User login
PAH care turns corner with new therapies, intensified monitoring
Aggressive up-front combination therapy, more lofty treatment goals, and earlier and more frequent reassessments to guide treatment are improving care of patients with pulmonary arterial hypertension (PAH) while at the same time making it more complex.
A larger number of oral and generic treatment options have in some respects ushered in more management ease. But overall, “I don’t know if management of these patients has ever been more complicated, given the treatment options and strategies,” said Murali M. Chakinala, MD, professor of medicine at Washington University, St. Louis. “We’re always thinking through approaches.”

Diagnosis continues to be challenging given the rarity of PAH and its nonspecific presentation – and in some cases it’s now harder. Experts such as Dr. Chakinala are seeing increasing number of aging patients with left heart disease, chronic kidney disease, and other comorbidities who have significant precapillary pulmonary hypertension and who exhibit hemodynamics consistent with PAH, or group 1 PH.
The question experts face is, do such patients have “true PAH,” as do a reported 25-50 people per million, or do they have another type of PH in the classification schema – or a mixture?
Deciding which patients “really fit into group 1 and should be managed like group 1,” Dr. Chakinala said, requires clinical acumen and has important implications, as patients with PAH are the main beneficiaries of vasodilator therapy. Most other patients with PH will not respond to or tolerate such treatment.
“These older patients may be getting PAH through different mechanisms than our younger patients, but because we define PAH through hemodynamic criteria and by ruling out other obvious explanations, they all get lumped together,” said Dr. Chakinala. “We need to parse these patients out better in the future, much like our oncology colleagues are doing.”
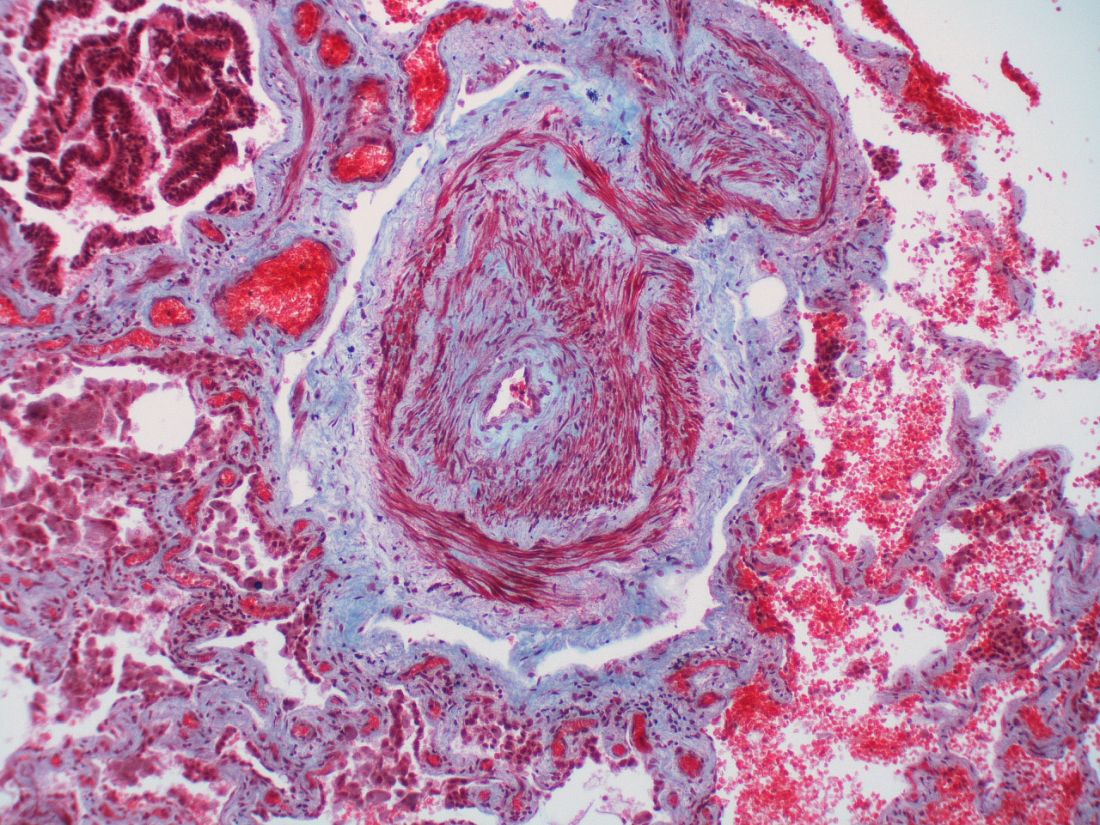
Personalized medicine hopefully is the next horizon for this condition, characterized by severe remodeling of the distal pulmonary arteries. Researchers are pushing to achieve deep phenotyping, identify biomarkers and improve risk assessment tools.
And with 80 or so centers now accredited by the Pulmonary Hypertension Association as Pulmonary Hypertension Care Centers, referred patients are accessing clinical trials of new nonvasodilatory drugs. Currently available therapies improve hemodynamics and symptoms, and can slow disease progression, but are not truly disease modifying, sources say.
“The endothelin, nitric oxide, and prostacyclin pathways have been exhaustively studied and we now have great drugs for those pathways,” said Dr. Chakinala, who leads the PHA’s scientific leadership council. But “we’re not going to put a greater dent into this disease until we have new drugs that work on different biologic pathways.”
Diagnostic challenges
The diagnosis of PAH – a remarkably heterogeneous condition that encompasses heritable forms and idiopathic forms, and that comprises a broad mix of predisposing conditions and exposures, from scleroderma to methamphetamine use – is still too often missed or delayed. Delayed diagnoses and misdiagnoses of PAH and other types of PH have been reported in up to 85% of at-risk patients, according to a 2016 literature review.

Being able to pivot from thinking about common pulmonary ailments or heart failure to considering PAH is a key part of earlier diagnosis and better treatment outcomes. “If someone has unexplained dyspnea or if they’re treated for other lung diseases and are not improving, think about a screening echocardiogram,” said Timothy L. Williamson, MD, vice president of quality and safety and a pulmonary and critical care physician at the University of Kansas Health Center, Kansas City.
One of the most common reasons Dr. Chakinala sees for missed diagnoses are right heart catheterizations that are incomplete or misinterpreted. (Right heart catheterizations are required to confirm the diagnosis.) “One can’t simply measure pressures and stop,” he said. “We need the full hemodynamic profile to know that it’s truly precapillary PAH ... and we need proper interpretation of [elements like] the waveforms.”
The 2019 World Symposium on Pulmonary Hypertension shifted the definition of PH from an arbitrarily defined mean pulmonary arterial pressure of at least 25 mm Hg at rest (as measured by right heart catheterization) to a more scientifically determined mPAP of at least 20 mm Hg.
The classification document also requires pulmonary vascular resistance (PVR) of at least 3 Wood units in the definition of all forms of precapillary PH. PAH specifically is defined as the presence of mPAP of at least 20 mm Hg, PVR of at least 3 Wood units, and pulmonary arterial wedge pressure 15 mm Hg or less.
Trends in treatment
The value of initial combination therapy with an endothelin receptor antagonist (ERA) and a phosphodiesterase-5 (PDE5) inhibitor in treatment-naive PAH was cemented in 2015 by the AMBITION trial. The primary endpoint (death, PAH hospitalization, or unsatisfactory clinical response) occurred in 18%, 34%, and 28% of patients who were randomized, respectively, to combination therapy, monotherapy with the ERA ambrisentan, or monotherapy with the PDE-5 inhibitor tadalafil – and in 31% of the two monotherapy groups combined.
The trial reported a 50% reduction in the primary endpoint in the combination-therapy group versus the pooled monotherapy group, as well as greater reductions in N-terminal of the prohormone brain natriuretic peptide levels, more satisfactory clinical response and greater improvement in 6-minute walking distance.
In practice, a minority of patients – typically older patients with multiple comorbidities – still receive initial monotherapy with sequential add-on therapies based on tolerance, but “for the most part PAH patients will start on combination therapy, most commonly with a ERA and PDE5 inhibitor,” Dr. Chakinala said.
For patients who are not improving on the ERA-PDE5 inhibitor approach – typically those who remain in the intermediate-risk category for intermediate-term mortality – substitution of the PDE5 inhibitor with the soluble guanylate cyclase stimulator riociguat may be considered, he and Dr. Williamson said. Clinical improvement with this substitution was demonstrated in the REPLACE trial.
Experts at PH care centers are also utilizing triple therapy for patients who do not improve to low-risk status after 2-4 months of dual combination therapy. The availability of oral prostacyclin analogues (selexipag and treprostinil) makes it easier to consider adding these agents early on, Dr. Chakinala and Dr. Richardson said.
Patients who fall into the high-risk category, at any point, are still best managed with parenteral prostacyclin analogues, Dr. Chakinala said.
In general, said Dr. Williamson, who also directs the University of Kansas Pulmonary Hypertension Comprehensive Care Center, “the PH community tends to be fairly aggressive up front, and with a low threshold for using prostacyclin analogues.”
The agents are “always part of the picture for someone who is really ill, in functional class IV, or has really impaired right ventricular function,” he said. “And we’re finding increased roles in patients who are not as ill but still have decompensated right ventricular dysfunction. It’s something we now consider.”
Recently published research on up-front oral triple therapy suggests possible benefit for some patients – but it’s far from conclusive, said Dr. Chakinala. The TRITON study randomized treatment-naive patients to the traditional ERA-PDE5 combination and either oral selexipag (a selective prostacyclin receptor agonist) or placebo as a third agent. It found no significant difference in reduction in PVR, the primary outcome, at week 26. However, the authors reported a “possible signal” for improved long-term outcomes with triple therapy.
“Based on this best evidence from a randomized clinical trial, I think it’s unfair to say that all patients should be on triple combination therapy right out of the gate,” he said. “Having said that, more recent [European] data showed that two drugs fell short of the mark in some patients, with high rates of clinical progression. And even in AMBITION, there were a number of patients in the combination arm who didn’t have a robust response.”
A 2021 retrospective analysis from the French Pulmonary Hypertension Registry – one of the European studies – assessed survival with monotherapy, dual therapy, or triple-combination therapy (two orals with a parenteral prostacyclin), and found no difference between monotherapy and dual therapy in high-risk patients.
Experts have been upping the ante, therefore, on early assessment and frequent reassessment of treatment response. Not long ago, patients were typically reassessed 6-12 months after the initiation of treatment. Now, experts at the PH care centers want to assess patients at 3-4 months and adjust or intensify treatment regimens for those who don’t yet qualify as low risk using a multidimensional risk score calculator.
The REVEAL (Registry to Evaluate Early and Long-Term PAH Management) risk score calculator, for instance, predicts the probability of 1-year survival and assigns patients to a strata of risk level based on either 12 or 6 variables (for the full or “lite” versions).]
Even better monitoring and risk assessment is needed, however, to “help sift out which patients are not improving enough on initial therapy or who are starting to fall off after being on a regimen for a period of time,” Dr. Chakinala said.
Today, with a network of accredited centers of expertise and a desire and need for many patients to remain close to home, Dr. Chakinala encourages finding a balance. Well-resourced clinicians can strive for early diagnosis and management – potentially initiating ERA–PDE-5 inhibitor combination therapy – but still should collaborate with PH experts.
“It’s a good idea to comanage these patients and let the experts see them periodically to help you determine when your patient may be declining,” he said. “The timetable for reassessment, the complexity of the reassessment, and the need to escalate to more advanced therapies has never been more important.”
Research highlights
Therapies that target inflammation and altered metabolism – including metformin – are among those being investigated for PAH. So are therapies targeting dysfunctional bone morphogenetic protein pathway signaling, which has been shown to be associated with hereditary, idiopathic, and likely other forms of PAH; one such drug, called sotatercept, is currently at the phase 3 trial stage.

Most promising for PAH may be the research efforts involving deep phenotyping, said Andrew J. Sweatt, MD, of Stanford (Calif.) University and the Vera Moulton Wall Center for Pulmonary Vascular Disease.
“It’s where a lot of research is headed – deep phenotyping to deconstruct the molecular and clinical heterogeneity that exists within PAH ... to detect distinct subphenotypes of patients who would respond to particular therapies,” said Dr. Sweatt, who led a review of PH clinical research presented at the 2020 American Thoracic Society International Conference
“Right now, we largely treat all patients the same ... [while] we know that patients have a wide response to therapies and there’s a lot of clinical heterogeneity in how their disease evolves over time,” he said.

Data from a large National Institutes of Health–funded multicenter phenotyping study of PH is being analyzed and should yield findings and publications starting this year, said Anna R. Hemnes, MD, associate professor of medicine at Vanderbilt University Medical Center, Nashville, Tenn., and an investigator with the initiative, coined “Redefining Pulmonary Hypertension through Pulmonary Disease Phenomics (PVDOMICS).”
Patients have undergone advanced imaging (for example, echocardiography, cardiac MRI, chest CT, ventilation/perfusion scans), advanced testing through right heart catheterization, body composition testing, quality of life questionnaires, and blood draws that have been analyzed for DNA and RNA expression, proteomics, and metabolomics, said Dr. Hemnes, assistant director of Vanderbilt’s Pulmonary Vascular Center.
The initiative aims to refine the classification of all kinds of PH and “to bring precision medicine to the field so we’re no longer characterizing somebody [based on imaging] and right heart catheterization, but we also incorporating molecular pieces and biomarkers into the diagnostic evaluation,” she said.
In the short term, the results of deep phenotyping should “allow us to be more effective with our therapy recommendations,” Dr. Hemnes said. “Then hopefully in the longer term, [identified biomarkers] will help us to develop new, more effective therapies.”
Dr. Sweatt and Dr. Williamson reported that they have no relevant financial disclosures. Dr. Hemnes reported that she holds stock in Tenax (which is studying a tyrosine kinase inhibitor for PAH) and serves as a consultant for Acceleron, Bayer, GossamerBio, United Therapeutics, and Janssen. She also receives research funding from Imara. Dr. Chakinala reported that he is an investigator on clinical trials for a number of pharmaceutical companies. He also serves on advisory boards for Phase Bio, Liquidia/Rare Gen, Bayer, Janssen, Trio Health Analytics, and Aerovate.
Aggressive up-front combination therapy, more lofty treatment goals, and earlier and more frequent reassessments to guide treatment are improving care of patients with pulmonary arterial hypertension (PAH) while at the same time making it more complex.
A larger number of oral and generic treatment options have in some respects ushered in more management ease. But overall, “I don’t know if management of these patients has ever been more complicated, given the treatment options and strategies,” said Murali M. Chakinala, MD, professor of medicine at Washington University, St. Louis. “We’re always thinking through approaches.”
Diagnosis continues to be challenging given the rarity of PAH and its nonspecific presentation – and in some cases it’s now harder. Experts such as Dr. Chakinala are seeing increasing number of aging patients with left heart disease, chronic kidney disease, and other comorbidities who have significant precapillary pulmonary hypertension and who exhibit hemodynamics consistent with PAH, or group 1 PH.
The question experts face is, do such patients have “true PAH,” as do a reported 25-50 people per million, or do they have another type of PH in the classification schema – or a mixture?
Deciding which patients “really fit into group 1 and should be managed like group 1,” Dr. Chakinala said, requires clinical acumen and has important implications, as patients with PAH are the main beneficiaries of vasodilator therapy. Most other patients with PH will not respond to or tolerate such treatment.
“These older patients may be getting PAH through different mechanisms than our younger patients, but because we define PAH through hemodynamic criteria and by ruling out other obvious explanations, they all get lumped together,” said Dr. Chakinala. “We need to parse these patients out better in the future, much like our oncology colleagues are doing.”
Personalized medicine hopefully is the next horizon for this condition, characterized by severe remodeling of the distal pulmonary arteries. Researchers are pushing to achieve deep phenotyping, identify biomarkers and improve risk assessment tools.
And with 80 or so centers now accredited by the Pulmonary Hypertension Association as Pulmonary Hypertension Care Centers, referred patients are accessing clinical trials of new nonvasodilatory drugs. Currently available therapies improve hemodynamics and symptoms, and can slow disease progression, but are not truly disease modifying, sources say.
“The endothelin, nitric oxide, and prostacyclin pathways have been exhaustively studied and we now have great drugs for those pathways,” said Dr. Chakinala, who leads the PHA’s scientific leadership council. But “we’re not going to put a greater dent into this disease until we have new drugs that work on different biologic pathways.”
Diagnostic challenges
The diagnosis of PAH – a remarkably heterogeneous condition that encompasses heritable forms and idiopathic forms, and that comprises a broad mix of predisposing conditions and exposures, from scleroderma to methamphetamine use – is still too often missed or delayed. Delayed diagnoses and misdiagnoses of PAH and other types of PH have been reported in up to 85% of at-risk patients, according to a 2016 literature review.
Being able to pivot from thinking about common pulmonary ailments or heart failure to considering PAH is a key part of earlier diagnosis and better treatment outcomes. “If someone has unexplained dyspnea or if they’re treated for other lung diseases and are not improving, think about a screening echocardiogram,” said Timothy L. Williamson, MD, vice president of quality and safety and a pulmonary and critical care physician at the University of Kansas Health Center, Kansas City.
One of the most common reasons Dr. Chakinala sees for missed diagnoses are right heart catheterizations that are incomplete or misinterpreted. (Right heart catheterizations are required to confirm the diagnosis.) “One can’t simply measure pressures and stop,” he said. “We need the full hemodynamic profile to know that it’s truly precapillary PAH ... and we need proper interpretation of [elements like] the waveforms.”
The 2019 World Symposium on Pulmonary Hypertension shifted the definition of PH from an arbitrarily defined mean pulmonary arterial pressure of at least 25 mm Hg at rest (as measured by right heart catheterization) to a more scientifically determined mPAP of at least 20 mm Hg.
The classification document also requires pulmonary vascular resistance (PVR) of at least 3 Wood units in the definition of all forms of precapillary PH. PAH specifically is defined as the presence of mPAP of at least 20 mm Hg, PVR of at least 3 Wood units, and pulmonary arterial wedge pressure 15 mm Hg or less.
Trends in treatment
The value of initial combination therapy with an endothelin receptor antagonist (ERA) and a phosphodiesterase-5 (PDE5) inhibitor in treatment-naive PAH was cemented in 2015 by the AMBITION trial. The primary endpoint (death, PAH hospitalization, or unsatisfactory clinical response) occurred in 18%, 34%, and 28% of patients who were randomized, respectively, to combination therapy, monotherapy with the ERA ambrisentan, or monotherapy with the PDE-5 inhibitor tadalafil – and in 31% of the two monotherapy groups combined.
The trial reported a 50% reduction in the primary endpoint in the combination-therapy group versus the pooled monotherapy group, as well as greater reductions in N-terminal of the prohormone brain natriuretic peptide levels, more satisfactory clinical response and greater improvement in 6-minute walking distance.
In practice, a minority of patients – typically older patients with multiple comorbidities – still receive initial monotherapy with sequential add-on therapies based on tolerance, but “for the most part PAH patients will start on combination therapy, most commonly with a ERA and PDE5 inhibitor,” Dr. Chakinala said.
For patients who are not improving on the ERA-PDE5 inhibitor approach – typically those who remain in the intermediate-risk category for intermediate-term mortality – substitution of the PDE5 inhibitor with the soluble guanylate cyclase stimulator riociguat may be considered, he and Dr. Williamson said. Clinical improvement with this substitution was demonstrated in the REPLACE trial.
Experts at PH care centers are also utilizing triple therapy for patients who do not improve to low-risk status after 2-4 months of dual combination therapy. The availability of oral prostacyclin analogues (selexipag and treprostinil) makes it easier to consider adding these agents early on, Dr. Chakinala and Dr. Richardson said.
Patients who fall into the high-risk category, at any point, are still best managed with parenteral prostacyclin analogues, Dr. Chakinala said.
In general, said Dr. Williamson, who also directs the University of Kansas Pulmonary Hypertension Comprehensive Care Center, “the PH community tends to be fairly aggressive up front, and with a low threshold for using prostacyclin analogues.”
The agents are “always part of the picture for someone who is really ill, in functional class IV, or has really impaired right ventricular function,” he said. “And we’re finding increased roles in patients who are not as ill but still have decompensated right ventricular dysfunction. It’s something we now consider.”
Recently published research on up-front oral triple therapy suggests possible benefit for some patients – but it’s far from conclusive, said Dr. Chakinala. The TRITON study randomized treatment-naive patients to the traditional ERA-PDE5 combination and either oral selexipag (a selective prostacyclin receptor agonist) or placebo as a third agent. It found no significant difference in reduction in PVR, the primary outcome, at week 26. However, the authors reported a “possible signal” for improved long-term outcomes with triple therapy.
“Based on this best evidence from a randomized clinical trial, I think it’s unfair to say that all patients should be on triple combination therapy right out of the gate,” he said. “Having said that, more recent [European] data showed that two drugs fell short of the mark in some patients, with high rates of clinical progression. And even in AMBITION, there were a number of patients in the combination arm who didn’t have a robust response.”
A 2021 retrospective analysis from the French Pulmonary Hypertension Registry – one of the European studies – assessed survival with monotherapy, dual therapy, or triple-combination therapy (two orals with a parenteral prostacyclin), and found no difference between monotherapy and dual therapy in high-risk patients.
Experts have been upping the ante, therefore, on early assessment and frequent reassessment of treatment response. Not long ago, patients were typically reassessed 6-12 months after the initiation of treatment. Now, experts at the PH care centers want to assess patients at 3-4 months and adjust or intensify treatment regimens for those who don’t yet qualify as low risk using a multidimensional risk score calculator.
The REVEAL (Registry to Evaluate Early and Long-Term PAH Management) risk score calculator, for instance, predicts the probability of 1-year survival and assigns patients to a strata of risk level based on either 12 or 6 variables (for the full or “lite” versions).]
Even better monitoring and risk assessment is needed, however, to “help sift out which patients are not improving enough on initial therapy or who are starting to fall off after being on a regimen for a period of time,” Dr. Chakinala said.
