failed triple therapy and leflunomide; is doing poorly; has a history of breast cancer treated successfully 3 years ago by lumpectomy, radiation therapy and chemotherapy; and asks for a change in therapy?
According to the ACR guidelines, any biologic agent can be used in a patient with solid malignancy or nonmelanoma skin cancer that was treated more than 5 years ago, while rituximab should be considered in those treated for a solid malignancy within the last 5 years, those treated with nonmelanoma skin cancer within the last 5 years, those treated for skin melanoma, and those treated for lymphoproliferative malignancy.
The British Society of Rheumatology guidelines are "broadly similar but less restrictive on the use of rituximab, and less restrictive of the time duration from solid malignancy to decision making," Dr. McInnes said at Perspectives in Rheumatic Diseases 2013.
A number of studies looking at whether TNF inhibitors are associated with increased cancer risk have been published and, basically, "the more one looks at the data, the less concerned one becomes," he said.
In a large meta-analysis that included 74 randomized controlled studies, more than 15,400 patients treated with TNF inhibitors, and nearly 7,500 controls, investigators found no evidence to support or refute a link between TNF inhibitors treatment and short-term risk of all cancers except nonmelanoma skin cancer. The risk of nonmelanoma skin cancer, however, was nearly more than doubled in the treatment group (Pharmacoedpidemiol. Drug Saf. 2011;20:119-30).
A 2012 study demonstrated a very low risk of malignancy among patients on TNF inhibitors across several registries (Rheum. Dis. Clin. North Am. 2012;38:761-70).
Also, a study published earlier this year showed that, in nearly 40,000 patients with various types of rheumatic diseases, treatment with anti-TNF drugs was not associated with a short-term increase in the risk of cancer, compared with commonly used therapies for immune-mediated chronic inflammatory diseases (Arthritis Rheum. 2013;65:48-58).
Another large population-based cohort study published this year (BMJ 2013;346:f1939), showed that RA patients treated with TNF inhibitors are not at increased overall risk for cancer, but they do have a 50% increased relative risk of invasive melanoma. The increase in absolute melanoma risk is small, however, and "may not markedly shift the overall risk-benefit balance of TNF inhibitors as used in clinical practice but might do so in patients at high risk of melanoma for other reasons."
Notably, another large study published in 2011 linked data from multiple Swedish clinical registries of RA with nationwide data on hospitalization and outpatient visits for RA, showing that cancers that occur in patients taking TNF inhibitors are not characterized by any distinction, such as a later stage at presentation or worse survival.
"This is true whether you look at overall cancer or lung, colorectal, or hematologic cancers. You can reassure your patient," Dr. McInnes said.
As for RA patients with a history of prior malignancy, findings from a 2010 analysis of data from the British Society for Rheumatology Biologics Register showed that, in 117 patients on a DMARD and 177 on an anti-TNF drug, the risk of cancer was actually greater among those on a DMARD, with a rate of incident malignancy per 1,000 person-years of 25.3 in the anti-TNF patients, compared with 38.3 in the DMARD patients (Arthritis Care Res. 2010;62:755-63).
"When you look at the numbers they are, by and large, reassuring, and this is the message you should give to patients, but make decisions on an individual-patient basis," Dr. McInnes said.
Dr. McInnes disclosed that he has been a speaker, adviser, and/or investigator for Janssen, Roche, Pfizer, BMS, and Novartis. The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
EXPERT ANALYSIS FROM PERSPECTIVES IN RHEUMATIC DISEASES 2013

Product News: 10 2013
Botox Cosmetic
Allergan, Inc, obtains US Food and Drug Administration approval of Botox Cosmetic (onabotulinumtoxinA) for temporary improvement of moderate to severe lateral canthal lines (crow’s-feet) in adults. It blocks nerve impulses and reduces muscle movements around the eyes. This indication will allow physicians to treat both crow’s-feet and frown lines (approved in 2002 for this latter indication) with little downtime for patients. For more information, visit www.allergan.com.
Fabior Foam 0.1%
Stiefel, a GSK company, receives US Food and Drug Administration approval of Fabior (tazarotene) Foam 0.1% for the treatment of acne vulgaris in patients 12 years and older. Fabior Foam is applied once daily before bedtime. For more information, visit www.fabiorfoam.com.
NIA24 Intensive Retinol Repair
Niadyne, Inc, introduces NIA24 Intensive Retinol Repair for photodamage. It targets the major signs of UV damage including wrinkles, hyperpigmentation, lack of firmness, and uneven texture and tone. Formulated with ProNiacin and retinol, NIA24 Intensive Retinol Repair strengthens the skin barrier and increases collagen. It is an alternative for patients who cannot tolerate retinoic acid or traditional retinol treatments. A prescription is not required, and it can be applied daily. For more information, visit www.NIA24.com.
Stelara
Janssen Biotech Inc obtains US Food and Drug Administration approval of Stelara (ustekinumab) to treat patients with active psoriatic arthritis, alone or in combination with methotrexate. Stelara targets the cytokines IL-12 and IL-23 to control joint pain, swelling, and stiffness associated with psoriatic arthritis, in addition to psoriasis plaque thickness, scaling, and redness. Stelara is administered every 12 weeks after 2 starter doses for the treatment of psoriatic arthritis. For more information, visit www.stelarainfo.com.
