User login
Drug produces similar results in older and younger ALL patients

Photo from MD Anderson
Data from two phase 2 studies suggests single-agent blinatumomab produces similar outcomes in adults with relapsed/refractory acute lymphoblastic leukemia (ALL), regardless of age.
Patients age 65 and older had similar hematologic response rates and relapse-free survival rates as patients younger than 65.
The incidence of grade 3 or higher adverse events (AEs) was similar between the age groups as well.
Older patients did have more serious AEs, however. And they had more neurologic events, but these were reversible.
Hagop M. Kantarjian, MD, of the University of Texas MD Anderson Cancer Center in Houston, and his colleagues reported these results in Cancer. The research was funded by Amgen Inc., makers of blinatumomab.
Patients
The researchers examined 261 adults with relapsed/refractory ALL who were enrolled in 2 different studies. There were 36 patients who were 65 or older and 225 patients who were younger than 65. The median ages were 70 (range, 65-79) and 34 (range, 18-64), respectively.
Among the older patients, 14% had primary refractory disease, 67% had 1 prior relapse, 14% had 2 prior relapses, and 6% had 3 or more. Among the younger patients, 9% had primary refractory disease, 55% had 1 prior relapse, 26% had 2 prior relapses, and 10% had 3 or more.
The younger patients were more likely to have received an allogeneic hematopoietic stem cell transplant (allo-HSCT) than the older patients—37% and 11%, respectively.
But older patients were more likely to have mild renal impairment (42% vs 13%) or moderate renal impairment (22% vs 1%).
Treatment
All patients received blinatumomab, and stepwise dosing was used to reduce the risk of cytokine release syndrome. A treatment cycle consisted of 4 weeks of continuous intravenous infusions, followed by a 2-week treatment-free interval.
The patients received 2 initial cycles. If they achieved a complete remission (CR) or CR with partial hematologic recovery (CRh) at this point, they could receive an additional 3 cycles as consolidation, unless they were scheduled to receive an allo-HSCT.
Patients also received intrathecal prophylaxis with dexamethasone and/or steroids, cytarabine, and methotrexate. And patients with a high blast percentage at baseline received a pre-phase treatment with dexamethasone and/or cyclophosphamide.
Older patients received a median of 2 cycles of blinatumomab (range, 1-6), as did the younger patients (range, 1-7).
Response and survival
Fifty-six percent of the older patients (20/36) achieved a CR/CRh during the first 2 cycles of blinatumomab, as did 46% of the younger patients (46/225). There were 14 CRs among the older patients (39%) and 78 CRs among the younger patients (35%).
There were 12 complete minimal residual disease responses among older patients (60% of responders) and 73 among the younger patients (70% of responders).
Of the responders, 3 older patients (15%) and 61 younger patients (59%) went on to allo-HSCT. Most of the patients received a transplant while in remission. However, 1 of the older patients and 8 of the younger patients went to transplant after an initial response to blinatumomab that was followed by a relapse.
The median relapse-free survival was 7.4 months for both age groups. The median overall survival was 5.5 months for older patients and 7.6 months for younger patients.
Safety
All of the older patients had at least 1 AE, and all but 1 of the younger patients had at least 1 AE. Older patients had higher rates of peripheral edema (42% vs 24%), fatigue (28% vs 18%), and dizziness (25% vs 11%) of any grade.
The incidence of grade 3 or higher AEs was similar between the groups—86% in the older group and 80% in the younger group. The same was true for AEs leading to treatment discontinuation—22% and 19%, respectively.
However, there was a higher incidence of serious AEs in the older patients (72% vs 64%). Device-related infection and encephalopathy were more common among older patients than younger patients (both 11% vs 3%).
The incidence of cytokine release syndrome was higher in the older group than the younger group—19% and 10%, respectively.
Older patients also had more neurologic events of any grade (72% vs 48%) and more grade 3 or higher neurologic events (28% vs 13%). However, all neurologic events were reversed by temporarily or permanently discontinuing blinatumomab.
There were 7 fatal treatment-emergent AEs in the older adults, including pneumonia (n=3), B-cell lymphoma (n=1), and disease progression (n=3). None of the fatal AEs were considered treatment-related. And none of the patients who were in remission died during treatment with blinatumomab. ![]()
Photo from MD Anderson
Data from two phase 2 studies suggests single-agent blinatumomab produces similar outcomes in adults with relapsed/refractory acute lymphoblastic leukemia (ALL), regardless of age.
Patients age 65 and older had similar hematologic response rates and relapse-free survival rates as patients younger than 65.
The incidence of grade 3 or higher adverse events (AEs) was similar between the age groups as well.
Older patients did have more serious AEs, however. And they had more neurologic events, but these were reversible.
Hagop M. Kantarjian, MD, of the University of Texas MD Anderson Cancer Center in Houston, and his colleagues reported these results in Cancer. The research was funded by Amgen Inc., makers of blinatumomab.
Patients
The researchers examined 261 adults with relapsed/refractory ALL who were enrolled in 2 different studies. There were 36 patients who were 65 or older and 225 patients who were younger than 65. The median ages were 70 (range, 65-79) and 34 (range, 18-64), respectively.
Among the older patients, 14% had primary refractory disease, 67% had 1 prior relapse, 14% had 2 prior relapses, and 6% had 3 or more. Among the younger patients, 9% had primary refractory disease, 55% had 1 prior relapse, 26% had 2 prior relapses, and 10% had 3 or more.
The younger patients were more likely to have received an allogeneic hematopoietic stem cell transplant (allo-HSCT) than the older patients—37% and 11%, respectively.
But older patients were more likely to have mild renal impairment (42% vs 13%) or moderate renal impairment (22% vs 1%).
Treatment
All patients received blinatumomab, and stepwise dosing was used to reduce the risk of cytokine release syndrome. A treatment cycle consisted of 4 weeks of continuous intravenous infusions, followed by a 2-week treatment-free interval.
The patients received 2 initial cycles. If they achieved a complete remission (CR) or CR with partial hematologic recovery (CRh) at this point, they could receive an additional 3 cycles as consolidation, unless they were scheduled to receive an allo-HSCT.
Patients also received intrathecal prophylaxis with dexamethasone and/or steroids, cytarabine, and methotrexate. And patients with a high blast percentage at baseline received a pre-phase treatment with dexamethasone and/or cyclophosphamide.
Older patients received a median of 2 cycles of blinatumomab (range, 1-6), as did the younger patients (range, 1-7).
Response and survival
Fifty-six percent of the older patients (20/36) achieved a CR/CRh during the first 2 cycles of blinatumomab, as did 46% of the younger patients (46/225). There were 14 CRs among the older patients (39%) and 78 CRs among the younger patients (35%).
There were 12 complete minimal residual disease responses among older patients (60% of responders) and 73 among the younger patients (70% of responders).
Of the responders, 3 older patients (15%) and 61 younger patients (59%) went on to allo-HSCT. Most of the patients received a transplant while in remission. However, 1 of the older patients and 8 of the younger patients went to transplant after an initial response to blinatumomab that was followed by a relapse.
The median relapse-free survival was 7.4 months for both age groups. The median overall survival was 5.5 months for older patients and 7.6 months for younger patients.
Safety
All of the older patients had at least 1 AE, and all but 1 of the younger patients had at least 1 AE. Older patients had higher rates of peripheral edema (42% vs 24%), fatigue (28% vs 18%), and dizziness (25% vs 11%) of any grade.
The incidence of grade 3 or higher AEs was similar between the groups—86% in the older group and 80% in the younger group. The same was true for AEs leading to treatment discontinuation—22% and 19%, respectively.
However, there was a higher incidence of serious AEs in the older patients (72% vs 64%). Device-related infection and encephalopathy were more common among older patients than younger patients (both 11% vs 3%).
The incidence of cytokine release syndrome was higher in the older group than the younger group—19% and 10%, respectively.
Older patients also had more neurologic events of any grade (72% vs 48%) and more grade 3 or higher neurologic events (28% vs 13%). However, all neurologic events were reversed by temporarily or permanently discontinuing blinatumomab.
There were 7 fatal treatment-emergent AEs in the older adults, including pneumonia (n=3), B-cell lymphoma (n=1), and disease progression (n=3). None of the fatal AEs were considered treatment-related. And none of the patients who were in remission died during treatment with blinatumomab. ![]()
Photo from MD Anderson
Data from two phase 2 studies suggests single-agent blinatumomab produces similar outcomes in adults with relapsed/refractory acute lymphoblastic leukemia (ALL), regardless of age.
Patients age 65 and older had similar hematologic response rates and relapse-free survival rates as patients younger than 65.
The incidence of grade 3 or higher adverse events (AEs) was similar between the age groups as well.
Older patients did have more serious AEs, however. And they had more neurologic events, but these were reversible.
Hagop M. Kantarjian, MD, of the University of Texas MD Anderson Cancer Center in Houston, and his colleagues reported these results in Cancer. The research was funded by Amgen Inc., makers of blinatumomab.
Patients
The researchers examined 261 adults with relapsed/refractory ALL who were enrolled in 2 different studies. There were 36 patients who were 65 or older and 225 patients who were younger than 65. The median ages were 70 (range, 65-79) and 34 (range, 18-64), respectively.
Among the older patients, 14% had primary refractory disease, 67% had 1 prior relapse, 14% had 2 prior relapses, and 6% had 3 or more. Among the younger patients, 9% had primary refractory disease, 55% had 1 prior relapse, 26% had 2 prior relapses, and 10% had 3 or more.
The younger patients were more likely to have received an allogeneic hematopoietic stem cell transplant (allo-HSCT) than the older patients—37% and 11%, respectively.
But older patients were more likely to have mild renal impairment (42% vs 13%) or moderate renal impairment (22% vs 1%).
Treatment
All patients received blinatumomab, and stepwise dosing was used to reduce the risk of cytokine release syndrome. A treatment cycle consisted of 4 weeks of continuous intravenous infusions, followed by a 2-week treatment-free interval.
The patients received 2 initial cycles. If they achieved a complete remission (CR) or CR with partial hematologic recovery (CRh) at this point, they could receive an additional 3 cycles as consolidation, unless they were scheduled to receive an allo-HSCT.
Patients also received intrathecal prophylaxis with dexamethasone and/or steroids, cytarabine, and methotrexate. And patients with a high blast percentage at baseline received a pre-phase treatment with dexamethasone and/or cyclophosphamide.
Older patients received a median of 2 cycles of blinatumomab (range, 1-6), as did the younger patients (range, 1-7).
Response and survival
Fifty-six percent of the older patients (20/36) achieved a CR/CRh during the first 2 cycles of blinatumomab, as did 46% of the younger patients (46/225). There were 14 CRs among the older patients (39%) and 78 CRs among the younger patients (35%).
There were 12 complete minimal residual disease responses among older patients (60% of responders) and 73 among the younger patients (70% of responders).
Of the responders, 3 older patients (15%) and 61 younger patients (59%) went on to allo-HSCT. Most of the patients received a transplant while in remission. However, 1 of the older patients and 8 of the younger patients went to transplant after an initial response to blinatumomab that was followed by a relapse.
The median relapse-free survival was 7.4 months for both age groups. The median overall survival was 5.5 months for older patients and 7.6 months for younger patients.
Safety
All of the older patients had at least 1 AE, and all but 1 of the younger patients had at least 1 AE. Older patients had higher rates of peripheral edema (42% vs 24%), fatigue (28% vs 18%), and dizziness (25% vs 11%) of any grade.
The incidence of grade 3 or higher AEs was similar between the groups—86% in the older group and 80% in the younger group. The same was true for AEs leading to treatment discontinuation—22% and 19%, respectively.
However, there was a higher incidence of serious AEs in the older patients (72% vs 64%). Device-related infection and encephalopathy were more common among older patients than younger patients (both 11% vs 3%).
The incidence of cytokine release syndrome was higher in the older group than the younger group—19% and 10%, respectively.
Older patients also had more neurologic events of any grade (72% vs 48%) and more grade 3 or higher neurologic events (28% vs 13%). However, all neurologic events were reversed by temporarily or permanently discontinuing blinatumomab.
There were 7 fatal treatment-emergent AEs in the older adults, including pneumonia (n=3), B-cell lymphoma (n=1), and disease progression (n=3). None of the fatal AEs were considered treatment-related. And none of the patients who were in remission died during treatment with blinatumomab. ![]()
CAR T-cell therapy granted orphan designation

The US Food and Drug Administration (FDA) has granted orphan drug designation for the chimeric antigen receptor (CAR) T-cell therapy KTE-C19 as a treatment for several hematologic malignancies.
This includes primary mediastinal B-cell lymphoma (PMBCL), mantle cell lymphoma (MCL), follicular lymphoma (FL), acute lymphoblastic leukemia (ALL), and chronic lymphocytic leukemia (CLL).
KTE-C19 previously received orphan designation from the FDA for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity.
KTE-C19 also has breakthrough therapy designation from the FDA as a treatment for DLBCL, PMBCL, and transformed FL.
About KTE-C19
KTE-C19 is an investigational therapy in which a patient’s T cells are genetically modified to express a CAR designed to target CD19. The product is being developed by Kite Pharma, Inc.
In a study published in the Journal of Clinical Oncology, researchers evaluated KTE-C19 in 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of KTE-C19. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. The overall response rate was 92%. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with DLBCL, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Three of the CRs were ongoing at the time of publication, with the duration ranging from 9 months to 22 months.
Of the 4 patients with CLL, 3 had a CR, and 1 had a PR. All 3 CRs were ongoing at the time of publication, with the duration ranging from 14 months to 23 months.
Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR. The duration of the CR was 11 months at the time of publication.
