User login
Pruritic Eruption on the Trunk and Extremities
THE DIAGNOSIS:
Acquired Perforating Disorder of Renal Disease
A papule with the central plug removed left a pitlike depression, representing Kyrle disease (Figure 1). A punch biopsy of the left forearm revealed epidermal hyperplasia (Figure 2A) surrounding a keratin plug that contained degenerated basophilic material (Figure 2B), confirming the diagnosis of acquired perforating disorder of renal disease (APDRD), classically described as Kyrle disease.
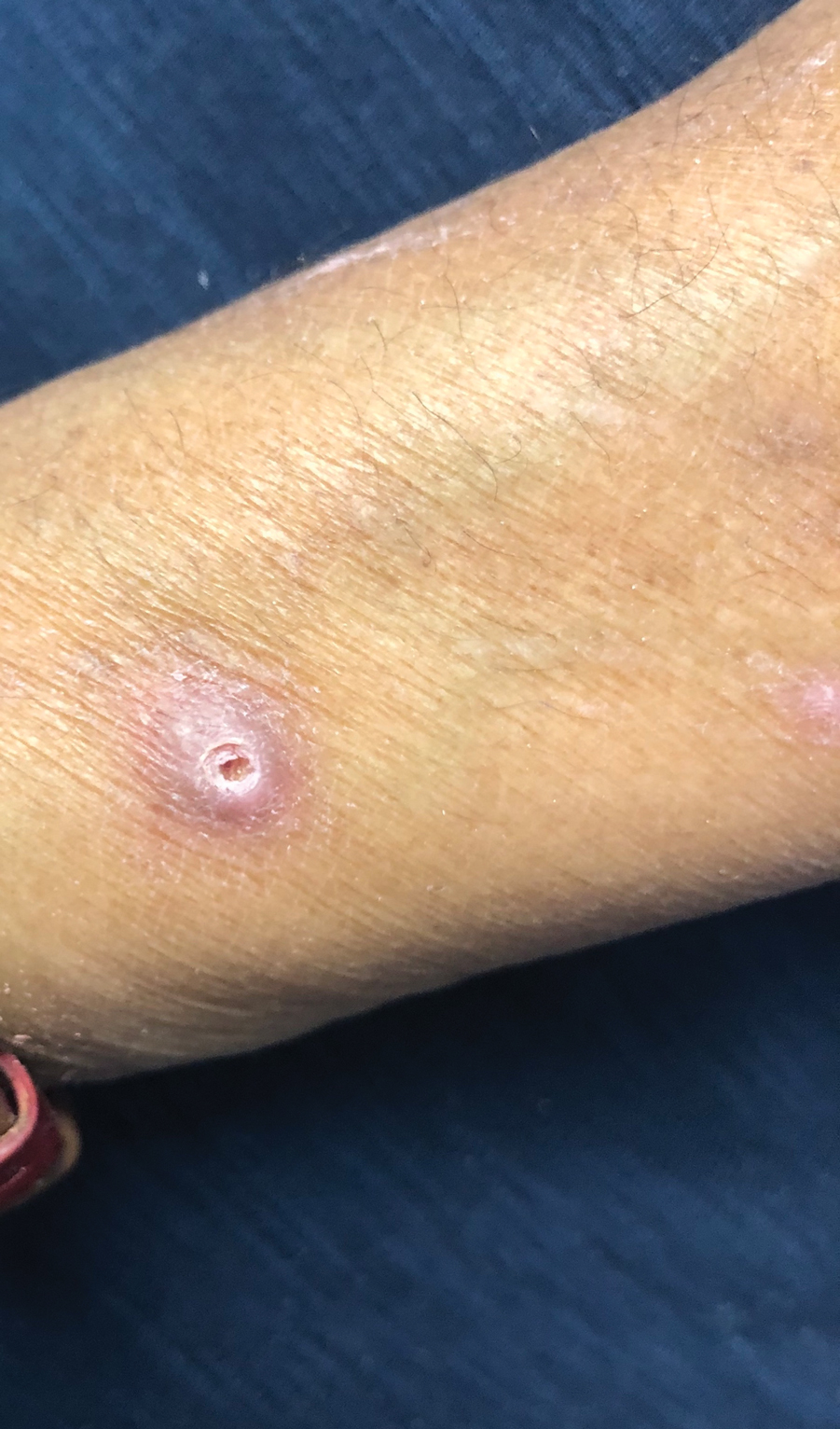
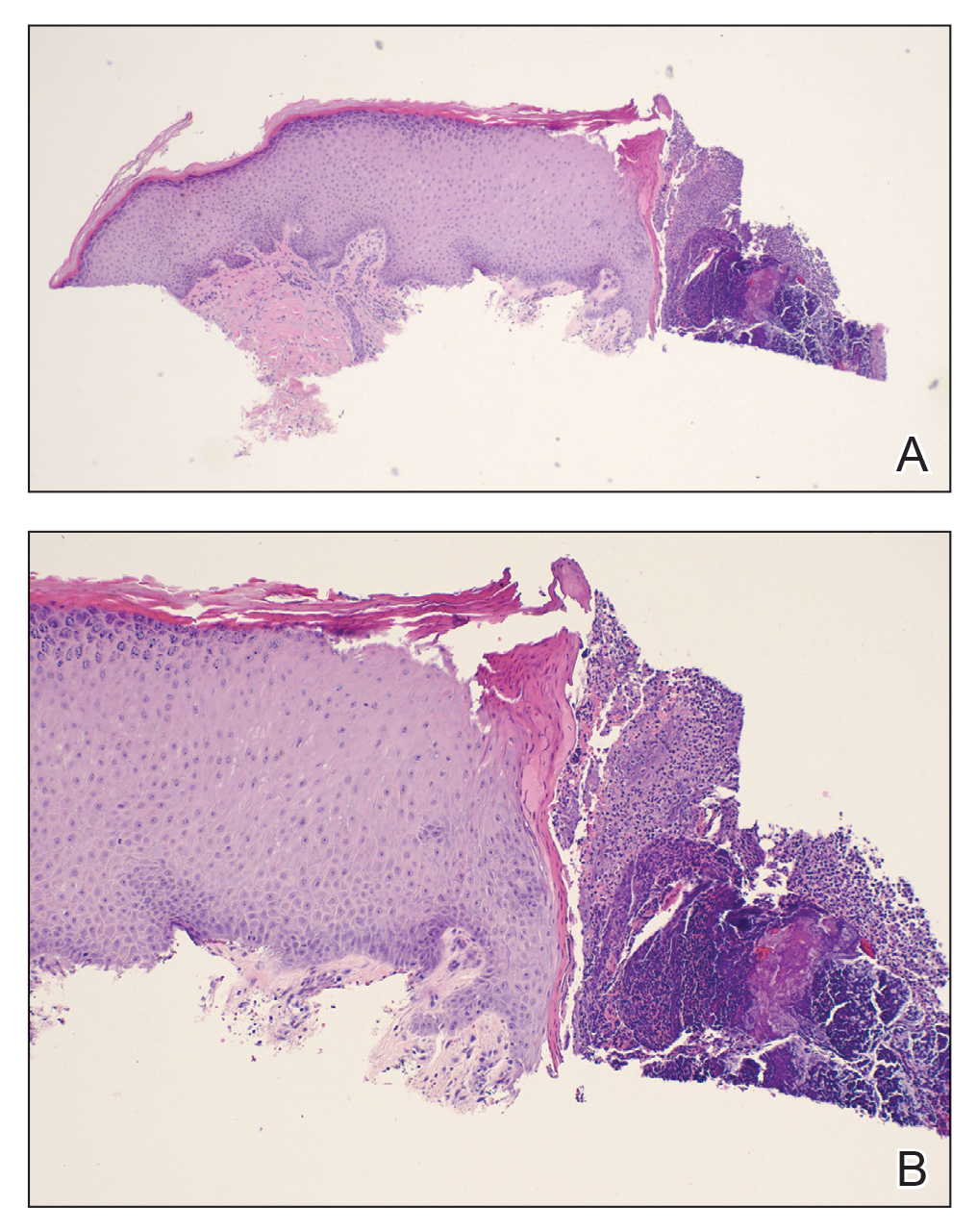
Acquired perforating disorder of renal disease is an uncommon condition in the general population. It is associated with systemic disease, commonly diabetes mellitus and chronic renal failure, and is seen in up to 10% patients receiving hemodialysis.1 The underlying etiology and pathogenesis of APDRD remains unknown. It has been proposed to be a variant of prurigo nodularis, representing end-stage excoriated folliculitis.1 Given that most cases appear in patients with systemic disease and metabolic abnormalities, APDRD also has been classified under the spectrum of acquired perforating dermatoses, a group of disorders defined by transepithelial elimination of dermal connective tissue. Elevated levels of serum and tissue fibronectin, uremia, and hyperphosphatemia have been observed in patients with APDRD.1,2 Fibronectin stimulates epithelial migration and proliferation and may lead to expulsion of keratin. Furthermore, dermal deposition of excess urea and/or phosphate could initiate transepithelial elimination of material. Alternative hypotheses implicate abnormal keratinization or an imbalance between the rates of epidermal proliferation/ differentiation and keratin production, whereby keratin production outpaces the former. Keratin deposited within the dermis subsequently elicits an inflammatory response along with alterations in the local dermis and connective tissue. These components become intermixed and are extruded through the plug opening.3 Lastly, immune dysregulation resulting from systemic disease could contribute to APDRD through increased expression of IL-31, a cytokine thought to play a role in several pruritic inflammatory skin diseases.4
Although standardized treatment guidelines for APDRD have not been established, the mainstay of therapy is control of the underlying systemic disorder. Intense pruritus and repeated scratching may contribute to microtrauma and subsequent koebnerization of new lesions.3 Thus, ameliorating pruritus can provide both symptomatic relief and prevent the development of new lesions. Retinoids, UV light, oral antibiotics, antihistamines, corticosteroids, keratolytic agents, and immunosuppressants (eg, allopurinol, tacrolimus) have shown some benefit.4
The differential diagnoses for APDRD include arthropod hypersensitivity reactions, eruptive keratoacanthomas, keratosis pilaris, and prurigo nodularis. Arthropod hypersensitivity reactions are seen in patients with a history of a bite or sting from arthropods such as bees, fleas, mites, ticks, and spiders. These reactions cause symptoms of pain, burning, or pruritus and present heterogeneously. They can be edematous and appear as single or multiple papules, pustules, plaques, vesicles, and/or bullae. A central punctum or crusting also may be present. Eruptive keratoacanthomas are seen in Grzybowski syndrome and Ferguson-Smith disease. Grzybowski syndrome arises in the fifth to seventh decades of life and is characterized by the eruptive onset of hundreds to thousands of pruritic, dome-shaped, follicular papules with or without central keratin plugs. Ectropion, mucosal lesions, and masklike facies are other clinical characteristics of Grzybowski syndrome. Ferguson-Smith disease begins in the second decade of life. The eruption of multiple keratoacanthomas and/or squamous cell carcinomas occurs in crops, rapidly growing over 2 to 4 weeks, and then self-resolves. This disease is inherited in an autosomal-dominant manner and is associated with chromosome 9q22. Keratosis pilaris is a benign condition of follicular hyperkeratosis that can appear in any age group and usually is absent of symptoms. It is not associated with any systemic disease. Clinically, the condition appears as folliculocentric keratotic papules with varying degrees of perifollicular erythema located along the extensor surfaces. Keratosis pilaris and APDRD share features of a follicular hyperkeratosis and dilated infundibulum; however, perforation is absent in keratosis pilaris. Lastly, prurigo nodularis is another intensely pruritic dermatosis associated with renal disease that presents as papulonodules on the extensor surfaces of the arms and legs. A biopsy can help to distinguish prurigo nodularis from APDRD.
- Rice AS, Zedek D. Kyrle disease. StatPearls [internet]. StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK532886/
- McKinley-Grant L, Peebles J. Renal disease. In: Kelly A, Taylor SC, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. McGraw-Hill; 2016
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Forouzandeh M, Stratman S, Yosipovitch G. The treatment of Kyrle’s disease: a systematic review. J Eur Acad Dermatol Venereol. 2020;34:1457-1463. doi:10.1111/jdv.16182
THE DIAGNOSIS:
Acquired Perforating Disorder of Renal Disease
A papule with the central plug removed left a pitlike depression, representing Kyrle disease (Figure 1). A punch biopsy of the left forearm revealed epidermal hyperplasia (Figure 2A) surrounding a keratin plug that contained degenerated basophilic material (Figure 2B), confirming the diagnosis of acquired perforating disorder of renal disease (APDRD), classically described as Kyrle disease.
Acquired perforating disorder of renal disease is an uncommon condition in the general population. It is associated with systemic disease, commonly diabetes mellitus and chronic renal failure, and is seen in up to 10% patients receiving hemodialysis.1 The underlying etiology and pathogenesis of APDRD remains unknown. It has been proposed to be a variant of prurigo nodularis, representing end-stage excoriated folliculitis.1 Given that most cases appear in patients with systemic disease and metabolic abnormalities, APDRD also has been classified under the spectrum of acquired perforating dermatoses, a group of disorders defined by transepithelial elimination of dermal connective tissue. Elevated levels of serum and tissue fibronectin, uremia, and hyperphosphatemia have been observed in patients with APDRD.1,2 Fibronectin stimulates epithelial migration and proliferation and may lead to expulsion of keratin. Furthermore, dermal deposition of excess urea and/or phosphate could initiate transepithelial elimination of material. Alternative hypotheses implicate abnormal keratinization or an imbalance between the rates of epidermal proliferation/ differentiation and keratin production, whereby keratin production outpaces the former. Keratin deposited within the dermis subsequently elicits an inflammatory response along with alterations in the local dermis and connective tissue. These components become intermixed and are extruded through the plug opening.3 Lastly, immune dysregulation resulting from systemic disease could contribute to APDRD through increased expression of IL-31, a cytokine thought to play a role in several pruritic inflammatory skin diseases.4
Although standardized treatment guidelines for APDRD have not been established, the mainstay of therapy is control of the underlying systemic disorder. Intense pruritus and repeated scratching may contribute to microtrauma and subsequent koebnerization of new lesions.3 Thus, ameliorating pruritus can provide both symptomatic relief and prevent the development of new lesions. Retinoids, UV light, oral antibiotics, antihistamines, corticosteroids, keratolytic agents, and immunosuppressants (eg, allopurinol, tacrolimus) have shown some benefit.4
The differential diagnoses for APDRD include arthropod hypersensitivity reactions, eruptive keratoacanthomas, keratosis pilaris, and prurigo nodularis. Arthropod hypersensitivity reactions are seen in patients with a history of a bite or sting from arthropods such as bees, fleas, mites, ticks, and spiders. These reactions cause symptoms of pain, burning, or pruritus and present heterogeneously. They can be edematous and appear as single or multiple papules, pustules, plaques, vesicles, and/or bullae. A central punctum or crusting also may be present. Eruptive keratoacanthomas are seen in Grzybowski syndrome and Ferguson-Smith disease. Grzybowski syndrome arises in the fifth to seventh decades of life and is characterized by the eruptive onset of hundreds to thousands of pruritic, dome-shaped, follicular papules with or without central keratin plugs. Ectropion, mucosal lesions, and masklike facies are other clinical characteristics of Grzybowski syndrome. Ferguson-Smith disease begins in the second decade of life. The eruption of multiple keratoacanthomas and/or squamous cell carcinomas occurs in crops, rapidly growing over 2 to 4 weeks, and then self-resolves. This disease is inherited in an autosomal-dominant manner and is associated with chromosome 9q22. Keratosis pilaris is a benign condition of follicular hyperkeratosis that can appear in any age group and usually is absent of symptoms. It is not associated with any systemic disease. Clinically, the condition appears as folliculocentric keratotic papules with varying degrees of perifollicular erythema located along the extensor surfaces. Keratosis pilaris and APDRD share features of a follicular hyperkeratosis and dilated infundibulum; however, perforation is absent in keratosis pilaris. Lastly, prurigo nodularis is another intensely pruritic dermatosis associated with renal disease that presents as papulonodules on the extensor surfaces of the arms and legs. A biopsy can help to distinguish prurigo nodularis from APDRD.
THE DIAGNOSIS:
Acquired Perforating Disorder of Renal Disease
A papule with the central plug removed left a pitlike depression, representing Kyrle disease (Figure 1). A punch biopsy of the left forearm revealed epidermal hyperplasia (Figure 2A) surrounding a keratin plug that contained degenerated basophilic material (Figure 2B), confirming the diagnosis of acquired perforating disorder of renal disease (APDRD), classically described as Kyrle disease.
Acquired perforating disorder of renal disease is an uncommon condition in the general population. It is associated with systemic disease, commonly diabetes mellitus and chronic renal failure, and is seen in up to 10% patients receiving hemodialysis.1 The underlying etiology and pathogenesis of APDRD remains unknown. It has been proposed to be a variant of prurigo nodularis, representing end-stage excoriated folliculitis.1 Given that most cases appear in patients with systemic disease and metabolic abnormalities, APDRD also has been classified under the spectrum of acquired perforating dermatoses, a group of disorders defined by transepithelial elimination of dermal connective tissue. Elevated levels of serum and tissue fibronectin, uremia, and hyperphosphatemia have been observed in patients with APDRD.1,2 Fibronectin stimulates epithelial migration and proliferation and may lead to expulsion of keratin. Furthermore, dermal deposition of excess urea and/or phosphate could initiate transepithelial elimination of material. Alternative hypotheses implicate abnormal keratinization or an imbalance between the rates of epidermal proliferation/ differentiation and keratin production, whereby keratin production outpaces the former. Keratin deposited within the dermis subsequently elicits an inflammatory response along with alterations in the local dermis and connective tissue. These components become intermixed and are extruded through the plug opening.3 Lastly, immune dysregulation resulting from systemic disease could contribute to APDRD through increased expression of IL-31, a cytokine thought to play a role in several pruritic inflammatory skin diseases.4
Although standardized treatment guidelines for APDRD have not been established, the mainstay of therapy is control of the underlying systemic disorder. Intense pruritus and repeated scratching may contribute to microtrauma and subsequent koebnerization of new lesions.3 Thus, ameliorating pruritus can provide both symptomatic relief and prevent the development of new lesions. Retinoids, UV light, oral antibiotics, antihistamines, corticosteroids, keratolytic agents, and immunosuppressants (eg, allopurinol, tacrolimus) have shown some benefit.4
The differential diagnoses for APDRD include arthropod hypersensitivity reactions, eruptive keratoacanthomas, keratosis pilaris, and prurigo nodularis. Arthropod hypersensitivity reactions are seen in patients with a history of a bite or sting from arthropods such as bees, fleas, mites, ticks, and spiders. These reactions cause symptoms of pain, burning, or pruritus and present heterogeneously. They can be edematous and appear as single or multiple papules, pustules, plaques, vesicles, and/or bullae. A central punctum or crusting also may be present. Eruptive keratoacanthomas are seen in Grzybowski syndrome and Ferguson-Smith disease. Grzybowski syndrome arises in the fifth to seventh decades of life and is characterized by the eruptive onset of hundreds to thousands of pruritic, dome-shaped, follicular papules with or without central keratin plugs. Ectropion, mucosal lesions, and masklike facies are other clinical characteristics of Grzybowski syndrome. Ferguson-Smith disease begins in the second decade of life. The eruption of multiple keratoacanthomas and/or squamous cell carcinomas occurs in crops, rapidly growing over 2 to 4 weeks, and then self-resolves. This disease is inherited in an autosomal-dominant manner and is associated with chromosome 9q22. Keratosis pilaris is a benign condition of follicular hyperkeratosis that can appear in any age group and usually is absent of symptoms. It is not associated with any systemic disease. Clinically, the condition appears as folliculocentric keratotic papules with varying degrees of perifollicular erythema located along the extensor surfaces. Keratosis pilaris and APDRD share features of a follicular hyperkeratosis and dilated infundibulum; however, perforation is absent in keratosis pilaris. Lastly, prurigo nodularis is another intensely pruritic dermatosis associated with renal disease that presents as papulonodules on the extensor surfaces of the arms and legs. A biopsy can help to distinguish prurigo nodularis from APDRD.
- Rice AS, Zedek D. Kyrle disease. StatPearls [internet]. StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK532886/
- McKinley-Grant L, Peebles J. Renal disease. In: Kelly A, Taylor SC, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. McGraw-Hill; 2016
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Forouzandeh M, Stratman S, Yosipovitch G. The treatment of Kyrle’s disease: a systematic review. J Eur Acad Dermatol Venereol. 2020;34:1457-1463. doi:10.1111/jdv.16182
- Rice AS, Zedek D. Kyrle disease. StatPearls [internet]. StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK532886/
- McKinley-Grant L, Peebles J. Renal disease. In: Kelly A, Taylor SC, Lim HW, et al, eds. Taylor and Kelly’s Dermatology for Skin of Color. 2nd ed. McGraw-Hill; 2016
- Patterson JW. The perforating disorders. J Am Acad Dermatol. 1984;10:561-581. doi:10.1016/s0190-9622(84)80259-5
- Forouzandeh M, Stratman S, Yosipovitch G. The treatment of Kyrle’s disease: a systematic review. J Eur Acad Dermatol Venereol. 2020;34:1457-1463. doi:10.1111/jdv.16182
A 74-year-old woman with a 30-year history of type 2 diabetes mellitus presented to our dermatology clinic with a pruritic eruption on the trunk, arms, and legs of 2 months’ duration. Several over-the-counter moisturizers had been used without improvement, and the pruritus was notably impacting her sleep. Physical examination revealed discrete, hyperkeratotic, predominantly follicular, eruptive papules with hyperkeratotic plugs diffusely distributed on the trunk, arms, and legs.
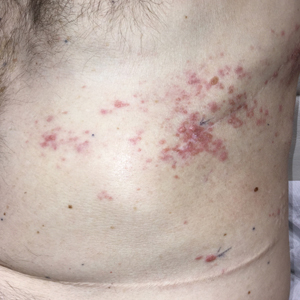
Zosteriform Eruption on the Chest and Abdomen
THE DIAGNOSIS:
Cutaneous Metastatic Mesothelioma
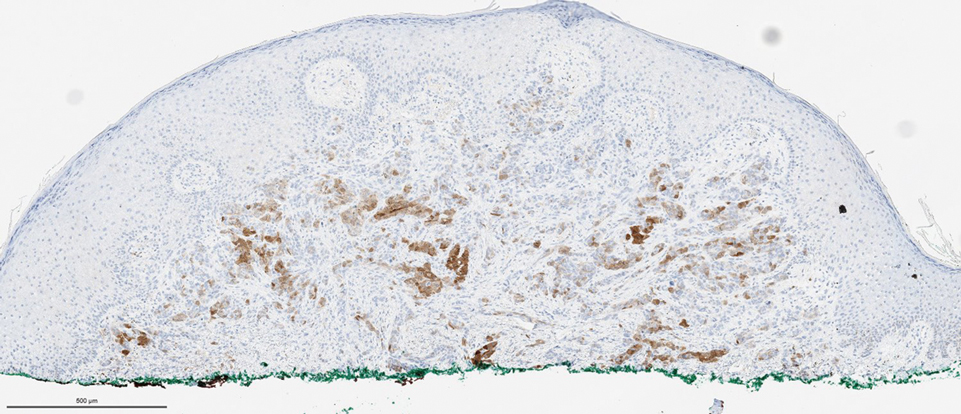
Biopsies of the larger erythematous papules revealed an infiltrate of atypical tumor cells with mitoses (Figure 1) that were immunoreactive for calretinin (Figure 2) and lacked nuclear BRCA1 associated protein-1, BAP1, expression (not shown). The patient’s prior mesothelioma was re-reviewed, and the cutaneous tumor cells were similar to the primary mesothelioma. A diagnosis of cutaneous metastatic mesothelioma (CMM) was made.
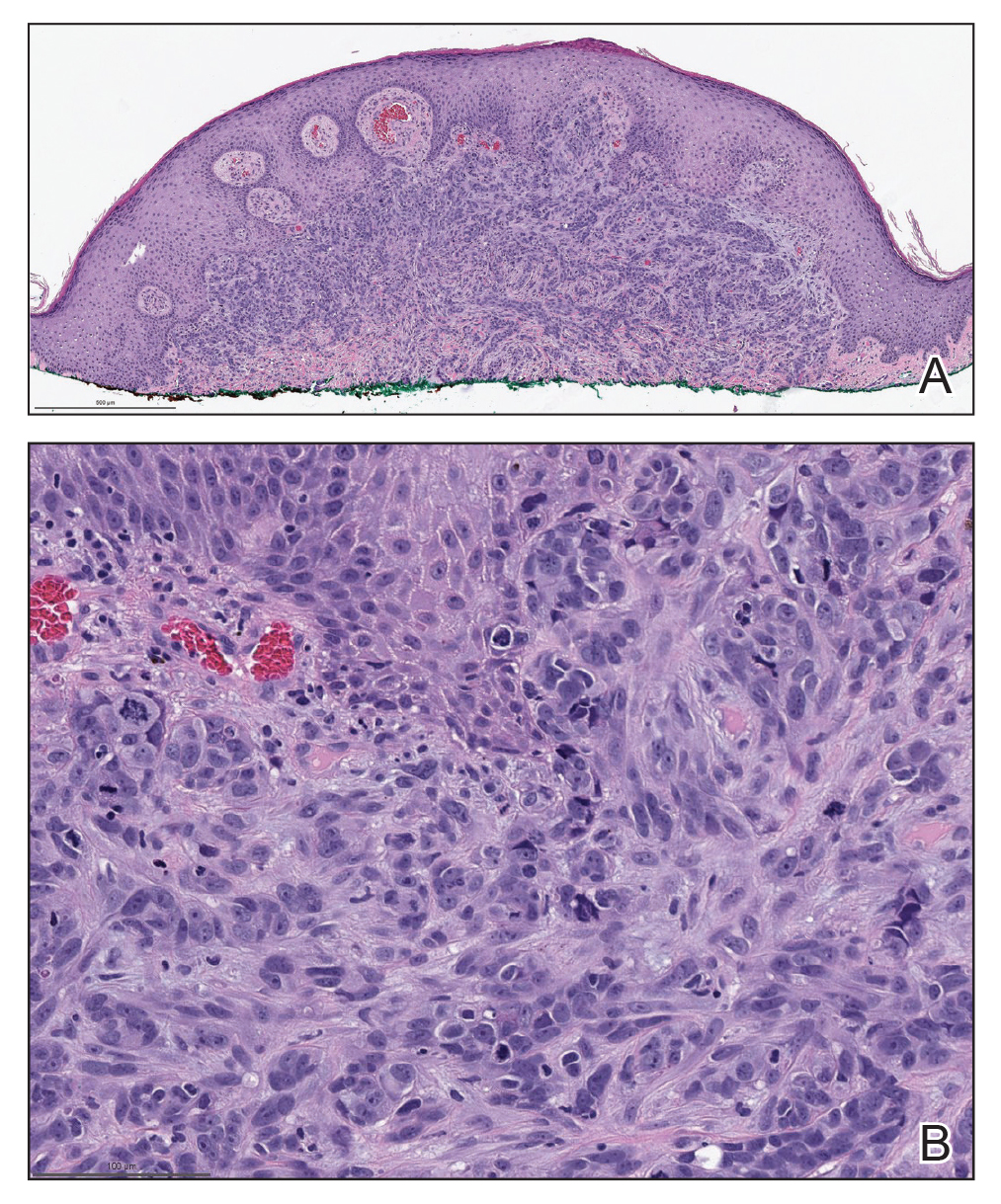
Mesothelioma is a rare neoplasm arising from the pleura, pericardium, peritoneum, and tunica vaginalis,1 with an estimated annual incidence of 2500 cases.2 The predominant risk factor for the development of pleural mesothelioma is asbestos exposure, which has been identified in up to 90% of cases. Mesothelioma can give rise to local and less frequently distant hematogenous metastases. Cutaneous involvement of mesothelioma is rare.3 More than 80% of CMM cases are attributed to seeding the skin at procedure sites or by direct infiltration of scars. Distant CMM is rare and typically presents as subcutaneous nodules.4 Few cases of inflammatory CMM have been published,1,4,5 with even fewer mimicking herpes zoster infection (HZI), as seen in our patient.
The most specific stain for mesothelioma is calretinin, which strongly and diffusely stains both the nucleus and cytoplasm. Other markers include Wilms tumor 1, cytokeratin 5/6, thrombomodulin, and HBME-1. Immunohistochemistry to detect the loss of BAP1 staining in the nucleus is important for differentiating between mesothelioma and mesothelial hyperplasia.3
Cutaneous metastases occur in 0.7% to 9% of patients with internal malignant disease. Most commonly, cutaneous metastases present as cutaneous nodules, though other reported inflammatory presentations include erysipeloides, generalized erythematous patches, telangiectasia, and zosteriform distributions.6 Zosteriform distributions are particularly rare and most commonly are due to breast carcinomas or lymphomas. The mechanism of zosteriform metastasis is unknown, but theories include tumoral spread along vessels, invasion of the thoracic perineural sheaths, localized spread of tumor cells from a surgical site, or a Koebner-like reaction at the site of an existing HZI. Regardless of primary tumor type or presentation, cutaneous metastasis is a poor prognostic sign, with survival rates varying based on primary tumor type.7
Other differential diagnoses include herpes zoster granulomatous dermatitis, radiation recall dermatitis, cutaneous Rosai-Dorfman disease, and zosteriform lichen planus, all of which have been reported after HZI.8-10 Herpes zoster granulomatous dermatitis typically presents weeks to years after acute HZI with erythematous to violaceous papules and plaques at the site of the prior HZI. A biopsy reveals interstitial granulomatous dermatitis and multinucleated giant cells.8 Radiation recall dermatitis is a cutaneous inflammatory reaction limited to regions of prior radiation exposure after the administration of a triggering medication. Radiation recall dermatitis can present days to many years after the completion of treatment.9 Although the eruption in our patient was at the site of prior radiation, the pathologic and clinical presentation was not consistent with radiation recall dermatitis. Cutaneous Rosai-Dorfman disease is a non-Langerhans cell histiocytosis that may present as either solitary or numerous papules, plaques, or nodules and has been reported to occur after HZI. Biopsy reveals a diffuse dermal histiocytic infiltration with plasma cells and lymphocytes. In contrast to metastatic disease, mitoses and nuclear atypia are rare in cutaneous RosaiDorfman disease.11 Lichen planus is an inflammatory disease of unknown etiology presenting as flat-topped, violaceous, pruritic papules12 that may present in a zosteriform pattern.13
Although it is uncommon, metastatic spread should be considered in patients with known malignancy presenting with zosteriform eruptions.2 Our patient remained on treatment with immunotherapy, as he was unable to undergo additional radiation and had failed multiple other lines of therapy. He died 3 months after presentation.
- Klebanov N, Reddy BY, Husain S, et al. Cutaneous presentation of mesothelioma with a sarcomatoid transformation. Am J Dermatopathol. 2018;40:378-382.
- Patel SC, Dowell JE. Modern management of malignant pleural mesothelioma. Lung Cancer (Auckl). 2016;7:63-72.
- Ward RE, Ali SA, Kuhar M. Epithelioid malignant mesothelioma metastatic to the skin: a case report and review of the literature. J Cutan Pathol. 2017;44:1057-1063.
- Prieto VG, Kenet BJ, Varghese M. Malignant mesothelioma metastatic to the skin, presenting as inflammatory carcinoma. Am J Dermatopathol. 1997;19:261-265.
- Gaudy-Marqueste C, Dales JP, Collet-Villette AM, et al. Cutaneous metastasis of pleural mesothelioma: two cases [in French]. Ann Dermatol Venereol. 2003;130:455-459.
- Chiang A, Salomon N, Gaikwad R, et al. A case of cutaneous metastasis mimicking herpes zoster rash. IDCases. 2018;12:167-168.
- Thomaidou E, Armon G, Klapholz L, et al. Zosteriform cutaneous metastases. Clin Exp Dermatol. 2018;43:734-736.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Carrasco L, Pastor MA, Izquierdo MJ, et al. Drug eruption secondary to acyclovir with recall phenomenon in a dermatome previously affected by herpes zoster. Clin Exp Dermatol. 2002;27:132-134.
- Malviya N, Marzuka A, Maamed-Tayeb M, et al. Cutaneous involvement of pre-existing Rosai-Dorfman disease via post-herpetic isotopic response. J Cutan Pathol. 2016;43:1211-1214.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review. Exp Ther Med. 2015;9:1389-1392.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Fink-Puches R, Hofmann-Wellenhof R, Smolle J. Zosteriform lichen planus. Dermatology. 1996;192:375-377.
THE DIAGNOSIS:
Cutaneous Metastatic Mesothelioma
Biopsies of the larger erythematous papules revealed an infiltrate of atypical tumor cells with mitoses (Figure 1) that were immunoreactive for calretinin (Figure 2) and lacked nuclear BRCA1 associated protein-1, BAP1, expression (not shown). The patient’s prior mesothelioma was re-reviewed, and the cutaneous tumor cells were similar to the primary mesothelioma. A diagnosis of cutaneous metastatic mesothelioma (CMM) was made.
Mesothelioma is a rare neoplasm arising from the pleura, pericardium, peritoneum, and tunica vaginalis,1 with an estimated annual incidence of 2500 cases.2 The predominant risk factor for the development of pleural mesothelioma is asbestos exposure, which has been identified in up to 90% of cases. Mesothelioma can give rise to local and less frequently distant hematogenous metastases. Cutaneous involvement of mesothelioma is rare.3 More than 80% of CMM cases are attributed to seeding the skin at procedure sites or by direct infiltration of scars. Distant CMM is rare and typically presents as subcutaneous nodules.4 Few cases of inflammatory CMM have been published,1,4,5 with even fewer mimicking herpes zoster infection (HZI), as seen in our patient.
The most specific stain for mesothelioma is calretinin, which strongly and diffusely stains both the nucleus and cytoplasm. Other markers include Wilms tumor 1, cytokeratin 5/6, thrombomodulin, and HBME-1. Immunohistochemistry to detect the loss of BAP1 staining in the nucleus is important for differentiating between mesothelioma and mesothelial hyperplasia.3
Cutaneous metastases occur in 0.7% to 9% of patients with internal malignant disease. Most commonly, cutaneous metastases present as cutaneous nodules, though other reported inflammatory presentations include erysipeloides, generalized erythematous patches, telangiectasia, and zosteriform distributions.6 Zosteriform distributions are particularly rare and most commonly are due to breast carcinomas or lymphomas. The mechanism of zosteriform metastasis is unknown, but theories include tumoral spread along vessels, invasion of the thoracic perineural sheaths, localized spread of tumor cells from a surgical site, or a Koebner-like reaction at the site of an existing HZI. Regardless of primary tumor type or presentation, cutaneous metastasis is a poor prognostic sign, with survival rates varying based on primary tumor type.7
Other differential diagnoses include herpes zoster granulomatous dermatitis, radiation recall dermatitis, cutaneous Rosai-Dorfman disease, and zosteriform lichen planus, all of which have been reported after HZI.8-10 Herpes zoster granulomatous dermatitis typically presents weeks to years after acute HZI with erythematous to violaceous papules and plaques at the site of the prior HZI. A biopsy reveals interstitial granulomatous dermatitis and multinucleated giant cells.8 Radiation recall dermatitis is a cutaneous inflammatory reaction limited to regions of prior radiation exposure after the administration of a triggering medication. Radiation recall dermatitis can present days to many years after the completion of treatment.9 Although the eruption in our patient was at the site of prior radiation, the pathologic and clinical presentation was not consistent with radiation recall dermatitis. Cutaneous Rosai-Dorfman disease is a non-Langerhans cell histiocytosis that may present as either solitary or numerous papules, plaques, or nodules and has been reported to occur after HZI. Biopsy reveals a diffuse dermal histiocytic infiltration with plasma cells and lymphocytes. In contrast to metastatic disease, mitoses and nuclear atypia are rare in cutaneous RosaiDorfman disease.11 Lichen planus is an inflammatory disease of unknown etiology presenting as flat-topped, violaceous, pruritic papules12 that may present in a zosteriform pattern.13
Although it is uncommon, metastatic spread should be considered in patients with known malignancy presenting with zosteriform eruptions.2 Our patient remained on treatment with immunotherapy, as he was unable to undergo additional radiation and had failed multiple other lines of therapy. He died 3 months after presentation.
THE DIAGNOSIS:
Cutaneous Metastatic Mesothelioma
Biopsies of the larger erythematous papules revealed an infiltrate of atypical tumor cells with mitoses (Figure 1) that were immunoreactive for calretinin (Figure 2) and lacked nuclear BRCA1 associated protein-1, BAP1, expression (not shown). The patient’s prior mesothelioma was re-reviewed, and the cutaneous tumor cells were similar to the primary mesothelioma. A diagnosis of cutaneous metastatic mesothelioma (CMM) was made.
Mesothelioma is a rare neoplasm arising from the pleura, pericardium, peritoneum, and tunica vaginalis,1 with an estimated annual incidence of 2500 cases.2 The predominant risk factor for the development of pleural mesothelioma is asbestos exposure, which has been identified in up to 90% of cases. Mesothelioma can give rise to local and less frequently distant hematogenous metastases. Cutaneous involvement of mesothelioma is rare.3 More than 80% of CMM cases are attributed to seeding the skin at procedure sites or by direct infiltration of scars. Distant CMM is rare and typically presents as subcutaneous nodules.4 Few cases of inflammatory CMM have been published,1,4,5 with even fewer mimicking herpes zoster infection (HZI), as seen in our patient.
The most specific stain for mesothelioma is calretinin, which strongly and diffusely stains both the nucleus and cytoplasm. Other markers include Wilms tumor 1, cytokeratin 5/6, thrombomodulin, and HBME-1. Immunohistochemistry to detect the loss of BAP1 staining in the nucleus is important for differentiating between mesothelioma and mesothelial hyperplasia.3
Cutaneous metastases occur in 0.7% to 9% of patients with internal malignant disease. Most commonly, cutaneous metastases present as cutaneous nodules, though other reported inflammatory presentations include erysipeloides, generalized erythematous patches, telangiectasia, and zosteriform distributions.6 Zosteriform distributions are particularly rare and most commonly are due to breast carcinomas or lymphomas. The mechanism of zosteriform metastasis is unknown, but theories include tumoral spread along vessels, invasion of the thoracic perineural sheaths, localized spread of tumor cells from a surgical site, or a Koebner-like reaction at the site of an existing HZI. Regardless of primary tumor type or presentation, cutaneous metastasis is a poor prognostic sign, with survival rates varying based on primary tumor type.7
Other differential diagnoses include herpes zoster granulomatous dermatitis, radiation recall dermatitis, cutaneous Rosai-Dorfman disease, and zosteriform lichen planus, all of which have been reported after HZI.8-10 Herpes zoster granulomatous dermatitis typically presents weeks to years after acute HZI with erythematous to violaceous papules and plaques at the site of the prior HZI. A biopsy reveals interstitial granulomatous dermatitis and multinucleated giant cells.8 Radiation recall dermatitis is a cutaneous inflammatory reaction limited to regions of prior radiation exposure after the administration of a triggering medication. Radiation recall dermatitis can present days to many years after the completion of treatment.9 Although the eruption in our patient was at the site of prior radiation, the pathologic and clinical presentation was not consistent with radiation recall dermatitis. Cutaneous Rosai-Dorfman disease is a non-Langerhans cell histiocytosis that may present as either solitary or numerous papules, plaques, or nodules and has been reported to occur after HZI. Biopsy reveals a diffuse dermal histiocytic infiltration with plasma cells and lymphocytes. In contrast to metastatic disease, mitoses and nuclear atypia are rare in cutaneous RosaiDorfman disease.11 Lichen planus is an inflammatory disease of unknown etiology presenting as flat-topped, violaceous, pruritic papules12 that may present in a zosteriform pattern.13
Although it is uncommon, metastatic spread should be considered in patients with known malignancy presenting with zosteriform eruptions.2 Our patient remained on treatment with immunotherapy, as he was unable to undergo additional radiation and had failed multiple other lines of therapy. He died 3 months after presentation.
