User login
Too many infants with congenital hypothyroidism go undetected, untreated
DENVER – An alarming percentage of infants born in Utah from 2006 to 2015 with primary congenital hypothyroidism were either lost to follow-up or inadequately treated.
If such a thing can happen in Utah with its highly functioning public health system, it probably can happen in the rest of the United States as well, Joel Ehrenkranz, MD, said at the American Thyroid Association annual meeting. “We just have not looked for it yet.”
Screening for and treating congenital hypothyroidism in infants is one of the great public health successes of the 20th century in the United States. It deserves to have the same level of importance as eradication of polio and smallpox in this country, noted Dr. Ehrenkranz, an endocrinologist in private practice in Glenwood Springs, Colo. At the time of this research, Dr. Ehrenkranz was with Intermountain Healthcare in Murray, Utah.
The American Academy of Pediatrics recommends diagnosis of congenital hypothyroidism by the 14th day of life and that the baby be biochemically euthyroid by week 6 (Pediatrics. 2006 June. doi: 10.1542/peds.2006-0915), he said.
The cohort included 4,394 children from birth to 24 months of age. The screening test was done by a third-generation bioluminescence serum TSH assay, not dried blood blot. Of these infants, 2% (82 babies) had a TSH level greater than or equal to 20 mIU/L at their initial test. That TSH was still high by day 14 in 42 infants (23 girls). But of all the babies with primary congenital hypothyroidism, 50% had a delayed diagnosis, he reported.
Twelve children (15%) were never retested; 34% reached AAP goals of having a TSH level less than 5 mIU/L within 28 days after starting treatment; half of the children with primary congenital hypothyropidism did not meet the treatment goal: “They were inadequately treated,” he said.
Of particular interest were 16 infants who had a TSH level of less than 20 mIU/L but on retesting had one of 20 or higher. Three of these infants had multiple TSH levels greater than 20, perhaps representing a subset with a late onset form of the disorder.
“We are not doing as well as we could do,” he said; 50% of affected babies have a delayed diagnosis with consequences of a delayed maturation of the pituitary thyroid axis. Logistics are a challenge. And 50% of babies did not meet treatment guidelines.
In comparison to the state of screening and management in Utah, moderator Alex S. Stagnaro-Green, MD, noted that many pediatricians and endocrinologists operate under the presumption that screening for primary congenital hypothyroidism is “a well-oiled machine and that these babies are being taken care of.”
Utah has a very functional public health infrastructure. Of note, Utah’s birth rate is the highest in the country. The birth rates in several of its counties rival the highest rates found in the world, according to Dr. Ehrenkranz.
“So I think we have a very significant problem nationwide that just hasn’t been looked at,” he noted in response to the question. He undertook looking at newborn TSH levels in the first place as part of a project with the Food and Drug Administration. It was only then that he and his associates were struck by how many babies had low serum TSH levels, he said.
He had no relevant financial relationships to disclose.
DENVER – An alarming percentage of infants born in Utah from 2006 to 2015 with primary congenital hypothyroidism were either lost to follow-up or inadequately treated.
If such a thing can happen in Utah with its highly functioning public health system, it probably can happen in the rest of the United States as well, Joel Ehrenkranz, MD, said at the American Thyroid Association annual meeting. “We just have not looked for it yet.”
Screening for and treating congenital hypothyroidism in infants is one of the great public health successes of the 20th century in the United States. It deserves to have the same level of importance as eradication of polio and smallpox in this country, noted Dr. Ehrenkranz, an endocrinologist in private practice in Glenwood Springs, Colo. At the time of this research, Dr. Ehrenkranz was with Intermountain Healthcare in Murray, Utah.
The American Academy of Pediatrics recommends diagnosis of congenital hypothyroidism by the 14th day of life and that the baby be biochemically euthyroid by week 6 (Pediatrics. 2006 June. doi: 10.1542/peds.2006-0915), he said.
The cohort included 4,394 children from birth to 24 months of age. The screening test was done by a third-generation bioluminescence serum TSH assay, not dried blood blot. Of these infants, 2% (82 babies) had a TSH level greater than or equal to 20 mIU/L at their initial test. That TSH was still high by day 14 in 42 infants (23 girls). But of all the babies with primary congenital hypothyroidism, 50% had a delayed diagnosis, he reported.
Twelve children (15%) were never retested; 34% reached AAP goals of having a TSH level less than 5 mIU/L within 28 days after starting treatment; half of the children with primary congenital hypothyropidism did not meet the treatment goal: “They were inadequately treated,” he said.
Of particular interest were 16 infants who had a TSH level of less than 20 mIU/L but on retesting had one of 20 or higher. Three of these infants had multiple TSH levels greater than 20, perhaps representing a subset with a late onset form of the disorder.
“We are not doing as well as we could do,” he said; 50% of affected babies have a delayed diagnosis with consequences of a delayed maturation of the pituitary thyroid axis. Logistics are a challenge. And 50% of babies did not meet treatment guidelines.
In comparison to the state of screening and management in Utah, moderator Alex S. Stagnaro-Green, MD, noted that many pediatricians and endocrinologists operate under the presumption that screening for primary congenital hypothyroidism is “a well-oiled machine and that these babies are being taken care of.”
Utah has a very functional public health infrastructure. Of note, Utah’s birth rate is the highest in the country. The birth rates in several of its counties rival the highest rates found in the world, according to Dr. Ehrenkranz.
“So I think we have a very significant problem nationwide that just hasn’t been looked at,” he noted in response to the question. He undertook looking at newborn TSH levels in the first place as part of a project with the Food and Drug Administration. It was only then that he and his associates were struck by how many babies had low serum TSH levels, he said.
He had no relevant financial relationships to disclose.
DENVER – An alarming percentage of infants born in Utah from 2006 to 2015 with primary congenital hypothyroidism were either lost to follow-up or inadequately treated.
If such a thing can happen in Utah with its highly functioning public health system, it probably can happen in the rest of the United States as well, Joel Ehrenkranz, MD, said at the American Thyroid Association annual meeting. “We just have not looked for it yet.”
Screening for and treating congenital hypothyroidism in infants is one of the great public health successes of the 20th century in the United States. It deserves to have the same level of importance as eradication of polio and smallpox in this country, noted Dr. Ehrenkranz, an endocrinologist in private practice in Glenwood Springs, Colo. At the time of this research, Dr. Ehrenkranz was with Intermountain Healthcare in Murray, Utah.
The American Academy of Pediatrics recommends diagnosis of congenital hypothyroidism by the 14th day of life and that the baby be biochemically euthyroid by week 6 (Pediatrics. 2006 June. doi: 10.1542/peds.2006-0915), he said.
The cohort included 4,394 children from birth to 24 months of age. The screening test was done by a third-generation bioluminescence serum TSH assay, not dried blood blot. Of these infants, 2% (82 babies) had a TSH level greater than or equal to 20 mIU/L at their initial test. That TSH was still high by day 14 in 42 infants (23 girls). But of all the babies with primary congenital hypothyroidism, 50% had a delayed diagnosis, he reported.
Twelve children (15%) were never retested; 34% reached AAP goals of having a TSH level less than 5 mIU/L within 28 days after starting treatment; half of the children with primary congenital hypothyropidism did not meet the treatment goal: “They were inadequately treated,” he said.
Of particular interest were 16 infants who had a TSH level of less than 20 mIU/L but on retesting had one of 20 or higher. Three of these infants had multiple TSH levels greater than 20, perhaps representing a subset with a late onset form of the disorder.
“We are not doing as well as we could do,” he said; 50% of affected babies have a delayed diagnosis with consequences of a delayed maturation of the pituitary thyroid axis. Logistics are a challenge. And 50% of babies did not meet treatment guidelines.
In comparison to the state of screening and management in Utah, moderator Alex S. Stagnaro-Green, MD, noted that many pediatricians and endocrinologists operate under the presumption that screening for primary congenital hypothyroidism is “a well-oiled machine and that these babies are being taken care of.”
Utah has a very functional public health infrastructure. Of note, Utah’s birth rate is the highest in the country. The birth rates in several of its counties rival the highest rates found in the world, according to Dr. Ehrenkranz.
“So I think we have a very significant problem nationwide that just hasn’t been looked at,” he noted in response to the question. He undertook looking at newborn TSH levels in the first place as part of a project with the Food and Drug Administration. It was only then that he and his associates were struck by how many babies had low serum TSH levels, he said.
He had no relevant financial relationships to disclose.
AT THE ATA ANNUAL MEETING
Key clinical point: A large percentage of babies with congenital hypothyroidism are falling through the cracks in Utah, and likely throughout the United States.
Major finding: Almost 2% of 4,395 babies had TSH levels equal to or above 20 mIU/L when assessed after birth; of those, a significant share were lost to follow-up or inadequately treated.
Data source: A review of TSH measurements in all babies born in Utah between 2006 and 2015.
Disclosures: Dr. Ehrenkranz had no relevant financial disclosures.
The thyroid takes a beating during PCI in about 3% of patients
DENVER – Undergoing percutaneous coronary intervention (PCI) may impair thyroid function and change the gland’s morphology, probably because of the cumulative effects of handling and exposure to radiation and iodine in the contrast dye, Samir Naim Assaad, MD, said during a poster presentation at the annual meeting of the American Thyroid Association.
Cardiologists should inform their patients of these possible effects as part of their pre- and post-PCI counseling so that they won’t be alarmed by the changes in how they feel, Dr. Assaad, chief of the division of endocrinology at Alexandria (Egypt) University, said in an interview.

Similarly, when a formerly euthyroid patient presents to an endocrinologist with sudden-onset hyperthyroidism, “Have you had a PCI recently?” should be one of the first questions asked. If the answer is yes, then further testing and imaging should be delayed at least 3 months, he noted.
Dr. Assaad and his associates examined 113 clinically euthyroid patients both before and several months after they underwent PCI for management of stable coronary artery disease. The cohort included 93 men, and patients’ ages ranged from 32 years to 73 years.
All the patients underwent a series of tests immediately before PCI, 24 hours after, and 3 months later. Those tests included serum free triiodothyronine (FT3), free thyroxine (FT4), thyroid-stimulating hormone (TSH), free T3/T4 ratio, antithyroperoxidase (anti-TPO), and high-sensitivity C-reactive protein.
The gland’s morphology, including volume, vascularity, nodules, and echogenicity, were assessed on the same timetable using ultrasonography.
One day after PCI, there was a significant increase in serum FT3 (5.2-0.5 vs. 3.3-0.7 pg/mL, P less than .001), and serum FT4 (1.3 – 0.5 vs 1.2 – 0.3 ng/dL, P = .04), with no significant change in serum TSH.
Three months after PCI, there was a further significant increase in serum FT4 (1.5 – 0.3 ng/dL), decrease in serum FT3 returning to baseline (3.2 – 1.3 pg/mL), and a significant increase in serum TSH, compared with just before PCI (mean, 3.2-5 vs. 1.5-2.1 mIU/L, P less than .001). Serum anti-TPO (AU/mL) showed a significant increase 3 months after PCI.
There was a significant increase in thyroid gland volume 3 months after PCI (13.6-3.9 vs. 13.1-3.5 cm3, P = .02). The measured echogenicity of the thyroid gland showed a significant decrease 3 months after PCI (67.1-10.9 vs. 88.7-25.6 GWE, P less than .001).
Thyroid radiation had a negative effect on serum TSH, anti-TPO, FT3, and FT3/FT4 ratio, and an inverse correlation of dye injection time with serum anti-TPO and TSH, judging from the findings of a regression analysis model.
Dr. Assaad was not included on the list of presenters with relevant financial disclosures that was provided by the American Thyroid Association.
DENVER – Undergoing percutaneous coronary intervention (PCI) may impair thyroid function and change the gland’s morphology, probably because of the cumulative effects of handling and exposure to radiation and iodine in the contrast dye, Samir Naim Assaad, MD, said during a poster presentation at the annual meeting of the American Thyroid Association.
Cardiologists should inform their patients of these possible effects as part of their pre- and post-PCI counseling so that they won’t be alarmed by the changes in how they feel, Dr. Assaad, chief of the division of endocrinology at Alexandria (Egypt) University, said in an interview.
Similarly, when a formerly euthyroid patient presents to an endocrinologist with sudden-onset hyperthyroidism, “Have you had a PCI recently?” should be one of the first questions asked. If the answer is yes, then further testing and imaging should be delayed at least 3 months, he noted.
Dr. Assaad and his associates examined 113 clinically euthyroid patients both before and several months after they underwent PCI for management of stable coronary artery disease. The cohort included 93 men, and patients’ ages ranged from 32 years to 73 years.