Today, with a network of accredited centers of expertise and a desire and need for many patients to remain close to home, Dr. Chakinala encourages finding a balance. Well-resourced clinicians can strive for early diagnosis and management – potentially initiating ERA–PDE-5 inhibitor combination therapy – but still should collaborate with PH experts.
“It’s a good idea to comanage these patients and let the experts see them periodically to help you determine when your patient may be declining,” he said. “The timetable for reassessment, the complexity of the reassessment, and the need to escalate to more advanced therapies has never been more important.”
Research highlights
Therapies that target inflammation and altered metabolism – including metformin – are among those being investigated for PAH. So are therapies targeting dysfunctional bone morphogenetic protein pathway signaling, which has been shown to be associated with hereditary, idiopathic, and likely other forms of PAH; one such drug, called sotatercept, is currently at the phase 3 trial stage.
Most promising for PAH may be the research efforts involving deep phenotyping, said Andrew J. Sweatt, MD, of Stanford (Calif.) University and the Vera Moulton Wall Center for Pulmonary Vascular Disease.
“It’s where a lot of research is headed – deep phenotyping to deconstruct the molecular and clinical heterogeneity that exists within PAH ... to detect distinct subphenotypes of patients who would respond to particular therapies,” said Dr. Sweatt, who led a review of PH clinical research presented at the 2020 American Thoracic Society International Conference
“Right now, we largely treat all patients the same ... [while] we know that patients have a wide response to therapies and there’s a lot of clinical heterogeneity in how their disease evolves over time,” he said.
Data from a large National Institutes of Health–funded multicenter phenotyping study of PH is being analyzed and should yield findings and publications starting this year, said Anna R. Hemnes, MD, associate professor of medicine at Vanderbilt University Medical Center, Nashville, Tenn., and an investigator with the initiative, coined “Redefining Pulmonary Hypertension through Pulmonary Disease Phenomics (PVDOMICS).”
Patients have undergone advanced imaging (for example, echocardiography, cardiac MRI, chest CT, ventilation/perfusion scans), advanced testing through right heart catheterization, body composition testing, quality of life questionnaires, and blood draws that have been analyzed for DNA and RNA expression, proteomics, and metabolomics, said Dr. Hemnes, assistant director of Vanderbilt’s Pulmonary Vascular Center.
The initiative aims to refine the classification of all kinds of PH and “to bring precision medicine to the field so we’re no longer characterizing somebody [based on imaging] and right heart catheterization, but we also incorporating molecular pieces and biomarkers into the diagnostic evaluation,” she said.
In the short term, the results of deep phenotyping should “allow us to be more effective with our therapy recommendations,” Dr. Hemnes said. “Then hopefully in the longer term, [identified biomarkers] will help us to develop new, more effective therapies.”
Dr. Sweatt and Dr. Williamson reported that they have no relevant financial disclosures. Dr. Hemnes reported that she holds stock in Tenax (which is studying a tyrosine kinase inhibitor for PAH) and serves as a consultant for Acceleron, Bayer, GossamerBio, United Therapeutics, and Janssen. She also receives research funding from Imara. Dr. Chakinala reported that he is an investigator on clinical trials for a number of pharmaceutical companies. He also serves on advisory boards for Phase Bio, Liquidia/Rare Gen, Bayer, Janssen, Trio Health Analytics, and Aerovate.
Aggressive up-front combination therapy, more lofty treatment goals, and earlier and more frequent reassessments to guide treatment are improving care of patients with pulmonary arterial hypertension (PAH) while at the same time making it more complex.
A larger number of oral and generic treatment options have in some respects ushered in more management ease. But overall, “I don’t know if management of these patients has ever been more complicated, given the treatment options and strategies,” said Murali M. Chakinala, MD, professor of medicine at Washington University, St. Louis. “We’re always thinking through approaches.”
Diagnosis continues to be challenging given the rarity of PAH and its nonspecific presentation – and in some cases it’s now harder. Experts such as Dr. Chakinala are seeing increasing number of aging patients with left heart disease, chronic kidney disease, and other comorbidities who have significant precapillary pulmonary hypertension and who exhibit hemodynamics consistent with PAH, or group 1 PH.
The question experts face is, do such patients have “true PAH,” as do a reported 25-50 people per million, or do they have another type of PH in the classification schema – or a mixture?
Deciding which patients “really fit into group 1 and should be managed like group 1,” Dr. Chakinala said, requires clinical acumen and has important implications, as patients with PAH are the main beneficiaries of vasodilator therapy. Most other patients with PH will not respond to or tolerate such treatment.
“These older patients may be getting PAH through different mechanisms than our younger patients, but because we define PAH through hemodynamic criteria and by ruling out other obvious explanations, they all get lumped together,” said Dr. Chakinala. “We need to parse these patients out better in the future, much like our oncology colleagues are doing.”
Personalized medicine hopefully is the next horizon for this condition, characterized by severe remodeling of the distal pulmonary arteries. Researchers are pushing to achieve deep phenotyping, identify biomarkers and improve risk assessment tools.
And with 80 or so centers now accredited by the Pulmonary Hypertension Association as Pulmonary Hypertension Care Centers, referred patients are accessing clinical trials of new nonvasodilatory drugs. Currently available therapies improve hemodynamics and symptoms, and can slow disease progression, but are not truly disease modifying, sources say.
“The endothelin, nitric oxide, and prostacyclin pathways have been exhaustively studied and we now have great drugs for those pathways,” said Dr. Chakinala, who leads the PHA’s scientific leadership council. But “we’re not going to put a greater dent into this disease until we have new drugs that work on different biologic pathways.”
Diagnostic challenges
The diagnosis of PAH – a remarkably heterogeneous condition that encompasses heritable forms and idiopathic forms, and that comprises a broad mix of predisposing conditions and exposures, from scleroderma to methamphetamine use – is still too often missed or delayed. Delayed diagnoses and misdiagnoses of PAH and other types of PH have been reported in up to 85% of at-risk patients, according to a 2016 literature review.
Being able to pivot from thinking about common pulmonary ailments or heart failure to considering PAH is a key part of earlier diagnosis and better treatment outcomes. “If someone has unexplained dyspnea or if they’re treated for other lung diseases and are not improving, think about a screening echocardiogram,” said Timothy L. Williamson, MD, vice president of quality and safety and a pulmonary and critical care physician at the University of Kansas Health Center, Kansas City.
One of the most common reasons Dr. Chakinala sees for missed diagnoses are right heart catheterizations that are incomplete or misinterpreted. (Right heart catheterizations are required to confirm the diagnosis.) “One can’t simply measure pressures and stop,” he said. “We need the full hemodynamic profile to know that it’s truly precapillary PAH ... and we need proper interpretation of [elements like] the waveforms.”
The 2019 World Symposium on Pulmonary Hypertension shifted the definition of PH from an arbitrarily defined mean pulmonary arterial pressure of at least 25 mm Hg at rest (as measured by right heart catheterization) to a more scientifically determined mPAP of at least 20 mm Hg.
The classification document also requires pulmonary vascular resistance (PVR) of at least 3 Wood units in the definition of all forms of precapillary PH. PAH specifically is defined as the presence of mPAP of at least 20 mm Hg, PVR of at least 3 Wood units, and pulmonary arterial wedge pressure 15 mm Hg or less.
Trends in treatment
The value of initial combination therapy with an endothelin receptor antagonist (ERA) and a phosphodiesterase-5 (PDE5) inhibitor in treatment-naive PAH was cemented in 2015 by the AMBITION trial. The primary endpoint (death, PAH hospitalization, or unsatisfactory clinical response) occurred in 18%, 34%, and 28% of patients who were randomized, respectively, to combination therapy, monotherapy with the ERA ambrisentan, or monotherapy with the PDE-5 inhibitor tadalafil – and in 31% of the two monotherapy groups combined.
The trial reported a 50% reduction in the primary endpoint in the combination-therapy group versus the pooled monotherapy group, as well as greater reductions in N-terminal of the prohormone brain natriuretic peptide levels, more satisfactory clinical response and greater improvement in 6-minute walking distance.
In practice, a minority of patients – typically older patients with multiple comorbidities – still receive initial monotherapy with sequential add-on therapies based on tolerance, but “for the most part PAH patients will start on combination therapy, most commonly with a ERA and PDE5 inhibitor,” Dr. Chakinala said.
For patients who are not improving on the ERA-PDE5 inhibitor approach – typically those who remain in the intermediate-risk category for intermediate-term mortality – substitution of the PDE5 inhibitor with the soluble guanylate cyclase stimulator riociguat may be considered, he and Dr. Williamson said. Clinical improvement with this substitution was demonstrated in the REPLACE trial.
Experts at PH care centers are also utilizing triple therapy for patients who do not improve to low-risk status after 2-4 months of dual combination therapy. The availability of oral prostacyclin analogues (selexipag and treprostinil) makes it easier to consider adding these agents early on, Dr. Chakinala and Dr. Richardson said.
Patients who fall into the high-risk category, at any point, are still best managed with parenteral prostacyclin analogues, Dr. Chakinala said.
In general, said Dr. Williamson, who also directs the University of Kansas Pulmonary Hypertension Comprehensive Care Center, “the PH community tends to be fairly aggressive up front, and with a low threshold for using prostacyclin analogues.”
The agents are “always part of the picture for someone who is really ill, in functional class IV, or has really impaired right ventricular function,” he said. “And we’re finding increased roles in patients who are not as ill but still have decompensated right ventricular dysfunction. It’s something we now consider.”
Recently published research on up-front oral triple therapy suggests possible benefit for some patients – but it’s far from conclusive, said Dr. Chakinala. The TRITON study randomized treatment-naive patients to the traditional ERA-PDE5 combination and either oral selexipag (a selective prostacyclin receptor agonist) or placebo as a third agent. It found no significant difference in reduction in PVR, the primary outcome, at week 26. However, the authors reported a “possible signal” for improved long-term outcomes with triple therapy.
“Based on this best evidence from a randomized clinical trial, I think it’s unfair to say that all patients should be on triple combination therapy right out of the gate,” he said. “Having said that, more recent [European] data showed that two drugs fell short of the mark in some patients, with high rates of clinical progression. And even in AMBITION, there were a number of patients in the combination arm who didn’t have a robust response.”
A 2021 retrospective analysis from the French Pulmonary Hypertension Registry – one of the European studies – assessed survival with monotherapy, dual therapy, or triple-combination therapy (two orals with a parenteral prostacyclin), and found no difference between monotherapy and dual therapy in high-risk patients.
Experts have been upping the ante, therefore, on early assessment and frequent reassessment of treatment response. Not long ago, patients were typically reassessed 6-12 months after the initiation of treatment. Now, experts at the PH care centers want to assess patients at 3-4 months and adjust or intensify treatment regimens for those who don’t yet qualify as low risk using a multidimensional risk score calculator.
The REVEAL (Registry to Evaluate Early and Long-Term PAH Management) risk score calculator, for instance, predicts the probability of 1-year survival and assigns patients to a strata of risk level based on either 12 or 6 variables (for the full or “lite” versions).]
Even better monitoring and risk assessment is needed, however, to “help sift out which patients are not improving enough on initial therapy or who are starting to fall off after being on a regimen for a period of time,” Dr. Chakinala said.
Today, with a network of accredited centers of expertise and a desire and need for many patients to remain close to home, Dr. Chakinala encourages finding a balance. Well-resourced clinicians can strive for early diagnosis and management – potentially initiating ERA–PDE-5 inhibitor combination therapy – but still should collaborate with PH experts.
“It’s a good idea to comanage these patients and let the experts see them periodically to help you determine when your patient may be declining,” he said. “The timetable for reassessment, the complexity of the reassessment, and the need to escalate to more advanced therapies has never been more important.”
Research highlights
Therapies that target inflammation and altered metabolism – including metformin – are among those being investigated for PAH. So are therapies targeting dysfunctional bone morphogenetic protein pathway signaling, which has been shown to be associated with hereditary, idiopathic, and likely other forms of PAH; one such drug, called sotatercept, is currently at the phase 3 trial stage.
Most promising for PAH may be the research efforts involving deep phenotyping, said Andrew J. Sweatt, MD, of Stanford (Calif.) University and the Vera Moulton Wall Center for Pulmonary Vascular Disease.
“It’s where a lot of research is headed – deep phenotyping to deconstruct the molecular and clinical heterogeneity that exists within PAH ... to detect distinct subphenotypes of patients who would respond to particular therapies,” said Dr. Sweatt, who led a review of PH clinical research presented at the 2020 American Thoracic Society International Conference
“Right now, we largely treat all patients the same ... [while] we know that patients have a wide response to therapies and there’s a lot of clinical heterogeneity in how their disease evolves over time,” he said.
Data from a large National Institutes of Health–funded multicenter phenotyping study of PH is being analyzed and should yield findings and publications starting this year, said Anna R. Hemnes, MD, associate professor of medicine at Vanderbilt University Medical Center, Nashville, Tenn., and an investigator with the initiative, coined “Redefining Pulmonary Hypertension through Pulmonary Disease Phenomics (PVDOMICS).”
Patients have undergone advanced imaging (for example, echocardiography, cardiac MRI, chest CT, ventilation/perfusion scans), advanced testing through right heart catheterization, body composition testing, quality of life questionnaires, and blood draws that have been analyzed for DNA and RNA expression, proteomics, and metabolomics, said Dr. Hemnes, assistant director of Vanderbilt’s Pulmonary Vascular Center.
The initiative aims to refine the classification of all kinds of PH and “to bring precision medicine to the field so we’re no longer characterizing somebody [based on imaging] and right heart catheterization, but we also incorporating molecular pieces and biomarkers into the diagnostic evaluation,” she said.
In the short term, the results of deep phenotyping should “allow us to be more effective with our therapy recommendations,” Dr. Hemnes said. “Then hopefully in the longer term, [identified biomarkers] will help us to develop new, more effective therapies.”
Dr. Sweatt and Dr. Williamson reported that they have no relevant financial disclosures. Dr. Hemnes reported that she holds stock in Tenax (which is studying a tyrosine kinase inhibitor for PAH) and serves as a consultant for Acceleron, Bayer, GossamerBio, United Therapeutics, and Janssen. She also receives research funding from Imara. Dr. Chakinala reported that he is an investigator on clinical trials for a number of pharmaceutical companies. He also serves on advisory boards for Phase Bio, Liquidia/Rare Gen, Bayer, Janssen, Trio Health Analytics, and Aerovate.
Absolute increase in Kawasaki CV risk remains small in long-term follow-up
Vasculitis of the coronary arteries is a well-recognized acute complication of Kawasaki disease, but the long-term risk of cardiovascular (CV) sequelae does not appear to be clinically meaningful for most patients, according to results from an analysis of data presented at the annual meeting of the Canadian Rheumatology Association.
For patients and parents, these data provide “a message of reassurance,” according to Jennifer J.Y. Lee, MD, a pediatric rheumatologist affiliated with the Hospital for Sick Children, Toronto.
The long-term outcomes were characterized as reassuring even though rates of hypertension, major adverse cardiac events (MACE), and death from CV events were higher in patients with Kawasaki disease relative to controls in a retrospective data-linkage study. In fact, these differences were highly statistically significant, but the absolute differences were extremely small.
For this analysis, the 1,174 patients diagnosed with Kawasaki disease at Dr. Lee’s institution between 1991 and 2008 were compared in a 10:1 ratio to 11,740 controls matched for factors such as age, sex, ethnicity, and geographic region. The median follow-up period was 20 years, and the maximum was 28 years.
Adjusted CV risks are significant
In an adjusted Cox proportional hazard ratio model, patients in the Kawasaki group had a more than twofold increase in risk for hypertension (aHR, 2.3; P < .0001) and all-cause mortality (aHR, 2.5; P = .009). They also had more than a 10-fold increase in risk for MACE (aHR, 10.3; P < .0001).
These statistics belie the clinical relevance, according to Dr. Lee. Because of the very low rates of all the measured events in both groups, there was just one more case of hypertension per 1,250 patient-years of follow-up, one more case of MACE per 833 patient-years of follow-up, and one more death for 3,846 patient years of follow-up.
Moreover, when these outcomes were graphed over time, most events occurred during the acute period or in the initial years of follow-up.
“There was not a constant increase in risk of these outcomes over time for patients with Kawasaki disease relative to the controls,” Dr. Lee reported. “The long-term prognosis for Kawasaki patients remains favorable.”
European group reports similar results
Similar results from a single-center experience were published 3 years ago. In that study, 207 Kawasaki patients treated at the University of Lausanne (Switzerland) were followed for 30 years. Complications after the acute phase were characterized as “rare.”
For example, only three patients (1.4%) had a subsequent episode of myocardial ischemia. All three had developed a coronary aneurysm during the acute phase of Kawasaki disease. The authors of that study reported that children who had not received immunoglobulins during the acute phase or who developed Kawasaki disease outside of the usual age range were more likely to have subsequent events, such as disease recurrence.
Other studies of long-term CV outcomes in patients with Kawasaki disease generally show similar data, according to James T. Gaensbauer, MD, a pediatric infectious disease specialist at the Mayo Clinic, Rochester, Minn.
“I generally agree with the premise that major complications are rare when you compare a cohort of patients with Kawasaki disease with the general population,” Dr. Gaensbauer said. However, he added, “I do not think you can say no one needs to worry.”
Severity of acute disease might matter
During the acute phase of Kawasaki disease, the arterial damage varies. As suggested in the University of Lausanne follow-up, patients with significant coronary aneurysms do appear to be at greater risk of long-term complications. Dr. Gaensbauer cited a statement from the American Heart Association that noted a higher risk of CV sequelae from Kawasaki disease with a greater or more severe coronary aneurysm or in the face of other evidence of damage to the arterial tree.
“The clinical course within the first 2 years of Kawasaki disease appears to be important for risk of CV complications after this time,” Dr. Gaensbauer said.
The absolute risk of CV events in patients with a more complicated acute course of Kawasaki disease remains incompletely understood, but Dr. Gaensbauer said that there are several sets of data, including these new data from the Hospital for Sick Children, that suggest that the overall prognosis is good. However, he cautioned that this reassurance does not necessarily apply to children with a difficult acute course.
According to the 2017 AHA statement on Kawasaki disease, risk stratification based on echocardiography and other measures after the acute phase of Kawasaki disease are reasonable to determine if long-term follow-up is needed. In those without abnormalities, it is reasonable to forgo further cardiology assessment.
Dr. Lee and Dr. Gaensbauer reported having no potential conflicts of interest.
Vasculitis of the coronary arteries is a well-recognized acute complication of Kawasaki disease, but the long-term risk of cardiovascular (CV) sequelae does not appear to be clinically meaningful for most patients, according to results from an analysis of data presented at the annual meeting of the Canadian Rheumatology Association.
For patients and parents, these data provide “a message of reassurance,” according to Jennifer J.Y. Lee, MD, a pediatric rheumatologist affiliated with the Hospital for Sick Children, Toronto.
The long-term outcomes were characterized as reassuring even though rates of hypertension, major adverse cardiac events (MACE), and death from CV events were higher in patients with Kawasaki disease relative to controls in a retrospective data-linkage study. In fact, these differences were highly statistically significant, but the absolute differences were extremely small.
For this analysis, the 1,174 patients diagnosed with Kawasaki disease at Dr. Lee’s institution between 1991 and 2008 were compared in a 10:1 ratio to 11,740 controls matched for factors such as age, sex, ethnicity, and geographic region. The median follow-up period was 20 years, and the maximum was 28 years.
Adjusted CV risks are significant
In an adjusted Cox proportional hazard ratio model, patients in the Kawasaki group had a more than twofold increase in risk for hypertension (aHR, 2.3; P < .0001) and all-cause mortality (aHR, 2.5; P = .009). They also had more than a 10-fold increase in risk for MACE (aHR, 10.3; P < .0001).
These statistics belie the clinical relevance, according to Dr. Lee. Because of the very low rates of all the measured events in both groups, there was just one more case of hypertension per 1,250 patient-years of follow-up, one more case of MACE per 833 patient-years of follow-up, and one more death for 3,846 patient years of follow-up.
Moreover, when these outcomes were graphed over time, most events occurred during the acute period or in the initial years of follow-up.
“There was not a constant increase in risk of these outcomes over time for patients with Kawasaki disease relative to the controls,” Dr. Lee reported. “The long-term prognosis for Kawasaki patients remains favorable.”
European group reports similar results
Similar results from a single-center experience were published 3 years ago. In that study, 207 Kawasaki patients treated at the University of Lausanne (Switzerland) were followed for 30 years. Complications after the acute phase were characterized as “rare.”
For example, only three patients (1.4%) had a subsequent episode of myocardial ischemia. All three had developed a coronary aneurysm during the acute phase of Kawasaki disease. The authors of that study reported that children who had not received immunoglobulins during the acute phase or who developed Kawasaki disease outside of the usual age range were more likely to have subsequent events, such as disease recurrence.
Other studies of long-term CV outcomes in patients with Kawasaki disease generally show similar data, according to James T. Gaensbauer, MD, a pediatric infectious disease specialist at the Mayo Clinic, Rochester, Minn.
“I generally agree with the premise that major complications are rare when you compare a cohort of patients with Kawasaki disease with the general population,” Dr. Gaensbauer said. However, he added, “I do not think you can say no one needs to worry.”
Severity of acute disease might matter
During the acute phase of Kawasaki disease, the arterial damage varies. As suggested in the University of Lausanne follow-up, patients with significant coronary aneurysms do appear to be at greater risk of long-term complications. Dr. Gaensbauer cited a statement from the American Heart Association that noted a higher risk of CV sequelae from Kawasaki disease with a greater or more severe coronary aneurysm or in the face of other evidence of damage to the arterial tree.
“The clinical course within the first 2 years of Kawasaki disease appears to be important for risk of CV complications after this time,” Dr. Gaensbauer said.
The absolute risk of CV events in patients with a more complicated acute course of Kawasaki disease remains incompletely understood, but Dr. Gaensbauer said that there are several sets of data, including these new data from the Hospital for Sick Children, that suggest that the overall prognosis is good. However, he cautioned that this reassurance does not necessarily apply to children with a difficult acute course.