- Klebanov N, Reddy BY, Husain S, et al. Cutaneous presentation of mesothelioma with a sarcomatoid transformation. Am J Dermatopathol. 2018;40:378-382.
- Patel SC, Dowell JE. Modern management of malignant pleural mesothelioma. Lung Cancer (Auckl). 2016;7:63-72.
- Ward RE, Ali SA, Kuhar M. Epithelioid malignant mesothelioma metastatic to the skin: a case report and review of the literature. J Cutan Pathol. 2017;44:1057-1063.
- Prieto VG, Kenet BJ, Varghese M. Malignant mesothelioma metastatic to the skin, presenting as inflammatory carcinoma. Am J Dermatopathol. 1997;19:261-265.
- Gaudy-Marqueste C, Dales JP, Collet-Villette AM, et al. Cutaneous metastasis of pleural mesothelioma: two cases [in French]. Ann Dermatol Venereol. 2003;130:455-459.
- Chiang A, Salomon N, Gaikwad R, et al. A case of cutaneous metastasis mimicking herpes zoster rash. IDCases. 2018;12:167-168.
- Thomaidou E, Armon G, Klapholz L, et al. Zosteriform cutaneous metastases. Clin Exp Dermatol. 2018;43:734-736.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Carrasco L, Pastor MA, Izquierdo MJ, et al. Drug eruption secondary to acyclovir with recall phenomenon in a dermatome previously affected by herpes zoster. Clin Exp Dermatol. 2002;27:132-134.
- Malviya N, Marzuka A, Maamed-Tayeb M, et al. Cutaneous involvement of pre-existing Rosai-Dorfman disease via post-herpetic isotopic response. J Cutan Pathol. 2016;43:1211-1214.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review. Exp Ther Med. 2015;9:1389-1392.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Fink-Puches R, Hofmann-Wellenhof R, Smolle J. Zosteriform lichen planus. Dermatology. 1996;192:375-377.
- Klebanov N, Reddy BY, Husain S, et al. Cutaneous presentation of mesothelioma with a sarcomatoid transformation. Am J Dermatopathol. 2018;40:378-382.
- Patel SC, Dowell JE. Modern management of malignant pleural mesothelioma. Lung Cancer (Auckl). 2016;7:63-72.
- Ward RE, Ali SA, Kuhar M. Epithelioid malignant mesothelioma metastatic to the skin: a case report and review of the literature. J Cutan Pathol. 2017;44:1057-1063.
- Prieto VG, Kenet BJ, Varghese M. Malignant mesothelioma metastatic to the skin, presenting as inflammatory carcinoma. Am J Dermatopathol. 1997;19:261-265.
- Gaudy-Marqueste C, Dales JP, Collet-Villette AM, et al. Cutaneous metastasis of pleural mesothelioma: two cases [in French]. Ann Dermatol Venereol. 2003;130:455-459.
- Chiang A, Salomon N, Gaikwad R, et al. A case of cutaneous metastasis mimicking herpes zoster rash. IDCases. 2018;12:167-168.
- Thomaidou E, Armon G, Klapholz L, et al. Zosteriform cutaneous metastases. Clin Exp Dermatol. 2018;43:734-736.
- Ferenczi K, Rosenberg AS, McCalmont TH, et al. Herpes zoster granulomatous dermatitis: histopathologic findings in a case series. J Cutan Pathol. 2015;42:739-745.
- Carrasco L, Pastor MA, Izquierdo MJ, et al. Drug eruption secondary to acyclovir with recall phenomenon in a dermatome previously affected by herpes zoster. Clin Exp Dermatol. 2002;27:132-134.
- Malviya N, Marzuka A, Maamed-Tayeb M, et al. Cutaneous involvement of pre-existing Rosai-Dorfman disease via post-herpetic isotopic response. J Cutan Pathol. 2016;43:1211-1214.
- Fang S, Chen AJ. Facial cutaneous Rosai-Dorfman disease: a case report and literature review. Exp Ther Med. 2015;9:1389-1392.
- Le Cleach L, Chosidow O. Clinical practice. lichen planus. N Engl J Med. 2012;366:723-732.
- Fink-Puches R, Hofmann-Wellenhof R, Smolle J. Zosteriform lichen planus. Dermatology. 1996;192:375-377.
A 50-year-old man presented with erythematous macules and papules with a dermatomal distribution on the left thoracic region with associated pain of 3 weeks’ duration. The lesions persisted after treatment for herpes zoster. His medical history was notable for mesothelioma that was diagnosed 6 years prior and was treated with ipilimumab and nivolumab following multiple lines of chemotherapy and investigational agents, left thoracotomy, extrapleural pneumonectomy, diaphragmatic reconstruction, and left chest radiation. His medical history also included Hodgkin lymphoma diagnosed 36 years prior that was treated with an appendectomy, splenectomy, systemic chemotherapy, and radiation. Three weeks prior to the current presentation, he was treated by oncology with valacyclovir 1 g 3 times daily for 7 days for presumed herpes zoster without improvement. Physical examination revealed the absence of vesicles, as well as firm, 1- to 6-mm, erythematous papules and plaques, including a few outside of the most affected dermatomes.
Vegetative Plaques on the Face
THE DIAGNOSIS: Vegetative Majocchi Granuloma
A biopsy and tissue culture showed acute dermal inflammation with granulomatous features and numerous fungal hyphae within the stratum corneum (Figure 1A), which were confirmed on GrocottGomori methenamine-silver staining (Figure 1B). Gram and Fite stains were negative for bacteria. A tissue culture speciated Trichophyton rubrum, which led to a diagnosis of deep dermatophyte infection (Majocchi granuloma) with a highly unusual clinical presentation of vegetative plaques. Predisposing factors included treatment with topical corticosteroids and possibly poor health and nutritional status at baseline. Our patient was treated with fluconazole 200 mg daily for 6 weeks, with near resolution of lesions at 3-week follow-up (Figure 2).
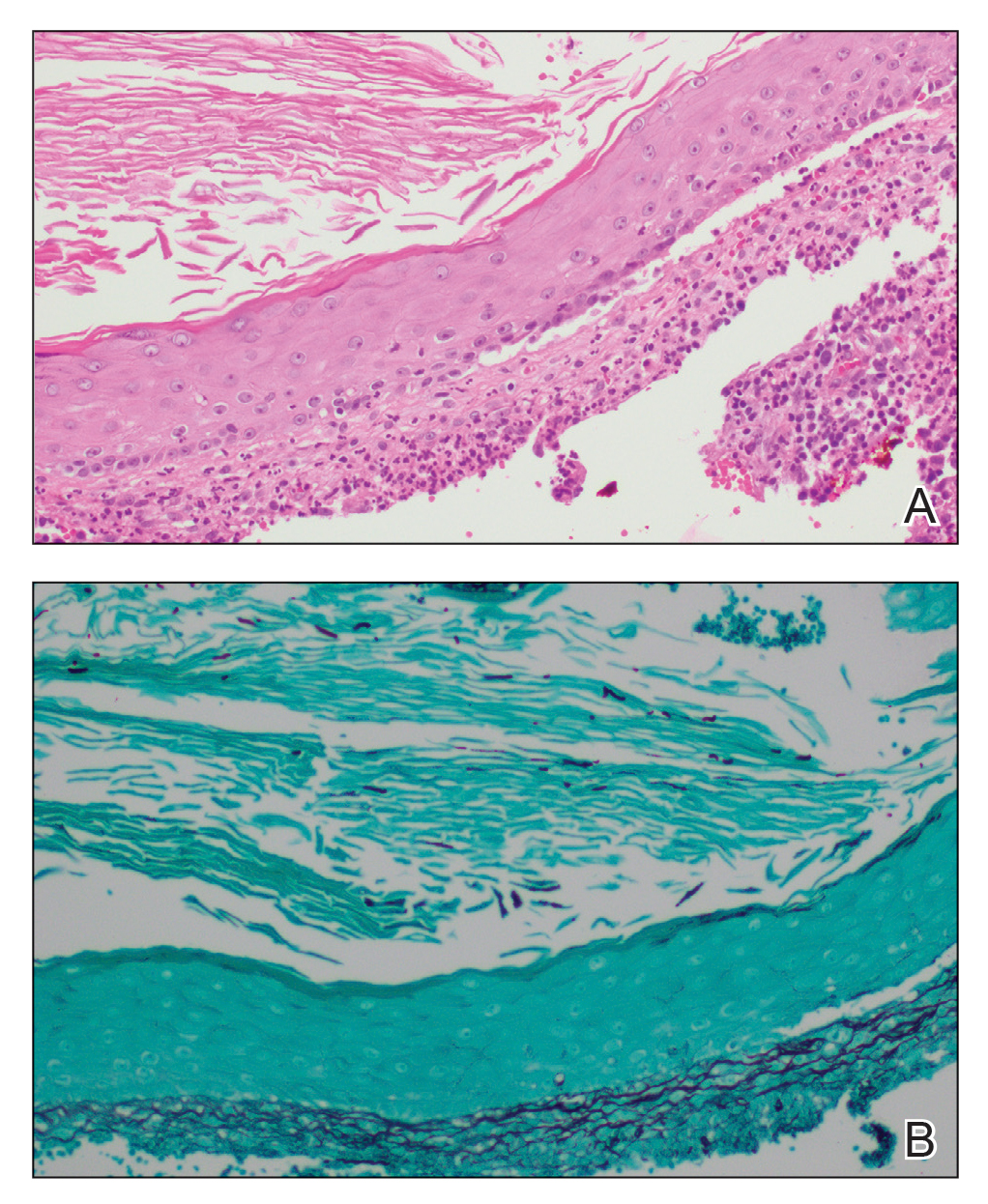
Dermatophytes are a common cause of superficial skin infections. The classic morphology consists of an annular scaly plaque; however, a wide variety of presentations have been observed (eg, verrucous, vesicular, pustular, granulomatous). Therefore, dermatophyte infections often mimic other dermatologic conditions, including atopic dermatitis, rosacea, psoriasis, bacterial abscess, erythema gyratum repens, lupus, granuloma annulare, cutaneous lymphoma, Hailey-Hailey disease, scarring alopecia, and syphilis.1
Notably, when dermatophytes grow downward along hair follicles causing deeper infection, disruption of the follicular wall can lead to an excessive inflammatory response with granulomatous features.2 Risk factors include cutaneous trauma, long-standing infection, immunocompromise, and treatment with topical corticosteroids.3 This disease evolution clinically appears as a nodule or infiltrated plaque, often without scale. The most well-known example is a kerion on the scalp. Elsewhere on the body, lesions often are termed Majocchi granulomas.2
Vegetative plaques, as seen in our patient, are a highly unusual morphology for deep tinea infection. Guanziroli et al4 reported a case of vegetative lesions on the forearm of a 67-year-old immunocompromised man that were successfully treated with a 3-month course of oral terbinafine after Trichophyton verrucosum was isolated. Skorepova et al5 reported a case of pyoderma vegetans triggered by recurrent Trichophyton mentagrophytes on the dorsal hands of a 64-year-old man with immunoglobulin deficiency of unknown etiology. The lesions were successfully treated with a prolonged course of doxycycline, topical triamcinolone, and intravenous immunoglobulin following 2 initial courses of terbinafine.
The differential diagnosis for vegetative lesions includes pemphigus vegetans, a vegetative variant of pyoderma gangrenosum; halogenoderma; and a variety of infections, including dimorphic fungi (histoplasmosis, blastomycosis), blastomycosislike pyoderma (bacterial), and candidiasis.6 These conditions usually can be distinguished based on histopathology. Clinically, pemphigus vegetans presents with pustules and vegetative lesions, as in our patient, but usually is more diffuse and favors the intertriginous areas. Histology likely would reveal foci of acantholysis and eosinophils. Vegetative pyoderma gangrenosum favors the trunk, particularly in sites of surgical trauma. In our patient, no lesions were present near the abdominal surgical sites, and there was no antecedent cribriform ulceration. Halogenoderma was a strong initial consideration given the localization, presence of large pustules, and history of numerous contrast computed tomography studies; however, our patient’s iodine levels were normal. Infectious etiologies including dimorphic fungi and blastomycosislike pyoderma generally are not restricted to the head and neck, and tissue culture helps exclude them. Vegetative lesions may occur in the setting of other infections, and tissue culture may be necessary to differentiate them if histopathology is not suggestive.
Deep dermatophyte infections require treatment with oral antifungals, as topicals do not penetrate adequately into the hair follicles. Exact regimens vary, but generally oral terbinafine or an oral azole (except ketoconazole) is administered for 2 to 6 weeks, with immunocompromise necessitating longer courses.
We present a rare case of vegetative Majocchi granuloma secondary to T rubrum infection. A dermatophyte infection should be included in the differential for vegetative lesions, especially in dense hair-bearing areas such as the beard. Treatment generally is straightforward with oral antifungals.
- Atzori L, Pau M, Aste N, et al. Dermatophyte infections mimicking other skin diseases: a 154-person case survey of tinea atypica in the district of Cagliari (Italy). Int J Dermatol. 2012;51:410-415.
- Ilkit M, Durdu M, Karakas M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Jevremovic L, Ilijin I, Kostic K, et al. Pyoderma vegetans—a case report. Serbian J Dermatol Venereol. 2017;9:22-28.
- Guanziroli E, Pavia G, Guttadauro A, et al. Deep dermatophytosis caused by Trichophyton verrucosum in an immunosuppressed patient: successful outcome with terbinafine. Mycopathologia. 2019;184:543-545.
- Skorepová M, Stuchlík D. Chronic pyoderma vegetans triggered by Trichophyton mentagrophytes. Mycoses. 2006;49:143-144.
- Reinholz M, Hermans C, Dietrich A, et al. A case of cutaneous vegetating candidiasis in a patient with keratitis-ichthyosis-deafness syndrome. J Eur Acad Dermatol Venereol. 2016;30:537-539.
THE DIAGNOSIS: Vegetative Majocchi Granuloma
A biopsy and tissue culture showed acute dermal inflammation with granulomatous features and numerous fungal hyphae within the stratum corneum (Figure 1A), which were confirmed on GrocottGomori methenamine-silver staining (Figure 1B). Gram and Fite stains were negative for bacteria. A tissue culture speciated Trichophyton rubrum, which led to a diagnosis of deep dermatophyte infection (Majocchi granuloma) with a highly unusual clinical presentation of vegetative plaques. Predisposing factors included treatment with topical corticosteroids and possibly poor health and nutritional status at baseline. Our patient was treated with fluconazole 200 mg daily for 6 weeks, with near resolution of lesions at 3-week follow-up (Figure 2).
Dermatophytes are a common cause of superficial skin infections. The classic morphology consists of an annular scaly plaque; however, a wide variety of presentations have been observed (eg, verrucous, vesicular, pustular, granulomatous). Therefore, dermatophyte infections often mimic other dermatologic conditions, including atopic dermatitis, rosacea, psoriasis, bacterial abscess, erythema gyratum repens, lupus, granuloma annulare, cutaneous lymphoma, Hailey-Hailey disease, scarring alopecia, and syphilis.1
Notably, when dermatophytes grow downward along hair follicles causing deeper infection, disruption of the follicular wall can lead to an excessive inflammatory response with granulomatous features.2 Risk factors include cutaneous trauma, long-standing infection, immunocompromise, and treatment with topical corticosteroids.3 This disease evolution clinically appears as a nodule or infiltrated plaque, often without scale. The most well-known example is a kerion on the scalp. Elsewhere on the body, lesions often are termed Majocchi granulomas.2
Vegetative plaques, as seen in our patient, are a highly unusual morphology for deep tinea infection. Guanziroli et al4 reported a case of vegetative lesions on the forearm of a 67-year-old immunocompromised man that were successfully treated with a 3-month course of oral terbinafine after Trichophyton verrucosum was isolated. Skorepova et al5 reported a case of pyoderma vegetans triggered by recurrent Trichophyton mentagrophytes on the dorsal hands of a 64-year-old man with immunoglobulin deficiency of unknown etiology. The lesions were successfully treated with a prolonged course of doxycycline, topical triamcinolone, and intravenous immunoglobulin following 2 initial courses of terbinafine.
The differential diagnosis for vegetative lesions includes pemphigus vegetans, a vegetative variant of pyoderma gangrenosum; halogenoderma; and a variety of infections, including dimorphic fungi (histoplasmosis, blastomycosis), blastomycosislike pyoderma (bacterial), and candidiasis.6 These conditions usually can be distinguished based on histopathology. Clinically, pemphigus vegetans presents with pustules and vegetative lesions, as in our patient, but usually is more diffuse and favors the intertriginous areas. Histology likely would reveal foci of acantholysis and eosinophils. Vegetative pyoderma gangrenosum favors the trunk, particularly in sites of surgical trauma. In our patient, no lesions were present near the abdominal surgical sites, and there was no antecedent cribriform ulceration. Halogenoderma was a strong initial consideration given the localization, presence of large pustules, and history of numerous contrast computed tomography studies; however, our patient’s iodine levels were normal. Infectious etiologies including dimorphic fungi and blastomycosislike pyoderma generally are not restricted to the head and neck, and tissue culture helps exclude them. Vegetative lesions may occur in the setting of other infections, and tissue culture may be necessary to differentiate them if histopathology is not suggestive.
Deep dermatophyte infections require treatment with oral antifungals, as topicals do not penetrate adequately into the hair follicles. Exact regimens vary, but generally oral terbinafine or an oral azole (except ketoconazole) is administered for 2 to 6 weeks, with immunocompromise necessitating longer courses.
We present a rare case of vegetative Majocchi granuloma secondary to T rubrum infection. A dermatophyte infection should be included in the differential for vegetative lesions, especially in dense hair-bearing areas such as the beard. Treatment generally is straightforward with oral antifungals.
THE DIAGNOSIS: Vegetative Majocchi Granuloma
A biopsy and tissue culture showed acute dermal inflammation with granulomatous features and numerous fungal hyphae within the stratum corneum (Figure 1A), which were confirmed on GrocottGomori methenamine-silver staining (Figure 1B). Gram and Fite stains were negative for bacteria. A tissue culture speciated Trichophyton rubrum, which led to a diagnosis of deep dermatophyte infection (Majocchi granuloma) with a highly unusual clinical presentation of vegetative plaques. Predisposing factors included treatment with topical corticosteroids and possibly poor health and nutritional status at baseline. Our patient was treated with fluconazole 200 mg daily for 6 weeks, with near resolution of lesions at 3-week follow-up (Figure 2).
Dermatophytes are a common cause of superficial skin infections. The classic morphology consists of an annular scaly plaque; however, a wide variety of presentations have been observed (eg, verrucous, vesicular, pustular, granulomatous). Therefore, dermatophyte infections often mimic other dermatologic conditions, including atopic dermatitis, rosacea, psoriasis, bacterial abscess, erythema gyratum repens, lupus, granuloma annulare, cutaneous lymphoma, Hailey-Hailey disease, scarring alopecia, and syphilis.1
Notably, when dermatophytes grow downward along hair follicles causing deeper infection, disruption of the follicular wall can lead to an excessive inflammatory response with granulomatous features.2 Risk factors include cutaneous trauma, long-standing infection, immunocompromise, and treatment with topical corticosteroids.3 This disease evolution clinically appears as a nodule or infiltrated plaque, often without scale. The most well-known example is a kerion on the scalp. Elsewhere on the body, lesions often are termed Majocchi granulomas.2
Vegetative plaques, as seen in our patient, are a highly unusual morphology for deep tinea infection. Guanziroli et al4 reported a case of vegetative lesions on the forearm of a 67-year-old immunocompromised man that were successfully treated with a 3-month course of oral terbinafine after Trichophyton verrucosum was isolated. Skorepova et al5 reported a case of pyoderma vegetans triggered by recurrent Trichophyton mentagrophytes on the dorsal hands of a 64-year-old man with immunoglobulin deficiency of unknown etiology. The lesions were successfully treated with a prolonged course of doxycycline, topical triamcinolone, and intravenous immunoglobulin following 2 initial courses of terbinafine.
The differential diagnosis for vegetative lesions includes pemphigus vegetans, a vegetative variant of pyoderma gangrenosum; halogenoderma; and a variety of infections, including dimorphic fungi (histoplasmosis, blastomycosis), blastomycosislike pyoderma (bacterial), and candidiasis.6 These conditions usually can be distinguished based on histopathology. Clinically, pemphigus vegetans presents with pustules and vegetative lesions, as in our patient, but usually is more diffuse and favors the intertriginous areas. Histology likely would reveal foci of acantholysis and eosinophils. Vegetative pyoderma gangrenosum favors the trunk, particularly in sites of surgical trauma. In our patient, no lesions were present near the abdominal surgical sites, and there was no antecedent cribriform ulceration. Halogenoderma was a strong initial consideration given the localization, presence of large pustules, and history of numerous contrast computed tomography studies; however, our patient’s iodine levels were normal. Infectious etiologies including dimorphic fungi and blastomycosislike pyoderma generally are not restricted to the head and neck, and tissue culture helps exclude them. Vegetative lesions may occur in the setting of other infections, and tissue culture may be necessary to differentiate them if histopathology is not suggestive.
Deep dermatophyte infections require treatment with oral antifungals, as topicals do not penetrate adequately into the hair follicles. Exact regimens vary, but generally oral terbinafine or an oral azole (except ketoconazole) is administered for 2 to 6 weeks, with immunocompromise necessitating longer courses.
We present a rare case of vegetative Majocchi granuloma secondary to T rubrum infection. A dermatophyte infection should be included in the differential for vegetative lesions, especially in dense hair-bearing areas such as the beard. Treatment generally is straightforward with oral antifungals.
- Atzori L, Pau M, Aste N, et al. Dermatophyte infections mimicking other skin diseases: a 154-person case survey of tinea atypica in the district of Cagliari (Italy). Int J Dermatol. 2012;51:410-415.
- Ilkit M, Durdu M, Karakas M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Jevremovic L, Ilijin I, Kostic K, et al. Pyoderma vegetans—a case report. Serbian J Dermatol Venereol. 2017;9:22-28.
- Guanziroli E, Pavia G, Guttadauro A, et al. Deep dermatophytosis caused by Trichophyton verrucosum in an immunosuppressed patient: successful outcome with terbinafine. Mycopathologia. 2019;184:543-545.
- Skorepová M, Stuchlík D. Chronic pyoderma vegetans triggered by Trichophyton mentagrophytes. Mycoses. 2006;49:143-144.
- Reinholz M, Hermans C, Dietrich A, et al. A case of cutaneous vegetating candidiasis in a patient with keratitis-ichthyosis-deafness syndrome. J Eur Acad Dermatol Venereol. 2016;30:537-539.
- Atzori L, Pau M, Aste N, et al. Dermatophyte infections mimicking other skin diseases: a 154-person case survey of tinea atypica in the district of Cagliari (Italy). Int J Dermatol. 2012;51:410-415.
- Ilkit M, Durdu M, Karakas M. Majocchi’s granuloma: a symptom complex caused by fungal pathogens. Med Mycol. 2012;50:449-457.
- Jevremovic L, Ilijin I, Kostic K, et al. Pyoderma vegetans—a case report. Serbian J Dermatol Venereol. 2017;9:22-28.
- Guanziroli E, Pavia G, Guttadauro A, et al. Deep dermatophytosis caused by Trichophyton verrucosum in an immunosuppressed patient: successful outcome with terbinafine. Mycopathologia. 2019;184:543-545.
- Skorepová M, Stuchlík D. Chronic pyoderma vegetans triggered by Trichophyton mentagrophytes. Mycoses. 2006;49:143-144.
- Reinholz M, Hermans C, Dietrich A, et al. A case of cutaneous vegetating candidiasis in a patient with keratitis-ichthyosis-deafness syndrome. J Eur Acad Dermatol Venereol. 2016;30:537-539.
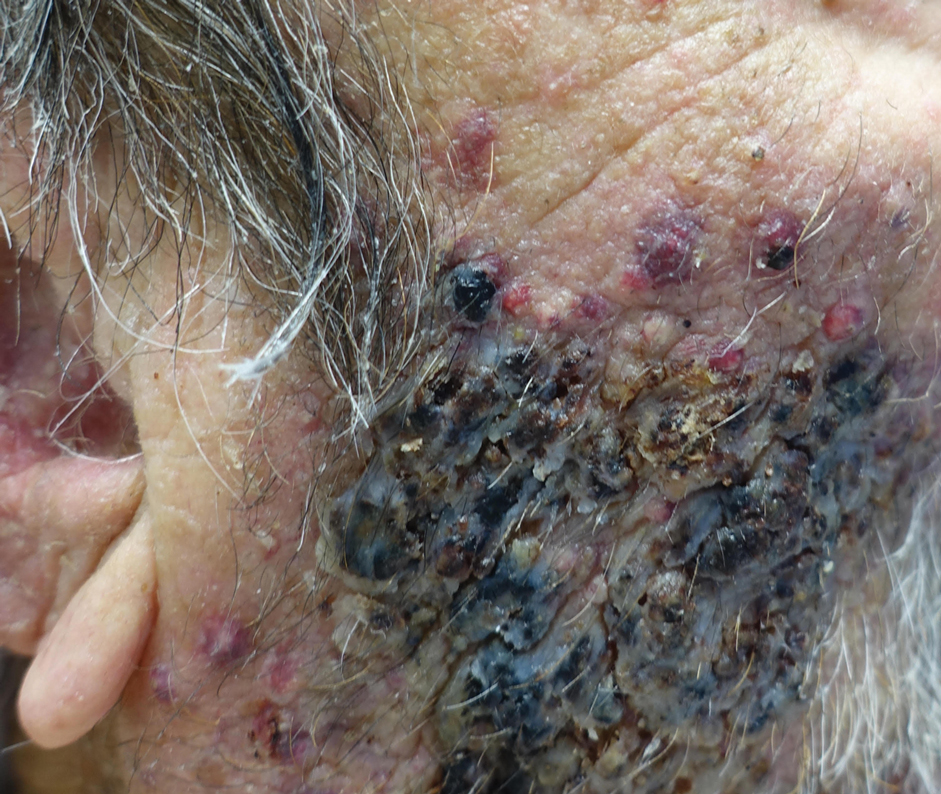
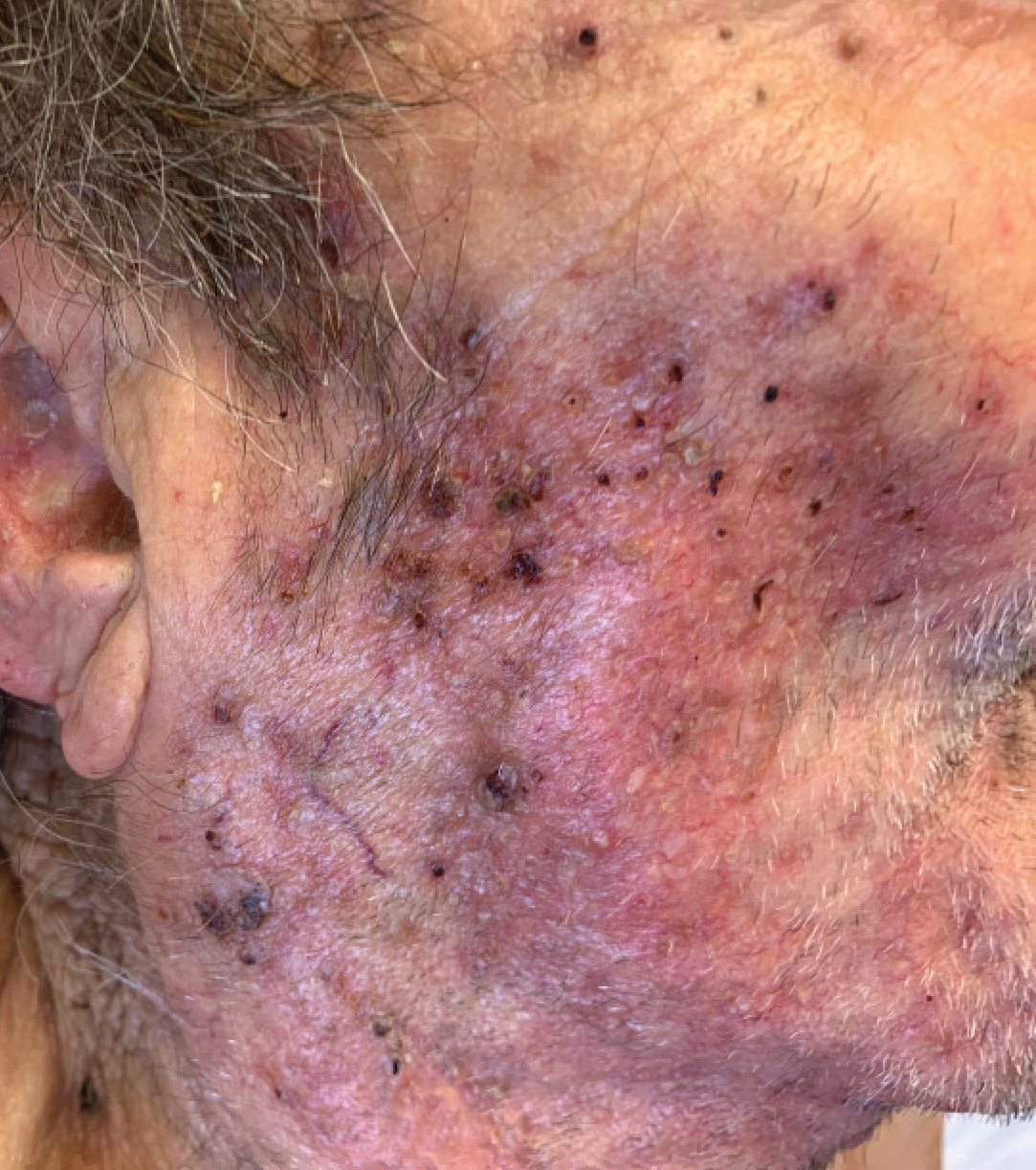
An 86-year-old man was admitted to the hospital for sigmoid colon perforation secondary to ischemic colitis. His medical history consisted of sequelae from atherosclerotic vascular disease. He had no known personal or family history of skin disease. His bowel perforation was surgically repaired, and his clinical status was stabilized, enabling transfer to a transitional care hospital. His course was complicated by delayed healing of the midline abdominal surgical wounds, leading to multiple computed tomography studies with iodinated contrast. One week following arrival at the transitional care hospital, he was noted to have a pustular rash on the face. He was empirically treated with topical steroids, mupirocin, and sulfacetamide. The rash did not improve, and the appearance changed, at which point dermatology was consulted. On evaluation, the patient was afebrile with a normal white blood cell count. Physical examination revealed gray-brown, moist, vegetative plaques on the cheeks with a few large pustules as well as similar-appearing lesions on the neck and upper chest. Attempted removal of a portion of the plaque left an erosion.