All the patients underwent a series of tests immediately before PCI, 24 hours after, and 3 months later. Those tests included serum free triiodothyronine (FT3), free thyroxine (FT4), thyroid-stimulating hormone (TSH), free T3/T4 ratio, antithyroperoxidase (anti-TPO), and high-sensitivity C-reactive protein.
The gland’s morphology, including volume, vascularity, nodules, and echogenicity, were assessed on the same timetable using ultrasonography.
One day after PCI, there was a significant increase in serum FT3 (5.2-0.5 vs. 3.3-0.7 pg/mL, P less than .001), and serum FT4 (1.3 – 0.5 vs 1.2 – 0.3 ng/dL, P = .04), with no significant change in serum TSH.
Three months after PCI, there was a further significant increase in serum FT4 (1.5 – 0.3 ng/dL), decrease in serum FT3 returning to baseline (3.2 – 1.3 pg/mL), and a significant increase in serum TSH, compared with just before PCI (mean, 3.2-5 vs. 1.5-2.1 mIU/L, P less than .001). Serum anti-TPO (AU/mL) showed a significant increase 3 months after PCI.
There was a significant increase in thyroid gland volume 3 months after PCI (13.6-3.9 vs. 13.1-3.5 cm3, P = .02). The measured echogenicity of the thyroid gland showed a significant decrease 3 months after PCI (67.1-10.9 vs. 88.7-25.6 GWE, P less than .001).
Thyroid radiation had a negative effect on serum TSH, anti-TPO, FT3, and FT3/FT4 ratio, and an inverse correlation of dye injection time with serum anti-TPO and TSH, judging from the findings of a regression analysis model.
Dr. Assaad was not included on the list of presenters with relevant financial disclosures that was provided by the American Thyroid Association.
DENVER – Undergoing percutaneous coronary intervention (PCI) may impair thyroid function and change the gland’s morphology, probably because of the cumulative effects of handling and exposure to radiation and iodine in the contrast dye, Samir Naim Assaad, MD, said during a poster presentation at the annual meeting of the American Thyroid Association.
Cardiologists should inform their patients of these possible effects as part of their pre- and post-PCI counseling so that they won’t be alarmed by the changes in how they feel, Dr. Assaad, chief of the division of endocrinology at Alexandria (Egypt) University, said in an interview.
Similarly, when a formerly euthyroid patient presents to an endocrinologist with sudden-onset hyperthyroidism, “Have you had a PCI recently?” should be one of the first questions asked. If the answer is yes, then further testing and imaging should be delayed at least 3 months, he noted.
Dr. Assaad and his associates examined 113 clinically euthyroid patients both before and several months after they underwent PCI for management of stable coronary artery disease. The cohort included 93 men, and patients’ ages ranged from 32 years to 73 years.
All the patients underwent a series of tests immediately before PCI, 24 hours after, and 3 months later. Those tests included serum free triiodothyronine (FT3), free thyroxine (FT4), thyroid-stimulating hormone (TSH), free T3/T4 ratio, antithyroperoxidase (anti-TPO), and high-sensitivity C-reactive protein.
The gland’s morphology, including volume, vascularity, nodules, and echogenicity, were assessed on the same timetable using ultrasonography.
One day after PCI, there was a significant increase in serum FT3 (5.2-0.5 vs. 3.3-0.7 pg/mL, P less than .001), and serum FT4 (1.3 – 0.5 vs 1.2 – 0.3 ng/dL, P = .04), with no significant change in serum TSH.
Three months after PCI, there was a further significant increase in serum FT4 (1.5 – 0.3 ng/dL), decrease in serum FT3 returning to baseline (3.2 – 1.3 pg/mL), and a significant increase in serum TSH, compared with just before PCI (mean, 3.2-5 vs. 1.5-2.1 mIU/L, P less than .001). Serum anti-TPO (AU/mL) showed a significant increase 3 months after PCI.
There was a significant increase in thyroid gland volume 3 months after PCI (13.6-3.9 vs. 13.1-3.5 cm3, P = .02). The measured echogenicity of the thyroid gland showed a significant decrease 3 months after PCI (67.1-10.9 vs. 88.7-25.6 GWE, P less than .001).
Thyroid radiation had a negative effect on serum TSH, anti-TPO, FT3, and FT3/FT4 ratio, and an inverse correlation of dye injection time with serum anti-TPO and TSH, judging from the findings of a regression analysis model.
Dr. Assaad was not included on the list of presenters with relevant financial disclosures that was provided by the American Thyroid Association.
AT THE ATA ANNUAL MEETING
Key clinical point: The thyroid function and morphology of any patient undergoing PCI may be altered by the procedure; but most changes normalize within several months.
Major finding: Thyroid function changes in close to 3% of patients undergoing PCI.
Data source: A study of 113 euthyroid patients who had PCI for coronary artery disease.
Disclosures: Dr. Assaad was not included on the list of presenters with relevant financial disclosures provided by the American Thyroid Association.

Lenvatinib sparked or worsened hypertension in patients with RAI-resistant thyroid cancer
DENVER – Patients put on lenvatinib for the management of radioactive iodine–resistant differentiated thyroid cancer need to be taught how to monitor their blood pressure, be given a cuff with which to do so, and be called daily by someone on the medical staff for at least the first 2 weeks, according to Sina A. Jasim, MD, reporting on real world use of the drug since its approval in February 2015.
For now, oncologists prescribe and manage this drug. But as lenvatinib (Lenvima, Eisai) becomes more widely used, endocrinologists can expect to be the ones prescribing it sometimes and counseling patients in the practical aspects of using this drug, Dr. Jasim of the Mayo Clinic, Rochester, Minn., said in an interview.

It was with endocrinologists in mind that Dr. Jasim and her associates compiled postapproval data on adverse events and patient quality of life. To date, no such data – including those from Mayo – have been published, she said during her presentation at the American Thyroid Association’s annual meeting.
While lenvatinib seems to be a promising therapeutic agent, adverse events are common with its use and occur early. Patients treated with it at Mayo get called by someone on the medical staff daily for the first 2 weeks of therapy, are given a blood pressure cuff, and are taught how to use it. They also receive the cell phone number of a member of the medical staff to consult with about sudden symptoms.
This retrospective analysis involved 25 sequentially treated patients given lenvatinib for RAI-resistant differentiated thyroid cancer (14 papillary, 7 poorly differentiated, 3 Hürthle cell, and 1 follicular). While all had received RAI, 11 also had received radiotherapy, 8 had been given at least one other kinase inhibitor previously, and 3 had received two. Fourteen were on an antihypertensive medication at baseline.
All patients initiated lenvatinib at the full dose, but it was reduced in four patients because of old age, renal impairment, or prior colitis. Twenty-one patients developed adverse events within the first month of being on the drug. Hypertension occurred in 16. Six of these required either a raising of the dose of antihypertensive drug they were on at baseline or initiation of antihypertensive therapy.
Adverse events were pronounced enough that the lenvatinib dose had to be lowered in 11 within a median 33 days of starting the drug. Drug treatment had to be interrupted for at least 3 weeks in four patients (two cases of cholecystitis, one case of diverticulitis, and one case of skin lesions).
Patients reported that their quality of life was stable at 2 months, but that their fatigue was worse.
The mean duration of lenvatinib therapy was 6.5 months. Twenty patients are alive at the time of this report.
The study was sponsored by the Mayo Clinic. Dr. Jasim reported that she had no relevant financial disclosures.
DENVER – Patients put on lenvatinib for the management of radioactive iodine–resistant differentiated thyroid cancer need to be taught how to monitor their blood pressure, be given a cuff with which to do so, and be called daily by someone on the medical staff for at least the first 2 weeks, according to Sina A. Jasim, MD, reporting on real world use of the drug since its approval in February 2015.
For now, oncologists prescribe and manage this drug. But as lenvatinib (Lenvima, Eisai) becomes more widely used, endocrinologists can expect to be the ones prescribing it sometimes and counseling patients in the practical aspects of using this drug, Dr. Jasim of the Mayo Clinic, Rochester, Minn., said in an interview.
It was with endocrinologists in mind that Dr. Jasim and her associates compiled postapproval data on adverse events and patient quality of life. To date, no such data – including those from Mayo – have been published, she said during her presentation at the American Thyroid Association’s annual meeting.
While lenvatinib seems to be a promising therapeutic agent, adverse events are common with its use and occur early. Patients treated with it at Mayo get called by someone on the medical staff daily for the first 2 weeks of therapy, are given a blood pressure cuff, and are taught how to use it. They also receive the cell phone number of a member of the medical staff to consult with about sudden symptoms.
This retrospective analysis involved 25 sequentially treated patients given lenvatinib for RAI-resistant differentiated thyroid cancer (14 papillary, 7 poorly differentiated, 3 Hürthle cell, and 1 follicular). While all had received RAI, 11 also had received radiotherapy, 8 had been given at least one other kinase inhibitor previously, and 3 had received two. Fourteen were on an antihypertensive medication at baseline.
All patients initiated lenvatinib at the full dose, but it was reduced in four patients because of old age, renal impairment, or prior colitis. Twenty-one patients developed adverse events within the first month of being on the drug. Hypertension occurred in 16. Six of these required either a raising of the dose of antihypertensive drug they were on at baseline or initiation of antihypertensive therapy.
Adverse events were pronounced enough that the lenvatinib dose had to be lowered in 11 within a median 33 days of starting the drug. Drug treatment had to be interrupted for at least 3 weeks in four patients (two cases of cholecystitis, one case of diverticulitis, and one case of skin lesions).
Patients reported that their quality of life was stable at 2 months, but that their fatigue was worse.
The mean duration of lenvatinib therapy was 6.5 months. Twenty patients are alive at the time of this report.
The study was sponsored by the Mayo Clinic. Dr. Jasim reported that she had no relevant financial disclosures.
DENVER – Patients put on lenvatinib for the management of radioactive iodine–resistant differentiated thyroid cancer need to be taught how to monitor their blood pressure, be given a cuff with which to do so, and be called daily by someone on the medical staff for at least the first 2 weeks, according to Sina A. Jasim, MD, reporting on real world use of the drug since its approval in February 2015.
For now, oncologists prescribe and manage this drug. But as lenvatinib (Lenvima, Eisai) becomes more widely used, endocrinologists can expect to be the ones prescribing it sometimes and counseling patients in the practical aspects of using this drug, Dr. Jasim of the Mayo Clinic, Rochester, Minn., said in an interview.
It was with endocrinologists in mind that Dr. Jasim and her associates compiled postapproval data on adverse events and patient quality of life. To date, no such data – including those from Mayo – have been published, she said during her presentation at the American Thyroid Association’s annual meeting.
While lenvatinib seems to be a promising therapeutic agent, adverse events are common with its use and occur early. Patients treated with it at Mayo get called by someone on the medical staff daily for the first 2 weeks of therapy, are given a blood pressure cuff, and are taught how to use it. They also receive the cell phone number of a member of the medical staff to consult with about sudden symptoms.
This retrospective analysis involved 25 sequentially treated patients given lenvatinib for RAI-resistant differentiated thyroid cancer (14 papillary, 7 poorly differentiated, 3 Hürthle cell, and 1 follicular). While all had received RAI, 11 also had received radiotherapy, 8 had been given at least one other kinase inhibitor previously, and 3 had received two. Fourteen were on an antihypertensive medication at baseline.
All patients initiated lenvatinib at the full dose, but it was reduced in four patients because of old age, renal impairment, or prior colitis. Twenty-one patients developed adverse events within the first month of being on the drug. Hypertension occurred in 16. Six of these required either a raising of the dose of antihypertensive drug they were on at baseline or initiation of antihypertensive therapy.
Adverse events were pronounced enough that the lenvatinib dose had to be lowered in 11 within a median 33 days of starting the drug. Drug treatment had to be interrupted for at least 3 weeks in four patients (two cases of cholecystitis, one case of diverticulitis, and one case of skin lesions).
Patients reported that their quality of life was stable at 2 months, but that their fatigue was worse.
The mean duration of lenvatinib therapy was 6.5 months. Twenty patients are alive at the time of this report.
The study was sponsored by the Mayo Clinic. Dr. Jasim reported that she had no relevant financial disclosures.
AT THE ATA ANNUAL MEETING
Key clinical point: Hypertension appeared suddenly or worsened abruptly in more than half of one group of patients with RAI-resistant differentiated thyroid cancer after initiation of treatment with lenvatinib.
Major finding: Of 25 patients in whom lenvatinib was initiated, 21 developed adverse events in the first month. Of those were 16 who developed hypertension, often requiring dose reduction.
Data source: A retrospective report on 25 consecutively treated patients with RAI-resistant differentiated thyroid cancer, who were given lenvatinib between February 2015 and May 2016.