According to the 2017 AHA statement on Kawasaki disease, risk stratification based on echocardiography and other measures after the acute phase of Kawasaki disease are reasonable to determine if long-term follow-up is needed. In those without abnormalities, it is reasonable to forgo further cardiology assessment.
Dr. Lee and Dr. Gaensbauer reported having no potential conflicts of interest.
Vasculitis of the coronary arteries is a well-recognized acute complication of Kawasaki disease, but the long-term risk of cardiovascular (CV) sequelae does not appear to be clinically meaningful for most patients, according to results from an analysis of data presented at the annual meeting of the Canadian Rheumatology Association.
For patients and parents, these data provide “a message of reassurance,” according to Jennifer J.Y. Lee, MD, a pediatric rheumatologist affiliated with the Hospital for Sick Children, Toronto.
The long-term outcomes were characterized as reassuring even though rates of hypertension, major adverse cardiac events (MACE), and death from CV events were higher in patients with Kawasaki disease relative to controls in a retrospective data-linkage study. In fact, these differences were highly statistically significant, but the absolute differences were extremely small.
For this analysis, the 1,174 patients diagnosed with Kawasaki disease at Dr. Lee’s institution between 1991 and 2008 were compared in a 10:1 ratio to 11,740 controls matched for factors such as age, sex, ethnicity, and geographic region. The median follow-up period was 20 years, and the maximum was 28 years.
Adjusted CV risks are significant
In an adjusted Cox proportional hazard ratio model, patients in the Kawasaki group had a more than twofold increase in risk for hypertension (aHR, 2.3; P < .0001) and all-cause mortality (aHR, 2.5; P = .009). They also had more than a 10-fold increase in risk for MACE (aHR, 10.3; P < .0001).
These statistics belie the clinical relevance, according to Dr. Lee. Because of the very low rates of all the measured events in both groups, there was just one more case of hypertension per 1,250 patient-years of follow-up, one more case of MACE per 833 patient-years of follow-up, and one more death for 3,846 patient years of follow-up.
Moreover, when these outcomes were graphed over time, most events occurred during the acute period or in the initial years of follow-up.
“There was not a constant increase in risk of these outcomes over time for patients with Kawasaki disease relative to the controls,” Dr. Lee reported. “The long-term prognosis for Kawasaki patients remains favorable.”
European group reports similar results
Similar results from a single-center experience were published 3 years ago. In that study, 207 Kawasaki patients treated at the University of Lausanne (Switzerland) were followed for 30 years. Complications after the acute phase were characterized as “rare.”
For example, only three patients (1.4%) had a subsequent episode of myocardial ischemia. All three had developed a coronary aneurysm during the acute phase of Kawasaki disease. The authors of that study reported that children who had not received immunoglobulins during the acute phase or who developed Kawasaki disease outside of the usual age range were more likely to have subsequent events, such as disease recurrence.
Other studies of long-term CV outcomes in patients with Kawasaki disease generally show similar data, according to James T. Gaensbauer, MD, a pediatric infectious disease specialist at the Mayo Clinic, Rochester, Minn.
“I generally agree with the premise that major complications are rare when you compare a cohort of patients with Kawasaki disease with the general population,” Dr. Gaensbauer said. However, he added, “I do not think you can say no one needs to worry.”
Severity of acute disease might matter
During the acute phase of Kawasaki disease, the arterial damage varies. As suggested in the University of Lausanne follow-up, patients with significant coronary aneurysms do appear to be at greater risk of long-term complications. Dr. Gaensbauer cited a statement from the American Heart Association that noted a higher risk of CV sequelae from Kawasaki disease with a greater or more severe coronary aneurysm or in the face of other evidence of damage to the arterial tree.
“The clinical course within the first 2 years of Kawasaki disease appears to be important for risk of CV complications after this time,” Dr. Gaensbauer said.
The absolute risk of CV events in patients with a more complicated acute course of Kawasaki disease remains incompletely understood, but Dr. Gaensbauer said that there are several sets of data, including these new data from the Hospital for Sick Children, that suggest that the overall prognosis is good. However, he cautioned that this reassurance does not necessarily apply to children with a difficult acute course.
According to the 2017 AHA statement on Kawasaki disease, risk stratification based on echocardiography and other measures after the acute phase of Kawasaki disease are reasonable to determine if long-term follow-up is needed. In those without abnormalities, it is reasonable to forgo further cardiology assessment.
Dr. Lee and Dr. Gaensbauer reported having no potential conflicts of interest.
FROM THE ANNUAL MEETING OF THE CANADIAN RHEUMATOLOGY ASSOCIATION
Lilly calls it quits on baricitinib’s development for lupus
The company is also in talks with the FDA about how to move forward with the drug’s development for atopic dermatitis.
from two pivotal phase 3 trials, SLE-BRAVE-I and II, the company announced Jan. 28.
Lilly said that the primary endpoint of the SLE-BRAVE-I trial, the proportion of adults with active SLE who met criteria for response on the SLE Responder Index-4 at week 52, was significantly greater among patients treated with 4 mg baricitinib daily than with placebo. However, this endpoint was not met in SLE-BRAVE-II, and no key secondary endpoints were met in either trial. In the announcement, Lilly noted that safety was not a reason for discontinuation because data from these trials were consistent with those previously seen with baricitinib.
The company statement said that it will work with investigators on concluding the combined long-term extension study of the trials.
Baricitinib, a Janus kinase (JAK) inhibitor, had previously shown promising results in a phase 2 trial in patients with SLE. It is approved by the U.S. Food and Drug Administration for treating adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more tumor necrosis factor blockers at a dose of 2 mg once daily and has an emergency use authorization for the treatment of hospitalized patients with COVID-19.
The decision to stop baricitinib’s development for SLE will not affect other research efforts with the drug, the company said.
Development for atopic dermatitis
Lilly also noted that it is in discussion with the FDA about the status of a supplemental new drug application of baricitinib for the treatment of adults with moderate to severe atopic dermatitis (AD). In its press release, Lilly said, “At this point, the company does not have alignment with the FDA on the indicated population. Given the agency’s position, there is a possibility that this could lead to a Complete Response Letter (CRL). The efficacy and safety profile of Olumiant was evaluated in eight atopic dermatitis clinical trials (six double-blind, randomized, placebo-controlled studies and two long-term extension studies) inclusive of patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. The safety profile in these trials was consistent with previously published Olumiant data.”
Baricitinib was the first JAK inhibitor approved to treat patients with moderate to severe AD who have an inadequate response to topical treatments in the European Union and Japan.
The Lilly announcement was made with Incyte, the company that discovered baricitinib.
The company is also in talks with the FDA about how to move forward with the drug’s development for atopic dermatitis.
The company is also in talks with the FDA about how to move forward with the drug’s development for atopic dermatitis.
from two pivotal phase 3 trials, SLE-BRAVE-I and II, the company announced Jan. 28.
Lilly said that the primary endpoint of the SLE-BRAVE-I trial, the proportion of adults with active SLE who met criteria for response on the SLE Responder Index-4 at week 52, was significantly greater among patients treated with 4 mg baricitinib daily than with placebo. However, this endpoint was not met in SLE-BRAVE-II, and no key secondary endpoints were met in either trial. In the announcement, Lilly noted that safety was not a reason for discontinuation because data from these trials were consistent with those previously seen with baricitinib.
The company statement said that it will work with investigators on concluding the combined long-term extension study of the trials.
Baricitinib, a Janus kinase (JAK) inhibitor, had previously shown promising results in a phase 2 trial in patients with SLE. It is approved by the U.S. Food and Drug Administration for treating adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more tumor necrosis factor blockers at a dose of 2 mg once daily and has an emergency use authorization for the treatment of hospitalized patients with COVID-19.
The decision to stop baricitinib’s development for SLE will not affect other research efforts with the drug, the company said.
Development for atopic dermatitis
Lilly also noted that it is in discussion with the FDA about the status of a supplemental new drug application of baricitinib for the treatment of adults with moderate to severe atopic dermatitis (AD). In its press release, Lilly said, “At this point, the company does not have alignment with the FDA on the indicated population. Given the agency’s position, there is a possibility that this could lead to a Complete Response Letter (CRL). The efficacy and safety profile of Olumiant was evaluated in eight atopic dermatitis clinical trials (six double-blind, randomized, placebo-controlled studies and two long-term extension studies) inclusive of patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. The safety profile in these trials was consistent with previously published Olumiant data.”
Baricitinib was the first JAK inhibitor approved to treat patients with moderate to severe AD who have an inadequate response to topical treatments in the European Union and Japan.
The Lilly announcement was made with Incyte, the company that discovered baricitinib.
from two pivotal phase 3 trials, SLE-BRAVE-I and II, the company announced Jan. 28.
Lilly said that the primary endpoint of the SLE-BRAVE-I trial, the proportion of adults with active SLE who met criteria for response on the SLE Responder Index-4 at week 52, was significantly greater among patients treated with 4 mg baricitinib daily than with placebo. However, this endpoint was not met in SLE-BRAVE-II, and no key secondary endpoints were met in either trial. In the announcement, Lilly noted that safety was not a reason for discontinuation because data from these trials were consistent with those previously seen with baricitinib.
The company statement said that it will work with investigators on concluding the combined long-term extension study of the trials.
Baricitinib, a Janus kinase (JAK) inhibitor, had previously shown promising results in a phase 2 trial in patients with SLE. It is approved by the U.S. Food and Drug Administration for treating adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more tumor necrosis factor blockers at a dose of 2 mg once daily and has an emergency use authorization for the treatment of hospitalized patients with COVID-19.
The decision to stop baricitinib’s development for SLE will not affect other research efforts with the drug, the company said.
Development for atopic dermatitis
Lilly also noted that it is in discussion with the FDA about the status of a supplemental new drug application of baricitinib for the treatment of adults with moderate to severe atopic dermatitis (AD). In its press release, Lilly said, “At this point, the company does not have alignment with the FDA on the indicated population. Given the agency’s position, there is a possibility that this could lead to a Complete Response Letter (CRL). The efficacy and safety profile of Olumiant was evaluated in eight atopic dermatitis clinical trials (six double-blind, randomized, placebo-controlled studies and two long-term extension studies) inclusive of patients whose disease is not adequately controlled with topical prescription therapies or when those therapies are not advisable. The safety profile in these trials was consistent with previously published Olumiant data.”
Baricitinib was the first JAK inhibitor approved to treat patients with moderate to severe AD who have an inadequate response to topical treatments in the European Union and Japan.
The Lilly announcement was made with Incyte, the company that discovered baricitinib.
Rituximab and COVID-19 vaccines: Studies begin to answer key questions
Rituximab has presented something of a conundrum for patients taking the monoclonal antibody during the COVID-19 pandemic.
Used to manage a variety of autoimmune diseases and cancers, rituximab acts against CD20 proteins expressed on the surface of B cells, causing B-cell depletion. However, it is this B-cell depletion that may put these patients at greater risk of COVID-19 development, progression to more severe disease, and in-hospital mortality. Evidence for this appears to be mixed, with studies showing both that patients using rituximab to manage various diseases are and are not at increased risk for SARS-CoV-2 infection, COVID-19 progression, and mortality.
As COVID-19 vaccine rollouts take place across the world, more questions have been raised about the relationship between B-cell depletion from anti-CD20 therapies and COVID-19 vaccines. Do rituximab and other anti-CD20 therapies affect a patient’s response to COVID-19 vaccines? If this is the case, does the timing of anti-CD20 treatment matter to maximize B-cell levels and improve the vaccine’s effectiveness? And how do COVID-19 vaccine booster doses factor into the equation?
Humoral and cell-mediated responses following COVID-19 vaccination
First, the bad news: The vaccine is unquestionably safe to administer in patients taking rituximab, but one thing that has been well established is that antibody response to COVID-19 vaccination in these individuals does is reduced. This isn’t entirely unprecedented, as previous studies have shown a weakened immune response to pneumococcal polysaccharide and keyhole limpet hemocyanin vaccines among patients taking rituximab.

“Compromised immunogenicity to the SARS-CoV-2 vaccines has been demonstrated in rituximab-treated patients, which is of particular concern given the observation that B-cell–depleting therapies may be associated with worse COVID outcomes,” Robert F. Spiera, MD, director of the Scleroderma, Vasculitis, and Myositis Center at the Hospital for Special Surgery in New York, said in an interview.
For example, in a recent study from the Medical University of Vienna, 29 (39%) of 74 patients receiving rituximab (43% as monotherapy, 57% with conventional-synthetic disease-modifying antirheumatic drugs) who were vaccinated with either the Comirnaty (Pfizer-BioNTech) or Spikevax (Moderna) COVID-19 vaccine achieved seroconversion, compared with 100% of patients in a healthy control group, and all but 1 patient without detectable CD19+ peripheral B cells did not develop anti–SARS-CoV-2 receptor-binding domain antibodies.
“There is an increasing number of studies in this field, and they confirm that patients treated with rituximab and other anti-CD20 agents have severely reduced serological responses to COVID-19 vaccines,” Ingrid Jyssum, MD, of the division of rheumatology and research at Diakonhjemmet Hospital in Oslo, said in an interview.

One silver lining is that patients treated with anti-CD20 therapies appear to have a cell-mediated response following vaccination even if they don’t develop SARS-CoV-2 antibodies. “Studies that also investigate T-cell responses are starting to emerge, and so far, they show that, even if the patients do not have antibodies, they may have T-cell responses,” Dr. Jyssum said.
One study of 24 patients with autoimmune diseases taking rituximab that evaluated humoral and T-cell responses following vaccination with the Comirnaty vaccine found that none had a humoral response to the vaccine, but the T-cell response from that group did not significantly differ from 35 patients receiving other immunosuppressants and 26 patients in a healthy control group. In another study of rituximab- or ocrelizumab-treated patients who received mRNA-based COVID-19 vaccines, 69.4% developed SARS-CoV-2–specific antibodies, compared with a control group, but 96.2% of patients taking ocrelizumab and 81.8% of patients taking rituximab mounted a spike-specific CD8+ T-cell response, compared with 66.7% in the control group, and there were comparable rates (85%-90%) of spike-specific CD4+ T cells in all groups. In the study from the Medical University of Vienna, T-cell response was detected in rituximab-treated patients who both did and did not mount an antibody response.
The clinical relevance of how a blunted humoral immune response but a respectable T-cell response to COVID-19 vaccines affects patients treated with anti-CD20 therapies isn’t currently known, Dr. Jyssum said.
While these data are reassuring, they’re also incomplete, Dr. Spiera noted. “The ultimate outcome of relevance to assess vaccine efficacy is protection from COVID and from severe outcomes of COVID infection (i.e., hospitalization, mechanical ventilation, death). That data will require assessment of very large numbers of rituximab-treated vaccinated patients to be compared with rituximab-treated unvaccinated patients, and is unlikely to be forthcoming in the very near future.
“In the meantime, however, achieving serologic positivity, meaning having evidence of serologic as well as cellular immunity following vaccination, is a desired outcome, and likely implies more robust immunity.”
Does treatment timing impact COVID-19 vaccine response?
Given enough time, B-cell reconstitution will occur in patients taking rituximab. With that in mind, is it beneficial to wait a certain amount of time after a patient has stopped rituximab therapy or time since their last dose before giving them a COVID-19 vaccine? In their guidance on COVID-19 vaccines for patients with rheumatic and musculoskeletal diseases, the American College of Rheumatology said there is moderate evidence to consider “optimal timing of dosing and vaccination with the rheumatology provider before proceeding.”
“Guidelines and preliminary studies of serologic response to COVID vaccine in rituximab-treated patients have suggested that longer time from last rituximab exposure is associated with a greater likelihood of a serologic response,” Dr. Spiera said.
In a brief report published in Arthritis & Rheumatology, Dr. Spiera and colleagues performed a retrospective chart review of 56 patients with varying levels of last exposure to rituximab who received a COVID-19 vaccine. Their results showed that, when patients were vaccinated 6-12 months after the last rituximab dose, 55% were seronegative, and when this was more than 12 months, only 13% were seronegative, compared with seronegativity in 86% who were vaccinated less than 6 months after their last rituximab dose.
The RituxiVac trial, conducted by researchers in Switzerland, also examined vaccine responses of 96 rituximab-treated patients who received Comirnaty or Spikevax; results recently published in The Lancet Rheumatology showed findings similar to other studies, with reduced humoral and cell-mediated responses. In the RituxiVac trial, the median time to last anti-CD20 treatment was 1.07 years.
“The typical interval between rituximab doses [for treatment of rheumatoid arthritis, as well as for remission maintenance in antineutrophil cytoplasmic antibody–associated vasculitis] is typically 6 months, and this has become widely used as the interval from last rituximab to time of COVID vaccination, with a recommendation to wait 4 weeks (if possible) from time of vaccination until the next rituximab administration,” Dr. Spiera explained. However, this window seems to vary depending on the study.
Recent research published in Arthritis & Rheumatology indicates B-cell levels could be a relevant indicator for humoral and cell-mediated response in patients with rheumatic diseases treated with rituximab, with a level of 10 B cells/mcL (0.4% of lymphocytes) identified as one potential marker for likely seroconversion following COVID-19 vaccination.
“In some smaller case series, it has been further recognized that rituximab-treated patients who were beginning to reconstitute peripheral B cells were most likely to respond serologically. Our present study confirmed those findings, demonstrating that the presence of detectable B cells was strongly associated with vaccine responsiveness, and affords complementary information to time from last [rituximab dose] in informing the likelihood of a vaccine response,” Dr. Spiera said.
However, the literature is limited in this area, and an exact cutoff for B-cell counts in these patients isn’t currently known, Dr. Jyssum said. A better metric is time away from anti-CD20 therapies, with CD19 cell count being highly correlated with last infusion.
Dr. Spiera agreed that there is no consistent B-cell percentage that works as a cutoff. “In our study, we looked at it as a binary variable, although we did find that a higher percentage of B cells in the peripheral lymphocyte population was associated with a higher likelihood of seroconversion. We did not, however, identify a ‘threshold’ for vaccine serologic responsiveness.”
Should clinicians measure antibodies?
The Food and Drug Administration and the Centers for Disease Control and Prevention have recommended that health care providers and the public not use COVID-19 antibody tests as a way to gauge immunity after exposure to SARS-CoV-2 and after receiving a COVID-19 vaccination. The ACR’s guidance on COVID-19 vaccination for patients with rheumatic and musculoskeletal diseases strongly recommends against ordering antibody tests for patients with autoimmune inflammatory rheumatic diseases as a way to measure immunity.
“Generally, such measurements are not recommended as the clinical correlate of various antibody levels are not known,” Dr. Jyssum said. “With regular infusions of rituximab or other anti-CD20 agents, one cannot expect that these patients will develop significant levels of antibodies.”
However, she said there might be situations where it’s useful to know whether a patient has developed antibodies at all. “Assessing the significance of specific antibody levels is difficult, and the subject of scientific studies. Patients lacking a humoral vaccine response are left to rely on their T-cell responses and on infectious control measures to prevent disease.”
Dr. Spiera said he disagreed with guidelines recommending against checking antibody levels after vaccination, “particularly in patients treated with immunosuppressive medications that might be expected to blunt their serologic response to the vaccines.
“Although we cannot be sure what level of measurable antibodies offer what level of protection, most clinicians would agree that patients who demonstrate no detectable antibodies (which is a common finding in rituximab-treated patients) should be considered at higher risk,” he said. “Indeed, recommendations regarding booster vaccine administration in general was initially based on the observation of declining antibody levels with longer time from vaccination.”
Do COVID-19 vaccine boosters help patients on anti-CD20 therapy?
As of January 2022, the FDA and CDC have recommended a third primary series shot of COVID-19 vaccines for some moderately to severely immunocompromised patients as young as 5 years old (for Comirnaty vaccine) or a booster shot of either Comirnaty or Spikevax for everyone aged 12 years and older, including immunocompromised people, while the ACR goes into more detail and recommends clinicians time a patient’s booster shot with temporary treatment interruption.
In The Lancet Rheumatology, Dr. Jyssum and colleagues recently published results from the prospective Nor-vaC study examining the humoral and cell-mediated immune responses of 87 patients with RA being treated with rituximab who received the Comirnaty, Spikevax, or Vaxzevria (AstraZeneca) COVID-19 vaccines; of these, 49 patients received a booster dose at a median of 70 days after completing their primary series. The results showed 19 patients (28.1%) had a serologic response after their primary series, while 8 of 49 patients (16.3%) who received their booster dose had a serologic response.
All patients who received a third dose in the study had a T-cell response, Dr. Jyssum said. “This is reassuring for patients and clinicians. T cells have been found to be important in countering COVID-19 disease, but whether we can rely on the T-cell response alone in the absence of antibodies to protect patients from infection or from serious COVID disease is still not determined,” she said.
When asked if she would recommend COVID-19 vaccine booster doses for patients on rituximab, Dr. Jyssum replied: “Absolutely.”
Another study, recently published in Annals of the Rheumatic Diseases, examined heterologous and homologous booster doses for 60 patients receiving rituximab without seroconversion after their COVID-19 vaccine primary series. The results showed no significant difference in new seroconversion at 4 weeks based on whether the patient received a vector or mRNA vaccine (22% vs. 32%), but all patients who received a booster dose with a vector vaccine had specific T-cell responses, compared with 81% of patients who received an mRNA vaccine booster. There was a new humoral and/or cellular response in 9 of 11 patients (82%), and most patients with peripheral B cells (12 of 18 patients; 67%) achieved seroconversion.
“Our data show that a cellular and/or humoral immune response can be achieved on a third COVID-19 vaccination in most of the patients who initially developed neither a humoral nor a cellular immune response,” the researchers concluded. “The efficacy data together with the safety data seen in our trial provide a favorable risk/benefit ratio and support the implementation of a third vaccination for nonseroconverted high-risk autoimmune disease patients treated with B-cell–depleting agents.”
Dr. Spiera said booster doses are an important part of the equation, and “it is important to consider factors that would be associated with a greater likelihood of achieving a serologic response, particularly in those patients who did not demonstrate a serologic response to the initial vaccines series.
“Preliminary data shows that the beginnings of B-cell reconstitution is also associated with a positive serologic response following a booster of the COVID-19 vaccine,” he said.