Tender Subcutaneous Nodule in a Prepubescent Boy
The Diagnosis: Dermatomyofibroma
Dermatomyofibroma is an uncommon, benign, cutaneous mesenchymal neoplasm composed of fibroblasts and myofibroblasts.1-3 This skin tumor was first described in 1991 by Hugel4 in the German literature as plaquelike fibromatosis. Pediatric dermatomyofibromas are exceedingly rare, with pediatric patients ranging in age from infants to teenagers.1
Clinically, dermatomyofibromas appear as long-standing, isolated, ill-demarcated, flesh-colored, slightly hyperpigmented or erythematous nodules or plaques that may be raised or indurated.1 Dermatomyofibromas may present with constant mild pain or pruritus, though in most cases the lesions are asymptomatic.1,3 The clinical presentation of dermatomyofibroma has a few distinct differences in children compared to adults. In adulthood, dermatomyofibroma has a strong female predominance and most commonly is located on the shoulder and adjacent upper body regions, including the axilla, neck, upper arm, and upper trunk.1-3 In childhood, the majority of dermatomyofibromas occur in young boys and usually are located on the neck with other upper body regions occurring less frequently.1,2 A shared characteristic includes the tendency for dermatomyofibromas to have an initial period of enlargement followed by stabilization or slow growth.1 Reported pediatric lesions have ranged in size from 4 to 60 mm with an average size of 14.9 mm (median, 12 mm).2
The diagnosis of dermatomyofibroma is based on histopathologic features in addition to clinical presentation. Histology from punch biopsy usually reveals a noninvasive dermal proliferation of bland, uniform, slender spindle cells oriented parallel to the overlying epidermis with increased and fragmented elastic fibers.1,3 Infiltration into the mid or deep dermis is common. The adnexal structures usually are spared; the stroma contains collagen and increased small blood vessels; and there typically is no inflammatory infiltrate, except for occasional scattered mast cells.2 Cytologically, the monomorphic spindleshaped tumor cells have an ill-defined, pale, eosinophilic cytoplasm and nuclei that are elongated with tapered edges.3 Dermatomyofibroma has a variable immunohistochemical profile, as it may stain focally positive for CD34 or smooth muscle actin, with occasional staining of factor XIIIa, desmin, calponin, or vimentin.1-3 Normal to increased levels of often fragmented elastic fibers is a helpful clue in distinguishing dermatomyofibroma from dermatofibroma, hypertrophic scar, dermatofibrosarcoma protuberans, and pilar leiomyoma, in which elastic fibers typically are reduced.3 Differential diagnoses of mesenchymal tumors in children include desmoid fibromatosis, connective tissue nevus, myofibromatosis, and smooth muscle hamartoma.1
A punch biopsy with clinical observation and followup is recommended for the management of lesions in cosmetically sensitive areas or in very young children who may not tolerate surgery. In symptomatic or cosmetically unappealing cases of dermatomyofibroma, simple surgical excision remains a viable treatment option. Recurrence is uncommon, even if only partially excised, and no instances of metastasis have been reported.1-5
Dermatomyofibromas may be mistaken for several other entities both benign and malignant. For example, the benign dermatofibroma is the second most common fibrohistiocytic tumor of the skin and presents as a firm, nontender, minimally elevated to dome-shaped papule that usually measures less than or equal to 1 cm in diameter with or without overlying skin changes.5,6 It primarily is seen in adults with a slight female predominance and favors the lower extremities.5 Patients usually are asymptomatic but often report a history of local trauma at the lesion site.6 Histologically, dermatofibroma is characterized by a nodular dermal proliferation of spindleshaped fibrous cells and histiocytes in a storiform pattern (Figure 1).6 Epidermal induction with acanthosis overlying the tumor often is found with occasional basilar hyperpigmentation.5 Dermatofibroma also characteristically has trapped collagen (“collagen balls”) seen at the periphery.5,6
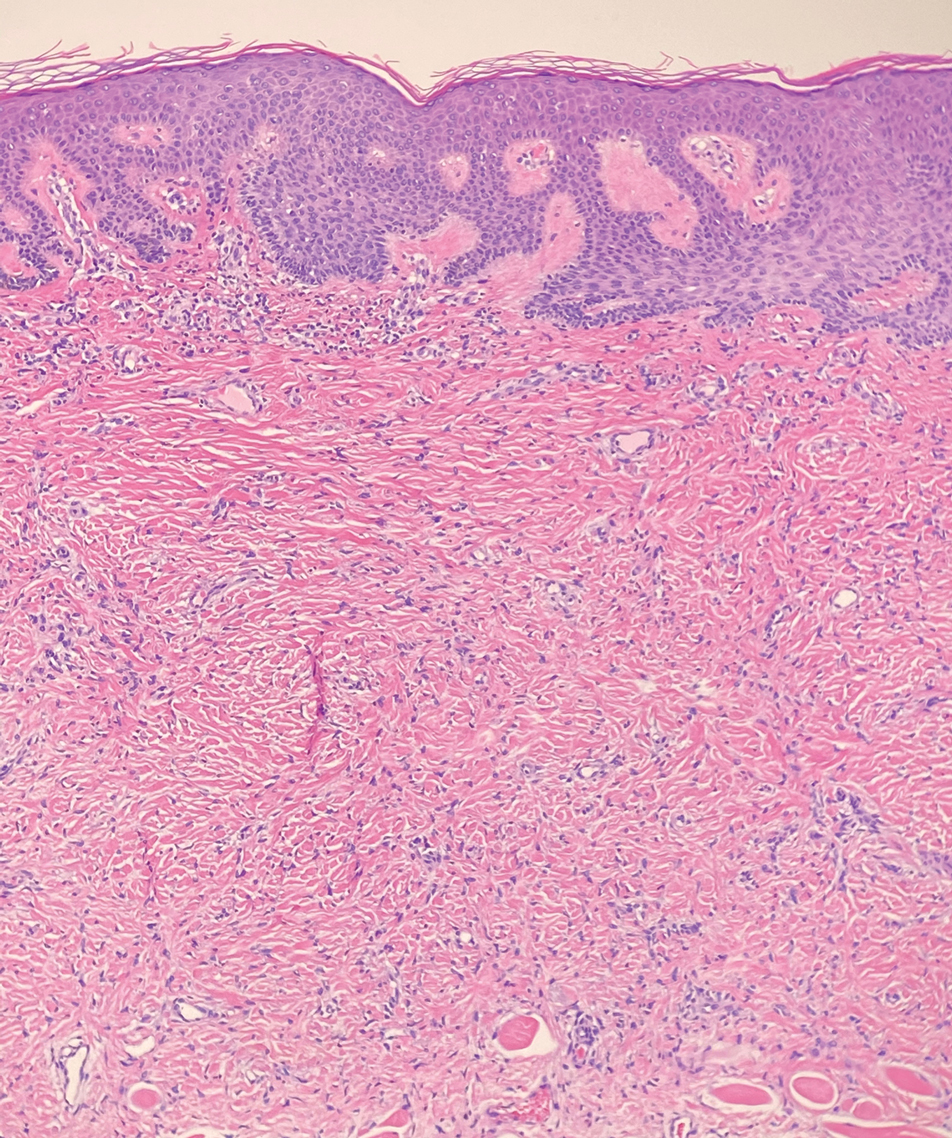
Piloleiomyomas are benign smooth muscle tumors arising from arrector pili muscles that may be solitary or multiple.5 Clinically, they typically present as firm, reddish-brown to flesh-colored papules or nodules that develop more commonly in adulthood.5,7 Piloleiomyomas favor the extremities and trunk, particularly the shoulder, and can be associated with spontaneous or induced pain. Histologically, piloleiomyomas are well circumscribed and centered within the reticular dermis situated closely to hair follicles (Figure 2).5 They are composed of numerous interlacing fascicles or whorls of smooth muscle cells with abundant eosinophilic cytoplasm and blunt-ended, cigar-shaped nuclei.5,7
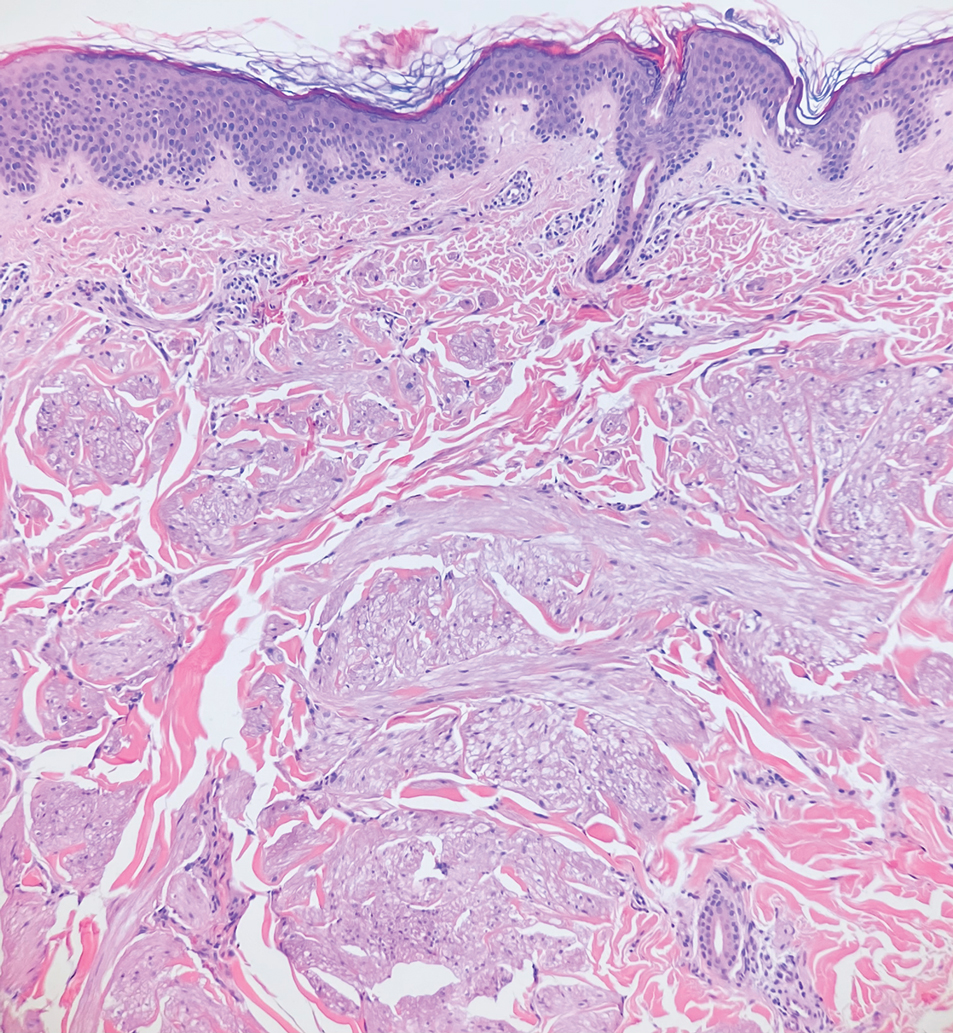
Solitary cutaneous myofibroma is a benign fibrous tumor found in adolescents and adults and is the counterpart to infantile myofibromatosis.8 Clinically, myofibromas typically present as painless, slow-growing, firm nodules with an occasional bluish hue. Histologically, solitary cutaneous myofibromas appear in a biphasic pattern, with hemangiopericytomatous components as well as spindle cells arranged in short bundles and fascicles resembling leiomyoma (Figure 3). The spindle cells also have abundant eosinophilic cytoplasm with short plump nuclei; the random, irregularly intersecting angles can be used to help differentiate myofibromas from smooth muscle lesions.8 Solitary cutaneous myofibroma is in the differential diagnosis for dermatomyofibroma because of their shared myofibroblastic nature.9
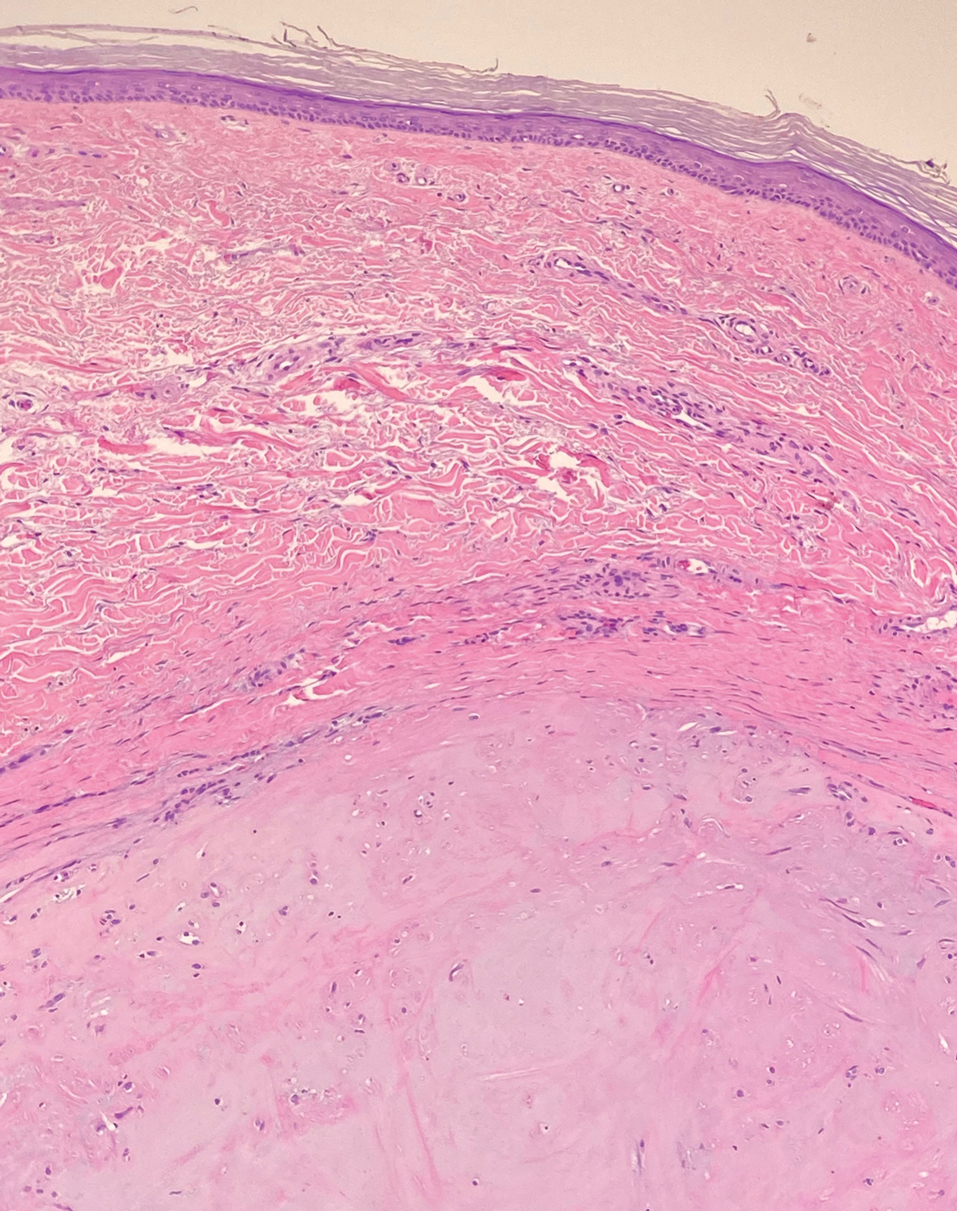
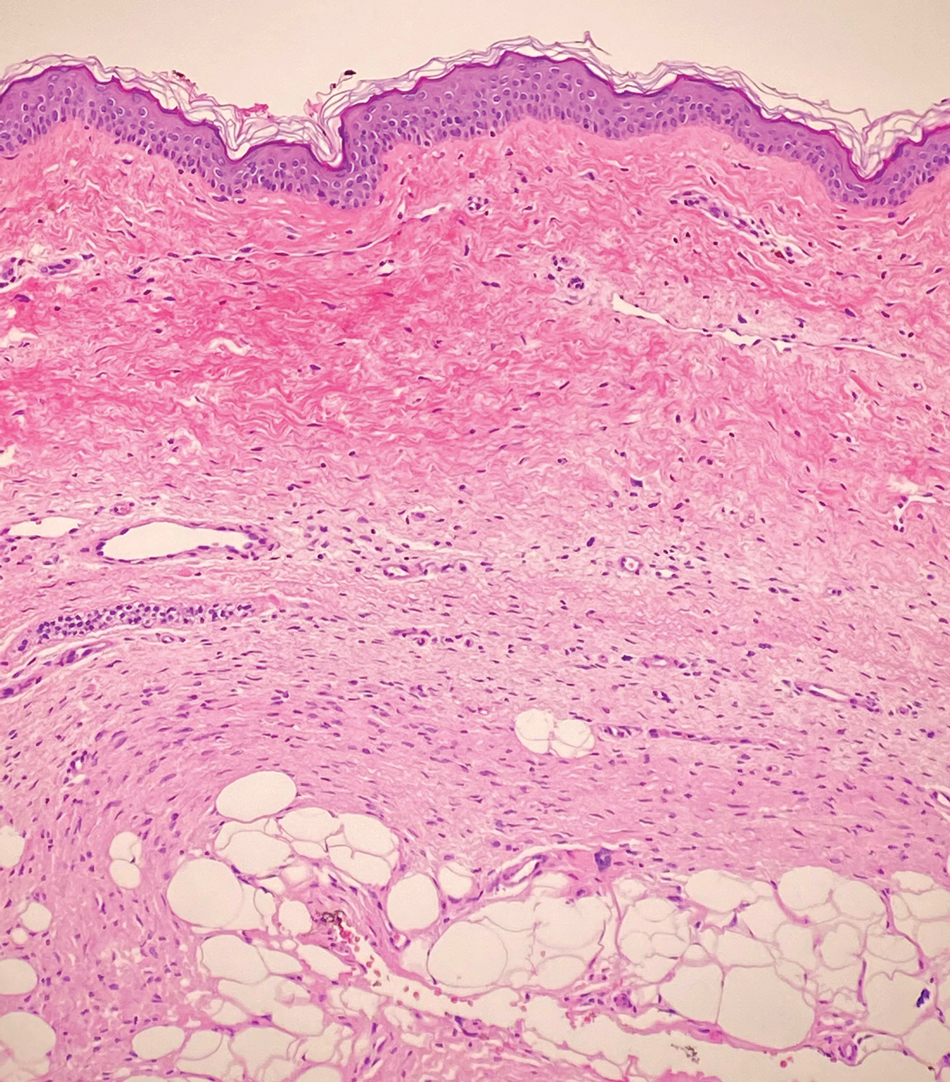
Dermatofibrosarcoma protuberans (DFSP) is an uncommon, locally invasive sarcoma with a high recurrence rate that favors young to middle-aged adults, with rare childhood onset reported.5,10,11 Clinically, DFSP typically presents as an asymptomatic, slow-growing, firm, flesh-colored, indurated plaque that develops into a violaceous to reddish-brown nodule.5 The atrophic variant of DFSP is characterized by a nonprotuberant lesion and can be especially difficult to distinguish from other entities such as dermatomyofibroma.11 The majority of DFSP lesions occur on the trunk, particularly in the shoulder or pelvic region.5 Histologically, early plaque lesions are comprised of monomorphic spindle cells arranged in long fascicles (parallel to the skin surface), infiltrating adnexal structures, and subcutaneous adipocytes in a multilayered honeycomb pattern; the spindle cells of late nodular lesions are arranged in short fascicles in a matted or storiform pattern (Figure 4).5,10 Early stages of DFSP as well as variations in childhood-onset DFSP can easily be misdiagnosed and incompletely excised.5
- Ma JE, Wieland CN, Tollefson MM. Dermatomyofibromas arising in children: report of two new cases and review of the literature. Pediatr Dermatol. 2017;34:347-351.
- Tardio JC, Azorin D, Hernandez-Nunez A, et al. Dermatomyofibromas presenting in pediatric patients: clinicopathologic characteristics and differential diagnosis. J Cutan Pathol. 2011;38:967-972.
- Mentzel T, Kutzner H. Dermatomyofibroma: clinicopathologic and immunohistochemical analysis of 56 cases and reappraisal of a rare and distinct cutaneous neoplasm. Am J Dermatopathol. 2009;31:44-49.
- Hugel H. Plaque-like dermal fibromatosis. Hautarzt. 1991;42:223-226.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. WB Saunders Co; 2012.
- Myers DJ, Fillman EP. Dermatofibroma. StatPearls [Internet]. StatPearls Publishing; 2020.
- Dilek N, Yuksel D, Sehitoglu I, et al. Cutaneous leiomyoma in a child: a case report. Oncol Lett. 2013;5:1163-1164.
- Roh HS, Paek JO, Yu HJ, et al. Solitary cutaneous myofibroma on the sole: an unusual localization. Ann Dermatol. 2012;24:220-222.
- Weedon D, Strutton G, Rubin AI, et al. Weedon’s Skin Pathology. Churchill Livingstone/Elsevier; 2010.
- Mendenhall WM, Zlotecki RA, Scarborough MT. Dermatofibrosarcoma protuberans. Cancer. 2004;101:2503-2508.
- Akay BN, Unlu E, Erdem C, et al. Dermatoscopic findings of atrophic dermatofibrosarcoma protuberans. Dermatol Pract Concept. 2015;5:71-73.
The Diagnosis: Dermatomyofibroma
Dermatomyofibroma is an uncommon, benign, cutaneous mesenchymal neoplasm composed of fibroblasts and myofibroblasts.1-3 This skin tumor was first described in 1991 by Hugel4 in the German literature as plaquelike fibromatosis. Pediatric dermatomyofibromas are exceedingly rare, with pediatric patients ranging in age from infants to teenagers.1
Clinically, dermatomyofibromas appear as long-standing, isolated, ill-demarcated, flesh-colored, slightly hyperpigmented or erythematous nodules or plaques that may be raised or indurated.1 Dermatomyofibromas may present with constant mild pain or pruritus, though in most cases the lesions are asymptomatic.1,3 The clinical presentation of dermatomyofibroma has a few distinct differences in children compared to adults. In adulthood, dermatomyofibroma has a strong female predominance and most commonly is located on the shoulder and adjacent upper body regions, including the axilla, neck, upper arm, and upper trunk.1-3 In childhood, the majority of dermatomyofibromas occur in young boys and usually are located on the neck with other upper body regions occurring less frequently.1,2 A shared characteristic includes the tendency for dermatomyofibromas to have an initial period of enlargement followed by stabilization or slow growth.1 Reported pediatric lesions have ranged in size from 4 to 60 mm with an average size of 14.9 mm (median, 12 mm).2
The diagnosis of dermatomyofibroma is based on histopathologic features in addition to clinical presentation. Histology from punch biopsy usually reveals a noninvasive dermal proliferation of bland, uniform, slender spindle cells oriented parallel to the overlying epidermis with increased and fragmented elastic fibers.1,3 Infiltration into the mid or deep dermis is common. The adnexal structures usually are spared; the stroma contains collagen and increased small blood vessels; and there typically is no inflammatory infiltrate, except for occasional scattered mast cells.2 Cytologically, the monomorphic spindleshaped tumor cells have an ill-defined, pale, eosinophilic cytoplasm and nuclei that are elongated with tapered edges.3 Dermatomyofibroma has a variable immunohistochemical profile, as it may stain focally positive for CD34 or smooth muscle actin, with occasional staining of factor XIIIa, desmin, calponin, or vimentin.1-3 Normal to increased levels of often fragmented elastic fibers is a helpful clue in distinguishing dermatomyofibroma from dermatofibroma, hypertrophic scar, dermatofibrosarcoma protuberans, and pilar leiomyoma, in which elastic fibers typically are reduced.3 Differential diagnoses of mesenchymal tumors in children include desmoid fibromatosis, connective tissue nevus, myofibromatosis, and smooth muscle hamartoma.1
A punch biopsy with clinical observation and followup is recommended for the management of lesions in cosmetically sensitive areas or in very young children who may not tolerate surgery. In symptomatic or cosmetically unappealing cases of dermatomyofibroma, simple surgical excision remains a viable treatment option. Recurrence is uncommon, even if only partially excised, and no instances of metastasis have been reported.1-5
Dermatomyofibromas may be mistaken for several other entities both benign and malignant. For example, the benign dermatofibroma is the second most common fibrohistiocytic tumor of the skin and presents as a firm, nontender, minimally elevated to dome-shaped papule that usually measures less than or equal to 1 cm in diameter with or without overlying skin changes.5,6 It primarily is seen in adults with a slight female predominance and favors the lower extremities.5 Patients usually are asymptomatic but often report a history of local trauma at the lesion site.6 Histologically, dermatofibroma is characterized by a nodular dermal proliferation of spindleshaped fibrous cells and histiocytes in a storiform pattern (Figure 1).6 Epidermal induction with acanthosis overlying the tumor often is found with occasional basilar hyperpigmentation.5 Dermatofibroma also characteristically has trapped collagen (“collagen balls”) seen at the periphery.5,6
Piloleiomyomas are benign smooth muscle tumors arising from arrector pili muscles that may be solitary or multiple.5 Clinically, they typically present as firm, reddish-brown to flesh-colored papules or nodules that develop more commonly in adulthood.5,7 Piloleiomyomas favor the extremities and trunk, particularly the shoulder, and can be associated with spontaneous or induced pain. Histologically, piloleiomyomas are well circumscribed and centered within the reticular dermis situated closely to hair follicles (Figure 2).5 They are composed of numerous interlacing fascicles or whorls of smooth muscle cells with abundant eosinophilic cytoplasm and blunt-ended, cigar-shaped nuclei.5,7
Solitary cutaneous myofibroma is a benign fibrous tumor found in adolescents and adults and is the counterpart to infantile myofibromatosis.8 Clinically, myofibromas typically present as painless, slow-growing, firm nodules with an occasional bluish hue. Histologically, solitary cutaneous myofibromas appear in a biphasic pattern, with hemangiopericytomatous components as well as spindle cells arranged in short bundles and fascicles resembling leiomyoma (Figure 3). The spindle cells also have abundant eosinophilic cytoplasm with short plump nuclei; the random, irregularly intersecting angles can be used to help differentiate myofibromas from smooth muscle lesions.8 Solitary cutaneous myofibroma is in the differential diagnosis for dermatomyofibroma because of their shared myofibroblastic nature.9
Dermatofibrosarcoma protuberans (DFSP) is an uncommon, locally invasive sarcoma with a high recurrence rate that favors young to middle-aged adults, with rare childhood onset reported.5,10,11 Clinically, DFSP typically presents as an asymptomatic, slow-growing, firm, flesh-colored, indurated plaque that develops into a violaceous to reddish-brown nodule.5 The atrophic variant of DFSP is characterized by a nonprotuberant lesion and can be especially difficult to distinguish from other entities such as dermatomyofibroma.11 The majority of DFSP lesions occur on the trunk, particularly in the shoulder or pelvic region.5 Histologically, early plaque lesions are comprised of monomorphic spindle cells arranged in long fascicles (parallel to the skin surface), infiltrating adnexal structures, and subcutaneous adipocytes in a multilayered honeycomb pattern; the spindle cells of late nodular lesions are arranged in short fascicles in a matted or storiform pattern (Figure 4).5,10 Early stages of DFSP as well as variations in childhood-onset DFSP can easily be misdiagnosed and incompletely excised.5
The Diagnosis: Dermatomyofibroma
Dermatomyofibroma is an uncommon, benign, cutaneous mesenchymal neoplasm composed of fibroblasts and myofibroblasts.1-3 This skin tumor was first described in 1991 by Hugel4 in the German literature as plaquelike fibromatosis. Pediatric dermatomyofibromas are exceedingly rare, with pediatric patients ranging in age from infants to teenagers.1
Clinically, dermatomyofibromas appear as long-standing, isolated, ill-demarcated, flesh-colored, slightly hyperpigmented or erythematous nodules or plaques that may be raised or indurated.1 Dermatomyofibromas may present with constant mild pain or pruritus, though in most cases the lesions are asymptomatic.1,3 The clinical presentation of dermatomyofibroma has a few distinct differences in children compared to adults. In adulthood, dermatomyofibroma has a strong female predominance and most commonly is located on the shoulder and adjacent upper body regions, including the axilla, neck, upper arm, and upper trunk.1-3 In childhood, the majority of dermatomyofibromas occur in young boys and usually are located on the neck with other upper body regions occurring less frequently.1,2 A shared characteristic includes the tendency for dermatomyofibromas to have an initial period of enlargement followed by stabilization or slow growth.1 Reported pediatric lesions have ranged in size from 4 to 60 mm with an average size of 14.9 mm (median, 12 mm).2
The diagnosis of dermatomyofibroma is based on histopathologic features in addition to clinical presentation. Histology from punch biopsy usually reveals a noninvasive dermal proliferation of bland, uniform, slender spindle cells oriented parallel to the overlying epidermis with increased and fragmented elastic fibers.1,3 Infiltration into the mid or deep dermis is common. The adnexal structures usually are spared; the stroma contains collagen and increased small blood vessels; and there typically is no inflammatory infiltrate, except for occasional scattered mast cells.2 Cytologically, the monomorphic spindleshaped tumor cells have an ill-defined, pale, eosinophilic cytoplasm and nuclei that are elongated with tapered edges.3 Dermatomyofibroma has a variable immunohistochemical profile, as it may stain focally positive for CD34 or smooth muscle actin, with occasional staining of factor XIIIa, desmin, calponin, or vimentin.1-3 Normal to increased levels of often fragmented elastic fibers is a helpful clue in distinguishing dermatomyofibroma from dermatofibroma, hypertrophic scar, dermatofibrosarcoma protuberans, and pilar leiomyoma, in which elastic fibers typically are reduced.3 Differential diagnoses of mesenchymal tumors in children include desmoid fibromatosis, connective tissue nevus, myofibromatosis, and smooth muscle hamartoma.1
A punch biopsy with clinical observation and followup is recommended for the management of lesions in cosmetically sensitive areas or in very young children who may not tolerate surgery. In symptomatic or cosmetically unappealing cases of dermatomyofibroma, simple surgical excision remains a viable treatment option. Recurrence is uncommon, even if only partially excised, and no instances of metastasis have been reported.1-5
Dermatomyofibromas may be mistaken for several other entities both benign and malignant. For example, the benign dermatofibroma is the second most common fibrohistiocytic tumor of the skin and presents as a firm, nontender, minimally elevated to dome-shaped papule that usually measures less than or equal to 1 cm in diameter with or without overlying skin changes.5,6 It primarily is seen in adults with a slight female predominance and favors the lower extremities.5 Patients usually are asymptomatic but often report a history of local trauma at the lesion site.6 Histologically, dermatofibroma is characterized by a nodular dermal proliferation of spindleshaped fibrous cells and histiocytes in a storiform pattern (Figure 1).6 Epidermal induction with acanthosis overlying the tumor often is found with occasional basilar hyperpigmentation.5 Dermatofibroma also characteristically has trapped collagen (“collagen balls”) seen at the periphery.5,6
Piloleiomyomas are benign smooth muscle tumors arising from arrector pili muscles that may be solitary or multiple.5 Clinically, they typically present as firm, reddish-brown to flesh-colored papules or nodules that develop more commonly in adulthood.5,7 Piloleiomyomas favor the extremities and trunk, particularly the shoulder, and can be associated with spontaneous or induced pain. Histologically, piloleiomyomas are well circumscribed and centered within the reticular dermis situated closely to hair follicles (Figure 2).5 They are composed of numerous interlacing fascicles or whorls of smooth muscle cells with abundant eosinophilic cytoplasm and blunt-ended, cigar-shaped nuclei.5,7
Solitary cutaneous myofibroma is a benign fibrous tumor found in adolescents and adults and is the counterpart to infantile myofibromatosis.8 Clinically, myofibromas typically present as painless, slow-growing, firm nodules with an occasional bluish hue. Histologically, solitary cutaneous myofibromas appear in a biphasic pattern, with hemangiopericytomatous components as well as spindle cells arranged in short bundles and fascicles resembling leiomyoma (Figure 3). The spindle cells also have abundant eosinophilic cytoplasm with short plump nuclei; the random, irregularly intersecting angles can be used to help differentiate myofibromas from smooth muscle lesions.8 Solitary cutaneous myofibroma is in the differential diagnosis for dermatomyofibroma because of their shared myofibroblastic nature.9
Dermatofibrosarcoma protuberans (DFSP) is an uncommon, locally invasive sarcoma with a high recurrence rate that favors young to middle-aged adults, with rare childhood onset reported.5,10,11 Clinically, DFSP typically presents as an asymptomatic, slow-growing, firm, flesh-colored, indurated plaque that develops into a violaceous to reddish-brown nodule.5 The atrophic variant of DFSP is characterized by a nonprotuberant lesion and can be especially difficult to distinguish from other entities such as dermatomyofibroma.11 The majority of DFSP lesions occur on the trunk, particularly in the shoulder or pelvic region.5 Histologically, early plaque lesions are comprised of monomorphic spindle cells arranged in long fascicles (parallel to the skin surface), infiltrating adnexal structures, and subcutaneous adipocytes in a multilayered honeycomb pattern; the spindle cells of late nodular lesions are arranged in short fascicles in a matted or storiform pattern (Figure 4).5,10 Early stages of DFSP as well as variations in childhood-onset DFSP can easily be misdiagnosed and incompletely excised.5
- Ma JE, Wieland CN, Tollefson MM. Dermatomyofibromas arising in children: report of two new cases and review of the literature. Pediatr Dermatol. 2017;34:347-351.
- Tardio JC, Azorin D, Hernandez-Nunez A, et al. Dermatomyofibromas presenting in pediatric patients: clinicopathologic characteristics and differential diagnosis. J Cutan Pathol. 2011;38:967-972.
- Mentzel T, Kutzner H. Dermatomyofibroma: clinicopathologic and immunohistochemical analysis of 56 cases and reappraisal of a rare and distinct cutaneous neoplasm. Am J Dermatopathol. 2009;31:44-49.
- Hugel H. Plaque-like dermal fibromatosis. Hautarzt. 1991;42:223-226.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. WB Saunders Co; 2012.
- Myers DJ, Fillman EP. Dermatofibroma. StatPearls [Internet]. StatPearls Publishing; 2020.
- Dilek N, Yuksel D, Sehitoglu I, et al. Cutaneous leiomyoma in a child: a case report. Oncol Lett. 2013;5:1163-1164.
- Roh HS, Paek JO, Yu HJ, et al. Solitary cutaneous myofibroma on the sole: an unusual localization. Ann Dermatol. 2012;24:220-222.
- Weedon D, Strutton G, Rubin AI, et al. Weedon’s Skin Pathology. Churchill Livingstone/Elsevier; 2010.
- Mendenhall WM, Zlotecki RA, Scarborough MT. Dermatofibrosarcoma protuberans. Cancer. 2004;101:2503-2508.
- Akay BN, Unlu E, Erdem C, et al. Dermatoscopic findings of atrophic dermatofibrosarcoma protuberans. Dermatol Pract Concept. 2015;5:71-73.
- Ma JE, Wieland CN, Tollefson MM. Dermatomyofibromas arising in children: report of two new cases and review of the literature. Pediatr Dermatol. 2017;34:347-351.
- Tardio JC, Azorin D, Hernandez-Nunez A, et al. Dermatomyofibromas presenting in pediatric patients: clinicopathologic characteristics and differential diagnosis. J Cutan Pathol. 2011;38:967-972.
- Mentzel T, Kutzner H. Dermatomyofibroma: clinicopathologic and immunohistochemical analysis of 56 cases and reappraisal of a rare and distinct cutaneous neoplasm. Am J Dermatopathol. 2009;31:44-49.
- Hugel H. Plaque-like dermal fibromatosis. Hautarzt. 1991;42:223-226.
- Bolognia JL, Jorizzo JL, Schaffer JV, eds. Dermatology. WB Saunders Co; 2012.
- Myers DJ, Fillman EP. Dermatofibroma. StatPearls [Internet]. StatPearls Publishing; 2020.
- Dilek N, Yuksel D, Sehitoglu I, et al. Cutaneous leiomyoma in a child: a case report. Oncol Lett. 2013;5:1163-1164.
- Roh HS, Paek JO, Yu HJ, et al. Solitary cutaneous myofibroma on the sole: an unusual localization. Ann Dermatol. 2012;24:220-222.
- Weedon D, Strutton G, Rubin AI, et al. Weedon’s Skin Pathology. Churchill Livingstone/Elsevier; 2010.
- Mendenhall WM, Zlotecki RA, Scarborough MT. Dermatofibrosarcoma protuberans. Cancer. 2004;101:2503-2508.
- Akay BN, Unlu E, Erdem C, et al. Dermatoscopic findings of atrophic dermatofibrosarcoma protuberans. Dermatol Pract Concept. 2015;5:71-73.
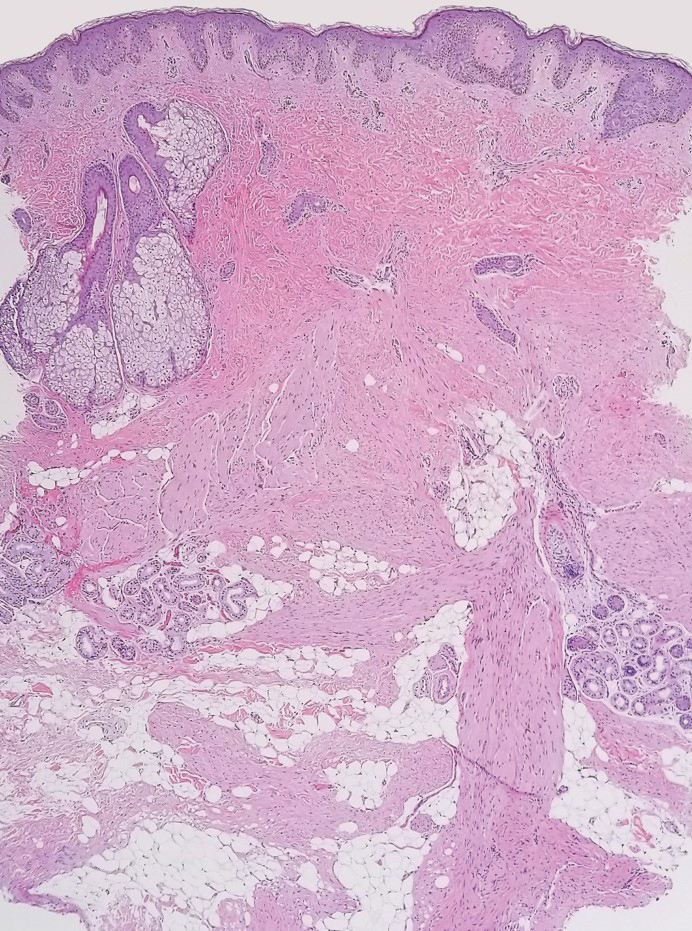

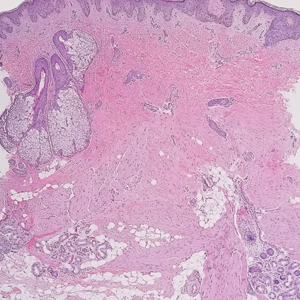
A 12-year-old boy with olive skin presented with a tender subcutaneous nodule on the back of 6 months’ duration. He reported the lesion initially grew rapidly with increasing pain for approximately 3 months with subsequent stabilization in size and modest resolution of his symptoms. Physical examination revealed a solitary, 15-mm, ill-defined, indurated, tender, subcutaneous nodule with subtle overlying hyperpigmentation on the left side of the upper back. Hematoxylin and eosin staining of a 4-mm punch biopsy revealed a nonencapsulated mass of monomorphic eosinophilic spindle cells organized into fascicles arranged predominantly parallel to the skin surface. The mass extended from the mid reticular dermis to the upper subcutis, sparing adnexal structures.
Periungual Papules in an Elderly Woman
The Diagnosis: Multicentric Reticulohistiocytosis
Te patient presented with pink papules coalescing into plaques on the upper chest and lower back (Figure 1) as well as a characteristic finding of periungual papules with a coral bead appearance. Histopathologic examination revealed a dense infiltrate of epithelioid histiocytes with amphophilic ground-glass cytoplasm in a nodular configuration (Figure 2). This pattern in conjunction with the clinical features seen in our patient was consistent with a diagnosis of multicentric reticulohistiocytosis (MRH).1-3 The cutaneous symptoms were managed with triamcinolone ointment 0.1% twice daily and oral hydroxyzine 10 mg 3 times daily as needed for itching with moderate improvement. She was referred to rheumatology for arthritis management, and the initial cancer screening was negative.
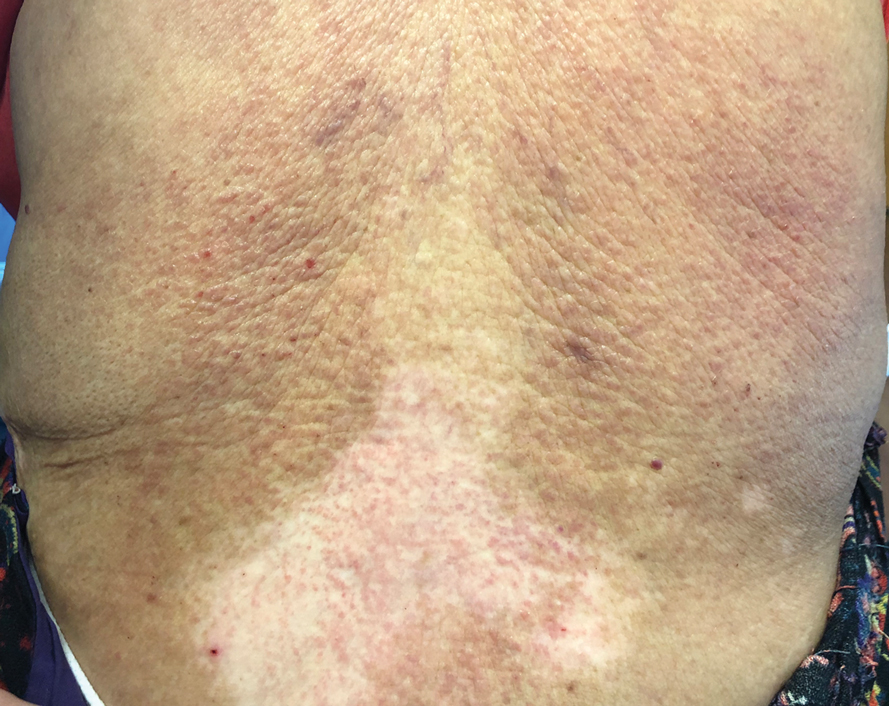
Multicentric reticulohistiocytosis is a rare granulomatous disease characterized by papulonodular cutaneous lesions and severe erosive arthritis. It has an insidious onset and most commonly affects middle-aged women.1 Multicentric reticulohistiocytosis typically presents as rounded pruritic papules or nodules that may be pink, red, or brown primarily affecting the face and distal upper extremities.1,3 Mucosal involvement occurs in more than half of patients and is characterized by multiple erythematous papules and nodules on the oral and nasopharyngeal mucosae that rarely can produce leonine facies.2 A hallmark feature of MRH is the presence of multiple shiny erythematous papules along the proximal and lateral nail folds that take on a coral bead appearance.1,3,4 Furthermore, nail changes such as atrophy, longitudinal ridging, brittleness, and hyperpigmentation can occur secondary to a synovial reaction that disturbs the nail matrix.4,5
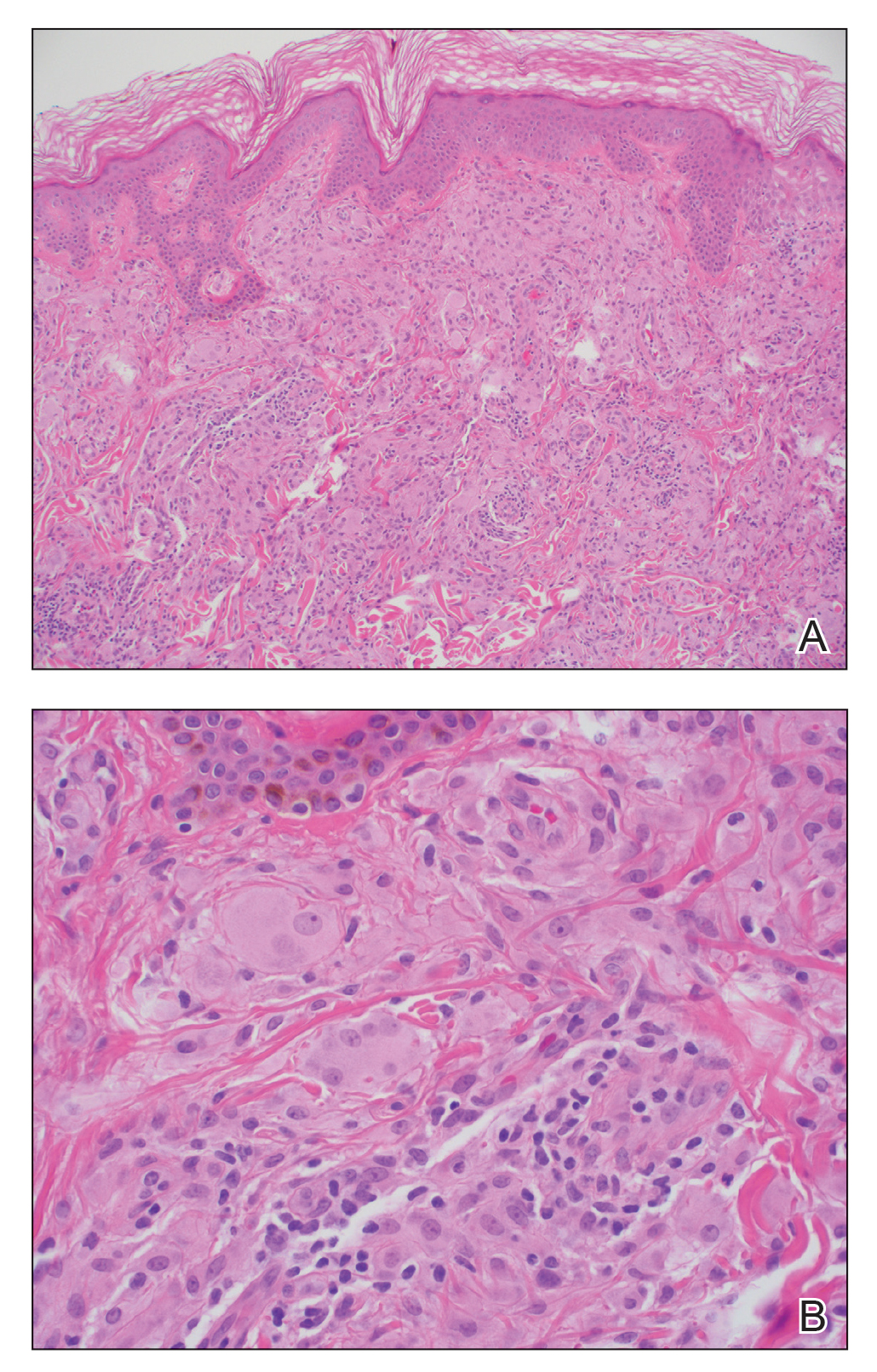
Joint involvement precedes cutaneous involvement in most cases of MRH.1,5 Multicentric reticulohistiocytosis is associated with a symmetric destructive arthritis affecting the hands, knees, shoulders, and hips that often is associated with pain, stiffness, and swelling.1,3 The arthritis rapidly progresses in the early stages of the disease but then becomes less active over the subsequent 8 to 10 years.1 It has the potential to develop into arthritis mutilans, an end-stage form of arthritis also seen in psoriatic and rheumatoid arthritis that leads to severe joint deformity and debilitation.1,2
The etiology of MRH still is unknown, but it has an association with underlying malignancy in up to 25% of patients.6 Multicentric reticulohistiocytosis has been reported in the context of a wide variety of malignancies including melanoma; sarcoma; lymphoma; leukemia; and carcinomas of the breast, colon, and lung. In some cases, the diagnosis of MRH may even precede the diagnosis of cancer.3 Multicentric reticulohistiocytosis also may be associated with autoimmune conditions,3 as seen in our patient who had a history of both hypothyroidism and vitiligo.