Disclosures: The study was sponsored by the Mayo Clinic. Dr. Jasim reported that she had no relevant financial disclosures.

High free T4 levels linked to sudden cardiac death
Higher levels of free thyroxine are associated with an increased risk of sudden cardiac death, even in euthyroid adults, according to a report published online Sept. 6 in Circulation.
Thyroid dysfunction, even in the subclinical range, is known to correlate with increased cardiovascular disease, but until now a possible link between free thyroxine levels and sudden cardiac death (SCD) has never been explored in the general population. Any factors that could improve prediction of SCD in the general population would be helpful because almost half of these cases are the first indication that the patient had heart disease, said Layal Chaker, MD, of the Rotterdam Thyroid Center and the departments of internal medicine and epidemiology, Erasmus University, Rotterdam, and her associates.
They assessed SCD among 10,318 participants in the Rotterdam Study, a prospective population-based cohort study examining endocrine, cardiovascular, neurologic, ophthalmologic, and psychiatric diseases in middle-aged and older adults in the Netherlands. Men and women aged 45-106 years who had thyroid testing at baseline were followed for a median of 9.2 years (range, 4-21 years) for the development of SCD. There were 261 cases of SCD, and 231 of these occurred in euthyroid participants.
Higher levels of free thyroxine (T4) were associated with an increased risk of SCD, with a hazard ratio of 1.87 for every 1 ng/dL increase in free T4. When the analysis was confined to the 231 euthyroid participants, this association was even stronger, with an HR of 2.26, the investigators said (Circulation 2016 Sept 6. doi: 10.1161/CirculationAHA.115.020789).
The findings were similar in several sensitivity analyses, including one that excluded participants who had an unwitnessed SCD. In addition, adjustment of the data to account for the presence or absence of diabetes, as well as exclusion of patients who had heart failure, did not alter the risk estimates significantly. The results also were consistent across all age groups and both sexes, Dr. Chaker and her associates said.
The exact mechanism for the association between free thyroxine and SCD is not yet known but appears to be independent of traditional cardiovascular risk factors. “Bigger sample size and more detailed data are needed to determine whether these associations share the same or have distinct pathways,” they added.
The Netherlands Organisation for Health Research and Development and Erasmus Medical Center supported the study. Dr. Chaker and her associates reported having no relevant financial disclosures.
Higher levels of free thyroxine are associated with an increased risk of sudden cardiac death, even in euthyroid adults, according to a report published online Sept. 6 in Circulation.
Thyroid dysfunction, even in the subclinical range, is known to correlate with increased cardiovascular disease, but until now a possible link between free thyroxine levels and sudden cardiac death (SCD) has never been explored in the general population. Any factors that could improve prediction of SCD in the general population would be helpful because almost half of these cases are the first indication that the patient had heart disease, said Layal Chaker, MD, of the Rotterdam Thyroid Center and the departments of internal medicine and epidemiology, Erasmus University, Rotterdam, and her associates.
They assessed SCD among 10,318 participants in the Rotterdam Study, a prospective population-based cohort study examining endocrine, cardiovascular, neurologic, ophthalmologic, and psychiatric diseases in middle-aged and older adults in the Netherlands. Men and women aged 45-106 years who had thyroid testing at baseline were followed for a median of 9.2 years (range, 4-21 years) for the development of SCD. There were 261 cases of SCD, and 231 of these occurred in euthyroid participants.
Higher levels of free thyroxine (T4) were associated with an increased risk of SCD, with a hazard ratio of 1.87 for every 1 ng/dL increase in free T4. When the analysis was confined to the 231 euthyroid participants, this association was even stronger, with an HR of 2.26, the investigators said (Circulation 2016 Sept 6. doi: 10.1161/CirculationAHA.115.020789).
The findings were similar in several sensitivity analyses, including one that excluded participants who had an unwitnessed SCD. In addition, adjustment of the data to account for the presence or absence of diabetes, as well as exclusion of patients who had heart failure, did not alter the risk estimates significantly. The results also were consistent across all age groups and both sexes, Dr. Chaker and her associates said.
The exact mechanism for the association between free thyroxine and SCD is not yet known but appears to be independent of traditional cardiovascular risk factors. “Bigger sample size and more detailed data are needed to determine whether these associations share the same or have distinct pathways,” they added.
The Netherlands Organisation for Health Research and Development and Erasmus Medical Center supported the study. Dr. Chaker and her associates reported having no relevant financial disclosures.
Higher levels of free thyroxine are associated with an increased risk of sudden cardiac death, even in euthyroid adults, according to a report published online Sept. 6 in Circulation.
Thyroid dysfunction, even in the subclinical range, is known to correlate with increased cardiovascular disease, but until now a possible link between free thyroxine levels and sudden cardiac death (SCD) has never been explored in the general population. Any factors that could improve prediction of SCD in the general population would be helpful because almost half of these cases are the first indication that the patient had heart disease, said Layal Chaker, MD, of the Rotterdam Thyroid Center and the departments of internal medicine and epidemiology, Erasmus University, Rotterdam, and her associates.
They assessed SCD among 10,318 participants in the Rotterdam Study, a prospective population-based cohort study examining endocrine, cardiovascular, neurologic, ophthalmologic, and psychiatric diseases in middle-aged and older adults in the Netherlands. Men and women aged 45-106 years who had thyroid testing at baseline were followed for a median of 9.2 years (range, 4-21 years) for the development of SCD. There were 261 cases of SCD, and 231 of these occurred in euthyroid participants.
Higher levels of free thyroxine (T4) were associated with an increased risk of SCD, with a hazard ratio of 1.87 for every 1 ng/dL increase in free T4. When the analysis was confined to the 231 euthyroid participants, this association was even stronger, with an HR of 2.26, the investigators said (Circulation 2016 Sept 6. doi: 10.1161/CirculationAHA.115.020789).
The findings were similar in several sensitivity analyses, including one that excluded participants who had an unwitnessed SCD. In addition, adjustment of the data to account for the presence or absence of diabetes, as well as exclusion of patients who had heart failure, did not alter the risk estimates significantly. The results also were consistent across all age groups and both sexes, Dr. Chaker and her associates said.
The exact mechanism for the association between free thyroxine and SCD is not yet known but appears to be independent of traditional cardiovascular risk factors. “Bigger sample size and more detailed data are needed to determine whether these associations share the same or have distinct pathways,” they added.
The Netherlands Organisation for Health Research and Development and Erasmus Medical Center supported the study. Dr. Chaker and her associates reported having no relevant financial disclosures.
FROM CIRCULATION
Key clinical point: High levels of free thyroxine are associated with an increased risk of sudden cardiac death, even in euthyroid adults.
Major finding: Higher levels of free thyroxine (T4) were associated with an increased risk of SCD, with a hazard ratio of 1.87 for every 1 ng/dL increase in free T4.
Data source: A prospective population-based cohort study involving 10,318 older adults in the Netherlands followed for a median of 9 years.
Disclosures: The Netherlands Organisation for Health Research and Development and Erasmus Medical Center supported the study. Dr. Chaker and her associates reported having no relevant financial disclosures.
Primary care physicians diagnose most pediatric thyroid conditions
Primary care physicians can play an important role in managing thyroid disease in children and teens by proactive screening and evaluation, based on data from a literature review of 83 articles published between Jan. 1, 2010, and Dec. 31, 2015. The review was published online Aug. 29 in JAMA Pediatrics.
“Early diagnosis and treatment of thyroid hormone deficiency is crucial to ensure normal development and cognition,” wrote Dr. Patrick Hanley of the Children’s Hospital of Philadelphia and his colleagues.
Thyroid dysgenesis accounts for 80%-85% of cases of primary congenital hypothyroidism, and many newborns with the condition are asymptomatic at birth because of protection by maternal thyroid hormones. Early signs of thyroid problems include a hoarse cry, prolonged jaundice, lethargy, poor feeding, and constipation, the researchers said (JAMA Pediatr. 2016. doi:10.1001/jamapediatrics.2016.0486).
“Once the diagnosis has been made, additional testing can be considered to determine the etiology of the hypothyroidism so that the family can receive anticipatory guidance in regard to the potential need for lifelong thyroid hormone replacement therapy,” the researchers wrote.
The treatment of choice for congenital hypothyroidism is levothyroxine at a starting dose of 10-15 mcg/kg once daily, they noted.
Read the full study here: http://archpedi.jamanetwork.com/article.aspx?doi=10.1001/jamapediatrics.2016.0486.
Primary care physicians can play an important role in managing thyroid disease in children and teens by proactive screening and evaluation, based on data from a literature review of 83 articles published between Jan. 1, 2010, and Dec. 31, 2015. The review was published online Aug. 29 in JAMA Pediatrics.
“Early diagnosis and treatment of thyroid hormone deficiency is crucial to ensure normal development and cognition,” wrote Dr. Patrick Hanley of the Children’s Hospital of Philadelphia and his colleagues.
Thyroid dysgenesis accounts for 80%-85% of cases of primary congenital hypothyroidism, and many newborns with the condition are asymptomatic at birth because of protection by maternal thyroid hormones. Early signs of thyroid problems include a hoarse cry, prolonged jaundice, lethargy, poor feeding, and constipation, the researchers said (JAMA Pediatr. 2016. doi:10.1001/jamapediatrics.2016.0486).
“Once the diagnosis has been made, additional testing can be considered to determine the etiology of the hypothyroidism so that the family can receive anticipatory guidance in regard to the potential need for lifelong thyroid hormone replacement therapy,” the researchers wrote.
The treatment of choice for congenital hypothyroidism is levothyroxine at a starting dose of 10-15 mcg/kg once daily, they noted.
Read the full study here: http://archpedi.jamanetwork.com/article.aspx?doi=10.1001/jamapediatrics.2016.0486.
Primary care physicians can play an important role in managing thyroid disease in children and teens by proactive screening and evaluation, based on data from a literature review of 83 articles published between Jan. 1, 2010, and Dec. 31, 2015. The review was published online Aug. 29 in JAMA Pediatrics.
“Early diagnosis and treatment of thyroid hormone deficiency is crucial to ensure normal development and cognition,” wrote Dr. Patrick Hanley of the Children’s Hospital of Philadelphia and his colleagues.
Thyroid dysgenesis accounts for 80%-85% of cases of primary congenital hypothyroidism, and many newborns with the condition are asymptomatic at birth because of protection by maternal thyroid hormones. Early signs of thyroid problems include a hoarse cry, prolonged jaundice, lethargy, poor feeding, and constipation, the researchers said (JAMA Pediatr. 2016. doi:10.1001/jamapediatrics.2016.0486).
“Once the diagnosis has been made, additional testing can be considered to determine the etiology of the hypothyroidism so that the family can receive anticipatory guidance in regard to the potential need for lifelong thyroid hormone replacement therapy,” the researchers wrote.
The treatment of choice for congenital hypothyroidism is levothyroxine at a starting dose of 10-15 mcg/kg once daily, they noted.
Read the full study here: http://archpedi.jamanetwork.com/article.aspx?doi=10.1001/jamapediatrics.2016.0486.
FROM JAMA PEDIATRICS
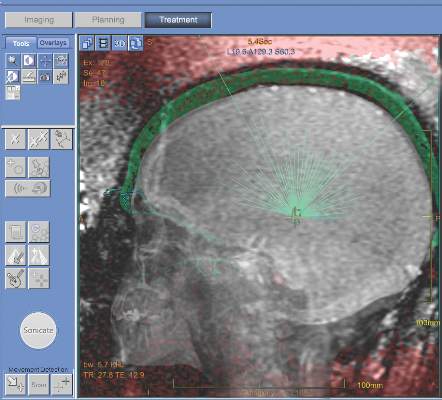
MRI-guided thalamotomy significantly reduces hand tremors
MRI-guided focused ultrasound thalamotomy can significantly mitigate the severity of hand tremors in patients suffering from essential tremor, the most common type of movement disorder, according to a new study published in the New England Journal of Medicine.
“The use of ultrasound energy for the creation of discrete intracranial lesions... has been of interest since the middle of the 20th century,” wrote the investigators, led by W. Jeffrey Elias, MD, of the University of Virginia, Charlottesville. “Prospective pilot trials of focused ultrasound thalamotomy with magnetic resonance imaging (MRI) guidance in patients with essential tremor have shown reductions in hand tremor, improvements in quality of life, and minimal procedural morbidity.”

The trial enrolled a total of 76 patients with a mean age of 71 years and mean disease duration of nearly 17 years; 68% were men and 75% were white. At a 3:1 ratio, they were randomized into one of two cohorts: one underwent thalamotomy and the other received a “sham” procedure. The subjects were unaware which they received for the first 3 months. The Clinical Rating Scale for Tremor (CRST) and the Quality of Life in Essential Tremor Questionnaire (QUEST) was used to determine the severity of tremors at baseline, and at follow-ups conducted at 1, 3, 6, and 12 months post-procedure (N Engl J Med. 2016;375[8]:730-9).
