User login
Pruritic Nodules on the Breast
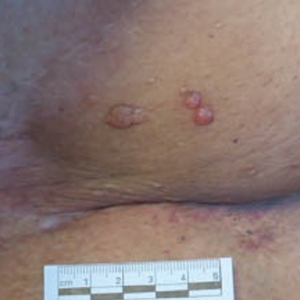
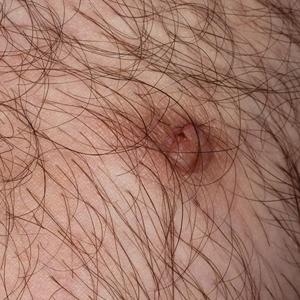
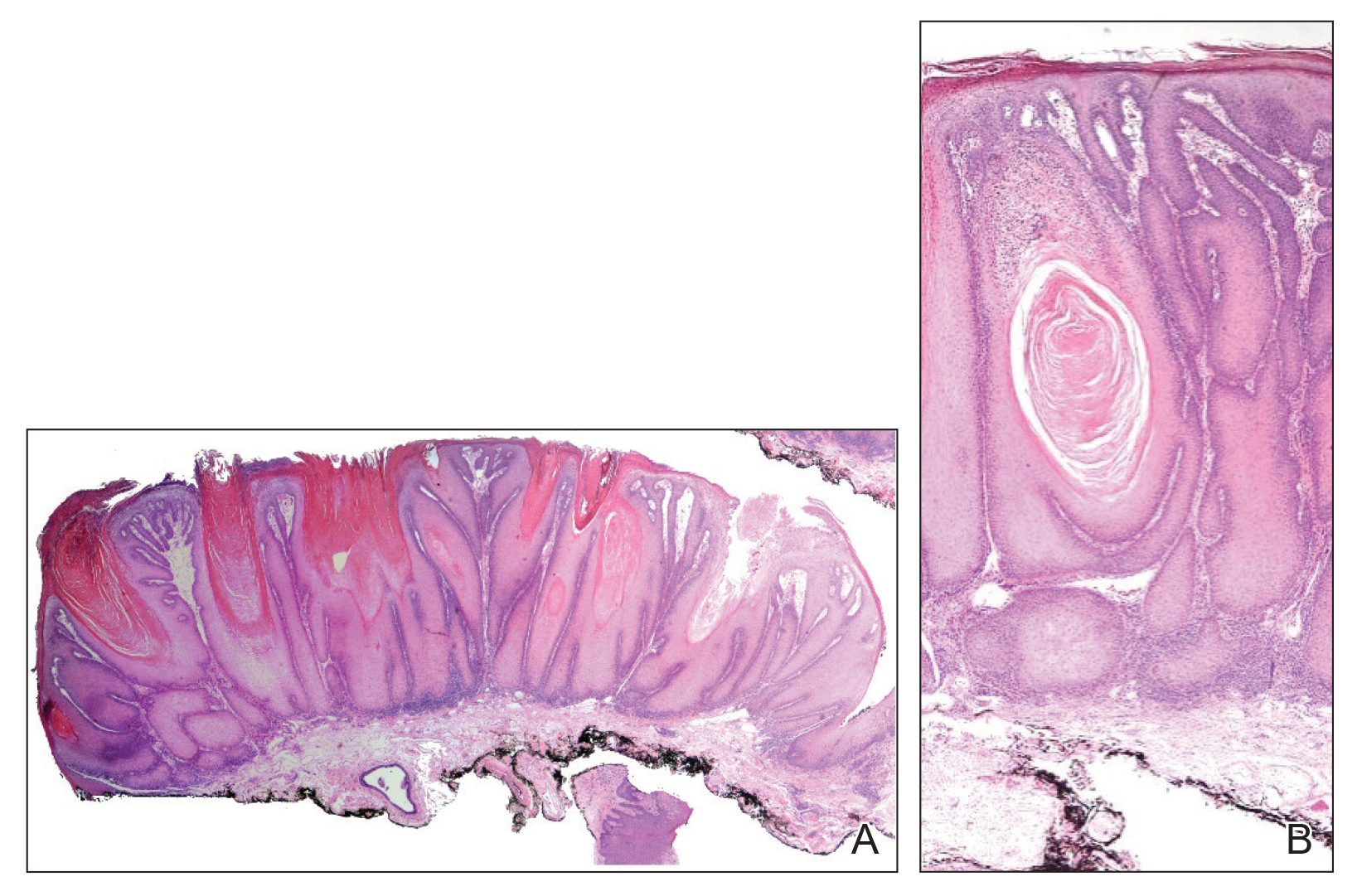
Microcystic lymphatic malformations, also known as lymphangioma circumscriptum, are rare hamartomatous lesions comprised of dilated lymphatic channels that can be both congenital and acquired.1 They often present as translucent or hemorrhagic vesicles of varying sizes that may contain lymphatic fluid and often can cluster together and appear verrucous (Figure 1). The differential diagnosis for microcystic lymphatic malformations commonly includes molluscum contagiosum, squamous cell carcinoma, verruca vulgaris, or condylomas, as well as atypical vascular lesions. They most often are found in children as congenital lesions but also may be acquired. Most acquired cases are due to chronic inflammatory and scarring processes that damage lymphatic structures, including surgery, radiation, infections, and even Crohn disease.2,3 Because the differential diagnosis is so broad and the disease can clinically mimic other common disease processes, biopsies often are performed to determine the diagnosis. On biopsy, pathologic examination revealed well-circumscribed nodular lesions with large lymphatic channels often in a background of connective tissue stroma. Increased eosinophilic material, including mast cells, also was seen (Figure 2A). On immunohistochemistry, staining showed D2-40 positivity (Figure 2B).
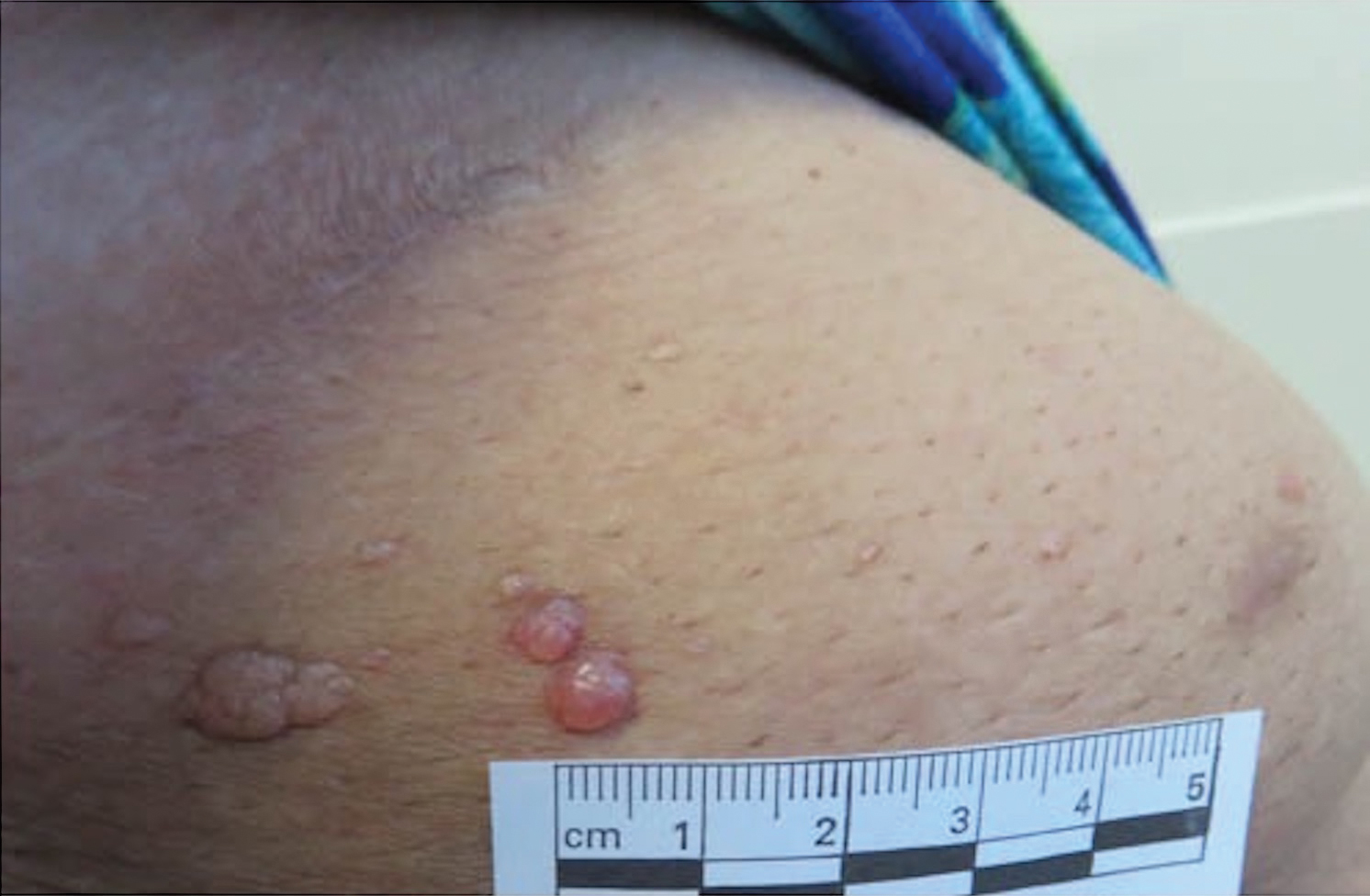
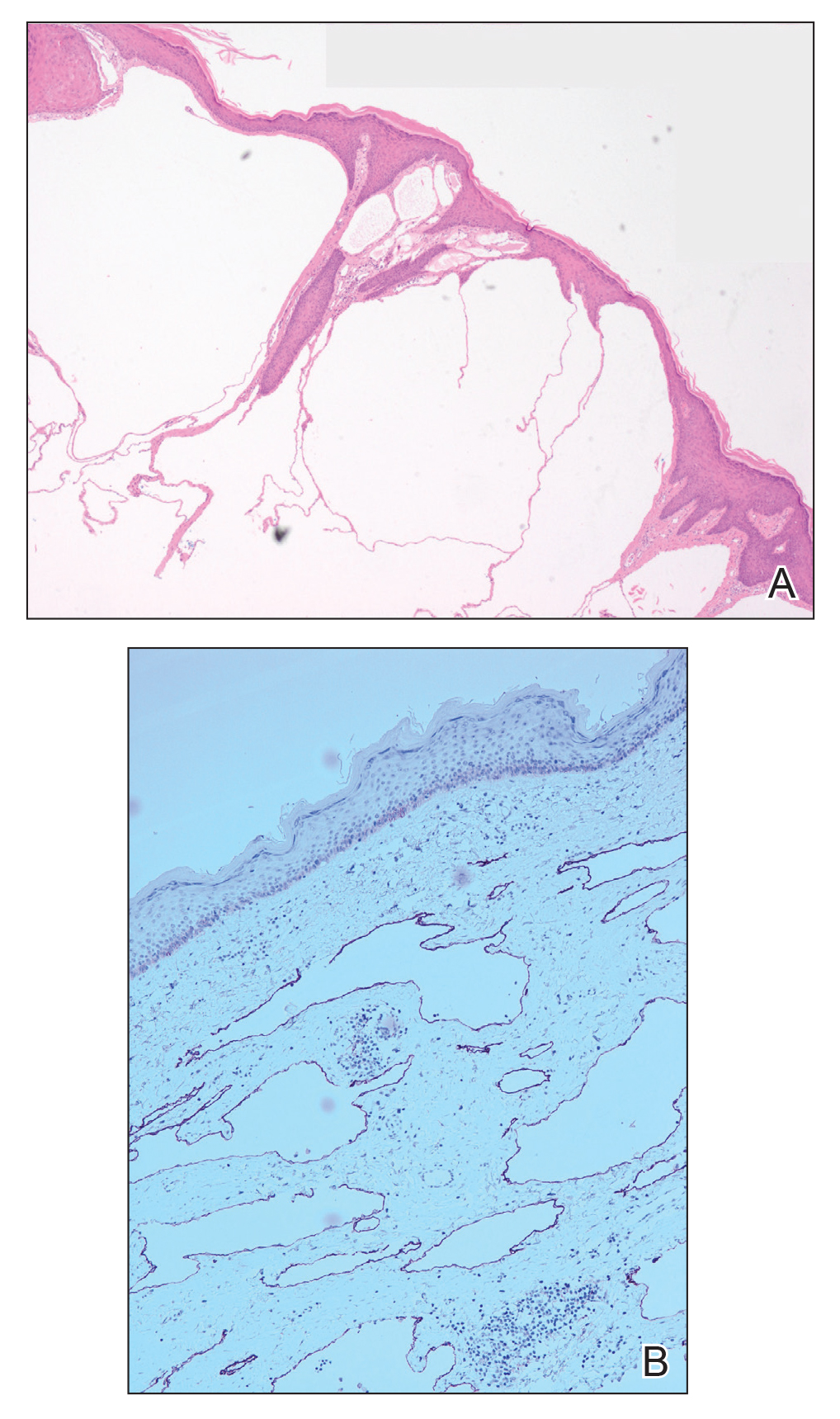
Damage to lymphatics from radiation and postsurgical excision of tumors are well-described causes of microcystic lymphatic malformations, as in our patient, with most instances in the literature occurring secondary to treatment of breast or cervical cancer.4-6 In these acquired cases, the pathogenesis is thought to be due to destruction and fibrosis at the layer of the reticular dermis, which causes lymphatic obstruction and subsequent dilation of superficial lymphatic channels.6
Microcystic lymphatic malformations can be difficult to distinguish from atypical vascular lesions, another common postradiation lesion. Both are benign well-circumscribed lesions that histologically do not extend into surrounding subcutaneous tissues and do not have multilayering of cells, mitosis, or hemorrhage.7 Although lymphatic lesions tend to form vesicles, atypical vascular lesions arising after radiation treatment present as erythematous or flesh-colored patches or papules. They also tend to be fairly superficial and often only involve the superficial to mid dermis. On histology they show thin-walled channels without erythrocytes that are lined by typical endothelial cells.7 Despite these differences, both clinically and histopathologically these lesions can appear similar to acquired microcystic lymphatic malformations. It is important to differentiate between these two entities, as atypical vascular lesions have a slightly higher rate of transformation into malignant tumors such as angiosarcomas.
Although angiosarcomas clinically may present as erythematous patches, plaques, or nodules similar to benign postradiation lesions, they tend to be more edematous than their benign counterparts.7,8 Two other clinical factors that can help determine if a postradiation lesion is benign or malignant are the size and time of onset of the lesion. Angiosarcomas tend to be much larger than benign postradiation lesions (median size, 7.5 cm) and tend to be more multifocal in nature.8,9 They also tend to arise on average 5 to 7 years after the initial radiation treatment, while benign lesions arise sooner.9
Small, asymptomatic, acquired microcystic lymphatic malformations can be followed clinically without treatment, but these lesions do not commonly regress spontaneously. Even when asymptomatic, many clinicians will opt for treatment to prevent secondary complications such as infections, drainage, and pain. Moreover, these lesions can have notable psychosocial impacts on patients due to poor cosmetic appearance. Unfortunately, there is no gold standard of treatment, and recurrence is common, even after treatment. Historically, surgical excision was the treatment of choice, but this option carries a high risk for scarring, invasiveness, and recurrence. Recurrence rates of up to 23.1% have been reported with decreased effectiveness of resection, particularly in areas of deeper involvement.10 For these deeper lesions, CO2 laser therapy is a promising evolving therapy. It can penetrate up to the mid dermis and seems to destroy the lymphatic channels between deep and surface lymphatics, preventing the cutaneous manifestations of the disease. It has the added benefit of minimal invasiveness and fewer side effects than complete excision, with most studies reporting hyperpigmentation and scarring as the most common side effects.11 Additional emerging therapies including sclerotherapy and isotretinoin have shown benefits in case studies. Sclerotherapy causes local tissue destruction and thrombosis leading to destruction of vessel lumens and fibrosis that halts disease progression and clears existing lesions.12 Oral therapy with isotretinoin appears to work by inhibiting certain cytokines and acting as an antiangiogenic factor.13 Given the rarity of microcystic lymphatic malformations, further research must be done to determine definitive treatment.
Acquired microcystic lymphatic malformation is an important sequela of radiation therapy and surgical excision of malignancy. Despite its striking clinical appearance, it is sometimes difficult to diagnose given its rarity. It is important that clinicians are able to recognize it clinically and understand common treatment options to prevent both the mental stigma and complications, including secondary infections, drainage, and pain.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473.
- Vlastos AT, Malpica A, Follen M. Lymphangioma circumscriptum of the vulva: a review of the literature. Obstet Gynecol. 2003;101:946-954.
- Papalas JA, Robboy SJ, Burchette JL, et al. Acquired vulvar lymphangioma circumscriptum: a comparison of 12 cases with Crohn’s associated lesions or radiation therapy induced tumors. J Cutan Pathol. 2010;37:958-965.
- Kaya TI, Kokturk A, Polat A, et al. A case of cutaneous lymphangiectasis secondary to breast cancer treatment. Int J Dermatol. 2001;40:760-761.
- Ambrojo P, Cogolluda EF, Aguilar A, et al. Cutaneous lymphangiectases after therapy for carcinoma of the cervix. Clin Exp Dermatol. 1990;15:57-59.
- Tasdelen I, Gokgoz S, Paksoy E, et al. Acquired lymphangiectasis after breast conservation treatment for breast cancer: report of a case. Dermatol Online J. 2004;10:9.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584-1598.
- Ghaemmaghami F, Karimi Zarchi M, Mousavi A. Major labiaectomy as surgical management of vulvar lymphangioma circumscriptum: three cases and a review of the literature. Arch Gynecol Obstet. 2008;278:57-60.
- Savas J. Carbon dioxide laser for the treatment of microcystic lymphatic malformations (lymphangioma circumscriptum): a systematic review. Dermatol Surg. 2013;39:1147-1157.
- Al Ghamdi KM, Mubki TF. Treatment of lymphangioma circumscriptum with sclerotherapy: an ignored effective remedy. J Cosmet Dermatol. 2011;10:156-158.
- Ayhan E. Lymphangioma circumscriptum: good clinical response to isotretinoin therapy. Pediatr Dermatol. 2016;33:E208-E209.
Microcystic lymphatic malformations, also known as lymphangioma circumscriptum, are rare hamartomatous lesions comprised of dilated lymphatic channels that can be both congenital and acquired.1 They often present as translucent or hemorrhagic vesicles of varying sizes that may contain lymphatic fluid and often can cluster together and appear verrucous (Figure 1). The differential diagnosis for microcystic lymphatic malformations commonly includes molluscum contagiosum, squamous cell carcinoma, verruca vulgaris, or condylomas, as well as atypical vascular lesions. They most often are found in children as congenital lesions but also may be acquired. Most acquired cases are due to chronic inflammatory and scarring processes that damage lymphatic structures, including surgery, radiation, infections, and even Crohn disease.2,3 Because the differential diagnosis is so broad and the disease can clinically mimic other common disease processes, biopsies often are performed to determine the diagnosis. On biopsy, pathologic examination revealed well-circumscribed nodular lesions with large lymphatic channels often in a background of connective tissue stroma. Increased eosinophilic material, including mast cells, also was seen (Figure 2A). On immunohistochemistry, staining showed D2-40 positivity (Figure 2B).