KTE-C19 elicited a number of adverse events, including fever, hypotension, delirium, and other neurologic toxicities. All but 2 patients experienced grade 3/4 adverse events.
Three patients developed unexpected neurologic abnormalities. One patient experienced aphasia and right-sided facial paresis. One patient developed aphasia, confusion, and severe, generalized myoclonus. And 1 patient had aphasia, confusion, hemifacial spasms, apraxia, and gait disturbances.
KTE-C19 is currently under investigation in a phase 2 trial of refractory DLBCL, PMBCL, and transformed FL (ZUMA-1), a phase 2 trial of relapsed/refractory MCL (ZUMA-2), a phase 1/2 trial of relapsed/refractory adult ALL (ZUMA-3), and a phase 1/2 trial of relapsed/refractory pediatric ALL (ZUMA-4).
Results from ZUMA-1 were recently presented at the 2016 AACR Annual Meeting (abstract CT135). ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the chimeric antigen receptor (CAR) T-cell therapy KTE-C19 as a treatment for several hematologic malignancies.
This includes primary mediastinal B-cell lymphoma (PMBCL), mantle cell lymphoma (MCL), follicular lymphoma (FL), acute lymphoblastic leukemia (ALL), and chronic lymphocytic leukemia (CLL).
KTE-C19 previously received orphan designation from the FDA for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity.
KTE-C19 also has breakthrough therapy designation from the FDA as a treatment for DLBCL, PMBCL, and transformed FL.
About KTE-C19
KTE-C19 is an investigational therapy in which a patient’s T cells are genetically modified to express a CAR designed to target CD19. The product is being developed by Kite Pharma, Inc.
In a study published in the Journal of Clinical Oncology, researchers evaluated KTE-C19 in 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of KTE-C19. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. The overall response rate was 92%. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with DLBCL, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Three of the CRs were ongoing at the time of publication, with the duration ranging from 9 months to 22 months.
Of the 4 patients with CLL, 3 had a CR, and 1 had a PR. All 3 CRs were ongoing at the time of publication, with the duration ranging from 14 months to 23 months.
Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR. The duration of the CR was 11 months at the time of publication.
KTE-C19 elicited a number of adverse events, including fever, hypotension, delirium, and other neurologic toxicities. All but 2 patients experienced grade 3/4 adverse events.
Three patients developed unexpected neurologic abnormalities. One patient experienced aphasia and right-sided facial paresis. One patient developed aphasia, confusion, and severe, generalized myoclonus. And 1 patient had aphasia, confusion, hemifacial spasms, apraxia, and gait disturbances.
KTE-C19 is currently under investigation in a phase 2 trial of refractory DLBCL, PMBCL, and transformed FL (ZUMA-1), a phase 2 trial of relapsed/refractory MCL (ZUMA-2), a phase 1/2 trial of relapsed/refractory adult ALL (ZUMA-3), and a phase 1/2 trial of relapsed/refractory pediatric ALL (ZUMA-4).
Results from ZUMA-1 were recently presented at the 2016 AACR Annual Meeting (abstract CT135). ![]()
The US Food and Drug Administration (FDA) has granted orphan drug designation for the chimeric antigen receptor (CAR) T-cell therapy KTE-C19 as a treatment for several hematologic malignancies.
This includes primary mediastinal B-cell lymphoma (PMBCL), mantle cell lymphoma (MCL), follicular lymphoma (FL), acute lymphoblastic leukemia (ALL), and chronic lymphocytic leukemia (CLL).
KTE-C19 previously received orphan designation from the FDA for the treatment of diffuse large B-cell lymphoma (DLBCL).
The FDA grants orphan designation to drugs and biologics intended to treat, diagnose, or prevent diseases/disorders that affect fewer than 200,000 people in the US.
The designation provides incentives for sponsors to develop products for rare diseases. This may include tax credits toward the cost of clinical trials, prescription drug user fee waivers, and 7 years of market exclusivity.
KTE-C19 also has breakthrough therapy designation from the FDA as a treatment for DLBCL, PMBCL, and transformed FL.
About KTE-C19
KTE-C19 is an investigational therapy in which a patient’s T cells are genetically modified to express a CAR designed to target CD19. The product is being developed by Kite Pharma, Inc.
In a study published in the Journal of Clinical Oncology, researchers evaluated KTE-C19 in 15 patients with advanced B-cell malignancies.
The patients received a conditioning regimen of cyclophosphamide and fludarabine, followed 1 day later by a single infusion of KTE-C19. The researchers noted that the conditioning regimen is known to be active against B-cell malignancies and could have made a direct contribution to patient responses.
Thirteen patients were evaluable for response. The overall response rate was 92%. Eight patients achieved a complete response (CR), and 4 had a partial response (PR).
Of the 7 patients with DLBCL, 4 achieved a CR, 2 achieved a PR, and 1 had stable disease. Three of the CRs were ongoing at the time of publication, with the duration ranging from 9 months to 22 months.
Of the 4 patients with CLL, 3 had a CR, and 1 had a PR. All 3 CRs were ongoing at the time of publication, with the duration ranging from 14 months to 23 months.
Among the 2 patients with indolent lymphomas, 1 achieved a CR, and 1 had a PR. The duration of the CR was 11 months at the time of publication.
KTE-C19 elicited a number of adverse events, including fever, hypotension, delirium, and other neurologic toxicities. All but 2 patients experienced grade 3/4 adverse events.
Three patients developed unexpected neurologic abnormalities. One patient experienced aphasia and right-sided facial paresis. One patient developed aphasia, confusion, and severe, generalized myoclonus. And 1 patient had aphasia, confusion, hemifacial spasms, apraxia, and gait disturbances.
KTE-C19 is currently under investigation in a phase 2 trial of refractory DLBCL, PMBCL, and transformed FL (ZUMA-1), a phase 2 trial of relapsed/refractory MCL (ZUMA-2), a phase 1/2 trial of relapsed/refractory adult ALL (ZUMA-3), and a phase 1/2 trial of relapsed/refractory pediatric ALL (ZUMA-4).
Results from ZUMA-1 were recently presented at the 2016 AACR Annual Meeting (abstract CT135). ![]()
On the road to harnessing CRISPR gene editing to treat cancer
CRISPR technology, a simple, yet incredibly powerful tool for genetic engineering, is not only allowing cancer researchers to screen for drug targets more efficiently, but is also opening the door for direct cancer treatment through gene interference or activation.
“The pace at which this technology is developing is astounding and almost every cancer research lab is now using some version of it in their studies,” Dr. Scott A. Armstrong, director of Memorial Sloan Kettering Leukemia Center, New York, said in an interview.

Ancient defense mechanism
While the term CRISPR is now synonymous with the editing of human genes, it actually refers to a sort of primitive immune system used by bacteria for billions of years. CRISPR is an acronym for Clustered Regularly Interspaced Short Palindromic Repeats, but the complex terminology belies an elegantly simple mechanism by which bacterial cells destroy invading pathogens.
The bacterial genome contains regions with short repetitive stretches of DNA that are separated by spacers. Researchers made the startling discovery that the spacers are often composed of bits of foreign DNA and it transpired that bacteria use it as a molecular memory of prior infection.
When the same pathogen is encountered again, the stretches of repeats and spacers are transcribed to form CRISPR RNAs (crRNA). Together with a transactivating RNA (tracrRNA), it forms a kind of GPS system for a series of CRISPR-associated (Cas) proteins that function like molecular scissors, destroying the target DNA sequence in the invader’s genome.
There are three CRISPR systems – type I, II, and III – that are associated with different sets of Cas proteins and each employ unique methods for achieving the same ultimate function. The type II system that pairs with Cas9 has received the most attention.
Cut and paste gene editing
The discovery that CRISPR could be exploited as a tool for genetic manipulation in mammalian cells sparked a revolution in the genome editing field. “The CRISPR-Cas9 system has been adapted to specifically edit the genomes of mammalian cells, allowing one to make targeted changes to almost any gene,” Dr. Armstrong said.
The use of CRISPR-Cas9 as a genome editing tool is simplified by joining the crRNA and tracrRNA together so they are transcribed in a single guide RNA (gRNA). The GPS coordinates of the gRNA can be preprogrammed to target a gene of interest, specifically directing the co-transcribed Cas9 protein to cut at that location, and introducing a double-strand break (DSB) in the DNA. Cells employ a number of different mechanisms to repair DSBs and these can then be exploited for genome editing purposes, allowing researchers to introduce changes to the DNA as it is repaired.
The CRISPR-Cas9 system excels in its simplicity – allowing alterations to be made to the genome much more easily, quickly, and cheaply than ever before, plagued by far fewer off-target effects. It also allows researchers to examine the function of multiple genes at once, where before they were mostly limited to a single gene.

“Cancer genomics has identified a large number of genes that are mutated in human cancer,” Tyler Jacks, Ph.D., director of the Koch Institute for Integrative Cancer Research at MIT, Boston, said in an interview. “CRISPR allows us to study these genes in cancer cells and in whole animals much more efficiently than the methods that were in use just a few years ago.”
But the potential of the CRISPR-Cas9 system doesn’t stop there. “At the moment, a particularly exciting application is the use of this approach to inactive genes in very specific fashion to assess the function of a given protein in a cancer cell, which should speed the identification of proteins that are important for cancer cells and thus potentially aid drug discovery efforts,” Dr. Armstrong said.
CRISPR at AACR
The latest developments in the use of the CRISPR-Cas9 system were highlighted at the annual meeting of the American Association of Cancer Research. Dr. David Sabatini, professor of biology at MIT, Boston, described his own lab’s method for using CRISPR-Cas9 to seek out the essential genes involved in different types of cancers. In a study recently published in Science, he and his colleagues employed this method in chronic myelogenous leukemia and Burkitt’s lymphoma cell lines. The gRNA library targeted just over 18,000 genes and roughly 10% of these proved to be essential. Mostly, these genes were linked to key cellular processes (Science 2015;350[6264]:1096-1101).
Dr. Christopher Vakoc of Cold Spring Harbor (N.Y.) Laboratory, presented a slightly different kind of CRISPR screen for drug targets. Most commonly, CRISPR introduces edits at the start of the gene, which may or may not change the DNA enough to produce a nonfunctional protein. Dr. Vakoc’s lab has developed a system that instead edits functional protein domains, which present ideal drug targets. Mutated domains can be identified that are essential for cancer cell survival and small molecule inhibitors designed that bind to them to kill cancer cells.
The technique has already been used to identify such a domain on the BRD4 protein and inhibitors that bind to this domain had significant antitumor activity in leukemia, Dr. Vakoc reported. A screen targeting 192 chromatin regulatory domains expressed in mouse acute myeloid leukemia cells was subsequently performed and identified 25 domains that impacted survival, 6 that are already being therapeutically targeted, and 19 novel potential targets.
Another development in CRISPR-Cas9 technology creates an inactive version of the Cas9 enzyme, one that has lost the ability to cut DNA. Though it seems counterintuitive, this has opened up a wealth of new possible uses. Jonathan S. Weissman, Ph.D., professor of cellular and molecular pharmacology, University of California, San Francisco, part of the group to develop this ‘dead’ Cas9 (dCas9), published a description of the use of two new tools dubbed CRISPR interference and CRISPR activation (Cell 2013;152[5]:1173-83).
Essentially, by fusing dCas9 with different proteins, such as epigenetic modifiers or transcriptional activators or repressors, it can be used as a delivery system to fine-tune gene expression, instead of editing the gene sequence.
Treating cancer?
Ultimately, CRISPR-Cas9 could be used to treat cancers by cutting out defective genes and replacing them with a wild-type version, or by repairing mutations, though for the time being this is theoretical. Studies have suggested it is possible with other types of diseases, however.
“It is not clear exactly how the CRISPR system would be used to directly treat cancer, but the discoveries that come from its use will likely lead to new ways to treat cancer,” said Dr Armstrong.
Dr Jacks highlighted the technical challenges that will need to be overcome first. “In principle, CRISPR-based genome editing could be used to correct cancer-causing mutations in tumors in vivo or to inactivate activated cancer genes,” he said. “At this point, however, we lack the technology necessary to deliver the CRISPR system to all cancer cells in the body. Improvements in this so-called ‘delivery problem’ may allow CRISPR to become a powerful anticancer therapy strategy.”
CRISPR technology, a simple, yet incredibly powerful tool for genetic engineering, is not only allowing cancer researchers to screen for drug targets more efficiently, but is also opening the door for direct cancer treatment through gene interference or activation.
“The pace at which this technology is developing is astounding and almost every cancer research lab is now using some version of it in their studies,” Dr. Scott A. Armstrong, director of Memorial Sloan Kettering Leukemia Center, New York, said in an interview.
Ancient defense mechanism
While the term CRISPR is now synonymous with the editing of human genes, it actually refers to a sort of primitive immune system used by bacteria for billions of years. CRISPR is an acronym for Clustered Regularly Interspaced Short Palindromic Repeats, but the complex terminology belies an elegantly simple mechanism by which bacterial cells destroy invading pathogens.
The bacterial genome contains regions with short repetitive stretches of DNA that are separated by spacers. Researchers made the startling discovery that the spacers are often composed of bits of foreign DNA and it transpired that bacteria use it as a molecular memory of prior infection.
When the same pathogen is encountered again, the stretches of repeats and spacers are transcribed to form CRISPR RNAs (crRNA). Together with a transactivating RNA (tracrRNA), it forms a kind of GPS system for a series of CRISPR-associated (Cas) proteins that function like molecular scissors, destroying the target DNA sequence in the invader’s genome.
There are three CRISPR systems – type I, II, and III – that are associated with different sets of Cas proteins and each employ unique methods for achieving the same ultimate function. The type II system that pairs with Cas9 has received the most attention.