The authors of the cited studies reported numerous relevant financial disclosures. Dr. Spiera and Dr. Jyssum reported no relevant financial disclosures.
Rituximab has presented something of a conundrum for patients taking the monoclonal antibody during the COVID-19 pandemic.
Used to manage a variety of autoimmune diseases and cancers, rituximab acts against CD20 proteins expressed on the surface of B cells, causing B-cell depletion. However, it is this B-cell depletion that may put these patients at greater risk of COVID-19 development, progression to more severe disease, and in-hospital mortality. Evidence for this appears to be mixed, with studies showing both that patients using rituximab to manage various diseases are and are not at increased risk for SARS-CoV-2 infection, COVID-19 progression, and mortality.
As COVID-19 vaccine rollouts take place across the world, more questions have been raised about the relationship between B-cell depletion from anti-CD20 therapies and COVID-19 vaccines. Do rituximab and other anti-CD20 therapies affect a patient’s response to COVID-19 vaccines? If this is the case, does the timing of anti-CD20 treatment matter to maximize B-cell levels and improve the vaccine’s effectiveness? And how do COVID-19 vaccine booster doses factor into the equation?
Humoral and cell-mediated responses following COVID-19 vaccination
First, the bad news: The vaccine is unquestionably safe to administer in patients taking rituximab, but one thing that has been well established is that antibody response to COVID-19 vaccination in these individuals does is reduced. This isn’t entirely unprecedented, as previous studies have shown a weakened immune response to pneumococcal polysaccharide and keyhole limpet hemocyanin vaccines among patients taking rituximab.
“Compromised immunogenicity to the SARS-CoV-2 vaccines has been demonstrated in rituximab-treated patients, which is of particular concern given the observation that B-cell–depleting therapies may be associated with worse COVID outcomes,” Robert F. Spiera, MD, director of the Scleroderma, Vasculitis, and Myositis Center at the Hospital for Special Surgery in New York, said in an interview.
For example, in a recent study from the Medical University of Vienna, 29 (39%) of 74 patients receiving rituximab (43% as monotherapy, 57% with conventional-synthetic disease-modifying antirheumatic drugs) who were vaccinated with either the Comirnaty (Pfizer-BioNTech) or Spikevax (Moderna) COVID-19 vaccine achieved seroconversion, compared with 100% of patients in a healthy control group, and all but 1 patient without detectable CD19+ peripheral B cells did not develop anti–SARS-CoV-2 receptor-binding domain antibodies.
“There is an increasing number of studies in this field, and they confirm that patients treated with rituximab and other anti-CD20 agents have severely reduced serological responses to COVID-19 vaccines,” Ingrid Jyssum, MD, of the division of rheumatology and research at Diakonhjemmet Hospital in Oslo, said in an interview.
One silver lining is that patients treated with anti-CD20 therapies appear to have a cell-mediated response following vaccination even if they don’t develop SARS-CoV-2 antibodies. “Studies that also investigate T-cell responses are starting to emerge, and so far, they show that, even if the patients do not have antibodies, they may have T-cell responses,” Dr. Jyssum said.
One study of 24 patients with autoimmune diseases taking rituximab that evaluated humoral and T-cell responses following vaccination with the Comirnaty vaccine found that none had a humoral response to the vaccine, but the T-cell response from that group did not significantly differ from 35 patients receiving other immunosuppressants and 26 patients in a healthy control group. In another study of rituximab- or ocrelizumab-treated patients who received mRNA-based COVID-19 vaccines, 69.4% developed SARS-CoV-2–specific antibodies, compared with a control group, but 96.2% of patients taking ocrelizumab and 81.8% of patients taking rituximab mounted a spike-specific CD8+ T-cell response, compared with 66.7% in the control group, and there were comparable rates (85%-90%) of spike-specific CD4+ T cells in all groups. In the study from the Medical University of Vienna, T-cell response was detected in rituximab-treated patients who both did and did not mount an antibody response.
The clinical relevance of how a blunted humoral immune response but a respectable T-cell response to COVID-19 vaccines affects patients treated with anti-CD20 therapies isn’t currently known, Dr. Jyssum said.
While these data are reassuring, they’re also incomplete, Dr. Spiera noted. “The ultimate outcome of relevance to assess vaccine efficacy is protection from COVID and from severe outcomes of COVID infection (i.e., hospitalization, mechanical ventilation, death). That data will require assessment of very large numbers of rituximab-treated vaccinated patients to be compared with rituximab-treated unvaccinated patients, and is unlikely to be forthcoming in the very near future.
“In the meantime, however, achieving serologic positivity, meaning having evidence of serologic as well as cellular immunity following vaccination, is a desired outcome, and likely implies more robust immunity.”
Does treatment timing impact COVID-19 vaccine response?
Given enough time, B-cell reconstitution will occur in patients taking rituximab. With that in mind, is it beneficial to wait a certain amount of time after a patient has stopped rituximab therapy or time since their last dose before giving them a COVID-19 vaccine? In their guidance on COVID-19 vaccines for patients with rheumatic and musculoskeletal diseases, the American College of Rheumatology said there is moderate evidence to consider “optimal timing of dosing and vaccination with the rheumatology provider before proceeding.”
“Guidelines and preliminary studies of serologic response to COVID vaccine in rituximab-treated patients have suggested that longer time from last rituximab exposure is associated with a greater likelihood of a serologic response,” Dr. Spiera said.
In a brief report published in Arthritis & Rheumatology, Dr. Spiera and colleagues performed a retrospective chart review of 56 patients with varying levels of last exposure to rituximab who received a COVID-19 vaccine. Their results showed that, when patients were vaccinated 6-12 months after the last rituximab dose, 55% were seronegative, and when this was more than 12 months, only 13% were seronegative, compared with seronegativity in 86% who were vaccinated less than 6 months after their last rituximab dose.
The RituxiVac trial, conducted by researchers in Switzerland, also examined vaccine responses of 96 rituximab-treated patients who received Comirnaty or Spikevax; results recently published in The Lancet Rheumatology showed findings similar to other studies, with reduced humoral and cell-mediated responses. In the RituxiVac trial, the median time to last anti-CD20 treatment was 1.07 years.
“The typical interval between rituximab doses [for treatment of rheumatoid arthritis, as well as for remission maintenance in antineutrophil cytoplasmic antibody–associated vasculitis] is typically 6 months, and this has become widely used as the interval from last rituximab to time of COVID vaccination, with a recommendation to wait 4 weeks (if possible) from time of vaccination until the next rituximab administration,” Dr. Spiera explained. However, this window seems to vary depending on the study.
Recent research published in Arthritis & Rheumatology indicates B-cell levels could be a relevant indicator for humoral and cell-mediated response in patients with rheumatic diseases treated with rituximab, with a level of 10 B cells/mcL (0.4% of lymphocytes) identified as one potential marker for likely seroconversion following COVID-19 vaccination.
“In some smaller case series, it has been further recognized that rituximab-treated patients who were beginning to reconstitute peripheral B cells were most likely to respond serologically. Our present study confirmed those findings, demonstrating that the presence of detectable B cells was strongly associated with vaccine responsiveness, and affords complementary information to time from last [rituximab dose] in informing the likelihood of a vaccine response,” Dr. Spiera said.
However, the literature is limited in this area, and an exact cutoff for B-cell counts in these patients isn’t currently known, Dr. Jyssum said. A better metric is time away from anti-CD20 therapies, with CD19 cell count being highly correlated with last infusion.
Dr. Spiera agreed that there is no consistent B-cell percentage that works as a cutoff. “In our study, we looked at it as a binary variable, although we did find that a higher percentage of B cells in the peripheral lymphocyte population was associated with a higher likelihood of seroconversion. We did not, however, identify a ‘threshold’ for vaccine serologic responsiveness.”
Should clinicians measure antibodies?
The Food and Drug Administration and the Centers for Disease Control and Prevention have recommended that health care providers and the public not use COVID-19 antibody tests as a way to gauge immunity after exposure to SARS-CoV-2 and after receiving a COVID-19 vaccination. The ACR’s guidance on COVID-19 vaccination for patients with rheumatic and musculoskeletal diseases strongly recommends against ordering antibody tests for patients with autoimmune inflammatory rheumatic diseases as a way to measure immunity.
“Generally, such measurements are not recommended as the clinical correlate of various antibody levels are not known,” Dr. Jyssum said. “With regular infusions of rituximab or other anti-CD20 agents, one cannot expect that these patients will develop significant levels of antibodies.”
However, she said there might be situations where it’s useful to know whether a patient has developed antibodies at all. “Assessing the significance of specific antibody levels is difficult, and the subject of scientific studies. Patients lacking a humoral vaccine response are left to rely on their T-cell responses and on infectious control measures to prevent disease.”
Dr. Spiera said he disagreed with guidelines recommending against checking antibody levels after vaccination, “particularly in patients treated with immunosuppressive medications that might be expected to blunt their serologic response to the vaccines.
“Although we cannot be sure what level of measurable antibodies offer what level of protection, most clinicians would agree that patients who demonstrate no detectable antibodies (which is a common finding in rituximab-treated patients) should be considered at higher risk,” he said. “Indeed, recommendations regarding booster vaccine administration in general was initially based on the observation of declining antibody levels with longer time from vaccination.”
Do COVID-19 vaccine boosters help patients on anti-CD20 therapy?
As of January 2022, the FDA and CDC have recommended a third primary series shot of COVID-19 vaccines for some moderately to severely immunocompromised patients as young as 5 years old (for Comirnaty vaccine) or a booster shot of either Comirnaty or Spikevax for everyone aged 12 years and older, including immunocompromised people, while the ACR goes into more detail and recommends clinicians time a patient’s booster shot with temporary treatment interruption.
In The Lancet Rheumatology, Dr. Jyssum and colleagues recently published results from the prospective Nor-vaC study examining the humoral and cell-mediated immune responses of 87 patients with RA being treated with rituximab who received the Comirnaty, Spikevax, or Vaxzevria (AstraZeneca) COVID-19 vaccines; of these, 49 patients received a booster dose at a median of 70 days after completing their primary series. The results showed 19 patients (28.1%) had a serologic response after their primary series, while 8 of 49 patients (16.3%) who received their booster dose had a serologic response.
All patients who received a third dose in the study had a T-cell response, Dr. Jyssum said. “This is reassuring for patients and clinicians. T cells have been found to be important in countering COVID-19 disease, but whether we can rely on the T-cell response alone in the absence of antibodies to protect patients from infection or from serious COVID disease is still not determined,” she said.
When asked if she would recommend COVID-19 vaccine booster doses for patients on rituximab, Dr. Jyssum replied: “Absolutely.”
Another study, recently published in Annals of the Rheumatic Diseases, examined heterologous and homologous booster doses for 60 patients receiving rituximab without seroconversion after their COVID-19 vaccine primary series. The results showed no significant difference in new seroconversion at 4 weeks based on whether the patient received a vector or mRNA vaccine (22% vs. 32%), but all patients who received a booster dose with a vector vaccine had specific T-cell responses, compared with 81% of patients who received an mRNA vaccine booster. There was a new humoral and/or cellular response in 9 of 11 patients (82%), and most patients with peripheral B cells (12 of 18 patients; 67%) achieved seroconversion.
“Our data show that a cellular and/or humoral immune response can be achieved on a third COVID-19 vaccination in most of the patients who initially developed neither a humoral nor a cellular immune response,” the researchers concluded. “The efficacy data together with the safety data seen in our trial provide a favorable risk/benefit ratio and support the implementation of a third vaccination for nonseroconverted high-risk autoimmune disease patients treated with B-cell–depleting agents.”
Dr. Spiera said booster doses are an important part of the equation, and “it is important to consider factors that would be associated with a greater likelihood of achieving a serologic response, particularly in those patients who did not demonstrate a serologic response to the initial vaccines series.
“Preliminary data shows that the beginnings of B-cell reconstitution is also associated with a positive serologic response following a booster of the COVID-19 vaccine,” he said.
The authors of the cited studies reported numerous relevant financial disclosures. Dr. Spiera and Dr. Jyssum reported no relevant financial disclosures.
Rituximab has presented something of a conundrum for patients taking the monoclonal antibody during the COVID-19 pandemic.
Used to manage a variety of autoimmune diseases and cancers, rituximab acts against CD20 proteins expressed on the surface of B cells, causing B-cell depletion. However, it is this B-cell depletion that may put these patients at greater risk of COVID-19 development, progression to more severe disease, and in-hospital mortality. Evidence for this appears to be mixed, with studies showing both that patients using rituximab to manage various diseases are and are not at increased risk for SARS-CoV-2 infection, COVID-19 progression, and mortality.
As COVID-19 vaccine rollouts take place across the world, more questions have been raised about the relationship between B-cell depletion from anti-CD20 therapies and COVID-19 vaccines. Do rituximab and other anti-CD20 therapies affect a patient’s response to COVID-19 vaccines? If this is the case, does the timing of anti-CD20 treatment matter to maximize B-cell levels and improve the vaccine’s effectiveness? And how do COVID-19 vaccine booster doses factor into the equation?
Humoral and cell-mediated responses following COVID-19 vaccination
First, the bad news: The vaccine is unquestionably safe to administer in patients taking rituximab, but one thing that has been well established is that antibody response to COVID-19 vaccination in these individuals does is reduced. This isn’t entirely unprecedented, as previous studies have shown a weakened immune response to pneumococcal polysaccharide and keyhole limpet hemocyanin vaccines among patients taking rituximab.
“Compromised immunogenicity to the SARS-CoV-2 vaccines has been demonstrated in rituximab-treated patients, which is of particular concern given the observation that B-cell–depleting therapies may be associated with worse COVID outcomes,” Robert F. Spiera, MD, director of the Scleroderma, Vasculitis, and Myositis Center at the Hospital for Special Surgery in New York, said in an interview.
For example, in a recent study from the Medical University of Vienna, 29 (39%) of 74 patients receiving rituximab (43% as monotherapy, 57% with conventional-synthetic disease-modifying antirheumatic drugs) who were vaccinated with either the Comirnaty (Pfizer-BioNTech) or Spikevax (Moderna) COVID-19 vaccine achieved seroconversion, compared with 100% of patients in a healthy control group, and all but 1 patient without detectable CD19+ peripheral B cells did not develop anti–SARS-CoV-2 receptor-binding domain antibodies.
“There is an increasing number of studies in this field, and they confirm that patients treated with rituximab and other anti-CD20 agents have severely reduced serological responses to COVID-19 vaccines,” Ingrid Jyssum, MD, of the division of rheumatology and research at Diakonhjemmet Hospital in Oslo, said in an interview.
One silver lining is that patients treated with anti-CD20 therapies appear to have a cell-mediated response following vaccination even if they don’t develop SARS-CoV-2 antibodies. “Studies that also investigate T-cell responses are starting to emerge, and so far, they show that, even if the patients do not have antibodies, they may have T-cell responses,” Dr. Jyssum said.
One study of 24 patients with autoimmune diseases taking rituximab that evaluated humoral and T-cell responses following vaccination with the Comirnaty vaccine found that none had a humoral response to the vaccine, but the T-cell response from that group did not significantly differ from 35 patients receiving other immunosuppressants and 26 patients in a healthy control group. In another study of rituximab- or ocrelizumab-treated patients who received mRNA-based COVID-19 vaccines, 69.4% developed SARS-CoV-2–specific antibodies, compared with a control group, but 96.2% of patients taking ocrelizumab and 81.8% of patients taking rituximab mounted a spike-specific CD8+ T-cell response, compared with 66.7% in the control group, and there were comparable rates (85%-90%) of spike-specific CD4+ T cells in all groups. In the study from the Medical University of Vienna, T-cell response was detected in rituximab-treated patients who both did and did not mount an antibody response.
The clinical relevance of how a blunted humoral immune response but a respectable T-cell response to COVID-19 vaccines affects patients treated with anti-CD20 therapies isn’t currently known, Dr. Jyssum said.
While these data are reassuring, they’re also incomplete, Dr. Spiera noted. “The ultimate outcome of relevance to assess vaccine efficacy is protection from COVID and from severe outcomes of COVID infection (i.e., hospitalization, mechanical ventilation, death). That data will require assessment of very large numbers of rituximab-treated vaccinated patients to be compared with rituximab-treated unvaccinated patients, and is unlikely to be forthcoming in the very near future.
“In the meantime, however, achieving serologic positivity, meaning having evidence of serologic as well as cellular immunity following vaccination, is a desired outcome, and likely implies more robust immunity.”
Does treatment timing impact COVID-19 vaccine response?
Given enough time, B-cell reconstitution will occur in patients taking rituximab. With that in mind, is it beneficial to wait a certain amount of time after a patient has stopped rituximab therapy or time since their last dose before giving them a COVID-19 vaccine? In their guidance on COVID-19 vaccines for patients with rheumatic and musculoskeletal diseases, the American College of Rheumatology said there is moderate evidence to consider “optimal timing of dosing and vaccination with the rheumatology provider before proceeding.”
“Guidelines and preliminary studies of serologic response to COVID vaccine in rituximab-treated patients have suggested that longer time from last rituximab exposure is associated with a greater likelihood of a serologic response,” Dr. Spiera said.
In a brief report published in Arthritis & Rheumatology, Dr. Spiera and colleagues performed a retrospective chart review of 56 patients with varying levels of last exposure to rituximab who received a COVID-19 vaccine. Their results showed that, when patients were vaccinated 6-12 months after the last rituximab dose, 55% were seronegative, and when this was more than 12 months, only 13% were seronegative, compared with seronegativity in 86% who were vaccinated less than 6 months after their last rituximab dose.
The RituxiVac trial, conducted by researchers in Switzerland, also examined vaccine responses of 96 rituximab-treated patients who received Comirnaty or Spikevax; results recently published in The Lancet Rheumatology showed findings similar to other studies, with reduced humoral and cell-mediated responses. In the RituxiVac trial, the median time to last anti-CD20 treatment was 1.07 years.
“The typical interval between rituximab doses [for treatment of rheumatoid arthritis, as well as for remission maintenance in antineutrophil cytoplasmic antibody–associated vasculitis] is typically 6 months, and this has become widely used as the interval from last rituximab to time of COVID vaccination, with a recommendation to wait 4 weeks (if possible) from time of vaccination until the next rituximab administration,” Dr. Spiera explained. However, this window seems to vary depending on the study.
Recent research published in Arthritis & Rheumatology indicates B-cell levels could be a relevant indicator for humoral and cell-mediated response in patients with rheumatic diseases treated with rituximab, with a level of 10 B cells/mcL (0.4% of lymphocytes) identified as one potential marker for likely seroconversion following COVID-19 vaccination.
“In some smaller case series, it has been further recognized that rituximab-treated patients who were beginning to reconstitute peripheral B cells were most likely to respond serologically. Our present study confirmed those findings, demonstrating that the presence of detectable B cells was strongly associated with vaccine responsiveness, and affords complementary information to time from last [rituximab dose] in informing the likelihood of a vaccine response,” Dr. Spiera said.
However, the literature is limited in this area, and an exact cutoff for B-cell counts in these patients isn’t currently known, Dr. Jyssum said. A better metric is time away from anti-CD20 therapies, with CD19 cell count being highly correlated with last infusion.
Dr. Spiera agreed that there is no consistent B-cell percentage that works as a cutoff. “In our study, we looked at it as a binary variable, although we did find that a higher percentage of B cells in the peripheral lymphocyte population was associated with a higher likelihood of seroconversion. We did not, however, identify a ‘threshold’ for vaccine serologic responsiveness.”
Should clinicians measure antibodies?
The Food and Drug Administration and the Centers for Disease Control and Prevention have recommended that health care providers and the public not use COVID-19 antibody tests as a way to gauge immunity after exposure to SARS-CoV-2 and after receiving a COVID-19 vaccination. The ACR’s guidance on COVID-19 vaccination for patients with rheumatic and musculoskeletal diseases strongly recommends against ordering antibody tests for patients with autoimmune inflammatory rheumatic diseases as a way to measure immunity.
“Generally, such measurements are not recommended as the clinical correlate of various antibody levels are not known,” Dr. Jyssum said. “With regular infusions of rituximab or other anti-CD20 agents, one cannot expect that these patients will develop significant levels of antibodies.”
However, she said there might be situations where it’s useful to know whether a patient has developed antibodies at all. “Assessing the significance of specific antibody levels is difficult, and the subject of scientific studies. Patients lacking a humoral vaccine response are left to rely on their T-cell responses and on infectious control measures to prevent disease.”
Dr. Spiera said he disagreed with guidelines recommending against checking antibody levels after vaccination, “particularly in patients treated with immunosuppressive medications that might be expected to blunt their serologic response to the vaccines.
“Although we cannot be sure what level of measurable antibodies offer what level of protection, most clinicians would agree that patients who demonstrate no detectable antibodies (which is a common finding in rituximab-treated patients) should be considered at higher risk,” he said. “Indeed, recommendations regarding booster vaccine administration in general was initially based on the observation of declining antibody levels with longer time from vaccination.”
Do COVID-19 vaccine boosters help patients on anti-CD20 therapy?
As of January 2022, the FDA and CDC have recommended a third primary series shot of COVID-19 vaccines for some moderately to severely immunocompromised patients as young as 5 years old (for Comirnaty vaccine) or a booster shot of either Comirnaty or Spikevax for everyone aged 12 years and older, including immunocompromised people, while the ACR goes into more detail and recommends clinicians time a patient’s booster shot with temporary treatment interruption.
In The Lancet Rheumatology, Dr. Jyssum and colleagues recently published results from the prospective Nor-vaC study examining the humoral and cell-mediated immune responses of 87 patients with RA being treated with rituximab who received the Comirnaty, Spikevax, or Vaxzevria (AstraZeneca) COVID-19 vaccines; of these, 49 patients received a booster dose at a median of 70 days after completing their primary series. The results showed 19 patients (28.1%) had a serologic response after their primary series, while 8 of 49 patients (16.3%) who received their booster dose had a serologic response.