XTRAC Velocity 7
PhotoMedex Inc introduces XTRAC Velocity 7 with an advanced user interface for psoriasis and vitiligo. Using UVB light, the XTRAC excimer laser treats areas of the skin affected by psoriasis or vitiligo without harming the surrounding tissue. XTRAC Velocity 7 offers increased efficiency, with the rate of output increasing the speed for delivery of treatment. Treatment guidelines and suggestions based on body area are provided using the touch screen. It can be used on hard-to-reach areas such as the elbows, knees, and scalp. Before and after photographs can be stored to show progression of resolution, enhancing patient compliance. The manufacturer also offers a patient advocacy program for patients to call and obtain answers to product and insurance questions from a live operator; patients also can book appointments with a participating physician faster using the XTRAC TeleCare Center. For more information, visit www.xtracnow.com
Botox Cosmetic
Allergan, Inc, obtains US Food and Drug Administration approval of Botox Cosmetic (onabotulinumtoxinA) for temporary improvement of moderate to severe lateral canthal lines (crow’s-feet) in adults. It blocks nerve impulses and reduces muscle movements around the eyes. This indication will allow physicians to treat both crow’s-feet and frown lines (approved in 2002 for this latter indication) with little downtime for patients. For more information, visit www.allergan.com.
Fabior Foam 0.1%
Stiefel, a GSK company, receives US Food and Drug Administration approval of Fabior (tazarotene) Foam 0.1% for the treatment of acne vulgaris in patients 12 years and older. Fabior Foam is applied once daily before bedtime. For more information, visit www.fabiorfoam.com.
NIA24 Intensive Retinol Repair
Niadyne, Inc, introduces NIA24 Intensive Retinol Repair for photodamage. It targets the major signs of UV damage including wrinkles, hyperpigmentation, lack of firmness, and uneven texture and tone. Formulated with ProNiacin and retinol, NIA24 Intensive Retinol Repair strengthens the skin barrier and increases collagen. It is an alternative for patients who cannot tolerate retinoic acid or traditional retinol treatments. A prescription is not required, and it can be applied daily. For more information, visit www.NIA24.com.
Stelara
Janssen Biotech Inc obtains US Food and Drug Administration approval of Stelara (ustekinumab) to treat patients with active psoriatic arthritis, alone or in combination with methotrexate. Stelara targets the cytokines IL-12 and IL-23 to control joint pain, swelling, and stiffness associated with psoriatic arthritis, in addition to psoriasis plaque thickness, scaling, and redness. Stelara is administered every 12 weeks after 2 starter doses for the treatment of psoriatic arthritis. For more information, visit www.stelarainfo.com.
XTRAC Velocity 7
PhotoMedex Inc introduces XTRAC Velocity 7 with an advanced user interface for psoriasis and vitiligo. Using UVB light, the XTRAC excimer laser treats areas of the skin affected by psoriasis or vitiligo without harming the surrounding tissue. XTRAC Velocity 7 offers increased efficiency, with the rate of output increasing the speed for delivery of treatment. Treatment guidelines and suggestions based on body area are provided using the touch screen. It can be used on hard-to-reach areas such as the elbows, knees, and scalp. Before and after photographs can be stored to show progression of resolution, enhancing patient compliance. The manufacturer also offers a patient advocacy program for patients to call and obtain answers to product and insurance questions from a live operator; patients also can book appointments with a participating physician faster using the XTRAC TeleCare Center. For more information, visit www.xtracnow.com
Botox Cosmetic
Allergan, Inc, obtains US Food and Drug Administration approval of Botox Cosmetic (onabotulinumtoxinA) for temporary improvement of moderate to severe lateral canthal lines (crow’s-feet) in adults. It blocks nerve impulses and reduces muscle movements around the eyes. This indication will allow physicians to treat both crow’s-feet and frown lines (approved in 2002 for this latter indication) with little downtime for patients. For more information, visit www.allergan.com.
Fabior Foam 0.1%
Stiefel, a GSK company, receives US Food and Drug Administration approval of Fabior (tazarotene) Foam 0.1% for the treatment of acne vulgaris in patients 12 years and older. Fabior Foam is applied once daily before bedtime. For more information, visit www.fabiorfoam.com.
NIA24 Intensive Retinol Repair
Niadyne, Inc, introduces NIA24 Intensive Retinol Repair for photodamage. It targets the major signs of UV damage including wrinkles, hyperpigmentation, lack of firmness, and uneven texture and tone. Formulated with ProNiacin and retinol, NIA24 Intensive Retinol Repair strengthens the skin barrier and increases collagen. It is an alternative for patients who cannot tolerate retinoic acid or traditional retinol treatments. A prescription is not required, and it can be applied daily. For more information, visit www.NIA24.com.
Stelara
Janssen Biotech Inc obtains US Food and Drug Administration approval of Stelara (ustekinumab) to treat patients with active psoriatic arthritis, alone or in combination with methotrexate. Stelara targets the cytokines IL-12 and IL-23 to control joint pain, swelling, and stiffness associated with psoriatic arthritis, in addition to psoriasis plaque thickness, scaling, and redness. Stelara is administered every 12 weeks after 2 starter doses for the treatment of psoriatic arthritis. For more information, visit www.stelarainfo.com.
