User login
Impact of NSAID Use on Bleeding Rates for Patients Taking Rivaroxaban or Apixaban
Impact of NSAID Use on Bleeding Rates for Patients Taking Rivaroxaban or Apixaban
Clinical practice has shifted from vitamin K antagonists to direct oral anticoagulants (DOACs) for atrial fibrillation treatment due to their more favorable risk-benefit profile and less lifestyle modification required.1,2 However, the advantage of a lower bleeding risk with DOACs could be compromised by potentially problematic pharmacokinetic interactions like those conferred by antiplatelets or nonsteroidal anti-inflammatory drugs (NSAIDs).3,4 Treating a patient needing anticoagulation with a DOAC who has comorbidities may introduce unavoidable drug-drug interactions. This particularly happens with over-the-counter and prescription NSAIDs used for the management of pain and inflammatory conditions.5
NSAIDs primarily affect 2 cyclooxygenase (COX) enzyme isomers, COX-1 and COX-2.6 COX-1 helps maintain gastrointestinal (GI) mucosa integrity and platelet aggregation processes, whereas COX-2 is engaged in pain signaling and inflammation mediation. COX-1 inhibition is associated with more bleeding-related adverse events (AEs), especially in the GI tract. COX-2 inhibition is thought to provide analgesia and anti-inflammatory properties without elevating bleeding risk. This premise is responsible for the preferential use of celecoxib, a COX-2 selective NSAID, which should confer a lower bleeding risk compared to nonselective NSAIDs such as ibuprofen and naproxen.7 NSAIDs have been documented as independent risk factors for bleeding. NSAID users are about 3 times as likely to develop GI AEs compared to nonNSAID users.8
Many clinicians aim to further mitigate NSAID-associated bleeding risk by coprescribing a proton pump inhibitor (PPI). PPIs provide gastroprotection against NSAID-induced mucosal injury and sequential complication of GI bleeding. In a multicenter randomized control trial, patients who received concomitant PPI therapy while undergoing chronic NSAID therapy—including nonselective and COX-2 selective NSAIDs—had a significantly lower risk of GI ulcer development (placebo, 17.0%; 20 mg esomeprazole, 5.2%; 40 mg esomeprazole, 4.6%).9 Current clinical guidelines for preventing NSAIDassociated bleeding complications recommend using a COX-2 selective NSAID in combination with PPI therapy for patients at high risk for GI-related bleeding, including the concomitant use of anticoagulants.10
There is evidence suggesting an increased bleeding risk with NSAIDs when used in combination with vitamin K antagonists such as warfarin.11,12 A systematic review of warfarin and concomitant NSAID use found an increased risk of overall bleeding with NSAID use in combination with warfarin (odds ratio 1.58; 95% CI, 1.18-2.12), compared to warfarin alone.12
Posthoc analyses of randomized clinical trials have also demonstrated an increased bleeding risk with oral anticoagulation and concomitant NSAID use.13,14 In the RE-LY trial, NSAID users on warfarin or dabigatran had a statistically significant increased risk of major bleeding compared to non-NSAID users (hazard ratio [HR] 1.68; 95% CI, 1.40- 2.02; P < .001).13 In the ARISTOTLE trial, patients on warfarin or apixaban who were incident NSAID users were found to have an increased risk of major bleeding (HR 1.61; 95% CI, 1.11-2.33) and clinically relevant nonmajor bleeding (HR 1.70; 95% CI, 1.16- 2.48).14 These trials found a statistically significant increased bleeding risk associated with NSAID use, though the populations evaluated included patients taking warfarin and patients taking DOACs. These trials did not evaluate the bleeding risk of concomitant NSAID use among DOACs alone.
Evidence on NSAID-associated bleeding risk with DOACs is lacking in settings where the patient population, prescribing practices, and monitoring levels are variable. Within the Veterans Health Administration, clinical pharmacist practitioners (CPPs) in anticoagulation clinics oversee DOAC therapy management. CPPs monitor safety and efficacy of DOAC therapies through a population health management tool, the DOAC Dashboard.15 The DOAC Dashboard creates alerts for patients who may require an intervention based on certain clinical parameters, such as drug-drug interactions.16 Whenever a patient on a DOAC is prescribed an NSAID, an alert is generated on the DOAC Dashboard to flag the CPPs for the potential need for an intervention. If NSAID therapy remains clinically indicated, CPPs may recommend risk reduction strategies such as a COX-2 selective NSAID or coprescribing a PPI.10
The DOAC Dashboard provides an ideal setting for investigating the effects of NSAID use, NSAID selectivity, and PPI coprescribing on DOAC bleeding rates. With an increasing population of patients receiving anticoagulation therapy with a DOAC, more guidance regarding the bleeding risk of concomitant NSAID use with DOACs is needed. Studies evaluating the bleeding risk with concomitant NSAID use in patients on a DOAC alone are limited. This is the first study to date to compare bleeding risk with concomitant NSAID use between DOACs. This study provides information on bleeding risk with NSAID use among commonly prescribed DOACs, rivaroxaban and apixaban, and the potential impacts of current risk reduction strategies.
METHODS
This single-center retrospective cohort review was performed using the electronic health records (EHRs) of patients enrolled in the US Department of Veterans Affairs (VA) Mountain Home Healthcare System who received rivaroxaban or apixaban from December 2020 to December 2022. This study received approval from the East Tennessee State University/VA Institutional Review Board committee.
Patients were identified through the DOAC Dashboard, aged 21 to 100 years, and received rivaroxaban or apixaban at a therapeutic dose: rivaroxaban 10 to 20 mg daily or apixaban 2.5 to 5 mg twice daily. Patients were excluded if they were prescribed dual antiplatelet therapy, received rivaroxaban at dosing indicated for peripheral vascular disease, were undergoing dialysis, had evidence of moderate to severe hepatic impairment or any hepatic disease with coagulopathy, were undergoing chemotherapy or radiation, or had hematological conditions with predisposed bleeding risk. These patients were excluded to mitigate the potential confounding impact from nontherapeutic DOAC dosing strategies and conditions associated with an increased bleeding risk.
Eligible patients were stratified based on NSAID use. NSAID users were defined as patients prescribed an oral NSAID, including both acute and chronic courses, at any point during the study time frame while actively on a DOAC. Bleeding events were reviewed to evaluate rates between rivaroxaban and apixaban among NSAID and nonNSAID users. Identified NSAID users were further assessed for NSAID selectivity and PPI coprescribing as a subgroup analysis for the secondary assessment.
Data Collection
Baseline data were collected, including age, body mass index, anticoagulation indication, DOAC agent, DOAC dose, and DOAC total daily dose. Baseline serum creatinine levels, liver function tests, hemoglobin levels, and platelet counts were collected from the most recent data available immediately prior to the bleeding event, if applicable.
The DOAC Dashboard was reviewed for active and dismissed drug interaction alerts to identify patients taking rivaroxaban or apixaban who were prescribed an NSAID. Patients were categorized in the NSAID group if an interacting drug alert with an NSAID was reported during the study time frame. Data available through the interacting drug alerts on NSAID use were limited to the interacting drug name and date of the reported flag. Manual EHR review was required to confirm dates of NSAID therapy initiation and NSAID discontinuation, if applicable.
Data regarding concomitant antiplatelet use were obtained through review of the active and dismissed drug interaction alerts on the DOAC Dashboard. Concomitant antiplatelet use was defined as the prescribing of a single antiplatelet agent at any point while receiving DOAC therapy. Data on concomitant antiplatelets were collected regardless of NSAID status.
Data on coprescribed PPI therapy were obtained through manual EHR review of identified NSAID users. Coprescribed PPI therapy was defined as the prescribing of a PPI at any point during NSAID therapy. Data regarding PPI use among non-NSAID users were not collected because the secondary endpoint was designed to assess PPI use only among patients coprescribed a DOAC and NSAID.
Outcomes
Bleeding events were identified through an outcomes report generated by the DOAC Dashboard based on International Classification of Diseases, Tenth Revision diagnosis codes associated with a bleeding event. The outcomes report captures diagnoses from the outpatient and inpatient care settings. Reported bleeding events were limited to patients who received a DOAC at any point in the 6 months prior to the event and excluded patients with recent DOAC initiation within 7 days of the event, as these patients are not captured on the DOAC Dashboard.
All reported bleeding events were manually reviewed in the EHR and categorized as a major or clinically relevant nonmajor bleed, according to International Society of Thrombosis and Haemostasis criteria. Validated bleeding events were then crossreferenced with the interacting drug alerts report to identify events with potentially overlapping NSAID therapy at the time of the event. Overlapping NSAID therapy was defined as the prescribing of an NSAID at any point in the 6 months prior to the event. All events with potential overlapping NSAID therapies were manually reviewed for confirmation of NSAID status at the time of the event.
The primary endpoint was a composite of any bleeding event per International Society of Thrombosis and Haemostasis criteria. The secondary endpoint evaluated the potential impact of NSAID selectivity or PPI coprescribing on the bleeding rate among the NSAID user groups.
Statistical Analysis
Analyses were performed consistent with the methods used in the ARISTOTLE and RE-LY trials. It was determined that a sample size of 504 patients, with ≥ 168 patients in each group, would provide 80% power using a 2-sided a of 0.05. HRs with 95% CIs and respective P values were calculated using a SPSS-adapted online calculator.
RESULTS
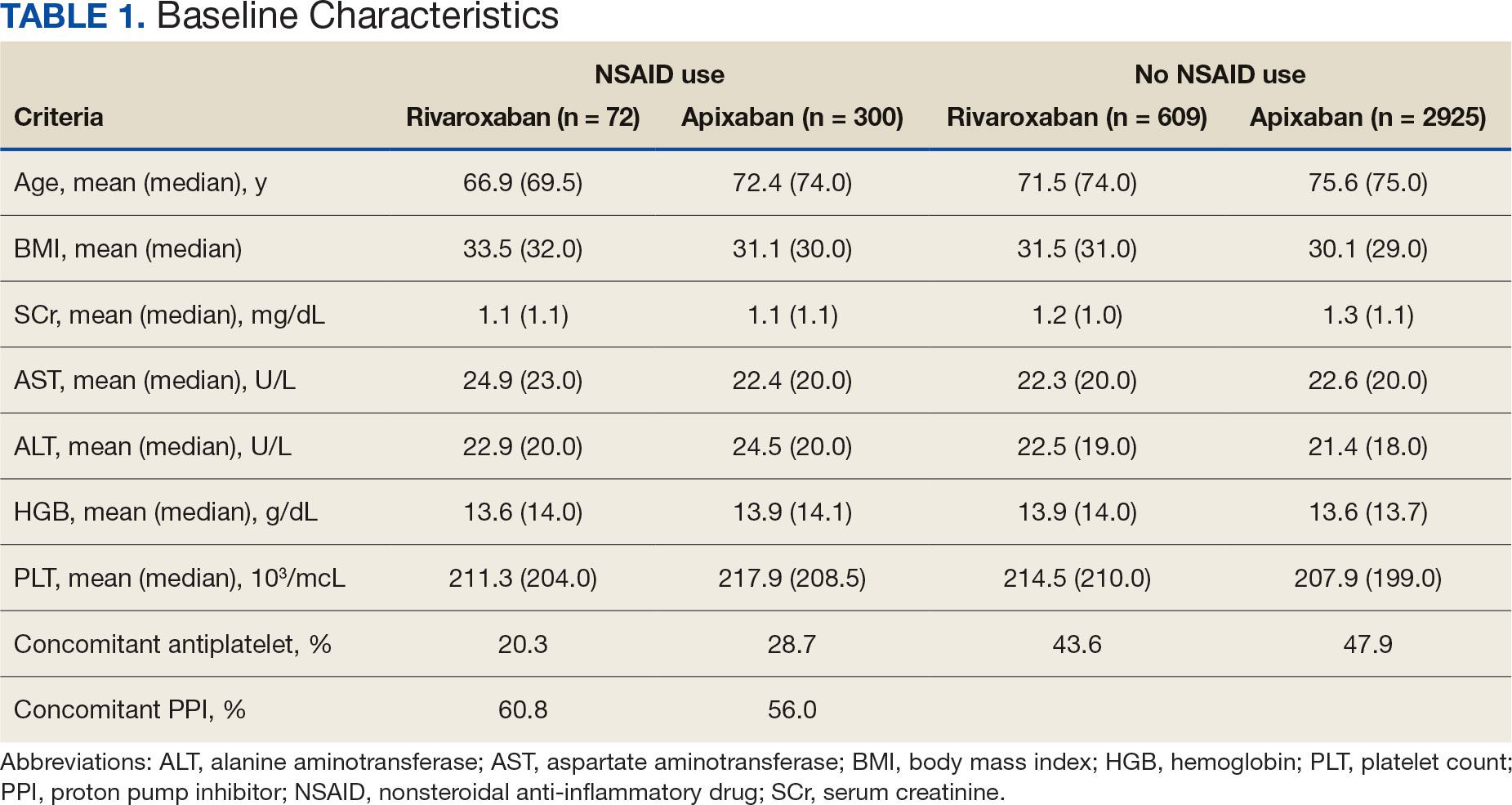
The DOAC Dashboard identified 681 patients on rivaroxaban and 3225 patients on apixaban; 72 patients on rivaroxaban (10.6%) and 300 patients on apixaban (9.3%) were NSAID users. The mean age of NSAID users was 66.9 years in the rivaroxaban group and 72.4 years in the apixaban group. The mean age of non-NSAID users was 71.5 years in the rivaroxaban group and 75.6 years in the apixaban group. No appreciable differences were observed among subgroups in body mass index, renal function, hepatic function, hemoglobin, or platelet counts, and no statistically significant differences were identified (Table 1). Antiplatelet agents identified included aspirin, clopidogrel, prasugrel, and ticagrelor. Fifteen patients (20.3%) in the rivaroxaban group and 87 patients (28.7%) in the apixaban group had concomitant antiplatelet and NSAID use. Forty-five patients on rivaroxaban (60.8%) and 170 (55.9%) on apixaban were prescribed concomitant PPI and NSAID at baseline. Among non-NSAID users, there was concomitant antiplatelet use for 265 patients (43.6%) in the rivaroxaban group and 1401 patients (47.9%) in the apixaban group. Concomitant PPI use was identified among 63 patients (60.0%) taking selective NSAIDs and 182 (57.2%) taking nonselective NSAIDs.
A total of 423 courses of NSAIDs were identified: 85 NSAID courses in the rivaroxaban group and 338 NSAID courses in the apixaban group. Most NSAID courses involved a nonselective NSAID in the rivaroxaban and apixaban NSAID user groups: 75.2% (n = 318) aggregately compared to 71.8% (n = 61) and 76.0% (n = 257) in the rivaroxaban and apixaban groups, respectively. The most frequent NSAID courses identified were meloxicam (26.7%; n = 113), celecoxib (24.8%; n = 105), ibuprofen (19.1%; n = 81), and naproxen (13.5%; n = 57). Data regarding NSAID therapy initiation and discontinuation dates were not readily available. As a result, the duration of NSAID courses was not captured.
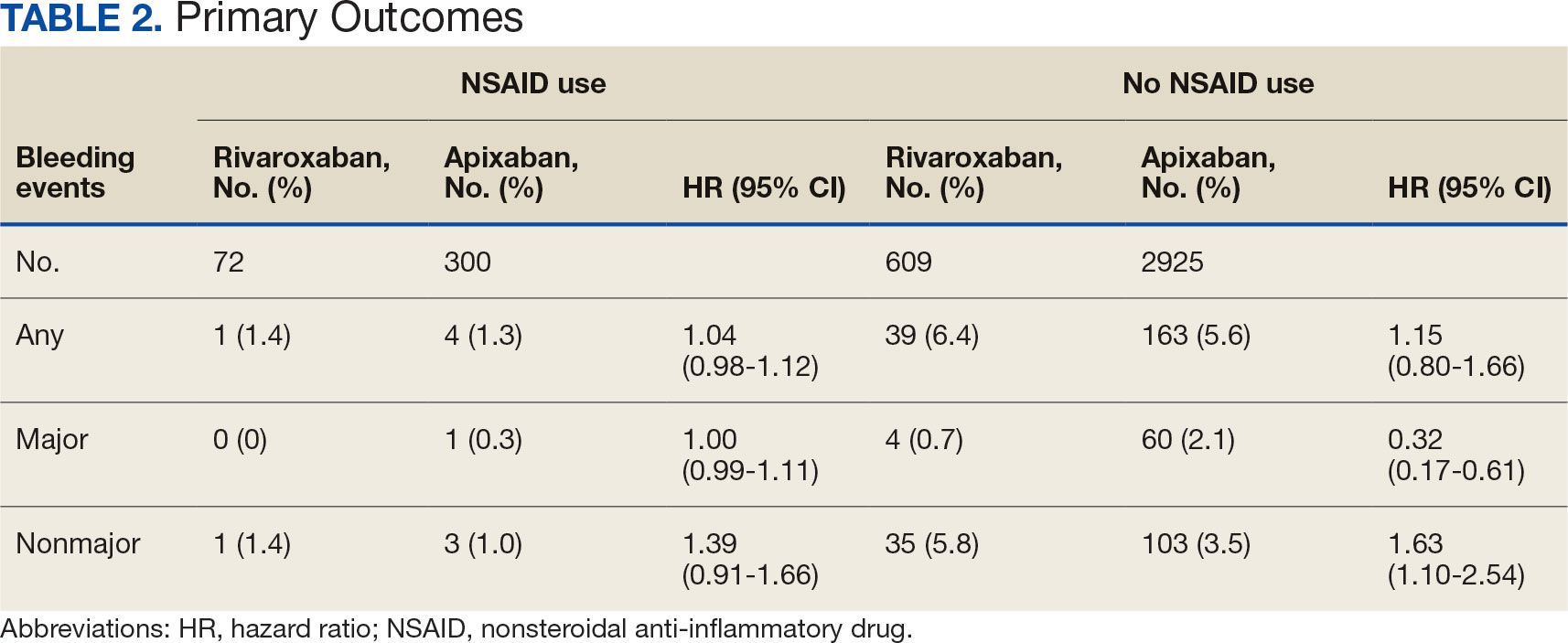
There was no statistically significant difference in bleeding rates between rivaroxaban and apixaban among NSAID users (HR 1.04; 95% CI, 0.98-1.12) or non-NSAID users (HR 1.15; 95% CI, 0.80-1.66) (Table 2). Apixaban non-NSAID users had a higher rate of major bleeds (HR 0.32; 95% CI, 0.17-0.61) while rivaroxaban non-NSAID users had a higher rate of clinically relevant nonmajor bleeds (HR 1.63; 95% CI, 1.10-2.54).
The sample size for the secondary endpoint consisted of bleeding events that were confirmed to have had an overlapping NSAID prescribed at the time of the event. For this secondary assessment, there was 1 rivaroxaban NSAID user bleeding event and 4 apixaban NSAID user bleeding events. For the rivaroxaban NSAID user bleeding event, the NSAID was nonselective and a PPI was not coprescribed. For the apixaban NSAID user bleeding events, 2 NSAIDs were nonselective and 2 were selective. All patients with apixaban and NSAID bleeding events had a coprescribed PPI. There was no clinically significant difference in the bleeding rates observed for NSAID selectivity or PPI coprescribing among the NSAID user subgroups.
DISCUSSION
This study found that there was no statistically significant difference for bleeding rates of major and nonmajor bleeding events between rivaroxaban and apixaban among NSAID users and non-NSAID users. This study did not identify a clinically significant impact on bleeding rates from NSAID selectivity or PPI coprescribing among the NSAID users.
There were notable but not statistically significant differences in baseline characteristics observed between the NSAID and non-NSAID user groups. On average, the rivaroxaban and apixaban NSAID users were younger compared with those not taking NSAIDs. NSAIDs, specifically nonselective NSAIDs, are recognized as potentially inappropriate medications for older adults given that this population is at an increased risk for GI ulcer development and/or GI bleeding.17 The non-NSAID user group likely consisted of older patients compared to the NSAID user group as clinicians may avoid prescribing NSAIDs to older adults regardless of concomitant DOAC therapy.
In addition to having an older patient population, non-NSAID users were more frequently prescribed a concomitant antiplatelet when compared with NSAID users. This prescribing pattern may be due to clinicians avoiding the use of NSAIDs in patients receiving DOAC therapy in combination with antiplatelet therapy, as these patients have been found to have an increased bleeding rate compared to DOAC therapy alone.18
Non-NSAID users had an overall higher bleeding rate for both major and nonmajor bleeding events. Based on this observation, it could be hypothesized that antiplatelet agents have a higher risk of bleeding in comparison to NSAIDs. In a subanalysis of the EXPAND study evaluating risk factors of major bleeding in patients receiving rivaroxaban, concomitant use of antiplatelet agents demonstrated a statistically significant increased risk of bleeding (HR 1.6; 95% CI, 1.2-2.3; P = .003) while concomitant use of NSAIDs did not (HR 0.8; 95% CI, 0.3-2.2; P = .67).19
In assessing PPI status at baseline, a majority of both rivaroxaban and apixaban NSAID users were coprescribed a PPI. This trend aligns with current clinical guideline recommendations for the prescribing of PPI therapy for GI protection in high-risk patients, such as those on DOAC therapy and concomitant NSAID therapy.10 Given the high proportion of NSAID users coprescribed a PPI at baseline, it may be possible that the true incidence of NSAID-associated bleeding events was higher than what this study found. This observation may reflect the impact from timely implementation of risk mitigation strategies by CPPs in the anticoagulation clinic. However, this study was not constructed to assess the efficacy of PPI use in this manner.
It is important to note the patients included in this study were followed by a pharmacist in an anticoagulation clinic using the DOAC Dashboard.15 This population management tool allows CPPs to make proactive interventions when a patient taking a DOAC receives an NSAID prescription, such as recommending the coprescribing of a PPI or use of a selective NSAID.10,16 These standards of care may have contributed to an overall reduced bleeding rate among the NSAID user group and may not be reflective of private practice.
The planned analysis of this study was modeled after the posthoc analysis of the RE-LY and ARISTOTLE trials. Both trials demonstrated an increased risk of bleeding with oral anticoagulation, including DOAC and warfarin, in combination with NSAID use. However, both trials found that NSAID use in patients treated with a DOAC was not independently associated with increased bleeding events compared with warfarin.13,14 The results of this study are comparable to the RE-LY and ARISTOTLE findings that NSAID use among patients treated with rivaroxaban or apixaban did not demonstrate a statistically significant increased bleeding risk.
Studies of NSAID use in combination with DOAC therapy have been limited to patient populations consisting of both DOAC and warfarin. Evidence from these trials outlines the increased bleeding risk associated with NSAID use in combination with oral anticoagulation; however, these patient populations include those on a DOAC and warfarin.13,14,19,20 Given the limited evidence on NSAID use among DOACs alone, it is assumed NSAID use in combination with DOACs has a similar risk of bleeding as warfarin use. This may cause clinicians to automatically exclude NSAID therapy as a treatment option for patients on a DOAC who are otherwise clinically appropriate candidates, such as those with underlying inflammatory conditions. Avoiding NSAID therapy in this patient population may lead to suboptimal pain management and increase the risk of patient harm from methods such as inappropriate opioid therapy prescribing.
DOAC therapy should not be a universal limitation to the use of NSAIDs. Although the risk of bleeding with NSAID therapy is always present, deliberate NSAID prescribing in addition to the timely implementation of risk mitigation strategies may provide an avenue for safe NSAID prescribing in patients receiving a DOAC. A population health-based approach to DOAC management, such as the DOAC Dashboard, appears to be effective at preventing patient harm when NSAIDs are prescribed in conjunction with DOACs.
Limitations
The DOAC Dashboard has been shown to be effective and efficient at monitoring DOAC therapy from a population-based approach.16 Reports generated through the DOAC Dashboard provide convenient access to patient data which allows for timely interventions; however, there are limits to its use for data collection. All the data elements necessary to properly assess bleeding risk with validated tools, such as HAS-BLED (hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile international normalized ratio, elderly, drugs/ alcohol concomitantly), are not available on DOAC Dashboard reports. Due to this constraint, bleeding risk assessments were not conducted at baseline and this study was unable to include risk modeling. Additionally, data elements like initiation and discontinuation dates and duration of therapies were not readily available. As a result, this study was unable to incorporate time as a data point.
This was a retrospective study that relied on manual review of chart documentation to verify bleeding events, but data obtained through the DOAC Dashboard were transferred directly from the EHR.15 Bleeding events available for evaluation were restricted to those that occurred at a VA facility. Additionally, the sample size within the rivaroxaban NSAID user group did not reach the predefined sample size required to reach power and may have been too small to detect a difference if one did exist. The secondary assessment had a low sample size of NSAID user bleeding events, making it difficult to fully assess its impact on NSAID selectivity and PPI coprescribing on bleeding rates. All courses of NSAIDs were equally valued regardless of the dose or therapy duration; however, this is consistent with how NSAID use was defined in the RE-LY and ARISTOTLE trials.