All patients who received a third dose in the study had a T-cell response, Dr. Jyssum said. “This is reassuring for patients and clinicians. T cells have been found to be important in countering COVID-19 disease, but whether we can rely on the T-cell response alone in the absence of antibodies to protect patients from infection or from serious COVID disease is still not determined,” she said.
When asked if she would recommend COVID-19 vaccine booster doses for patients on rituximab, Dr. Jyssum replied: “Absolutely.”
Another study, recently published in Annals of the Rheumatic Diseases, examined heterologous and homologous booster doses for 60 patients receiving rituximab without seroconversion after their COVID-19 vaccine primary series. The results showed no significant difference in new seroconversion at 4 weeks based on whether the patient received a vector or mRNA vaccine (22% vs. 32%), but all patients who received a booster dose with a vector vaccine had specific T-cell responses, compared with 81% of patients who received an mRNA vaccine booster. There was a new humoral and/or cellular response in 9 of 11 patients (82%), and most patients with peripheral B cells (12 of 18 patients; 67%) achieved seroconversion.
“Our data show that a cellular and/or humoral immune response can be achieved on a third COVID-19 vaccination in most of the patients who initially developed neither a humoral nor a cellular immune response,” the researchers concluded. “The efficacy data together with the safety data seen in our trial provide a favorable risk/benefit ratio and support the implementation of a third vaccination for nonseroconverted high-risk autoimmune disease patients treated with B-cell–depleting agents.”
Dr. Spiera said booster doses are an important part of the equation, and “it is important to consider factors that would be associated with a greater likelihood of achieving a serologic response, particularly in those patients who did not demonstrate a serologic response to the initial vaccines series.
“Preliminary data shows that the beginnings of B-cell reconstitution is also associated with a positive serologic response following a booster of the COVID-19 vaccine,” he said.
The authors of the cited studies reported numerous relevant financial disclosures. Dr. Spiera and Dr. Jyssum reported no relevant financial disclosures.
One-third of trials for connective tissue diseases go unpublished
Approximately one-third of registered randomized, controlled trials for connective tissue diseases are incomplete or unpublished, based on data from 175 studies.
“The failure to complete a trial is a waste of time and money, and a missed opportunity to contribute to patient’s health,” Alejandro Brigante, MD, of the Internal Medicine–Rheumatology service at Güemes Sanitorium in Buenos Aires, and colleagues wrote.
Patients with connective tissue diseases (CTDs) experience high levels of disability, poor quality of life, and poor survival, and more randomized, controlled trials are needed to explore treatment options, they said.
In a study published in Arthritis Care & Research, the researchers examined factors leading to the failure of CTD studies. They identified 175 studies of CTDs registered at clinicaltrials.gov since 2000. Most of the studies were phase 3, placebo-controlled trials involving pharmacologic treatments; 117 (67%) were identified as completed, 58 (33%) were identified as discontinued. Approximately half (51%) of the studies involved systemic lupus erythematosus, and half were funded by industry. The median sample size planned for the studies was 101 patients, and 83 studies stated a plan to recruit less than 100 patients.
Of the 58 discontinued trials, 12 were withdrawn, 33 were terminated, and 13 had an unknown status. These trials represented a potential enrollment of 11,389 patients, 31% of the estimated number of patients across all 175 studies.
The researchers found identified reasons for discontinuation for 39 of the 58 discontinued trials. The main reasons included insufficient patient accrual in 11 trials, interim results showing futility (8 trials), safety concerns (5 trials), funding issues (5 trials), conduct problems (4 trials), company decisions (2 trials), administrative reasons (2 trials), and departure of the principal investigator (1 trial); the reason for discontinuation was unclear in 1 trial. Discontinuation rates were not significantly different across disease types.
“By subtracting from the 58 discontinued trials the 13 studies for which early termination was justified (e.g., discontinuation for futility or safety concerns), we considered 45 (26%) trials prematurely terminated,” the researchers wrote. Overall, completed studies were less likely than discontinued studies to have a placebo group, and they had longer treatment periods to evaluate primary outcomes. A sample size of less than 100 patients was the only factor significantly associated with early study termination (odds ratio, 2.1), after controlling for multiple variables.
The researchers checked the publication status of 130 studies, including 94 completed and 36 discontinued randomized, controlled trials. Of these, 44 were unpublished and 86 were published in a peer-reviewed journal at a median of 24 months after study completion. The publication rate was significantly higher for completed studies, compared with discontinued studies (81% vs. 22%), and the rates were not significantly different among diseases. The main reasons for nonpublication included poor recruitment, study rejection and preparation for resubmission, lack of time, low priority, and the fact that the study was ongoing. A sample size of less than 100 patients was the main barrier to publication for completed studies.
The study findings were limited by several factors including selection bias and inability to study factors, such as study complexity or the nature of interventions that might have affected trial completion, the researchers noted. Other limitations include a lack of data on negative results and the possible missed publication of some of the studies.
However, the results illustrate the waste of resources in CTD trials, which are needed to identify effective treatments for these patients, the researchers said. “A better understanding of the factors leading to waste will guide future allocation of resources and could help to maximize the successful conduct of RCTs.”
More research is needed to determine the most effective interventions and reduce the risk of trial noncompletion and nonpublication, they concluded.
The study received no outside funding. The researchers had no financial conflicts to disclose.
Approximately one-third of registered randomized, controlled trials for connective tissue diseases are incomplete or unpublished, based on data from 175 studies.
“The failure to complete a trial is a waste of time and money, and a missed opportunity to contribute to patient’s health,” Alejandro Brigante, MD, of the Internal Medicine–Rheumatology service at Güemes Sanitorium in Buenos Aires, and colleagues wrote.
Patients with connective tissue diseases (CTDs) experience high levels of disability, poor quality of life, and poor survival, and more randomized, controlled trials are needed to explore treatment options, they said.
In a study published in Arthritis Care & Research, the researchers examined factors leading to the failure of CTD studies. They identified 175 studies of CTDs registered at clinicaltrials.gov since 2000. Most of the studies were phase 3, placebo-controlled trials involving pharmacologic treatments; 117 (67%) were identified as completed, 58 (33%) were identified as discontinued. Approximately half (51%) of the studies involved systemic lupus erythematosus, and half were funded by industry. The median sample size planned for the studies was 101 patients, and 83 studies stated a plan to recruit less than 100 patients.
Of the 58 discontinued trials, 12 were withdrawn, 33 were terminated, and 13 had an unknown status. These trials represented a potential enrollment of 11,389 patients, 31% of the estimated number of patients across all 175 studies.
The researchers found identified reasons for discontinuation for 39 of the 58 discontinued trials. The main reasons included insufficient patient accrual in 11 trials, interim results showing futility (8 trials), safety concerns (5 trials), funding issues (5 trials), conduct problems (4 trials), company decisions (2 trials), administrative reasons (2 trials), and departure of the principal investigator (1 trial); the reason for discontinuation was unclear in 1 trial. Discontinuation rates were not significantly different across disease types.
“By subtracting from the 58 discontinued trials the 13 studies for which early termination was justified (e.g., discontinuation for futility or safety concerns), we considered 45 (26%) trials prematurely terminated,” the researchers wrote. Overall, completed studies were less likely than discontinued studies to have a placebo group, and they had longer treatment periods to evaluate primary outcomes. A sample size of less than 100 patients was the only factor significantly associated with early study termination (odds ratio, 2.1), after controlling for multiple variables.
The researchers checked the publication status of 130 studies, including 94 completed and 36 discontinued randomized, controlled trials. Of these, 44 were unpublished and 86 were published in a peer-reviewed journal at a median of 24 months after study completion. The publication rate was significantly higher for completed studies, compared with discontinued studies (81% vs. 22%), and the rates were not significantly different among diseases. The main reasons for nonpublication included poor recruitment, study rejection and preparation for resubmission, lack of time, low priority, and the fact that the study was ongoing. A sample size of less than 100 patients was the main barrier to publication for completed studies.
The study findings were limited by several factors including selection bias and inability to study factors, such as study complexity or the nature of interventions that might have affected trial completion, the researchers noted. Other limitations include a lack of data on negative results and the possible missed publication of some of the studies.
However, the results illustrate the waste of resources in CTD trials, which are needed to identify effective treatments for these patients, the researchers said. “A better understanding of the factors leading to waste will guide future allocation of resources and could help to maximize the successful conduct of RCTs.”
More research is needed to determine the most effective interventions and reduce the risk of trial noncompletion and nonpublication, they concluded.
The study received no outside funding. The researchers had no financial conflicts to disclose.
Approximately one-third of registered randomized, controlled trials for connective tissue diseases are incomplete or unpublished, based on data from 175 studies.
“The failure to complete a trial is a waste of time and money, and a missed opportunity to contribute to patient’s health,” Alejandro Brigante, MD, of the Internal Medicine–Rheumatology service at Güemes Sanitorium in Buenos Aires, and colleagues wrote.
Patients with connective tissue diseases (CTDs) experience high levels of disability, poor quality of life, and poor survival, and more randomized, controlled trials are needed to explore treatment options, they said.
In a study published in Arthritis Care & Research, the researchers examined factors leading to the failure of CTD studies. They identified 175 studies of CTDs registered at clinicaltrials.gov since 2000. Most of the studies were phase 3, placebo-controlled trials involving pharmacologic treatments; 117 (67%) were identified as completed, 58 (33%) were identified as discontinued. Approximately half (51%) of the studies involved systemic lupus erythematosus, and half were funded by industry. The median sample size planned for the studies was 101 patients, and 83 studies stated a plan to recruit less than 100 patients.
Of the 58 discontinued trials, 12 were withdrawn, 33 were terminated, and 13 had an unknown status. These trials represented a potential enrollment of 11,389 patients, 31% of the estimated number of patients across all 175 studies.
The researchers found identified reasons for discontinuation for 39 of the 58 discontinued trials. The main reasons included insufficient patient accrual in 11 trials, interim results showing futility (8 trials), safety concerns (5 trials), funding issues (5 trials), conduct problems (4 trials), company decisions (2 trials), administrative reasons (2 trials), and departure of the principal investigator (1 trial); the reason for discontinuation was unclear in 1 trial. Discontinuation rates were not significantly different across disease types.
“By subtracting from the 58 discontinued trials the 13 studies for which early termination was justified (e.g., discontinuation for futility or safety concerns), we considered 45 (26%) trials prematurely terminated,” the researchers wrote. Overall, completed studies were less likely than discontinued studies to have a placebo group, and they had longer treatment periods to evaluate primary outcomes. A sample size of less than 100 patients was the only factor significantly associated with early study termination (odds ratio, 2.1), after controlling for multiple variables.
The researchers checked the publication status of 130 studies, including 94 completed and 36 discontinued randomized, controlled trials. Of these, 44 were unpublished and 86 were published in a peer-reviewed journal at a median of 24 months after study completion. The publication rate was significantly higher for completed studies, compared with discontinued studies (81% vs. 22%), and the rates were not significantly different among diseases. The main reasons for nonpublication included poor recruitment, study rejection and preparation for resubmission, lack of time, low priority, and the fact that the study was ongoing. A sample size of less than 100 patients was the main barrier to publication for completed studies.
The study findings were limited by several factors including selection bias and inability to study factors, such as study complexity or the nature of interventions that might have affected trial completion, the researchers noted. Other limitations include a lack of data on negative results and the possible missed publication of some of the studies.
However, the results illustrate the waste of resources in CTD trials, which are needed to identify effective treatments for these patients, the researchers said. “A better understanding of the factors leading to waste will guide future allocation of resources and could help to maximize the successful conduct of RCTs.”
More research is needed to determine the most effective interventions and reduce the risk of trial noncompletion and nonpublication, they concluded.
The study received no outside funding. The researchers had no financial conflicts to disclose.
FROM ARTHRITIS CARE & RESEARCH
Low BMI, weight loss predict mortality risk in ILD
A low body mass index (BMI) indicative of being underweight as well as a weight loss of 2 kg or more over the course of 1 year were both independently associated with a higher mortality risk in the following year in patients with fibrotic interstitial lung disease (ILD). In contrast, being both overweight and obese appeared to be protective against mortality at the same 1-year endpoint, according to the results of an observational, retrospective cohort study.
Compared with patients with a normal BMI, patients who were underweight at a BMI of less than 18.5 kg/m2 were over three times more likely to die at 1 year, at a hazard ratio of 3.19 (P < .001), senior author Christopher Ryerson, MD, University of British Columbia, Vancouver, and colleagues reported in the journal Chest.
In contrast, patients who were overweight with a BMI of 25-29 had roughly half the mortality risk as those who were underweight, at an HR of 0.52 (P < .001). Results were roughly similar among the patients with obesity with a BMI in excess of 30, among whom the HR for mortality at 1 year was 0.55 (P < .001), compared with those who were underweight.
“All patients with fibrotic ILD should still engage in exercise and eat an appropriate diet and it is still okay if you are obese and lose weight as a consequence of these lifestyle choices,” Dr. Ryerson told this news organization. “But physicians should be concerned about patients who have severe ILD and who start to lose weight unintentionally since this often represents end-stage fibrosis or some other major comorbidity such as cancer.”
Two large cohorts
Patients from two large cohorts, including the six-center Canadian Registry for Pulmonary Fibrosis (CARE-PF) and the ILD registry at the University of California, San Francisco, were enrolled in the study. A total of 1,786 patients were included from the CARE-PF registry, which served as the derivation cohort, while another 1,779 patients from the UCSF registry served as the validation cohort. In the CARE-PF cohort, 21% of all ILD patients experienced a weight loss of at least 1 kg in the first year of follow-up, including 31% of patients with idiopathic pulmonary fibrosis (IPF).
“Fewer patients experienced a weight loss of at least 1 kg during the first year of the study period in the UCSF cohort,” the authors noted, at only 12% of all ILD patients, some 14% of those with IPF losing at least 1 kg of weight over the course of the year. At 2 years’ follow-up, 35% of all ILD patients had lost at least 1 kg, as had 46% of all IPF patients. Looking at BMI, “a higher value was associated with decreased 1-year mortality in both cohorts on unadjusted analysis,” the investigators observed.
In the CARE-PF cohort, the HR for 1-year mortality was 0.96 per unit difference in BMI (P < .001), while in the UCSF cohort, the HR for 1-year mortality was exactly the same, at 0.96 per unit difference in BMI (P < .001). The authors then adjusted findings for the ILD-GAP index, which included gender, age, and physiology index. After adjusting for this index, the HR for 1-year mortality in the CARE-PF cohort was 0.93 per unit change in BMI (95% CI, 0.90-0.967; P < .001), while in the UCSF cohort, the HR was 0.96 per unit change in BMI (95% CI, 0.94-0.98; P = .001).
Indeed, each 1-kg change above a BMI of 30, adjusted for the ILD-GAP index, was associated with a reduced risk of mortality at 1 year in both cohorts, at an HR of 0.98 (P = .001) in the CARE-PF cohort and an HR of 0.98 (P < .001) in the UCSF cohort. In contrast, patients who experienced a BMI weight loss of 2 kg or more within 1 year had a 41% increased risk of death in the subsequent year after adjusting for the ILD-GAP index and baseline BMI category, at an HR of 1.41 (P = .04). “The absolute change in mortality is much smaller than this,” Dr. Ryerson acknowledged.
“However, the magnitude [in mortality risk] did impress us and this illustrates how weight loss is a frequent consequence of end-stage disease which is something that we have all observed clinically as well,” he added.
Mortality risk plateaued in patients with a greater weight loss, the investigators observed, and there was no association between weight and subsequent 1-year mortality in either cohort on unadjusted analysis.
On the other hand, being underweight was associated with between a 13% and 16% higher mortality risk at 1 year after adjusting for the ILD-GAP, at an HR of 0.84 per 10 kg (P = .001) in the CARE-PF cohort and an HR of 0.87 per 10 kg (P < .001) in the UCSF cohort. “Results were similar in the two studied cohorts, suggesting a robust and generalizable association of both low BMI and weight loss with mortality,” the authors emphasized.
“Together these studies highlight the potential link between obesity and ILD pathogenesis and further suggest the possibility that nutritional support may have a more specific and important role in the management of fibrotic ILD,” the authors wrote. Dr. Ryerson in turn noted that being able to determine mortality risk more accurately than current mortality risk prediction models are able to do is very helpful when dealing with what are sometimes life-and-death decisions.
He also said that having more insight into a patient’s prognosis can change how physicians manage patients with respect to either transplantation or palliation and potentially the need to be more aggressive with pharmacotherapy as well.
Addressing weight loss
Asked to comment on the findings, Elizabeth Volkmann, MD, associate professor of medicine, University of California, Los Angeles, said that this was a very important study and something that she feels does not get adequate attention in clinical practice.
“Weight loss and malnutrition occur in many patients with ILD due to various factors such as gastrointestinal side effects from antifibrotic therapies, decreased oral intake due to psychosocial issues including depression, and increased caloric requirements due to increased work of breathing,” she said in an interview. That said, weight loss and malnutrition are still often underaddressed during clinical encounters for patients with ILD where the focus is on lung health.
“This study illuminates the importance of addressing weight loss in all patients with ILD as it can contribute to heightened risk of mortality,” Dr. Volkmann reemphasized. Dr. Volkmann and colleagues themselves recently reported that radiographic progression of scleroderma lung disease over the course of 1-2 years is associated with an increased risk of long-term mortality, based on two independent studies of systemic sclerosis–interstitial lung disease with extensive follow-up.
Over 8 years of follow-up, patients in the Scleroderma Lung Study II who exhibited an increase of 2% or more in the QILD score – a score that reflects the sum of all abnormally classified scores, including those for fibrosis, ground glass opacity, and honeycombing – for the whole lung at 24 months had an almost fourfold increased risk in mortality, which was significant (P = .014).
The association of an increase in the QILD of at least 2% at 12 months was suggestive in its association with mortality in the SLS I cohort at 12 years of follow-up, a finding that suggests that radiographic progression measured at 2 years is a better predictor of long-term mortality than at 1 year, as the authors concluded.
The CARR-PF is funded by Boehringer Ingelheim. Dr. Ryerson reported receiving personal fees from Boehringer Ingelheim. Dr. Volkmann consults or has received speaker fees from Boehringer Ingelheim and has received grant support from Kadmon and Horizon Therapeutics.
A version of this article first appeared on Medscape.com.
A low body mass index (BMI) indicative of being underweight as well as a weight loss of 2 kg or more over the course of 1 year were both independently associated with a higher mortality risk in the following year in patients with fibrotic interstitial lung disease (ILD). In contrast, being both overweight and obese appeared to be protective against mortality at the same 1-year endpoint, according to the results of an observational, retrospective cohort study.
Compared with patients with a normal BMI, patients who were underweight at a BMI of less than 18.5 kg/m2 were over three times more likely to die at 1 year, at a hazard ratio of 3.19 (P < .001), senior author Christopher Ryerson, MD, University of British Columbia, Vancouver, and colleagues reported in the journal Chest.
In contrast, patients who were overweight with a BMI of 25-29 had roughly half the mortality risk as those who were underweight, at an HR of 0.52 (P < .001). Results were roughly similar among the patients with obesity with a BMI in excess of 30, among whom the HR for mortality at 1 year was 0.55 (P < .001), compared with those who were underweight.
“All patients with fibrotic ILD should still engage in exercise and eat an appropriate diet and it is still okay if you are obese and lose weight as a consequence of these lifestyle choices,” Dr. Ryerson told this news organization. “But physicians should be concerned about patients who have severe ILD and who start to lose weight unintentionally since this often represents end-stage fibrosis or some other major comorbidity such as cancer.”
Two large cohorts
Patients from two large cohorts, including the six-center Canadian Registry for Pulmonary Fibrosis (CARE-PF) and the ILD registry at the University of California, San Francisco, were enrolled in the study. A total of 1,786 patients were included from the CARE-PF registry, which served as the derivation cohort, while another 1,779 patients from the UCSF registry served as the validation cohort. In the CARE-PF cohort, 21% of all ILD patients experienced a weight loss of at least 1 kg in the first year of follow-up, including 31% of patients with idiopathic pulmonary fibrosis (IPF).
“Fewer patients experienced a weight loss of at least 1 kg during the first year of the study period in the UCSF cohort,” the authors noted, at only 12% of all ILD patients, some 14% of those with IPF losing at least 1 kg of weight over the course of the year. At 2 years’ follow-up, 35% of all ILD patients had lost at least 1 kg, as had 46% of all IPF patients. Looking at BMI, “a higher value was associated with decreased 1-year mortality in both cohorts on unadjusted analysis,” the investigators observed.
In the CARE-PF cohort, the HR for 1-year mortality was 0.96 per unit difference in BMI (P < .001), while in the UCSF cohort, the HR for 1-year mortality was exactly the same, at 0.96 per unit difference in BMI (P < .001). The authors then adjusted findings for the ILD-GAP index, which included gender, age, and physiology index. After adjusting for this index, the HR for 1-year mortality in the CARE-PF cohort was 0.93 per unit change in BMI (95% CI, 0.90-0.967; P < .001), while in the UCSF cohort, the HR was 0.96 per unit change in BMI (95% CI, 0.94-0.98; P = .001).
Indeed, each 1-kg change above a BMI of 30, adjusted for the ILD-GAP index, was associated with a reduced risk of mortality at 1 year in both cohorts, at an HR of 0.98 (P = .001) in the CARE-PF cohort and an HR of 0.98 (P < .001) in the UCSF cohort. In contrast, patients who experienced a BMI weight loss of 2 kg or more within 1 year had a 41% increased risk of death in the subsequent year after adjusting for the ILD-GAP index and baseline BMI category, at an HR of 1.41 (P = .04). “The absolute change in mortality is much smaller than this,” Dr. Ryerson acknowledged.
“However, the magnitude [in mortality risk] did impress us and this illustrates how weight loss is a frequent consequence of end-stage disease which is something that we have all observed clinically as well,” he added.
Mortality risk plateaued in patients with a greater weight loss, the investigators observed, and there was no association between weight and subsequent 1-year mortality in either cohort on unadjusted analysis.