Cut and paste gene editing
The discovery that CRISPR could be exploited as a tool for genetic manipulation in mammalian cells sparked a revolution in the genome editing field. “The CRISPR-Cas9 system has been adapted to specifically edit the genomes of mammalian cells, allowing one to make targeted changes to almost any gene,” Dr. Armstrong said.
The use of CRISPR-Cas9 as a genome editing tool is simplified by joining the crRNA and tracrRNA together so they are transcribed in a single guide RNA (gRNA). The GPS coordinates of the gRNA can be preprogrammed to target a gene of interest, specifically directing the co-transcribed Cas9 protein to cut at that location, and introducing a double-strand break (DSB) in the DNA. Cells employ a number of different mechanisms to repair DSBs and these can then be exploited for genome editing purposes, allowing researchers to introduce changes to the DNA as it is repaired.
The CRISPR-Cas9 system excels in its simplicity – allowing alterations to be made to the genome much more easily, quickly, and cheaply than ever before, plagued by far fewer off-target effects. It also allows researchers to examine the function of multiple genes at once, where before they were mostly limited to a single gene.
“Cancer genomics has identified a large number of genes that are mutated in human cancer,” Tyler Jacks, Ph.D., director of the Koch Institute for Integrative Cancer Research at MIT, Boston, said in an interview. “CRISPR allows us to study these genes in cancer cells and in whole animals much more efficiently than the methods that were in use just a few years ago.”
But the potential of the CRISPR-Cas9 system doesn’t stop there. “At the moment, a particularly exciting application is the use of this approach to inactive genes in very specific fashion to assess the function of a given protein in a cancer cell, which should speed the identification of proteins that are important for cancer cells and thus potentially aid drug discovery efforts,” Dr. Armstrong said.
CRISPR at AACR
The latest developments in the use of the CRISPR-Cas9 system were highlighted at the annual meeting of the American Association of Cancer Research. Dr. David Sabatini, professor of biology at MIT, Boston, described his own lab’s method for using CRISPR-Cas9 to seek out the essential genes involved in different types of cancers. In a study recently published in Science, he and his colleagues employed this method in chronic myelogenous leukemia and Burkitt’s lymphoma cell lines. The gRNA library targeted just over 18,000 genes and roughly 10% of these proved to be essential. Mostly, these genes were linked to key cellular processes (Science 2015;350[6264]:1096-1101).
Dr. Christopher Vakoc of Cold Spring Harbor (N.Y.) Laboratory, presented a slightly different kind of CRISPR screen for drug targets. Most commonly, CRISPR introduces edits at the start of the gene, which may or may not change the DNA enough to produce a nonfunctional protein. Dr. Vakoc’s lab has developed a system that instead edits functional protein domains, which present ideal drug targets. Mutated domains can be identified that are essential for cancer cell survival and small molecule inhibitors designed that bind to them to kill cancer cells.
The technique has already been used to identify such a domain on the BRD4 protein and inhibitors that bind to this domain had significant antitumor activity in leukemia, Dr. Vakoc reported. A screen targeting 192 chromatin regulatory domains expressed in mouse acute myeloid leukemia cells was subsequently performed and identified 25 domains that impacted survival, 6 that are already being therapeutically targeted, and 19 novel potential targets.
Another development in CRISPR-Cas9 technology creates an inactive version of the Cas9 enzyme, one that has lost the ability to cut DNA. Though it seems counterintuitive, this has opened up a wealth of new possible uses. Jonathan S. Weissman, Ph.D., professor of cellular and molecular pharmacology, University of California, San Francisco, part of the group to develop this ‘dead’ Cas9 (dCas9), published a description of the use of two new tools dubbed CRISPR interference and CRISPR activation (Cell 2013;152[5]:1173-83).
Essentially, by fusing dCas9 with different proteins, such as epigenetic modifiers or transcriptional activators or repressors, it can be used as a delivery system to fine-tune gene expression, instead of editing the gene sequence.
Treating cancer?
Ultimately, CRISPR-Cas9 could be used to treat cancers by cutting out defective genes and replacing them with a wild-type version, or by repairing mutations, though for the time being this is theoretical. Studies have suggested it is possible with other types of diseases, however.
“It is not clear exactly how the CRISPR system would be used to directly treat cancer, but the discoveries that come from its use will likely lead to new ways to treat cancer,” said Dr Armstrong.
Dr Jacks highlighted the technical challenges that will need to be overcome first. “In principle, CRISPR-based genome editing could be used to correct cancer-causing mutations in tumors in vivo or to inactivate activated cancer genes,” he said. “At this point, however, we lack the technology necessary to deliver the CRISPR system to all cancer cells in the body. Improvements in this so-called ‘delivery problem’ may allow CRISPR to become a powerful anticancer therapy strategy.”
CRISPR technology, a simple, yet incredibly powerful tool for genetic engineering, is not only allowing cancer researchers to screen for drug targets more efficiently, but is also opening the door for direct cancer treatment through gene interference or activation.
“The pace at which this technology is developing is astounding and almost every cancer research lab is now using some version of it in their studies,” Dr. Scott A. Armstrong, director of Memorial Sloan Kettering Leukemia Center, New York, said in an interview.
Ancient defense mechanism
While the term CRISPR is now synonymous with the editing of human genes, it actually refers to a sort of primitive immune system used by bacteria for billions of years. CRISPR is an acronym for Clustered Regularly Interspaced Short Palindromic Repeats, but the complex terminology belies an elegantly simple mechanism by which bacterial cells destroy invading pathogens.
The bacterial genome contains regions with short repetitive stretches of DNA that are separated by spacers. Researchers made the startling discovery that the spacers are often composed of bits of foreign DNA and it transpired that bacteria use it as a molecular memory of prior infection.
When the same pathogen is encountered again, the stretches of repeats and spacers are transcribed to form CRISPR RNAs (crRNA). Together with a transactivating RNA (tracrRNA), it forms a kind of GPS system for a series of CRISPR-associated (Cas) proteins that function like molecular scissors, destroying the target DNA sequence in the invader’s genome.
There are three CRISPR systems – type I, II, and III – that are associated with different sets of Cas proteins and each employ unique methods for achieving the same ultimate function. The type II system that pairs with Cas9 has received the most attention.
Cut and paste gene editing
The discovery that CRISPR could be exploited as a tool for genetic manipulation in mammalian cells sparked a revolution in the genome editing field. “The CRISPR-Cas9 system has been adapted to specifically edit the genomes of mammalian cells, allowing one to make targeted changes to almost any gene,” Dr. Armstrong said.
The use of CRISPR-Cas9 as a genome editing tool is simplified by joining the crRNA and tracrRNA together so they are transcribed in a single guide RNA (gRNA). The GPS coordinates of the gRNA can be preprogrammed to target a gene of interest, specifically directing the co-transcribed Cas9 protein to cut at that location, and introducing a double-strand break (DSB) in the DNA. Cells employ a number of different mechanisms to repair DSBs and these can then be exploited for genome editing purposes, allowing researchers to introduce changes to the DNA as it is repaired.
The CRISPR-Cas9 system excels in its simplicity – allowing alterations to be made to the genome much more easily, quickly, and cheaply than ever before, plagued by far fewer off-target effects. It also allows researchers to examine the function of multiple genes at once, where before they were mostly limited to a single gene.
“Cancer genomics has identified a large number of genes that are mutated in human cancer,” Tyler Jacks, Ph.D., director of the Koch Institute for Integrative Cancer Research at MIT, Boston, said in an interview. “CRISPR allows us to study these genes in cancer cells and in whole animals much more efficiently than the methods that were in use just a few years ago.”
But the potential of the CRISPR-Cas9 system doesn’t stop there. “At the moment, a particularly exciting application is the use of this approach to inactive genes in very specific fashion to assess the function of a given protein in a cancer cell, which should speed the identification of proteins that are important for cancer cells and thus potentially aid drug discovery efforts,” Dr. Armstrong said.
CRISPR at AACR
The latest developments in the use of the CRISPR-Cas9 system were highlighted at the annual meeting of the American Association of Cancer Research. Dr. David Sabatini, professor of biology at MIT, Boston, described his own lab’s method for using CRISPR-Cas9 to seek out the essential genes involved in different types of cancers. In a study recently published in Science, he and his colleagues employed this method in chronic myelogenous leukemia and Burkitt’s lymphoma cell lines. The gRNA library targeted just over 18,000 genes and roughly 10% of these proved to be essential. Mostly, these genes were linked to key cellular processes (Science 2015;350[6264]:1096-1101).
Dr. Christopher Vakoc of Cold Spring Harbor (N.Y.) Laboratory, presented a slightly different kind of CRISPR screen for drug targets. Most commonly, CRISPR introduces edits at the start of the gene, which may or may not change the DNA enough to produce a nonfunctional protein. Dr. Vakoc’s lab has developed a system that instead edits functional protein domains, which present ideal drug targets. Mutated domains can be identified that are essential for cancer cell survival and small molecule inhibitors designed that bind to them to kill cancer cells.
The technique has already been used to identify such a domain on the BRD4 protein and inhibitors that bind to this domain had significant antitumor activity in leukemia, Dr. Vakoc reported. A screen targeting 192 chromatin regulatory domains expressed in mouse acute myeloid leukemia cells was subsequently performed and identified 25 domains that impacted survival, 6 that are already being therapeutically targeted, and 19 novel potential targets.
Another development in CRISPR-Cas9 technology creates an inactive version of the Cas9 enzyme, one that has lost the ability to cut DNA. Though it seems counterintuitive, this has opened up a wealth of new possible uses. Jonathan S. Weissman, Ph.D., professor of cellular and molecular pharmacology, University of California, San Francisco, part of the group to develop this ‘dead’ Cas9 (dCas9), published a description of the use of two new tools dubbed CRISPR interference and CRISPR activation (Cell 2013;152[5]:1173-83).
Essentially, by fusing dCas9 with different proteins, such as epigenetic modifiers or transcriptional activators or repressors, it can be used as a delivery system to fine-tune gene expression, instead of editing the gene sequence.
Treating cancer?
Ultimately, CRISPR-Cas9 could be used to treat cancers by cutting out defective genes and replacing them with a wild-type version, or by repairing mutations, though for the time being this is theoretical. Studies have suggested it is possible with other types of diseases, however.
“It is not clear exactly how the CRISPR system would be used to directly treat cancer, but the discoveries that come from its use will likely lead to new ways to treat cancer,” said Dr Armstrong.
Dr Jacks highlighted the technical challenges that will need to be overcome first. “In principle, CRISPR-based genome editing could be used to correct cancer-causing mutations in tumors in vivo or to inactivate activated cancer genes,” he said. “At this point, however, we lack the technology necessary to deliver the CRISPR system to all cancer cells in the body. Improvements in this so-called ‘delivery problem’ may allow CRISPR to become a powerful anticancer therapy strategy.”

Study links radon and hematologic cancers in women

New research suggests there is a significant positive association between high levels of residential radon and the risk of hematologic malignancies in women.
The study is the first prospective, population-based study of residential radon exposure and hematologic malignancy risk.
Therefore, the researchers caution that it requires replication to better understand the association and whether it truly differs by sex.
Lauren Teras, PhD, of the American Cancer Society in Atlanta, Georgia, and her colleagues conducted this study and reported the results in Environmental Research.
Radon is a naturally occurring byproduct of the decay of radium and is a known human lung carcinogen. It is the second-leading cause of lung cancer in the US.
Modeling studies have shown that radon delivers a non-negligible dose of alpha radiation to the bone marrow and therefore could increase the risk of hematologic malignancies. However, studies investigating the link between radon and hematologic malignancies have produced inconsistent results.
For the current study, Dr Teras and her colleagues used data from the American Cancer Society Cancer Prevention Study-II Nutrition Cohort to examine the association between county-level residential radon exposure and the risk of hematologic cancer.
The analysis included 140,652 participants, including 3019 who had hematologic malignancies during 19 years of follow-up (1992 to 2011).
The researchers found that women living in counties with the highest mean radon concentration (> 148 Bq/m3) had a significantly higher risk of developing a hematologic malignancy than women living in counties with the lowest radon levels (< 74 Bq/m3).
The adjusted hazard ratio (adjusted for age, race, family history of hematologic malignancy, etc.) was 1.63 (P=0.0010).
The researchers also found evidence of a dose-response relationship, with an adjusted hazard ratio of 1.38 (P=0.001).
The team said there was evidence of a positive exposure-response relationship between radon concentration and the risk of all lymphoid malignancy subtypes in women. But the highest risk was observed for follicular lymphoma, with an adjusted hazard ratio of 2.74 (P=0.02).
On the other hand, there was a non-significant inverse association between radon and myeloid leukemias in women.
There was no association between hematologic malignancy and radon exposure among the men.
The researchers said a possible explanation for this finding is that men may have a higher baseline risk of hematologic malignancy, possibly because of more exposure to occupational or other risk factors, which would reduce the impact of any additional risk from residential radon.
In women, who have a smaller baseline risk, residential radon exposure might be a larger contributor to overall risk.
Another reason for the sex difference observed in this study may be that the women of this generation spent more time in their homes, so they had more residential exposure than men.
“The overall lifetime risk of hematological cancers in the United States is about 2%, so even a 60% relative increase would still mean a relatively small absolute risk,” Dr Teras noted.
“Nonetheless, radon is already associated with lung cancer, and if other studies confirm the link to blood cancers, we think it would warrant strengthened public health efforts to mitigate residential radon risks.” ![]()
New research suggests there is a significant positive association between high levels of residential radon and the risk of hematologic malignancies in women.
The study is the first prospective, population-based study of residential radon exposure and hematologic malignancy risk.
Therefore, the researchers caution that it requires replication to better understand the association and whether it truly differs by sex.