XTRAC Velocity 7
PhotoMedex Inc introduces XTRAC Velocity 7 with an advanced user interface for psoriasis and vitiligo. Using UVB light, the XTRAC excimer laser treats areas of the skin affected by psoriasis or vitiligo without harming the surrounding tissue. XTRAC Velocity 7 offers increased efficiency, with the rate of output increasing the speed for delivery of treatment. Treatment guidelines and suggestions based on body area are provided using the touch screen. It can be used on hard-to-reach areas such as the elbows, knees, and scalp. Before and after photographs can be stored to show progression of resolution, enhancing patient compliance. The manufacturer also offers a patient advocacy program for patients to call and obtain answers to product and insurance questions from a live operator; patients also can book appointments with a participating physician faster using the XTRAC TeleCare Center. For more information, visit www.xtracnow.com

Nodular Fasciitis: A Possible Side Effect of Etanercept?
Palmar-plantar psoriasis? Anti-TNF therapy may be culprit
LAS VEGAS – Psoriasiform eruptions are increasingly recognized as a "real event" among patients treated with anti–tumor necrosis factor therapy, according to Dr. Kenneth B. Gordon.
At Perspectives in Rheumatic Diseases 2013, Dr. Gordon of Northwestern University in Chicago presented the case of a 56-year-old woman with Crohn’s disease who developed a painful rash with fissures on both hands about 8 months after starting adalimumab therapy.
The patient first presented with Crohn’s disease at age 28 and was initially treated with mesalamine and systemic corticosteroids for flares. She became prednisone dependent and was started on azathioprine, but could not tolerate the drug, which caused pancreatitis. She was switched to adalimumab at a dose of 80 mg at week 1, 40 mg at week 2, and then 40 mg every other week thereafter.

The patient had an excellent response, including resolution of the gastrointestinal symptoms she experienced on azathioprine, but the rash, which involved well-demarcated plaques suggestive of a psoriasiform structure, was debilitating, Dr. Gordon said.
Another case, presented by Dr. Iain McInnes of Glasgow University, Scotland, involved a patient undergoing anti–tumor necrosis factor (anti-TNF) therapy for rheumatoid arthritis. That patient also developed palmar-plantar pustular psoriasis, which is the most common presentation of psoriasis induced by anti-TNF agents.
Data on the frequency of this condition are lacking, but Dr. Gordon said his "best guess" based on the available literature is that it occurs in about 1%-2% of patients on TNF blockers.
Dr. McInnes described the frequency as "sufficiently common to know it happens and reassure patients about it; sufficiently uncommon enough not to influence decision making" about prescribing anti-TNF therapy.
According to a 2010 systematic literature review by Dr. Angelique N. Collamer and Dr. Daniel F. Battafarano of the rheumatology service at Brooke Army Medical Center in Fort Sam Houston, Tex., pustular psoriasis occurs in 56% of cases, plaque psoriasis occurs in 50%, and guttate psoriasis occurs in 12% of cases. Most (85%) are new-onset cases, and 15% represent an exacerbation of existing psoriasis. Also, patients who are being treated for a variety of diseases – including rheumatoid arthritis, seronegative spondyloarthropathy, inflammatory bowel disease, and psoriasis – may be affected, and all anti-TNF therapies have been implicated (Semin. Arthritis Rheum. 2010;40:233-40).
However, treatment does not necessarily require stopping the TNF blockers, which is important because many patients don’t want to stop – they’re pleased with the effects of treatment on their underlying disease, Dr. Gordon noted.
He said that the first-line treatment is topical therapy, such as a potent topical corticosteroid, but retinoids may also be needed. Other options include phototherapy and switching to another anti-TNF therapy.
"My philosophy is to try to treat the patient while maintaining [anti-TNF] treatment," he said, noting that stopping anti-TNF therapy is a last resort.
Although the patient Dr. Gordon discussed was not a smoker, smoking is a risk factor for worsening of palm psoriasis. Patients with this condition should be advised to quit smoking, and many smokers will get better just by quitting smoking, Dr. Gordon noted.
Dr. McInnes also outlined his approach to patients presenting with possible anti-TNF therapy–induced psoriasis.
He stressed the importance of a good differential diagnosis, noting that infection may be the actual cause.
A biopsy can sometimes help with the diagnosis, he said.
In general, his treatment approach in cases of true anti-TNF–induced psoriasis is to:
• Continue the anti-TNF therapy when less than 5% of body surface area is affected.
• Change to a different anti-TNF drug if the psoriasis is tolerable but appears "a little more severe or is a palmar-pustular variant." The timing of the change will depend on how the patient responds to cutaneous management.
• Stop the drug if the psoriasis is intolerable, and treat the psoriasis aggressively.
In any of these circumstances, a change in the mode of action of treatment may be warranted, he said at the meeting.