Histopathologic examination is essential in distinguishing MRH from other autoimmune disorders associated with hand lesions, rash, and arthralgia. Erythema elevatum diutinum is associated with symmetric, violaceous, red or brown papules and plaques located on the extensor surfaces of the extremities and hands; however, histology reveals a leukocytoclastic vasculitis with a mixture of polymorphonuclear leukocytes and lymphocytes.7 Dermatomyositis may present with arthralgia, flattopped, erythematous (Gottron) papules localized over the proximal interphalangeal and distal interphalangeal joints, as well as proximal nail findings. The latter generally presents with periungual erythema associated with dilated capillary loops rather than the discrete orderly papules seen in MRH. Histologic examination of dermatomyositis shows mild epidermal atrophy, vacuolar changes in the basal keratinocyte layer, and a dermal perivascular lymphocytic infiltrate.8 Because MRH initially can present with joint symptoms and hand nodules, it may be confused with rheumatoid arthritis. However, rheumatoid arthritis typically is associated with severe osteopenia and tends to affect the metacarpophalangeal and proximal interphalangeal joints rather than the distal interphalangeal joints that most often are affected in MRH.1 Histologic examination of rheumatoid nodules reveals palisading granulomas surrounding a central area of fibrinoid necrosis.9 Sarcoidosis is a multisystem disease that can present with cutaneous involvement including erythema nodosum, skin plaques, subcutaneous nodules, and papular eruptions in addition to joint lesions.10 Sarcoidosis most frequently involves the lungs, manifesting as diffuse interstitial lung disease with bilateral hilar lymphadenopathy. Furthermore, histologic examination of lesions demonstrates classic noncaseating granulomas containing epithelioid cells, multinucleated giant cells with inclusion bodies, and lymphocytes.11
A skin biopsy is required to establish the diagnosis of MRH. In general, patients with MRH and no underlying malignancy have a good prognosis and respond to anti-inflammatory therapies such as nonsteroidal antiinflammatory drugs and corticosteroids. Other agents including methotrexate, cyclophosphamide, and tumor necrosis factor α inhibitors also have been effective in more severe cases.1,3,12 Finally, in addition to treating the cutaneous manifestations of MRH, it is important to screen patients for underlying malignancies and other autoimmune conditions.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). an erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Barrow MV. The nails in multicentric reticulohistiocytosis. (lipoid dermato-arthritis). Arch Dermatol. 1967;95:200-201.
- Barrow MV, Holubar K. Multicentric reticulohistiocytosis. a review of 33 patients. Medicine (Baltimore). 1969;48:287-305.
- Snow JL, Muller SA. Malignancy-associated multicentric reticulohistiocytosis: a clinical, histological and immunophenotypic study. Br J Dermatol. 1995;133:71-76.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Smith ES, Hallman JR, DeLuca AM, et al. Dermatomyositis: a clinicopathological study of 40 patients. Am J Dermatopathol. 2009; 31:61-67.
- Athanasou NA, Quinn J, Woods CG, et al. Immunohistology of rheumatoid nodules and rheumatoid synovium. Ann Rheum Dis. 1988;47:398-403.
- Yanardag H, Pamuk ON, Karayel T. Cutaneous involvement in sarcoidosis: analysis of the features in 170 patients. Respir Med. 2003;97:978-982.
- Ma Y, Gal A, Koss MN. The pathology of pulmonary sarcoidosis: update. Semin Diagn Pathol. 2007;24:150-161.
- Kovach BT, Calamia KT, Walsh JS, et al. Treatment of multicentric reticulohistiocytosis with etanercept. Arch Dermatol. 2004;140:919-921.
The Diagnosis: Multicentric Reticulohistiocytosis
Te patient presented with pink papules coalescing into plaques on the upper chest and lower back (Figure 1) as well as a characteristic finding of periungual papules with a coral bead appearance. Histopathologic examination revealed a dense infiltrate of epithelioid histiocytes with amphophilic ground-glass cytoplasm in a nodular configuration (Figure 2). This pattern in conjunction with the clinical features seen in our patient was consistent with a diagnosis of multicentric reticulohistiocytosis (MRH).1-3 The cutaneous symptoms were managed with triamcinolone ointment 0.1% twice daily and oral hydroxyzine 10 mg 3 times daily as needed for itching with moderate improvement. She was referred to rheumatology for arthritis management, and the initial cancer screening was negative.
Multicentric reticulohistiocytosis is a rare granulomatous disease characterized by papulonodular cutaneous lesions and severe erosive arthritis. It has an insidious onset and most commonly affects middle-aged women.1 Multicentric reticulohistiocytosis typically presents as rounded pruritic papules or nodules that may be pink, red, or brown primarily affecting the face and distal upper extremities.1,3 Mucosal involvement occurs in more than half of patients and is characterized by multiple erythematous papules and nodules on the oral and nasopharyngeal mucosae that rarely can produce leonine facies.2 A hallmark feature of MRH is the presence of multiple shiny erythematous papules along the proximal and lateral nail folds that take on a coral bead appearance.1,3,4 Furthermore, nail changes such as atrophy, longitudinal ridging, brittleness, and hyperpigmentation can occur secondary to a synovial reaction that disturbs the nail matrix.4,5
Joint involvement precedes cutaneous involvement in most cases of MRH.1,5 Multicentric reticulohistiocytosis is associated with a symmetric destructive arthritis affecting the hands, knees, shoulders, and hips that often is associated with pain, stiffness, and swelling.1,3 The arthritis rapidly progresses in the early stages of the disease but then becomes less active over the subsequent 8 to 10 years.1 It has the potential to develop into arthritis mutilans, an end-stage form of arthritis also seen in psoriatic and rheumatoid arthritis that leads to severe joint deformity and debilitation.1,2
The etiology of MRH still is unknown, but it has an association with underlying malignancy in up to 25% of patients.6 Multicentric reticulohistiocytosis has been reported in the context of a wide variety of malignancies including melanoma; sarcoma; lymphoma; leukemia; and carcinomas of the breast, colon, and lung. In some cases, the diagnosis of MRH may even precede the diagnosis of cancer.3 Multicentric reticulohistiocytosis also may be associated with autoimmune conditions,3 as seen in our patient who had a history of both hypothyroidism and vitiligo.
Histopathologic examination is essential in distinguishing MRH from other autoimmune disorders associated with hand lesions, rash, and arthralgia. Erythema elevatum diutinum is associated with symmetric, violaceous, red or brown papules and plaques located on the extensor surfaces of the extremities and hands; however, histology reveals a leukocytoclastic vasculitis with a mixture of polymorphonuclear leukocytes and lymphocytes.7 Dermatomyositis may present with arthralgia, flattopped, erythematous (Gottron) papules localized over the proximal interphalangeal and distal interphalangeal joints, as well as proximal nail findings. The latter generally presents with periungual erythema associated with dilated capillary loops rather than the discrete orderly papules seen in MRH. Histologic examination of dermatomyositis shows mild epidermal atrophy, vacuolar changes in the basal keratinocyte layer, and a dermal perivascular lymphocytic infiltrate.8 Because MRH initially can present with joint symptoms and hand nodules, it may be confused with rheumatoid arthritis. However, rheumatoid arthritis typically is associated with severe osteopenia and tends to affect the metacarpophalangeal and proximal interphalangeal joints rather than the distal interphalangeal joints that most often are affected in MRH.1 Histologic examination of rheumatoid nodules reveals palisading granulomas surrounding a central area of fibrinoid necrosis.9 Sarcoidosis is a multisystem disease that can present with cutaneous involvement including erythema nodosum, skin plaques, subcutaneous nodules, and papular eruptions in addition to joint lesions.10 Sarcoidosis most frequently involves the lungs, manifesting as diffuse interstitial lung disease with bilateral hilar lymphadenopathy. Furthermore, histologic examination of lesions demonstrates classic noncaseating granulomas containing epithelioid cells, multinucleated giant cells with inclusion bodies, and lymphocytes.11
A skin biopsy is required to establish the diagnosis of MRH. In general, patients with MRH and no underlying malignancy have a good prognosis and respond to anti-inflammatory therapies such as nonsteroidal antiinflammatory drugs and corticosteroids. Other agents including methotrexate, cyclophosphamide, and tumor necrosis factor α inhibitors also have been effective in more severe cases.1,3,12 Finally, in addition to treating the cutaneous manifestations of MRH, it is important to screen patients for underlying malignancies and other autoimmune conditions.
The Diagnosis: Multicentric Reticulohistiocytosis
Te patient presented with pink papules coalescing into plaques on the upper chest and lower back (Figure 1) as well as a characteristic finding of periungual papules with a coral bead appearance. Histopathologic examination revealed a dense infiltrate of epithelioid histiocytes with amphophilic ground-glass cytoplasm in a nodular configuration (Figure 2). This pattern in conjunction with the clinical features seen in our patient was consistent with a diagnosis of multicentric reticulohistiocytosis (MRH).1-3 The cutaneous symptoms were managed with triamcinolone ointment 0.1% twice daily and oral hydroxyzine 10 mg 3 times daily as needed for itching with moderate improvement. She was referred to rheumatology for arthritis management, and the initial cancer screening was negative.
Multicentric reticulohistiocytosis is a rare granulomatous disease characterized by papulonodular cutaneous lesions and severe erosive arthritis. It has an insidious onset and most commonly affects middle-aged women.1 Multicentric reticulohistiocytosis typically presents as rounded pruritic papules or nodules that may be pink, red, or brown primarily affecting the face and distal upper extremities.1,3 Mucosal involvement occurs in more than half of patients and is characterized by multiple erythematous papules and nodules on the oral and nasopharyngeal mucosae that rarely can produce leonine facies.2 A hallmark feature of MRH is the presence of multiple shiny erythematous papules along the proximal and lateral nail folds that take on a coral bead appearance.1,3,4 Furthermore, nail changes such as atrophy, longitudinal ridging, brittleness, and hyperpigmentation can occur secondary to a synovial reaction that disturbs the nail matrix.4,5
Joint involvement precedes cutaneous involvement in most cases of MRH.1,5 Multicentric reticulohistiocytosis is associated with a symmetric destructive arthritis affecting the hands, knees, shoulders, and hips that often is associated with pain, stiffness, and swelling.1,3 The arthritis rapidly progresses in the early stages of the disease but then becomes less active over the subsequent 8 to 10 years.1 It has the potential to develop into arthritis mutilans, an end-stage form of arthritis also seen in psoriatic and rheumatoid arthritis that leads to severe joint deformity and debilitation.1,2
The etiology of MRH still is unknown, but it has an association with underlying malignancy in up to 25% of patients.6 Multicentric reticulohistiocytosis has been reported in the context of a wide variety of malignancies including melanoma; sarcoma; lymphoma; leukemia; and carcinomas of the breast, colon, and lung. In some cases, the diagnosis of MRH may even precede the diagnosis of cancer.3 Multicentric reticulohistiocytosis also may be associated with autoimmune conditions,3 as seen in our patient who had a history of both hypothyroidism and vitiligo.
Histopathologic examination is essential in distinguishing MRH from other autoimmune disorders associated with hand lesions, rash, and arthralgia. Erythema elevatum diutinum is associated with symmetric, violaceous, red or brown papules and plaques located on the extensor surfaces of the extremities and hands; however, histology reveals a leukocytoclastic vasculitis with a mixture of polymorphonuclear leukocytes and lymphocytes.7 Dermatomyositis may present with arthralgia, flattopped, erythematous (Gottron) papules localized over the proximal interphalangeal and distal interphalangeal joints, as well as proximal nail findings. The latter generally presents with periungual erythema associated with dilated capillary loops rather than the discrete orderly papules seen in MRH. Histologic examination of dermatomyositis shows mild epidermal atrophy, vacuolar changes in the basal keratinocyte layer, and a dermal perivascular lymphocytic infiltrate.8 Because MRH initially can present with joint symptoms and hand nodules, it may be confused with rheumatoid arthritis. However, rheumatoid arthritis typically is associated with severe osteopenia and tends to affect the metacarpophalangeal and proximal interphalangeal joints rather than the distal interphalangeal joints that most often are affected in MRH.1 Histologic examination of rheumatoid nodules reveals palisading granulomas surrounding a central area of fibrinoid necrosis.9 Sarcoidosis is a multisystem disease that can present with cutaneous involvement including erythema nodosum, skin plaques, subcutaneous nodules, and papular eruptions in addition to joint lesions.10 Sarcoidosis most frequently involves the lungs, manifesting as diffuse interstitial lung disease with bilateral hilar lymphadenopathy. Furthermore, histologic examination of lesions demonstrates classic noncaseating granulomas containing epithelioid cells, multinucleated giant cells with inclusion bodies, and lymphocytes.11
A skin biopsy is required to establish the diagnosis of MRH. In general, patients with MRH and no underlying malignancy have a good prognosis and respond to anti-inflammatory therapies such as nonsteroidal antiinflammatory drugs and corticosteroids. Other agents including methotrexate, cyclophosphamide, and tumor necrosis factor α inhibitors also have been effective in more severe cases.1,3,12 Finally, in addition to treating the cutaneous manifestations of MRH, it is important to screen patients for underlying malignancies and other autoimmune conditions.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). an erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Barrow MV. The nails in multicentric reticulohistiocytosis. (lipoid dermato-arthritis). Arch Dermatol. 1967;95:200-201.
- Barrow MV, Holubar K. Multicentric reticulohistiocytosis. a review of 33 patients. Medicine (Baltimore). 1969;48:287-305.
- Snow JL, Muller SA. Malignancy-associated multicentric reticulohistiocytosis: a clinical, histological and immunophenotypic study. Br J Dermatol. 1995;133:71-76.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Smith ES, Hallman JR, DeLuca AM, et al. Dermatomyositis: a clinicopathological study of 40 patients. Am J Dermatopathol. 2009; 31:61-67.
- Athanasou NA, Quinn J, Woods CG, et al. Immunohistology of rheumatoid nodules and rheumatoid synovium. Ann Rheum Dis. 1988;47:398-403.
- Yanardag H, Pamuk ON, Karayel T. Cutaneous involvement in sarcoidosis: analysis of the features in 170 patients. Respir Med. 2003;97:978-982.
- Ma Y, Gal A, Koss MN. The pathology of pulmonary sarcoidosis: update. Semin Diagn Pathol. 2007;24:150-161.
- Kovach BT, Calamia KT, Walsh JS, et al. Treatment of multicentric reticulohistiocytosis with etanercept. Arch Dermatol. 2004;140:919-921.
- Tajirian AL, Malik MK, Robinson-Bostom L, et al. Multicentric reticulohistiocytosis. Clin Dermatol. 2006;24:486-492.
- Gold RH, Metzger AL, Mirra JM, et al. Multicentric reticulohistiocytosis (lipoid dermato-arthritis). an erosive polyarthritis with distinctive clinical, roentgenographic and pathologic features. Am J Roentgenol Radium Ther Nucl Med. 1975;124:610-624.
- Luz FB, Gaspar TAP, Kalil-Gaspar N, et al. Multicentric reticulohistiocytosis. J Eur Acad Dermatol Venereol. 2001;15:524-531.
- Barrow MV. The nails in multicentric reticulohistiocytosis. (lipoid dermato-arthritis). Arch Dermatol. 1967;95:200-201.
- Barrow MV, Holubar K. Multicentric reticulohistiocytosis. a review of 33 patients. Medicine (Baltimore). 1969;48:287-305.
- Snow JL, Muller SA. Malignancy-associated multicentric reticulohistiocytosis: a clinical, histological and immunophenotypic study. Br J Dermatol. 1995;133:71-76.
- Yiannias JA, el-Azhary RA, Gibson LE. Erythema elevatum diutinum: a clinical and histopathologic study of 13 patients. J Am Acad Dermatol. 1992;26:38-44.
- Smith ES, Hallman JR, DeLuca AM, et al. Dermatomyositis: a clinicopathological study of 40 patients. Am J Dermatopathol. 2009; 31:61-67.
- Athanasou NA, Quinn J, Woods CG, et al. Immunohistology of rheumatoid nodules and rheumatoid synovium. Ann Rheum Dis. 1988;47:398-403.
- Yanardag H, Pamuk ON, Karayel T. Cutaneous involvement in sarcoidosis: analysis of the features in 170 patients. Respir Med. 2003;97:978-982.
- Ma Y, Gal A, Koss MN. The pathology of pulmonary sarcoidosis: update. Semin Diagn Pathol. 2007;24:150-161.
- Kovach BT, Calamia KT, Walsh JS, et al. Treatment of multicentric reticulohistiocytosis with etanercept. Arch Dermatol. 2004;140:919-921.
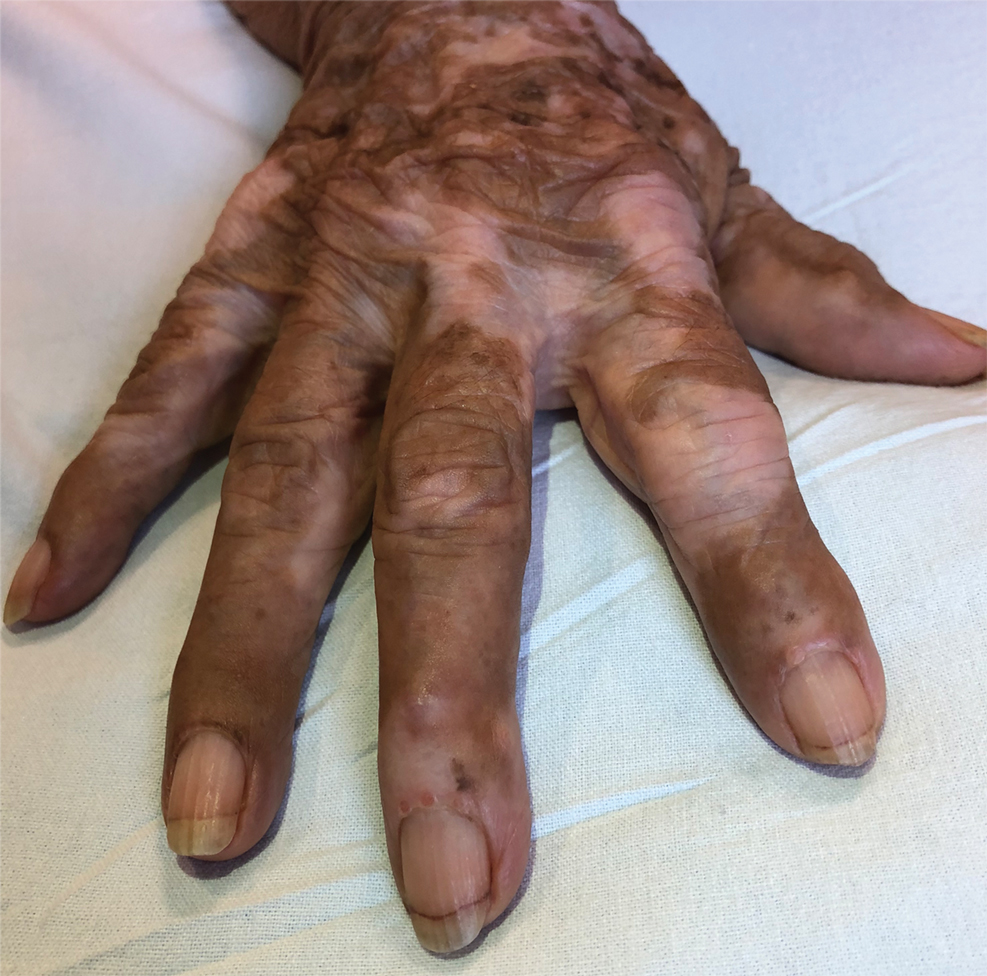
A 79-year-old woman presented with pruritic papules and plaques on the chest, back, arms, hands, legs, and feet of 1 year’s duration. She reported a history of hypothyroidism, arthritis, and vitiligo but denied a history of cancer. Physical examination showed pink papules coalescing into plaques on the upper chest and lower back as well as lichenified plaques on the forearms and knees. Erythematous papules on the proximal nail folds of the right first and second digits also were noted. Multiple depigmented patches on the hands, wrists, arms, and lower back also were present, and deformities of the hands and bulbous-appearing knees were observed. Results from a complete blood cell count and blood chemistry analyses showed mild anemia but were otherwise normal. Radiography of the right knee showed degenerative changes and periarticular radiolucencies consistent with an inflammatory arthropathy. A 4-mm punch biopsy specimen from the back was obtained for histopathologic examination.
A Starter Guide to Immunofluorescence Testing in Dermatology
Direct immunofluorescence (DIF) is the go-to diagnostic test when evaluating vesiculobullous eruptions, connective tissue disease, and vasculitis. This specialized test allows visualization of autoantibodies and their reaction products in the epidermis and dermis (skin) and epithelium and subepithelium (mucosa). Indirect immunofluorescence (IIF) and enzyme-linked immunosorbent assay (ELISA) are additional tests that can help in the diagnosis of autoimmune blistering disease. In the blistering autoimmune diseases, the autoantibodies target components in skin and mucous membranes that are essential for cell-cell and cell-matrix adhesion causing separation within or beneath the epidermis, depending on where the target components are located. This article is intended to serve as a helpful primer for immunofluorescence testing in dermatology, with an overview of the tests available as well as pragmatic tips for optimal biopsy sites and specimen transport.
Direct Immunofluorescence
Immunofluorescence techniques date back to 1941 when Albert Coons, an American physician, pathologist, and immunologist, fluorescently labelled antibodies to visualize pneumococcal antigens in infected tissues.1-3 In dermatology, similar methodology was used to visualize the deposition of immunoglobulins and complement in the skin of patients with systemic lupus erythematosus in 1963.4 Basement membrane zone antibodies were first visualized via DIF in bullous pemphigoid in 1967.5 This elegant test utilizes specific antibodies labeled with fluorophores that are then incubated with the patient’s tissue, ultimately forming antibody-antigen conjugates that can be visualized with a fluorescent microscope. Antibodies usually include IgG, IgM, IgA, fibrinogen, and C3. Some institutions also evaluate for IgG4.
Transport medium is critical for proper evaluation of tissues using DIF. Inappropriate storage of tissue can degrade the antigen and confuse the interpretation of specimens. An acceptable medium for DIF includes Michel transport medium, which allows tissue to be stored for days while being transported at ambient temperature without loss of signal.6,7 Zeus medium also can be used and is more readily available. Alternatively, biopsy tissue can be snap frozen using liquid nitrogen. Specimens also may be stored on saline gauze but should be analyzed within 24 to 48 hours.8 Most importantly, do not place the specimen in formalin; even a brief soak in formalin can greatly alter results, especially when trying to diagnose pemphigus.9 Proper transport conditions are critical to prevent autolysis, mitigate putrefaction, and preserve morphology while maintaining antigenicity.10
Indirect Immunofluorescence
Indirect immunofluorescence can be helpful for detecting antibodies circulating in patient serum. Indirect immunofluorescence can be used to help diagnose pemphigoid, pemphigus, epidermolysis bullosa acquisita, bullous lupus erythematosus, and dermatitis herpetiformis. Serum testing also can be a helpful alternative when obtaining tissue is difficult, such as in children.
Indirect immunofluorescence is a 2-part technique that takes a bit longer to assay than DIF.11 The first step involves incubating prepared tissue substrates with patient serum. Unlabeled antibodies in the patient serum are allowed to bind to antigens in the substrate tissue for about 30 minutes. Doubling dilutions of patient serum can be performed to titer antibody levels. The second step uses fluorescein-labeled antihuman antibodies to recognize the antigen-antibody conjugates. Normal whole tissues (eg, monkey esophagus for pemphigus vulgaris, rat bladder for paraneoplastic pemphigus, salt-split normal human skin substrate for pemphigoid and epidermolysis bullosa) are the usual substrates for testing.11,12 Again, this test requires serum and should be collected in a red-top tube or serum-separator tube. Usually, a minimum of 0.5 mL is required for testing, but check with your preferred immunodermatology send-out laboratory before collecting.13
Indirect immunofluorescence usually involves an initial screening panel using 1 or 2 tissue substrates followed by individual antigen-specific assays that correspond to the clinical suspicion and IIF screening results.11 Salt-split skin is used to localize basement membrane zone autoantibodies to either the epidermal (roof) or dermal (floor) side. Although many dermatopathology laboratories offer DIF testing, IIF is more specialized and may be a send-out test at your institution.
Enzyme-linked Immunosorbent Assays
Another tool in the immunodermatology armamentarium is ELISA. Commercial ELISA systems are available for the detection of autoantibodies against bullous pemphigoid (BP) antigen 180, BP230, type VII collagen, desmoglein (Dsg) 1, Dsg3, and envoplakin.11 This test allows semiquantitative measurement of antibody levels and thus can be used to monitor response to treatment or identify relapse and treatment failure.11 For example, in BP, significantly increased baseline anti-BP180 IgG levels correlate with 1-year mortality rates (P=.001) and relapse rates (P=.041).14,15 Numerous additional studies support the observation that monitoring anti-BP180 as a potential marker of disease relapse can be helpful.16,17 In pemphigus, the presence or increase of autoantibodies at remission, either anti-Dsg3 or anti-Dsg1, may be a useful tool in predicting disease relapse.18 It is important for physicians to be aware of this to be able to offer guidance on prognosis.
Where Should I Biopsy?
Knowing where to biopsy can be confusing when beginning residency. But the short answer is, it depends. Let your clinical suspicion guide your specimen site. The Figure provides a quick reference for which location will give you the highest yield for a specific diagnosis.

A few cardinal rules should guide which site is biopsied. Avoid obtaining specimens from the lower extremities as much as possible, as this site has been linked with false-negative results, especially in bullous pemphigoid.19,20 As a dependent area prone to stasis, this site gets a lot of abuse and inflammatory changes secondary to everyday insults that can theoretically alter DIF findings, especially fibrinogen deposition.
Although tissue sent for hematoxylin and eosin staining should be lesional, biopsy for DIF ideally should not contain a new or active blister, ulcer, erosion, or bulla. Immunoreactants are more likely to be degraded in these areas, and DIF may be falsely negative.21
It is worthwhile to briefly discuss the definitions of the terms perilesional and nonlesional. Perilesional skin most frequently refers to skin adjacent to a bulla or vesicle. This skin can be erythematous/inflamed or appear normal. When obtaining tissue for a diagnosis of blistering disease, the general recommendation is to obtain the biopsy from lesional nonbullous skin or perilesional uninvolved skin within 1 cm of the bulla.22-24 The only exception to this is dermatitis herpetiformis, which is best diagnosed on tissue obtained from normal-appearing perilesional skin within 1 cm of an active lesion.25 Additionally, if your patient has oral disease, the recommendation is to obtain the biopsy from nonlesional buccal mucosa, especially if there is desquamative gingivitis.26,27
The ideal biopsy size is 4 or 5 mm. If considering both DIF and histopathology, it is best to procure 2 separate specimens. One larger biopsy can be carefully bisected in 2 but often is subject to more handling artifacts, which can affect findings. In the case of 1 biopsy bisected into 2 specimens, the punch should be at least 6 mm. Shave biopsies also can be performed as long as they extend into the reticular dermis.23
For vasculitis, biopsies for DIF should be taken from lesions that are less than 24 hours old for highest yield, as the level of tissue immunoreactants tends to decline over time.28 This guideline does differ from hematoxylin and eosin specimens sent for evaluation of vasculitis, which ideally should be lesional tissue over 72 hours old. When evaluating for lupus (including subacute cutaneous lupus, discoid lupus, and systemic lupus), DIF is more likely to be positive in well-established, active lesions.
Which Test Should I Order?
The answer to this question depends, but the use of all 3 tests has a specificity close to 100% when evaluating for autoantibody-associated diseases.23 For autoimmune blistering disease, DIF is considered the diagnostic standard. The sensitivity of DIF for diagnosing BP is in the range of 82% to 90.5%, while specificity is 98%.29-31 Other autoimmune blistering diseases, such as pemphigus or dermatitis herpetiformis, have even higher sensitivities and specificities. Direct immunofluorescence often is used as a screening test, but false negatives do occur.32,33 Although rare, false positives also can occur, especially in cases of infection, and should be suspected when there is a lack of clinicopathologic correlation.34 If DIF is negative but clinical suspicion remains high, IIF should be ordered to directly evaluate a patient’s serum for autoantibodies.
In acute cutaneous lupus, subacute cutaneous lupus, and discoid lupus, DIF of active lesions may be helpful if histopathologic examination of a cutaneous lupus erythematosus lesion is nondiagnostic. However, histopathologic examination of formalin-fixed tissue remains the standard for these diagnoses. In vasculitis, while DIF is not used for diagnosis, it is useful to evaluate for IgA deposition. This is important in adults, as IgA deposition has been associated with a greater risk for developing end-stage renal disease.35
Final Thoughts
This is an overview of the tests available for diagnosing autoimmune blistering diseases. Residents should keep in mind that these tests are just one part of the puzzle when it comes to diagnosing these diseases. Results of DIF, IIF, and ELISA testing should be considered in conjunction with patient history and physical examination as well as histopathologic examination of lesional tissue when evaluating for dermatologic diseases with autoantibodies.
- Arthur G. Albert Coons: harnessing the power of the antibody. Lancet Respir Med. 2016;4:181-182.
- Coons AH, Creech HJ, Jones RN. Immunological properties of an antibody containing a fluorescent group. Proc Soc Exp Biol Med. 1941;47:200-202.
- Coons AH, Creech HJ, Jones RN, et al. The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody. J Immunol. 1942;45:159-170.
- Burnham TK, Neblett TR, Fine G. The application of the fluorescent antibody technic to the investigation of lupus erythematosus and various dermatoses. J Invest Dermatol. 1963;41:451-456.
- Jordon RE, Beutner EH, Witebsky E, et al. Basement zone antibodies in bullous pemphigoid. JAMA. 1967;200:751-756.
- Vaughan Jones SA, Salas J, McGrath JA, et al. A retrospective analysis of tissue-fixed immunoreactants from skin biopsies maintained in Michel’s medium. Dermatology. 1994;189(suppl 1):131-132.
- Kim RH, Brinster NK. Practical direct immunofluorescence. Am J Dermatopathol. 2020;42:75-85.
- Vodegel RM, de Jong MC, Meijer HJ, et al. Enhanced diagnostic immunofluorescence using biopsies transported in saline. BMC Dermatol. 2004;4:10.
- Arbesman J, Grover R, Helm TN, et al. Can direct immunofluorescence testing still be accurate if performed on biopsy specimens after brief inadvertent immersion in formalin? J Am Acad Dermatol. 2011;65:106-111.
- Im K, Mareninov S, Diaz MFP, et al. An introduction to performing immunofluorescence staining. Methods Mol Biol. 2019;1897:299-311.
- Saschenbrecker S, Karl I, Komorowski L, et al. Serological diagnosis of autoimmune bullous skin diseases. Front Immunol. 2019;10:1974.
- Baum S, Sakka N, Artsi O, et al. Diagnosis and classification of autoimmune blistering diseases. Autoimmun Rev. 2014;13:482-489.
- Immunobullous disease panel, epithelial. ARUP Laboratories website. Accessed November 22, 2021. https://ltd.aruplab.com/Tests/Pub/3001409
- Monshi B, Gulz L, Piringer B, et al. Anti-BP180 autoantibody levels at diagnosis correlate with 1-year mortality rates in patients with bullous pemphigoid. J Eur Acad Dermatol Venereol. 2020;34:1583-1589.
- Koga H, Teye K, Ishii N, et al. High index values of enzyme-linked immunosorbent assay for BP180 at baseline predict relapse in patients with bullous pemphigoid. Front Med (Lausanne). 2018;5:139.
- Fichel F, Barbe C, Joly P, et al. Clinical and immunologic factors associated with bullous pemphigoid relapse during the first year of treatment: a multicenter, prospective study. JAMA Dermatol. 2014;150:25-33.
- Cai SC, Lim YL, Li W, et al. Anti-BP180 NC16A IgG titres as an indicator of disease activity and outcome in Asian patients with bullous pemphigoid. Ann Acad Med Singap. 2015;44:119-126.
- Genovese G, Maronese CA, Casazza G, et al. Clinical and serological predictors of relapse in pemphigus: a study of 143 patients [published online July 20, 2021]. Clin Exp Dermatol. doi:10.1111/ced.14854
- Weigand DA. Effect of anatomic region on immunofluorescence diagnosis of bullous pemphigoid. J Am Acad Dermatol. 1985;12(2, pt 1):274-278.
- Weigand DA, Clements MK. Direct immunofluorescence in bullous pemphigoid: effects of extent and location of lesions. J Am Acad Dermatol. 1989;20:437-440.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822; quiz 822-824.
- Sladden C, Kirchhof MG, Crawford RI. Biopsy location for direct immunofluorescence in patients with suspected bullous pemphigoid impacts probability of a positive test result. J Cutan Med Surg. 2014;18:392-396.
- Elston DM, Stratman EJ, Miller SJ. Skin biopsy: biopsy issues in specific diseases. J Am Acad Dermatol. 2016;74:1-16; quiz 17-18.
- Seishima M, Izumi T, Kitajima Y. Antibody to bullous pemphigoid antigen 1 binds to the antigen at perilesional but not uninvolved skin, in localized bullous pemphigoid. Eur J Dermatol. 1999;9:39-42.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol. 1996;132:912-918.
- Kamaguchi M, Iwata H, Ujiie I, et al. Direct immunofluorescence using non-lesional buccal mucosa in mucous membrane pemphigoid. Front Med (Lausanne). 2018;5:20.
- Carey B, Joshi S, Abdelghani A, et al. The optimal oral biopsy site for diagnosis of mucous membrane pemphigoid and pemphigus vulgaris. Br J Dermatol. 2020;182:747-753.
- Kulthanan K, Pinkaew S, Jiamton S, et al. Cutaneous leukocytoclastic vasculitis: the yield of direct immunofluorescence study. J Med Assoc Thai. 2004;87:531-535.
- Chaidemenos GC, Maltezos E, Chrysomallis F, et al. Value of routine diagnostic criteria of bullous pemphigoid. Int J Dermatol. 1998;37:206-210.
- Mysorekar VV, Sumathy TK, Shyam Prasad AL. Role of direct immunofluorescence in dermatological disorders. Indian Dermatol Online J. 2015;6:172-180.
- Fudge JG, Crawford RI. Bullous pemphigoid: a 10-year study of discordant results on direct immunofluorescence. J Cutan Med Surg. 2018;22:472-475.
- Sárdy M, Kostaki D, Varga R, et al. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol. 2013;69:748-753.
- Buch AC, Kumar H, Panicker N, et al. A cross-sectional study of direct immunofluorescence in the diagnosis of immunobullous dermatoses. Indian J Dermatol. 2014;59:364-368.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
- Cao R, Lau S, Tan V, et al. Adult Henoch-Schönlein purpura: clinical and histopathological predictors of systemic disease and profound renal disease. Indian J Dermatol Venereol Leprol. 2017;83:577-582.
Direct immunofluorescence (DIF) is the go-to diagnostic test when evaluating vesiculobullous eruptions, connective tissue disease, and vasculitis. This specialized test allows visualization of autoantibodies and their reaction products in the epidermis and dermis (skin) and epithelium and subepithelium (mucosa). Indirect immunofluorescence (IIF) and enzyme-linked immunosorbent assay (ELISA) are additional tests that can help in the diagnosis of autoimmune blistering disease. In the blistering autoimmune diseases, the autoantibodies target components in skin and mucous membranes that are essential for cell-cell and cell-matrix adhesion causing separation within or beneath the epidermis, depending on where the target components are located. This article is intended to serve as a helpful primer for immunofluorescence testing in dermatology, with an overview of the tests available as well as pragmatic tips for optimal biopsy sites and specimen transport.
Direct Immunofluorescence
Immunofluorescence techniques date back to 1941 when Albert Coons, an American physician, pathologist, and immunologist, fluorescently labelled antibodies to visualize pneumococcal antigens in infected tissues.1-3 In dermatology, similar methodology was used to visualize the deposition of immunoglobulins and complement in the skin of patients with systemic lupus erythematosus in 1963.4 Basement membrane zone antibodies were first visualized via DIF in bullous pemphigoid in 1967.5 This elegant test utilizes specific antibodies labeled with fluorophores that are then incubated with the patient’s tissue, ultimately forming antibody-antigen conjugates that can be visualized with a fluorescent microscope. Antibodies usually include IgG, IgM, IgA, fibrinogen, and C3. Some institutions also evaluate for IgG4.