Lauren Teras, PhD, of the American Cancer Society in Atlanta, Georgia, and her colleagues conducted this study and reported the results in Environmental Research.
Radon is a naturally occurring byproduct of the decay of radium and is a known human lung carcinogen. It is the second-leading cause of lung cancer in the US.
Modeling studies have shown that radon delivers a non-negligible dose of alpha radiation to the bone marrow and therefore could increase the risk of hematologic malignancies. However, studies investigating the link between radon and hematologic malignancies have produced inconsistent results.
For the current study, Dr Teras and her colleagues used data from the American Cancer Society Cancer Prevention Study-II Nutrition Cohort to examine the association between county-level residential radon exposure and the risk of hematologic cancer.
The analysis included 140,652 participants, including 3019 who had hematologic malignancies during 19 years of follow-up (1992 to 2011).
The researchers found that women living in counties with the highest mean radon concentration (> 148 Bq/m3) had a significantly higher risk of developing a hematologic malignancy than women living in counties with the lowest radon levels (< 74 Bq/m3).
The adjusted hazard ratio (adjusted for age, race, family history of hematologic malignancy, etc.) was 1.63 (P=0.0010).
The researchers also found evidence of a dose-response relationship, with an adjusted hazard ratio of 1.38 (P=0.001).
The team said there was evidence of a positive exposure-response relationship between radon concentration and the risk of all lymphoid malignancy subtypes in women. But the highest risk was observed for follicular lymphoma, with an adjusted hazard ratio of 2.74 (P=0.02).
On the other hand, there was a non-significant inverse association between radon and myeloid leukemias in women.
There was no association between hematologic malignancy and radon exposure among the men.
The researchers said a possible explanation for this finding is that men may have a higher baseline risk of hematologic malignancy, possibly because of more exposure to occupational or other risk factors, which would reduce the impact of any additional risk from residential radon.
In women, who have a smaller baseline risk, residential radon exposure might be a larger contributor to overall risk.
Another reason for the sex difference observed in this study may be that the women of this generation spent more time in their homes, so they had more residential exposure than men.
“The overall lifetime risk of hematological cancers in the United States is about 2%, so even a 60% relative increase would still mean a relatively small absolute risk,” Dr Teras noted.
“Nonetheless, radon is already associated with lung cancer, and if other studies confirm the link to blood cancers, we think it would warrant strengthened public health efforts to mitigate residential radon risks.” ![]()
New research suggests there is a significant positive association between high levels of residential radon and the risk of hematologic malignancies in women.
The study is the first prospective, population-based study of residential radon exposure and hematologic malignancy risk.
Therefore, the researchers caution that it requires replication to better understand the association and whether it truly differs by sex.
Lauren Teras, PhD, of the American Cancer Society in Atlanta, Georgia, and her colleagues conducted this study and reported the results in Environmental Research.
Radon is a naturally occurring byproduct of the decay of radium and is a known human lung carcinogen. It is the second-leading cause of lung cancer in the US.
Modeling studies have shown that radon delivers a non-negligible dose of alpha radiation to the bone marrow and therefore could increase the risk of hematologic malignancies. However, studies investigating the link between radon and hematologic malignancies have produced inconsistent results.
For the current study, Dr Teras and her colleagues used data from the American Cancer Society Cancer Prevention Study-II Nutrition Cohort to examine the association between county-level residential radon exposure and the risk of hematologic cancer.
The analysis included 140,652 participants, including 3019 who had hematologic malignancies during 19 years of follow-up (1992 to 2011).
The researchers found that women living in counties with the highest mean radon concentration (> 148 Bq/m3) had a significantly higher risk of developing a hematologic malignancy than women living in counties with the lowest radon levels (< 74 Bq/m3).
The adjusted hazard ratio (adjusted for age, race, family history of hematologic malignancy, etc.) was 1.63 (P=0.0010).
The researchers also found evidence of a dose-response relationship, with an adjusted hazard ratio of 1.38 (P=0.001).
The team said there was evidence of a positive exposure-response relationship between radon concentration and the risk of all lymphoid malignancy subtypes in women. But the highest risk was observed for follicular lymphoma, with an adjusted hazard ratio of 2.74 (P=0.02).
On the other hand, there was a non-significant inverse association between radon and myeloid leukemias in women.
There was no association between hematologic malignancy and radon exposure among the men.
The researchers said a possible explanation for this finding is that men may have a higher baseline risk of hematologic malignancy, possibly because of more exposure to occupational or other risk factors, which would reduce the impact of any additional risk from residential radon.
In women, who have a smaller baseline risk, residential radon exposure might be a larger contributor to overall risk.
Another reason for the sex difference observed in this study may be that the women of this generation spent more time in their homes, so they had more residential exposure than men.
“The overall lifetime risk of hematological cancers in the United States is about 2%, so even a 60% relative increase would still mean a relatively small absolute risk,” Dr Teras noted.
“Nonetheless, radon is already associated with lung cancer, and if other studies confirm the link to blood cancers, we think it would warrant strengthened public health efforts to mitigate residential radon risks.” ![]()
CHMP recommends approving drug to treat FL

The European Medicine Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that obinutuzumab (Gazyvaro), an anti-CD20 monoclonal antibody, be approved for use in patients with follicular lymphoma (FL).
The recommended indication is for obinutuzumab to be given first in combination with bendamustine and then as maintenance therapy in FL patients who did not respond to, progressed during, or progressed up to 6 months after treatment with rituximab or a rituximab-containing regimen.
Based on the CHMP’s recommendation, a final decision regarding the approval of obinutuzumab in FL is expected from the European Commission in the coming months.
Obinutuzumab is already approved in the European Union for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia and comorbidities that make them unsuitable for full-dose fludarabine-based therapy.
Obinutuzumab is being developed by Roche.
GADOLIN trial
The CHMP’s recommendation to approve obinutuzumab in FL is based on results from the phase 3 GADOLIN trial. The study included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
The best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% for those receiving bendamustine alone (18.7% CR, 56% PR), as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%). ![]()
The European Medicine Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that obinutuzumab (Gazyvaro), an anti-CD20 monoclonal antibody, be approved for use in patients with follicular lymphoma (FL).
The recommended indication is for obinutuzumab to be given first in combination with bendamustine and then as maintenance therapy in FL patients who did not respond to, progressed during, or progressed up to 6 months after treatment with rituximab or a rituximab-containing regimen.
Based on the CHMP’s recommendation, a final decision regarding the approval of obinutuzumab in FL is expected from the European Commission in the coming months.
Obinutuzumab is already approved in the European Union for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia and comorbidities that make them unsuitable for full-dose fludarabine-based therapy.
Obinutuzumab is being developed by Roche.
GADOLIN trial
The CHMP’s recommendation to approve obinutuzumab in FL is based on results from the phase 3 GADOLIN trial. The study included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
The best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% for those receiving bendamustine alone (18.7% CR, 56% PR), as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%). ![]()
The European Medicine Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended that obinutuzumab (Gazyvaro), an anti-CD20 monoclonal antibody, be approved for use in patients with follicular lymphoma (FL).
The recommended indication is for obinutuzumab to be given first in combination with bendamustine and then as maintenance therapy in FL patients who did not respond to, progressed during, or progressed up to 6 months after treatment with rituximab or a rituximab-containing regimen.
Based on the CHMP’s recommendation, a final decision regarding the approval of obinutuzumab in FL is expected from the European Commission in the coming months.
Obinutuzumab is already approved in the European Union for use in combination with chlorambucil to treat patients with previously untreated chronic lymphocytic leukemia and comorbidities that make them unsuitable for full-dose fludarabine-based therapy.
Obinutuzumab is being developed by Roche.
GADOLIN trial
The CHMP’s recommendation to approve obinutuzumab in FL is based on results from the phase 3 GADOLIN trial. The study included 413 patients with rituximab-refractory non-Hodgkin lymphoma, including 321 patients with FL, 46 with marginal zone lymphoma, and 28 with small lymphocytic lymphoma.
The patients were randomized to receive bendamustine alone (control arm) or a combination of bendamustine and obinutuzumab followed by obinutuzumab maintenance (every 2 months for 2 years or until progression).
The primary endpoint of the study was progression-free survival (PFS), as assessed by an independent review committee (IRC). The secondary endpoints were PFS assessed by investigator review, best overall response, complete response (CR), partial response (PR), duration of response, overall survival, and safety profile.
Among patients with FL, the obinutuzumab regimen improved PFS compared to bendamustine alone, as assessed by IRC (hazard ratio [HR]=0.48, P<0.0001). The median PFS was not reached in patients receiving the obinutuzumab regimen but was 13.8 months in those receiving bendamustine alone.
Investigator-assessed PFS was consistent with IRC-assessed PFS. Investigators said the median PFS with the obinutuzumab regimen was more than double that with bendamustine alone—29.2 months vs 13.7 months (HR=0.48, P<0.0001).
The best overall response for patients receiving the obinutuzumab regimen was 78.7% (15.5% CR, 63.2% PR), compared to 74.7% for those receiving bendamustine alone (18.7% CR, 56% PR), as assessed by the IRC.
The median duration of response was not reached for patients receiving the obinutuzumab regimen and was 11.6 months for those receiving bendamustine alone.
The median overall survival has not yet been reached in either study arm.
The most common grade 3/4 adverse events observed in patients receiving the obinutuzumab regimen were neutropenia (33%), infusion reactions (11%), and thrombocytopenia (10%).
The most common adverse events of any grade were infusion reactions (69%), neutropenia (35%), nausea (54%), fatigue (39%), cough (26%), diarrhea (27%), constipation (19%), fever (18%), thrombocytopenia (15%), vomiting (22%), upper respiratory tract infection (13%), decreased appetite (18%), joint or muscle pain (12%), sinusitis (12%), anemia (12%), general weakness (11%), and urinary tract infection (10%). ![]()
Search is on for cases of aggressive, ruxolitinib-associated skin cancers
ORLANDO – The hematologic cancer drug ruxolitinib seems to be associated with cases of aggressive nonmelanoma skin cancer.
After treating a very aggressive squamous cell carcinoma in a 55-year-old man treated with ruxolitinib for polycythemia vera, and hearing firsthand of three other similar cases, Dr. Fiona Zwald is collecting additional data on the association. She intends to publish these cases in a monograph as a warning to dermatologists, hematologists, oncologists, and other physicians who manage patients with hematologic malignancies, she said at the annual meeting of the American College of Mohs Surgery.
The prescribing information for ruxolitinib (Jakafi, Incyte Pharmaceuticals; Jakavi, Novartis) was updated in 2014 to warn that patients taking the drug face an increased risk of nonmelanoma skin cancers. The label also recommends that physicians inspect the skin regularly and urge patients to be alert for and report any new or changing lesions.
Despite the warnings and recommendations, cases are occurring – and some are quite serious, said Dr. Zwald, a Mohs surgeon in Atlanta.
“People should know this is actually happening. If you have experience with this medication, please let us know so we can compile this report. We are trying to assess the number of skin cancers before and after initiating this medication,” she said.
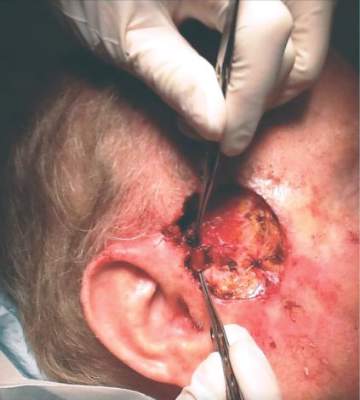
Ruxolitinib is an inhibitor of Janus kinase with a special affinity for the JAK1 and JAK2 subtypes. Like other cytokine-signaling molecules, their function depends on cell context; it may inhibit cell growth in one setting, and, in another, stimulate it. Ruxolitinib was initially approved in 2011 for the treatment of intermediate- and high-risk myelofibrosis, including primary myelofibrosis, post–polycythemia vera myelofibrosis, and post–essential thrombocythemia myelofibrosis.
In 2014, indications for ruxolitinib were expanded to include treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
Dr. Zwald’s patient had a 10-year history of polycythemia vera. He was initially well controlled on the standard hydroxyurea treatment. In the meantime, he began working as a caddy at a major U.S. golf club. He developed many facial squamous cell carcinomas that were treated with excision and radiation. A year before he presented to Dr. Zwald, he stopped responding to hydroxyurea and was placed on ruxolitinib.
The patient presented with a 4-cm ulcerated lesion over part of his right temple and to the right helical crus; the lesion had developed over 3 months. Dr. Zwald consulted with the patient’s medical oncologist; treatment with ruxolitinib continued, albeit at a reduced dosage in light of recent events.
She performed Mohs surgery on the patient. It was a challenging case, she said, not the least because adequate anesthesia could not be achieved with local anesthetic. Preoperative staging showed no nodal spread.
“He did, unfortunately demonstrate a large, indurated mass located over one branch of the superficial temporal artery. At the helical crus there was an area of bound-down, fixed tumor. Knowing that I would not be able to fully resect this, I passed him on to the operating room,” Dr. Zwald said. “This tumor was found to extend down to the parotid capsule, but margins were clear.” The surgical defect was successfully repaired with a split-thickness skin graft.
The tumor recurred about 3 months later, and the patient underwent another surgery.
“This time we could not get clear surgical margins, and the tumor was approaching the external auditory meatus. Surgery was abandoned due to fears of complications to that area,” she said.
She presented the case at tumor board, during which she and her colleagues discussed adjuvant radiation. They initially abandoned this idea because he had already had so much radiation to his face. After the second surgery, they decide to proceed with radiation. “The next conversation we have will be whether to add another adjuvant therapy to treatment.”