Damage to lymphatics from radiation and postsurgical excision of tumors are well-described causes of microcystic lymphatic malformations, as in our patient, with most instances in the literature occurring secondary to treatment of breast or cervical cancer.4-6 In these acquired cases, the pathogenesis is thought to be due to destruction and fibrosis at the layer of the reticular dermis, which causes lymphatic obstruction and subsequent dilation of superficial lymphatic channels.6
Microcystic lymphatic malformations can be difficult to distinguish from atypical vascular lesions, another common postradiation lesion. Both are benign well-circumscribed lesions that histologically do not extend into surrounding subcutaneous tissues and do not have multilayering of cells, mitosis, or hemorrhage.7 Although lymphatic lesions tend to form vesicles, atypical vascular lesions arising after radiation treatment present as erythematous or flesh-colored patches or papules. They also tend to be fairly superficial and often only involve the superficial to mid dermis. On histology they show thin-walled channels without erythrocytes that are lined by typical endothelial cells.7 Despite these differences, both clinically and histopathologically these lesions can appear similar to acquired microcystic lymphatic malformations. It is important to differentiate between these two entities, as atypical vascular lesions have a slightly higher rate of transformation into malignant tumors such as angiosarcomas.
Although angiosarcomas clinically may present as erythematous patches, plaques, or nodules similar to benign postradiation lesions, they tend to be more edematous than their benign counterparts.7,8 Two other clinical factors that can help determine if a postradiation lesion is benign or malignant are the size and time of onset of the lesion. Angiosarcomas tend to be much larger than benign postradiation lesions (median size, 7.5 cm) and tend to be more multifocal in nature.8,9 They also tend to arise on average 5 to 7 years after the initial radiation treatment, while benign lesions arise sooner.9
Small, asymptomatic, acquired microcystic lymphatic malformations can be followed clinically without treatment, but these lesions do not commonly regress spontaneously. Even when asymptomatic, many clinicians will opt for treatment to prevent secondary complications such as infections, drainage, and pain. Moreover, these lesions can have notable psychosocial impacts on patients due to poor cosmetic appearance. Unfortunately, there is no gold standard of treatment, and recurrence is common, even after treatment. Historically, surgical excision was the treatment of choice, but this option carries a high risk for scarring, invasiveness, and recurrence. Recurrence rates of up to 23.1% have been reported with decreased effectiveness of resection, particularly in areas of deeper involvement.10 For these deeper lesions, CO2 laser therapy is a promising evolving therapy. It can penetrate up to the mid dermis and seems to destroy the lymphatic channels between deep and surface lymphatics, preventing the cutaneous manifestations of the disease. It has the added benefit of minimal invasiveness and fewer side effects than complete excision, with most studies reporting hyperpigmentation and scarring as the most common side effects.11 Additional emerging therapies including sclerotherapy and isotretinoin have shown benefits in case studies. Sclerotherapy causes local tissue destruction and thrombosis leading to destruction of vessel lumens and fibrosis that halts disease progression and clears existing lesions.12 Oral therapy with isotretinoin appears to work by inhibiting certain cytokines and acting as an antiangiogenic factor.13 Given the rarity of microcystic lymphatic malformations, further research must be done to determine definitive treatment.
Acquired microcystic lymphatic malformation is an important sequela of radiation therapy and surgical excision of malignancy. Despite its striking clinical appearance, it is sometimes difficult to diagnose given its rarity. It is important that clinicians are able to recognize it clinically and understand common treatment options to prevent both the mental stigma and complications, including secondary infections, drainage, and pain.
Microcystic lymphatic malformations, also known as lymphangioma circumscriptum, are rare hamartomatous lesions comprised of dilated lymphatic channels that can be both congenital and acquired.1 They often present as translucent or hemorrhagic vesicles of varying sizes that may contain lymphatic fluid and often can cluster together and appear verrucous (Figure 1). The differential diagnosis for microcystic lymphatic malformations commonly includes molluscum contagiosum, squamous cell carcinoma, verruca vulgaris, or condylomas, as well as atypical vascular lesions. They most often are found in children as congenital lesions but also may be acquired. Most acquired cases are due to chronic inflammatory and scarring processes that damage lymphatic structures, including surgery, radiation, infections, and even Crohn disease.2,3 Because the differential diagnosis is so broad and the disease can clinically mimic other common disease processes, biopsies often are performed to determine the diagnosis. On biopsy, pathologic examination revealed well-circumscribed nodular lesions with large lymphatic channels often in a background of connective tissue stroma. Increased eosinophilic material, including mast cells, also was seen (Figure 2A). On immunohistochemistry, staining showed D2-40 positivity (Figure 2B).
Damage to lymphatics from radiation and postsurgical excision of tumors are well-described causes of microcystic lymphatic malformations, as in our patient, with most instances in the literature occurring secondary to treatment of breast or cervical cancer.4-6 In these acquired cases, the pathogenesis is thought to be due to destruction and fibrosis at the layer of the reticular dermis, which causes lymphatic obstruction and subsequent dilation of superficial lymphatic channels.6
Microcystic lymphatic malformations can be difficult to distinguish from atypical vascular lesions, another common postradiation lesion. Both are benign well-circumscribed lesions that histologically do not extend into surrounding subcutaneous tissues and do not have multilayering of cells, mitosis, or hemorrhage.7 Although lymphatic lesions tend to form vesicles, atypical vascular lesions arising after radiation treatment present as erythematous or flesh-colored patches or papules. They also tend to be fairly superficial and often only involve the superficial to mid dermis. On histology they show thin-walled channels without erythrocytes that are lined by typical endothelial cells.7 Despite these differences, both clinically and histopathologically these lesions can appear similar to acquired microcystic lymphatic malformations. It is important to differentiate between these two entities, as atypical vascular lesions have a slightly higher rate of transformation into malignant tumors such as angiosarcomas.
Although angiosarcomas clinically may present as erythematous patches, plaques, or nodules similar to benign postradiation lesions, they tend to be more edematous than their benign counterparts.7,8 Two other clinical factors that can help determine if a postradiation lesion is benign or malignant are the size and time of onset of the lesion. Angiosarcomas tend to be much larger than benign postradiation lesions (median size, 7.5 cm) and tend to be more multifocal in nature.8,9 They also tend to arise on average 5 to 7 years after the initial radiation treatment, while benign lesions arise sooner.9
Small, asymptomatic, acquired microcystic lymphatic malformations can be followed clinically without treatment, but these lesions do not commonly regress spontaneously. Even when asymptomatic, many clinicians will opt for treatment to prevent secondary complications such as infections, drainage, and pain. Moreover, these lesions can have notable psychosocial impacts on patients due to poor cosmetic appearance. Unfortunately, there is no gold standard of treatment, and recurrence is common, even after treatment. Historically, surgical excision was the treatment of choice, but this option carries a high risk for scarring, invasiveness, and recurrence. Recurrence rates of up to 23.1% have been reported with decreased effectiveness of resection, particularly in areas of deeper involvement.10 For these deeper lesions, CO2 laser therapy is a promising evolving therapy. It can penetrate up to the mid dermis and seems to destroy the lymphatic channels between deep and surface lymphatics, preventing the cutaneous manifestations of the disease. It has the added benefit of minimal invasiveness and fewer side effects than complete excision, with most studies reporting hyperpigmentation and scarring as the most common side effects.11 Additional emerging therapies including sclerotherapy and isotretinoin have shown benefits in case studies. Sclerotherapy causes local tissue destruction and thrombosis leading to destruction of vessel lumens and fibrosis that halts disease progression and clears existing lesions.12 Oral therapy with isotretinoin appears to work by inhibiting certain cytokines and acting as an antiangiogenic factor.13 Given the rarity of microcystic lymphatic malformations, further research must be done to determine definitive treatment.
Acquired microcystic lymphatic malformation is an important sequela of radiation therapy and surgical excision of malignancy. Despite its striking clinical appearance, it is sometimes difficult to diagnose given its rarity. It is important that clinicians are able to recognize it clinically and understand common treatment options to prevent both the mental stigma and complications, including secondary infections, drainage, and pain.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473.
- Vlastos AT, Malpica A, Follen M. Lymphangioma circumscriptum of the vulva: a review of the literature. Obstet Gynecol. 2003;101:946-954.
- Papalas JA, Robboy SJ, Burchette JL, et al. Acquired vulvar lymphangioma circumscriptum: a comparison of 12 cases with Crohn’s associated lesions or radiation therapy induced tumors. J Cutan Pathol. 2010;37:958-965.
- Kaya TI, Kokturk A, Polat A, et al. A case of cutaneous lymphangiectasis secondary to breast cancer treatment. Int J Dermatol. 2001;40:760-761.
- Ambrojo P, Cogolluda EF, Aguilar A, et al. Cutaneous lymphangiectases after therapy for carcinoma of the cervix. Clin Exp Dermatol. 1990;15:57-59.
- Tasdelen I, Gokgoz S, Paksoy E, et al. Acquired lymphangiectasis after breast conservation treatment for breast cancer: report of a case. Dermatol Online J. 2004;10:9.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584-1598.
- Ghaemmaghami F, Karimi Zarchi M, Mousavi A. Major labiaectomy as surgical management of vulvar lymphangioma circumscriptum: three cases and a review of the literature. Arch Gynecol Obstet. 2008;278:57-60.
- Savas J. Carbon dioxide laser for the treatment of microcystic lymphatic malformations (lymphangioma circumscriptum): a systematic review. Dermatol Surg. 2013;39:1147-1157.
- Al Ghamdi KM, Mubki TF. Treatment of lymphangioma circumscriptum with sclerotherapy: an ignored effective remedy. J Cosmet Dermatol. 2011;10:156-158.
- Ayhan E. Lymphangioma circumscriptum: good clinical response to isotretinoin therapy. Pediatr Dermatol. 2016;33:E208-E209.
- Whimster IW. The pathology of lymphangioma circumscriptum. Br J Dermatol. 1976;94:473.
- Vlastos AT, Malpica A, Follen M. Lymphangioma circumscriptum of the vulva: a review of the literature. Obstet Gynecol. 2003;101:946-954.
- Papalas JA, Robboy SJ, Burchette JL, et al. Acquired vulvar lymphangioma circumscriptum: a comparison of 12 cases with Crohn’s associated lesions or radiation therapy induced tumors. J Cutan Pathol. 2010;37:958-965.
- Kaya TI, Kokturk A, Polat A, et al. A case of cutaneous lymphangiectasis secondary to breast cancer treatment. Int J Dermatol. 2001;40:760-761.
- Ambrojo P, Cogolluda EF, Aguilar A, et al. Cutaneous lymphangiectases after therapy for carcinoma of the cervix. Clin Exp Dermatol. 1990;15:57-59.
- Tasdelen I, Gokgoz S, Paksoy E, et al. Acquired lymphangiectasis after breast conservation treatment for breast cancer: report of a case. Dermatol Online J. 2004;10:9.
- Lucas DR. Angiosarcoma, radiation-associated angiosarcoma, and atypical vascular lesion. Arch Pathol Lab Med. 2009;133:1804-1809.
- Brenn T, Fletcher CD. Radiation-associated cutaneous atypical vascular lesions and angiosarcoma: clinicopathologic analysis of 42 cases. Am J Surg Pathol. 2005;29:983-996.
- Gengler C, Coindre JM, Leroux A. Vascular proliferations of the skin after radiation therapy for breast cancer: clinicopathologic analysis of a series in favor of a benign process: a study from the French Sarcoma Group. Cancer. 2007;109:1584-1598.
- Ghaemmaghami F, Karimi Zarchi M, Mousavi A. Major labiaectomy as surgical management of vulvar lymphangioma circumscriptum: three cases and a review of the literature. Arch Gynecol Obstet. 2008;278:57-60.
- Savas J. Carbon dioxide laser for the treatment of microcystic lymphatic malformations (lymphangioma circumscriptum): a systematic review. Dermatol Surg. 2013;39:1147-1157.
- Al Ghamdi KM, Mubki TF. Treatment of lymphangioma circumscriptum with sclerotherapy: an ignored effective remedy. J Cosmet Dermatol. 2011;10:156-158.
- Ayhan E. Lymphangioma circumscriptum: good clinical response to isotretinoin therapy. Pediatr Dermatol. 2016;33:E208-E209.
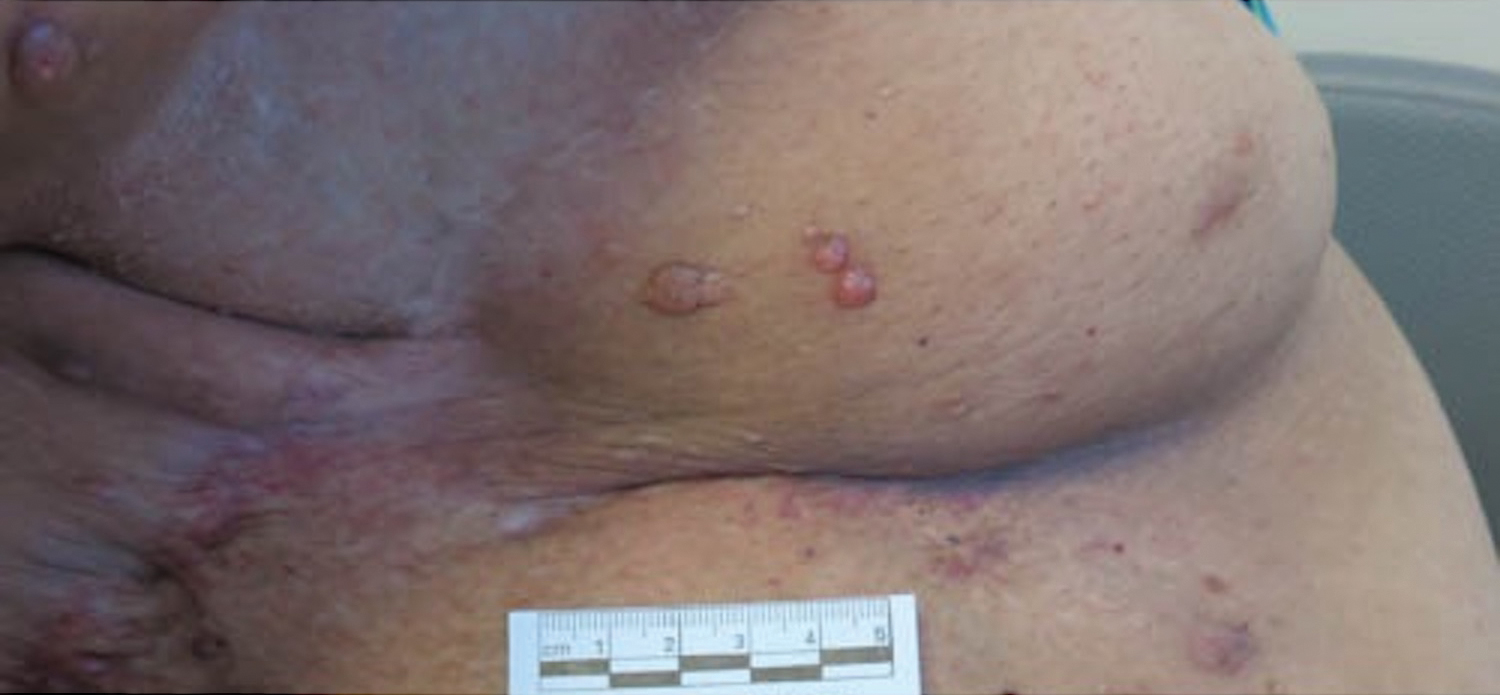
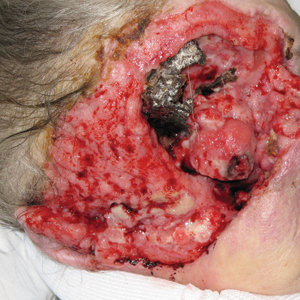
A 51-year-old woman with a history of bilateral breast cancer presented for evaluation of lesions on the underside of the right breast. She was first diagnosed with stage II cancer of the right breast that was subsequently treated with a mastectomy and adjuvant chemotherapy 7 years prior to presentation. One year later, she developed stage IIIC adenocarcinoma of the left breast and was treated with a modified radical mastectomy, adjuvant chemotherapy, and radiation. She had been followed closely by her oncologist with regular surveillance imaging (last at 7 months prior to presentation) that had all been negative for recurrent breast cancer. She presented to our dermatology clinic for evaluation of lesions on the underside of the right breast that were pruritic and occasionally painful with a burning quality. These lesions had recently begun to bleed when scratched but were not otherwise growing or spreading. On physical examination she was afebrile with stable vital signs. Skin examination was notable for numerous violaceous and translucent papules and nodules underneath the right breast and axilla overlying a well-healed mastectomy scar. No lymphadenopathy was present. Shave biopsies were performed and showed well-circumscribed nodular lesions with ectatic vascular channels separated by thin fibrous walls and filled with eosinophilic proteinaceous material and scattered red blood cells. Immunohistochemical staining also showed positivity for D2-40.