On the other hand, being underweight was associated with between a 13% and 16% higher mortality risk at 1 year after adjusting for the ILD-GAP, at an HR of 0.84 per 10 kg (P = .001) in the CARE-PF cohort and an HR of 0.87 per 10 kg (P < .001) in the UCSF cohort. “Results were similar in the two studied cohorts, suggesting a robust and generalizable association of both low BMI and weight loss with mortality,” the authors emphasized.
“Together these studies highlight the potential link between obesity and ILD pathogenesis and further suggest the possibility that nutritional support may have a more specific and important role in the management of fibrotic ILD,” the authors wrote. Dr. Ryerson in turn noted that being able to determine mortality risk more accurately than current mortality risk prediction models are able to do is very helpful when dealing with what are sometimes life-and-death decisions.
He also said that having more insight into a patient’s prognosis can change how physicians manage patients with respect to either transplantation or palliation and potentially the need to be more aggressive with pharmacotherapy as well.
Addressing weight loss
Asked to comment on the findings, Elizabeth Volkmann, MD, associate professor of medicine, University of California, Los Angeles, said that this was a very important study and something that she feels does not get adequate attention in clinical practice.
“Weight loss and malnutrition occur in many patients with ILD due to various factors such as gastrointestinal side effects from antifibrotic therapies, decreased oral intake due to psychosocial issues including depression, and increased caloric requirements due to increased work of breathing,” she said in an interview. That said, weight loss and malnutrition are still often underaddressed during clinical encounters for patients with ILD where the focus is on lung health.
“This study illuminates the importance of addressing weight loss in all patients with ILD as it can contribute to heightened risk of mortality,” Dr. Volkmann reemphasized. Dr. Volkmann and colleagues themselves recently reported that radiographic progression of scleroderma lung disease over the course of 1-2 years is associated with an increased risk of long-term mortality, based on two independent studies of systemic sclerosis–interstitial lung disease with extensive follow-up.
Over 8 years of follow-up, patients in the Scleroderma Lung Study II who exhibited an increase of 2% or more in the QILD score – a score that reflects the sum of all abnormally classified scores, including those for fibrosis, ground glass opacity, and honeycombing – for the whole lung at 24 months had an almost fourfold increased risk in mortality, which was significant (P = .014).
The association of an increase in the QILD of at least 2% at 12 months was suggestive in its association with mortality in the SLS I cohort at 12 years of follow-up, a finding that suggests that radiographic progression measured at 2 years is a better predictor of long-term mortality than at 1 year, as the authors concluded.
The CARR-PF is funded by Boehringer Ingelheim. Dr. Ryerson reported receiving personal fees from Boehringer Ingelheim. Dr. Volkmann consults or has received speaker fees from Boehringer Ingelheim and has received grant support from Kadmon and Horizon Therapeutics.
A version of this article first appeared on Medscape.com.
A low body mass index (BMI) indicative of being underweight as well as a weight loss of 2 kg or more over the course of 1 year were both independently associated with a higher mortality risk in the following year in patients with fibrotic interstitial lung disease (ILD). In contrast, being both overweight and obese appeared to be protective against mortality at the same 1-year endpoint, according to the results of an observational, retrospective cohort study.
Compared with patients with a normal BMI, patients who were underweight at a BMI of less than 18.5 kg/m2 were over three times more likely to die at 1 year, at a hazard ratio of 3.19 (P < .001), senior author Christopher Ryerson, MD, University of British Columbia, Vancouver, and colleagues reported in the journal Chest.
In contrast, patients who were overweight with a BMI of 25-29 had roughly half the mortality risk as those who were underweight, at an HR of 0.52 (P < .001). Results were roughly similar among the patients with obesity with a BMI in excess of 30, among whom the HR for mortality at 1 year was 0.55 (P < .001), compared with those who were underweight.
“All patients with fibrotic ILD should still engage in exercise and eat an appropriate diet and it is still okay if you are obese and lose weight as a consequence of these lifestyle choices,” Dr. Ryerson told this news organization. “But physicians should be concerned about patients who have severe ILD and who start to lose weight unintentionally since this often represents end-stage fibrosis or some other major comorbidity such as cancer.”
Two large cohorts
Patients from two large cohorts, including the six-center Canadian Registry for Pulmonary Fibrosis (CARE-PF) and the ILD registry at the University of California, San Francisco, were enrolled in the study. A total of 1,786 patients were included from the CARE-PF registry, which served as the derivation cohort, while another 1,779 patients from the UCSF registry served as the validation cohort. In the CARE-PF cohort, 21% of all ILD patients experienced a weight loss of at least 1 kg in the first year of follow-up, including 31% of patients with idiopathic pulmonary fibrosis (IPF).
“Fewer patients experienced a weight loss of at least 1 kg during the first year of the study period in the UCSF cohort,” the authors noted, at only 12% of all ILD patients, some 14% of those with IPF losing at least 1 kg of weight over the course of the year. At 2 years’ follow-up, 35% of all ILD patients had lost at least 1 kg, as had 46% of all IPF patients. Looking at BMI, “a higher value was associated with decreased 1-year mortality in both cohorts on unadjusted analysis,” the investigators observed.
In the CARE-PF cohort, the HR for 1-year mortality was 0.96 per unit difference in BMI (P < .001), while in the UCSF cohort, the HR for 1-year mortality was exactly the same, at 0.96 per unit difference in BMI (P < .001). The authors then adjusted findings for the ILD-GAP index, which included gender, age, and physiology index. After adjusting for this index, the HR for 1-year mortality in the CARE-PF cohort was 0.93 per unit change in BMI (95% CI, 0.90-0.967; P < .001), while in the UCSF cohort, the HR was 0.96 per unit change in BMI (95% CI, 0.94-0.98; P = .001).
Indeed, each 1-kg change above a BMI of 30, adjusted for the ILD-GAP index, was associated with a reduced risk of mortality at 1 year in both cohorts, at an HR of 0.98 (P = .001) in the CARE-PF cohort and an HR of 0.98 (P < .001) in the UCSF cohort. In contrast, patients who experienced a BMI weight loss of 2 kg or more within 1 year had a 41% increased risk of death in the subsequent year after adjusting for the ILD-GAP index and baseline BMI category, at an HR of 1.41 (P = .04). “The absolute change in mortality is much smaller than this,” Dr. Ryerson acknowledged.
“However, the magnitude [in mortality risk] did impress us and this illustrates how weight loss is a frequent consequence of end-stage disease which is something that we have all observed clinically as well,” he added.
Mortality risk plateaued in patients with a greater weight loss, the investigators observed, and there was no association between weight and subsequent 1-year mortality in either cohort on unadjusted analysis.
On the other hand, being underweight was associated with between a 13% and 16% higher mortality risk at 1 year after adjusting for the ILD-GAP, at an HR of 0.84 per 10 kg (P = .001) in the CARE-PF cohort and an HR of 0.87 per 10 kg (P < .001) in the UCSF cohort. “Results were similar in the two studied cohorts, suggesting a robust and generalizable association of both low BMI and weight loss with mortality,” the authors emphasized.
“Together these studies highlight the potential link between obesity and ILD pathogenesis and further suggest the possibility that nutritional support may have a more specific and important role in the management of fibrotic ILD,” the authors wrote. Dr. Ryerson in turn noted that being able to determine mortality risk more accurately than current mortality risk prediction models are able to do is very helpful when dealing with what are sometimes life-and-death decisions.
He also said that having more insight into a patient’s prognosis can change how physicians manage patients with respect to either transplantation or palliation and potentially the need to be more aggressive with pharmacotherapy as well.
Addressing weight loss
Asked to comment on the findings, Elizabeth Volkmann, MD, associate professor of medicine, University of California, Los Angeles, said that this was a very important study and something that she feels does not get adequate attention in clinical practice.
“Weight loss and malnutrition occur in many patients with ILD due to various factors such as gastrointestinal side effects from antifibrotic therapies, decreased oral intake due to psychosocial issues including depression, and increased caloric requirements due to increased work of breathing,” she said in an interview. That said, weight loss and malnutrition are still often underaddressed during clinical encounters for patients with ILD where the focus is on lung health.
“This study illuminates the importance of addressing weight loss in all patients with ILD as it can contribute to heightened risk of mortality,” Dr. Volkmann reemphasized. Dr. Volkmann and colleagues themselves recently reported that radiographic progression of scleroderma lung disease over the course of 1-2 years is associated with an increased risk of long-term mortality, based on two independent studies of systemic sclerosis–interstitial lung disease with extensive follow-up.
Over 8 years of follow-up, patients in the Scleroderma Lung Study II who exhibited an increase of 2% or more in the QILD score – a score that reflects the sum of all abnormally classified scores, including those for fibrosis, ground glass opacity, and honeycombing – for the whole lung at 24 months had an almost fourfold increased risk in mortality, which was significant (P = .014).
The association of an increase in the QILD of at least 2% at 12 months was suggestive in its association with mortality in the SLS I cohort at 12 years of follow-up, a finding that suggests that radiographic progression measured at 2 years is a better predictor of long-term mortality than at 1 year, as the authors concluded.
The CARR-PF is funded by Boehringer Ingelheim. Dr. Ryerson reported receiving personal fees from Boehringer Ingelheim. Dr. Volkmann consults or has received speaker fees from Boehringer Ingelheim and has received grant support from Kadmon and Horizon Therapeutics.
A version of this article first appeared on Medscape.com.
FROM CHEST
FDA approves new myasthenia gravis drug
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” Billy Dunn, MD, director, office of neuroscience, FDA Center for Drug Evaluation and Research, said in a news release.
This approval represents “an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases,” Dr. Dunn added.
Effective, well tolerated
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have IgG antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
As previously reported, efgartigimod was effective and well tolerated in the phase 3, randomized, placebo-controlled ADAPT trial, which enrolled 187 adults with gMG regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis–Activities of Daily Living score of at least 5 (>50% nonocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed, depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
Treatment with efgartigimod reduced disease burden and improved strength and quality of life in patients with gMG across four MG-specific scales. In addition, these benefits were observed early and were reproducible and durable.
The results were published in Lancet Neurology.
‘Important new advance’
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work; and the side-effect profile is much like placebo,” said principal investigator James Howard Jr., MD, department of neurology, University of North Carolina at Chapel Hill.
The FDA granted efgartigimod fast track and orphan drug designation.
“People living with gMG have been in need of new treatment options that are targeted to the underlying pathogenesis of the disease and supported by clinical data,” Dr. Howard said in a company news release issued upon approval.
This approval “represents an important new advance for gMG patients and families affected by this debilitating disease. This therapy has the potential to reduce the disease burden of gMG and transform the way we treat this disease,” Dr. Howard added.
A version of this article first appeared on Medscape.com.
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” Billy Dunn, MD, director, office of neuroscience, FDA Center for Drug Evaluation and Research, said in a news release.
This approval represents “an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases,” Dr. Dunn added.
Effective, well tolerated
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have IgG antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
As previously reported, efgartigimod was effective and well tolerated in the phase 3, randomized, placebo-controlled ADAPT trial, which enrolled 187 adults with gMG regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis–Activities of Daily Living score of at least 5 (>50% nonocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed, depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
Treatment with efgartigimod reduced disease burden and improved strength and quality of life in patients with gMG across four MG-specific scales. In addition, these benefits were observed early and were reproducible and durable.
The results were published in Lancet Neurology.
‘Important new advance’
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work; and the side-effect profile is much like placebo,” said principal investigator James Howard Jr., MD, department of neurology, University of North Carolina at Chapel Hill.
The FDA granted efgartigimod fast track and orphan drug designation.
“People living with gMG have been in need of new treatment options that are targeted to the underlying pathogenesis of the disease and supported by clinical data,” Dr. Howard said in a company news release issued upon approval.
This approval “represents an important new advance for gMG patients and families affected by this debilitating disease. This therapy has the potential to reduce the disease burden of gMG and transform the way we treat this disease,” Dr. Howard added.
A version of this article first appeared on Medscape.com.
“There are significant unmet medical needs for people living with myasthenia gravis, as with many other rare diseases,” Billy Dunn, MD, director, office of neuroscience, FDA Center for Drug Evaluation and Research, said in a news release.
This approval represents “an important step in providing a novel therapy option for patients and underscores the agency’s commitment to help make new treatment options available for people living with rare diseases,” Dr. Dunn added.
Effective, well tolerated
The rare and chronic autoimmune neuromuscular disorder of gMG causes debilitating and potentially life-threatening muscle weakness and significantly impaired independence and quality of life. Most patients with gMG have IgG antibodies, which are most often directed against skeletal muscle nicotinic acetylcholine receptors.
Efgartigimod is an antibody fragment designed to reduce pathogenic IgG antibodies and block the IgG recycling process in patients with gMG.
The novel agent binds to the neonatal Fc receptor (FcRn), which is widely expressed throughout the body and plays a central role in rescuing IgG antibodies from degradation. Blocking FcRn reduces IgG antibody levels.
As previously reported, efgartigimod was effective and well tolerated in the phase 3, randomized, placebo-controlled ADAPT trial, which enrolled 187 adults with gMG regardless of acetylcholine receptor antibody status. All had a Myasthenia Gravis–Activities of Daily Living score of at least 5 (>50% nonocular) on a background of a stable dose of at least one MG drug.
For 26 weeks, 84 patients were randomly assigned to receive efgartigimod 10 mg/kg and 83 to receive matching placebo. Both treatments were administered as four infusions per cycle at one infusion per week. The process was repeated as needed, depending on clinical response no sooner than 8 weeks after initiation of the previous cycle.
Treatment with efgartigimod reduced disease burden and improved strength and quality of life in patients with gMG across four MG-specific scales. In addition, these benefits were observed early and were reproducible and durable.
The results were published in Lancet Neurology.
‘Important new advance’
Efgartigimod is a “very rapidly acting drug relative to other treatments that may take 4, 6, sometimes 10 months before they start to work; and the side-effect profile is much like placebo,” said principal investigator James Howard Jr., MD, department of neurology, University of North Carolina at Chapel Hill.
The FDA granted efgartigimod fast track and orphan drug designation.
“People living with gMG have been in need of new treatment options that are targeted to the underlying pathogenesis of the disease and supported by clinical data,” Dr. Howard said in a company news release issued upon approval.
This approval “represents an important new advance for gMG patients and families affected by this debilitating disease. This therapy has the potential to reduce the disease burden of gMG and transform the way we treat this disease,” Dr. Howard added.
A version of this article first appeared on Medscape.com.
A 22-year-old presented with erythematous papules on her fingers and toes
than men. Clinically, distal extremities such as toes, fingertips and heels, as well as the rims of the ears or nose develop erythematous to purple plaques. Lesions may be painful or pruritic. Over time, lesions may develop atrophy and resemble those of discoid lupus. While the pathogenesis is unknown, exposure to cold or wet environments can precipitate lesions.

Histopathology reveals a deep and superficial lymphocytic infiltrate with perieccrine involvement and fibrin deposition in vessels. Dermal edema is often present. Direct immunofluorescence shows an interface dermatitis positive for IgM, IgA, and C3.
The Mayo Clinic developed diagnostic criteria for diagnosing chilblains lupus. Two major criteria are acral skin lesions induced by cold exposure and evidence of lupus erythematosus in skin lesions (histopathologically or by direct immunofluorescence). Three minor criteria are the coexistence of systemic lupus erythematosus or discoid lupus erythematosus, response to antilupus treatment, and negative cryoglobulin and cold agglutinin studies.
Chilblains, or perniosis, has a similar clinical presentation to chilblain lupus erythematosus. However, serologic evidence of lupus, such as a positive antinuclear antibody (ANA), will be absent. Lupus pernio (Besnier-Tenneson syndrome) is a form of sarcoidosis that tends to favor the nose. These lesions are not precipitated by cold. It can be differentiated on histology. “COVID toes” is an entity described during the coronavirus pandemic, during which dermatologists noted pernio-like lesions in patients testing positive for coronavirus.
The patient’s labs revealed a positive ANA at 1:320 in a nucleolar speckled pattern, elevated double-stranded DNA, low C3 and C4 levels, elevated cardiolipin IgM Ab, and elevated sedimentation rate. COVID-19 antigen testing and COVID-19 antibodies were negative. A serum protein electrophoresis was negative. Cryoglobulins were negative.
Treatment includes protection from cold. Smoking cessation should be discussed. Topical steroids and topical calcineurin inhibitors are first-line treatments for mild disease. Antimalarials, such as hydroxychloroquine can be helpful. Systemic calcium channel blockers, systemic steroids, mycophenolate mofetil, and tacrolimus have all been reported as treatments. This patient responded well to hydroxychloroquine and topical steroids with full resolution of lesions.
This case was submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
Su WP et al. Cutis. 1994 Dec;54(6):395-9.
Werth V and Newman S. Chilblain lupus (SLE pernio). Dermatology Advisor. 2017.
than men. Clinically, distal extremities such as toes, fingertips and heels, as well as the rims of the ears or nose develop erythematous to purple plaques. Lesions may be painful or pruritic. Over time, lesions may develop atrophy and resemble those of discoid lupus. While the pathogenesis is unknown, exposure to cold or wet environments can precipitate lesions.
Histopathology reveals a deep and superficial lymphocytic infiltrate with perieccrine involvement and fibrin deposition in vessels. Dermal edema is often present. Direct immunofluorescence shows an interface dermatitis positive for IgM, IgA, and C3.
The Mayo Clinic developed diagnostic criteria for diagnosing chilblains lupus. Two major criteria are acral skin lesions induced by cold exposure and evidence of lupus erythematosus in skin lesions (histopathologically or by direct immunofluorescence). Three minor criteria are the coexistence of systemic lupus erythematosus or discoid lupus erythematosus, response to antilupus treatment, and negative cryoglobulin and cold agglutinin studies.
Chilblains, or perniosis, has a similar clinical presentation to chilblain lupus erythematosus. However, serologic evidence of lupus, such as a positive antinuclear antibody (ANA), will be absent. Lupus pernio (Besnier-Tenneson syndrome) is a form of sarcoidosis that tends to favor the nose. These lesions are not precipitated by cold. It can be differentiated on histology. “COVID toes” is an entity described during the coronavirus pandemic, during which dermatologists noted pernio-like lesions in patients testing positive for coronavirus.
The patient’s labs revealed a positive ANA at 1:320 in a nucleolar speckled pattern, elevated double-stranded DNA, low C3 and C4 levels, elevated cardiolipin IgM Ab, and elevated sedimentation rate. COVID-19 antigen testing and COVID-19 antibodies were negative. A serum protein electrophoresis was negative. Cryoglobulins were negative.
Treatment includes protection from cold. Smoking cessation should be discussed. Topical steroids and topical calcineurin inhibitors are first-line treatments for mild disease. Antimalarials, such as hydroxychloroquine can be helpful. Systemic calcium channel blockers, systemic steroids, mycophenolate mofetil, and tacrolimus have all been reported as treatments. This patient responded well to hydroxychloroquine and topical steroids with full resolution of lesions.
This case was submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
Su WP et al. Cutis. 1994 Dec;54(6):395-9.
Werth V and Newman S. Chilblain lupus (SLE pernio). Dermatology Advisor. 2017.
than men. Clinically, distal extremities such as toes, fingertips and heels, as well as the rims of the ears or nose develop erythematous to purple plaques. Lesions may be painful or pruritic. Over time, lesions may develop atrophy and resemble those of discoid lupus. While the pathogenesis is unknown, exposure to cold or wet environments can precipitate lesions.
Histopathology reveals a deep and superficial lymphocytic infiltrate with perieccrine involvement and fibrin deposition in vessels. Dermal edema is often present. Direct immunofluorescence shows an interface dermatitis positive for IgM, IgA, and C3.
The Mayo Clinic developed diagnostic criteria for diagnosing chilblains lupus. Two major criteria are acral skin lesions induced by cold exposure and evidence of lupus erythematosus in skin lesions (histopathologically or by direct immunofluorescence). Three minor criteria are the coexistence of systemic lupus erythematosus or discoid lupus erythematosus, response to antilupus treatment, and negative cryoglobulin and cold agglutinin studies.
Chilblains, or perniosis, has a similar clinical presentation to chilblain lupus erythematosus. However, serologic evidence of lupus, such as a positive antinuclear antibody (ANA), will be absent. Lupus pernio (Besnier-Tenneson syndrome) is a form of sarcoidosis that tends to favor the nose. These lesions are not precipitated by cold. It can be differentiated on histology. “COVID toes” is an entity described during the coronavirus pandemic, during which dermatologists noted pernio-like lesions in patients testing positive for coronavirus.
The patient’s labs revealed a positive ANA at 1:320 in a nucleolar speckled pattern, elevated double-stranded DNA, low C3 and C4 levels, elevated cardiolipin IgM Ab, and elevated sedimentation rate. COVID-19 antigen testing and COVID-19 antibodies were negative. A serum protein electrophoresis was negative. Cryoglobulins were negative.
Treatment includes protection from cold. Smoking cessation should be discussed. Topical steroids and topical calcineurin inhibitors are first-line treatments for mild disease. Antimalarials, such as hydroxychloroquine can be helpful. Systemic calcium channel blockers, systemic steroids, mycophenolate mofetil, and tacrolimus have all been reported as treatments. This patient responded well to hydroxychloroquine and topical steroids with full resolution of lesions.
This case was submitted by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to dermnews@mdedge.com.
References
Su WP et al. Cutis. 1994 Dec;54(6):395-9.
Werth V and Newman S. Chilblain lupus (SLE pernio). Dermatology Advisor. 2017.

The patient denied any arthralgias, alopecia, oral ulcers, or photosensitivity. She denied any recent illness.
Giant cell arteritis fast-track clinics pave way for greater ultrasound use in U.S.
Temporal artery biopsy has been the standard for diagnosing giant cell arteritis (GCA), but vascular ultrasound, a procedure that’s less invasive, less time-intensive, less expensive, and more convenient, has gained widespread use in Europe, and now clinics in the United States are adopting this approach and moving toward having rheumatologists take on the role of ultrasonographer.
However, directors at these clinics – known as GCA fast-track clinics – caution that the bar can be high for adopting vascular ultrasound (VUS) as a tool to diagnose GCA. Ultrasonographers need specialized training to perform the task and an adequate caseload to maintain their skills. Clinics also need to be outfitted with high-definition ultrasound machines.