CONCLUSIONS
This retrospective cohort review found no statistically significant difference in the composite bleeding rates between rivaroxaban and apixaban among NSAID users and non-NSAID users. Moreover, there was no clinically significant impact observed for bleeding rates in regard to NSAID selectivity and PPI coprescribing among NSAID users. However, coprescribing of PPI therapy to patients on a DOAC who are clinically indicated for an NSAID may reduce the risk of bleeding. Population health management tools, such as the DOAC Dashboard, may also allow clinicians to safely prescribe NSAIDs to patients on a DOAC. Further large-scale observational studies are needed to quantify the real-world risk of bleeding with concomitant NSAID use among DOACs alone and to evaluate the impact from NSAID selectivity or PPI coprescribing.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. doi:10.1016/S0140-6736(13)62343-0
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e44S-e88S. doi:10.1378/chest.11-2292
- Eikelboom J, Merli G. Bleeding with direct oral anticoagulants vs warfarin: clinical experience. Am J Med. 2016;129(11S):S33-S40. doi:10.1016/j.amjmed.2016.06.003
- Vranckx P, Valgimigli M, Heidbuchel H. The significance of drug-drug and drug-food interactions of oral anticoagulation. Arrhythm Electrophysiol Rev. 2018;7(1):55-61. doi:10.15420/aer.2017.50.1
- Davis JS, Lee HY, Kim J, et al. Use of non-steroidal antiinflammatory drugs in US adults: changes over time and by demographic. Open Heart. 2017;4(1):e000550. doi:10.1136/openhrt-2016-000550
- Schafer AI. Effects of nonsteroidal antiinflammatory drugs on platelet function and systemic hemostasis. J Clin Pharmacol. 1995;35(3):209-219. doi:10.1002/j.1552-4604.1995.tb04050.x
- Al-Saeed A. Gastrointestinal and cardiovascular risk of nonsteroidal anti-inflammatory drugs. Oman Med J. 2011;26(6):385-391. doi:10.5001/omj.2011.101
- Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. Ann Intern Med. 1991;115(10):787-796. doi:10.7326/0003-4819-115-10-787
- Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101(4):701-710. doi:10.1111/j.1572-0241.2006.00499.x
- Freedberg DE, Kim LS, Yang YX. The risks and benefits of long-term use of proton pump inhibitors: expert review and best practice advice from the American Gastroenterological Association. Gastroenterology. 2017;152(4):706-715. doi:10.1053/j.gastro.2017.01.031
- Lamberts M, Lip GYH, Hansen ML, et al. Relation of nonsteroidal anti-inflammatory drugs to serious bleeding and thromboembolism risk in patients with atrial fibrillation receiving antithrombotic therapy: a nationwide cohort study. Ann Intern Med. 2014;161(10):690-698. doi:10.7326/M13-1581
- Villa Zapata L, Hansten PD, Panic J, et al. Risk of bleeding with exposure to warfarin and nonsteroidal anti-inflammatory drugs: a systematic review and metaanalysis. Thromb Haemost. 2020;120(7):1066-1074. doi:10.1055/s-0040-1710592
- Kent AP, Brueckmann M, Fraessdorf M, et al. Concomitant oral anticoagulant and nonsteroidal anti-inflammatory drug therapy in patients with atrial fibrillation. J Am Coll Cardiol. 2018;72(3):255-267. doi:10.1016/j.jacc.2018.04.063
- Dalgaard F, Mulder H, Wojdyla DM, et al. Patients with atrial fibrillation taking nonsteroidal antiinflammatory drugs and oral anticoagulants in the ARISTOTLE Trial. Circulation. 2020;141(1):10-20. doi:10.1161/CIRCULATIONAHA.119.041296
- Allen AL, Lucas J, Parra D, et al. Shifting the paradigm: a population health approach to the management of direct oral anticoagulants. J Am Heart Asssoc. 2021;10(24):e022758. doi:10.1161/JAHA.121.022758
- . Valencia D, Spoutz P, Stoppi J, et al. Impact of a direct oral anticoagulant population management tool on anticoagulation therapy monitoring in clinical practice. Ann Pharmacother. 2019;53(8):806-811. doi:10.1177/1060028019835843
- By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 Updated AGS Beers Criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052-2081. doi:10.1111/jgs.18372
- Kumar S, Danik SB, Altman RK, et al. Non-vitamin K antagonist oral anticoagulants and antiplatelet therapy for stroke prevention in patients with atrial fibrillation. Cardiol Rev. 2016;24(5):218-223. doi:10.1097/CRD.0000000000000088
- Sakuma I, Uchiyama S, Atarashi H, et al. Clinical risk factors of stroke and major bleeding in patients with nonvalvular atrial fibrillation under rivaroxaban: the EXPAND study sub-analysis. Heart Vessels. 2019;34(11):1839-1851. doi:10.1007/s00380-019-01425-x
- Davidson BL, Verheijen S, Lensing AWA, et al. Bleeding risk of patients with acute venous thromboembolism taking nonsteroidal anti-inflammatory drugs or aspirin. JAMA Intern Med. 2014;174(6):947-953. doi:10.1001/jamainternmed.2014.946
Clinical practice has shifted from vitamin K antagonists to direct oral anticoagulants (DOACs) for atrial fibrillation treatment due to their more favorable risk-benefit profile and less lifestyle modification required.1,2 However, the advantage of a lower bleeding risk with DOACs could be compromised by potentially problematic pharmacokinetic interactions like those conferred by antiplatelets or nonsteroidal anti-inflammatory drugs (NSAIDs).3,4 Treating a patient needing anticoagulation with a DOAC who has comorbidities may introduce unavoidable drug-drug interactions. This particularly happens with over-the-counter and prescription NSAIDs used for the management of pain and inflammatory conditions.5
NSAIDs primarily affect 2 cyclooxygenase (COX) enzyme isomers, COX-1 and COX-2.6 COX-1 helps maintain gastrointestinal (GI) mucosa integrity and platelet aggregation processes, whereas COX-2 is engaged in pain signaling and inflammation mediation. COX-1 inhibition is associated with more bleeding-related adverse events (AEs), especially in the GI tract. COX-2 inhibition is thought to provide analgesia and anti-inflammatory properties without elevating bleeding risk. This premise is responsible for the preferential use of celecoxib, a COX-2 selective NSAID, which should confer a lower bleeding risk compared to nonselective NSAIDs such as ibuprofen and naproxen.7 NSAIDs have been documented as independent risk factors for bleeding. NSAID users are about 3 times as likely to develop GI AEs compared to nonNSAID users.8
Many clinicians aim to further mitigate NSAID-associated bleeding risk by coprescribing a proton pump inhibitor (PPI). PPIs provide gastroprotection against NSAID-induced mucosal injury and sequential complication of GI bleeding. In a multicenter randomized control trial, patients who received concomitant PPI therapy while undergoing chronic NSAID therapy—including nonselective and COX-2 selective NSAIDs—had a significantly lower risk of GI ulcer development (placebo, 17.0%; 20 mg esomeprazole, 5.2%; 40 mg esomeprazole, 4.6%).9 Current clinical guidelines for preventing NSAIDassociated bleeding complications recommend using a COX-2 selective NSAID in combination with PPI therapy for patients at high risk for GI-related bleeding, including the concomitant use of anticoagulants.10
There is evidence suggesting an increased bleeding risk with NSAIDs when used in combination with vitamin K antagonists such as warfarin.11,12 A systematic review of warfarin and concomitant NSAID use found an increased risk of overall bleeding with NSAID use in combination with warfarin (odds ratio 1.58; 95% CI, 1.18-2.12), compared to warfarin alone.12
Posthoc analyses of randomized clinical trials have also demonstrated an increased bleeding risk with oral anticoagulation and concomitant NSAID use.13,14 In the RE-LY trial, NSAID users on warfarin or dabigatran had a statistically significant increased risk of major bleeding compared to non-NSAID users (hazard ratio [HR] 1.68; 95% CI, 1.40- 2.02; P < .001).13 In the ARISTOTLE trial, patients on warfarin or apixaban who were incident NSAID users were found to have an increased risk of major bleeding (HR 1.61; 95% CI, 1.11-2.33) and clinically relevant nonmajor bleeding (HR 1.70; 95% CI, 1.16- 2.48).14 These trials found a statistically significant increased bleeding risk associated with NSAID use, though the populations evaluated included patients taking warfarin and patients taking DOACs. These trials did not evaluate the bleeding risk of concomitant NSAID use among DOACs alone.
Evidence on NSAID-associated bleeding risk with DOACs is lacking in settings where the patient population, prescribing practices, and monitoring levels are variable. Within the Veterans Health Administration, clinical pharmacist practitioners (CPPs) in anticoagulation clinics oversee DOAC therapy management. CPPs monitor safety and efficacy of DOAC therapies through a population health management tool, the DOAC Dashboard.15 The DOAC Dashboard creates alerts for patients who may require an intervention based on certain clinical parameters, such as drug-drug interactions.16 Whenever a patient on a DOAC is prescribed an NSAID, an alert is generated on the DOAC Dashboard to flag the CPPs for the potential need for an intervention. If NSAID therapy remains clinically indicated, CPPs may recommend risk reduction strategies such as a COX-2 selective NSAID or coprescribing a PPI.10
The DOAC Dashboard provides an ideal setting for investigating the effects of NSAID use, NSAID selectivity, and PPI coprescribing on DOAC bleeding rates. With an increasing population of patients receiving anticoagulation therapy with a DOAC, more guidance regarding the bleeding risk of concomitant NSAID use with DOACs is needed. Studies evaluating the bleeding risk with concomitant NSAID use in patients on a DOAC alone are limited. This is the first study to date to compare bleeding risk with concomitant NSAID use between DOACs. This study provides information on bleeding risk with NSAID use among commonly prescribed DOACs, rivaroxaban and apixaban, and the potential impacts of current risk reduction strategies.
METHODS
This single-center retrospective cohort review was performed using the electronic health records (EHRs) of patients enrolled in the US Department of Veterans Affairs (VA) Mountain Home Healthcare System who received rivaroxaban or apixaban from December 2020 to December 2022. This study received approval from the East Tennessee State University/VA Institutional Review Board committee.
Patients were identified through the DOAC Dashboard, aged 21 to 100 years, and received rivaroxaban or apixaban at a therapeutic dose: rivaroxaban 10 to 20 mg daily or apixaban 2.5 to 5 mg twice daily. Patients were excluded if they were prescribed dual antiplatelet therapy, received rivaroxaban at dosing indicated for peripheral vascular disease, were undergoing dialysis, had evidence of moderate to severe hepatic impairment or any hepatic disease with coagulopathy, were undergoing chemotherapy or radiation, or had hematological conditions with predisposed bleeding risk. These patients were excluded to mitigate the potential confounding impact from nontherapeutic DOAC dosing strategies and conditions associated with an increased bleeding risk.
Eligible patients were stratified based on NSAID use. NSAID users were defined as patients prescribed an oral NSAID, including both acute and chronic courses, at any point during the study time frame while actively on a DOAC. Bleeding events were reviewed to evaluate rates between rivaroxaban and apixaban among NSAID and nonNSAID users. Identified NSAID users were further assessed for NSAID selectivity and PPI coprescribing as a subgroup analysis for the secondary assessment.
Data Collection
Baseline data were collected, including age, body mass index, anticoagulation indication, DOAC agent, DOAC dose, and DOAC total daily dose. Baseline serum creatinine levels, liver function tests, hemoglobin levels, and platelet counts were collected from the most recent data available immediately prior to the bleeding event, if applicable.
The DOAC Dashboard was reviewed for active and dismissed drug interaction alerts to identify patients taking rivaroxaban or apixaban who were prescribed an NSAID. Patients were categorized in the NSAID group if an interacting drug alert with an NSAID was reported during the study time frame. Data available through the interacting drug alerts on NSAID use were limited to the interacting drug name and date of the reported flag. Manual EHR review was required to confirm dates of NSAID therapy initiation and NSAID discontinuation, if applicable.
Data regarding concomitant antiplatelet use were obtained through review of the active and dismissed drug interaction alerts on the DOAC Dashboard. Concomitant antiplatelet use was defined as the prescribing of a single antiplatelet agent at any point while receiving DOAC therapy. Data on concomitant antiplatelets were collected regardless of NSAID status.
Data on coprescribed PPI therapy were obtained through manual EHR review of identified NSAID users. Coprescribed PPI therapy was defined as the prescribing of a PPI at any point during NSAID therapy. Data regarding PPI use among non-NSAID users were not collected because the secondary endpoint was designed to assess PPI use only among patients coprescribed a DOAC and NSAID.
Outcomes
Bleeding events were identified through an outcomes report generated by the DOAC Dashboard based on International Classification of Diseases, Tenth Revision diagnosis codes associated with a bleeding event. The outcomes report captures diagnoses from the outpatient and inpatient care settings. Reported bleeding events were limited to patients who received a DOAC at any point in the 6 months prior to the event and excluded patients with recent DOAC initiation within 7 days of the event, as these patients are not captured on the DOAC Dashboard.
All reported bleeding events were manually reviewed in the EHR and categorized as a major or clinically relevant nonmajor bleed, according to International Society of Thrombosis and Haemostasis criteria. Validated bleeding events were then crossreferenced with the interacting drug alerts report to identify events with potentially overlapping NSAID therapy at the time of the event. Overlapping NSAID therapy was defined as the prescribing of an NSAID at any point in the 6 months prior to the event. All events with potential overlapping NSAID therapies were manually reviewed for confirmation of NSAID status at the time of the event.
The primary endpoint was a composite of any bleeding event per International Society of Thrombosis and Haemostasis criteria. The secondary endpoint evaluated the potential impact of NSAID selectivity or PPI coprescribing on the bleeding rate among the NSAID user groups.
Statistical Analysis
Analyses were performed consistent with the methods used in the ARISTOTLE and RE-LY trials. It was determined that a sample size of 504 patients, with ≥ 168 patients in each group, would provide 80% power using a 2-sided a of 0.05. HRs with 95% CIs and respective P values were calculated using a SPSS-adapted online calculator.
RESULTS
The DOAC Dashboard identified 681 patients on rivaroxaban and 3225 patients on apixaban; 72 patients on rivaroxaban (10.6%) and 300 patients on apixaban (9.3%) were NSAID users. The mean age of NSAID users was 66.9 years in the rivaroxaban group and 72.4 years in the apixaban group. The mean age of non-NSAID users was 71.5 years in the rivaroxaban group and 75.6 years in the apixaban group. No appreciable differences were observed among subgroups in body mass index, renal function, hepatic function, hemoglobin, or platelet counts, and no statistically significant differences were identified (Table 1). Antiplatelet agents identified included aspirin, clopidogrel, prasugrel, and ticagrelor. Fifteen patients (20.3%) in the rivaroxaban group and 87 patients (28.7%) in the apixaban group had concomitant antiplatelet and NSAID use. Forty-five patients on rivaroxaban (60.8%) and 170 (55.9%) on apixaban were prescribed concomitant PPI and NSAID at baseline. Among non-NSAID users, there was concomitant antiplatelet use for 265 patients (43.6%) in the rivaroxaban group and 1401 patients (47.9%) in the apixaban group. Concomitant PPI use was identified among 63 patients (60.0%) taking selective NSAIDs and 182 (57.2%) taking nonselective NSAIDs.
A total of 423 courses of NSAIDs were identified: 85 NSAID courses in the rivaroxaban group and 338 NSAID courses in the apixaban group. Most NSAID courses involved a nonselective NSAID in the rivaroxaban and apixaban NSAID user groups: 75.2% (n = 318) aggregately compared to 71.8% (n = 61) and 76.0% (n = 257) in the rivaroxaban and apixaban groups, respectively. The most frequent NSAID courses identified were meloxicam (26.7%; n = 113), celecoxib (24.8%; n = 105), ibuprofen (19.1%; n = 81), and naproxen (13.5%; n = 57). Data regarding NSAID therapy initiation and discontinuation dates were not readily available. As a result, the duration of NSAID courses was not captured.
There was no statistically significant difference in bleeding rates between rivaroxaban and apixaban among NSAID users (HR 1.04; 95% CI, 0.98-1.12) or non-NSAID users (HR 1.15; 95% CI, 0.80-1.66) (Table 2). Apixaban non-NSAID users had a higher rate of major bleeds (HR 0.32; 95% CI, 0.17-0.61) while rivaroxaban non-NSAID users had a higher rate of clinically relevant nonmajor bleeds (HR 1.63; 95% CI, 1.10-2.54).
The sample size for the secondary endpoint consisted of bleeding events that were confirmed to have had an overlapping NSAID prescribed at the time of the event. For this secondary assessment, there was 1 rivaroxaban NSAID user bleeding event and 4 apixaban NSAID user bleeding events. For the rivaroxaban NSAID user bleeding event, the NSAID was nonselective and a PPI was not coprescribed. For the apixaban NSAID user bleeding events, 2 NSAIDs were nonselective and 2 were selective. All patients with apixaban and NSAID bleeding events had a coprescribed PPI. There was no clinically significant difference in the bleeding rates observed for NSAID selectivity or PPI coprescribing among the NSAID user subgroups.
DISCUSSION
This study found that there was no statistically significant difference for bleeding rates of major and nonmajor bleeding events between rivaroxaban and apixaban among NSAID users and non-NSAID users. This study did not identify a clinically significant impact on bleeding rates from NSAID selectivity or PPI coprescribing among the NSAID users.
There were notable but not statistically significant differences in baseline characteristics observed between the NSAID and non-NSAID user groups. On average, the rivaroxaban and apixaban NSAID users were younger compared with those not taking NSAIDs. NSAIDs, specifically nonselective NSAIDs, are recognized as potentially inappropriate medications for older adults given that this population is at an increased risk for GI ulcer development and/or GI bleeding.17 The non-NSAID user group likely consisted of older patients compared to the NSAID user group as clinicians may avoid prescribing NSAIDs to older adults regardless of concomitant DOAC therapy.
In addition to having an older patient population, non-NSAID users were more frequently prescribed a concomitant antiplatelet when compared with NSAID users. This prescribing pattern may be due to clinicians avoiding the use of NSAIDs in patients receiving DOAC therapy in combination with antiplatelet therapy, as these patients have been found to have an increased bleeding rate compared to DOAC therapy alone.18
Non-NSAID users had an overall higher bleeding rate for both major and nonmajor bleeding events. Based on this observation, it could be hypothesized that antiplatelet agents have a higher risk of bleeding in comparison to NSAIDs. In a subanalysis of the EXPAND study evaluating risk factors of major bleeding in patients receiving rivaroxaban, concomitant use of antiplatelet agents demonstrated a statistically significant increased risk of bleeding (HR 1.6; 95% CI, 1.2-2.3; P = .003) while concomitant use of NSAIDs did not (HR 0.8; 95% CI, 0.3-2.2; P = .67).19
In assessing PPI status at baseline, a majority of both rivaroxaban and apixaban NSAID users were coprescribed a PPI. This trend aligns with current clinical guideline recommendations for the prescribing of PPI therapy for GI protection in high-risk patients, such as those on DOAC therapy and concomitant NSAID therapy.10 Given the high proportion of NSAID users coprescribed a PPI at baseline, it may be possible that the true incidence of NSAID-associated bleeding events was higher than what this study found. This observation may reflect the impact from timely implementation of risk mitigation strategies by CPPs in the anticoagulation clinic. However, this study was not constructed to assess the efficacy of PPI use in this manner.
It is important to note the patients included in this study were followed by a pharmacist in an anticoagulation clinic using the DOAC Dashboard.15 This population management tool allows CPPs to make proactive interventions when a patient taking a DOAC receives an NSAID prescription, such as recommending the coprescribing of a PPI or use of a selective NSAID.10,16 These standards of care may have contributed to an overall reduced bleeding rate among the NSAID user group and may not be reflective of private practice.
The planned analysis of this study was modeled after the posthoc analysis of the RE-LY and ARISTOTLE trials. Both trials demonstrated an increased risk of bleeding with oral anticoagulation, including DOAC and warfarin, in combination with NSAID use. However, both trials found that NSAID use in patients treated with a DOAC was not independently associated with increased bleeding events compared with warfarin.13,14 The results of this study are comparable to the RE-LY and ARISTOTLE findings that NSAID use among patients treated with rivaroxaban or apixaban did not demonstrate a statistically significant increased bleeding risk.
Studies of NSAID use in combination with DOAC therapy have been limited to patient populations consisting of both DOAC and warfarin. Evidence from these trials outlines the increased bleeding risk associated with NSAID use in combination with oral anticoagulation; however, these patient populations include those on a DOAC and warfarin.13,14,19,20 Given the limited evidence on NSAID use among DOACs alone, it is assumed NSAID use in combination with DOACs has a similar risk of bleeding as warfarin use. This may cause clinicians to automatically exclude NSAID therapy as a treatment option for patients on a DOAC who are otherwise clinically appropriate candidates, such as those with underlying inflammatory conditions. Avoiding NSAID therapy in this patient population may lead to suboptimal pain management and increase the risk of patient harm from methods such as inappropriate opioid therapy prescribing.
DOAC therapy should not be a universal limitation to the use of NSAIDs. Although the risk of bleeding with NSAID therapy is always present, deliberate NSAID prescribing in addition to the timely implementation of risk mitigation strategies may provide an avenue for safe NSAID prescribing in patients receiving a DOAC. A population health-based approach to DOAC management, such as the DOAC Dashboard, appears to be effective at preventing patient harm when NSAIDs are prescribed in conjunction with DOACs.
Limitations
The DOAC Dashboard has been shown to be effective and efficient at monitoring DOAC therapy from a population-based approach.16 Reports generated through the DOAC Dashboard provide convenient access to patient data which allows for timely interventions; however, there are limits to its use for data collection. All the data elements necessary to properly assess bleeding risk with validated tools, such as HAS-BLED (hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile international normalized ratio, elderly, drugs/ alcohol concomitantly), are not available on DOAC Dashboard reports. Due to this constraint, bleeding risk assessments were not conducted at baseline and this study was unable to include risk modeling. Additionally, data elements like initiation and discontinuation dates and duration of therapies were not readily available. As a result, this study was unable to incorporate time as a data point.
This was a retrospective study that relied on manual review of chart documentation to verify bleeding events, but data obtained through the DOAC Dashboard were transferred directly from the EHR.15 Bleeding events available for evaluation were restricted to those that occurred at a VA facility. Additionally, the sample size within the rivaroxaban NSAID user group did not reach the predefined sample size required to reach power and may have been too small to detect a difference if one did exist. The secondary assessment had a low sample size of NSAID user bleeding events, making it difficult to fully assess its impact on NSAID selectivity and PPI coprescribing on bleeding rates. All courses of NSAIDs were equally valued regardless of the dose or therapy duration; however, this is consistent with how NSAID use was defined in the RE-LY and ARISTOTLE trials.
CONCLUSIONS
This retrospective cohort review found no statistically significant difference in the composite bleeding rates between rivaroxaban and apixaban among NSAID users and non-NSAID users. Moreover, there was no clinically significant impact observed for bleeding rates in regard to NSAID selectivity and PPI coprescribing among NSAID users. However, coprescribing of PPI therapy to patients on a DOAC who are clinically indicated for an NSAID may reduce the risk of bleeding. Population health management tools, such as the DOAC Dashboard, may also allow clinicians to safely prescribe NSAIDs to patients on a DOAC. Further large-scale observational studies are needed to quantify the real-world risk of bleeding with concomitant NSAID use among DOACs alone and to evaluate the impact from NSAID selectivity or PPI coprescribing.
Clinical practice has shifted from vitamin K antagonists to direct oral anticoagulants (DOACs) for atrial fibrillation treatment due to their more favorable risk-benefit profile and less lifestyle modification required.1,2 However, the advantage of a lower bleeding risk with DOACs could be compromised by potentially problematic pharmacokinetic interactions like those conferred by antiplatelets or nonsteroidal anti-inflammatory drugs (NSAIDs).3,4 Treating a patient needing anticoagulation with a DOAC who has comorbidities may introduce unavoidable drug-drug interactions. This particularly happens with over-the-counter and prescription NSAIDs used for the management of pain and inflammatory conditions.5
NSAIDs primarily affect 2 cyclooxygenase (COX) enzyme isomers, COX-1 and COX-2.6 COX-1 helps maintain gastrointestinal (GI) mucosa integrity and platelet aggregation processes, whereas COX-2 is engaged in pain signaling and inflammation mediation. COX-1 inhibition is associated with more bleeding-related adverse events (AEs), especially in the GI tract. COX-2 inhibition is thought to provide analgesia and anti-inflammatory properties without elevating bleeding risk. This premise is responsible for the preferential use of celecoxib, a COX-2 selective NSAID, which should confer a lower bleeding risk compared to nonselective NSAIDs such as ibuprofen and naproxen.7 NSAIDs have been documented as independent risk factors for bleeding. NSAID users are about 3 times as likely to develop GI AEs compared to nonNSAID users.8
Many clinicians aim to further mitigate NSAID-associated bleeding risk by coprescribing a proton pump inhibitor (PPI). PPIs provide gastroprotection against NSAID-induced mucosal injury and sequential complication of GI bleeding. In a multicenter randomized control trial, patients who received concomitant PPI therapy while undergoing chronic NSAID therapy—including nonselective and COX-2 selective NSAIDs—had a significantly lower risk of GI ulcer development (placebo, 17.0%; 20 mg esomeprazole, 5.2%; 40 mg esomeprazole, 4.6%).9 Current clinical guidelines for preventing NSAIDassociated bleeding complications recommend using a COX-2 selective NSAID in combination with PPI therapy for patients at high risk for GI-related bleeding, including the concomitant use of anticoagulants.10
There is evidence suggesting an increased bleeding risk with NSAIDs when used in combination with vitamin K antagonists such as warfarin.11,12 A systematic review of warfarin and concomitant NSAID use found an increased risk of overall bleeding with NSAID use in combination with warfarin (odds ratio 1.58; 95% CI, 1.18-2.12), compared to warfarin alone.12
Posthoc analyses of randomized clinical trials have also demonstrated an increased bleeding risk with oral anticoagulation and concomitant NSAID use.13,14 In the RE-LY trial, NSAID users on warfarin or dabigatran had a statistically significant increased risk of major bleeding compared to non-NSAID users (hazard ratio [HR] 1.68; 95% CI, 1.40- 2.02; P < .001).13 In the ARISTOTLE trial, patients on warfarin or apixaban who were incident NSAID users were found to have an increased risk of major bleeding (HR 1.61; 95% CI, 1.11-2.33) and clinically relevant nonmajor bleeding (HR 1.70; 95% CI, 1.16- 2.48).14 These trials found a statistically significant increased bleeding risk associated with NSAID use, though the populations evaluated included patients taking warfarin and patients taking DOACs. These trials did not evaluate the bleeding risk of concomitant NSAID use among DOACs alone.
Evidence on NSAID-associated bleeding risk with DOACs is lacking in settings where the patient population, prescribing practices, and monitoring levels are variable. Within the Veterans Health Administration, clinical pharmacist practitioners (CPPs) in anticoagulation clinics oversee DOAC therapy management. CPPs monitor safety and efficacy of DOAC therapies through a population health management tool, the DOAC Dashboard.15 The DOAC Dashboard creates alerts for patients who may require an intervention based on certain clinical parameters, such as drug-drug interactions.16 Whenever a patient on a DOAC is prescribed an NSAID, an alert is generated on the DOAC Dashboard to flag the CPPs for the potential need for an intervention. If NSAID therapy remains clinically indicated, CPPs may recommend risk reduction strategies such as a COX-2 selective NSAID or coprescribing a PPI.10
The DOAC Dashboard provides an ideal setting for investigating the effects of NSAID use, NSAID selectivity, and PPI coprescribing on DOAC bleeding rates. With an increasing population of patients receiving anticoagulation therapy with a DOAC, more guidance regarding the bleeding risk of concomitant NSAID use with DOACs is needed. Studies evaluating the bleeding risk with concomitant NSAID use in patients on a DOAC alone are limited. This is the first study to date to compare bleeding risk with concomitant NSAID use between DOACs. This study provides information on bleeding risk with NSAID use among commonly prescribed DOACs, rivaroxaban and apixaban, and the potential impacts of current risk reduction strategies.
METHODS
This single-center retrospective cohort review was performed using the electronic health records (EHRs) of patients enrolled in the US Department of Veterans Affairs (VA) Mountain Home Healthcare System who received rivaroxaban or apixaban from December 2020 to December 2022. This study received approval from the East Tennessee State University/VA Institutional Review Board committee.