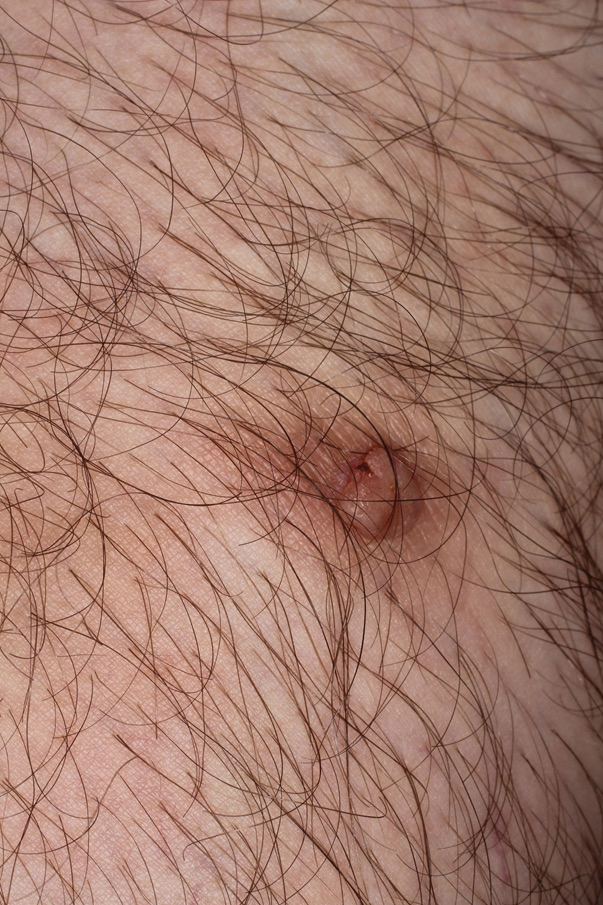
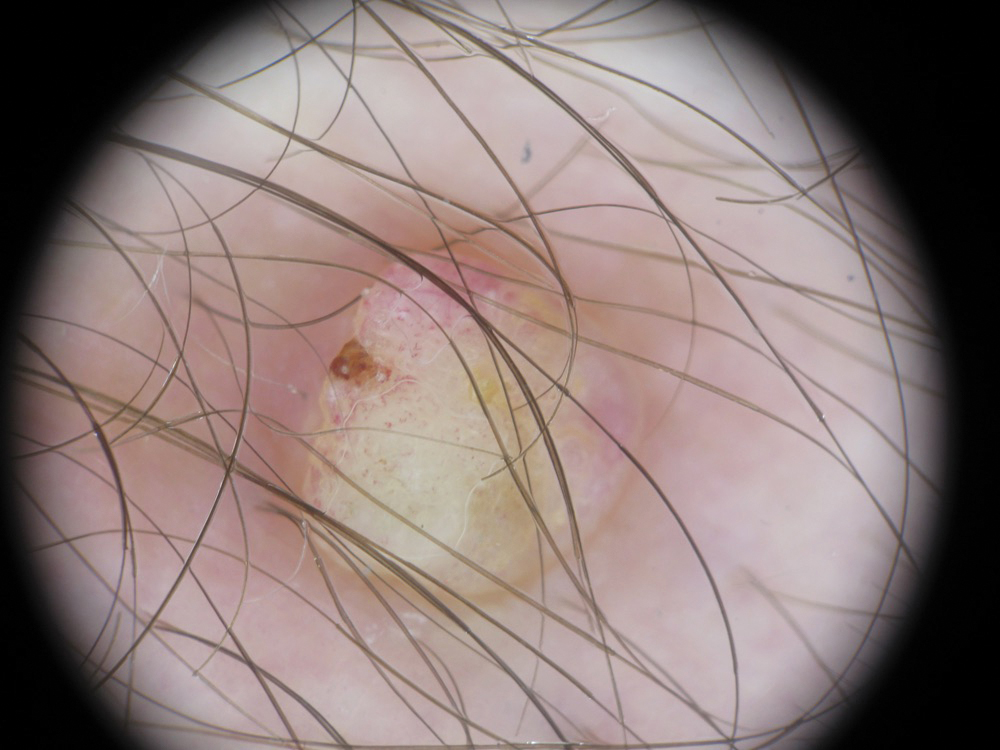
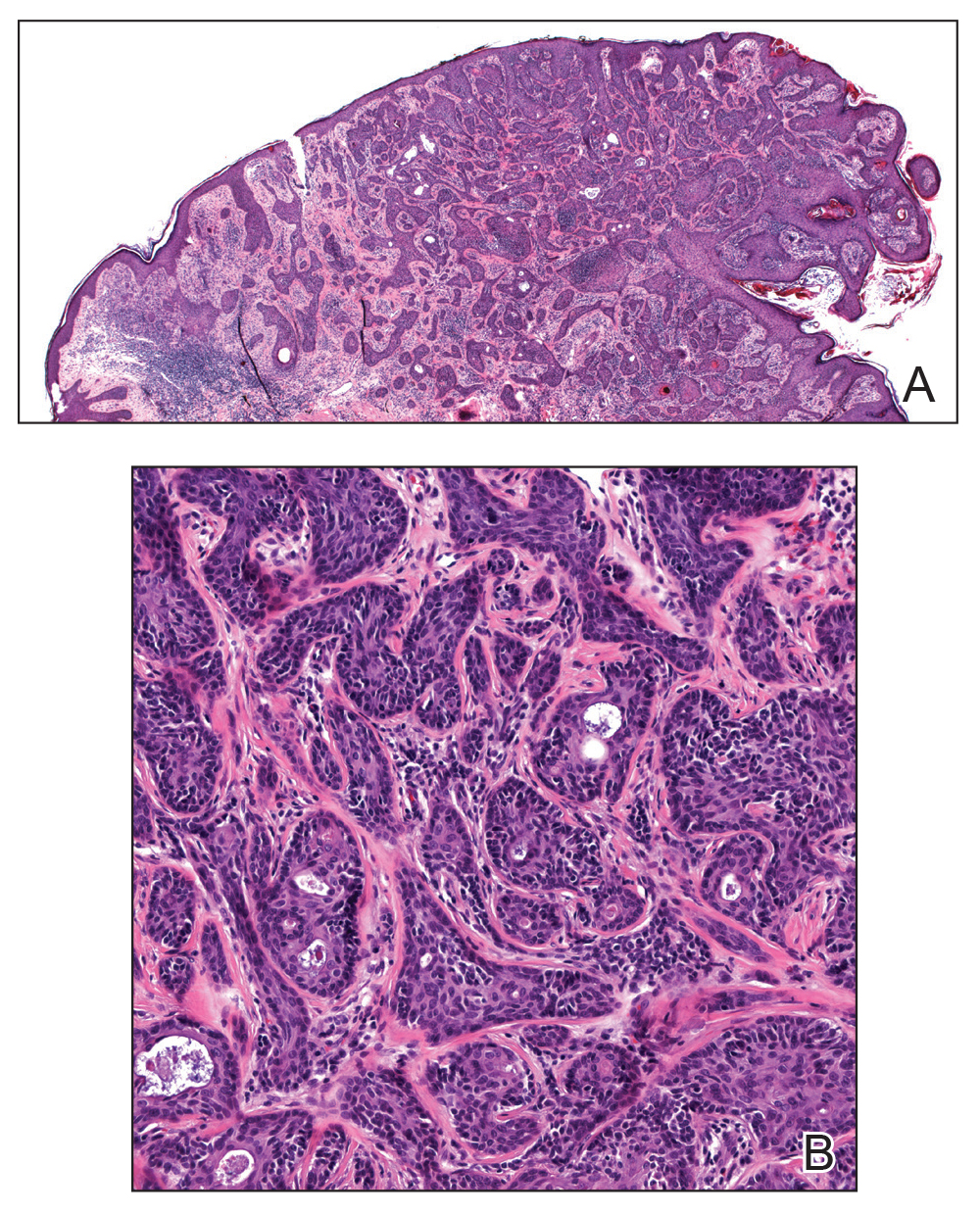
Eccrine Porocarcinoma Presenting as a Recurrent Wart
Eccrine porocarcinoma (EPC), originally described by Pinkus and Mehregan1 in 1963, is an exceedingly rare sweat gland tumor most commonly seen in older patients. Fewer than 300 cases have been reported in the literature, and it is believed to represent only 0.005% to 0.01% of cutaneous malignancies.2 In the absence of established guidelines, wide local excision (WLE) has traditionally been considered the standard of treatment; however, local recurrence and nodal metastasis rates associated with WLE have been reported as high as 20%.3 More recently, a number of case reports and small case series have demonstrated higher cure rates with Mohs micrographic surgery (MMS), though follow-up is limited.3-5 We describe a case of EPC presenting as a recurrent wart in a 36-year-old man that was successfully treated with MMS.
Case Report
A 36-year-old man with no notable medical history presented with a 0.5×0.5-cm, asymptomatic, flesh-colored, hyperkeratotic, polypoid papule on the right medial thigh (Figure 1). The lesion was diagnosed as a wart and treated with cryotherapy by another dermatologist several years prior to presentation. Dermatoscopic examination at the current presentation showed a homogenous yellow center with a few peripheral vessels and a faint pink-tan halo (Figure 2). Our differential diagnosis included a recurrent wart, fibrosed pyogenic granuloma, irritated intradermal nevus, skin tag, and adnexal neoplasm. A shave biopsy was performed. Histopathologic analysis revealed multiple aggregations of mildly pleomorphic epithelial cells emanating from the epidermis, with many aggregations containing ductal structures (Figure 3). Rare necrotic and pyknotic cells were present, but no mitotic figures or lymphovascular invasion were identified. Immunohistochemical staining was positive for carcinoembryonic antigen and epithelial membrane antigen but negative for Ber-EP4. These findings were consistent with a well-differentiated EPC.
The patient was offered MMS or WLE, with or without sentinel lymph node biopsy (SLNB). He opted for MMS. The initial 1-cm margin taken during MMS was sufficient to achieve complete tumor extirpation, and the final 3.7×2.5-cm defect was closed primarily. The MMS debulking specimen was sent for permanent sectioning and showed a small focus of residual tumor cells, but no mitoses or lymphovascular invasion were seen. The patient was referred to surgical oncology to discuss the option of SLNB, which he ultimately declined. He also was offered regional or whole-body positron emission tomography–computed tomography (PET-CT) to rule out metastatic disease, which he also declined. There was no evidence of recurrence or lymphadenopathy 19 months postoperatively.
Comment
Eccrine porocarcinoma is an exceptionally rare adnexal neoplasm that most commonly affects older adults. The average age at diagnosis is 71 years in men and 75 years in women.2 Our case is rare because of the patient’s age. Benign eccrine poromas occur most frequently on the palms, soles, axillae, and forehead where eccrine density is highest; EPC occurs most frequently on the lower extremities.6 It may arise de novo or from malignant transformation of a preexisting benign poroma. Clinically, EPC may present as an asymptomatic pink-brown papule, plaque, or nodule and may have a polypoid or verrucous appearance, as in our patient. Ulceration is common.7 The differential diagnosis often includes nodular basal cell carcinoma, squamous cell carcinoma, pyogenic granuloma, and seborrheic keratosis.
Histologically, EPCs are characterized by aggregations of cohesive basaloid epithelial cells forming eccrine ductal structures.2 Cellular atypia may be extremely subtle but, if present, can be helpful in differentiating malignant from benign lesions. Features of basal and squamous cell carcinoma also may be present. Definitive diagnosis is frequently based on the overall invasive architectural pattern.5 Robson et al2 examined 69 cases of EPC for high-risk histologic features and concluded that tumor depth greater than 7 mm, mitoses greater than 14 per high-power field, and the presence of lymphovascular invasion were independently predictive of mortality. Moreover, after adjusting for mitosis and depth, an infiltrative border vs a pushing border was strongly predictive of local recurrence.2 Immunohistochemical stains, although not necessary for diagnosis, may have utility as adjunctive tools. Cells lining the ducts within EPCs commonly stain positive for carcinoembryonic antigen, though glandular myoepithelial cells stain positive for S-100. Negative Ber-EP4 staining helps to differentiate EPC from basal cell carcinoma. Abnormal expression of p53 and overexpression of p16 also has been described.4
The rarity of EPC has precluded the development of any evidence-based management guidelines. Historically, the standard of care has been WLE with 2- to 3-cm margins. A review of 105 cases of EPC treated with WLE showed 20% local recurrence, 20% regional metastases, and 12% distant metastasis rates.8 Mohs micrographic surgery, which allows examination of 100% of the surgical margin vs less than 1% for WLE with the standard bread-loafing technique, might be expected to achieve higher cure rates. A review of 29 cases treated with MMS monotherapy demonstrated no local recurrences, distant metastasis, or disease-specific deaths with follow-up ranging from 19 months to 6 years.5 One case was associated with regional lymph node metastases that were treated with completion lymphadenectomy and adjuvant radiation therapy.7 The high mortality rate of patients with nodal disease has led some to recommend PET-CT and SLNB for patients with EPC. However, the prognostic value of such procedures has not been clearly defined and there is no demonstrated survival benefit for treatment of widespread disease. Our patient declined both SLNB and PET-CT, and our plan was to follow him clinically with symptom-directed imaging only.
Conclusion
Patients with EPC generally have a favorable prognosis with prompt diagnosis and complete surgical excision. Although most commonly seen in elderly patients, EPC may present in younger patients and may be clinically and histologically nondescript with little cytologic atypia. Based on a small but growing body of literature, MMS appears to be at least as effective as WLE as a primary treatment modality for EPC, while offering the advantage of tissue sparing in cosmetically or functionally important areas.
- Pinkus H, Mehregan AH. Epidermatropic eccrine carcinoma. a case combining eccrine poroma and Paget’s dermatoses. Arch Dermatol. 1963;88:597-606.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Tolkachjov SN, Hocker TL, Camilleri MJ, et al. Treatment of porocarcinoma with Mohs micrographic surgery: The Mayo Clinic Experience. Dermatol Surg. 2016;42:745-750.
- Tidwell WJ, Mayer JE, Malone J, et al. Treatment of eccrine porocarcinoma with Mohs micrographic surgery: a cases series and literature review. Int J Dermatol. 2015;54:1078-1083.
- Xu YG, Aylward J, Longley BJ, et al. Eccrine porocarcinoma treated by Mohs micrographic surgery: over 6-year follow-up of 12 cases and literature review. Dermatol Surg. 2015;41:685-692.
- D’Ambrosia RA, Ward H, Parry E. Eccrine porocarcinoma of the eyelid treated with Mohs micrographic surgery. Dermatol Surg. 2004;30:4:570-571.
- Vleugels FR, Girouard SD, Schmults CD, et al. Metastatic eccrine porocarcinoma after Mohs micrographic surgery: a case report. J Clin Oncol. 2012;30:188-191.
- Snow SN, Reizner GT. Eccrine porocarcinoma of the face. J Am Acad Dermatol. 1992;27:306-311.
Eccrine porocarcinoma (EPC), originally described by Pinkus and Mehregan1 in 1963, is an exceedingly rare sweat gland tumor most commonly seen in older patients. Fewer than 300 cases have been reported in the literature, and it is believed to represent only 0.005% to 0.01% of cutaneous malignancies.2 In the absence of established guidelines, wide local excision (WLE) has traditionally been considered the standard of treatment; however, local recurrence and nodal metastasis rates associated with WLE have been reported as high as 20%.3 More recently, a number of case reports and small case series have demonstrated higher cure rates with Mohs micrographic surgery (MMS), though follow-up is limited.3-5 We describe a case of EPC presenting as a recurrent wart in a 36-year-old man that was successfully treated with MMS.
Case Report
A 36-year-old man with no notable medical history presented with a 0.5×0.5-cm, asymptomatic, flesh-colored, hyperkeratotic, polypoid papule on the right medial thigh (Figure 1). The lesion was diagnosed as a wart and treated with cryotherapy by another dermatologist several years prior to presentation. Dermatoscopic examination at the current presentation showed a homogenous yellow center with a few peripheral vessels and a faint pink-tan halo (Figure 2). Our differential diagnosis included a recurrent wart, fibrosed pyogenic granuloma, irritated intradermal nevus, skin tag, and adnexal neoplasm. A shave biopsy was performed. Histopathologic analysis revealed multiple aggregations of mildly pleomorphic epithelial cells emanating from the epidermis, with many aggregations containing ductal structures (Figure 3). Rare necrotic and pyknotic cells were present, but no mitotic figures or lymphovascular invasion were identified. Immunohistochemical staining was positive for carcinoembryonic antigen and epithelial membrane antigen but negative for Ber-EP4. These findings were consistent with a well-differentiated EPC.
The patient was offered MMS or WLE, with or without sentinel lymph node biopsy (SLNB). He opted for MMS. The initial 1-cm margin taken during MMS was sufficient to achieve complete tumor extirpation, and the final 3.7×2.5-cm defect was closed primarily. The MMS debulking specimen was sent for permanent sectioning and showed a small focus of residual tumor cells, but no mitoses or lymphovascular invasion were seen. The patient was referred to surgical oncology to discuss the option of SLNB, which he ultimately declined. He also was offered regional or whole-body positron emission tomography–computed tomography (PET-CT) to rule out metastatic disease, which he also declined. There was no evidence of recurrence or lymphadenopathy 19 months postoperatively.
Comment
Eccrine porocarcinoma is an exceptionally rare adnexal neoplasm that most commonly affects older adults. The average age at diagnosis is 71 years in men and 75 years in women.2 Our case is rare because of the patient’s age. Benign eccrine poromas occur most frequently on the palms, soles, axillae, and forehead where eccrine density is highest; EPC occurs most frequently on the lower extremities.6 It may arise de novo or from malignant transformation of a preexisting benign poroma. Clinically, EPC may present as an asymptomatic pink-brown papule, plaque, or nodule and may have a polypoid or verrucous appearance, as in our patient. Ulceration is common.7 The differential diagnosis often includes nodular basal cell carcinoma, squamous cell carcinoma, pyogenic granuloma, and seborrheic keratosis.
Histologically, EPCs are characterized by aggregations of cohesive basaloid epithelial cells forming eccrine ductal structures.2 Cellular atypia may be extremely subtle but, if present, can be helpful in differentiating malignant from benign lesions. Features of basal and squamous cell carcinoma also may be present. Definitive diagnosis is frequently based on the overall invasive architectural pattern.5 Robson et al2 examined 69 cases of EPC for high-risk histologic features and concluded that tumor depth greater than 7 mm, mitoses greater than 14 per high-power field, and the presence of lymphovascular invasion were independently predictive of mortality. Moreover, after adjusting for mitosis and depth, an infiltrative border vs a pushing border was strongly predictive of local recurrence.2 Immunohistochemical stains, although not necessary for diagnosis, may have utility as adjunctive tools. Cells lining the ducts within EPCs commonly stain positive for carcinoembryonic antigen, though glandular myoepithelial cells stain positive for S-100. Negative Ber-EP4 staining helps to differentiate EPC from basal cell carcinoma. Abnormal expression of p53 and overexpression of p16 also has been described.4
The rarity of EPC has precluded the development of any evidence-based management guidelines. Historically, the standard of care has been WLE with 2- to 3-cm margins. A review of 105 cases of EPC treated with WLE showed 20% local recurrence, 20% regional metastases, and 12% distant metastasis rates.8 Mohs micrographic surgery, which allows examination of 100% of the surgical margin vs less than 1% for WLE with the standard bread-loafing technique, might be expected to achieve higher cure rates. A review of 29 cases treated with MMS monotherapy demonstrated no local recurrences, distant metastasis, or disease-specific deaths with follow-up ranging from 19 months to 6 years.5 One case was associated with regional lymph node metastases that were treated with completion lymphadenectomy and adjuvant radiation therapy.7 The high mortality rate of patients with nodal disease has led some to recommend PET-CT and SLNB for patients with EPC. However, the prognostic value of such procedures has not been clearly defined and there is no demonstrated survival benefit for treatment of widespread disease. Our patient declined both SLNB and PET-CT, and our plan was to follow him clinically with symptom-directed imaging only.
Conclusion
Patients with EPC generally have a favorable prognosis with prompt diagnosis and complete surgical excision. Although most commonly seen in elderly patients, EPC may present in younger patients and may be clinically and histologically nondescript with little cytologic atypia. Based on a small but growing body of literature, MMS appears to be at least as effective as WLE as a primary treatment modality for EPC, while offering the advantage of tissue sparing in cosmetically or functionally important areas.
Eccrine porocarcinoma (EPC), originally described by Pinkus and Mehregan1 in 1963, is an exceedingly rare sweat gland tumor most commonly seen in older patients. Fewer than 300 cases have been reported in the literature, and it is believed to represent only 0.005% to 0.01% of cutaneous malignancies.2 In the absence of established guidelines, wide local excision (WLE) has traditionally been considered the standard of treatment; however, local recurrence and nodal metastasis rates associated with WLE have been reported as high as 20%.3 More recently, a number of case reports and small case series have demonstrated higher cure rates with Mohs micrographic surgery (MMS), though follow-up is limited.3-5 We describe a case of EPC presenting as a recurrent wart in a 36-year-old man that was successfully treated with MMS.