“It definitely takes adequate training and learning of how to adjust the settings on the ultrasound machine to be able to visualize the findings appropriately,” said Minna Kohler, MD, director of the rheumatology and musculoskeletal ultrasound program at Massachusetts General Hospital in Boston, which has its own GCA fast-track clinic.

“And the clinical context is very important,” she said. “If you have a high suspicion for someone with temporal arteritis, or GCA, and the patient has been on steroids for weeks before you see them, the ultrasound findings may not show signs of the disease. In those cases in which the imaging is equivocal, we would still pursue a biopsy.”
The idea of the fast-track clinic is as the name implies: to quickly confirm the presence of GCA in a matter of hours, or days at the most, in an outpatient setting without the hassles of a biopsy. Temporal artery biopsy (TAB), by comparison, “is more costly because it requires operating room time, a surgical consultation, and surgery time, whereas ultrasound is a very inexpensive exam since it’s done in the clinic by the rheumatologist,” Dr. Kohler said.
European experience
Use of VUS to diagnose GCA is supplanting TAB in Europe and other countries. In Denmark alone – with a population of 6 million – three outpatient fast-track clinics are operating. The United States, with a population more than 50 times larger than Denmark’s, has six.
Stavros Chrysidis, MD, PhD, chief of rheumatology at the Hospital South West Jutland, one of the fast-track clinic sites in Denmark, led a recent multicenter study, known as EUREKA, of VUS in patients with suspected GCA. He and his colleagues reported in The Lancet Rheumatology that the sensitivity and specificity of VUS was superior to TAB in confirming a diagnosis of GCA. Dr. Chrysidis has instructed U.S. rheumatologists and ultrasonographers in performing and interpreting VUS for GCA.
The study emphasizes the importance of training for ultrasonographers, said Dr. Chrysidis, who regularly performs VUS at his institution. “The most important finding is that, when we apply VUS by systematically trained ultrasonographers using appropriate equipment in appropriate settings, it has excellent diagnostic accuracy on GCA,” he told this news organization.
He noted that The Lancet Rheumatology report is the first multicenter study of VUS for diagnosing GCA in which all the ultrasonographers participated in a standardized training program, which his group developed. “Ultrasound is very operator dependent,” he said. “That’s why the training is very important.”
The training occurred over a year and included two workshops consisting of 5 days of theoretical training on VUS; supervised hands-on evaluation of healthy individuals and patients with known GCA; and evaluation of ultrasound images. Over the course of the year, trainees performed at least 50 VUS evaluations, half of which were in patients with confirmed GCA. During the training period, an external rheumatologist with broad experience in VUS made the final diagnosis.
“The equipment and settings are very important because ultrasound can be very time consuming if you are not educated well and if your equipment is not adjusted well,” Dr. Chrysidis said. The equipment must be calibrated beforehand “so you don’t spend time on adjustments.”
For diagnosing temporal artery anomalies, the ultrasound equipment must have a resolution of 0.3-0.4 mm, he said. “When you have a transducer of 10 MHz, you cannot visualize changes smaller than 5 mm.”
The EUREKA study stated that VUS could replace TAB as a first-line diagnostic tool for GCA – provided the ultrasonographers are systematically trained and the equipment and settings are appropriate. In the Jutland clinic, VUS already has replaced TAB, Dr. Chrysidis said.
“In my department since 2017, when we started the fast-track clinic after the EUREKA study was terminated, we have performed three temporal artery biopsies in the last 4 years, and we screen 60-70 patients per year because we use ultrasound as the primary imaging,” he said. In cases when the results are inconclusive, they order a PET scan. “We don’t perform biopsies anymore,” he said.
U.S. fast-track clinic models
The fast-track clinic models in the United States vary. Results of a survey of the U.S. clinics were presented as an abstract at the 2021 American College of Rheumatology annual meeting. Some centers have a vasculitis specialist obtain and interpret the imaging. At others, a vasculitis specialist refers patients to a VUS-trained rheumatologist to perform and interpret the test. Another approach is to have vasculitis specialists refer patients to ultrasound technicians trained in VUS, with a vascular surgeon interpreting the images and either a VUS-trained rheumatologist or vascular medicine specialist verifying the images.
The take-home message of that survey is that “ultrasound evaluation should be considered in the hands of experts, realizing that not everyone has that skill set, but if it is available, it’s a way to expedite diagnosis and it can be helpful in managing the GCA patient in an appropriate way, quicker than trying to schedule cross-sectional imaging,” said Massachusetts General’s Dr. Kohler, who is a coauthor of the abstract. “Certainly, cross-sectional imaging also plays an important role, but when it comes to confirming whether to continue with treatment or not for a very serious condition, ultrasound is a quick way to get the answer.”
In addition to the fast-track clinic at Massachusetts General, the survey included fast-track clinics at the University of Washington, Seattle; Brigham and Women’s Hospital, Boston; Loma Linda (Calif.) University; University of California, Los Angeles; and at a private practice, Arthritis and Rheumatism Associates in the Washington area.
Advantages of VUS vs. TAB
At Massachusetts General, some of the rheumatologists are trained to perform VUS. The rheumatologists also perform the clinical evaluation of suspected cases of GCA. The advantage of VUS, Dr. Kohler said, is that the answer is “right there”; that is, the imaging yields a diagnosis almost instantaneously whereas a biopsy must be sent to a lab for analysis.
“Since a lot of patients with suspected vasculitis may already come to us on steroid therapy, and if there’s a low probability or low suspicion for vasculitis, [VUS] actually confirms that, and we’re able to taper prednisone or steroids quickly rather than keep them on a prolonged course.”
Alison Bays, MD, MPH, of the department of rheumatology at the University of Washington, said that the advantages of avoiding biopsy aren’t to be overlooked. “Temporal artery biopsies are invasive and carry surgical risks, especially as many of our patients are elderly,” she told this news organization.

“These patients occasionally refuse biopsy, but the acceptance of ultrasound is high,” Dr. Bays said. “Scheduling surgery can be more complicated, resulting in delays to biopsy and potentially higher rates of false negatives. Additionally, ultrasound has resulted in a higher rate of diagnosis with GCA as TAB misses large-vessel involvement.” The fast-track clinic at the university has evaluated 250 patients since it opened in 2017.
Dr. Bays and colleagues published the first United States study of a fast-track protocol using vascular sonographers. “Our group has demonstrated that fast-track clinics can rapidly and effectively evaluate patients, and we demonstrated a different method of evaluation using vascular sonographers rather than rheumatologists to do the vascular ultrasound,” she said. “It utilizes the familiarity vascular sonographers already have with imaging blood vessels.”
She added that the TABUL study in the United Kingdom in 2016 demonstrated that VUS yielded a savings of $686 (£484 as reported in the study), compared with TAB. “Further studies need to be done in the United States,” she said. “Beyond direct comparison of costs, reduction in steroid burden due to quick evaluation and diagnosis may carry additional benefits.”
At Brigham and Women’s Hospital, the division of rheumatology and the vascular section of the cardiology division collaborate on the GCA evaluation, said Sara Tedeschi, MD, MPH, codirector of the fast-track clinic there. Trained vascular technologists credentialed in the procedure and specifically trained in using VUS for evaluating arteritis perform the VUS. Cardiologists with a subspecialty in vascular medicine interpret the studies.

VUS patients have a rheumatology evaluation just before they have the ultrasound. “The rheumatology evaluation is then able to incorporate information from the VUS together with laboratory data, the patient’s history, and physical examination,” Dr. Tedeschi said.
“If the rheumatologist recommends a temporal artery biopsy as a next step, we arrange this with vascular surgery,” she said. “If the rheumatologist recommends other imaging such as MRA [magnetic resonance angiography] or PET-CT, we frequently review the images together with our colleagues in cardiovascular radiology and/or nuclear medicine.”
But in the United States, it will take some time for GCA fast-track clinics to become the standard, Dr. Tedeschi said. “Temporal artery biopsy may be faster to arrange in certain practice settings if VUS is not already being employed for giant cell arteritis evaluation,” she said.
Dr. Bays recognized this limitation, saying, “We are hoping that in the future, the American College of Rheumatology will consider vascular ultrasound as a first-line diagnostic test in diagnosis as rheumatologists and vascular sonographers gain familiarity over time.”
But that would mean that centers performing VUS for GCA would have to meet rigorous standards for the procedure. “With that, standardization of a protocol, high-quality equipment, and adequate training are necessary to ensure quality and reduce the chance of false-positive or false-negative results,” she said.
Dr. Chrysidis, Dr. Bays, and Dr. Tedeschi have disclosed no relevant financial relationships. Dr. Kohler is a board member of Ultrasound School of North American Rheumatologists.
A version of this article first appeared on Medscape.com.
Temporal artery biopsy has been the standard for diagnosing giant cell arteritis (GCA), but vascular ultrasound, a procedure that’s less invasive, less time-intensive, less expensive, and more convenient, has gained widespread use in Europe, and now clinics in the United States are adopting this approach and moving toward having rheumatologists take on the role of ultrasonographer.
However, directors at these clinics – known as GCA fast-track clinics – caution that the bar can be high for adopting vascular ultrasound (VUS) as a tool to diagnose GCA. Ultrasonographers need specialized training to perform the task and an adequate caseload to maintain their skills. Clinics also need to be outfitted with high-definition ultrasound machines.
“It definitely takes adequate training and learning of how to adjust the settings on the ultrasound machine to be able to visualize the findings appropriately,” said Minna Kohler, MD, director of the rheumatology and musculoskeletal ultrasound program at Massachusetts General Hospital in Boston, which has its own GCA fast-track clinic.
“And the clinical context is very important,” she said. “If you have a high suspicion for someone with temporal arteritis, or GCA, and the patient has been on steroids for weeks before you see them, the ultrasound findings may not show signs of the disease. In those cases in which the imaging is equivocal, we would still pursue a biopsy.”
The idea of the fast-track clinic is as the name implies: to quickly confirm the presence of GCA in a matter of hours, or days at the most, in an outpatient setting without the hassles of a biopsy. Temporal artery biopsy (TAB), by comparison, “is more costly because it requires operating room time, a surgical consultation, and surgery time, whereas ultrasound is a very inexpensive exam since it’s done in the clinic by the rheumatologist,” Dr. Kohler said.
European experience
Use of VUS to diagnose GCA is supplanting TAB in Europe and other countries. In Denmark alone – with a population of 6 million – three outpatient fast-track clinics are operating. The United States, with a population more than 50 times larger than Denmark’s, has six.
Stavros Chrysidis, MD, PhD, chief of rheumatology at the Hospital South West Jutland, one of the fast-track clinic sites in Denmark, led a recent multicenter study, known as EUREKA, of VUS in patients with suspected GCA. He and his colleagues reported in The Lancet Rheumatology that the sensitivity and specificity of VUS was superior to TAB in confirming a diagnosis of GCA. Dr. Chrysidis has instructed U.S. rheumatologists and ultrasonographers in performing and interpreting VUS for GCA.
The study emphasizes the importance of training for ultrasonographers, said Dr. Chrysidis, who regularly performs VUS at his institution. “The most important finding is that, when we apply VUS by systematically trained ultrasonographers using appropriate equipment in appropriate settings, it has excellent diagnostic accuracy on GCA,” he told this news organization.
He noted that The Lancet Rheumatology report is the first multicenter study of VUS for diagnosing GCA in which all the ultrasonographers participated in a standardized training program, which his group developed. “Ultrasound is very operator dependent,” he said. “That’s why the training is very important.”
The training occurred over a year and included two workshops consisting of 5 days of theoretical training on VUS; supervised hands-on evaluation of healthy individuals and patients with known GCA; and evaluation of ultrasound images. Over the course of the year, trainees performed at least 50 VUS evaluations, half of which were in patients with confirmed GCA. During the training period, an external rheumatologist with broad experience in VUS made the final diagnosis.
“The equipment and settings are very important because ultrasound can be very time consuming if you are not educated well and if your equipment is not adjusted well,” Dr. Chrysidis said. The equipment must be calibrated beforehand “so you don’t spend time on adjustments.”
For diagnosing temporal artery anomalies, the ultrasound equipment must have a resolution of 0.3-0.4 mm, he said. “When you have a transducer of 10 MHz, you cannot visualize changes smaller than 5 mm.”
The EUREKA study stated that VUS could replace TAB as a first-line diagnostic tool for GCA – provided the ultrasonographers are systematically trained and the equipment and settings are appropriate. In the Jutland clinic, VUS already has replaced TAB, Dr. Chrysidis said.
“In my department since 2017, when we started the fast-track clinic after the EUREKA study was terminated, we have performed three temporal artery biopsies in the last 4 years, and we screen 60-70 patients per year because we use ultrasound as the primary imaging,” he said. In cases when the results are inconclusive, they order a PET scan. “We don’t perform biopsies anymore,” he said.
U.S. fast-track clinic models
The fast-track clinic models in the United States vary. Results of a survey of the U.S. clinics were presented as an abstract at the 2021 American College of Rheumatology annual meeting. Some centers have a vasculitis specialist obtain and interpret the imaging. At others, a vasculitis specialist refers patients to a VUS-trained rheumatologist to perform and interpret the test. Another approach is to have vasculitis specialists refer patients to ultrasound technicians trained in VUS, with a vascular surgeon interpreting the images and either a VUS-trained rheumatologist or vascular medicine specialist verifying the images.
The take-home message of that survey is that “ultrasound evaluation should be considered in the hands of experts, realizing that not everyone has that skill set, but if it is available, it’s a way to expedite diagnosis and it can be helpful in managing the GCA patient in an appropriate way, quicker than trying to schedule cross-sectional imaging,” said Massachusetts General’s Dr. Kohler, who is a coauthor of the abstract. “Certainly, cross-sectional imaging also plays an important role, but when it comes to confirming whether to continue with treatment or not for a very serious condition, ultrasound is a quick way to get the answer.”
In addition to the fast-track clinic at Massachusetts General, the survey included fast-track clinics at the University of Washington, Seattle; Brigham and Women’s Hospital, Boston; Loma Linda (Calif.) University; University of California, Los Angeles; and at a private practice, Arthritis and Rheumatism Associates in the Washington area.
Advantages of VUS vs. TAB
At Massachusetts General, some of the rheumatologists are trained to perform VUS. The rheumatologists also perform the clinical evaluation of suspected cases of GCA. The advantage of VUS, Dr. Kohler said, is that the answer is “right there”; that is, the imaging yields a diagnosis almost instantaneously whereas a biopsy must be sent to a lab for analysis.
“Since a lot of patients with suspected vasculitis may already come to us on steroid therapy, and if there’s a low probability or low suspicion for vasculitis, [VUS] actually confirms that, and we’re able to taper prednisone or steroids quickly rather than keep them on a prolonged course.”
Alison Bays, MD, MPH, of the department of rheumatology at the University of Washington, said that the advantages of avoiding biopsy aren’t to be overlooked. “Temporal artery biopsies are invasive and carry surgical risks, especially as many of our patients are elderly,” she told this news organization.
“These patients occasionally refuse biopsy, but the acceptance of ultrasound is high,” Dr. Bays said. “Scheduling surgery can be more complicated, resulting in delays to biopsy and potentially higher rates of false negatives. Additionally, ultrasound has resulted in a higher rate of diagnosis with GCA as TAB misses large-vessel involvement.” The fast-track clinic at the university has evaluated 250 patients since it opened in 2017.
Dr. Bays and colleagues published the first United States study of a fast-track protocol using vascular sonographers. “Our group has demonstrated that fast-track clinics can rapidly and effectively evaluate patients, and we demonstrated a different method of evaluation using vascular sonographers rather than rheumatologists to do the vascular ultrasound,” she said. “It utilizes the familiarity vascular sonographers already have with imaging blood vessels.”
She added that the TABUL study in the United Kingdom in 2016 demonstrated that VUS yielded a savings of $686 (£484 as reported in the study), compared with TAB. “Further studies need to be done in the United States,” she said. “Beyond direct comparison of costs, reduction in steroid burden due to quick evaluation and diagnosis may carry additional benefits.”
At Brigham and Women’s Hospital, the division of rheumatology and the vascular section of the cardiology division collaborate on the GCA evaluation, said Sara Tedeschi, MD, MPH, codirector of the fast-track clinic there. Trained vascular technologists credentialed in the procedure and specifically trained in using VUS for evaluating arteritis perform the VUS. Cardiologists with a subspecialty in vascular medicine interpret the studies.
VUS patients have a rheumatology evaluation just before they have the ultrasound. “The rheumatology evaluation is then able to incorporate information from the VUS together with laboratory data, the patient’s history, and physical examination,” Dr. Tedeschi said.
“If the rheumatologist recommends a temporal artery biopsy as a next step, we arrange this with vascular surgery,” she said. “If the rheumatologist recommends other imaging such as MRA [magnetic resonance angiography] or PET-CT, we frequently review the images together with our colleagues in cardiovascular radiology and/or nuclear medicine.”
But in the United States, it will take some time for GCA fast-track clinics to become the standard, Dr. Tedeschi said. “Temporal artery biopsy may be faster to arrange in certain practice settings if VUS is not already being employed for giant cell arteritis evaluation,” she said.
Dr. Bays recognized this limitation, saying, “We are hoping that in the future, the American College of Rheumatology will consider vascular ultrasound as a first-line diagnostic test in diagnosis as rheumatologists and vascular sonographers gain familiarity over time.”
But that would mean that centers performing VUS for GCA would have to meet rigorous standards for the procedure. “With that, standardization of a protocol, high-quality equipment, and adequate training are necessary to ensure quality and reduce the chance of false-positive or false-negative results,” she said.
Dr. Chrysidis, Dr. Bays, and Dr. Tedeschi have disclosed no relevant financial relationships. Dr. Kohler is a board member of Ultrasound School of North American Rheumatologists.
A version of this article first appeared on Medscape.com.
Temporal artery biopsy has been the standard for diagnosing giant cell arteritis (GCA), but vascular ultrasound, a procedure that’s less invasive, less time-intensive, less expensive, and more convenient, has gained widespread use in Europe, and now clinics in the United States are adopting this approach and moving toward having rheumatologists take on the role of ultrasonographer.
However, directors at these clinics – known as GCA fast-track clinics – caution that the bar can be high for adopting vascular ultrasound (VUS) as a tool to diagnose GCA. Ultrasonographers need specialized training to perform the task and an adequate caseload to maintain their skills. Clinics also need to be outfitted with high-definition ultrasound machines.
“It definitely takes adequate training and learning of how to adjust the settings on the ultrasound machine to be able to visualize the findings appropriately,” said Minna Kohler, MD, director of the rheumatology and musculoskeletal ultrasound program at Massachusetts General Hospital in Boston, which has its own GCA fast-track clinic.
“And the clinical context is very important,” she said. “If you have a high suspicion for someone with temporal arteritis, or GCA, and the patient has been on steroids for weeks before you see them, the ultrasound findings may not show signs of the disease. In those cases in which the imaging is equivocal, we would still pursue a biopsy.”
The idea of the fast-track clinic is as the name implies: to quickly confirm the presence of GCA in a matter of hours, or days at the most, in an outpatient setting without the hassles of a biopsy. Temporal artery biopsy (TAB), by comparison, “is more costly because it requires operating room time, a surgical consultation, and surgery time, whereas ultrasound is a very inexpensive exam since it’s done in the clinic by the rheumatologist,” Dr. Kohler said.
European experience
Use of VUS to diagnose GCA is supplanting TAB in Europe and other countries. In Denmark alone – with a population of 6 million – three outpatient fast-track clinics are operating. The United States, with a population more than 50 times larger than Denmark’s, has six.
Stavros Chrysidis, MD, PhD, chief of rheumatology at the Hospital South West Jutland, one of the fast-track clinic sites in Denmark, led a recent multicenter study, known as EUREKA, of VUS in patients with suspected GCA. He and his colleagues reported in The Lancet Rheumatology that the sensitivity and specificity of VUS was superior to TAB in confirming a diagnosis of GCA. Dr. Chrysidis has instructed U.S. rheumatologists and ultrasonographers in performing and interpreting VUS for GCA.
The study emphasizes the importance of training for ultrasonographers, said Dr. Chrysidis, who regularly performs VUS at his institution. “The most important finding is that, when we apply VUS by systematically trained ultrasonographers using appropriate equipment in appropriate settings, it has excellent diagnostic accuracy on GCA,” he told this news organization.
He noted that The Lancet Rheumatology report is the first multicenter study of VUS for diagnosing GCA in which all the ultrasonographers participated in a standardized training program, which his group developed. “Ultrasound is very operator dependent,” he said. “That’s why the training is very important.”
The training occurred over a year and included two workshops consisting of 5 days of theoretical training on VUS; supervised hands-on evaluation of healthy individuals and patients with known GCA; and evaluation of ultrasound images. Over the course of the year, trainees performed at least 50 VUS evaluations, half of which were in patients with confirmed GCA. During the training period, an external rheumatologist with broad experience in VUS made the final diagnosis.
“The equipment and settings are very important because ultrasound can be very time consuming if you are not educated well and if your equipment is not adjusted well,” Dr. Chrysidis said. The equipment must be calibrated beforehand “so you don’t spend time on adjustments.”
For diagnosing temporal artery anomalies, the ultrasound equipment must have a resolution of 0.3-0.4 mm, he said. “When you have a transducer of 10 MHz, you cannot visualize changes smaller than 5 mm.”
The EUREKA study stated that VUS could replace TAB as a first-line diagnostic tool for GCA – provided the ultrasonographers are systematically trained and the equipment and settings are appropriate. In the Jutland clinic, VUS already has replaced TAB, Dr. Chrysidis said.
“In my department since 2017, when we started the fast-track clinic after the EUREKA study was terminated, we have performed three temporal artery biopsies in the last 4 years, and we screen 60-70 patients per year because we use ultrasound as the primary imaging,” he said. In cases when the results are inconclusive, they order a PET scan. “We don’t perform biopsies anymore,” he said.