Patients were identified through the DOAC Dashboard, aged 21 to 100 years, and received rivaroxaban or apixaban at a therapeutic dose: rivaroxaban 10 to 20 mg daily or apixaban 2.5 to 5 mg twice daily. Patients were excluded if they were prescribed dual antiplatelet therapy, received rivaroxaban at dosing indicated for peripheral vascular disease, were undergoing dialysis, had evidence of moderate to severe hepatic impairment or any hepatic disease with coagulopathy, were undergoing chemotherapy or radiation, or had hematological conditions with predisposed bleeding risk. These patients were excluded to mitigate the potential confounding impact from nontherapeutic DOAC dosing strategies and conditions associated with an increased bleeding risk.
Eligible patients were stratified based on NSAID use. NSAID users were defined as patients prescribed an oral NSAID, including both acute and chronic courses, at any point during the study time frame while actively on a DOAC. Bleeding events were reviewed to evaluate rates between rivaroxaban and apixaban among NSAID and nonNSAID users. Identified NSAID users were further assessed for NSAID selectivity and PPI coprescribing as a subgroup analysis for the secondary assessment.
Data Collection
Baseline data were collected, including age, body mass index, anticoagulation indication, DOAC agent, DOAC dose, and DOAC total daily dose. Baseline serum creatinine levels, liver function tests, hemoglobin levels, and platelet counts were collected from the most recent data available immediately prior to the bleeding event, if applicable.
The DOAC Dashboard was reviewed for active and dismissed drug interaction alerts to identify patients taking rivaroxaban or apixaban who were prescribed an NSAID. Patients were categorized in the NSAID group if an interacting drug alert with an NSAID was reported during the study time frame. Data available through the interacting drug alerts on NSAID use were limited to the interacting drug name and date of the reported flag. Manual EHR review was required to confirm dates of NSAID therapy initiation and NSAID discontinuation, if applicable.
Data regarding concomitant antiplatelet use were obtained through review of the active and dismissed drug interaction alerts on the DOAC Dashboard. Concomitant antiplatelet use was defined as the prescribing of a single antiplatelet agent at any point while receiving DOAC therapy. Data on concomitant antiplatelets were collected regardless of NSAID status.
Data on coprescribed PPI therapy were obtained through manual EHR review of identified NSAID users. Coprescribed PPI therapy was defined as the prescribing of a PPI at any point during NSAID therapy. Data regarding PPI use among non-NSAID users were not collected because the secondary endpoint was designed to assess PPI use only among patients coprescribed a DOAC and NSAID.
Outcomes
Bleeding events were identified through an outcomes report generated by the DOAC Dashboard based on International Classification of Diseases, Tenth Revision diagnosis codes associated with a bleeding event. The outcomes report captures diagnoses from the outpatient and inpatient care settings. Reported bleeding events were limited to patients who received a DOAC at any point in the 6 months prior to the event and excluded patients with recent DOAC initiation within 7 days of the event, as these patients are not captured on the DOAC Dashboard.
All reported bleeding events were manually reviewed in the EHR and categorized as a major or clinically relevant nonmajor bleed, according to International Society of Thrombosis and Haemostasis criteria. Validated bleeding events were then crossreferenced with the interacting drug alerts report to identify events with potentially overlapping NSAID therapy at the time of the event. Overlapping NSAID therapy was defined as the prescribing of an NSAID at any point in the 6 months prior to the event. All events with potential overlapping NSAID therapies were manually reviewed for confirmation of NSAID status at the time of the event.
The primary endpoint was a composite of any bleeding event per International Society of Thrombosis and Haemostasis criteria. The secondary endpoint evaluated the potential impact of NSAID selectivity or PPI coprescribing on the bleeding rate among the NSAID user groups.
Statistical Analysis
Analyses were performed consistent with the methods used in the ARISTOTLE and RE-LY trials. It was determined that a sample size of 504 patients, with ≥ 168 patients in each group, would provide 80% power using a 2-sided a of 0.05. HRs with 95% CIs and respective P values were calculated using a SPSS-adapted online calculator.
RESULTS
The DOAC Dashboard identified 681 patients on rivaroxaban and 3225 patients on apixaban; 72 patients on rivaroxaban (10.6%) and 300 patients on apixaban (9.3%) were NSAID users. The mean age of NSAID users was 66.9 years in the rivaroxaban group and 72.4 years in the apixaban group. The mean age of non-NSAID users was 71.5 years in the rivaroxaban group and 75.6 years in the apixaban group. No appreciable differences were observed among subgroups in body mass index, renal function, hepatic function, hemoglobin, or platelet counts, and no statistically significant differences were identified (Table 1). Antiplatelet agents identified included aspirin, clopidogrel, prasugrel, and ticagrelor. Fifteen patients (20.3%) in the rivaroxaban group and 87 patients (28.7%) in the apixaban group had concomitant antiplatelet and NSAID use. Forty-five patients on rivaroxaban (60.8%) and 170 (55.9%) on apixaban were prescribed concomitant PPI and NSAID at baseline. Among non-NSAID users, there was concomitant antiplatelet use for 265 patients (43.6%) in the rivaroxaban group and 1401 patients (47.9%) in the apixaban group. Concomitant PPI use was identified among 63 patients (60.0%) taking selective NSAIDs and 182 (57.2%) taking nonselective NSAIDs.
A total of 423 courses of NSAIDs were identified: 85 NSAID courses in the rivaroxaban group and 338 NSAID courses in the apixaban group. Most NSAID courses involved a nonselective NSAID in the rivaroxaban and apixaban NSAID user groups: 75.2% (n = 318) aggregately compared to 71.8% (n = 61) and 76.0% (n = 257) in the rivaroxaban and apixaban groups, respectively. The most frequent NSAID courses identified were meloxicam (26.7%; n = 113), celecoxib (24.8%; n = 105), ibuprofen (19.1%; n = 81), and naproxen (13.5%; n = 57). Data regarding NSAID therapy initiation and discontinuation dates were not readily available. As a result, the duration of NSAID courses was not captured.
There was no statistically significant difference in bleeding rates between rivaroxaban and apixaban among NSAID users (HR 1.04; 95% CI, 0.98-1.12) or non-NSAID users (HR 1.15; 95% CI, 0.80-1.66) (Table 2). Apixaban non-NSAID users had a higher rate of major bleeds (HR 0.32; 95% CI, 0.17-0.61) while rivaroxaban non-NSAID users had a higher rate of clinically relevant nonmajor bleeds (HR 1.63; 95% CI, 1.10-2.54).
The sample size for the secondary endpoint consisted of bleeding events that were confirmed to have had an overlapping NSAID prescribed at the time of the event. For this secondary assessment, there was 1 rivaroxaban NSAID user bleeding event and 4 apixaban NSAID user bleeding events. For the rivaroxaban NSAID user bleeding event, the NSAID was nonselective and a PPI was not coprescribed. For the apixaban NSAID user bleeding events, 2 NSAIDs were nonselective and 2 were selective. All patients with apixaban and NSAID bleeding events had a coprescribed PPI. There was no clinically significant difference in the bleeding rates observed for NSAID selectivity or PPI coprescribing among the NSAID user subgroups.
DISCUSSION
This study found that there was no statistically significant difference for bleeding rates of major and nonmajor bleeding events between rivaroxaban and apixaban among NSAID users and non-NSAID users. This study did not identify a clinically significant impact on bleeding rates from NSAID selectivity or PPI coprescribing among the NSAID users.
There were notable but not statistically significant differences in baseline characteristics observed between the NSAID and non-NSAID user groups. On average, the rivaroxaban and apixaban NSAID users were younger compared with those not taking NSAIDs. NSAIDs, specifically nonselective NSAIDs, are recognized as potentially inappropriate medications for older adults given that this population is at an increased risk for GI ulcer development and/or GI bleeding.17 The non-NSAID user group likely consisted of older patients compared to the NSAID user group as clinicians may avoid prescribing NSAIDs to older adults regardless of concomitant DOAC therapy.
In addition to having an older patient population, non-NSAID users were more frequently prescribed a concomitant antiplatelet when compared with NSAID users. This prescribing pattern may be due to clinicians avoiding the use of NSAIDs in patients receiving DOAC therapy in combination with antiplatelet therapy, as these patients have been found to have an increased bleeding rate compared to DOAC therapy alone.18
Non-NSAID users had an overall higher bleeding rate for both major and nonmajor bleeding events. Based on this observation, it could be hypothesized that antiplatelet agents have a higher risk of bleeding in comparison to NSAIDs. In a subanalysis of the EXPAND study evaluating risk factors of major bleeding in patients receiving rivaroxaban, concomitant use of antiplatelet agents demonstrated a statistically significant increased risk of bleeding (HR 1.6; 95% CI, 1.2-2.3; P = .003) while concomitant use of NSAIDs did not (HR 0.8; 95% CI, 0.3-2.2; P = .67).19
In assessing PPI status at baseline, a majority of both rivaroxaban and apixaban NSAID users were coprescribed a PPI. This trend aligns with current clinical guideline recommendations for the prescribing of PPI therapy for GI protection in high-risk patients, such as those on DOAC therapy and concomitant NSAID therapy.10 Given the high proportion of NSAID users coprescribed a PPI at baseline, it may be possible that the true incidence of NSAID-associated bleeding events was higher than what this study found. This observation may reflect the impact from timely implementation of risk mitigation strategies by CPPs in the anticoagulation clinic. However, this study was not constructed to assess the efficacy of PPI use in this manner.
It is important to note the patients included in this study were followed by a pharmacist in an anticoagulation clinic using the DOAC Dashboard.15 This population management tool allows CPPs to make proactive interventions when a patient taking a DOAC receives an NSAID prescription, such as recommending the coprescribing of a PPI or use of a selective NSAID.10,16 These standards of care may have contributed to an overall reduced bleeding rate among the NSAID user group and may not be reflective of private practice.
The planned analysis of this study was modeled after the posthoc analysis of the RE-LY and ARISTOTLE trials. Both trials demonstrated an increased risk of bleeding with oral anticoagulation, including DOAC and warfarin, in combination with NSAID use. However, both trials found that NSAID use in patients treated with a DOAC was not independently associated with increased bleeding events compared with warfarin.13,14 The results of this study are comparable to the RE-LY and ARISTOTLE findings that NSAID use among patients treated with rivaroxaban or apixaban did not demonstrate a statistically significant increased bleeding risk.
Studies of NSAID use in combination with DOAC therapy have been limited to patient populations consisting of both DOAC and warfarin. Evidence from these trials outlines the increased bleeding risk associated with NSAID use in combination with oral anticoagulation; however, these patient populations include those on a DOAC and warfarin.13,14,19,20 Given the limited evidence on NSAID use among DOACs alone, it is assumed NSAID use in combination with DOACs has a similar risk of bleeding as warfarin use. This may cause clinicians to automatically exclude NSAID therapy as a treatment option for patients on a DOAC who are otherwise clinically appropriate candidates, such as those with underlying inflammatory conditions. Avoiding NSAID therapy in this patient population may lead to suboptimal pain management and increase the risk of patient harm from methods such as inappropriate opioid therapy prescribing.
DOAC therapy should not be a universal limitation to the use of NSAIDs. Although the risk of bleeding with NSAID therapy is always present, deliberate NSAID prescribing in addition to the timely implementation of risk mitigation strategies may provide an avenue for safe NSAID prescribing in patients receiving a DOAC. A population health-based approach to DOAC management, such as the DOAC Dashboard, appears to be effective at preventing patient harm when NSAIDs are prescribed in conjunction with DOACs.
Limitations
The DOAC Dashboard has been shown to be effective and efficient at monitoring DOAC therapy from a population-based approach.16 Reports generated through the DOAC Dashboard provide convenient access to patient data which allows for timely interventions; however, there are limits to its use for data collection. All the data elements necessary to properly assess bleeding risk with validated tools, such as HAS-BLED (hypertension, abnormal renal/liver function, stroke, bleeding history or predisposition, labile international normalized ratio, elderly, drugs/ alcohol concomitantly), are not available on DOAC Dashboard reports. Due to this constraint, bleeding risk assessments were not conducted at baseline and this study was unable to include risk modeling. Additionally, data elements like initiation and discontinuation dates and duration of therapies were not readily available. As a result, this study was unable to incorporate time as a data point.
This was a retrospective study that relied on manual review of chart documentation to verify bleeding events, but data obtained through the DOAC Dashboard were transferred directly from the EHR.15 Bleeding events available for evaluation were restricted to those that occurred at a VA facility. Additionally, the sample size within the rivaroxaban NSAID user group did not reach the predefined sample size required to reach power and may have been too small to detect a difference if one did exist. The secondary assessment had a low sample size of NSAID user bleeding events, making it difficult to fully assess its impact on NSAID selectivity and PPI coprescribing on bleeding rates. All courses of NSAIDs were equally valued regardless of the dose or therapy duration; however, this is consistent with how NSAID use was defined in the RE-LY and ARISTOTLE trials.
CONCLUSIONS
This retrospective cohort review found no statistically significant difference in the composite bleeding rates between rivaroxaban and apixaban among NSAID users and non-NSAID users. Moreover, there was no clinically significant impact observed for bleeding rates in regard to NSAID selectivity and PPI coprescribing among NSAID users. However, coprescribing of PPI therapy to patients on a DOAC who are clinically indicated for an NSAID may reduce the risk of bleeding. Population health management tools, such as the DOAC Dashboard, may also allow clinicians to safely prescribe NSAIDs to patients on a DOAC. Further large-scale observational studies are needed to quantify the real-world risk of bleeding with concomitant NSAID use among DOACs alone and to evaluate the impact from NSAID selectivity or PPI coprescribing.
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. doi:10.1016/S0140-6736(13)62343-0
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e44S-e88S. doi:10.1378/chest.11-2292
- Eikelboom J, Merli G. Bleeding with direct oral anticoagulants vs warfarin: clinical experience. Am J Med. 2016;129(11S):S33-S40. doi:10.1016/j.amjmed.2016.06.003
- Vranckx P, Valgimigli M, Heidbuchel H. The significance of drug-drug and drug-food interactions of oral anticoagulation. Arrhythm Electrophysiol Rev. 2018;7(1):55-61. doi:10.15420/aer.2017.50.1
- Davis JS, Lee HY, Kim J, et al. Use of non-steroidal antiinflammatory drugs in US adults: changes over time and by demographic. Open Heart. 2017;4(1):e000550. doi:10.1136/openhrt-2016-000550
- Schafer AI. Effects of nonsteroidal antiinflammatory drugs on platelet function and systemic hemostasis. J Clin Pharmacol. 1995;35(3):209-219. doi:10.1002/j.1552-4604.1995.tb04050.x
- Al-Saeed A. Gastrointestinal and cardiovascular risk of nonsteroidal anti-inflammatory drugs. Oman Med J. 2011;26(6):385-391. doi:10.5001/omj.2011.101
- Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. Ann Intern Med. 1991;115(10):787-796. doi:10.7326/0003-4819-115-10-787
- Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101(4):701-710. doi:10.1111/j.1572-0241.2006.00499.x
- Freedberg DE, Kim LS, Yang YX. The risks and benefits of long-term use of proton pump inhibitors: expert review and best practice advice from the American Gastroenterological Association. Gastroenterology. 2017;152(4):706-715. doi:10.1053/j.gastro.2017.01.031
- Lamberts M, Lip GYH, Hansen ML, et al. Relation of nonsteroidal anti-inflammatory drugs to serious bleeding and thromboembolism risk in patients with atrial fibrillation receiving antithrombotic therapy: a nationwide cohort study. Ann Intern Med. 2014;161(10):690-698. doi:10.7326/M13-1581
- Villa Zapata L, Hansten PD, Panic J, et al. Risk of bleeding with exposure to warfarin and nonsteroidal anti-inflammatory drugs: a systematic review and metaanalysis. Thromb Haemost. 2020;120(7):1066-1074. doi:10.1055/s-0040-1710592
- Kent AP, Brueckmann M, Fraessdorf M, et al. Concomitant oral anticoagulant and nonsteroidal anti-inflammatory drug therapy in patients with atrial fibrillation. J Am Coll Cardiol. 2018;72(3):255-267. doi:10.1016/j.jacc.2018.04.063
- Dalgaard F, Mulder H, Wojdyla DM, et al. Patients with atrial fibrillation taking nonsteroidal antiinflammatory drugs and oral anticoagulants in the ARISTOTLE Trial. Circulation. 2020;141(1):10-20. doi:10.1161/CIRCULATIONAHA.119.041296
- Allen AL, Lucas J, Parra D, et al. Shifting the paradigm: a population health approach to the management of direct oral anticoagulants. J Am Heart Asssoc. 2021;10(24):e022758. doi:10.1161/JAHA.121.022758
- . Valencia D, Spoutz P, Stoppi J, et al. Impact of a direct oral anticoagulant population management tool on anticoagulation therapy monitoring in clinical practice. Ann Pharmacother. 2019;53(8):806-811. doi:10.1177/1060028019835843
- By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 Updated AGS Beers Criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052-2081. doi:10.1111/jgs.18372
- Kumar S, Danik SB, Altman RK, et al. Non-vitamin K antagonist oral anticoagulants and antiplatelet therapy for stroke prevention in patients with atrial fibrillation. Cardiol Rev. 2016;24(5):218-223. doi:10.1097/CRD.0000000000000088
- Sakuma I, Uchiyama S, Atarashi H, et al. Clinical risk factors of stroke and major bleeding in patients with nonvalvular atrial fibrillation under rivaroxaban: the EXPAND study sub-analysis. Heart Vessels. 2019;34(11):1839-1851. doi:10.1007/s00380-019-01425-x
- Davidson BL, Verheijen S, Lensing AWA, et al. Bleeding risk of patients with acute venous thromboembolism taking nonsteroidal anti-inflammatory drugs or aspirin. JAMA Intern Med. 2014;174(6):947-953. doi:10.1001/jamainternmed.2014.946
- Ruff CT, Giugliano RP, Braunwald E, et al. Comparison of the efficacy and safety of new oral anticoagulants with warfarin in patients with atrial fibrillation: a meta-analysis of randomised trials. Lancet. 2014;383(9921):955-962. doi:10.1016/S0140-6736(13)62343-0
- Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e44S-e88S. doi:10.1378/chest.11-2292
- Eikelboom J, Merli G. Bleeding with direct oral anticoagulants vs warfarin: clinical experience. Am J Med. 2016;129(11S):S33-S40. doi:10.1016/j.amjmed.2016.06.003
- Vranckx P, Valgimigli M, Heidbuchel H. The significance of drug-drug and drug-food interactions of oral anticoagulation. Arrhythm Electrophysiol Rev. 2018;7(1):55-61. doi:10.15420/aer.2017.50.1
- Davis JS, Lee HY, Kim J, et al. Use of non-steroidal antiinflammatory drugs in US adults: changes over time and by demographic. Open Heart. 2017;4(1):e000550. doi:10.1136/openhrt-2016-000550
- Schafer AI. Effects of nonsteroidal antiinflammatory drugs on platelet function and systemic hemostasis. J Clin Pharmacol. 1995;35(3):209-219. doi:10.1002/j.1552-4604.1995.tb04050.x
- Al-Saeed A. Gastrointestinal and cardiovascular risk of nonsteroidal anti-inflammatory drugs. Oman Med J. 2011;26(6):385-391. doi:10.5001/omj.2011.101
- Gabriel SE, Jaakkimainen L, Bombardier C. Risk for serious gastrointestinal complications related to use of nonsteroidal anti-inflammatory drugs. Ann Intern Med. 1991;115(10):787-796. doi:10.7326/0003-4819-115-10-787
- Scheiman JM, Yeomans ND, Talley NJ, et al. Prevention of ulcers by esomeprazole in at-risk patients using non-selective NSAIDs and COX-2 inhibitors. Am J Gastroenterol. 2006;101(4):701-710. doi:10.1111/j.1572-0241.2006.00499.x
- Freedberg DE, Kim LS, Yang YX. The risks and benefits of long-term use of proton pump inhibitors: expert review and best practice advice from the American Gastroenterological Association. Gastroenterology. 2017;152(4):706-715. doi:10.1053/j.gastro.2017.01.031
- Lamberts M, Lip GYH, Hansen ML, et al. Relation of nonsteroidal anti-inflammatory drugs to serious bleeding and thromboembolism risk in patients with atrial fibrillation receiving antithrombotic therapy: a nationwide cohort study. Ann Intern Med. 2014;161(10):690-698. doi:10.7326/M13-1581
- Villa Zapata L, Hansten PD, Panic J, et al. Risk of bleeding with exposure to warfarin and nonsteroidal anti-inflammatory drugs: a systematic review and metaanalysis. Thromb Haemost. 2020;120(7):1066-1074. doi:10.1055/s-0040-1710592
- Kent AP, Brueckmann M, Fraessdorf M, et al. Concomitant oral anticoagulant and nonsteroidal anti-inflammatory drug therapy in patients with atrial fibrillation. J Am Coll Cardiol. 2018;72(3):255-267. doi:10.1016/j.jacc.2018.04.063
- Dalgaard F, Mulder H, Wojdyla DM, et al. Patients with atrial fibrillation taking nonsteroidal antiinflammatory drugs and oral anticoagulants in the ARISTOTLE Trial. Circulation. 2020;141(1):10-20. doi:10.1161/CIRCULATIONAHA.119.041296
- Allen AL, Lucas J, Parra D, et al. Shifting the paradigm: a population health approach to the management of direct oral anticoagulants. J Am Heart Asssoc. 2021;10(24):e022758. doi:10.1161/JAHA.121.022758
- . Valencia D, Spoutz P, Stoppi J, et al. Impact of a direct oral anticoagulant population management tool on anticoagulation therapy monitoring in clinical practice. Ann Pharmacother. 2019;53(8):806-811. doi:10.1177/1060028019835843
- By the 2023 American Geriatrics Society Beers Criteria® Update Expert Panel. American Geriatrics Society 2023 Updated AGS Beers Criteria® for potentially inappropriate medication use in older adults. J Am Geriatr Soc. 2023;71(7):2052-2081. doi:10.1111/jgs.18372
- Kumar S, Danik SB, Altman RK, et al. Non-vitamin K antagonist oral anticoagulants and antiplatelet therapy for stroke prevention in patients with atrial fibrillation. Cardiol Rev. 2016;24(5):218-223. doi:10.1097/CRD.0000000000000088
- Sakuma I, Uchiyama S, Atarashi H, et al. Clinical risk factors of stroke and major bleeding in patients with nonvalvular atrial fibrillation under rivaroxaban: the EXPAND study sub-analysis. Heart Vessels. 2019;34(11):1839-1851. doi:10.1007/s00380-019-01425-x
- Davidson BL, Verheijen S, Lensing AWA, et al. Bleeding risk of patients with acute venous thromboembolism taking nonsteroidal anti-inflammatory drugs or aspirin. JAMA Intern Med. 2014;174(6):947-953. doi:10.1001/jamainternmed.2014.946
Impact of NSAID Use on Bleeding Rates for Patients Taking Rivaroxaban or Apixaban
Impact of NSAID Use on Bleeding Rates for Patients Taking Rivaroxaban or Apixaban
Skin Cancer Risk Elevated Among Blood, Marrow Transplant Survivors
TOPLINE:
with a cumulative incidence of 27.4% over 30 years, according to the results of a cohort study.
METHODOLOGY:
- The retrospective cohort study included 3880 BMT survivors (median age, 44 years; 55.8% men; 4.9% Black, 12.1 Hispanic, and 74.7% non-Hispanic White individuals) who underwent transplant between 1974 to 2014.
- Participants completed the BMT Survivor Study survey and were followed up for a median of 9.5 years.
- The primary outcomes were the development of subsequent cutaneous malignant neoplasms (BCC, SCC, or melanoma).
TAKEAWAY:
- The 30-year cumulative incidence of any cutaneous malignant neoplasm was 27.4% — 18% for BCC, 9.8% for SCC, and 3.7% for melanoma.
- A higher risk for skin cancer was reported for patients aged 50 years or more (subdistribution hazard ratio [SHR], 2.23; 95% CI, 1.83-2.71), and men (SHR, 1.40; 95% CI, 1.18-1.65).
- Allogeneic BMT with chronic graft-vs-host disease (cGVHD) increased the risk for skin cancer (SHR, 1.84; 95% CI, 1.37-2.47), compared with autologous BMT, while post-BMT immunosuppression increased risk for all types (overall SHR, 1.53; 95% CI, 1.26-1.86).
- The risk for any skin cancer was significantly lower in Black individuals (SHR, 0.14; 95% CI, 0.05-0.37), Hispanic individuals (SHR, 0.29; 95%CI, 0.20-0.62), and patients of other races or who were multiracial (SHR, 0.22; 95% CI, 0.13-0.37) than in non-Hispanic White patients.
IN PRACTICE:
In the study, “risk factors for post-BMT cutaneous malignant neoplasms included pretransplant treatment with a monoclonal antibody, cGVHD, and posttransplant immunosuppression,” the authors wrote, adding that the findings “could inform targeted surveillance of BMT survivors.” Most BMT survivors, “do not undergo routine dermatologic surveillance, highlighting the need to understand risk factors and incorporate risk-informed dermatologic surveillance into survivorship care plans.”
SOURCE:
The study was led by Kristy K. Broman, MD, MPH, University of Alabama at Birmingham, and was published online on December 18 in JAMA Dermatology.
LIMITATIONS:
Limitations included self-reported data and possible underreporting of melanoma cases in the SEER database. Additionally, the study did not capture other risk factors for cutaneous malignant neoplasms such as skin phototype, ultraviolet light exposure, or family history. The duration of posttransplant immunosuppression was not collected, and surveys were administered at variable intervals, though all were completed more than 2 years post BMT.
DISCLOSURES:
The study was supported by the National Cancer Institute (NCI) and the Leukemia and Lymphoma Society. Broman received grants from NCI, the National Center for Advancing Translational Sciences, the American Society of Clinical Oncology, and the American College of Surgeons. Another author reported receiving grants outside this work.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
with a cumulative incidence of 27.4% over 30 years, according to the results of a cohort study.
METHODOLOGY:
- The retrospective cohort study included 3880 BMT survivors (median age, 44 years; 55.8% men; 4.9% Black, 12.1 Hispanic, and 74.7% non-Hispanic White individuals) who underwent transplant between 1974 to 2014.
- Participants completed the BMT Survivor Study survey and were followed up for a median of 9.5 years.
- The primary outcomes were the development of subsequent cutaneous malignant neoplasms (BCC, SCC, or melanoma).
TAKEAWAY:
- The 30-year cumulative incidence of any cutaneous malignant neoplasm was 27.4% — 18% for BCC, 9.8% for SCC, and 3.7% for melanoma.
- A higher risk for skin cancer was reported for patients aged 50 years or more (subdistribution hazard ratio [SHR], 2.23; 95% CI, 1.83-2.71), and men (SHR, 1.40; 95% CI, 1.18-1.65).