The trial’s primary outcome of between-group difference in the change in tremor score from baseline to 3 months significantly favored thalamotomy (8.5-point improvement, from 18.1 to 9.6) over the sham procedure (0.2-point improvement, from 16.0 to 15.8). The mean between-group difference in the change in score of 8.3 points at 3 months decreased slightly to 7.2 points at 12 months. The tremor score (range, 0-32) was derived from part A of the CRST (three items: resting, postural, and action or intention components of hand tremor), and part B of the CRST (five tasks involving handwriting, drawing, and pouring), in the hand contralateral to the thalamotomy.
Thalamotomy patients also reported 46% better quality of life on QUEST at 3 months, compared with 3% better among sham-procedure patients.
There were adverse events in the thalamotomy cohort. At the 3-month follow-up, 36% of subjects experienced gait disturbance, 38% experienced paresthesias or some kind of numbness. The rates of these adverse events dropped to 9% and 14%, respectively, at the 12-month follow-up.
“Deep-brain stimulation is currently the surgical standard for medication-refractory essential tremor [but] a control group of patients undergoing deep-brain stimulation was not included in this trial; the two technologies were not compared,” the authors noted, indicating that such comparison could potentially be the next step for this research.
This study was supported by InSightec, the Focused Ultrasound Foundation, and the Binational Industrial Research and Development Foundation. Dr. Elias disclosed receiving grant support from InSightec and the Focused Ultrasound Foundation. Other coauthors disclosed receiving similar support.
There are several important concerns about this study. Its 12-month follow-up period is relatively short, so the sustained benefit at 2 years, 3 years, and 5 or more years is unknown. The tremor score of patients who underwent focused ultrasound thalamotomy increased by 23% from 1 month to 12 months, and it’s unclear whether this loss of efficacy is due to disease progression or tolerance. The typical estimates of the rate of disease progression in essential tremor make tolerance less likely. The loss of efficacy is also seen to some extent with deep-brain stimulation. Furthermore, some patients who underwent thalamotomy did not achieve large improvements in tremor. The percentage change was less than 20% in 9 of 56 patients.
It’s also worthwhile to note that focused ultrasound thalamotomy creates a fixed brain lesion, whereas with deep-brain stimulation there is the potential to adjust stimulator settings in order to obtain further therapeutic gains. The procedure also is not suitable for all patients, such as those with particularly thick skulls. The most common side effect involved altered sensation, which remained permanent in 14% of patients.
The procedure will take its place among other surgical procedures for medically refractory essential tremor. A head-to-head comparison with deep-brain stimulation would facilitate the direct comparison of the two approaches.
Elan D. Louis, MD, is the chief of the division of movement disorders and professor of neurology and epidemiology (chronic diseases) at Yale University, New Haven, Conn. His comments were taken from his editorial accompanying the report by Dr. Elias and his colleagues (N Engl J Med. 2016;375[8]:792-3).
There are several important concerns about this study. Its 12-month follow-up period is relatively short, so the sustained benefit at 2 years, 3 years, and 5 or more years is unknown. The tremor score of patients who underwent focused ultrasound thalamotomy increased by 23% from 1 month to 12 months, and it’s unclear whether this loss of efficacy is due to disease progression or tolerance. The typical estimates of the rate of disease progression in essential tremor make tolerance less likely. The loss of efficacy is also seen to some extent with deep-brain stimulation. Furthermore, some patients who underwent thalamotomy did not achieve large improvements in tremor. The percentage change was less than 20% in 9 of 56 patients.
It’s also worthwhile to note that focused ultrasound thalamotomy creates a fixed brain lesion, whereas with deep-brain stimulation there is the potential to adjust stimulator settings in order to obtain further therapeutic gains. The procedure also is not suitable for all patients, such as those with particularly thick skulls. The most common side effect involved altered sensation, which remained permanent in 14% of patients.
The procedure will take its place among other surgical procedures for medically refractory essential tremor. A head-to-head comparison with deep-brain stimulation would facilitate the direct comparison of the two approaches.
Elan D. Louis, MD, is the chief of the division of movement disorders and professor of neurology and epidemiology (chronic diseases) at Yale University, New Haven, Conn. His comments were taken from his editorial accompanying the report by Dr. Elias and his colleagues (N Engl J Med. 2016;375[8]:792-3).
There are several important concerns about this study. Its 12-month follow-up period is relatively short, so the sustained benefit at 2 years, 3 years, and 5 or more years is unknown. The tremor score of patients who underwent focused ultrasound thalamotomy increased by 23% from 1 month to 12 months, and it’s unclear whether this loss of efficacy is due to disease progression or tolerance. The typical estimates of the rate of disease progression in essential tremor make tolerance less likely. The loss of efficacy is also seen to some extent with deep-brain stimulation. Furthermore, some patients who underwent thalamotomy did not achieve large improvements in tremor. The percentage change was less than 20% in 9 of 56 patients.
It’s also worthwhile to note that focused ultrasound thalamotomy creates a fixed brain lesion, whereas with deep-brain stimulation there is the potential to adjust stimulator settings in order to obtain further therapeutic gains. The procedure also is not suitable for all patients, such as those with particularly thick skulls. The most common side effect involved altered sensation, which remained permanent in 14% of patients.
The procedure will take its place among other surgical procedures for medically refractory essential tremor. A head-to-head comparison with deep-brain stimulation would facilitate the direct comparison of the two approaches.
Elan D. Louis, MD, is the chief of the division of movement disorders and professor of neurology and epidemiology (chronic diseases) at Yale University, New Haven, Conn. His comments were taken from his editorial accompanying the report by Dr. Elias and his colleagues (N Engl J Med. 2016;375[8]:792-3).
MRI-guided focused ultrasound thalamotomy can significantly mitigate the severity of hand tremors in patients suffering from essential tremor, the most common type of movement disorder, according to a new study published in the New England Journal of Medicine.
“The use of ultrasound energy for the creation of discrete intracranial lesions... has been of interest since the middle of the 20th century,” wrote the investigators, led by W. Jeffrey Elias, MD, of the University of Virginia, Charlottesville. “Prospective pilot trials of focused ultrasound thalamotomy with magnetic resonance imaging (MRI) guidance in patients with essential tremor have shown reductions in hand tremor, improvements in quality of life, and minimal procedural morbidity.”
The trial enrolled a total of 76 patients with a mean age of 71 years and mean disease duration of nearly 17 years; 68% were men and 75% were white. At a 3:1 ratio, they were randomized into one of two cohorts: one underwent thalamotomy and the other received a “sham” procedure. The subjects were unaware which they received for the first 3 months. The Clinical Rating Scale for Tremor (CRST) and the Quality of Life in Essential Tremor Questionnaire (QUEST) was used to determine the severity of tremors at baseline, and at follow-ups conducted at 1, 3, 6, and 12 months post-procedure (N Engl J Med. 2016;375[8]:730-9).
The trial’s primary outcome of between-group difference in the change in tremor score from baseline to 3 months significantly favored thalamotomy (8.5-point improvement, from 18.1 to 9.6) over the sham procedure (0.2-point improvement, from 16.0 to 15.8). The mean between-group difference in the change in score of 8.3 points at 3 months decreased slightly to 7.2 points at 12 months. The tremor score (range, 0-32) was derived from part A of the CRST (three items: resting, postural, and action or intention components of hand tremor), and part B of the CRST (five tasks involving handwriting, drawing, and pouring), in the hand contralateral to the thalamotomy.
Thalamotomy patients also reported 46% better quality of life on QUEST at 3 months, compared with 3% better among sham-procedure patients.
There were adverse events in the thalamotomy cohort. At the 3-month follow-up, 36% of subjects experienced gait disturbance, 38% experienced paresthesias or some kind of numbness. The rates of these adverse events dropped to 9% and 14%, respectively, at the 12-month follow-up.
“Deep-brain stimulation is currently the surgical standard for medication-refractory essential tremor [but] a control group of patients undergoing deep-brain stimulation was not included in this trial; the two technologies were not compared,” the authors noted, indicating that such comparison could potentially be the next step for this research.
This study was supported by InSightec, the Focused Ultrasound Foundation, and the Binational Industrial Research and Development Foundation. Dr. Elias disclosed receiving grant support from InSightec and the Focused Ultrasound Foundation. Other coauthors disclosed receiving similar support.
MRI-guided focused ultrasound thalamotomy can significantly mitigate the severity of hand tremors in patients suffering from essential tremor, the most common type of movement disorder, according to a new study published in the New England Journal of Medicine.
“The use of ultrasound energy for the creation of discrete intracranial lesions... has been of interest since the middle of the 20th century,” wrote the investigators, led by W. Jeffrey Elias, MD, of the University of Virginia, Charlottesville. “Prospective pilot trials of focused ultrasound thalamotomy with magnetic resonance imaging (MRI) guidance in patients with essential tremor have shown reductions in hand tremor, improvements in quality of life, and minimal procedural morbidity.”
The trial enrolled a total of 76 patients with a mean age of 71 years and mean disease duration of nearly 17 years; 68% were men and 75% were white. At a 3:1 ratio, they were randomized into one of two cohorts: one underwent thalamotomy and the other received a “sham” procedure. The subjects were unaware which they received for the first 3 months. The Clinical Rating Scale for Tremor (CRST) and the Quality of Life in Essential Tremor Questionnaire (QUEST) was used to determine the severity of tremors at baseline, and at follow-ups conducted at 1, 3, 6, and 12 months post-procedure (N Engl J Med. 2016;375[8]:730-9).
The trial’s primary outcome of between-group difference in the change in tremor score from baseline to 3 months significantly favored thalamotomy (8.5-point improvement, from 18.1 to 9.6) over the sham procedure (0.2-point improvement, from 16.0 to 15.8). The mean between-group difference in the change in score of 8.3 points at 3 months decreased slightly to 7.2 points at 12 months. The tremor score (range, 0-32) was derived from part A of the CRST (three items: resting, postural, and action or intention components of hand tremor), and part B of the CRST (five tasks involving handwriting, drawing, and pouring), in the hand contralateral to the thalamotomy.
Thalamotomy patients also reported 46% better quality of life on QUEST at 3 months, compared with 3% better among sham-procedure patients.
There were adverse events in the thalamotomy cohort. At the 3-month follow-up, 36% of subjects experienced gait disturbance, 38% experienced paresthesias or some kind of numbness. The rates of these adverse events dropped to 9% and 14%, respectively, at the 12-month follow-up.
“Deep-brain stimulation is currently the surgical standard for medication-refractory essential tremor [but] a control group of patients undergoing deep-brain stimulation was not included in this trial; the two technologies were not compared,” the authors noted, indicating that such comparison could potentially be the next step for this research.
This study was supported by InSightec, the Focused Ultrasound Foundation, and the Binational Industrial Research and Development Foundation. Dr. Elias disclosed receiving grant support from InSightec and the Focused Ultrasound Foundation. Other coauthors disclosed receiving similar support.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: The severity of hand tremors in patients with essential tremor can be significantly reduced via use of MRI-guided focused ultrasound thalamotomy.
Major finding: Greater improvement was observed in the ultrasound thalamotomy cohort than in the control (sham treatment) cohort: 18.1 to 9.6 hand tremor score vs. 16.0 to 15.8, respectively.
Data source: A double-blind, randomized, sham-controlled cohort study of 76 patients with moderate-to-severe essential tremor.
Disclosures: Study supported by InSightec, the Focused Ultrasound Foundation, and the Binational Industrial Research and Development Foundation. Some coauthors reported potentially relevant disclosures.
Expert shares new insights on the pathophysiology of rosacea
NEWPORT BEACH, CALIF. – In the clinical opinion of Richard L. Gallo, MD, PhD, current nomenclature for the diagnosis of rosacea could use a makeover.
“Currently, we’re still operating with an almost 20-year-old set of diagnostic subtypes of rosacea,” Dr. Gallo said at the annual meeting of the Pacific Dermatologic Association. He plans to participate in consensus meeting of experts who will convene this fall in an effort to update and modify these diagnostic criteria.

According to current nomenclature, subtype 1 is erythematotelangiectatic rosacea characterized by facial redness; subtype 2 is papulopustular, marked by bumps and pimples; subtype 3 is phymatous, characterized by enlargement of the nose, and subtype 4 is ocular, marked by eye irritation. Dr. Gallo pointed out that it’s rare to see just one of these subtypes in rosacea patients, with the exception of the erythematotelangiectatic rosacea (ETR). “There is a large population with ETR alone,” he said.” Most patients with the papulopustular subtype have aspects of ETR. There is a mix of subtypes of rosacea and we clearly need to modify our diagnostic criteria.”