Case Report
A 36-year-old man with no notable medical history presented with a 0.5×0.5-cm, asymptomatic, flesh-colored, hyperkeratotic, polypoid papule on the right medial thigh (Figure 1). The lesion was diagnosed as a wart and treated with cryotherapy by another dermatologist several years prior to presentation. Dermatoscopic examination at the current presentation showed a homogenous yellow center with a few peripheral vessels and a faint pink-tan halo (Figure 2). Our differential diagnosis included a recurrent wart, fibrosed pyogenic granuloma, irritated intradermal nevus, skin tag, and adnexal neoplasm. A shave biopsy was performed. Histopathologic analysis revealed multiple aggregations of mildly pleomorphic epithelial cells emanating from the epidermis, with many aggregations containing ductal structures (Figure 3). Rare necrotic and pyknotic cells were present, but no mitotic figures or lymphovascular invasion were identified. Immunohistochemical staining was positive for carcinoembryonic antigen and epithelial membrane antigen but negative for Ber-EP4. These findings were consistent with a well-differentiated EPC.
The patient was offered MMS or WLE, with or without sentinel lymph node biopsy (SLNB). He opted for MMS. The initial 1-cm margin taken during MMS was sufficient to achieve complete tumor extirpation, and the final 3.7×2.5-cm defect was closed primarily. The MMS debulking specimen was sent for permanent sectioning and showed a small focus of residual tumor cells, but no mitoses or lymphovascular invasion were seen. The patient was referred to surgical oncology to discuss the option of SLNB, which he ultimately declined. He also was offered regional or whole-body positron emission tomography–computed tomography (PET-CT) to rule out metastatic disease, which he also declined. There was no evidence of recurrence or lymphadenopathy 19 months postoperatively.
Comment
Eccrine porocarcinoma is an exceptionally rare adnexal neoplasm that most commonly affects older adults. The average age at diagnosis is 71 years in men and 75 years in women.2 Our case is rare because of the patient’s age. Benign eccrine poromas occur most frequently on the palms, soles, axillae, and forehead where eccrine density is highest; EPC occurs most frequently on the lower extremities.6 It may arise de novo or from malignant transformation of a preexisting benign poroma. Clinically, EPC may present as an asymptomatic pink-brown papule, plaque, or nodule and may have a polypoid or verrucous appearance, as in our patient. Ulceration is common.7 The differential diagnosis often includes nodular basal cell carcinoma, squamous cell carcinoma, pyogenic granuloma, and seborrheic keratosis.
Histologically, EPCs are characterized by aggregations of cohesive basaloid epithelial cells forming eccrine ductal structures.2 Cellular atypia may be extremely subtle but, if present, can be helpful in differentiating malignant from benign lesions. Features of basal and squamous cell carcinoma also may be present. Definitive diagnosis is frequently based on the overall invasive architectural pattern.5 Robson et al2 examined 69 cases of EPC for high-risk histologic features and concluded that tumor depth greater than 7 mm, mitoses greater than 14 per high-power field, and the presence of lymphovascular invasion were independently predictive of mortality. Moreover, after adjusting for mitosis and depth, an infiltrative border vs a pushing border was strongly predictive of local recurrence.2 Immunohistochemical stains, although not necessary for diagnosis, may have utility as adjunctive tools. Cells lining the ducts within EPCs commonly stain positive for carcinoembryonic antigen, though glandular myoepithelial cells stain positive for S-100. Negative Ber-EP4 staining helps to differentiate EPC from basal cell carcinoma. Abnormal expression of p53 and overexpression of p16 also has been described.4
The rarity of EPC has precluded the development of any evidence-based management guidelines. Historically, the standard of care has been WLE with 2- to 3-cm margins. A review of 105 cases of EPC treated with WLE showed 20% local recurrence, 20% regional metastases, and 12% distant metastasis rates.8 Mohs micrographic surgery, which allows examination of 100% of the surgical margin vs less than 1% for WLE with the standard bread-loafing technique, might be expected to achieve higher cure rates. A review of 29 cases treated with MMS monotherapy demonstrated no local recurrences, distant metastasis, or disease-specific deaths with follow-up ranging from 19 months to 6 years.5 One case was associated with regional lymph node metastases that were treated with completion lymphadenectomy and adjuvant radiation therapy.7 The high mortality rate of patients with nodal disease has led some to recommend PET-CT and SLNB for patients with EPC. However, the prognostic value of such procedures has not been clearly defined and there is no demonstrated survival benefit for treatment of widespread disease. Our patient declined both SLNB and PET-CT, and our plan was to follow him clinically with symptom-directed imaging only.
Conclusion
Patients with EPC generally have a favorable prognosis with prompt diagnosis and complete surgical excision. Although most commonly seen in elderly patients, EPC may present in younger patients and may be clinically and histologically nondescript with little cytologic atypia. Based on a small but growing body of literature, MMS appears to be at least as effective as WLE as a primary treatment modality for EPC, while offering the advantage of tissue sparing in cosmetically or functionally important areas.
- Pinkus H, Mehregan AH. Epidermatropic eccrine carcinoma. a case combining eccrine poroma and Paget’s dermatoses. Arch Dermatol. 1963;88:597-606.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Tolkachjov SN, Hocker TL, Camilleri MJ, et al. Treatment of porocarcinoma with Mohs micrographic surgery: The Mayo Clinic Experience. Dermatol Surg. 2016;42:745-750.
- Tidwell WJ, Mayer JE, Malone J, et al. Treatment of eccrine porocarcinoma with Mohs micrographic surgery: a cases series and literature review. Int J Dermatol. 2015;54:1078-1083.
- Xu YG, Aylward J, Longley BJ, et al. Eccrine porocarcinoma treated by Mohs micrographic surgery: over 6-year follow-up of 12 cases and literature review. Dermatol Surg. 2015;41:685-692.
- D’Ambrosia RA, Ward H, Parry E. Eccrine porocarcinoma of the eyelid treated with Mohs micrographic surgery. Dermatol Surg. 2004;30:4:570-571.
- Vleugels FR, Girouard SD, Schmults CD, et al. Metastatic eccrine porocarcinoma after Mohs micrographic surgery: a case report. J Clin Oncol. 2012;30:188-191.
- Snow SN, Reizner GT. Eccrine porocarcinoma of the face. J Am Acad Dermatol. 1992;27:306-311.
- Pinkus H, Mehregan AH. Epidermatropic eccrine carcinoma. a case combining eccrine poroma and Paget’s dermatoses. Arch Dermatol. 1963;88:597-606.
- Robson A, Greene J, Ansari N, et al. Eccrine porocarcinoma (malignant eccrine poroma): a clinicopathologic study of 69 cases. Am J Surg Pathol. 2001;25:710-720.
- Tolkachjov SN, Hocker TL, Camilleri MJ, et al. Treatment of porocarcinoma with Mohs micrographic surgery: The Mayo Clinic Experience. Dermatol Surg. 2016;42:745-750.
- Tidwell WJ, Mayer JE, Malone J, et al. Treatment of eccrine porocarcinoma with Mohs micrographic surgery: a cases series and literature review. Int J Dermatol. 2015;54:1078-1083.
- Xu YG, Aylward J, Longley BJ, et al. Eccrine porocarcinoma treated by Mohs micrographic surgery: over 6-year follow-up of 12 cases and literature review. Dermatol Surg. 2015;41:685-692.
- D’Ambrosia RA, Ward H, Parry E. Eccrine porocarcinoma of the eyelid treated with Mohs micrographic surgery. Dermatol Surg. 2004;30:4:570-571.
- Vleugels FR, Girouard SD, Schmults CD, et al. Metastatic eccrine porocarcinoma after Mohs micrographic surgery: a case report. J Clin Oncol. 2012;30:188-191.
- Snow SN, Reizner GT. Eccrine porocarcinoma of the face. J Am Acad Dermatol. 1992;27:306-311.
Practice Points
- Eccrine porocarcinoma is more common in older patients (age range, 71–75 years).
- Local recurrence and nodal metastasis are reported as high as 20% with wide local excision.
- Higher cure rates recently have been reported with Mohs micrographic surgery.
CLL, GVHD may raise risk for skin cancer after allo-HCT
Previously unknown risk factors for secondary skin cancer linked with allogeneic hematopoietic cell transplantation (HCT) have been identified, researchers report after a retrospective analysis.
“We confirmed [graft-versus-host disease] as a risk factor, identified [chronic lymphocytic leukemia] as an additional risk factor, and found that patients who received myeloablative transplants in adulthood had fewer [basal cell carcinomas] than their counterparts,” Peggy A. Wu, MD, of the Beth Israel Deaconess Medical Center in Boston, and her colleagues wrote in the Journal of Investigative Dermatology.
The team analyzed 1,974 patients who underwent transplantation for various types of hematologic cancer and survived for a minimum of 100 days following transplant. Among this cohort, 119 patients developed various forms of skin cancer, including basal and squamous cell carcinoma.
Reports of skin malignancy were confirmed using physician records and pathology reports. Dr. Wu and her colleagues excluded patients whose indication for transplant was a primary immunodeficiency or Fanconi anemia.
“Reflecting advances that allow older patients to be eligible for HCT, the median age at transplantation of our cohort was one of the oldest (51.1 years) in the literature,” the researchers wrote.
In univariable models, the researchers found that prior chronic lymphocytic leukemia (CLL) (hazard ratio, 2.2; 95% CI, 1.3-3.7), chronic graft-versus-host disease (GVHD) (HR, 3.1; 95% CI, 1.7-5.4), and age at transplant of more than 60 years (HR, 10.8; 95% CI, 3.3-35.6) were all linked to an increased risk for squamous cell carcinomas. A multivariable analysis found that these factors continued as significant risk factors.
For basal cell carcinomas, the risk factors identified were prior CLL (HR, 3.5; 95% CI, 2.0-6.4), acute GVHD (HR, 1.9; 95% CI, 1.1-3.3), and chronic GVHD (HR, 3.2; 95% CI, 1.6-6.5) using univariable models. These factors all continued to be significant in multivariable analysis.
Additionally, the researchers found that a myeloablative conditioning regimen and total body irradiation were protective against development of basal cell carcinomas in univariable models. However, the protective effect continued for myeloablative condition in the multivariable model only.
“To our knowledge, previously unreported risk factors in this contemporary cohort include prior CLL for squamous cell carcinoma and basal cell carcinoma and reduced-intensity conditioning for basal cell carcinoma,” the researchers wrote.
The study was supported by the Skin Cancer Foundation, Women’s Dermatologic Society, Harvard Catalyst, and Harvard University. The authors reported having no conflicts of interest.
SOURCE: Wu PA et al. J Invest Dermatol. 2019 Mar;139(3):591-9.
Previously unknown risk factors for secondary skin cancer linked with allogeneic hematopoietic cell transplantation (HCT) have been identified, researchers report after a retrospective analysis.
“We confirmed [graft-versus-host disease] as a risk factor, identified [chronic lymphocytic leukemia] as an additional risk factor, and found that patients who received myeloablative transplants in adulthood had fewer [basal cell carcinomas] than their counterparts,” Peggy A. Wu, MD, of the Beth Israel Deaconess Medical Center in Boston, and her colleagues wrote in the Journal of Investigative Dermatology.
The team analyzed 1,974 patients who underwent transplantation for various types of hematologic cancer and survived for a minimum of 100 days following transplant. Among this cohort, 119 patients developed various forms of skin cancer, including basal and squamous cell carcinoma.
Reports of skin malignancy were confirmed using physician records and pathology reports. Dr. Wu and her colleagues excluded patients whose indication for transplant was a primary immunodeficiency or Fanconi anemia.
“Reflecting advances that allow older patients to be eligible for HCT, the median age at transplantation of our cohort was one of the oldest (51.1 years) in the literature,” the researchers wrote.
In univariable models, the researchers found that prior chronic lymphocytic leukemia (CLL) (hazard ratio, 2.2; 95% CI, 1.3-3.7), chronic graft-versus-host disease (GVHD) (HR, 3.1; 95% CI, 1.7-5.4), and age at transplant of more than 60 years (HR, 10.8; 95% CI, 3.3-35.6) were all linked to an increased risk for squamous cell carcinomas. A multivariable analysis found that these factors continued as significant risk factors.
For basal cell carcinomas, the risk factors identified were prior CLL (HR, 3.5; 95% CI, 2.0-6.4), acute GVHD (HR, 1.9; 95% CI, 1.1-3.3), and chronic GVHD (HR, 3.2; 95% CI, 1.6-6.5) using univariable models. These factors all continued to be significant in multivariable analysis.
Additionally, the researchers found that a myeloablative conditioning regimen and total body irradiation were protective against development of basal cell carcinomas in univariable models. However, the protective effect continued for myeloablative condition in the multivariable model only.
“To our knowledge, previously unreported risk factors in this contemporary cohort include prior CLL for squamous cell carcinoma and basal cell carcinoma and reduced-intensity conditioning for basal cell carcinoma,” the researchers wrote.
The study was supported by the Skin Cancer Foundation, Women’s Dermatologic Society, Harvard Catalyst, and Harvard University. The authors reported having no conflicts of interest.
SOURCE: Wu PA et al. J Invest Dermatol. 2019 Mar;139(3):591-9.
Previously unknown risk factors for secondary skin cancer linked with allogeneic hematopoietic cell transplantation (HCT) have been identified, researchers report after a retrospective analysis.
“We confirmed [graft-versus-host disease] as a risk factor, identified [chronic lymphocytic leukemia] as an additional risk factor, and found that patients who received myeloablative transplants in adulthood had fewer [basal cell carcinomas] than their counterparts,” Peggy A. Wu, MD, of the Beth Israel Deaconess Medical Center in Boston, and her colleagues wrote in the Journal of Investigative Dermatology.
The team analyzed 1,974 patients who underwent transplantation for various types of hematologic cancer and survived for a minimum of 100 days following transplant. Among this cohort, 119 patients developed various forms of skin cancer, including basal and squamous cell carcinoma.
Reports of skin malignancy were confirmed using physician records and pathology reports. Dr. Wu and her colleagues excluded patients whose indication for transplant was a primary immunodeficiency or Fanconi anemia.
“Reflecting advances that allow older patients to be eligible for HCT, the median age at transplantation of our cohort was one of the oldest (51.1 years) in the literature,” the researchers wrote.
In univariable models, the researchers found that prior chronic lymphocytic leukemia (CLL) (hazard ratio, 2.2; 95% CI, 1.3-3.7), chronic graft-versus-host disease (GVHD) (HR, 3.1; 95% CI, 1.7-5.4), and age at transplant of more than 60 years (HR, 10.8; 95% CI, 3.3-35.6) were all linked to an increased risk for squamous cell carcinomas. A multivariable analysis found that these factors continued as significant risk factors.
For basal cell carcinomas, the risk factors identified were prior CLL (HR, 3.5; 95% CI, 2.0-6.4), acute GVHD (HR, 1.9; 95% CI, 1.1-3.3), and chronic GVHD (HR, 3.2; 95% CI, 1.6-6.5) using univariable models. These factors all continued to be significant in multivariable analysis.
Additionally, the researchers found that a myeloablative conditioning regimen and total body irradiation were protective against development of basal cell carcinomas in univariable models. However, the protective effect continued for myeloablative condition in the multivariable model only.
“To our knowledge, previously unreported risk factors in this contemporary cohort include prior CLL for squamous cell carcinoma and basal cell carcinoma and reduced-intensity conditioning for basal cell carcinoma,” the researchers wrote.
The study was supported by the Skin Cancer Foundation, Women’s Dermatologic Society, Harvard Catalyst, and Harvard University. The authors reported having no conflicts of interest.
SOURCE: Wu PA et al. J Invest Dermatol. 2019 Mar;139(3):591-9.
FROM THE JOURNAL OF INVESTIGATIVE DERMATOLOGY
Growing Painful Nodule on the Lower Lip
The Diagnosis: Verrucous Carcinoma
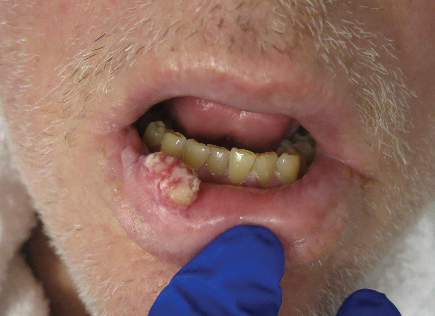
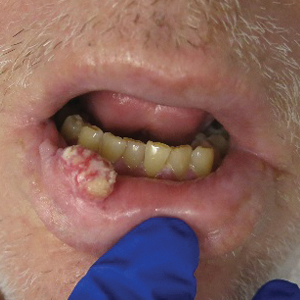
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
The Diagnosis: Verrucous Carcinoma
An excisional biopsy revealed an endophytic and exophytic squamous proliferation with a papillomatous growth pattern, bulbous pushing border, and confluent parakeratosis (Figure). No fungal organisms were seen. Due to clinical and histological findings, a diagnosis of verrucous carcinoma (VC) was made.
Verrucous carcinoma is a rare variant of squamous cell carcinoma (SCC) with specific clinical and histological features.1 These tumors have a slow and localized growth pattern but can be locally aggressive. Metastasis of VC is rare, giving VC an overall good prognosis, with a 5-year survival rate greater than 75%.2 Verrucous carcinoma typically occurs in 1 of 3 locations: the oropharynx, genitals, or soles of the feet. Depending on the site of involvement, various names have been used in the literature to describe this entity, including Ackerman tumor (solitary oral mucosal lesion), Buschke-Lowenstein tumor (genital involvement), florid oral papillomatosis (multiple oral lesions), and carcinoma cuniculatum (sole of the foot).3 The most common sites for VC in the oral cavity are the buccal mucosa and gingiva.4
Verrucous carcinoma occurs more often among men in the sixth decade of life.3 The etiology of oral VC remains unclear; however, use of chewing tobacco, chemical carcinogens, chronic irritation, human papillomavirus (HPV), and poor oral hygiene have been reported as predisposing risk factors.4,5 The role of HPV in the pathogenesis of VC remains controversial, but both low-risk types HPV-6 and HPV-11 and high-risk types HPV-16 and HPV-18 have been found in association with VC.5,6
Clinically, oral VC lesions most often present as pink-white erythematous papules or plaques with exophytic cauliflowerlike surface alterations. Although the tumors are slow growing with little risk for metastasis, they may be locally invasive with deep involvement of the surrounding
structures.1 Histopathologically, VC displays proliferation of the epithelium with downward growth into the connective tissue but usually without a pattern of true invasion. The epithelium is well differentiated and displays little pleomorphism or mitoses.5,7 Obtaining a generous biopsy specimen is essential to view the diagnostic architecture of VC and rule out other entities, such as viral verruca, blastomycosis, SCC, and verruciform xanthoma. Squamous cell carcinoma characteristically has a more infiltrative border as opposed to the bulbous border of VC. In addition, the distribution of p53 and Ki-67 staining differs between SCC and VC. Squamous cell carcinoma shows positive p53 and Ki-67 staining for the full thickness of the epidermis, while VC has positive staining only in the lower third of the epidermis.5
Surgical resection is considered the first-line treatment of VC through excision or Mohs micrographic surgery. Radiation therapy is controversial due to the risk for anaplastic transformation. When surgery is not ideal due to the tumor size or location or the patient’s preference, other treatment modalities with reported success include intralesional interferon alfa; cryosurgery; topical imiquimod; and topical or systemic cytostatic agents such as bleomycin, 5-fluorouracil, cisplatin, or methotrexate.1,2
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
- Pattee SF, Bordeaux J, Mahalingam M, et al. Verrucous carcinoma of the scalp. J Am Acad Dermatol. 2006;56:506-508.