In the review by Dr. Collamer and Dr. Battafarano, 24% of patients resolved off anti-TNF therapy, 5% had partial or no resolution off anti-TNF therapy, 25% had partial resolution on anti-TNF therapy, and 6% had no recurrence with change of anti-TNF. Only 1% had no resolution on anti-TNF therapy, and only 2% who resolved after anti-TNF discontinuation then had recurrence after reintroduction of anti-TNF therapy, while 6% resolved off anti-TNF therapy and then had recurrence with introduction of a different anti-TNF. The outcome was unknown in 5% of patients. A Mayo Clinic series showed a similar breakdown (J. Am. Acad. Dermatol. 2012;67:e179-85).
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
Dr. Gordon reported receiving research support and/or honoraria from AbbVie, Amgen, and other companies. Dr. McInnes reported serving as a speaker, adviser, and/or researcher for Janssen, Roche, and other companies.
LAS VEGAS – Psoriasiform eruptions are increasingly recognized as a "real event" among patients treated with anti–tumor necrosis factor therapy, according to Dr. Kenneth B. Gordon.
At Perspectives in Rheumatic Diseases 2013, Dr. Gordon of Northwestern University in Chicago presented the case of a 56-year-old woman with Crohn’s disease who developed a painful rash with fissures on both hands about 8 months after starting adalimumab therapy.
The patient first presented with Crohn’s disease at age 28 and was initially treated with mesalamine and systemic corticosteroids for flares. She became prednisone dependent and was started on azathioprine, but could not tolerate the drug, which caused pancreatitis. She was switched to adalimumab at a dose of 80 mg at week 1, 40 mg at week 2, and then 40 mg every other week thereafter.
The patient had an excellent response, including resolution of the gastrointestinal symptoms she experienced on azathioprine, but the rash, which involved well-demarcated plaques suggestive of a psoriasiform structure, was debilitating, Dr. Gordon said.
Another case, presented by Dr. Iain McInnes of Glasgow University, Scotland, involved a patient undergoing anti–tumor necrosis factor (anti-TNF) therapy for rheumatoid arthritis. That patient also developed palmar-plantar pustular psoriasis, which is the most common presentation of psoriasis induced by anti-TNF agents.
Data on the frequency of this condition are lacking, but Dr. Gordon said his "best guess" based on the available literature is that it occurs in about 1%-2% of patients on TNF blockers.
Dr. McInnes described the frequency as "sufficiently common to know it happens and reassure patients about it; sufficiently uncommon enough not to influence decision making" about prescribing anti-TNF therapy.
According to a 2010 systematic literature review by Dr. Angelique N. Collamer and Dr. Daniel F. Battafarano of the rheumatology service at Brooke Army Medical Center in Fort Sam Houston, Tex., pustular psoriasis occurs in 56% of cases, plaque psoriasis occurs in 50%, and guttate psoriasis occurs in 12% of cases. Most (85%) are new-onset cases, and 15% represent an exacerbation of existing psoriasis. Also, patients who are being treated for a variety of diseases – including rheumatoid arthritis, seronegative spondyloarthropathy, inflammatory bowel disease, and psoriasis – may be affected, and all anti-TNF therapies have been implicated (Semin. Arthritis Rheum. 2010;40:233-40).
However, treatment does not necessarily require stopping the TNF blockers, which is important because many patients don’t want to stop – they’re pleased with the effects of treatment on their underlying disease, Dr. Gordon noted.
He said that the first-line treatment is topical therapy, such as a potent topical corticosteroid, but retinoids may also be needed. Other options include phototherapy and switching to another anti-TNF therapy.
"My philosophy is to try to treat the patient while maintaining [anti-TNF] treatment," he said, noting that stopping anti-TNF therapy is a last resort.
Although the patient Dr. Gordon discussed was not a smoker, smoking is a risk factor for worsening of palm psoriasis. Patients with this condition should be advised to quit smoking, and many smokers will get better just by quitting smoking, Dr. Gordon noted.
Dr. McInnes also outlined his approach to patients presenting with possible anti-TNF therapy–induced psoriasis.
He stressed the importance of a good differential diagnosis, noting that infection may be the actual cause.
A biopsy can sometimes help with the diagnosis, he said.
In general, his treatment approach in cases of true anti-TNF–induced psoriasis is to:
• Continue the anti-TNF therapy when less than 5% of body surface area is affected.
• Change to a different anti-TNF drug if the psoriasis is tolerable but appears "a little more severe or is a palmar-pustular variant." The timing of the change will depend on how the patient responds to cutaneous management.
• Stop the drug if the psoriasis is intolerable, and treat the psoriasis aggressively.
In any of these circumstances, a change in the mode of action of treatment may be warranted, he said at the meeting.
In the review by Dr. Collamer and Dr. Battafarano, 24% of patients resolved off anti-TNF therapy, 5% had partial or no resolution off anti-TNF therapy, 25% had partial resolution on anti-TNF therapy, and 6% had no recurrence with change of anti-TNF. Only 1% had no resolution on anti-TNF therapy, and only 2% who resolved after anti-TNF discontinuation then had recurrence after reintroduction of anti-TNF therapy, while 6% resolved off anti-TNF therapy and then had recurrence with introduction of a different anti-TNF. The outcome was unknown in 5% of patients. A Mayo Clinic series showed a similar breakdown (J. Am. Acad. Dermatol. 2012;67:e179-85).