Secondary rosacea features may include burning or stinging, plaque, dry appearance and scale, edema, ocular manifestations, peripheral location, and phymatous changes. Work by several researchers in recent years has shed light on the pathophysiology of rosacea. “We’re learning that there are many aspects to this disease that both trigger it and result in progression of the disease,” said Dr. Gallo, professor and chair of the department of dermatology at the University of California, San Diego. “It seems to have biological triggers that exist both in the environment and initiate from internal sources. We’re understanding more about the nature of those, at least specific molecules externally from microbes and so forth. Internally we understand more about the unique inflammatory signals.”
For example, he and other researchers began to look at the innate immune system patients with rosacea and identified LL37, a multifunction peptide that plays a role in a number of skin diseases, as something that can promote inflammation (Nat Med. 2007;13[8]:975-80). “It also promotes the vascular changes [that occur with the disease],” Dr. Gallo said. “We’re now learning how a dysregulation of enzymes in the skin contributes to making too much of these types of peptides. Therefore, treatment approaches that might modify enzymatic activity become useful.” Researchers have also discovered that some of the innate recognition molecules like toll-like receptor 2 (TLR2) are overexpressed in rosacea patients. “Similarly, in terms of the vascular signals, a number of labs are identifying some of the newer vascular transmitters that seem to be uniquely elevated in rosacea, so there’s great reason to be optimistic that given the increased specificity and understanding of what uniquely makes this disease happen, we’ll be able to target it in a safe way.”
A number of published studies have supported these notions, including an analysis of 275 twin pairs (JAMA Dermatol. 2015;151[11]:1213-9). The researchers found that compared with fraternal twins, identical twins had a higher association of National Rosacea Society scores (P = .04), “supporting the concept that there are fundamental genetic factors that are influencing disease,” Dr. Gallo said. Environmental factors found to be associated with rosacea include lifetime UV exposure, smoking, obesity, and alcohol use.
In an assessment of the genetic basis of rosacea by genome-wide association study, researchers identified one confirmed single-nucleotide polymorphism that could be associated with rosacea (J Invest Dermatol. 2015;135[6]:1548-55). It was located in an intergenic region between HLA-DRA and BTNL2, “which is consistent with the overall concept that there is perhaps a genetic abnormality that is leading to increased amino modulation of difficulties,” Dr. Gallo said. For another recent study, researchers analyzed 14 randomized or case control trials involving rosacea patients (Int J Med Sci. 2015;12[5]:387-96). They concluded that vasculature, chronic inflammatory responses, environmental triggers, food and chemicals ingested, and microorganisms either alone or in combination are responsible for rosacea.
Dr. Gallo went on to highlight findings from several studies that link rosacea to an increased risk for certain comorbidities. One, a nationwide case-control study from Taiwan that comprised 35,553 rosacea patients, found that the disease was significantly associated with a risk of certain cardiovascular comorbidities (J Acad Dermatol. 2015;73:249-54). These included dyslipidemia (OR 1.41), coronary artery disease (OR 1.35), and hypertension (OR 1.17). A separate analysis, based 93,314 participants in the Nurse’s Health Study, found that rosacea was significantly associated with a risk of coronary artery disease (OR 2.2). The researchers also observed that comorbidities seemed to increase with duration of the disease (Clin Gastroenterol Hepatol. 2016;14[2]:220-5). Another smaller case-control study of 65 patients and 65 controls found an increased risk with rosacea for coronary artery disease, hyperlipidemia, hypertension, and gastroesophageal reflux disease (J Am Acad Dermatol. 2015;73[4]:604-8).
A recent analysis of 75,088 participants in the Nurses’ Health Study found that women with rosacea faced an increased risk of thyroid cancer (HR 1.59) and basal cell carcinoma (HR 1.50; Br J Cancer 2015;113[3]:520-3). Rosacea may also impact one’s risk for developing certain neurological conditions. One study found an increased risk for dementia (HR 1.42) and Alzheimer’s disease (HR 1.92; Ann Neurol. 2016;79:921-8), while another found an increased risk for Parkinson’s disease (an adjusted incident ratio of 1.71 in patients with rosacea, compared with the referent population; JAMA Neurol. 2016;73[5]:529-34).
As for therapy, a recent Cochrane systematic review found strong evidence supporting benefits of several therapies over placebo, including metronidazole, azelaic acid, brimonidine, tetracycline, doxycycline 40 mg, ivermectin, and isotretinoin (Br J Dermatol. 2015;173[3]:651-2). A separate, 7-year retrospective study of 275 adults with rosacea published online in The Journal of Dermatology on Oct. 28, 2015, found that patients with the PPR subtype had a better overall prognosis, compared with their counterparts with the other subtypes. Overall, the median time to complete remission was 56 months. Complete remission was achieved in 46% of those with PPR subtype, compared with 19% of those with mixed subtype and 11% of those with ETR subtype.
Dr. Gallo disclosed that he has received research grants from the National Institutes of Health, Allergan, L’Oreal, Colgate-Palmolive, Regeneron, GSK, Galderma, and Bayer. He is a consultant for Allergan, Colgate-Palmolive, Sente, Matrisys, Dermata, Alnylam, Abbvie, Roche, and Promius.
NEWPORT BEACH, CALIF. – In the clinical opinion of Richard L. Gallo, MD, PhD, current nomenclature for the diagnosis of rosacea could use a makeover.
“Currently, we’re still operating with an almost 20-year-old set of diagnostic subtypes of rosacea,” Dr. Gallo said at the annual meeting of the Pacific Dermatologic Association. He plans to participate in consensus meeting of experts who will convene this fall in an effort to update and modify these diagnostic criteria.
According to current nomenclature, subtype 1 is erythematotelangiectatic rosacea characterized by facial redness; subtype 2 is papulopustular, marked by bumps and pimples; subtype 3 is phymatous, characterized by enlargement of the nose, and subtype 4 is ocular, marked by eye irritation. Dr. Gallo pointed out that it’s rare to see just one of these subtypes in rosacea patients, with the exception of the erythematotelangiectatic rosacea (ETR). “There is a large population with ETR alone,” he said.” Most patients with the papulopustular subtype have aspects of ETR. There is a mix of subtypes of rosacea and we clearly need to modify our diagnostic criteria.”
Secondary rosacea features may include burning or stinging, plaque, dry appearance and scale, edema, ocular manifestations, peripheral location, and phymatous changes. Work by several researchers in recent years has shed light on the pathophysiology of rosacea. “We’re learning that there are many aspects to this disease that both trigger it and result in progression of the disease,” said Dr. Gallo, professor and chair of the department of dermatology at the University of California, San Diego. “It seems to have biological triggers that exist both in the environment and initiate from internal sources. We’re understanding more about the nature of those, at least specific molecules externally from microbes and so forth. Internally we understand more about the unique inflammatory signals.”
For example, he and other researchers began to look at the innate immune system patients with rosacea and identified LL37, a multifunction peptide that plays a role in a number of skin diseases, as something that can promote inflammation (Nat Med. 2007;13[8]:975-80). “It also promotes the vascular changes [that occur with the disease],” Dr. Gallo said. “We’re now learning how a dysregulation of enzymes in the skin contributes to making too much of these types of peptides. Therefore, treatment approaches that might modify enzymatic activity become useful.” Researchers have also discovered that some of the innate recognition molecules like toll-like receptor 2 (TLR2) are overexpressed in rosacea patients. “Similarly, in terms of the vascular signals, a number of labs are identifying some of the newer vascular transmitters that seem to be uniquely elevated in rosacea, so there’s great reason to be optimistic that given the increased specificity and understanding of what uniquely makes this disease happen, we’ll be able to target it in a safe way.”
A number of published studies have supported these notions, including an analysis of 275 twin pairs (JAMA Dermatol. 2015;151[11]:1213-9). The researchers found that compared with fraternal twins, identical twins had a higher association of National Rosacea Society scores (P = .04), “supporting the concept that there are fundamental genetic factors that are influencing disease,” Dr. Gallo said. Environmental factors found to be associated with rosacea include lifetime UV exposure, smoking, obesity, and alcohol use.
In an assessment of the genetic basis of rosacea by genome-wide association study, researchers identified one confirmed single-nucleotide polymorphism that could be associated with rosacea (J Invest Dermatol. 2015;135[6]:1548-55). It was located in an intergenic region between HLA-DRA and BTNL2, “which is consistent with the overall concept that there is perhaps a genetic abnormality that is leading to increased amino modulation of difficulties,” Dr. Gallo said. For another recent study, researchers analyzed 14 randomized or case control trials involving rosacea patients (Int J Med Sci. 2015;12[5]:387-96). They concluded that vasculature, chronic inflammatory responses, environmental triggers, food and chemicals ingested, and microorganisms either alone or in combination are responsible for rosacea.
Dr. Gallo went on to highlight findings from several studies that link rosacea to an increased risk for certain comorbidities. One, a nationwide case-control study from Taiwan that comprised 35,553 rosacea patients, found that the disease was significantly associated with a risk of certain cardiovascular comorbidities (J Acad Dermatol. 2015;73:249-54). These included dyslipidemia (OR 1.41), coronary artery disease (OR 1.35), and hypertension (OR 1.17). A separate analysis, based 93,314 participants in the Nurse’s Health Study, found that rosacea was significantly associated with a risk of coronary artery disease (OR 2.2). The researchers also observed that comorbidities seemed to increase with duration of the disease (Clin Gastroenterol Hepatol. 2016;14[2]:220-5). Another smaller case-control study of 65 patients and 65 controls found an increased risk with rosacea for coronary artery disease, hyperlipidemia, hypertension, and gastroesophageal reflux disease (J Am Acad Dermatol. 2015;73[4]:604-8).
A recent analysis of 75,088 participants in the Nurses’ Health Study found that women with rosacea faced an increased risk of thyroid cancer (HR 1.59) and basal cell carcinoma (HR 1.50; Br J Cancer 2015;113[3]:520-3). Rosacea may also impact one’s risk for developing certain neurological conditions. One study found an increased risk for dementia (HR 1.42) and Alzheimer’s disease (HR 1.92; Ann Neurol. 2016;79:921-8), while another found an increased risk for Parkinson’s disease (an adjusted incident ratio of 1.71 in patients with rosacea, compared with the referent population; JAMA Neurol. 2016;73[5]:529-34).
As for therapy, a recent Cochrane systematic review found strong evidence supporting benefits of several therapies over placebo, including metronidazole, azelaic acid, brimonidine, tetracycline, doxycycline 40 mg, ivermectin, and isotretinoin (Br J Dermatol. 2015;173[3]:651-2). A separate, 7-year retrospective study of 275 adults with rosacea published online in The Journal of Dermatology on Oct. 28, 2015, found that patients with the PPR subtype had a better overall prognosis, compared with their counterparts with the other subtypes. Overall, the median time to complete remission was 56 months. Complete remission was achieved in 46% of those with PPR subtype, compared with 19% of those with mixed subtype and 11% of those with ETR subtype.
Dr. Gallo disclosed that he has received research grants from the National Institutes of Health, Allergan, L’Oreal, Colgate-Palmolive, Regeneron, GSK, Galderma, and Bayer. He is a consultant for Allergan, Colgate-Palmolive, Sente, Matrisys, Dermata, Alnylam, Abbvie, Roche, and Promius.
NEWPORT BEACH, CALIF. – In the clinical opinion of Richard L. Gallo, MD, PhD, current nomenclature for the diagnosis of rosacea could use a makeover.
“Currently, we’re still operating with an almost 20-year-old set of diagnostic subtypes of rosacea,” Dr. Gallo said at the annual meeting of the Pacific Dermatologic Association. He plans to participate in consensus meeting of experts who will convene this fall in an effort to update and modify these diagnostic criteria.
According to current nomenclature, subtype 1 is erythematotelangiectatic rosacea characterized by facial redness; subtype 2 is papulopustular, marked by bumps and pimples; subtype 3 is phymatous, characterized by enlargement of the nose, and subtype 4 is ocular, marked by eye irritation. Dr. Gallo pointed out that it’s rare to see just one of these subtypes in rosacea patients, with the exception of the erythematotelangiectatic rosacea (ETR). “There is a large population with ETR alone,” he said.” Most patients with the papulopustular subtype have aspects of ETR. There is a mix of subtypes of rosacea and we clearly need to modify our diagnostic criteria.”