- Nikkels AF, Thirion L, Quatresooz P, et al. Photodynamic therapy for cutaneous verrucous carcinoma. J Am Acad Dermatol. 2007;57:516-519.
- Ho J, Diven DG, Butler PJ, et al. An ulcerating verrucous plaque on the foot. Arch Dermatol. 2000;136:547-552.
- Sonalika WG, Anand T. Oral verrucous carcinoma: a retrospective analysis for clinicopathologic features. J Cancer Res Ther. 2016;12:142-145.
- Dubina M, Goldenberg G. Viral-associated nonmelanoma skin cancers: a review. Am J Dermatopathol. 2009;31:561-573.
- Geusau A, Heinz-Peer G, Volc-Platzer B, et al. Regression of deeply infiltrating giant condyloma (Buschke-Lowenstein tumor) following long-term intralesional interferon alpha therapy. Arch Dermatol. 2000;136:707-710.
- Ansai S, Kimura T, Hayashi M. Fatal genital verrucous carcinoma. Am J Dermatopathol. 2007;29:68-71.
Pembrolizumab extends Merkel cell PFS, OS
Patients with the aggressive skin cancer Merkel cell carcinoma who were treated with the immune checkpoint inhibitor pembrolizumab (Keytruda) in the first line had higher complete response rates, better progression-free survival, and longer overall survival than historical controls treated with cytotoxic chemotherapy.
Among 50 adults with advanced Merkel cell carcinoma (MCC) with no prior systemic therapy who received pembrolizumab 2 mg/kg every 3 weeks for up to 2 years in a phase 2 clinical trial (NCT02267603), 24% had a complete response and 32% a partial response, for an overall response rate of 56%.
The 24-month overall survival rate was 68.7%, with median overall survival not reached after a median follow-up time of 14.9 months. In contrast, a retrospective study of 67 patients with MCC treated with first-line chemotherapy showed an ORR of 29.4%, a median OS of 10.5 months, and a 24-month OS of 24.5% (Future Oncol. 2017 Aug;13(19):1699-1710).
Similarly, a second retrospective study showed that, among 62 patients treated with first-line chemotherapy, the ORR was 55%, median OS was 9.5 months, and 24-month OS was 20% (Cancer Med. 2016 Sep;5(9):2294-2301), reported Paul Nghiem, MD, PhD, from the University of Washington and Fred Hutchinson Cancer Research Center in Seattle, and his colleagues.
The rationale for using a checkpoint inhibitor for advanced MCC is that “[m]ultiple lines of evidence support the notion that MCC is an immunogenic cancer, including the fact that MCC incidence is greater than 10-fold higher in chronically immunosuppressed persons,” they wrote in the Journal of Clinical Oncology.
The current National Comprehensive Cancer Network guideline on Merkel cell carcinoma recommends the use of the programmed death–1/programmed death–ligand 1 (PD-1/PD-L1) inhibitors pembrolizumab, avelumab (Bavencio), or nivolumab (Opdivo) as preferred first-line systemic therapy for patients with disseminated disease, Dr. Nghiem and his colleagues noted.
In the current report, they presented data on the longest follow-up to date of patients with advance MCC who received a PD-1 inhibitor in the first line.
In the multicenter, phase 2 trial, 50 patients with a median age of 70.5 years were treated. Of this group, 64% had tumors positive for the Merkel cell polyomavirus and 49% had PD-L1 expression on tumor cells.
Of the 50 total patients, 28 had an objective response according to Response Evaluation Criteria in Solid Tumors version 1.1, including 12 with a complete response and 16 with a partial response. A total of 5 patients had stable disease, 16 had progressive disease, and 1 patient died before the first on-treatment scan for assessment.
After a median follow-up of 4.9 months, the 24-month progression-free survival rate (PFS) was 48.3% months, with a median PFS of 16.8 months.
As noted before, the 24-month OS rate was 68.7% and the median OS had not been reached at the time of the analysis.
There were no significant differences in PFS or OS between patients with tumors positive or negative for the Merkel polyomavirus, and there was a nonsignificant trend toward better PFS and OS for patients whose tumors had PD-L1 expression greater than 1%.
In all, 48 of the 50 patients had a treatment-related adverse event of any kind, and 14 had grade 3 or greater events. Treatment-related events led to discontinuation of pembrolizumab for seven patients, and one patient, a 73-year-old man with metastatic MCC and atrial fibrillation, developed pericardial and pleural effusions 1 day after receiving a single pembrolizumab infusion. The patient died 10 days after receiving pembrolizumab, and his death was deemed to be related to the drug.
The investigators noted that the drug’s efficacy in patients with both polyomavirus- and UV-induced subtypes of MCC “provides compelling evidence that both the quality and quantity of tumor antigens are important factors driving antitumor immunity and tumor rejection.”
The study was supported by grants from the National Cancer Institute, the Merkel cell carcinoma (MCC) patient gift fund at University of Washington, the Kelsey Dickson MCC Challenge Grant from the Prostate Cancer Foundation, and Merck, which provided pembrolizumab and partial funding. Dr. Nghiem reported receiving honoraria, travel expenses, and a consulting or advisory role from/for Merck and others. Multiple coauthors reported similar relations with Merck and/or other companies.
SOURCE: Nghiem P et al. J Clin Oncol. 2019 Feb 6. doi: 10.1200/JCO.18.01896.
Patients with the aggressive skin cancer Merkel cell carcinoma who were treated with the immune checkpoint inhibitor pembrolizumab (Keytruda) in the first line had higher complete response rates, better progression-free survival, and longer overall survival than historical controls treated with cytotoxic chemotherapy.
Among 50 adults with advanced Merkel cell carcinoma (MCC) with no prior systemic therapy who received pembrolizumab 2 mg/kg every 3 weeks for up to 2 years in a phase 2 clinical trial (NCT02267603), 24% had a complete response and 32% a partial response, for an overall response rate of 56%.
The 24-month overall survival rate was 68.7%, with median overall survival not reached after a median follow-up time of 14.9 months. In contrast, a retrospective study of 67 patients with MCC treated with first-line chemotherapy showed an ORR of 29.4%, a median OS of 10.5 months, and a 24-month OS of 24.5% (Future Oncol. 2017 Aug;13(19):1699-1710).
Similarly, a second retrospective study showed that, among 62 patients treated with first-line chemotherapy, the ORR was 55%, median OS was 9.5 months, and 24-month OS was 20% (Cancer Med. 2016 Sep;5(9):2294-2301), reported Paul Nghiem, MD, PhD, from the University of Washington and Fred Hutchinson Cancer Research Center in Seattle, and his colleagues.
The rationale for using a checkpoint inhibitor for advanced MCC is that “[m]ultiple lines of evidence support the notion that MCC is an immunogenic cancer, including the fact that MCC incidence is greater than 10-fold higher in chronically immunosuppressed persons,” they wrote in the Journal of Clinical Oncology.
The current National Comprehensive Cancer Network guideline on Merkel cell carcinoma recommends the use of the programmed death–1/programmed death–ligand 1 (PD-1/PD-L1) inhibitors pembrolizumab, avelumab (Bavencio), or nivolumab (Opdivo) as preferred first-line systemic therapy for patients with disseminated disease, Dr. Nghiem and his colleagues noted.
In the current report, they presented data on the longest follow-up to date of patients with advance MCC who received a PD-1 inhibitor in the first line.
In the multicenter, phase 2 trial, 50 patients with a median age of 70.5 years were treated. Of this group, 64% had tumors positive for the Merkel cell polyomavirus and 49% had PD-L1 expression on tumor cells.
Of the 50 total patients, 28 had an objective response according to Response Evaluation Criteria in Solid Tumors version 1.1, including 12 with a complete response and 16 with a partial response. A total of 5 patients had stable disease, 16 had progressive disease, and 1 patient died before the first on-treatment scan for assessment.
After a median follow-up of 4.9 months, the 24-month progression-free survival rate (PFS) was 48.3% months, with a median PFS of 16.8 months.
As noted before, the 24-month OS rate was 68.7% and the median OS had not been reached at the time of the analysis.
There were no significant differences in PFS or OS between patients with tumors positive or negative for the Merkel polyomavirus, and there was a nonsignificant trend toward better PFS and OS for patients whose tumors had PD-L1 expression greater than 1%.
In all, 48 of the 50 patients had a treatment-related adverse event of any kind, and 14 had grade 3 or greater events. Treatment-related events led to discontinuation of pembrolizumab for seven patients, and one patient, a 73-year-old man with metastatic MCC and atrial fibrillation, developed pericardial and pleural effusions 1 day after receiving a single pembrolizumab infusion. The patient died 10 days after receiving pembrolizumab, and his death was deemed to be related to the drug.
The investigators noted that the drug’s efficacy in patients with both polyomavirus- and UV-induced subtypes of MCC “provides compelling evidence that both the quality and quantity of tumor antigens are important factors driving antitumor immunity and tumor rejection.”
The study was supported by grants from the National Cancer Institute, the Merkel cell carcinoma (MCC) patient gift fund at University of Washington, the Kelsey Dickson MCC Challenge Grant from the Prostate Cancer Foundation, and Merck, which provided pembrolizumab and partial funding. Dr. Nghiem reported receiving honoraria, travel expenses, and a consulting or advisory role from/for Merck and others. Multiple coauthors reported similar relations with Merck and/or other companies.
SOURCE: Nghiem P et al. J Clin Oncol. 2019 Feb 6. doi: 10.1200/JCO.18.01896.
Patients with the aggressive skin cancer Merkel cell carcinoma who were treated with the immune checkpoint inhibitor pembrolizumab (Keytruda) in the first line had higher complete response rates, better progression-free survival, and longer overall survival than historical controls treated with cytotoxic chemotherapy.
Among 50 adults with advanced Merkel cell carcinoma (MCC) with no prior systemic therapy who received pembrolizumab 2 mg/kg every 3 weeks for up to 2 years in a phase 2 clinical trial (NCT02267603), 24% had a complete response and 32% a partial response, for an overall response rate of 56%.
The 24-month overall survival rate was 68.7%, with median overall survival not reached after a median follow-up time of 14.9 months. In contrast, a retrospective study of 67 patients with MCC treated with first-line chemotherapy showed an ORR of 29.4%, a median OS of 10.5 months, and a 24-month OS of 24.5% (Future Oncol. 2017 Aug;13(19):1699-1710).
Similarly, a second retrospective study showed that, among 62 patients treated with first-line chemotherapy, the ORR was 55%, median OS was 9.5 months, and 24-month OS was 20% (Cancer Med. 2016 Sep;5(9):2294-2301), reported Paul Nghiem, MD, PhD, from the University of Washington and Fred Hutchinson Cancer Research Center in Seattle, and his colleagues.
The rationale for using a checkpoint inhibitor for advanced MCC is that “[m]ultiple lines of evidence support the notion that MCC is an immunogenic cancer, including the fact that MCC incidence is greater than 10-fold higher in chronically immunosuppressed persons,” they wrote in the Journal of Clinical Oncology.
The current National Comprehensive Cancer Network guideline on Merkel cell carcinoma recommends the use of the programmed death–1/programmed death–ligand 1 (PD-1/PD-L1) inhibitors pembrolizumab, avelumab (Bavencio), or nivolumab (Opdivo) as preferred first-line systemic therapy for patients with disseminated disease, Dr. Nghiem and his colleagues noted.
In the current report, they presented data on the longest follow-up to date of patients with advance MCC who received a PD-1 inhibitor in the first line.
In the multicenter, phase 2 trial, 50 patients with a median age of 70.5 years were treated. Of this group, 64% had tumors positive for the Merkel cell polyomavirus and 49% had PD-L1 expression on tumor cells.
Of the 50 total patients, 28 had an objective response according to Response Evaluation Criteria in Solid Tumors version 1.1, including 12 with a complete response and 16 with a partial response. A total of 5 patients had stable disease, 16 had progressive disease, and 1 patient died before the first on-treatment scan for assessment.
After a median follow-up of 4.9 months, the 24-month progression-free survival rate (PFS) was 48.3% months, with a median PFS of 16.8 months.
As noted before, the 24-month OS rate was 68.7% and the median OS had not been reached at the time of the analysis.
There were no significant differences in PFS or OS between patients with tumors positive or negative for the Merkel polyomavirus, and there was a nonsignificant trend toward better PFS and OS for patients whose tumors had PD-L1 expression greater than 1%.
In all, 48 of the 50 patients had a treatment-related adverse event of any kind, and 14 had grade 3 or greater events. Treatment-related events led to discontinuation of pembrolizumab for seven patients, and one patient, a 73-year-old man with metastatic MCC and atrial fibrillation, developed pericardial and pleural effusions 1 day after receiving a single pembrolizumab infusion. The patient died 10 days after receiving pembrolizumab, and his death was deemed to be related to the drug.
The investigators noted that the drug’s efficacy in patients with both polyomavirus- and UV-induced subtypes of MCC “provides compelling evidence that both the quality and quantity of tumor antigens are important factors driving antitumor immunity and tumor rejection.”
The study was supported by grants from the National Cancer Institute, the Merkel cell carcinoma (MCC) patient gift fund at University of Washington, the Kelsey Dickson MCC Challenge Grant from the Prostate Cancer Foundation, and Merck, which provided pembrolizumab and partial funding. Dr. Nghiem reported receiving honoraria, travel expenses, and a consulting or advisory role from/for Merck and others. Multiple coauthors reported similar relations with Merck and/or other companies.
SOURCE: Nghiem P et al. J Clin Oncol. 2019 Feb 6. doi: 10.1200/JCO.18.01896.
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point: A programmed death–1/programmed death–ligand 1 inhibitor is preferred in the first line for disseminated Merkel cell carcinoma.
Major finding: Overall survival after 24 months was 68.7%, with the median overall survival not reached.
Study details: A follow-up of a phase 2, open-label trial in 50 patients with advanced Merkel cell carcinoma.
Disclosures: The study was supported by supported by grants from the National Cancer Institute, the Merkel cell carcinoma (MCC) patient gift fund at University of Washington, the Kelsey Dickson MCC Challenge Grant from the Prostate Cancer Foundation, and Merck, which provided pembrolizumab and partial funding. Dr. Nghiem reported receiving honoraria, travel expenses, and a consulting or advisory role from/for Merck and others. Multiple coauthors reported similar relations with Merck and/or other companies.
Source: Nghiem P et al. J Clin Oncol. 2019 Feb 6. doi: 10.1200/JCO.18.01896.
BCL expression intensity key in distinguishing FL lesions
Intensity of BCL2 expression, and to a lesser extent expression of t(14;18), may help distinguish common and indolent cutaneous lymphomas from poorer-prognosis cutaneous lesions secondary to systemic follicular lymphomas, results of a recent investigation show.
Strong expression of BCL2 was almost always associated with secondary cutaneous follicular lymphoma (SCFL), and infrequently associated with primary cutaneous follicular center-cell lymphoma (PCFCL), according to the study results.
The translocation t(14;18) was likewise linked to secondary lesions, occurring less frequently in PCFCL in the study, reported recently in the Journal of Cutaneous Pathology.
“BCL2 expression intensity is the single most valuable clue in differentiating PCFCL from SCFL cases on histopathological grounds,” said Ramon M. Pujol, MD, PhD, of Hospital del Mar, Barcelona, Spain, and colleagues.
One of the main cutaneous B-cell lymphoma subtypes, PCFCL is marked by frequent relapses, but little incidence of systemic spread, meaning that conservative, skin-based therapies are usually warranted. By contrast, patients with SCFLs have a poor prognosis and may require systemic therapy, the investigators noted in their report.
Previous investigations have yielded conflicting results on the role of BCL2 expression, CD10 expression, and presence of t(14;18) translocation in distinguishing PCFCL from SCFL.
While early studies suggested most PCFCLs were negative for these markers, some recent reports suggested BCL positivity in PCFCLs is as high as 86%, the investigators said.
Accordingly, Dr. Pujol and colleagues evaluated clinicopathologic and genetic features in a large series of patients, including 59 with PCFCL and 22 with SCFL.
Significant BCL2 expression was seen in 69% of PCFCLs and in 100% of SCFLs (P = .003) in this patient series; however, when looking at BCL2 intensity, investigators found strong expression almost exclusively in SCFL. Strong expression was seen in 46% of those patients with secondary lymphomas, versus just 4%, or two cases, in the PCFCL group (P = .001).
The t(14;18) translocation was seen in 64% of SCFLs and only 9.1% of PCFCLs (P = .001).
Similar to what was seen for BCL2, expression of CD10 was observed in 66% of PCFCLs and 91% of SCFLs, and again, intensity differences mattered. Strong CD10 expression was seen in 62% of secondary lymphomas and 16% of PCFCLs (P = .01). But the high number of positive PCFCLs made this marker less useful than BCL2, the investigators said.
“We believe that differences in BCL2 and CD10 expression between our results and older previous studies could reflect the improvement of antigen retrieval laboratory techniques,” they said.
The investigators did not report disclosures related to the research.
SOURCE: Servitje O et al. J Cutan Pathol. 2019;46:182-9.
Intensity of BCL2 expression, and to a lesser extent expression of t(14;18), may help distinguish common and indolent cutaneous lymphomas from poorer-prognosis cutaneous lesions secondary to systemic follicular lymphomas, results of a recent investigation show.
Strong expression of BCL2 was almost always associated with secondary cutaneous follicular lymphoma (SCFL), and infrequently associated with primary cutaneous follicular center-cell lymphoma (PCFCL), according to the study results.
The translocation t(14;18) was likewise linked to secondary lesions, occurring less frequently in PCFCL in the study, reported recently in the Journal of Cutaneous Pathology.
“BCL2 expression intensity is the single most valuable clue in differentiating PCFCL from SCFL cases on histopathological grounds,” said Ramon M. Pujol, MD, PhD, of Hospital del Mar, Barcelona, Spain, and colleagues.
One of the main cutaneous B-cell lymphoma subtypes, PCFCL is marked by frequent relapses, but little incidence of systemic spread, meaning that conservative, skin-based therapies are usually warranted. By contrast, patients with SCFLs have a poor prognosis and may require systemic therapy, the investigators noted in their report.
Previous investigations have yielded conflicting results on the role of BCL2 expression, CD10 expression, and presence of t(14;18) translocation in distinguishing PCFCL from SCFL.