She sent out the case and requests for feedback to the International Transplant Skin Cancer Collaborative, an 800-member consortium of dermatologists and Mohs surgeons who take care of transplant patients. She received information on three additional cases of aggressive squamous cell carcinoma (SCC) associated with ruxolitinib treatment:
• A patient with myelodysplastic syndrome with aggressive scalp SCC with cutaneous metastases.
• A patient with undifferentiated pleomorphic sarcoma of the scalp, several cutaneous SCCs.
• A patient with a myelodysplastic syndrome with in-transit metastases and explosive cutaneous SCCs. The patient has had the ruxolitinib dose reduced and may be switched to capecitabine.
Dr. Zwald noted that her patient was at risk for aggressive skin cancers for reasons in addition to ruxolitinib treatment.
“He was already immunosuppressed from his malignancy. He was on hydroxyurea, a drug that’s a cumulative phototoxin, and he’s out in the sun playing golf every day, and then was put on ruxolitinib. But the question we face now is how to try and stop this medication so we can get better treatment for him which will, of course, be very difficult.”
To contribute to Dr. Zwald’s case series, please email her at Fiona.Zwald@gmail.com.
She had no relevant financial disclosures.
On Twitter @Alz_Gal
ORLANDO – The hematologic cancer drug ruxolitinib seems to be associated with cases of aggressive nonmelanoma skin cancer.
After treating a very aggressive squamous cell carcinoma in a 55-year-old man treated with ruxolitinib for polycythemia vera, and hearing firsthand of three other similar cases, Dr. Fiona Zwald is collecting additional data on the association. She intends to publish these cases in a monograph as a warning to dermatologists, hematologists, oncologists, and other physicians who manage patients with hematologic malignancies, she said at the annual meeting of the American College of Mohs Surgery.
The prescribing information for ruxolitinib (Jakafi, Incyte Pharmaceuticals; Jakavi, Novartis) was updated in 2014 to warn that patients taking the drug face an increased risk of nonmelanoma skin cancers. The label also recommends that physicians inspect the skin regularly and urge patients to be alert for and report any new or changing lesions.
Despite the warnings and recommendations, cases are occurring – and some are quite serious, said Dr. Zwald, a Mohs surgeon in Atlanta.
“People should know this is actually happening. If you have experience with this medication, please let us know so we can compile this report. We are trying to assess the number of skin cancers before and after initiating this medication,” she said.
Ruxolitinib is an inhibitor of Janus kinase with a special affinity for the JAK1 and JAK2 subtypes. Like other cytokine-signaling molecules, their function depends on cell context; it may inhibit cell growth in one setting, and, in another, stimulate it. Ruxolitinib was initially approved in 2011 for the treatment of intermediate- and high-risk myelofibrosis, including primary myelofibrosis, post–polycythemia vera myelofibrosis, and post–essential thrombocythemia myelofibrosis.
In 2014, indications for ruxolitinib were expanded to include treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
Dr. Zwald’s patient had a 10-year history of polycythemia vera. He was initially well controlled on the standard hydroxyurea treatment. In the meantime, he began working as a caddy at a major U.S. golf club. He developed many facial squamous cell carcinomas that were treated with excision and radiation. A year before he presented to Dr. Zwald, he stopped responding to hydroxyurea and was placed on ruxolitinib.
The patient presented with a 4-cm ulcerated lesion over part of his right temple and to the right helical crus; the lesion had developed over 3 months. Dr. Zwald consulted with the patient’s medical oncologist; treatment with ruxolitinib continued, albeit at a reduced dosage in light of recent events.
She performed Mohs surgery on the patient. It was a challenging case, she said, not the least because adequate anesthesia could not be achieved with local anesthetic. Preoperative staging showed no nodal spread.
“He did, unfortunately demonstrate a large, indurated mass located over one branch of the superficial temporal artery. At the helical crus there was an area of bound-down, fixed tumor. Knowing that I would not be able to fully resect this, I passed him on to the operating room,” Dr. Zwald said. “This tumor was found to extend down to the parotid capsule, but margins were clear.” The surgical defect was successfully repaired with a split-thickness skin graft.
The tumor recurred about 3 months later, and the patient underwent another surgery.
“This time we could not get clear surgical margins, and the tumor was approaching the external auditory meatus. Surgery was abandoned due to fears of complications to that area,” she said.
She presented the case at tumor board, during which she and her colleagues discussed adjuvant radiation. They initially abandoned this idea because he had already had so much radiation to his face. After the second surgery, they decide to proceed with radiation. “The next conversation we have will be whether to add another adjuvant therapy to treatment.”
She sent out the case and requests for feedback to the International Transplant Skin Cancer Collaborative, an 800-member consortium of dermatologists and Mohs surgeons who take care of transplant patients. She received information on three additional cases of aggressive squamous cell carcinoma (SCC) associated with ruxolitinib treatment:
• A patient with myelodysplastic syndrome with aggressive scalp SCC with cutaneous metastases.
• A patient with undifferentiated pleomorphic sarcoma of the scalp, several cutaneous SCCs.
• A patient with a myelodysplastic syndrome with in-transit metastases and explosive cutaneous SCCs. The patient has had the ruxolitinib dose reduced and may be switched to capecitabine.
Dr. Zwald noted that her patient was at risk for aggressive skin cancers for reasons in addition to ruxolitinib treatment.
“He was already immunosuppressed from his malignancy. He was on hydroxyurea, a drug that’s a cumulative phototoxin, and he’s out in the sun playing golf every day, and then was put on ruxolitinib. But the question we face now is how to try and stop this medication so we can get better treatment for him which will, of course, be very difficult.”
To contribute to Dr. Zwald’s case series, please email her at Fiona.Zwald@gmail.com.
She had no relevant financial disclosures.
On Twitter @Alz_Gal
ORLANDO – The hematologic cancer drug ruxolitinib seems to be associated with cases of aggressive nonmelanoma skin cancer.
After treating a very aggressive squamous cell carcinoma in a 55-year-old man treated with ruxolitinib for polycythemia vera, and hearing firsthand of three other similar cases, Dr. Fiona Zwald is collecting additional data on the association. She intends to publish these cases in a monograph as a warning to dermatologists, hematologists, oncologists, and other physicians who manage patients with hematologic malignancies, she said at the annual meeting of the American College of Mohs Surgery.
The prescribing information for ruxolitinib (Jakafi, Incyte Pharmaceuticals; Jakavi, Novartis) was updated in 2014 to warn that patients taking the drug face an increased risk of nonmelanoma skin cancers. The label also recommends that physicians inspect the skin regularly and urge patients to be alert for and report any new or changing lesions.
Despite the warnings and recommendations, cases are occurring – and some are quite serious, said Dr. Zwald, a Mohs surgeon in Atlanta.
“People should know this is actually happening. If you have experience with this medication, please let us know so we can compile this report. We are trying to assess the number of skin cancers before and after initiating this medication,” she said.
Ruxolitinib is an inhibitor of Janus kinase with a special affinity for the JAK1 and JAK2 subtypes. Like other cytokine-signaling molecules, their function depends on cell context; it may inhibit cell growth in one setting, and, in another, stimulate it. Ruxolitinib was initially approved in 2011 for the treatment of intermediate- and high-risk myelofibrosis, including primary myelofibrosis, post–polycythemia vera myelofibrosis, and post–essential thrombocythemia myelofibrosis.
In 2014, indications for ruxolitinib were expanded to include treatment of patients with polycythemia vera who have had an inadequate response to or are intolerant of hydroxyurea.
Dr. Zwald’s patient had a 10-year history of polycythemia vera. He was initially well controlled on the standard hydroxyurea treatment. In the meantime, he began working as a caddy at a major U.S. golf club. He developed many facial squamous cell carcinomas that were treated with excision and radiation. A year before he presented to Dr. Zwald, he stopped responding to hydroxyurea and was placed on ruxolitinib.
The patient presented with a 4-cm ulcerated lesion over part of his right temple and to the right helical crus; the lesion had developed over 3 months. Dr. Zwald consulted with the patient’s medical oncologist; treatment with ruxolitinib continued, albeit at a reduced dosage in light of recent events.
She performed Mohs surgery on the patient. It was a challenging case, she said, not the least because adequate anesthesia could not be achieved with local anesthetic. Preoperative staging showed no nodal spread.
“He did, unfortunately demonstrate a large, indurated mass located over one branch of the superficial temporal artery. At the helical crus there was an area of bound-down, fixed tumor. Knowing that I would not be able to fully resect this, I passed him on to the operating room,” Dr. Zwald said. “This tumor was found to extend down to the parotid capsule, but margins were clear.” The surgical defect was successfully repaired with a split-thickness skin graft.
The tumor recurred about 3 months later, and the patient underwent another surgery.
“This time we could not get clear surgical margins, and the tumor was approaching the external auditory meatus. Surgery was abandoned due to fears of complications to that area,” she said.
She presented the case at tumor board, during which she and her colleagues discussed adjuvant radiation. They initially abandoned this idea because he had already had so much radiation to his face. After the second surgery, they decide to proceed with radiation. “The next conversation we have will be whether to add another adjuvant therapy to treatment.”
She sent out the case and requests for feedback to the International Transplant Skin Cancer Collaborative, an 800-member consortium of dermatologists and Mohs surgeons who take care of transplant patients. She received information on three additional cases of aggressive squamous cell carcinoma (SCC) associated with ruxolitinib treatment:
• A patient with myelodysplastic syndrome with aggressive scalp SCC with cutaneous metastases.
• A patient with undifferentiated pleomorphic sarcoma of the scalp, several cutaneous SCCs.
• A patient with a myelodysplastic syndrome with in-transit metastases and explosive cutaneous SCCs. The patient has had the ruxolitinib dose reduced and may be switched to capecitabine.
Dr. Zwald noted that her patient was at risk for aggressive skin cancers for reasons in addition to ruxolitinib treatment.
“He was already immunosuppressed from his malignancy. He was on hydroxyurea, a drug that’s a cumulative phototoxin, and he’s out in the sun playing golf every day, and then was put on ruxolitinib. But the question we face now is how to try and stop this medication so we can get better treatment for him which will, of course, be very difficult.”
To contribute to Dr. Zwald’s case series, please email her at Fiona.Zwald@gmail.com.
She had no relevant financial disclosures.
On Twitter @Alz_Gal
AT THE ACMS ANNUAL MEETING
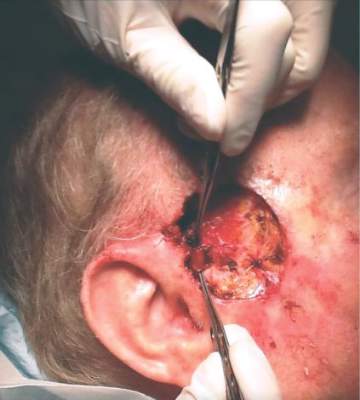
Cancer diagnosis linked to mental health disorders

A recent cancer diagnosis is associated with an increased risk for mental health disorders and increased use of psychiatric medications, according to a large, nationwide study conducted in Sweden.
Overall, there was an increased risk of mental health disorders from 10 months before a cancer diagnosis that peaked during the first week after diagnosis and decreased after that, although the risk remained elevated at 10 years after diagnosis.
In addition, there was an increased use of psychiatric medications from 1 month before cancer diagnosis that peaked at about 3 months after diagnosis and remained elevated 2 years after diagnosis.
Donghao Lu, MD, of the Karolinska Institutet in Stockholm, Sweden and colleagues conducted this study and reported the results in JAMA Oncology.
The study included 304,118 patients with cancer and 3,041,174 cancer-free individuals randomly selected from the Swedish population for comparison.
The researchers investigated changes in risk for several common and potentially stress-related mental disorders—including depression, anxiety, substance abuse, somatoform/conversion disorder, and stress reaction/adjustment disorder—from the cancer diagnostic workup through to post-diagnosis.
They found the relative rate for all of the mental disorders studied started to increase from 10 months before cancer diagnosis, with a hazard ratio [HR] of 1.1 (95%CI, 1.1-1.2).
The rate peaked during the first week after diagnosis, with an HR of 6.7 (95%CI, 6.1-7.4). It decreased rapidly thereafter but was still elevated 10 years after diagnosis, with an HR of 1.1 (95%CI, 1.1-1.2).
The rate elevation was clear for all of the main cancers, including hematologic malignancies, except for nonmelanoma skin cancer.
Among the cancer patients, the mental disorder with the highest cumulative incidence was depression. This was followed by anxiety and stress reaction/adjustment disorder.
When compared to controls, the cancer patients had a higher cumulative incidence of most of the mental disorders. The exception was somatoform/conversion disorder.
The researchers also examined the use of psychiatric medications for patients with cancer to assess milder mental health conditions and symptoms.
The team found an increased use of psychiatric medications in cancer patients compared to controls, from 1 month before diagnosis—12.2% vs 11.7% (P=0.04)—that peaked at about 3 months after diagnosis—18.1% vs 11.9% (P<0.001)—and was still elevated 2 years after diagnosis—15.4% vs 12.7% (P<0.001).
The researchers said the results of this study support the existing guidelines of integrating psychological management into cancer care and call for extended vigilance for multiple mental disorders starting from the time of the cancer diagnostic workup. ![]()
A recent cancer diagnosis is associated with an increased risk for mental health disorders and increased use of psychiatric medications, according to a large, nationwide study conducted in Sweden.
Overall, there was an increased risk of mental health disorders from 10 months before a cancer diagnosis that peaked during the first week after diagnosis and decreased after that, although the risk remained elevated at 10 years after diagnosis.
In addition, there was an increased use of psychiatric medications from 1 month before cancer diagnosis that peaked at about 3 months after diagnosis and remained elevated 2 years after diagnosis.