Secondary rosacea features may include burning or stinging, plaque, dry appearance and scale, edema, ocular manifestations, peripheral location, and phymatous changes. Work by several researchers in recent years has shed light on the pathophysiology of rosacea. “We’re learning that there are many aspects to this disease that both trigger it and result in progression of the disease,” said Dr. Gallo, professor and chair of the department of dermatology at the University of California, San Diego. “It seems to have biological triggers that exist both in the environment and initiate from internal sources. We’re understanding more about the nature of those, at least specific molecules externally from microbes and so forth. Internally we understand more about the unique inflammatory signals.”
For example, he and other researchers began to look at the innate immune system patients with rosacea and identified LL37, a multifunction peptide that plays a role in a number of skin diseases, as something that can promote inflammation (Nat Med. 2007;13[8]:975-80). “It also promotes the vascular changes [that occur with the disease],” Dr. Gallo said. “We’re now learning how a dysregulation of enzymes in the skin contributes to making too much of these types of peptides. Therefore, treatment approaches that might modify enzymatic activity become useful.” Researchers have also discovered that some of the innate recognition molecules like toll-like receptor 2 (TLR2) are overexpressed in rosacea patients. “Similarly, in terms of the vascular signals, a number of labs are identifying some of the newer vascular transmitters that seem to be uniquely elevated in rosacea, so there’s great reason to be optimistic that given the increased specificity and understanding of what uniquely makes this disease happen, we’ll be able to target it in a safe way.”
A number of published studies have supported these notions, including an analysis of 275 twin pairs (JAMA Dermatol. 2015;151[11]:1213-9). The researchers found that compared with fraternal twins, identical twins had a higher association of National Rosacea Society scores (P = .04), “supporting the concept that there are fundamental genetic factors that are influencing disease,” Dr. Gallo said. Environmental factors found to be associated with rosacea include lifetime UV exposure, smoking, obesity, and alcohol use.
In an assessment of the genetic basis of rosacea by genome-wide association study, researchers identified one confirmed single-nucleotide polymorphism that could be associated with rosacea (J Invest Dermatol. 2015;135[6]:1548-55). It was located in an intergenic region between HLA-DRA and BTNL2, “which is consistent with the overall concept that there is perhaps a genetic abnormality that is leading to increased amino modulation of difficulties,” Dr. Gallo said. For another recent study, researchers analyzed 14 randomized or case control trials involving rosacea patients (Int J Med Sci. 2015;12[5]:387-96). They concluded that vasculature, chronic inflammatory responses, environmental triggers, food and chemicals ingested, and microorganisms either alone or in combination are responsible for rosacea.
Dr. Gallo went on to highlight findings from several studies that link rosacea to an increased risk for certain comorbidities. One, a nationwide case-control study from Taiwan that comprised 35,553 rosacea patients, found that the disease was significantly associated with a risk of certain cardiovascular comorbidities (J Acad Dermatol. 2015;73:249-54). These included dyslipidemia (OR 1.41), coronary artery disease (OR 1.35), and hypertension (OR 1.17). A separate analysis, based 93,314 participants in the Nurse’s Health Study, found that rosacea was significantly associated with a risk of coronary artery disease (OR 2.2). The researchers also observed that comorbidities seemed to increase with duration of the disease (Clin Gastroenterol Hepatol. 2016;14[2]:220-5). Another smaller case-control study of 65 patients and 65 controls found an increased risk with rosacea for coronary artery disease, hyperlipidemia, hypertension, and gastroesophageal reflux disease (J Am Acad Dermatol. 2015;73[4]:604-8).
A recent analysis of 75,088 participants in the Nurses’ Health Study found that women with rosacea faced an increased risk of thyroid cancer (HR 1.59) and basal cell carcinoma (HR 1.50; Br J Cancer 2015;113[3]:520-3). Rosacea may also impact one’s risk for developing certain neurological conditions. One study found an increased risk for dementia (HR 1.42) and Alzheimer’s disease (HR 1.92; Ann Neurol. 2016;79:921-8), while another found an increased risk for Parkinson’s disease (an adjusted incident ratio of 1.71 in patients with rosacea, compared with the referent population; JAMA Neurol. 2016;73[5]:529-34).
As for therapy, a recent Cochrane systematic review found strong evidence supporting benefits of several therapies over placebo, including metronidazole, azelaic acid, brimonidine, tetracycline, doxycycline 40 mg, ivermectin, and isotretinoin (Br J Dermatol. 2015;173[3]:651-2). A separate, 7-year retrospective study of 275 adults with rosacea published online in The Journal of Dermatology on Oct. 28, 2015, found that patients with the PPR subtype had a better overall prognosis, compared with their counterparts with the other subtypes. Overall, the median time to complete remission was 56 months. Complete remission was achieved in 46% of those with PPR subtype, compared with 19% of those with mixed subtype and 11% of those with ETR subtype.
Dr. Gallo disclosed that he has received research grants from the National Institutes of Health, Allergan, L’Oreal, Colgate-Palmolive, Regeneron, GSK, Galderma, and Bayer. He is a consultant for Allergan, Colgate-Palmolive, Sente, Matrisys, Dermata, Alnylam, Abbvie, Roche, and Promius.
EXPERT ANALYSIS AT PDA 2016

Intraoperative nerve stimulation reduces risk of shoulder pain from neck dissection
SEATTLE – Direct, intraoperative electrical stimulation of the spinal accessory nerve reduced shoulder pain and dysfunction from oncologic neck dissection in a small, randomized trial.
Shoulder problems are common after neck dissection because of traction and compression of the spinal accessory nerve. Although brief electrical stimulation (BES) has been shown before to improve regeneration and recovery of injured peripheral nerves, it hasn’t been shown until now to help patients recover from neck surgery, said investigator Brittany Barber, MD, a fifth-year resident at the University of Alberta, Edmonton.

After neck dissection in 21 patients, the investigators wrapped a small electrode (Automatic Periodic Stimulation [APS] electrode, Medtronic) around the spinal accessory nerve at the base of the skull on the side of the neck with the most extensive nodal burden; the electrode delivered 100-msec pulses at 20 Hz and 3-5 V for an hour, and then the neck was closed. The team compared outcomes with 20 controls who had neck dissections without BES.
At 12 months, the BES group had an 8.4 point drop from baseline on the 100-point Constant Murley Shoulder Outcome Score, while the controls lost a mean of 29.4 points. The Murley score measures shoulder pain, performance of daily tasks, range of motion, and strength; higher scores are better. Similarly, BES patients lost a mean of 16.2 points on the 50-point Neck Dissection Impairment Index, while controls lost 30.1 points, and controls performed markedly worse on nerve conduction studies. In short, BES patients “were less likely to have clinically significant shoulder dysfunction” after surgery, Dr. Barber said at the International Conference on Head and Neck Cancer, held by the American Head and Neck Society.
The APS electrode is a tiny silicone cuff with a metal conductor. The device was originally designed to monitor recurrent laryngeal nerve function during thyroid surgery. “We had [Medtronic] write a software patch” so it could be used for stimulation, she said.
The team is planning a larger, multicenter trial to shore up their findings, and also plans to test the device for hypoglossal nerve preservation after resection.
Transcutaneous nerve stimulation is another option, but it’s a bit uncomfortable and patients often don’t complete their sessions. “Compliance is not as good as with a single intraoperative procedure,” and the results aren’t that great. “We thought this might be a better alternative,” Dr. Barber said.
The two groups were well matched: Mean age was about 60 years and most patients had advanced-stage tumors. There was no difference in preop shoulder problems or risks for poor postop shoulder outcomes, and no difference in the number of level 5 neck dissections or mean number of lymph nodes removed during surgery.
There was no outside funding for the work. Dr. Barber had no disclosures; a coinvestigator was a Medtronic consultant.
SEATTLE – Direct, intraoperative electrical stimulation of the spinal accessory nerve reduced shoulder pain and dysfunction from oncologic neck dissection in a small, randomized trial.
Shoulder problems are common after neck dissection because of traction and compression of the spinal accessory nerve. Although brief electrical stimulation (BES) has been shown before to improve regeneration and recovery of injured peripheral nerves, it hasn’t been shown until now to help patients recover from neck surgery, said investigator Brittany Barber, MD, a fifth-year resident at the University of Alberta, Edmonton.
After neck dissection in 21 patients, the investigators wrapped a small electrode (Automatic Periodic Stimulation [APS] electrode, Medtronic) around the spinal accessory nerve at the base of the skull on the side of the neck with the most extensive nodal burden; the electrode delivered 100-msec pulses at 20 Hz and 3-5 V for an hour, and then the neck was closed. The team compared outcomes with 20 controls who had neck dissections without BES.
At 12 months, the BES group had an 8.4 point drop from baseline on the 100-point Constant Murley Shoulder Outcome Score, while the controls lost a mean of 29.4 points. The Murley score measures shoulder pain, performance of daily tasks, range of motion, and strength; higher scores are better. Similarly, BES patients lost a mean of 16.2 points on the 50-point Neck Dissection Impairment Index, while controls lost 30.1 points, and controls performed markedly worse on nerve conduction studies. In short, BES patients “were less likely to have clinically significant shoulder dysfunction” after surgery, Dr. Barber said at the International Conference on Head and Neck Cancer, held by the American Head and Neck Society.
The APS electrode is a tiny silicone cuff with a metal conductor. The device was originally designed to monitor recurrent laryngeal nerve function during thyroid surgery. “We had [Medtronic] write a software patch” so it could be used for stimulation, she said.
The team is planning a larger, multicenter trial to shore up their findings, and also plans to test the device for hypoglossal nerve preservation after resection.
Transcutaneous nerve stimulation is another option, but it’s a bit uncomfortable and patients often don’t complete their sessions. “Compliance is not as good as with a single intraoperative procedure,” and the results aren’t that great. “We thought this might be a better alternative,” Dr. Barber said.
The two groups were well matched: Mean age was about 60 years and most patients had advanced-stage tumors. There was no difference in preop shoulder problems or risks for poor postop shoulder outcomes, and no difference in the number of level 5 neck dissections or mean number of lymph nodes removed during surgery.
There was no outside funding for the work. Dr. Barber had no disclosures; a coinvestigator was a Medtronic consultant.
SEATTLE – Direct, intraoperative electrical stimulation of the spinal accessory nerve reduced shoulder pain and dysfunction from oncologic neck dissection in a small, randomized trial.
Shoulder problems are common after neck dissection because of traction and compression of the spinal accessory nerve. Although brief electrical stimulation (BES) has been shown before to improve regeneration and recovery of injured peripheral nerves, it hasn’t been shown until now to help patients recover from neck surgery, said investigator Brittany Barber, MD, a fifth-year resident at the University of Alberta, Edmonton.
After neck dissection in 21 patients, the investigators wrapped a small electrode (Automatic Periodic Stimulation [APS] electrode, Medtronic) around the spinal accessory nerve at the base of the skull on the side of the neck with the most extensive nodal burden; the electrode delivered 100-msec pulses at 20 Hz and 3-5 V for an hour, and then the neck was closed. The team compared outcomes with 20 controls who had neck dissections without BES.
At 12 months, the BES group had an 8.4 point drop from baseline on the 100-point Constant Murley Shoulder Outcome Score, while the controls lost a mean of 29.4 points. The Murley score measures shoulder pain, performance of daily tasks, range of motion, and strength; higher scores are better. Similarly, BES patients lost a mean of 16.2 points on the 50-point Neck Dissection Impairment Index, while controls lost 30.1 points, and controls performed markedly worse on nerve conduction studies. In short, BES patients “were less likely to have clinically significant shoulder dysfunction” after surgery, Dr. Barber said at the International Conference on Head and Neck Cancer, held by the American Head and Neck Society.
The APS electrode is a tiny silicone cuff with a metal conductor. The device was originally designed to monitor recurrent laryngeal nerve function during thyroid surgery. “We had [Medtronic] write a software patch” so it could be used for stimulation, she said.
The team is planning a larger, multicenter trial to shore up their findings, and also plans to test the device for hypoglossal nerve preservation after resection.
Transcutaneous nerve stimulation is another option, but it’s a bit uncomfortable and patients often don’t complete their sessions. “Compliance is not as good as with a single intraoperative procedure,” and the results aren’t that great. “We thought this might be a better alternative,” Dr. Barber said.
The two groups were well matched: Mean age was about 60 years and most patients had advanced-stage tumors. There was no difference in preop shoulder problems or risks for poor postop shoulder outcomes, and no difference in the number of level 5 neck dissections or mean number of lymph nodes removed during surgery.
There was no outside funding for the work. Dr. Barber had no disclosures; a coinvestigator was a Medtronic consultant.
AT AHNS 2016
Key clinical point: Direct, intraoperative electrical stimulation of the spinal accessory nerve reduces shoulder pain and dysfunction from oncologic neck dissection.