While early studies suggested most PCFCLs were negative for these markers, some recent reports suggested BCL positivity in PCFCLs is as high as 86%, the investigators said.
Accordingly, Dr. Pujol and colleagues evaluated clinicopathologic and genetic features in a large series of patients, including 59 with PCFCL and 22 with SCFL.
Significant BCL2 expression was seen in 69% of PCFCLs and in 100% of SCFLs (P = .003) in this patient series; however, when looking at BCL2 intensity, investigators found strong expression almost exclusively in SCFL. Strong expression was seen in 46% of those patients with secondary lymphomas, versus just 4%, or two cases, in the PCFCL group (P = .001).
The t(14;18) translocation was seen in 64% of SCFLs and only 9.1% of PCFCLs (P = .001).
Similar to what was seen for BCL2, expression of CD10 was observed in 66% of PCFCLs and 91% of SCFLs, and again, intensity differences mattered. Strong CD10 expression was seen in 62% of secondary lymphomas and 16% of PCFCLs (P = .01). But the high number of positive PCFCLs made this marker less useful than BCL2, the investigators said.
“We believe that differences in BCL2 and CD10 expression between our results and older previous studies could reflect the improvement of antigen retrieval laboratory techniques,” they said.
The investigators did not report disclosures related to the research.
SOURCE: Servitje O et al. J Cutan Pathol. 2019;46:182-9.
Intensity of BCL2 expression, and to a lesser extent expression of t(14;18), may help distinguish common and indolent cutaneous lymphomas from poorer-prognosis cutaneous lesions secondary to systemic follicular lymphomas, results of a recent investigation show.
Strong expression of BCL2 was almost always associated with secondary cutaneous follicular lymphoma (SCFL), and infrequently associated with primary cutaneous follicular center-cell lymphoma (PCFCL), according to the study results.
The translocation t(14;18) was likewise linked to secondary lesions, occurring less frequently in PCFCL in the study, reported recently in the Journal of Cutaneous Pathology.
“BCL2 expression intensity is the single most valuable clue in differentiating PCFCL from SCFL cases on histopathological grounds,” said Ramon M. Pujol, MD, PhD, of Hospital del Mar, Barcelona, Spain, and colleagues.
One of the main cutaneous B-cell lymphoma subtypes, PCFCL is marked by frequent relapses, but little incidence of systemic spread, meaning that conservative, skin-based therapies are usually warranted. By contrast, patients with SCFLs have a poor prognosis and may require systemic therapy, the investigators noted in their report.
Previous investigations have yielded conflicting results on the role of BCL2 expression, CD10 expression, and presence of t(14;18) translocation in distinguishing PCFCL from SCFL.
While early studies suggested most PCFCLs were negative for these markers, some recent reports suggested BCL positivity in PCFCLs is as high as 86%, the investigators said.
Accordingly, Dr. Pujol and colleagues evaluated clinicopathologic and genetic features in a large series of patients, including 59 with PCFCL and 22 with SCFL.
Significant BCL2 expression was seen in 69% of PCFCLs and in 100% of SCFLs (P = .003) in this patient series; however, when looking at BCL2 intensity, investigators found strong expression almost exclusively in SCFL. Strong expression was seen in 46% of those patients with secondary lymphomas, versus just 4%, or two cases, in the PCFCL group (P = .001).
The t(14;18) translocation was seen in 64% of SCFLs and only 9.1% of PCFCLs (P = .001).
Similar to what was seen for BCL2, expression of CD10 was observed in 66% of PCFCLs and 91% of SCFLs, and again, intensity differences mattered. Strong CD10 expression was seen in 62% of secondary lymphomas and 16% of PCFCLs (P = .01). But the high number of positive PCFCLs made this marker less useful than BCL2, the investigators said.
“We believe that differences in BCL2 and CD10 expression between our results and older previous studies could reflect the improvement of antigen retrieval laboratory techniques,” they said.
The investigators did not report disclosures related to the research.
SOURCE: Servitje O et al. J Cutan Pathol. 2019;46:182-9.
FROM THE JOURNAL OF CUTANEOUS PATHOLOGY
Key clinical point:
Major finding: Strong BCL2 expression was seen in 46% of secondary lymphomas, versus just 4% of primary cutaneous follicular center-cell lymphomas (P = .001).
Study details: A comparative study evaluating clinicopathologic and genetic features in a series of patients, including 59 with PCFCL and 22 with SCFL.
Disclosures: Investigators did not report disclosures related to the research.
Source: Servitje O et al. J Cutan Pathol. 2019;46:182-9.
Fine-tune staging for better SCC risk stratification
ORLANDO – When caring for individuals with sun-damaged skin, dermatologists need comfort with the full spectrum of photo-related skin disease. From assessment and treatment of actinic keratoses (AKs) and field cancerization, to long-term follow-up of cutaneous squamous cell carcinomas (SCCs), appropriate treatment and staging can improve patient quality of life and reduce health care costs, Vishal Patel, MD, said at the Orlando Dermatology Aesthetic and Clinical Conference.

said Dr. Patel, director of cutaneous oncology at George Washington University Cancer Center, Washington. On the other hand, he added, “field disease can be a marker for invasive squamous cell carcinoma risk, and it requires field treatment.” Treatment that reduces field disease is primary prevention because it decreases the formation of invasive SCC, he noted.
“But this level of disease – AKs and SCC in situ – doesn’t kill people,” he emphasized. “I want to leave you with an ability to stage this disease,” said Dr. Patel, noting that SCC mortality may eventually surpass melanoma mortality as deaths from the latter decline and numbers of older Americans with high ultraviolet light exposure and other risk factors climb.
While the majority of AKs regress within 5 years, he looks at the total burden of AKs as a marker for field cancerization “because having less than five in situ or actinic lesions puts you at less than a 1% risk of squamous cell carcinoma formation. Having more than 20 increases that risk 20-fold to 20%,” he said. “That’s the way we need to start thinking about this: Is this a disease – or a symptom?”
Rather than thinking of each AK or SCC in situ as a separate disease event, “the disease we need to be focusing on and treating is field cancerization,” he continued. Within this context, “we should not be thinking that … we need to be aggressive in our management,” which is what results in high costs.
“The reality is that this is a big quality of life issue for our patients. So what do we do?” Field treatment is appropriate for field disease, he said. Dr. Patel said that at GW only field treatment is used; destructive treatment for AKs and SCC in situ is not used. In the absence of patient and lesion characteristics that elevate risk,“surgery is really not the standard of care for in situ lesions for us,” he commented.
“We start by discerning the field disease from the invasive disease” with an initial round of field treatment and, if needed, adjunctive oral chemoprophylaxis. “We lather, rinse, and repeat” the field therapy, continuously if needed, Dr. Patel said.
“We like to do that because we can then identify those specific lesions we want to go after. No cryosurgery, no destructive therapy, because we run the risk of burying those tumors under the scar. They may recur and make it more difficult to accurately stage them in the future,” he noted.
“I like to be more sophisticated in thinking about our approach to the outcomes of these individual lesions,” he said. When it comes to excising lesions that have been biopsied and show invasive SCC, “disc excision may be a more cost-effective way to treat many low-risk SCCs,” he noted. In any case, “removal with clear surgical margins is key.”
Primary tumors with such low-risk attributes as diameter under a centimeter and thickness under 2 mm; well-defined borders; location on the trunk, neck, or extremities; well-differentiated histology; and lack of perineural invasion can all be considered for a disc technique, especially if the patient is immunocompetent without background chronic inflammation or a history of prior radiation therapy.
Staging SCCs, said Dr. Patel, is where things really get tricky. Older staging systems for SCC “led us to overtreat nonaggressive disease and undertreat aggressive disease. I think we have the responsibility to lead the charge to having a more sophisticated approach.” For example, patients whose tumors were staged T2 in the American Joint Commission on Cancer (AJCC) 7 classification system were most likely to have poor outcomes – in part because so few tumors were staged higher – which meant AJCC 7 didn’t provide adequate differentiation for useful risk prognostication.
A group of researchers at the Brigham and Women’s Hospital (BWH), Boston, “came up with a better system to better differentiate those T2 tumors into a high-risk and a low-risk subtype,” according to Dr. Patel.
With use of validated risk factors, the investigators applied a long list of risk factors to 2,000 tumors to see which risk factors, taken individually, were really contributing to poor outcomes. Eventually, four risk factors that made the most difference were identified: size greater than 2 cm, poor tumor differentiation, perineural invasion greater than 0.1 mm in diameter, and tumor invasion beyond subcutaneous fat. “I really want to highlight the size portion of those risk factors,” said Dr. Patel. “Something I’d like you to do in your clinical practice is to measure and document the size of the lesion. … That really, clearly helps” with risk prognostication.
These four factors were then used to break out a T2a stage for tumors with one risk factor and a T2b stage for tumors with two or three risk factors. Tumors with no risk factors are stage T1, and those with all four risk factors are stage T3. In situ SCC is T0.
Applying this new staging system to a 2,000-patient cohort with SCC yielded clear separation in outcomes including recurrence, nodal metastasis, disease-specific death, and overall survival between patients with the T2a and T2b tumors (P less than .001 for all; J Clin Oncol. 2014 Feb 1;32[4]:327-34).
While AJCC 8 is “significantly better” than AJCC 7 in its incorporation of meaningful risk factors into the SCC staging system, “it still underperforms in comparison” with the BWH staging system using the 2000 patient cohort, he said. Recent work has shown the BWH classification system to have superior specificity and positive predictive value in detecting nodal metastasis and disease-specific death in higher-grade tumors. But both BWH and AJCC 8 need further refinement.
“So what are the staging pearls to take home?” Dr. Patel asked. “First, utilize a staging system.” “Staging of SCC utilizing should be done routinely. Most data seems to suggest that the BWH system appears to outperform AJCC 8, and it is what we currently use routinely at GW,” he said.
Patients who are T1 by BWH criteria, with no risk factors, are at low or even no risk, he noted. He pointed out that of the nearly 1,400 patients who met T1 criteria, there were just eight local recurrences, one nodal metastasis, and no distant metastases or deaths. Knowing this should guide physicians on a treatment path that will reduce costs and provide patients with peace of mind, he said.
In the BWH schema, T2a patients fared almost as well, with a 2% risk of nodal metastasis and an overall 1% risk of disease-specific death. “T2a disease is low risk, in my mind. Most of these patients will go on to do well,” he said.
By contrast, “there may be a number of tumors that you are missing” that are candidates for close follow-up if the BWH criteria are not being used, said Dr. Patel. These are the T2b tumors. “For those patients, we want to aggressively follow them and think about a more aggressive management plan.”
The bottom line is that BWH T2b and T3 tumors are both high risk, and management needs to acknowledge this, he said. The current protocol in our cutaneous oncology program includes using routine radiologic nodal staging in patients with BWH stage 2b and above SCCs and considering sentinel lymph node biopsy for certain individuals.
For patients with BWH T2b and T3 tumors, dermatologists should give consideration to tertiary care or cancer center referrals so they have access to the full spectrum of diagnostic and therapeutic modalities and the opportunity to participate in clinical trials, Dr. Patel said.
Dr. Patel reported that he is a speaker for Regeneron/Sanofi and a cofounder of the Skin Cancer Outcomes (SCOUT) consortium.
This article was updated 2/9/2019
ORLANDO – When caring for individuals with sun-damaged skin, dermatologists need comfort with the full spectrum of photo-related skin disease. From assessment and treatment of actinic keratoses (AKs) and field cancerization, to long-term follow-up of cutaneous squamous cell carcinomas (SCCs), appropriate treatment and staging can improve patient quality of life and reduce health care costs, Vishal Patel, MD, said at the Orlando Dermatology Aesthetic and Clinical Conference.
said Dr. Patel, director of cutaneous oncology at George Washington University Cancer Center, Washington. On the other hand, he added, “field disease can be a marker for invasive squamous cell carcinoma risk, and it requires field treatment.” Treatment that reduces field disease is primary prevention because it decreases the formation of invasive SCC, he noted.
“But this level of disease – AKs and SCC in situ – doesn’t kill people,” he emphasized. “I want to leave you with an ability to stage this disease,” said Dr. Patel, noting that SCC mortality may eventually surpass melanoma mortality as deaths from the latter decline and numbers of older Americans with high ultraviolet light exposure and other risk factors climb.
While the majority of AKs regress within 5 years, he looks at the total burden of AKs as a marker for field cancerization “because having less than five in situ or actinic lesions puts you at less than a 1% risk of squamous cell carcinoma formation. Having more than 20 increases that risk 20-fold to 20%,” he said. “That’s the way we need to start thinking about this: Is this a disease – or a symptom?”
Rather than thinking of each AK or SCC in situ as a separate disease event, “the disease we need to be focusing on and treating is field cancerization,” he continued. Within this context, “we should not be thinking that … we need to be aggressive in our management,” which is what results in high costs.
“The reality is that this is a big quality of life issue for our patients. So what do we do?” Field treatment is appropriate for field disease, he said. Dr. Patel said that at GW only field treatment is used; destructive treatment for AKs and SCC in situ is not used. In the absence of patient and lesion characteristics that elevate risk,“surgery is really not the standard of care for in situ lesions for us,” he commented.
“We start by discerning the field disease from the invasive disease” with an initial round of field treatment and, if needed, adjunctive oral chemoprophylaxis. “We lather, rinse, and repeat” the field therapy, continuously if needed, Dr. Patel said.
“We like to do that because we can then identify those specific lesions we want to go after. No cryosurgery, no destructive therapy, because we run the risk of burying those tumors under the scar. They may recur and make it more difficult to accurately stage them in the future,” he noted.
“I like to be more sophisticated in thinking about our approach to the outcomes of these individual lesions,” he said. When it comes to excising lesions that have been biopsied and show invasive SCC, “disc excision may be a more cost-effective way to treat many low-risk SCCs,” he noted. In any case, “removal with clear surgical margins is key.”
Primary tumors with such low-risk attributes as diameter under a centimeter and thickness under 2 mm; well-defined borders; location on the trunk, neck, or extremities; well-differentiated histology; and lack of perineural invasion can all be considered for a disc technique, especially if the patient is immunocompetent without background chronic inflammation or a history of prior radiation therapy.
Staging SCCs, said Dr. Patel, is where things really get tricky. Older staging systems for SCC “led us to overtreat nonaggressive disease and undertreat aggressive disease. I think we have the responsibility to lead the charge to having a more sophisticated approach.” For example, patients whose tumors were staged T2 in the American Joint Commission on Cancer (AJCC) 7 classification system were most likely to have poor outcomes – in part because so few tumors were staged higher – which meant AJCC 7 didn’t provide adequate differentiation for useful risk prognostication.
A group of researchers at the Brigham and Women’s Hospital (BWH), Boston, “came up with a better system to better differentiate those T2 tumors into a high-risk and a low-risk subtype,” according to Dr. Patel.
With use of validated risk factors, the investigators applied a long list of risk factors to 2,000 tumors to see which risk factors, taken individually, were really contributing to poor outcomes. Eventually, four risk factors that made the most difference were identified: size greater than 2 cm, poor tumor differentiation, perineural invasion greater than 0.1 mm in diameter, and tumor invasion beyond subcutaneous fat. “I really want to highlight the size portion of those risk factors,” said Dr. Patel. “Something I’d like you to do in your clinical practice is to measure and document the size of the lesion. … That really, clearly helps” with risk prognostication.
These four factors were then used to break out a T2a stage for tumors with one risk factor and a T2b stage for tumors with two or three risk factors. Tumors with no risk factors are stage T1, and those with all four risk factors are stage T3. In situ SCC is T0.
Applying this new staging system to a 2,000-patient cohort with SCC yielded clear separation in outcomes including recurrence, nodal metastasis, disease-specific death, and overall survival between patients with the T2a and T2b tumors (P less than .001 for all; J Clin Oncol. 2014 Feb 1;32[4]:327-34).
While AJCC 8 is “significantly better” than AJCC 7 in its incorporation of meaningful risk factors into the SCC staging system, “it still underperforms in comparison” with the BWH staging system using the 2000 patient cohort, he said. Recent work has shown the BWH classification system to have superior specificity and positive predictive value in detecting nodal metastasis and disease-specific death in higher-grade tumors. But both BWH and AJCC 8 need further refinement.
“So what are the staging pearls to take home?” Dr. Patel asked. “First, utilize a staging system.” “Staging of SCC utilizing should be done routinely. Most data seems to suggest that the BWH system appears to outperform AJCC 8, and it is what we currently use routinely at GW,” he said.
Patients who are T1 by BWH criteria, with no risk factors, are at low or even no risk, he noted. He pointed out that of the nearly 1,400 patients who met T1 criteria, there were just eight local recurrences, one nodal metastasis, and no distant metastases or deaths. Knowing this should guide physicians on a treatment path that will reduce costs and provide patients with peace of mind, he said.
In the BWH schema, T2a patients fared almost as well, with a 2% risk of nodal metastasis and an overall 1% risk of disease-specific death. “T2a disease is low risk, in my mind. Most of these patients will go on to do well,” he said.
By contrast, “there may be a number of tumors that you are missing” that are candidates for close follow-up if the BWH criteria are not being used, said Dr. Patel. These are the T2b tumors. “For those patients, we want to aggressively follow them and think about a more aggressive management plan.”
The bottom line is that BWH T2b and T3 tumors are both high risk, and management needs to acknowledge this, he said. The current protocol in our cutaneous oncology program includes using routine radiologic nodal staging in patients with BWH stage 2b and above SCCs and considering sentinel lymph node biopsy for certain individuals.
For patients with BWH T2b and T3 tumors, dermatologists should give consideration to tertiary care or cancer center referrals so they have access to the full spectrum of diagnostic and therapeutic modalities and the opportunity to participate in clinical trials, Dr. Patel said.
Dr. Patel reported that he is a speaker for Regeneron/Sanofi and a cofounder of the Skin Cancer Outcomes (SCOUT) consortium.
This article was updated 2/9/2019
ORLANDO – When caring for individuals with sun-damaged skin, dermatologists need comfort with the full spectrum of photo-related skin disease. From assessment and treatment of actinic keratoses (AKs) and field cancerization, to long-term follow-up of cutaneous squamous cell carcinomas (SCCs), appropriate treatment and staging can improve patient quality of life and reduce health care costs, Vishal Patel, MD, said at the Orlando Dermatology Aesthetic and Clinical Conference.
said Dr. Patel, director of cutaneous oncology at George Washington University Cancer Center, Washington. On the other hand, he added, “field disease can be a marker for invasive squamous cell carcinoma risk, and it requires field treatment.” Treatment that reduces field disease is primary prevention because it decreases the formation of invasive SCC, he noted.
“But this level of disease – AKs and SCC in situ – doesn’t kill people,” he emphasized. “I want to leave you with an ability to stage this disease,” said Dr. Patel, noting that SCC mortality may eventually surpass melanoma mortality as deaths from the latter decline and numbers of older Americans with high ultraviolet light exposure and other risk factors climb.
While the majority of AKs regress within 5 years, he looks at the total burden of AKs as a marker for field cancerization “because having less than five in situ or actinic lesions puts you at less than a 1% risk of squamous cell carcinoma formation. Having more than 20 increases that risk 20-fold to 20%,” he said. “That’s the way we need to start thinking about this: Is this a disease – or a symptom?”