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
Dr. Gordon reported receiving research support and/or honoraria from AbbVie, Amgen, and other companies. Dr. McInnes reported serving as a speaker, adviser, and/or researcher for Janssen, Roche, and other companies.
LAS VEGAS – Psoriasiform eruptions are increasingly recognized as a "real event" among patients treated with anti–tumor necrosis factor therapy, according to Dr. Kenneth B. Gordon.
At Perspectives in Rheumatic Diseases 2013, Dr. Gordon of Northwestern University in Chicago presented the case of a 56-year-old woman with Crohn’s disease who developed a painful rash with fissures on both hands about 8 months after starting adalimumab therapy.
The patient first presented with Crohn’s disease at age 28 and was initially treated with mesalamine and systemic corticosteroids for flares. She became prednisone dependent and was started on azathioprine, but could not tolerate the drug, which caused pancreatitis. She was switched to adalimumab at a dose of 80 mg at week 1, 40 mg at week 2, and then 40 mg every other week thereafter.
The patient had an excellent response, including resolution of the gastrointestinal symptoms she experienced on azathioprine, but the rash, which involved well-demarcated plaques suggestive of a psoriasiform structure, was debilitating, Dr. Gordon said.
Another case, presented by Dr. Iain McInnes of Glasgow University, Scotland, involved a patient undergoing anti–tumor necrosis factor (anti-TNF) therapy for rheumatoid arthritis. That patient also developed palmar-plantar pustular psoriasis, which is the most common presentation of psoriasis induced by anti-TNF agents.
Data on the frequency of this condition are lacking, but Dr. Gordon said his "best guess" based on the available literature is that it occurs in about 1%-2% of patients on TNF blockers.
Dr. McInnes described the frequency as "sufficiently common to know it happens and reassure patients about it; sufficiently uncommon enough not to influence decision making" about prescribing anti-TNF therapy.
According to a 2010 systematic literature review by Dr. Angelique N. Collamer and Dr. Daniel F. Battafarano of the rheumatology service at Brooke Army Medical Center in Fort Sam Houston, Tex., pustular psoriasis occurs in 56% of cases, plaque psoriasis occurs in 50%, and guttate psoriasis occurs in 12% of cases. Most (85%) are new-onset cases, and 15% represent an exacerbation of existing psoriasis. Also, patients who are being treated for a variety of diseases – including rheumatoid arthritis, seronegative spondyloarthropathy, inflammatory bowel disease, and psoriasis – may be affected, and all anti-TNF therapies have been implicated (Semin. Arthritis Rheum. 2010;40:233-40).
However, treatment does not necessarily require stopping the TNF blockers, which is important because many patients don’t want to stop – they’re pleased with the effects of treatment on their underlying disease, Dr. Gordon noted.
He said that the first-line treatment is topical therapy, such as a potent topical corticosteroid, but retinoids may also be needed. Other options include phototherapy and switching to another anti-TNF therapy.
"My philosophy is to try to treat the patient while maintaining [anti-TNF] treatment," he said, noting that stopping anti-TNF therapy is a last resort.
Although the patient Dr. Gordon discussed was not a smoker, smoking is a risk factor for worsening of palm psoriasis. Patients with this condition should be advised to quit smoking, and many smokers will get better just by quitting smoking, Dr. Gordon noted.
Dr. McInnes also outlined his approach to patients presenting with possible anti-TNF therapy–induced psoriasis.
He stressed the importance of a good differential diagnosis, noting that infection may be the actual cause.
A biopsy can sometimes help with the diagnosis, he said.
In general, his treatment approach in cases of true anti-TNF–induced psoriasis is to:
• Continue the anti-TNF therapy when less than 5% of body surface area is affected.
• Change to a different anti-TNF drug if the psoriasis is tolerable but appears "a little more severe or is a palmar-pustular variant." The timing of the change will depend on how the patient responds to cutaneous management.
• Stop the drug if the psoriasis is intolerable, and treat the psoriasis aggressively.
In any of these circumstances, a change in the mode of action of treatment may be warranted, he said at the meeting.
In the review by Dr. Collamer and Dr. Battafarano, 24% of patients resolved off anti-TNF therapy, 5% had partial or no resolution off anti-TNF therapy, 25% had partial resolution on anti-TNF therapy, and 6% had no recurrence with change of anti-TNF. Only 1% had no resolution on anti-TNF therapy, and only 2% who resolved after anti-TNF discontinuation then had recurrence after reintroduction of anti-TNF therapy, while 6% resolved off anti-TNF therapy and then had recurrence with introduction of a different anti-TNF. The outcome was unknown in 5% of patients. A Mayo Clinic series showed a similar breakdown (J. Am. Acad. Dermatol. 2012;67:e179-85).
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
Dr. Gordon reported receiving research support and/or honoraria from AbbVie, Amgen, and other companies. Dr. McInnes reported serving as a speaker, adviser, and/or researcher for Janssen, Roche, and other companies.