Major finding: At 12 months, the BES group had an 8.4 point drop from baseline on the 100-point Constant Murley Shoulder Score, while the controls lost a mean of 29.4 points.
Data source: Randomized trial with 41 patients.
Disclosures: There was no outside funding for the work. The presenter had no disclosures; a co-investigator was a Medtronic consultant.

Psoriatic arthritis patients face more endocrine comorbidities
MIAMI – Diabetes mellitus, hypothyroidism, Cushing’s disease, and osteoporosis occur more frequently in people with psoriatic arthritis than in controls, a large cohort study reveals. Prevalence of these endocrine conditions was greater in a group of 3,161 patients with psoriatic arthritis, compared with 31,610 matched controls.
“We recommend that physicians should be aware of comorbid associations to provide comprehensive medical care to patients with psoriatic arthritis,” said Amir Haddad, MD, of the department of rheumatology at Carmel Medical Center in Haifa, Israel.
Dr. Haddad and his colleagues, however, found no significant differences in the prevalence of hyperthyroidism, hypo- and hyperparathyroidism, hyperprolactinemia, Addison’s disease, diabetes insipidus, pituitary adenoma, or acromegaly between groups in this retrospective, cross-sectional study.
They identified 1,474 men and 1,687 women diagnosed with psoriatic disease from 2000 to 2013 using the Clalit health services database in Israel. This group was a mean of 58 years old and 53% were women. Each patient was matched with 10 age- and gender-matched controls without psoriatic disease for the study.
“This is, to our knowledge, one of the largest real-life cohorts of psoriatic patient registries,” Dr. Haddad said at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA).
In the psoriatic arthritis group versus controls, diabetes mellitus prevalence was 27.9% vs. 20.7%; for hypothyroidism it was 12.7% vs. 8.6%; and for Cushing’s disease it was 0.3% vs. 0.1%. All these differences were statically significant (P less than 0.0001). Osteoporosis prevalence also differed significantly between the psoriatic arthritis and control groups: 13.2% vs. 9.1% (P less than 0.001).
Greater awareness of nonskin and nonjoint comorbidities is important, Dr. Haddad said, because it can influence choice of therapy and management of patients with psoriatic arthritis.
The investigators also conducted univariate and multivariate regression analyses. Compared with controls, the results suggest psoriatic arthritis patients have a higher risk for diabetes mellitus (odds ratio, 1.48), hypothyroidism (OR, 1.56), and osteoporosis (OR, 1.52). The risk for Cushing’s disease was notably higher (OR, 5.31) in the univariate analysis.
Risks for these endocrine conditions remained higher for the psoriatic arthritis patients in a multivariate regression analysis as well. For example, risk for diabetes mellitus (OR, 1.30) remained after adjusting for age, gender, smoking, obesity, and steroid use. Risk of hypothyroidism (OR, 1.61) remained after adjusting for age and gender; risk of osteoporosis (OR, 1.50) after adjusting for age, gender, steroid use, and smoking; and risk of Cushing’s disease (OR, 3.79) after adjustment for age, gender, and steroid use.
The large, population-based cohort is a strength of the study. “We are now going back to see how many of these patients were seen by rheumatologists,” Dr. Haddad said. A lack of association with disease burden is a potential limitation, he added.
Thirty percent of patients were treated with biologics and about 67% with steroids. “That number treated with steroids seems high,” a meeting attendee commented. Dr. Haddad explained that it is the percentage ever treated with steroids, not necessarily currently on steroids.
In a separate session at the GRAPPA meeting addressing psoriatic disease treatment recommendations, an attendee asked about specific recommendations for comorbidities. For now, GRAPPA plans to include comorbidities within its overall recommendations, as it did in its most recent update, released in January 2016. A limited amount of data is a primary reason.
“As the evidence on comorbidities gets better, we may someday have separate recommendations for comorbidities,” said Laura Coates, MD, a clinical lecturer in rheumatology at the University of Leeds (England).
“The comorbidities are very important,” said Arthur F. Kavanaugh, MD, professor of medicine at the University of California, San Diego. “That’s trickier and deals with the international nature of GRAPPA. It’s hard to say, ‘Go see this specialist,’ because that might not be standard of care in that country.”
Dr. Haddad, Dr. Coates, and Dr. Kavanaugh reported having no relevant financial disclosures.
MIAMI – Diabetes mellitus, hypothyroidism, Cushing’s disease, and osteoporosis occur more frequently in people with psoriatic arthritis than in controls, a large cohort study reveals. Prevalence of these endocrine conditions was greater in a group of 3,161 patients with psoriatic arthritis, compared with 31,610 matched controls.
“We recommend that physicians should be aware of comorbid associations to provide comprehensive medical care to patients with psoriatic arthritis,” said Amir Haddad, MD, of the department of rheumatology at Carmel Medical Center in Haifa, Israel.
Dr. Haddad and his colleagues, however, found no significant differences in the prevalence of hyperthyroidism, hypo- and hyperparathyroidism, hyperprolactinemia, Addison’s disease, diabetes insipidus, pituitary adenoma, or acromegaly between groups in this retrospective, cross-sectional study.
They identified 1,474 men and 1,687 women diagnosed with psoriatic disease from 2000 to 2013 using the Clalit health services database in Israel. This group was a mean of 58 years old and 53% were women. Each patient was matched with 10 age- and gender-matched controls without psoriatic disease for the study.
“This is, to our knowledge, one of the largest real-life cohorts of psoriatic patient registries,” Dr. Haddad said at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA).
In the psoriatic arthritis group versus controls, diabetes mellitus prevalence was 27.9% vs. 20.7%; for hypothyroidism it was 12.7% vs. 8.6%; and for Cushing’s disease it was 0.3% vs. 0.1%. All these differences were statically significant (P less than 0.0001). Osteoporosis prevalence also differed significantly between the psoriatic arthritis and control groups: 13.2% vs. 9.1% (P less than 0.001).
Greater awareness of nonskin and nonjoint comorbidities is important, Dr. Haddad said, because it can influence choice of therapy and management of patients with psoriatic arthritis.
The investigators also conducted univariate and multivariate regression analyses. Compared with controls, the results suggest psoriatic arthritis patients have a higher risk for diabetes mellitus (odds ratio, 1.48), hypothyroidism (OR, 1.56), and osteoporosis (OR, 1.52). The risk for Cushing’s disease was notably higher (OR, 5.31) in the univariate analysis.
Risks for these endocrine conditions remained higher for the psoriatic arthritis patients in a multivariate regression analysis as well. For example, risk for diabetes mellitus (OR, 1.30) remained after adjusting for age, gender, smoking, obesity, and steroid use. Risk of hypothyroidism (OR, 1.61) remained after adjusting for age and gender; risk of osteoporosis (OR, 1.50) after adjusting for age, gender, steroid use, and smoking; and risk of Cushing’s disease (OR, 3.79) after adjustment for age, gender, and steroid use.
The large, population-based cohort is a strength of the study. “We are now going back to see how many of these patients were seen by rheumatologists,” Dr. Haddad said. A lack of association with disease burden is a potential limitation, he added.
Thirty percent of patients were treated with biologics and about 67% with steroids. “That number treated with steroids seems high,” a meeting attendee commented. Dr. Haddad explained that it is the percentage ever treated with steroids, not necessarily currently on steroids.
In a separate session at the GRAPPA meeting addressing psoriatic disease treatment recommendations, an attendee asked about specific recommendations for comorbidities. For now, GRAPPA plans to include comorbidities within its overall recommendations, as it did in its most recent update, released in January 2016. A limited amount of data is a primary reason.
“As the evidence on comorbidities gets better, we may someday have separate recommendations for comorbidities,” said Laura Coates, MD, a clinical lecturer in rheumatology at the University of Leeds (England).
“The comorbidities are very important,” said Arthur F. Kavanaugh, MD, professor of medicine at the University of California, San Diego. “That’s trickier and deals with the international nature of GRAPPA. It’s hard to say, ‘Go see this specialist,’ because that might not be standard of care in that country.”
Dr. Haddad, Dr. Coates, and Dr. Kavanaugh reported having no relevant financial disclosures.
MIAMI – Diabetes mellitus, hypothyroidism, Cushing’s disease, and osteoporosis occur more frequently in people with psoriatic arthritis than in controls, a large cohort study reveals. Prevalence of these endocrine conditions was greater in a group of 3,161 patients with psoriatic arthritis, compared with 31,610 matched controls.
“We recommend that physicians should be aware of comorbid associations to provide comprehensive medical care to patients with psoriatic arthritis,” said Amir Haddad, MD, of the department of rheumatology at Carmel Medical Center in Haifa, Israel.
Dr. Haddad and his colleagues, however, found no significant differences in the prevalence of hyperthyroidism, hypo- and hyperparathyroidism, hyperprolactinemia, Addison’s disease, diabetes insipidus, pituitary adenoma, or acromegaly between groups in this retrospective, cross-sectional study.
They identified 1,474 men and 1,687 women diagnosed with psoriatic disease from 2000 to 2013 using the Clalit health services database in Israel. This group was a mean of 58 years old and 53% were women. Each patient was matched with 10 age- and gender-matched controls without psoriatic disease for the study.
“This is, to our knowledge, one of the largest real-life cohorts of psoriatic patient registries,” Dr. Haddad said at the annual meeting of the Group for Research and Assessment of Psoriasis and Psoriatic Arthritis (GRAPPA).
In the psoriatic arthritis group versus controls, diabetes mellitus prevalence was 27.9% vs. 20.7%; for hypothyroidism it was 12.7% vs. 8.6%; and for Cushing’s disease it was 0.3% vs. 0.1%. All these differences were statically significant (P less than 0.0001). Osteoporosis prevalence also differed significantly between the psoriatic arthritis and control groups: 13.2% vs. 9.1% (P less than 0.001).
Greater awareness of nonskin and nonjoint comorbidities is important, Dr. Haddad said, because it can influence choice of therapy and management of patients with psoriatic arthritis.
The investigators also conducted univariate and multivariate regression analyses. Compared with controls, the results suggest psoriatic arthritis patients have a higher risk for diabetes mellitus (odds ratio, 1.48), hypothyroidism (OR, 1.56), and osteoporosis (OR, 1.52). The risk for Cushing’s disease was notably higher (OR, 5.31) in the univariate analysis.
Risks for these endocrine conditions remained higher for the psoriatic arthritis patients in a multivariate regression analysis as well. For example, risk for diabetes mellitus (OR, 1.30) remained after adjusting for age, gender, smoking, obesity, and steroid use. Risk of hypothyroidism (OR, 1.61) remained after adjusting for age and gender; risk of osteoporosis (OR, 1.50) after adjusting for age, gender, steroid use, and smoking; and risk of Cushing’s disease (OR, 3.79) after adjustment for age, gender, and steroid use.
The large, population-based cohort is a strength of the study. “We are now going back to see how many of these patients were seen by rheumatologists,” Dr. Haddad said. A lack of association with disease burden is a potential limitation, he added.
Thirty percent of patients were treated with biologics and about 67% with steroids. “That number treated with steroids seems high,” a meeting attendee commented. Dr. Haddad explained that it is the percentage ever treated with steroids, not necessarily currently on steroids.
In a separate session at the GRAPPA meeting addressing psoriatic disease treatment recommendations, an attendee asked about specific recommendations for comorbidities. For now, GRAPPA plans to include comorbidities within its overall recommendations, as it did in its most recent update, released in January 2016. A limited amount of data is a primary reason.
“As the evidence on comorbidities gets better, we may someday have separate recommendations for comorbidities,” said Laura Coates, MD, a clinical lecturer in rheumatology at the University of Leeds (England).
“The comorbidities are very important,” said Arthur F. Kavanaugh, MD, professor of medicine at the University of California, San Diego. “That’s trickier and deals with the international nature of GRAPPA. It’s hard to say, ‘Go see this specialist,’ because that might not be standard of care in that country.”
Dr. Haddad, Dr. Coates, and Dr. Kavanaugh reported having no relevant financial disclosures.
AT 2016 GRAPPA ANNUAL MEETING
Key clinical point:Patients with psoriatic disease had a significantly higher prevalence of diabetes mellitus and some other endocrine comorbidities.
Major finding: In a univariate analysis, the risk for Cushing’s disease was notably higher among psoriatic arthritis patients, compared with controls (odds ratio, 5.31).
Data source: Retrospective, cross-sectional comparison of 3,161 patients with psoriatic arthritis and 31,610 matched controls.
Disclosures: Dr. Haddad, Dr. Coates, and Dr. Kavanaugh reported having no relevant financial disclosures.