- Allogeneic BMT with chronic graft-vs-host disease (cGVHD) increased the risk for skin cancer (SHR, 1.84; 95% CI, 1.37-2.47), compared with autologous BMT, while post-BMT immunosuppression increased risk for all types (overall SHR, 1.53; 95% CI, 1.26-1.86).
- The risk for any skin cancer was significantly lower in Black individuals (SHR, 0.14; 95% CI, 0.05-0.37), Hispanic individuals (SHR, 0.29; 95%CI, 0.20-0.62), and patients of other races or who were multiracial (SHR, 0.22; 95% CI, 0.13-0.37) than in non-Hispanic White patients.
IN PRACTICE:
In the study, “risk factors for post-BMT cutaneous malignant neoplasms included pretransplant treatment with a monoclonal antibody, cGVHD, and posttransplant immunosuppression,” the authors wrote, adding that the findings “could inform targeted surveillance of BMT survivors.” Most BMT survivors, “do not undergo routine dermatologic surveillance, highlighting the need to understand risk factors and incorporate risk-informed dermatologic surveillance into survivorship care plans.”
SOURCE:
The study was led by Kristy K. Broman, MD, MPH, University of Alabama at Birmingham, and was published online on December 18 in JAMA Dermatology.
LIMITATIONS:
Limitations included self-reported data and possible underreporting of melanoma cases in the SEER database. Additionally, the study did not capture other risk factors for cutaneous malignant neoplasms such as skin phototype, ultraviolet light exposure, or family history. The duration of posttransplant immunosuppression was not collected, and surveys were administered at variable intervals, though all were completed more than 2 years post BMT.
DISCLOSURES:
The study was supported by the National Cancer Institute (NCI) and the Leukemia and Lymphoma Society. Broman received grants from NCI, the National Center for Advancing Translational Sciences, the American Society of Clinical Oncology, and the American College of Surgeons. Another author reported receiving grants outside this work.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
with a cumulative incidence of 27.4% over 30 years, according to the results of a cohort study.
METHODOLOGY:
- The retrospective cohort study included 3880 BMT survivors (median age, 44 years; 55.8% men; 4.9% Black, 12.1 Hispanic, and 74.7% non-Hispanic White individuals) who underwent transplant between 1974 to 2014.
- Participants completed the BMT Survivor Study survey and were followed up for a median of 9.5 years.
- The primary outcomes were the development of subsequent cutaneous malignant neoplasms (BCC, SCC, or melanoma).
TAKEAWAY:
- The 30-year cumulative incidence of any cutaneous malignant neoplasm was 27.4% — 18% for BCC, 9.8% for SCC, and 3.7% for melanoma.
- A higher risk for skin cancer was reported for patients aged 50 years or more (subdistribution hazard ratio [SHR], 2.23; 95% CI, 1.83-2.71), and men (SHR, 1.40; 95% CI, 1.18-1.65).
- Allogeneic BMT with chronic graft-vs-host disease (cGVHD) increased the risk for skin cancer (SHR, 1.84; 95% CI, 1.37-2.47), compared with autologous BMT, while post-BMT immunosuppression increased risk for all types (overall SHR, 1.53; 95% CI, 1.26-1.86).
- The risk for any skin cancer was significantly lower in Black individuals (SHR, 0.14; 95% CI, 0.05-0.37), Hispanic individuals (SHR, 0.29; 95%CI, 0.20-0.62), and patients of other races or who were multiracial (SHR, 0.22; 95% CI, 0.13-0.37) than in non-Hispanic White patients.
IN PRACTICE:
In the study, “risk factors for post-BMT cutaneous malignant neoplasms included pretransplant treatment with a monoclonal antibody, cGVHD, and posttransplant immunosuppression,” the authors wrote, adding that the findings “could inform targeted surveillance of BMT survivors.” Most BMT survivors, “do not undergo routine dermatologic surveillance, highlighting the need to understand risk factors and incorporate risk-informed dermatologic surveillance into survivorship care plans.”
SOURCE:
The study was led by Kristy K. Broman, MD, MPH, University of Alabama at Birmingham, and was published online on December 18 in JAMA Dermatology.
LIMITATIONS:
Limitations included self-reported data and possible underreporting of melanoma cases in the SEER database. Additionally, the study did not capture other risk factors for cutaneous malignant neoplasms such as skin phototype, ultraviolet light exposure, or family history. The duration of posttransplant immunosuppression was not collected, and surveys were administered at variable intervals, though all were completed more than 2 years post BMT.
DISCLOSURES:
The study was supported by the National Cancer Institute (NCI) and the Leukemia and Lymphoma Society. Broman received grants from NCI, the National Center for Advancing Translational Sciences, the American Society of Clinical Oncology, and the American College of Surgeons. Another author reported receiving grants outside this work.
This article was created using several editorial tools, including artificial intelligence, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Iron Overload: The Silent Bone Breaker
TOPLINE:
Patients with serum ferritin levels higher than 1000 μg/L show a 91% increased risk for any fracture, with a doubled risk for vertebral and humerus fractures compared with those without iron overload.
METHODOLOGY:
- Iron overload’s association with decreased bone mineral density is established, but its relationship to osteoporotic fracture risk has remained understudied and inconsistent across fracture sites.
- Researchers conducted a population-based cohort study using a UK general practice database to evaluate the fracture risk in 20,264 patients with iron overload and 192,956 matched controls without elevated ferritin (mean age, 57 years; about 40% women).
- Patients with iron overload were identified as those with laboratory-confirmed iron overload (serum ferritin levels > 1000 μg/L; n = 13,510) or a diagnosis of an iron overloading disorder, such as thalassemia major, sickle cell disease, or hemochromatosis (n = 6754).
- The primary outcome of interest was the first occurrence of an osteoporotic fracture after the diagnosis of iron overload or first record of high ferritin.
- A sensitivity analysis was conducted to check the impact of laboratory-confirmed iron overload on the risk for osteoporotic fracture compared with a diagnosis code without elevated ferritin.
TAKEAWAY:
- In the overall cohort, patients with iron overload had a 55% higher risk for any osteoporotic fracture than control individuals (adjusted hazard ratio [aHR], 1.55; 95% CI, 1.42-1.68), with the highest risk observed for vertebral fractures (aHR, 1.97; 95% CI, 1.63-2.37) and humerus fractures (aHR, 1.91; 95% CI, 1.61-2.26).
- Patients with laboratory-confirmed iron overload showed a 91% increased risk for any fracture (aHR, 1.91; 95% CI, 1.73-2.10), with a 2.5-fold higher risk observed for vertebral fractures (aHR, 2.51; 95% CI, 2.01-3.12), followed by humerus fractures (aHR, 2.41; 95% CI, 1.96-2.95).
- There was no increased risk for fracture at any site in patients with a diagnosis of an iron overloading disorder but no laboratory-confirmed iron overload.
- No sex-specific differences were identified in the association between iron overload and fracture risk.
IN PRACTICE:
“The main clinical message from our findings is that clinicians should consider iron overloading as a risk factor for fracture. Importantly, among high-risk patients presenting with serum ferritin values exceeding 1000 μg/L, osteoporosis screening and treatment strategies should be initiated in accordance with the guidelines for patients with hepatic disease,” the authors wrote.
SOURCE:
The study was led by Andrea Michelle Burden, PhD, Institute of Pharmaceutical Sciences, Department of Chemistry and Applied Biosciences, ETH Zürich in Switzerland, and was published online in The Journal of Clinical Endocrinology & Metabolism.
LIMITATIONS:
The study could not assess the duration of iron overload on fracture risk, and thus, patients could enter the cohort with a single elevated serum ferritin value that may not have reflected systemic iron overload. The authors also acknowledged potential exposure misclassification among matched control individuals because only 2.9% had a serum ferritin value available at baseline. Also, researchers were unable to adjust for inflammation status due to the limited availability of C-reactive protein measurements and the lack of leukocyte count data in primary care settings.
DISCLOSURES:
This study received support through grants from the German Research Foundation. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Patients with serum ferritin levels higher than 1000 μg/L show a 91% increased risk for any fracture, with a doubled risk for vertebral and humerus fractures compared with those without iron overload.
METHODOLOGY:
- Iron overload’s association with decreased bone mineral density is established, but its relationship to osteoporotic fracture risk has remained understudied and inconsistent across fracture sites.
- Researchers conducted a population-based cohort study using a UK general practice database to evaluate the fracture risk in 20,264 patients with iron overload and 192,956 matched controls without elevated ferritin (mean age, 57 years; about 40% women).
- Patients with iron overload were identified as those with laboratory-confirmed iron overload (serum ferritin levels > 1000 μg/L; n = 13,510) or a diagnosis of an iron overloading disorder, such as thalassemia major, sickle cell disease, or hemochromatosis (n = 6754).
- The primary outcome of interest was the first occurrence of an osteoporotic fracture after the diagnosis of iron overload or first record of high ferritin.
- A sensitivity analysis was conducted to check the impact of laboratory-confirmed iron overload on the risk for osteoporotic fracture compared with a diagnosis code without elevated ferritin.
TAKEAWAY:
- In the overall cohort, patients with iron overload had a 55% higher risk for any osteoporotic fracture than control individuals (adjusted hazard ratio [aHR], 1.55; 95% CI, 1.42-1.68), with the highest risk observed for vertebral fractures (aHR, 1.97; 95% CI, 1.63-2.37) and humerus fractures (aHR, 1.91; 95% CI, 1.61-2.26).
- Patients with laboratory-confirmed iron overload showed a 91% increased risk for any fracture (aHR, 1.91; 95% CI, 1.73-2.10), with a 2.5-fold higher risk observed for vertebral fractures (aHR, 2.51; 95% CI, 2.01-3.12), followed by humerus fractures (aHR, 2.41; 95% CI, 1.96-2.95).
- There was no increased risk for fracture at any site in patients with a diagnosis of an iron overloading disorder but no laboratory-confirmed iron overload.
- No sex-specific differences were identified in the association between iron overload and fracture risk.
IN PRACTICE:
“The main clinical message from our findings is that clinicians should consider iron overloading as a risk factor for fracture. Importantly, among high-risk patients presenting with serum ferritin values exceeding 1000 μg/L, osteoporosis screening and treatment strategies should be initiated in accordance with the guidelines for patients with hepatic disease,” the authors wrote.
SOURCE:
The study was led by Andrea Michelle Burden, PhD, Institute of Pharmaceutical Sciences, Department of Chemistry and Applied Biosciences, ETH Zürich in Switzerland, and was published online in The Journal of Clinical Endocrinology & Metabolism.
LIMITATIONS:
The study could not assess the duration of iron overload on fracture risk, and thus, patients could enter the cohort with a single elevated serum ferritin value that may not have reflected systemic iron overload. The authors also acknowledged potential exposure misclassification among matched control individuals because only 2.9% had a serum ferritin value available at baseline. Also, researchers were unable to adjust for inflammation status due to the limited availability of C-reactive protein measurements and the lack of leukocyte count data in primary care settings.
DISCLOSURES:
This study received support through grants from the German Research Foundation. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
TOPLINE:
Patients with serum ferritin levels higher than 1000 μg/L show a 91% increased risk for any fracture, with a doubled risk for vertebral and humerus fractures compared with those without iron overload.
METHODOLOGY:
- Iron overload’s association with decreased bone mineral density is established, but its relationship to osteoporotic fracture risk has remained understudied and inconsistent across fracture sites.
- Researchers conducted a population-based cohort study using a UK general practice database to evaluate the fracture risk in 20,264 patients with iron overload and 192,956 matched controls without elevated ferritin (mean age, 57 years; about 40% women).
- Patients with iron overload were identified as those with laboratory-confirmed iron overload (serum ferritin levels > 1000 μg/L; n = 13,510) or a diagnosis of an iron overloading disorder, such as thalassemia major, sickle cell disease, or hemochromatosis (n = 6754).
- The primary outcome of interest was the first occurrence of an osteoporotic fracture after the diagnosis of iron overload or first record of high ferritin.
- A sensitivity analysis was conducted to check the impact of laboratory-confirmed iron overload on the risk for osteoporotic fracture compared with a diagnosis code without elevated ferritin.
TAKEAWAY:
- In the overall cohort, patients with iron overload had a 55% higher risk for any osteoporotic fracture than control individuals (adjusted hazard ratio [aHR], 1.55; 95% CI, 1.42-1.68), with the highest risk observed for vertebral fractures (aHR, 1.97; 95% CI, 1.63-2.37) and humerus fractures (aHR, 1.91; 95% CI, 1.61-2.26).
- Patients with laboratory-confirmed iron overload showed a 91% increased risk for any fracture (aHR, 1.91; 95% CI, 1.73-2.10), with a 2.5-fold higher risk observed for vertebral fractures (aHR, 2.51; 95% CI, 2.01-3.12), followed by humerus fractures (aHR, 2.41; 95% CI, 1.96-2.95).
- There was no increased risk for fracture at any site in patients with a diagnosis of an iron overloading disorder but no laboratory-confirmed iron overload.
- No sex-specific differences were identified in the association between iron overload and fracture risk.
IN PRACTICE:
“The main clinical message from our findings is that clinicians should consider iron overloading as a risk factor for fracture. Importantly, among high-risk patients presenting with serum ferritin values exceeding 1000 μg/L, osteoporosis screening and treatment strategies should be initiated in accordance with the guidelines for patients with hepatic disease,” the authors wrote.
SOURCE:
The study was led by Andrea Michelle Burden, PhD, Institute of Pharmaceutical Sciences, Department of Chemistry and Applied Biosciences, ETH Zürich in Switzerland, and was published online in The Journal of Clinical Endocrinology & Metabolism.
LIMITATIONS:
The study could not assess the duration of iron overload on fracture risk, and thus, patients could enter the cohort with a single elevated serum ferritin value that may not have reflected systemic iron overload. The authors also acknowledged potential exposure misclassification among matched control individuals because only 2.9% had a serum ferritin value available at baseline. Also, researchers were unable to adjust for inflammation status due to the limited availability of C-reactive protein measurements and the lack of leukocyte count data in primary care settings.
DISCLOSURES:
This study received support through grants from the German Research Foundation. The authors declared no conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
New Gel Stops Severe Bleeding in Seconds
This video transcript has been edited for clarity.
Robert D. Glatter, MD: Hi and welcome. I’m Dr. Robert Glatter, medical adviser for Medscape Emergency Medicine. Joining me today to discuss a novel, plant-based approach to stopping moderate to severe bleeding is Joe Landolina, CEO and cofounder of Cresilon. Welcome, Joe.
Joe Landolina, MS: Thank you so much for taking the time. It’s great to be here.
Educational Background and Inception of Cresilon
Glatter: It’s a pleasure to have you join me, and I want to congratulate you on your recent 510(k) FDA clearance for your novel product to save lives and stop bleeding. To begin with, can you explain how the idea for launching your company came about?
Landolina: The way that Cresilon came about was a little bit unorthodox, because I was 17 years old when I invented the technology behind the product that eventually became Traumagel®.
My grandfather was an ex-pharmaceutical executive, who later in life started a vineyard. I grew up on a vineyard with a winery chemistry lab across the street from my house and a grandfather who learned lab safety in the 60s. So, that meant that the day I learned how to walk, I was tossed into a lab and I fell head over heels in love with lab research.
That started experimentation and my academic pursuits. That led to discovering a blend of two plant-based polymers derived from algae that stop bleeding on contact, effectively creating a mechanical barrier and allowing anything from a gunshot wound to anything quite a bit more minor to stop in a matter of seconds.
Glatter: Your background is in biomedical engineering. How is it that you started tinkering and doing all this type of work?
Landolina: That’s correct. I did my undergrad in chemical engineering, and my graduate studies were in biomedical engineering. For me, that was supposed to be a pathway into medical school. I always wanted to be a surgeon myself, and I love the field of medicine.
As a freshman in college at NYU Engineering, I had this idea. I entered it into NYU’s business plan competition, and we won at the engineering school. That gave us just enough capital to start developing and researching Traumagel more, and Cresilon was born out of that research.
Techniques for Stopping Hemorrhage
Glatter: In terms of stopping hemorrhage, which takes so many lives in the United States and globally — certainly, uncontrolled hemorrhage — what are the techniques that you see, prior to the arrival of your product, as being effective? Can you elucidate some of these techniques?
Landolina: In emergency medicine, the primary mode of controlling hemorrhage is passive. It’s what, in Brooklyn, we like to call “pressure and a prayer”, where you have a material that’s either gauze or an impregnated gauze in most cases, where the mode of action is absorbing blood, with the adjunct of pressure by the first responder or by the clinician who’s providing aid.
These types of technologies are widespread. There are many versions of this technology carried by EMS agencies, trauma bays, US military soldiers, and soldiers across NATO countries. But these types of technologies tend to be relatively inefficient, meaning that they’re very difficult to get into wounds because of the gauze or the powder form of the devices, and it’s very hard to get them in contact with the form of bleeding.
On top of that, if the patient is clotting compromised or immunocompromised in some way, the ability to create a durable clot that will not be ripped off when you remove the product at the next level of care is also of concern. And so, this type of technology or the type of treatment of massive hemorrhage hasn’t changed in decades.
Current Applications and Potential Use
Glatter: I envision this product will be carried by paramedics, used on the battlefield at some point after your FDA clearance, and recently it went through.
Do you see any possibility that this could be an AED equivalent to Stop the Bleed? In other words, could the average lay person be trained to use your product if kits are available?
Landolina: To be very clear, Traumagel today is only approved or cleared under a “prescription-only” indication, which means that it will not initially be available OTC. However, that is our goal. Our goal is to make this product available and usable by someone with no medical training whatsoever.
The form factor of being a gel in a syringe lends itself well to that, meaning that we try to make it as easy as point and shoot to control hemorrhage, where there’s not as much technique to be learned in the application of a product like Traumagel as there is in current hemorrhage control techniques.
Mechanism of Action and Physiology
Glatter: Once you apply Traumagel, can you explain what happens to the product after it’s applied and the bleeding has stopped? Does it get reabsorbed by the body? What’s the process here?
Landolina: Under Traumagel’s indication, because it’s used in traumatic injury, it must be removed within 24 hours.
One of the big benefits of Traumagel is that when the patient produces a blood clot underneath Traumagel, it doesn’t become incorporated within the gel itself. To contrast that with the use of gauze, gauze is porous. The clot ends up wrapped around the fibers of the gauze, so if you peel the gauze away, it’s very likely that clot is coming off with it. The surgeon or the clinician at the next level of care is going to have to deal with the re-bleed.
You can remove Traumagel cleanly and entirely without disturbing the underlying clot. That’s a major benefit, not only to the patient but also to the next level of care, to the next clinician or physician that is required to remove the product.
Glatter: How is it possible to remove the substance without disturbing the clot? Can you explain in more detail?
Landolina: That’s one of the hallmarks of these plant-based polymers and the way that we design Traumagel itself. Traumagel is completely nonporous, and it has no fibrous nature to it. What that means is when the patient produces a blood clot or fibrin next to or on top of Traumagel, that fibrin ends up not incorporated within the polymers of Traumagel itself.
Over time, because Traumagel is a hydrogel, meaning that by weight it’s mostly water, you end up having less adhesion to the clot over time. When it’s time to remove Traumagel from the injury, it has lost almost all of its adhesive capabilities, meaning that when you peel it away, that clot is going to stick better to tissue than it will to the gel itself.
Glatter: Can you explain a little bit about the matrix that’s formed, the physiology, and how the polymers work to form this matrix?
Landolina: Sure. Traumagel is made of two polysaccharides that are plant derived. One polysaccharide is polyanionic, and the other is polycationic, meaning one has negative charges and the other has positive charges, which together create almost a Lego block effect, where when the material comes in contact with tissue, it adheres strongly and allows for itself to effectively create a mechanical barrier against bleeding.
Courtesy of Cresilon
Landolina: Even in the face of major arterial blood flow, Traumagel will stay where it needs to stay, and it’s not going to get washed away. This means that it is much more easily appliable to these types of surfaces and will allow the patient to produce their own endogenous fibrin clot at that location.
Like I mentioned before, when that fibrin clot is formed, because the gel itself has no pores or fibers, it doesn’t become incorporated within the fibrin clot. You can take the gel away, leaving that clot behind without the chance of a rebleed.
Testing With Major Bleeds
Glatter: In terms of bleeding itself, have you tested your product with major aortic bleeds or carotid bleeds in preclinical work?
Landolina: We have used the US military’s model for lethal hemorrhage, and the idea there is to create a model that is just that — lethal. These are the worst types of bleeds that you can possibly imagine, where the patients are clotting compromised, and where you have, in most cases, a very strong arterial component, so something like a femoral artery bleed.
We’ve also tested in carotid artery, aortic applications, as well as combinations of venous and arterial bleeds. The idea here is to show the use of the product in the absolute worst-case scenario so that when this translates into the clinic, the models that we’ve used for evaluation, hopefully, are worse than what actually rolls into the trauma bay.
Glatter: Excellent. What’s the mean time to stop an arterial vs a venous bleed? Are we talking a matter of seconds?
Landolina: In the case of a healthy patient, meaning a patient without clotting compromise, you’re in a matter of seconds. It’s less than 10 seconds.
In the case where you have clotting compromise, a deep, complicated wound geometry, we recommend holding a pressure bandage on for 3 minutes just because it increases the chance of Traumagel coming into contact with the bleed, especially when you can’t visualize the bleed in the bleed source. Because of that pressure time, that becomes the mean. But again, it’s highly dependent on the type of bleed and the style of application.
Failure Rates and Effectiveness
Glatter: As a segue to that, what is the failure rate based on your studies and internal research using Traumagel? Have there been cases where bleeding has not been able to be stopped?
Landolina: It depends on the study, but the failure rates are incredibly low with Traumagel, assuming that it’s correctly used. That’s one of the benefits to this product, where with proper technique, with overwrap with gauze, you nearly always get control of hemorrhage with a product like this.
Glatter: Is manual pressure required in that sense? From what you described earlier, manual pressure would not be required.
Landolina: It depends on the injury. What we recommend is that, if you have a very deep wound where you cannot visualize the source of bleed, you use pressure to seat Traumagel into the source of bleeding, meaning that you’re following Committee on Tactical Combat Casualty Care (Co-TCCC) regulations or requirements, where you’re over wrapping with gauze, and you’re providing a pressure wrapping to ensure that the Traumagel is in contact with the bleed while it’s doing what it’s doing.
In most cases, it doesn’t hurt to apply pressure on top of Traumagel as well. In more surface level bleeds, you don’t need pressure at all.
Applications Beyond Trauma
Glatter: Interesting. In terms of further applications (eg, nose bleeds or GYN bleeding, which are life-threatening), do you see this coming as an application for the future?
Landolina: That’s where we’re working. Traumagel is the successor to an animal health product called Vetigel. The formulations of the gel behind Vetigel and Traumagel are identical. Vetigel has a full surgical indication, and that’s everything from epistaxis to neuro and spine procedures, into cardiovascular and soft tissue surgeries, orthopedic medicine, and so on.
Cresilon’s goal is to eventually expand the indication of our technology to include surgical indications and other indications where we can help any patient that’s bleeding.
Glatter: That’s important, because we use prehospital whole blood, low titer, specifically, when patients have life-threatening hemorrhage. With your product, that would reduce the amount of blood products that would need to be administered. This could be a real game changer.
Landolina: Definitely, that’s the goal we’re working on.
Infection Risks and Biocompatibility
Glatter: In terms of any risk for infection, has that been studied as well? Does Traumagel in any way lead to increased rates of infection?
Landolina: Traumagel is biocompatible. It’s a sterile product. We’ve done the full suite of biocompatibility testing as required by FDA. On top of that, remember that Vetigel, which is the same formulation, is an implantable product. As a result, that has even extended biocompatibility testing beyond what would be necessary for an external product.
In Vetigel’s use case, which has been used now in over 60,000 patients, primarily companion animals, dogs and cats, we haven’t seen instances of infection. There’s no reason to believe that we would see that clinically with Traumagel.
Research Collaborations and Future Applications
Glatter: In terms of other research that your company’s embarked on preclinically, I understand there were some studies done at Walter Reed Army Institute of Research. I was wondering if you could expand on these, specifically, in terms of traumatic brain injury (TBI) and hemorrhage related to that. For example, with shrapnel or even a gunshot wound.
Landolina: The Walter Reed collaboration with Cresilon is something that I’m particularly excited about, because it marks Cresilon’s first project that’s outside the scope of just hemostasis. Walter Reed came to us with this proposal where there’s a big challenge in a subset of TBI called penetrating ballistic-like brain injury, where the brain has been penetrated by a bullet, shrapnel, or some other projectile, and there’s an injury that exposes the brain to the outside.
Today, there is no standard of care to treat patients with those types of injuries. In many cases, mortality is caused through swelling of the brain, or collapse of the brain. What they came to us with was the potential of using our technology, not primarily as a hemostatic agent, but to be able to stabilize that patient enough to get to the next level of care to be treated by a neurosurgeon.
That study Walter Reed did was just a pilot that was done in small animals. In that pilot, they showed that over the period of treatment, there was no negative change in vital signs, no increase in edema or in swelling, or in any of the biomarkers that were being monitored at that time.
At the very least, this is not full indication that this indication will work for Cresilon, but it shows that there’s promise. It’s something that we’re working on and hopefully we’ll be able to bring to market soon.
Glatter: Certainly, maintaining intracranial pressure and cerebral perfusion pressures are very critical. In the future, do you think this product would be able to be deployed endovascularly? Imagine this in terms of stopping bleeding from some source, whether it’s from a stroke or another intracranial source.
Landolina: That’s been an area of interest for us. We have no evidence to prove that indication works at this point, but there’s also nothing to say that it wouldn’t be possible for our technology. At this point, we’ve only looked at a cursory level at those indications.
Glatter: Does the use of Traumagel obviate the need for a more definitive repair (eg, with sutures) or something that’s more permanent?
Landolina: I always say that Traumagel — and Vetigel, for that matter — is not a replacement for good surgical technique. The surgeon always needs to make his or her best judgment when reviewing the patient. That doesn’t mean that there won’t need to be sutures or vascular repair in most of these cases, especially in major trauma.
Final Takeaways
Glatter: Do you have some bullet points or pearls you could give our audience as a takeaway?
Landolina: When Cresilon looks at Traumagel — and for us, Traumagel is the next generation of hemostatic agent, especially in trauma care and in emergency medicine — it allows for a far-simplified application of the product and much faster control of hemorrhage with better patient outcomes.
As we roll this out through EMS agencies, trauma hospitals, military agencies, and eventually to the general public through a future indication, it’s something we’re very excited about. Personally, I started this business 14 years ago, and so it’s great to see our mission of saving lives transitioning to saving human lives.