Donghao Lu, MD, of the Karolinska Institutet in Stockholm, Sweden and colleagues conducted this study and reported the results in JAMA Oncology.
The study included 304,118 patients with cancer and 3,041,174 cancer-free individuals randomly selected from the Swedish population for comparison.
The researchers investigated changes in risk for several common and potentially stress-related mental disorders—including depression, anxiety, substance abuse, somatoform/conversion disorder, and stress reaction/adjustment disorder—from the cancer diagnostic workup through to post-diagnosis.
They found the relative rate for all of the mental disorders studied started to increase from 10 months before cancer diagnosis, with a hazard ratio [HR] of 1.1 (95%CI, 1.1-1.2).
The rate peaked during the first week after diagnosis, with an HR of 6.7 (95%CI, 6.1-7.4). It decreased rapidly thereafter but was still elevated 10 years after diagnosis, with an HR of 1.1 (95%CI, 1.1-1.2).
The rate elevation was clear for all of the main cancers, including hematologic malignancies, except for nonmelanoma skin cancer.
Among the cancer patients, the mental disorder with the highest cumulative incidence was depression. This was followed by anxiety and stress reaction/adjustment disorder.
When compared to controls, the cancer patients had a higher cumulative incidence of most of the mental disorders. The exception was somatoform/conversion disorder.
The researchers also examined the use of psychiatric medications for patients with cancer to assess milder mental health conditions and symptoms.
The team found an increased use of psychiatric medications in cancer patients compared to controls, from 1 month before diagnosis—12.2% vs 11.7% (P=0.04)—that peaked at about 3 months after diagnosis—18.1% vs 11.9% (P<0.001)—and was still elevated 2 years after diagnosis—15.4% vs 12.7% (P<0.001).
The researchers said the results of this study support the existing guidelines of integrating psychological management into cancer care and call for extended vigilance for multiple mental disorders starting from the time of the cancer diagnostic workup. ![]()
A recent cancer diagnosis is associated with an increased risk for mental health disorders and increased use of psychiatric medications, according to a large, nationwide study conducted in Sweden.
Overall, there was an increased risk of mental health disorders from 10 months before a cancer diagnosis that peaked during the first week after diagnosis and decreased after that, although the risk remained elevated at 10 years after diagnosis.
In addition, there was an increased use of psychiatric medications from 1 month before cancer diagnosis that peaked at about 3 months after diagnosis and remained elevated 2 years after diagnosis.
Donghao Lu, MD, of the Karolinska Institutet in Stockholm, Sweden and colleagues conducted this study and reported the results in JAMA Oncology.
The study included 304,118 patients with cancer and 3,041,174 cancer-free individuals randomly selected from the Swedish population for comparison.
The researchers investigated changes in risk for several common and potentially stress-related mental disorders—including depression, anxiety, substance abuse, somatoform/conversion disorder, and stress reaction/adjustment disorder—from the cancer diagnostic workup through to post-diagnosis.
They found the relative rate for all of the mental disorders studied started to increase from 10 months before cancer diagnosis, with a hazard ratio [HR] of 1.1 (95%CI, 1.1-1.2).
The rate peaked during the first week after diagnosis, with an HR of 6.7 (95%CI, 6.1-7.4). It decreased rapidly thereafter but was still elevated 10 years after diagnosis, with an HR of 1.1 (95%CI, 1.1-1.2).
The rate elevation was clear for all of the main cancers, including hematologic malignancies, except for nonmelanoma skin cancer.
Among the cancer patients, the mental disorder with the highest cumulative incidence was depression. This was followed by anxiety and stress reaction/adjustment disorder.
When compared to controls, the cancer patients had a higher cumulative incidence of most of the mental disorders. The exception was somatoform/conversion disorder.
The researchers also examined the use of psychiatric medications for patients with cancer to assess milder mental health conditions and symptoms.
The team found an increased use of psychiatric medications in cancer patients compared to controls, from 1 month before diagnosis—12.2% vs 11.7% (P=0.04)—that peaked at about 3 months after diagnosis—18.1% vs 11.9% (P<0.001)—and was still elevated 2 years after diagnosis—15.4% vs 12.7% (P<0.001).
The researchers said the results of this study support the existing guidelines of integrating psychological management into cancer care and call for extended vigilance for multiple mental disorders starting from the time of the cancer diagnostic workup. ![]()
Costs for orally administered cancer drugs on the rise

Photo courtesy of the CDC
New orally administered cancer drugs are much more expensive in their first year on the market than such drugs launched about 15 years ago, according to a study published in JAMA Oncology.
The research showed that a month of treatment with orally administered cancer drugs introduced in 2014 was, on average, 6 times more expensive at launch than monthly treatment costs for such drugs introduced in 2000, after adjusting for inflation.
In addition, most existing therapies had substantial price increases from the time they were launched to 2014.
“The major trend here is that these products are just getting more expensive over time,” said study author Stacie Dusetzina, PhD, of the University of North Carolina at Chapel Hill.
For this study, Dr Dusetzina evaluated what commercial health insurance companies and patients paid for prescription fills—before rebates and discounts—for 32 orally administered cancer drugs from 2000 to 2014. The information came from the TruvenHealth MarketScan Commercial Claims and Encounters database.
The data showed that orally administered drugs approved in 2000 cost an average of $1869 (95% CI, $1648-$2121) per month, compared to $11,325 (95% CI, $10 989-$11 671) for those approved in 2014.
When Dr Dusetzina compared changes in spending by year from a product’s launch to 2014, she observed increases in most of the drugs studied.
The drugs with the largest increases in monthly spending were thalidomide, which increased from $1869 to $7564 ($5695) and imatinib, which increased from $3346 to $8479 ($5133).
However, 2 drugs showed decreases in mean monthly spending between their launch and 2014. Monthly spending for lenalidomide decreased from $10,109 to $9640 ($469), and monthly spending for vorinostat decreased from $9755 to $7592 ($2163).
Dr Dusetzina pointed out that the amount patients pay for these drugs depends on their healthcare benefits. However, the high prices are being passed along to patients more and more, potentially affecting the patients’ access to these drugs.
“Patients are increasingly taking on the burden of paying for these high-cost specialty drugs as plans move toward use of higher deductibles and co-insurance—where a patient will pay a percentage of the drug cost rather than a flat copay,” Dr Dusetzina said.
She noted that while this study did account for payments by commercial health plans, it did not account for spending by Medicaid and Medicare, which may differ. In addition, only the products that were dispensed and reimbursed by commercial health plans were included, which may have excluded rarely used or recently approved products. ![]()
Photo courtesy of the CDC
New orally administered cancer drugs are much more expensive in their first year on the market than such drugs launched about 15 years ago, according to a study published in JAMA Oncology.
The research showed that a month of treatment with orally administered cancer drugs introduced in 2014 was, on average, 6 times more expensive at launch than monthly treatment costs for such drugs introduced in 2000, after adjusting for inflation.
In addition, most existing therapies had substantial price increases from the time they were launched to 2014.
“The major trend here is that these products are just getting more expensive over time,” said study author Stacie Dusetzina, PhD, of the University of North Carolina at Chapel Hill.
For this study, Dr Dusetzina evaluated what commercial health insurance companies and patients paid for prescription fills—before rebates and discounts—for 32 orally administered cancer drugs from 2000 to 2014. The information came from the TruvenHealth MarketScan Commercial Claims and Encounters database.
The data showed that orally administered drugs approved in 2000 cost an average of $1869 (95% CI, $1648-$2121) per month, compared to $11,325 (95% CI, $10 989-$11 671) for those approved in 2014.
When Dr Dusetzina compared changes in spending by year from a product’s launch to 2014, she observed increases in most of the drugs studied.
The drugs with the largest increases in monthly spending were thalidomide, which increased from $1869 to $7564 ($5695) and imatinib, which increased from $3346 to $8479 ($5133).
However, 2 drugs showed decreases in mean monthly spending between their launch and 2014. Monthly spending for lenalidomide decreased from $10,109 to $9640 ($469), and monthly spending for vorinostat decreased from $9755 to $7592 ($2163).
Dr Dusetzina pointed out that the amount patients pay for these drugs depends on their healthcare benefits. However, the high prices are being passed along to patients more and more, potentially affecting the patients’ access to these drugs.
“Patients are increasingly taking on the burden of paying for these high-cost specialty drugs as plans move toward use of higher deductibles and co-insurance—where a patient will pay a percentage of the drug cost rather than a flat copay,” Dr Dusetzina said.
She noted that while this study did account for payments by commercial health plans, it did not account for spending by Medicaid and Medicare, which may differ. In addition, only the products that were dispensed and reimbursed by commercial health plans were included, which may have excluded rarely used or recently approved products. ![]()
Photo courtesy of the CDC
New orally administered cancer drugs are much more expensive in their first year on the market than such drugs launched about 15 years ago, according to a study published in JAMA Oncology.
The research showed that a month of treatment with orally administered cancer drugs introduced in 2014 was, on average, 6 times more expensive at launch than monthly treatment costs for such drugs introduced in 2000, after adjusting for inflation.
In addition, most existing therapies had substantial price increases from the time they were launched to 2014.
“The major trend here is that these products are just getting more expensive over time,” said study author Stacie Dusetzina, PhD, of the University of North Carolina at Chapel Hill.
For this study, Dr Dusetzina evaluated what commercial health insurance companies and patients paid for prescription fills—before rebates and discounts—for 32 orally administered cancer drugs from 2000 to 2014. The information came from the TruvenHealth MarketScan Commercial Claims and Encounters database.
The data showed that orally administered drugs approved in 2000 cost an average of $1869 (95% CI, $1648-$2121) per month, compared to $11,325 (95% CI, $10 989-$11 671) for those approved in 2014.
When Dr Dusetzina compared changes in spending by year from a product’s launch to 2014, she observed increases in most of the drugs studied.
The drugs with the largest increases in monthly spending were thalidomide, which increased from $1869 to $7564 ($5695) and imatinib, which increased from $3346 to $8479 ($5133).
However, 2 drugs showed decreases in mean monthly spending between their launch and 2014. Monthly spending for lenalidomide decreased from $10,109 to $9640 ($469), and monthly spending for vorinostat decreased from $9755 to $7592 ($2163).
Dr Dusetzina pointed out that the amount patients pay for these drugs depends on their healthcare benefits. However, the high prices are being passed along to patients more and more, potentially affecting the patients’ access to these drugs.
“Patients are increasingly taking on the burden of paying for these high-cost specialty drugs as plans move toward use of higher deductibles and co-insurance—where a patient will pay a percentage of the drug cost rather than a flat copay,” Dr Dusetzina said.
She noted that while this study did account for payments by commercial health plans, it did not account for spending by Medicaid and Medicare, which may differ. In addition, only the products that were dispensed and reimbursed by commercial health plans were included, which may have excluded rarely used or recently approved products. ![]()
‘Universal’ CAR T cell may overcome limitations
FROM THE AACR ANNUAL MEETING
A differently engineered chimeric antigen receptor (CAR) T cell promises to overcome major limitations of current CAR T cell therapies. Rather than engineer the CAR T cells to have a receptor that recognizes specific tumor antigens one at a time and requiring different CAR T cells for every antigen, this technique engineers a T cell receptor that can bind one invariant end of a bifunctional molecule. The molecule is constructed such that the other end can bind to whatever tumor cell surface marker is of interest. In this way, the CAR T cells can be constructed once and be directed to various tumor markers.
Standard CAR T cells are engineered to express on their surfaces receptors that recognize a specific antigen. These cells have been used up to now to recognize and kill tumor cells – for example, B cell leukemias carrying the pan-B cell marker CD19. The CAR T cells and their progeny, including memory T cells, remain in the body and continue to carry out their functions, potentially providing immune surveillance in case cancer cells arise again. But they uniquely recognize just CD19 – a problem, in that they kill even normal B cells, so-called off-target toxicity.
Beyond the unique specificity of standard CAR T cells, Philip Low, Ph.D., director of the Center for Drug Discovery at Purdue University in West Lafayette, Indiana, said these cells have three major limitations. First, they may lyse tumor cells so rapidly that a systemic tumor lysis syndrome or “cytokine storm” occurs. Second, the persisting CAR T cells can kill normal cells – for example, ones directed against CD19 killing normal B cells. Third, tumor cells have unstable genomes, leading to tumor heterogeneity, with some cells potentially losing the targeted antigens and therefore becoming “invisible” to the CAR T cells.
“So what we have done is basically designed a solution to all three, and we call it a universal CAR T cell because of its ability, with the help of an adapter molecule, to recognize all of these mutated tumor cells within a heterogeneous tumor,” he said at the annual meeting of the American Association for Cancer Research. The key was to make a CAR T cell with a surface receptor that binds to the dye fluorescein. Then fluorescein is coupled through a short linker to a molecule that binds specifically to a molecule expressed on tumor cells. In this way the CAR T cell can be made to interact with any tumor cell, depending on what is coupled to the fluorescein. The technique is analogous to a socket wrench. Every socket has the same size hole that the ratchet handle fits into regardless of the size of the “business end” of the sockets, which recognize different size nuts.
Dr. Low gave an example of folic acid, for which he says a receptor is overexpressed on about 40% of human tumors but almost never on normal cells. “We link fluorescein to the vitamin folic acid,” he said. CAR T cells are injected into an animal, and nothing happens unless a folate-fluorescein conjugate is also injected. “As soon as we inject folate-fluorescein, the folate binds to the tumor cell surface, the fluorescein part of the folate-fluorescein binds to the CAR T cell, this forces a very tight interaction between the engineered T cell and the cancer cell, and we found it leads to melting away of the tumor,” he said.