Transport medium is critical for proper evaluation of tissues using DIF. Inappropriate storage of tissue can degrade the antigen and confuse the interpretation of specimens. An acceptable medium for DIF includes Michel transport medium, which allows tissue to be stored for days while being transported at ambient temperature without loss of signal.6,7 Zeus medium also can be used and is more readily available. Alternatively, biopsy tissue can be snap frozen using liquid nitrogen. Specimens also may be stored on saline gauze but should be analyzed within 24 to 48 hours.8 Most importantly, do not place the specimen in formalin; even a brief soak in formalin can greatly alter results, especially when trying to diagnose pemphigus.9 Proper transport conditions are critical to prevent autolysis, mitigate putrefaction, and preserve morphology while maintaining antigenicity.10
Indirect Immunofluorescence
Indirect immunofluorescence can be helpful for detecting antibodies circulating in patient serum. Indirect immunofluorescence can be used to help diagnose pemphigoid, pemphigus, epidermolysis bullosa acquisita, bullous lupus erythematosus, and dermatitis herpetiformis. Serum testing also can be a helpful alternative when obtaining tissue is difficult, such as in children.
Indirect immunofluorescence is a 2-part technique that takes a bit longer to assay than DIF.11 The first step involves incubating prepared tissue substrates with patient serum. Unlabeled antibodies in the patient serum are allowed to bind to antigens in the substrate tissue for about 30 minutes. Doubling dilutions of patient serum can be performed to titer antibody levels. The second step uses fluorescein-labeled antihuman antibodies to recognize the antigen-antibody conjugates. Normal whole tissues (eg, monkey esophagus for pemphigus vulgaris, rat bladder for paraneoplastic pemphigus, salt-split normal human skin substrate for pemphigoid and epidermolysis bullosa) are the usual substrates for testing.11,12 Again, this test requires serum and should be collected in a red-top tube or serum-separator tube. Usually, a minimum of 0.5 mL is required for testing, but check with your preferred immunodermatology send-out laboratory before collecting.13
Indirect immunofluorescence usually involves an initial screening panel using 1 or 2 tissue substrates followed by individual antigen-specific assays that correspond to the clinical suspicion and IIF screening results.11 Salt-split skin is used to localize basement membrane zone autoantibodies to either the epidermal (roof) or dermal (floor) side. Although many dermatopathology laboratories offer DIF testing, IIF is more specialized and may be a send-out test at your institution.
Enzyme-linked Immunosorbent Assays
Another tool in the immunodermatology armamentarium is ELISA. Commercial ELISA systems are available for the detection of autoantibodies against bullous pemphigoid (BP) antigen 180, BP230, type VII collagen, desmoglein (Dsg) 1, Dsg3, and envoplakin.11 This test allows semiquantitative measurement of antibody levels and thus can be used to monitor response to treatment or identify relapse and treatment failure.11 For example, in BP, significantly increased baseline anti-BP180 IgG levels correlate with 1-year mortality rates (P=.001) and relapse rates (P=.041).14,15 Numerous additional studies support the observation that monitoring anti-BP180 as a potential marker of disease relapse can be helpful.16,17 In pemphigus, the presence or increase of autoantibodies at remission, either anti-Dsg3 or anti-Dsg1, may be a useful tool in predicting disease relapse.18 It is important for physicians to be aware of this to be able to offer guidance on prognosis.
Where Should I Biopsy?
Knowing where to biopsy can be confusing when beginning residency. But the short answer is, it depends. Let your clinical suspicion guide your specimen site. The Figure provides a quick reference for which location will give you the highest yield for a specific diagnosis.
A few cardinal rules should guide which site is biopsied. Avoid obtaining specimens from the lower extremities as much as possible, as this site has been linked with false-negative results, especially in bullous pemphigoid.19,20 As a dependent area prone to stasis, this site gets a lot of abuse and inflammatory changes secondary to everyday insults that can theoretically alter DIF findings, especially fibrinogen deposition.
Although tissue sent for hematoxylin and eosin staining should be lesional, biopsy for DIF ideally should not contain a new or active blister, ulcer, erosion, or bulla. Immunoreactants are more likely to be degraded in these areas, and DIF may be falsely negative.21
It is worthwhile to briefly discuss the definitions of the terms perilesional and nonlesional. Perilesional skin most frequently refers to skin adjacent to a bulla or vesicle. This skin can be erythematous/inflamed or appear normal. When obtaining tissue for a diagnosis of blistering disease, the general recommendation is to obtain the biopsy from lesional nonbullous skin or perilesional uninvolved skin within 1 cm of the bulla.22-24 The only exception to this is dermatitis herpetiformis, which is best diagnosed on tissue obtained from normal-appearing perilesional skin within 1 cm of an active lesion.25 Additionally, if your patient has oral disease, the recommendation is to obtain the biopsy from nonlesional buccal mucosa, especially if there is desquamative gingivitis.26,27
The ideal biopsy size is 4 or 5 mm. If considering both DIF and histopathology, it is best to procure 2 separate specimens. One larger biopsy can be carefully bisected in 2 but often is subject to more handling artifacts, which can affect findings. In the case of 1 biopsy bisected into 2 specimens, the punch should be at least 6 mm. Shave biopsies also can be performed as long as they extend into the reticular dermis.23
For vasculitis, biopsies for DIF should be taken from lesions that are less than 24 hours old for highest yield, as the level of tissue immunoreactants tends to decline over time.28 This guideline does differ from hematoxylin and eosin specimens sent for evaluation of vasculitis, which ideally should be lesional tissue over 72 hours old. When evaluating for lupus (including subacute cutaneous lupus, discoid lupus, and systemic lupus), DIF is more likely to be positive in well-established, active lesions.
Which Test Should I Order?
The answer to this question depends, but the use of all 3 tests has a specificity close to 100% when evaluating for autoantibody-associated diseases.23 For autoimmune blistering disease, DIF is considered the diagnostic standard. The sensitivity of DIF for diagnosing BP is in the range of 82% to 90.5%, while specificity is 98%.29-31 Other autoimmune blistering diseases, such as pemphigus or dermatitis herpetiformis, have even higher sensitivities and specificities. Direct immunofluorescence often is used as a screening test, but false negatives do occur.32,33 Although rare, false positives also can occur, especially in cases of infection, and should be suspected when there is a lack of clinicopathologic correlation.34 If DIF is negative but clinical suspicion remains high, IIF should be ordered to directly evaluate a patient’s serum for autoantibodies.
In acute cutaneous lupus, subacute cutaneous lupus, and discoid lupus, DIF of active lesions may be helpful if histopathologic examination of a cutaneous lupus erythematosus lesion is nondiagnostic. However, histopathologic examination of formalin-fixed tissue remains the standard for these diagnoses. In vasculitis, while DIF is not used for diagnosis, it is useful to evaluate for IgA deposition. This is important in adults, as IgA deposition has been associated with a greater risk for developing end-stage renal disease.35
Final Thoughts
This is an overview of the tests available for diagnosing autoimmune blistering diseases. Residents should keep in mind that these tests are just one part of the puzzle when it comes to diagnosing these diseases. Results of DIF, IIF, and ELISA testing should be considered in conjunction with patient history and physical examination as well as histopathologic examination of lesional tissue when evaluating for dermatologic diseases with autoantibodies.
Direct immunofluorescence (DIF) is the go-to diagnostic test when evaluating vesiculobullous eruptions, connective tissue disease, and vasculitis. This specialized test allows visualization of autoantibodies and their reaction products in the epidermis and dermis (skin) and epithelium and subepithelium (mucosa). Indirect immunofluorescence (IIF) and enzyme-linked immunosorbent assay (ELISA) are additional tests that can help in the diagnosis of autoimmune blistering disease. In the blistering autoimmune diseases, the autoantibodies target components in skin and mucous membranes that are essential for cell-cell and cell-matrix adhesion causing separation within or beneath the epidermis, depending on where the target components are located. This article is intended to serve as a helpful primer for immunofluorescence testing in dermatology, with an overview of the tests available as well as pragmatic tips for optimal biopsy sites and specimen transport.
Direct Immunofluorescence
Immunofluorescence techniques date back to 1941 when Albert Coons, an American physician, pathologist, and immunologist, fluorescently labelled antibodies to visualize pneumococcal antigens in infected tissues.1-3 In dermatology, similar methodology was used to visualize the deposition of immunoglobulins and complement in the skin of patients with systemic lupus erythematosus in 1963.4 Basement membrane zone antibodies were first visualized via DIF in bullous pemphigoid in 1967.5 This elegant test utilizes specific antibodies labeled with fluorophores that are then incubated with the patient’s tissue, ultimately forming antibody-antigen conjugates that can be visualized with a fluorescent microscope. Antibodies usually include IgG, IgM, IgA, fibrinogen, and C3. Some institutions also evaluate for IgG4.
Transport medium is critical for proper evaluation of tissues using DIF. Inappropriate storage of tissue can degrade the antigen and confuse the interpretation of specimens. An acceptable medium for DIF includes Michel transport medium, which allows tissue to be stored for days while being transported at ambient temperature without loss of signal.6,7 Zeus medium also can be used and is more readily available. Alternatively, biopsy tissue can be snap frozen using liquid nitrogen. Specimens also may be stored on saline gauze but should be analyzed within 24 to 48 hours.8 Most importantly, do not place the specimen in formalin; even a brief soak in formalin can greatly alter results, especially when trying to diagnose pemphigus.9 Proper transport conditions are critical to prevent autolysis, mitigate putrefaction, and preserve morphology while maintaining antigenicity.10
Indirect Immunofluorescence
Indirect immunofluorescence can be helpful for detecting antibodies circulating in patient serum. Indirect immunofluorescence can be used to help diagnose pemphigoid, pemphigus, epidermolysis bullosa acquisita, bullous lupus erythematosus, and dermatitis herpetiformis. Serum testing also can be a helpful alternative when obtaining tissue is difficult, such as in children.
Indirect immunofluorescence is a 2-part technique that takes a bit longer to assay than DIF.11 The first step involves incubating prepared tissue substrates with patient serum. Unlabeled antibodies in the patient serum are allowed to bind to antigens in the substrate tissue for about 30 minutes. Doubling dilutions of patient serum can be performed to titer antibody levels. The second step uses fluorescein-labeled antihuman antibodies to recognize the antigen-antibody conjugates. Normal whole tissues (eg, monkey esophagus for pemphigus vulgaris, rat bladder for paraneoplastic pemphigus, salt-split normal human skin substrate for pemphigoid and epidermolysis bullosa) are the usual substrates for testing.11,12 Again, this test requires serum and should be collected in a red-top tube or serum-separator tube. Usually, a minimum of 0.5 mL is required for testing, but check with your preferred immunodermatology send-out laboratory before collecting.13
Indirect immunofluorescence usually involves an initial screening panel using 1 or 2 tissue substrates followed by individual antigen-specific assays that correspond to the clinical suspicion and IIF screening results.11 Salt-split skin is used to localize basement membrane zone autoantibodies to either the epidermal (roof) or dermal (floor) side. Although many dermatopathology laboratories offer DIF testing, IIF is more specialized and may be a send-out test at your institution.
Enzyme-linked Immunosorbent Assays
Another tool in the immunodermatology armamentarium is ELISA. Commercial ELISA systems are available for the detection of autoantibodies against bullous pemphigoid (BP) antigen 180, BP230, type VII collagen, desmoglein (Dsg) 1, Dsg3, and envoplakin.11 This test allows semiquantitative measurement of antibody levels and thus can be used to monitor response to treatment or identify relapse and treatment failure.11 For example, in BP, significantly increased baseline anti-BP180 IgG levels correlate with 1-year mortality rates (P=.001) and relapse rates (P=.041).14,15 Numerous additional studies support the observation that monitoring anti-BP180 as a potential marker of disease relapse can be helpful.16,17 In pemphigus, the presence or increase of autoantibodies at remission, either anti-Dsg3 or anti-Dsg1, may be a useful tool in predicting disease relapse.18 It is important for physicians to be aware of this to be able to offer guidance on prognosis.
Where Should I Biopsy?
Knowing where to biopsy can be confusing when beginning residency. But the short answer is, it depends. Let your clinical suspicion guide your specimen site. The Figure provides a quick reference for which location will give you the highest yield for a specific diagnosis.
A few cardinal rules should guide which site is biopsied. Avoid obtaining specimens from the lower extremities as much as possible, as this site has been linked with false-negative results, especially in bullous pemphigoid.19,20 As a dependent area prone to stasis, this site gets a lot of abuse and inflammatory changes secondary to everyday insults that can theoretically alter DIF findings, especially fibrinogen deposition.
Although tissue sent for hematoxylin and eosin staining should be lesional, biopsy for DIF ideally should not contain a new or active blister, ulcer, erosion, or bulla. Immunoreactants are more likely to be degraded in these areas, and DIF may be falsely negative.21
It is worthwhile to briefly discuss the definitions of the terms perilesional and nonlesional. Perilesional skin most frequently refers to skin adjacent to a bulla or vesicle. This skin can be erythematous/inflamed or appear normal. When obtaining tissue for a diagnosis of blistering disease, the general recommendation is to obtain the biopsy from lesional nonbullous skin or perilesional uninvolved skin within 1 cm of the bulla.22-24 The only exception to this is dermatitis herpetiformis, which is best diagnosed on tissue obtained from normal-appearing perilesional skin within 1 cm of an active lesion.25 Additionally, if your patient has oral disease, the recommendation is to obtain the biopsy from nonlesional buccal mucosa, especially if there is desquamative gingivitis.26,27
The ideal biopsy size is 4 or 5 mm. If considering both DIF and histopathology, it is best to procure 2 separate specimens. One larger biopsy can be carefully bisected in 2 but often is subject to more handling artifacts, which can affect findings. In the case of 1 biopsy bisected into 2 specimens, the punch should be at least 6 mm. Shave biopsies also can be performed as long as they extend into the reticular dermis.23
For vasculitis, biopsies for DIF should be taken from lesions that are less than 24 hours old for highest yield, as the level of tissue immunoreactants tends to decline over time.28 This guideline does differ from hematoxylin and eosin specimens sent for evaluation of vasculitis, which ideally should be lesional tissue over 72 hours old. When evaluating for lupus (including subacute cutaneous lupus, discoid lupus, and systemic lupus), DIF is more likely to be positive in well-established, active lesions.
Which Test Should I Order?
The answer to this question depends, but the use of all 3 tests has a specificity close to 100% when evaluating for autoantibody-associated diseases.23 For autoimmune blistering disease, DIF is considered the diagnostic standard. The sensitivity of DIF for diagnosing BP is in the range of 82% to 90.5%, while specificity is 98%.29-31 Other autoimmune blistering diseases, such as pemphigus or dermatitis herpetiformis, have even higher sensitivities and specificities. Direct immunofluorescence often is used as a screening test, but false negatives do occur.32,33 Although rare, false positives also can occur, especially in cases of infection, and should be suspected when there is a lack of clinicopathologic correlation.34 If DIF is negative but clinical suspicion remains high, IIF should be ordered to directly evaluate a patient’s serum for autoantibodies.
In acute cutaneous lupus, subacute cutaneous lupus, and discoid lupus, DIF of active lesions may be helpful if histopathologic examination of a cutaneous lupus erythematosus lesion is nondiagnostic. However, histopathologic examination of formalin-fixed tissue remains the standard for these diagnoses. In vasculitis, while DIF is not used for diagnosis, it is useful to evaluate for IgA deposition. This is important in adults, as IgA deposition has been associated with a greater risk for developing end-stage renal disease.35
Final Thoughts
This is an overview of the tests available for diagnosing autoimmune blistering diseases. Residents should keep in mind that these tests are just one part of the puzzle when it comes to diagnosing these diseases. Results of DIF, IIF, and ELISA testing should be considered in conjunction with patient history and physical examination as well as histopathologic examination of lesional tissue when evaluating for dermatologic diseases with autoantibodies.
- Arthur G. Albert Coons: harnessing the power of the antibody. Lancet Respir Med. 2016;4:181-182.
- Coons AH, Creech HJ, Jones RN. Immunological properties of an antibody containing a fluorescent group. Proc Soc Exp Biol Med. 1941;47:200-202.
- Coons AH, Creech HJ, Jones RN, et al. The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody. J Immunol. 1942;45:159-170.
- Burnham TK, Neblett TR, Fine G. The application of the fluorescent antibody technic to the investigation of lupus erythematosus and various dermatoses. J Invest Dermatol. 1963;41:451-456.
- Jordon RE, Beutner EH, Witebsky E, et al. Basement zone antibodies in bullous pemphigoid. JAMA. 1967;200:751-756.
- Vaughan Jones SA, Salas J, McGrath JA, et al. A retrospective analysis of tissue-fixed immunoreactants from skin biopsies maintained in Michel’s medium. Dermatology. 1994;189(suppl 1):131-132.
- Kim RH, Brinster NK. Practical direct immunofluorescence. Am J Dermatopathol. 2020;42:75-85.
- Vodegel RM, de Jong MC, Meijer HJ, et al. Enhanced diagnostic immunofluorescence using biopsies transported in saline. BMC Dermatol. 2004;4:10.
- Arbesman J, Grover R, Helm TN, et al. Can direct immunofluorescence testing still be accurate if performed on biopsy specimens after brief inadvertent immersion in formalin? J Am Acad Dermatol. 2011;65:106-111.
- Im K, Mareninov S, Diaz MFP, et al. An introduction to performing immunofluorescence staining. Methods Mol Biol. 2019;1897:299-311.
- Saschenbrecker S, Karl I, Komorowski L, et al. Serological diagnosis of autoimmune bullous skin diseases. Front Immunol. 2019;10:1974.
- Baum S, Sakka N, Artsi O, et al. Diagnosis and classification of autoimmune blistering diseases. Autoimmun Rev. 2014;13:482-489.
- Immunobullous disease panel, epithelial. ARUP Laboratories website. Accessed November 22, 2021. https://ltd.aruplab.com/Tests/Pub/3001409
- Monshi B, Gulz L, Piringer B, et al. Anti-BP180 autoantibody levels at diagnosis correlate with 1-year mortality rates in patients with bullous pemphigoid. J Eur Acad Dermatol Venereol. 2020;34:1583-1589.
- Koga H, Teye K, Ishii N, et al. High index values of enzyme-linked immunosorbent assay for BP180 at baseline predict relapse in patients with bullous pemphigoid. Front Med (Lausanne). 2018;5:139.
- Fichel F, Barbe C, Joly P, et al. Clinical and immunologic factors associated with bullous pemphigoid relapse during the first year of treatment: a multicenter, prospective study. JAMA Dermatol. 2014;150:25-33.
- Cai SC, Lim YL, Li W, et al. Anti-BP180 NC16A IgG titres as an indicator of disease activity and outcome in Asian patients with bullous pemphigoid. Ann Acad Med Singap. 2015;44:119-126.
- Genovese G, Maronese CA, Casazza G, et al. Clinical and serological predictors of relapse in pemphigus: a study of 143 patients [published online July 20, 2021]. Clin Exp Dermatol. doi:10.1111/ced.14854
- Weigand DA. Effect of anatomic region on immunofluorescence diagnosis of bullous pemphigoid. J Am Acad Dermatol. 1985;12(2, pt 1):274-278.
- Weigand DA, Clements MK. Direct immunofluorescence in bullous pemphigoid: effects of extent and location of lesions. J Am Acad Dermatol. 1989;20:437-440.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822; quiz 822-824.
- Sladden C, Kirchhof MG, Crawford RI. Biopsy location for direct immunofluorescence in patients with suspected bullous pemphigoid impacts probability of a positive test result. J Cutan Med Surg. 2014;18:392-396.
- Elston DM, Stratman EJ, Miller SJ. Skin biopsy: biopsy issues in specific diseases. J Am Acad Dermatol. 2016;74:1-16; quiz 17-18.
- Seishima M, Izumi T, Kitajima Y. Antibody to bullous pemphigoid antigen 1 binds to the antigen at perilesional but not uninvolved skin, in localized bullous pemphigoid. Eur J Dermatol. 1999;9:39-42.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol. 1996;132:912-918.
- Kamaguchi M, Iwata H, Ujiie I, et al. Direct immunofluorescence using non-lesional buccal mucosa in mucous membrane pemphigoid. Front Med (Lausanne). 2018;5:20.
- Carey B, Joshi S, Abdelghani A, et al. The optimal oral biopsy site for diagnosis of mucous membrane pemphigoid and pemphigus vulgaris. Br J Dermatol. 2020;182:747-753.
- Kulthanan K, Pinkaew S, Jiamton S, et al. Cutaneous leukocytoclastic vasculitis: the yield of direct immunofluorescence study. J Med Assoc Thai. 2004;87:531-535.
- Chaidemenos GC, Maltezos E, Chrysomallis F, et al. Value of routine diagnostic criteria of bullous pemphigoid. Int J Dermatol. 1998;37:206-210.
- Mysorekar VV, Sumathy TK, Shyam Prasad AL. Role of direct immunofluorescence in dermatological disorders. Indian Dermatol Online J. 2015;6:172-180.
- Fudge JG, Crawford RI. Bullous pemphigoid: a 10-year study of discordant results on direct immunofluorescence. J Cutan Med Surg. 2018;22:472-475.
- Sárdy M, Kostaki D, Varga R, et al. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol. 2013;69:748-753.
- Buch AC, Kumar H, Panicker N, et al. A cross-sectional study of direct immunofluorescence in the diagnosis of immunobullous dermatoses. Indian J Dermatol. 2014;59:364-368.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
- Cao R, Lau S, Tan V, et al. Adult Henoch-Schönlein purpura: clinical and histopathological predictors of systemic disease and profound renal disease. Indian J Dermatol Venereol Leprol. 2017;83:577-582.
- Arthur G. Albert Coons: harnessing the power of the antibody. Lancet Respir Med. 2016;4:181-182.
- Coons AH, Creech HJ, Jones RN. Immunological properties of an antibody containing a fluorescent group. Proc Soc Exp Biol Med. 1941;47:200-202.
- Coons AH, Creech HJ, Jones RN, et al. The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody. J Immunol. 1942;45:159-170.
- Burnham TK, Neblett TR, Fine G. The application of the fluorescent antibody technic to the investigation of lupus erythematosus and various dermatoses. J Invest Dermatol. 1963;41:451-456.
- Jordon RE, Beutner EH, Witebsky E, et al. Basement zone antibodies in bullous pemphigoid. JAMA. 1967;200:751-756.
- Vaughan Jones SA, Salas J, McGrath JA, et al. A retrospective analysis of tissue-fixed immunoreactants from skin biopsies maintained in Michel’s medium. Dermatology. 1994;189(suppl 1):131-132.
- Kim RH, Brinster NK. Practical direct immunofluorescence. Am J Dermatopathol. 2020;42:75-85.
- Vodegel RM, de Jong MC, Meijer HJ, et al. Enhanced diagnostic immunofluorescence using biopsies transported in saline. BMC Dermatol. 2004;4:10.
- Arbesman J, Grover R, Helm TN, et al. Can direct immunofluorescence testing still be accurate if performed on biopsy specimens after brief inadvertent immersion in formalin? J Am Acad Dermatol. 2011;65:106-111.
- Im K, Mareninov S, Diaz MFP, et al. An introduction to performing immunofluorescence staining. Methods Mol Biol. 2019;1897:299-311.
- Saschenbrecker S, Karl I, Komorowski L, et al. Serological diagnosis of autoimmune bullous skin diseases. Front Immunol. 2019;10:1974.
- Baum S, Sakka N, Artsi O, et al. Diagnosis and classification of autoimmune blistering diseases. Autoimmun Rev. 2014;13:482-489.
- Immunobullous disease panel, epithelial. ARUP Laboratories website. Accessed November 22, 2021. https://ltd.aruplab.com/Tests/Pub/3001409
- Monshi B, Gulz L, Piringer B, et al. Anti-BP180 autoantibody levels at diagnosis correlate with 1-year mortality rates in patients with bullous pemphigoid. J Eur Acad Dermatol Venereol. 2020;34:1583-1589.
- Koga H, Teye K, Ishii N, et al. High index values of enzyme-linked immunosorbent assay for BP180 at baseline predict relapse in patients with bullous pemphigoid. Front Med (Lausanne). 2018;5:139.
- Fichel F, Barbe C, Joly P, et al. Clinical and immunologic factors associated with bullous pemphigoid relapse during the first year of treatment: a multicenter, prospective study. JAMA Dermatol. 2014;150:25-33.
- Cai SC, Lim YL, Li W, et al. Anti-BP180 NC16A IgG titres as an indicator of disease activity and outcome in Asian patients with bullous pemphigoid. Ann Acad Med Singap. 2015;44:119-126.
- Genovese G, Maronese CA, Casazza G, et al. Clinical and serological predictors of relapse in pemphigus: a study of 143 patients [published online July 20, 2021]. Clin Exp Dermatol. doi:10.1111/ced.14854
- Weigand DA. Effect of anatomic region on immunofluorescence diagnosis of bullous pemphigoid. J Am Acad Dermatol. 1985;12(2, pt 1):274-278.
- Weigand DA, Clements MK. Direct immunofluorescence in bullous pemphigoid: effects of extent and location of lesions. J Am Acad Dermatol. 1989;20:437-440.
- Mutasim DF, Adams BB. Immunofluorescence in dermatology. J Am Acad Dermatol. 2001;45:803-822; quiz 822-824.
- Sladden C, Kirchhof MG, Crawford RI. Biopsy location for direct immunofluorescence in patients with suspected bullous pemphigoid impacts probability of a positive test result. J Cutan Med Surg. 2014;18:392-396.
- Elston DM, Stratman EJ, Miller SJ. Skin biopsy: biopsy issues in specific diseases. J Am Acad Dermatol. 2016;74:1-16; quiz 17-18.
- Seishima M, Izumi T, Kitajima Y. Antibody to bullous pemphigoid antigen 1 binds to the antigen at perilesional but not uninvolved skin, in localized bullous pemphigoid. Eur J Dermatol. 1999;9:39-42.
- Zone JJ, Meyer LJ, Petersen MJ. Deposition of granular IgA relative to clinical lesions in dermatitis herpetiformis. Arch Dermatol. 1996;132:912-918.
- Kamaguchi M, Iwata H, Ujiie I, et al. Direct immunofluorescence using non-lesional buccal mucosa in mucous membrane pemphigoid. Front Med (Lausanne). 2018;5:20.
- Carey B, Joshi S, Abdelghani A, et al. The optimal oral biopsy site for diagnosis of mucous membrane pemphigoid and pemphigus vulgaris. Br J Dermatol. 2020;182:747-753.
- Kulthanan K, Pinkaew S, Jiamton S, et al. Cutaneous leukocytoclastic vasculitis: the yield of direct immunofluorescence study. J Med Assoc Thai. 2004;87:531-535.
- Chaidemenos GC, Maltezos E, Chrysomallis F, et al. Value of routine diagnostic criteria of bullous pemphigoid. Int J Dermatol. 1998;37:206-210.
- Mysorekar VV, Sumathy TK, Shyam Prasad AL. Role of direct immunofluorescence in dermatological disorders. Indian Dermatol Online J. 2015;6:172-180.
- Fudge JG, Crawford RI. Bullous pemphigoid: a 10-year study of discordant results on direct immunofluorescence. J Cutan Med Surg. 2018;22:472-475.
- Sárdy M, Kostaki D, Varga R, et al. Comparative study of direct and indirect immunofluorescence and of bullous pemphigoid 180 and 230 enzyme-linked immunosorbent assays for diagnosis of bullous pemphigoid. J Am Acad Dermatol. 2013;69:748-753.
- Buch AC, Kumar H, Panicker N, et al. A cross-sectional study of direct immunofluorescence in the diagnosis of immunobullous dermatoses. Indian J Dermatol. 2014;59:364-368.
- Miller DD, Bhawan J. Bullous tinea pedis with direct immunofluorescence positivity: when is a positive result not autoimmune bullous disease? Am J Dermatopathol. 2013;35:587-594.
- Cao R, Lau S, Tan V, et al. Adult Henoch-Schönlein purpura: clinical and histopathological predictors of systemic disease and profound renal disease. Indian J Dermatol Venereol Leprol. 2017;83:577-582.
Resident Pearl
- Direct immunofluorescence, indirect immunofluorescence, and enzyme-linked immunosorbent assay are important tests for residents to have in their diagnostic tool box, especially when evaluating patients with blistering diseases.

Erythematous Nodule With Central Erosions on the Calf
The Diagnosis: Osteoma Cutis
Osteoma cutis is the heterotopic development of cutaneous ossifications in the dermis or subcutaneous fat and presents as plaquelike, stony, hard nodules. It can manifest as either a primary or secondary condition based on the presence or absence of a prior skin insult at the lesion site. Primary osteoma cutis occurs in 15% of patients and arises either de novo or in association with any of several inflammatory, neoplastic, and metabolic diseases that provide a favorable environment for abnormal mesenchymal stem cell commitment to osteoid,1 including Albright hereditary osteodystrophy, myositis ossificans progressiva, and progressive osseous heteroplasia, which are all associated with mutations in the heterotrimeric G-protein alpha subunit encoding gene, GNAS. 1,2 It is suggested that an insufficiency of Gsα leads to uncontrolled negative regulation of nonosseous connective tissue differentiation, forming osteoid.3 Additionally, diseases involving gain-of-function mutations in the activin A receptor type 1 encoding gene, ACVR1, such as fibrodysplasia ossificans progressiva, have been associated with osteoma cutis.4 These mutations lead to decreased receptor affinity to molecular safeguards of bone morphogenetic protein signaling, ultimately contributing to progressive ectopic bone formation.5 Secondary osteoma cutis occurs in 85% of patients and develops at the site of prior skin damage due to inflammation, neoplasm, or trauma.6 It is believed that tissue damage and degeneration lead to mesenchymal stem cell proliferation and skeletogenicinducing factor recruitment forming cartilaginous tissue, later replaced by bone through endochondral ossification.7
Although osteoma cutis previously was believed to be rare, more recent radiologic studies suggest otherwise, detecting cutaneous osteomas in up to 42.1% of patients.8 Consequently, it is likely that osteoma cutis is underdiagnosed due to its subclinical nature. Our patient, however, presented with a solitary osteoma cutis with perforation of the epidermis, a rare phenomenon.9-12
A shave biopsy in our patient revealed moderate to focally marked, irregular epidermal hyperplasia with a large focus of moderate, compact, parakeratotic crust overlying the epidermis in the center of the specimen. The papillary dermis in the center of the specimen revealed large foci of dark pink to purple bone fragments surrounded by moderate lymphocytic infiltrate with few foci perforating through the overlying epidermis (Figure, A). These findings were characteristic of osteoma cutis with perforation through the overlying epidermis.
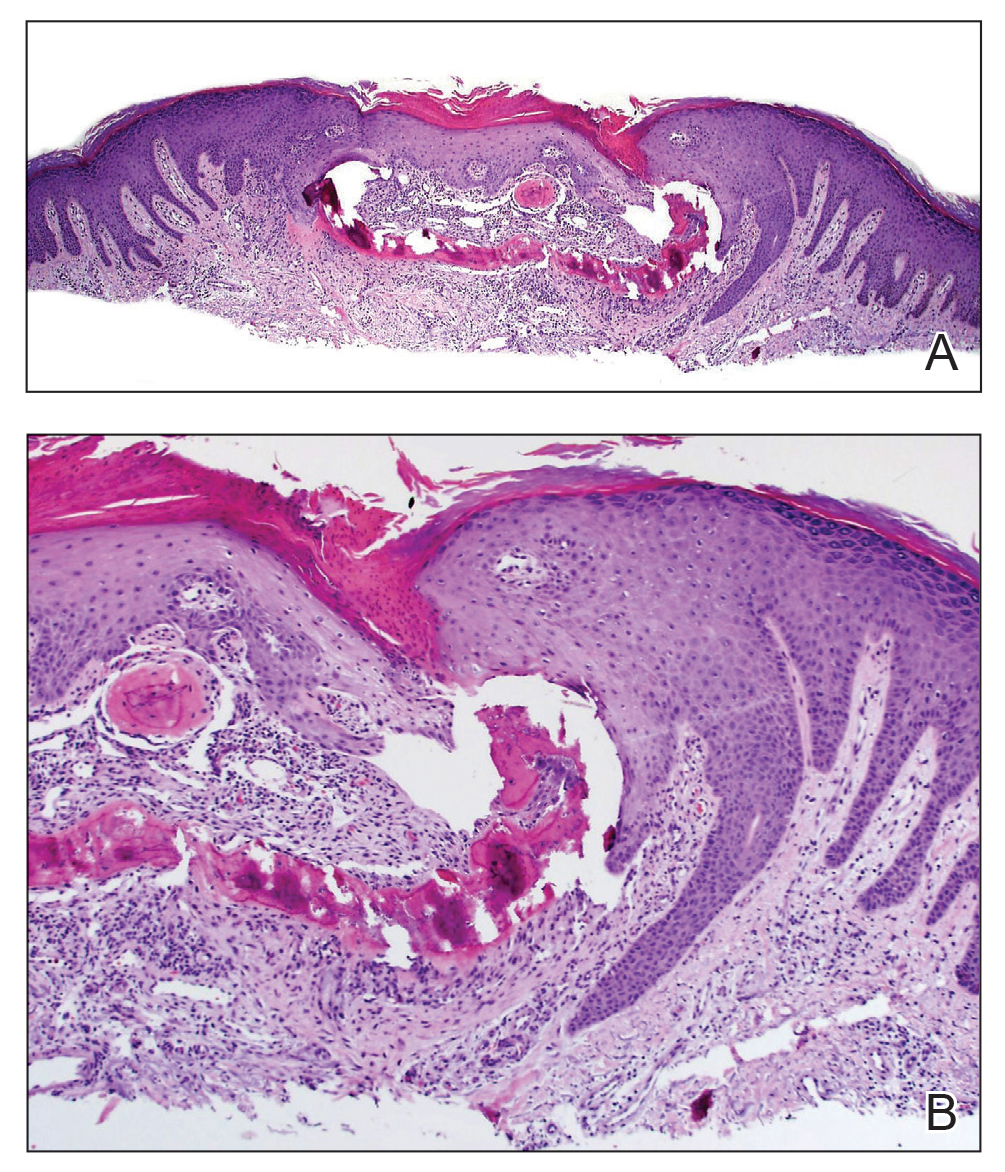
The diagnosis of osteoma cutis at the age of 62 years suggested that the lesion was not primary in association with previously described diseases. Furthermore, the lack of phenotypic features of these diseases including obesity, developmental disability, and high parathyroid hormone levels essentially excluded this possibility. The presence of the lesion on the lower extremities initially may have suggested osteoma cutis secondary to chronic venous insufficiency13; however, the absence of visible varicose veins or obvious signs of stasis disease made this unlikely. No further cutaneous disorders at or around the lesion site clinically and histologically suggested that our patient’s lesion was primary and of idiopathic nature. Dermatofibroma can present similarly in appearance but would characteristically dimple centrally when pinched. Keratoacanthoma presents with central ulceration and keratin plugging. Pilomatricoma more commonly presents on the head and neck and less frequently as a firm nodule. Lastly, prurigo nodularis more commonly presents as a symmetrically diffuse rash compared to an isolated nodule.
Osteoma cutis is a cutaneous ossification that may be primary or secondary in nature and less rare than originally thought. Workup for potentially associated inflammatory, neoplastic, and metabolic diseases should be considered in patients with this condition. Perforating osteoma cutis is a rare variant that presents as solitary or multiple nodules with central erosion and crust. The mechanism of transepidermal elimination leading to skin perforation is hypothesized to involve epidermal hyperproliferation leading to upward movement.14 Shave biopsy establishes a definitive histopathologic diagnosis and often is curative. Given that lesions of osteoma cutis themselves are benign, removal may not be necessary.
- Falsey RR, Ackerman L. Eruptive, hard cutaneous nodules in a 61-yearold woman. osteoma cutis in a patient with Albright hereditary osteodystrophy (AHO). JAMA Dermatol. 2013;149:975-976.
- Martin J, Tucker M, Browning JC. Infantile osteoma cutis as a presentation of a GNAS mutation. Pediatr Dermatol. 2012;29:483-484.
- Shore EM, Ahn J, de Beur SJ, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99-106.
- Kaplan FS, Le Merrer M, Glaser DL, et al. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191-205.
- Song GA, Kim HJ, Woo KM, et al. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem. 2010;285:22542-22553.
- Roth SI, Stowell RE, Helwig EB, et al. Cutaneous ossification. report of 120 cases and review of the literature. Arch Pathol. 1963;76:44-54.
- Shimono K, Uchibe K, Kuboki T, et al. The pathophysiology of heterotopic ossification: current treatment considerations in dentistry. Japanese Dental Science Review. 2014;50:1-8.
- Kim D, Franco GA, Shigehara H, et al. Benign miliary osteoma cutis of the face: a common incidental CT finding. AJNR Am J Neuroradiol. 2017;38:789-794.
- Basu P, Erickson CP, Calame A, et al. Osteoma cutis: an adverse event following tattoo placement. Cureus. 2019;11:E4323.
- Cohen PR. Perforating osteoma cutis: case report and literature review of patients with a solitary perforating osteoma cutis lesion. Dermatol Online J. 2018;24:13030/qt6kt5n92w.
- Hong SH, Kang HY. A case of perforating osteoma cutis. Ann Dermatol. 2003;15:153-155.
- Kim BK, Ahn SK. Acquired perforating osteoma cutis. Ann Dermatol. 2015;27:452-453.
- Lippmann HI, Goldin RR. Subcutaneous ossification of the legs in chronic venous insufficiency. Radiology. 1960;74:279-288.
- Haro R, Revelles JM, Angulo J, et al. Plaque-like osteoma cutis with transepidermal elimination. J Cutan Pathol. 2009;36:591-593.
The Diagnosis: Osteoma Cutis
Osteoma cutis is the heterotopic development of cutaneous ossifications in the dermis or subcutaneous fat and presents as plaquelike, stony, hard nodules. It can manifest as either a primary or secondary condition based on the presence or absence of a prior skin insult at the lesion site. Primary osteoma cutis occurs in 15% of patients and arises either de novo or in association with any of several inflammatory, neoplastic, and metabolic diseases that provide a favorable environment for abnormal mesenchymal stem cell commitment to osteoid,1 including Albright hereditary osteodystrophy, myositis ossificans progressiva, and progressive osseous heteroplasia, which are all associated with mutations in the heterotrimeric G-protein alpha subunit encoding gene, GNAS. 1,2 It is suggested that an insufficiency of Gsα leads to uncontrolled negative regulation of nonosseous connective tissue differentiation, forming osteoid.3 Additionally, diseases involving gain-of-function mutations in the activin A receptor type 1 encoding gene, ACVR1, such as fibrodysplasia ossificans progressiva, have been associated with osteoma cutis.4 These mutations lead to decreased receptor affinity to molecular safeguards of bone morphogenetic protein signaling, ultimately contributing to progressive ectopic bone formation.5 Secondary osteoma cutis occurs in 85% of patients and develops at the site of prior skin damage due to inflammation, neoplasm, or trauma.6 It is believed that tissue damage and degeneration lead to mesenchymal stem cell proliferation and skeletogenicinducing factor recruitment forming cartilaginous tissue, later replaced by bone through endochondral ossification.7
Although osteoma cutis previously was believed to be rare, more recent radiologic studies suggest otherwise, detecting cutaneous osteomas in up to 42.1% of patients.8 Consequently, it is likely that osteoma cutis is underdiagnosed due to its subclinical nature. Our patient, however, presented with a solitary osteoma cutis with perforation of the epidermis, a rare phenomenon.9-12
A shave biopsy in our patient revealed moderate to focally marked, irregular epidermal hyperplasia with a large focus of moderate, compact, parakeratotic crust overlying the epidermis in the center of the specimen. The papillary dermis in the center of the specimen revealed large foci of dark pink to purple bone fragments surrounded by moderate lymphocytic infiltrate with few foci perforating through the overlying epidermis (Figure, A). These findings were characteristic of osteoma cutis with perforation through the overlying epidermis.