Glatter: I look forward to seeing this product in the emergency department, but also in other settings, such as in the operating room where we can really help patients who are dying from hemorrhage, certainly on the battlefield, and the lay public. If someone were to come upon a patient who’s bleeding out, this could be certainly a game changer and a lifesaver.
I want to thank you for your time. This is a really important product that’s transformed the lives of so many animals, but also people in the future.
Dr. Glatter is an assistant professor of emergency medicine at Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York. He reported no relevant conflicts of interest. Mr. Landolina is the CEO and co-founder of Cresilon, a biotechnology company specializing in plant-based solutions for emergency bleeding control.
A version of this article first appeared on Medscape.com.
This video transcript has been edited for clarity.
Robert D. Glatter, MD: Hi and welcome. I’m Dr. Robert Glatter, medical adviser for Medscape Emergency Medicine. Joining me today to discuss a novel, plant-based approach to stopping moderate to severe bleeding is Joe Landolina, CEO and cofounder of Cresilon. Welcome, Joe.
Joe Landolina, MS: Thank you so much for taking the time. It’s great to be here.
Educational Background and Inception of Cresilon
Glatter: It’s a pleasure to have you join me, and I want to congratulate you on your recent 510(k) FDA clearance for your novel product to save lives and stop bleeding. To begin with, can you explain how the idea for launching your company came about?
Landolina: The way that Cresilon came about was a little bit unorthodox, because I was 17 years old when I invented the technology behind the product that eventually became Traumagel®.
My grandfather was an ex-pharmaceutical executive, who later in life started a vineyard. I grew up on a vineyard with a winery chemistry lab across the street from my house and a grandfather who learned lab safety in the 60s. So, that meant that the day I learned how to walk, I was tossed into a lab and I fell head over heels in love with lab research.
That started experimentation and my academic pursuits. That led to discovering a blend of two plant-based polymers derived from algae that stop bleeding on contact, effectively creating a mechanical barrier and allowing anything from a gunshot wound to anything quite a bit more minor to stop in a matter of seconds.
Glatter: Your background is in biomedical engineering. How is it that you started tinkering and doing all this type of work?
Landolina: That’s correct. I did my undergrad in chemical engineering, and my graduate studies were in biomedical engineering. For me, that was supposed to be a pathway into medical school. I always wanted to be a surgeon myself, and I love the field of medicine.
As a freshman in college at NYU Engineering, I had this idea. I entered it into NYU’s business plan competition, and we won at the engineering school. That gave us just enough capital to start developing and researching Traumagel more, and Cresilon was born out of that research.
Techniques for Stopping Hemorrhage
Glatter: In terms of stopping hemorrhage, which takes so many lives in the United States and globally — certainly, uncontrolled hemorrhage — what are the techniques that you see, prior to the arrival of your product, as being effective? Can you elucidate some of these techniques?
Landolina: In emergency medicine, the primary mode of controlling hemorrhage is passive. It’s what, in Brooklyn, we like to call “pressure and a prayer”, where you have a material that’s either gauze or an impregnated gauze in most cases, where the mode of action is absorbing blood, with the adjunct of pressure by the first responder or by the clinician who’s providing aid.
These types of technologies are widespread. There are many versions of this technology carried by EMS agencies, trauma bays, US military soldiers, and soldiers across NATO countries. But these types of technologies tend to be relatively inefficient, meaning that they’re very difficult to get into wounds because of the gauze or the powder form of the devices, and it’s very hard to get them in contact with the form of bleeding.
On top of that, if the patient is clotting compromised or immunocompromised in some way, the ability to create a durable clot that will not be ripped off when you remove the product at the next level of care is also of concern. And so, this type of technology or the type of treatment of massive hemorrhage hasn’t changed in decades.
Current Applications and Potential Use
Glatter: I envision this product will be carried by paramedics, used on the battlefield at some point after your FDA clearance, and recently it went through.
Do you see any possibility that this could be an AED equivalent to Stop the Bleed? In other words, could the average lay person be trained to use your product if kits are available?
Landolina: To be very clear, Traumagel today is only approved or cleared under a “prescription-only” indication, which means that it will not initially be available OTC. However, that is our goal. Our goal is to make this product available and usable by someone with no medical training whatsoever.
The form factor of being a gel in a syringe lends itself well to that, meaning that we try to make it as easy as point and shoot to control hemorrhage, where there’s not as much technique to be learned in the application of a product like Traumagel as there is in current hemorrhage control techniques.
Mechanism of Action and Physiology
Glatter: Once you apply Traumagel, can you explain what happens to the product after it’s applied and the bleeding has stopped? Does it get reabsorbed by the body? What’s the process here?
Landolina: Under Traumagel’s indication, because it’s used in traumatic injury, it must be removed within 24 hours.
One of the big benefits of Traumagel is that when the patient produces a blood clot underneath Traumagel, it doesn’t become incorporated within the gel itself. To contrast that with the use of gauze, gauze is porous. The clot ends up wrapped around the fibers of the gauze, so if you peel the gauze away, it’s very likely that clot is coming off with it. The surgeon or the clinician at the next level of care is going to have to deal with the re-bleed.
You can remove Traumagel cleanly and entirely without disturbing the underlying clot. That’s a major benefit, not only to the patient but also to the next level of care, to the next clinician or physician that is required to remove the product.
Glatter: How is it possible to remove the substance without disturbing the clot? Can you explain in more detail?
Landolina: That’s one of the hallmarks of these plant-based polymers and the way that we design Traumagel itself. Traumagel is completely nonporous, and it has no fibrous nature to it. What that means is when the patient produces a blood clot or fibrin next to or on top of Traumagel, that fibrin ends up not incorporated within the polymers of Traumagel itself.
Over time, because Traumagel is a hydrogel, meaning that by weight it’s mostly water, you end up having less adhesion to the clot over time. When it’s time to remove Traumagel from the injury, it has lost almost all of its adhesive capabilities, meaning that when you peel it away, that clot is going to stick better to tissue than it will to the gel itself.
Glatter: Can you explain a little bit about the matrix that’s formed, the physiology, and how the polymers work to form this matrix?
Landolina: Sure. Traumagel is made of two polysaccharides that are plant derived. One polysaccharide is polyanionic, and the other is polycationic, meaning one has negative charges and the other has positive charges, which together create almost a Lego block effect, where when the material comes in contact with tissue, it adheres strongly and allows for itself to effectively create a mechanical barrier against bleeding.
Courtesy of Cresilon
Landolina: Even in the face of major arterial blood flow, Traumagel will stay where it needs to stay, and it’s not going to get washed away. This means that it is much more easily appliable to these types of surfaces and will allow the patient to produce their own endogenous fibrin clot at that location.
Like I mentioned before, when that fibrin clot is formed, because the gel itself has no pores or fibers, it doesn’t become incorporated within the fibrin clot. You can take the gel away, leaving that clot behind without the chance of a rebleed.
Testing With Major Bleeds
Glatter: In terms of bleeding itself, have you tested your product with major aortic bleeds or carotid bleeds in preclinical work?
Landolina: We have used the US military’s model for lethal hemorrhage, and the idea there is to create a model that is just that — lethal. These are the worst types of bleeds that you can possibly imagine, where the patients are clotting compromised, and where you have, in most cases, a very strong arterial component, so something like a femoral artery bleed.
We’ve also tested in carotid artery, aortic applications, as well as combinations of venous and arterial bleeds. The idea here is to show the use of the product in the absolute worst-case scenario so that when this translates into the clinic, the models that we’ve used for evaluation, hopefully, are worse than what actually rolls into the trauma bay.
Glatter: Excellent. What’s the mean time to stop an arterial vs a venous bleed? Are we talking a matter of seconds?
Landolina: In the case of a healthy patient, meaning a patient without clotting compromise, you’re in a matter of seconds. It’s less than 10 seconds.
In the case where you have clotting compromise, a deep, complicated wound geometry, we recommend holding a pressure bandage on for 3 minutes just because it increases the chance of Traumagel coming into contact with the bleed, especially when you can’t visualize the bleed in the bleed source. Because of that pressure time, that becomes the mean. But again, it’s highly dependent on the type of bleed and the style of application.
Failure Rates and Effectiveness
Glatter: As a segue to that, what is the failure rate based on your studies and internal research using Traumagel? Have there been cases where bleeding has not been able to be stopped?
Landolina: It depends on the study, but the failure rates are incredibly low with Traumagel, assuming that it’s correctly used. That’s one of the benefits to this product, where with proper technique, with overwrap with gauze, you nearly always get control of hemorrhage with a product like this.
Glatter: Is manual pressure required in that sense? From what you described earlier, manual pressure would not be required.
Landolina: It depends on the injury. What we recommend is that, if you have a very deep wound where you cannot visualize the source of bleed, you use pressure to seat Traumagel into the source of bleeding, meaning that you’re following Committee on Tactical Combat Casualty Care (Co-TCCC) regulations or requirements, where you’re over wrapping with gauze, and you’re providing a pressure wrapping to ensure that the Traumagel is in contact with the bleed while it’s doing what it’s doing.
In most cases, it doesn’t hurt to apply pressure on top of Traumagel as well. In more surface level bleeds, you don’t need pressure at all.
Applications Beyond Trauma
Glatter: Interesting. In terms of further applications (eg, nose bleeds or GYN bleeding, which are life-threatening), do you see this coming as an application for the future?
Landolina: That’s where we’re working. Traumagel is the successor to an animal health product called Vetigel. The formulations of the gel behind Vetigel and Traumagel are identical. Vetigel has a full surgical indication, and that’s everything from epistaxis to neuro and spine procedures, into cardiovascular and soft tissue surgeries, orthopedic medicine, and so on.
Cresilon’s goal is to eventually expand the indication of our technology to include surgical indications and other indications where we can help any patient that’s bleeding.
Glatter: That’s important, because we use prehospital whole blood, low titer, specifically, when patients have life-threatening hemorrhage. With your product, that would reduce the amount of blood products that would need to be administered. This could be a real game changer.
Landolina: Definitely, that’s the goal we’re working on.
Infection Risks and Biocompatibility
Glatter: In terms of any risk for infection, has that been studied as well? Does Traumagel in any way lead to increased rates of infection?
Landolina: Traumagel is biocompatible. It’s a sterile product. We’ve done the full suite of biocompatibility testing as required by FDA. On top of that, remember that Vetigel, which is the same formulation, is an implantable product. As a result, that has even extended biocompatibility testing beyond what would be necessary for an external product.
In Vetigel’s use case, which has been used now in over 60,000 patients, primarily companion animals, dogs and cats, we haven’t seen instances of infection. There’s no reason to believe that we would see that clinically with Traumagel.
Research Collaborations and Future Applications
Glatter: In terms of other research that your company’s embarked on preclinically, I understand there were some studies done at Walter Reed Army Institute of Research. I was wondering if you could expand on these, specifically, in terms of traumatic brain injury (TBI) and hemorrhage related to that. For example, with shrapnel or even a gunshot wound.
Landolina: The Walter Reed collaboration with Cresilon is something that I’m particularly excited about, because it marks Cresilon’s first project that’s outside the scope of just hemostasis. Walter Reed came to us with this proposal where there’s a big challenge in a subset of TBI called penetrating ballistic-like brain injury, where the brain has been penetrated by a bullet, shrapnel, or some other projectile, and there’s an injury that exposes the brain to the outside.
Today, there is no standard of care to treat patients with those types of injuries. In many cases, mortality is caused through swelling of the brain, or collapse of the brain. What they came to us with was the potential of using our technology, not primarily as a hemostatic agent, but to be able to stabilize that patient enough to get to the next level of care to be treated by a neurosurgeon.
That study Walter Reed did was just a pilot that was done in small animals. In that pilot, they showed that over the period of treatment, there was no negative change in vital signs, no increase in edema or in swelling, or in any of the biomarkers that were being monitored at that time.
At the very least, this is not full indication that this indication will work for Cresilon, but it shows that there’s promise. It’s something that we’re working on and hopefully we’ll be able to bring to market soon.
Glatter: Certainly, maintaining intracranial pressure and cerebral perfusion pressures are very critical. In the future, do you think this product would be able to be deployed endovascularly? Imagine this in terms of stopping bleeding from some source, whether it’s from a stroke or another intracranial source.
Landolina: That’s been an area of interest for us. We have no evidence to prove that indication works at this point, but there’s also nothing to say that it wouldn’t be possible for our technology. At this point, we’ve only looked at a cursory level at those indications.
Glatter: Does the use of Traumagel obviate the need for a more definitive repair (eg, with sutures) or something that’s more permanent?
Landolina: I always say that Traumagel — and Vetigel, for that matter — is not a replacement for good surgical technique. The surgeon always needs to make his or her best judgment when reviewing the patient. That doesn’t mean that there won’t need to be sutures or vascular repair in most of these cases, especially in major trauma.
Final Takeaways
Glatter: Do you have some bullet points or pearls you could give our audience as a takeaway?
Landolina: When Cresilon looks at Traumagel — and for us, Traumagel is the next generation of hemostatic agent, especially in trauma care and in emergency medicine — it allows for a far-simplified application of the product and much faster control of hemorrhage with better patient outcomes.
As we roll this out through EMS agencies, trauma hospitals, military agencies, and eventually to the general public through a future indication, it’s something we’re very excited about. Personally, I started this business 14 years ago, and so it’s great to see our mission of saving lives transitioning to saving human lives.
Glatter: I look forward to seeing this product in the emergency department, but also in other settings, such as in the operating room where we can really help patients who are dying from hemorrhage, certainly on the battlefield, and the lay public. If someone were to come upon a patient who’s bleeding out, this could be certainly a game changer and a lifesaver.
I want to thank you for your time. This is a really important product that’s transformed the lives of so many animals, but also people in the future.
Dr. Glatter is an assistant professor of emergency medicine at Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York. He reported no relevant conflicts of interest. Mr. Landolina is the CEO and co-founder of Cresilon, a biotechnology company specializing in plant-based solutions for emergency bleeding control.
A version of this article first appeared on Medscape.com.
This video transcript has been edited for clarity.
Robert D. Glatter, MD: Hi and welcome. I’m Dr. Robert Glatter, medical adviser for Medscape Emergency Medicine. Joining me today to discuss a novel, plant-based approach to stopping moderate to severe bleeding is Joe Landolina, CEO and cofounder of Cresilon. Welcome, Joe.
Joe Landolina, MS: Thank you so much for taking the time. It’s great to be here.
Educational Background and Inception of Cresilon
Glatter: It’s a pleasure to have you join me, and I want to congratulate you on your recent 510(k) FDA clearance for your novel product to save lives and stop bleeding. To begin with, can you explain how the idea for launching your company came about?
Landolina: The way that Cresilon came about was a little bit unorthodox, because I was 17 years old when I invented the technology behind the product that eventually became Traumagel®.
My grandfather was an ex-pharmaceutical executive, who later in life started a vineyard. I grew up on a vineyard with a winery chemistry lab across the street from my house and a grandfather who learned lab safety in the 60s. So, that meant that the day I learned how to walk, I was tossed into a lab and I fell head over heels in love with lab research.
That started experimentation and my academic pursuits. That led to discovering a blend of two plant-based polymers derived from algae that stop bleeding on contact, effectively creating a mechanical barrier and allowing anything from a gunshot wound to anything quite a bit more minor to stop in a matter of seconds.
Glatter: Your background is in biomedical engineering. How is it that you started tinkering and doing all this type of work?
Landolina: That’s correct. I did my undergrad in chemical engineering, and my graduate studies were in biomedical engineering. For me, that was supposed to be a pathway into medical school. I always wanted to be a surgeon myself, and I love the field of medicine.
As a freshman in college at NYU Engineering, I had this idea. I entered it into NYU’s business plan competition, and we won at the engineering school. That gave us just enough capital to start developing and researching Traumagel more, and Cresilon was born out of that research.
Techniques for Stopping Hemorrhage
Glatter: In terms of stopping hemorrhage, which takes so many lives in the United States and globally — certainly, uncontrolled hemorrhage — what are the techniques that you see, prior to the arrival of your product, as being effective? Can you elucidate some of these techniques?
Landolina: In emergency medicine, the primary mode of controlling hemorrhage is passive. It’s what, in Brooklyn, we like to call “pressure and a prayer”, where you have a material that’s either gauze or an impregnated gauze in most cases, where the mode of action is absorbing blood, with the adjunct of pressure by the first responder or by the clinician who’s providing aid.
These types of technologies are widespread. There are many versions of this technology carried by EMS agencies, trauma bays, US military soldiers, and soldiers across NATO countries. But these types of technologies tend to be relatively inefficient, meaning that they’re very difficult to get into wounds because of the gauze or the powder form of the devices, and it’s very hard to get them in contact with the form of bleeding.
On top of that, if the patient is clotting compromised or immunocompromised in some way, the ability to create a durable clot that will not be ripped off when you remove the product at the next level of care is also of concern. And so, this type of technology or the type of treatment of massive hemorrhage hasn’t changed in decades.
Current Applications and Potential Use
Glatter: I envision this product will be carried by paramedics, used on the battlefield at some point after your FDA clearance, and recently it went through.
Do you see any possibility that this could be an AED equivalent to Stop the Bleed? In other words, could the average lay person be trained to use your product if kits are available?
Landolina: To be very clear, Traumagel today is only approved or cleared under a “prescription-only” indication, which means that it will not initially be available OTC. However, that is our goal. Our goal is to make this product available and usable by someone with no medical training whatsoever.
The form factor of being a gel in a syringe lends itself well to that, meaning that we try to make it as easy as point and shoot to control hemorrhage, where there’s not as much technique to be learned in the application of a product like Traumagel as there is in current hemorrhage control techniques.
Mechanism of Action and Physiology
Glatter: Once you apply Traumagel, can you explain what happens to the product after it’s applied and the bleeding has stopped? Does it get reabsorbed by the body? What’s the process here?
Landolina: Under Traumagel’s indication, because it’s used in traumatic injury, it must be removed within 24 hours.
One of the big benefits of Traumagel is that when the patient produces a blood clot underneath Traumagel, it doesn’t become incorporated within the gel itself. To contrast that with the use of gauze, gauze is porous. The clot ends up wrapped around the fibers of the gauze, so if you peel the gauze away, it’s very likely that clot is coming off with it. The surgeon or the clinician at the next level of care is going to have to deal with the re-bleed.
You can remove Traumagel cleanly and entirely without disturbing the underlying clot. That’s a major benefit, not only to the patient but also to the next level of care, to the next clinician or physician that is required to remove the product.
Glatter: How is it possible to remove the substance without disturbing the clot? Can you explain in more detail?
Landolina: That’s one of the hallmarks of these plant-based polymers and the way that we design Traumagel itself. Traumagel is completely nonporous, and it has no fibrous nature to it. What that means is when the patient produces a blood clot or fibrin next to or on top of Traumagel, that fibrin ends up not incorporated within the polymers of Traumagel itself.
Over time, because Traumagel is a hydrogel, meaning that by weight it’s mostly water, you end up having less adhesion to the clot over time. When it’s time to remove Traumagel from the injury, it has lost almost all of its adhesive capabilities, meaning that when you peel it away, that clot is going to stick better to tissue than it will to the gel itself.
Glatter: Can you explain a little bit about the matrix that’s formed, the physiology, and how the polymers work to form this matrix?
Landolina: Sure. Traumagel is made of two polysaccharides that are plant derived. One polysaccharide is polyanionic, and the other is polycationic, meaning one has negative charges and the other has positive charges, which together create almost a Lego block effect, where when the material comes in contact with tissue, it adheres strongly and allows for itself to effectively create a mechanical barrier against bleeding.
Courtesy of Cresilon
Landolina: Even in the face of major arterial blood flow, Traumagel will stay where it needs to stay, and it’s not going to get washed away. This means that it is much more easily appliable to these types of surfaces and will allow the patient to produce their own endogenous fibrin clot at that location.
Like I mentioned before, when that fibrin clot is formed, because the gel itself has no pores or fibers, it doesn’t become incorporated within the fibrin clot. You can take the gel away, leaving that clot behind without the chance of a rebleed.
Testing With Major Bleeds
Glatter: In terms of bleeding itself, have you tested your product with major aortic bleeds or carotid bleeds in preclinical work?
Landolina: We have used the US military’s model for lethal hemorrhage, and the idea there is to create a model that is just that — lethal. These are the worst types of bleeds that you can possibly imagine, where the patients are clotting compromised, and where you have, in most cases, a very strong arterial component, so something like a femoral artery bleed.
We’ve also tested in carotid artery, aortic applications, as well as combinations of venous and arterial bleeds. The idea here is to show the use of the product in the absolute worst-case scenario so that when this translates into the clinic, the models that we’ve used for evaluation, hopefully, are worse than what actually rolls into the trauma bay.
Glatter: Excellent. What’s the mean time to stop an arterial vs a venous bleed? Are we talking a matter of seconds?
Landolina: In the case of a healthy patient, meaning a patient without clotting compromise, you’re in a matter of seconds. It’s less than 10 seconds.
In the case where you have clotting compromise, a deep, complicated wound geometry, we recommend holding a pressure bandage on for 3 minutes just because it increases the chance of Traumagel coming into contact with the bleed, especially when you can’t visualize the bleed in the bleed source. Because of that pressure time, that becomes the mean. But again, it’s highly dependent on the type of bleed and the style of application.
Failure Rates and Effectiveness
Glatter: As a segue to that, what is the failure rate based on your studies and internal research using Traumagel? Have there been cases where bleeding has not been able to be stopped?
Landolina: It depends on the study, but the failure rates are incredibly low with Traumagel, assuming that it’s correctly used. That’s one of the benefits to this product, where with proper technique, with overwrap with gauze, you nearly always get control of hemorrhage with a product like this.
Glatter: Is manual pressure required in that sense? From what you described earlier, manual pressure would not be required.
Landolina: It depends on the injury. What we recommend is that, if you have a very deep wound where you cannot visualize the source of bleed, you use pressure to seat Traumagel into the source of bleeding, meaning that you’re following Committee on Tactical Combat Casualty Care (Co-TCCC) regulations or requirements, where you’re over wrapping with gauze, and you’re providing a pressure wrapping to ensure that the Traumagel is in contact with the bleed while it’s doing what it’s doing.
In most cases, it doesn’t hurt to apply pressure on top of Traumagel as well. In more surface level bleeds, you don’t need pressure at all.
Applications Beyond Trauma
Glatter: Interesting. In terms of further applications (eg, nose bleeds or GYN bleeding, which are life-threatening), do you see this coming as an application for the future?
Landolina: That’s where we’re working. Traumagel is the successor to an animal health product called Vetigel. The formulations of the gel behind Vetigel and Traumagel are identical. Vetigel has a full surgical indication, and that’s everything from epistaxis to neuro and spine procedures, into cardiovascular and soft tissue surgeries, orthopedic medicine, and so on.
Cresilon’s goal is to eventually expand the indication of our technology to include surgical indications and other indications where we can help any patient that’s bleeding.
Glatter: That’s important, because we use prehospital whole blood, low titer, specifically, when patients have life-threatening hemorrhage. With your product, that would reduce the amount of blood products that would need to be administered. This could be a real game changer.
Landolina: Definitely, that’s the goal we’re working on.
Infection Risks and Biocompatibility
Glatter: In terms of any risk for infection, has that been studied as well? Does Traumagel in any way lead to increased rates of infection?
Landolina: Traumagel is biocompatible. It’s a sterile product. We’ve done the full suite of biocompatibility testing as required by FDA. On top of that, remember that Vetigel, which is the same formulation, is an implantable product. As a result, that has even extended biocompatibility testing beyond what would be necessary for an external product.
In Vetigel’s use case, which has been used now in over 60,000 patients, primarily companion animals, dogs and cats, we haven’t seen instances of infection. There’s no reason to believe that we would see that clinically with Traumagel.
Research Collaborations and Future Applications
Glatter: In terms of other research that your company’s embarked on preclinically, I understand there were some studies done at Walter Reed Army Institute of Research. I was wondering if you could expand on these, specifically, in terms of traumatic brain injury (TBI) and hemorrhage related to that. For example, with shrapnel or even a gunshot wound.
Landolina: The Walter Reed collaboration with Cresilon is something that I’m particularly excited about, because it marks Cresilon’s first project that’s outside the scope of just hemostasis. Walter Reed came to us with this proposal where there’s a big challenge in a subset of TBI called penetrating ballistic-like brain injury, where the brain has been penetrated by a bullet, shrapnel, or some other projectile, and there’s an injury that exposes the brain to the outside.
Today, there is no standard of care to treat patients with those types of injuries. In many cases, mortality is caused through swelling of the brain, or collapse of the brain. What they came to us with was the potential of using our technology, not primarily as a hemostatic agent, but to be able to stabilize that patient enough to get to the next level of care to be treated by a neurosurgeon.
That study Walter Reed did was just a pilot that was done in small animals. In that pilot, they showed that over the period of treatment, there was no negative change in vital signs, no increase in edema or in swelling, or in any of the biomarkers that were being monitored at that time.
At the very least, this is not full indication that this indication will work for Cresilon, but it shows that there’s promise. It’s something that we’re working on and hopefully we’ll be able to bring to market soon.
Glatter: Certainly, maintaining intracranial pressure and cerebral perfusion pressures are very critical. In the future, do you think this product would be able to be deployed endovascularly? Imagine this in terms of stopping bleeding from some source, whether it’s from a stroke or another intracranial source.
Landolina: That’s been an area of interest for us. We have no evidence to prove that indication works at this point, but there’s also nothing to say that it wouldn’t be possible for our technology. At this point, we’ve only looked at a cursory level at those indications.
Glatter: Does the use of Traumagel obviate the need for a more definitive repair (eg, with sutures) or something that’s more permanent?
Landolina: I always say that Traumagel — and Vetigel, for that matter — is not a replacement for good surgical technique. The surgeon always needs to make his or her best judgment when reviewing the patient. That doesn’t mean that there won’t need to be sutures or vascular repair in most of these cases, especially in major trauma.
Final Takeaways
Glatter: Do you have some bullet points or pearls you could give our audience as a takeaway?
Landolina: When Cresilon looks at Traumagel — and for us, Traumagel is the next generation of hemostatic agent, especially in trauma care and in emergency medicine — it allows for a far-simplified application of the product and much faster control of hemorrhage with better patient outcomes.
As we roll this out through EMS agencies, trauma hospitals, military agencies, and eventually to the general public through a future indication, it’s something we’re very excited about. Personally, I started this business 14 years ago, and so it’s great to see our mission of saving lives transitioning to saving human lives.
Glatter: I look forward to seeing this product in the emergency department, but also in other settings, such as in the operating room where we can really help patients who are dying from hemorrhage, certainly on the battlefield, and the lay public. If someone were to come upon a patient who’s bleeding out, this could be certainly a game changer and a lifesaver.
I want to thank you for your time. This is a really important product that’s transformed the lives of so many animals, but also people in the future.