This technique addresses the three major problems with standard CAR T cell therapy. By titrating the binding affinity, concentration, and rate of administration of the fluorescein conjugate, the rate of tumor killing can be controlled, mitigating tumor lysis syndrome. Plus, normal cells may be spared if the parameters are adjusted so that the conjugate binds only to cells with high levels of the target molecule, such as tumor cells.
Because its low molecular weight, the bi-specific conjugate rapidly disappears from the circulation, and the cell killing can be terminated, allowing normal cells to regenerate – for example, in the case of normal B cells that carry CD19. Since CAR T cells generate progeny that stay in the body, the progeny remain “dormant” but are ready to be activated again by addition of the conjugate to attack tumor cells if they arise.
A major issue is dealing with tumor heterogeneity; Dr. Low’s method seems to address that, as well. “We have tumor-specific ligands for over 90% of all human cancers,” he said. “Within another couple of months we’ll have them for 100%.”
Tumors typically contain lots of hypoxic cells, and hypoxic cells overexpress carbonic anhydrase-9. “Virtually every tumor has large fractions of the tumor mass that overexpress carbonic anhydrase-9, and we have a ligand that binds very specifically to that,” Dr. Low said.
To address the problem of tumor heterogeneity, with different mutations within different areas of the tumor or over time because of genetic instability in the cells, Dr. Low said, “We have a cocktail of about five of these small molecules… they are inexpensive to produce… and they clear very rapidly… and with the cocktail we can hit nearly all cancer cells, even in heterogeneous cancers.”
One limitation, as with standard CAR T cell therapy, is that the technique will still depend on using an individual patient’s T cells to modify through use of a lentiviral vector, so there would not be a universal, off-the-shelf T cell to use for everyone.
The technique and materials have been tested only in animals so far, using tumor-specific ligands for the folate receptor, a prostate-specific membrane antigen, and an antigen overexpressed on neuroendocrine tumors. Dr. Low has intentions to move the technology into human trials. He said the bridging molecules exist in highly purified form, and CAR T cell technology has already been developed by others. “Today we see great success in animal models and have no reason to believe that it won’t translate at least to a good extent to the clinic,” he said. Still, he expects some obstacles along the way and is willing to partner with others working on similar problems as well as large pharmaceutical companies.
The research has been supported by Endocyte, a company that Dr. Low founded and for which he is Chief Scientific Officer and a member of the board of directors. He has filed two patents on the technology, which are held by Purdue University and licensed to Endocyte.
FROM THE AACR ANNUAL MEETING
A differently engineered chimeric antigen receptor (CAR) T cell promises to overcome major limitations of current CAR T cell therapies. Rather than engineer the CAR T cells to have a receptor that recognizes specific tumor antigens one at a time and requiring different CAR T cells for every antigen, this technique engineers a T cell receptor that can bind one invariant end of a bifunctional molecule. The molecule is constructed such that the other end can bind to whatever tumor cell surface marker is of interest. In this way, the CAR T cells can be constructed once and be directed to various tumor markers.
Standard CAR T cells are engineered to express on their surfaces receptors that recognize a specific antigen. These cells have been used up to now to recognize and kill tumor cells – for example, B cell leukemias carrying the pan-B cell marker CD19. The CAR T cells and their progeny, including memory T cells, remain in the body and continue to carry out their functions, potentially providing immune surveillance in case cancer cells arise again. But they uniquely recognize just CD19 – a problem, in that they kill even normal B cells, so-called off-target toxicity.
Beyond the unique specificity of standard CAR T cells, Philip Low, Ph.D., director of the Center for Drug Discovery at Purdue University in West Lafayette, Indiana, said these cells have three major limitations. First, they may lyse tumor cells so rapidly that a systemic tumor lysis syndrome or “cytokine storm” occurs. Second, the persisting CAR T cells can kill normal cells – for example, ones directed against CD19 killing normal B cells. Third, tumor cells have unstable genomes, leading to tumor heterogeneity, with some cells potentially losing the targeted antigens and therefore becoming “invisible” to the CAR T cells.
“So what we have done is basically designed a solution to all three, and we call it a universal CAR T cell because of its ability, with the help of an adapter molecule, to recognize all of these mutated tumor cells within a heterogeneous tumor,” he said at the annual meeting of the American Association for Cancer Research. The key was to make a CAR T cell with a surface receptor that binds to the dye fluorescein. Then fluorescein is coupled through a short linker to a molecule that binds specifically to a molecule expressed on tumor cells. In this way the CAR T cell can be made to interact with any tumor cell, depending on what is coupled to the fluorescein. The technique is analogous to a socket wrench. Every socket has the same size hole that the ratchet handle fits into regardless of the size of the “business end” of the sockets, which recognize different size nuts.
Dr. Low gave an example of folic acid, for which he says a receptor is overexpressed on about 40% of human tumors but almost never on normal cells. “We link fluorescein to the vitamin folic acid,” he said. CAR T cells are injected into an animal, and nothing happens unless a folate-fluorescein conjugate is also injected. “As soon as we inject folate-fluorescein, the folate binds to the tumor cell surface, the fluorescein part of the folate-fluorescein binds to the CAR T cell, this forces a very tight interaction between the engineered T cell and the cancer cell, and we found it leads to melting away of the tumor,” he said.
This technique addresses the three major problems with standard CAR T cell therapy. By titrating the binding affinity, concentration, and rate of administration of the fluorescein conjugate, the rate of tumor killing can be controlled, mitigating tumor lysis syndrome. Plus, normal cells may be spared if the parameters are adjusted so that the conjugate binds only to cells with high levels of the target molecule, such as tumor cells.
Because its low molecular weight, the bi-specific conjugate rapidly disappears from the circulation, and the cell killing can be terminated, allowing normal cells to regenerate – for example, in the case of normal B cells that carry CD19. Since CAR T cells generate progeny that stay in the body, the progeny remain “dormant” but are ready to be activated again by addition of the conjugate to attack tumor cells if they arise.
A major issue is dealing with tumor heterogeneity; Dr. Low’s method seems to address that, as well. “We have tumor-specific ligands for over 90% of all human cancers,” he said. “Within another couple of months we’ll have them for 100%.”
Tumors typically contain lots of hypoxic cells, and hypoxic cells overexpress carbonic anhydrase-9. “Virtually every tumor has large fractions of the tumor mass that overexpress carbonic anhydrase-9, and we have a ligand that binds very specifically to that,” Dr. Low said.
To address the problem of tumor heterogeneity, with different mutations within different areas of the tumor or over time because of genetic instability in the cells, Dr. Low said, “We have a cocktail of about five of these small molecules… they are inexpensive to produce… and they clear very rapidly… and with the cocktail we can hit nearly all cancer cells, even in heterogeneous cancers.”
One limitation, as with standard CAR T cell therapy, is that the technique will still depend on using an individual patient’s T cells to modify through use of a lentiviral vector, so there would not be a universal, off-the-shelf T cell to use for everyone.
The technique and materials have been tested only in animals so far, using tumor-specific ligands for the folate receptor, a prostate-specific membrane antigen, and an antigen overexpressed on neuroendocrine tumors. Dr. Low has intentions to move the technology into human trials. He said the bridging molecules exist in highly purified form, and CAR T cell technology has already been developed by others. “Today we see great success in animal models and have no reason to believe that it won’t translate at least to a good extent to the clinic,” he said. Still, he expects some obstacles along the way and is willing to partner with others working on similar problems as well as large pharmaceutical companies.
The research has been supported by Endocyte, a company that Dr. Low founded and for which he is Chief Scientific Officer and a member of the board of directors. He has filed two patents on the technology, which are held by Purdue University and licensed to Endocyte.
FROM THE AACR ANNUAL MEETING
A differently engineered chimeric antigen receptor (CAR) T cell promises to overcome major limitations of current CAR T cell therapies. Rather than engineer the CAR T cells to have a receptor that recognizes specific tumor antigens one at a time and requiring different CAR T cells for every antigen, this technique engineers a T cell receptor that can bind one invariant end of a bifunctional molecule. The molecule is constructed such that the other end can bind to whatever tumor cell surface marker is of interest. In this way, the CAR T cells can be constructed once and be directed to various tumor markers.
Standard CAR T cells are engineered to express on their surfaces receptors that recognize a specific antigen. These cells have been used up to now to recognize and kill tumor cells – for example, B cell leukemias carrying the pan-B cell marker CD19. The CAR T cells and their progeny, including memory T cells, remain in the body and continue to carry out their functions, potentially providing immune surveillance in case cancer cells arise again. But they uniquely recognize just CD19 – a problem, in that they kill even normal B cells, so-called off-target toxicity.
Beyond the unique specificity of standard CAR T cells, Philip Low, Ph.D., director of the Center for Drug Discovery at Purdue University in West Lafayette, Indiana, said these cells have three major limitations. First, they may lyse tumor cells so rapidly that a systemic tumor lysis syndrome or “cytokine storm” occurs. Second, the persisting CAR T cells can kill normal cells – for example, ones directed against CD19 killing normal B cells. Third, tumor cells have unstable genomes, leading to tumor heterogeneity, with some cells potentially losing the targeted antigens and therefore becoming “invisible” to the CAR T cells.
“So what we have done is basically designed a solution to all three, and we call it a universal CAR T cell because of its ability, with the help of an adapter molecule, to recognize all of these mutated tumor cells within a heterogeneous tumor,” he said at the annual meeting of the American Association for Cancer Research. The key was to make a CAR T cell with a surface receptor that binds to the dye fluorescein. Then fluorescein is coupled through a short linker to a molecule that binds specifically to a molecule expressed on tumor cells. In this way the CAR T cell can be made to interact with any tumor cell, depending on what is coupled to the fluorescein. The technique is analogous to a socket wrench. Every socket has the same size hole that the ratchet handle fits into regardless of the size of the “business end” of the sockets, which recognize different size nuts.
Dr. Low gave an example of folic acid, for which he says a receptor is overexpressed on about 40% of human tumors but almost never on normal cells. “We link fluorescein to the vitamin folic acid,” he said. CAR T cells are injected into an animal, and nothing happens unless a folate-fluorescein conjugate is also injected. “As soon as we inject folate-fluorescein, the folate binds to the tumor cell surface, the fluorescein part of the folate-fluorescein binds to the CAR T cell, this forces a very tight interaction between the engineered T cell and the cancer cell, and we found it leads to melting away of the tumor,” he said.
This technique addresses the three major problems with standard CAR T cell therapy. By titrating the binding affinity, concentration, and rate of administration of the fluorescein conjugate, the rate of tumor killing can be controlled, mitigating tumor lysis syndrome. Plus, normal cells may be spared if the parameters are adjusted so that the conjugate binds only to cells with high levels of the target molecule, such as tumor cells.
Because its low molecular weight, the bi-specific conjugate rapidly disappears from the circulation, and the cell killing can be terminated, allowing normal cells to regenerate – for example, in the case of normal B cells that carry CD19. Since CAR T cells generate progeny that stay in the body, the progeny remain “dormant” but are ready to be activated again by addition of the conjugate to attack tumor cells if they arise.
A major issue is dealing with tumor heterogeneity; Dr. Low’s method seems to address that, as well. “We have tumor-specific ligands for over 90% of all human cancers,” he said. “Within another couple of months we’ll have them for 100%.”
Tumors typically contain lots of hypoxic cells, and hypoxic cells overexpress carbonic anhydrase-9. “Virtually every tumor has large fractions of the tumor mass that overexpress carbonic anhydrase-9, and we have a ligand that binds very specifically to that,” Dr. Low said.
To address the problem of tumor heterogeneity, with different mutations within different areas of the tumor or over time because of genetic instability in the cells, Dr. Low said, “We have a cocktail of about five of these small molecules… they are inexpensive to produce… and they clear very rapidly… and with the cocktail we can hit nearly all cancer cells, even in heterogeneous cancers.”
One limitation, as with standard CAR T cell therapy, is that the technique will still depend on using an individual patient’s T cells to modify through use of a lentiviral vector, so there would not be a universal, off-the-shelf T cell to use for everyone.
The technique and materials have been tested only in animals so far, using tumor-specific ligands for the folate receptor, a prostate-specific membrane antigen, and an antigen overexpressed on neuroendocrine tumors. Dr. Low has intentions to move the technology into human trials. He said the bridging molecules exist in highly purified form, and CAR T cell technology has already been developed by others. “Today we see great success in animal models and have no reason to believe that it won’t translate at least to a good extent to the clinic,” he said. Still, he expects some obstacles along the way and is willing to partner with others working on similar problems as well as large pharmaceutical companies.
The research has been supported by Endocyte, a company that Dr. Low founded and for which he is Chief Scientific Officer and a member of the board of directors. He has filed two patents on the technology, which are held by Purdue University and licensed to Endocyte.
CAR T-cell trial explores new territory

Photo courtesy of
Fred Hutch News Service
Researchers say they have conducted the first trial of CD19-directed chimeric antigen receptor (CAR) T-cell therapy in which CD4+ and CD8+ cells were administered in equal proportions.
And the assurance that each patient received the same mixture of cells allowed the team to draw clear conclusions about the effects of administering CAR T-cell therapy at different doses.
The researchers detailed these conclusions in The Journal of Clinical Investigation.
This phase 1/2 trial (NCT01865617) was funded, in part, by Juno Therapeutics, the company developing the CAR T-cell therapy as JCAR014.
The work was also funded by the National Cancer Institute, private philanthropists, and the Life Sciences Discovery Fund.
Patients and treatment
The researchers reported data on 32 patients who had relapsed or refractory, CD19+ B-cell acute lymphoblastic leukemia and a median age of 40 (range, 20–73). Two of these patients were excluded due to complications prior to receiving treatment.