U.S. fast-track clinic models
The fast-track clinic models in the United States vary. Results of a survey of the U.S. clinics were presented as an abstract at the 2021 American College of Rheumatology annual meeting. Some centers have a vasculitis specialist obtain and interpret the imaging. At others, a vasculitis specialist refers patients to a VUS-trained rheumatologist to perform and interpret the test. Another approach is to have vasculitis specialists refer patients to ultrasound technicians trained in VUS, with a vascular surgeon interpreting the images and either a VUS-trained rheumatologist or vascular medicine specialist verifying the images.
The take-home message of that survey is that “ultrasound evaluation should be considered in the hands of experts, realizing that not everyone has that skill set, but if it is available, it’s a way to expedite diagnosis and it can be helpful in managing the GCA patient in an appropriate way, quicker than trying to schedule cross-sectional imaging,” said Massachusetts General’s Dr. Kohler, who is a coauthor of the abstract. “Certainly, cross-sectional imaging also plays an important role, but when it comes to confirming whether to continue with treatment or not for a very serious condition, ultrasound is a quick way to get the answer.”
In addition to the fast-track clinic at Massachusetts General, the survey included fast-track clinics at the University of Washington, Seattle; Brigham and Women’s Hospital, Boston; Loma Linda (Calif.) University; University of California, Los Angeles; and at a private practice, Arthritis and Rheumatism Associates in the Washington area.
Advantages of VUS vs. TAB
At Massachusetts General, some of the rheumatologists are trained to perform VUS. The rheumatologists also perform the clinical evaluation of suspected cases of GCA. The advantage of VUS, Dr. Kohler said, is that the answer is “right there”; that is, the imaging yields a diagnosis almost instantaneously whereas a biopsy must be sent to a lab for analysis.
“Since a lot of patients with suspected vasculitis may already come to us on steroid therapy, and if there’s a low probability or low suspicion for vasculitis, [VUS] actually confirms that, and we’re able to taper prednisone or steroids quickly rather than keep them on a prolonged course.”
Alison Bays, MD, MPH, of the department of rheumatology at the University of Washington, said that the advantages of avoiding biopsy aren’t to be overlooked. “Temporal artery biopsies are invasive and carry surgical risks, especially as many of our patients are elderly,” she told this news organization.
“These patients occasionally refuse biopsy, but the acceptance of ultrasound is high,” Dr. Bays said. “Scheduling surgery can be more complicated, resulting in delays to biopsy and potentially higher rates of false negatives. Additionally, ultrasound has resulted in a higher rate of diagnosis with GCA as TAB misses large-vessel involvement.” The fast-track clinic at the university has evaluated 250 patients since it opened in 2017.
Dr. Bays and colleagues published the first United States study of a fast-track protocol using vascular sonographers. “Our group has demonstrated that fast-track clinics can rapidly and effectively evaluate patients, and we demonstrated a different method of evaluation using vascular sonographers rather than rheumatologists to do the vascular ultrasound,” she said. “It utilizes the familiarity vascular sonographers already have with imaging blood vessels.”
She added that the TABUL study in the United Kingdom in 2016 demonstrated that VUS yielded a savings of $686 (£484 as reported in the study), compared with TAB. “Further studies need to be done in the United States,” she said. “Beyond direct comparison of costs, reduction in steroid burden due to quick evaluation and diagnosis may carry additional benefits.”
At Brigham and Women’s Hospital, the division of rheumatology and the vascular section of the cardiology division collaborate on the GCA evaluation, said Sara Tedeschi, MD, MPH, codirector of the fast-track clinic there. Trained vascular technologists credentialed in the procedure and specifically trained in using VUS for evaluating arteritis perform the VUS. Cardiologists with a subspecialty in vascular medicine interpret the studies.
VUS patients have a rheumatology evaluation just before they have the ultrasound. “The rheumatology evaluation is then able to incorporate information from the VUS together with laboratory data, the patient’s history, and physical examination,” Dr. Tedeschi said.
“If the rheumatologist recommends a temporal artery biopsy as a next step, we arrange this with vascular surgery,” she said. “If the rheumatologist recommends other imaging such as MRA [magnetic resonance angiography] or PET-CT, we frequently review the images together with our colleagues in cardiovascular radiology and/or nuclear medicine.”
But in the United States, it will take some time for GCA fast-track clinics to become the standard, Dr. Tedeschi said. “Temporal artery biopsy may be faster to arrange in certain practice settings if VUS is not already being employed for giant cell arteritis evaluation,” she said.
Dr. Bays recognized this limitation, saying, “We are hoping that in the future, the American College of Rheumatology will consider vascular ultrasound as a first-line diagnostic test in diagnosis as rheumatologists and vascular sonographers gain familiarity over time.”
But that would mean that centers performing VUS for GCA would have to meet rigorous standards for the procedure. “With that, standardization of a protocol, high-quality equipment, and adequate training are necessary to ensure quality and reduce the chance of false-positive or false-negative results,” she said.
Dr. Chrysidis, Dr. Bays, and Dr. Tedeschi have disclosed no relevant financial relationships. Dr. Kohler is a board member of Ultrasound School of North American Rheumatologists.
A version of this article first appeared on Medscape.com.
Validity of commercial serologic tests for dermatomyositis still questionable
, according to Jeffrey P. Callen, MD.

That’s because the validity and reproducibility of testing in commercial laboratories remain questionable, Dr. Callen, professor of medicine and chief of the division of dermatology at the University of Louisville, Ky., said during MedscapeLive’s annual Las Vegas Dermatology Seminar. “The testing in research laboratories is not widely available and the results are often delayed by weeks to months,” he said.
In addition, while the associations between antibody results and risks of malignancy or pulmonary disease are “statistically valid,” he said, “there are patients with disease in whom antibodies are not present and those without associated disease in whom the testing was positive.” For example, there are patients positive for anti–transition initiation factor (TIF)-1gamma but don’t have a malignancy, “and the ones with anti-MDA-5 tend to have pulmonary disease, but there are patients with anti-MDA-5 who don’t have pulmonary disease.”
Compared with patients with systemic lupus erythematosus, patients with dermatomyositis tend to have more itching and they tend of have fewer serologic abnormalities, such as anti-Ro/SS-A antibody, “but there is overlap,” Dr. Callen said. “The reason to differentiate cutaneous lupus erythematosus from dermatomyositis is because we think that patients who have amyopathic dermatomyositis still have an increased risk of having or developing an internal malignancy,” he added. Another differentiating point that is substantive is the presence of Gottron papules.
In a recent development related to antibody testing, researchers demonstrated that the IgG2 isotype of anti-TIF-1gamma antibodies is a biomarker of cancer and mortality in adult dermatomyositis.
According to population-based studies, about 20%-25% of dermatomyositis patients have had, have, or will develop a cancer (Lancet 2001;357: 96-100). Amyopathic dermatomyositis patients may also have cancer. Polymyositis patients generally have lower rates and their risk of subsequent malignancy is much closer to that of the general population, suggesting that the presence of the association is due to a “diagnostic suspicion bias,” Dr. Callen said.
A large-scale multicenter cohort study that set out to identify the risk factors and prognosis of patients with cancer-associated myositis found that ovarian cancer seems to be overrepresented. The only serologic abnormality that was statistically significant was anti-TIF-1gamma antibody (P less than .001). Patients with cancer-associated myositis also have less overall survival compared with those with non–cancer-associated myositis (P = .004), with malignancy being the primary cause of death (P less than .001).
In what is believed to be the largest study of its kind, Dr. Callen and colleagues retrospectively examined the prevalence of malignancy and screening practices in 400 dermatomyositis patients. Of the 400 patients, 48 (12%) had malignancies, and 21 cancers (40%) were diagnosed within 1 year of the dermatomyositis diagnosis. Both classic dermatomyositis and amyopathic dermatomyositis were associated with cancer, and 27 patients (6.8%) had a cancer at the time of diagnosis. Of those, 59% were asymptomatic; their cancers were discovered with CT scans, suggesting that “blind” screening is effective in identifying cancers in DM patients.
Dr. Callen’s malignancy evaluation includes chest x-ray, CT of the chest and abdomen, stool Hematest in all dermatomyositis patients; a mammogram, pelvic ultrasound and/or CT of the pelvis in women; and age, race or ethnicity-related testing. “I generally reevaluate patients annually for 3 years, because data from epidemiologic studies suggest that after 3 years [from the initial diagnosis], the rates of malignancy return toward normal,” he said. “I also evaluate any new symptom that might be suggestive of malignancy. The remaining issue is how to handle a patient in remission for several years, but who develops a relapse. What I do is perform another malignancy assessment.”
According to results from a meta-analysis of risk factors and systematic review of screening approaches, factors that increase malignancy risk include dermatomyositis subtype (risk ratio, 2.21), older age (weighted mean difference 11.19), male gender (RR, 1.53), dysphagia (RR, 2.09), cutaneous necrosis (RR, 2.73), and positive anti-TIF-1gamma (RR, 4.41).
Factors associated with a decreased risk of malignancy include polymyositis (RR, 0.49), clinically amyopathic dermatomyositis subtypes (RR, 0.44), Raynaud’s phenomenon (RR, 0.61), interstitial lung disease (RR, 0.49), very high serum creatine kinase (WMD –1189.96) or lactate dehydrogenase levels (WMD –336.53), and anti-Jo1 (RR, 0.45) or anti-EJ (RR, 0.17) positivity.
The analysis also found that CT scanning of the thorax, abdomen and pelvis appeared to yield a high proportion of underlying asymptomatic cancers. Limited evidence relating to the utility of tumor markers and 18F-FDG PET/CT was available.
As for treatment, the use of tofacitinib for cutaneous lesions of dermatomyositis has been suggested in various studies. In a recent open-label study of 10 patients with dermatomyositis who took extended release the JAK inhibitor tofacitinib 11 mg daily for 12 weeks, half experienced moderate improvement in disease activity, and the other half experienced minimal improvement. JAK inhibitors have been used in patients with juvenile dermatomyositis.
Dr. Callen’s treatment approach with dermatomyositis patients includes recommendations for sunscreens and protective clothing, plus assessment of vitamin D levels. “I will use topical emollients, corticosteroids, and calcineurin inhibitors,” he said. “Antimalarials might be used. I generally reach for methotrexate or mycophenolate mofetil relatively early. IVIG has also been studied.” Off-label therapies that have been used include dapsone, thalidomide, leflunomide, sirolimus, chlorambucil, etanercept, infliximab, rituximab, apremilast, tofacitinib, lenabasum, and low-dose naltrexone.
Dr. Callen disclosed that he is a consultant to Genentech and is a member of the safety monitoring committee for Principia Biopharma. He holds equity in Celgene, Pfizer, 3M, Johnson & Johnson, Merck, Abbott Laboratories, AbbVie, Procter & Gamble, Gilead, Allergen, and Amgen.
MedscapeLive and this news organization are owned by the same parent company.
, according to Jeffrey P. Callen, MD.
That’s because the validity and reproducibility of testing in commercial laboratories remain questionable, Dr. Callen, professor of medicine and chief of the division of dermatology at the University of Louisville, Ky., said during MedscapeLive’s annual Las Vegas Dermatology Seminar. “The testing in research laboratories is not widely available and the results are often delayed by weeks to months,” he said.
In addition, while the associations between antibody results and risks of malignancy or pulmonary disease are “statistically valid,” he said, “there are patients with disease in whom antibodies are not present and those without associated disease in whom the testing was positive.” For example, there are patients positive for anti–transition initiation factor (TIF)-1gamma but don’t have a malignancy, “and the ones with anti-MDA-5 tend to have pulmonary disease, but there are patients with anti-MDA-5 who don’t have pulmonary disease.”
Compared with patients with systemic lupus erythematosus, patients with dermatomyositis tend to have more itching and they tend of have fewer serologic abnormalities, such as anti-Ro/SS-A antibody, “but there is overlap,” Dr. Callen said. “The reason to differentiate cutaneous lupus erythematosus from dermatomyositis is because we think that patients who have amyopathic dermatomyositis still have an increased risk of having or developing an internal malignancy,” he added. Another differentiating point that is substantive is the presence of Gottron papules.
In a recent development related to antibody testing, researchers demonstrated that the IgG2 isotype of anti-TIF-1gamma antibodies is a biomarker of cancer and mortality in adult dermatomyositis.
According to population-based studies, about 20%-25% of dermatomyositis patients have had, have, or will develop a cancer (Lancet 2001;357: 96-100). Amyopathic dermatomyositis patients may also have cancer. Polymyositis patients generally have lower rates and their risk of subsequent malignancy is much closer to that of the general population, suggesting that the presence of the association is due to a “diagnostic suspicion bias,” Dr. Callen said.
A large-scale multicenter cohort study that set out to identify the risk factors and prognosis of patients with cancer-associated myositis found that ovarian cancer seems to be overrepresented. The only serologic abnormality that was statistically significant was anti-TIF-1gamma antibody (P less than .001). Patients with cancer-associated myositis also have less overall survival compared with those with non–cancer-associated myositis (P = .004), with malignancy being the primary cause of death (P less than .001).
In what is believed to be the largest study of its kind, Dr. Callen and colleagues retrospectively examined the prevalence of malignancy and screening practices in 400 dermatomyositis patients. Of the 400 patients, 48 (12%) had malignancies, and 21 cancers (40%) were diagnosed within 1 year of the dermatomyositis diagnosis. Both classic dermatomyositis and amyopathic dermatomyositis were associated with cancer, and 27 patients (6.8%) had a cancer at the time of diagnosis. Of those, 59% were asymptomatic; their cancers were discovered with CT scans, suggesting that “blind” screening is effective in identifying cancers in DM patients.
Dr. Callen’s malignancy evaluation includes chest x-ray, CT of the chest and abdomen, stool Hematest in all dermatomyositis patients; a mammogram, pelvic ultrasound and/or CT of the pelvis in women; and age, race or ethnicity-related testing. “I generally reevaluate patients annually for 3 years, because data from epidemiologic studies suggest that after 3 years [from the initial diagnosis], the rates of malignancy return toward normal,” he said. “I also evaluate any new symptom that might be suggestive of malignancy. The remaining issue is how to handle a patient in remission for several years, but who develops a relapse. What I do is perform another malignancy assessment.”
According to results from a meta-analysis of risk factors and systematic review of screening approaches, factors that increase malignancy risk include dermatomyositis subtype (risk ratio, 2.21), older age (weighted mean difference 11.19), male gender (RR, 1.53), dysphagia (RR, 2.09), cutaneous necrosis (RR, 2.73), and positive anti-TIF-1gamma (RR, 4.41).
Factors associated with a decreased risk of malignancy include polymyositis (RR, 0.49), clinically amyopathic dermatomyositis subtypes (RR, 0.44), Raynaud’s phenomenon (RR, 0.61), interstitial lung disease (RR, 0.49), very high serum creatine kinase (WMD –1189.96) or lactate dehydrogenase levels (WMD –336.53), and anti-Jo1 (RR, 0.45) or anti-EJ (RR, 0.17) positivity.
The analysis also found that CT scanning of the thorax, abdomen and pelvis appeared to yield a high proportion of underlying asymptomatic cancers. Limited evidence relating to the utility of tumor markers and 18F-FDG PET/CT was available.
As for treatment, the use of tofacitinib for cutaneous lesions of dermatomyositis has been suggested in various studies. In a recent open-label study of 10 patients with dermatomyositis who took extended release the JAK inhibitor tofacitinib 11 mg daily for 12 weeks, half experienced moderate improvement in disease activity, and the other half experienced minimal improvement. JAK inhibitors have been used in patients with juvenile dermatomyositis.
Dr. Callen’s treatment approach with dermatomyositis patients includes recommendations for sunscreens and protective clothing, plus assessment of vitamin D levels. “I will use topical emollients, corticosteroids, and calcineurin inhibitors,” he said. “Antimalarials might be used. I generally reach for methotrexate or mycophenolate mofetil relatively early. IVIG has also been studied.” Off-label therapies that have been used include dapsone, thalidomide, leflunomide, sirolimus, chlorambucil, etanercept, infliximab, rituximab, apremilast, tofacitinib, lenabasum, and low-dose naltrexone.
Dr. Callen disclosed that he is a consultant to Genentech and is a member of the safety monitoring committee for Principia Biopharma. He holds equity in Celgene, Pfizer, 3M, Johnson & Johnson, Merck, Abbott Laboratories, AbbVie, Procter & Gamble, Gilead, Allergen, and Amgen.
MedscapeLive and this news organization are owned by the same parent company.
, according to Jeffrey P. Callen, MD.
That’s because the validity and reproducibility of testing in commercial laboratories remain questionable, Dr. Callen, professor of medicine and chief of the division of dermatology at the University of Louisville, Ky., said during MedscapeLive’s annual Las Vegas Dermatology Seminar. “The testing in research laboratories is not widely available and the results are often delayed by weeks to months,” he said.
In addition, while the associations between antibody results and risks of malignancy or pulmonary disease are “statistically valid,” he said, “there are patients with disease in whom antibodies are not present and those without associated disease in whom the testing was positive.” For example, there are patients positive for anti–transition initiation factor (TIF)-1gamma but don’t have a malignancy, “and the ones with anti-MDA-5 tend to have pulmonary disease, but there are patients with anti-MDA-5 who don’t have pulmonary disease.”
Compared with patients with systemic lupus erythematosus, patients with dermatomyositis tend to have more itching and they tend of have fewer serologic abnormalities, such as anti-Ro/SS-A antibody, “but there is overlap,” Dr. Callen said. “The reason to differentiate cutaneous lupus erythematosus from dermatomyositis is because we think that patients who have amyopathic dermatomyositis still have an increased risk of having or developing an internal malignancy,” he added. Another differentiating point that is substantive is the presence of Gottron papules.
In a recent development related to antibody testing, researchers demonstrated that the IgG2 isotype of anti-TIF-1gamma antibodies is a biomarker of cancer and mortality in adult dermatomyositis.
According to population-based studies, about 20%-25% of dermatomyositis patients have had, have, or will develop a cancer (Lancet 2001;357: 96-100). Amyopathic dermatomyositis patients may also have cancer. Polymyositis patients generally have lower rates and their risk of subsequent malignancy is much closer to that of the general population, suggesting that the presence of the association is due to a “diagnostic suspicion bias,” Dr. Callen said.
A large-scale multicenter cohort study that set out to identify the risk factors and prognosis of patients with cancer-associated myositis found that ovarian cancer seems to be overrepresented. The only serologic abnormality that was statistically significant was anti-TIF-1gamma antibody (P less than .001). Patients with cancer-associated myositis also have less overall survival compared with those with non–cancer-associated myositis (P = .004), with malignancy being the primary cause of death (P less than .001).
In what is believed to be the largest study of its kind, Dr. Callen and colleagues retrospectively examined the prevalence of malignancy and screening practices in 400 dermatomyositis patients. Of the 400 patients, 48 (12%) had malignancies, and 21 cancers (40%) were diagnosed within 1 year of the dermatomyositis diagnosis. Both classic dermatomyositis and amyopathic dermatomyositis were associated with cancer, and 27 patients (6.8%) had a cancer at the time of diagnosis. Of those, 59% were asymptomatic; their cancers were discovered with CT scans, suggesting that “blind” screening is effective in identifying cancers in DM patients.
Dr. Callen’s malignancy evaluation includes chest x-ray, CT of the chest and abdomen, stool Hematest in all dermatomyositis patients; a mammogram, pelvic ultrasound and/or CT of the pelvis in women; and age, race or ethnicity-related testing. “I generally reevaluate patients annually for 3 years, because data from epidemiologic studies suggest that after 3 years [from the initial diagnosis], the rates of malignancy return toward normal,” he said. “I also evaluate any new symptom that might be suggestive of malignancy. The remaining issue is how to handle a patient in remission for several years, but who develops a relapse. What I do is perform another malignancy assessment.”
According to results from a meta-analysis of risk factors and systematic review of screening approaches, factors that increase malignancy risk include dermatomyositis subtype (risk ratio, 2.21), older age (weighted mean difference 11.19), male gender (RR, 1.53), dysphagia (RR, 2.09), cutaneous necrosis (RR, 2.73), and positive anti-TIF-1gamma (RR, 4.41).
Factors associated with a decreased risk of malignancy include polymyositis (RR, 0.49), clinically amyopathic dermatomyositis subtypes (RR, 0.44), Raynaud’s phenomenon (RR, 0.61), interstitial lung disease (RR, 0.49), very high serum creatine kinase (WMD –1189.96) or lactate dehydrogenase levels (WMD –336.53), and anti-Jo1 (RR, 0.45) or anti-EJ (RR, 0.17) positivity.
The analysis also found that CT scanning of the thorax, abdomen and pelvis appeared to yield a high proportion of underlying asymptomatic cancers. Limited evidence relating to the utility of tumor markers and 18F-FDG PET/CT was available.
As for treatment, the use of tofacitinib for cutaneous lesions of dermatomyositis has been suggested in various studies. In a recent open-label study of 10 patients with dermatomyositis who took extended release the JAK inhibitor tofacitinib 11 mg daily for 12 weeks, half experienced moderate improvement in disease activity, and the other half experienced minimal improvement. JAK inhibitors have been used in patients with juvenile dermatomyositis.
Dr. Callen’s treatment approach with dermatomyositis patients includes recommendations for sunscreens and protective clothing, plus assessment of vitamin D levels. “I will use topical emollients, corticosteroids, and calcineurin inhibitors,” he said. “Antimalarials might be used. I generally reach for methotrexate or mycophenolate mofetil relatively early. IVIG has also been studied.” Off-label therapies that have been used include dapsone, thalidomide, leflunomide, sirolimus, chlorambucil, etanercept, infliximab, rituximab, apremilast, tofacitinib, lenabasum, and low-dose naltrexone.
Dr. Callen disclosed that he is a consultant to Genentech and is a member of the safety monitoring committee for Principia Biopharma. He holds equity in Celgene, Pfizer, 3M, Johnson & Johnson, Merck, Abbott Laboratories, AbbVie, Procter & Gamble, Gilead, Allergen, and Amgen.
MedscapeLive and this news organization are owned by the same parent company.
FROM THE MEDSCAPELIVE LAS VEGAS DERMATOLOGY SEMINAR