Study identifies important predictors for PC/PGL
BALTIMORE – Tumor size and the presence of mutations of the succinate dehydrogenase complex subunit B (SDHB) gene may be reliable indicators of prognosis after surgery for pheochromocytoma and abdominal paraganglioma, investigators in a National Cancer Institute–funded study have reported.
“The staging of pheochromocytoma and abdominal paraganglioma can be difficult, but it is critical for optimal patient care,” Yasmine Assadipour, MD, of the National Cancer Institute, Bethesda, Md., and the George Washington University Hospital, Washington, reported at the annual meeting of the American Association of Endocrine Surgeons.

“Any clinically relevant grading or prognostic system should include SDHB mutation status and primary tumor size as prime features of scoring,” Dr. Assadipour said. “Histologic features such as Ki-67 or mitotic index may not be as useful for prognostic information in patients with pheochromocytoma and abdominal paraganglioma, particularly in the setting of SDHB mutation.”
Dr. Assadipour and her coinvestigators focused their investigation on mutations of the SDHB (succinate dehydrogenase complex subunit B) gene, which codes for one of four subunits comprising a mitochondrial protein.
They also considered primary tumor size, functionality, pathology, surgical approach, and histologic features including Ki-67 index and mitotic index. The study was a retrospective analysis of 84 patients who had surgery for PC [pheochromocytoma] or PGL [paraganglioma] and had germ line genetic testing. Of the 84 patients, 35 patients had sporadic disease and 49 had germ line SDHB mutation. The study analyzed tumor samples for Ki-67/MIB-1 staining and mitotic index.
“In a univariate analysis, SDHB mutation, tumor size and surgical approach were associated with local regional recurrence,” Dr. Assadipour said. “In a multivariate analysis, the only independent risk factors were SDHB mutation status and tumor size; Ki-67 and mitotic index did not have any association with recurrence.”
The researchers found similar results when they looked at distant metastasis. “SDHB mutation, tumor size, abdominal paraganglioma and surgical approach were associated with distant metastasis,” Dr. Assadipour said. “Again, Ki-67 and mitotic index were not.”
In the multivariate analysis, again, only patient SDHB status and tumor size were independently associated with metastasis.”
The incidence of local recurrence in patients with the SDHB mutation was 47.6% vs. 9.1% in those without the gene mutation, Dr. Assadipour said. The rates of distant metastasis showed a similar disparity: 56.5% and 9.1%, respectively.
Patients with the SDHB mutation presented at a younger age than those without the mutation, 33 vs. 49.6 years old. Among the 65 patients who underwent R0 primary tumor resection, those with the SDHB mutation, paraganglioma, and larger tumor size had a shorter disease-free survival, Dr. Assadipour said.
In analyzing tumor size, Dr. Assadipour said two stratifications were studied: evaluating tumors sized 0-3 cm, 3-6 cm and 6 cm and larger; and 0-5 cm and 5 cm and larger. “Tumors over 6 cm had the shortest disease-free survival, and even when we applied the under-5 cm and over-5 cm scale, we clearly saw a difference in disease-free survival,” she said. Ki-67 and mitotic index were not related to disease-free survival.
The presence of a SDHB mutation had a hazard ratio of 16.2, while tumor diameter greater than 6 cm had a HR of 15.4, Dr. Assadipour said. These were the only independent risk factors for local recurrence, distant metastases and shorter disease-free interval found in the study.
During the discussion, Lawrence T. Kim, MD, of the University of North Carolina asked if the researchers found any differences in outcomes related to the surgical approach. “We were unable to identify whether any surgical approach improved or worsened outcomes on multivariate analysis” Dr. Assadipour said.
Thomas J. Fahey, MD, of New York asked what she would recommend for surgical approaches for patients with PC and PGL.
“Our general recommendation is that an adrenal pheochromocytoma that is over 6 cm carries a higher risk of recurrence and distant metastasis so an open approach with lymph node dissection ensuring negative surgical margins should be considered,” Dr. Assadipour said. “For abdominal paragangliomas, unless they are quite small and in a favorable location, we would generally recommend an open approach.”
The study was supported by the intramural program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. Dr. Assadipour and her coauthors had no financial relationships to disclose.
BALTIMORE – Tumor size and the presence of mutations of the succinate dehydrogenase complex subunit B (SDHB) gene may be reliable indicators of prognosis after surgery for pheochromocytoma and abdominal paraganglioma, investigators in a National Cancer Institute–funded study have reported.
“The staging of pheochromocytoma and abdominal paraganglioma can be difficult, but it is critical for optimal patient care,” Yasmine Assadipour, MD, of the National Cancer Institute, Bethesda, Md., and the George Washington University Hospital, Washington, reported at the annual meeting of the American Association of Endocrine Surgeons.
“Any clinically relevant grading or prognostic system should include SDHB mutation status and primary tumor size as prime features of scoring,” Dr. Assadipour said. “Histologic features such as Ki-67 or mitotic index may not be as useful for prognostic information in patients with pheochromocytoma and abdominal paraganglioma, particularly in the setting of SDHB mutation.”
Dr. Assadipour and her coinvestigators focused their investigation on mutations of the SDHB (succinate dehydrogenase complex subunit B) gene, which codes for one of four subunits comprising a mitochondrial protein.
They also considered primary tumor size, functionality, pathology, surgical approach, and histologic features including Ki-67 index and mitotic index. The study was a retrospective analysis of 84 patients who had surgery for PC [pheochromocytoma] or PGL [paraganglioma] and had germ line genetic testing. Of the 84 patients, 35 patients had sporadic disease and 49 had germ line SDHB mutation. The study analyzed tumor samples for Ki-67/MIB-1 staining and mitotic index.
“In a univariate analysis, SDHB mutation, tumor size and surgical approach were associated with local regional recurrence,” Dr. Assadipour said. “In a multivariate analysis, the only independent risk factors were SDHB mutation status and tumor size; Ki-67 and mitotic index did not have any association with recurrence.”
The researchers found similar results when they looked at distant metastasis. “SDHB mutation, tumor size, abdominal paraganglioma and surgical approach were associated with distant metastasis,” Dr. Assadipour said. “Again, Ki-67 and mitotic index were not.”
In the multivariate analysis, again, only patient SDHB status and tumor size were independently associated with metastasis.”
The incidence of local recurrence in patients with the SDHB mutation was 47.6% vs. 9.1% in those without the gene mutation, Dr. Assadipour said. The rates of distant metastasis showed a similar disparity: 56.5% and 9.1%, respectively.
Patients with the SDHB mutation presented at a younger age than those without the mutation, 33 vs. 49.6 years old. Among the 65 patients who underwent R0 primary tumor resection, those with the SDHB mutation, paraganglioma, and larger tumor size had a shorter disease-free survival, Dr. Assadipour said.
In analyzing tumor size, Dr. Assadipour said two stratifications were studied: evaluating tumors sized 0-3 cm, 3-6 cm and 6 cm and larger; and 0-5 cm and 5 cm and larger. “Tumors over 6 cm had the shortest disease-free survival, and even when we applied the under-5 cm and over-5 cm scale, we clearly saw a difference in disease-free survival,” she said. Ki-67 and mitotic index were not related to disease-free survival.
The presence of a SDHB mutation had a hazard ratio of 16.2, while tumor diameter greater than 6 cm had a HR of 15.4, Dr. Assadipour said. These were the only independent risk factors for local recurrence, distant metastases and shorter disease-free interval found in the study.
During the discussion, Lawrence T. Kim, MD, of the University of North Carolina asked if the researchers found any differences in outcomes related to the surgical approach. “We were unable to identify whether any surgical approach improved or worsened outcomes on multivariate analysis” Dr. Assadipour said.
Thomas J. Fahey, MD, of New York asked what she would recommend for surgical approaches for patients with PC and PGL.
“Our general recommendation is that an adrenal pheochromocytoma that is over 6 cm carries a higher risk of recurrence and distant metastasis so an open approach with lymph node dissection ensuring negative surgical margins should be considered,” Dr. Assadipour said. “For abdominal paragangliomas, unless they are quite small and in a favorable location, we would generally recommend an open approach.”
The study was supported by the intramural program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. Dr. Assadipour and her coauthors had no financial relationships to disclose.
BALTIMORE – Tumor size and the presence of mutations of the succinate dehydrogenase complex subunit B (SDHB) gene may be reliable indicators of prognosis after surgery for pheochromocytoma and abdominal paraganglioma, investigators in a National Cancer Institute–funded study have reported.
“The staging of pheochromocytoma and abdominal paraganglioma can be difficult, but it is critical for optimal patient care,” Yasmine Assadipour, MD, of the National Cancer Institute, Bethesda, Md., and the George Washington University Hospital, Washington, reported at the annual meeting of the American Association of Endocrine Surgeons.
“Any clinically relevant grading or prognostic system should include SDHB mutation status and primary tumor size as prime features of scoring,” Dr. Assadipour said. “Histologic features such as Ki-67 or mitotic index may not be as useful for prognostic information in patients with pheochromocytoma and abdominal paraganglioma, particularly in the setting of SDHB mutation.”
Dr. Assadipour and her coinvestigators focused their investigation on mutations of the SDHB (succinate dehydrogenase complex subunit B) gene, which codes for one of four subunits comprising a mitochondrial protein.
They also considered primary tumor size, functionality, pathology, surgical approach, and histologic features including Ki-67 index and mitotic index. The study was a retrospective analysis of 84 patients who had surgery for PC [pheochromocytoma] or PGL [paraganglioma] and had germ line genetic testing. Of the 84 patients, 35 patients had sporadic disease and 49 had germ line SDHB mutation. The study analyzed tumor samples for Ki-67/MIB-1 staining and mitotic index.
“In a univariate analysis, SDHB mutation, tumor size and surgical approach were associated with local regional recurrence,” Dr. Assadipour said. “In a multivariate analysis, the only independent risk factors were SDHB mutation status and tumor size; Ki-67 and mitotic index did not have any association with recurrence.”
The researchers found similar results when they looked at distant metastasis. “SDHB mutation, tumor size, abdominal paraganglioma and surgical approach were associated with distant metastasis,” Dr. Assadipour said. “Again, Ki-67 and mitotic index were not.”
In the multivariate analysis, again, only patient SDHB status and tumor size were independently associated with metastasis.”
The incidence of local recurrence in patients with the SDHB mutation was 47.6% vs. 9.1% in those without the gene mutation, Dr. Assadipour said. The rates of distant metastasis showed a similar disparity: 56.5% and 9.1%, respectively.
Patients with the SDHB mutation presented at a younger age than those without the mutation, 33 vs. 49.6 years old. Among the 65 patients who underwent R0 primary tumor resection, those with the SDHB mutation, paraganglioma, and larger tumor size had a shorter disease-free survival, Dr. Assadipour said.
In analyzing tumor size, Dr. Assadipour said two stratifications were studied: evaluating tumors sized 0-3 cm, 3-6 cm and 6 cm and larger; and 0-5 cm and 5 cm and larger. “Tumors over 6 cm had the shortest disease-free survival, and even when we applied the under-5 cm and over-5 cm scale, we clearly saw a difference in disease-free survival,” she said. Ki-67 and mitotic index were not related to disease-free survival.
The presence of a SDHB mutation had a hazard ratio of 16.2, while tumor diameter greater than 6 cm had a HR of 15.4, Dr. Assadipour said. These were the only independent risk factors for local recurrence, distant metastases and shorter disease-free interval found in the study.
During the discussion, Lawrence T. Kim, MD, of the University of North Carolina asked if the researchers found any differences in outcomes related to the surgical approach. “We were unable to identify whether any surgical approach improved or worsened outcomes on multivariate analysis” Dr. Assadipour said.
Thomas J. Fahey, MD, of New York asked what she would recommend for surgical approaches for patients with PC and PGL.
“Our general recommendation is that an adrenal pheochromocytoma that is over 6 cm carries a higher risk of recurrence and distant metastasis so an open approach with lymph node dissection ensuring negative surgical margins should be considered,” Dr. Assadipour said. “For abdominal paragangliomas, unless they are quite small and in a favorable location, we would generally recommend an open approach.”
The study was supported by the intramural program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. Dr. Assadipour and her coauthors had no financial relationships to disclose.
AT AAES 2016
Key clinical point: SDHB mutation and tumor size may be better predictors of outcomes in patients with pheochromocytoma and abdominal paraganglioma than are other previously identified predictors.
Major finding: The incidence of local recurrence in patients with the SDHB mutation was 47.6% vs. 9.1% in those without the gene mutation.
Data source: Retrospective analysis of 84 patients with PC/PGL evaluated by the Surgical Endocrine Oncology branch at George Washington University Hospital from 1998-2015.
Disclosures: The study was supported by the intramural program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. Dr. Assadipour and her coauthors reported having no financial disclosures.