Rather than thinking of each AK or SCC in situ as a separate disease event, “the disease we need to be focusing on and treating is field cancerization,” he continued. Within this context, “we should not be thinking that … we need to be aggressive in our management,” which is what results in high costs.
“The reality is that this is a big quality of life issue for our patients. So what do we do?” Field treatment is appropriate for field disease, he said. Dr. Patel said that at GW only field treatment is used; destructive treatment for AKs and SCC in situ is not used. In the absence of patient and lesion characteristics that elevate risk,“surgery is really not the standard of care for in situ lesions for us,” he commented.
“We start by discerning the field disease from the invasive disease” with an initial round of field treatment and, if needed, adjunctive oral chemoprophylaxis. “We lather, rinse, and repeat” the field therapy, continuously if needed, Dr. Patel said.
“We like to do that because we can then identify those specific lesions we want to go after. No cryosurgery, no destructive therapy, because we run the risk of burying those tumors under the scar. They may recur and make it more difficult to accurately stage them in the future,” he noted.
“I like to be more sophisticated in thinking about our approach to the outcomes of these individual lesions,” he said. When it comes to excising lesions that have been biopsied and show invasive SCC, “disc excision may be a more cost-effective way to treat many low-risk SCCs,” he noted. In any case, “removal with clear surgical margins is key.”
Primary tumors with such low-risk attributes as diameter under a centimeter and thickness under 2 mm; well-defined borders; location on the trunk, neck, or extremities; well-differentiated histology; and lack of perineural invasion can all be considered for a disc technique, especially if the patient is immunocompetent without background chronic inflammation or a history of prior radiation therapy.
Staging SCCs, said Dr. Patel, is where things really get tricky. Older staging systems for SCC “led us to overtreat nonaggressive disease and undertreat aggressive disease. I think we have the responsibility to lead the charge to having a more sophisticated approach.” For example, patients whose tumors were staged T2 in the American Joint Commission on Cancer (AJCC) 7 classification system were most likely to have poor outcomes – in part because so few tumors were staged higher – which meant AJCC 7 didn’t provide adequate differentiation for useful risk prognostication.
A group of researchers at the Brigham and Women’s Hospital (BWH), Boston, “came up with a better system to better differentiate those T2 tumors into a high-risk and a low-risk subtype,” according to Dr. Patel.
With use of validated risk factors, the investigators applied a long list of risk factors to 2,000 tumors to see which risk factors, taken individually, were really contributing to poor outcomes. Eventually, four risk factors that made the most difference were identified: size greater than 2 cm, poor tumor differentiation, perineural invasion greater than 0.1 mm in diameter, and tumor invasion beyond subcutaneous fat. “I really want to highlight the size portion of those risk factors,” said Dr. Patel. “Something I’d like you to do in your clinical practice is to measure and document the size of the lesion. … That really, clearly helps” with risk prognostication.
These four factors were then used to break out a T2a stage for tumors with one risk factor and a T2b stage for tumors with two or three risk factors. Tumors with no risk factors are stage T1, and those with all four risk factors are stage T3. In situ SCC is T0.
Applying this new staging system to a 2,000-patient cohort with SCC yielded clear separation in outcomes including recurrence, nodal metastasis, disease-specific death, and overall survival between patients with the T2a and T2b tumors (P less than .001 for all; J Clin Oncol. 2014 Feb 1;32[4]:327-34).
While AJCC 8 is “significantly better” than AJCC 7 in its incorporation of meaningful risk factors into the SCC staging system, “it still underperforms in comparison” with the BWH staging system using the 2000 patient cohort, he said. Recent work has shown the BWH classification system to have superior specificity and positive predictive value in detecting nodal metastasis and disease-specific death in higher-grade tumors. But both BWH and AJCC 8 need further refinement.
“So what are the staging pearls to take home?” Dr. Patel asked. “First, utilize a staging system.” “Staging of SCC utilizing should be done routinely. Most data seems to suggest that the BWH system appears to outperform AJCC 8, and it is what we currently use routinely at GW,” he said.
Patients who are T1 by BWH criteria, with no risk factors, are at low or even no risk, he noted. He pointed out that of the nearly 1,400 patients who met T1 criteria, there were just eight local recurrences, one nodal metastasis, and no distant metastases or deaths. Knowing this should guide physicians on a treatment path that will reduce costs and provide patients with peace of mind, he said.
In the BWH schema, T2a patients fared almost as well, with a 2% risk of nodal metastasis and an overall 1% risk of disease-specific death. “T2a disease is low risk, in my mind. Most of these patients will go on to do well,” he said.
By contrast, “there may be a number of tumors that you are missing” that are candidates for close follow-up if the BWH criteria are not being used, said Dr. Patel. These are the T2b tumors. “For those patients, we want to aggressively follow them and think about a more aggressive management plan.”
The bottom line is that BWH T2b and T3 tumors are both high risk, and management needs to acknowledge this, he said. The current protocol in our cutaneous oncology program includes using routine radiologic nodal staging in patients with BWH stage 2b and above SCCs and considering sentinel lymph node biopsy for certain individuals.
For patients with BWH T2b and T3 tumors, dermatologists should give consideration to tertiary care or cancer center referrals so they have access to the full spectrum of diagnostic and therapeutic modalities and the opportunity to participate in clinical trials, Dr. Patel said.
Dr. Patel reported that he is a speaker for Regeneron/Sanofi and a cofounder of the Skin Cancer Outcomes (SCOUT) consortium.
This article was updated 2/9/2019
EXPERT ANALYSIS FROM ODAC 2019
Increased risk of second cancers in mycosis fungoides
LA JOLLA, CALIF. – A retrospective study suggests patients with mycosis fungoides (MF) have an increased risk of developing hematologic and solid tumor malignancies.

Researchers found the risk of second malignancy was highest among MF patients aged 30 to 50 years and patients who had tumor stage or advanced stage MF.
The increased risk was present during the entire period after MF diagnosis, but it was greatest in the first 6 months after diagnosis and roughly a dozen years later.
Amrita Goyal, MD, of the University of Minnesota in Minneapolis, and her colleagues presented these findings at the annual T-cell Lymphoma Forum.
The researchers first assessed the risk of second malignancy in 172 MF patients treated at UMN from 2005 to 2017, comparing this cohort to a control group of 172 patients with seborrheic dermatitis.
Second malignancies occurred in 24 MF patients and three controls, which was a significant difference (P = .0045). The most common second malignancies among the MF patients were melanoma (n = 4), prostate cancer (n = 3), and renal cell carcinoma (n = 3).
Further analyses revealed that MF patients were more likely to develop a second malignancy if they had tumor stage disease (P = .0024) or stage IIB or higher disease (P = .03).
To corroborate and expand upon these results, Dr. Goyal and her colleagues analyzed data from the Surveillance, Epidemiology, and End Results (SEER) database on patients diagnosed with MF from 2000 to 2014.
Among the 6,196 MF patients in this cohort, there were 514 second cancers.
“We found that MF patients were, overall, 10 times more likely to develop a second malignancy [compared with the general population],” Dr. Goyal said.
Specifically, the standardized incidence ratio was 10.15 for all malignancies, 7.33 for solid tumors, and 41.72 for hematologic malignancies.
Standardized incidence ratios for individual malignancies were:
- 69.8 for Hodgkin lymphoma.
- 46.5 for non-Hodgkin lymphoma.
- 8.6 for leukemia.
- 7.2 for melanoma.
- 6.2 for lung cancer.
- 7.9 for female breast cancer.
- 5.2 for colon cancer.
- 4.1 for prostate cancer.
- 3.9 for renal cell carcinoma.
- 3.8 for pancreatic cancer.
- 3.6 for bladder cancer.
“We found there is an increased risk [of second malignancy] during the first 6 months after diagnosis of MF, likely related to patients being in contact with the health care system more,” Dr. Goyal said. “Over time, patients have about a 7- to 10-fold increased risk over baseline, until they reach about 12 or 13 years after diagnosis, at which point, there is an increase in risk.”
The researchers found the greatest risk of second malignancy was among patients aged 30 to 50 years, although there was an increased risk for all age groups.
“The reason we think patients are experiencing an increased risk of cancers is we believe this may be due to immune suppression secondary to the mycosis fungoides, although further studies need to be performed to determine if that’s accurate,” Dr. Goyal said.
To that end, she and her colleagues are planning gene expression studies in patients from the UMN cohort. The researchers plan to examine genes involved in the pathogenesis of second malignancies and MF progression in tissue samples from 36 MF patients, 12 who developed second malignancies and 24 who did not.
The current research was funded by the American Society of Hematology. Dr. Goyal reported having no relevant financial disclosures. The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – A retrospective study suggests patients with mycosis fungoides (MF) have an increased risk of developing hematologic and solid tumor malignancies.
Researchers found the risk of second malignancy was highest among MF patients aged 30 to 50 years and patients who had tumor stage or advanced stage MF.
The increased risk was present during the entire period after MF diagnosis, but it was greatest in the first 6 months after diagnosis and roughly a dozen years later.
Amrita Goyal, MD, of the University of Minnesota in Minneapolis, and her colleagues presented these findings at the annual T-cell Lymphoma Forum.
The researchers first assessed the risk of second malignancy in 172 MF patients treated at UMN from 2005 to 2017, comparing this cohort to a control group of 172 patients with seborrheic dermatitis.
Second malignancies occurred in 24 MF patients and three controls, which was a significant difference (P = .0045). The most common second malignancies among the MF patients were melanoma (n = 4), prostate cancer (n = 3), and renal cell carcinoma (n = 3).
Further analyses revealed that MF patients were more likely to develop a second malignancy if they had tumor stage disease (P = .0024) or stage IIB or higher disease (P = .03).
To corroborate and expand upon these results, Dr. Goyal and her colleagues analyzed data from the Surveillance, Epidemiology, and End Results (SEER) database on patients diagnosed with MF from 2000 to 2014.
Among the 6,196 MF patients in this cohort, there were 514 second cancers.
“We found that MF patients were, overall, 10 times more likely to develop a second malignancy [compared with the general population],” Dr. Goyal said.
Specifically, the standardized incidence ratio was 10.15 for all malignancies, 7.33 for solid tumors, and 41.72 for hematologic malignancies.
Standardized incidence ratios for individual malignancies were:
- 69.8 for Hodgkin lymphoma.
- 46.5 for non-Hodgkin lymphoma.
- 8.6 for leukemia.
- 7.2 for melanoma.
- 6.2 for lung cancer.
- 7.9 for female breast cancer.
- 5.2 for colon cancer.
- 4.1 for prostate cancer.
- 3.9 for renal cell carcinoma.
- 3.8 for pancreatic cancer.
- 3.6 for bladder cancer.
“We found there is an increased risk [of second malignancy] during the first 6 months after diagnosis of MF, likely related to patients being in contact with the health care system more,” Dr. Goyal said. “Over time, patients have about a 7- to 10-fold increased risk over baseline, until they reach about 12 or 13 years after diagnosis, at which point, there is an increase in risk.”
The researchers found the greatest risk of second malignancy was among patients aged 30 to 50 years, although there was an increased risk for all age groups.
“The reason we think patients are experiencing an increased risk of cancers is we believe this may be due to immune suppression secondary to the mycosis fungoides, although further studies need to be performed to determine if that’s accurate,” Dr. Goyal said.
To that end, she and her colleagues are planning gene expression studies in patients from the UMN cohort. The researchers plan to examine genes involved in the pathogenesis of second malignancies and MF progression in tissue samples from 36 MF patients, 12 who developed second malignancies and 24 who did not.
The current research was funded by the American Society of Hematology. Dr. Goyal reported having no relevant financial disclosures. The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – A retrospective study suggests patients with mycosis fungoides (MF) have an increased risk of developing hematologic and solid tumor malignancies.
Researchers found the risk of second malignancy was highest among MF patients aged 30 to 50 years and patients who had tumor stage or advanced stage MF.
The increased risk was present during the entire period after MF diagnosis, but it was greatest in the first 6 months after diagnosis and roughly a dozen years later.
Amrita Goyal, MD, of the University of Minnesota in Minneapolis, and her colleagues presented these findings at the annual T-cell Lymphoma Forum.
The researchers first assessed the risk of second malignancy in 172 MF patients treated at UMN from 2005 to 2017, comparing this cohort to a control group of 172 patients with seborrheic dermatitis.
Second malignancies occurred in 24 MF patients and three controls, which was a significant difference (P = .0045). The most common second malignancies among the MF patients were melanoma (n = 4), prostate cancer (n = 3), and renal cell carcinoma (n = 3).
Further analyses revealed that MF patients were more likely to develop a second malignancy if they had tumor stage disease (P = .0024) or stage IIB or higher disease (P = .03).
To corroborate and expand upon these results, Dr. Goyal and her colleagues analyzed data from the Surveillance, Epidemiology, and End Results (SEER) database on patients diagnosed with MF from 2000 to 2014.
Among the 6,196 MF patients in this cohort, there were 514 second cancers.
“We found that MF patients were, overall, 10 times more likely to develop a second malignancy [compared with the general population],” Dr. Goyal said.
Specifically, the standardized incidence ratio was 10.15 for all malignancies, 7.33 for solid tumors, and 41.72 for hematologic malignancies.
Standardized incidence ratios for individual malignancies were:
- 69.8 for Hodgkin lymphoma.
- 46.5 for non-Hodgkin lymphoma.
- 8.6 for leukemia.
- 7.2 for melanoma.
- 6.2 for lung cancer.
- 7.9 for female breast cancer.
- 5.2 for colon cancer.
- 4.1 for prostate cancer.
- 3.9 for renal cell carcinoma.
- 3.8 for pancreatic cancer.
- 3.6 for bladder cancer.
“We found there is an increased risk [of second malignancy] during the first 6 months after diagnosis of MF, likely related to patients being in contact with the health care system more,” Dr. Goyal said. “Over time, patients have about a 7- to 10-fold increased risk over baseline, until they reach about 12 or 13 years after diagnosis, at which point, there is an increase in risk.”
The researchers found the greatest risk of second malignancy was among patients aged 30 to 50 years, although there was an increased risk for all age groups.
“The reason we think patients are experiencing an increased risk of cancers is we believe this may be due to immune suppression secondary to the mycosis fungoides, although further studies need to be performed to determine if that’s accurate,” Dr. Goyal said.
To that end, she and her colleagues are planning gene expression studies in patients from the UMN cohort. The researchers plan to examine genes involved in the pathogenesis of second malignancies and MF progression in tissue samples from 36 MF patients, 12 who developed second malignancies and 24 who did not.
The current research was funded by the American Society of Hematology. Dr. Goyal reported having no relevant financial disclosures. The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019
Key clinical point:
Major finding: In a cohort of MF patients from the SEER database, the standardized incidence ratio was 10.15 for all malignancies, 7.33 for solid tumors, and 41.72 for hematologic malignancies.
Study details: Retrospective study of 6,196 MF patients from the SEER database, and a single-center cohort of 172 MF patients who were matched to 172 patients with seborrheic dermatitis.
Disclosures: This research was funded by the American Society of Hematology. Dr. Goyal reported having no relevant financial disclosures.

Cerdulatinib yields ‘encouraging’ results in CTCL, PTCL
LA JOLLA, CALIF. – The spleen tyrosine kinase/Janus kinase inhibitor cerdulatinib has demonstrated activity against relapsed and refractory T-cell lymphomas.

In a phase 2 trial, cerdulatinib produced responses in 34% of patients with peripheral T-cell lymphoma (PTCL) and 26% of those with cutaneous T-cell lymphoma (CTCL).
The best responders were patients with angioimmunoblastic T-cell lymphoma, half of whom achieved a complete response (CR).
The most common grade 3 or higher adverse events (AEs) were amylase increase and lipase increase. However, these increases resolved with dose reduction or interruption, and there were no cases of clinical pancreatitis.
“The data is very encouraging,” said Tatyana Feldman, MD, of the John Theurer Cancer Center in Hackensack, N.J.
Dr. Feldman and her colleagues previously presented results from the phase 2 trial of cerdulatinib (NCT01994382) at the 2018 annual congress of the European Hematology Association.
Dr. Feldman and her colleagues presented data from expansion cohorts of the ongoing trial at the annual T-cell Lymphoma Forum. The cohorts included patients with PTCL or CTCL who had received at least one prior systemic therapy.
PTCL cohort
The 45 PTCL patients had a median age of 65 years (range, 21-84). They had received a median of 3 (range, 1-12) prior therapeutic regimens, 51% were refractory to their last therapy, and 27% had undergone stem cell transplant (SCT).
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 41 patients were evaluable for response.
The overall response rate was 34% (n = 14). Eleven patients had a CR, three had a partial response (PR), and nine had stable disease.
Responses according to subtype were as follows:
- 7 CRs and 1 PR in angioimmunoblastic T-cell lymphoma.
- 2 CRs in PTCL not otherwise specified.
- 1 CR in gamma-delta T-cell lymphoma.
- 1 PR in ALK-negative anaplastic large-cell lymphoma.
- 1 CR and 1 PR in adult T-cell leukemia/lymphoma.
Eight responders have remained on cerdulatinib for anywhere from 3 months to more than 12 months. Five patients have had a response lasting at least 6 months. One patient went on to SCT after achieving a CR.
The most common grade 3 or higher AEs observed in PTCL patients were amylase increase (n = 8), lipase increase (n = 6), pneumonia/lung infection (n = 5), neutropenia (n = 4), diarrhea (n = 4), febrile neutropenia (n = 4), abdominal pain (n = 4), sepsis/bacteremia (n = 3), anemia (n = 3), fatigue (n = 2), and pain (n = 1).
There were two grade 5 AEs – acute respiratory distress syndrome and pneumonia.
CTCL cohort
The 29 CTCL patients had a median age of 62 years (range, 24-79). They had received a median of 4 (range, 1-13) prior therapies, 55% were refractory to their last therapy, and 3% had undergone SCT.
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 27 were evaluable for response.
The overall response rate was 26% (n = 7). Two patients achieved a CR, five achieved a PR, and nine had stable disease. Responses occurred in mycosis fungoides and Sézary syndrome.
Eleven of 23 patients (48%) achieved at least a 50% reduction in skin lesions, and the researchers observed rapid improvements in pruritus.
“I saw patients who would take the first pill, and they would call me and say, ‘I no longer itch,’ ” Dr. Feldman said.
The most common grade 3 or higher AEs in CTCL patients were lipase increase (n = 11), amylase increase (n = 5), sepsis/bacteremia (n = 3), pain (n = 2), fatigue (n = 1), neutropenia (n = 1), and diarrhea (n = 1).
“It’s a very well-tolerated drug,” Dr. Feldman said, adding that there were “really no severe side effects which would prohibit the use of the drug.”
She noted that cerdulatinib’s “favorable” side effect profile might make it a promising candidate for use in combination regimens.
“I think it will be possible to combine it with other drugs in development in T-cell lymphoma. … immunological checkpoint inhibitors, epigenetic modulators such as HDAC [histone deacetylase] inhibitors, methylating agents, and PI3 kinase inhibitors,” Dr. Feldman said.