The diagnosis of osteoma cutis at the age of 62 years suggested that the lesion was not primary in association with previously described diseases. Furthermore, the lack of phenotypic features of these diseases including obesity, developmental disability, and high parathyroid hormone levels essentially excluded this possibility. The presence of the lesion on the lower extremities initially may have suggested osteoma cutis secondary to chronic venous insufficiency13; however, the absence of visible varicose veins or obvious signs of stasis disease made this unlikely. No further cutaneous disorders at or around the lesion site clinically and histologically suggested that our patient’s lesion was primary and of idiopathic nature. Dermatofibroma can present similarly in appearance but would characteristically dimple centrally when pinched. Keratoacanthoma presents with central ulceration and keratin plugging. Pilomatricoma more commonly presents on the head and neck and less frequently as a firm nodule. Lastly, prurigo nodularis more commonly presents as a symmetrically diffuse rash compared to an isolated nodule.
Osteoma cutis is a cutaneous ossification that may be primary or secondary in nature and less rare than originally thought. Workup for potentially associated inflammatory, neoplastic, and metabolic diseases should be considered in patients with this condition. Perforating osteoma cutis is a rare variant that presents as solitary or multiple nodules with central erosion and crust. The mechanism of transepidermal elimination leading to skin perforation is hypothesized to involve epidermal hyperproliferation leading to upward movement.14 Shave biopsy establishes a definitive histopathologic diagnosis and often is curative. Given that lesions of osteoma cutis themselves are benign, removal may not be necessary.
The Diagnosis: Osteoma Cutis
Osteoma cutis is the heterotopic development of cutaneous ossifications in the dermis or subcutaneous fat and presents as plaquelike, stony, hard nodules. It can manifest as either a primary or secondary condition based on the presence or absence of a prior skin insult at the lesion site. Primary osteoma cutis occurs in 15% of patients and arises either de novo or in association with any of several inflammatory, neoplastic, and metabolic diseases that provide a favorable environment for abnormal mesenchymal stem cell commitment to osteoid,1 including Albright hereditary osteodystrophy, myositis ossificans progressiva, and progressive osseous heteroplasia, which are all associated with mutations in the heterotrimeric G-protein alpha subunit encoding gene, GNAS. 1,2 It is suggested that an insufficiency of Gsα leads to uncontrolled negative regulation of nonosseous connective tissue differentiation, forming osteoid.3 Additionally, diseases involving gain-of-function mutations in the activin A receptor type 1 encoding gene, ACVR1, such as fibrodysplasia ossificans progressiva, have been associated with osteoma cutis.4 These mutations lead to decreased receptor affinity to molecular safeguards of bone morphogenetic protein signaling, ultimately contributing to progressive ectopic bone formation.5 Secondary osteoma cutis occurs in 85% of patients and develops at the site of prior skin damage due to inflammation, neoplasm, or trauma.6 It is believed that tissue damage and degeneration lead to mesenchymal stem cell proliferation and skeletogenicinducing factor recruitment forming cartilaginous tissue, later replaced by bone through endochondral ossification.7
Although osteoma cutis previously was believed to be rare, more recent radiologic studies suggest otherwise, detecting cutaneous osteomas in up to 42.1% of patients.8 Consequently, it is likely that osteoma cutis is underdiagnosed due to its subclinical nature. Our patient, however, presented with a solitary osteoma cutis with perforation of the epidermis, a rare phenomenon.9-12
A shave biopsy in our patient revealed moderate to focally marked, irregular epidermal hyperplasia with a large focus of moderate, compact, parakeratotic crust overlying the epidermis in the center of the specimen. The papillary dermis in the center of the specimen revealed large foci of dark pink to purple bone fragments surrounded by moderate lymphocytic infiltrate with few foci perforating through the overlying epidermis (Figure, A). These findings were characteristic of osteoma cutis with perforation through the overlying epidermis.
The diagnosis of osteoma cutis at the age of 62 years suggested that the lesion was not primary in association with previously described diseases. Furthermore, the lack of phenotypic features of these diseases including obesity, developmental disability, and high parathyroid hormone levels essentially excluded this possibility. The presence of the lesion on the lower extremities initially may have suggested osteoma cutis secondary to chronic venous insufficiency13; however, the absence of visible varicose veins or obvious signs of stasis disease made this unlikely. No further cutaneous disorders at or around the lesion site clinically and histologically suggested that our patient’s lesion was primary and of idiopathic nature. Dermatofibroma can present similarly in appearance but would characteristically dimple centrally when pinched. Keratoacanthoma presents with central ulceration and keratin plugging. Pilomatricoma more commonly presents on the head and neck and less frequently as a firm nodule. Lastly, prurigo nodularis more commonly presents as a symmetrically diffuse rash compared to an isolated nodule.
Osteoma cutis is a cutaneous ossification that may be primary or secondary in nature and less rare than originally thought. Workup for potentially associated inflammatory, neoplastic, and metabolic diseases should be considered in patients with this condition. Perforating osteoma cutis is a rare variant that presents as solitary or multiple nodules with central erosion and crust. The mechanism of transepidermal elimination leading to skin perforation is hypothesized to involve epidermal hyperproliferation leading to upward movement.14 Shave biopsy establishes a definitive histopathologic diagnosis and often is curative. Given that lesions of osteoma cutis themselves are benign, removal may not be necessary.
- Falsey RR, Ackerman L. Eruptive, hard cutaneous nodules in a 61-yearold woman. osteoma cutis in a patient with Albright hereditary osteodystrophy (AHO). JAMA Dermatol. 2013;149:975-976.
- Martin J, Tucker M, Browning JC. Infantile osteoma cutis as a presentation of a GNAS mutation. Pediatr Dermatol. 2012;29:483-484.
- Shore EM, Ahn J, de Beur SJ, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99-106.
- Kaplan FS, Le Merrer M, Glaser DL, et al. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191-205.
- Song GA, Kim HJ, Woo KM, et al. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem. 2010;285:22542-22553.
- Roth SI, Stowell RE, Helwig EB, et al. Cutaneous ossification. report of 120 cases and review of the literature. Arch Pathol. 1963;76:44-54.
- Shimono K, Uchibe K, Kuboki T, et al. The pathophysiology of heterotopic ossification: current treatment considerations in dentistry. Japanese Dental Science Review. 2014;50:1-8.
- Kim D, Franco GA, Shigehara H, et al. Benign miliary osteoma cutis of the face: a common incidental CT finding. AJNR Am J Neuroradiol. 2017;38:789-794.
- Basu P, Erickson CP, Calame A, et al. Osteoma cutis: an adverse event following tattoo placement. Cureus. 2019;11:E4323.
- Cohen PR. Perforating osteoma cutis: case report and literature review of patients with a solitary perforating osteoma cutis lesion. Dermatol Online J. 2018;24:13030/qt6kt5n92w.
- Hong SH, Kang HY. A case of perforating osteoma cutis. Ann Dermatol. 2003;15:153-155.
- Kim BK, Ahn SK. Acquired perforating osteoma cutis. Ann Dermatol. 2015;27:452-453.
- Lippmann HI, Goldin RR. Subcutaneous ossification of the legs in chronic venous insufficiency. Radiology. 1960;74:279-288.
- Haro R, Revelles JM, Angulo J, et al. Plaque-like osteoma cutis with transepidermal elimination. J Cutan Pathol. 2009;36:591-593.
- Falsey RR, Ackerman L. Eruptive, hard cutaneous nodules in a 61-yearold woman. osteoma cutis in a patient with Albright hereditary osteodystrophy (AHO). JAMA Dermatol. 2013;149:975-976.
- Martin J, Tucker M, Browning JC. Infantile osteoma cutis as a presentation of a GNAS mutation. Pediatr Dermatol. 2012;29:483-484.
- Shore EM, Ahn J, de Beur SJ, et al. Paternally inherited inactivating mutations of the GNAS1 gene in progressive osseous heteroplasia. N Engl J Med. 2002;346:99-106.
- Kaplan FS, Le Merrer M, Glaser DL, et al. Fibrodysplasia ossificans progressiva. Best Pract Res Clin Rheumatol. 2008;22:191-205.
- Song GA, Kim HJ, Woo KM, et al. Molecular consequences of the ACVR1(R206H) mutation of fibrodysplasia ossificans progressiva. J Biol Chem. 2010;285:22542-22553.
- Roth SI, Stowell RE, Helwig EB, et al. Cutaneous ossification. report of 120 cases and review of the literature. Arch Pathol. 1963;76:44-54.
- Shimono K, Uchibe K, Kuboki T, et al. The pathophysiology of heterotopic ossification: current treatment considerations in dentistry. Japanese Dental Science Review. 2014;50:1-8.
- Kim D, Franco GA, Shigehara H, et al. Benign miliary osteoma cutis of the face: a common incidental CT finding. AJNR Am J Neuroradiol. 2017;38:789-794.
- Basu P, Erickson CP, Calame A, et al. Osteoma cutis: an adverse event following tattoo placement. Cureus. 2019;11:E4323.
- Cohen PR. Perforating osteoma cutis: case report and literature review of patients with a solitary perforating osteoma cutis lesion. Dermatol Online J. 2018;24:13030/qt6kt5n92w.
- Hong SH, Kang HY. A case of perforating osteoma cutis. Ann Dermatol. 2003;15:153-155.
- Kim BK, Ahn SK. Acquired perforating osteoma cutis. Ann Dermatol. 2015;27:452-453.
- Lippmann HI, Goldin RR. Subcutaneous ossification of the legs in chronic venous insufficiency. Radiology. 1960;74:279-288.
- Haro R, Revelles JM, Angulo J, et al. Plaque-like osteoma cutis with transepidermal elimination. J Cutan Pathol. 2009;36:591-593.
![]()
A 62-year-old woman presented with an irregular, erythematous, 4-mm nodule with central erosions on the left proximal calf of 2 months’ duration. The patient had a history of actinic keratoses and dysplastic nevi. She had no other notable medical history. She was not taking any medications and reported no history of trauma to the area. A shave biopsy of the lesion (encircled by black ink) was performed.
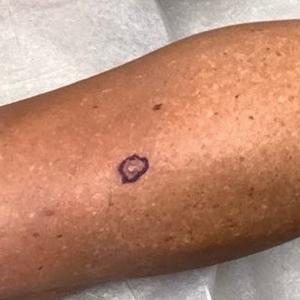
Firm Digital Papulonodules in an Infant
The Diagnosis: Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF) is a rare benign neoplasm of infancy prone to recurrence after resection but not to metastasis. It usually is limited to the fingers and toes.1 One-third of cases occur at birth. Most patients develop clinical symptoms within the first year of life, but presentation can occur in adolescents and adults. The exact etiology and pathogenesis of IDF remain unclear, but trauma is thought to be a trigger.
Physical examination reveals single or multiple smooth, round, pink papules or nodules confined to the sides and backs of the fingers, sparing the thumb and first toe.2,3 The nodules typically are firm, less than 2 cm in diameter, and often painless. Infantile digital fibromatosis exhibits an indolent progression followed by a rapid growth phase during several months, which may lead to functional impairment and joint deformities.4,5 Histopathology displays spindle cells with eosinophilic cytoplasmic inclusions that range from round to oval with uneven distribution, lack of refraction, and a large size difference (3–15 μm).6 The inclusions are deep red with Masson trichrome staining and can express smooth muscle actin and calponin. Tumor cells usually express vimentin, smooth muscle actin, calponin, and desmin but fail to express S-100 protein. The Ki67 proliferation index is 2% to 15%.6,7
Nonsurgical treatments for IDF include topical imiquimod, topical or intradermal injection of glucocorticoids, and intradermal injection of 5-fluorouracil. Complete resection should be reserved for cases with invasive growth that may lead to joint deformities, tendon or ligament involvement, digit or contracture deformity, and complications such as decreased joint mobility. Although there is a recurrence rate of up to 50% after excision, most lesions eventually will spontaneously regress and will leave no scar.8-10
The clinical and histopathologic differential diagnoses of IDF include other cutaneous diseases that occur in the digits. A dermatofibroma is a round, firm, fibrohistiocytic nodule that mainly occurs on the extensor limbs. Histopathology includes both fibrous and cellular types.11 Histologic analysis shows an ill-defined dermal proliferation of spindled fibroblasts with pale eosinophilic cytoplasm and bland fusiform nuclei growing in bands or fascicles that trap collagen fibers at the periphery (Figure 1). Generally, dermatofibromas have marked epidermal hyperplasia, which differs from IDF.
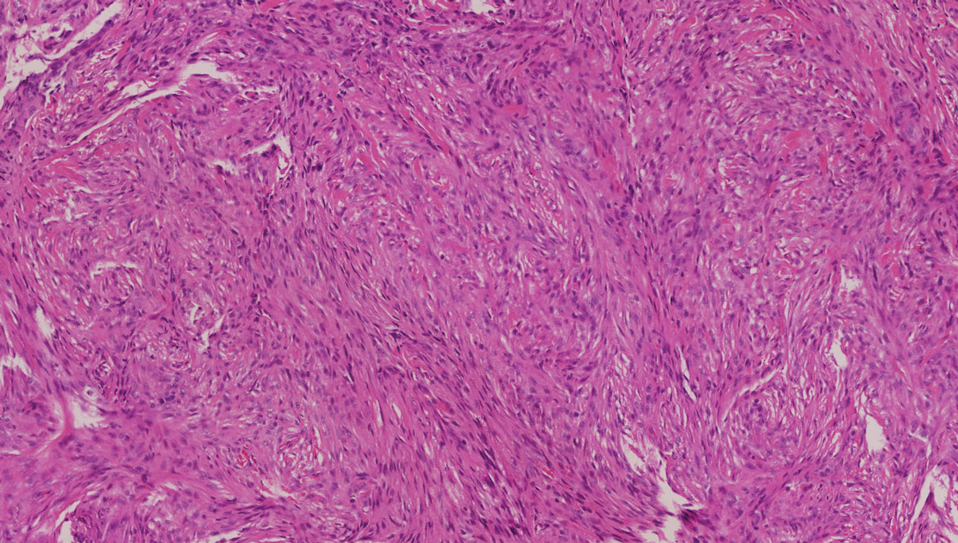
A digital myxoid cyst is characterized by a fleshcolored, hemispherical, and translucent cystic nodule that arises from the dorsum of the distal interphalangeal joint.12 It commonly is associated with injury and chronic pressure. Translucent viscous liquid may flow out when the cyst is punctured, a hallmark feature of this entity. Clinical variants of myxoid cyst include myxomatous and ganglion types. Histopathology reveals excessive mucin deposited in the dermis, and the surrounding collagen is compressed to form the pseudocyst (Figure 2).
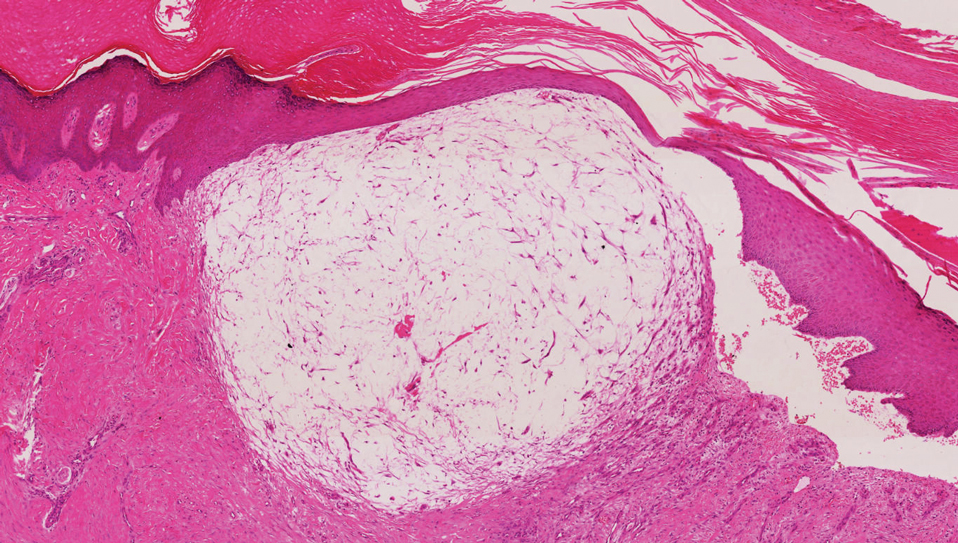
A giant cell tumor of the tendon sheath presents with asymptomatic nodules or lumps. Lesions frequently are localized to the tendon sheath, especially on the fingers and wrists, with no malignant tendency or propensity for spontaneous regression.13 The local recurrence rate is as high as 45%, which is related to surgical resection insufficiency.14 Histopathologic examination shows lobulated tumor tissue surrounded by dense fibrosis. The tumor cells are histiocytic with scattered giant cells (Figure 3). The characteristic osteoclastlike giant cells have eosinophilic cytoplasm and irregularly arranged nuclei in varying numbers.
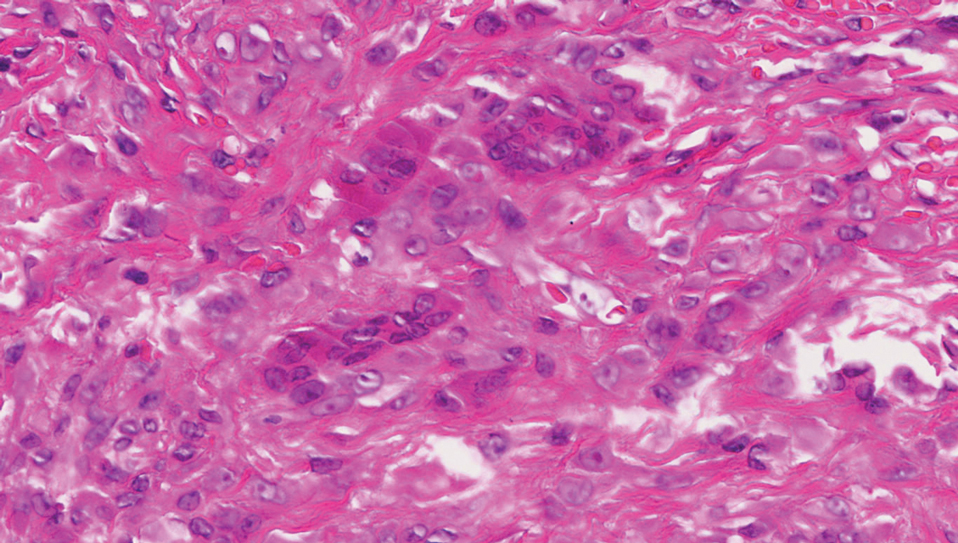
Keloids are connective tissue hyperplasias caused by skin injury. Histopathologically, keloids are characterized by nodules of thick hyalinized collagen bundles and whorled fibroblasts (Figure 4). No inclusions in the fibroblasts and a history of trauma can differentiate keloids from IDF.

- Marks E, Ewart M. Infantile digital fibroma: a rare fibromatosis. Arch Pathol Lab Med. 2016;140:1153‐1156.
- Botelho LF, Matsushigue T, Enokihara MM, et al. Case for diagnosis. An Bras Dermatol. 2012;87:493-494.
- Paloni G, Mattei I, Salmaso R, et al. Infantile digital fibromatosis. Arch Dis Child. 2013;98:308.
- Girgenti V, Restano L, Arcangeli F, et al. Infantile digital fibromatosis: a rare tumour of infancy. report of five cases. Australas J Dermatol. 2012;53:285-287.
- Eypper EH, Lee JC, Tarasen AJ, et al. An algorithmic approach to the management of infantile digital fibromatosis: review of literature and a case report. Eplasty. 2018;18:E19.
- Laskin WB, Miettinen M, Fetsch JF. Infantile digital fibroma /fibromatosis: a clinicopathologic and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up. Am J Surg Pathol. 2009;33:1-13.
- Henderson H, Peng YJ, Salter DM. Anti-calponin 1 antibodies highlight intracytoplasmic inclusions of infantile digital fibromatosis. Histopathology. 2014,64:752-755.
- Campbell LB, Petrick MG. Mohs micrographic surgery for a problematic infantile digital fibroma. Dermatol Surg. 2007;33:385-387.
- Ochi H, Puhaindran ME, Tan KW. Firm digital papulonodules in a young boy. Int J Dermatol. 2019;58:91-92.
- Albertini JG, Welsch MJ, Conger LA, et al. Infantile digital fibroma treated with Mohs micrography surgery. Dermatol Surg. 2002;28:959-961.
- Alves JV, Matos DM, Barreiros HF, et al. Variants of dermatofibroma— a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Meyers AL, Fallahi AKM. Digital Mucous Cyst. StatPearls Publishing; 2020.
- Zhao Q, Lu H. Giant cell tumor of tendon sheath in the wrist that damaged the extensor indicis proprius tendon: a case report and literature review. BMC Cancer. 2019;19:1057.
- DiGrazia S, Succi G, Fragetta F, et al. Giant cell tumor of tendon sheath: study of 64 cases and review of literature. G Chir. 2013;34:149-152.
The Diagnosis: Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF) is a rare benign neoplasm of infancy prone to recurrence after resection but not to metastasis. It usually is limited to the fingers and toes.1 One-third of cases occur at birth. Most patients develop clinical symptoms within the first year of life, but presentation can occur in adolescents and adults. The exact etiology and pathogenesis of IDF remain unclear, but trauma is thought to be a trigger.
Physical examination reveals single or multiple smooth, round, pink papules or nodules confined to the sides and backs of the fingers, sparing the thumb and first toe.2,3 The nodules typically are firm, less than 2 cm in diameter, and often painless. Infantile digital fibromatosis exhibits an indolent progression followed by a rapid growth phase during several months, which may lead to functional impairment and joint deformities.4,5 Histopathology displays spindle cells with eosinophilic cytoplasmic inclusions that range from round to oval with uneven distribution, lack of refraction, and a large size difference (3–15 μm).6 The inclusions are deep red with Masson trichrome staining and can express smooth muscle actin and calponin. Tumor cells usually express vimentin, smooth muscle actin, calponin, and desmin but fail to express S-100 protein. The Ki67 proliferation index is 2% to 15%.6,7
Nonsurgical treatments for IDF include topical imiquimod, topical or intradermal injection of glucocorticoids, and intradermal injection of 5-fluorouracil. Complete resection should be reserved for cases with invasive growth that may lead to joint deformities, tendon or ligament involvement, digit or contracture deformity, and complications such as decreased joint mobility. Although there is a recurrence rate of up to 50% after excision, most lesions eventually will spontaneously regress and will leave no scar.8-10
The clinical and histopathologic differential diagnoses of IDF include other cutaneous diseases that occur in the digits. A dermatofibroma is a round, firm, fibrohistiocytic nodule that mainly occurs on the extensor limbs. Histopathology includes both fibrous and cellular types.11 Histologic analysis shows an ill-defined dermal proliferation of spindled fibroblasts with pale eosinophilic cytoplasm and bland fusiform nuclei growing in bands or fascicles that trap collagen fibers at the periphery (Figure 1). Generally, dermatofibromas have marked epidermal hyperplasia, which differs from IDF.
A digital myxoid cyst is characterized by a fleshcolored, hemispherical, and translucent cystic nodule that arises from the dorsum of the distal interphalangeal joint.12 It commonly is associated with injury and chronic pressure. Translucent viscous liquid may flow out when the cyst is punctured, a hallmark feature of this entity. Clinical variants of myxoid cyst include myxomatous and ganglion types. Histopathology reveals excessive mucin deposited in the dermis, and the surrounding collagen is compressed to form the pseudocyst (Figure 2).
A giant cell tumor of the tendon sheath presents with asymptomatic nodules or lumps. Lesions frequently are localized to the tendon sheath, especially on the fingers and wrists, with no malignant tendency or propensity for spontaneous regression.13 The local recurrence rate is as high as 45%, which is related to surgical resection insufficiency.14 Histopathologic examination shows lobulated tumor tissue surrounded by dense fibrosis. The tumor cells are histiocytic with scattered giant cells (Figure 3). The characteristic osteoclastlike giant cells have eosinophilic cytoplasm and irregularly arranged nuclei in varying numbers.
Keloids are connective tissue hyperplasias caused by skin injury. Histopathologically, keloids are characterized by nodules of thick hyalinized collagen bundles and whorled fibroblasts (Figure 4). No inclusions in the fibroblasts and a history of trauma can differentiate keloids from IDF.
The Diagnosis: Infantile Digital Fibromatosis
Infantile digital fibromatosis (IDF) is a rare benign neoplasm of infancy prone to recurrence after resection but not to metastasis. It usually is limited to the fingers and toes.1 One-third of cases occur at birth. Most patients develop clinical symptoms within the first year of life, but presentation can occur in adolescents and adults. The exact etiology and pathogenesis of IDF remain unclear, but trauma is thought to be a trigger.
Physical examination reveals single or multiple smooth, round, pink papules or nodules confined to the sides and backs of the fingers, sparing the thumb and first toe.2,3 The nodules typically are firm, less than 2 cm in diameter, and often painless. Infantile digital fibromatosis exhibits an indolent progression followed by a rapid growth phase during several months, which may lead to functional impairment and joint deformities.4,5 Histopathology displays spindle cells with eosinophilic cytoplasmic inclusions that range from round to oval with uneven distribution, lack of refraction, and a large size difference (3–15 μm).6 The inclusions are deep red with Masson trichrome staining and can express smooth muscle actin and calponin. Tumor cells usually express vimentin, smooth muscle actin, calponin, and desmin but fail to express S-100 protein. The Ki67 proliferation index is 2% to 15%.6,7
Nonsurgical treatments for IDF include topical imiquimod, topical or intradermal injection of glucocorticoids, and intradermal injection of 5-fluorouracil. Complete resection should be reserved for cases with invasive growth that may lead to joint deformities, tendon or ligament involvement, digit or contracture deformity, and complications such as decreased joint mobility. Although there is a recurrence rate of up to 50% after excision, most lesions eventually will spontaneously regress and will leave no scar.8-10
The clinical and histopathologic differential diagnoses of IDF include other cutaneous diseases that occur in the digits. A dermatofibroma is a round, firm, fibrohistiocytic nodule that mainly occurs on the extensor limbs. Histopathology includes both fibrous and cellular types.11 Histologic analysis shows an ill-defined dermal proliferation of spindled fibroblasts with pale eosinophilic cytoplasm and bland fusiform nuclei growing in bands or fascicles that trap collagen fibers at the periphery (Figure 1). Generally, dermatofibromas have marked epidermal hyperplasia, which differs from IDF.
A digital myxoid cyst is characterized by a fleshcolored, hemispherical, and translucent cystic nodule that arises from the dorsum of the distal interphalangeal joint.12 It commonly is associated with injury and chronic pressure. Translucent viscous liquid may flow out when the cyst is punctured, a hallmark feature of this entity. Clinical variants of myxoid cyst include myxomatous and ganglion types. Histopathology reveals excessive mucin deposited in the dermis, and the surrounding collagen is compressed to form the pseudocyst (Figure 2).
A giant cell tumor of the tendon sheath presents with asymptomatic nodules or lumps. Lesions frequently are localized to the tendon sheath, especially on the fingers and wrists, with no malignant tendency or propensity for spontaneous regression.13 The local recurrence rate is as high as 45%, which is related to surgical resection insufficiency.14 Histopathologic examination shows lobulated tumor tissue surrounded by dense fibrosis. The tumor cells are histiocytic with scattered giant cells (Figure 3). The characteristic osteoclastlike giant cells have eosinophilic cytoplasm and irregularly arranged nuclei in varying numbers.
Keloids are connective tissue hyperplasias caused by skin injury. Histopathologically, keloids are characterized by nodules of thick hyalinized collagen bundles and whorled fibroblasts (Figure 4). No inclusions in the fibroblasts and a history of trauma can differentiate keloids from IDF.
- Marks E, Ewart M. Infantile digital fibroma: a rare fibromatosis. Arch Pathol Lab Med. 2016;140:1153‐1156.
- Botelho LF, Matsushigue T, Enokihara MM, et al. Case for diagnosis. An Bras Dermatol. 2012;87:493-494.
- Paloni G, Mattei I, Salmaso R, et al. Infantile digital fibromatosis. Arch Dis Child. 2013;98:308.
- Girgenti V, Restano L, Arcangeli F, et al. Infantile digital fibromatosis: a rare tumour of infancy. report of five cases. Australas J Dermatol. 2012;53:285-287.
- Eypper EH, Lee JC, Tarasen AJ, et al. An algorithmic approach to the management of infantile digital fibromatosis: review of literature and a case report. Eplasty. 2018;18:E19.
- Laskin WB, Miettinen M, Fetsch JF. Infantile digital fibroma /fibromatosis: a clinicopathologic and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up. Am J Surg Pathol. 2009;33:1-13.
- Henderson H, Peng YJ, Salter DM. Anti-calponin 1 antibodies highlight intracytoplasmic inclusions of infantile digital fibromatosis. Histopathology. 2014,64:752-755.
- Campbell LB, Petrick MG. Mohs micrographic surgery for a problematic infantile digital fibroma. Dermatol Surg. 2007;33:385-387.
- Ochi H, Puhaindran ME, Tan KW. Firm digital papulonodules in a young boy. Int J Dermatol. 2019;58:91-92.
- Albertini JG, Welsch MJ, Conger LA, et al. Infantile digital fibroma treated with Mohs micrography surgery. Dermatol Surg. 2002;28:959-961.
- Alves JV, Matos DM, Barreiros HF, et al. Variants of dermatofibroma— a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Meyers AL, Fallahi AKM. Digital Mucous Cyst. StatPearls Publishing; 2020.
- Zhao Q, Lu H. Giant cell tumor of tendon sheath in the wrist that damaged the extensor indicis proprius tendon: a case report and literature review. BMC Cancer. 2019;19:1057.
- DiGrazia S, Succi G, Fragetta F, et al. Giant cell tumor of tendon sheath: study of 64 cases and review of literature. G Chir. 2013;34:149-152.
- Marks E, Ewart M. Infantile digital fibroma: a rare fibromatosis. Arch Pathol Lab Med. 2016;140:1153‐1156.
- Botelho LF, Matsushigue T, Enokihara MM, et al. Case for diagnosis. An Bras Dermatol. 2012;87:493-494.
- Paloni G, Mattei I, Salmaso R, et al. Infantile digital fibromatosis. Arch Dis Child. 2013;98:308.
- Girgenti V, Restano L, Arcangeli F, et al. Infantile digital fibromatosis: a rare tumour of infancy. report of five cases. Australas J Dermatol. 2012;53:285-287.
- Eypper EH, Lee JC, Tarasen AJ, et al. An algorithmic approach to the management of infantile digital fibromatosis: review of literature and a case report. Eplasty. 2018;18:E19.
- Laskin WB, Miettinen M, Fetsch JF. Infantile digital fibroma /fibromatosis: a clinicopathologic and immunohistochemical study of 69 tumors from 57 patients with long-term follow-up. Am J Surg Pathol. 2009;33:1-13.
- Henderson H, Peng YJ, Salter DM. Anti-calponin 1 antibodies highlight intracytoplasmic inclusions of infantile digital fibromatosis. Histopathology. 2014,64:752-755.
- Campbell LB, Petrick MG. Mohs micrographic surgery for a problematic infantile digital fibroma. Dermatol Surg. 2007;33:385-387.
- Ochi H, Puhaindran ME, Tan KW. Firm digital papulonodules in a young boy. Int J Dermatol. 2019;58:91-92.
- Albertini JG, Welsch MJ, Conger LA, et al. Infantile digital fibroma treated with Mohs micrography surgery. Dermatol Surg. 2002;28:959-961.
- Alves JV, Matos DM, Barreiros HF, et al. Variants of dermatofibroma— a histopathological study. An Bras Dermatol. 2014;89:472-477.
- Meyers AL, Fallahi AKM. Digital Mucous Cyst. StatPearls Publishing; 2020.
- Zhao Q, Lu H. Giant cell tumor of tendon sheath in the wrist that damaged the extensor indicis proprius tendon: a case report and literature review. BMC Cancer. 2019;19:1057.
- DiGrazia S, Succi G, Fragetta F, et al. Giant cell tumor of tendon sheath: study of 64 cases and review of literature. G Chir. 2013;34:149-152.
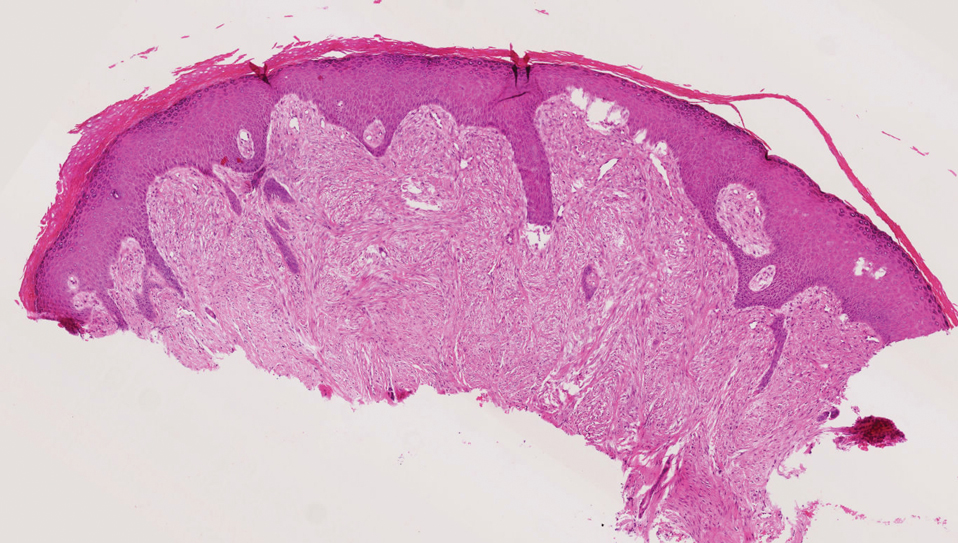
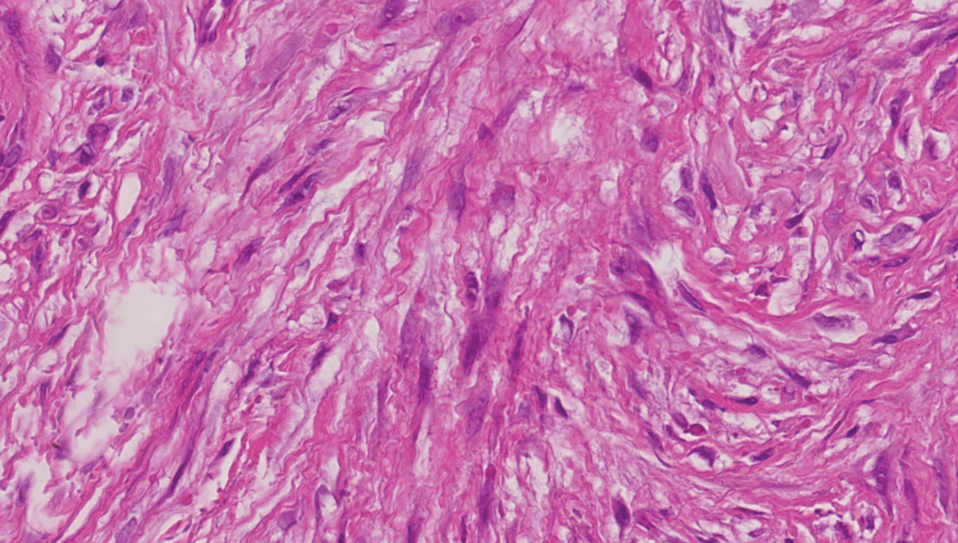
A 3-month-old girl presented with papulonodules on the distal left ring finger. Initially the lesions were thought to be insect bites but became firm over the course of 3 weeks and then gradually increased in size over 2 months. Physical examination revealed a 0.5×0.5-cm firm nodule and a 0.2×0.3-cm firm papule on the radial aspect of the left ring finger over the distal interphalangeal joint. There was no deformity or dysfunction of the finger. Radiography showed soft tissue swelling without bony abnormalities. The lesions were excised; however, a new fleshy nodule reappeared 1 month postoperatively on the radial aspect of the left ring finger over the distal interphalangeal joint. The patient did not seem bothered by the lesions and was in good general health.
Advocacy Update: Is Your Practice Equipped to Handle Looming Changes in Dermatopathology?
The proposed 2022 Medicare physician fee schedule and quality payment program (QPP) regulations were released on July 13, 2021.1 Final regulations are expected to be released on or around November 1, 2021, but they may be delayed. Multiple national medical organizations, including the College of American Pathologists (CAP), the American Society of Dermatopathology, the American Academy of Dermatology Association (AADA), and the American Medical Association (AMA) Physicians’ Grassroots Network all work together to engage with the Centers for Medicare & Medicaid Services (CMS) to influence these regulations. Stated advocacy priorities include protecting the value of dermatopathology services, mobilizing dermatopathologists for political action, ensuring dermatopathologists can participate in new payment models, strengthening the profession with advocacy on a state level, and conducting socioeconomic research. Is your practice aware and prepared to handle the changes coming in 2022?