Dr. Glatter is an assistant professor of emergency medicine at Zucker School of Medicine at Hofstra/Northwell in Hempstead, New York. He reported no relevant conflicts of interest. Mr. Landolina is the CEO and co-founder of Cresilon, a biotechnology company specializing in plant-based solutions for emergency bleeding control.
A version of this article first appeared on Medscape.com.
Revumenib Approved for R/R Acute Leukemia With KMT2A Translocation
The approval makes the oral small-molecule menin inhibitor the first pharmaceutical to carry the indication. It blocks the binding of menin to mutated KMT2A fusion proteins, tamping down the process that leads to the disease.
Although a relatively uncommon form of leukemia, KMT2A rearrangements are a major driver of acute leukemia in infants.
Approval was based on a single-arm of the open-label AUGMENT-101 trial with 104 adult and pediatric patients with the mutation. Pediatric patients were at least 30 days old.
The rate of complete remission (CR) plus CR with partial hematologic recovery was 21.2% (22 patients) with a median duration of 6.4 months. The median time to remission was 1.9 months.
Eighty-three patients required blood cell and/or platelet transfusions at baseline; 12 (14%) did not need transfusions for 56 days afterward. Of the 21 who were transfusion free at baseline, 10 (48%) remained so over the same period.
The most common adverse reactions in 20% or more of patients were hemorrhage, nausea, increased phosphate, musculoskeletal pain, infection, increased aspartate aminotransferase, febrile neutropenia, increased alanine aminotransferase, increased intact parathyroid hormone, bacterial infection, diarrhea, differentiation syndrome, electrocardiogram QT prolonged, decreased phosphate, increased triglycerides, decreased potassium, decreased appetite, constipation, edema, viral infection, fatigue, and increased alkaline phosphatase.
The recommended dose varies by weight and concomitant use of strong CYP3A4 inhibitors. Because of an anticipated delay in commercial availability, the lowest strength dose of revumenib will be available through an expanded access program for patients who weigh < 40 kg.
A version of this article appeared on Medscape.com.
The approval makes the oral small-molecule menin inhibitor the first pharmaceutical to carry the indication. It blocks the binding of menin to mutated KMT2A fusion proteins, tamping down the process that leads to the disease.
Although a relatively uncommon form of leukemia, KMT2A rearrangements are a major driver of acute leukemia in infants.
Approval was based on a single-arm of the open-label AUGMENT-101 trial with 104 adult and pediatric patients with the mutation. Pediatric patients were at least 30 days old.
The rate of complete remission (CR) plus CR with partial hematologic recovery was 21.2% (22 patients) with a median duration of 6.4 months. The median time to remission was 1.9 months.
Eighty-three patients required blood cell and/or platelet transfusions at baseline; 12 (14%) did not need transfusions for 56 days afterward. Of the 21 who were transfusion free at baseline, 10 (48%) remained so over the same period.
The most common adverse reactions in 20% or more of patients were hemorrhage, nausea, increased phosphate, musculoskeletal pain, infection, increased aspartate aminotransferase, febrile neutropenia, increased alanine aminotransferase, increased intact parathyroid hormone, bacterial infection, diarrhea, differentiation syndrome, electrocardiogram QT prolonged, decreased phosphate, increased triglycerides, decreased potassium, decreased appetite, constipation, edema, viral infection, fatigue, and increased alkaline phosphatase.
The recommended dose varies by weight and concomitant use of strong CYP3A4 inhibitors. Because of an anticipated delay in commercial availability, the lowest strength dose of revumenib will be available through an expanded access program for patients who weigh < 40 kg.
A version of this article appeared on Medscape.com.
The approval makes the oral small-molecule menin inhibitor the first pharmaceutical to carry the indication. It blocks the binding of menin to mutated KMT2A fusion proteins, tamping down the process that leads to the disease.
Although a relatively uncommon form of leukemia, KMT2A rearrangements are a major driver of acute leukemia in infants.
Approval was based on a single-arm of the open-label AUGMENT-101 trial with 104 adult and pediatric patients with the mutation. Pediatric patients were at least 30 days old.
The rate of complete remission (CR) plus CR with partial hematologic recovery was 21.2% (22 patients) with a median duration of 6.4 months. The median time to remission was 1.9 months.
Eighty-three patients required blood cell and/or platelet transfusions at baseline; 12 (14%) did not need transfusions for 56 days afterward. Of the 21 who were transfusion free at baseline, 10 (48%) remained so over the same period.
The most common adverse reactions in 20% or more of patients were hemorrhage, nausea, increased phosphate, musculoskeletal pain, infection, increased aspartate aminotransferase, febrile neutropenia, increased alanine aminotransferase, increased intact parathyroid hormone, bacterial infection, diarrhea, differentiation syndrome, electrocardiogram QT prolonged, decreased phosphate, increased triglycerides, decreased potassium, decreased appetite, constipation, edema, viral infection, fatigue, and increased alkaline phosphatase.
The recommended dose varies by weight and concomitant use of strong CYP3A4 inhibitors. Because of an anticipated delay in commercial availability, the lowest strength dose of revumenib will be available through an expanded access program for patients who weigh < 40 kg.
A version of this article appeared on Medscape.com.
Thrombocytosis and Cancer Risk: Management in Primary Care
This transcript has been edited for clarity.
In this podcast, I’m going to talk about unexplained high platelet counts, or thrombocytosis, and the risk for cancer in primary care. Let’s start with a typical case we all might see in primary care.
Louisa is 47 years old and is the chief financial officer for a tech startup company. She presents to us in primary care feeling tired all the time — a very common presentation in primary care — with associated reduced appetite. Past medical history includes irritable bowel syndrome, and she’s an ex-smoker.
Systemic inquiry is unremarkable. Specifically, there is no history of weight loss. Louisa has not been prescribed any medication and uses over-the-counter remedies for her irritable bowel syndrome. Examination is also unremarkable. Blood tests were checked, which were all reassuring, except for a platelet count of 612 × 109 cells/L (usual normal range, about 150-450).
What do we do next? Do we refer for an urgent chest x-ray to exclude lung cancer? Do we check a quantitative immunohistochemical fecal occult blood test (qFIT) to identify any occult bleeding in her stool? Do we refer for a routine upper gastrointestinal endoscopy or pelvic ultrasound scan to exclude any upper gastrointestinal or endometrial malignancy?
Do we simply repeat the bloods? If so, do we repeat them routinely or urgently, and indeed, which ones should we recheck?
Louisa has an unexplained thrombocytosis. How do we manage this in primary care? Thrombocytosis is generally defined as a raised platelet count over 450. Importantly, thrombocytosis is a common incidental finding in around 2% of those over 40 years of age attending primary care. Reassuringly, 80%-90% of thrombocytosis is reactive, secondary to acute blood loss, infection, or inflammation, and the majority of cases resolve within 3 months.
Why the concern with Louisa then? Although most cases are reactive, clinical guidance (for example, NICE suspected cancer guidance in the UK and Scottish suspected cancer guidance in Scotland) reminds us that unexplained thrombocytosis is a risk marker for some solid-tumor malignancies.
Previous studies have demonstrated that unexplained thrombocytosis is associated with a 1-year cancer incidence of 11.6% in males and 6.2% in females, well exceeding the standard 3% threshold warranting investigation for underlying malignancy. However, thrombocytosis should not be used as a stand-alone diagnostic or screening test for cancer, or indeed to rule out cancer.
Instead, unexplained thrombocytosis should prompt us to think cancer. The Scottish suspected cancer referral guidelines include thrombocytosis in the investigation criteria for what they call the LEGO-C cancers — L for lung, E for endometrial, G for gastric, O for oesophageal, and C for colorectal, which is a useful reminder for us all.
What further history, examination, and investigations might we consider in primary care if we identify an unexplained high platelet count? As always, we should use our clinical judgment and trust our clinical acumen.
We should consider all the possible underlying causes, including infection, inflammation, and blood loss, including menstrual blood loss in women; myeloproliferative disorders such as polycythemia rubra vera, chronic myeloid leukemia, and essential thrombocythemia; and, of course, underlying malignancy. If a likely underlying reversible cause is present (for example, a recent lower respiratory tract infection), simply repeating the full blood count in 4-6 weeks is quite appropriate to see if the thrombocytosis has resolved.
Remember, 80%-90% of cases are reactive thrombocytosis, and most cases resolve within 3 months. If thrombocytosis is unexplained or not resolving, consider checking ferritin levels to exclude iron deficiency. Consider checking C-reactive protein (CRP) levels to exclude any inflammation, and also consider checking a blood film to exclude any hematologic disorders, in addition, of course, to more detailed history-taking and examination to elicit any red flags.
We can also consider a JAK2 gene mutation test, if it is available to you locally, or a hematology referral if we suspect a myeloproliferative disorder. JAK2 is a genetic mutation that may be present in people with essential thrombocythemia and can indicate a diagnosis of polycythemia rubra vera.
Subsequent to this, and again using our clinical judgment, we then need to exclude the LEGO-C cancers. Consider urgent chest x-ray to exclude lung cancer or pelvic ultrasound in women to exclude endometrial cancer. Also, we should consider an upper gastrointestinal endoscopy, particularly in those individuals who have associated upper gastrointestinal symptoms and/or weight loss.
Finally, consider a qFIT to identify any occult bleeding in the stool, again if it’s available to you, or certainly if not, urgent lower gastrointestinal investigations to exclude colorectal cancer.
Alongside these possible investigations, as always, we should safety-net appropriately within agreed timeframes and check for resolution of the thrombocytosis according to the condition being suspected. Remember, most cases resolve within 3 months.
Returning to Louisa, what did I do? After seeing a platelet count of 600, I subsequently telephoned her and reexplored her history, which yielded nil else of note. Specifically, there was no history of unexplained weight loss, no history of upper or lower gastrointestinal symptoms, and certainly nothing significantly different from her usual irritable bowel syndrome symptoms. There were also no respiratory or genitourinary symptoms of note.
I did arrange for Louisa to undergo a chest x-ray over the next few days, though, as she was an ex-smoker. This was subsequently reported as normal. I appreciate chest x-rays have poor sensitivity for detecting lung cancer, as highlighted in a number of recent papers, but it was mutually agreed with Louisa that we would simply repeat her blood test in around 6 weeks. As well as repeating the full blood count, I arranged to check her ferritin, CRP, and a blood film, and then I was planning to reassess her clinically in person.
These bloods and my subsequent clinical review were reassuring. In fact, her platelet count had normalized after that 6 weeks had elapsed. Her thrombocytosis had resolved.
I didn’t arrange any further follow-up for her, but I did give her the usual safety netting advice to re-present to me or one of my colleagues if she does develop any worrying symptoms or signs.
I appreciate these scenarios are not always this straightforward, but I wanted to outline what investigations and referrals we may need to consider in primary care if we encounter an unexplained high platelet count.
There are a couple of quality-improvement activities for us all to consider in primary care. Consider as a team how we would respond to an incidental finding of thrombocytosis on a full blood count. Also consider what are our safety-netting options for those found to have raised platelet counts but no other symptoms or risk factors for underlying malignancy.
Finally, I’ve produced a Medscape UK primary care hack or clinical aide-memoire on managing unexplained thrombocytosis and associated cancer risk in primary care for all healthcare professionals working in primary care. This can be found online. I hope you find this resource helpful.
Dr. Kevin Fernando, General practitioner partner with specialist interests in cardiovascular, renal, and metabolic medicine, North Berwick Group Practice in Scotland, has disclosed relevant financial relationships with Amarin, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Dexcom, Lilly, Menarini, Novartis, Novo Nordisk, Roche Diagnostics, Embecta, Roche Diabetes Care, Sanofi Menarini, and Daiichi Sankyo.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
In this podcast, I’m going to talk about unexplained high platelet counts, or thrombocytosis, and the risk for cancer in primary care. Let’s start with a typical case we all might see in primary care.
Louisa is 47 years old and is the chief financial officer for a tech startup company. She presents to us in primary care feeling tired all the time — a very common presentation in primary care — with associated reduced appetite. Past medical history includes irritable bowel syndrome, and she’s an ex-smoker.
Systemic inquiry is unremarkable. Specifically, there is no history of weight loss. Louisa has not been prescribed any medication and uses over-the-counter remedies for her irritable bowel syndrome. Examination is also unremarkable. Blood tests were checked, which were all reassuring, except for a platelet count of 612 × 109 cells/L (usual normal range, about 150-450).
What do we do next? Do we refer for an urgent chest x-ray to exclude lung cancer? Do we check a quantitative immunohistochemical fecal occult blood test (qFIT) to identify any occult bleeding in her stool? Do we refer for a routine upper gastrointestinal endoscopy or pelvic ultrasound scan to exclude any upper gastrointestinal or endometrial malignancy?
Do we simply repeat the bloods? If so, do we repeat them routinely or urgently, and indeed, which ones should we recheck?
Louisa has an unexplained thrombocytosis. How do we manage this in primary care? Thrombocytosis is generally defined as a raised platelet count over 450. Importantly, thrombocytosis is a common incidental finding in around 2% of those over 40 years of age attending primary care. Reassuringly, 80%-90% of thrombocytosis is reactive, secondary to acute blood loss, infection, or inflammation, and the majority of cases resolve within 3 months.
Why the concern with Louisa then? Although most cases are reactive, clinical guidance (for example, NICE suspected cancer guidance in the UK and Scottish suspected cancer guidance in Scotland) reminds us that unexplained thrombocytosis is a risk marker for some solid-tumor malignancies.
Previous studies have demonstrated that unexplained thrombocytosis is associated with a 1-year cancer incidence of 11.6% in males and 6.2% in females, well exceeding the standard 3% threshold warranting investigation for underlying malignancy. However, thrombocytosis should not be used as a stand-alone diagnostic or screening test for cancer, or indeed to rule out cancer.
Instead, unexplained thrombocytosis should prompt us to think cancer. The Scottish suspected cancer referral guidelines include thrombocytosis in the investigation criteria for what they call the LEGO-C cancers — L for lung, E for endometrial, G for gastric, O for oesophageal, and C for colorectal, which is a useful reminder for us all.
What further history, examination, and investigations might we consider in primary care if we identify an unexplained high platelet count? As always, we should use our clinical judgment and trust our clinical acumen.
We should consider all the possible underlying causes, including infection, inflammation, and blood loss, including menstrual blood loss in women; myeloproliferative disorders such as polycythemia rubra vera, chronic myeloid leukemia, and essential thrombocythemia; and, of course, underlying malignancy. If a likely underlying reversible cause is present (for example, a recent lower respiratory tract infection), simply repeating the full blood count in 4-6 weeks is quite appropriate to see if the thrombocytosis has resolved.
Remember, 80%-90% of cases are reactive thrombocytosis, and most cases resolve within 3 months. If thrombocytosis is unexplained or not resolving, consider checking ferritin levels to exclude iron deficiency. Consider checking C-reactive protein (CRP) levels to exclude any inflammation, and also consider checking a blood film to exclude any hematologic disorders, in addition, of course, to more detailed history-taking and examination to elicit any red flags.
We can also consider a JAK2 gene mutation test, if it is available to you locally, or a hematology referral if we suspect a myeloproliferative disorder. JAK2 is a genetic mutation that may be present in people with essential thrombocythemia and can indicate a diagnosis of polycythemia rubra vera.
Subsequent to this, and again using our clinical judgment, we then need to exclude the LEGO-C cancers. Consider urgent chest x-ray to exclude lung cancer or pelvic ultrasound in women to exclude endometrial cancer. Also, we should consider an upper gastrointestinal endoscopy, particularly in those individuals who have associated upper gastrointestinal symptoms and/or weight loss.
Finally, consider a qFIT to identify any occult bleeding in the stool, again if it’s available to you, or certainly if not, urgent lower gastrointestinal investigations to exclude colorectal cancer.
Alongside these possible investigations, as always, we should safety-net appropriately within agreed timeframes and check for resolution of the thrombocytosis according to the condition being suspected. Remember, most cases resolve within 3 months.
Returning to Louisa, what did I do? After seeing a platelet count of 600, I subsequently telephoned her and reexplored her history, which yielded nil else of note. Specifically, there was no history of unexplained weight loss, no history of upper or lower gastrointestinal symptoms, and certainly nothing significantly different from her usual irritable bowel syndrome symptoms. There were also no respiratory or genitourinary symptoms of note.
I did arrange for Louisa to undergo a chest x-ray over the next few days, though, as she was an ex-smoker. This was subsequently reported as normal. I appreciate chest x-rays have poor sensitivity for detecting lung cancer, as highlighted in a number of recent papers, but it was mutually agreed with Louisa that we would simply repeat her blood test in around 6 weeks. As well as repeating the full blood count, I arranged to check her ferritin, CRP, and a blood film, and then I was planning to reassess her clinically in person.
These bloods and my subsequent clinical review were reassuring. In fact, her platelet count had normalized after that 6 weeks had elapsed. Her thrombocytosis had resolved.
I didn’t arrange any further follow-up for her, but I did give her the usual safety netting advice to re-present to me or one of my colleagues if she does develop any worrying symptoms or signs.
I appreciate these scenarios are not always this straightforward, but I wanted to outline what investigations and referrals we may need to consider in primary care if we encounter an unexplained high platelet count.
There are a couple of quality-improvement activities for us all to consider in primary care. Consider as a team how we would respond to an incidental finding of thrombocytosis on a full blood count. Also consider what are our safety-netting options for those found to have raised platelet counts but no other symptoms or risk factors for underlying malignancy.
Finally, I’ve produced a Medscape UK primary care hack or clinical aide-memoire on managing unexplained thrombocytosis and associated cancer risk in primary care for all healthcare professionals working in primary care. This can be found online. I hope you find this resource helpful.
Dr. Kevin Fernando, General practitioner partner with specialist interests in cardiovascular, renal, and metabolic medicine, North Berwick Group Practice in Scotland, has disclosed relevant financial relationships with Amarin, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Dexcom, Lilly, Menarini, Novartis, Novo Nordisk, Roche Diagnostics, Embecta, Roche Diabetes Care, Sanofi Menarini, and Daiichi Sankyo.
A version of this article first appeared on Medscape.com.
This transcript has been edited for clarity.
In this podcast, I’m going to talk about unexplained high platelet counts, or thrombocytosis, and the risk for cancer in primary care. Let’s start with a typical case we all might see in primary care.
Louisa is 47 years old and is the chief financial officer for a tech startup company. She presents to us in primary care feeling tired all the time — a very common presentation in primary care — with associated reduced appetite. Past medical history includes irritable bowel syndrome, and she’s an ex-smoker.
Systemic inquiry is unremarkable. Specifically, there is no history of weight loss. Louisa has not been prescribed any medication and uses over-the-counter remedies for her irritable bowel syndrome. Examination is also unremarkable. Blood tests were checked, which were all reassuring, except for a platelet count of 612 × 109 cells/L (usual normal range, about 150-450).
What do we do next? Do we refer for an urgent chest x-ray to exclude lung cancer? Do we check a quantitative immunohistochemical fecal occult blood test (qFIT) to identify any occult bleeding in her stool? Do we refer for a routine upper gastrointestinal endoscopy or pelvic ultrasound scan to exclude any upper gastrointestinal or endometrial malignancy?
Do we simply repeat the bloods? If so, do we repeat them routinely or urgently, and indeed, which ones should we recheck?
Louisa has an unexplained thrombocytosis. How do we manage this in primary care? Thrombocytosis is generally defined as a raised platelet count over 450. Importantly, thrombocytosis is a common incidental finding in around 2% of those over 40 years of age attending primary care. Reassuringly, 80%-90% of thrombocytosis is reactive, secondary to acute blood loss, infection, or inflammation, and the majority of cases resolve within 3 months.
Why the concern with Louisa then? Although most cases are reactive, clinical guidance (for example, NICE suspected cancer guidance in the UK and Scottish suspected cancer guidance in Scotland) reminds us that unexplained thrombocytosis is a risk marker for some solid-tumor malignancies.
Previous studies have demonstrated that unexplained thrombocytosis is associated with a 1-year cancer incidence of 11.6% in males and 6.2% in females, well exceeding the standard 3% threshold warranting investigation for underlying malignancy. However, thrombocytosis should not be used as a stand-alone diagnostic or screening test for cancer, or indeed to rule out cancer.
Instead, unexplained thrombocytosis should prompt us to think cancer. The Scottish suspected cancer referral guidelines include thrombocytosis in the investigation criteria for what they call the LEGO-C cancers — L for lung, E for endometrial, G for gastric, O for oesophageal, and C for colorectal, which is a useful reminder for us all.
What further history, examination, and investigations might we consider in primary care if we identify an unexplained high platelet count? As always, we should use our clinical judgment and trust our clinical acumen.
We should consider all the possible underlying causes, including infection, inflammation, and blood loss, including menstrual blood loss in women; myeloproliferative disorders such as polycythemia rubra vera, chronic myeloid leukemia, and essential thrombocythemia; and, of course, underlying malignancy. If a likely underlying reversible cause is present (for example, a recent lower respiratory tract infection), simply repeating the full blood count in 4-6 weeks is quite appropriate to see if the thrombocytosis has resolved.
Remember, 80%-90% of cases are reactive thrombocytosis, and most cases resolve within 3 months. If thrombocytosis is unexplained or not resolving, consider checking ferritin levels to exclude iron deficiency. Consider checking C-reactive protein (CRP) levels to exclude any inflammation, and also consider checking a blood film to exclude any hematologic disorders, in addition, of course, to more detailed history-taking and examination to elicit any red flags.
We can also consider a JAK2 gene mutation test, if it is available to you locally, or a hematology referral if we suspect a myeloproliferative disorder. JAK2 is a genetic mutation that may be present in people with essential thrombocythemia and can indicate a diagnosis of polycythemia rubra vera.
Subsequent to this, and again using our clinical judgment, we then need to exclude the LEGO-C cancers. Consider urgent chest x-ray to exclude lung cancer or pelvic ultrasound in women to exclude endometrial cancer. Also, we should consider an upper gastrointestinal endoscopy, particularly in those individuals who have associated upper gastrointestinal symptoms and/or weight loss.
Finally, consider a qFIT to identify any occult bleeding in the stool, again if it’s available to you, or certainly if not, urgent lower gastrointestinal investigations to exclude colorectal cancer.
Alongside these possible investigations, as always, we should safety-net appropriately within agreed timeframes and check for resolution of the thrombocytosis according to the condition being suspected. Remember, most cases resolve within 3 months.
Returning to Louisa, what did I do? After seeing a platelet count of 600, I subsequently telephoned her and reexplored her history, which yielded nil else of note. Specifically, there was no history of unexplained weight loss, no history of upper or lower gastrointestinal symptoms, and certainly nothing significantly different from her usual irritable bowel syndrome symptoms. There were also no respiratory or genitourinary symptoms of note.
I did arrange for Louisa to undergo a chest x-ray over the next few days, though, as she was an ex-smoker. This was subsequently reported as normal. I appreciate chest x-rays have poor sensitivity for detecting lung cancer, as highlighted in a number of recent papers, but it was mutually agreed with Louisa that we would simply repeat her blood test in around 6 weeks. As well as repeating the full blood count, I arranged to check her ferritin, CRP, and a blood film, and then I was planning to reassess her clinically in person.
These bloods and my subsequent clinical review were reassuring. In fact, her platelet count had normalized after that 6 weeks had elapsed. Her thrombocytosis had resolved.
I didn’t arrange any further follow-up for her, but I did give her the usual safety netting advice to re-present to me or one of my colleagues if she does develop any worrying symptoms or signs.
I appreciate these scenarios are not always this straightforward, but I wanted to outline what investigations and referrals we may need to consider in primary care if we encounter an unexplained high platelet count.
There are a couple of quality-improvement activities for us all to consider in primary care. Consider as a team how we would respond to an incidental finding of thrombocytosis on a full blood count. Also consider what are our safety-netting options for those found to have raised platelet counts but no other symptoms or risk factors for underlying malignancy.
Finally, I’ve produced a Medscape UK primary care hack or clinical aide-memoire on managing unexplained thrombocytosis and associated cancer risk in primary care for all healthcare professionals working in primary care. This can be found online. I hope you find this resource helpful.
Dr. Kevin Fernando, General practitioner partner with specialist interests in cardiovascular, renal, and metabolic medicine, North Berwick Group Practice in Scotland, has disclosed relevant financial relationships with Amarin, Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Dexcom, Lilly, Menarini, Novartis, Novo Nordisk, Roche Diagnostics, Embecta, Roche Diabetes Care, Sanofi Menarini, and Daiichi Sankyo.
A version of this article first appeared on Medscape.com.
Rituximab Not Inferior to Cyclophosphamide in Pediatric Vasculitis
TOPLINE:
and those who received rituximab required a significantly lower steroid dose than those who received cyclophosphamide or a combination therapy.
METHODOLOGY:
- Researchers evaluated the efficacy of rituximab, cyclophosphamide, or a combination of both in pediatric patients diagnosed with granulomatosis with polyangiitis (GPA) or microscopic polyangiitis.
- A total of 104 patients (median age at diagnosis, 14 years; 67% girls) were included from A Registry of Childhood Vasculitis; the majority had a diagnosis of GPA (81%) and renal involvement (87%). Overall, induction therapy involved rituximab for 43%, cyclophosphamide for 46%, and a combination of both for 11% patients.
- The primary endpoint was the rate of achieving remission (Pediatric Vasculitis Activity Score [PVAS] of 0) or low disease activity (PVAS ≤ 2) at the post-induction visit (4-6 months after diagnosis).
- The secondary endpoints were the degree of disease-related damage at 12- and 24-month visits and rates of drug-related hospitalization occurring between the diagnosis and post-induction visits.
TAKEAWAY:
- At the post-induction visit, 63% patients achieved remission or low disease activity, with the rates being similar between patients who received rituximab and those who received cyclophosphamide (64% vs 62%).
- Patients treated with rituximab required a significantly lower median steroid dose (0.13 mg/kg per day) than those treated with cyclophosphamide (0.3 mg/kg per day) or the combination therapy (0.3 mg/kg per day; P < .001) at the post-induction visit.
- Overall, 61% and 56% patients receiving rituximab and cyclophosphamide, respectively, had disease-related damage measure on the Pediatric Vasculitis Damage Index at the 12-month visit; however, the degree of damage was low.
- The percentage of patients requiring hospitalization was higher in the rituximab group than in the cyclophosphamide group (22% vs 10%), primarily stemming from drug- or infection-related causes (11% vs 2%).
IN PRACTICE:
“The results of this study may assist with current clinical decision-making with regard to the choice of induction medications in childhood-onset AAV and will complement the ongoing [Childhood Arthritis and Rheumatology Research Alliance] prospective [consensus treatment plans] study,” the authors wrote.