EXPERT ANALYSIS FROM PERSPECTIVES IN RHEUMATIC DISEASES 2013

Psoriasis Treatment With Tumor Necrosis Factor Inhibitors

Autoantibodies play role in myositis classification, treatment
LAS VEGAS – Autoantibodies and autoantibody subsets can be particularly helpful for classifying and treating patients with myositis, including those with myositis-associated interstitial lung disease, according to Dr. Chester V. Oddis.
Many autoantibody subsets are phenotypically and clinically well defined, and have clinical relevance, said Dr. Oddis, professor of medicine and associate director of the rheumatology fellowship training program at the University of Pittsburgh.
Serological classification isn’t always accurate or routinely available for all clinicians, but this may change in the near future as improved techniques for autoantibody detection become available, he said at Perspectives in Rheumatic Diseases 2013.
Anti-MDA-5 and interstitial lung disease
One autoantibody that has gained attention in recent years is anti-MDA-5, also known as anti-CADM-140, which is often seen in patients with amyopathic dermatomyositis (ADM).
Patients with ADM represent a subset of dermatomyositis patients who have cutaneous manifestations of dermatomyositis for 6 months or longer and have no clinical evidence of proximal muscle weakness but may have mild serum muscle enzyme abnormalities. More extensive muscle testing in these patients generally demonstrates no or minimal abnormalities. However, these patients should not be considered to have simply a benign cutaneous form of disease; in fact, they have a frequency of malignancy similar to that of patients with classic dermatomyositis (14% in one series of nearly 300 patients, compared with 15% in classic dermatomyositis).
In addition, ADM patients also have a relatively high frequency of lung disease, Dr. Oddis said. In a published review of the literature of nearly 200 patients with ADM, 10% had interstitial lung disease (ILD) – an important point given that the rash of dermatomyositis may be subtle and missed, he noted.
The Asian population seems to be particularly at risk for this complication. Two studies in recent years have demonstrated that Japanese ADM patients with anti-MDA-5 present with rapidly progressive ILD. A 2011 study showed an increased incidence of acute or subacute interstitial pneumonitis in Chinese patients. Other studies have shown similar findings in Korean and other Asian populations, Dr. Oddis noted.
The presence of anti-MDA-5 represents a novel cutaneous phenotype involving palmar papules and cutaneous ulcerations, severe vasculopathy, and rapidly progressive ILD. The target autoantigen in these cases is MDA-5, which is involved in innate immune defense against viruses, he explained, noting that this supports the possibility that a viral trigger plays a role in the disease.
"I think this complication is filtering into the U.S. population, as we have seen it in our myositis cohort," Dr. Oddis said of the anti-MDA-5 association with ADM and severe ILD. He noted that he recently cared for a 70-year-old white male with "double pneumonia" (a finding that "should always raise suspicion of autoimmune ILD") in June of 2012, a rash of dermatomyositis in September of 2012, and vasculitic skin changes in January of 2013. He presented without muscle weakness.
Anti-synthetase syndrome and ILD
Another autoantibody myositis subset involves the anti-synthetases, including PL-7, PL-12, EJ, and Jo-1.
Patients with anti-synthetase syndrome are generally a clinically homogeneous patient population characterized by fever, myositis, arthritis, Raynaud’s phenomenon, mechanic’s hands, and ILD. About 30%-40% of myositis patients have ILD, which is a significant contributor to morbidity and mortality.
Anti-Jo-1 is found in 50%-75% of these patients, and the coexistence of Ro52 may portend worse prognosis, Dr. Oddis said.
There are certain clinical features of ILD in polymyositis and dermatomyositis, including progressive dyspnea with or without nonproductive cough, lack of digital clubbing (unlike in idiopathic pulmonary fibrosis), and lack of pleuritis and pleural effusion in most cases (unlike in systemic lupus erythematosus). However, presentation can be variable, with about one-third of patients developing ILD before muscle or skin manifestations are apparent. Some patients present with acute disease (acute respiratory distress syndrome), and others present with subacute disease that is chronic and slowly progressing or asymptomatic.
It is important to understand when making a diagnosis of autoimmune ILD that not all patients will present with the classic anti-synthetase syndrome, Dr. Oddis said.
In some cases, patients will have an "incomplete" clinical syndrome with ILD alone or ILD with only subtle connective tissue disease findings, myositis-specific autoantibodies in the absence of myositis, and/or a negative antinuclear antibody (ANA) test, he explained.
The initial symptoms in patients may vary depending on the anti-synthetase autoantibody that is present. In a University of Pittsburgh study, for example, Jo-1 was found in 60% of cases; non-Jo-1 synthetase positive cases more often experienced Raynaud’s as their initial symptom, less often experienced muscle and joint problems as their initial symptom, and had a longer delay in diagnosis, compared with Jo-1 patients. Survival was also decreased, compared with Jo-1 patients.
As for ANA, about half of patients tested positive, whereas 72% demonstrated positive anti-cytoplasmic staining on indirect immunofluorescence.
The diagnosis of autoimmune ILD can be missed when there is a failure to recognize "incomplete" clinical syndromes, when there is a failure to order or detect myositis-specific autoantibodies – even in patients without myositis – and when a negative ANA is considered to be reassuring, Dr. Oddis said.
Dr. Oddis has served on an advisory board for Questcor.