The recent revisions and revaluations of the outpatient evaluation and management (E/M) codes2 resulted in a considerable redistribution of Medicare dollars in 2021, negatively impacting dermatopathologists and other specialties and services due to budget neutrality required by law (Figure). Important steps were taken to mitigate the 2021 Medicare cuts for all non–office-based dermatopathology services (eg, pathology, surgical services, emergency department).1,3 Direct engagement by the CAP, American Society of Dermatopathology, and AADA, along with the AMA Physicians’ Grassroots Network resulted in legislative action on December 27, 2020, which directed Medicare to make a 3.75% positive adjustment to the 2021 physician payments. Additionally, the CMS updated the 2021 physician conversion factor to $34.8931, a 3.3% reduction from the 2020 conversion factor rather than $32.41, or a 10.20% decrease. The 2% payment adjustment (sequestration) through December 21, 2021, also was suspended, and Congress and the Biden administration mandated delayed implementation of the inherent complexity add-on code for E/M services (G2211) until 2024.1,3
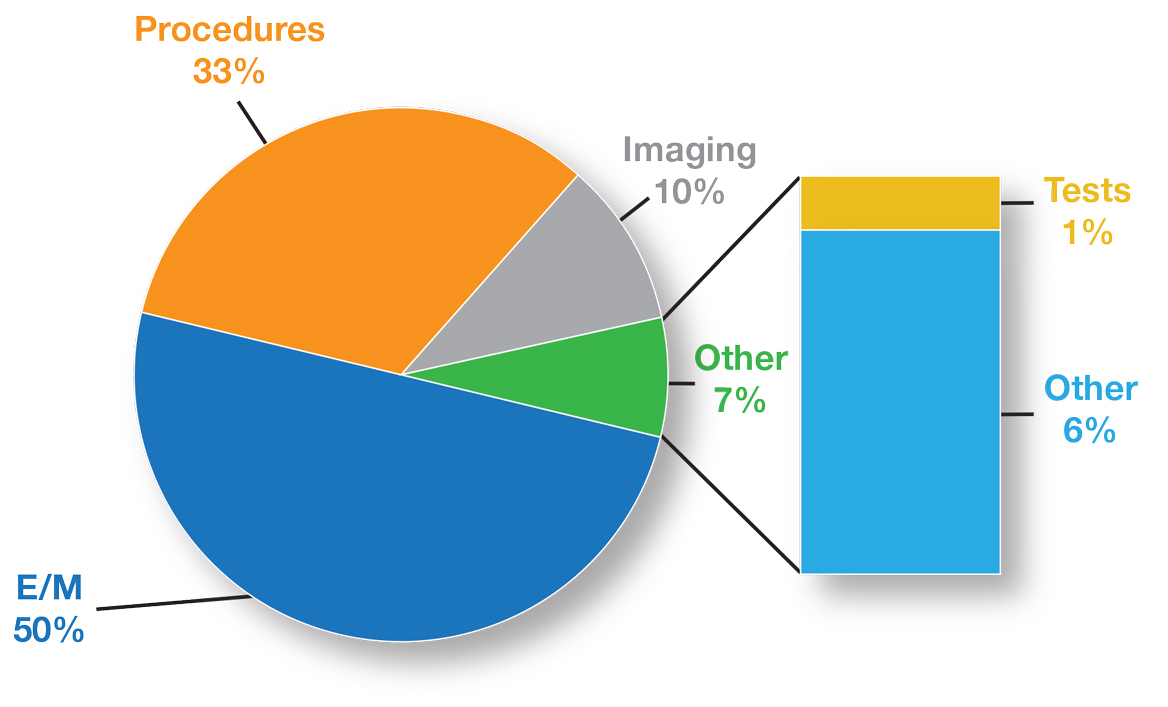
Threat of Medicare Cuts in 2022
Based on dermatopathology utilization data, the overall impact on reimbursement for 2022 represents an approximately 5% decrease from 2021 dermatopathology payments (Table 1).1,4 This represents a 3.75% cut from revaluation of E/M services, and a 1% cut due to changes in practice expense pricing. The estimated change in reimbursement for independent laboratories is a 6% decrease. Advocacy groups have been working to mitigate the 2022 cuts by engaging with Congress and urging them to act before these changes go into effect next year. Keep in mind that approximately half of all pathology Current Procedural Terminology (CPT) codes have been targeted for evaluation by the CMS since 2006.1,4
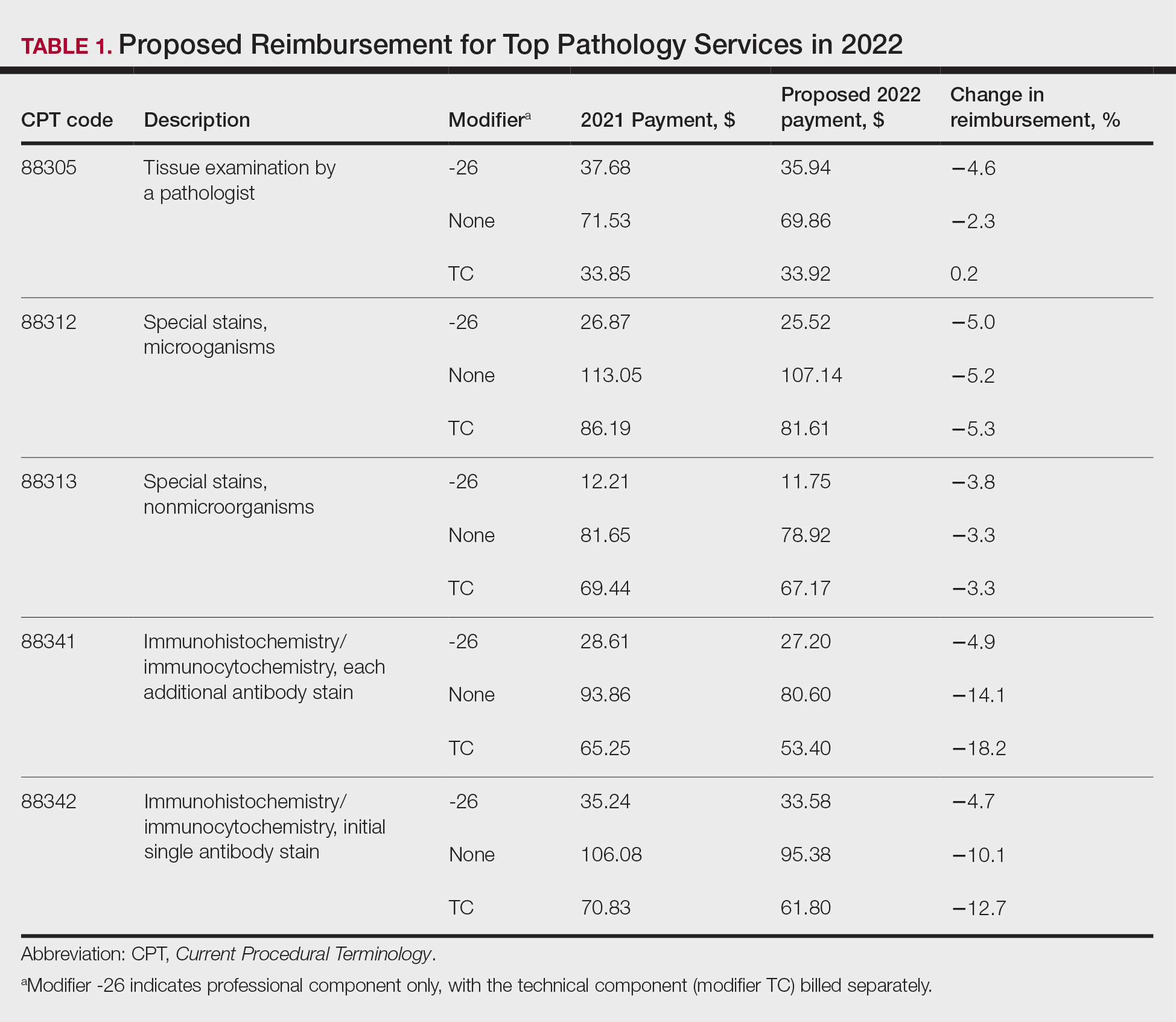
The current clinical pathology consultation services (CPT codes 80500 and 80502) previously were identified as potentially misvalued for review by the AMA Relative Value Scale Update Committee’s (RUC’s) relativity assessment workgroup.4 Consequently, the CAP worked with the AMA’s CPT Editorial Panel to delete codes 80500 and 80502, as well as to modernize and create the 4 new clinical pathology consultation codes: 80XX0, 80XX1, 80XX2, and 80XX3. Then the CAP worked with the RUC to develop physician work and practice expense values for the new clinical pathology consultation codes. Once the fee schedule is finalized, pathologists can begin using the new codes to bill these services in 2022 (Table 2).4

According to CPT, clinical pathology consultation services may be reported when the following criteria have been met: (1) the pathologist renders a clinical pathology consultation at the request of a physician or qualified health care professional at the same or another institution; (2) the pathology clinical consultation request relating to pathology and laboratory findings or other relevant clinical or diagnostic information requiring additional medical interpretative judgment is made; and (3) these codes are not reported in conjunction with codes 88321, 88323, and 88325.4
Proposed 2022 Medicare QPP Requirements
On July 13, 2021, the CMS also published its proposed 2022 QPP proposals that will take effect next year.4 According to the proposed regulation, nearly all dermatopathologists will be required to participate in Medicare’s QPP, either through advanced alternative payment models (APMs) or the Merit-based Incentive Payment System (MIPS). The CAP has long advocated for reducing MIPS reporting burdens for dermatopathologists. In this regulation, the CMS is proposing key program changes that move the program forward but also introduce additional complexities; for example, the CMS will move forward with a new participation pathway called MIPS Value Pathways (MVPs). The CMS proposed 7 specific MVPs that align with certain clinical topics; however, it will not implement these MVPs until the 2023 MIPS performance period.
In 2022, dermatopathologists who are eligible for MIPS will have to take action to avoid penalties that reduce future Medicare Part B payments for their services. Performance in MIPS in 2022 affects Medicare Part B payments in 2024 by an increase of 9% to a decrease of 9%.
In its proposed 2022 QPP regulations, the CMS proposed an increase of the performance threshold from 60 MIPS points to 75 MIPS points. It also proposed an increase of the exceptional Performance Threshold from 85 MIPS points to 89 MIPS points.
The CMS also proposed notable scoring changes for quality measures, including removing the 3-point floor for measures that can be scored against a benchmark. These measures would receive 1 to 10 points. Measures without a benchmark or that do not meet case requirements would earn 0 points, with an exception for small practices. The CMS also proposed removing bonus points for reporting additional outcomes and high-priority measures beyond the 1 that is required, as well as establishing a 5-point floor for the first 2 performance periods for new measures, which is in line with the CAP’s advocacy.
The Pathology Specialty Measure Set will remain the same as the 2021 set containing 6 quality measures, including the AADA-stewarded quality measure #440 (skin cancer: biopsy reporting time—pathologist to clinician). Although the CAP recognizes the importance of prompt turnaround of biopsy reports, it also is working with the CMS and the AADA to mitigate the operational challenges dermatopathologists encounter when using this measure.
Due to advocacy from the CAP, the CMS included a CAP-proposed improvement activity on implementation of a laboratory preparedness plan to support continued or expanded patient care during the COVID-19 pandemic or another public health emergency. This plan should address how the laboratory would maintain or expand access to improve beneficiary health outcomes and reduce health care disparities.
The CAP has actively worked with the CMS to demonstrate the need for more appropriate and alternative measures and improvement activities so that pathologists can more fully participate in MIPS.
Alternative Payment Models—For those dermatopathologists who practice in an APM, the proposed 2022 QPP makes minimal changes to the advanced APM track while adding transition time for accountable care organizations in the Medicare Shared Savings Program to report on certain quality measures and increasing flexibility related to the program’s quality performance standard.
Cures Act 2021: To Do No Harm
The 21st Century Cures Act (Cures Act) was signed into federal law in 2016. The Office of the National Coordinator for Health Information Technology (ONC) laid the groundwork for patients to have easier access to and control of their health information.5 The ONC’s final rule, which went into effect on April 5, 2021, requires that all providers make their office notes, laboratory results, and other diagnostic reports (including dermatopathology reports) available to patients as soon as the physician’s office receives an electronic copy. Penalty for noncompliance has not been determined.
There are information-blocking exceptions, but delaying access to a patient’s report so that a provider can review the result before the patient receives it is not considered an exception.6 The exceptions are situational and must be evaluated by the referring clinician or their employer. Documentation of the exception is critical. The specific facts and circumstances associated with your decision to use an exception will be important to include in your documentation. Information blocking necessary to prevent “harm” to a patient or another person requires a reasonable belief that the practice will substantially reduce the risk of harm.6
The AMA passed a resolution in June 2021 calling for changes to this rule to allow for a delay of pathology results, advocating to the Office for Civil Rights to revise the harm exception to include psychological distress.6 In August 2021, the AADA met with senior officials at the ONC also asking to revise its definition of harm, sharing examples of emotional strain that resulted from receiving results without clinical context.7 California enacted a law requiring a delay before a patient receives the result of a malignant diagnosis, giving the clinician time to contact the patient before they see their report.8
The Cures Act requirements are about patients accessing their health care information. Always consider what is best for the patient and ensure that your policies and procedures reflect this.5
Final Thoughts
It is important to learn and support advocacy priorities and efforts and to join forces to protect your practice. Physician advocacy is no longer an elective pursuit. We need to be involved and engaged through our medical societies to help patients, communities, and ourselves.
- Centers for Medicare & Medicaid Services. Calendar Year (CY) 2022 Medicare Physician Fee Schedule Proposed Rule. Published July 13, 2021. Accessed October 22, 2021. https://www.cms.gov/newsroom/fact-sheets/calendar-year-cy-2022-medicare-physician-fee-schedule-proposed-rule
- Healthcare spending and the Medicare program. Medicare Payment Advisory Commission; July 2020. Accessed October 25, 2021.http://www.medpac.gov/docs/default-source/data-book/july2020_databook_entirereport_sec.pdf
- Frieden J. 2021 Medicare fee schedule includes 10.2% cut in conversion factor. MedPage Today website. Published December 2, 2020. Accessed October 22, 2021. https://www.medpagetoday.com/practicemanagement/reimbursement/89970
- Advocacy. College of American Pathologists website. Accessed October 13, 2021. https://www.cap.org/advocacy
- ONC’s Cures Act Final Rule. The Office of the National Coordinator for Health Information Technology website. Accessed October 13, 2021. https://www.healthit.gov/curesrule/
- Nelson H. Delegates call AMA to advocate for provider info-blocking flexibility. Published June 18, 2021. Accessed October 13, 2021. https://ehrintelligence.com/news/delegates-call-ama-to-advocate-for-provider-info-blocking-flexibility
- Rosamilia LL. Immediate Pathology report release to patients—is the 21st Century Cures Act worse than the disease? American Academy of Dermatology website. Published August 25, 2021. Accessed October 22, 2021. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2021/cures-act-immediate-pathology-report-release-to-patients
- Purington K, Alfreds ST, Pritts J, et al; The National Academy for State Health Policy. Electronic release of clinical laboratory results: a review of state and federal policy. Published January 2010. Accessed October 13, 2021. https://www.nashp.org/wp-content/uploads/2010/02/ElectronicLabResultsExchangePolicy.pdf
The proposed 2022 Medicare physician fee schedule and quality payment program (QPP) regulations were released on July 13, 2021.1 Final regulations are expected to be released on or around November 1, 2021, but they may be delayed. Multiple national medical organizations, including the College of American Pathologists (CAP), the American Society of Dermatopathology, the American Academy of Dermatology Association (AADA), and the American Medical Association (AMA) Physicians’ Grassroots Network all work together to engage with the Centers for Medicare & Medicaid Services (CMS) to influence these regulations. Stated advocacy priorities include protecting the value of dermatopathology services, mobilizing dermatopathologists for political action, ensuring dermatopathologists can participate in new payment models, strengthening the profession with advocacy on a state level, and conducting socioeconomic research. Is your practice aware and prepared to handle the changes coming in 2022?
The recent revisions and revaluations of the outpatient evaluation and management (E/M) codes2 resulted in a considerable redistribution of Medicare dollars in 2021, negatively impacting dermatopathologists and other specialties and services due to budget neutrality required by law (Figure). Important steps were taken to mitigate the 2021 Medicare cuts for all non–office-based dermatopathology services (eg, pathology, surgical services, emergency department).1,3 Direct engagement by the CAP, American Society of Dermatopathology, and AADA, along with the AMA Physicians’ Grassroots Network resulted in legislative action on December 27, 2020, which directed Medicare to make a 3.75% positive adjustment to the 2021 physician payments. Additionally, the CMS updated the 2021 physician conversion factor to $34.8931, a 3.3% reduction from the 2020 conversion factor rather than $32.41, or a 10.20% decrease. The 2% payment adjustment (sequestration) through December 21, 2021, also was suspended, and Congress and the Biden administration mandated delayed implementation of the inherent complexity add-on code for E/M services (G2211) until 2024.1,3
Threat of Medicare Cuts in 2022
Based on dermatopathology utilization data, the overall impact on reimbursement for 2022 represents an approximately 5% decrease from 2021 dermatopathology payments (Table 1).1,4 This represents a 3.75% cut from revaluation of E/M services, and a 1% cut due to changes in practice expense pricing. The estimated change in reimbursement for independent laboratories is a 6% decrease. Advocacy groups have been working to mitigate the 2022 cuts by engaging with Congress and urging them to act before these changes go into effect next year. Keep in mind that approximately half of all pathology Current Procedural Terminology (CPT) codes have been targeted for evaluation by the CMS since 2006.1,4
The current clinical pathology consultation services (CPT codes 80500 and 80502) previously were identified as potentially misvalued for review by the AMA Relative Value Scale Update Committee’s (RUC’s) relativity assessment workgroup.4 Consequently, the CAP worked with the AMA’s CPT Editorial Panel to delete codes 80500 and 80502, as well as to modernize and create the 4 new clinical pathology consultation codes: 80XX0, 80XX1, 80XX2, and 80XX3. Then the CAP worked with the RUC to develop physician work and practice expense values for the new clinical pathology consultation codes. Once the fee schedule is finalized, pathologists can begin using the new codes to bill these services in 2022 (Table 2).4
According to CPT, clinical pathology consultation services may be reported when the following criteria have been met: (1) the pathologist renders a clinical pathology consultation at the request of a physician or qualified health care professional at the same or another institution; (2) the pathology clinical consultation request relating to pathology and laboratory findings or other relevant clinical or diagnostic information requiring additional medical interpretative judgment is made; and (3) these codes are not reported in conjunction with codes 88321, 88323, and 88325.4
Proposed 2022 Medicare QPP Requirements
On July 13, 2021, the CMS also published its proposed 2022 QPP proposals that will take effect next year.4 According to the proposed regulation, nearly all dermatopathologists will be required to participate in Medicare’s QPP, either through advanced alternative payment models (APMs) or the Merit-based Incentive Payment System (MIPS). The CAP has long advocated for reducing MIPS reporting burdens for dermatopathologists. In this regulation, the CMS is proposing key program changes that move the program forward but also introduce additional complexities; for example, the CMS will move forward with a new participation pathway called MIPS Value Pathways (MVPs). The CMS proposed 7 specific MVPs that align with certain clinical topics; however, it will not implement these MVPs until the 2023 MIPS performance period.
In 2022, dermatopathologists who are eligible for MIPS will have to take action to avoid penalties that reduce future Medicare Part B payments for their services. Performance in MIPS in 2022 affects Medicare Part B payments in 2024 by an increase of 9% to a decrease of 9%.
In its proposed 2022 QPP regulations, the CMS proposed an increase of the performance threshold from 60 MIPS points to 75 MIPS points. It also proposed an increase of the exceptional Performance Threshold from 85 MIPS points to 89 MIPS points.
The CMS also proposed notable scoring changes for quality measures, including removing the 3-point floor for measures that can be scored against a benchmark. These measures would receive 1 to 10 points. Measures without a benchmark or that do not meet case requirements would earn 0 points, with an exception for small practices. The CMS also proposed removing bonus points for reporting additional outcomes and high-priority measures beyond the 1 that is required, as well as establishing a 5-point floor for the first 2 performance periods for new measures, which is in line with the CAP’s advocacy.
The Pathology Specialty Measure Set will remain the same as the 2021 set containing 6 quality measures, including the AADA-stewarded quality measure #440 (skin cancer: biopsy reporting time—pathologist to clinician). Although the CAP recognizes the importance of prompt turnaround of biopsy reports, it also is working with the CMS and the AADA to mitigate the operational challenges dermatopathologists encounter when using this measure.
Due to advocacy from the CAP, the CMS included a CAP-proposed improvement activity on implementation of a laboratory preparedness plan to support continued or expanded patient care during the COVID-19 pandemic or another public health emergency. This plan should address how the laboratory would maintain or expand access to improve beneficiary health outcomes and reduce health care disparities.
The CAP has actively worked with the CMS to demonstrate the need for more appropriate and alternative measures and improvement activities so that pathologists can more fully participate in MIPS.
Alternative Payment Models—For those dermatopathologists who practice in an APM, the proposed 2022 QPP makes minimal changes to the advanced APM track while adding transition time for accountable care organizations in the Medicare Shared Savings Program to report on certain quality measures and increasing flexibility related to the program’s quality performance standard.
Cures Act 2021: To Do No Harm
The 21st Century Cures Act (Cures Act) was signed into federal law in 2016. The Office of the National Coordinator for Health Information Technology (ONC) laid the groundwork for patients to have easier access to and control of their health information.5 The ONC’s final rule, which went into effect on April 5, 2021, requires that all providers make their office notes, laboratory results, and other diagnostic reports (including dermatopathology reports) available to patients as soon as the physician’s office receives an electronic copy. Penalty for noncompliance has not been determined.
There are information-blocking exceptions, but delaying access to a patient’s report so that a provider can review the result before the patient receives it is not considered an exception.6 The exceptions are situational and must be evaluated by the referring clinician or their employer. Documentation of the exception is critical. The specific facts and circumstances associated with your decision to use an exception will be important to include in your documentation. Information blocking necessary to prevent “harm” to a patient or another person requires a reasonable belief that the practice will substantially reduce the risk of harm.6
The AMA passed a resolution in June 2021 calling for changes to this rule to allow for a delay of pathology results, advocating to the Office for Civil Rights to revise the harm exception to include psychological distress.6 In August 2021, the AADA met with senior officials at the ONC also asking to revise its definition of harm, sharing examples of emotional strain that resulted from receiving results without clinical context.7 California enacted a law requiring a delay before a patient receives the result of a malignant diagnosis, giving the clinician time to contact the patient before they see their report.8
The Cures Act requirements are about patients accessing their health care information. Always consider what is best for the patient and ensure that your policies and procedures reflect this.5
Final Thoughts
It is important to learn and support advocacy priorities and efforts and to join forces to protect your practice. Physician advocacy is no longer an elective pursuit. We need to be involved and engaged through our medical societies to help patients, communities, and ourselves.
The proposed 2022 Medicare physician fee schedule and quality payment program (QPP) regulations were released on July 13, 2021.1 Final regulations are expected to be released on or around November 1, 2021, but they may be delayed. Multiple national medical organizations, including the College of American Pathologists (CAP), the American Society of Dermatopathology, the American Academy of Dermatology Association (AADA), and the American Medical Association (AMA) Physicians’ Grassroots Network all work together to engage with the Centers for Medicare & Medicaid Services (CMS) to influence these regulations. Stated advocacy priorities include protecting the value of dermatopathology services, mobilizing dermatopathologists for political action, ensuring dermatopathologists can participate in new payment models, strengthening the profession with advocacy on a state level, and conducting socioeconomic research. Is your practice aware and prepared to handle the changes coming in 2022?
The recent revisions and revaluations of the outpatient evaluation and management (E/M) codes2 resulted in a considerable redistribution of Medicare dollars in 2021, negatively impacting dermatopathologists and other specialties and services due to budget neutrality required by law (Figure). Important steps were taken to mitigate the 2021 Medicare cuts for all non–office-based dermatopathology services (eg, pathology, surgical services, emergency department).1,3 Direct engagement by the CAP, American Society of Dermatopathology, and AADA, along with the AMA Physicians’ Grassroots Network resulted in legislative action on December 27, 2020, which directed Medicare to make a 3.75% positive adjustment to the 2021 physician payments. Additionally, the CMS updated the 2021 physician conversion factor to $34.8931, a 3.3% reduction from the 2020 conversion factor rather than $32.41, or a 10.20% decrease. The 2% payment adjustment (sequestration) through December 21, 2021, also was suspended, and Congress and the Biden administration mandated delayed implementation of the inherent complexity add-on code for E/M services (G2211) until 2024.1,3
Threat of Medicare Cuts in 2022
Based on dermatopathology utilization data, the overall impact on reimbursement for 2022 represents an approximately 5% decrease from 2021 dermatopathology payments (Table 1).1,4 This represents a 3.75% cut from revaluation of E/M services, and a 1% cut due to changes in practice expense pricing. The estimated change in reimbursement for independent laboratories is a 6% decrease. Advocacy groups have been working to mitigate the 2022 cuts by engaging with Congress and urging them to act before these changes go into effect next year. Keep in mind that approximately half of all pathology Current Procedural Terminology (CPT) codes have been targeted for evaluation by the CMS since 2006.1,4
The current clinical pathology consultation services (CPT codes 80500 and 80502) previously were identified as potentially misvalued for review by the AMA Relative Value Scale Update Committee’s (RUC’s) relativity assessment workgroup.4 Consequently, the CAP worked with the AMA’s CPT Editorial Panel to delete codes 80500 and 80502, as well as to modernize and create the 4 new clinical pathology consultation codes: 80XX0, 80XX1, 80XX2, and 80XX3. Then the CAP worked with the RUC to develop physician work and practice expense values for the new clinical pathology consultation codes. Once the fee schedule is finalized, pathologists can begin using the new codes to bill these services in 2022 (Table 2).4
According to CPT, clinical pathology consultation services may be reported when the following criteria have been met: (1) the pathologist renders a clinical pathology consultation at the request of a physician or qualified health care professional at the same or another institution; (2) the pathology clinical consultation request relating to pathology and laboratory findings or other relevant clinical or diagnostic information requiring additional medical interpretative judgment is made; and (3) these codes are not reported in conjunction with codes 88321, 88323, and 88325.4
Proposed 2022 Medicare QPP Requirements
On July 13, 2021, the CMS also published its proposed 2022 QPP proposals that will take effect next year.4 According to the proposed regulation, nearly all dermatopathologists will be required to participate in Medicare’s QPP, either through advanced alternative payment models (APMs) or the Merit-based Incentive Payment System (MIPS). The CAP has long advocated for reducing MIPS reporting burdens for dermatopathologists. In this regulation, the CMS is proposing key program changes that move the program forward but also introduce additional complexities; for example, the CMS will move forward with a new participation pathway called MIPS Value Pathways (MVPs). The CMS proposed 7 specific MVPs that align with certain clinical topics; however, it will not implement these MVPs until the 2023 MIPS performance period.
In 2022, dermatopathologists who are eligible for MIPS will have to take action to avoid penalties that reduce future Medicare Part B payments for their services. Performance in MIPS in 2022 affects Medicare Part B payments in 2024 by an increase of 9% to a decrease of 9%.
In its proposed 2022 QPP regulations, the CMS proposed an increase of the performance threshold from 60 MIPS points to 75 MIPS points. It also proposed an increase of the exceptional Performance Threshold from 85 MIPS points to 89 MIPS points.
The CMS also proposed notable scoring changes for quality measures, including removing the 3-point floor for measures that can be scored against a benchmark. These measures would receive 1 to 10 points. Measures without a benchmark or that do not meet case requirements would earn 0 points, with an exception for small practices. The CMS also proposed removing bonus points for reporting additional outcomes and high-priority measures beyond the 1 that is required, as well as establishing a 5-point floor for the first 2 performance periods for new measures, which is in line with the CAP’s advocacy.
The Pathology Specialty Measure Set will remain the same as the 2021 set containing 6 quality measures, including the AADA-stewarded quality measure #440 (skin cancer: biopsy reporting time—pathologist to clinician). Although the CAP recognizes the importance of prompt turnaround of biopsy reports, it also is working with the CMS and the AADA to mitigate the operational challenges dermatopathologists encounter when using this measure.
Due to advocacy from the CAP, the CMS included a CAP-proposed improvement activity on implementation of a laboratory preparedness plan to support continued or expanded patient care during the COVID-19 pandemic or another public health emergency. This plan should address how the laboratory would maintain or expand access to improve beneficiary health outcomes and reduce health care disparities.
The CAP has actively worked with the CMS to demonstrate the need for more appropriate and alternative measures and improvement activities so that pathologists can more fully participate in MIPS.
Alternative Payment Models—For those dermatopathologists who practice in an APM, the proposed 2022 QPP makes minimal changes to the advanced APM track while adding transition time for accountable care organizations in the Medicare Shared Savings Program to report on certain quality measures and increasing flexibility related to the program’s quality performance standard.
Cures Act 2021: To Do No Harm
The 21st Century Cures Act (Cures Act) was signed into federal law in 2016. The Office of the National Coordinator for Health Information Technology (ONC) laid the groundwork for patients to have easier access to and control of their health information.5 The ONC’s final rule, which went into effect on April 5, 2021, requires that all providers make their office notes, laboratory results, and other diagnostic reports (including dermatopathology reports) available to patients as soon as the physician’s office receives an electronic copy. Penalty for noncompliance has not been determined.
There are information-blocking exceptions, but delaying access to a patient’s report so that a provider can review the result before the patient receives it is not considered an exception.6 The exceptions are situational and must be evaluated by the referring clinician or their employer. Documentation of the exception is critical. The specific facts and circumstances associated with your decision to use an exception will be important to include in your documentation. Information blocking necessary to prevent “harm” to a patient or another person requires a reasonable belief that the practice will substantially reduce the risk of harm.6
The AMA passed a resolution in June 2021 calling for changes to this rule to allow for a delay of pathology results, advocating to the Office for Civil Rights to revise the harm exception to include psychological distress.6 In August 2021, the AADA met with senior officials at the ONC also asking to revise its definition of harm, sharing examples of emotional strain that resulted from receiving results without clinical context.7 California enacted a law requiring a delay before a patient receives the result of a malignant diagnosis, giving the clinician time to contact the patient before they see their report.8
The Cures Act requirements are about patients accessing their health care information. Always consider what is best for the patient and ensure that your policies and procedures reflect this.5
Final Thoughts
It is important to learn and support advocacy priorities and efforts and to join forces to protect your practice. Physician advocacy is no longer an elective pursuit. We need to be involved and engaged through our medical societies to help patients, communities, and ourselves.
- Centers for Medicare & Medicaid Services. Calendar Year (CY) 2022 Medicare Physician Fee Schedule Proposed Rule. Published July 13, 2021. Accessed October 22, 2021. https://www.cms.gov/newsroom/fact-sheets/calendar-year-cy-2022-medicare-physician-fee-schedule-proposed-rule
- Healthcare spending and the Medicare program. Medicare Payment Advisory Commission; July 2020. Accessed October 25, 2021.http://www.medpac.gov/docs/default-source/data-book/july2020_databook_entirereport_sec.pdf
- Frieden J. 2021 Medicare fee schedule includes 10.2% cut in conversion factor. MedPage Today website. Published December 2, 2020. Accessed October 22, 2021. https://www.medpagetoday.com/practicemanagement/reimbursement/89970
- Advocacy. College of American Pathologists website. Accessed October 13, 2021. https://www.cap.org/advocacy
- ONC’s Cures Act Final Rule. The Office of the National Coordinator for Health Information Technology website. Accessed October 13, 2021. https://www.healthit.gov/curesrule/
- Nelson H. Delegates call AMA to advocate for provider info-blocking flexibility. Published June 18, 2021. Accessed October 13, 2021. https://ehrintelligence.com/news/delegates-call-ama-to-advocate-for-provider-info-blocking-flexibility
- Rosamilia LL. Immediate Pathology report release to patients—is the 21st Century Cures Act worse than the disease? American Academy of Dermatology website. Published August 25, 2021. Accessed October 22, 2021. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2021/cures-act-immediate-pathology-report-release-to-patients
- Purington K, Alfreds ST, Pritts J, et al; The National Academy for State Health Policy. Electronic release of clinical laboratory results: a review of state and federal policy. Published January 2010. Accessed October 13, 2021. https://www.nashp.org/wp-content/uploads/2010/02/ElectronicLabResultsExchangePolicy.pdf
- Centers for Medicare & Medicaid Services. Calendar Year (CY) 2022 Medicare Physician Fee Schedule Proposed Rule. Published July 13, 2021. Accessed October 22, 2021. https://www.cms.gov/newsroom/fact-sheets/calendar-year-cy-2022-medicare-physician-fee-schedule-proposed-rule
- Healthcare spending and the Medicare program. Medicare Payment Advisory Commission; July 2020. Accessed October 25, 2021.http://www.medpac.gov/docs/default-source/data-book/july2020_databook_entirereport_sec.pdf
- Frieden J. 2021 Medicare fee schedule includes 10.2% cut in conversion factor. MedPage Today website. Published December 2, 2020. Accessed October 22, 2021. https://www.medpagetoday.com/practicemanagement/reimbursement/89970
- Advocacy. College of American Pathologists website. Accessed October 13, 2021. https://www.cap.org/advocacy
- ONC’s Cures Act Final Rule. The Office of the National Coordinator for Health Information Technology website. Accessed October 13, 2021. https://www.healthit.gov/curesrule/
- Nelson H. Delegates call AMA to advocate for provider info-blocking flexibility. Published June 18, 2021. Accessed October 13, 2021. https://ehrintelligence.com/news/delegates-call-ama-to-advocate-for-provider-info-blocking-flexibility
- Rosamilia LL. Immediate Pathology report release to patients—is the 21st Century Cures Act worse than the disease? American Academy of Dermatology website. Published August 25, 2021. Accessed October 22, 2021. https://www.aad.org/dw/dw-insights-and-inquiries/archive/2021/cures-act-immediate-pathology-report-release-to-patients
- Purington K, Alfreds ST, Pritts J, et al; The National Academy for State Health Policy. Electronic release of clinical laboratory results: a review of state and federal policy. Published January 2010. Accessed October 13, 2021. https://www.nashp.org/wp-content/uploads/2010/02/ElectronicLabResultsExchangePolicy.pdf
Practice Points
- A proposed 2022 fee schedule negatively impacting dermatopathology practices has been published by the Centers for Medicare & Medicaid Services (CMS) in July 2021.
- New pathology consultation codes with new payment rates proposed by CMS can be used starting January 1, 2022.
- The 21st Century Cures Act Final Rule has information blocking provisions.
Early Pilomatrix Carcinoma: A Case Report With Emphasis on Molecular Pathology and Review of the Literature
Pilomatrix carcinoma is a rare adnexal tumor with origin from the germinative matrical cells of the hair follicle. Clinically, it presents as a solitary lesion commonly found in the head and neck region as well as the upper back. The tumors cannot be distinguished by their clinical appearance only and frequently are mistaken for cysts. Histopathologic examination provides the definitive diagnosis in most cases. These carcinomas are aggressive neoplasms with a high probability of local recurrence and distant metastasis. Assessment of the Wnt signaling pathway components such as β-catenin, lymphoid enhancer-binding factor 1 (LEF-1), and caudal-related homeobox transcription factor 2 (CDX-2) potentially can be used for diagnostic purposes and targeted therapy.
We report a rare and unique case of early pilomatrix carcinoma with intralesional melanocytes. We review the molecular pathology and pathogenesis of these carcinomas as well as the significance of early diagnosis.
Case Report
A 73-year-old man with a history of extensive sun exposure presented with a 1-cm, raised, rapidly growing, slightly irregular, purple lesion on the right forearm of 3 months’ duration with tendency to bleed. He did not have a history of skin cancers and was otherwise healthy. Excision was recommended due to the progressive and rapid growth of the lesion.
Histopathologic Findings—Gross examination revealed a 0.9×0.7-cm, raised, slightly irregular lesion located 1 mm away from the closest peripheral margin. Histologically, the lesion was a relatively circumscribed, dermal-based basaloid neoplasm with slightly ill-defined edges involving the superficial and deep dermis (Figure 1A). The neoplasm was formed predominantly of sheets of basaloid cells and small nests of ghost cells, in addition to some squamoid and transitional cells (Figure 1B). The basaloid cells exhibited severe nuclear atypia, pleomorphism, increased nuclear to cytoplasmic ratio (Figure 1C), minimal to moderate amounts of eosinophilic cytoplasm, enlarged nuclei, prominent nucleoli, and coarse chromatin pattern. Abundant mitotic activity and apoptotic bodies were present as well as focal area of central necrosis (Figure 1C). Also, melanophages and a multinucleated giant cell reaction was noted. Elastic trichrome special stain highlighted focal infiltration of the neoplastic cells into the adjacent desmoplastic stroma. Melanin stain was negative for melanin pigment within the neoplasm. Given the presence of severely atypical basaloid cells along with ghost cells indicating matrical differentiation, a diagnosis of pilomatrix carcinoma was rendered.
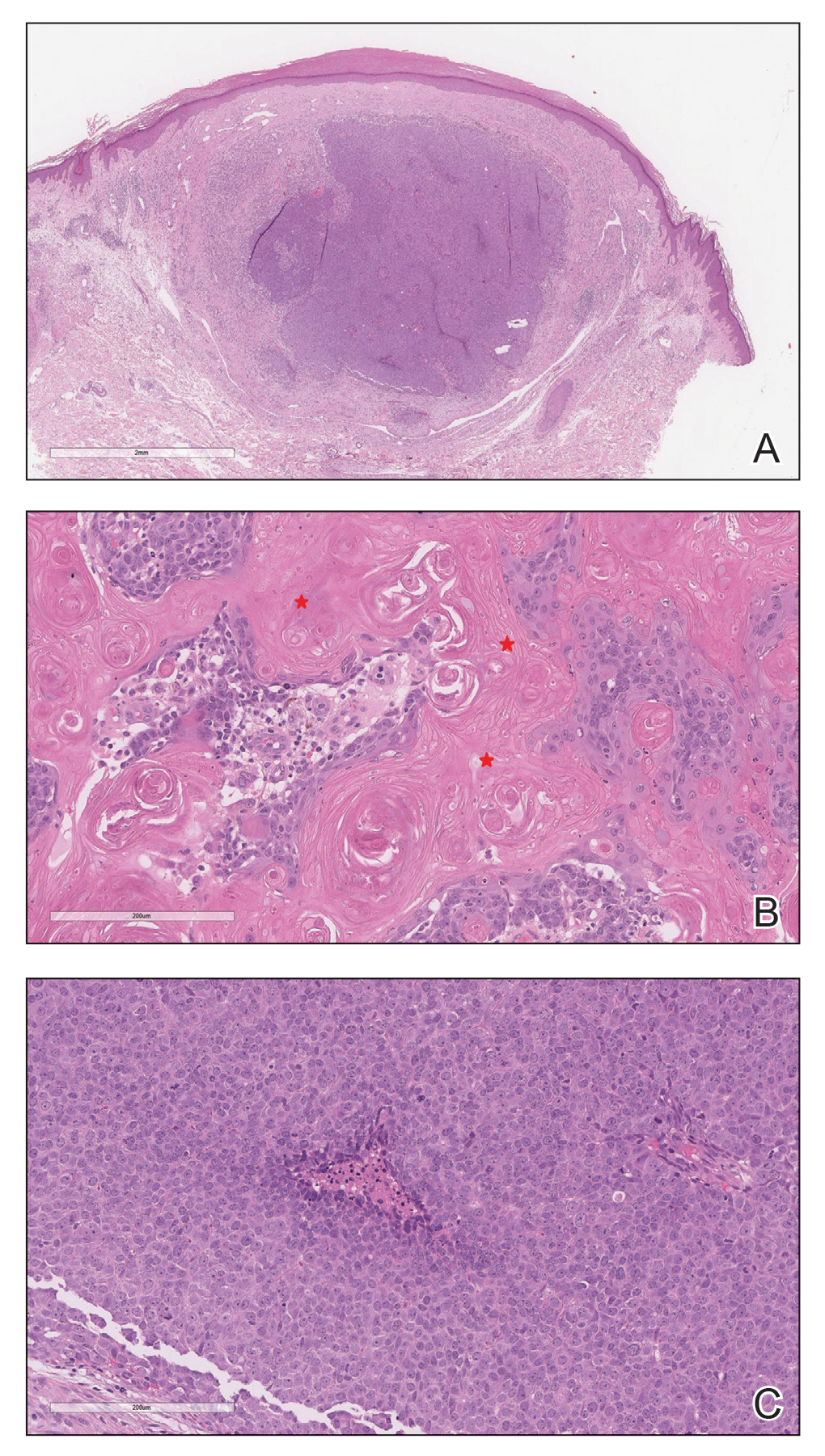
Immunohistochemistry—The neoplastic cells were diffusely positive for p63, CDX-2 (Figure 2A), β-catenin (Figure 2B), and CD10 (Figure 2C), and focally and weakly positive for cytokeratin (CK) 5, BerEP4 (staining the tumor periphery), androgen receptor, and CK18 (a low-molecular-weight keratin). They were negative for monoclonal carcinoembryonic antigen, epithelial membrane antigen, CK7, CK20, CD34, SOX-10, CD56, synaptophysin, and chromogranin. Cytokeratin 14 was positive in the squamoid cells but negative in the basaloid cells. SOX-10 and melanoma cocktail immunostains demonstrated few intralesional dendritic melanocytes.