The 30 patients who were treated in this study had received a median of 3 prior intensive chemotherapy regimens (range, 1–11). Eleven patients had failed a hematopoietic stem cell transplant (HSCT). And all patients had detectable disease in the bone marrow, extramedullary sites, or cerebrospinal fluid at baseline.
To create the CAR T-cell therapy, the researchers modified CD8+ and CD4+ T-cell subsets separately to express a CD19-targeted CAR incorporating 4-1BB and CD3ζ signaling domains. The cells were then formulated in a defined ratio of CD4+:CD8+ CAR T cells.
Patients underwent lymphodepletion with a cyclophosphamide-based regimen (alone or with fludarabine or etoposide) and then received CAR T cells 48 to 96 hours later. The CAR T cells were given at 3 dose levels—2 × 105/kg (DL1), 2 × 106/kg (DL2), and 2 × 107/kg (DL3).
Toxicity
The first 2 patients who received CAR T-cell therapy at DL3 developed severe toxicities, including 1 patient who died. So DL3 was not given to any subsequent patients.
Two patients died after CAR T-cell infusion. One patient had severe cytokine release syndrome (CRS) and multiorgan failure and died on day 3. The other patient had transient severe CRS with irreversible neurologic toxicity and died on day 122.
The most common adverse event observed in the first 14 days after CAR T-cell infusion was CRS, which occurred in 25 patients. Seven patients had severe CRS that put them in the intensive care unit.
However, for the patients treated at DL1 and DL2, dexamethasone alone or with tocilizumab resolved CRS.
Fifteen patients developed severe neurotoxicity (grade 3 or higher), either with CRS or after it resolved. For all but 1 patient (the aforementioned patient who died), neurologic symptoms and signs resolved.
Response
One patient died before response assessment, so 29 patients were evaluable. Twenty-seven of these patients (93%) achieved bone marrow remission by flow cytometry.
Of the 2 patients who did not achieve a complete response (CR), 1 underwent an allogeneic HSCT after receiving CAR T cells. After HSCT, she relapsed, was re-enrolled on the trial, and achieved a CR after receiving a higher dose of CAR T cells (DL2).
Twenty-five patients (86%) achieved a CR without evidence of minimal residual disease by flow cytometry and conventional karyotyping, FISH, or QPCR.
“Patients who come onto the trial have really limited options for treatment,” said study author Cameron Turtle, MBBS, PhD, of the Fred Hutchinson Cancer Research Center in Seattle, Washington.
“They have refractory acute leukemia. So the fact that we’re getting so many into remission is giving these people a way forward.”
Unfortunately, not all patients stayed in CR. Some relapsed and were treated again with CAR T cells, and 2 patients relapsed with leukemias that were immune to the CAR T cells. The researchers said it is still too early to know the long-term outcomes of the therapy.
“This is just the beginning,” Dr Turtle said. “It sounds fantastic to say that we get over 90% remissions, but there’s so much more work to do make sure they’re durable remissions, to work out who’s going to benefit the most, and extend this work to other diseases.”
Lessons learned
The researchers said that, because these CAR T cells had a defined CD4+:CD8+ composition, this study provides the first clear evidence of the relationships between the CAR T-cell dose patients receive and their outcomes after infusion.
The team found that high doses of CAR T cells and high tumor burden increase the risks of severe CRS and neurotoxicity. However, certain biomarkers can identify patients at the highest risk of toxicity.
Levels of IL-6, IFN-γ, and TNF-α on the first day after CAR T-cell infusion were significantly higher in patients who later developed severe neurotoxicity. Levels of these biomarkers were also higher in patients who were later sent to the intensive care unit.
Furthermore, risk-stratified CAR T-cell dosing based on bone marrow disease burden decreased toxicity.
The researchers also said they observed CD8+ T-cell-mediated anti-CAR transgene product immune responses in some patients, which limited CAR T-cell persistence and increased the risk of relapse.
And including fludarabine in the lymphodepletion regimen resulted in better CAR T-cell persistence and disease-free survival than when cyclophosphamide was given alone or with etoposide. ![]()
Photo courtesy of
Fred Hutch News Service
Researchers say they have conducted the first trial of CD19-directed chimeric antigen receptor (CAR) T-cell therapy in which CD4+ and CD8+ cells were administered in equal proportions.
And the assurance that each patient received the same mixture of cells allowed the team to draw clear conclusions about the effects of administering CAR T-cell therapy at different doses.
The researchers detailed these conclusions in The Journal of Clinical Investigation.
This phase 1/2 trial (NCT01865617) was funded, in part, by Juno Therapeutics, the company developing the CAR T-cell therapy as JCAR014.
The work was also funded by the National Cancer Institute, private philanthropists, and the Life Sciences Discovery Fund.
Patients and treatment
The researchers reported data on 32 patients who had relapsed or refractory, CD19+ B-cell acute lymphoblastic leukemia and a median age of 40 (range, 20–73). Two of these patients were excluded due to complications prior to receiving treatment.
The 30 patients who were treated in this study had received a median of 3 prior intensive chemotherapy regimens (range, 1–11). Eleven patients had failed a hematopoietic stem cell transplant (HSCT). And all patients had detectable disease in the bone marrow, extramedullary sites, or cerebrospinal fluid at baseline.
To create the CAR T-cell therapy, the researchers modified CD8+ and CD4+ T-cell subsets separately to express a CD19-targeted CAR incorporating 4-1BB and CD3ζ signaling domains. The cells were then formulated in a defined ratio of CD4+:CD8+ CAR T cells.
Patients underwent lymphodepletion with a cyclophosphamide-based regimen (alone or with fludarabine or etoposide) and then received CAR T cells 48 to 96 hours later. The CAR T cells were given at 3 dose levels—2 × 105/kg (DL1), 2 × 106/kg (DL2), and 2 × 107/kg (DL3).
Toxicity
The first 2 patients who received CAR T-cell therapy at DL3 developed severe toxicities, including 1 patient who died. So DL3 was not given to any subsequent patients.
Two patients died after CAR T-cell infusion. One patient had severe cytokine release syndrome (CRS) and multiorgan failure and died on day 3. The other patient had transient severe CRS with irreversible neurologic toxicity and died on day 122.
The most common adverse event observed in the first 14 days after CAR T-cell infusion was CRS, which occurred in 25 patients. Seven patients had severe CRS that put them in the intensive care unit.
However, for the patients treated at DL1 and DL2, dexamethasone alone or with tocilizumab resolved CRS.
Fifteen patients developed severe neurotoxicity (grade 3 or higher), either with CRS or after it resolved. For all but 1 patient (the aforementioned patient who died), neurologic symptoms and signs resolved.
Response
One patient died before response assessment, so 29 patients were evaluable. Twenty-seven of these patients (93%) achieved bone marrow remission by flow cytometry.
Of the 2 patients who did not achieve a complete response (CR), 1 underwent an allogeneic HSCT after receiving CAR T cells. After HSCT, she relapsed, was re-enrolled on the trial, and achieved a CR after receiving a higher dose of CAR T cells (DL2).
Twenty-five patients (86%) achieved a CR without evidence of minimal residual disease by flow cytometry and conventional karyotyping, FISH, or QPCR.
“Patients who come onto the trial have really limited options for treatment,” said study author Cameron Turtle, MBBS, PhD, of the Fred Hutchinson Cancer Research Center in Seattle, Washington.
“They have refractory acute leukemia. So the fact that we’re getting so many into remission is giving these people a way forward.”
Unfortunately, not all patients stayed in CR. Some relapsed and were treated again with CAR T cells, and 2 patients relapsed with leukemias that were immune to the CAR T cells. The researchers said it is still too early to know the long-term outcomes of the therapy.
“This is just the beginning,” Dr Turtle said. “It sounds fantastic to say that we get over 90% remissions, but there’s so much more work to do make sure they’re durable remissions, to work out who’s going to benefit the most, and extend this work to other diseases.”
Lessons learned
The researchers said that, because these CAR T cells had a defined CD4+:CD8+ composition, this study provides the first clear evidence of the relationships between the CAR T-cell dose patients receive and their outcomes after infusion.
The team found that high doses of CAR T cells and high tumor burden increase the risks of severe CRS and neurotoxicity. However, certain biomarkers can identify patients at the highest risk of toxicity.
Levels of IL-6, IFN-γ, and TNF-α on the first day after CAR T-cell infusion were significantly higher in patients who later developed severe neurotoxicity. Levels of these biomarkers were also higher in patients who were later sent to the intensive care unit.
Furthermore, risk-stratified CAR T-cell dosing based on bone marrow disease burden decreased toxicity.
The researchers also said they observed CD8+ T-cell-mediated anti-CAR transgene product immune responses in some patients, which limited CAR T-cell persistence and increased the risk of relapse.
And including fludarabine in the lymphodepletion regimen resulted in better CAR T-cell persistence and disease-free survival than when cyclophosphamide was given alone or with etoposide. ![]()
Photo courtesy of
Fred Hutch News Service
Researchers say they have conducted the first trial of CD19-directed chimeric antigen receptor (CAR) T-cell therapy in which CD4+ and CD8+ cells were administered in equal proportions.
And the assurance that each patient received the same mixture of cells allowed the team to draw clear conclusions about the effects of administering CAR T-cell therapy at different doses.
The researchers detailed these conclusions in The Journal of Clinical Investigation.
This phase 1/2 trial (NCT01865617) was funded, in part, by Juno Therapeutics, the company developing the CAR T-cell therapy as JCAR014.
The work was also funded by the National Cancer Institute, private philanthropists, and the Life Sciences Discovery Fund.
Patients and treatment
The researchers reported data on 32 patients who had relapsed or refractory, CD19+ B-cell acute lymphoblastic leukemia and a median age of 40 (range, 20–73). Two of these patients were excluded due to complications prior to receiving treatment.
The 30 patients who were treated in this study had received a median of 3 prior intensive chemotherapy regimens (range, 1–11). Eleven patients had failed a hematopoietic stem cell transplant (HSCT). And all patients had detectable disease in the bone marrow, extramedullary sites, or cerebrospinal fluid at baseline.
To create the CAR T-cell therapy, the researchers modified CD8+ and CD4+ T-cell subsets separately to express a CD19-targeted CAR incorporating 4-1BB and CD3ζ signaling domains. The cells were then formulated in a defined ratio of CD4+:CD8+ CAR T cells.
Patients underwent lymphodepletion with a cyclophosphamide-based regimen (alone or with fludarabine or etoposide) and then received CAR T cells 48 to 96 hours later. The CAR T cells were given at 3 dose levels—2 × 105/kg (DL1), 2 × 106/kg (DL2), and 2 × 107/kg (DL3).
Toxicity
The first 2 patients who received CAR T-cell therapy at DL3 developed severe toxicities, including 1 patient who died. So DL3 was not given to any subsequent patients.
Two patients died after CAR T-cell infusion. One patient had severe cytokine release syndrome (CRS) and multiorgan failure and died on day 3. The other patient had transient severe CRS with irreversible neurologic toxicity and died on day 122.
The most common adverse event observed in the first 14 days after CAR T-cell infusion was CRS, which occurred in 25 patients. Seven patients had severe CRS that put them in the intensive care unit.
However, for the patients treated at DL1 and DL2, dexamethasone alone or with tocilizumab resolved CRS.
Fifteen patients developed severe neurotoxicity (grade 3 or higher), either with CRS or after it resolved. For all but 1 patient (the aforementioned patient who died), neurologic symptoms and signs resolved.
Response
One patient died before response assessment, so 29 patients were evaluable. Twenty-seven of these patients (93%) achieved bone marrow remission by flow cytometry.
Of the 2 patients who did not achieve a complete response (CR), 1 underwent an allogeneic HSCT after receiving CAR T cells. After HSCT, she relapsed, was re-enrolled on the trial, and achieved a CR after receiving a higher dose of CAR T cells (DL2).
Twenty-five patients (86%) achieved a CR without evidence of minimal residual disease by flow cytometry and conventional karyotyping, FISH, or QPCR.
“Patients who come onto the trial have really limited options for treatment,” said study author Cameron Turtle, MBBS, PhD, of the Fred Hutchinson Cancer Research Center in Seattle, Washington.
“They have refractory acute leukemia. So the fact that we’re getting so many into remission is giving these people a way forward.”
Unfortunately, not all patients stayed in CR. Some relapsed and were treated again with CAR T cells, and 2 patients relapsed with leukemias that were immune to the CAR T cells. The researchers said it is still too early to know the long-term outcomes of the therapy.
“This is just the beginning,” Dr Turtle said. “It sounds fantastic to say that we get over 90% remissions, but there’s so much more work to do make sure they’re durable remissions, to work out who’s going to benefit the most, and extend this work to other diseases.”
Lessons learned
The researchers said that, because these CAR T cells had a defined CD4+:CD8+ composition, this study provides the first clear evidence of the relationships between the CAR T-cell dose patients receive and their outcomes after infusion.
The team found that high doses of CAR T cells and high tumor burden increase the risks of severe CRS and neurotoxicity. However, certain biomarkers can identify patients at the highest risk of toxicity.
Levels of IL-6, IFN-γ, and TNF-α on the first day after CAR T-cell infusion were significantly higher in patients who later developed severe neurotoxicity. Levels of these biomarkers were also higher in patients who were later sent to the intensive care unit.
Furthermore, risk-stratified CAR T-cell dosing based on bone marrow disease burden decreased toxicity.
The researchers also said they observed CD8+ T-cell-mediated anti-CAR transgene product immune responses in some patients, which limited CAR T-cell persistence and increased the risk of relapse.
And including fludarabine in the lymphodepletion regimen resulted in better CAR T-cell persistence and disease-free survival than when cyclophosphamide was given alone or with etoposide.