She reported having no disclosures relevant to this study. The trial is sponsored by Portola Pharmaceuticals.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – The spleen tyrosine kinase/Janus kinase inhibitor cerdulatinib has demonstrated activity against relapsed and refractory T-cell lymphomas.
In a phase 2 trial, cerdulatinib produced responses in 34% of patients with peripheral T-cell lymphoma (PTCL) and 26% of those with cutaneous T-cell lymphoma (CTCL).
The best responders were patients with angioimmunoblastic T-cell lymphoma, half of whom achieved a complete response (CR).
The most common grade 3 or higher adverse events (AEs) were amylase increase and lipase increase. However, these increases resolved with dose reduction or interruption, and there were no cases of clinical pancreatitis.
“The data is very encouraging,” said Tatyana Feldman, MD, of the John Theurer Cancer Center in Hackensack, N.J.
Dr. Feldman and her colleagues previously presented results from the phase 2 trial of cerdulatinib (NCT01994382) at the 2018 annual congress of the European Hematology Association.
Dr. Feldman and her colleagues presented data from expansion cohorts of the ongoing trial at the annual T-cell Lymphoma Forum. The cohorts included patients with PTCL or CTCL who had received at least one prior systemic therapy.
PTCL cohort
The 45 PTCL patients had a median age of 65 years (range, 21-84). They had received a median of 3 (range, 1-12) prior therapeutic regimens, 51% were refractory to their last therapy, and 27% had undergone stem cell transplant (SCT).
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 41 patients were evaluable for response.
The overall response rate was 34% (n = 14). Eleven patients had a CR, three had a partial response (PR), and nine had stable disease.
Responses according to subtype were as follows:
- 7 CRs and 1 PR in angioimmunoblastic T-cell lymphoma.
- 2 CRs in PTCL not otherwise specified.
- 1 CR in gamma-delta T-cell lymphoma.
- 1 PR in ALK-negative anaplastic large-cell lymphoma.
- 1 CR and 1 PR in adult T-cell leukemia/lymphoma.
Eight responders have remained on cerdulatinib for anywhere from 3 months to more than 12 months. Five patients have had a response lasting at least 6 months. One patient went on to SCT after achieving a CR.
The most common grade 3 or higher AEs observed in PTCL patients were amylase increase (n = 8), lipase increase (n = 6), pneumonia/lung infection (n = 5), neutropenia (n = 4), diarrhea (n = 4), febrile neutropenia (n = 4), abdominal pain (n = 4), sepsis/bacteremia (n = 3), anemia (n = 3), fatigue (n = 2), and pain (n = 1).
There were two grade 5 AEs – acute respiratory distress syndrome and pneumonia.
CTCL cohort
The 29 CTCL patients had a median age of 62 years (range, 24-79). They had received a median of 4 (range, 1-13) prior therapies, 55% were refractory to their last therapy, and 3% had undergone SCT.
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 27 were evaluable for response.
The overall response rate was 26% (n = 7). Two patients achieved a CR, five achieved a PR, and nine had stable disease. Responses occurred in mycosis fungoides and Sézary syndrome.
Eleven of 23 patients (48%) achieved at least a 50% reduction in skin lesions, and the researchers observed rapid improvements in pruritus.
“I saw patients who would take the first pill, and they would call me and say, ‘I no longer itch,’ ” Dr. Feldman said.
The most common grade 3 or higher AEs in CTCL patients were lipase increase (n = 11), amylase increase (n = 5), sepsis/bacteremia (n = 3), pain (n = 2), fatigue (n = 1), neutropenia (n = 1), and diarrhea (n = 1).
“It’s a very well-tolerated drug,” Dr. Feldman said, adding that there were “really no severe side effects which would prohibit the use of the drug.”
She noted that cerdulatinib’s “favorable” side effect profile might make it a promising candidate for use in combination regimens.
“I think it will be possible to combine it with other drugs in development in T-cell lymphoma. … immunological checkpoint inhibitors, epigenetic modulators such as HDAC [histone deacetylase] inhibitors, methylating agents, and PI3 kinase inhibitors,” Dr. Feldman said.
She reported having no disclosures relevant to this study. The trial is sponsored by Portola Pharmaceuticals.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
LA JOLLA, CALIF. – The spleen tyrosine kinase/Janus kinase inhibitor cerdulatinib has demonstrated activity against relapsed and refractory T-cell lymphomas.
In a phase 2 trial, cerdulatinib produced responses in 34% of patients with peripheral T-cell lymphoma (PTCL) and 26% of those with cutaneous T-cell lymphoma (CTCL).
The best responders were patients with angioimmunoblastic T-cell lymphoma, half of whom achieved a complete response (CR).
The most common grade 3 or higher adverse events (AEs) were amylase increase and lipase increase. However, these increases resolved with dose reduction or interruption, and there were no cases of clinical pancreatitis.
“The data is very encouraging,” said Tatyana Feldman, MD, of the John Theurer Cancer Center in Hackensack, N.J.
Dr. Feldman and her colleagues previously presented results from the phase 2 trial of cerdulatinib (NCT01994382) at the 2018 annual congress of the European Hematology Association.
Dr. Feldman and her colleagues presented data from expansion cohorts of the ongoing trial at the annual T-cell Lymphoma Forum. The cohorts included patients with PTCL or CTCL who had received at least one prior systemic therapy.
PTCL cohort
The 45 PTCL patients had a median age of 65 years (range, 21-84). They had received a median of 3 (range, 1-12) prior therapeutic regimens, 51% were refractory to their last therapy, and 27% had undergone stem cell transplant (SCT).
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 41 patients were evaluable for response.
The overall response rate was 34% (n = 14). Eleven patients had a CR, three had a partial response (PR), and nine had stable disease.
Responses according to subtype were as follows:
- 7 CRs and 1 PR in angioimmunoblastic T-cell lymphoma.
- 2 CRs in PTCL not otherwise specified.
- 1 CR in gamma-delta T-cell lymphoma.
- 1 PR in ALK-negative anaplastic large-cell lymphoma.
- 1 CR and 1 PR in adult T-cell leukemia/lymphoma.
Eight responders have remained on cerdulatinib for anywhere from 3 months to more than 12 months. Five patients have had a response lasting at least 6 months. One patient went on to SCT after achieving a CR.
The most common grade 3 or higher AEs observed in PTCL patients were amylase increase (n = 8), lipase increase (n = 6), pneumonia/lung infection (n = 5), neutropenia (n = 4), diarrhea (n = 4), febrile neutropenia (n = 4), abdominal pain (n = 4), sepsis/bacteremia (n = 3), anemia (n = 3), fatigue (n = 2), and pain (n = 1).
There were two grade 5 AEs – acute respiratory distress syndrome and pneumonia.
CTCL cohort
The 29 CTCL patients had a median age of 62 years (range, 24-79). They had received a median of 4 (range, 1-13) prior therapies, 55% were refractory to their last therapy, and 3% had undergone SCT.
The patients received cerdulatinib at 30 mg orally twice a day until progression or intolerance, and 27 were evaluable for response.
The overall response rate was 26% (n = 7). Two patients achieved a CR, five achieved a PR, and nine had stable disease. Responses occurred in mycosis fungoides and Sézary syndrome.
Eleven of 23 patients (48%) achieved at least a 50% reduction in skin lesions, and the researchers observed rapid improvements in pruritus.
“I saw patients who would take the first pill, and they would call me and say, ‘I no longer itch,’ ” Dr. Feldman said.
The most common grade 3 or higher AEs in CTCL patients were lipase increase (n = 11), amylase increase (n = 5), sepsis/bacteremia (n = 3), pain (n = 2), fatigue (n = 1), neutropenia (n = 1), and diarrhea (n = 1).
“It’s a very well-tolerated drug,” Dr. Feldman said, adding that there were “really no severe side effects which would prohibit the use of the drug.”
She noted that cerdulatinib’s “favorable” side effect profile might make it a promising candidate for use in combination regimens.
“I think it will be possible to combine it with other drugs in development in T-cell lymphoma. … immunological checkpoint inhibitors, epigenetic modulators such as HDAC [histone deacetylase] inhibitors, methylating agents, and PI3 kinase inhibitors,” Dr. Feldman said.
She reported having no disclosures relevant to this study. The trial is sponsored by Portola Pharmaceuticals.
The T-cell Lymphoma Forum is organized by Jonathan Wood & Associates, which is owned by the same company as this news organization.
REPORTING FROM TCLF 2019
Key clinical point:
Major finding: The overall response rate was 34% in patients with peripheral T-cell lymphoma (PTCL) and 26% in patients with cutaneous T-cell lymphoma (CTCL).
Study details: Expansion cohorts of a phase 2 trial including 45 PTCL patients and 29 CTCL patients
Disclosures: The study was funded by Portola Pharmaceuticals. The investigator reported having no relevant conflicts.

Locally Destructive Metastatic Basal Cell Carcinoma
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
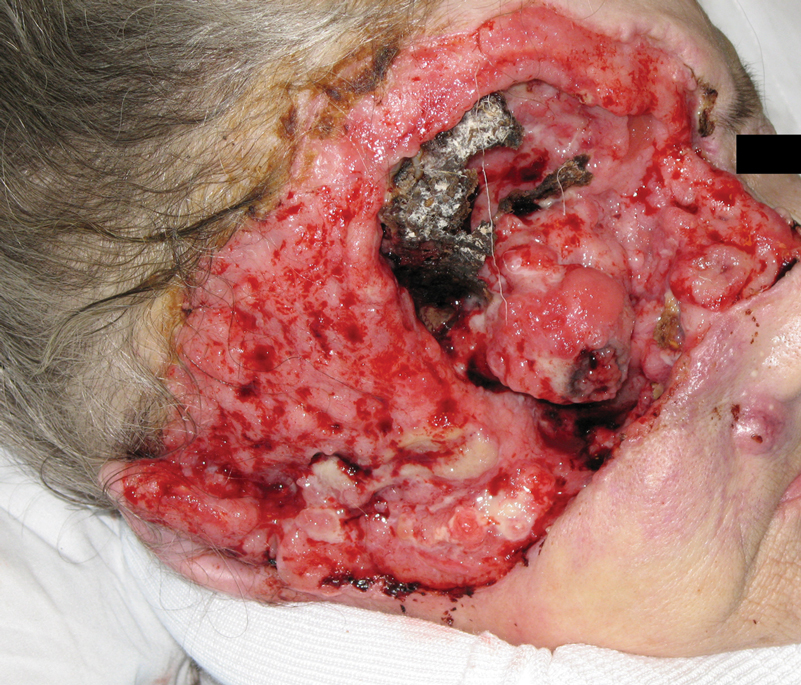
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
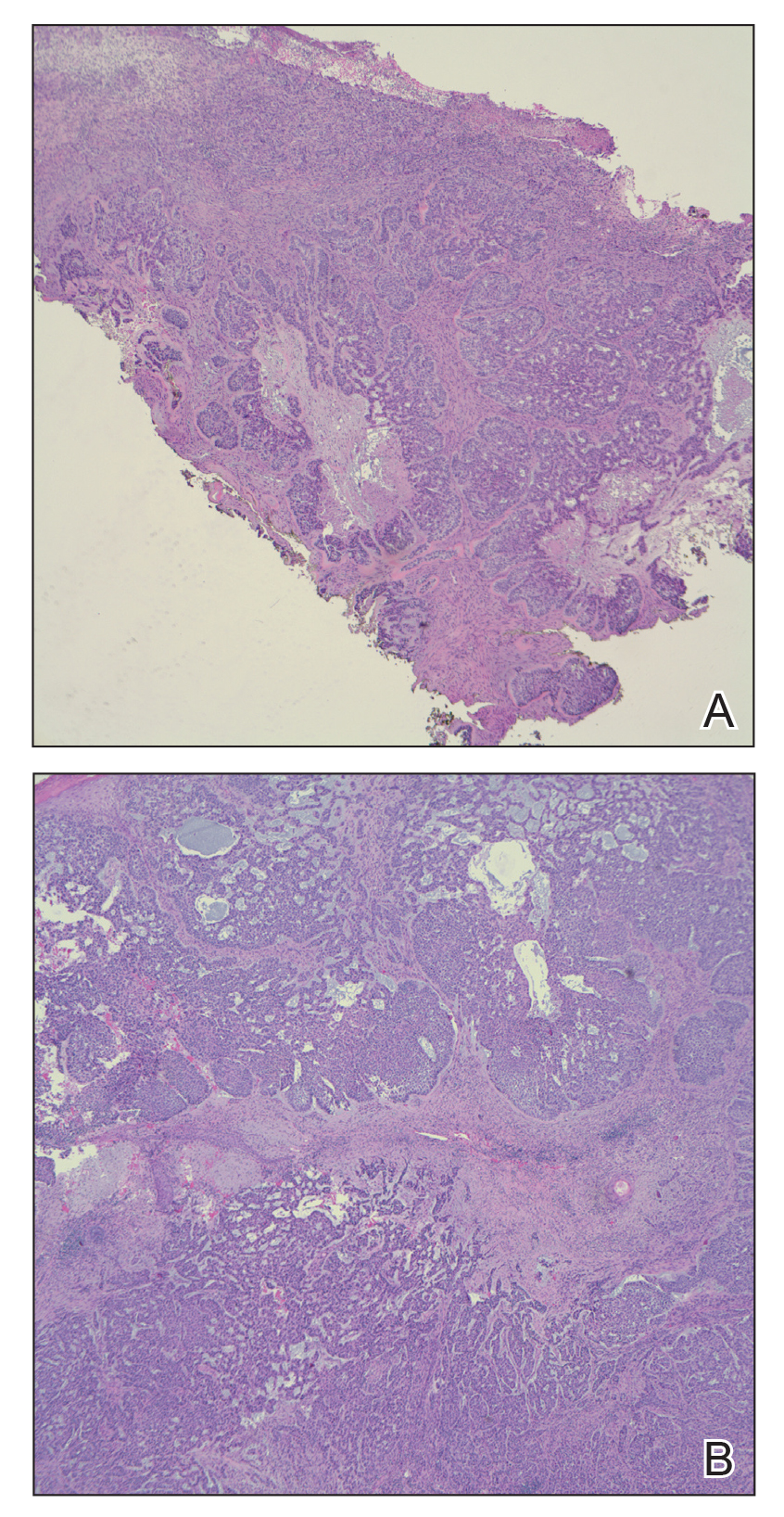
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
To the Editor:
A 60-year-old woman with a history of lymphoma presented to the emergency department for evaluation of intermittent diarrhea and vomiting of 2 weeks’ duration. On presentation, a rather large dressing covering the entire right half of the face was noted. Removal of the bandage revealed a necrotic, extensively destructive, right-sided facial lesion with a fully exposed ocular globe (Figure 1). The patient lived alone and was accompanied by a neighbor, who disclosed that the lesion had been neglected and enlarged over the last 15 years. Moreover, the neighbor reported that the patient had recently experienced several episodes of vertigo and frequent falls.
On admission to the hospital, dermatology was consulted and initial workup included computed tomography (CT) scan of the head and maxillofacial region, which showed a destructive process involving the right frontotemporal bone, maxillofacial region, sphenoid, and skull base with exposure of intracranial contents (Figure 2). An aggressive wound care regimen was instituted. Biopsy of the wound margin revealed nodular and focally infiltrative basal cell carcinoma (BCC) (Figure 3).
and focally infiltrative basal cell carcinoma (H&E, original magnifications×4 and ×10).
Several days into her hospitalization, the patient developed radicular pain in both arms and weakness in all 4 extremities. A CT scan of the neck revealed a pathologic fracture of the C7 vertebrae. Several medical and surgical services as well as psychiatry were consulted. Given the extensive nature of the disease involvement with limited treatment options, the patient sought to forego further interventions and was discharged to hospice care.
Basal cell carcinomas rarely metastasize, with a reported incidence of 0.0028% to 0.5%.1 The likelihood of metastasis is most closely related to tumor size and depth of invasion. Tumors greater than 3 cm in diameter have a 2% incidence of metastatic spread and/or death. The incidence of metastatic spread and/or death is estimated to be 25% for tumors with a diameter of 5 cm and 50% for tumors with a diameter of 10 cm or greater.2 Other risk factors for metastatic spread include long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.1 In one review, the median interval between onset of BCC and metastasis was 9 years.3 In our case, 15 years of neglect most likely led to the aggressiveness of the tumor. Although the workup in our patient was limited per her request, there was no evidence that her lymphoma had recurred or that she was in any other way immunocompromised. Unfortunately, in this patient’s case, the local destructiveness of the carcinoma with subsequent bony invasion and necrosis was complicated with secondary Rhizopus infection. A PubMed search of articles indexed for MEDLINE using the terms basal cell carcinoma and mucormycosis revealed no other reported cases of BCC associated with mucormycosis; therefore, our case represents a rare presentation of this association. Rhinocerebral mucormycosis is the most common manifestation of mucormycosis and more commonly occurs in diabetics with ketoacidosis and in severely debilitated or immunosuppressed individuals.4 The extensive bony destruction, especially of the nasal region, of our patient’s tumor likely led to secondary infection with Rhizopus.
Approximately 85% of all metastatic BCCs originate in the head and neck region, with lymph nodes being the first site of metastasis and involved in approximately half of all cases.1,4 Metastases to the lungs, bone, liver, and other viscera can occur with advanced disease. Metastasis generally portends a poor prognosis, with survival rarely exceeding 1.5 years. Until recently, therapeutic options for metastatic disease were limited, with marginal response to chemotherapy with methotrexate, fluorouracil, bleomycin, and cisplatin.4 Vismodegib, a novel smoothened receptor inhibitor that blocks the sonic hedgehog pathway implicated in BCC carcinogenesis, offers a new promising treatment for management and control of advanced disease.5
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
- Junior W, Ribeiro SC, Vieira SC, et al. Metastatic basal cell carcinoma: a case report. Dermatol Online J. 2003;9:18.
- Snow SN, Sahl WJ, Lo J, et al. Metastatic basal cell carcinoma: report of 5 cases. Cancer. 1994;73:328-335.
- Von Domarus H, Stevens PJ. Metastatic basal cell carcinoma: report of five cases and review of 170 cases in the literature. J Am Acad Dermatol. 1984;10:1043-1060.
- Kolekar JS. Rhinocerebral mucormycosis: a retrospective study. Indian J Otolaryngol Head Neck Surg. 2015;67:93-96.
- Koehlblinger P, Lang R. New developments in the treatment of basal cell carcinoma: update on current and emerging treatment options with a focus on vismodegib. Onc Targets Ther. 2018;11:8327-8340.
Practice Points
- Risk factors associated with metastatic spread of basal cell carcinoma (BCC) include larger tumor size, greater depth of invasion, long duration of disease, failure to respond to conventional treatment, and prior radiation treatment in the affected area.
- The median interval between onset of BCC and metastasis has been shown to be approximately 9 years.
- Vismodegib can be an effective oral therapy for patients with metastatic BCC, locally advanced BCC, recurrence following surgery, or those who are not candidates for surgery or radiation.