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
LAS VEGAS – Autoantibodies and autoantibody subsets can be particularly helpful for classifying and treating patients with myositis, including those with myositis-associated interstitial lung disease, according to Dr. Chester V. Oddis.
Many autoantibody subsets are phenotypically and clinically well defined, and have clinical relevance, said Dr. Oddis, professor of medicine and associate director of the rheumatology fellowship training program at the University of Pittsburgh.
Serological classification isn’t always accurate or routinely available for all clinicians, but this may change in the near future as improved techniques for autoantibody detection become available, he said at Perspectives in Rheumatic Diseases 2013.
Anti-MDA-5 and interstitial lung disease
One autoantibody that has gained attention in recent years is anti-MDA-5, also known as anti-CADM-140, which is often seen in patients with amyopathic dermatomyositis (ADM).
Patients with ADM represent a subset of dermatomyositis patients who have cutaneous manifestations of dermatomyositis for 6 months or longer and have no clinical evidence of proximal muscle weakness but may have mild serum muscle enzyme abnormalities. More extensive muscle testing in these patients generally demonstrates no or minimal abnormalities. However, these patients should not be considered to have simply a benign cutaneous form of disease; in fact, they have a frequency of malignancy similar to that of patients with classic dermatomyositis (14% in one series of nearly 300 patients, compared with 15% in classic dermatomyositis).
In addition, ADM patients also have a relatively high frequency of lung disease, Dr. Oddis said. In a published review of the literature of nearly 200 patients with ADM, 10% had interstitial lung disease (ILD) – an important point given that the rash of dermatomyositis may be subtle and missed, he noted.
The Asian population seems to be particularly at risk for this complication. Two studies in recent years have demonstrated that Japanese ADM patients with anti-MDA-5 present with rapidly progressive ILD. A 2011 study showed an increased incidence of acute or subacute interstitial pneumonitis in Chinese patients. Other studies have shown similar findings in Korean and other Asian populations, Dr. Oddis noted.
The presence of anti-MDA-5 represents a novel cutaneous phenotype involving palmar papules and cutaneous ulcerations, severe vasculopathy, and rapidly progressive ILD. The target autoantigen in these cases is MDA-5, which is involved in innate immune defense against viruses, he explained, noting that this supports the possibility that a viral trigger plays a role in the disease.
"I think this complication is filtering into the U.S. population, as we have seen it in our myositis cohort," Dr. Oddis said of the anti-MDA-5 association with ADM and severe ILD. He noted that he recently cared for a 70-year-old white male with "double pneumonia" (a finding that "should always raise suspicion of autoimmune ILD") in June of 2012, a rash of dermatomyositis in September of 2012, and vasculitic skin changes in January of 2013. He presented without muscle weakness.
Anti-synthetase syndrome and ILD
Another autoantibody myositis subset involves the anti-synthetases, including PL-7, PL-12, EJ, and Jo-1.
Patients with anti-synthetase syndrome are generally a clinically homogeneous patient population characterized by fever, myositis, arthritis, Raynaud’s phenomenon, mechanic’s hands, and ILD. About 30%-40% of myositis patients have ILD, which is a significant contributor to morbidity and mortality.
Anti-Jo-1 is found in 50%-75% of these patients, and the coexistence of Ro52 may portend worse prognosis, Dr. Oddis said.
There are certain clinical features of ILD in polymyositis and dermatomyositis, including progressive dyspnea with or without nonproductive cough, lack of digital clubbing (unlike in idiopathic pulmonary fibrosis), and lack of pleuritis and pleural effusion in most cases (unlike in systemic lupus erythematosus). However, presentation can be variable, with about one-third of patients developing ILD before muscle or skin manifestations are apparent. Some patients present with acute disease (acute respiratory distress syndrome), and others present with subacute disease that is chronic and slowly progressing or asymptomatic.
It is important to understand when making a diagnosis of autoimmune ILD that not all patients will present with the classic anti-synthetase syndrome, Dr. Oddis said.
In some cases, patients will have an "incomplete" clinical syndrome with ILD alone or ILD with only subtle connective tissue disease findings, myositis-specific autoantibodies in the absence of myositis, and/or a negative antinuclear antibody (ANA) test, he explained.
The initial symptoms in patients may vary depending on the anti-synthetase autoantibody that is present. In a University of Pittsburgh study, for example, Jo-1 was found in 60% of cases; non-Jo-1 synthetase positive cases more often experienced Raynaud’s as their initial symptom, less often experienced muscle and joint problems as their initial symptom, and had a longer delay in diagnosis, compared with Jo-1 patients. Survival was also decreased, compared with Jo-1 patients.
As for ANA, about half of patients tested positive, whereas 72% demonstrated positive anti-cytoplasmic staining on indirect immunofluorescence.
The diagnosis of autoimmune ILD can be missed when there is a failure to recognize "incomplete" clinical syndromes, when there is a failure to order or detect myositis-specific autoantibodies – even in patients without myositis – and when a negative ANA is considered to be reassuring, Dr. Oddis said.
Dr. Oddis has served on an advisory board for Questcor.