Comment
Pilomatrix carcinoma is a rare malignant cutaneous adnexal neoplasm with origin from the germinative matrix of the hair bulb region of hair follicles. Pilomatrix carcinoma was first reported in 1980.1,2 These tumors are characterized by rapid growth and aggressive behavior. Their benign counterpart, pilomatrixoma, is a slow-growing, dermal or subcutaneous tumor that rarely recurs after complete excision.
As with pilomatrixoma, pilomatrix carcinomas are asymptomatic and present as solitary dermal or subcutaneous masses3,4 that most commonly are found in the posterior neck, upper back, and preauricular regions of middle-aged or elderly adults with male predominance.5 They range in size from 0.5 to 20 cm with a mean of 4 cm that is slightly larger than pilomatrixoma. Pilomatrix carcinomas predominantly are firm tumors with or without cystic components, and they exhibit a high probability of recurrence and have risk for distant metastasis.6-15
The differential diagnosis includes epidermal cysts, pilomatrixoma, basal cell carcinoma with matrical differentiation, trichoblastoma/trichoblastic carcinoma, and trichilemmal carcinoma. Pilomatrix carcinomas frequently are mistaken for epidermal cysts on clinical examination. Such a distinction can be easily resolved by histopathologic evaluation. The more challenging differential diagnosis is with pilomatrixoma. Histologically, pilomatrixomas consist of a distinct population of cells including basaloid, squamoid, transitional, and shadow cells in variable proportions. The basaloid cells transition to shadow cells in an organized zonal fashion.16 Compared to pilomatrixomas, pilomatrix carcinomas often show predominance of the basaloid cells; marked cytologic atypia and pleomorphism; numerous mitotic figures; deep infiltrative pattern into subcutaneous fat, fascia, and skeletal muscle; stromal desmoplasia; necrosis; and neurovascular invasion (Tables 1 and 2). Furthermore, the shadow cells tend to form a small nested pattern in pilomatrix carcinoma instead of the flat sheetlike pattern usually observed in pilomatrixoma.16 Basal cell carcinoma with matrical differentiation can pose a diagnostic challenge in the differential diagnosis; basal cell carcinoma usually exhibits a peripheral palisade of the basaloid cells accompanied by retraction spaces separating the tumor from the stroma. Trichoblastoma/trichoblastic carcinoma with matrical differentiation can be distinguished by its exuberant stroma, prominent primitive hair follicles, and papillary mesenchymal bodies. Trichilemmal carcinomas are recognized by their connection to the overlying epidermis, peripheral palisading, and presence of clear cells, while pilomatrix carcinoma lacks connection to the surface epithelium.

Immunohistochemical stains have little to no role in the differential diagnosis, and morphology is the mainstay in making the diagnosis. Rarely, pilomatrix carcinoma can be confused with poorly differentiated sebaceous carcinoma and poorly differentiated squamous cell carcinoma. Although careful scrutiny of the histologic features may help identify mature sebocytes in sebaceous carcinoma, evidence of keratinization in squamous cell carcinoma and ghost cells in pilomatrix carcinoma, using a panel of immunohistochemical stains can be helpful in reaching the final diagnosis (Table 3).

The development of hair matrix tumors have been known to harbor mutations in exon 3 of the catenin beta-1 gene, CTNNB1, that encodes for β-catenin, a downstream effector in the Wnt signaling pathway responsible for differentiation, proliferation, and adhesion of epithelial stem cells.17-21 In a study conducted by Kazakov et al,22 DNA was extracted from 86 lesions: 4 were pilomatrixomas and 1 was a pilomatrix carcinoma. A polymerase chain reaction assay revealed 8 pathogenic variants of the β-catenin gene. D32Y (CTNNB1):c.94G>T (p.Asp32Tyr) and G34R (CTNNB1):c.100G>C (p.Gly34Arg) were the mutations present in pilomatrixoma and pilomatrix carcinoma, respectively.22 In addition, there are several proteins that are part of the Wnt pathway in addition to β-catenin—LEF-1 and CDX-2.
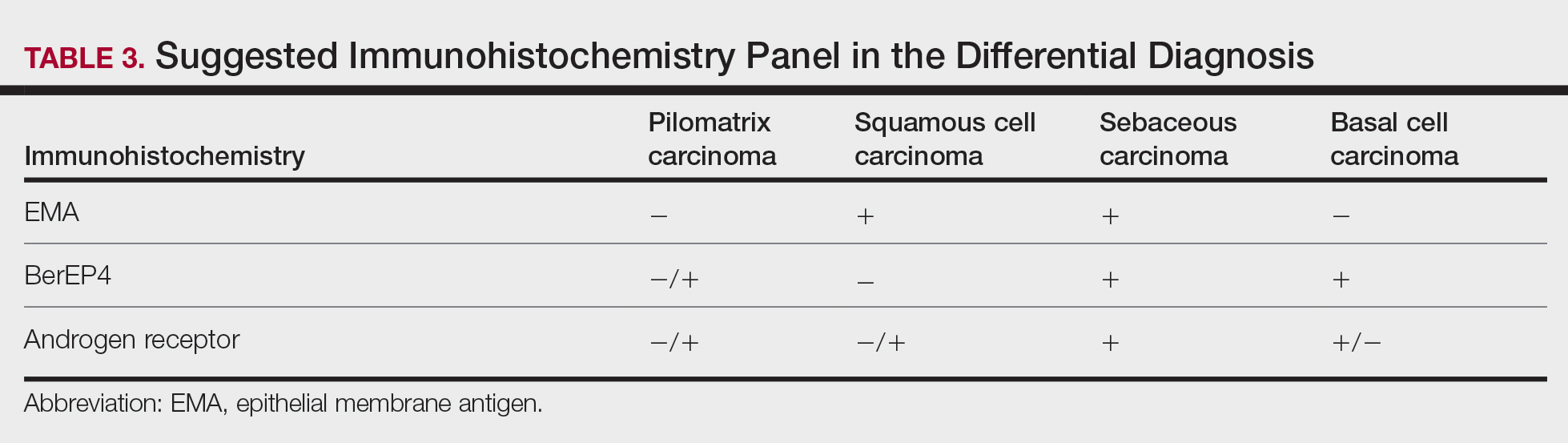
Tumminello and Hosler23 found that pilomatrixomas and pilomatrix carcinomas were positive for CDX-2, β-catenin, and LEF-1 by immunohistochemistry. These downstream molecules in the Wnt signaling pathway could have the potential to be used as diagnostic and prognostic markers.2,13,15,23
Although the pathogenesis is unclear, there are 2 possible mechanisms by which pilomatrix carcinomas develop. They can either arise as de novo tumors, or it is possible that initial mutations in β-catenin result in the formation of pilomatrixomas at an early age that may undergo malignant transformation in elderly patients over time with additional mutations.2
Our case was strongly and diffusely positive for β-catenin in a nuclear and cytoplasmic pattern and CDX-2 in a nuclear pattern, supporting the role of the Wnt signaling pathway in such tumors. Furthermore, our case demonstrated the presence of few intralesional normal dendritic melanocytes, a rare finding1,24,25 but not unexpected, as melanocytes normally are present within the hair follicle matrix.
Pilomatrix carcinomas are aggressive tumors with a high risk for local recurrence and tendency for metastasis. In a study of 13 cases of pilomatrix carcinomas, Herrmann et al13 found that metastasis was significantly associated with local tumor recurrence (P<.0413). They concluded that the combination of overall high local recurrence and metastatic rates of pilomatrix carcinoma as well as documented tumor-related deaths would warrant continued patient follow-up, especially for recurrent tumors.13 Rapid growth of a tumor, either de novo or following several months of stable size, should alert physicians to perform a diagnostic biopsy.
Management options of pilomatrix carcinoma include surgery or radiation with close follow-up. The most widely reported treatment of pilomatrix carcinoma is wide local excision with histologically confirmed clear margins. Mohs micrographic surgery is an excellent treatment option.2,13-15 Adjuvant radiation therapy may be necessary following excision. Currently there is no consensus on surgical management, and standard excisional margins have not been defined.26 Jones et al2 concluded that complete excision with wide margins likely is curative, with decreased rates of recurrence, and better awareness of this carcinoma would lead to appropriate treatment while avoiding unnecessary diagnostic tests.2
Conclusion
We report an exceptionally unique case of early pilomatrix carcinoma with a discussion on the pathogenesis and molecular pathology of hair matrix tumors. A large cohort of patients with longer follow-up periods and better molecular characterization is essential in drawing accurate information about their prognosis, identifying molecular markers that can be used as therapeutic targets, and determining ideal management strategy.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38.
- Forbis R, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606.
- Elder D, Elenitsas R, Ragsdale BD. Tumors of epidermal appendages. In: Elder D, Elenitsas R, Jaworsky C, eds. Lever’s Histopathology of the Skin. 8th ed. Lippincott Raven; 1997:757-759.
- Aherne NJ, Fitzpatrick DA, Gibbons D, et al. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol. 2008;3:47.
- Papadakis M, de Bree E, Floros N, et al. Pilomatrix carcinoma: more malignant biological behavior than was considered in the past. Mol Clin Oncol. 2017;6:415-418.
- LeBoit PE, Parslow TG, Choy SH. Hair matrix differentiation: occurrence in lesions other than pilomatricoma. Am J Dermatopathol. 1987;9:399-405.
- Campoy F, Stiefel P, Stiefel E, et al. Pilomatrix carcinoma: role played by MR imaging. Neuroradiology. 1989;31:196-198.
- Tateyama H, Eimoto T, Tada T, et al. Malignant pilomatricoma: an immunohistochemical study with antihair keratin antibody. Cancer. 1992;69:127-132.
- O’Donovan DG, Freemont AJ, Adams JE, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1993;23:385-386.
- Cross P, Richmond I, Wells S, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1994;24:499-500.
- Niedermeyer HP, Peris K, Höfler H. Pilomatrix carcinoma with multiple visceral metastases: report of a case. Cancer. 1996;77:1311-1314.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Xing L, Marzolf SA, Vandergriff T, et al. Facial pilomatrix carcinomas treated with Mohs micrographic surgery. JAAD Case Rep. 2018;4:253-255.
- Fernandez-Flores A, Cassarino DS. Sarcomatoid pilomatrix carcinoma. J Cutan Pathol. 2018;45:508-514.
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498.
- Chan E, Gat U, McNiff JM, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410-413.
- Huelsken J, Vogel R, Erdmann B, et al. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533-545.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225-229.
- Durand M, Moles J. Beta-catenin mutations in a common skin cancer: pilomatricoma. Bull Cancer. 1999;86:725-726.
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Kazakov DV, Sima R, Vanecek T, et al. Mutations in exon 3 of the CTNNB1 gene (β-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol. 2009;31:248-255.
- Tumminello K, Hosler GA. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J Cutan Pathol. 2018;45:318-324.
- Rodic´ N, Taube JM, Manson P, et al Locally invasive dermal squamomelanocytic tumor with matrical differentiation: a peculiar case with review of the literature. Am J Dermatopathol. 2013;35:E72-E76.
- Perez C, Debbaneh M, Cassarino D. Preference for the term pilomatrical carcinoma with melanocytic hyperplasia: letter to the editor. J Cutan Pathol. 2017;44:655-657.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
Pilomatrix carcinoma is a rare adnexal tumor with origin from the germinative matrical cells of the hair follicle. Clinically, it presents as a solitary lesion commonly found in the head and neck region as well as the upper back. The tumors cannot be distinguished by their clinical appearance only and frequently are mistaken for cysts. Histopathologic examination provides the definitive diagnosis in most cases. These carcinomas are aggressive neoplasms with a high probability of local recurrence and distant metastasis. Assessment of the Wnt signaling pathway components such as β-catenin, lymphoid enhancer-binding factor 1 (LEF-1), and caudal-related homeobox transcription factor 2 (CDX-2) potentially can be used for diagnostic purposes and targeted therapy.
We report a rare and unique case of early pilomatrix carcinoma with intralesional melanocytes. We review the molecular pathology and pathogenesis of these carcinomas as well as the significance of early diagnosis.
Case Report
A 73-year-old man with a history of extensive sun exposure presented with a 1-cm, raised, rapidly growing, slightly irregular, purple lesion on the right forearm of 3 months’ duration with tendency to bleed. He did not have a history of skin cancers and was otherwise healthy. Excision was recommended due to the progressive and rapid growth of the lesion.
Histopathologic Findings—Gross examination revealed a 0.9×0.7-cm, raised, slightly irregular lesion located 1 mm away from the closest peripheral margin. Histologically, the lesion was a relatively circumscribed, dermal-based basaloid neoplasm with slightly ill-defined edges involving the superficial and deep dermis (Figure 1A). The neoplasm was formed predominantly of sheets of basaloid cells and small nests of ghost cells, in addition to some squamoid and transitional cells (Figure 1B). The basaloid cells exhibited severe nuclear atypia, pleomorphism, increased nuclear to cytoplasmic ratio (Figure 1C), minimal to moderate amounts of eosinophilic cytoplasm, enlarged nuclei, prominent nucleoli, and coarse chromatin pattern. Abundant mitotic activity and apoptotic bodies were present as well as focal area of central necrosis (Figure 1C). Also, melanophages and a multinucleated giant cell reaction was noted. Elastic trichrome special stain highlighted focal infiltration of the neoplastic cells into the adjacent desmoplastic stroma. Melanin stain was negative for melanin pigment within the neoplasm. Given the presence of severely atypical basaloid cells along with ghost cells indicating matrical differentiation, a diagnosis of pilomatrix carcinoma was rendered.
Immunohistochemistry—The neoplastic cells were diffusely positive for p63, CDX-2 (Figure 2A), β-catenin (Figure 2B), and CD10 (Figure 2C), and focally and weakly positive for cytokeratin (CK) 5, BerEP4 (staining the tumor periphery), androgen receptor, and CK18 (a low-molecular-weight keratin). They were negative for monoclonal carcinoembryonic antigen, epithelial membrane antigen, CK7, CK20, CD34, SOX-10, CD56, synaptophysin, and chromogranin. Cytokeratin 14 was positive in the squamoid cells but negative in the basaloid cells. SOX-10 and melanoma cocktail immunostains demonstrated few intralesional dendritic melanocytes.
Comment
Pilomatrix carcinoma is a rare malignant cutaneous adnexal neoplasm with origin from the germinative matrix of the hair bulb region of hair follicles. Pilomatrix carcinoma was first reported in 1980.1,2 These tumors are characterized by rapid growth and aggressive behavior. Their benign counterpart, pilomatrixoma, is a slow-growing, dermal or subcutaneous tumor that rarely recurs after complete excision.
As with pilomatrixoma, pilomatrix carcinomas are asymptomatic and present as solitary dermal or subcutaneous masses3,4 that most commonly are found in the posterior neck, upper back, and preauricular regions of middle-aged or elderly adults with male predominance.5 They range in size from 0.5 to 20 cm with a mean of 4 cm that is slightly larger than pilomatrixoma. Pilomatrix carcinomas predominantly are firm tumors with or without cystic components, and they exhibit a high probability of recurrence and have risk for distant metastasis.6-15
The differential diagnosis includes epidermal cysts, pilomatrixoma, basal cell carcinoma with matrical differentiation, trichoblastoma/trichoblastic carcinoma, and trichilemmal carcinoma. Pilomatrix carcinomas frequently are mistaken for epidermal cysts on clinical examination. Such a distinction can be easily resolved by histopathologic evaluation. The more challenging differential diagnosis is with pilomatrixoma. Histologically, pilomatrixomas consist of a distinct population of cells including basaloid, squamoid, transitional, and shadow cells in variable proportions. The basaloid cells transition to shadow cells in an organized zonal fashion.16 Compared to pilomatrixomas, pilomatrix carcinomas often show predominance of the basaloid cells; marked cytologic atypia and pleomorphism; numerous mitotic figures; deep infiltrative pattern into subcutaneous fat, fascia, and skeletal muscle; stromal desmoplasia; necrosis; and neurovascular invasion (Tables 1 and 2). Furthermore, the shadow cells tend to form a small nested pattern in pilomatrix carcinoma instead of the flat sheetlike pattern usually observed in pilomatrixoma.16 Basal cell carcinoma with matrical differentiation can pose a diagnostic challenge in the differential diagnosis; basal cell carcinoma usually exhibits a peripheral palisade of the basaloid cells accompanied by retraction spaces separating the tumor from the stroma. Trichoblastoma/trichoblastic carcinoma with matrical differentiation can be distinguished by its exuberant stroma, prominent primitive hair follicles, and papillary mesenchymal bodies. Trichilemmal carcinomas are recognized by their connection to the overlying epidermis, peripheral palisading, and presence of clear cells, while pilomatrix carcinoma lacks connection to the surface epithelium.
Immunohistochemical stains have little to no role in the differential diagnosis, and morphology is the mainstay in making the diagnosis. Rarely, pilomatrix carcinoma can be confused with poorly differentiated sebaceous carcinoma and poorly differentiated squamous cell carcinoma. Although careful scrutiny of the histologic features may help identify mature sebocytes in sebaceous carcinoma, evidence of keratinization in squamous cell carcinoma and ghost cells in pilomatrix carcinoma, using a panel of immunohistochemical stains can be helpful in reaching the final diagnosis (Table 3).
The development of hair matrix tumors have been known to harbor mutations in exon 3 of the catenin beta-1 gene, CTNNB1, that encodes for β-catenin, a downstream effector in the Wnt signaling pathway responsible for differentiation, proliferation, and adhesion of epithelial stem cells.17-21 In a study conducted by Kazakov et al,22 DNA was extracted from 86 lesions: 4 were pilomatrixomas and 1 was a pilomatrix carcinoma. A polymerase chain reaction assay revealed 8 pathogenic variants of the β-catenin gene. D32Y (CTNNB1):c.94G>T (p.Asp32Tyr) and G34R (CTNNB1):c.100G>C (p.Gly34Arg) were the mutations present in pilomatrixoma and pilomatrix carcinoma, respectively.22 In addition, there are several proteins that are part of the Wnt pathway in addition to β-catenin—LEF-1 and CDX-2.
Tumminello and Hosler23 found that pilomatrixomas and pilomatrix carcinomas were positive for CDX-2, β-catenin, and LEF-1 by immunohistochemistry. These downstream molecules in the Wnt signaling pathway could have the potential to be used as diagnostic and prognostic markers.2,13,15,23
Although the pathogenesis is unclear, there are 2 possible mechanisms by which pilomatrix carcinomas develop. They can either arise as de novo tumors, or it is possible that initial mutations in β-catenin result in the formation of pilomatrixomas at an early age that may undergo malignant transformation in elderly patients over time with additional mutations.2
Our case was strongly and diffusely positive for β-catenin in a nuclear and cytoplasmic pattern and CDX-2 in a nuclear pattern, supporting the role of the Wnt signaling pathway in such tumors. Furthermore, our case demonstrated the presence of few intralesional normal dendritic melanocytes, a rare finding1,24,25 but not unexpected, as melanocytes normally are present within the hair follicle matrix.
Pilomatrix carcinomas are aggressive tumors with a high risk for local recurrence and tendency for metastasis. In a study of 13 cases of pilomatrix carcinomas, Herrmann et al13 found that metastasis was significantly associated with local tumor recurrence (P<.0413). They concluded that the combination of overall high local recurrence and metastatic rates of pilomatrix carcinoma as well as documented tumor-related deaths would warrant continued patient follow-up, especially for recurrent tumors.13 Rapid growth of a tumor, either de novo or following several months of stable size, should alert physicians to perform a diagnostic biopsy.
Management options of pilomatrix carcinoma include surgery or radiation with close follow-up. The most widely reported treatment of pilomatrix carcinoma is wide local excision with histologically confirmed clear margins. Mohs micrographic surgery is an excellent treatment option.2,13-15 Adjuvant radiation therapy may be necessary following excision. Currently there is no consensus on surgical management, and standard excisional margins have not been defined.26 Jones et al2 concluded that complete excision with wide margins likely is curative, with decreased rates of recurrence, and better awareness of this carcinoma would lead to appropriate treatment while avoiding unnecessary diagnostic tests.2
Conclusion
We report an exceptionally unique case of early pilomatrix carcinoma with a discussion on the pathogenesis and molecular pathology of hair matrix tumors. A large cohort of patients with longer follow-up periods and better molecular characterization is essential in drawing accurate information about their prognosis, identifying molecular markers that can be used as therapeutic targets, and determining ideal management strategy.
Pilomatrix carcinoma is a rare adnexal tumor with origin from the germinative matrical cells of the hair follicle. Clinically, it presents as a solitary lesion commonly found in the head and neck region as well as the upper back. The tumors cannot be distinguished by their clinical appearance only and frequently are mistaken for cysts. Histopathologic examination provides the definitive diagnosis in most cases. These carcinomas are aggressive neoplasms with a high probability of local recurrence and distant metastasis. Assessment of the Wnt signaling pathway components such as β-catenin, lymphoid enhancer-binding factor 1 (LEF-1), and caudal-related homeobox transcription factor 2 (CDX-2) potentially can be used for diagnostic purposes and targeted therapy.
We report a rare and unique case of early pilomatrix carcinoma with intralesional melanocytes. We review the molecular pathology and pathogenesis of these carcinomas as well as the significance of early diagnosis.
Case Report
A 73-year-old man with a history of extensive sun exposure presented with a 1-cm, raised, rapidly growing, slightly irregular, purple lesion on the right forearm of 3 months’ duration with tendency to bleed. He did not have a history of skin cancers and was otherwise healthy. Excision was recommended due to the progressive and rapid growth of the lesion.
Histopathologic Findings—Gross examination revealed a 0.9×0.7-cm, raised, slightly irregular lesion located 1 mm away from the closest peripheral margin. Histologically, the lesion was a relatively circumscribed, dermal-based basaloid neoplasm with slightly ill-defined edges involving the superficial and deep dermis (Figure 1A). The neoplasm was formed predominantly of sheets of basaloid cells and small nests of ghost cells, in addition to some squamoid and transitional cells (Figure 1B). The basaloid cells exhibited severe nuclear atypia, pleomorphism, increased nuclear to cytoplasmic ratio (Figure 1C), minimal to moderate amounts of eosinophilic cytoplasm, enlarged nuclei, prominent nucleoli, and coarse chromatin pattern. Abundant mitotic activity and apoptotic bodies were present as well as focal area of central necrosis (Figure 1C). Also, melanophages and a multinucleated giant cell reaction was noted. Elastic trichrome special stain highlighted focal infiltration of the neoplastic cells into the adjacent desmoplastic stroma. Melanin stain was negative for melanin pigment within the neoplasm. Given the presence of severely atypical basaloid cells along with ghost cells indicating matrical differentiation, a diagnosis of pilomatrix carcinoma was rendered.
Immunohistochemistry—The neoplastic cells were diffusely positive for p63, CDX-2 (Figure 2A), β-catenin (Figure 2B), and CD10 (Figure 2C), and focally and weakly positive for cytokeratin (CK) 5, BerEP4 (staining the tumor periphery), androgen receptor, and CK18 (a low-molecular-weight keratin). They were negative for monoclonal carcinoembryonic antigen, epithelial membrane antigen, CK7, CK20, CD34, SOX-10, CD56, synaptophysin, and chromogranin. Cytokeratin 14 was positive in the squamoid cells but negative in the basaloid cells. SOX-10 and melanoma cocktail immunostains demonstrated few intralesional dendritic melanocytes.
Comment
Pilomatrix carcinoma is a rare malignant cutaneous adnexal neoplasm with origin from the germinative matrix of the hair bulb region of hair follicles. Pilomatrix carcinoma was first reported in 1980.1,2 These tumors are characterized by rapid growth and aggressive behavior. Their benign counterpart, pilomatrixoma, is a slow-growing, dermal or subcutaneous tumor that rarely recurs after complete excision.
As with pilomatrixoma, pilomatrix carcinomas are asymptomatic and present as solitary dermal or subcutaneous masses3,4 that most commonly are found in the posterior neck, upper back, and preauricular regions of middle-aged or elderly adults with male predominance.5 They range in size from 0.5 to 20 cm with a mean of 4 cm that is slightly larger than pilomatrixoma. Pilomatrix carcinomas predominantly are firm tumors with or without cystic components, and they exhibit a high probability of recurrence and have risk for distant metastasis.6-15
The differential diagnosis includes epidermal cysts, pilomatrixoma, basal cell carcinoma with matrical differentiation, trichoblastoma/trichoblastic carcinoma, and trichilemmal carcinoma. Pilomatrix carcinomas frequently are mistaken for epidermal cysts on clinical examination. Such a distinction can be easily resolved by histopathologic evaluation. The more challenging differential diagnosis is with pilomatrixoma. Histologically, pilomatrixomas consist of a distinct population of cells including basaloid, squamoid, transitional, and shadow cells in variable proportions. The basaloid cells transition to shadow cells in an organized zonal fashion.16 Compared to pilomatrixomas, pilomatrix carcinomas often show predominance of the basaloid cells; marked cytologic atypia and pleomorphism; numerous mitotic figures; deep infiltrative pattern into subcutaneous fat, fascia, and skeletal muscle; stromal desmoplasia; necrosis; and neurovascular invasion (Tables 1 and 2). Furthermore, the shadow cells tend to form a small nested pattern in pilomatrix carcinoma instead of the flat sheetlike pattern usually observed in pilomatrixoma.16 Basal cell carcinoma with matrical differentiation can pose a diagnostic challenge in the differential diagnosis; basal cell carcinoma usually exhibits a peripheral palisade of the basaloid cells accompanied by retraction spaces separating the tumor from the stroma. Trichoblastoma/trichoblastic carcinoma with matrical differentiation can be distinguished by its exuberant stroma, prominent primitive hair follicles, and papillary mesenchymal bodies. Trichilemmal carcinomas are recognized by their connection to the overlying epidermis, peripheral palisading, and presence of clear cells, while pilomatrix carcinoma lacks connection to the surface epithelium.
Immunohistochemical stains have little to no role in the differential diagnosis, and morphology is the mainstay in making the diagnosis. Rarely, pilomatrix carcinoma can be confused with poorly differentiated sebaceous carcinoma and poorly differentiated squamous cell carcinoma. Although careful scrutiny of the histologic features may help identify mature sebocytes in sebaceous carcinoma, evidence of keratinization in squamous cell carcinoma and ghost cells in pilomatrix carcinoma, using a panel of immunohistochemical stains can be helpful in reaching the final diagnosis (Table 3).
The development of hair matrix tumors have been known to harbor mutations in exon 3 of the catenin beta-1 gene, CTNNB1, that encodes for β-catenin, a downstream effector in the Wnt signaling pathway responsible for differentiation, proliferation, and adhesion of epithelial stem cells.17-21 In a study conducted by Kazakov et al,22 DNA was extracted from 86 lesions: 4 were pilomatrixomas and 1 was a pilomatrix carcinoma. A polymerase chain reaction assay revealed 8 pathogenic variants of the β-catenin gene. D32Y (CTNNB1):c.94G>T (p.Asp32Tyr) and G34R (CTNNB1):c.100G>C (p.Gly34Arg) were the mutations present in pilomatrixoma and pilomatrix carcinoma, respectively.22 In addition, there are several proteins that are part of the Wnt pathway in addition to β-catenin—LEF-1 and CDX-2.
Tumminello and Hosler23 found that pilomatrixomas and pilomatrix carcinomas were positive for CDX-2, β-catenin, and LEF-1 by immunohistochemistry. These downstream molecules in the Wnt signaling pathway could have the potential to be used as diagnostic and prognostic markers.2,13,15,23
Although the pathogenesis is unclear, there are 2 possible mechanisms by which pilomatrix carcinomas develop. They can either arise as de novo tumors, or it is possible that initial mutations in β-catenin result in the formation of pilomatrixomas at an early age that may undergo malignant transformation in elderly patients over time with additional mutations.2
Our case was strongly and diffusely positive for β-catenin in a nuclear and cytoplasmic pattern and CDX-2 in a nuclear pattern, supporting the role of the Wnt signaling pathway in such tumors. Furthermore, our case demonstrated the presence of few intralesional normal dendritic melanocytes, a rare finding1,24,25 but not unexpected, as melanocytes normally are present within the hair follicle matrix.
Pilomatrix carcinomas are aggressive tumors with a high risk for local recurrence and tendency for metastasis. In a study of 13 cases of pilomatrix carcinomas, Herrmann et al13 found that metastasis was significantly associated with local tumor recurrence (P<.0413). They concluded that the combination of overall high local recurrence and metastatic rates of pilomatrix carcinoma as well as documented tumor-related deaths would warrant continued patient follow-up, especially for recurrent tumors.13 Rapid growth of a tumor, either de novo or following several months of stable size, should alert physicians to perform a diagnostic biopsy.
Management options of pilomatrix carcinoma include surgery or radiation with close follow-up. The most widely reported treatment of pilomatrix carcinoma is wide local excision with histologically confirmed clear margins. Mohs micrographic surgery is an excellent treatment option.2,13-15 Adjuvant radiation therapy may be necessary following excision. Currently there is no consensus on surgical management, and standard excisional margins have not been defined.26 Jones et al2 concluded that complete excision with wide margins likely is curative, with decreased rates of recurrence, and better awareness of this carcinoma would lead to appropriate treatment while avoiding unnecessary diagnostic tests.2
Conclusion
We report an exceptionally unique case of early pilomatrix carcinoma with a discussion on the pathogenesis and molecular pathology of hair matrix tumors. A large cohort of patients with longer follow-up periods and better molecular characterization is essential in drawing accurate information about their prognosis, identifying molecular markers that can be used as therapeutic targets, and determining ideal management strategy.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38.
- Forbis R, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606.
- Elder D, Elenitsas R, Ragsdale BD. Tumors of epidermal appendages. In: Elder D, Elenitsas R, Jaworsky C, eds. Lever’s Histopathology of the Skin. 8th ed. Lippincott Raven; 1997:757-759.
- Aherne NJ, Fitzpatrick DA, Gibbons D, et al. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol. 2008;3:47.
- Papadakis M, de Bree E, Floros N, et al. Pilomatrix carcinoma: more malignant biological behavior than was considered in the past. Mol Clin Oncol. 2017;6:415-418.
- LeBoit PE, Parslow TG, Choy SH. Hair matrix differentiation: occurrence in lesions other than pilomatricoma. Am J Dermatopathol. 1987;9:399-405.
- Campoy F, Stiefel P, Stiefel E, et al. Pilomatrix carcinoma: role played by MR imaging. Neuroradiology. 1989;31:196-198.
- Tateyama H, Eimoto T, Tada T, et al. Malignant pilomatricoma: an immunohistochemical study with antihair keratin antibody. Cancer. 1992;69:127-132.
- O’Donovan DG, Freemont AJ, Adams JE, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1993;23:385-386.
- Cross P, Richmond I, Wells S, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1994;24:499-500.
- Niedermeyer HP, Peris K, Höfler H. Pilomatrix carcinoma with multiple visceral metastases: report of a case. Cancer. 1996;77:1311-1314.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Xing L, Marzolf SA, Vandergriff T, et al. Facial pilomatrix carcinomas treated with Mohs micrographic surgery. JAAD Case Rep. 2018;4:253-255.
- Fernandez-Flores A, Cassarino DS. Sarcomatoid pilomatrix carcinoma. J Cutan Pathol. 2018;45:508-514.
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498.
- Chan E, Gat U, McNiff JM, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410-413.
- Huelsken J, Vogel R, Erdmann B, et al. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533-545.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225-229.
- Durand M, Moles J. Beta-catenin mutations in a common skin cancer: pilomatricoma. Bull Cancer. 1999;86:725-726.
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Kazakov DV, Sima R, Vanecek T, et al. Mutations in exon 3 of the CTNNB1 gene (β-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol. 2009;31:248-255.
- Tumminello K, Hosler GA. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J Cutan Pathol. 2018;45:318-324.
- Rodic´ N, Taube JM, Manson P, et al Locally invasive dermal squamomelanocytic tumor with matrical differentiation: a peculiar case with review of the literature. Am J Dermatopathol. 2013;35:E72-E76.
- Perez C, Debbaneh M, Cassarino D. Preference for the term pilomatrical carcinoma with melanocytic hyperplasia: letter to the editor. J Cutan Pathol. 2017;44:655-657.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Jani P, Chetty R, Ghazarian DM. An unusual composite pilomatrix carcinoma with intralesional melanocytes: differential diagnosis, immunohistochemical evaluation, and review of the literature. Am J Dermatopathol. 2008;30:174-177.
- Jones C, Twoon M, Ho W, et al. Pilomatrix carcinoma: 12-year experience and review of the literature. J Cutan Pathol. 2018;45:33-38.
- Forbis R, Helwig EB. Pilomatrixoma (calcifying epithelioma). Arch Dermatol. 1961;83:606.
- Elder D, Elenitsas R, Ragsdale BD. Tumors of epidermal appendages. In: Elder D, Elenitsas R, Jaworsky C, eds. Lever’s Histopathology of the Skin. 8th ed. Lippincott Raven; 1997:757-759.
- Aherne NJ, Fitzpatrick DA, Gibbons D, et al. Pilomatrix carcinoma presenting as an extra axial mass: clinicopathological features. Diagn Pathol. 2008;3:47.
- Papadakis M, de Bree E, Floros N, et al. Pilomatrix carcinoma: more malignant biological behavior than was considered in the past. Mol Clin Oncol. 2017;6:415-418.
- LeBoit PE, Parslow TG, Choy SH. Hair matrix differentiation: occurrence in lesions other than pilomatricoma. Am J Dermatopathol. 1987;9:399-405.
- Campoy F, Stiefel P, Stiefel E, et al. Pilomatrix carcinoma: role played by MR imaging. Neuroradiology. 1989;31:196-198.
- Tateyama H, Eimoto T, Tada T, et al. Malignant pilomatricoma: an immunohistochemical study with antihair keratin antibody. Cancer. 1992;69:127-132.
- O’Donovan DG, Freemont AJ, Adams JE, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1993;23:385-386.
- Cross P, Richmond I, Wells S, et al. Malignant pilomatrixoma with bone metastasis. Histopathology. 1994;24:499-500.
- Niedermeyer HP, Peris K, Höfler H. Pilomatrix carcinoma with multiple visceral metastases: report of a case. Cancer. 1996;77:1311-1314.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
- Xing L, Marzolf SA, Vandergriff T, et al. Facial pilomatrix carcinomas treated with Mohs micrographic surgery. JAAD Case Rep. 2018;4:253-255.
- Fernandez-Flores A, Cassarino DS. Sarcomatoid pilomatrix carcinoma. J Cutan Pathol. 2018;45:508-514.
- Sau P, Lupton GP, Graham JH. Pilomatrix carcinoma. Cancer. 1993;71:2491-2498.
- Chan E, Gat U, McNiff JM, et al. A common human skin tumour is caused by activating mutations in β-catenin. Nat Genet. 1999;21:410-413.
- Huelsken J, Vogel R, Erdmann B, et al. β-catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell. 2001;105:533-545.
- Kikuchi A. Tumor formation by genetic mutations in the components of the Wnt signaling pathway. Cancer Sci. 2003;94:225-229.
- Durand M, Moles J. Beta-catenin mutations in a common skin cancer: pilomatricoma. Bull Cancer. 1999;86:725-726.
- Lazar AJF, Calonje E, Grayson W, et al. Pilomatrix carcinomas contain mutations in CTNNB1, the gene encoding beta-catenin. J Cutan Pathol. 2005;32:148-157.
- Kazakov DV, Sima R, Vanecek T, et al. Mutations in exon 3 of the CTNNB1 gene (β-catenin gene) in cutaneous adnexal tumors. Am J Dermatopathol. 2009;31:248-255.
- Tumminello K, Hosler GA. CDX2 and LEF-1 expression in pilomatrical tumors and their utility in the diagnosis of pilomatrical carcinoma. J Cutan Pathol. 2018;45:318-324.
- Rodic´ N, Taube JM, Manson P, et al Locally invasive dermal squamomelanocytic tumor with matrical differentiation: a peculiar case with review of the literature. Am J Dermatopathol. 2013;35:E72-E76.
- Perez C, Debbaneh M, Cassarino D. Preference for the term pilomatrical carcinoma with melanocytic hyperplasia: letter to the editor. J Cutan Pathol. 2017;44:655-657.
- Herrmann JL, Allan A, Trapp KM, et al. Pilomatrix carcinoma: 13 new cases and review of the literature with emphasis on predictors of metastasis. J Am Acad Dermatol. 2014;71:38-43.
Practice Points
- Clinicians and pathologists should be aware of pilomatrix carcinoma to facilitate early detection.
- Early diagnosis and prompt treatment of pilomatrix carcinoma is crucial in lowering recurrence rate and avoiding a poor outcome.
- Caudal-related homeobox transcription factor 2 and β-catenin components of the Wnt signaling pathway play an important role in the pathogenesis of pilomatrix carcinoma.
- Although controversial, wide local excision is the treatment of choice for pilomatrix carcinoma.