SOURCE:
This study was led by Samuel J. Gagne, MD, Children’s Hospital of Pittsburgh, University of Pittsburgh Medical Center in Pennsylvania, and was published online in Arthritis Care & Research.
LIMITATIONS:
Study limitations included the inconsistencies in glucocorticoid dosing, which may have affected remission rates. Moreover, data on the adverse events not requiring hospitalization and long-term adverse events were not captured.
DISCLOSURES:
This study received funding through a Nationwide Children’s Hospital intramural grant award. The authors reported no potential conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
and those who received rituximab required a significantly lower steroid dose than those who received cyclophosphamide or a combination therapy.
METHODOLOGY:
- Researchers evaluated the efficacy of rituximab, cyclophosphamide, or a combination of both in pediatric patients diagnosed with granulomatosis with polyangiitis (GPA) or microscopic polyangiitis.
- A total of 104 patients (median age at diagnosis, 14 years; 67% girls) were included from A Registry of Childhood Vasculitis; the majority had a diagnosis of GPA (81%) and renal involvement (87%). Overall, induction therapy involved rituximab for 43%, cyclophosphamide for 46%, and a combination of both for 11% patients.
- The primary endpoint was the rate of achieving remission (Pediatric Vasculitis Activity Score [PVAS] of 0) or low disease activity (PVAS ≤ 2) at the post-induction visit (4-6 months after diagnosis).
- The secondary endpoints were the degree of disease-related damage at 12- and 24-month visits and rates of drug-related hospitalization occurring between the diagnosis and post-induction visits.
TAKEAWAY:
- At the post-induction visit, 63% patients achieved remission or low disease activity, with the rates being similar between patients who received rituximab and those who received cyclophosphamide (64% vs 62%).
- Patients treated with rituximab required a significantly lower median steroid dose (0.13 mg/kg per day) than those treated with cyclophosphamide (0.3 mg/kg per day) or the combination therapy (0.3 mg/kg per day; P < .001) at the post-induction visit.
- Overall, 61% and 56% patients receiving rituximab and cyclophosphamide, respectively, had disease-related damage measure on the Pediatric Vasculitis Damage Index at the 12-month visit; however, the degree of damage was low.
- The percentage of patients requiring hospitalization was higher in the rituximab group than in the cyclophosphamide group (22% vs 10%), primarily stemming from drug- or infection-related causes (11% vs 2%).
IN PRACTICE:
“The results of this study may assist with current clinical decision-making with regard to the choice of induction medications in childhood-onset AAV and will complement the ongoing [Childhood Arthritis and Rheumatology Research Alliance] prospective [consensus treatment plans] study,” the authors wrote.
SOURCE:
This study was led by Samuel J. Gagne, MD, Children’s Hospital of Pittsburgh, University of Pittsburgh Medical Center in Pennsylvania, and was published online in Arthritis Care & Research.
LIMITATIONS:
Study limitations included the inconsistencies in glucocorticoid dosing, which may have affected remission rates. Moreover, data on the adverse events not requiring hospitalization and long-term adverse events were not captured.
DISCLOSURES:
This study received funding through a Nationwide Children’s Hospital intramural grant award. The authors reported no potential conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
TOPLINE:
and those who received rituximab required a significantly lower steroid dose than those who received cyclophosphamide or a combination therapy.
METHODOLOGY:
- Researchers evaluated the efficacy of rituximab, cyclophosphamide, or a combination of both in pediatric patients diagnosed with granulomatosis with polyangiitis (GPA) or microscopic polyangiitis.
- A total of 104 patients (median age at diagnosis, 14 years; 67% girls) were included from A Registry of Childhood Vasculitis; the majority had a diagnosis of GPA (81%) and renal involvement (87%). Overall, induction therapy involved rituximab for 43%, cyclophosphamide for 46%, and a combination of both for 11% patients.
- The primary endpoint was the rate of achieving remission (Pediatric Vasculitis Activity Score [PVAS] of 0) or low disease activity (PVAS ≤ 2) at the post-induction visit (4-6 months after diagnosis).
- The secondary endpoints were the degree of disease-related damage at 12- and 24-month visits and rates of drug-related hospitalization occurring between the diagnosis and post-induction visits.
TAKEAWAY:
- At the post-induction visit, 63% patients achieved remission or low disease activity, with the rates being similar between patients who received rituximab and those who received cyclophosphamide (64% vs 62%).
- Patients treated with rituximab required a significantly lower median steroid dose (0.13 mg/kg per day) than those treated with cyclophosphamide (0.3 mg/kg per day) or the combination therapy (0.3 mg/kg per day; P < .001) at the post-induction visit.
- Overall, 61% and 56% patients receiving rituximab and cyclophosphamide, respectively, had disease-related damage measure on the Pediatric Vasculitis Damage Index at the 12-month visit; however, the degree of damage was low.
- The percentage of patients requiring hospitalization was higher in the rituximab group than in the cyclophosphamide group (22% vs 10%), primarily stemming from drug- or infection-related causes (11% vs 2%).
IN PRACTICE:
“The results of this study may assist with current clinical decision-making with regard to the choice of induction medications in childhood-onset AAV and will complement the ongoing [Childhood Arthritis and Rheumatology Research Alliance] prospective [consensus treatment plans] study,” the authors wrote.
SOURCE:
This study was led by Samuel J. Gagne, MD, Children’s Hospital of Pittsburgh, University of Pittsburgh Medical Center in Pennsylvania, and was published online in Arthritis Care & Research.
LIMITATIONS:
Study limitations included the inconsistencies in glucocorticoid dosing, which may have affected remission rates. Moreover, data on the adverse events not requiring hospitalization and long-term adverse events were not captured.
DISCLOSURES:
This study received funding through a Nationwide Children’s Hospital intramural grant award. The authors reported no potential conflicts of interest.
This article was created using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article first appeared on Medscape.com.
Scurvy: A Diagnosis Still Relevant Today
“Petechial rash often prompts further investigation into hematological, dermatological, or vasculitis causes. However, if the above investigations are negative and skin biopsy has not revealed a cause, there is a Renaissance-era diagnosis that is often overlooked but is easily investigated and treated,” wrote Andrew Dermawan, MD, and colleagues from Sir Charles Gairdner Hospital in Nedlands, Australia, in BMJ Case Reports. The diagnosis they highlight is scurvy, a disease that has faded from common medical concern but is reemerging, partly because of the rise in bariatric surgery.
Diagnosing Scurvy in the 2020s
In their article, Dermawan and colleagues present the case of a 50-year-old man with a bilateral petechial rash on his lower limbs, without any history of trauma. The patient, who exhibited no infectious symptoms, also had gross hematuria, microcytic anemia, mild neutropenia, and lymphopenia. Tests for autoimmune and hematological diseases were negative, as were abdominal and leg CT scans, ruling out abdominal hemorrhage and vasculitis. Additionally, a skin biopsy showed no causative findings.
The doctors noted that the patient had undergone sleeve gastrectomy, prompting them to inquire about his diet. They discovered that, because of financial difficulties, his diet primarily consisted of processed foods with little to no fruits or vegetables, and he had stopped taking supplements recommended by his gastroenterologist. Further tests revealed a vitamin D deficiency and a severe deficiency in vitamin C. With the diagnosis of scurvy confirmed, the doctors treated the patient with 1000 mg of ascorbic acid daily, along with cholecalciferol, folic acid, and a multivitamin complex, leading to a complete resolution of his symptoms.
Risk Factors Then and Now
It can cause mucosal and gastric hemorrhages, and if left untreated, it can lead to fatal bleeding.
Historically known as “sailors’ disease,” scurvy plagued men on long voyages who lacked access to fresh fruits or vegetables and thus did not get enough vitamin C. In 1747, James Lind, a British physician in the Royal Navy, demonstrated that the consumption of oranges and lemons could combat scurvy.
Today’s risk factors for scurvy include malnutrition, gastrointestinal disorders (eg, chronic inflammatory bowel diseases), alcohol and tobacco use, eating disorders, psychiatric illnesses, dialysis, and the use of medications that reduce the absorption of ascorbic acid (such as corticosteroids and proton pump inhibitors).
Scurvy remains more common among individuals with unfavorable socioeconomic conditions. The authors of the study emphasize how the rising cost of living — specifically in Australia but applicable elsewhere — is changing eating habits, leading to a high consumption of low-cost, nutritionally poor foods.
Poverty has always been a risk factor for scurvy, but today there may be an additional cause: bariatric surgery. Patients undergoing these procedures are at a risk for deficiencies in fat-soluble vitamins A, D, E, and K, and if their diet is inadequate, they may also experience a vitamin C deficiency. Awareness of this can facilitate the timely diagnosis of scurvy in these patients.
This story was translated from Univadis Italy using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
“Petechial rash often prompts further investigation into hematological, dermatological, or vasculitis causes. However, if the above investigations are negative and skin biopsy has not revealed a cause, there is a Renaissance-era diagnosis that is often overlooked but is easily investigated and treated,” wrote Andrew Dermawan, MD, and colleagues from Sir Charles Gairdner Hospital in Nedlands, Australia, in BMJ Case Reports. The diagnosis they highlight is scurvy, a disease that has faded from common medical concern but is reemerging, partly because of the rise in bariatric surgery.
Diagnosing Scurvy in the 2020s
In their article, Dermawan and colleagues present the case of a 50-year-old man with a bilateral petechial rash on his lower limbs, without any history of trauma. The patient, who exhibited no infectious symptoms, also had gross hematuria, microcytic anemia, mild neutropenia, and lymphopenia. Tests for autoimmune and hematological diseases were negative, as were abdominal and leg CT scans, ruling out abdominal hemorrhage and vasculitis. Additionally, a skin biopsy showed no causative findings.
The doctors noted that the patient had undergone sleeve gastrectomy, prompting them to inquire about his diet. They discovered that, because of financial difficulties, his diet primarily consisted of processed foods with little to no fruits or vegetables, and he had stopped taking supplements recommended by his gastroenterologist. Further tests revealed a vitamin D deficiency and a severe deficiency in vitamin C. With the diagnosis of scurvy confirmed, the doctors treated the patient with 1000 mg of ascorbic acid daily, along with cholecalciferol, folic acid, and a multivitamin complex, leading to a complete resolution of his symptoms.
Risk Factors Then and Now
It can cause mucosal and gastric hemorrhages, and if left untreated, it can lead to fatal bleeding.
Historically known as “sailors’ disease,” scurvy plagued men on long voyages who lacked access to fresh fruits or vegetables and thus did not get enough vitamin C. In 1747, James Lind, a British physician in the Royal Navy, demonstrated that the consumption of oranges and lemons could combat scurvy.
Today’s risk factors for scurvy include malnutrition, gastrointestinal disorders (eg, chronic inflammatory bowel diseases), alcohol and tobacco use, eating disorders, psychiatric illnesses, dialysis, and the use of medications that reduce the absorption of ascorbic acid (such as corticosteroids and proton pump inhibitors).
Scurvy remains more common among individuals with unfavorable socioeconomic conditions. The authors of the study emphasize how the rising cost of living — specifically in Australia but applicable elsewhere — is changing eating habits, leading to a high consumption of low-cost, nutritionally poor foods.
Poverty has always been a risk factor for scurvy, but today there may be an additional cause: bariatric surgery. Patients undergoing these procedures are at a risk for deficiencies in fat-soluble vitamins A, D, E, and K, and if their diet is inadequate, they may also experience a vitamin C deficiency. Awareness of this can facilitate the timely diagnosis of scurvy in these patients.
This story was translated from Univadis Italy using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
“Petechial rash often prompts further investigation into hematological, dermatological, or vasculitis causes. However, if the above investigations are negative and skin biopsy has not revealed a cause, there is a Renaissance-era diagnosis that is often overlooked but is easily investigated and treated,” wrote Andrew Dermawan, MD, and colleagues from Sir Charles Gairdner Hospital in Nedlands, Australia, in BMJ Case Reports. The diagnosis they highlight is scurvy, a disease that has faded from common medical concern but is reemerging, partly because of the rise in bariatric surgery.
Diagnosing Scurvy in the 2020s
In their article, Dermawan and colleagues present the case of a 50-year-old man with a bilateral petechial rash on his lower limbs, without any history of trauma. The patient, who exhibited no infectious symptoms, also had gross hematuria, microcytic anemia, mild neutropenia, and lymphopenia. Tests for autoimmune and hematological diseases were negative, as were abdominal and leg CT scans, ruling out abdominal hemorrhage and vasculitis. Additionally, a skin biopsy showed no causative findings.
The doctors noted that the patient had undergone sleeve gastrectomy, prompting them to inquire about his diet. They discovered that, because of financial difficulties, his diet primarily consisted of processed foods with little to no fruits or vegetables, and he had stopped taking supplements recommended by his gastroenterologist. Further tests revealed a vitamin D deficiency and a severe deficiency in vitamin C. With the diagnosis of scurvy confirmed, the doctors treated the patient with 1000 mg of ascorbic acid daily, along with cholecalciferol, folic acid, and a multivitamin complex, leading to a complete resolution of his symptoms.
Risk Factors Then and Now
It can cause mucosal and gastric hemorrhages, and if left untreated, it can lead to fatal bleeding.
Historically known as “sailors’ disease,” scurvy plagued men on long voyages who lacked access to fresh fruits or vegetables and thus did not get enough vitamin C. In 1747, James Lind, a British physician in the Royal Navy, demonstrated that the consumption of oranges and lemons could combat scurvy.
Today’s risk factors for scurvy include malnutrition, gastrointestinal disorders (eg, chronic inflammatory bowel diseases), alcohol and tobacco use, eating disorders, psychiatric illnesses, dialysis, and the use of medications that reduce the absorption of ascorbic acid (such as corticosteroids and proton pump inhibitors).
Scurvy remains more common among individuals with unfavorable socioeconomic conditions. The authors of the study emphasize how the rising cost of living — specifically in Australia but applicable elsewhere — is changing eating habits, leading to a high consumption of low-cost, nutritionally poor foods.
Poverty has always been a risk factor for scurvy, but today there may be an additional cause: bariatric surgery. Patients undergoing these procedures are at a risk for deficiencies in fat-soluble vitamins A, D, E, and K, and if their diet is inadequate, they may also experience a vitamin C deficiency. Awareness of this can facilitate the timely diagnosis of scurvy in these patients.
This story was translated from Univadis Italy using several editorial tools, including AI, as part of the process. Human editors reviewed this content before publication. A version of this article appeared on Medscape.com.
On Second Thought: Aspirin for Primary Prevention — What We Really Know
This transcript has been edited for clarity.
Our recommendations vis-à-vis aspirin have evolved at a dizzying pace. The young’uns watching us right now don’t know what things were like in the 1980s. The Reagan era was a wild, heady time where nuclear war was imminent and we didn’t prescribe aspirin to patients.
That only started in 1988, which was a banner year in human history. Not because a number of doves were incinerated by the lighting of the Olympic torch at the Seoul Olympics — look it up if you don’t know what I’m talking about — but because 1988 saw the publication of the ISIS-2 trial, which first showed a mortality benefit to prescribing aspirin post–myocardial infarction (MI).
Giving patients aspirin during or after a heart attack is not controversial. It’s one of the few things in this business that isn’t, but that’s secondary prevention — treating somebody after they develop a disease. Primary prevention, treating them before they have their incident event, is a very different ballgame. Here, things are messy.
For one thing, the doses used have been very inconsistent. We should point out that the reason for 81 mg of aspirin is very arbitrary and is rooted in the old apothecary system of weights and measurements. A standard dose of aspirin was 5 grains, where 20 grains made 1 scruple, 3 scruples made 1 dram, 8 drams made 1 oz, and 12 oz made 1 lb - because screw you, metric system. Therefore, 5 grains was 325 mg of aspirin, and 1 quarter of the standard dose became 81 mg if you rounded out the decimal.
People have tried all kinds of dosing structures with aspirin prophylaxis. The Physicians’ Health Study used a full-dose aspirin, 325 mg every 2 days, while the Hypertension Optimal Treatment (HOT) trial tested 75 mg daily and the Women’s Health Study tested 100 mg, but every other day.
Ironically, almost no one has studied 81 mg every day, which is weird if you think about it. The bigger problem here is not the variability of doses used, but the discrepancy when you look at older vs newer studies.
Older studies, like the Physicians’ Health Study, did show a benefit, at least in the subgroup of patients over age 50 years, which is probably where the “everybody over 50 should be taking an aspirin” idea comes from, at least as near as I can tell.
More recent studies, like the Women’s Health Study, ASPREE, or ASPIRE, didn’t show a benefit. I know what you’re thinking: Newer stuff is always better. That’s why you should never trust anybody over age 40 years. The context of primary prevention studies has changed. In the ‘80s and ‘90s, people smoked more and we didn’t have the same medications that we have today. We talked about all this in the beta-blocker video to explain why beta-blockers don’t seem to have a benefit post MI.
We have a similar issue here. The magnitude of the benefit with aspirin primary prevention has decreased because we’re all just healthier overall. So, yay! Progress! Here’s where the numbers matter. No one is saying that aspirin doesn’t help. It does.
If we look at the 2019 meta-analysis published in JAMA, there is a cardiovascular benefit. The numbers bear that out. I know you’re all here for the math, so here we go. Aspirin reduced the composite cardiovascular endpoint from 65.2 to 60.2 events per 10,000 patient-years; or to put it more meaningfully in absolute risk reduction terms, because that’s my jam, an absolute risk reduction of 0.41%, which means a number needed to treat of 241, which is okay-ish. It’s not super-great, but it may be justifiable for something that costs next to nothing.
The tradeoff is bleeding. Major bleeding increased from 16.4 to 23.1 bleeds per 10,000 patient-years, or an absolute risk increase of 0.47%, which is a number needed to harm of 210. That’s the problem. Aspirin does prevent heart disease. The benefit is small, for sure, but the real problem is that it’s outweighed by the risk of bleeding, so you’re not really coming out ahead.
The real tragedy here is that the public is locked into this idea of everyone over age 50 years should be taking an aspirin. Even today, even though guidelines have recommended against aspirin for primary prevention for some time, data from the National Health Interview Survey sample found that nearly one in three older adults take aspirin for primary prevention when they shouldn’t be. That’s a large number of people. That’s millions of Americans — and Canadians, but nobody cares about us. It’s fine.
That’s the point. We’re not debunking aspirin. It does work. The benefits are just really small in a primary prevention population and offset by the admittedly also really small risks of bleeding. It’s a tradeoff that doesn’t really work in your favor.
But that’s aspirin for cardiovascular disease. When it comes to cancer or DVT prophylaxis, that’s another really interesting story. We might have to save that for another time. Do I know how to tease a sequel or what?
Labos, a cardiologist at Kirkland Medical Center, Montreal, Quebec, Canada, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Our recommendations vis-à-vis aspirin have evolved at a dizzying pace. The young’uns watching us right now don’t know what things were like in the 1980s. The Reagan era was a wild, heady time where nuclear war was imminent and we didn’t prescribe aspirin to patients.
That only started in 1988, which was a banner year in human history. Not because a number of doves were incinerated by the lighting of the Olympic torch at the Seoul Olympics — look it up if you don’t know what I’m talking about — but because 1988 saw the publication of the ISIS-2 trial, which first showed a mortality benefit to prescribing aspirin post–myocardial infarction (MI).
Giving patients aspirin during or after a heart attack is not controversial. It’s one of the few things in this business that isn’t, but that’s secondary prevention — treating somebody after they develop a disease. Primary prevention, treating them before they have their incident event, is a very different ballgame. Here, things are messy.
For one thing, the doses used have been very inconsistent. We should point out that the reason for 81 mg of aspirin is very arbitrary and is rooted in the old apothecary system of weights and measurements. A standard dose of aspirin was 5 grains, where 20 grains made 1 scruple, 3 scruples made 1 dram, 8 drams made 1 oz, and 12 oz made 1 lb - because screw you, metric system. Therefore, 5 grains was 325 mg of aspirin, and 1 quarter of the standard dose became 81 mg if you rounded out the decimal.
People have tried all kinds of dosing structures with aspirin prophylaxis. The Physicians’ Health Study used a full-dose aspirin, 325 mg every 2 days, while the Hypertension Optimal Treatment (HOT) trial tested 75 mg daily and the Women’s Health Study tested 100 mg, but every other day.
Ironically, almost no one has studied 81 mg every day, which is weird if you think about it. The bigger problem here is not the variability of doses used, but the discrepancy when you look at older vs newer studies.
Older studies, like the Physicians’ Health Study, did show a benefit, at least in the subgroup of patients over age 50 years, which is probably where the “everybody over 50 should be taking an aspirin” idea comes from, at least as near as I can tell.
More recent studies, like the Women’s Health Study, ASPREE, or ASPIRE, didn’t show a benefit. I know what you’re thinking: Newer stuff is always better. That’s why you should never trust anybody over age 40 years. The context of primary prevention studies has changed. In the ‘80s and ‘90s, people smoked more and we didn’t have the same medications that we have today. We talked about all this in the beta-blocker video to explain why beta-blockers don’t seem to have a benefit post MI.
We have a similar issue here. The magnitude of the benefit with aspirin primary prevention has decreased because we’re all just healthier overall. So, yay! Progress! Here’s where the numbers matter. No one is saying that aspirin doesn’t help. It does.
If we look at the 2019 meta-analysis published in JAMA, there is a cardiovascular benefit. The numbers bear that out. I know you’re all here for the math, so here we go. Aspirin reduced the composite cardiovascular endpoint from 65.2 to 60.2 events per 10,000 patient-years; or to put it more meaningfully in absolute risk reduction terms, because that’s my jam, an absolute risk reduction of 0.41%, which means a number needed to treat of 241, which is okay-ish. It’s not super-great, but it may be justifiable for something that costs next to nothing.
The tradeoff is bleeding. Major bleeding increased from 16.4 to 23.1 bleeds per 10,000 patient-years, or an absolute risk increase of 0.47%, which is a number needed to harm of 210. That’s the problem. Aspirin does prevent heart disease. The benefit is small, for sure, but the real problem is that it’s outweighed by the risk of bleeding, so you’re not really coming out ahead.
The real tragedy here is that the public is locked into this idea of everyone over age 50 years should be taking an aspirin. Even today, even though guidelines have recommended against aspirin for primary prevention for some time, data from the National Health Interview Survey sample found that nearly one in three older adults take aspirin for primary prevention when they shouldn’t be. That’s a large number of people. That’s millions of Americans — and Canadians, but nobody cares about us. It’s fine.
That’s the point. We’re not debunking aspirin. It does work. The benefits are just really small in a primary prevention population and offset by the admittedly also really small risks of bleeding. It’s a tradeoff that doesn’t really work in your favor.
But that’s aspirin for cardiovascular disease. When it comes to cancer or DVT prophylaxis, that’s another really interesting story. We might have to save that for another time. Do I know how to tease a sequel or what?
Labos, a cardiologist at Kirkland Medical Center, Montreal, Quebec, Canada, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
This transcript has been edited for clarity.
Our recommendations vis-à-vis aspirin have evolved at a dizzying pace. The young’uns watching us right now don’t know what things were like in the 1980s. The Reagan era was a wild, heady time where nuclear war was imminent and we didn’t prescribe aspirin to patients.
That only started in 1988, which was a banner year in human history. Not because a number of doves were incinerated by the lighting of the Olympic torch at the Seoul Olympics — look it up if you don’t know what I’m talking about — but because 1988 saw the publication of the ISIS-2 trial, which first showed a mortality benefit to prescribing aspirin post–myocardial infarction (MI).
Giving patients aspirin during or after a heart attack is not controversial. It’s one of the few things in this business that isn’t, but that’s secondary prevention — treating somebody after they develop a disease. Primary prevention, treating them before they have their incident event, is a very different ballgame. Here, things are messy.
For one thing, the doses used have been very inconsistent. We should point out that the reason for 81 mg of aspirin is very arbitrary and is rooted in the old apothecary system of weights and measurements. A standard dose of aspirin was 5 grains, where 20 grains made 1 scruple, 3 scruples made 1 dram, 8 drams made 1 oz, and 12 oz made 1 lb - because screw you, metric system. Therefore, 5 grains was 325 mg of aspirin, and 1 quarter of the standard dose became 81 mg if you rounded out the decimal.
People have tried all kinds of dosing structures with aspirin prophylaxis. The Physicians’ Health Study used a full-dose aspirin, 325 mg every 2 days, while the Hypertension Optimal Treatment (HOT) trial tested 75 mg daily and the Women’s Health Study tested 100 mg, but every other day.
Ironically, almost no one has studied 81 mg every day, which is weird if you think about it. The bigger problem here is not the variability of doses used, but the discrepancy when you look at older vs newer studies.
Older studies, like the Physicians’ Health Study, did show a benefit, at least in the subgroup of patients over age 50 years, which is probably where the “everybody over 50 should be taking an aspirin” idea comes from, at least as near as I can tell.
More recent studies, like the Women’s Health Study, ASPREE, or ASPIRE, didn’t show a benefit. I know what you’re thinking: Newer stuff is always better. That’s why you should never trust anybody over age 40 years. The context of primary prevention studies has changed. In the ‘80s and ‘90s, people smoked more and we didn’t have the same medications that we have today. We talked about all this in the beta-blocker video to explain why beta-blockers don’t seem to have a benefit post MI.
We have a similar issue here. The magnitude of the benefit with aspirin primary prevention has decreased because we’re all just healthier overall. So, yay! Progress! Here’s where the numbers matter. No one is saying that aspirin doesn’t help. It does.
If we look at the 2019 meta-analysis published in JAMA, there is a cardiovascular benefit. The numbers bear that out. I know you’re all here for the math, so here we go. Aspirin reduced the composite cardiovascular endpoint from 65.2 to 60.2 events per 10,000 patient-years; or to put it more meaningfully in absolute risk reduction terms, because that’s my jam, an absolute risk reduction of 0.41%, which means a number needed to treat of 241, which is okay-ish. It’s not super-great, but it may be justifiable for something that costs next to nothing.
The tradeoff is bleeding. Major bleeding increased from 16.4 to 23.1 bleeds per 10,000 patient-years, or an absolute risk increase of 0.47%, which is a number needed to harm of 210. That’s the problem. Aspirin does prevent heart disease. The benefit is small, for sure, but the real problem is that it’s outweighed by the risk of bleeding, so you’re not really coming out ahead.
The real tragedy here is that the public is locked into this idea of everyone over age 50 years should be taking an aspirin. Even today, even though guidelines have recommended against aspirin for primary prevention for some time, data from the National Health Interview Survey sample found that nearly one in three older adults take aspirin for primary prevention when they shouldn’t be. That’s a large number of people. That’s millions of Americans — and Canadians, but nobody cares about us. It’s fine.
That’s the point. We’re not debunking aspirin. It does work. The benefits are just really small in a primary prevention population and offset by the admittedly also really small risks of bleeding. It’s a tradeoff that doesn’t really work in your favor.