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
LAS VEGAS – Autoantibodies and autoantibody subsets can be particularly helpful for classifying and treating patients with myositis, including those with myositis-associated interstitial lung disease, according to Dr. Chester V. Oddis.
Many autoantibody subsets are phenotypically and clinically well defined, and have clinical relevance, said Dr. Oddis, professor of medicine and associate director of the rheumatology fellowship training program at the University of Pittsburgh.
Serological classification isn’t always accurate or routinely available for all clinicians, but this may change in the near future as improved techniques for autoantibody detection become available, he said at Perspectives in Rheumatic Diseases 2013.
Anti-MDA-5 and interstitial lung disease
One autoantibody that has gained attention in recent years is anti-MDA-5, also known as anti-CADM-140, which is often seen in patients with amyopathic dermatomyositis (ADM).
Patients with ADM represent a subset of dermatomyositis patients who have cutaneous manifestations of dermatomyositis for 6 months or longer and have no clinical evidence of proximal muscle weakness but may have mild serum muscle enzyme abnormalities. More extensive muscle testing in these patients generally demonstrates no or minimal abnormalities. However, these patients should not be considered to have simply a benign cutaneous form of disease; in fact, they have a frequency of malignancy similar to that of patients with classic dermatomyositis (14% in one series of nearly 300 patients, compared with 15% in classic dermatomyositis).
In addition, ADM patients also have a relatively high frequency of lung disease, Dr. Oddis said. In a published review of the literature of nearly 200 patients with ADM, 10% had interstitial lung disease (ILD) – an important point given that the rash of dermatomyositis may be subtle and missed, he noted.
The Asian population seems to be particularly at risk for this complication. Two studies in recent years have demonstrated that Japanese ADM patients with anti-MDA-5 present with rapidly progressive ILD. A 2011 study showed an increased incidence of acute or subacute interstitial pneumonitis in Chinese patients. Other studies have shown similar findings in Korean and other Asian populations, Dr. Oddis noted.
The presence of anti-MDA-5 represents a novel cutaneous phenotype involving palmar papules and cutaneous ulcerations, severe vasculopathy, and rapidly progressive ILD. The target autoantigen in these cases is MDA-5, which is involved in innate immune defense against viruses, he explained, noting that this supports the possibility that a viral trigger plays a role in the disease.
"I think this complication is filtering into the U.S. population, as we have seen it in our myositis cohort," Dr. Oddis said of the anti-MDA-5 association with ADM and severe ILD. He noted that he recently cared for a 70-year-old white male with "double pneumonia" (a finding that "should always raise suspicion of autoimmune ILD") in June of 2012, a rash of dermatomyositis in September of 2012, and vasculitic skin changes in January of 2013. He presented without muscle weakness.
Anti-synthetase syndrome and ILD
Another autoantibody myositis subset involves the anti-synthetases, including PL-7, PL-12, EJ, and Jo-1.
Patients with anti-synthetase syndrome are generally a clinically homogeneous patient population characterized by fever, myositis, arthritis, Raynaud’s phenomenon, mechanic’s hands, and ILD. About 30%-40% of myositis patients have ILD, which is a significant contributor to morbidity and mortality.
Anti-Jo-1 is found in 50%-75% of these patients, and the coexistence of Ro52 may portend worse prognosis, Dr. Oddis said.
There are certain clinical features of ILD in polymyositis and dermatomyositis, including progressive dyspnea with or without nonproductive cough, lack of digital clubbing (unlike in idiopathic pulmonary fibrosis), and lack of pleuritis and pleural effusion in most cases (unlike in systemic lupus erythematosus). However, presentation can be variable, with about one-third of patients developing ILD before muscle or skin manifestations are apparent. Some patients present with acute disease (acute respiratory distress syndrome), and others present with subacute disease that is chronic and slowly progressing or asymptomatic.
It is important to understand when making a diagnosis of autoimmune ILD that not all patients will present with the classic anti-synthetase syndrome, Dr. Oddis said.
In some cases, patients will have an "incomplete" clinical syndrome with ILD alone or ILD with only subtle connective tissue disease findings, myositis-specific autoantibodies in the absence of myositis, and/or a negative antinuclear antibody (ANA) test, he explained.
The initial symptoms in patients may vary depending on the anti-synthetase autoantibody that is present. In a University of Pittsburgh study, for example, Jo-1 was found in 60% of cases; non-Jo-1 synthetase positive cases more often experienced Raynaud’s as their initial symptom, less often experienced muscle and joint problems as their initial symptom, and had a longer delay in diagnosis, compared with Jo-1 patients. Survival was also decreased, compared with Jo-1 patients.
As for ANA, about half of patients tested positive, whereas 72% demonstrated positive anti-cytoplasmic staining on indirect immunofluorescence.
The diagnosis of autoimmune ILD can be missed when there is a failure to recognize "incomplete" clinical syndromes, when there is a failure to order or detect myositis-specific autoantibodies – even in patients without myositis – and when a negative ANA is considered to be reassuring, Dr. Oddis said.
Dr. Oddis has served on an advisory board for Questcor.
The meeting was held by Global Academy for Medical Education. GAME and this news organization are owned by Frontline Medical Communications.
AT PERSPECTIVES IN RHEUMATIC DISEASES 2013