But that’s aspirin for cardiovascular disease. When it comes to cancer or DVT prophylaxis, that’s another really interesting story. We might have to save that for another time. Do I know how to tease a sequel or what?
Labos, a cardiologist at Kirkland Medical Center, Montreal, Quebec, Canada, has disclosed no relevant financial relationships.
A version of this article appeared on Medscape.com.
Outpatient CAR T: Safe, Effective, Accessible
In one recent study, an industry-funded phase 2 trial, researchers found similar outcomes from outpatient and inpatient CAR T-cell therapy for relapsed/refractory large B-cell lymphoma with lisocabtagene maraleucel (Breyanzi).
Another recent study reported that outpatient treatment of B cell non-Hodgkin lymphoma with tisagenlecleucel (Kymriah) had similar efficacy to inpatient treatment. Meanwhile, a 2023 review of CAR T-cell therapy in various settings found similar outcomes in outpatient and inpatient treatment.
“The future of CAR T-cell therapy lies in balancing safety with accessibility,” said Rayne Rouce, MD, a pediatric oncologist at Texas Children’s Cancer Center in Houston, Texas, in an interview. “Expanding CAR T-cell therapy beyond large medical centers is a critical next step.”
Great Outcomes, Low Access
Since 2017, the FDA has approved six CAR T-cell therapies, which target cancer by harnessing the power of a patient’s own T cells. As an Oregon Health & Sciences University/Knight Cancer Center website explains, T cells are removed from the patient’s body, “genetically modified to make the chimeric antigen receptor, or CAR, [which] protein binds to specific proteins on the surface of cancer cells.”
Modified cells are grown and then infused back into the body, where they “multiply and may be able to destroy all the cancer cells.”
As Rouce puts it, “CAR T-cells have revolutionized the treatment of relapsed or refractory blood cancers.” One or more of the therapies have been approved to treat types of lymphoblastic leukemia, B-cell lymphoma, follicular lymphoma, mantle cell lymphoma, and multiple myeloma.
A 2023 review of clinical trial data reported complete response rates of 40%-54% in aggressive B-cell lymphoma, 67% in mantle cell lymphoma, and 69%-74% in indolent B cell lymphoma.
“Commercialization of CAR T-cell therapy brought hope that access would expand beyond the major academic medical centers with the highly specialized infrastructure and advanced laboratories required to manufacture and ultimately treat patients,” Rouce said. “However, it quickly became clear that patients who are underinsured or uninsured — or who live outside the network of the well-resourced institutions that house these therapies — are still unable to access these potentially life-saving therapies.”
A 2024 report estimated the cost of CAR T-cell therapy as $700,000-$1 million and said only a small percentage of those who could benefit from the treatment actually get it. For example, an estimated 10,000 patients with diffuse large B-cell lymphoma alone could benefit from CAR T therapy annually, but a survey of 200 US healthcare centers in 2021 found that 1900 procedures were performed overall for all indications.
Distance to Treatment Is a Major Obstacle
Even if patients have insurance plans willing to cover CAR T-cell therapy, they may not be able get care. While more than 150 US centers are certified to administer the therapy, “distance to major medical centers with CAR T capabilities is a major obstacle,” Yuliya Linhares, MD, chief of lymphoma at Miami Cancer Institute in Miami, Florida, said in an interview.
“I have had patients who chose to not proceed with CAR T therapy due to inability to travel the distance to the medical center for pre-CAR T appointments and assessments and a lack of caretakers who are available to stay nearby,” Linhares said.
Indeed, the challenges facing patients in rural and underserved urban areas can be overwhelming, Hoda Badr, PhD, professor of medicine at Baylor College of Medicine in Houston, Texas, said in an interview.
“They must take time off work, arrange accommodations near treatment sites, and manage travel costs, all of which strain limited financial resources. The inability to afford these additional expenses can lead to delays in receiving care or patients forgoing the treatment altogether,” Badr said. She added that “the psychological and social burden of being away from family and community support systems during treatment can intensify the stress of an already difficult situation.”
A statistic tells the story of the urban/community divide. CAR T-cell therapy administration at academic centers after leukapheresis — the separation and collection of white blood cells — is reported to be at around 90%, while it’s only 47% in community-based practices that have to refer patients elsewhere, Linhares noted.
Researchers Explore CAR T-Cell Therapy in the Community
Linhares is lead author of the phase 2 trial that explored administration of lisocabtagene maraleucel in 82 patients with relapsed/refractory large B-cell lymphoma. The findings were published Sept. 30 in Blood Advances.
The OUTREACH trial, funded by Juno/Bristol-Myers Squibb, treated patients in the third line and beyond at community medical centers (outpatient-monitored, 70%; inpatient-monitored, 30%). The trial didn’t require facilities to be certified by the Foundation for the Accreditation of Cellular Therapy (FACT); all had to be non-tertiary cancer centers that weren’t associated with a university. In order to administer therapy on the outpatient basis, the centers had to have phase 1 or hematopoietic stem cell transplant capabilities.
As Linhares explained, 72% of participating centers hadn’t provided CAR T-cell therapy before, and 44% did not have FACT accreditation. “About 32% of patients received CAR T at CAR T naive sites, while 70% of patients received CAR T as outpatients. Investigators had to decide whether patients qualified for the outpatient observation or had to be admitted for the inpatient observation,” she noted.
Community Outcomes Were Comparable to Major Trial
As for the results, grade 3 or higher adverse events occurred at a similar frequency among outpatients and inpatients at 74% and 76%, Linhares said. There were no grade 5 adverse events, and 25% of patients treated as outpatients were never hospitalized.
Response rates were similar to those in the major TRANSCEND trial with the objective response rates rate of 80% and complete response rates of 54%.
“Overall,” Linhares said, “our study demonstrated that with the availability of standard operating procedures, specially trained staff and a multidisciplinary team trained in CAR T toxicity management, inpatient and outpatient CAR T administration is feasible at specialized community medical centers.”
In 2023, another study examined patients with B-cell non-Hodgkin lymphoma who were treated on an outpatient basis with tisagenlecleucel. Researchers reported that outpatient therapy was “feasible and associated with similar efficacy outcomes as inpatient treatment.”
And a 2023 systematic literature review identified 11 studies that reported outpatient vs inpatient outcomes in CAR T-cell therapy and found “comparable response rates (80-82% in outpatient and 72-80% in inpatient).” Costs were cheaper in the outpatient setting.
Research findings like these are good news, Baylor College of Medicine’s Badr said. “Outpatient administration could help to scale the availability of this therapy to a broader range of healthcare settings, including those serving underserved populations. Findings indicate promising safety profiles, which is encouraging for expanding access.”
Not Every Patient Can Tolerate Outpatient Care
Linhares noted that the patients who received outpatient care in the lisocabtagene maraleucel study were in better shape than those in the inpatient group. Those selected for inpatient care had “higher disease risk characteristics, including high grade B cell lymphoma histology, higher disease burden, and having received bridging therapy. This points to the fact that the investigators properly selected patients who were at a higher risk of complications for inpatient observation. Additionally, some patients stayed as inpatient due to social factors, which increases length of stay independently of disease characteristics.”
Specifically, reasons for inpatient monitoring were disease characteristics (48%) including tumor burden and risk of adverse events; psychosocial factors (32%) including lack of caregiver support or transportation; COVID-19 precautions (8%); pre-infusion adverse events (8%) of fever and vasovagal reaction; and principal investigator decision (4%) due to limited hospital experience with CAR T-cell therapy.
Texas Children’s Cancer Center’s Rouce said “certain patients, particularly those with higher risk for complications or those who require intensive monitoring, may not be suited for outpatient CAR T-cell therapy. This may be due to other comorbidities or baseline factors known to predispose to CAR T-related toxicities. However, evidence-based risk mitigation algorithms may still allow closely monitored outpatient treatment, with recognition that hospital admission for incipient side effects may be necessary.”
What’s Next for Access to Therapy?
Rouce noted that her institution, like many others, is offering CAR T-cell therapy on an outpatient basis. “Additionally, continued scientific innovation, such as immediately available, off-the-shelf cell therapies and inducible safety switches, will ultimately improve access,” she said.
Linhares noted a recent advance and highlighted research that’s now in progress. “CAR Ts now have an indication as a second-line therapy in relapsed/refractory large B-cell lymphoma, and there are ongoing clinical trials that will potentially move CAR Ts into the first line,” she said. “Some trials are exploring allogeneic, readily available off-the-shelf CAR T for the treatment of minimal residual disease positive large B-cell lymphoma after completion of first-line therapy.”
These potential advances “are increasing the need for CAR T-capable medical centers,” Linhares noted. “More and more medical centers with expert hematology teams are becoming CAR T-certified, with more patients having access to CAR T.”
Still, she said, “I don’t think access is nearly as good as it should be. Many patients in rural areas are still unable to get this life-saving treatment. “However, “it is very possible that other novel targeted therapies, such as bispecific antibodies, will be used in place of CAR T in areas with poor CAR T access. Bispecific antibody efficacy in various B cell lymphoma histologies are being currently explored.”
Rouce discloses relationships with Novartis and Pfizer. Linhares reports ties with Kyowa Kirin, AbbVie, ADC, BeiGene, Genentech, Gilead, GlaxoSmithKline, Seagen, and TG. Badr has no disclosures.
A version of this article appeared on Medscape.com.
In one recent study, an industry-funded phase 2 trial, researchers found similar outcomes from outpatient and inpatient CAR T-cell therapy for relapsed/refractory large B-cell lymphoma with lisocabtagene maraleucel (Breyanzi).
Another recent study reported that outpatient treatment of B cell non-Hodgkin lymphoma with tisagenlecleucel (Kymriah) had similar efficacy to inpatient treatment. Meanwhile, a 2023 review of CAR T-cell therapy in various settings found similar outcomes in outpatient and inpatient treatment.
“The future of CAR T-cell therapy lies in balancing safety with accessibility,” said Rayne Rouce, MD, a pediatric oncologist at Texas Children’s Cancer Center in Houston, Texas, in an interview. “Expanding CAR T-cell therapy beyond large medical centers is a critical next step.”
Great Outcomes, Low Access
Since 2017, the FDA has approved six CAR T-cell therapies, which target cancer by harnessing the power of a patient’s own T cells. As an Oregon Health & Sciences University/Knight Cancer Center website explains, T cells are removed from the patient’s body, “genetically modified to make the chimeric antigen receptor, or CAR, [which] protein binds to specific proteins on the surface of cancer cells.”
Modified cells are grown and then infused back into the body, where they “multiply and may be able to destroy all the cancer cells.”
As Rouce puts it, “CAR T-cells have revolutionized the treatment of relapsed or refractory blood cancers.” One or more of the therapies have been approved to treat types of lymphoblastic leukemia, B-cell lymphoma, follicular lymphoma, mantle cell lymphoma, and multiple myeloma.
A 2023 review of clinical trial data reported complete response rates of 40%-54% in aggressive B-cell lymphoma, 67% in mantle cell lymphoma, and 69%-74% in indolent B cell lymphoma.
“Commercialization of CAR T-cell therapy brought hope that access would expand beyond the major academic medical centers with the highly specialized infrastructure and advanced laboratories required to manufacture and ultimately treat patients,” Rouce said. “However, it quickly became clear that patients who are underinsured or uninsured — or who live outside the network of the well-resourced institutions that house these therapies — are still unable to access these potentially life-saving therapies.”
A 2024 report estimated the cost of CAR T-cell therapy as $700,000-$1 million and said only a small percentage of those who could benefit from the treatment actually get it. For example, an estimated 10,000 patients with diffuse large B-cell lymphoma alone could benefit from CAR T therapy annually, but a survey of 200 US healthcare centers in 2021 found that 1900 procedures were performed overall for all indications.
Distance to Treatment Is a Major Obstacle
Even if patients have insurance plans willing to cover CAR T-cell therapy, they may not be able get care. While more than 150 US centers are certified to administer the therapy, “distance to major medical centers with CAR T capabilities is a major obstacle,” Yuliya Linhares, MD, chief of lymphoma at Miami Cancer Institute in Miami, Florida, said in an interview.
“I have had patients who chose to not proceed with CAR T therapy due to inability to travel the distance to the medical center for pre-CAR T appointments and assessments and a lack of caretakers who are available to stay nearby,” Linhares said.
Indeed, the challenges facing patients in rural and underserved urban areas can be overwhelming, Hoda Badr, PhD, professor of medicine at Baylor College of Medicine in Houston, Texas, said in an interview.
“They must take time off work, arrange accommodations near treatment sites, and manage travel costs, all of which strain limited financial resources. The inability to afford these additional expenses can lead to delays in receiving care or patients forgoing the treatment altogether,” Badr said. She added that “the psychological and social burden of being away from family and community support systems during treatment can intensify the stress of an already difficult situation.”
A statistic tells the story of the urban/community divide. CAR T-cell therapy administration at academic centers after leukapheresis — the separation and collection of white blood cells — is reported to be at around 90%, while it’s only 47% in community-based practices that have to refer patients elsewhere, Linhares noted.
Researchers Explore CAR T-Cell Therapy in the Community
Linhares is lead author of the phase 2 trial that explored administration of lisocabtagene maraleucel in 82 patients with relapsed/refractory large B-cell lymphoma. The findings were published Sept. 30 in Blood Advances.
The OUTREACH trial, funded by Juno/Bristol-Myers Squibb, treated patients in the third line and beyond at community medical centers (outpatient-monitored, 70%; inpatient-monitored, 30%). The trial didn’t require facilities to be certified by the Foundation for the Accreditation of Cellular Therapy (FACT); all had to be non-tertiary cancer centers that weren’t associated with a university. In order to administer therapy on the outpatient basis, the centers had to have phase 1 or hematopoietic stem cell transplant capabilities.
As Linhares explained, 72% of participating centers hadn’t provided CAR T-cell therapy before, and 44% did not have FACT accreditation. “About 32% of patients received CAR T at CAR T naive sites, while 70% of patients received CAR T as outpatients. Investigators had to decide whether patients qualified for the outpatient observation or had to be admitted for the inpatient observation,” she noted.
Community Outcomes Were Comparable to Major Trial
As for the results, grade 3 or higher adverse events occurred at a similar frequency among outpatients and inpatients at 74% and 76%, Linhares said. There were no grade 5 adverse events, and 25% of patients treated as outpatients were never hospitalized.
Response rates were similar to those in the major TRANSCEND trial with the objective response rates rate of 80% and complete response rates of 54%.
“Overall,” Linhares said, “our study demonstrated that with the availability of standard operating procedures, specially trained staff and a multidisciplinary team trained in CAR T toxicity management, inpatient and outpatient CAR T administration is feasible at specialized community medical centers.”
In 2023, another study examined patients with B-cell non-Hodgkin lymphoma who were treated on an outpatient basis with tisagenlecleucel. Researchers reported that outpatient therapy was “feasible and associated with similar efficacy outcomes as inpatient treatment.”
And a 2023 systematic literature review identified 11 studies that reported outpatient vs inpatient outcomes in CAR T-cell therapy and found “comparable response rates (80-82% in outpatient and 72-80% in inpatient).” Costs were cheaper in the outpatient setting.
Research findings like these are good news, Baylor College of Medicine’s Badr said. “Outpatient administration could help to scale the availability of this therapy to a broader range of healthcare settings, including those serving underserved populations. Findings indicate promising safety profiles, which is encouraging for expanding access.”
Not Every Patient Can Tolerate Outpatient Care
Linhares noted that the patients who received outpatient care in the lisocabtagene maraleucel study were in better shape than those in the inpatient group. Those selected for inpatient care had “higher disease risk characteristics, including high grade B cell lymphoma histology, higher disease burden, and having received bridging therapy. This points to the fact that the investigators properly selected patients who were at a higher risk of complications for inpatient observation. Additionally, some patients stayed as inpatient due to social factors, which increases length of stay independently of disease characteristics.”
Specifically, reasons for inpatient monitoring were disease characteristics (48%) including tumor burden and risk of adverse events; psychosocial factors (32%) including lack of caregiver support or transportation; COVID-19 precautions (8%); pre-infusion adverse events (8%) of fever and vasovagal reaction; and principal investigator decision (4%) due to limited hospital experience with CAR T-cell therapy.
Texas Children’s Cancer Center’s Rouce said “certain patients, particularly those with higher risk for complications or those who require intensive monitoring, may not be suited for outpatient CAR T-cell therapy. This may be due to other comorbidities or baseline factors known to predispose to CAR T-related toxicities. However, evidence-based risk mitigation algorithms may still allow closely monitored outpatient treatment, with recognition that hospital admission for incipient side effects may be necessary.”
What’s Next for Access to Therapy?
Rouce noted that her institution, like many others, is offering CAR T-cell therapy on an outpatient basis. “Additionally, continued scientific innovation, such as immediately available, off-the-shelf cell therapies and inducible safety switches, will ultimately improve access,” she said.
Linhares noted a recent advance and highlighted research that’s now in progress. “CAR Ts now have an indication as a second-line therapy in relapsed/refractory large B-cell lymphoma, and there are ongoing clinical trials that will potentially move CAR Ts into the first line,” she said. “Some trials are exploring allogeneic, readily available off-the-shelf CAR T for the treatment of minimal residual disease positive large B-cell lymphoma after completion of first-line therapy.”
These potential advances “are increasing the need for CAR T-capable medical centers,” Linhares noted. “More and more medical centers with expert hematology teams are becoming CAR T-certified, with more patients having access to CAR T.”
Still, she said, “I don’t think access is nearly as good as it should be. Many patients in rural areas are still unable to get this life-saving treatment. “However, “it is very possible that other novel targeted therapies, such as bispecific antibodies, will be used in place of CAR T in areas with poor CAR T access. Bispecific antibody efficacy in various B cell lymphoma histologies are being currently explored.”
Rouce discloses relationships with Novartis and Pfizer. Linhares reports ties with Kyowa Kirin, AbbVie, ADC, BeiGene, Genentech, Gilead, GlaxoSmithKline, Seagen, and TG. Badr has no disclosures.
A version of this article appeared on Medscape.com.
In one recent study, an industry-funded phase 2 trial, researchers found similar outcomes from outpatient and inpatient CAR T-cell therapy for relapsed/refractory large B-cell lymphoma with lisocabtagene maraleucel (Breyanzi).
Another recent study reported that outpatient treatment of B cell non-Hodgkin lymphoma with tisagenlecleucel (Kymriah) had similar efficacy to inpatient treatment. Meanwhile, a 2023 review of CAR T-cell therapy in various settings found similar outcomes in outpatient and inpatient treatment.
“The future of CAR T-cell therapy lies in balancing safety with accessibility,” said Rayne Rouce, MD, a pediatric oncologist at Texas Children’s Cancer Center in Houston, Texas, in an interview. “Expanding CAR T-cell therapy beyond large medical centers is a critical next step.”
Great Outcomes, Low Access
Since 2017, the FDA has approved six CAR T-cell therapies, which target cancer by harnessing the power of a patient’s own T cells. As an Oregon Health & Sciences University/Knight Cancer Center website explains, T cells are removed from the patient’s body, “genetically modified to make the chimeric antigen receptor, or CAR, [which] protein binds to specific proteins on the surface of cancer cells.”
Modified cells are grown and then infused back into the body, where they “multiply and may be able to destroy all the cancer cells.”
As Rouce puts it, “CAR T-cells have revolutionized the treatment of relapsed or refractory blood cancers.” One or more of the therapies have been approved to treat types of lymphoblastic leukemia, B-cell lymphoma, follicular lymphoma, mantle cell lymphoma, and multiple myeloma.
A 2023 review of clinical trial data reported complete response rates of 40%-54% in aggressive B-cell lymphoma, 67% in mantle cell lymphoma, and 69%-74% in indolent B cell lymphoma.
“Commercialization of CAR T-cell therapy brought hope that access would expand beyond the major academic medical centers with the highly specialized infrastructure and advanced laboratories required to manufacture and ultimately treat patients,” Rouce said. “However, it quickly became clear that patients who are underinsured or uninsured — or who live outside the network of the well-resourced institutions that house these therapies — are still unable to access these potentially life-saving therapies.”
A 2024 report estimated the cost of CAR T-cell therapy as $700,000-$1 million and said only a small percentage of those who could benefit from the treatment actually get it. For example, an estimated 10,000 patients with diffuse large B-cell lymphoma alone could benefit from CAR T therapy annually, but a survey of 200 US healthcare centers in 2021 found that 1900 procedures were performed overall for all indications.
Distance to Treatment Is a Major Obstacle
Even if patients have insurance plans willing to cover CAR T-cell therapy, they may not be able get care. While more than 150 US centers are certified to administer the therapy, “distance to major medical centers with CAR T capabilities is a major obstacle,” Yuliya Linhares, MD, chief of lymphoma at Miami Cancer Institute in Miami, Florida, said in an interview.
“I have had patients who chose to not proceed with CAR T therapy due to inability to travel the distance to the medical center for pre-CAR T appointments and assessments and a lack of caretakers who are available to stay nearby,” Linhares said.
Indeed, the challenges facing patients in rural and underserved urban areas can be overwhelming, Hoda Badr, PhD, professor of medicine at Baylor College of Medicine in Houston, Texas, said in an interview.
“They must take time off work, arrange accommodations near treatment sites, and manage travel costs, all of which strain limited financial resources. The inability to afford these additional expenses can lead to delays in receiving care or patients forgoing the treatment altogether,” Badr said. She added that “the psychological and social burden of being away from family and community support systems during treatment can intensify the stress of an already difficult situation.”
A statistic tells the story of the urban/community divide. CAR T-cell therapy administration at academic centers after leukapheresis — the separation and collection of white blood cells — is reported to be at around 90%, while it’s only 47% in community-based practices that have to refer patients elsewhere, Linhares noted.
Researchers Explore CAR T-Cell Therapy in the Community
Linhares is lead author of the phase 2 trial that explored administration of lisocabtagene maraleucel in 82 patients with relapsed/refractory large B-cell lymphoma. The findings were published Sept. 30 in Blood Advances.
The OUTREACH trial, funded by Juno/Bristol-Myers Squibb, treated patients in the third line and beyond at community medical centers (outpatient-monitored, 70%; inpatient-monitored, 30%). The trial didn’t require facilities to be certified by the Foundation for the Accreditation of Cellular Therapy (FACT); all had to be non-tertiary cancer centers that weren’t associated with a university. In order to administer therapy on the outpatient basis, the centers had to have phase 1 or hematopoietic stem cell transplant capabilities.
As Linhares explained, 72% of participating centers hadn’t provided CAR T-cell therapy before, and 44% did not have FACT accreditation. “About 32% of patients received CAR T at CAR T naive sites, while 70% of patients received CAR T as outpatients. Investigators had to decide whether patients qualified for the outpatient observation or had to be admitted for the inpatient observation,” she noted.
Community Outcomes Were Comparable to Major Trial
As for the results, grade 3 or higher adverse events occurred at a similar frequency among outpatients and inpatients at 74% and 76%, Linhares said. There were no grade 5 adverse events, and 25% of patients treated as outpatients were never hospitalized.
Response rates were similar to those in the major TRANSCEND trial with the objective response rates rate of 80% and complete response rates of 54%.
“Overall,” Linhares said, “our study demonstrated that with the availability of standard operating procedures, specially trained staff and a multidisciplinary team trained in CAR T toxicity management, inpatient and outpatient CAR T administration is feasible at specialized community medical centers.”
In 2023, another study examined patients with B-cell non-Hodgkin lymphoma who were treated on an outpatient basis with tisagenlecleucel. Researchers reported that outpatient therapy was “feasible and associated with similar efficacy outcomes as inpatient treatment.”
And a 2023 systematic literature review identified 11 studies that reported outpatient vs inpatient outcomes in CAR T-cell therapy and found “comparable response rates (80-82% in outpatient and 72-80% in inpatient).” Costs were cheaper in the outpatient setting.
Research findings like these are good news, Baylor College of Medicine’s Badr said. “Outpatient administration could help to scale the availability of this therapy to a broader range of healthcare settings, including those serving underserved populations. Findings indicate promising safety profiles, which is encouraging for expanding access.”
Not Every Patient Can Tolerate Outpatient Care
Linhares noted that the patients who received outpatient care in the lisocabtagene maraleucel study were in better shape than those in the inpatient group. Those selected for inpatient care had “higher disease risk characteristics, including high grade B cell lymphoma histology, higher disease burden, and having received bridging therapy. This points to the fact that the investigators properly selected patients who were at a higher risk of complications for inpatient observation. Additionally, some patients stayed as inpatient due to social factors, which increases length of stay independently of disease characteristics.”
Specifically, reasons for inpatient monitoring were disease characteristics (48%) including tumor burden and risk of adverse events; psychosocial factors (32%) including lack of caregiver support or transportation; COVID-19 precautions (8%); pre-infusion adverse events (8%) of fever and vasovagal reaction; and principal investigator decision (4%) due to limited hospital experience with CAR T-cell therapy.
Texas Children’s Cancer Center’s Rouce said “certain patients, particularly those with higher risk for complications or those who require intensive monitoring, may not be suited for outpatient CAR T-cell therapy. This may be due to other comorbidities or baseline factors known to predispose to CAR T-related toxicities. However, evidence-based risk mitigation algorithms may still allow closely monitored outpatient treatment, with recognition that hospital admission for incipient side effects may be necessary.”
What’s Next for Access to Therapy?
Rouce noted that her institution, like many others, is offering CAR T-cell therapy on an outpatient basis. “Additionally, continued scientific innovation, such as immediately available, off-the-shelf cell therapies and inducible safety switches, will ultimately improve access,” she said.
Linhares noted a recent advance and highlighted research that’s now in progress. “CAR Ts now have an indication as a second-line therapy in relapsed/refractory large B-cell lymphoma, and there are ongoing clinical trials that will potentially move CAR Ts into the first line,” she said. “Some trials are exploring allogeneic, readily available off-the-shelf CAR T for the treatment of minimal residual disease positive large B-cell lymphoma after completion of first-line therapy.”
These potential advances “are increasing the need for CAR T-capable medical centers,” Linhares noted. “More and more medical centers with expert hematology teams are becoming CAR T-certified, with more patients having access to CAR T.”
Still, she said, “I don’t think access is nearly as good as it should be. Many patients in rural areas are still unable to get this life-saving treatment. “However, “it is very possible that other novel targeted therapies, such as bispecific antibodies, will be used in place of CAR T in areas with poor CAR T access. Bispecific antibody efficacy in various B cell lymphoma histologies are being currently explored.”
Rouce discloses relationships with Novartis and Pfizer. Linhares reports ties with Kyowa Kirin, AbbVie, ADC, BeiGene, Genentech, Gilead, GlaxoSmithKline, Seagen, and TG. Badr has no disclosures.
A version of this article appeared on Medscape.com.